Kineret 100 Mg
SOUHRN ÚDAJŮ O PŘÍPRAVKU
PŘÍLOHA I
Kineret 100 mg, injekční roztok v předplněné injekční stříkačce.
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna předplněná injekční stříkačka obsahuje 100 mg anakinrum* (anakinra) v 0,67 ml (150 mg/ml).
* Antagonista humánního receptoru pro interleukin-1 (r-metHuIL-1ra) produkovaný v buňkách Escherichia coli pomocí rekombinantní DNA technologie.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok (injekce).
Čirý, bezbarvý až bělavý injekční roztok, který může obsahovat průsvitné až bílé amorfní částice léku.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Kineret je indikován k léčbě známek a symptomů revmatoidní artritidy (RA) v kombinaci s methotrexatem u dospělých pacientů s nedostatečnou odpovědí na samotný methotrexat.
4.2 Dávkování a způsob podání
Léčbu Kineretem mají zahajovat a sledovat specializovaní lékaři se zkušenostmi v diagnostice a léčbě revmatoidní artritidy.
Dávkování
Doporučená dávka Kineretu je 100 mg jednou denně v subkutánní injekci. Dávka by měla být aplikována každý den přibližně ve stejnou dobu.
Starší_populace (> 65 let)
Není zapotřebí žádná úprava dávek. Dávkování a způsob podání jsou stejné jako pro dospělé osoby ve věku od 18 do 64 let.
Pediatrická _populace (< 18 let)
Bezpečnost a účinnost Kineretu u dětí s RA (JIA) ve věku od 0 do 18 let nebyla stanovena.
Porucha _ funkce _ jater
U pacientů se středně závažnou poruchou funkce jater (Child-Pughova klasifikace B) není zapotřebí žádná úprava dávek. U pacientů s vážnou poruchou funkce jater by měl být Kineret používán s opatrností.
Porucha funkce ledvin
Pacientům s těžkou poruchou funkce ledvin (CLcr < 30 ml/min) nesmí být Kineret podáván (viz bod 4.3). U pacientů s mírnou poruchou funkce ledvin (CLcr 50 až 80 ml/min) není nutná žádná úprava dávek. Pacientům se střední poruchou funkce ledvin (CLcr 30 až 50 ml/min) by měl být Kineret podáván s nezbytnou opatrností, vzhledem k tomu, že příslušné údaje chybí.
Způsob podání
Kineret se podává subkutánní injekcí.
Kineret se dodává připravený k okamžitému použití v předplněné injekční stříkačce. Předplněnou stříkačkou netřepejte. Pokyny pro použití přípravku a zacházení s ním jsou uvedeny v bodě 6.6.
Aby se předešlo nepříjemným pocitům v místě vpichu, doporučuje se měnit místa aplikace. Příznaky a symptomy v místě vpichu může zmírnit chlazení místa vpichu, ohřátí vstřikované kapaliny, použití chladicích balíčků (před a po injekci) a použití topických kortikosteroidů a antihistaminik po vpichu.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1 nebo na proteiny pocházející z E. coli.
Pacientům s vážnou poruchou funkce ledvin (CLCT < 30 ml/min) nesmí být Kineret podáván (viz bod 4.2).
U pacientů s neutropenií (ANC < 1,5 x 109/l) nesmí být léčba Kineretem zahajována (viz bod 4.4).
4.4 Zvláštní upozornění a opatření pro použití
Alergické reakce
Alergické reakce, včetně anafylaktických reakcí či angioedému, byly hlášeny méně často. Ve většině případů se jednalo o makulopapulární nebo urtikariální kožní exantém. V případě výskytu těžké alergické reakce by mělo být podávání Kineretu přerušeno a měla by být zahájena odpovídající léčba.
Jaterní příhody
V klinických studiích byla u pacientů s RA a CAPS pozorována méně často přechodná zvýšení jaterních enzymů. Tato zvýšení nebyla spojena s příznaky nebo symptomy buněčného poškození jater. Při postmarketingovém používání byl hlášen ojedinělý případ indikující neinfekční hepatitidu. Jaterní příhody při postmarketingovém používání byly hlavně hlášeny u pacientů s predispozicí, např. zvýšenými transaminázami v anamnéze před zahájením léčby Kineretem.
Účinnost a bezpečnost Kineretu u pacientů s AST/ALT >1,5x zvýšenou hladinou nad normál nebyly hodnoceny.
Vážné infekce
Podávání Kineretu bylo provázeno zvýšením incidence vážných infekcí v porovnání s placebem (1,8 % vs. 0,7 %). U malého počtu pacientů s astmatem byla incidence závažných infekcí vyšší u pacientů léčených Kineretem (4,5 %) při porovnání s pacienty užívajícími placebo (0 %), tyto infekce se týkaly hlavně dýchacích cest.
Bezpečnost a účinnost Kineretu u pacientů s chronickými infekcemi nebyla hodnocena.
Léčba Kineretem by neměla být zahajována u pacientů s aktivními infekcemi. Při rozvinutí těžké infekce by měla být léčba Kineretem přerušena.
K podávání Kineretu pacientům s anamnézou rekurentních infekcí nebo se základním onemocněním, které může být predisponujícím faktorem infekce, by měl lékař přistupovat s nezbytnou obezřelostí.
Bezpečnost Kineretu není u jedinců s latentní tuberkulózou známa. U pacientů užívajících několik biologických protizánětlivých léčebných režimů byla hlášena tuberkulóza. Před zahájením léčby Kineretem by měli být pacienti vyšetřeni na latentní tuberkulózu. Také by měla být brána v úvahu dostupná lékařská doporučení.
S reaktivací hepatitidy B byla spojena další antirevmatická terapie. Proto by měla být i před zahájením léčby Kineretem prováděna vyšetření na virovou hepatitidu v souladu s vydanými pokyny.
Neutropenie
V placebem kontrolovaných studiích RA byl Kineret často spojován s neutropenií (ANC < 1,5 x 109/l). Pro více informací o neutropenii viz část 4.8.
U pacientů s neutropenií (absolutní počet neutrofilů < 1,5 x 109/l) by léčba Kineretem neměla být zahajována. Počet neutrofilů se doporučuje vyšetřit před zahájením terapie pomocí Kineretu, jednou za měsíc během prvních 6 měsíců léčby a dále čtvrtletně. U pacientů, u kterých se neutropenie objeví (absolutní počet neutrofilů < 1,5 x 109/l), by měla být léčba Kineretem přerušena a absolutní počet neutrofilů by měl být pečlivě sledován. Bezpečnost a účinnost Kineretu u pacientů s neutropenií nebyla hodnocena.
Imunosuprese
Účinky léčby Kineretem na preexistující maligní nádorové onemocnění nebyly studovány.
Podávání Kineretu pacientům s preexistujícím maligním nádorovým onemocněním se proto nedoporučuje.
Vakcinace
V placebem kontrolovaném klinickém hodnocení (n = 126) nebyl zjištěn žádný rozdíl v protilátkové odpovědi na podání antitetanového séra mezi skupinami pacientů léčených Kineretem a placebem, kdy byla vakcína obsahující tetanový/difterický toxoid aplikována současně s Kineretem. Nejsou k dispozici žádné údaje o účinnosti očkování pacientů užívajících Kineret jinými inaktivovanými antigeny.
Nejsou k dispozici žádné údaje o účincích vakcinace živou očkovací látkou ani o sekundárním přenosu infekce živou očkovací látkou u pacientů užívajících Kineret. Pacienti užívající Kineret by proto neměli být současně očkováni živou vakcínou.
Starší populace (> 65 let)
V klinických studiích bylo sledováno celkem 752 pacientů > 65 let, včetně 163 pacientů > 75 let. Mezi těmito pacienty a mladšími pacienty nebyl zaznamenán žádný celkový rozdíl s ohledem na bezpečnost anebo účinnost. Vzhledem k tomu, že u starší populace je incidence infekčních onemocnění obecně vyšší, léčbu starších pacientů je třeba provádět s nezbytnou opatrností.
Současná léčba Kineretem a TNF antagonisty
Současná léčba Kineretem a etanerceptem je spojena se zvýšeným rizikem vážných infekcí a s rizikem neutropenie v porovnání s léčbou etanerceptem samotným. Tato léčebná kombinace neprokázala vyšší klinický přínos.
Současné podávání Kineretu a etanerceptu nebo dalších TNF antagonistů se nedoporučuje (viz bod 4.5).
Tento léčivý přípravek obsahuje méně než 1 mmol sodíku (23 mg) na 100 mg dávky, tj. „je v podstatě bez sodíku“.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Interakce mezi Kineretem a jinými léčivými přípravky nebyly ve formálně prováděných studiích sledovány. V klinických studiích nebyly pozorovány interakce mezi Kineretem a ostatními léčivými přípravky (včetně nesteroidních protizánětlivých léčivých přípravků, kortikosteroidů a DMARD).
Současná léčba Kineretem a TNF antagonisty
V klinické studii s pacienty, jejichž základní léčbou byl methotrexat, byl u pacientů léčených Kineretem a etanerceptem zaznamenán vyšší počet vážných infekcí (7 %) a neutropenií než u pacientů léčených etanerceptem samotným. Tento počet byt rovněž vyšší než v předchozích studiích, ve kterých byl Kineret podáván samotný. Současné podávání Kineretu a etanerceptu neprokázalo vyšší klinický přínos.
Současné podávání Kineretu s etanerceptem nebo jinými TNF antagonisty se nedoporučuje (viz bod 4.4).
Substráty cvtochromu P450
Tvorba enzymů CYP450 je při chronickém zánětu potlačena zvýšenými hladinami cytokinů (např. IL-1). Proto lze očekávat, že by antagonista receptoru IL-1, např. anakinra, mohl během léčby normalizovat tvorbu enzymů CYP450. To by bylo klinicky relevantní pro substráty CYP450 s úzkým terapeutickým indexem (např. warfarin a fenytoin). Na začátku nebo konci léčby Kineretem u pacientů léčených těmito léčivými přípravky může být relevantní zvážit, terapeutické sledování účinku nebo koncentrace těchto přípravků a může být zapotřebí upravit individuální dávku léčivého přípravku.
Informace o vakcinaci jsou uvedeny v bodě 4.4.
4.6 Fertilita, těhotenství a kojení
Údaje týkající se užívání látky anakinra u těhotných žen jsou omezené. Reprodukční studie s Kineretem však byly provedeny u potkanů a králíků při dávkách až 100krát vyšších, než jsou dávky u člověka s RA, a nebyl odhalen žádný důkaz narušení fertility ani poškození plodu.
Podávání Kineretu se v těhotenství a u žen v reprodukčním věku, které nepoužívají antikoncepci, nedoporučuje.
Není známo, zda se anakinra/metabolity vylučují do lidského mateřského mléka. Riziko pro kojené novorozence/děti nelze vyloučit. Kojení má být během léčby Kineretem přerušeno.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Není relevantní.
4.8 Nežádoucí účinky
Ve studiích kontrolovaných placebem u pacientů s RA byly nejčastěji hlášenými nežádoucími účinky spojenými s podáváním Kineretu reakce v místě vpichu (ISR), jež dosáhly u většiny pacientů mírné až střední intenzity. Nejčastějším důvodem přerušení účasti pacientů léčených Kineretem v klinické studii byla reakce v místě vpichu. Incidence vážných nežádoucích účinků u subjektů při aplikaci doporučené dávky Kineretu (100 mg/den) byla srovnatelná s placebem (7,1 % v porovnání s 6,5 % v placebové skupině). Incidence vážných infekcí byla u pacientů léčených Kineretem vyšší než u pacientů na placebu (1,8 % vs. 0,7 %). U pacientů léčených Kineretem se v porovnání s pacienty, kteří dostávali placebo, častěji vyskytl pokles počtu neutrofilů.
Nežádoucí účinky jsou řazeny podle tříd orgánových systémů MedDRA a kategorie četností.
Kategorie četností jsou definovány za použití následující konvence: velmi časté (> 1/10); časté (> 1/100 až < 1/10); méně časté (> 1/1 000 až < 1/100); vzácné (> 1/10 000 až < 1/1 000); velmi vzácné (<1/10 000); není známo (z dostupných údajů nelze určit). V každé kategorii četností jsou nežádoucí účinky řazeny podle klesající závažnosti.
|
Orgánový systém dle MedDRA |
F rekvence |
Nežádoucí účinek |
|
Infekce a infestace |
Časté (> 1/100 až < 1/10) |
Závažné infekce |
|
Poruchy krve a lymfatického systému |
Časté (> 1/100 až < 1/10) |
Neutropenie T rombocytopenie |
|
Poruchy imunitního systému |
Méně časté (> 1/1 000 až < 1/100) |
Alergické reakce zahrnující anafylaktické reakce, angioedém, kopřivku a pruritus |
|
Poruchy nervového systému |
Velmi časté (> 1/10) |
Bolesti hlavy |
|
Poruchy jater a žlučových cest |
Méně časté (> 1/1 000 až < 1/100) |
Zvýšené jaterní enzymy |
|
Není známo (z dostupných údajů nelze hodnotit) |
Neinfekční hepatitida | |
|
Poruchy kůže a podkožní tkáně |
Velmi časté (> 1/10) |
Reakce v místě vpichu |
|
Méně časté (> 1/1 000 až < 1/100) | ||
|
Vyšetření |
Velmi časté (> 1/10) |
Zvýšení hladiny cholesterolu v krvi |
Vážné infekce
Incidence vážných infekcí ve studiích RA s aplikací doporučené dávky (100 mg/den) byla 1,8 % ve skupině pacientů léčených Kineretem a 0,7 % ve skupině pacientů léčených placebem. V průběhu až tříletého období sledování zůstával počet závažných infekcí stabilní. Pozorované infekce byly především bakteriálního původu jako například celulitida, pneumonie či infekční onemocnění kostí a kloubů. Po zhojení infekce pokračovala většina pacientů ve studii léčivým přípravkem.
Ve studiích RA se nevyskytla žádná úmrtí z důvodu závažných infekcí.
V průběhu klinických hodnocení a postmarketingového období byly pozorovány vzácné případy infekcí oportunními mikroorganismy včetně plísní, mykobakterií, bakterií a virů. Infekce byly zjištěny u všech orgánových systémů a byly zaznamenány u pacientů užívajících Kineret
v monoterapii i v kombinaci s imunosupresivy.
Neutropenie
V placebem kontrolovaných studiích s Kineretem byla léčba doprovázena malým snížením průměrných hodnot celkového počtu leukocytů a absolutního počtu neutrofilů. Podávání Kineretu bylo spojeno s neutropenií (absolutní počet neutrofilů < 1,5 x 109/l) u 2,4 % pacientů v porovnání s 0,4 %
pacientů na placebu. Žádný z těchto pacientů neměl závažné infekce spojené s neutropenií.
T rombocytopenie
V klinických studiích prováděných u pacientů s RA byla trombocytopenie zjištěna u 1,9 % léčených pacientů v porovnání s 0,3 % v placebové skupině. Trombocytopenie byly mírné, tj. počty krevních destiček byly >75 x109/l. Mírná trombocytopenie byla pozorována i u pacientů s CAPS.
Při postmarketingovém používání Kineretu byla zjištěna trombocytopenie, včetně občasných hlášení o případech těžké trombocytopenie (tj. počty krevních destiček<10 x109/l).
Malignity
U pacientů s RA může být vyšší riziko (v průměru 2- až 3násobné) vzniku lymfomů. Při klinických hodnoceních u pacientů léčených Kineretem byla zjištěna incidence lymfomů vyšší, než se předpokládá v běžné populaci; tento počet však odpovídá počtům uváděným obecně pro pacienty s RA.
Incidence maligních nádorových onemocnění byla při klinických hodnoceních stejná u pacientů léčených Kineretem jako ve skupině užívající placebo a nelišila se od běžné populace. Kromě toho se celková incidence malignit u pacientů nezvyšovala ani během tříleté léčby Kineretem.
Alergické reakce
Méně často byly u Kineretu hlášeny alergické reakce, včetně anafylaktických reakcí, angioedému, kopřivky, vyrážky a pruritu. Většina těchto reakcí byly makulopapulární nebo urtikariální vyrážky.
Imunogenicita
V klinických studiích měla až 3 % dospělých pacientů alespoň jednou pozitivní výsledek
v sérologickém testu v průběhu studie na protilátky schopné neutralizovat biologické účinky anakinry. Výskyt protilátek byl většinou přechodný a neměl souvislost s výskytem nežádoucích účinků nebo snížením účinnosti.
Kromě toho bylo v klinické studii 6 % pediatrických pacientů testováno seropozitivně alespoň jednou v průběhu studie na protilátky schopné neutralizovat biologické účinky anakinry.
Jaterní příhody
V klinických studiích byla u pacientů s RA pozorována méně často přechodná zvýšení jaterních enzymů. Tato zvýšení nebyla spojena s příznaky nebo symptomy buněčného poškození jater. Při postmarketingovém používání byl hlášen ojedinělý případ indikující neinfekční hepatitidu. Jaterní příhody při postmarketingovém používání byly hlavně hlášeny u pacientů s predispozicí, např. zvýšenými transaminázami v anamnéze před zahájením léčby Kineretem.
Reakce v místě vpichu
Nejčastěji a nejdůsledněji hlášeným nežádoucím účinkem spojeným s léčbou Kineretem byla reakce v místě vpichu injekce (ISR). Většina (95 %) z těchto hlášených reakcí měla lehkou až středně těžkou intenzitu. Reakce typicky zahrnovaly jeden nebo více z následujících projevů: erytém, ekchymózu, zánětlivé projevy a bolestivost. Při aplikaci dávky 100 mg/den došlo ke vzniku reakce v místě vpichu u 71 % pacientů, ve srovnání s 28 % pacientů, kteří dostávali placebo. ISR se obvykle objeví během 2. týdne léčby a během 4-6 týdnů zmizí. Výskyt reakce v místě vpichu po uplynutí prvního měsíce léčby u pacientů, u kterých se tato reakce neobjevila ani v minulosti, nebyl častý.
Zvýšení hladiny cholesterolu v krvi
V klinických studiích RA se 775 pacienty denně léčenými dávkami Kineretu o síle 30 mg, 75 mg,
150 mg, 1 mg/kg nebo 2 mg/kg došlo k nárustu o 2,4 % až 5,3 % celkových hladin cholesterolu 2 týdny po zahájení léčby Kineretem, bez vzájemného vztahu mezi dávkou a odezvou. Podobný vzorec se opakoval po 24 týdnech léčby Kineretem. Léčba placebem (n=213) vedla ke snížení celkové hladiny cholesterolu přibližně o 2,2 % ve 2. týdnu a 2,3 % ve 24. týdnu. Nejsou k dispozici žádné údaje o cholesterolu LDL nebo HDL.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Během klinických studií u pacientů s RA nebyly zaznamenány žádné známky toxicity, které by si vynutily úpravu dávek.
Ve studiích, kde byla sledována sepse, dostávalo Kineret 1015 pacientů, a to v dávkách až do 2 mg/kg/hod i.v. (~35 násobku doporučené dávky při RA) během 72 hodinové léčebné periody. Profil nežádoucích příhod zaznamenaných v těchto studiích se celkově nijak nelišil od údajů získaných ve studiích s revmatoidní artritidou.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Imunosupresiva, inhibitory interleukinu, ATC kód: L04AC03
Anakinra neutralizuje biologickou aktivitu interleukinu-1a (IL-1a) a interleukinu-ip (IL-1P) kompetitivní inhibicí jejich vazby na receptor typu I pro interleukin-1 (IL-1RI). Interleukin-1 (IL-1) je klíčovým prozánětlivým cytokinem, který zprostředkovává řadu buněčných odpovědí, včetně reakcí významných při zánětlivých procesech synoviální tkáně.
Anakinra inhibuje reakce vyvolané prostřednictvím IL-1 in vitro, včetně indukce oxidu dusnatého a prostaglandinu E2 a/nebo produkce kolagenázy synoviálními buňkami, fibroblasty a chondrocyty.
Klinická účinnost a bezpečnost
Bezpečnost a účinnost anakinry v kombinaci s methotrexatem byla prokázána u 1790 pacientů s RA > 18 let s různým stupněm intenzity onemocnění.
Klinická odpověď na léčbu anakinrou se většinou objevila během 2 týdnů po zahájení léčby a trvala s pokračující aplikací anakinry. Maximální klinická odezva byla obvykle zaznamenána během 12 týdnů po zahájení terapie.
Kombinovaná léčba anakinrou a methotrexatem prokázala statisticky a klinicky signifikantní snížení intenzity známek a symptomů revmatoidní artritidy u pacientů, u kterých odpověď na léčbu samotným methotrexatem nebyla dostatečná (38 % vs. 22 % pozitivních odpovědí na léčbu, měřeno pomocí kritérií ACR20). Významné zlepšení se týká bolestivosti, počtu bolestivých kloubů, tělesných funkcí (HAQ skóre), reaktantů akutní fáze a celkového hodnocení stavu lékařem i pacientem.
V jedné klinické studii s anakinrou byla prováděna RTG vyšetření. Tato vyšetření neprokázala žádný škodlivý účinek anakinry na kloubní chrupavku.
Bezpečnost u pediatrických pacientů s RA (JIA)
Kineret byl zkoumán v jediné randomizované, zaslepené, multicentrické studii s 86 pacienty s polyartikulárním průběhem juvenilní revmatoidní artritidy (JRA; věk 2-17 let) přijímajícími podkožně dávku 1 mg/kg denně, až do dávky 100 mg. 50 pacientů, kteří dosáhli klinické odpovědi po 12 týdnech odslepené zaváděcí fáze, bylo randomizováno mezi Kineret (25 pacientů) a placebo (25 pacientů), podávané denně po dalších 16 týdnů. Podskupina těchto pacientů pokračovala v odslepené léčbě s Kineretem až 1 rok ve společné navazující studii. V těchto studiích byl pozorovaný profil nežádoucích účinků podobný těm, které lze vidět u dospělých pacientů s RA. Údaje v těchto studiích jsou nedostatečné pro prokázání účinnosti, a tudíž se Kineret nedoporučuje pro použití u pediatrických pacientů s juvenilní revmatoidní artritidou.
Imunogenicita Viz bod 4.8
5.2 Farmakokinetické vlastnosti
Absolutní biologická dostupnost anakinry po subkutánním podání dávky 70 mg zdravým subjektům (n = 11) je 95 %. Proces absorpce je faktorem limitujícím rychlost vymizení anakinry z plasmy po subkutánní injekci. U pacientů s RA bylo maximální plasmatické koncentrace anakinry dosaženo za 3 až 7 hodin po subkutánní aplikaci anakinry v klinicky relevantních dávkách (1 až 2 mg/kg; n = 18). Koncentrace v plazmě se snižovala bez patrné distribuční fáze a terminální poločas se pohyboval od 4 do 6 hodin. U pacientů s RA nebyla zaznamenána žádná neočekávaná kumulace anakinry po každodenní subkutánní aplikaci dávek po dobu až 24 týdnů. Průměrné (SD) hodnoty clearance (CL/F) a objemu distribuce (Vd/F) získané analýzou populace z údajů ze dvou farmakokinetických studií na 35 pacientech s RA byly 105 (27) ml/min nebo 18,5 (11) l. Údaje od lidí a zvířat prokázaly, že hlavní orgán zodpovědný za odstranění anakinry jsou ledviny. Clearance anakinry u pacientů s RA se zvyšovala s rostoucí clearancí kreatininu.
Vliv demografických kovariancí na farmakokinetiku anakinry byl hodnocen na základě populační farmakokinetické analýzy vzorku 341 pacientů, kteří dostávali denně subkutánní injekce anakinry v dávkách 30, 75 a 150 mg po dobu až 24 týdnů. Předběžně určená clearance anakinry vzrůstala se stoupající clearance kreatininu a tělesnou hmotností. Populační farmakokinetická analýza prokázala, že průměrná hodnota plasmatické clearance po podání subkutánního bolusu byla přibližně o 14 % vyšší u mužů než u žen a přibližně o 10 % vyšší u subjektů < 65 let než u osob > 65 let. Nicméně po úpravě s ohledem na clearanci kreatininu a tělesnou hmotnost nebyly pohlaví ani věk významnými faktory působícími na průměrnou plazmatickou clearanci. Není třeba žádná úprava dávky podle věku nebo pohlaví.
Jatemí poškození
Byla provedena studie zahrnující 12 pacientů s poruchou funkce jater (Child-Pughova klasifikace B), ve které byla podána jednorázová intravenózní dávka 1mg/kg. Farmakokinetické parametry se od zdravých dobrovolníků podstatně nelišily, mimo snížení clearance o 30 % v porovnání s údaji ze studie se zdravými dobrovolníky. V populaci s jaterním selháním bylo pozorováno odpovídající snížení clearance kreatininu. V souladu s tím se nejpravděpodobnějším vysvětlením zdálo snížení clearance snížením renálních funkcí u této populace. Tyto údaje dokládají, že u pacientů s jaterní dysfunkcí B dle Child-Pughovy klasifikace není třeba žádná úprava dávkování. Viz bod 4.2.
Porucha funkce ledvin
Průměrná plazmatická clearance Kineretu u subjektů s mírnou (clearance kreatininu 50-80 ml/min) a střední (clearance kreatininu 30-49 ml/min) poruchou funkce ledvin se snížila na 16 % a 50 %. U závažné poruchy funkce ledvin a v konečném stádiu onemocnění ledvin (clearance kreatininu < 30 ml/min), se průměrná plazmatická clearance snížila o 70 % a 75 %. Hemodialýzou nebo kontinuální ambulantní peritoneální dialýzou bylo odstraněno méně než 2,5 % podané dávky Kineretu. Tyto údaje dokládají, že u pacientů s mírnou poruchou funkce ledvin (CLcr 50 až 80 ml/min) není nutná žádná úprava dávek. Viz bod 4.2.
5.3 Předklinické údaje vztahující se k bezpečnosti
Nebyly pozorovány žádné účinky anakinry na plodnost, časný vývoj, vývoj embrya a plodu ani na peri- a postnatální vývoj u potkanů v dávkách až stonásobně vyšších, než je dávka pro člověka. Nebyly rovněž pozorovány žádné účinky anakinry na vývoj embrya a plodu u králíka v dávkách až stonásobně vyšších, než je dávka pro člověka.
Ve standardní sérii testů určených k identifikaci rizika s ohledem na DNA, anakinra nevyvolávala genové mutace bakteriálních ani savčích buněk. Anakinra rovněž nezvyšovala výskyt chromozomálních abnormalit či mikrojader v buňkách kostní dřeně u myší. Dlouhodobé studie, které by hodnotily karcinogenní potenciál anakinry, nebyly provedeny. Údaje získané od myší s nadměrnou expresí IL-1ra a od mutantních „knock-out“ myší pro IL-1ra nenaznačovaly zvýšené riziko vzniku nádorového bujení.
Formální toxikologické a toxikokinetické interakční studie u potkanů nezjistily žádný důkaz toho, že by Kineret ovlivňoval toxikologický nebo farmakokinetický profil methotrexatu.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Bezvodá kyselina citronová Chlorid sodný Dihydrát dinatrium-edetátu Polysorbát 80 Hydroxid sodný Voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti
3 roky.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Pro účely ambulantního použití smí být Kineret přemístěn z chladničky do prostředí o teplotě do 25 °C maximálně na období 12 hodin. Datum expirace nesmí být překročeno. Na konci tohoto období přípravek nesmí být vrácen do ledničky a musí být zlikvidován.
6.5 Druh obalu a obsah balení
0,67 ml injekčního roztoku v předplněné injekční stříkačce (sklo typu I) se zarážkou pístu (bromobutylová pryž) a jehlou 29 G. Předplněná injekční stříkačka má vnější tuhý plastový chránič jehly připevněný k vnitřnímu krytu jehly. Žádná ze složek stříkačky a chrániče jehly není vyrobena z přírodní pryže latexu.
Velikost balení 1, 7 nebo 28 (balení obsahující 4 balíčky po 7 předplněných stříkačkách).
Na trhu nemusí být k dispozici všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Kineret je sterilní roztok bez konzervačních přísad. Je určen pouze pro jednorázové použití.
Neprotřepávejte. Před injekčním podáním ponechte předplněnou injekční stříkačku dosáhnout pokojové teploty.
Před podáním vizuálně zkontrolujte přítomnost částic a zabarvení. Podán může být pouze čirý, bezbarvý až bílý roztok, který může obsahovat průsvitné až bílé amorfní částice léku.
Přítomnost těchto částic neovlivňuje kvalitu léku.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Swedish Orphan Biovitrum AB (publ)
SE-112 76 Stockholm Švédsko
8. REGISTRAČNÍ ČÍSLO/A
EU/1/02/203/001 - 1 ks EU/1/02/203/002 - 7 ks EU/1/02/203/003 - 28 ks
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 8. března 2002
Datum posledního prodloužení registrace: 20. března 2007
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu
NÁZEV PŘÍPRAVKU
1.
Kineret 100 mg/0,67 ml injekční roztok v předplněné injekční stříkačce.
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna předplněná injekční stříkačka se stupnicí obsahuje anakinrum* 100 mg v 0,67 ml (150 mg/ml).
* Antagonista humánního receptoru pro interleukin-1 (r-metHuIL-1ra) produkovaný v buňkách Escherichia coli pomocí rekombinantní DNA technologie.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok (injekce).
Čirý, bezbarvý až bělavý injekční roztok, který může obsahovat průsvitné až bílé amorfní částice léku.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Kineret je indikován v kombinaci s methotrexatem k léčbě známek a symptomů revmatoidní artritidy (RA) u dospělých pacientů s nedostatečnou odpovědí na samotný methotrexat.
Kineret je indikován dospělým, dospívajícím, dětem a kojencům ve věku 8 měsíců a starším s hmotností 10 kg nebo větší k léčbě kryopyrin-asociovaných periodických syndromů (CAPS), včetně:
- multisystémového zánětlivého onemocnění se začátkem v novorozeneckém věku (NOMID) / chronického infantilního neurologického kožního a kloubního syndromu (CINCA)
- Muckle-Wellsova syndromu (MWS)
- familiárního chladového autozánětlivého syndromu (FCAS)
4.2 Dávkování a způsob podání
Léčbu Kineretem mají zahajovat a sledovat specializovaní lékaři se zkušenostmi v diagnostice a léčbě revmatoidní artritidy a CAPS.
Dávkování
RA: dospělí
Doporučená dávka Kineretu je 100 mg jednou denně v subkutánní injekci. Dávka by měla být aplikována každý den přibližně ve stejnou dobu.
CAPS: dospělí, dospívající, děti a kojenci ve věku 8 měsíců nebo starší s hmotností 10 kg nebo větší: Počáteční dávka:
Doporučená počáteční dávka pro všechny subtypy CAPS je 1-2 mg/kg/den podkožní injekcí. Terapeutická odpověď se primárně projeví snížením klinických příznaků, jako je horečka, vyrážka, bolesti kloubů a bolest hlavy, ale také snížením zánětlivých markerů v séru (hladiny CRP/SAA) nebo snížením výskytu exacerbací.
Udržovací dávka u mírných CAPS (FCAS, mírný MWS):
Pacienti jsou obvykle dobře kontrolováni udržováním doporučené počáteční dávky (1-2 mg/kg/den). Udržovací dávka u závažných CAPS (MWS a NOMID/CINCA):
Zvyšování dávek může být na základě terapeutické odpovědi během 1-2 měsíců nezbytné. Obvyklá udržovací dávka u těžkých CAPS je 3-4 mg/kg/den, a může být upravena až na maximálně 8 mg/kg/den.
Kromě hodnocení klinických symptomů a zánětlivých markerů u těžkých CAPS se doporučuje po počátečních 3 měsících léčby a poté každých 6 měsíců, dokud není stanovena účinná léčebná dávka, hodnocení zánětů CNS, včetně vnitřního ucha (MRI nebo CT, lumbální punkce a audiologie) a očí (oftalmologická hodnocení). Když jsou pacienti dobře klinicky kontrolováni, lze provádět CNS a oftalmologická sledování ročně.
Starší _populace (> 65 let)
U pacientů s RA není zapotřebí žádná úprava dávek. Dávkování a způsob podání jsou stejné jako pro dospělé osoby ve věku od 18 do 64 let.
Údaje o starších pacientech s CAPS jsou omezené. Nepředpokládá se, že by byla nutná úprava dávek.
Pediatrická populace (< 18 let)
RA: Účinnost Kineretu u dětí s RA (JIA) ve věku od 0 do 18 let nebyla stanovena.
CAPS: Dávkování a způsob podání u dětí a kojenců ve věku 8 měsíců a starších s hmotností 10 kg nebo větší jsou stejné jako pro dospělé pacienty s CAPS, na základě tělesné hmotnosti. Pro děti mladší 8 měsíců nejsou k dispozici žádné údaje.
Porucha _ funkce _ jater
U pacientů se středně závažnou poruchou funkce jater (Child-Pughova klasifikace B) není zapotřebí žádná úprava dávek. U pacientů s vážnou poruchou funkce jater by měl být Kineret používán s opatrností.
Porucha _ funkce ledvin
Pacientům s těžkou poruchou funkce ledvin (CLcr < 30 ml/min) nesmí být Kineret podáván (viz bod 4.3).U pacientů s mírnou poruchou funkce ledvin (CLcr 50 až 80 ml/min) není nutná žádná úprava dávek. Pacientům se středně závažnou poruchou funkce ledvin (CLcr 30 až 50 ml/min) by měl být Kineret podáván s nezbytnou opatrností, vzhledem k tomu, že příslušné údaje chybí.
Způsob podání
Kineret se podává subkutánní injekcí.
Kineret se dodává připravený k okamžitému použití v předplněné injekční stříkačce se stupnicí. Předplněná injekční stříkačka se stupnicí umožňuje dávkování od 20 do 100 mg. Vzhledem k tomu, že je minimální dávka 20 mg, není stříkačka vhodná pro pediatrické pacienty s hmotností nižší než 10 kg. Předplněnou stříkačkou netřepejte. Pokyny pro použití přípravku a zacházení s ním jsou uvedeny v bodě 6.6.
Aby se předešlo nepříjemným pocitům v místě vpichu, doporučuje se měnit místa aplikace. Příznaky a symptomy v místě vpichu může zmírnit chlazení místa vpichu, ohřátí vstřikované kapaliny, použití chladicích balíčků (před a po injekci) a použití topických kortikosteroidů a antihistaminik po vpichu.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1 nebo na proteiny pocházející z E. coli.
Pacientům s vážnou poruchou funkce ledvin (CLcr < 30 ml/min) nesmí být Kineret podáván (viz bod 4.2).
U pacientů s neutropenií (ANC < 1,5 x 109/l) nesmí být léčba Kineretem zahajována (viz bod 4.4).
4.4 Zvláštní upozornění a opatření pro použití
Alergické reakce
Alergické reakce, včetně anafylaktických reakcí či angioedému, byly hlášeny méně často. Ve většině případů se jednalo o makulopapulární nebo urtikariální kožní exantém.
V případě výskytu těžké alergické reakce by mělo být podávání Kineretu přerušeno a měla by být zahájena odpovídající léčba.
Jaterní příhody
V klinických studiích byla u pacientů s RA a CAPS pozorována méně často přechodná zvýšení jaterních enzymů. Tato zvýšení nebyla spojena s příznaky nebo symptomy buněčného poškození jater. Při postmarketingovém používání byl hlášen ojedinělý případ indikující neinfekční hepatitidu. Jaterní příhody při postmarketingovém používání byly hlavně hlášeny u pacientů s predispozicí, např. zvýšenými transaminázami v anamnéze před zahájením léčby Kineretem.
Účinnost a bezpečnost Kineretu u pacientů s AST/ALT >1,5x zvýšenou hladinou nad normál nebyly hodnoceny.
Vážné infekce
Podávání Kineretu u pacientů s RA bylo provázeno zvýšením incidence vážných infekcí v porovnání s placebem (1,8 % vs. 0,7 %). U malého počtu pacientů s astmatem byla incidence závažných infekcí vyšší u pacientů léčených Kineretem (4,5 %) při porovnání s pacienty užívajícími placebo (0 %), tyto infekce se týkaly hlavně dýchacích cest.
Bezpečnost a účinnost léčby Kineretem u pacientů s chronickými a vážnými infekcemi nebyla hodnocena.
Léčba Kineretem by neměla být zahajována u pacientů s aktivními infekcemi. U pacientů s RA, u nichž se rozvine těžká infekce, by měla být léčba Kineretem ukončena. U pacientů s CAPS léčených Kineretem existuje riziko vzplanutí nemoci, když ukončí léčbu Kineretem. To je třeba to vzít v úvahu při rozhodování o ukončení léčby Kineretem během závažné infekce.
K podávání Kineretu pacientům s anamnézou rekurentních infekcí nebo se základním onemocněním, které může být predisponujícím faktorem infekce, by měl lékař přistupovat s nezbytnou obezřelostí.
Bezpečnost Kineretu není u jedinců s latentní tuberkulózou známa. U pacientů užívajících několik biologických protizánětlivých léčebných režimů byla hlášena tuberkulóza. Před zahájením léčby Kineretem by měli být pacienti vyšetřeni na latentní tuberkulózu. Také by měla být brána v úvahu dostupná lékařská doporučení.
S reaktivací hepatitidy B byla spojena další antirevmatická terapie. Proto by měla být i před zahájením léčby Kineretem prováděna vyšetření na virovou hepatitidu v souladu s vydanými pokyny.
Neutropenie
V placebem kontrolovaných studiích RA byl Kineret často spojován s neutropenií (ANC < 1,5 x 109/l) a u pacientů s CAPS byly pozorovány případy neutropenie. Pro více informací o neutropenii viz část 4.8.
U pacientů s neutropenií (absolutní počet neutrofilů < 1,5 x 109/l) by léčba Kineretem neměla být zahajována. Počet neutrofilů se doporučuje vyšetřit před zahájením terapie pomocí Kineretu, jednou za měsíc během prvních 6 měsíců léčby a dále čtvrtletně. U pacientů, u kterých se neutropenie objeví (absolutní počet neutrofilů < 1,5 x 109/l), by měla být léčba Kineretem přerušena a absolutní počet neutrofilů by měl být pečlivě sledován. Bezpečnost a účinnost Kineretu u pacientů s neutropenií nebyla hodnocena.
Imunosuprese
Účinky léčby Kineretem na preexistující maligní nádorové onemocnění nebyly studovány. Podávání Kineretu pacientům s preexistujícím maligním nádorovým onemocněním se proto nedoporučuje.
Vakcinace
V placebem kontrolovaném klinickém hodnocení (n = 126) nebyl zjištěn žádný rozdíl v protilátkové odpovědi na podání antitetanového séra mezi skupinami pacientů léčených Kineretem a placebem, kdy byla vakcína obsahující tetanový/difterický toxoid aplikována současně s Kineretem. Nejsou k dispozici žádné údaje o účinnosti očkování pacientů užívajících Kineret jinými inaktivovanými antigeny.
Nejsou k dispozici žádné údaje o účincích vakcinace živou očkovací látkou ani o sekundárním přenosu infekce živou očkovací látkou u pacientů užívajících Kineret. Pacienti užívající Kineret by proto neměli být současně očkováni živou vakcínou.
Starší populace (> 65 let)
V klinických studiích bylo sledováno celkem 752 pacientů s RA > 65 let, včetně 163 pacientů > 75 let. Mezi těmito pacienty a mladšími pacienty nebyl zaznamenán žádný celkový rozdíl s ohledem na bezpečnost anebo účinnost. S léčbou starších pacientů s CAPS jsou omezené zkušenosti. Vzhledem k tomu, že u starší populace je incidence infekčních onemocnění obecně vyšší, je třeba léčbu starších pacientů provádět s nezbytnou opatrností.
Současná léčba Kineretem a TNF antagonisty
Současná léčba Kineretem a etanerceptem je spojena se zvýšeným rizikem vážných infekcí a s rizikem neutropenie v porovnání s léčbou etanerceptem samotným u pacientů s RA. Tato léčebná kombinace neprokázala vyšší klinický přínos.
Současné podávání Kineretu a etanerceptu nebo dalších TNF antagonistů se nedoporučuje (viz bod 4.5).
Tento léčivý přípravek obsahuje méně než 1 mmol sodíku (23 mg) na 100 mg dávky, tj. „je v podstatě bez sodíku“.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Interakce mezi Kineretem a jinými léčivými přípravky nebyly ve formálně prováděných studiích sledovány. V klinických studiích nebyly pozorovány interakce mezi Kineretem a ostatními léčivými přípravky (včetně nesteroidních protizánětlivých, léčivých přípravků, kortikosteroidů a DMARD).
Současná léčba Kineretem a TNF antagonisty
V klinické studii s pacienty s RA, jejichž základní léčbou byl methotrexat, byl u pacientů léčených Kineretem a etanerceptem zaznamenán vyšší počet vážných infekcí (7 %) a neutropenií než u pacientů léčených etanerceptem samotným. Tento počet byt rovněž vyšší než v předchozích studiích, ve kterých byl Kineret podáván samotný. Současné podávání Kineretu a etanerceptu neprokázalo vyšší klinický přínos.
Současné podávání Kineretu s etanerceptem nebo jinými TNF antagonisty se nedoporučuje (viz bod 4.4).
Substráty cytochromu P450
Tvorba enzymů CYP450 je při chronickém zánětu potlačena zvýšenými hladinami cytokinů (např. IL-1). Proto lze očekávat, že by antagonista receptoru IL-1, např. anakinra, mohl během léčby normalizovat tvorbu enzymů CYP450. To by bylo klinicky relevantní pro substráty CYP450 s úzkým terapeutickým indexem (např. warfarin a fenytoin). Na začátku nebo konci léčby Kineretem u pacientů léčených těmito léčivými přípravky může být relevantní zvážit, terapeutické sledování účinku nebo koncentrace těchto přípravků a může být zapotřebí upravit individuální dávku léčivého přípravku.
Informace o vakcinaci jsou uvedeny v bodě 4.4.
4.6 Fertilita, těhotenství a kojení
Údaje týkající se užívání látky anakinra u těhotných žen jsou omezené. Reprodukční studie s Kineretem však byly provedeny u potkanů a králíků při dávkách až 100krát vyšších, než jsou dávky u člověka s RA, a nebyl odhalen žádný důkaz narušení fertility ani poškození plodu.
Podávání Kineretu se v těhotenství a u žen v reprodukčním věku, které nepoužívají antikoncepci, nedoporučuje.
Není známo, zda se anakinra/metabolity vylučují do lidského mateřského mléka. Riziko pro kojené novorozence/děti nelze vyloučit. Kojení má být během léčby Kineretem přerušeno.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Není relevantní.
4.8 Nežádoucí účinky
Ve studiích kontrolovaných placebem u pacientů s RA byly nejčastěji hlášenými nežádoucími účinky spojenými s podáváním Kineretu reakce v místě vpichu (ISR), jež dosáhly u většiny pacientů mírné až střední intenzity. Nejčastějším důvodem přerušení účasti pacientů s RA léčených Kineretem v klinické studii byla reakce v místě vpichu. Incidence vážných nežádoucích účinků ve studiích RA při aplikaci doporučené dávky Kineretu (100 mg/den) byla srovnatelná s placebem (7,1 % v porovnání s 6,5 % v placebové skupině). Incidence vážných infekcí byla u pacientů léčených Kineretem vyšší než u pacientů na placebu (1,8 % vs. 0,7 %). U pacientů léčených Kineretem se v porovnání s pacienty, kteří dostávali placebo, častěji vyskytl pokles počtu neutrofilů.
Údaje o nežádoucích účincích u pacientů s CAPS jsou založeny na otevřené studii 43 pacientů s NOMID/CINCA léčených Kineretem až 5 let, s celkovou expozicí Kineretu 159,8 pacientoroků. Během 5leté studie hlásilo 14 pacientů (32,6 %) 24 závažných účinků. O jedenácti závažných účincích 4 (9,3 %) pacientů se soudilo, že souvisely s léčbou Kineretem. Žádný pacient z důvodu nežádoucích účinků z léčby Kineretem neodstoupil. Ani z této studie, ani z hlášení postmarketingových nežádoucích účinků neexistují žádné náznaky, že se celkový profil pacientů s CAPS liší od pacientů s RA. Proto lze aplikovat tabulku nežádoucích účinků níže na léčbu Kineretem pro pacienty s RA i s CAPS.
Nežádoucí účinky jsou řazeny podle tříd orgánových systémů MedDRA a kategorie četností.
Kategorie četností jsou definovány za použití následující konvence: velmi časté (> 1/10); časté (> 1/100 až < 1/10); méně časté (> 1/1 000 až < 1/100); vzácné (> 1/10 000 až < 1/1 000); velmi vzácné (<1/10 000); není známo (z dostupných údajů nelze určit). V každé kategorii četností jsou nežádoucí účinky řazeny podle klesající závažnosti.
|
Orgánový systém dle MedDRA |
F rekvence |
Nežádoucí účinek |
|
Infekce a infestace |
Časté (> 1/100 až < 1/10) |
Závažné infekce |
|
Poruchy krve a lymfatického systému |
Časté (> 1/100 až < 1/10) |
Neutropenie T rombocvtopenie |
|
Poruchy imunitního systému |
Méně časté (> 1/1 000 až < 1/100) |
Alergické reakce zahrnující anafylaktické reakce, angioedém, kopřivku a pruritus |
|
Poruchy nervového systému |
Velmi časté (> 1/10) |
Bolesti hlavy |
|
Poruchy jater a žlučových cest |
Méně časté (> 1/1 000 až < 1/100) |
Zvýšené jaterní enzymy |
|
Není známo |
Neinfekční hepatitida |
|
(z dostupných údajů nelze hodnotit) | ||
|
Poruchy kůže a podkožní tkáně |
Velmi časté (> 1/10) |
Reakce v místě vpichu |
|
Méně časté (> 1/1 000 až < 1/100) | ||
|
Vyšetření |
Velmi časté (> 1/10) |
Zvýšení hladiny cholesterolu v krvi |
Vážné infekce
Incidence vážných infekcí ve studiích RA s aplikací doporučené dávky (100 mg/den) byla 1,8 % ve skupině pacientů léčených Kineretem a 0,7 % ve skupině pacientů léčených placebem. V průběhu až tříletého období sledování zůstával počet závažných infekcí stabilní. Pozorované infekce byly především bakteriálního původu jako například celulitida, pneumonie či infekční onemocnění kostí a kloubů. Po zhojení infekce pokračovala většina pacientů ve studii léčivým přípravkem.
U 43 pacientů s CAPS sledovaných až 5 let byla frekvence závažných infekcí 0,1/rok, nejčastější byly zápal plic a gastroenteritida. U jednoho pacienta bylo užívání Kineretu dočasně přerušeno, všichni ostatní pacienti během infekcí pokračovali v jeho užívání.
Ve studiích RA nebo CAPS se nevyskytla žádná úmrtí z důvodu závažných infekcí.
V klinických studiích RA a postmarketingových zkušenostech byly pozorovány vzácné případy infekcí oportunními mikroorganismy včetně plísní, mykobakterií, bakterií a virů. Infekce byly zjištěny u všech orgánových systémů a byly zaznamenány u pacientů užívajících Kineret v monoterapii i
v kombinaci s imunosupresivy.
Neutropenie
V placebem kontrolovaných studiích RA s Kineretem byla léčba doprovázena malým snížením průměrných hodnot celkového počtu leukocytů a absolutního počtu neutrofilů. Podávání Kineretu bylo spojeno s neutropenií (absolutní počet neutrofilů < 1,5 x 109/l) u 2,4 % pacientů v porovnání s 0,4 % pacientů na placebu. Žádný z těchto pacientů neměl závažné infekce spojené s neutropenií.
Ze 43 pacientů s CAPS sledovaných až 5 let byla neutropenie hlášena u 2 pacientů. Obě epizody neutropenie byly v průběhu času vyřešeny při pokračující léčbě Kineretem.
T rombocytopenie
V klinických studiích prováděných u pacientů s RA byla trombocytopenie zjištěna u 1,9 % léčených pacientů v porovnání s 0,3 % v placebové skupině. Trombocytopenie byly mírné, tj. počty krevních destiček byly >75 x109/l. Mírná trombocytopenie byla pozorována i u pacientů s CAPS.
Při postmarketingovém používání Kineretu byla zjištěna trombocytopenie, včetně občasných hlášení o případech těžké trombocytopenie (tj. počty krevních destiček<10 x109/l).
Malignity
U pacientů s RA může být vyšší riziko (v průměru 2- až 3násobné) vzniku lymfomů. Při klinických hodnoceních u pacientů léčených Kineretem byla zjištěna incidence lymfomů vyšší, než se předpokládá v běžné populaci; tento počet však odpovídá počtům uváděným obecně pro pacienty s RA.
Incidence maligních nádorových onemocnění byla při klinických hodnoceních stejná u pacientů léčených Kineretem jako ve skupině užívající placebo a nelišila se od běžné populace. Kromě toho se celková incidence malignit u pacientů nezvyšovala ani během tříleté léčby Kineretem.
Alergické reakce
Méně často byly u Kineretu hlášeny alergické reakce, včetně anafylaktických reakcí, angioedému, kopřivky, vyrážky a pruritu. Většina těchto reakcí byly makulopapulární nebo urtikariální vyrážky.
U 43 pacientů s CAPS sledovaných až 5 let nebyla žádná alergická reakce závažná a žádná událost nevyžadovala ukončení léčby Kineretem.
Imunogenicita
V klinických studiích RA měla až 3 % dospělých pacientů alespoň jednou pozitivní výsledek
v sérologickém testu v průběhu studie na protilátky schopné neutralizovat biologické účinky anakinry. Výskyt protilátek byl většinou přechodný a neměl souvislost s výskytem nežádoucích účinků nebo snížením účinnosti. Kromě toho bylo v klinické studii 6 % pediatrických pacientů testováno seropozitivně alespoň jednou v průběhu studie na protilátky schopné neutralizovat biologické účinky anakinry.
U většiny pacientů s CAPS se ve studii 03-AR-0298 vyvinuly protilátky proti anakinře. Nebylo to spojováno s žádnými klinicky významnými účinky na farmakokinetiku, účinnost nebo bezpečnost.
Jaterní příhody
V klinických studiích byla u pacientů s RA a CAPS pozorována méně často přechodná zvýšení jaterních enzymů. Tato zvýšení nebyla spojena s příznaky nebo symptomy buněčného poškození jater. Při postmarketingovém používání byl hlášen ojedinělý případ indikující neinfekční hepatitidu. Jaterní příhody při postmarketingovém používání byly hlavně hlášeny u pacientů s predispozicí, např. zvýšenými transaminázami v anamnéze před zahájením léčby Kineretem.
Reakce v místě vpichu
Nejčastěji a nejdůsledněji hlášeným nežádoucím účinkem spojeným s léčbou Kineretem u pacientů s RA byla reakce v místě vpichu injekce (ISR). Většina (95 %) z těchto hlášených reakcí měla lehkou až středně těžkou intenzitu. Reakce typicky zahrnovaly jeden nebo více z následujících projevů: erytém, ekchymózu, zánětlivé projevy a bolestivost. Při aplikaci dávky 100 mg/den došlo ke vzniku reakce v místě vpichu u 71 % pacientů s RA, ve srovnání s 28 % pacientů, kteří dostávali placebo. U 43 pacientů s CAPS sledovaných až 5 let neukončil ani dočasně nepřerušil léčbu Kineretem z důvodu reakce v místě vpichu žádný pacient. ISR se obvykle objeví během 2. týdne léčby a během 4-6 týdnů zmizí. Výskyt reakce v místě vpichu po uplynutí prvního měsíce léčby u pacientů, u kterých se tato reakce neobjevila ani v minulosti, nebyl častý.
Zvýšení hladiny cholesterolu v krvi
V klinických studiích RA se 775 pacienty denně léčenými dávkami Kineret o síle 30 mg, 75 mg,
150 mg, 1 mg/kg nebo 2 mg/kg došlo k nárustu o 2,4 % až 5,3 % celkových hladin cholesterolu 2 týdny po zahájení léčby Kineretem, bez vzájemného vztahu mezi dávkou a odezvou. Podobný vzorec se opakoval po 24 týdnech léčby Kineretem. Léčba placebem (n=213) vedla ke snížení celkové hladiny cholesterolu přibližně o 2,2 % ve 2. týdnu a 2,3 % ve 24. týdnu. Nejsou k dispozici žádné údaje o cholesterolu LDL nebo HDL.
Pediatrická _ populace
Kineret byl až 5 let studován na 36 pacientech s CAPS ve věku 8 měsíců až < 18 let. S výjimkou infekcí a s nimi souvisejících symptomů, které byly častěji hlášeny u pacientů < 2 let, byl bezpečnostní profil podobný v celé pediatrické věkové skupině. Bezpečnostní profil byl u pediatrických pacientů podobný tomu v dospělých populacích a nebyly pozorovány žádné klinicky relevantní nové nežádoucí reakce.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Během klinických studií u pacientů s RA nebo CAPS nebyly zaznamenány žádné známky toxicity, které by si vynutily úpravu dávek. Ve studiích, kde byla sledována sepse, dostávalo Kineret 1015 pacientů, a to v dávkách až do 2 mg/kg/hod i.v. (~35 násobku doporučené dávky při RA) během 72 hodinové léčebné periody. Profil nežádoucích příhod zaznamenaných v těchto studiích se celkově nijak nelišil od údajů získaných ve studiích s revmatoidní artritidou.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Imunosupresiva, inhibitory interleukinu, ATC kód: L04AC03
Anakinra neutralizuje biologickou aktivitu interleukinu-1a (IL-1a) a interleukinu-1p (IL-1P) kompetitivní inhibicí jejich vazby na receptor typu I pro interleukin-1 (IL-1RI). Interleukin-1 (IL-1) je klíčovým prozánětlivým cytokinem, který zprostředkovává řadu buněčných odpovědí, včetně reakcí významných při zánětlivých procesech synoviální tkáně.
IL-1 se nachází v plasmě a synoviální tekutině pacientů s revmatoidní artritidou a byla popsána korelace mezi plasmatickými koncentracemi IL-1 a aktivitou onemocnění.
Anakinra inhibuje reakce vyvolané prostřednictvím IL-1 in vitro, včetně indukce oxidu dusnatého a prostaglandinu E2 a/nebo produkce kolagenázy synoviálními buňkami, fibroblasty a chondrocyty.
U většiny pacientů s CAPS byla zjištěna spontánní mutace v genu CIAS1/NLRP3. CIAS1/NLRP3 kódují kryopyrin, složku inflamazomu. Aktivovaný inflamazom způsobuje proteolytickou mutaci a sekreci IL-ip, který má široké spektrum účinků, včetně systémového zánětu. Neléčené pacienty s CAPS charakterizuje zvýšené CRP, SAA a IL-6 vzhledem k normálním hladinám séra. Podávání Kineretu vede ke snížení reaktantů akutní fáze a bylo pozorováno snížení hladiny exprese IL-6.
Během prvních týdnů léčby byly zaznamenány snížené hladiny proteinů akutní fáze.
Klinická účinnost a bezpečnost při RA
Bezpečnost a účinnost anakinry v kombinaci s methotrexatem byla prokázána u 1790 pacientů s RA > 18 let s různým stupněm intenzity onemocnění.
Klinická odpověď na léčbu anakinrou se většinou objevila během 2 týdnů po zahájení léčby a trvala s pokračující aplikací anakinry. Maximální klinická odezva byla obvykle zaznamenána během 12 týdnů po zahájení terapie.
Kombinovaná léčba anakinrou a methotrexatem prokázala statisticky a klinicky signifikantní snížení intenzity známek a symptomů revmatoidní artritidy u pacientů, u kterých odpověď na léčbu samotným methotrexatem nebyla dostatečná (38 % vs. 22 % pozitivních odpovědí na léčbu, měřeno pomocí kritérií ACR20). Významné zlepšení se týká bolestivosti, počtu bolestivých kloubů, tělesných funkcí (HAQ skóre), reaktantů akutní fáze a celkového hodnocení stavu lékařem i pacientem.
V jedné klinické studii s anakinrou byla prováděna RTG vyšetření. Tato vyšetření neprokázala žádný škodlivý účinek anakinry na kloubní chrupavku.
Klinická účinnost a bezpečnost při CAPS
Bezpečnost a účinnost Kineretu byla prokázána u pacientů s CAPS s různými stupni závažnosti onemocnění. V klinické studii zahrnující 43 dospělých a pediatrických pacientů (36 pacientů ve věku 8 měsíců až < 18 let) se závažným CAPS (NOMID/CINCA a MWS) byla u všech pacientů pozorována klinická odezva na anakinru během 10 dnů a byla udržována až 5 let při pokračujícím podávání Kineretu.
Léčba Kineretem významně snižuje projevy CAPS, včetně snížení často se vyskytujících symptomů, jako je horečka, vyrážka, bolesti kloubů, hlavy, únava a zarudnutí očí. Byl pozorován rychlý a trvalý pokles hladin zánětlivých biomarkerů, sérového amyloidu A (SAA), C reaktivního proteinu (CRP) a rychlosti sedimentace eryrocytů (ESR) a normalizace zánětlivých hematologických změn. U těžkých forem CAPS zlepšuje dlouhodobá léčby projevy systémového zánětu orgánů, očí, vnitřního ucha a CNS. Ostrost slyšení a vidění se při léčbě anakinrou dále nezhoršovala.
Analýza nežádoucích účinků (AE) vyžadujících léčbu, klasifikovaná přítomností mutace CIAS1, ukázala, že mezi skupinami CIAS1 a non-CIAS 1 nebyly velké rozdíly v celkových četnostech hlášení AE, jež byly 7,4 a 9,2. Podobné četnosti byly získány pro skupiny na hladině SOC, s výjimkou poruch zraku - 55 AE (četnost 0,5), z čehož bylo 35 očních hyperémií (což by mohl být také symptom CAPS) ve skupině CIAS1 a 4 AE ve skupině non-CIAS1 (četnost 0,1).
Pediatrická populace
Celkově j sou účinnost a bezpečnostní profil Kineretu srovnatelné u dospělých a pediatrických pacientů s CAPS.
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s Kineretem u jedné nebo více podskupin pediatrické populace s CAPS a RA (JIA), (informace o použití u dětí viz bod 4.2).
Bezpečnost u pediatrických pacientů s RA (JIA)
Kineret byl zkoumán v jediné randomizované, zaslepené, multicentrické studii s 86 pacienty s polyartikulárním průběhem juvenilní revmatoidní artritidy (JRA; věk 2-17 let) přijímajícími podkožně dávku 1 mg/kg denně, až do dávky 100 mg. 50 pacientů, kteří dosáhli klinické odpovědi po 12 týdnech odslepené zaváděcí fáze, bylo randomizováno mezi Kineret (25 pacientů) a placebo (25 pacientů), podávané denně po dalších 16 týdnů. Podskupina těchto pacientů pokračovala v odslepené léčbě s Kineretem až 1 rok ve společné navazující studii. V těchto studiích byl pozorovaný profil nežádoucích účinků podobný těm, které lze vidět u dospělých pacientů s RA. Údaje v těchto studiích jsou nedostatečné pro prokázání účinnosti, a tudíž se Kineret nedoporučuje pro použití u pediatrických pacientů s juvenilní revmatoidní artritidou.
Imunogenicita Viz bod 4.8
5.2 Farmakokinetické vlastnosti
Absolutní biologická dostupnost anakinry po subkutánním podání dávky 70 mg zdravým subjektům (n = 11) je 95 %. Faktorem limitujícím rychlost vymizení anakinry z plasmy po subkutánní injekci je absorpční proces. U subjektů s RA bylo maximální plasmatické koncentrace anakinry dosaženo za
3 až 7 hodin po subkutánní aplikaci anakinry v klinicky relevantních dávkách (1 až 2 mg/kg; n = 18). Koncentrace v plazmě se snižovala bez patrné distribuční fáze a terminální poločas se pohyboval od
4 do 6 hodin. U pacientů s RA nebyla zaznamenána žádná neočekávaná kumulace anakinry po každodenní subkutánní aplikaci dávek po dobu až 24 týdnů. Průměrné (SD) hodnoty clearance (CL/F) a objemu distribuce (Vd/F) získané analýzou populace z údajů ze dvou farmakokinetických studií na 35 pacientech s RA byly 105 (27) ml/min nebo 18,5 (11) l. Údaje od lidí a zvířat prokázaly, že hlavní orgán zodpovědný za odstranění anakinry jsou ledviny. Clearance anakinry u pacientů s RA se zvyšovala s rostoucí clearancí kreatininu.
Vliv demografických kovariancí na farmakokinetiku anakinry byl hodnocen na základě populační farmakokinetické analýzy vzorku 341 pacientů, kteří dostávali denně subkutánní injekce anakinry v dávkách 30, 75 a 150 mg po dobu až 24 týdnů. Předběžně určená clearance anakinry vzrůstala se stoupající clearance kreatininu a tělesnou hmotností. Populační farmakokinetická analýza prokázala, že průměrná hodnota plasmatické clearance po podání subkutánního bolusu byla přibližně o 14 % vyšší u mužů než u žen a přibližně o 10 % vyšší u subjektů < 65 let než u subjektů > 65 let. Nicméně po úpravě s ohledem na clearanci kreatininu a tělesnou hmotnost nebyly pohlaví ani věk významnými faktory působícími na průměrnou plazmatickou clearanci. Není třeba žádná úprava dávky podle věku nebo pohlaví.
Farmakokinetika u pacientů s CAPS je obecně podobná té u pacientů s RA. U pacientů s CAPS byla zaznamenána přibližná linearita dávky s mírným sklonem k vyšším než přímo úměrným hodnotám. Chybí farmakokinetické údaje u dětí < 4 let, ale je k dispozici klinická zkušenost u dětí od věku 8 měsíců a při zahájení na doporučené denní dávce 1 -2 mg/kg nebyly zjištěny žádné obavy o bezpečnost. U starších pacientů s CAPS farmakokinetické údaje chybí. Byla prokázána distribuce do mozkomíšního moku.
Jatemí poškození
Byla provedena studie zahrnující 12 pacientů s poruchou funkce jater (Child-Pughova klasifikace B), ve které byla podána jednorázová intravenózní dávka 1mg/kg. Farmakokinetické parametry se od zdravých dobrovolníků podstatně nelišily, mimo snížení clearance o 30 % v porovnání s údaji ze studie se zdravými dobrovolníky. V populaci s jaterním selháním bylo pozorováno odpovídající snížení clearance kreatininu. V souladu s tím se nejpravděpodobnějším vysvětlením zdálo snížení clearance snížením renálních funkcí u této populace. Tyto údaje dokládají, že u pacientů s jaterní dysfunkcí B dle Child-Pughovy klasifikace není třeba žádná úprava dávkování. Viz bod 4.2.
Porucha funkce ledvin
Průměrná plazmatická clearance Kineretu u subjektů s mírnou (clearance kreatininu 50-80 ml/min) a střední (clearance kreatininu 30-49 ml/min) poruchou funkce ledvin se snížila na 16 % a 50 %. U závažné poruchy funkce ledvin a v konečném stádiu onemocnění ledvin (clearance kreatininu < 30 ml/min), se průměrná plazmatická clearance snížila o 70 % a 75 %. Hemodialýzou nebo kontinuální ambulantní peritoneální dialýzou bylo odstraněno méně než 2,5 % podané dávky Kineretu. Tyto údaje dokládají, že u pacientů s mírnou poruchou funkce ledvin (CLcr 50 až 80 ml/min) není nutná žádná úprava dávek. Viz bod 4.2.
5.3 Předklinické údaje vztahující se k bezpečnosti
Nebyly pozorovány žádné účinky anakinry na plodnost, časný vývoj, vývoj embrya a plodu ani na peri- a postnatální vývoj u potkanů v dávkách až stonásobně vyšších, než je dávka pro člověka. Nebyly rovněž pozorovány žádné účinky anakinry na vývoj embrya a plodu u králíka v dávkách až stonásobně vyšších, než je dávka pro člověka.
Ve standardní sérii testů určených k identifikaci rizika s ohledem na DNA, anakinra nevyvolávala genové mutace bakteriálních ani savčích buněk. Anakinra rovněž nezvyšovala výskyt chromozomálních abnormalit či mikrojader v buňkách kostní dřeně u myší. Dlouhodobé studie, které by hodnotily karcinogenní potenciál anakinry, nebyly provedeny. Údaje získané od myší s nadměrnou expresí IL-1ra a od mutantních „knock-out“ myší pro IL-1ra nenaznačovaly zvýšené riziko vzniku nádorového bujení.
Formální toxikologické a toxikokinetické interakční studie u potkanů nezjistily žádný důkaz toho, že by Kineret ovlivňoval toxikologický nebo farmakokinetický profil methotrexatu.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Bezvodá kyselina citronová Chlorid sodný Dihydrát dinatrium-edetátu Polysorbát 80 Hydroxid sodný Voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti 3 roky.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Pro účely ambulantního použití smí být Kineret přemístěn z chladničky do prostředí o teplotě do 25 °C maximálně na období 12 hodin. Datum expirace nesmí být překročeno. Na konci tohoto období přípravek nesmí být vrácen do ledničky a musí být zlikvidován.
6.5 Druh obalu a obsah balení
0,67 ml injekčního roztoku v předplněné injekční stříkačce se stupnicí (sklo typu I) se zarážkou pístu (bromobutylová pryž) a jehlou 29 G. Předplněná injekční stříkačka má vnější tuhý plastový chránič jehly připevněný k vnitřnímu krytu jehly. Žádná ze složek stříkačky a chrániče jehly není vyrobena z přírodní pryže latexu.
Velikost balení 1, 7 nebo 28 předplněných stříkaček (balení obsahující 4 balíčky po 7 předplněných stříkačkách).
Na trhu nemusí být k dispozici všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Kineret je sterilní roztok bez konzervačních přísad. Je určen pouze pro jednorázové použití.
Neprotřepávejte. Před injekčním podáním ponechte předplněnou injekční stříkačku dosáhnout pokojové teploty.
Před podáním vizuálně zkontrolujte přítomnost částic a zabarvení. Podán může být pouze čirý, bezbarvý až bílý roztok, který může obsahovat průsvitné až bílé amorfní částice léku.
Přítomnost těchto částic neovlivňuje kvalitu léku.
Předplněná stříkačka je pouze pro jednorázové použití. Veškerý nepoužitý léčivý přípravek zlikvidujte.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Swedish Orphan Biovitrum AB (publ)
SE-112 76 Stockholm Švédsko
8. REGISTRAČNÍ ČÍSLO/A
EU/1/02/203/005 - 1 balení EU/1/02/203/006 - 7 balení EU/1/02/203/007 - 28 balení
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 8. března 2002
Datum posledního prodloužení registrace: 20. března 2007
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http: //www .ema.europa.eu
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce biologické léčivé látky
Boehringer Ingelheim RCV GmbH & Co kg Dr. Boehringer-Gasse 5-11 A-1121 Vídeň Rakousko
Název a adresa výrobce odpovědného za propouštění šarží v EHP
Swedish Orphan Biovitrum AB (publ)
SE-112 76 Stockholm Švédsko
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ, TÝKAJÍCÍ SE BEZPEČNÉHO A ÚČINNÉHO
POUŽÍVÁNÍ TOHOTO LÉČIVA
• Plán řízení rizik (RPM)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky;
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
• Další opatření k minimalizaci rizik
Držitel rozhodnutí o registraci se dohodne na obsahu a formátu vzdělávacích materiálů s národními odpovědnými orgány v každém členském státu, kde se Kineret uvádí na trh a před uvedením na trh v jakémkoli dalším členském státu.
Držitel rozhodnutí o registraci musí zajistit, aby všem lékařům, kteří budou předepisovat KINERET, byly poskytovány následující položky:
• Vzdělávací materiál pro poskytovatele zdravotní péče
• Vzdělávací materiál pro pacienty a pečovatele
Vzdělávací materiál pro poskytovatele zdravotní péče musí obsahovat následující klíčové prvky:
• Důležitost vysvětlení pacientům nebo pečovatelům, jak používat novou injekční stříkačku se stupnicí a správnou injekční techniku
• Důležitost poskytovat pacientům nebo pečovatelům vzdělávací materiály
Vzdělávací materiál pro pacienty a pečovatele bude obsahovat následující klíčové prvky:
• Návod na použití stříkačky se stupnicí
• Návod na správné postupy vstřikování injekce a likvidace použitých stříkaček
• Jak zvládat reakce v místě vpichu
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Kineret 100 mg injekční roztok v předplněné injekční stříkačce Anakinrum
2 OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna předplněná injekční stříkačka o objemu 0,67 ml obsahuje 100 mg anakinry.
3. SEZNAM POMOCNÝCH LÁTEK
Pomocné látky: bezvodá kyselina citronová, chlorid sodný, dihydrát dinatrium-edetátu, polysorbát 80, hydroxid sodný, voda na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Injekční roztok v předplněné injekční stříkačce 1 předplněná injekční stříkačka 7 předplněných injekčních stříkaček Balení: 28 (4x7) předplněných injekčních stříkaček
5 ZPŮSOB A CESTA/CESTY PODÁNÍ
Jednorázové použití.
Subkutánní podání
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Swedish Orphan Biovitrum AB (publ)
SE-112 76 Stockholm
Švédsko
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/02/203/001 - 1 ks EU/1/02/203/002 - 7 ks EU/1/02/203/003 - 28 ks
13 ČÍSLO ŠARŽE
Č.š.
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ 16. INFORMACE V BRAILLOVE PÍSMU
Kineret 100 mg
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU
KRABIČKA OBSAHUJÍCÍ 7 PŘEDPLNĚNÝCH INJEKČNÍCH STŘÍKAČEK (BEZ BLUE BOXU)_
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Kineret 100 mg injekční roztok v předplněné injekční stříkačce Anakinrum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna předplněná injekční stříkačka o objemu 0,67 ml obsahuje 100 mg anakinry.
3. SEZNAM POMOCNÝCH LÁTEK
Pomocné látky: bezvodá kyselina citronová, chlorid sodný, dihydrát dinatrium-edetátu, polysorbát 80, hydroxid sodný, voda na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Injekční roztok v předplněné injekční stříkačce 7 předplněných injekčních stříkaček
Toto balení obsahující 7 předplněných injekčních stříkaček je součástí balení s 28 ks.
5 ZPŮSOB A CESTA/CESTY PODÁNÍ
Jednorázové použití.
Subkutánní podání
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Swedish Orphan Biovitrum AB (publ)
SE-112 76 Stockholm
Švédsko 12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/02/203/003 13. ČÍSLO ŠARŽE
Č.š.
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.

1 NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Kineret 100 mg injekce Anakinra
s.c.
2 ZPŮSOB PODÁNÍ
3 POUŽITELNOST
EXP.:
4 ČÍSLO ŠARŽE
Lot:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
0,67 ml
6. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Kineret 100 mg/0,67 ml injekční roztok v předplněné injekční stříkačce Anakinrum
2 OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna předplněná injekční stříkačka se stupnicí o objemu 0,67 ml obsahuje 100 mg anakinry.
3. SEZNAM POMOCNÝCH LÁTEK
Pomocné látky: bezvodá kyselina citronová, chlorid sodný, dihydrát dinatrium-edetátu, polysorbát 80, hydroxid sodný, voda na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Injekční roztok v předplněné injekční stříkačce 1 předplněná stříkačka SE STUPNICÍ 7 předplněných stříkaček SE STUPNICÍ Balení: 28 (4 x 7) předplněných stříkaček SE STUPNICÍ
5 ZPŮSOB A CESTA/CESTY PODÁNÍ
Jednorázové použití.
Subkutánní podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Swedish Orphan Biovitrum AB (publ)
SE-112 76 Stockholm
Švédsko
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/02/203/005 - 1 balení EU/1/02/203/006 - 7 balení EU/1/02/203/007 - 28 balení
13 ČÍSLO ŠARŽE
Č.š.
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ 16. INFORMACE V BRAILLOVE PÍSMU
Kineret 100 mg 0,67 ml
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU
KRABIČKA OBSAHUJÍCÍ 7 PŘEDPLNĚNÝCH INJEKČNÍCH STŘÍKAČEK (BEZ BLUE BOXU)_
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Kineret 100 mg/0,67 ml injekční roztok v předplněné injekční stříkačce Anakinrum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna předplněná injekční stříkačka se stupnicí o objemu 0,67 ml obsahuje 100 mg anakinry.
3. SEZNAM POMOCNÝCH LÁTEK
Pomocné látky: bezvodá kyselina citrónová, chlorid sodný, dihydrát dinatrium-edetátu, polysorbát 80, hydroxid sodný, voda na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Injekční roztok v předplněné injekční stříkačce 7 předplněných stříkaček SE STUPNICÍ
Toto balení obsahující 7 předplněných injekčních stříkaček je součástí balení s 28 ks.
5 ZPŮSOB A CESTA/CESTY PODÁNÍ
Jednorázové použití.
Subkutánní podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Swedish Orphan Biovitrum AB (publ)
SE-112 76 Stockholm
Švédsko 12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/02/203/007 13. ČÍSLO ŠARŽE
Č.š.
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.

1 NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Kineret 100 mg/0,67 ml injekce Anakinra
s.c.
2 ZPŮSOB PODÁNÍ
3 POUŽITELNOST
EXP
4. ČÍSLO SARZE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
0,67 ml
6. JINÉ
B. PŘÍBALOVÁ INFORMACE
Kineret 100 mg injekční roztok v předplněné injekční stříkačce
Anakinrum (anakinra)
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoliv další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci:
1. Co je Kineret a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete Kineret užívat
3. Jak se Kineret používá
4. Možné nežádoucí účinky
5. Jak Kineret uchovávat
6. Obsah balení a další informace
1. Co je Kineret a k čemu se používá
Léčivou látkou obsaženou v Kineretu je anakinra. Je to typ cytokinu (imunosupresivní látka), který se používá k léčbě revmatoidní artritidy. Cytokiny jsou bílkoviny produkované v lidském organismu, které koordinují vzájemné dorozumívání mezi buňkami a pomáhají usměrňovat aktivitu buněk. Při revmatoidní artritidě organismus tvoří nadměrné množství cytokinu, který se nazývá interleukin-1. To vede ke škodlivým účinkům, jako j sou otoky a poškození tkání. Za normálních okolností váš organismus produkuje bílkovinu, která škodlivý účinek interleukinu-1 blokuje. Léčivou látkou obsaženou v Kineretu je anakinra, která účinkuje stejně jako bílkovina přirozeně blokující interleukin-1. Anakinra je produkovaná DNA technologií za použití mikroorganismu E. coli.
Kineret se používá k léčbě známek a příznaků revmatoidní artritidy (RA) u dospělých pacientů (věk 18 let a více) v kombinaci s dalším lékem, který se nazývá methotrexat. Kineret se podává těm pacientům, jejichž odpověď na léčbu samotným methotrexatem ke zvládnutí revmatoidní artritidy nestačí.
2. Čemu musíte věnovat pozornost, než začnete Kineret užívat
Neužívejte Kineret:
- jestliže jste alergický(á) na anakinru nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
- jestliže jste alergický/á (přecitlivělý/á) na jiné přípravky vyrobené DNA technologií s použitím mikroorganismu E. coli.
- jestliže máte vážnou poruchu funkce ledvin (poškození ledvin).
- jestliže máte neutropenii (nízký počet leukocytů) stanovenou krevním testem.
Ihned se spojte se svým lékařem
- jestliže se po injekci Kineretu objeví vyrážka na celém těle, dýchavičnost, sípot při dýchání, zrychlení pulsu nebo pocení. Může se jednat o projevy přecitlivělosti na Kineret.
Upozornění a opatření
Poraďte se se svým lékařem:
- jestliže trpíte opakovanými infekcemi nebo pokud máte astma. Kineret může tato onemocnění zhoršit;
- jestliže máte zhoubné nádorové onemocnění. Váš lékař rozhodne, zda můžete Kineret užívat;
- jestliže máte v anamnéze zvýšené hladiny jaterních enzymů;
- jestliže potřebujete očkování. Během léčby Kineretem nesmíte být očkován(a) pomocí živých vakcín.
Děti a dospívající
Použití Kineretu u dětí ani dospívajících s revmatoidní artritidou nebylo dostatečně zhodnoceno, a proto jej nelze doporučit.
Další léčivé přípravky a přípravek Kineret
Informujte svého lékaře o všech lécích, které užíváte, které jste v nedávné době užíval/a nebo které možná budete užívat.
S Kineretem by se neměly používat léčivé přípravky zvané inhibitory faktoru způsobujícího nekrózu nádorů (TNF), například etanercept, protože se tím může zvyšovat riziko infekce.
Když začnete užívat Kineret, začne chronický zánět ve vašem těle ustupovat. To může vést k tomu, že bude potřeba upravit dávky některých dalších léčiv, např. warfarinu nebo fenytoinu.
Těhotenství a kojení
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem dříve, než začnete tento přípravek užívat.
Kineret nebyl testován u těhotných žen. Během těhotenství se nedoporučuje užívat Kineret, a pokud je žena v plodném věku, musí při užívání Kineretu užívat patřičnou antikoncepci.
Není známo, zda se anakinra vylučuje do lidského mateřského mléka. Pokud užíváte Kineret, musíte přestat kojit.
Kineret obsahuje sodík
Tento léčivý přípravek obsahuje méně než 1 mmol sodíku (23 mg) na 100 mg dávky, tj. v podstatě je bez sodíku.
3. Jak se Kineret používá
Vždy používejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem. Kineret se podává jednou denně v podkožní injekci (subkutánně). Je vhodné, abyste se vynasnažil(a) aplikovat injekci každý den ve stejnou dobu.
Podávání injekcí Kineretu pacientem samotným
Váš lékař může rozhodnout, že by pro Vás bylo nejvhodnější, abyste si sám (sama) podával(a) injekce Kineretu. Postup, jakým si sám (sama) budete injekce podávat, Vám ukáže lékař nebo sestra. Nepokoušejte si dát injekci sami, pokud jste k tomu nebyli vyškoleni.
Pokyny, jak si sám (sama) podávat injekce Kineretu, naleznete v odstavci na konci této příbalové informace.
Jestliže jste použil(a) více Kineretu, než jste měl(a):
Jestliže jste náhodně užil(a) více Kineretu, než potřebujete, nemělo by dojít k žádným závažným potížím. I přesto se, prosím, obraťte na svého lékaře, sestru nebo lékárníka, pokud k tomu dojde. V případě, že se v jakémkoliv ohledu nebudete cítit dobře, neprodleně vyhledejte svého lékaře nebo sestru.
Jestliže jste zapomněl(a) použít Kineret:
Jestliže jste si zapomněl(a) aplikovat dávku Kineretu, měl(a) byste se spojit se svým lékařem a dohodnout se, kdy byste si měl(a) vzít další dávku.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Dojde-li k výskytu kteréhokoli z následujících nežádoucích účinků, okamžitě informujte svého lékaře:
- Během léčby Kineretem se mohou objevit těžké infekce, jako je pneumonie (zápal plic) nebo infekce kůže. Příznaky mohou zahrnovat horečku a kašel nebo zarudnutí a citlivost kůže.
- Závažné alergické reakce jsou méně časté. Kterýkoli z následujících symptomů však může ukazovat na alergickou reakci na Kineret; proto objeví-li se u vás, okamžitě vyhledejte lékařskou pomoc. Přípravek Kineret dále neužívejte.
- Otok obličeje, j azyka nebo hrdla.
- Potíže s polykáním či dýcháním.
- Náhlý pocit rychlého tlukotu srdce či pocení.
- Svědění kůže či vyrážka.
Velmi časté nežádoucí účinky (mohou postihnout více než 1 člověka z 10):
- Zarudnutí, otok, podlitina nebo svědění v místě vpichu injekce. Tyto obtíže jsou obvykle mírné až střední intenzity a vyskytují se častěji na začátku léčby.
- Bolesti hlavy.
- Zvýšené celkové hladiny cholesterolu v krvi.
Časté nežádoucí účinky (mohou postihnout až 1 člověka z 10):
- Neutropenie (nízký počet bílých krvinek) zjištěná po vyšetření krve. Pokles počtu bílých krvinek může zvýšit riziko vzniku infekce. Příznaky infekce mohou zahrnovat horečku nebo bolest v krku.
- Těžké infekce, jako je například pneumonie (zápal plic) nebo infekce kůže.
- Trombocytopenie (nízký počet krevních destiček).
Méně časté nežádoucí účinky (mohou postihnout až 1 člověka ze 100):
- Vážné alergické reakce, včetně otoku obličeje, jazyka nebo hrdla, potíží při polykání či dýchání, náhlého pocitu rychlého tlukotu srdce či pocení, svědění kůže či vyrážky.
- Zvýšené hladiny jaterních enzymů stanovené po krevním testu.
Nežádoucí účinky s neznámou četností (četnost z dostupných údajů nešlo stanovit):
- Příznaky j aterních poruch, jako j sou žlutá kůže a oči, nevolnost, ztráta chuti k j ídlu, tmavě zbarvená moč a světle zbarvená stolice.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak Kineret uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti, uvedené na štítku a krabičce za Použitelné do. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v chladničce (2 °C - 8 °C). Chraňte před mrazem.
Uchovávejte v původní krabičce, aby byl přípravek chráněn před světlem.
Nepoužívejte Kineret, jestliže si myslíte, že mohlo dojít k jeho zmrznutí. Jestliže byla injekční stříkačka vyjmuta z chladničky a dosáhla pokojové teploty (do 25 °C), musí být buď použita do 12 hodin, nebo zlikvidována.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak máte naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co Kineret obsahuje
- Léčivou látkou je anakinra. Jedna předplněná injekční stříkačka obsahuje 100 mg anakinry.
- Pomocnými látkami jsou bezvodá kyselina citronová, chlorid sodný, dihydrát dinatrium-edetátu, polysorbát 80, hydroxid sodný a voda na injekci.
Jak Kineret vypadá a co obsahuje toto balení
Kineret je čirý, bezbarvý až bělavý injekční roztok a je dodáván v předplněných injekčních stříkačkách připravených pro použití. Může obsahovat průhledné až bílé částice bílkoviny. Přítomnost těchto částic neovlivňuje kvalitu léku.
Velikost balení 1, 7 nebo 28 (balení obsahující 4 balíčky po 7 předplněných stříkačkách) předplněných stříkaček.
Na trhu nemusí být všechny velikosti balení.
Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci a výrobce
Swedish Orphan Biovitrum AB (publ)
SE-112 76 Stockholm Švédsko
Tato příbalová informace byla naposledy revidována
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu
Pokyny k podávání injekce Kineretu pacientem samotným
Tento bod obsahuje pokyny, jak se postupuje při podávání injekce Kineretu pacientem samotným. Je důležité, abyste se nepokoušeli sami sobě podávat injekce, dokud Vám lékař, sestra nebo lékárník neposkytne odborný zácvik. Pokud máte jakékoliv další otázky, jak si podávat injekci, požádejte, prosím, o pomoc svého lékaře, sestru nebo lékárníka.
Jak použijete Vy nebo druhá osoba předplněnou stříkačku Kineretu?
Injekci je nutné aplikovat každý den ve stejnou dobu. Kineret se podává injekcí pod kůži. Tento způsob podání se označuje jako podkožní (subkutánní) injekce.
Vybavení:
k podání podkožní injekce budete potřebovat:
• novou předplněnou injekční stříkačku Kineretu;
• alkoholové tampóny nebo jim podobný prostředek.
Co máte udělat před podáním subkutánní injekce Kineretu?
1. Vyjměte předplněnou injekční stříkačku Kineretu z chladničky.
2. Předplněnou inj ekční stříkačku neprotřepávej te.
3. Zkontrolujte dobu použitelnosti na štítku předplněné injekční stříkačky (Použitelné do:). Nepoužívejte ji po uplynutí posledního dne uvedeného měsíce.
4. Zkontrolujte vzhled Kineretu. Musí jít o čirý, bezbarvý až bílý roztok. Roztok může obsahovat průhledné až bílé částice bílkoviny. Přítomnost těchto částic neovlivňuje kvalitu léku. Roztok se nesmí použít při změně barvy nebo při zákalu, nebo pokud obsahuje částice, které nejsou průhledné až bílé.
5. Aby byla injekce příjemnější, ponechte předplněnou injekční stříkačku po dobu 30 minut při pokojové teplotě nebo ji po dobu několika minut opatrně držte v ruce. Kineret neohřívejte žádným jiným způsobem (například jej neohřívejte v mikrovlnné troubě nebo v horké vodě).
6. Neodstraňujte kryt ze stříkačky dříve, než jste připraveni k injekci.
7. Důkladně si umyjte ruce.
8. Najděte si pohodlné, dobře osvětlené a čisté místo a vše, co potřebujete, si připravte na dosah. Jak budete postupovat při přípravě injekce Kineretu?
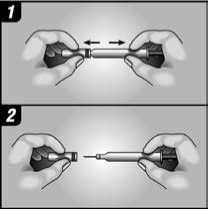
Před podáním injekce Kineretu musíte provést následující kroky:
1. Držte tělo stříkačky a j emně, bez kroucení, odstraňte kryt z j ehly.
Stahujte ho rovně, jak ukazuje obrázek 1 a 2. Nedotýkejte se jehly a nestlačujte píst.
2. V předplněné stříkačce mohou být malé bubliny vzduchu. Bubliny nemusíte před injekcí odstraňovat. Injekce roztoku s bublinami je neškodná.
3. Předplněnou injekční stříkačku můžete nyní použít.
Nejvhodnějšími místy k podávání injekcí jsou:
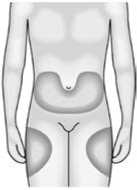
• horní část stehen a
• břicho, kromě oblasti v okolí pupku.
Pokaždé změňte místo podání injekce, abyste se vyhnuli vzniku bolestivosti v jedné oblasti. Pokud Vám injekci bude podávat někdo další, může použít také zadní stranu paží.
Jak budete postupovat při vpichu injekce?
1. Vydezinfikujte kůži pomocí alkoholového tampónu a uchopte záhyb kůže mezi palec a ukazováček, aniž byste ji tiskli silou.
2. Proveďte vpich jehlou do kůže tak, jak Vám to ukázala sestra nebo lékař.
3. Tekutinu injikujte pomalu a rovnoměrně; záhyb kůže stále držte.
4. Po dokončení injekce vyjměte jehlu z kůže a kůži uvolněte.
5. Jednu inj ekční stříkačku můžete použít pouze k jedné inj ekci.
Zapamatujte si
Pokud budete mít jakékoliv problémy, obraťte se bez obav s žádostí o radu či pomoc na svého lékaře nebo sestru.
Likvidace použitých injekčních stříkaček
• Nenasazujte kryt zpět na použité jehly.
• Použité injekční stříkačky uchovávejte mimo dosah a dohled dětí.
• Nikdy nevyhazujte použité předplněné injekční stříkačky do běžného domácího koše na odpadky.
• Použitá předplněná injekční stříkačka musí být zlikvidována v souladu s místními požadavky. Zeptejte se svého lékárníka, jak máte likvidovat přípravky, které již nepotřebujete. Tato opatření pomáhají chránit životní prostředí.
Kineret 100 mg/0,67 ml injekční roztok v předplněné injekční stříkačce
Anakinrum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoliv další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci:
1. Co je Kineret a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete Kineret užívat
3. Jak se Kineret používá
4. Možné nežádoucí účinky
5. Jak Kineret uchovávat
6. Obsah balení a další informace
1. Co je Kineret a k čemu se používá
Léčivou látkou obsaženou v Kineretu je anakinra. To je typ cytokinu (imunosupresivní látka), který se používá k léčbě:
• revmatoidní artritidy (RA)
• kryopyrin-asociovaných periodických syndromů (CAPS), které zahrnují následující autozánětlivá onemocnění:
- multisystémové zánětlivé onemocnění se začátkem v novorozeneckém věku (NOMID), také zvané chronický infantilní neurologický kožní a kloubní syndromu (CINCA),
- Muckle-Wellsův syndrom (MWS),
- familiární chladový autozánětlivý syndrom (FCAS)
Cytokiny jsou bílkoviny produkované v lidském organismu, které koordinují vzájemné dorozumívání mezi buňkami a pomáhají usměrňovat aktivitu buněk. Při revmatoidní artritidě a CAPS organismus tvoří nadměrné množství cytokinu, který se nazývá interleukin-1. To vede ke škodlivým účinkům, které způsobují zánět, a vyvolávají příznaky nemoci. Za normálních okolností váš organismus produkuje bílkovinu, která škodlivý účinek interleukinu-1 blokuje. Léčivou látkou obsaženou v Kineretu je anakinra, která účinkuje stejně jako bílkovina přirozeně blokující interleukin-1. Anakinra je produkovaná DNA technologií za použití mikroorganismu E. coli.
Kineret se používá k léčbě známek a příznaků revmatoidní artritidy (RA) u dospělých pacientů (věk 18 let a více) v kombinaci s dalším lékem, který se nazývá methotrexat. Kineret se podává těm pacientům, jejichž odpověď na léčbu samotným methotrexatem ke zvládnutí revmatoidní artritidy nestačí.
U kryopyrin-asociovaných periodických syndromů (CAPS) se Kineret používá k léčbě příznaků a známek zánětu spojených s onemocněním, jako je vyrážka. bolesti kloubů, horečka, bolest hlavy a únava u dospělých a dětí (ve věku 8 měsíců a starších).
2. Čemu musíte věnovat pozornost, než začnete Kineret užívat Neužívejte Kineret:
- jestliže j ste alergický(á) na anakinru nebo na kteroukoli další složku tohoto přípravku uvedenou v bodě 6;
- jestliže j ste alergický/á na jiné přípravky vyrobené DNA technologií s použitím mikroorganismu E. coli;
- jestliže máte vážnou poruchu funkce ledvin (poškození ledvin);
- jestliže máte neutropenii (nízký počet leukocytů) stanovenou krevním testem.
Ihned se spojte se svým lékařem
- jestliže se po injekci Kineretu objeví vyrážka na celém těle, dýchavičnost, sípot při dýchání, zrychlení pulsu nebo pocení. Může se jednat o projevy přecitlivělosti na Kineret.
Upozornění a opatření
Poraďte se se svým lékařem:
- jestliže trpíte opakovanými infekcemi nebo pokud máte astma. Kineret může tato onemocnění zhoršit;
- jestliže máte zhoubné nádorové onemocnění. Váš lékař rozhodne, zda můžete Kineret užívat;
- jestliže máte v anamnéze zvýšené hladiny jaterních enzymů;
- jestliže potřebujete očkování. Během léčby Kineretem nesmíte být očkován(a) pomocí živých vakcín.
Děti a dospívající
• RA: Použití Kineretu u dětí ani dospívajících s revmatoidní artritidou nebylo dostatečně zhodnoceno, a proto jej nelze doporučit.
• CAPS: Kineret se nedoporučuje pro děti mladší 8 měsíců, protože pro tuto skupinu nejsou žádné údaje.
Další léčivé přípravky a přípravek Kineret
Informujte svého lékaře o všech lécích, které užíváte, které jste v nedávné době užíval/a nebo které možná budete užívat.
S Kineretem by se neměly používat léčivé přípravky zvané inhibitory faktoru způsobujícího nekrózu nádorů (TNF), například etanercept, protože se tím může zvyšovat riziko infekce.
Když začnete užívat Kineret, začne chronický zánět ve vašem těle ustupovat. To může vést k tomu, že bude potřeba upravit dávky některých dalších léčiv, např. warfarinu nebo fenytoinu.
Těhotenství a kojení
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem dříve, než začnete tento přípravek užívat.
Kineret nebyl testován u těhotných žen. Během těhotenství se nedoporučuje užívat Kineret, a pokud je žena v plodném věku, musí při užívání Kineretu užívat patřičnou antikoncepci.
Není známo, zda se anakinra vylučuje do lidského mateřského mléka. Pokud užíváte Kineret, nesmíte kojit.
Kineret obsahuje sodík
Tento léčivý přípravek obsahuje méně než 1 mmol sodíku (23 mg) na 100 mg dávky, tj. v podstatě je bez sodíku.
Vždy používejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem. Kineret se podává denně v podkožní injekci (subkutánně). Je vhodné, abyste se vynasnažil(a) aplikovat injekci každý den ve stejnou dobu.
Doporučená dávka je buď 20 až 90 mg, nebo 100 mg. Váš lékař Vás bude informovat o dávce, kterou potřebujete, nebo zda potřebujete dávku vyšší než 100 mg.
Podávání injekcí Kineretu pacientem samotným
Váš lékař může rozhodnout, že by pro Vás bylo nejvhodnější, abyste si sám (sama) podával(a) injekce Kineretu. Postup, jakým si sám (sama) budete injekce podávat, Vám ukáže lékař nebo sestra. Nepokoušejte si dát injekci sami, pokud jste k tomu nebyli vyškoleni.
Pokyny, jak si sám (sama) podávat injekce Kineretu, naleznete v odstavci na konci této příbalové informace.
Pokyny, jak si sám (sama) nebo svému dítěti podávat injekce Kineretu, naleznete v odstavci „Pokyny pro přípravu a podávání injekce Kineretu“ na konci této příbalové informace.
Jestliže jste použil(a) více Kineretu, než jste měl(a):
Jestliže jste náhodně užil(a) více Kineretu, než potřebujete, nemělo by dojít k žádným závažným potížím. I přesto se, prosím, obraťte na svého lékaře, sestru nebo lékárníka, pokud k tomu dojde. V případě, že se v jakémkoliv ohledu nebudete cítit dobře, neprodleně vyhledejte svého lékaře nebo sestru.
Jestliže jste zapomněl(a) použít Kineret:
Jestliže jste si zapomněl(a) aplikovat dávku Kineretu, měl(a) byste se spojit se svým lékařem a dohodnout se, kdy byste si měl(a) vzít další dávku.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Bez ohledu na to, zda Kineretem léčíte RA nebo CAPS jsou možné nežádoucí účinky podobné. Dojde-li k výskytu kteréhokoli z následujících nežádoucích účinků, okamžitě informujte svého lékaře:
- Během léčby Kineretem se mohou objevit těžké infekce, jako je pneumonie (zápal plic) nebo infekce kůže. Příznaky mohou zahrnovat horečku a kašel nebo zarudnutí a citlivost kůže.
- Závažné alergické reakce jsou méně časté. Kterýkoli z následujících symptomů však může ukazovat na alergickou reakci na Kineret; proto objeví-li se u vás, okamžitě vyhledejte lékařskou pomoc. Přípravek Kineret dále neužívejte.
- Otok obličeje, j azyka nebo hrdla.
- Potíže s polykáním či dýcháním.
- Náhlý pocit rychlého tlukotu srdce či pocení.
- Svědění kůže či vyrážka.
Velmi časté nežádoucí účinky (mohou postihnout více než 1 člověka z 10):
- Zarudnutí, otok, podlitina nebo svědění v místě vpichu injekce. Tyto obtíže jsou obvykle mírné až střední intenzity a vyskytují se častěji na začátku léčby.
- Bolesti hlavy.
- Zvýšené celkové hladiny cholesterolu v krvi.
Časté nežádoucí účinky (mohou postihnout až 1 člověka z 10):
- Neutropenie (nízký počet bílých krvinek) zjištěná po vyšetření krve. Pokles počtu bílých krvinek může zvýšit riziko vzniku infekce. Příznaky infekce mohou zahrnovat horečku nebo bolest v krku.
- Těžké infekce, jako je například pneumonie (zápal plic) nebo infekce kůže.
- Trombocytopenie (nízký počet krevních destiček).
Méně časté nežádoucí účinky (mohou postihnout až 1 člověka ze 100):
- Vážné alergické reakce, včetně otoku obličeje, jazyka nebo hrdla, potíží při polykání či dýchání, náhlého pocitu rychlého tlukotu srdce či pocení, svědění kůže či vyrážky.
- Zvýšené hladiny jaterních enzymů stanovené po krevním testu.
Nežádoucí účinky s neznámou četností (četnost z dostupných údajů nešlo stanovit):
- Příznaky j aterních poruch, jako j sou žlutá kůže a oči, nevolnost, ztráta chuti k j ídlu, tmavě zbarvená moč a světle zbarvená stolice.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi.
Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové
informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení
nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k
získání více informací o bezpečnosti tohoto přípravku.
5. Jak Kineret uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti, uvedené na štítku a krabičce za Použitelné do. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v chladničce (2 °C až 8 °C). Chraňte před mrazem.
Uchovávejte v původní krabičce, aby byl přípravek chráněn před světlem.
Nepoužívejte Kineret, jestliže si myslíte, že mohlo dojít k jeho zmrznutí. Jestliže byla injekční stříkačka vyjmuta z chladničky a dosáhla pokojové teploty (do 25 °C), musí být buď použita do 12 hodin, nebo zlikvidována.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak máte naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co Kineret obsahuje
- Léčivou látkou je anakinrum. Jedna předplněná injekční stříkačka se stupnicí obsahuje anakinrum 100 mg.
- Dalšími složkami jsou kyselina citronová, chlorid sodný, dihydrát dinatrium-edetátu, polysorbát 80, hydroxid sodný a voda na injekci
Jak Kineret vypadá a co obsahuje toto balení
Kineret je čirý, bezbarvý až bělavý injekční roztok a je dodáván v předplněných injekčních stříkačkách připravených pro použití. Může obsahovat průhledné až bílé částice bílkoviny. Přítomnost těchto částic neovlivňuje kvalitu léku.
Držitel rozhodnutí o registraci a výrobce
Swedish Orphan Biovitrum AB (publ)
SE-112 76 Stockholm
Švédsko
Tato příbalová informace byla naposledy revidována
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu.
Tato část podává informace o tom, jak sobě nebo svému dítěti podávat injekce Kineretu. Je důležité, abyste se nepokoušeli sami sobě podávat injekce, dokud Vám lékař, sestra nebo lékárník neposkytne odborný zácvik. Pokud máte jakékoliv další otázky, jak si podávat injekci, požádejte, prosím, o pomoc svého lékaře, sestru nebo lékárníka.
Jak použijete Vy nebo druhá osoba předplněnou stříkačku Kineretu?
Je třeba, abyste si nebo svému dítěti aplikoval (aplikovala) injekci každý den ve stejnou dobu. Kineret se podává injekcí pod kůži. Tento způsob podání se označuje jako podkožní (subkutánní) injekce.
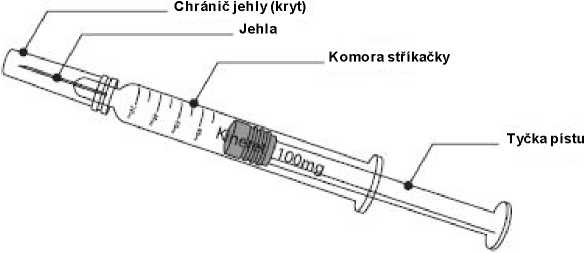
Vybavení:
K podání podkožní injekce sobě nebo svému dítěti budete potřebovat:
• předplněnou stříkačku Kineretu
• alkoholové tampóny nebo jim podobný prostředek a
• sterilní gázu nebo hadřík
Co máte udělat před podáním subkutánní injekce Kineretu sobě nebo svému dítěti?
1. Vyjměte předplněnou injekční stříkačku Kineretu z chladničky.
2. Předplněnou injekční stříkačku neprotřepávejte.
3. Zkontrolujte dobu použitelnosti na štítku předplněné injekční stříkačky (EXP:). Nepoužívejte ji po uplynutí posledního dne uvedeného měsíce.
4. Zkontrolujte vzhled Kineretu. Musí jít o čirý, bezbarvý až bílý roztok. Roztok může obsahovat průhledné až bílé částice bílkoviny. Přítomnost těchto částic neovlivňuje kvalitu léku. Roztok se nesmí použít při změně barvy nebo při zákalu, nebo pokud obsahuje částice, které nejsou průhledné až bílé.
5. Aby byla injekce příjemnější, ponechte předplněnou injekční stříkačku po dobu 30 minut při pokojové teplotě nebo ji po dobu několika minut opatrně držte v ruce. Kineret neohřívejte žádným jiným způsobem (například jej neohřívejte v mikrovlnné troubě nebo v horké vodě).
6. Neodstraňujte kryt ze stříkačky dříve, než jste připraveni k injekci.
7. Důkladně si umyjte ruce.
8. Najděte si pohodlné, dobře osvětlené a čisté místo a vše, co potřebujete, si připravte na dosah.
9. Ujistěte se, že víte, jakou dávku Kineretu vám lékař předepsal; 20 až 90 mg, 100 mg nebo vyšší.
• Pokud Váš lékař předepsal dávku 100 mg, přejděte k odstavci „Jak připravit dávku 100 mg“.
• Pokud Váš lékař předepsal nižší dávku, přejděte k odstavci „Jak připravit dávku 20 až
90 mg“.
Jak připravit dávku 100 mg
Před podáním injekce Kineretu musíte provést následující kroky:
Chránič jehly (kryt)
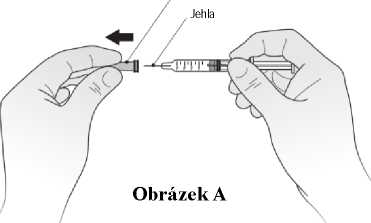
1. Držte komoru stříkačky a opatrně bez otáčení odstraňte kryt z jehly. Táhněte přímo, jak je vidět na obrázku A. Nedotýkejte se jehly a nestlačujte píst. Kryt jehly okamžitě zlikvidujte.
2. V předplněné stříkačce mohou být malé bubliny vzduchu. Bubliny nemusíte před injekcí odstraňovat. Injekce roztoku s bublinami je neškodná.
3. Předplněnou injekční stříkačku můžete nyní použít podle popisu v částech „Kam si budete injekce podávat?“ a „Jak budete postupovat při vpichu injekce?“
Jak připravit dávku 20 až 90 mg
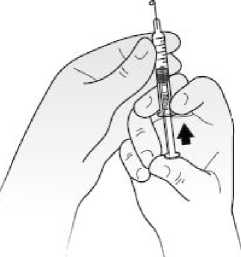
Před podáním injekce Kineretu musíte provést následující kroky:
1. Držte komoru stříkačky a opatrně bez otáčení odstraňte kryt z jehly. Táhněte přímo, jak je vidět na obrázku A. Nedotýkejte se jehly a nestlačujte píst. Kryt jehly okamžitě zlikvidujte.
2. Vezměte stříkačku jednou rukou s jehlou směřující přímo vzhůru, jak je znázorněno na obrázku B. Položte palec na píst a pomalu tlačte, dokud neuvidíte na špičce jehly kapičku tekutiny.
Obrázek C
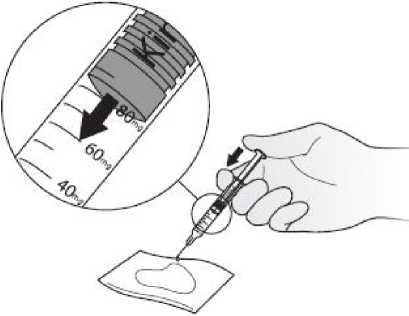
3. Otočte stříkačku tak, aby jehla nyní směřovala dolů. Dejte sterilní gázu nebo hadřík na rovnou plochu. Držte nad ním jehlu tak, aby směřovala ke gáze nebo hadříku, jak je vidět na obrázku C. Ujistěte se, že se jehla nedotýká gázy nebo hadříku.
4. Dejte palec na píst a pomalu tlačte, dokud přední část pístu nedosáhne značky stupnice s vaší dávkou Kineretu. (Váš doktor Vám řekne, jakou dávku budete užívat.) Vypuštěné množství kapaliny se vsákne do gázy nebo hadříku, jak je vidět na obrázku C.
5. Pokud se Vám nepovedlo nastavit správnou dávku, zlikvidujte stříkačku a vezměte si novou.
6. Předplněnou injekční stříkačku můžete nyní použít podle popisu v částech „Kam si budete injekce podávat?“ a „Jak budete postupovat při vpichu injekce?“.
Kam si budete injekce podávat?
Nejvhodnějšími místy k podávání injekcí Vám nebo Vašemu dítěti (viz obrázek D) jsou:
• břicho (kromě oblasti v okolí pupku)
• horní část stehen
• horní vnější oblast hýždí a
• vnější oblast horní části paží
Dítě
Dospělý
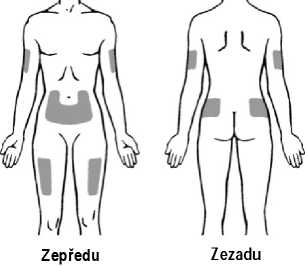
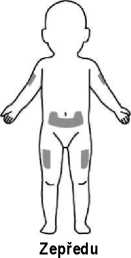
Pokaždé změňte místo podání injekce, abyste se vyhnuli vzniku bolestivosti v jedné oblasti. Pokud Vám injekci bude podávat někdo další, může použít také zadní stranu paží.
Jak budete postupovat při vpichu injekce?
1. Alkoholovým tampónem vydezinfikujte kůži a uchopte záhyb kůže mezi palec a ukazováček, aniž byste ji tiskli silou.
2. Proveďte vpich jehlou do kůže tak, jak Vám to ukázala sestra nebo lékař.
3. Tekutinu injikujte pomalu a rovnoměrně; záhyb kůže stále držte jako na obrázku E.
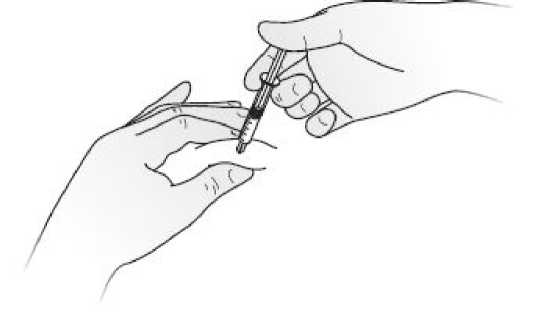
4. Po dokončení injekce vyjměte jehlu z kůže a kůži uvolněte.
5. Veškeré nepoužité léčivo se musí zlikvidovat. Jednu stříkačku použijte pouze k jedné injekci. Nepoužívejte stříkačku opakovaně, může to způsobit infekci.
Zapamatujte si
Pokud budete mít jakékoliv problémy, obraťte se bez obav s žádostí o radu či pomoc na svého lékaře nebo sestru.
u
Likvidace použitých stříkaček a dalšího materiál
• Nenasazujte kryt zpět na použité jehly.
• Použité injekční stříkačky uchovávejte mimo dosah a dohled dětí.
• Nikdy nevyhazujte použité předplněné injekční stříkačky do běžného domácího koše na odpadky.
• Pokud máte dávku nižší než 100 mg, bude vám řečeno, jak vypustit kapalinu ze stříkačky na gázu nebo hadřík. Po aplikaci injekce zlikvidujte vlhkou gázu nebo hadřík se svou stříkačkou a očistěte povrch čistým hadříkem.
• Použitá předplněná stříkačka a veškerá gáza nebo hadřík s roztokem Kineretu se musí zlikvidovat v souladu s místními předpisy. Zeptejte se svého lékárníka, jak máte likvidovat přípravky, které již nepotřebujete. Tato opatření pomáhají chránit životní prostředí.
54