Keytruda 50 Mg
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
KEYTRUDA 50 mg prášek pro koncentrát pro infuzní roztok.
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička s práškem obsahuje pembrolizumabum 50 mg.
Po rekonstituci obsahuje 1 ml koncentrátu pembrolizumabum 25 mg.
Pembrolizumab je humanizovaná monoklonální protilátka proti receptoru programované buněčné smrti PD-1 (programmed cell death-1) (izotyp IgG4/K se stabilizující změnou sekvence v Fc oblasti) vytvářená technologií rekombinantní DNA v buňkách vaječníků čínského křečíka.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek pro koncentrát pro infuzní roztok. Bílý až téměř bílý lyofilizovaný prášek.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek KEYTRUDA je v monoterapii indikován k léčbě pokročilého (neresekovatelného nebo metastazujícího) melanomu u dospělých.
Přípravek KEYTRUDA je indikován k léčbě lokálně pokročilého nebo metastazujícího nemalobuněčného karcinomu plic (NSCLC) u dospělých, jejichž nádory exprimují PD-L1 a kteří již byli léčeni nejméně jedním chemoterapeutickým režimem. Pacienti s pozitivními nádorovými mutacemi EGFR nebo ALK musí předtím, než dostanou přípravek KEYTRUDA, být rovněž léčeni terapií schválenou při těchto mutacích.
4.2 Dávkování a způsob podání
Léčbu musí zahájit a musí na ni dohlížet lékař specialista se zkušenostmi v onkologické léčbě. Testování na PD-L1 u pacientů s NSCLC
Pacienti s NSCLC musí být k léčbě vybráni na základě exprese PD-L1 nádorovými buňkami potvrzené validovaným testem (viz bod 5.1).
Dávkování
Doporučená dávka přípravku KEYTRUDA je 2 mg/kg podávaná intravenózně po dobu 30 minut každé 3 týdny. Pacienty je nutno přípravkem KEYTRUDA léčit do progrese nemoci nebo do vzniku nepřijatelné toxicity. Byly pozorovány atypické odpovědi (tj. počáteční přechodné zvětšení nádoru nebo vznik nových malých lézí během prvních několika měsíců, následované zmenšením nádoru).
Klinicky stabilní pacienty s počátečními známkami progrese nemoci se doporučuje léčit dál, dokud se prog rese nepotvrdí.
Odložení dávky nebo vysazení přípravku (viz také bod 4.4)
Tabulka 1: Doporučené úpravy léčby přípravkem KEYTRUDA
|
Imunitně zprostředkované nežádoucí účinky |
Závažnost |
Úprava léčby |
|
Pneumonitida |
Stupeň 2 |
Dočasně vysadit* |
|
Stupeň 3 nebo 4, nebo recidivující stupeň 2 |
Trvale vysadit | |
|
Kolitida |
Stupeň 2 nebo 3 |
Dočasně vysadit* |
|
Stupeň 4 |
Trvale vysadit | |
|
Nefritida |
Stupeň 2 s kreatininem >1,5 až < 3násobek horní hranice normálu (ULN) |
Dočasně vysadit* |
|
Stupeň > 3 s kreatininem > 3násobku ULN |
Trvale vysadit | |
|
Endokrinopatie |
Symptomatická hypofyzitida Diabetes typu 1 spojený s hyperglykemií stupně > 3 (glukóza >250 mg/dl nebo >13,9 mmol/l) nebo spojený s ketoacidózou Hypertyreóza stupně > 3 |
Dočasně vysadit* U pacientů s endokrinopatií stupně 3 nebo stupně 4, která se zlepšila na stupeň 2 nebo nižší a je kontrolována pomocí hormonální substituce, je možno, pokud je indikováno, zvážit v případě potřeby pokračování podávání pembrolizumabu následně po postupném vysazení kortikosteroidů. Jinak má být léčba ukončena. Hypotyreózu lze zvládnout substituční terapií bez přerušení léčby. |
|
Hepatitida |
Stupeň 2 s aspartátaminotransferázou (AST) nebo alaninaminotransferázou (ALT) >3 až 5násobek ULN nebo celkový bilirubin >1,5 až 3násobek ULN |
Dočasně vysadit* |
|
Stupeň > 3 s AST nebo ALT > 5násobek ULN nebo celkovým bilirubinem > 3násobek ULN |
Trvale vysadit | |
|
V případě jaterních metastáz s výchozí zvýšenou hodnotou AST nebo ALT stupně 2, hepatitida, kdy AST nebo ALT stoupne o > 50 % a trvá > 1 týden |
Trvale vysadit | |
|
Reakce spojené s infuzí |
Stupeň 3 nebo 4 |
Trvale vysadit |
Poznámka: stupně toxicity jsou v souladu s National Cancer Institute Common Terminology Criteria for Adverse Events Version 4.0 (NCI-CTCAE v.4).
* než se nežádoucí účinky zlepší na stupeň 0 - 1.
Přípravek KEYTRUDA je nutno trvale vysadit:
• při toxicitě stupně 4 kromě endokrinopatií, které jsou zvládnuty substitučními hormony
• pokud během 12 týdnů nelze snížit dávku kortikosteroidů na <10 mg prednisonu nebo jeho ekvivalentu za den
• pokud se toxicita související s léčbou během 12 týdnů po poslední dávce přípravku KEYTRUDA nesníží na stupeň 0 - 1
• pokud se podruhé objeví jakákoli příhoda závažnosti stupně > 3.
Pacientům léčeným přípravkem KEYTRUDA musí být poskytnuta karta s upozorněním pro pacienta a mají být informováni o rizicích přípravku KEYTRUDA (viz také příbalová informace).
Zvláštní populace Starší osoby
Mezi staršími (> 65 let) a mladšími pacienty (< 65 let) nebyly hlášeny žádné rozdíly týkající se bezpečnosti a účinnosti. U této populace není úprava dávkování nutná.
Porucha funkce ledvin
U pacientů s lehkou nebo středně těžkou poruchou funkce ledvin není úprava dávkování nutná. Přípravek KEYTRUDA nebyl hodnocen u pacientů s těžkou poruchou funkce ledvin (viz bod 5.2).
Porucha funkce jater
U pacientů s lehkou poruchou funkce jater není úprava dávkování nutná. Přípravek KEYTRUDA nebyl hodnocen u pacientů se středně těžkou nebo těžkou poruchou funkce jater (viz bod 5.2).
Oční melanom
O účinnosti a bezpečnosti přípravku KEYTRUDA u pacientů s očním melanomem jsou k dispozici jen omezené údaje (viz bod 5.1).
Pediatrická populace
Bezpečnost a účinnost přípravku KEYTRUDA u dětí ve věku do 18 let nebyla dosud stanovena. Nejsou dostupné žádné údaje.
Způsob podání
Přípravek KEYTRUDA se musí podávat intravenózní infuzí trvající 30 minut. Přípravek KEYTRUDA se nesmí podávat jako nitrožilní bolus nebo bolusová injekce.
Návod k rekonstituci a naředění léčivého přípravku před jeho podáním je uveden v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití Vyhodnocení stavu PD-L1
Při hodnocení stavu tumoru s ohledem na PD-L1 je důležité zvolit dobře validovanou a robustní metodiku, aby se minimalizovalo riziko falešně negativních nebo falešně pozitivních stanovení.
Imunitně zprostředkované nežádoucí účinky
Většina imunitně zprostředkovaných nežádoucích účinků, které se vyskytly během léčby pembrolizumabem, byla reverzibilní a zvládla se přerušením podávání pembrolizumabu, podáním kortikosteroidů a/nebo podpůrnou léčbou. Imunitně zprostředkované nežádoucí účinky se vyskytly také po podání poslední dávky pembrolizumabu.
Při podezření na imunitně zprostředkované nežádoucí účinky má být zajištěno odpovídající vyšetření, aby se etiologie potvrdila, nebo aby se vyloučily jiné příčiny. Na základě závažnosti nežádoucího účinku má být pembrolizumab vysazen a podávány kortikosteroidy. Po zlepšení na stupeň < 1 má být zahájeno postupné vysazování kortikosteroidů a ve vysazování se má pokračovat nejméně 1 měsíc. Na základě omezených údajů z klinických studií, u pacientů, jejichž imunitně zprostředkované nežádoucí účinky nemohly být kontrolovány použitím kortikosteroidů, může být zváženo podávání jiných systémových imunosupresiv.
Pembrolizumab může být znovu nasazen po 12 týdnech po poslední dávce přípravku KEYTRUDA, pokud nežádoucí účinek zůstává na stupni <1 a dávka kortikosteroidů byla redukována na < 10 mg prednisonu nebo jeho ekvivalentu za den.
Pembrolizumab musí být trvale vysazen při jakémkoli imunitně zprostředkovaném nežádoucím účinku stupně 3, který se opakuje nebo při jakékoli imunitně zprostředkované nežádoucí toxicitě stupně 4, kromě endokrinopatií, které jsou zvládnuty substitučními hormony (viz body 4.2 a 4.8).
Imunitně zprostředkovaná pneumonitida
U pacientů léčených pembrolizumabem byla hlášena pneumonitida, včetně fatálních případů (viz bod 4.8). Pacienti mají být sledováni na projevy a příznaky pneumonitidy. Podezření na pneumonitidu má být potvrzeno radiografickou zobrazovací metodou a mají být vyloučeny jiné příčiny. Při příhodách stupně > 2 mají být podávány kortikosteroidy (zahajovací dávka 1 - 2 mg/kg/den prednisonu nebo jeho ekvivalentu následovaná postupným vysazováním); při pneumonitidě stupně 2 má být pembrolizumab dočasně vysazen a při pneumonitidě stupně 3, stupně 4 nebo recidivující pneumonitidě stupně 2 má být pembrolizumab vysazen natrvalo (viz bod 4.2).
Imunitně zprostředkovaná kolitida
U pacientů léčených pembrolizumabem byla hlášena kolitida (viz bod 4.8). Pacienti mají být sledováni na projevy a příznaky kolitidy a mají být vyloučeny jiné příčiny. Při příhodách stupně > 2 mají být podávány kortikosteroidy (zahajovací dávka 1 - 2 mg/kg/den prednisonu nebo jeho ekvivalentu následovaná postupným vysazováním); při kolitidě stupně 2 nebo stupně 3 má být pembrolizumab dočasně vysazen a při kolitidě stupně 4 vysazen natrvalo (viz bod 4.2). Je nutno vzít v úvahu potenciální riziko vzniku gastrointestinální perforace.
Imunitně zprostředkovaná hepatitida
U pacientů léčených pembrolizumabem byla hlášena hepatitida (viz bod 4.8). Pacienti mají být sledováni na změny jaterních funkcí (na začátku léčby, pravidelně během léčby a dle klinického stavu) a příznaky hepatitidy a mají být vyloučeny jiné příčiny. Mají být podávány kortikosteroidy (zahajovací dávka 0,5 - 1 mg/kg/den (u příhod stupně 2) a 1 - 2 mg/kg/den (u příhod stupně > 3) prednisonu nebo jeho ekvivalentu následovaná postupným vysazováním) a na základě závažnosti zvýšení jaterních enzymů má být pembrolizumab dočasně nebo natrvalo vysazen (viz bod 4.2).
Imunitně zprostředkovaná nefritida
U pacientů léčených pembrolizumabem byla hlášena nefritida (viz bod 4.8). Pacienti mají být sledováni na změny renální funkce a mají být vyloučeny jiné příčiny renální dysfunkce. U příhod stupně > 2 mají být podávány kortikosteroidy (zahajovací dávka 1 - 2 mg/kg/den prednisonu nebo jeho ekvivalentu následovaná postupným vysazováním) a na základě závažnosti zvýšení kreatininu má být pembrolizumab při nefritidě stupně 2 dočasně vysazen a při nefritidě stupně 3 nebo 4 vysazen natrvalo (viz bod 4.2).
Imunitně zprostředkované endokrinopatie
Při léčbě pembrolizumabem byly pozorovány těžké endokrinopatie, včetně hypofyzitidy, diabetes mellitus typu 1, diabetické ketoacidózy, hypotyreózy a hypertyreózy.
V případech imunitně zprostředkovaných endokrinopatií může být nezbytná dlouhodobá substituční hormonální léčba.
U pacientů léčených pembrolizumabem byla hlášena hypofyzitida (viz bod 4.8). Pacienti mají být sledováni na projevy a příznaky hypofyzitidy (včetně hypopituitarismu a sekundární nedostatečnosti nadledvin) a mají být vyloučeny jiné příčiny. K léčbě sekundární nedostatečnosti nadledvin mají být podávány kortikosteroidy a další hormonální substituce podle klinické indikace, při symptomatické hypofyzitidě má být pembrolizumab dočasně vysazen dokud se příhoda nedostane pod kontrolu pomocí hormonální substituce. Pokud je potřeba, o pokračování v léčbě pembrolizumabem lze uvažovat po postupném vysazení kortikosteroidů (viz bod 4.2). Je nezbytné monitorovat funkci hypofýzy a hladiny hormonů, aby byla zajištěna vhodná hormonální substituce.
U pacientů léčených pembrolizumabem byl hlášen diabetes mellitus typu 1, včetně diabetické ketoacidózy (viz bod 4.8). Pacienti mají být sledováni na hyperglykemii nebo jiné projevy a příznaky diabetu. U diabetu typu 1 má být podáván inzulin a v případech hyperglykemie stupně 3 má být pembrolizumab dočasně vysazen dokud se metabolická situace nedostane pod kontrolu (viz bod 4.2).
U pacientů léčených pembrolizumabem byly hlášeny poruchy štítné žlázy, zahrnující hypotyreózu, hypertyreózu a tyreoiditidu, které se mohou objevit kdykoli během léčby; proto pacienti mají být sledováni na změny funkce štítné žlázy (na začátku léčby, pravidelně během léčby a dle klinického stavu) a na klinické projevy a příznaky poruch štítné žlázy. Hypotyreózu lze zvládnout substituční terapií bez přerušení léčby a bez kortikosteroidů. Hypertyreózu lze zvládnout symptomaticky. Při hypertyreóze stupně > 3 má být pembrolizumab dočasně vysazen dokud není znovu dosaženo stupně < 1. U pacientů s hypertyreózou stupně 3 nebo stupně 4, která se zlepšila na stupeň 2 nebo nižší, lze podle potřeby o pokračování v léčbě pembrolizumabem uvažovat po postupném vysazení kortikosteroidů (viz body 4.2 a 4.8). Je nezbytné monitorovat funkci štítné žlázy a hladiny hormonů, aby byla zajištěna vhodná hormonální substituce.
Další imunitně zprostředkované nežádoucí účinky
U pacientů léčených pembrolizumabem byly hlášeny následující další klinicky významné imunitně zprostředkované nežádoucí účinky: uveitida, artritida, myozitida, pankreatitida, těžké kožní reakce, Guillain-Barrého syndrom, myastenický syndrom, hemolytická anémie a parciální záchvaty vznikající u pacienta se zánětlivými ložisky v mozkovém parenchymu (viz bod 4.8).
Na základě závažnosti nežádoucího účinku má být pembrolizumab vysazen a podávány kortikosteroidy.
Pembrolizumab musí být znovu nasazen po 12 týdnech po poslední dávce přípravku KEYTRUDA, pokud nežádoucí účinek zůstává na stupni <1 a dávka kortikosteroidů byla redukována na < 10 mg prednisonu nebo jeho ekvivalentu za den.
Pembrolizumab má být trvale vysazen při jakémkoli imunitně zprostředkovaném nežádoucím účinku stupně 3, který se opakuje nebo při jakékoli imunitně zprostředkované nežádoucí toxicitě stupně 4 (viz body 4.2 a 4.8).
Reakce spojené s infuzí
U pacientů léčených pembrolizumabem byly hlášeny těžké reakce spojené s infuzí (viz bod 4.8). Při těžkých reakcích spojených s infuzí má být infuze ukončena a pembrolizumab natrvalo vysazen (viz bod 4.2). Pacienti s lehkou nebo středně těžkou reakcí spojenou s infuzí mohou pod pečlivým dohledem pembrolizumab nadále používat; lze uvažovat o premedikaci antipyretiky a antihistaminiky.
Pacienti vyloučení z klinických studií
Do klinických studií nebyli zařazeni pacienti s následujícími stavy: aktivní metastázy v CNS; HIV, infekce virem hepatitidy B nebo hepatitidy C; aktivní systémová autoimunitní nemoc; intersticiální plicní onemocnění; prodělaná pneumonitida vyžadující systémovou léčbu kortikosteroidy; těžká hypersenzitivita k jiné monoklonální protilátce v anamnéze; užívání imunosupresivní léčby; těžké imunitně zprostředkované nežádoucí účinky léčby ipilimumabem v anamnéze, definované jako jakákoli toxicita stupně 4 nebo toxicita stupně 3 vyžadující léčbu kortikosteroidy (> 10 mg/den prednisonu nebo jeho ekvivalentu) po dobu delší než 12 týdnů. Pacienti s aktivními infekcemi byli z klinických studií vyloučeni a předtím, než začali užívat pembrolizumab, se požadovalo, aby jejich infekce byla vyléčena. Pacienti s aktivními infekcemi vyskytujícími se během léčby pembrolizumabem byli příslušně léčeni. Pacienti s počátečními klinicky významnými renálními (kreatinin > 1,5 x ULN) nebo jaterními (bilirubin > 1,5 x ULN, ALT, AST > 2,5 x ULN při absenci jaterních metastáz) abnormalitami byli vyloučeni z klinických studií, proto jsou informace o pacientech s těžkou poruchou funkce ledvin a středně težkou až těžkou poruchou funkce jater omezeny.
U těchto pacientů lze pembrolizumab používat po pečlivém zvážení potenciálního zvýšeného rizika a za vhodné léčebné strategie.
Karta s upozorněním pro pacienta
Každý, kdo předepisuje přípravek KEYTRUDA musí být seznámen s informací pro lékaře a léčebnými strategiemi. Předepisující lékař musí s pacientem prodiskutovat rizika léčby přípravkem KEYTRUDA. Pacientovi bude poskytnuta karta s upozorněním pro pacienta s každým receptem.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
S pembrolizumabem nebyly provedeny žádné formální farmakokinetické studie lékových interakcí. Jelikož se pembrolizumab odstraňuje z oběhu katabolizací, žádné metabolické lékové interakce se nepředpokládají.
Před nasazením pembrolizumabu je nutno se vyhnout podávání systémových kortikosteroidů nebo imunosupresiv, a to kvůli jejich potenciálnímu vlivu na farmakodynamickou aktivitu a účinnost pembrolizumabu. Systémové kortikosteroidy nebo jiná imunosupresiva však lze používat po nasazení pembrolizumabu k léčbě imunitně zprostředkovaných nežádoucích účinků (viz bod 4.4)
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku.
Ženy ve fertilním věku musí během léčby pembrolizumabem a ještě nejméně 4 měsíce po poslední dávce pembrolizumabu používat účinnou antikoncepci.
Údaje o podávání pembrolizumabu těhotným ženám nejsou k dispozici. Reprodukční studie na zvířatech nebyly s pembrolizumabem provedeny; nicméně na modelech březích myší bylo prokázáno, že blokáda signálu zprostředkovaného receptorem PD-L1 narušuje toleranci vůči plodu a vede ke zvýšeným ztrátám plodů (viz bod 5.3). Tyto výsledky naznačují potenciální riziko, že by podávání pembrolizumabu během těhotenství mohlo způsobit poškození plodu, včetně zvýšené míry potratu nebo porodu mrtvého plodu. Je známo, že lidské imunoglobuliny G4 (IgG4) prostupují placentární bariérou; proto má pembrolizumab, který je IgG4, potenciál ktomu, aby byl přenesen z matky do vyvíjejícího se plodu. Pembrolizumab se v těhotenství nemá používat, ledaže by klinický stav ženy léčbu pembrolizumabem vyžadoval.
Kojení
Není známo, zda se pembrolizumab vylučuje do lidského mateřského mléka. Jelikož je ale známo, že protilátky mohou být vylučovány do lidského mateřského mléka, riziko pro novorozence/kojence nelze vyloučit. Je nutno se rozhodnout, zda přerušit kojení nebo vysadit pembrolizumab, přičemž se posoudí přínos kojení pro dítě a přínos léčby pembrolizumabem pro ženu.
Fertilita
O možném vlivu pembrolizumabu na fertilitu nejsou k dispozici žádné klinické údaje. Na základě jednoměsíční a šestiměsíční studie toxicity po opakovaných dávkách u opic nebyly zjištěny žádné pozorovatelné účinky na samčí ani samičí reprodukční orgány (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Pembrolizumab může mít mírný vliv na schopnost řídit a obsluhovat stroje. Po podání pembrolizumabu byla hlášena únava (viz bod 4.8).
Souhrn bezpečnostního profilu
Pembrolizumab je nejčastěji spojován s imunitně zprostředkovanými nežádoucími účinky. Většina z nich, včetně těžkých reakcí, byla vyřešena po zahájení příslušné léčby nebo vysazení pembrolizumabu (viz „Popis vybraných nežádoucích účinků“ níže).
Bezpečnost pembrolizumabu byla v klinických studiích hodnocena u 2799 pacientů s pokročilým melanomem nebo NSCLC ve třech dávkách (2 mg/kg každé 3 týdny nebo 10 mg/kg každé 2 nebo 3 týdny). U této populace pacientů byly nejčastějšími nežádoucími účinky (> 10 %) pembrolizumabu únava (24 %), vyrážka (19 %), pruritus (18 %), průjem (12 %), nauzea (11%) a artralgie (10 %). Většina hlášených nežádoucích účinků byla stupně závažnosti 1 nebo 2. Nejzávažnějšími nežádoucími účinky byly imunitně zprostředkované nežádoucí účinky a závažné nežádoucí účinky spojené s infuzí (viz bod 4.4).
Tabulkový seznam nežádoucích účinků
V tabulce 2 jsou hlášeny nežádoucí účinky hlášené u 2799 pacientů léčených pembrolizumabem v klinických studiích. Tyto nežádoucí účinky jsou uvedeny podle třídy orgánových systémů a četnosti. Četnosti jsou definovány následovně: velmi časté (> 1/10); časté (> 1/100 až < 1/10); méně časté (>
1/1 000 až < 1/100); vzácné (> 1/10 000 až < 1/1 000); velmi vzácné (< 1/10 000). V rámci každé skupiny četnosti jsou nežádoucí účinky seřazeny v pořadí klesající závažnosti.
Tabulka 2: Nežádoucí účinky u pacientů léčených pembrolizumabem v klinických studiích
|
Poruchy krve a lymfatického systému | |
|
Časté | |
|
Méně časté |
neutropenie, leukopenie, trombocytopenie, lymfopenie, eozinofilie |
|
Vzácné |
imunitní trombocytopenická purpura, hemolytická anémie |
|
Poruchy imunitního systému | |
|
Časté |
reakce spojená s infuzía |
|
Endokrinní poruchy | |
|
Časté |
hypertyreóza, hypotyreózab |
|
Méně časté |
hypofyzitidac, adrenální insuficience, tyreoiditida |
|
Poruchy metabolismu a výživy | |
|
Časté |
snížená chuť k jídlu |
|
Méně časté |
diabetes mellitus typu 1d, hyponatremie, hypokalemie, hypokalcemie |
|
Psychiatrické poruchy | |
|
Méně časté |
Insomnie |
|
Poruchy nervového systému | |
|
Časté |
bolest hlavy, závrať, dysgeuzie, |
|
Méně časté |
epilepsie, letargie, periferní neuropatie |
|
Vzácné |
Guillain-Barrého syndrom, myastenický syndrom |
|
Poruchy oka | |
|
Časté |
suché oko |
|
Méně časté |
uveitidae |
|
Cévní poruchy | |
|
Méně časté |
Hypertenze |
|
Respirační, hrudní a mediastinální poruchy | |
|
Časté | |
|
Gastrointestinální poruchy | |
|
Velmi časté | |
|
Časté |
kolitidag, zvracení, břišní bolesth, zácpa, suchá ústa |
|
Méně časté |
pankreatitidai |
|
Vzácné |
perforace tenkého střeva |
|
Poruchy jater a žlučových cest | |
|
Méně časté |
hepatitidaJ |
cytokinů)
|
Poruchy kůže a podkožní tkáně | |
|
Velmi časté |
vyrážkak, pruritus* |
|
Časté |
těžké kožní reakcem, vitiligon, akneiformní dermatitida, suchá kůže, erytém, ekzém |
|
Méně časté |
lišejová keratózao, psoriáza, alopecie, erythema nodosum, dermatitida, změny barvy vlasů, papula |
|
Poruchy svalové a kosterní |
soustavy a pojivové tkáně |
|
Velmi časté |
Artralgie |
|
Časté |
myozitidap, muskuloskeletální bolestq, bolest v končetině, artritida1 |
|
Méně časté |
tenosynovitidas |
|
Poruchy ledvin a močových cest | |
|
Méně časté |
nefritidat |
|
Celkové poruchy a reakce v místě aplikace | |
|
Velmi časté |
Únava |
|
Časté |
astenie, edému, pyrexie, onemocnění podobající se chřipce, třesavka |
|
Vyšetření | |
|
Časté |
zvýšená alaninaminotransferáza, zvýšená aspartátaminotransferáza, , zvýšená alkalická fosfatáza v krvi, zvýšený kreatinin v krvi |
|
Méně časté |
zvýšená amyláza, zvýšený bilirubin v krvi, hyperkalcemie |
“Následující pojmy představují skupinu příbuzných příhod, které spíše než jednotlivou příhodu popisují zdravotní stav. a. reakce spojené s infuzí (přecitlivělost na lék, anafylaktická reakce, hypersenzitivita a syndrom uvolňování
b. hypotyreóza (myxedém)
c. hypofyzitida (hypopituitarismus)
d. diabetes mellitus typu 1 (diabetická ketoacidóza)
e. uveitida (iritida a iridocyklitida)
f. pneumonitida (intersticiální plicní nemoc)
g. kolitida (mikroskopická kolitida a enterokolitida)
h. bolest břicha (břišní diskomfort, bolest horní poloviny břicha, bolest dolní poloviny břicha)
i. pankreatitida (autoimunitní pankreatitida a akutní pankreatitida)
j. hepatitida (autoimunitní hepatitida a lékové poškození jater)
k. vyrážka (erytematózní vyrážka, folikulární vyrážka, generalizovaná vyrážka, makulózní vyrážka, makulopapulózní vyrážka, papulózní vyrážka, svědící vyrážka, vezikulózní vyrážka a genitální vyrážka)
l. pruritus (kopřivka, papulózní kopřivka, generalizovaný pruritus a genitální pruritus)
m. závažné kožní reakce (exfoliativní dermatitida, erythema multiforme, exfoliativní vyrážka, pemfigoid, Stevens-Johnsonův syndrom a následující nežádoucí účinky stupně 3 a vyššího: pruritus, vyrážka, generalizovaná vyrážka a makulopapulózní vyrážka)
n. vitiligo (kožní depigmentace, kožní hypopigmentace a hypopigmentace očního víčka)
o. lišejová keratóza (lichen planus a lichen sclerosus)
p. myozitida (myalgie, myopatie, polymyalgia rheumatica a rabdomyolýza)
q. muskuloskeletální bolest (muskuloskeletální diskomfort, bolest zad, muskuloskeletální ztuhlost, muskuloskeletální bolesti na hrudi a tortikolis)
r. artritida (otok kloubů, polyartritida a kloubní výron)
s. tendosynovitida (tendinitida, synovitida a bolest šlach)
t. nefritida (autoimunitní nefritida, tubulointersticiální nefritida a selhání ledvin nebo akutní selhání ledvin se známkami nefritidy)
u. edém (periferní edém, generalizovaný edém, nadměrná zátěž tekutinami, retence tekutin, otok víček a otok rtů, otok obličeje, lokalizovaný edém, periorbitální edém)
Popis vybraných nežádoucích účinků
Údaje pro následující imunitně zprostředkované nežádoucí účinky jsou založeny na pacientech, kteří dostávali pembrolizumab ve třech dávkách (2 mg/kg každé 3 týdny nebo 10 mg/kg každé 2 nebo 3 týdny) v klinických studiích (viz bod 5.1). Léčebná strategie těchto nežádoucích účinků je popsána v bodě 4.4.
Imunitně zprostředkované nežádoucí účinky (viz bod 4.4)
Imunitně zprostředkovaná pneumonitida
Pneumonitida se vyskytla u 94 (3,4 %) pacientů léčených pembrolizumabem, včetně případů stupně 2 u 36 (1,3 %) pacientů, stupně 3 u 25 (0,9 %), stupně 4 u 7 (0,3 %) nebo stupně 5 u 4 (0,1 %) pacientů. Medián doby do nástupu pneumonitidy byl 3,3 měsíce (rozmezí 2 dny až 19,3 měsíce). Medián trvání
byl 1,5 měsíce (rozmezí 1 den až 17,2+ měsíce). Pneumonitida vedla k vysazení pembrolizumabu u 36 (1,3 %) pacientů. Pneumonitida vymizela u 55 pacientů.
Imunitně zprostředkovaná kolitida
Kolitida se vyskytla u 48 (1,7 %) pacientů léčených pembrolizumabem, včetně případů stupně 2 u 10 (0,4 %) pacientů, stupně 3 u 31(1,1%) pacientů a stupně 4 u 2 (< 0,1 %) pacientů. Medián doby do nástupu kolitidy byl 3,5 měsíce (rozmezí 10 dní až 16,2 měsíce). Medián trvání byl 1,3 měsíce (rozmezí 1 den až 8,7+ měsíce). Kolitida vedla k vysazení pembrolizumabu u 15 (0,5 %) pacientů. Kolitida vymizela u 41 pacientů.
Imunitně zprostředkovaná hepatitida
Hepatitida se vyskytla u 19 (0,7 %) pacientů léčených pembrolizumabem, včetně případů stupně 2 u 4 (0,1 %) pacientů, stupně 3 u 12 (0,4 %) pacientů a stupně 4 u 2 (< 0,1 %) pacientů. Medián doby do nástupu hepatitidy byl 1,3 měsíce (rozmezí 8 dní až 21,4 měsíce). Medián trvání byl 1,8 měsíce (rozmezí 8 dní až 20,9+ měsíce). Hepatitida vedla k vysazení pembrolizumabu u 6 (0,2 %) pacientů. Hepatitida vymizela u 15 pacientů.
Imunitně zprostředkovaná nefritida
Nefritida se vyskytla u 9 (0,3 %) pacientů léčených pembrolizumabem, včetně případů stupně 2 u 3 (0,1 %) pacientů, stupně 3 u 4(0,1%) pacientů a stupně 4 u 1 (< 0,1%) pacienta. Medián doby do nástupu nefritidy byl 5,1 měsíce (rozmezí 12 dní až 12,8 měsíce). Medián trvání byl 3,3 měsíce (rozmezí 12 dní až 8,9+ měsíce). Nefritida vedla k vysazení pembrolizumabu u 3 (0,1 %) pacientů. Nefritida vymizela u 5 pacientů.
Imunitně zprostředkované endokrinopatie
Hypofyzitida se vyskytla u 17 (0,6 %) pacientů léčených pembrolizumabem, včetně případů stupně 2 u 6 (0,2 %) pacientů, stupně 3 u 8 (0,3 %) pacientů a stupně 4 u 1 (< 0,1%) pacienta. Medián doby do nástupu hypofyzitidy byl 3,7 měsíce (rozmezí 1 den až 11,9 měsíce). Medián trvání byl 4,7 měsíce (rozmezí 8+ dní až 12,7+ měsíce). Hypofyzitida vedla k vysazení pembrolizumabu u 4 (0,1 %) pacientů. Hypofyzitida vymizela u 7 pacientů, 2 měli následky.
Hypertyreóza se vyskytla u 96 (3,4 %) pacientů léčených pembrolizumabem, včetně případů stupně 2 u 22 (0,8 %) pacientů a stupně 3 u 4 (0,1 %) pacientů. Medián doby do nástupu hypertyreózy byl
1.4 měsíce (rozmezí 1 den až 21,9 měsíce). Medián trvání byl 2,1 měsíce (rozmezí 3 dny až 15,0+ měsíce). Hypertyreóza vedla k vysazení pembrolizumabu u 2 (< 0,1 %) pacientů. Hypertyreóza vymizela u 71 (74 %) pacientů.
Hypotyreóza se vyskytla u 237 (8,5 %) pacientů léčených pembrolizumabem, včetně případů stupně 2 u 174 (6,2 %) pacientů a stupně 3 u 3 (0,1 %) pacientů. Medián doby do nástupu hypotyreózy byl
3.5 měsíce (rozmezí 1 den až 18,9 měsíce). Mediánu trvání nebylo dosaženo (rozmezí 2 dny až 27,7+ měsíce). Kvůli hypotyreóze pembrolizumab vysadil jeden pacient (< 0,1 %). Hypotyreóza vymizela u 48 (20 %) pacientů.
Imunogenita
V klinických studiích u pacientů léčených pembrolizumabem v dávce 2 mg/kg každé 3 týdny nebo 10 mg/kg každé 2 nebo 3 týdny bylo 19 (1,7 %) z 1087 vyhodnotitelných pacientů pozitivně testováno na protilátky proti pembrolizumabu, které se objevily během léčby. Při vzniku protilátek vážících pembrolizumab nebyl nalezen žádný důkaz změněné farmakokinetiky nebo bezpečnostního profilu.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
O předávkování pembrolizumabem nejsou žádné informace.
Při předávkování je nutno pacienty pečlivě sledovat na projevy nebo příznaky nežádoucích účinků a nasadit příslušnou symptomatickou léčbu.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: cytostatika, monoklonální protilátky. ATC kód: L01XC18 Mechanismus účinku
Přípravek KEYTRUDA je humánní monoklonální protilátka, která se váže na receptor programované buněčné smrti PD-1 (programmed cell death-1) a blokuje jeho interakci s ligandy PD-L1 a PD-L2. Receptor PD-1 je negativním regulátorem aktivity T-buněk, u kterého bylo prokázáno, že se podílí na regulaci T-buněčné imunitní odpovědi. Přípravek KEYTRUDA posiluje T-buněčnou odpověď, včetně protinádorové odpovědi prostřednictvím blokády vazby PD-1 na PD-L1 and PD-L2, které se exprimují v buňkách prezentujících antigeny a mohou být exprimovány nádorovými nebo jinými buňkami v mikrosprostředí nádoru.
Klinická účinnost a bezpečnost
Melanom
KEYNOTE-006: klinická studie _pacientů s melanomem, kteří dosud nebyli léčeni ipilimumabem Bezpečnost a účinnost pembrolizumabu byla hodnocena ve studii KEYNOTE-006, což byla multicentrická, kontrolovaná studie fáze III léčby pokročilého melanomu u pacientů, kteří dosud nebyli léčeni ipilimumabem. Pacienti byli randomizováni (1:1:1) do skupin léčených pembrolizumabem 10 mg/kg každé 2 (n=279) nebo 3 týdny (n=277) nebo ipilimumabem 3 mg/kg každé 3 týdny (n=278). U pacientů s melanomem s mutací BRAF V600E nebylo požadováno, aby předtím byli léčeni BRAF inhibitorem.
Pacienti byli léčeni pembrolizumabem do progrese nemoci nebo do nepřijatelné toxicity. Klinicky stabilním pacientům s počátečními známkami progrese nemoci bylo dovoleno setrvat na léčbě do potvrzení progrese nemoci. Vyhodnocení stavu nádoru bylo provedeno po 12 týdnech, poté každých 6 týdnů až do 48. týdne a následně každých 12 týdnů.
Z 834 pacientů bylo 60 % mužů, 44 % bylo ve věku > 65 let (medián věku byl 62 let [rozmezí 18-89]) a 98 % byli běloši. Stadium M1c mělo 65 % pacientů, 9 % mělo mozkové metastázy v anamnéze,
66 % nemělo žádnou a 34 % podstoupilo jednu přechozí léčbu. U 31 % byla hodnota výkonnostního stavu dle ECOG 1, 69 % mělo hodnotu výkonnostního stavu dle ECOG 0 a 32 % mělo zvýšené LDH. BRAF mutace byly hlášeny u 302 (36 %) pacientů. Z pacientů s BRAF mutací bylo 139 (46 %) již léčeno BRAF inhibitorem.
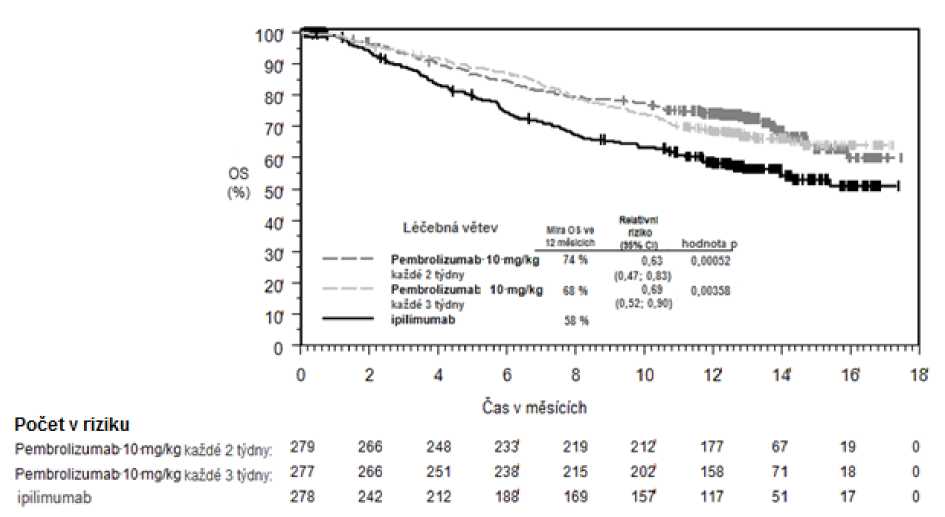
Primárními cíli bylo měření výsledků účinnosti v přežití bez progrese nemoci, hodnoceno pomocí Integrated Radiology and Oncology Assessment (IRO) s využitím Response Evaluation Criteria in Solid Tumours (RECIST), verze 1.1, a v celkovém přežití. Sekundárními cíli bylo měření výsledků účinnosti v míře celkového přežití a trvání odpovědi. Tabulka 3 shrnuje klíčová měření účinnosti u pacientů dosud neléčených ipilimumabem a na obrázcích 1 a 2 jsou uvedeny Kaplan-Meierovy křivky pro celkové přežití (OS) a přežití bez progrese nemoci (PFS).
Tabulka 3: Odpověď na pembrolizumab 10 mg/kg každé 2 nebo 3 týdny u pacientů s pokročilým melanomem neléčeným ipilimumabem ve studii KEYNOTE-006*
Obrázek 1: Kaplan-Meierova křivka celkového přežití podle léčebného ramene ve studii
KEYNOTE-006 (populace všech zařazených pacientů)
|
Cílový parametr |
Pembrolizumab 10 mg/kg každé 3 týdny n=277 |
Pembrolizumab 10 mg/kg každé 2 týdny n=279 |
Ipilimumab 3 mg/kg každé 3 týdny n=278 |
|
Celkové přežití (OS) | |||
|
Počet (%) pacientů s příhodou |
92 (33 %) |
85 (30 %) |
112 (40 %) |
|
Relativní riziko* (95% CI) |
0,69 (0,52; 0,90) |
0,63 (0,47; 0,83) |
--- |
|
Hodnota pT |
0,00358 |
0,00052 |
— |
|
Medián v měsících (95% CI) |
Nebylo dosaženo (NA, NA) |
Nebylo dosaženo (NA, NA) |
Nebylo dosaženo (13, NA) |
|
Přežití bez progrese nemoci (PFS) | |||
|
Počet (%) pacientů s příhodou |
157 (57 %) |
157 (56 %) |
188 (68 %) |
|
Relativní riziko* (95% CI) |
0,58 (0,47; 0,72) |
0,58 (0,46; 0,72) |
— |
|
Hodnota pT |
<0,00001 |
<0,00001 |
— |
|
Medián v měsících (95% CI) |
4,1 (2,9; 6,9) |
5,5 (3,4; 6,9) |
2,8 (2,8; 2,9) |
|
Nejlepší celková odpověď | |||
|
ORR % (95% CI) |
33 % (27, 39) |
34 % (28, 40) |
12 % (8, 16) |
|
Úplná odpověď % |
6% |
5 % |
1 % |
|
Částečná odpověď % |
27% |
29% |
10% |
|
Trvání odpovědi* | |||
|
Medián v měsících (rozmezí) |
Nebylo dosaženo (1,4+; 8,1+) |
8 3 (1,4+;’8,3+) |
Nebylo dosaženo (1,1+; 7,9+) |
|
% přetrvávající odpovědi |
97 % |
89 % |
88 % |
* Relativní riziko (pembrolizumab v porovnání s ipilimumabem) založeno na stratifikovaném Cox proporčním modelu rizik
* Založeno na stratifikovaném Log rank testu
* Založeno na pacientech s nejlepší celkovou odpovědí jako potvrzenou úplnou nebo částečnou odpovědí NA = není k dispozici
120 "
110 -
100
30
TQ
PFS o® .
40
Lecsbnivtis'.-
ním PFS* Min PFS v
laum * ■*! •** Mriniap
Přřft&iůJ«iw<wa& 10
klid# 2 fa dna
k!3d+ 3 fídň-p
ipilirrumjt
1r*~
i-
í :
:
*0.00001
44 %
27
iř %
-i -
ID.!*: 0.721
■;0.i7. 9.721
L._
<0.00001
10
Čas v mísících
Počet v riziku
PembicliiumaMOiTiBiligyaJdř Ztfdňh- 27$
PímbrclizumablCmaAaioíaf Jlýdny 277
ipiliiriumab $76
KEYNOTE-002: klinická studie pacientů s melanomem, kteří již byli léčeni ipilimumabem Bezpečnost a účinnost pembrolizumabu byla zjišťována ve studii KEYNOTE-002, což je multicentrická, kontrolovaná studie léčby pokročilého melanomu u pacientů, kteří byli léčeni ipilimumabem a pokud byli pozitivní na mutaci BRAF V600, léčeni BRAF nebo MEK inhibitory. Pacienti byli randomizováni (1:1:1) do skupin s pembrolizumabem v dávce 2 (n=180) nebo 10 mg/kg (n=181) každé 3 týdny nebo chemoterapií (n=179; včetně dakarbazinu, temozolomidu, karboplatiny, paklitaxelu nebo karboplatiny+paklitaxelu). Do studie nebyli zařazeni pacienti s autoimunitním onemocněním nebo pacienti léčení imunosupresivy; dalšími kritérii pro nezařazení byl těžký nebo život ohrožující imunitně zprostředkovaný nežádoucí účinek léčby ipilimumabem v anamnéze, definovaný jako jakákoli toxicita stupně 4 nebo toxicita stupně 3 vyžadující léčbu kortikosteroidem (> 10 mg/den prednisonu nebo ekvivalentní dávka) po dobu delší než 12 týdnů; probíhající nežádoucí účinky v důsledku předchozí léčby ipilimumabem stupně > 2; předchozí těžká hypersenzitivita na jiné monoklonální protilátky; pneumonitida nebo intersticiální plicní nemoc v anamnéze; HIV, infekce virem hepatitidy B nebo hepatitidy C a hodnota výkonnostního stavu dle ECOG > 2.
Pacienti byli léčeni pembrolizumabem do progrese nemoci nebo nepřijatelné toxicity. Klinicky stabilním pacientům s počátečními známkami progrese nemoci bylo dovoleno setrvat na léčbě do potvrzení progrese nemoci. Vyhodnocení stavu nádoru bylo provedeno po 12 týdnech, poté každých 6 týdnů až do 48. týdne a následně každých 12 týdnů. Pacientům na chemoterapii, u kterých po prvním plánovaném vyhodnocení nemoci došlo k nezávisle potvrzené progresi, byl umožněn přechod do dvojitě zaslepeného uspořádání, kde dostávali 2 mg/kg nebo 10 mg/kg pembrolizumabu každé 3 týdny.
Z 540 pacientů bylo 61 % mužů, 43 % mělo > 65 let (medián věku byl 62 let [rozmezí 15-89]) a 98 % byli běloši. Stadium M1c mělo 82 % pacientů, 73 % podstoupilo nejméně dvě a 32 % pacientů podstoupilo tři nebo více předchozích systémových terapií pokročilého melanomu. U 45 % byla hodnota výkonnostního stavu dle ECOG 1, 40 % mělo zvýšené LDH a 23 % mělo BRAF mutaci.
Primárními cíli bylo měření výsledků účinnosti v PFS hodnocené pomocí IRO s využitím RECIST verze 1.1a v OS. Sekundárními cíli bylo měření výsledků účinnosti v ORR a v trvání odpovědi. Tabulka 4 shrnuje klíčové cíle měření účinnosti u pacientů již léčených ipilimumabem, přičemž na obrázku 3 je uvedena Kaplan-Meierova křivka přežití bez progrese nemoci. V obou ramenech léčených pembrolizumabem bylo přežití bez progrese nemoci vyšší než při chemoterapii a nebyly zde žádné rozdíly mezi dávkami pembrolizumabu. Údaje o celkovém přežití nebyly v době analýzy přežití
bez progrese nemoci dostatečné. V předběžných analýzách celkového přežití, které nebyly upraveny na potenciálně matoucí vliv přechodu některých pacientů léčených ve skupině chemoterapií do skupiny léčených pembrolizumabem, nebyl rozdíl mezi pembrolizumabem a chemoterapií statisticky významný. Z pacientů randomizovaných do skupiny léčené chemoterapií přešlo do skupin léčených pembrolizumabem 48 %.
Tabulka 4: Odpověď na pembrolizumab 2 mg/kg nebo 10 mg/kg každé 3 týdny u pacientů s neresekovatelným nebo metastazujícím melanomem ve studii KEYNOTE-002
|
Cílový parametr |
Pembrolizumab 2 mg/kg každé 3 týdny n=180 |
Pembrolizumab 10 mg/kg každé 3 týdny n=181 |
Chemoterapie n=179 |
|
Přežití bez progrese nemoci (PFS) | |||
|
Počet (%) pacientů s příhodou |
129 (72 %) |
126 (70 %) |
155 (87 %) |
|
Relativní riziko* (95% CI) |
0,57 (0,45; 0,73) |
0,50 (0,39; 0,64) |
--- |
|
Hodnota pT |
<0,0001 |
< 0,0001 |
— |
|
Medián v měsících (95% CI) |
2,9 (2,8; 3,8) |
2,9 (2,8; 4,7) |
2,7 (2,5; 2,8) |
|
Celkové přežití (OS) | |||
|
Počet (%) pacientů s příhodou |
73 (41 %) |
69 (38 %) |
78 (44 %) |
|
Relativní riziko* (95% CI) |
0,88 (0,64; 1,22) |
0,78 (0,56; 1,08) |
--- |
|
Hodnota pT |
0,2294 |
0,0664 |
— |
|
Nejlepší celková odpověď | |||
|
Celková míra odpovědi % (95% CI) |
21 %(15, 28) |
25 % (19, 32) |
4 % (2, 9) |
|
Úplná odpověď % |
2% |
3 % |
0% |
|
Částečná odpověď % |
19% |
23 % |
4% |
|
Trvání odpovědi | |||
|
Medián v měsících (rozmezí) |
Nebylo dosaženo (1,4+; 11,5+) |
Nebylo dosaženo (1,2+; 11,1+) |
8,5 (1,6+; 9,5) |
|
% přetrvávající |
87% |
80% |
63 % |
* Relativní riziko (pembrolizumab v porovnání s chemoterapií) založeno na stratifikovaném Cox proporčním modelu rizik ^ Založeno na stratifikovaném Log rank testu
Lecebna vtlev
Miř* PF 5 v UimFFSv
90 -
Snřiicich .<saHCii Hodnota p
Pcmbrdiz.uín*j lumg ^
30 -
UlS* i tp ín>
Pumbrdi z.uíTiii 2 mg*1 Iq Vaidt J týdny
CN^OtřílCit
:: i
□,5Ů
O.Í7
lO.íí 5.13)
* G.DCOl
T0 -
'TO -
PFS
4Q -
30 -
Cas v měsících
Počet v riziku
PefTTtuulzumat? 10 n^JKo Pembratzumas 2 mgíKgi Chemoterapie
KEYNOTE-001: otevřená studie _pacientů s melanomem, kteří dosud nebyli léčeni ipilimumabem a kteří j již byli léčeni ipilimumabem
Bezpečnost a účinnost pembrolizumabu u pacientů s pokročilým melanomem byla hodnocena v nekontrolované otevřené studii KEYNOTE-001. Účinnost byla hodnocena u 276 pacientů ze dvou definovaných kohort, jedna zahrnovala pacienty již léčené ipilimumabem (a pokud byli pozitivní na mutaci BRAF V600, léčeni BRAF nebo MEK inhibitory) a druhá zahrnovala pacienty, kteří ipilimumabem dosud léčeni nebyli. Pacienti byli náhodně přiřazeni do skupin s pembrolizumabem v dávce 2 mg/kg každé 3 týdny nebo 10 mg/kg každé 3 týdny. Pacienti byli léčeni pembrolizumabem do progrese nemoci nebo nepřijatelné toxicity. Klinicky stabilním pacientům s počátečními známkami progrese nemoci bylo dovoleno setrvat na léčbě do potvrzení progrese nemoci. Vylučovací kritéria byla podobná jako ve studii KEYNOTE-002.
Z 89 pacientů léčených 2 mg/kg pembrolizumabu, kteří byli předtím léčeni ipilimumabem, bylo 53 % mužů, 33 % bylo ve věku > 65 let a medián věku byl 59 let (rozmezí 18 až 88). Kromě 2 byli všichni pacienti běloši. Stadium M1c mělo 84 % pacientů a 8 % mělo mozkové metastázy v anamnéze.
70 % podstoupilo nejméně dvě a 35 % pacientů podstoupilo tři nebo více předchozích terapií pokročilého melanomu. BRAF mutace byly hlášeny u 13 % hodnocené populace. Všichni pacienti s BRAF mutací byli předtím léčeni BRAF inhibitorem.
Z 51 pacientů léčených 2 mg/kg pembrolizumabu, kteří dosud nebyli léčeni ipilimumabem, bylo 63 % mužů, 35 % bylo ve věku > 65 let a medián věku byl 60 let (rozmezí 35 až 80). Kromě jednoho pacienta byli všichni běloši. Stadium M1c mělo 63 % pacientů a 2 % měla mozkové metastázy v anamnéze. 45 % procent nepodstoupilo žádné předchozí terapie pokročilého melanomu. BRAF mutace byly hlášeny u 20 (39 %) pacientů. Z pacientů s BRAF mutací bylo 10 (50 %) předtím léčeno BRAF inhibitorem.
Primárním cílem bylo měření výsledků účinnosti v celkové míře odpovědi vyhodnocené nezávislým vyšetřením pomocí RECIST 1.1. Sekundárními cíli bylo měření výsledků účinnosti v míře kontroly nemoci (DCR; včetně úplné odpovědi, částečné odpovědi a stabilizace onemocnění), v trvání odpovědi, v přežití bez progrese nemoci a v celkovém přežití. Odpověď nádoru byla hodnocena ve 12týdenních intervalech. Klíčové cíle měření účinnosti u již léčených pacientů nebo u pacientů dosud neléčených ipilimumabem, dostávajících pembrolizumab v doporučené dávce, uvádí tabulka 5.
Tabulka 5: Odpověď na pembrolizumab 2 mg/kg každé 3 týdny u pacientů s neresekovatelným nebo metastazujícím melanomem ve studii KEYNOTE-001
|
Cílový parametr |
Pembrolizumab 2 mg/kg každé 3 týdny u pacientů předtím léčených ipilimumabem n=89 |
Pembrolizumab 2 mg/kg každé 3 týdny u pacientů dosud ipilimumabem neléčených n=51 |
|
Nejlepší celková odpověď* stanovená pomocí IRO^ | ||
|
ORR %, (95% CI) |
25 % (16, 35) |
33 % (21, 48) |
|
Úplná odpověď |
3 % |
10% |
|
Částečná odpověď |
21 % |
24% |
|
Míra kontroly onemocnění %* |
49% |
49% |
|
Trvání odpovědi5 | ||
|
Medián v měsících (rozmezí) |
Nebylo dosaženo (2,8+; 14,3+) |
Nebylo dosaženo (1,6+; 13,8+) |
|
% přetrvávající |
86 %1 |
82 %# |
|
Přežití bez progrese nemoci (PFS) | ||
|
Medián v měsících (95% CI) |
4,9 (2,8; 8,3) |
5,5 (2,8; 14,0) |
|
Míra přežití bez progrese nemoci po 6 měsících |
43 % |
50% |
|
Celkové přežití (OS) | ||
|
Medián v měsících (95% CI) |
Nebylo dosaženo (11, není k dispozici) |
Nebylo dosaženo (14, není k dispozici) |
|
Míra celkového přežití po 12 měsících |
60% |
72% |
* Zahrnuje pacienty, kteří při vstupu nemají nezávislou radiologickou metodou zjištěnu žádnou měřitelnou nemoc t IRO = Integrated radiology and oncologist assessment (integrované rentgenové a onkologické vyhodnocení) s využitím
RECIST 1.1
í
§
1
#
Založeno na nejlepší odpovědi stabilizované nemoci nebo lepší
Založeno na pacientech s odpovědí potvrzenou nezávislou kontrolou, od data, kdy byla odpověď poprvé zaznamenána; n=22 u pacientů již léčených ipilimumabem; n=17 u pacientů dosud ipilimumabem neléčených Respondéři byli sledováni po dobu nejméně 12 měsíců po zahájení léčby Respondéři byli sledováni po dobu nejméně 15 měsíců po zahájení léčby
Výsledky byly u pacientů již léčených ipilimumabem (n=84) a pacientů dosud ipilimumabem neléčených (n=52), kteří dostávali 10 mg/kg pembrolizumabu každé 3 týdny, podobné jako výsledky pozorované u pacientů, kteří dostávali 2 mg/kg pembrolizumabu každé 3 týdny.
Analýzy subpopulací
Stav BRAF mutací u melanomu
Byla provedena analýza podskupin studie KEYNOTE-002 u pacientů, kteří měli gen BRAF divokého typu (n=415; 77 %) nebo BRAF mutaci bez předchozí léčby cílené na BRAF (n=125; 23 %). Relativní riziko přežití bez progrese nemoci (souhrnně pro pembrolizumab [2 mg/kg nebo 10 mg/kg každé 3 týdny] vs. chemoterapie) bylo 0,51 (95% CI: 0,41; 0,65) u genu BRAF divokého typu a 0,56 (95% CI: 0,37; 0,85) u BRAF mutace s předchozí léčbou cílenou na BRAF. Relativní riziko přežití bez progrese nemoci pro pembrolizumab 2 mg/kg každé 3 týdny vs. chemoterapie bylo 0,51 (95% CI:
0,39; 0,67) u genu BRAF divokého typu a 0,74 (95% CI: 0,46; 1,18) u BRAF mutace s předchozí léčbou cílenou na BRAF. Relativní riziko celkového přežití souhrnně pro pembrolizumab vs. chemoterapie bylo 0,83 (95% CI: 0,60; 1,15) u genu BRAF divokého typu a 0,82 (95% CI: 0,47; 1,43) u BRAF mutace s předchozí léčbou cílenou na BRAF. Relativní riziko celkového přežití pro pembrolizumab 2 mg/kg každé 3 týdny vs. chemoterapie bylo 0,80 (95% CI: 0,55; 1,18) u genu BRAF divokého typu a 1,03 (95% CI: 0,55; 1,91) u BRAF mutace s předchozí léčbou cílenou na BRAF. Celková míra odpovědi souhrnně pro pembrolizumab a pembrolizumab 2 mg/kg každé 3 týdny vs. chemoterapie byla 27 % a 25 % vs. 6 % u genu BRAF divokého typu a 12% a 9 % vs. 0% u BRAF mutace s předchozí léčbou cílenou na BRAF.
Byla provedena analýza podskupin studie KEYNOTE-006 u pacientů, kteří měli BRAF divokého typu (n=525; 63 %), BRAF mutaci bez předchozí léčby cílené na BRAF (n=163; 20 %) a BRAF mutaci s předchozí léčbou cílenou na BRAF (n=139; 17 %). Relativní riziko přežití bez progrese nemoci (souhrnně pro pembrolizumab [10 mg/kg každé 2 nebo 3 týdny] vs. ipilimumab) bylo 0,57 (95% CI: 0,45; 0,73) u genu BRAF divokého typu, 0,50 (95% CI: 0,32; 0,77) u BRAF mutace bez předchozí léčby cílené na BRAF a 0,73 (95% CI: 0,48; 1,11) u BRAF mutace s předchozí léčbou cílenou na BRAF. Relativní riziko celkového přežití souhrnně pro pembrolizumab vs. ipilimumab bylo 0,61 (95% CI: 0,46; 0,82) u genu BRAF divokého typu, 0,69 (95% CI: 0,33; 1,45) u BRAF mutace bez předchozí léčby cílené na BRAF a 0,75 (95% CI: 0,45; 1,26) u BRAF mutace s předchozí léčbou cílenou na BRAF. Celková míra odpovědi souhrnně pro pembrolizumab vs. ipilimumab byla 34 % vs. 13 % u genu BRAF divokého typu, 41% vs. 13% u BRAF mutace bez předchozí léčby cílené na BRAF a 21% vs. 6% u BRAF mutace spředchozí léčbou cílenou na BRAF.
Stav PD-L1 u melanomu
Byla provedena analýza podskupin studie KEYNOTE-002 u pacientů, kteří byli PD-L1 pozitivní (Allredovo histoskore > 2 vyjadřující membránovou expresi PD-L1 u > 1 % nádorových buněk) vs. PD-L1 negativní (Allredovo histoskore 0 nebo 1). Exprese PD-L1 byla testována retrospektivně imunohistochemickým stanovením pomocí 22C3 protilátky PD-L1. Z pacientů, u kterých byla měřitelná exprese PD-L1 (78%), bylo 69 % (n=291) PD-L1 pozitivních a 31 % (n=130) bylo PD-L1 negativních. Relativní riziko přežití bez progrese nemoci (souhrnně pro pembrolizumab [2 mg/kg nebo 10 mg/kg každé 3 týdny] vs. chemoterapie) bylo 0,52 (95% CI: 0,39; 0,68) u PD-L1 pozitivních pacientů a 0,60 (95% CI: 0,38; 0,94) u PD-L1 negativních pacientů. Relativní riziko přežití bez progrese nemoci pro pembrolizumab 2 mg/kg každé 3 týdny vs. chemoterapie bylo 0,54 (95% CI:
0,39; 0,75) u PD-L1 pozitivních pacientů a 0,89 (95% CI: 0,53; 1,50) u PD-L1 negativních pacientů. Relativní riziko celkového přežití souhrnně pro pembrolizumab vs. chemoterapie bylo 0,82 (95% CI: 0,55; 1,23) u PD-L1 pozitivních pacientů a 0,77 (95% CI: 0,43; 1,37) u PD-L1 negativních pacientů. Relativní riziko celkového přežití pro pembrolizumab 2 mg/kg každé 3 týdny vs. chemoterapie bylo 0,93 (95% CI: 0,58; 1,49) u PD-L1 pozitivních pacientů a 1,19 (95% CI: 0,58; 2,46) u PD-L1 negativních pacientů. Celková míra odpovědi souhrnně pro pembrolizumab a pembrolizumab 2 mg/kg každé 3 týdny vs. chemoterapie byla 26 % a 23 % vs. 4 % u PD-L1 pozitivních pacientů a 15 % a 11 % vs. 8 % u PD-L1 negativních pacientů.
Byla provedena analýza podskupin studie KEYNOTE-006 u pacientů, kteří byli PD-L1 pozitivní (n=671; 80 %) vs. PD-L1 negativní (n=150; 18 %). Z pacientů, u kterých byla měřitelná exprese PD-L1 (98 %), bylo 82 % PD-L1 pozitivních a 18 % bylo PD-L1 negativních. Relativní riziko přežití bez progrese nemoci (souhrnně pro pembrolizumab [10 mg/kg každé 2 nebo 3 týdny] vs. ipilimumab) bylo 0,53 (95% CI: 0,43; 0,65) u PD-L1 pozitivních pacientů a 0,73 (95% CI: 0,47; 1,11) u PD-L1 negativních pacientů. Relativní riziko celkového přežití souhrnně pro pembrolizumab vs. ipilimumab bylo 0,56 (95% CI: 0,43; 0,73) u PD-L1 pozitivních pacientů a 0,95 (95% CI: 0,56; 1,62) u PD-L1 negativních pacientů. Celková míra odpovědi souhrnně pro pembrolizumab vs. ipilimumabová skupina byla 37% vs. 12% u PD-L1 pozitivních pacientů a 18% vs. 11% u PD-L1 negativních pacientů.
Oční melanom
U 20 subjektů s očním melanomem zařazených do studie KEYNOTE-001 nebyly hlášeny žádné objektivní odpovědi; u 6 pacientů byla hlášena stabilizace onemocnění.
NSCLC
KEYNOTE-010: kontrolovaná studie u pacientů s NSCLC, kteří již byli léčeni chemoterapií Bezpečnost a účinnost pembrolizumabu byla hodnocena ve studii KEYNOTE-010, což byla multicentrická, otevřená, kontrolovaná studie léčby pokročilého NSCLC u pacientů, kteří již byli léčeni chemoterapií obsahující platinu. Pacienti byli pozitivní na expresi PD-L1 (skóre nádorového podílu (tumour proportion score - TPS) > 1 % získáno pomocí PD-L1 IHC 22C3 pharmDx™ Kit). U pacientů s mutací aktivující EGFR nebo translokací ALK před podáním pembrolizumabu také došlo k progresi nemoci při terapii schválené při těchto mutacích. Pacienti byli randomizováni (1:1:1) do skupiny léčené pembrolizumabem v dávce 2 (n=344) nebo 10 mg/kg (n=346) každé 3 týdny nebo docetaxelem v dávce 75 mg/m2 každé 3 týdny (n=343) do progrese nemoci nebo nepřijatelné toxicity. Do klinického hodnocení nebyli zařazeni pacienti s autoimunitním onemocněním; zdravotním stavem vyžadujícím imunosupresi ani pacienti, kteří byli v předchozích 26 týdnech na hrudníku ozářeni dávkou větší než 30 Gy. Vyhodnocení stavu nádoru bylo provedeno každých 9 týdnů.
Výchozí charakteristiky této populace byly: medián věku 63 let (42 % bylo ve věku 65 nebo více);
61 % byli muži; 72 % běloši a 21 % asiaté a 34 % mělo stav výkonnosti ECOG 0 a 66 % mělo stav výkonnosti ECOG 1. Charakteristiky nemoci byly skvamózní (21 %) a neskvamózní (70 %); M1 (91 %); stabilní metastázy v mozku (15 %) a incidence mutací byla EGFR (8 %) nebo ALK (1 %). Předchozí léčba zahrnovala režim s platinovým dubletem (100 %); pacienti předtím podstoupili jednu (69 %) nebo dvě nebo více léčebných linií (29 %).
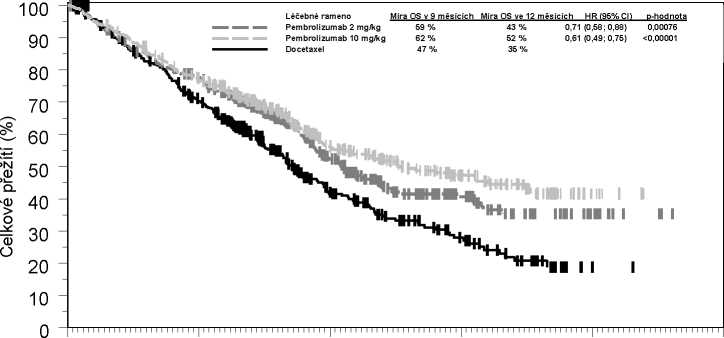
Primárními kritérii hodnocení účinnosti bylo celkové přežití a přežití bez progrese nemoci, které byly hodnoceny zaslepenou nezávislou centrální posudkovou komisí za využití kritérií RECIST 1.1. Sekundárními kritérii hodnocení účinnosti byla míra celkového přežití a trvání odpovědi. Tabulka 6 shrnuje klíčová měření účinnosti u celé populace (TPS > 1 %) a u pacientů s TPS > 50 %, přičemž Kaplan-Meierova křivka celkového přežití (TPS > 1 %) je uvedena na obrázku 4.
Tabulka 6: odpověď na pembrolizumab 2 nebo 10 mg/kg každé 3 týdny u pacientů s již léčeným NSCLC ve studii KEYNOTE-010
|
Cílový parametr |
Pembrolizumab 2 mg/kg každé 3 týdny |
Pembrolizumab 10 mg/kg každé 3 týdny |
Docetaxel 75 mg/m2 každé 3 týdny |
|
TPS > 1 % | |||
|
Počet pacientů |
344 |
346 |
343 |
|
Celkové přežití | |||
|
Počet (%) pacientů s příhodou |
172 (50 %) |
156 (45 %) |
193 (56 %) |
|
Poměr rizik* (95% CI) |
0,71 (0,58; 0,88) |
0,61 (0,49; 0,75) |
--- |
|
Hodnota p* |
<0,001* |
<0,001* |
--- |
|
Medián v měsících (95% CI) |
10,4 (9,4; 11,9) |
12,7 (10,0; 17,3) |
8,5 (7,5; 9,8) |
|
Přežití bez progrese nemoci5 | |||
|
Počet (%) pacientů s příhodou |
266 (77 %) |
255 (74 %) |
257 (75 %) |
|
Poměr rizik* (95% CI) |
0,88 (0,73; 1,04) |
0,79 (0,66; 0,94) |
--- |
|
Hodnota p* |
0,068 |
0,005 |
— |
|
Medián v měsících (95% CI) |
3,9 (3,1; 4,1) |
4,0 (2,6; 4,3) |
4,0 (3,1; 4,2) |
|
Celková míra odpovědi5 | |||
|
% celkové míry odpovědi1 (95% CI) |
18 % (14, 23) |
18 % (15,23) |
9 % (7, 13) |
|
Trvání odpovědi 5,#’P | |||
|
Medián v měsících (rozmezí) |
Nebylo dosaženo (0,7+; 20,1+) |
Nebylo dosaženo (2,1+; 17,8+) |
6,2 (1,4+; 8,8+) |
|
% přetrvávajících |
73 % |
72% |
34% |
|
TPS > 50% | |||
|
Počet pacientů |
139 |
151 |
152 |
|
Celkové přežití | |||
|
Počet (%) pacientů s příhodou |
58 (42 %) |
60 (40 %) |
86 (57 %) |
|
Poměr rizik* (95% CI) |
0,54 (0,38; 0,77) |
0,50 (0,36; 0,70) |
— |
|
Hodnota p* |
<0,001* |
<0,001* |
— |
|
Medián v měsících (95% CI) |
14,9 (10,4; NA) |
17,3 (11,8; NA) |
8,2 (6,4; 10,7) |
|
Přežití bez progrese nemoci5 | |||
|
Počet (%) pacientů s příhodou |
89 (64 %) |
97 (64 %) |
118 (78 %) |
|
Poměr rizik* (95% CI) |
0,58 (0,43, 0,77) |
0,59 (0,45, 0,78) |
— |
|
Hodnota p* |
<0,001* |
<0,001* |
— |
|
Medián v měsících (95% CI) |
5,2 (4,0; 6,5) |
5,2 (4,1; 8,1) |
4,1 (3,6; 4,3) |
|
Celková míra odpovědi5 | |||
|
% celkové míry odpovědi1 (95% CI) |
30 % (23; 39) |
29 % (22; 37) |
8 % (4; 13) |
|
Trvání odpovědi5’#’14 | |||
|
Medián v měsících (rozmezí) |
Nebylo dosaženo (0,7+; 16,8+) |
Nebylo dosaženo (2,1+; 17,8+) |
8,1 (2,1+; 8,8+) |
|
% přetrvávajících |
76% |
75 % |
33 % |
* Poměr rizik (pembrolizumab v porovnání s docetaxelem) na základě stratifikovaného
Coxova proporčního modelu rizik
* Na základě stratifikovaného Log rank testu
* Statisticky významné na základě předem specifikované a úrovně upravené na multiplicitu § Vyhodnoceno zaslepenou nezávislou centrální posudkovou komisí za využití kritérií
RECIST 1.1
1 Všechny odpovědi byly částečnými odpověďmi
* Na základě pacientů s nejlepší celkovou odpovědí jako potvrzenou úplnou nebo částečnou odpovědí
P Zahrnuje 30, 31 a 2 pacienty s odpovědí přetrvávající 6 nebo více měsíců v rameni léčeném
pembrolizumabem 2 mg/kg, pembrolizumabem 10 mg/kg a docetaxelem, v uvedeném pořadí
R Zahrnuje 22, 24 a 1 pacienta s odpovědí přetrvávající 6 nebo více měsíců v rameni léčeném pembrolizumabem 2 mg/kg, pembrolizumabem 10 mg/kg a docetaxelem, v uvedeném pořadí
Obrázek 4: Kaplan-Meierova křivka celkového přežití podle léčebného ramene ve studii KEYNOTE-010 (pacienti se skóre nádorového podílu exprimujícího PD-L1 > 1 %, populace
všech léčených pacientů)
|
Počet v riziku |
0 |
5 |
10 Čas v měsících |
15 |
20 |
25 |
|
Pembrolizumab 2 mg/kg: |
344 |
259 |
115 |
49 |
12 |
0 |
|
Pembrolizumab 10 mg/kg: |
346 |
255 |
124 |
56 |
6 |
0 |
|
Docetaxel: |
343 |
212 |
79 |
33 |
1 |
0 |
Výsledky účinnosti byly v ramenech léčených 2 mg/kg a 10 mg/kg pembrolizumabu podobné. Výsledky účinnosti ohledně celkového přežití byly na základě meziskupinového porovnání konzistentní bez ohledu na stáří vzorku nádoru (nový vs. archivní).
V analýzách podskupin byl u pacientů, kteří nikdy nebyli kuřáci nebo u pacientů s nádory nesoucími mutace aktivující EGFR, kteří dostávali alespoň chemoterapii na bázi platiny a inhibitor tyrosinkinázy, pozorován snížený přínos přežití u pembrolizumabu ve srovnání s docetaxelem; avšak vzhledem k nízkému počtu pacientů nemohou být z těchto údajů učiněny žádné konečné závěry.
Bezpečnost a účinnost pembrolizumabu u pacientů s nádory, které neexprimují PD-L1 nebyla stanovena.
Pediatrická populace
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s pembrolizumabem u jedné nebo více podskupin pediatrické populace při léčbě všech nemocí zařazených v kategorii maligních neoplazmat (kromě nervového systému, krvetvorné a lymfoidní tkáně) (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Farmakokinetika pembrolizumabu byla hodnocena u 2856 pacientů s metastazujícím nebo neresekovatelným melanomem, NSCLC nebo karcinomem, kteří dostávali dávky v rozmezí 1 až 10 mg/kg každé 2 nebo 3 týdny.
Absorpce
Pembrolizumab se podává intravenózní cestou a proto je okamžitě a zcela biologicky dostupný.
Distribuce
V souladu s omezenou extravaskulární distribucí je distribuční objem pembrolizumabu
v rovnovážném stavu malý (~7,4 l; CV: 19 %). Jak se u protilátky předpokládá, pembrolizumab se specificky neváže na plazmatické proteiny.
Biotransformace
Pembrolizumab se katabolizuje nespecifickými cestami; metabolismus k jeho clearance nepřispívá. Eliminace
Systémová clearance pembrolizumabu je přibližně 0,2 l/den (CV: 37 %) a terminální biologický poločas (t/2) je přibližně 27 dní (CV: 38 %).
Linearita/nelinearita
Expozice pembrolizumabu, vyjádřená jako maximální koncentrace (Cmax) nebo plocha pod křivkou průběhu plazmatických koncentrací v čase (AUC) se v rozmezí použitém pro zjišťování účinnosti zvyšovala v závislosti na dávce. Při opakovaném podání bylo zjištěno, že clearance pembrolizumabu je nezávislá na čase, přičemž systémová akumulace byla při podávání každé 3 týdny přibližně 2,2násobná. Téměř rovnovážné koncentrace pembrolizumabu byly dosaženy po 18 týdnech; medián Cmin po 18 týdnech byla během režimu podávání 2 mg/kg každé 3 týdny přibližně 24 ^g/ml.
Zvláštní populace
V analýzách populační farmakokinetiky byly hodnoceny vlivy různých proměnných
na farmakokinetiku pembrolizumabu. Clearance pembrolizumabu se zvyšovala se zvyšující se tělesnou hmotností; výsledné rozdíly v expozici byly odpovídajícím způsobem vyřešeny dávkováním na bázi mg/kg. Následující faktory neměly na clearance pembrolizumabu žádný klinicky významný vliv: věk (rozmezí 15 až 94 let), pohlaví, rasa, lehká nebo středně těžká porucha funkce ledvin, lehká porucha funkce jater a nádorová zátěž.
Porucha funkce ledvin
Vliv poruchy funkce ledvin na clearance pembrolizumabu byl vyhodnocen analýzami populační farmakokinetiky u pacientů s lehkou nebo středně těžkou poruchou funkce ledvin v porovnání s pacienty s normální funkcí ledvin. Mezi pacienty s lehkou nebo středně těžkou poruchou funkce ledvin a pacienty s normální funkcí ledvin nebyly ohledně clearance pembrolizumabu zjištěny žádné klinicky významné rozdíly. U pacientů s těžkou poruchou funkce ledvin nebyl pembrolizumab hodnocen.
Porucha funkce jater
Vliv poruchy funkce jater na clearance pembrolizumabu byl vyhodnocen analýzami populační farmakokinetiky u pacientů s lehkou poruchou funkce jater (definováno za použití kritérií hodnocení jaterní dysfunkce amerického National Cancer Institute) v porovnání s pacienty s normální funkcí jater. Mezi pacienty s lehkou poruchou funkce jater a pacienty s normální funkcí jater nebyly ohledně clearance pembrolizumabu nalezeny žádné klinicky významné rozdíly. U pacientů se středně těžkou nebo těžkou poruchou funkce jater nebyl pembrolizumab hodnocen (viz bod 4.2).
5.3 Předklinické údaje vztahující se k bezpečnosti
Bezpečnost pembrolizumabu byla hodnocena v 1měsíční a 6měsíční studii toxicity po opakovaném podávání na makacích jávských, kterým se podávaly intravenózně dávky 6, 40 a 200 mg/kg jednou týdně v 1měsíční studii a jednou každé 2 týdny v 6měsíční studii, následované 4měsíčním obdobím bez léčby. Nebyla zaznamenána žádná toxikologicky významná zjištění, přičemž hladina, při níž nebyly pozorovány žádné nežádoucí účinky (No Observed Adverse Effect Level - NOAEL) v obou studiích byla >200 mg/kg, což je 19násobek expozice u lidí při nejvyšší klinicky testované dávce (10 mg/kg).
Reprodukční studie na zvířatech nebyly s pembrolizumabem provedeny. Má se za to, že cesta PD-1/PD-L1 se podílí na udržování tolerance plodu v průběhu těhotenství. Na modelech březích myší bylo prokázáno, že blokáda signálů zprostředkovaných PD-L1 narušuje toleranci plodu a vede ke zvýšeným ztrátám plodů.
Studie fertility na zvířatech nebyly s pembrolizumabem provedeny. V 1měsíční a 6měsíční studií toxicity po opakovaném podávání na opicích nebyly na samčích a samičích reprodukčních orgánech zjištěny žádné pozorovatelné účinky; nicméně mnohá zvířata v těchto studiích nebyla pohlavně dospělá.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Histidin
Monohydrát histidin-hydrochloridu
Sacharóza
Polysorbát 80
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky s výjimkou těch, které jsou uvedeny v bodě 6.6.
6.3 Doba použitelnosti
Neotevřená injekční lahvička 2 roky.
Po rekonstituci
Chemická a fyzikální stabilita rekonstituovaného a naředěného roztoku byla prokázána na dobu 24 hodin při pokojové teplotě (25 °C nebo nižší). Z mikrobiologického hlediska musí být přípravek použit okamžitě. Rekonstituovaný nebo naředěný roztok nesmí být zmražen. Není-li použit okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou v odpovědnosti uživatele a nesmí být delší než celkem 24 hodin. Tento 24hodinový limit může zahrnovat až 6 hodin při pokojové teplotě (25 °C nebo nižší); veškerá další doba uchovávání musí být při teplotě 2 °C - 8 °C. Při uchovávání v chladničce musíte nechat injekční lahvičky a/nebo i.v. vaky před použitím ohřát na pokojovou teplotu.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C).
Podmínky uchovávání tohoto léčivého přípravku po jeho rekonstituci nebo naředění viz bod 6.3.
6.5 Druh obalu a obsah balení
15ml injekční lahvička ze skla třídy I s šedou brombutylovou zátkou a hliníkovým uzávěrem s odtrhovacím víčkem avokádové barvy obsahující 50 mg pembrolizumabu.
Jedna krabička obsahuje jednu injekční lahvičku.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Příprava a podání
• Před rekonstitucí může být injekční lahvička s lyofylizovaným práškem mimo chladničku (teplota 25 °C nebo nižší) na dobu až 24 hodin.
• Asepticky přidejte 2,3 ml vody na injekci, čímž získáte roztok přípravku KEYTRUDA
o koncentraci 25 mg/ml (pH 5,2 - 5,8). Každá injekční lahvička obsahuje dodatečných 10 mg (0,4 ml), aby se zajistilo, že se z injekční lahvičky získá 50 mg přípravku KEYTRUDA.
Po rekonstituci obsahuje 1 ml koncentrátu 25 mg pembrolizumabu.
• Aby se zamezilo vzniku pěny, vodu vstřikujte podél stěn injekční lahvičky, nikoli přímo do lyofilizovaného prášku.
• Injekční lahvičkou pomalu otáčejte, aby se umožnila rekonstituce lyofilizovaného prášku. Nechejte stát až 5 minut, aby zmizely bubliny. Injekční lahvičkou netřepejte.
• Parenterální léčivé přípravky je nutno před podáním vizuálně zkontrolovat na výskyt částic a změnu barvy. Rekonstituovaný přípravek KEYTRUDA je čirý až mírně opalizující, bezbarvý až slabě žlutý roztok. Injekční lahvičku zlikvidujte, pokud v ní jsou viditelné částice.
• Odeberte požadovaný objem až 2 ml (50 mg) přípravku KEYTRUDA a přeneste jej do intravenózního vaku obsahujícího roztok chloridu sodného o koncentraci 9 mg/ml (0,9%) nebo glukózy o koncentraci 50 mg/ml (5%), čímž připravíte naředěný roztok o konečné koncentraci v rozmezí od 1 do 10 mg/ml. Naředěný roztok promíchejte mírným obracením.
• Chemická a fyzikální stabilita rekonstituovaného a naředěného roztoku byla prokázána na dobu 24 hodin při pokojové teplotě (25 °C nebo nižší). Z mikrobiologického hlediska musí být přípravek použit okamžitě. Rekonstituovaný nebo naředěný roztok nesmí být zmražen. Není-li použit okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou
v odpovědnosti uživatele a nesmí být delší než celkem 24 hodin. Tento 24hodinový limit může zahrnovat až 6 hodin při pokojové teplotě (25 °C nebo nižší); veškerá další doba uchovávání musí být při teplotě 2 °C - 8 °C. Při uchovávání v chladničce musíte nechat injekční lahvičky a/nebo i.v. vaky před použitím ohřát na pokojovou teplotu. Infuzní roztok podávejte intravenózně po dobu 30 minut za použití sterilního, nepyrogenního in-line nebo add-on filtru málo vázajícího proteiny o velikosti pórů 0,2 až 5 ^m.
• Stejnou infuzní hadičkou nepodávejte současně jiné léčivé přípravky.
• Přípravek KEYTRUDA je určen k jednorázovému použití. Veškerý nepoužitý lék v injekční lahvičce zlikvidujte.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními
požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Merck Sharp & Dohme Limited
Hertford Road
Hoddesdon
Hertfordshire EN11 9BU Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/15/1024/001
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 17. července 2015
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
KEYTRUDA 25 mg/ml koncentrát pro infuzní roztok.
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička se 4 ml koncentrátu obsahuje pembrolizumabum 100 mg.
Jeden ml koncentrátu obsahuje pembrolizumabum 25 mg.
Pembrolizumab je humanizovaná monoklonální protilátka proti receptoru programované buněčné smrti PD-1 (programmed cell death-1) (izotyp IgG4/K se stabilizující změnou sekvence v Fc oblasti) vytvářená technologií rekombinantní DNA v buňkách vaječníků čínského křečíka.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Koncentrát pro infuzní roztok.
Čirý až lehce opalizující, bezbarvý až nažloutlý roztok, pH 5,2 - 5,8.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek KEYTRUDA je v monoterapii indikován k léčbě pokročilého (neresekovatelného nebo metastazujícího) melanomu u dospělých.
Přípravek KEYTRUDA je indikován k léčbě lokálně pokročilého nebo metastazujícího nemalobuněčného karcinomu plic (NSCLC) u dospělých, jejichž nádory exprimují PD-L1 a kteří již byli léčeni nejméně jedním chemoterapeutickým režimem. Pacienti s pozitivními nádorovými mutacemi EGFR nebo ALK musí předtím, než dostanou přípravek KEYTRUDA, být rovněž léčeni terapií schválenou při těchto mutacích.
4.2 Dávkování a způsob podání
Léčbu musí zahájit a musí na ni dohlížet lékař specialista se zkušenostmi v onkologické léčbě. Testování na PD-L1 u pacientů s NSCLC
Pacienti s NSCLC musí být k léčbě vybráni na základě exprese PD-L1 nádorovými buňkami potvrzené validovaným testem (viz bod 5.1).
Dávkování
Doporučená dávka přípravku KEYTRUDA je 2 mg/kg podávaná intravenózně po dobu 30 minut každé 3 týdny. Pacienty je nutno přípravkem KEYTRUDA léčit do progrese nemoci nebo do vzniku nepřijatelné toxicity. Byly pozorovány atypické odpovědi (tj. počáteční přechodné zvětšení nádoru nebo vznik nových malých lézí během prvních několika měsíců, následované zmenšením nádoru). Klinicky stabilní pacienty s počátečními známkami progrese nemoci se doporučuje léčit dál, dokud se progrese nepotvrdí.
Odložení dávky nebo vysazení přípravku (viz také bod 4.4)
Tabulka 1: Doporučené úpravy léčby přípravkem KEYTRUDA
|
Imunitně zprostředkované nežádoucí účinky |
Závažnost |
Úprava léčby |
|
Pneumonitida |
Stupeň 2 |
Dočasně vysadit* |
|
Stupeň 3 nebo 4, nebo recidivující stupeň 2 |
Trvale vysadit | |
|
Kolitida |
Stupeň 2 nebo 3 |
Dočasně vysadit* |
|
Stupeň 4 |
Trvale vysadit | |
|
Nefritida |
Stupeň 2 s kreatininem >1,5 až < 3násobek horní hranice normálu (ULN) |
Dočasně vysadit* |
|
Stupeň >3 s kreatininem > 3násobku ULN |
Trvale vysadit | |
|
Endokrinopatie |
Symptomatická hypofyzitida Diabetes typu 1 spojený s hyperglykemií stupně > 3 (glukóza >250 mg/dl nebo >13,9 mmol/l) nebo spojený s ketoacidózou Hypertyreóza stupně > 3 |
Dočasně vysadit* U pacientů s endokrinopatií stupně 3 nebo stupně 4, která se zlepšila na stupeň 2 nebo nižší a je kontrolována pomocí hormonální substituce, je možno, pokud je indikováno, zvážit v případě potřeby pokračování podávání pembrolizumabu následně po postupném vysazení kortikosteroidů. Jinak má být léčba ukončena. Hypotyreózu lze zvládnout substituční terapií bez přerušení léčby. |
|
Hepatitida |
Stupeň 2 s aspartátaminotransferázou (AST) nebo alaninaminotransferázou (ALT) >3 až 5násobek ULN nebo celkový bilirubin > 1,5 až 3násobek ULN |
Dočasně vysadit* |
|
Stupeň >3 s AST nebo ALT > 5násobek ULN nebo celkovým bilirubinem > 3násobek ULN |
Trvale vysadit | |
|
V případě jaterních metastáz s výchozí zvýšenou hodnotou AST nebo ALT stupně 2, hepatitida, kdy AST nebo ALT stoupne o > 50 % a trvá > 1 týden |
Trvale vysadit | |
|
Reakce spojené s infuzí |
Stupeň 3 nebo 4 |
Trvale vysadit |
Poznámka: stupně toxicity jsou v souladu s National Cancer Institute Common Terminology Criteria for Adverse Events Version 4.0 (NCI-CTCAE v.4).
* než se nežádoucí účinky zlepší na stupeň 0 - 1.
Přípravek KEYTRUDA je nutno trvale vysadit:
• při toxicitě stupně 4 kromě endokrinopatií, které jsou zvládnuty substitučními hormony
• pokud během 12 týdnů nelze snížit dávku kortikosteroidů na < 10 mg prednisonu nebo jeho ekvivalentu za den
• pokud se toxicita související s léčbou během 12 týdnů po poslední dávce přípravku KEYTRUDA nesníží na stupeň 0 - 1
• pokud se podruhé objeví jakákoli příhoda závažnosti stupně > 3.
Pacientům léčeným přípravkem KEYTRUDA musí být poskytnuta karta s upozorněním pro pacienta a mají být informováni o rizicích přípravku KEYTRUDA (viz také příbalová informace).
Zvláštní _ populace Starší osoby
Mezi staršími (> 65 let) a mladšími pacienty (< 65 let) nebyly hlášeny žádné rozdíly týkající se bezpečnosti a účinnosti. U této populace není úprava dávkování nutná.
Porucha funkce ledvin
U pacientů s lehkou nebo středně těžkou poruchou funkce ledvin není úprava dávkování nutná. Přípravek KEYTRUDA nebyl hodnocen u pacientů s těžkou poruchou funkce ledvin (viz bod 5.2).
Porucha funkce jater
U pacientů s lehkou poruchou funkce jater není úprava dávkování nutná. Přípravek KEYTRUDA nebyl hodnocen u pacientů se středně těžkou nebo těžkou poruchou funkce jater (viz bod 5.2).
Oční melanom
O účinnosti a bezpečnosti přípravku KEYTRUDA u pacientů s očním melanomem jsou k dispozici jen omezené údaje (viz bod 5.1).
Pediatrická populace
Bezpečnost a účinnost přípravku KEYTRUDA u dětí ve věku do 18 let nebyla dosud stanovena. Nejsou dostupné žádné údaje.
Způsob podání
Přípravek KEYTRUDA se musí podávat intravenózní infuzí trvající 30 minut. Přípravek KEYTRUDA se nesmí podávat jako nitrožilní bolus nebo bolusová injekce.
Návod k naředění léčivého přípravku před jeho podáním je uveden v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití Vyhodnocení stavu PD-L1
Při hodnocení stavu tumoru s ohledem na PD-L1 je důležité zvolit dobře validovanou a robustní metodiku, aby se minimalizovalo riziko falešně negativních nebo falešně pozitivních stanovení.
Imunitně zprostředkované nežádoucí účinky
Většina imunitně zprostředkovaných nežádoucích účinků, které se vyskytly během léčby pembrolizumabem, byla reverzibilní a zvládla se přerušením podávání pembrolizumabu, podáním kortikosteroidů a/nebo podpůrnou léčbou. Imunitně zprostředkované nežádoucí účinky se vyskytly také po podání poslední dávky pembrolizumabu.
Při podezření na imunitně zprostředkované nežádoucí účinky má být zajištěno odpovídající vyšetření, aby se etiologie potvrdila, nebo aby se vyloučily jiné příčiny. Na základě závažnosti nežádoucího účinku má být pembrolizumab vysazen a podávány kortikosteroidy. Po zlepšení na stupeň < 1 má být zahájeno postupné vysazování kortikosteroidů a ve vysazování se má pokračovat nejméně 1 měsíc. Na základě omezených údajů z klinických studií, u pacientů, jejichž imunitně zprostředkované nežádoucí účinky nemohly být kontrolovány použitím kortikosteroidů, může být zváženo podávání jiných systémových imunosupresiv.
Pembrolizumab může být znovu nasazen po 12 týdnech po poslední dávce přípravku KEYTRUDA, pokud nežádoucí účinek zůstává na stupni <1 a dávka kortikosteroidů byla redukována na < 10 mg prednisonu nebo jeho ekvivalentu za den.
Pembrolizumab musí být trvale vysazen při jakémkoli imunitně zprostředkovaném nežádoucím účinku stupně 3, který se opakuje nebo při jakékoli imunitně zprostředkované nežádoucí toxicitě stupně 4, kromě endokrinopatií, které jsou zvládnuty substitučními hormony (viz body 4.2 a 4.8).
Imunitně zprostředkovaná pneumonitida
U pacientů léčených pembrolizumabem byla hlášena pneumonitida, včetně fatálních případů (viz bod 4.8). Pacienti mají být sledováni na projevy a příznaky pneumonitidy. Podezření na pneumonitidu má být potvrzeno radiografickou zobrazovací metodou a mají být vyloučeny jiné příčiny. Při příhodách stupně > 2 mají být podávány kortikosteroidy (zahajovací dávka 1 - 2 mg/kg/den prednisonu nebo jeho ekvivalentu následovaná postupným vysazováním); při pneumonitidě stupně 2 má být pembrolizumab dočasně vysazen a při pneumonitidě stupně 3, stupně 4 nebo recidivující pneumonitidě stupně 2 má být pembrolizumab vysazen natrvalo (viz bod 4.2).
Imunitně zprostředkovaná kolitida
U pacientů léčených pembrolizumabem byla hlášena kolitida (viz bod 4.8). Pacienti mají být sledováni na projevy a příznaky kolitidy a mají být vyloučeny jiné příčiny. Při příhodách stupně > 2 mají být podávány kortikosteroidy (zahajovací dávka 1 - 2 mg/kg/den prednisonu nebo jeho ekvivalentu následovaná postupným vysazováním); při kolitidě stupně 2 nebo stupně 3 má být pembrolizumab dočasně vysazen a při kolitidě stupně 4 vysazen natrvalo (viz bod 4.2). Je nutno vzít v úvahu potenciální riziko vzniku gastrointestinální perforace.
Imunitně zprostředkovaná hepatitida
U pacientů léčených pembrolizumabem byla hlášena hepatitida (viz bod 4.8). Pacienti mají být sledováni na změny jaterních funkcí (na začátku léčby, pravidelně během léčby a dle klinického stavu) a příznaky hepatitidy a mají být vyloučeny jiné příčiny. Mají být podávány kortikosteroidy (zahajovací dávka 0,5 - 1 mg/kg/den (u příhod stupně 2) a 1 - 2 mg/kg/den (u příhod stupně > 3) prednisonu nebo jeho ekvivalentu následovaná postupným vysazováním) a na základě závažnosti zvýšení jaterních enzymů má být pembrolizumab dočasně nebo natrvalo vysazen (viz bod 4.2).
Imunitně zprostředkovaná nefritida
U pacientů léčených pembrolizumabem byla hlášena nefritida (viz bod 4.8). Pacienti mají být sledováni na změny renální funkce a mají být vyloučeny jiné příčiny renální dysfunkce. U příhod stupně > 2 mají být podávány kortikosteroidy (zahajovací dávka 1 - 2 mg/kg/den prednisonu nebo jeho ekvivalentu následovaná postupným vysazováním) a na základě závažnosti zvýšení kreatininu má být pembrolizumab při nefritidě stupně 2 dočasně vysazen a při nefritidě stupně 3 nebo 4 vysazen natrvalo (viz bod 4.2).
Imunitně zprostředkované endokrinopatie
Při léčbě pembrolizumabem byly pozorovány těžké endokrinopatie, včetně hypofyzitidy, diabetes mellitus typu 1, diabetické ketoacidózy, hypotyreózy a hypertyreózy.
V případech imunitně zprostředkovaných endokrinopatií může být nezbytná dlouhodobá substituční hormonální léčba.
U pacientů léčených pembrolizumabem byla hlášena hypofyzitida (viz bod 4.8). Pacienti mají být sledováni na projevy a příznaky hypofyzitidy (včetně hypopituitarismu a sekundární nedostatečnosti nadledvin) a mají být vyloučeny jiné příčiny. K léčbě sekundární nedostatečnosti nadledvin mají být podávány kortikosteroidy a další hormonální substituce podle klinické indikace, při symptomatické hypofyzitidě má být pembrolizumab dočasně vysazen dokud se příhoda nedostane pod kontrolu pomocí hormonální substituce. Pokud je potřeba, o pokračování v léčbě pembrolizumabem lze uvažovat po postupném vysazení kortikosteroidů (viz bod 4.2). Je nezbytné monitorovat funkci hypofýzy a hladiny hormonů, aby byla zajištěna vhodná hormonální substituce.
U pacientů léčených pembrolizumabem byl hlášen diabetes mellitus typu 1, včetně diabetické ketoacidózy (viz bod 4.8). Pacienti mají být sledováni na hyperglykemii nebo jiné projevy a příznaky diabetu. U diabetu typu 1 má být podáván inzulin a v případech hyperglykemie stupně 3 má být pembrolizumab dočasně vysazen dokud se metabolická situace nedostane pod kontrolu (viz bod 4.2).
U pacientů léčených pembrolizumabem byly hlášeny poruchy štítné žlázy, zahrnující hypotyreózu, hypertyreózu a tyreoiditidu, které se mohou objevit kdykoli během léčby; proto pacienti mají být sledováni na změny funkce štítné žlázy (na začátku léčby, pravidelně během léčby a dle klinického stavu) a na klinické projevy a příznaky poruch štítné žlázy. Hypotyreózu lze zvládnout substituční terapií bez přerušení léčby a bez kortikosteroidů. Hypertyreózu lze zvládnout symptomaticky. Při hypertyreóze stupně > 3 má být pembrolizumab dočasně vysazen dokud není znovu dosaženo stupně < 1. U pacientů s hypertyreózou stupně 3 nebo stupně 4, která se zlepšila na stupeň 2 nebo nižší, lze podle potřeby o pokračování v léčbě pembrolizumabem uvažovat po postupném vysazení kortikosteroidů (viz body 4.2 a 4.8). Je nezbytné monitorovat funkci štítné žlázy a hladiny hormonů, aby byla zajištěna vhodná hormonální substituce.
Další imunitně zprostředkované nežádoucí účinky
U pacientů léčených pembrolizumabem byly hlášeny následující další klinicky významné imunitně zprostředkované nežádoucí účinky: uveitida, artritida, myozitida, pankreatitida, těžké kožní reakce, Guillain-Barrého syndrom, myastenický syndrom, hemolytická anémie a parciální záchvaty vznikající u pacienta se zánětlivými ložisky v mozkovém parenchymu (viz bod 4.8).
Na základě závažnosti nežádoucího účinku má být pembrolizumab vysazen a podávány kortikosteroidy.
Pembrolizumab musí být znovu nasazen po 12 týdnech po poslední dávce přípravku KEYTRUDA, pokud nežádoucí účinek zůstává na stupni <1 a dávka kortikosteroidů byla redukována na < 10 mg prednisonu nebo jeho ekvivalentu za den.
Pembrolizumab má být trvale vysazen při jakémkoli imunitně zprostředkovaném nežádoucím účinku stupně 3, který se opakuje nebo při jakékoli imunitně zprostředkované nežádoucí toxicitě stupně 4 (viz body 4.2 a 4.8).
Reakce spojené s infuzí
U pacientů léčených pembrolizumabem byly hlášeny těžké reakce spojené s infuzí (viz bod 4.8). Při těžkých reakcích spojených s infuzí má být infuze ukončena a pembrolizumab natrvalo vysazen (viz bod 4.2). Pacienti s lehkou nebo středně těžkou reakcí spojenou s infuzí mohou pod pečlivým dohledem pembrolizumab nadále používat; lze uvažovat o premedikaci antipyretiky a antihistaminiky.
Pacienti vyloučení z klinických studií
Do klinických studií nebyli zařazeni pacienti s následujícími stavy: aktivní metastázy v CNS; HIV, infekce virem hepatitidy B nebo hepatitidy C; aktivní systémová autoimunitní nemoc; intersticiální plicní onemocnění; prodělaná pneumonitida vyžadující systémovou léčbu kortikosteroidy; těžká hypersenzitivita k jiné monoklonální protilátce v anamnéze; užívání imunosupresivní léčby; těžké imunitně zprostředkované nežádoucí účinky léčby ipilimumabem v anamnéze, definované jako jakákoli toxicita stupně 4 nebo toxicita stupně 3 vyžadující léčbu kortikosteroidy (> 10 mg/den prednisonu nebo jeho ekvivalentu) po dobu delší než 12 týdnů. Pacienti s aktivními infekcemi byli z klinických studií vyloučeni a předtím, než začali užívat pembrolizumab, se požadovalo, aby jejich infekce byla vyléčena. Pacienti s aktivními infekcemi vyskytujícími se během léčby pembrolizumabem byli příslušně léčeni. Pacienti s počátečními klinicky významnými renálními (kreatinin > 1,5 x ULN) nebo jaterními (bilirubin > 1,5 x ULN, ALT, AST > 2,5 x ULN při absenci jaterních metastáz) abnormalitami byli vyloučeni z klinických studií, proto jsou informace o pacientech s těžkou poruchou funkce ledvin a středně težkou až těžkou poruchou funkce jater omezeny.
U těchto pacientů lze pembrolizumab používat po pečlivém zvážení potenciálního zvýšeného rizika a za vhodné léčebné strategie.
Karta s upozorněním pro pacienta
Každý, kdo předepisuje přípravek KEYTRUDA musí být seznámen s informací pro lékaře a léčebnými strategiemi. Předepisující lékař musí s pacientem prodiskutovat rizika léčby přípravkem KEYTRUDA. Pacientovi bude poskytnuta karta s upozorněním pro pacienta s každým receptem.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
S pembrolizumabem nebyly provedeny žádné formální farmakokinetické studie lékových interakcí. Jelikož se pembrolizumab odstraňuje z oběhu katabolizací, žádné metabolické lékové interakce se nepředpokládají.
Před nasazením pembrolizumabu je nutno se vyhnout podávání systémových kortikosteroidů nebo imunosupresiv, a to kvůli jejich potenciálnímu vlivu na farmakodynamickou aktivitu a účinnost pembrolizumabu. Systémové kortikosteroidy nebo jiná imunosupresiva však lze používat po nasazení pembrolizumabu k léčbě imunitně zprostředkovaných nežádoucích účinků (viz bod 4.4)
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku
Ženy ve fertilním věku musí během léčby pembrolizumabem a ještě nejméně 4 měsíce po poslední dávce pembrolizumabu používat účinnou antikoncepci.
Údaje o podávání pembrolizumabu těhotným ženám nejsou k dispozici. Reprodukční studie na zvířatech nebyly s pembrolizumabem provedeny; nicméně na modelech březích myší bylo prokázáno, že blokáda signálu zprostředkovaného receptorem PD-L1 narušuje toleranci vůči plodu a vede ke zvýšeným ztrátám plodů (viz bod 5.3). Tyto výsledky naznačují potenciální riziko, že by podávání pembrolizumabu během těhotenství mohlo způsobit poškození plodu, včetně zvýšené míry potratu nebo porodu mrtvého plodu. Je známo, že lidské imunoglobuliny G4 (IgG4) prostupují placentární bariérou; proto má pembrolizumab, který je IgG4, potenciál ktomu, aby byl přenesen z matky do vyvíjejícího se plodu. Pembrolizumab se v těhotenství nemá používat, ledaže by klinický stav ženy léčbu pembrolizumabem vyžadoval.
Kojení
Není známo, zda se pembrolizumab vylučuje do lidského mateřského mléka. Jelikož je ale známo, že protilátky mohou být vylučovány do lidského mateřského mléka, riziko pro novorozence/kojence nelze vyloučit. Je nutno se rozhodnout, zda přerušit kojení nebo vysadit pembrolizumab, přičemž se posoudí přínos kojení pro dítě a přínos léčby pembrolizumabem pro ženu.
Fertilita
O možném vlivu pembrolizumabu na fertilitu nejsou k dispozici žádné klinické údaje. Na základě jednoměsíční a šestiměsíční studie toxicity po opakovaných dávkách u opic nebyly zjištěny žádné pozorovatelné účinky na samčí ani samičí reprodukční orgány (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Pembrolizumab může mít mírný vliv na schopnost řídit a obsluhovat stroje. Po podání pembrolizumabu byla hlášena únava (viz bod 4.8).
Souhrn bezpečnostního profilu
Pembrolizumab je nejčastěji spojován s imunitně zprostředkovanými nežádoucími účinky. Většina z nich, včetně těžkých reakcí, byla vyřešena po zahájení příslušné léčby nebo vysazení pembrolizumabu (viz „Popis vybraných nežádoucích účinků“ níže).
Bezpečnost pembrolizumabu byla v klinických studiích hodnocena u 2799 pacientů s pokročilým melanomem nebo NSCLC ve třech dávkách (2 mg/kg každé 3 týdny nebo 10 mg/kg každé 2 nebo 3 týdny). U této populace pacientů byly nejčastějšími nežádoucími účinky (> 10 %) pembrolizumabu únava (24 %), vyrážka (19 %), pruritus (18 %), průjem (12 %), nauzea (11 %) a artralgie (10 %). Většina hlášených nežádoucích účinků byla stupně závažnosti 1 nebo 2. Nejzávažnějšími nežádoucími účinky byly imunitně zprostředkované nežádoucí účinky a závažné nežádoucí účinky spojené s infuzí (viz bod 4.4).
Tabulkový seznam nežádoucích účinků
V tabulce 2 jsou hlášeny nežádoucí účinky hlášené u 2799 pacientů léčených pembrolizumabem v klinických studiích. Tyto nežádoucí účinky jsou uvedeny podle třídy orgánových systémů a četnosti. Četnosti jsou definovány následovně: velmi časté (> 1/10); časté (> 1/100 až < 1/10); méně časté (>
1/1 000 až < 1/100); vzácné (> 1/10 000 až < 1/1 000); velmi vzácné (< 1/10 000). V rámci každé skupiny četnosti jsou nežádoucí účinky seřazeny v pořadí klesající závažnosti.
Tabulka 2: Nežádoucí účinky u pacientů léčených pembrolizumabem v klinických studiích
|
Poruchy krve a lymfatického systému | |
|
Časté | |
|
Méně časté |
neutropenie, leukopenie, trombocytopenie, lymfopenie, eozinofilie |
|
Vzácné |
imunitní trombocytopenická purpura, hemolytická anémie |
|
Poruchy imunitního systému | |
|
Časté |
reakce spojená s infuzía |
|
Endokrinní poruchy | |
|
Časté |
hypertyreóza, hypotyreózab |
|
Méně časté |
hypofyzitidac, adrenální insuficience, tyreoiditida |
|
Poruchy metabolismu a výživy | |
|
Časté |
snížená chuť k jídlu |
|
Méně časté |
diabetes mellitus typu 1d, hyponatremie, hypokalemie, hypokalcemie |
|
Psychiatrické poruchy | |
|
Méně časté |
Insomnie |
|
Poruchy nervového systému | |
|
Časté |
bolest hlavy, závrať, dysgeuzie, |
|
Méně časté |
epilepsie, letargie, periferní neuropatie |
|
Vzácné |
Guillain-Barrého syndrom, myastenický syndrom |
|
Poruchy oka | |
|
Časté |
suché oko |
|
Méně časté |
uveitidae |
|
Cévní poruchy | |
|
Méně časté |
Hypertenze |
|
Respirační, hrudní a mediastinální poruchy | |
|
Časté | |
|
Gastrointestinální poruchy | |
|
Velmi časté | |
|
Časté |
kolitidag, zvracení, břišní bolesth, zácpa, suchá ústa |
|
Méně časté |
pankreatitidai |
|
Vzácné |
perforace tenkého střeva |
|
Poruchy jater a žlučových cest | |
|
Méně časté |
hepatitidaJ |
cytokinů)
|
Poruchy kůže a podkožní tkáně | |
|
Velmi časté |
vyrážkak, pruritus* |
|
Časté |
těžké kožní reakcem, vitiligon, akneiformní dermatitida, suchá kůže, erytém, ekzém |
|
Méně časté |
lišejová keratózao, psoriáza, alopecie, erythema nodosum, dermatitida, změny barvy vlasů, papula |
|
Poruchy svalové a kosterní |
soustavy a pojivové tkáně |
|
Velmi časté |
Artralgie |
|
Časté |
myozitidap, muskuloskeletální bolestq, bolest v končetině, artritida1 |
|
Méně časté |
tenosynovitidas |
|
Poruchy ledvin a močových cest | |
|
Méně časté |
nefritidat |
|
Celkové poruchy a reakce v místě aplikace | |
|
Velmi časté |
Únava |
|
Časté |
astenie, edému, pyrexie, onemocnění podobající se chřipce, třesavka |
|
Vyšetření | |
|
Časté |
zvýšená alaninaminotransferáza, zvýšená aspartátaminotransferáza, , zvýšená alkalická fosfatáza v krvi, zvýšený kreatinin v krvi |
|
Méně časté |
zvýšená amyláza, zvýšený bilirubin v krvi, hyperkalcemie |
“Následující pojmy představují skupinu příbuzných příhod, které spíše než jednotlivou příhodu popisují zdravotní stav. a. reakce spojené s infuzí (přecitlivělost na lék, anafylaktická reakce, hypersenzitivita a syndrom uvolňování
b. hypotyreóza (myxedém)
c. hypofyzitida (hypopituitarismus)
d. diabetes mellitus typu 1 (diabetická ketoacidóza)
e. uveitida (iritida a iridocyklitida)
f. pneumonitida (intersticiální plicní nemoc)
g. kolitida (mikroskopická kolitida a enterokolitida)
h. bolest břicha (břišní diskomfort, bolest horní poloviny břicha, bolest dolní poloviny břicha)
i. pankreatitida (autoimunitní pankreatitida a akutní pankreatitida)
j. hepatitida (autoimunitní hepatitida a lékové poškození jater)
k. vyrážka (erytematózní vyrážka, folikulární vyrážka, generalizovaná vyrážka, makulózní vyrážka, makulopapulózní vyrážka, papulózní vyrážka, svědící vyrážka, vezikulózní vyrážka a genitální vyrážka)
l. pruritus (kopřivka, papulózní kopřivka, generalizovaný pruritus a genitální pruritus)
m. závažné kožní reakce (exfoliativní dermatitida, erythema multiforme, exfoliativní vyrážka, pemfigoid, Stevens-Johnsonův syndrom a následující nežádoucí účinky stupně 3 a vyššího: pruritus, vyrážka, generalizovaná vyrážka a makulopapulózní vyrážka)
n. vitiligo (kožní depigmentace, kožní hypopigmentace a hypopigmentace očního víčka)
o. lišejová keratóza (lichen planus a lichen sclerosus)
p. myozitida (myalgie, myopatie, polymyalgia rheumatica a rabdomyolýza)
q. muskuloskeletální bolest (muskuloskeletální diskomfort, bolest zad, muskuloskeletální ztuhlost, muskuloskeletální bolesti na hrudi a tortikolis)
r. artritida (otok kloubů, polyartritida a kloubní výron)
s. tendosynovitida (tendinitida, synovitida a bolest šlach)
t. nefritida (autoimunitní nefritida, tubulointersticiální nefritida a selhání ledvin nebo akutní selhání ledvin se známkami nefritidy)
u. edém (periferní edém, generalizovaný edém, nadměrná zátěž tekutinami, retence tekutin, otok víček a otok rtů, otok obličeje, lokalizovaný edém, periorbitální edém)
Popis vybraných nežádoucích účinků
Údaje pro následující imunitně zprostředkované nežádoucí účinky jsou založeny na pacientech, kteří dostávali pembrolizumab ve třech dávkách (2 mg/kg každé 3 týdny nebo 10 mg/kg každé 2 nebo 3 týdny) v klinických studiích (viz bod 5.1). Léčebná strategie těchto nežádoucích účinků je popsána v bodě 4.4.
Imunitně zprostředkované nežádoucí účinky (viz bod 4.4)
Imunitně zprostředkovaná pneumonitida
Pneumonitida se vyskytla u 94 (3,4 %) pacientů léčených pembrolizumabem, včetně případů stupně 2 u 36 (1,3 %) pacientů, stupně 3 u 25 (0,9 %), stupně 4 u 7 (0,3 %) nebo stupně 5 u 4 (0,1 %) pacientů. Medián doby do nástupu pneumonitidy byl 3,3 měsíce (rozmezí 2 dny až 19,3 měsíce). Medián trvání
byl 1,5 měsíce (rozmezí 1 den až 17,2+ měsíce). Pneumonitida vedla k vysazení pembrolizumabu u 36 (1,3 %) pacientů. Pneumonitida vymizela u 55 pacientů.
Imunitně zprostředkovaná kolitida
Kolitida se vyskytla u 48 (1,7 %) pacientů léčených pembrolizumabem, včetně případů stupně 2 u 10 (0,4 %) pacientů, stupně 3 u 31(1,1%) pacientů a stupně 4 u 2 (< 0,1 %) pacientů. Medián doby do nástupu kolitidy byl 3,5 měsíce (rozmezí 10 dní až 16,2 měsíce). Medián trvání byl 1,3 měsíce (rozmezí 1 den až 8,7+ měsíce). Kolitida vedla k vysazení pembrolizumabu u 15 (0,5 %) pacientů. Kolitida vymizela u 41 pacientů.
Imunitně zprostředkovaná hepatitida
Hepatitida se vyskytla u 19 (0,7 %) pacientů léčených pembrolizumabem, včetně případů stupně 2 u 4 (0,1 %) pacientů, stupně 3 u 12 (0,4 %) pacientů a stupně 4 u 2 (< 0,1 %) pacientů. Medián doby do nástupu hepatitidy byl 1,3 měsíce (rozmezí 8 dní až 21,4 měsíce). Medián trvání byl 1,8 měsíce (rozmezí 8 dní až 20,9+ měsíce). Hepatitida vedla k vysazení pembrolizumabu u 6 (0,2 %) pacientů. Hepatitida vymizela u 15 pacientů.
Imunitně zprostředkovaná nefritida
Nefritida se vyskytla u 9 (0,3 %) pacientů léčených pembrolizumabem, včetně případů stupně 2 u 3 (0,1 %) pacientů, stupně 3 u 4(0,1%) pacientů a stupně 4 u 1 (< 0,1%) pacienta. Medián doby do nástupu nefritidy byl 5,1 měsíce (rozmezí 12 dní až 12,8 měsíce). Medián trvání byl 3,3 měsíce (rozmezí 12 dní až 8,9+ měsíce). Nefritida vedla k vysazení pembrolizumabu u 3 (0,1 %) pacientů. Nefritida vymizela u 5 pacientů.
Imunitně zprostředkované endokrinopatie
Hypofyzitida se vyskytla u 17 (0,6 %) pacientů léčených pembrolizumabem, včetně případů stupně 2 u 6 (0,2 %) pacientů, stupně 3 u 8 (0,3 %) pacientů a stupně 4 u 1 (< 0,1%) pacienta. Medián doby do nástupu hypofyzitidy byl 3,7 měsíce (rozmezí 1 den až 11,9 měsíce). Medián trvání byl 4,7 měsíce (rozmezí 8+ dní až 12,7+ měsíce). Hypofyzitida vedla k vysazení pembrolizumabu u 4 (0,1 %) pacientů. Hypofyzitida vymizela u 7 pacientů, 2 měli následky.
Hypertyreóza se vyskytla u 96 (3,4 %) pacientů léčených pembrolizumabem, včetně případů stupně 2 u 22 (0,8 %) pacientů a stupně 3 u 4 (0,1 %) pacientů. Medián doby do nástupu hypertyreózy byl
1.4 měsíce (rozmezí 1 den až 21,9 měsíce). Medián trvání byl 2,1 měsíce (rozmezí 3 dny až 15,0+ měsíce). Hypertyreóza vedla k vysazení pembrolizumabu u 2 (< 0,1 %) pacientů. Hypertyreóza vymizela u 71 (74 %) pacientů.
Hypotyreóza se vyskytla u 237 (8,5 %) pacientů léčených pembrolizumabem, včetně případů stupně 2 u 174 (6,2 %) pacientů a stupně 3 u 3 (0,1 %) pacientů. Medián doby do nástupu hypotyreózy byl
3.5 měsíce (rozmezí 1 den až 18,9 měsíce). Mediánu trvání nebylo dosaženo (rozmezí 2 dny až 27,7+ měsíce). Kvůli hypotyreóze pembrolizumab vysadil jeden pacient (< 0,1 %). Hypotyreóza vymizela u 48 (20 %) pacientů.
Imunogenita
V klinických studiích u pacientů léčených pembrolizumabem v dávce 2 mg/kg každé 3 týdny nebo 10 mg/kg každé 2 nebo 3 týdny bylo 19 (1,7 %) z 1087 vyhodnotitelných pacientů pozitivně testováno na protilátky proti pembrolizumabu, které se objevily během léčby. Při vzniku protilátek vážících pembrolizumab nebyl nalezen žádný důkaz změněné farmakokinetiky nebo bezpečnostního profilu.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
O předávkování pembrolizumabem nejsou žádné informace.
Při předávkování je nutno pacienty pečlivě sledovat na projevy nebo příznaky nežádoucích účinků a nasadit příslušnou symptomatickou léčbu.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: cytostatika, monoklonální protilátky. ATC kód: L01XC18 Mechanismus účinku
Přípravek KEYTRUDA je humánní monoklonální protilátka, která se váže na receptor programované buněčné smrti PD-1 (programmed cell death-1) a blokuje jeho interakci s ligandy PD-L1 a PD-L2. Receptor PD-1 je negativním regulátorem aktivity T-buněk, u kterého bylo prokázáno, že se podílí na regulaci T-buněčné imunitní odpovědi. Přípravek KEYTRUDA posiluje T-buněčnou odpověď, včetně protinádorové odpovědi prostřednictvím blokády vazby PD-1 na PD-L1 and PD-L2, které se exprimují v buňkách prezentujících antigeny a mohou být exprimovány nádorovými nebo jinými buňkami v mikrosprostředí nádoru.
Klinická účinnost a bezpečnost
Melanom
KEYNOTE-006: klinická studie _pacientů s melanomem, kteří dosud nebyli léčeni ipilimumabem Bezpečnost a účinnost pembrolizumabu byla hodnocena ve studii KEYNOTE-006, což byla multicentrická, kontrolovaná studie fáze III léčby pokročilého melanomu u pacientů, kteří dosud nebyli léčeni ipilimumabem. Pacienti byli randomizováni (1:1:1) do skupin léčených pembrolizumabem 10 mg/kg každé 2 (n=279) nebo 3 týdny (n=277) nebo ipilimumabem 3 mg/kg každé 3 týdny (n=278). U pacientů s melanomem s mutací BRAF V600E nebylo požadováno, aby předtím byli léčeni BRAF inhibitorem.
Pacienti byli léčeni pembrolizumabem do progrese nemoci nebo do nepřijatelné toxicity. Klinicky stabilním pacientům s počátečními známkami progrese nemoci bylo dovoleno setrvat na léčbě do potvrzení progrese nemoci. Vyhodnocení stavu nádoru bylo provedeno po 12 týdnech, poté každých 6 týdnů až do 48. týdne a následně každých 12 týdnů.
Z 834 pacientů bylo 60 % mužů, 44 % bylo ve věku > 65 let (medián věku byl 62 let [rozmezí 18-89]) a 98 % byli běloši. Stadium M1c mělo 65 % pacientů, 9 % mělo mozkové metastázy v anamnéze,
66 % nemělo žádnou a 34 % podstoupilo jednu přechozí léčbu. U 31 % byla hodnota výkonnostního stavu dle ECOG 1, 69 % mělo hodnotu výkonnostního stavu dle ECOG 0 a 32 % mělo zvýšené LDH. BRAF mutace byly hlášeny u 302 (36 %) pacientů. Z pacientů s BRAF mutací bylo 139 (46 %) již léčeno BRAF inhibitorem.
Primárními cíli bylo měření výsledků účinnosti v přežití bez progrese nemoci, hodnoceno pomocí Integrated Radiology and Oncology Assessment (IRO) s využitím Response Evaluation Criteria in Solid Tumours (RECIST), verze 1.1, a v celkovém přežití. Sekundárními cíli bylo měření výsledků účinnosti v míře celkového přežití a trvání odpovědi. Tabulka 3 shrnuje klíčová měření účinnosti u pacientů dosud neléčených ipilimumabem a na obrázcích 1 a 2 jsou uvedeny Kaplan-Meierovy křivky pro celkové přežití (OS) a přežití bez progrese nemoci (PFS).
Tabulka 3: Odpověď na pembrolizumab 10 mg/kg každé 2 nebo 3 týdny u pacientů s pokročilým melanomem neléčeným ipilimumabem ve studii KEYNOTE-006*
Obrázek 1: Kaplan-Meierova křivka celkového přežití podle léčebného ramene ve studii
KEYNOTE-006 (populace všech zařazených pacientů)
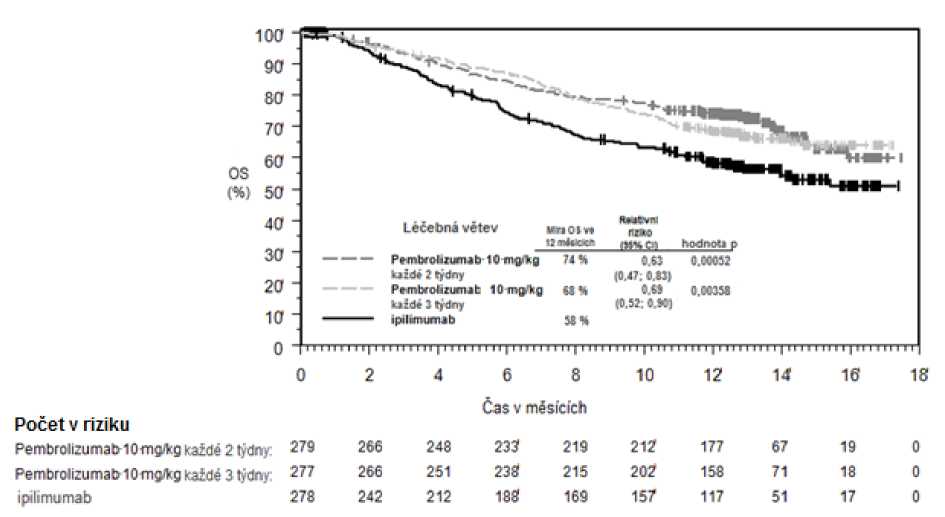
|
Cílový parametr |
Pembrolizumab 10 mg/kg každé 3 týdny n=277 |
Pembrolizumab 10 mg/kg každé 2 týdny n=279 |
Ipilimumab 3 mg/kg každé 3 týdny n=278 |
|
Celkové přežití (OS) | |||
|
Počet (%) pacientů s příhodou |
92 (33 %) |
85 (30 %) |
112 (40 %) |
|
Relativní riziko* (95% CI) |
0,69 (0,52; 0,90) |
0,63 (0,47; 0,83) |
--- |
|
Hodnota pT |
0,00358 |
0,00052 |
— |
|
Medián v měsících (95% CI) |
Nebylo dosaženo (NA, NA) |
Nebylo dosaženo (NA, NA) |
Nebylo dosaženo (13, NA) |
|
Přežití bez progrese nemoci (PFS) | |||
|
Počet (%) pacientů s příhodou |
157 (57 %) |
157 (56 %) |
188 (68 %) |
|
Relativní riziko* (95% CI) |
0,58 (0,47; 0,72) |
0,58 (0,46; 0,72) |
— |
|
Hodnota pT |
<0,00001 |
<0,00001 |
— |
|
Medián v měsících (95% CI) |
4,1 (2,9; 6,9) |
5,5 (3,4; 6,9) |
2,8 (2,8; 2,9) |
|
Nejlepší celková odpověď | |||
|
ORR % (95% CI) |
33 % (27, 39) |
34 % (28, 40) |
12 % (8, 16) |
|
Úplná odpověď % |
6% |
5 % |
1 % |
|
Částečná odpověď % |
27% |
29% |
10% |
|
Trvání odpovědi* | |||
|
Medián v měsících (rozmezí) |
Nebylo dosaženo (1,4+; 8,1+) |
8 3 (1,4+; 8,3+) |
Nebylo dosaženo (1,1+; 7,9+) |
|
% přetrvávající odpovědi |
97 % |
89 % |
88 % |
* Relativní riziko (pembrolizumab v porovnání s ipilimumabem) založeno na stratifikovaném Cox proporčním modelu rizik
* Založeno na stratifikovaném Log rank testu
* Založeno na pacientech s nejlepší celkovou odpovědí jako potvrzenou úplnou nebo částečnou odpovědí NA = není k dispozici
120 "
110 -
100
30
TQ
PFS o® .
40
Lecsbnivtis'.-
ním PFS* Min PFS v
laum * ■*! •** Mriniap
Přřft&iůJ«iw<wa& 10
klid# 2 fa dna
k!3d+ 3 fídň-p
ipilirrumjt
1r*~
i-
í :
:
*0.00001
44 %
27
iř %
-i -
ID.!*: 0.721
■;0.i7. 9.721
L._
<0.00001
10
Čas v mísících
Počet v riziku
PembicliiumaMOiTiBiligyaJdř Ztfdňh- 27$
PímbrclizumablCmaAaioíaf Jlýdny 277
ipiliiriumab $76
KEYNOTE-002: klinická studie _pacientů s melanomem, kteří _již byli léčeni ipilimumabem Bezpečnost a účinnost pembrolizumabu byla zjišťována ve studii KEYNOTE-002, což je multicentrická, kontrolovaná studie léčby pokročilého melanomu u pacientů, kteří byli léčeni ipilimumabem a pokud byli pozitivní na mutaci BRAF V600, léčeni BRAF nebo MEK inhibitory. Pacienti byli randomizováni (1:1:1) do skupin s pembrolizumabem v dávce 2 (n=180) nebo 10 mg/kg (n=181) každé 3 týdny nebo chemoterapií (n=179; včetně dakarbazinu, temozolomidu, karboplatiny, paklitaxelu nebo karboplatiny+paklitaxelu). Do studie nebyli zařazeni pacienti s autoimunitním onemocněním nebo pacienti léčení imunosupresivy; dalšími kritérii pro nezařazení byl těžký nebo život ohrožující imunitně zprostředkovaný nežádoucí účinek léčby ipilimumabem v anamnéze, definovaný jako jakákoli toxicita stupně 4 nebo toxicita stupně 3 vyžadující léčbu kortikosteroidem (> 10 mg/den prednisonu nebo ekvivalentní dávka) po dobu delší než 12 týdnů; probíhající nežádoucí účinky v důsledku předchozí léčby ipilimumabem stupně > 2; předchozí těžká hypersenzitivita na jiné monoklonální protilátky; pneumonitida nebo intersticiální plicní nemoc v anamnéze; HIV, infekce virem hepatitidy B nebo hepatitidy C a hodnota výkonnostního stavu dle ECOG > 2.
Pacienti byli léčeni pembrolizumabem do progrese nemoci nebo nepřijatelné toxicity. Klinicky stabilním pacientům s počátečními známkami progrese nemoci bylo dovoleno setrvat na léčbě do potvrzení progrese nemoci. Vyhodnocení stavu nádoru bylo provedeno po 12 týdnech, poté každých 6 týdnů až do 48. týdne a následně každých 12 týdnů. Pacientům na chemoterapii, u kterých po prvním plánovaném vyhodnocení nemoci došlo k nezávisle potvrzené progresi, byl umožněn přechod do dvojitě zaslepeného uspořádání, kde dostávali 2 mg/kg nebo 10 mg/kg pembrolizumabu každé 3 týdny.
Z 540 pacientů bylo 61 % mužů, 43 % mělo > 65 let (medián věku byl 62 let [rozmezí 15-89]) a 98 % byli běloši. Stadium M1c mělo 82 % pacientů, 73 % podstoupilo nejméně dvě a 32 % pacientů podstoupilo tři nebo více předchozích systémových terapií pokročilého melanomu. U 45 % byla hodnota výkonnostního stavu dle ECOG 1, 40 % mělo zvýšené LDH a 23 % mělo BRAF mutaci.
Primárními cíli bylo měření výsledků účinnosti v PFS hodnocené pomocí IRO s využitím RECIST verze 1.1 a v OS. Sekundárními cíli bylo měření výsledků účinnosti v ORR a v trvání odpovědi. Tabulka 4 shrnuje klíčové cíle měření účinnosti u pacientů již léčených ipilimumabem, přičemž na obrázku 3 je uvedena Kaplan-Meierova křivka přežití bez progrese nemoci. V obou ramenech léčených pembrolizumabem bylo přežití bez progrese nemoci vyšší než při chemoterapii a nebyly zde žádné rozdíly mezi dávkami pembrolizumabu. Údaje o celkovém přežití nebyly v době analýzy přežití
bez progrese nemoci dostatečné. V předběžných analýzách celkového přežití, které nebyly upraveny na potenciálně matoucí vliv přechodu některých pacientů léčených ve skupině chemoterapií do skupiny léčených pembrolizumabem, nebyl rozdíl mezi pembrolizumabem a chemoterapií statisticky významný. Z pacientů randomizovaných do skupiny léčené chemoterapií přešlo do skupin léčených pembrolizumabem 48 %.
Tabulka 4: Odpověď na pembrolizumab 2 mg/kg nebo 10 mg/kg každé 3 týdny u pacientů s neresekovatelným nebo metastazujícím melanomem ve studii KEYNOTE-002
|
Cílový parametr |
Pembrolizumab 2 mg/kg každé 3 týdny n=180 |
Pembrolizumab 10 mg/kg každé 3 týdny n=181 |
Chemoterapie n=179 |
|
Přežití bez progrese nemoci (PFS) | |||
|
Počet (%) pacientů s příhodou |
129(72 %) |
126 (70 %) |
155 (87 %) |
|
Relativní riziko* (95% CI) |
0,57 (0,45; 0,73) |
0,50 (0,39; 0,64) |
--- |
|
Hodnota pT |
< 0,0001 |
< 0,0001 |
— |
|
Medián v měsících (95% CI) |
2,9 (2,8; 3,8) |
2,9 (2,8; 4,7) |
2,7 (2,5; 2,8) |
|
Celkové přežití (OS) | |||
|
Počet (%) pacientů s příhodou |
73 (41 %) |
69 (38 %) |
78 (44 %) |
|
Relativní riziko* (95% CI) |
0,88 (0,64; 1,22) |
0,78 (0,56; 1,08) |
--- |
|
Hodnota pT |
0,2294 |
0,0664 |
— |
|
Nejlepší celková odpověď | |||
|
Celková míra odpovědi % (95% CI) |
21 %(15, 28) |
25 % (19, 32) |
4 % (2, 9) |
|
Úplná odpověď % |
2% |
3 % |
0% |
|
Částečná odpověď % |
19% |
23 % |
4% |
|
Trvání odpovědi | |||
|
Medián v měsících (rozmezí) |
Nebylo dosaženo (1,4+; 11,5+) |
Nebylo dosaženo (1,2+; 11,1+) |
8,5 (1,6+; 9,5) |
|
% přetrvávající |
87% |
80% |
63 % |
* Relativní riziko (pembrolizumab v porovnání s chemoterapií) založeno na stratifikovaném Cox proporčním modelu rizik ^ Založeno na stratifikovaném Log rank testu
Lecebna vtlev
Miř* PF 5 v UimFFSv
90 -
Snřiicich .<saHCii Hodnota p
Pcmbrdiz.uín*j lumg ^
30 -
UlS* i tp ín>
Pumbrdi z.uíTiii 2 mg*1 Iq Vaidt J týdny
CN^OtřílCit
:: i
□,5Ů
O.Í7
lO.íí 5.13)
* G.DCOl
T0 -
'TO -
PFS
4Q -
30 -
Cas v měsících
Počet v riziku
PefTTtuulzumat? 10 n^JKo Pembratzumas 2 mgíKgi Chemoterapie
KEYNOTE-001: otevřená studie _pacientů s melanomem, kteří dosud nebyli léčeni ipilimumabem a kteří j již byli léčeni ipilimumabem
Bezpečnost a účinnost pembrolizumabu u pacientů s pokročilým melanomem byla hodnocena v nekontrolované otevřené studii KEYNOTE-001. Účinnost byla hodnocena u 276 pacientů ze dvou definovaných kohort, jedna zahrnovala pacienty již léčené ipilimumabem (a pokud byli pozitivní na mutaci BRAF V600, léčeni BRAF nebo MEK inhibitory) a druhá zahrnovala pacienty, kteří ipilimumabem dosud léčeni nebyli. Pacienti byli náhodně přiřazeni do skupin s pembrolizumabem v dávce 2 mg/kg každé 3 týdny nebo 10 mg/kg každé 3 týdny. Pacienti byli léčeni pembrolizumabem do progrese nemoci nebo nepřijatelné toxicity. Klinicky stabilním pacientům s počátečními známkami progrese nemoci bylo dovoleno setrvat na léčbě do potvrzení progrese nemoci. Vylučovací kritéria byla podobná jako ve studii KEYNOTE-002.
Z 89 pacientů léčených 2 mg/kg pembrolizumabu, kteří byli předtím léčeni ipilimumabem, bylo 53 % mužů, 33 % bylo ve věku > 65 let a medián věku byl 59 let (rozmezí 18 až 88). Kromě 2 byli všichni pacienti běloši. Stadium M1c mělo 84 % pacientů a 8 % mělo mozkové metastázy v anamnéze.
70 % podstoupilo nejméně dvě a 35 % pacientů podstoupilo tři nebo více předchozích terapií pokročilého melanomu. BRAF mutace byly hlášeny u 13 % hodnocené populace. Všichni pacienti s BRAF mutací byli předtím léčeni BRAF inhibitorem.
Z 51 pacientů léčených 2 mg/kg pembrolizumabu, kteří dosud nebyli léčeni ipilimumabem, bylo 63 % mužů, 35 % bylo ve věku > 65 let a medián věku byl 60 let (rozmezí 35 až 80). Kromě jednoho pacienta byli všichni běloši. Stadium M1c mělo 63 % pacientů a 2 % měla mozkové metastázy v anamnéze. 45 % procent nepodstoupilo žádné předchozí terapie pokročilého melanomu. BRAF mutace byly hlášeny u 20 (39 %) pacientů. Z pacientů s BRAF mutací bylo 10 (50 %) předtím léčeno BRAF inhibitorem.
Primárním cílem bylo měření výsledků účinnosti v celkové míře odpovědi vyhodnocené nezávislým vyšetřením pomocí RECIST 1.1. Sekundárními cíli bylo měření výsledků účinnosti v míře kontroly nemoci (DCR; včetně úplné odpovědi, částečné odpovědi a stabilizace onemocnění), v trvání odpovědi, v přežití bez progrese nemoci a v celkovém přežití. Odpověď nádoru byla hodnocena ve 12týdenních intervalech. Klíčové cíle měření účinnosti u již léčených pacientů nebo u pacientů dosud neléčených ipilimumabem, dostávajících pembrolizumab v doporučené dávce, uvádí tabulka 5.
Tabulka 5: Odpověď na pembrolizumab 2 mg/kg každé 3 týdny u pacientů s neresekovatelným nebo metastazujícím melanomem ve studii KEYNOTE-001
|
Cílový parametr |
Pembrolizumab 2 mg/kg každé 3 týdny u pacientů předtím léčených ipilimumabem n=89 |
Pembrolizumab 2 mg/kg každé 3 týdny u pacientů dosud ipilimumabem neléčených n=51 |
|
Nejlepší celková odpověď* stanovená pomocí IRO^ | ||
|
ORR %, (95% CI) |
25 % (16, 35) |
33 % (21, 48) |
|
Úplná odpověď |
3 % |
10% |
|
Částečná odpověď |
21 % |
24% |
|
Míra kontroly onemocnění %* |
49% |
49% |
|
Trvání odpovědi5 | ||
|
Medián v měsících (rozmezí) |
Nebylo dosaženo (2,8+; 14,3+) |
Nebylo dosaženo (1,6+; 13,8+) |
|
% přetrvávající |
86 %1 |
82 %# |
|
Přežití bez progrese nemoci (PFS) | ||
|
Medián v měsících (95% CI) |
4,9 (2,8; 8,3) |
5,5 (2,8; 14,0) |
|
Míra přežití bez progrese nemoci po 6 měsících |
43 % |
50% |
|
Celkové přežití (OS) | ||
|
Medián v měsících (95% CI) |
Nebylo dosaženo (11, není k dispozici) |
Nebylo dosaženo (14, není k dispozici) |
|
Míra celkového přežití po 12 měsících |
60% |
72% |
* Zahrnuje pacienty, kteří při vstupu nemají nezávislou radiologickou metodou zjištěnu žádnou měřitelnou nemoc t IRO = Integrated radiology and oncologist assessment (integrované rentgenové a onkologické vyhodnocení) s využitím
RECIST 1.1
í
§
1
#
Založeno na nejlepší odpovědi stabilizované nemoci nebo lepší
Založeno na pacientech s odpovědí potvrzenou nezávislou kontrolou, od data, kdy byla odpověď poprvé zaznamenána; n=22 u pacientů již léčených ipilimumabem; n=17 u pacientů dosud ipilimumabem neléčených Respondéři byli sledováni po dobu nejméně 12 měsíců po zahájení léčby Respondéři byli sledováni po dobu nejméně 15 měsíců po zahájení léčby
Výsledky byly u pacientů již léčených ipilimumabem (n=84) a pacientů dosud ipilimumabem neléčených (n=52), kteří dostávali 10 mg/kg pembrolizumabu každé 3 týdny, podobné jako výsledky pozorované u pacientů, kteří dostávali 2 mg/kg pembrolizumabu každé 3 týdny.
Analýzy subpopulací
Stav BRAF mutací u melanomu
Byla provedena analýza podskupin studie KEYNOTE-002 u pacientů, kteří měli gen BRAF divokého typu (n=415; 77 %) nebo BRAF mutaci bez předchozí léčby cílené na BRAF (n=125; 23 %). Relativní riziko přežití bez progrese nemoci (souhrnně pro pembrolizumab [2 mg/kg nebo 10 mg/kg každé 3 týdny] vs. chemoterapie) bylo 0,51 (95% CI: 0,41; 0,65) u genu BRAF divokého typu a 0,56 (95% CI: 0,37; 0,85) u BRAF mutace s předchozí léčbou cílenou na BRAF. Relativní riziko přežití bez progrese nemoci pro pembrolizumab 2 mg/kg každé 3 týdny vs. chemoterapie bylo 0,51 (95% CI:
0,39; 0,67) u genu BRAF divokého typu a 0,74 (95% CI: 0,46; 1,18) u BRAF mutace s předchozí léčbou cílenou na BRAF. Relativní riziko celkového přežití souhrnně pro pembrolizumab vs. chemoterapie bylo 0,83 (95% CI: 0,60; 1,15) u genu BRAF divokého typu a 0,82 (95% CI: 0,47; 1,43) u BRAF mutace s předchozí léčbou cílenou na BRAF. Relativní riziko celkového přežití pro pembrolizumab 2 mg/kg každé 3 týdny vs. chemoterapie bylo 0,80 (95% CI: 0,55; 1,18) u genu BRAF divokého typu a 1,03 (95% CI: 0,55; 1,91) u BRAF mutace s předchozí léčbou cílenou na BRAF. Celková míra odpovědi souhrnně pro pembrolizumab a pembrolizumab 2 mg/kg každé 3 týdny vs. chemoterapie byla 27 % a 25 % vs. 6 % u genu BRAF divokého typu a 12% a 9 % vs. 0% u BRAF mutace s předchozí léčbou cílenou na BRAF.
Byla provedena analýza podskupin studie KEYNOTE-006 u pacientů, kteří měli BRAF divokého typu (n=525; 63 %), BRAF mutaci bez předchozí léčby cílené na BRAF (n=163; 20 %) a BRAF mutaci s předchozí léčbou cílenou na BRAF (n=139; 17 %). Relativní riziko přežití bez progrese nemoci (souhrnně pro pembrolizumab [10 mg/kg každé 2 nebo 3 týdny] vs. ipilimumab) bylo 0,57 (95% CI: 0,45; 0,73) u genu BRAF divokého typu, 0,50 (95% CI: 0,32; 0,77) u BRAF mutace bez předchozí léčby cílené na BRAF a 0,73 (95% CI: 0,48; 1,11) u BRAF mutace s předchozí léčbou cílenou na BRAF. Relativní riziko celkového přežití souhrnně pro pembrolizumab vs. ipilimumab bylo 0,61 (95% CI: 0,46; 0,82) u genu BRAF divokého typu, 0,69 (95% CI: 0,33; 1,45) u BRAF mutace bez předchozí léčby cílené na BRAF a 0,75 (95% CI: 0,45; 1,26) u BRAF mutace s předchozí léčbou cílenou na BRAF. Celková míra odpovědi souhrnně pro pembrolizumab vs. ipilimumab byla 34 % vs. 13 % u genu BRAF divokého typu, 41% vs. 13% u BRAF mutace bez předchozí léčby cílené na BRAF a 21% vs. 6% u BRAF mutace s předchozí léčbou cílenou na BRAF.
Stav PD-L1 u melanomu
Byla provedena analýza podskupin studie KEYNOTE-002 u pacientů, kteří byli PD-L1 pozitivní (Allredovo histoskore > 2 vyjadřující membránovou expresi PD-L1 u > 1 % nádorových buněk) vs. PD-L1 negativní (Allredovo histoskore 0 nebo 1). Exprese PD-L1 byla testována retrospektivně imunohistochemickým stanovením pomocí 22C3 protilátky PD-L1. Z pacientů, u kterých byla měřitelná exprese PD-L1 (78%), bylo 69 % (n=291) PD-L1 pozitivních a 31 % (n=130) bylo PD-L1 negativních. Relativní riziko přežití bez progrese nemoci (souhrnně pro pembrolizumab [2 mg/kg nebo 10 mg/kg každé 3 týdny] vs. chemoterapie) bylo 0,52 (95% CI: 0,39; 0,68) u PD-L1 pozitivních pacientů a 0,60 (95% CI: 0,38; 0,94) u PD-L1 negativních pacientů. Relativní riziko přežití bez progrese nemoci pro pembrolizumab 2 mg/kg každé 3 týdny vs. chemoterapie bylo 0,54 (95% CI:
0,39; 0,75) u PD-L1 pozitivních pacientů a 0,89 (95% CI: 0,53; 1,50) u PD-L1 negativních pacientů. Relativní riziko celkového přežití souhrnně pro pembrolizumab vs. chemoterapie bylo 0,82 (95% CI: 0,55; 1,23) u PD-L1 pozitivních pacientů a 0,77 (95% CI: 0,43; 1,37) u PD-L1 negativních pacientů. Relativní riziko celkového přežití pro pembrolizumab 2 mg/kg každé 3 týdny vs. chemoterapie bylo 0,93 (95% CI: 0,58; 1,49) u PD-L1 pozitivních pacientů a 1,19 (95% CI: 0,58; 2,46) u PD-L1 negativních pacientů. Celková míra odpovědi souhrnně pro pembrolizumab a pembrolizumab 2 mg/kg každé 3 týdny vs. chemoterapie byla 26 % a 23 % vs. 4 % u PD-L1 pozitivních pacientů a 15 % a 11 % vs. 8 % u PD-L1 negativních pacientů.
Byla provedena analýza podskupin studie KEYNOTE-006 u pacientů, kteří byli PD-L1 pozitivní (n=671; 80 %) vs. PD-L1 negativní (n=150; 18 %). Z pacientů, u kterých byla měřitelná exprese PD-L1 (98 %), bylo 82 % PD-L1 pozitivních a 18 % bylo PD-L1 negativních. Relativní riziko přežití bez progrese nemoci (souhrnně pro pembrolizumab [10 mg/kg každé 2 nebo 3 týdny] vs. ipilimumab) bylo 0,53 (95% CI: 0,43; 0,65) u PD-L1 pozitivních pacientů a 0,73 (95% CI: 0,47; 1,11) u PD-L1 negativních pacientů. Relativní riziko celkového přežití souhrnně pro pembrolizumab vs. ipilimumab bylo 0,56 (95% CI: 0,43; 0,73) u PD-L1 pozitivních pacientů a 0,95 (95% CI: 0,56; 1,62) u PD-L1 negativních pacientů. Celková míra odpovědi souhrnně pro pembrolizumab vs. ipilimumabová skupina byla 37% vs. 12% u PD-L1 pozitivních pacientů a 18% vs. 11% u PD-L1 negativních pacientů.
Oční melanom
U 20 subjektů s očním melanomem zařazených do studie KEYNOTE-001 nebyly hlášeny žádné objektivní odpovědi; u 6 pacientů byla hlášena stabilizace onemocnění.
NSCLC
KEYNOTE-010: kontrolovaná studie u pacientů s NSCLC, kteří již byli léčeni chemoterapií Bezpečnost a účinnost pembrolizumabu byla hodnocena ve studii KEYNOTE-010, což byla multicentrická, otevřená, kontrolovaná studie léčby pokročilého NSCLC u pacientů, kteří již byli léčeni chemoterapií obsahující platinu. Pacienti byli pozitivní na expresi PD-L1 (skóre nádorového podílu (tumour proportion score - TPS) > 1 % získáno pomocí PD-L1 IHC 22C3 pharmDx™ Kit). U pacientů s mutací aktivující EGFR nebo translokací ALK před podáním pembrolizumabu také došlo k progresi nemoci při terapii schválené při těchto mutacích. Pacienti byli randomizováni (1:1:1) do skupiny léčené pembrolizumabem v dávce 2 (n=344) nebo 10 mg/kg (n=346) každé 3 týdny nebo docetaxelem v dávce 75 mg/m2 každé 3 týdny (n=343) do progrese nemoci nebo nepřijatelné toxicity. Do klinického hodnocení nebyli zařazeni pacienti s autoimunitním onemocněním; zdravotním stavem vyžadujícím imunosupresi ani pacienti, kteří byli v předchozích 26 týdnech na hrudníku ozářeni dávkou větší než 30 Gy. Vyhodnocení stavu nádoru bylo provedeno každých 9 týdnů.
Výchozí charakteristiky této populace byly: medián věku 63 let (42 % bylo ve věku 65 nebo více);
61 % byli muži; 72 % běloši a 21 % asiaté a 34 % mělo stav výkonnosti ECOG 0 a 66 % mělo stav výkonnosti ECOG 1. Charakteristiky nemoci byly skvamózní (21 %) a neskvamózní (70 %); M1 (91 %); stabilní metastázy v mozku (15 %) a incidence mutací byla EGFR (8 %) nebo ALK (1 %). Předchozí léčba zahrnovala režim s platinovým dubletem (100 %); pacienti předtím podstoupili jednu (69 %) nebo dvě nebo více léčebných linií (29 %).
Primárními kritérii hodnocení účinnosti bylo celkové přežití a přežití bez progrese nemoci, které byly hodnoceny zaslepenou nezávislou centrální posudkovou komisí za využití kritérií RECIST 1.1. Sekundárními kritérii hodnocení účinnosti byla míra celkového přežití a trvání odpovědi. Tabulka 6 shrnuje klíčová měření účinnosti u celé populace (TPS > 1 %) a u pacientů s TPS > 50 %, přičemž Kaplan-Meierova křivka celkového přežití (TPS > 1 %) je uvedena na obrázku 4.
Tabulka 6: odpověď na pembrolizumab 2 nebo 10 mg/kg každé 3 týdny u pacientů s již léčeným NSCLC ve studii KEYNOTE-010
|
Cílový parametr |
Pembrolizumab 2 mg/kg každé 3 týdny |
Pembrolizumab 10 mg/kg každé 3 týdny |
Docetaxel 75 mg/m2 každé 3 týdny |
|
TPS > 1 % | |||
|
Počet pacientů |
344 |
346 |
343 |
|
Celkové přežití | |||
|
Počet (%) pacientů s příhodou |
172 (50 %) |
156 (45 %) |
193 (56 %) |
|
Poměr rizik* (95% CI) |
0,71 (0,58; 0,88) |
0,61 (0,49; 0,75) |
--- |
|
Hodnota p* |
<0,001* |
<0,001* |
--- |
|
Medián v měsících (95% CI) |
10,4 (9,4; 11,9) |
12,7 (10,0; 17,3) |
8,5 (7,5; 9,8) |
|
Přežití bez progrese nemoci5 | |||
|
Počet (%) pacientů s příhodou |
266(77 %) |
255 (74 %) |
257 (75 %) |
|
Poměr rizik* (95% CI) |
0,88 (0,73; 1,04) |
0,79 (0,66; 0,94) |
--- |
|
Hodnota p* |
0,068 |
0,005 |
— |
|
Medián v měsících (95% CI) |
3,9 (3,1; 4,1) |
4,0 (2,6; 4,3) |
4,0 (3,1; 4,2) |
|
Celková míra odpovědi5 | |||
|
% celkové míry odpovědi1 (95% CI) |
18 % (14, 23) |
18 % (15, 23) |
9 % (7, 13) |
|
Trvání odpovědi 5,#’P | |||
|
Medián v měsících (rozmezí) |
Nebylo dosaženo (0,7+; 20,1+) |
Nebylo dosaženo (2,1+; 17,8+) |
6,2 (1,4+; 8,8+) |
|
% přetrvávajících |
73 % |
72 % |
34 % |
|
TPS > 50% | |||
|
Počet pacientů |
139 |
151 |
152 |
|
Celkové přežití | |||
|
Počet (%) pacientů s příhodou |
58 (42 %) |
60 (40 %) |
86 (57 %) |
|
Poměr rizik* (95% CI) |
0,54 (0,38; 0,77) |
0,50 (0,36; 0,70) |
— |
|
Hodnota p* |
<0,001* |
<0,001* |
— |
|
Medián v měsících (95% CI) |
14,9 (10,4; NA) |
17,3 (11,8; NA) |
8,2 (6,4; 10,7) |
|
Přežití bez progrese nemoci5 | |||
|
Počet (%) pacientů s příhodou |
89 (64 %) |
97 (64 %) |
118 (78 %) |
|
Poměr rizik* (95% CI) |
0,58 (0,43, 0,77) |
0,59 (0,45, 0,78) |
— |
|
Hodnota p* |
<0,001* |
<0,001* |
— |
|
Medián v měsících (95% CI) |
5,2 (4,0; 6,5) |
5,2 (4,1; 8,1) |
4,1 (3,6; 4,3) |
|
Celková míra odpovědi5 | |||
|
% celkové míry odpovědi1 (95% CI) |
30 % (23; 39) |
29 % (22; 37) |
8 % (4; 13) |
|
Trvání odpovědi5’#’14 | |||
|
Medián v měsících (rozmezí) |
Nebylo dosaženo (0,7+; 16,8+) |
Nebylo dosaženo (2,1+; 17,8+) |
8,1 (2,1+; 8,8+) |
|
% přetrvávajících |
76 % |
75 % |
33 % |
* Poměr rizik (pembrolizumab v porovnání s docetaxelem) na základě stratifikovaného
Coxova proporčního modelu rizik
* Na základě stratifikovaného Log rank testu
* Statisticky významné na základě předem specifikované a úrovně upravené na multiplicitu § Vyhodnoceno zaslepenou nezávislou centrální posudkovou komisí za využití kritérií
RECIST 1.1
1 Všechny odpovědi byly částečnými odpověďmi
* Na základě pacientů s nejlepší celkovou odpovědí jako potvrzenou úplnou nebo částečnou odpovědí
P Zahrnuje 30, 31 a 2 pacienty s odpovědí přetrvávající 6 nebo více měsíců v rameni léčeném
pembrolizumabem 2 mg/kg, pembrolizumabem 10 mg/kg a docetaxelem, v uvedeném pořadí
R Zahrnuje 22, 24 a 1 pacienta s odpovědí přetrvávající 6 nebo více měsíců v rameni léčeném pembrolizumabem 2 mg/kg, pembrolizumabem 10 mg/kg a docetaxelem, v uvedeném pořadí
Obrázek 4: Kaplan-Meierova křivka celkového přežití podle léčebného ramene ve studii KEYNOTE-010 (pacienti se skóre nádorového podílu exprimujícího PD-L1 > 1 %, populace
všech léčených pacientů)

|
Počet v riziku |
0 |
5 |
10 Čas v měsících |
15 |
20 |
25 |
|
Pembrolizumab 2 mg/kg: |
344 |
259 |
115 |
49 |
12 |
0 |
|
Pembrolizumab 10 mg/kg: |
346 |
255 |
124 |
56 |
6 |
0 |
|
Docetaxel: |
343 |
212 |
79 |
33 |
1 |
0 |
Výsledky účinnosti byly v ramenech léčených 2 mg/kg a 10 mg/kg pembrolizumabu podobné. Výsledky účinnosti ohledně celkového přežití byly na základě meziskupinového porovnání konzistentní bez ohledu na stáří vzorku nádoru (nový vs. archivní).
V analýzách podskupin byl u pacientů, kteří nikdy nebyli kuřáci nebo u pacientů s nádory nesoucími mutace aktivující EGFR, kteří dostávali alespoň chemoterapii na bázi platiny a inhibitor tyrosinkinázy, pozorován snížený přínos přežití u pembrolizumabu ve srovnání s docetaxelem; avšak vzhledem k nízkému počtu pacientů nemohou být z těchto údajů učiněny žádné konečné závěry.
Bezpečnost a účinnost pembrolizumabu u pacientů s nádory, které neexprimují PD-L1 nebyla stanovena.
Pediatrická populace
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s pembrolizumabem u jedné nebo více podskupin pediatrické populace při léčbě všech nemocí zařazených v kategorii maligních neoplazmat (kromě nervového systému, krvetvorné a lymfoidní tkáně) (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Farmakokinetika pembrolizumabu byla hodnocena u 2856 pacientů s metastazujícím nebo neresekovatelným melanomem, NSCLC nebo karcinomem, kteří dostávali dávky v rozmezí 1 až 10 mg/kg každé 2 nebo 3 týdny.
Absorpce
Pembrolizumab se podává intravenózní cestou a proto je okamžitě a zcela biologicky dostupný.
Distribuce
V souladu s omezenou extravaskulární distribucí je distribuční objem pembrolizumabu
v rovnovážném stavu malý (~7,4 l; CV: 19 %). Jak se u protilátky předpokládá, pembrolizumab se specificky neváže na plazmatické proteiny.
Biotransformace
Pembrolizumab se katabolizuje nespecifickými cestami; metabolismus k jeho clearance nepřispívá. Eliminace
Systémová clearance pembrolizumabu je přibližně 0,2 l/den (CV: 37 %) a terminální biologický poločas (t/2) je přibližně 27 dní (CV: 38 %).
Linearita/nelinearita
Expozice pembrolizumabu, vyjádřená jako maximální koncentrace (Cmax) nebo plocha pod křivkou průběhu plazmatických koncentrací v čase (AUC) se v rozmezí použitém pro zjišťování účinnosti zvyšovala v závislosti na dávce. Při opakovaném podání bylo zjištěno, že clearance pembrolizumabu je nezávislá na čase, přičemž systémová akumulace byla při podávání každé 3 týdny přibližně 2,2násobná. Téměř rovnovážné koncentrace pembrolizumabu byly dosaženy po 18 týdnech; medián Cmin po 18 týdnech byla během režimu podávání 2 mg/kg každé 3 týdny přibližně 24 ^g/ml.
Zvláštní populace
V analýzách populační farmakokinetiky byly hodnoceny vlivy různých proměnných
na farmakokinetiku pembrolizumabu. Clearance pembrolizumabu se zvyšovala se zvyšující se tělesnou hmotností; výsledné rozdíly v expozici byly odpovídajícím způsobem vyřešeny dávkováním na bázi mg/kg. Následující faktory neměly na clearance pembrolizumabu žádný klinicky významný vliv: věk (rozmezí 15 až 94 let), pohlaví, rasa, lehká nebo středně těžká porucha funkce ledvin, lehká porucha funkce jater a nádorová zátěž.
Porucha funkce ledvin
Vliv poruchy funkce ledvin na clearance pembrolizumabu byl vyhodnocen analýzami populační farmakokinetiky u pacientů s lehkou nebo středně těžkou poruchou funkce ledvin v porovnání s pacienty s normální funkcí ledvin. Mezi pacienty s lehkou nebo středně těžkou poruchou funkce ledvin a pacienty s normální funkcí ledvin nebyly ohledně clearance pembrolizumabu zjištěny žádné klinicky významné rozdíly. U pacientů s těžkou poruchou funkce ledvin nebyl pembrolizumab hodnocen.
Porucha funkce jater
Vliv poruchy funkce jater na clearance pembrolizumabu byl vyhodnocen analýzami populační farmakokinetiky u pacientů s lehkou poruchou funkce jater (definováno za použití kritérií hodnocení jaterní dysfunkce amerického National Cancer Institute) v porovnání s pacienty s normální funkcí jater. Mezi pacienty s lehkou poruchou funkce jater a pacienty s normální funkcí jater nebyly ohledně clearance pembrolizumabu nalezeny žádné klinicky významné rozdíly. U pacientů se středně těžkou nebo těžkou poruchou funkce jater nebyl pembrolizumab hodnocen (viz bod 4.2).
5.3 Předklinické údaje vztahující se k bezpečnosti
Bezpečnost pembrolizumabu byla hodnocena v 1měsíční a 6měsíční studii toxicity po opakovaném podávání na makacích jávských, kterým se podávaly intravenózně dávky 6, 40 a 200 mg/kg jednou týdně v 1měsíční studii a jednou každé 2 týdny v 6měsíční studii, následované 4měsíčním obdobím bez léčby. Nebyla zaznamenána žádná toxikologicky významná zjištění, přičemž hladina, při níž nebyly pozorovány žádné nežádoucí účinky (No Observed Adverse Effect Level - NOAEL) v obou studiích byla >200 mg/kg, což je 19násobek expozice u lidí při nejvyšší klinicky testované dávce (10 mg/kg).
Reprodukční studie na zvířatech nebyly s pembrolizumabem provedeny. Má se za to, že cesta PD-1/PD-L1 se podílí na udržování tolerance plodu v průběhu těhotenství. Na modelech březích myší bylo prokázáno, že blokáda signálů zprostředkovaných PD-L1 narušuje toleranci plodu a vede ke zvýšeným ztrátám plodů.
Studie fertility na zvířatech nebyly s pembrolizumabem provedeny. V 1měsíční a 6měsíční studií toxicity po opakovaném podávání na opicích nebyly na samčích a samičích reprodukčních orgánech zjištěny žádné pozorovatelné účinky; nicméně mnohá zvířata v těchto studiích nebyla pohlavně dospělá.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Histidin
Monohydrát histidin-hydrochloridu
Sacharóza
Polysorbát 80
Voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky s výjimkou těch, které jsou uvedeny v bodě 6.6.
6.3 Doba použitelnosti
Neotevřená injekční lahvička 2 roky.
Po přípravě infuze
Chemická a fyzikální stabilita naředěného roztoku byla prokázána na dobu 24 hodin při pokojové teplotě (25 °C nebo nižší). Z mikrobiologického hlediska musí být přípravek použit okamžitě. Naředěný roztok nesmí být zmražen. Není-li použit okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou v odpovědnosti uživatele a nesmí být delší než celkem 24 hodin. Tento 24hodinový limit může zahrnovat až 6 hodin při pokojové teplotě (25 °C nebo nižší); veškerá další doba uchovávání musí být při teplotě 2 °C - 8 °C. Při uchovávání v chladničce musíte nechat injekční lahvičky a/nebo i.v. vaky před použitím ohřát na pokojovou teplotu.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Uchovávejte v původní krabičce, aby byl přípravek chráněn před světlem.
Ohledně podmínek uchovávání po naředění léčivého přípravku viz bod 6.3.
6.5 Druh obalu a obsah balení
4 ml koncentrátu v 10ml injekční lahvičce z čirého skla třídy I s potaženou šedou chlorobutylovou zátkou a hliníkovým uzávěrem s odtrhovacím víčkem tmavomodré barvy obsahující 100 mg pembrolizumabu.
Jedna krabička obsahuje jednu injekční lahvičku.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Příprava a podání infuze
• Injekční lahvičkou netřepejte.
• Injekční lahvičku ekvilibrujte na pokojovou teplotu (25 °C nebo nižší).
• Před naředěním může být injekční lahvička s tekutinou mimo chladničku (teplota 25 °C nebo nižší) na dobu až 24 hodin.
• Parenterální léčivé přípravky je nutno před podáním vizuálně zkontrolovat na výskyt částic a změnu barvy. Koncentrát je čirý až mírně opalizující, bezbarvý až slabě žlutý roztok. Injekční lahvičku zlikvidujte, pokud v ní jsou viditelné částice.
• Odeberte požadovaný objem až 4 ml (100 mg) koncentrátu a přeneste jej do intravenózního vaku obsahujícího roztok chloridu sodného o koncentraci 9 mg/ml (0,9%) nebo glukózy o koncentraci 50 mg/ml (5%), čímž připravíte naředěný roztok o konečné koncentraci v rozmezí od 1 do 10 mg/ml. Jedna injekční lahvička obsahuje ještě navíc 0,25 ml (celkový obsah
v jedné injekční lahvičce 4,25 ml), aby bylo zajištěno získání 4 ml koncentrátu. Naředěný roztok promíchejte mírným obracením.
• Chemická a fyzikální stabilita naředěného roztoku byla prokázána na dobu 24 hodin při pokojové teplotě (25 °C nebo nižší). Z mikrobiologického hlediska musí být přípravek použit okamžitě. Naředěný roztok nesmí být zmražen. Není-li použit okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou v odpovědnosti uživatele a nesmí být delší než celkem 24 hodin. Tento 24hodinový limit může zahrnovat až 6 hodin při pokojové teplotě (25 °C nebo nižší); veškerá další doba uchovávání musí být při teplotě 2 °C - 8 °C. Při uchovávání v chladničce musíte nechat injekční lahvičky a/nebo i.v. vaky před použitím ohřát na pokojovou teplotu. Infuzní roztok podávejte intravenózně po dobu 30 minut za použití sterilního, nepyrogenního in-line nebo add-on filtru málo vázajícího proteiny o velikosti pórů 0,2 až 5 ^m.
• Stejnou infuzní hadičkou nepodávejte současně jiné léčivé přípravky.
• Přípravek KEYTRUDA je určen k jednorázovému použití. Veškerý nepoužitý lék v injekční lahvičce zlikvidujte.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními
požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Merck Sharp & Dohme Limited
Hertford Road
Hoddesdon
Hertfordshire EN11 9BU Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/15/1024/002
10. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 17. července 2015
11. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA
PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce biologické léčivé látky
MedImmune, LLC Frederick Manufacturing Center (FMC) 633/636/660 Research Court Frederick MD 21703-8619, USA
Název a adresa výrobce odpovědného za propouštění šarží
Schering-Plough Labo NV Industriepark 30, Heist-op-den-Berg B-2220, Belgie
Boehringer Ingelheim (BIB)
Pharma GmbH & Co. KG Birkendorfer StraBe 65 88397 Biberach an der Riss Německo
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl.
107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
Držitel rozhodnutí o registraci předloží první pravidelně aktualizovanou zprávu o bezpečnosti pro tento léčivý přípravek do 6 měsíců od jeho registrace.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Další opatření k minimalizaci rizik
Před uvedením přípravku KEYTRUDA na trh musí držitel rozhodnutí o registraci v každém členském státě odsouhlasit obsah a formát edukačního programu, včetně komunikačních medií, způsobů distribuce a jakýchkoli dalších aspektů programu s národní kompetentní autoritou.
Edukační program je zaměřen na zvyšování povědomí lékařů o potenciálních:
• imunitně zprostředkovaných nežádoucích účincích
• nežádoucích účincích spojených s infuzí
spojených s používáním přípravku KEYTRUDA, a o tom, jak je zvládnout, a na zvyšování povědomí pacientů a/nebo jejich ošetřovatelů o projevech a příznacích relevantních pro časné rozpoznání/identifikaci těchto nežádoucích účinků.
Držitel rozhodnutí o registraci zajistí, že v každém členském státě, kde je přípravek KEYTRUDA uváděn na trh, všichni zdravotničtí pracovníci a pacienti/ošetřující, u kterých se dá předpokládat, že budou přípravek KEYTRUDA předepisovat a používat, mají přístup k/obdrží následující edukační balíček:
• Edukační materiál pro lékaře
• Edukační materiál pro pacienta
Edukační materiál pro lékaře musí obsahovat:
• Souhrn údajů o přípravku
• Brožuru pro zdravotnické pracovníky s nejčastějšími dotazy
Brožura pro zdravotnické pracovníky s nejčastějšími dotazy musí obsahovat následující klíčové body: Seznam důležitých imunitně zprostředkovaných nežádoucích účinků a jejich příznaků, včetně opatření a léčby, jak jsou uvedeny v bodě 4.4 souhrnu údajů o přípravku: o Imunitně zprostředkované nežádoucí účinky o Pneumonitida o Kolitida o Hepatitida o Nefritida
o Těžké endokrinopatie, včetně hypofyzitidy (zahrnující hypopituitarismus a sekundární nedostatečnost nadledvin), diabetes mellitus typu 1, diabetickou ketoacidózu, hypotyreózu, hypertyreózu a tyreoiditidu
o Další imunitně zprostředkované nežádoucí účinky zahrnující: uveitidu, myozitidu, pankreatitidu, těžké kožní reakce a Guillain-Barrého syndrom o Reakce související s infuzí
• Podrobnosti jak minimalizovat bezpečnostní obavy prostřednictvím vhodného sledování a léčebné strategie
• Připomínka, že je třeba rozdávat brožuru s informacemi pro pacienta a kartu pacienta.
Edukační materiál pro pacienta musí obsahovat:
• Brožuru s informacemi pro pacienta
• Kartu pacienta
Brožura s informacemi pro pacienty musí obsahovat následující klíčové body:
• Popis hlavních projevů a příznaků imunitně zprostředkovaných nežádoucích účinků a důležitost toho, aby při jejich výskytu byl ihned informován ošetřující lékař.
• Skutečnost, že je důležité, aby se žádné příznaky nepokoušeli léčit bez předchozí porady se svým lékařem.
• Skutečnost, že je důležité mít vždy u sebe kartu s upozorněním pro pacienta a ukazovat ji při všech návštěvách jiných lékařů, než je ošetřující lékař, který přípravek předepsal (např. na pohotovosti).
Karta pacientovi připomíná klíčové příznaky, které je třeba lékaři/zdravotní sestře ihned hlásit. Rovněž obsahuje kolonky k zapisování kontaktů na lékaře a upozornění dalším lékařům, že je pacient léčen přípravkem KEYTRUDA.
• Povinnost uskutečnit poregistrační opatření
Držitel rozhodnutí o registraci uskuteční v daném termínu níže uvedená opatření:
|
Popis |
Termín splnění |
|
1. Poregistrační studie účinnosti (Post-authorisation efficacy study - PAES): držitel rozhodnutí o registraci musí předložit konečnou zprávu ze studie P002: Randomized, Phase II Study of MK-3475 versus Chemotherapy in Patients with Advanced Melanoma - Final Study Report |
1Q 2017 |
|
2. Poregistrační studie účinnosti (Post-authorisation efficacy study - PAES): držitel rozhodnutí o registraci musí předložit konečnou zprávu ze studie P006: A Multicenter, Randomized, Controlled, Three-Arm, Phase III Study to Evaluate the Safety and Efficacy of Two Dosing Schedules of MK-3475 Compared to Ipilimumab in Patients with Advanced Melanoma - Final Study Report |
1Q 2017 |
|
3. Poregistrační studie účinnosti (Post-authorisation efficacy study - PAES): s cílem potvrdit přínosy u podskupin pacientů s mutací BRAF V600 a u PD-L1 negativních pacientů v doporučené dávce musí držitel rozhodnutí o registraci poskytnout aktualizované analýzy ze studie P001 a studie P002: • Aktualizované údaje o účinnosti u podskupin porovnávající 2 vs 10 mg/kg Q3W z konečné analýzy studie P002. • Údaje o účinnosti u podskupin porovnávající 2 vs 10 mg/kg Q3W ze studie P001 s využitím údajů s rozhodným datem 18. října 2014 z částí B2 a D studie P001 podle úrovně dávkování. |
1Q 2017 3Q 2015 |
|
4. Je nutno dále zkoumat hodnoty biomarkerů k predikci účinnosti pembrolizumabu, zvláště: I když stav PD-L1 má u pacientů s pokročilým melanomem prediktivní význam, pokud jde o odpověď, trvající odpovědi byly pozorovány i u PD-L1 negativních pacientů. Imunohistochemicky je nutno prošetřit i další biomarkery, než je stav exprese PD-L1 (např. PD-L2, RNA signatura, atd.), které jsou prediktivní ohledně účinnosti pembrolizumabu, spolu s dalšími informacemi týkajícími se vzoru exprese PD-L1 získaného v probíhajících studiích melanomu (P001, P002 a P006) a NSCLC studiích (P001, P010, P024 a P042): • srovnání mezi imunohistochemickým barvením PD-L1 v archivní tkáni a v nově získané tkáni (pouze studie melanomu) • srovnání imunohistochemie PD-L1 v nádorové tkáni před léčbou a po léčbě (pouze studie melanomu) • údaje o genové signatuře RNA získané pomocí technologie firmy Nanostring • imunohistochemické barvení PD-L2 • údaje o RNA a proteomovém sérovém profilu • údaje o profilu imunitních buněk (periferní krev) (pouze studie melanomu) |
1Q 2017 2Q 2020 |
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
KEYTRUDA 50 mg prášek pro koncentrát pro infuzní roztok pembrolizumabum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna injekční lahvička s práškem obsahuje pembrolizumabum 50 mg. Po rekonstituci obsahuje 1 ml koncentrátu pembrolizumabum 25 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Pomocné látky: histidin, monohydrát histidin-hydrochloridu, sacharóza, polysorbát 80.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
prášek pro koncentrát pro infuzní roztok 1 injekční lahvička
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Intravenózní podání.
Pouze pro jednorázové použití.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
Rekonstituované injekční lahvičky a/nebo naředěné intravenózní vaky lze v chladničce (2 °C - 8 °C) uchovávat po kumulativní dobu až 24 hodin.
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce (2 °C - 8 °C).
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Merck Sharp & Dohme Limited
Hertford Road
Hoddesdon
Hertfordshire EN11 9BU Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/15/1024/001 (1 injekční lahvička)
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
Nevyžaduje se - odůvodnění přijato
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
2D čárový kód s jedinečným identifikátorem.
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
PC:
SN:
NN:
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
KEYTRUDA 50 mg prášek pro koncentrát pro infuzní roztok
pembrolizumabum
Intravenózní podání
i.v.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
KEYTRUDA 25 mg/ml koncentrát pro infuzní roztok
pembrolizumabum
100 mg/4 ml
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna injekční lahvička se 4 ml obsahuje pembrolizumabum 100 mg. Jeden ml koncentrátu obsahuje pembrolizumabum 25 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Pomocné látky: histidin, monohydrát histidin-hydrochloridu, sacharóza, polysorbát 80, voda na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Koncentrát pro infuzní roztok 1 injekční lahvička
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Intravenózní podání po naředění.
Pouze pro jednorázové použití.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
Netřepejte.
8. POUŽITELNOST
EXP
Naředěný roztok lze uchovávat v chladničce (2 °C - 8 °C) po dobu až 24 hodin.
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Uchovávejte v původní krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Merck Sharp & Dohme Limited
Hertford Road
Hoddesdon
Hertfordshire EN11 9BU Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/15/1024/002 (1 injekční lahvička)
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
Nevyžaduje se - odůvodnění přijato
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
2D čárový kód s jedinečným identifikátorem.
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
PC:
SN:
NN:
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
KEYTRUDA 25 mg/ml koncentrát pro infuzní roztok
pembrolizumabum
100 mg/4 ml
i.v.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro pacienta
KEYTRUDA 50 mg prášek pro koncentrát pro infuzní roztok
pembrolizumabum
'VTento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než Vám bude tento přípravek podán, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Je důležité, abyste si v průběhu léčby u sebe uschovali kartu s upozorněním pro pacienta.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek KEYTRUDA a k čemu se používá
2. Čemu musíte věnovat pozornost, než Vám bude přípravek KEYTRUDA podán
3. Jak se přípravek KEYTRUDA podává
4. Možné nežádoucí účinky
5 Jak přípravek KEYTRUDA uchovávat
6. Obsah balení a další informace
1. Co je přípravek KEYTRUDA a k čemu se používá
Přípravek KEYTRUDA obsahuje léčivou látku pembrolizumab, což je monoklonální protilátka. Přípravek KEYTRUDA svým působením pomáhá imunitnímu systému bojovat s rakovinou.
Přípravek KEYTRUDA se používá u dospělých k léčbě:
• druhu rakoviny kůže nazývaného melanom
• druhu rakoviny plic nazývaného nemalobuněčný karcinom plic.
Přípravek KEYTRUDA podává tehdy, kdy se rakovina rozšířila nebo ji nelze odstranit chirurgicky.
2. Čemu musíte věnovat pozornost, než Vám bude přípravek KEYTRUDA podán Přípravek KEYTRUDA Vám nesmí být podán:
- jestliže jste alergický(á) na pembrolizumab nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6 „Obsah balení a další informace“). Pokud si nejste jistý(á), poraďte se se svým lékařem.
Upozornění a opatření
Před podáním přípravku KEYTRUDA se poraďte se svým lékařem nebo zdravotní sestrou.
Předtím, než Vám bude přípravek KEYTRUDA podán, svého lékaře informujte, pokud:
- máte autoimunitní onemocnění (stav, kdy tělo napadá své vlastní buňky)
- máte zánět plic nebo zánět plicní tkáně (nazývá se pneumonitida)
- Vám byl dříve podáván ipilimumab, jiný lék k léčbě melanomu a měl(a) jste kvůli tomuto léku
závažné nežádoucí účinky
- jste měl(a) alergickou reakci na jiné podání monoklonálních protilátek
- máte nebo jste měl(a) chronickou virovou infekci jater, zahrnující hepatitidu B (HBV)
nebo hepatitidu C (HCV)
- máte infekci HIV (virus lidské imunodeficience) nebo AIDS (syndrom získané
imunodeficience)
- máte poškození jater nebo jste prodělal(a) transplantaci jater
- máte poškození ledvin nebo jste prodělal(a) transplantaci ledvin
Je-li Vám podáván přípravek KEYTRUDA, můžete mít jisté závažné nežádoucí účinky.
Pokud se u Vás objeví některý z následujících stavů, ihned kontaktujte nebo navštivte svého lékaře. Lékař Vám může dát jiné léky, aby zabránil závažnějším komplikacím a omezil Vaše příznaky. Váš lékař může další dávku přípravku KEYTRUDA dočasně vysadit nebo Vaši léčbu přípravkem KEYTRUDA ukončit.
- zánět plic, což může zahrnovat dýchavičnost, bolest na hrudi nebo kašel
- zánět střev, což může zahrnovat průjem nebo častější vyprazdňování střev, než je obvyklé,
tmavou, dehtovitou, lepivou stolici nebo stolici s krví nebo hlenem, silnou bolest břicha nebo citlivost, pocit na zvracení, zvracení
- zánět jater, což může zahrnovat pocit na zvracení nebo zvracení, menší chuť k jídlu, bolest na
pravé straně břicha, zežloutnutí kůže nebo bělma očí, tmavou moč nebo krvácení nebo snadnější tvorbu modřin, než je obvyklé
- zánět ledvin, což může zahrnovat změny množství nebo barvy moči
- zánět hormonálních žláz (zvláště štítné žlázy, podvěsku mozkového a nadledvin), což může
zahrnovat rychlý tep srdce, snížení tělesné hmotnosti, zvýšené pocení, zvýšení tělesné hmotnosti, vypadávání vlasů, pocit chladu, zácpu, hlubší hlas, bolesti svalů, závrať nebo mdloby, bolesti hlavy, které nemizí nebo neobvyklé bolesti hlavy
- cukrovka typu 1, což může zahrnovat silnější pocit hladu nebo žízně než obvykle, častější
potřebu močení nebo snížení tělesné hmotnosti
- zánět očí, což může zahrnovat změny vidění
- zánět svalů, což může zahrnovat svalovou bolest nebo slabost
- zánět slinivky břišní, což může zahrnovat bolest břicha, pocit na zvracení a zvracení
- zánět kůže, což může zahrnovat vyrážku
- infuzní reakce, což může zahrnovat dýchavičnost, svědění nebo vyrážku, závrať nebo horečku Děti a dospívající
Přípravek KEYTRUDA se nemá používat u dětí a dospívajících mladších 18 let.
Další léčivé přípravky a přípravek KEYTRUDA
Informujte svého lékaře
- pokud užíváte jiné léky, které oslabují imunitní systém. Příklady takových léků mohou zahrnovat kortikosteroidy, jako je prednison. Tyto léky mohou zasahovat do účinku přípravku KEYTRUDA. Nicméně pokud jste léčen(a) přípravkem KEYTRUDA, může Vám lékař podat kortikosteroidy ke snížení nežádoucích účinků, které může vyvolat přípravek KEYTRUDA.
- o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Těhotenství
- Pokud jste těhotná, přípravek KEYTRUDA nesmíte používat, není-li to lékařem výslovně doporučeno.
- Pokud jste těhotná, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, informujte svého lékaře.
- Přípravek KEYTRUDA může nenarozené dítě poškodit nebo usmrtit.
- Pokud jste žena, která může otěhotnět, musíte během léčby přípravkem KEYTRUDA a nejméně 4 měsíce po poslední dávce používat odpovídající antikoncepci.
Kojení
- Pokud kojíte, informujte svého lékaře.
- Během používání přípravku KEYTRUDA nekojte.
- Není známo, zda přípravek KEYTRUDA prostupuje do mateřského mléka.
Řízení dopravních prostředků a obsluha strojů
Poté, co Vám byl podán přípravek KEYTRUDA, neřiďte ani neobsluhujte stroje, ledaže byste si byl(a) jistý(á), že je Vám dobře. Velmi častým nežádoucím účinkem přípravku KEYTRUDA je pocit únavy nebo slabosti. To může ovlivnit Vaši schopnost řídit nebo obsluhovat stroje.
3. Jak se přípravek KEYTRUDA podává
Přípravek KEYTRUDA Vám bude podáván v nemocnici nebo na klinice pod dohledem lékaře zkušeného v léčbě rakoviny.
- Lékař Vám bude podávat přípravek KEYTRUDA infuzí do žíly (i.v.) po dobu asi 30 minut každé 3 týdny.
- O tom, kolik léčebných cyklů budete potřebovat, rozhodne Váš lékař.
Doporučená dávka je 2 mg pembrolizumabu na kilogram tělesné hmotnosti.
Jestliže se nedostavíte na podání přípravku KEYTRUDA
- Ihned zavolejte svému lékaři, abyste si dohodl(a) jiný termín.
- Je velmi důležité, abyste dávku tohoto léku nevynechal(a).
Jestliže ukončíte léčbu přípravkem KEYTRUDA
Ukončení léčby může zastavit účinek tohoto léku. Léčbu přípravkem KEYTRUDA neukončujte, pokud jste to se svým lékařem neprodiskutoval(a).
Máte-li jakékoli další otázky týkající se léčby, zeptejte se svého lékaře.
Tyto informace naleznete také v Kartě pacienta, kterou jste obdržel(a) od svého lékaře. Je důležité, abyste tuto kartu s upozorněním pro pacienta měl(a) při sobě a ukázal(a) ji svému partnerovi nebo ošetřovateli.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Během léčby přípravkem KEYTRUDA můžete mít závažné nežádoucí účinky. Viz bod 2.
V klinických studiích byly hlášeny následující nežádoucí účinky:
Velmi časté (mohou postihnout více než 1 z 10 pacientů)
- průjem; pocit na zvracení
- svědění; kožní vyrážka
- bolest kloubů
- pocit únavy
Časté (mohou postihnout až 1 z 10 pacientů)
- pokles počtu červených krvinek
- problémy se štítnou žlázou; nával horka
- menší pocit hladu
- bolest hlavy; závrať; změna ve vnímání chuti
- zánět plic; dušnost; kašel
- zánět střev; suchá ústa
- suché oko
- bolest břicha; zácpa; zvracení
- červená, vystouplá vyrážka, někdy s puchýři; skvrny na kůži s odbarvením; problémy s kůží podobné akné; suchá, svědivá kůže
- bolest svalů, pobolívání nebo citlivost; bolest svalů a kostí; bolest rukou nebo nohou; bolest kloubů s otokem
- otok; neobvyklá únava nebo slabost; zimnice; příznaky podobné chřipce; horečka
- zvýšené hladiny jaterních enzymů v krvi; abnormální testy funkce ledvin
- reakce spojené s infuzí léku
Méně časté (mohou postihnout až 1 ze 100 pacientů)
- snížení počtu bílých krvinek (neutrofily, leukocyty, lymfocyty a eozinofily); snížení počtu krevních destiček (snadnější tvorba modřin nebo krvácení)
- zánět hypofýzy (podvěsku mozkového), která se nachází na spodní části mozku; snížená sekrece hormonů produkovaných nadledvinami, zánět štítné žlázy
- cukrovka typu 1; snížená hladina sodíku, draslíku a vápníku v krvi
- poruchy spánku
- epileptické záchvaty; nedostatek energie; zánět nervů způsobující necitlivost, slabost, brnění nebo palčivost horních a dolních končetin
- zánět očí; bolest oka, podráždění, svědění nebo začervenání; nepříjemná citlivost na světlo; vidění skvrn
- vysoký krevní tlak
- zánět slinivky břišní
- zánět jater
- zesílené, někdy šupinaté kožní výrůstky; vypadávání vlasů; citlivé červené boule pod kůží; zánět kůže; změny barvy vlasů; malé kožní rány, boule nebo vředy
- zánět pochvy, která obklopuje šlachy
- zánět ledvin
- zvýšená hladina amylázy, enzymu, který štěpí škrob; zvýšená hladina vápníku v krvi
Vzácné (mohou postihnout až 1 z 1000 pacientů)
- zánětlivá reakce proti krevním destičkám nebo červeným krvinkám
- dočasný zánět nervů způsobující bolest, slabost a ochrnutí končetin; stav ve kterém se svaly snadno oslabí a unaví
- proděravění tenkého střeva
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek KEYTRUDA uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a štítku injekční lahvičky za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v chladničce (2 °C - 8 °C).
Chemická a fyzikální stabilita rekonstituovaného a naředěného roztoku byla prokázána na dobu 24 hodin při pokojové teplotě (25 °C nebo nižší). Z mikrobiologického hlediska musí být přípravek použit okamžitě. Rekonstituovaný nebo naředěný roztok nesmí být zmražen. Není-li použit okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou v odpovědnosti uživatele a nesmí být delší než celkem 24 hodin. Tento 24hodinový limit může zahrnovat až 6 hodin při pokojové teplotě (25 °C nebo nižší); veškerá další doba uchovávání musí být při teplotě 2 °C - 8 °C. Při uchovávání v chladničce musíte nechat injekční lahvičky a/nebo i.v. vaky před použitím ohřát na pokojovou teplotu.
Žádnou nepoužitou část infuzního roztoku neuchovávejte k dalšímu použití. Veškerý nepoužitý léčivý přípravek nebo odpadní materiál musí být zlikvidován v souladu s místními požadavky.
6. Obsah balení a další informace Co přípravek KEYTRUDA obsahuje
Léčivou látkou je pembrolizumabum. Jedna injekční lahvička obsahuje pembrolizumabum 50 mg. Po rekonstituci obsahuje 1 ml koncentrátu pembrolizumabum 25 mg.
Pomocnými látkami jsou histidin, monohydrát histidin-hydrochloridu, sacharóza a polysorbát 80.
Jak přípravek KEYTRUDA vypadá a co obsahuje toto balení
Přípravek KEYTRUDA je bílý až téměř bílý lyofilizovaný prášek.
Přípravek je k dispozici v krabičkách obsahujících jednu skleněnou injekční lahvičku.
Držitel rozhodnutí o registraci
Merck Sharp & Dohme Limited
Hertford Road
Hoddesdon
Hertfordshire EN11 9BU Velká Británie
Výrobce
Schering-Plough Labo NV Industriepark 30 B-2220 Heist-op-den-Berg Belgie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgie/Belgique/Belgien
MSD Belgium BVBA/SPRL
Tél/Tel: 0800 38 693 (+32(0)27766211)
Eunrapnu
MepK fflapn h floyM EtnrapHa EOOfl Ten.: +359 2 819 3737 info-msdbg@merck.com
Česká republika
Merck Sharp & Dohme s.r.o.
Tel: +420 233 010 111 dpoc_czechslovak@merck.com
Danmark
MSD Danmark ApS Tlf: + 45 4482 4000 dkmail@merck.com
Lietuva
UAB Merck Sharp & Dohme Tel. + 370 5 278 02 47 msd_lietuva@merck.com
Luxembourg/Luxemburg
MSD Belgium BVBA/SPRL Tél/Tel: +32(0)27766211 dpoc_belux@merck.com
Magyarország
MSD Pharma Hungary Kft.
Tel.: +36 1 888 5300 hungary_msd@merck.com
Malta
Merck Sharp & Dohme Cyprus Limited Tel: 8007 4433 (+356 99917558) malta_info@merck.com
Deutschland
MSD SHARP & DOHME GMBH
Tel: 0800 673 673 673 (+49 (0) 89 4561 2612)
Eesti
Merck Sharp & Dohme OU Tel.: +372 6144 200 msdeesti@merck.com
EXXáSa
MSD A.O.B.E.E.
TpA,: +30 210 98 97 300 dpoc_greece@merck.com
Espaňa
Merck Sharp & Dohme de Espana, S.A. Tel: +34 91 321 06 00 msd_info@merck.com
France
MSD France
Tél: + 33 (0) 1 80 46 40 40
Hrvatska
Merck Sharp & Dohme d.o.o.
Tel: + 385 1 6611 333 croatia_info@merck.com
Ireland
Merck Sharp & Dohme Ireland (Human Health) Limited
Tel: +353 (0)1 2998700 medinfo_ireland@merck.com
Ísland
Vistor hf.
Sími: + 354 535 7000
Italia
MSD Italia S.r.l.
Tel: +39 06 361911 medicalinformation.it@merck.com
Kúrcpog
Merck Sharp & Dohme Cyprus Limited TpL: 800 00 673 (+357 22866700) cyprus_info@merck.com
Latvija
SIA Merck Sharp & Dohme Latvija Tel: + 371 67364224 msd_lv@merck.com
Nederland
Merck Sharp & Dohme BV Tel: 0800 9999000 (+31 23 5153153) medicalinfo.nl@merck.com
Norge
MSD (Norge) AS Tlf: +47 32 20 73 00 msdnorge@msd.no
Osterreich
Merck Sharp & Dohme Ges.m.b.H.
Tel: +43 (0) 1 26 044 msd-medizin@merck.com
Polska
MSD Polska Sp. z o.o.
Tel: +48 22 549 51 00 msdpolska@merck.com
Portugal
Merck Sharp & Dohme, Lda Tel: +351 21 4465700 clic@merck.com
Románia
Merck Sharp & Dohme Romania S.R.L.
Tel: +40 21 529 29 00 msdromania@merck.com
Slovenija
Merck Sharp & Dohme, inovativna zdravila d.o.o. Tel: +386 1 5204 201 msd.slovenia@merck.com
Slovenská republika
Merck Sharp & Dohme, s. r. o.
Tel: +421 2 58282010 dpoc_czechslovak@merck.com
Suomi/Finland
MSD Finland Oy Puh/Tel: +358 (0)9 804 650 info@msd.fi
Sverige
Merck Sharp & Dohme (Sweden) AB Tel: +46 77 5700488 medicinskinfo@merck.com
United Kingdom
Merck Sharp & Dohme Limited Tel: +44 (0) 1992 467272 medicalinformationuk@merck.com
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu.
Následující informace jsou určeny pouze pro zdravotnické pracovníky:
Příprava a podání
• Před rekonstitucí může být injekční lahvička s lyofylizovaným práškem mimo chladničku (teplota 25 °C nebo nižší) na dobu až 24 hodin.
• Asepticky přidejte 2,3 ml vody na injekci, čímž získáte roztok přípravku KEYTRUDA
o koncentraci 25 mg/ml (pH 5,2-5,8). Každá injekční lahvička obsahuje dodatečných 10 mg (0,4 ml), aby se zajistilo, že se z injekční lahvičky získá 50 mg přípravku KEYTRUDA. Po rekonstituci obsahuje 1 ml koncentrátu 25 mg pembrolizumabu.
• Aby se zamezilo vzniku pěny, vodu vstřikujte podél stěn injekční lahvičky, nikoli přímo do lyofilizovaného prášku.
• Injekční lahvičkou pomalu otáčejte, aby se umožnila rekonstituce lyofilizovaného prášku. Nechejte stát až 5 minut, aby zmizely bubliny. Injekční lahvičkou netřepejte.
• Parenterální léčivé přípravky je nutno před podáním vizuálně zkontrolovat na výskyt částic a změnu barvy. Rekonstituovaný přípravek KEYTRUDA je čirý až mírně opalizující, bezbarvý až slabě žlutý roztok. Rekonstituovanou injekční lahvičku zlikvidujte, pokud v ní jsou viditelné částice.
• Odeberte požadovaný objem až 2 ml (50 mg) přípravku KEYTRUDA a přeneste jej do intravenózního vaku obsahujícího roztok chloridu sodného o koncentraci 9 mg/ml (0,9%) nebo glukózy o koncentraci 50 mg/ml (5%), čímž připravíte naředěný roztok o konečné koncentraci v rozmezí od 1 do 10 mg/ml. Naředěný roztok promíchejte mírným obracením.
• Chemická a fyzikální stabilita rekonstituovaného a naředěného roztoku byla prokázána na dobu 24 hodin při pokojové teplotě (25 °C nebo nižší). Z mikrobiologického hlediska musí být přípravek použit okamžitě. Rekonstituovaný nebo naředěný roztok nesmí být zmražen. Není-li použit okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou
v odpovědnosti uživatele a nesmí být delší než celkem 24 hodin. Tento 24hodinový limit může zahrnovat až 6 hodin při pokojové teplotě (25 °C nebo nižší); veškerá další doba uchovávání musí být při teplotě 2 °C- 8 °C. Při uchovávání v chladničce musíte nechat injekční lahvičky a/nebo i.v. vaky před použitím ohřát na pokojovou teplotu. Infuzní roztok podávejte intravenózně po dobu 30 minut za použití sterilního, nepyrogenního in-line nebo add-on filtru málo vázajícího proteiny o velikosti pórů 0,2 až 5 ^m.
• Stejnou infuzní hadičkou nepodávejte současně jiné léčivé přípravky.
• Přípravek KEYTRUDA je pouze k jednorázovému použití. Veškerý nepoužitý lék v injekční lahvičce zlikvidujte.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními
požadavky.
KEYTRUDA 25 mg koncentrát pro infuzní roztok
pembrolizumabum
'VTento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než Vám bude tento přípravek podán, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Je důležité, abyste si v průběhu léčby u sebe uschovali kartu s upozorněním pro pacienta.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek KEYTRUDA a k čemu se používá
2. Čemu musíte věnovat pozornost, než Vám bude přípravek KEYTRUDA podán
3. Jak se přípravek KEYTRUDA podává
4. Možné nežádoucí účinky
5 Jak přípravek KEYTRUDA uchovávat
6. Obsah balení a další informace
1. Co je přípravek KEYTRUDA a k čemu se používá
Přípravek KEYTRUDA obsahuje léčivou látku pembrolizumab, což je monoklonální protilátka. Přípravek KEYTRUDA svým působením pomáhá imunitnímu systému bojovat s rakovinou.
Přípravek KEYTRUDA se používá u dospělých k léčbě:
• druhu rakoviny kůže nazývaného melanom
• druhu rakoviny plic nazývaného nemalobuněčný karcinom plic.
Přípravek KEYTRUDA podává tehdy, kdy se rakovina rozšířila nebo ji nelze odstranit chirurgicky.
2. Čemu musíte věnovat pozornost, než Vám bude přípravek KEYTRUDA podán Přípravek KEYTRUDA Vám nesmí být podán:
- jestliže jste alergický(á) na pembrolizumab nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6 „Obsah balení a další informace“). Pokud si nejste jistý(á), poraďte se se svým lékařem.
Upozornění a opatření
Před podáním přípravku KEYTRUDA se poraďte se svým lékařem nebo zdravotní sestrou.
Předtím, než Vám bude přípravek KEYTRUDA podán, svého lékaře informujte, pokud:
- máte autoimunitní onemocnění (stav, kdy tělo napadá své vlastní buňky)
- máte zánět plic nebo zánět plicní tkáně (nazývá se pneumonitida)
- Vám byl dříve podáván ipilimumab, jiný lék k léčbě melanomu a měl(a) jste kvůli tomuto léku
závažné nežádoucí účinky
- jste měl(a) alergickou reakci na jiné podání monoklonálních protilátek
- máte nebo jste měl(a) chronickou virovou infekci jater, zahrnující hepatitidu B (HBV)
nebo hepatitidu C (HCV)
- máte infekci HIV (virus lidské imunodeficience) nebo AIDS (syndrom získané
imunodeficience)
- máte poškození jater nebo jste prodělal(a) transplantaci jater
- máte poškození ledvin nebo jste prodělal(a) transplantaci ledvin
Je-li Vám podáván přípravek KEYTRUDA, můžete mít jisté závažné nežádoucí účinky.
Pokud se u Vás objeví některý z následujících stavů, ihned kontaktujte nebo navštivte svého lékaře. Lékař Vám může dát jiné léky, aby zabránil závažnějším komplikacím a omezil Vaše příznaky. Váš lékař může další dávku přípravku KEYTRUDA dočasně vysadit nebo Vaši léčbu přípravkem KEYTRUDA ukončit.
- zánět plic, což může zahrnovat dýchavičnost, bolest na hrudi nebo kašel
- zánět střev, což může zahrnovat průjem nebo častější vyprazdňování střev, než je obvyklé,
tmavou, dehtovitou, lepivou stolici nebo stolici s krví nebo hlenem, silnou bolest břicha nebo citlivost, pocit na zvracení, zvracení
- zánět jater, což může zahrnovat pocit na zvracení nebo zvracení, menší chuť k jídlu, bolest na
pravé straně břicha, zežloutnutí kůže nebo bělma očí, tmavou moč nebo krvácení nebo snadnější tvorbu modřin, než je obvyklé
- zánět ledvin, což může zahrnovat změny množství nebo barvy moči
- zánět hormonálních žláz (zvláště štítné žlázy, podvěsku mozkového a nadledvin), což může
zahrnovat rychlý tep srdce, snížení tělesné hmotnosti, zvýšené pocení, zvýšení tělesné hmotnosti, vypadávání vlasů, pocit chladu, zácpu, hlubší hlas, bolesti svalů, závrať nebo mdloby, bolesti hlavy, které nemizí nebo neobvyklé bolesti hlavy
- cukrovka typu 1, což může zahrnovat silnější pocit hladu nebo žízně než obvykle, častější
potřebu močení nebo snížení tělesné hmotnosti
- zánět očí, což může zahrnovat změny vidění
- zánět svalů, což může zahrnovat svalovou bolest nebo slabost
- zánět slinivky břišní, což může zahrnovat bolest břicha, pocit na zvracení a zvracení
- zánět kůže, což může zahrnovat vyrážku
- infuzní reakce, což může zahrnovat dýchavičnost, svědění nebo vyrážku, závrať nebo horečku Děti a dospívající
Přípravek KEYTRUDA se nemá používat u dětí a dospívajících mladších 18 let.
Další léčivé přípravky a přípravek KEYTRUDA
Informujte svého lékaře
- pokud užíváte jiné léky, které oslabují imunitní systém. Příklady takových léků mohou zahrnovat kortikosteroidy, jako je prednison. Tyto léky mohou zasahovat do účinku přípravku KEYTRUDA. Nicméně pokud jste léčen(a) přípravkem KEYTRUDA, může Vám lékař podat kortikosteroidy ke snížení nežádoucích účinků, které může vyvolat přípravek KEYTRUDA.
- o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Těhotenství
- Pokud jste těhotná, přípravek KEYTRUDA nesmíte používat, není-li to lékařem výslovně doporučeno.
- Pokud jste těhotná, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, informujte svého lékaře.
- Přípravek KEYTRUDA může nenarozené dítě poškodit nebo usmrtit.
- Pokud jste žena, která může otěhotnět, musíte během léčby přípravkem KEYTRUDA a nejméně 4 měsíce po poslední dávce používat odpovídající antikoncepci.
Kojení
- Pokud kojíte, informujte svého lékaře.
- Během používání přípravku KEYTRUDA nekojte.
- Není známo, zda přípravek KEYTRUDA prostupuje do mateřského mléka.
Řízení dopravních prostředků a obsluha strojů
Poté, co Vám byl podán přípravek KEYTRUDA, neřiďte ani neobsluhujte stroje, ledaže byste si byl(a) jistý(á), že je Vám dobře. Velmi častým nežádoucím účinkem přípravku KEYTRUDA je pocit únavy nebo slabosti. To může ovlivnit Vaši schopnost řídit nebo obsluhovat stroje.
3. Jak se přípravek KEYTRUDA podává
Přípravek KEYTRUDA Vám bude podáván v nemocnici nebo na klinice pod dohledem lékaře zkušeného v léčbě rakoviny.
- Lékař Vám bude podávat přípravek KEYTRUDA infuzí do žíly (i.v.) po dobu asi 30 minut každé 3 týdny.
- O tom, kolik léčebných cyklů budete potřebovat, rozhodne Váš lékař.
Doporučená dávka je 2 mg pembrolizumabu na kilogram tělesné hmotnosti.
Jestliže se nedostavíte na podání přípravku KEYTRUDA
- Ihned zavolejte svému lékaři, abyste si dohodl(a) jiný termín.
- Je velmi důležité, abyste dávku tohoto léku nevynechal(a).
Jestliže ukončíte léčbu přípravkem KEYTRUDA
Ukončení léčby může zastavit účinek tohoto léku. Léčbu přípravkem KEYTRUDA neukončujte, pokud jste to se svým lékařem neprodiskutoval(a).
Máte-li jakékoli další otázky týkající se léčby, zeptejte se svého lékaře.
Tyto informace naleznete také v Kartě pacienta, kterou jste obdržel(a) od svého lékaře. Je důležité, abyste tuto kartu s upozorněním pro pacienta měl(a) při sobě a ukázal(a) ji svému partnerovi nebo ošetřovateli.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Během léčby přípravkem KEYTRUDA můžete mít závažné nežádoucí účinky. Viz bod 2.
V klinických studiích byly hlášeny následující nežádoucí účinky:
Velmi časté (mohou postihnout více než 1 z 10 pacientů)
- průjem; pocit na zvracení
- svědění; kožní vyrážka
- bolest kloubů
- pocit únavy
Časté (mohou postihnout až 1 z 10 pacientů)
- pokles počtu červených krvinek
- problémy se štítnou žlázou; nával horka
- menší pocit hladu
- bolest hlavy; závrať; změna ve vnímání chuti
- zánět plic; dušnost; kašel
- zánět střev; suchá ústa
- suché oko
- bolest břicha; zácpa; zvracení
- červená, vystouplá vyrážka, někdy s puchýři; skvrny na kůži s odbarvením; problémy s kůží podobné akné; suchá, svědivá kůže
- bolest svalů, pobolívání nebo citlivost; bolest svalů a kostí; bolest rukou nebo nohou; bolest kloubů s otokem
- otok; neobvyklá únava nebo slabost; zimnice; příznaky podobné chřipce; horečka
- zvýšené hladiny jaterních enzymů v krvi; abnormální testy funkce ledvin
- reakce spojené s infuzí léku
Méně časté (mohou postihnout až 1 ze 100 pacientů)
- snížení počtu bílých krvinek (neutrofily, leukocyty, lymfocyty a eozinofily); snížení počtu krevních destiček (snadnější tvorba modřin nebo krvácení)
- zánět hypofýzy (podvěsku mozkového), která se nachází na spodní části mozku; snížená sekrece hormonů produkovaných nadledvinami, zánět štítné žlázy
- cukrovka typu 1; snížená hladina sodíku, draslíku a vápníku v krvi
- poruchy spánku
- epileptické záchvaty; nedostatek energie; zánět nervů způsobující necitlivost, slabost, brnění nebo palčivost horních a dolních končetin
- zánět očí; bolest oka, podráždění, svědění nebo začervenání; nepříjemná citlivost na světlo; vidění skvrn
- vysoký krevní tlak
- zánět slinivky břišní
- zánět jater
- zesílené, někdy šupinaté kožní výrůstky; vypadávání vlasů; citlivé červené boule pod kůží; zánět kůže; změny barvy vlasů; malé kožní rány, boule nebo vředy
- zánět pochvy, která obklopuje šlachy
- zánět ledvin
- zvýšená hladina amylázy, enzymu, který štěpí škrob; zvýšená hladina vápníku v krvi
Vzácné (mohou postihnout až 1 z 1000 pacientů)
- zánětlivá reakce proti krevním destičkám nebo červeným krvinkám
- dočasný zánět nervů způsobující bolest, slabost a ochrnutí končetin; stav ve kterém se svaly snadno oslabí a unaví
- proděravění tenkého střeva
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek KEYTRUDA uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a štítku injekční lahvičky za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Uchovávejte v původní krabičce, aby byl přípravek chráněn před světlem.
Chemická a fyzikální stabilita naředěného roztoku byla prokázána na dobu 24 hodin při pokojové teplotě (25 °C nebo nižší). Z mikrobiologického hlediska musí být přípravek použit okamžitě. Naředěný roztok nesmí být zmražen. Není-li použit okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou v odpovědnosti uživatele a nesmí být delší než celkem 24 hodin. Tento 24hodinový limit může zahrnovat až 6 hodin při pokojové teplotě (25 °C nebo nižší); veškerá další doba uchovávání musí být při teplotě 2 °C - 8 °C. Při uchovávání v chladničce musíte nechat injekční lahvičky a/nebo i.v. vaky před použitím ohřát na pokojovou teplotu.
Žádnou nepoužitou část infuzního roztoku neuchovávejte k dalšímu použití. Veškerý nepoužitý léčivý přípravek nebo odpadní materiál musí být zlikvidován v souladu s místními požadavky.
6. Obsah balení a další informace
Co přípravek KEYTRUDA obsahuje
Léčivou látkou je pembrolizumabum.
Jedna injekční lahvička se 4 ml obsahuje pembrolizumabum 100 mg.
Jeden ml koncentrátu obsahuje pembrolizumabum 25 mg.
Pomocnými látkami jsou histidin, monohydrát histidin-hydrochloridu, sacharóza, polysorbát 80 a voda na injekci.
Jak přípravek KEYTRUDA vypadá a co obsahuje toto balení
Přípravek KEYTRUDA je čirý až lehce opalizující, bezbarvý až nažloutlý roztok, pH 5,2 - 5,8. Přípravek je k dispozici v krabičkách obsahujících jednu skleněnou injekční lahvičku.
Držitel rozhodnutí o registraci
Merck Sharp & Dohme Limited
Hertford Road
Hoddesdon
Hertfordshire EN11 9BU Velká Británie
Výrobce
Schering-Plough Labo NV Industriepark 30 B-2220 Heist-op-den-Berg Belgie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci
Belgie/Belgique/Belgien
MSD Belgium BVBA/SPRL
Tél/Tel: 0800 38 693 (+32(0)27766211)
BtnrapHH
MepK fflapn h floyM Etnrapua EOOfl Ten.: +359 2 819 3737 info-msdbg@merck.com
Česká republika
Merck Sharp & Dohme s.r.o.
Tel: +420 233 010 111 dpoc_czechslovak@merck.com
Danmark
MSD Danmark ApS Tlf: + 45 4482 4000 dkmail@merck.com
Lietuva
UAB Merck Sharp & Dohme Tel. + 370 5 278 02 47 msd_lietuva@merck.com
Luxembourg/Luxemburg
MSD Belgium BVBA/SPRL Tél/Tel: +32(0)27766211 dpoc_belux@merck.com
Magyarország
MSD Pharma Hungary Kft.
Tel.: +36 1 888 5300 hungary_msd@merck.com
Malta
Merck Sharp & Dohme Cyprus Limited Tel: 8007 4433 (+356 99917558) malta_info@merck.com
Deutschland
MSD SHARP & DOHME GMBH
Tel: 0800 673 673 673 (+49 (0) 89 4561 2612)
Eesti
Merck Sharp & Dohme OU Tel.: +372 6144 200 msdeesti@merck.com
EXXáSa
MSD A.O.B.E.E.
TpA,: +30 210 98 97 300 dpoc_greece@merck.com
Espaňa
Merck Sharp & Dohme de Espana, S.A. Tel: +34 91 321 06 00 msd_info@merck.com
France
MSD France
Tél: + 33 (0) 1 80 46 40 40
Hrvatska
Merck Sharp & Dohme d.o.o.
Tel: + 385 1 6611 333 croatia_info@merck.com
Ireland
Merck Sharp & Dohme Ireland (Human Health) Limited
Tel: +353 (0)1 2998700 medinfo_ireland@merck.com
Ísland
Vistor hf.
Sími: + 354 535 7000
Italia
MSD Italia S.r.l.
Tel: +39 06 361911 medicalinformation.it@merck.com
Kúrcpog
Merck Sharp & Dohme Cyprus Limited TpL: 800 00 673 (+357 22866700) cyprus_info@merck.com
Latvija
SIA Merck Sharp & Dohme Latvija Tel: + 371 67364224 msd_lv@merck.com
Nederland
Merck Sharp & Dohme BV Tel: 0800 9999000 (+31 23 5153153) medicalinfo.nl@merck.com
Norge
MSD (Norge) AS Tlf: +47 32 20 73 00 msdnorge@msd.no
Osterreich
Merck Sharp & Dohme Ges.m.b.H.
Tel: +43 (0) 1 26 044 msd-medizin@merck.com
Polska
MSD Polska Sp. z o.o.
Tel: +48 22 549 51 00 msdpolska@merck.com
Portugal
Merck Sharp & Dohme, Lda Tel: +351 21 4465700 clic@merck.com
Románia
Merck Sharp & Dohme Romania S.R.L.
Tel: +40 21 529 29 00 msdromania@merck.com
Slovenija
Merck Sharp & Dohme, inovativna zdravila d.o.o. Tel: +386 1 5204 201 msd.slovenia@merck.com
Slovenská republika
Merck Sharp & Dohme, s. r. o.
Tel: +421 2 58282010 dpoc_czechslovak@merck.com
Suomi/Finland
MSD Finland Oy Puh/Tel: +358 (0)9 804 650 info@msd.fi
Sverige
Merck Sharp & Dohme (Sweden) AB Tel: +46 77 5700488 medicinskinfo@merck.com
United Kingdom
Merck Sharp & Dohme Limited Tel: +44 (0) 1992 467272 medicalinformationuk@merck.com
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu.
Následující informace jsou určeny pouze pro zdravotnické pracovníky:
Příprava a podání infuze
• Injekční lahvičkou netřepejte.
• Injekční lahvičku ekvilibrujte na pokojovou teplotu (25 °C nebo nižší).
• Před naředěním může být injekční lahvička s tekutinou mimo chladničku (teplota 25 °C nebo nižší) na dobu až 24 hodin.
• Parenterální léčivé přípravky je nutno před podáním vizuálně zkontrolovat na výskyt částic a změnu barvy. Koncentrát je čirý až mírně opalizují, bezbarvý až slabě žlutý roztok. Rekonstituovanou injekční lahvičku zlikvidujte, pokud v ní jsou viditelné částice.
• Odeberte požadovaný objem až 4 ml (100 mg) koncentrátu a přeneste jej do intravenózního vaku obsahujícího roztok chloridu sodného o koncentraci 9 mg/ml (9%) nebo glukózy o koncentraci 50 mg/ml (5%), čímž připravíte naředěný roztok o konečné koncentraci v rozmezí od 1 do 10 mg/ml. Jedna injekční lahvička obsahuje ještě navíc 0,25 ml (celkový obsah
v jedné injekční lahvičce 4,25 ml), aby bylo zajištěno získání 4 ml koncentrátu. Naředěný roztok promíchejte mírným obracením.
• Chemická a fyzikální stabilita rekonstituovaného a naředěného roztoku byla prokázána na dobu 24 hodin při pokojové teplotě (25 °C nebo nižší). Z mikrobiologického hlediska musí být přípravek použit okamžitě. Naředěný roztok nesmí být zmražen. Není-li použit okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou v odpovědnosti uživatele a nesmí být delší než celkem 24 hodin. Tento 24hodinový limit může zahrnovat až 6 hodin při pokojové teplotě (25 °C nebo nižší); veškerá další doba uchovávání musí být při teplotě
2 °C- 8 °C. Při uchovávání v chladničce musíte injekční lahvičky a/nebo i.v. vaky před použitím ohřát na pokojovou teplotu. Infuzní roztok podávejte intravenózně po dobu 30 minut za použití sterilního, nepyrogenního in-line nebo add-on filtru málo vázajícího proteiny o velikosti pórů 0,2 až 5 ^m.
• Stejnou infuzní hadičkou nepodávejte současně jiné léčivé přípravky.
• Přípravek KEYTRUDA je pouze k jednorázovému použití. Veškerý nepoužitý lék v injekční lahvičce zlikvidujte.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními
požadavky.
74