Kadcyla 160 Mg
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Kadcyla 100 mg prášek pro koncentrát pro infuzní roztok Kadcyla 160 mg prášek pro koncentrát pro infuzní roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Po rekonstituci se z jedné 100 mg injekční lahvičky pro jednorázové použití obsahující prášek pro koncentrát pro infuzní roztok získá trastuzumabum emtansinum 5 ml o koncentraci 20 mg/ml (viz bod 6.6).
Po rekonstituci se z jedné 160 mg injekční lahvičky pro jednorázové použití obsahující prášek pro koncentrát pro infuzní roztok získá trastuzumabum emtansinum 8 ml o koncentraci 20 mg/ml (viz bod 6.6).
Trastuzumab emtansin je konjugát protilátka-léčivo obsahující trastuzumab, humanizovanou monoklonální protilátku IgG1, která je produkována buněčnou suspenzní kulturou savčích buněk (z ovarií čínského křečka), s kovalentní vazbou na DM1, mikrotubulární inhibitor, pomocí stabilního thioéterového vazebného můstku MCC (4-[N-maleimidomethyl]cyklohexan-1-karboxylát).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek pro koncentrát pro infuzní roztok. Bílý až světle šedý lyofilizovaný prášek.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Kadcyla v monoterapii je indikován k léčbě dospělých pacientů s HER2-pozitivním neresekovatelným lokálně pokročilým nebo metastazujícím karcinomem prsu, kteří byli dříve léčeni trastuzumabem a taxanem, a to samostatně nebo v kombinaci. Pacienti buď:
• byli dříve léčeni pro lokálně pokročilé nebo metastazující onemocnění, nebo
• měli onemocnění, k jehož rekurenci došlo v průběhu adjuvantní léčby nebo do 6 měsíců po jejím ukončení.
4.2 Dávkování a způsob podání
Léčbu přípravkem Kadcyla může předepsat pouze lékař a podána musí být pod dohledem zdravotnického pracovníka, který má dostatečné zkušenosti s léčbou onkologických pacientů.
Pacienti léčení trastuzumabem emtansinem musí mít HER2-pozitivní nádor s HER2 pozitivitou definovanou jako skóre 3+ při imunohistochemickém (IHC) stanovení nebo poměr > 2,0 při stanovení in situ hybridizací (ISH) za použití zdravotnického prostředku pro in vitro diagnostiku s označením CE. Pokud není k dispozici zdravotnický prostředek pro in vitro diagnostiku s označením CE, může být stav HER2 stanoven jiným validovaným testem.
Aby se zabránilo chybám při podání léku, je nutné zkontrolovat označení na injekční lahvičce a ubezpečit se, že je připravován a podáván přípravek Kadcyla (trastuzumab emtansin) a nikoli Herceptin (trastuzumab).
Dávkování
Doporučená dávka trastuzumabu emtansinu je 3,6 mg/kg tělesné hmotnosti podávaného formou intravenózní infuze každé 3 týdny (21denní cyklus). Pacienti mají být léčení do progrese nemoci nebo nepřijatelné toxicity.
První dávka má být podána v intravenózní infuzi trvající 90 minut. Pacient má být sledován během infuze a nejméně 90 minut po podání první dávky k zachycení horečky, třesavky nebo jiných reakcí souvisejících s infuzí. Je třeba pečlivě sledovat místo infuze k zachycení případné podkožní infiltrace během podání (viz bod 4.8).
Pokud byla předchozí infuze dobře snášena, mohou být následné dávky trastuzumabu emtansinu podávány v infuzi trvající 30 minut. Pacient má být sledován během infuze a nejméně 30 minut po infuzi.
U pacientů s projevy příznaků souvisejících s infuzí (viz body 4.4 a 4.8) má být snížena rychlost infuze trastuzumabu emtansinu nebo má být infuze přerušena. Při život ohrožujících reakcích na infuzi má být podávání trastuzumabu emtansinu ukončeno.
K okamžitému použití mají být k dispozici léky pro léčbu alergie/anafylaktické reakce na infuzi a pohotovostní vybavení (viz bod 4.4).
Opoždění nebo vynechání dávky
Pokud je vynechána plánovaná dávka, má být podána co nejdříve; nečekejte až do příštího plánovaného cyklu. Režim podávání má být upraven tak, aby byl zachován interval 3 týdny mezi dávkami. Další dávka má být podána dle doporučeného dávkování (viz bod 4.2, Dávkování).
Úprava dávky
Ke zvládnutí symptomatických nežádoucích účinků může být nutné dočasné přerušení léčby, snížení dávky nebo ukončení léčby přípravkem Kadcyla dle doporučení uvedených v textu a v tabulkách 1 až
5.
Dávka přípravku Kadcyla nemá být po předchozím snížení zpětně zvyšována.
Tabulka 1 Schéma snižovaní dávky
|
Schéma snižovaní dávky (Úvodní dávka je 3,6 mg/kg) |
Dávka, která má být podána |
|
První snížení dávky |
3 mg/kg |
|
Druhé snížení dávky |
2,4 mg/kg |
|
Potřeba dalšího snížení dávky |
Ukončení léčby |
|
Stupeň 2 (> 2,5 až < 5násobek horní hranice normy) |
Stupeň 3 (> 5 až < 20násobek horní hranice normy) |
Stupeň 4 (> 20násobek horní hranice normy) |
|
Úprava dávky není nutná. |
Nepodávejte trastuzumab emtansin do úpravy AST/ALT na stupeň < 2 (>2,5 až <5násobek horní hranice normy), následně snižte dávku (viz tabulka 1). |
Ukončete podávání trastuzumabu emtansinu. |
ALT = alaninaminotransferáza; AST = aspartátaminotransferáza
Tabulka 3 Pokyny pro úpravu dávky při zvýšené hladině bilirubinu
|
Stupeň 2 (> 1,5 až < 3násobek horní hranice normy) |
Stupeň 3 (> 3 až < 10násobek horní hranice normy) |
Stupeň 4 (> 10násobek horní hranice normy) |
|
Nepodávejte trastuzumab emtansin do úpravy hladiny celkového bilirubinu na stupeň < 1 (>horní hranice normy až 1,5násobek horní hranice normy). Úprava dávky není nutná. |
Nepodávejte trastuzumab emtansin do úpravy hladiny celkového bilirubinu na stupeň < 1 (>horní hranice normy až 1,5násobek horní hranice normy), následně snižte dávku (viz tabulka 1). |
Ukončete podávání trastuzumabu emtansinu. |
Tabulka 4 Pokyny pro úpravu dávky při trombocytopenii
|
Stupeň 3 (Trombocyty: 25 000 až < 50 000/mm3) |
Stupeň 4 (Trombocyty: < 25 000/mm3) |
|
Nepodávejte trastuzumab emtansin, dokud se počet krevních destiček neupraví na stupeň < 1 (tj. trombocyty > 75 000/mm3). Úprava dávky není nutná. |
Nepodávejte trastuzumab emtansin, dokud se počet krevních destiček neupraví na stupeň < 1 (tj. trombocyty > 75 000/mm3), následně snižte dávku (viz tabulka 1). |
|
LVEF < 40 % |
LVEF > 45 % |
LVEF 40 % až < 45 % a pokles je < 10 procentních bodů od hodnoty před léčbou |
LVEF 40 % až < 45 % a pokles je > 10 procentních bodů od hodnoty před léčbou |
Symptomatické městnavé srdeční selhávání |
|
Nepodávejte |
Pokračujte |
Pokračujte |
Nepodávejte |
Ukončete léčbu |
|
trastuzumab |
v léčbě |
v léčbě |
trastuzumab |
trastuzumabem |
|
emtansin. Zopakujte vyšetření LVEF během 3 týdnů. Při potvrzení hodnoty LVEF < 40 % ukončete podávání trastuzumabu emtansinu. |
trastuzumabem emtansinem. |
trastuzumabem emtansinem. Zopakujte vyšetření LVEF během 3 týdnů. |
emtansin. Zopakujte vyšetření LVEF během 3 týdnů. Ukončete podávání trastuzumabu emtansinu, pokud nebude hodnota LVEF v rozmezí 10 procentních bodů od hodnoty před léčbou. |
emtansinem. |
LVEF = ejekční frakce levé komory
Periferní neuropatie
Pokud bude mít pacient projevy periferní neuropatie stupně 3 nebo 4, má být léčba trastuzumabem emtansinem dočasně přerušena do úpravy na stupeň < 2. Při obnovení léčby může být zvážena redukce dávky dle schématu redukce dávek (viz tabulka 1).
Starší pacienti
U pacientů ve věku > 65 let není nutná úprava dávky. Údaje pro stanovení bezpečnosti a účinnosti přípravku Kadcyla u pacientů ve věku > 75 let jsou vzhledem k omezeným datům pro tuto podskupinu nedostatečné. Analýzy populační farmakokinetiky ukazují, že věk nemá klinicky významný vliv na farmakokinetiku trastuzumabu emtansinu (viz body 5.1 a 5.2).
Porucha funkce ledvin
U pacientů s lehkou až středně těžkou poruchou funkce ledvin není nutné upravovat úvodní dávku (viz bod 5.2). Vzhledem k nedostatku údajů nelze stanovit případnou potřebu úpravy dávky u pacientů s těžkou poruchou funkce ledvin, a proto pacienti s těžkou poruchou funkce ledvin mají být pečlivě sledováni.
Porucha funkce jater
U pacientů s lehkou až středně těžkou poruchou funkce jater není třeba jakkoli upravovat úvodní dávku. Trastuzumab emtansin nebyl studován u pacientů s těžkou poruchou funkce jater. Při léčbě pacientů s poruchou funkce jater je nutná opatrnost, kvůli známé hepatotoxicitě pozorované u trastuzumabu emtansin (viz body 4.4 a 5.2).
Pediatrická populace
U dětí a adolescentů ve věku do 18 let nebyla bezpečnost a účinnost přípravku Kadcyla stanovena, protože použití přípravku Kadcyla v indikaci metastatického karcinomu prsu u pediatrické populace není relevantní.
Způsob podání
Trastuzumab emtansin musí být rekonstituován a naředěn zdravotnickým pracovníkem a podán jako intravenózní infuze. Nesmí být podán jako intravenózní injekce nebo bolus.
Návod k rekonstituci a naředění tohoto léčivého přípravku před jeho podáním je uveden v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Z důvodu snadnější zpětné dohledatelnosti biologických léčivých přípravků mají být obchodní název a číslo šarže podávaného přípravku zřetelně zaznamenány (nebo vyznačeny) v pacientově dokumentaci.
Aby se zabránilo chybám při podání léku, je nutné zkontrolovat označení na injekční lahvičce a ubezpečit se, že je připravován a podáván přípravek Kadcyla (trastuzumab emtansin) a nikoli přípravek Herceptin (trastuzumab).
Plicní toxicita
V klinických studiích s trastuzumabem emtansinem byly hlášeny případy intersticiální plicní nemoci včetně pneumonitidy, některé vedoucí k syndromu akutní dechové tísně nebo končící úmrtím (viz bod 4.8). Ke známkám a příznakům patří dušnost, kašel, únava a plicní infiltráty.
U pacientů s diagnostikovanou intersticiální plicní nemocí nebo pneumonitidou se doporučuje trvalé ukončení léčby trastuzumabem emtansinem.
Pacienti s klidovou dušností související s pokročilým nádorem nebo komorbiditami mohou mít vyšší riziko plicních příhod.
Hepatotoxicita
V klinických studiích byla při léčbě trastuzumabem emtansinem pozorována hepatotoxicita především ve formě zvýšení hladin transamináz v séru (stupně 1-4) (viz bod 4.8). Zvýšení hladiny transamináz bylo zpravidla přechodné s maximem osmý den po podání a s následnou úpravou na stupeň 1 nebo méně před dalším cyklem. Byl pozorován také kumulativní účinek transamináz (poměr pacientů
s odchylkami ALT/AST se stupněm 1-2 se zvyšuje s následujícími cykly).
U pacientů se zvýšenou hladinou transamináz došlo ke zlepšení na stupeň 1 nebo k normě ve většině případů do 30 dnů od poslední dávky trastuzumabu emtansinu (viz bod 4.8).
Při léčbě trastuzumabem emtansinem byly pozorovány případy závažných poruch jater a žlučových cest včetně nodulární regenerační hyperplazie jater a polékového poškození jater, v některých případech končícího úmrtím. Pozorované případy mohly být ovlivněny komorbiditami a/nebo souběžně podávanými léčivými přípravky s hepatotoxickým potenciálem.
Před zahájením léčby a před podáním každé dávky má být zkontrolována funkce jater. Pacienti se zvýšenou hladinou ALT před zahájením léčby (např. při jaterních metastázách) mohou být predisponováni k poškození jater s vyšším rizikem jaterních příhod nebo vzestupem hodnot funkčních jaterních testů stupně 3-5. Snížení dávky nebo ukončení léčby při zvýšení hladiny transamináz nebo celkového bilirubinu je uvedeno v bodě 4.2.
U pacientů léčených trastuzumabem emtansinem byly při biopsii jater zaznamenány případy nodulární regenerační hyperplazie (NRH). NRH je vzácné postižení jater charakterizované rozsáhlou benigní transformací jaterního parenchymu do malých regenerativních uzlů; NRH může vést k necirhotické portální hypertenzi. Diagnózu NRH lze potvrdit pouze histopatologicky. Na NRH je nutno pomýšlet u všech pacientů s klinickými známkami portální hypertenze a/nebo při obrazu připomínajícím cirhózu při vyšetření jater výpočetní tomografií (CT), ale s normální hladinou transamináz a bez dalších projevů cirhózy. Při diagnóze NRH je nutno léčbu trastuzumabem emtansinem trvale ukončit.
Trastuzumab emtansin nebyl hodnocen u pacientů s hladinou transamináz v séru > 2,5násobek horní hranice normy nebo celkovým bilirubinem > 1,5násobek horní hranice normy před zahájením léčby. Léčba trastuzumabem emtansinem má být trvale ukončena u pacientů s hladinou transamináz v séru > 3násobek horní hranice normy a se současnou hladinou celkového bilirubinu > 2násobek horní hranice normy. Při léčbě pacientů s poruchou funkce jater je nutná opatrnost (viz body 4.2 a 5.2).
Dysfunkce levé srdeční komory
Pacienti léčení trastuzumabem emtansinem mají zvýšené riziko vzniku dysfunkce levé srdeční komory. U pacientů léčených trastuzumabem emtansinem byl pozorován pokles ejekční frakce levé komory (LVEF) na < 40 %, a proto je možným rizikem symptomatické městnavé srdeční selhání (viz bod 4.8). Obecná rizika kardiální příhody a rizika identifikovaná v adjuvantních studiích karcinomu prsu při léčbě trastuzumabem zahrnují vyšší věk (> 50 let), nízkou vstupní hodnotu LVEF (< 55 %), nízkou hodnotu LVEF před podáním nebo po podání paklitaxelu v adjuvanci, předchozí nebo souběžné užívání antihypertenzních léčivých přípravků, předchozí léčbu antracykliny a vysoký index tělesné hmotnosti (BMI > 25 kg/m2).
Před zahájením léčby a v pravidelných intervalech (např. každé tři měsíce) během léčby má být prováděno standardní kardiologické vyšetření (echokardiogram nebo radionuklidová ventrikulografie (MUGA)). V klinických studiích měli pacienti před zahájením léčby LVEF > 50 %. Z klinických studií byli vyloučeni pacienti s anamnézou městnavého srdečního selhání, závažné arytmie vyžadující léčbu, anamnézou infarktu myokardu nebo nestabilní anginy pectoris v období 6 měsíců před randomizací nebo s klidovou dušností při pokročilém nádoru. V případě dysfunkce levé komory má být dle potřeby léčba odložena nebo ukončena (viz bod 4.2).
Reakce související s infuzí
Trastuzumab emtansin nebyl hodnocen u pacientů, u kterých byla trvale ukončena léčba trastuzumabem kvůli reakcím souvisejícím s infuzí, a proto se u těchto pacientů léčba přípravkem Kadcyla nedoporučuje. Pacienti mají být pečlivě sledováni na reakce související s infuzí, a to zejména při první infuzi.
Byly hlášeny reakce související s infuzí (v důsledku uvolnění cytokinů) charakterizované jedním nebo více z následujících příznaků: zrudnutí, třesavka, pyrexie, dušnost, hypotenze, sípání, bronchospazmus a tachykardie. Tyto příznaky zpravidla nebyly závažné (viz bod 4.8). U většiny pacientů došlo k ustoupení těchto účinků během několika hodin až jednoho dne po ukončení infuze. Při závažné reakci související s infuzí má být léčba přerušena, dokud příznaky neustoupí. Pokračování v léčbě má být zvažováno na základě klinického zhodnocení závažnosti reakce. V případě život ohrožující reakce související s infuzí musí být léčba trvale ukončena (viz bod 4.2).
Reakce přecitlivělosti
Trastuzumab emtansin nebyl hodnocen u pacientů, u kterých byla trvale ukončena léčba trastuzumabem z důvodu přecitlivělosti, a proto se u těchto pacientů léčba trastuzumabem emtansinem nedoporučuje.
Pacienti mají být pečlivě sledováni na reakce přecitlivělosti/alergii, které mohou mít stejné klinické projevy jako reakce související s infuzí. U pacientů léčených v klinických studiích trastuzumabem emtansinem byly pozorovány závažné anafylaktické reakce. K okamžitému použití mají být k dispozici léky i pohotovostní vybavení k léčbě takových reakcí. V případě skutečné reakce přecitlivělosti (při které se závažnost reakce zvyšuje s následujícími infuzemi) musí být léčba trastuzumabem emtansinem trvale ukončena.
Trombocytopenie
U pacientů léčených trastuzumabem emtansinem byla často hlášena trombocytopenie neboli pokles počtu trombocytů a byl to nej častější nežádoucí účinek vedoucí k ukončení léčby (viz bod 4.8).
V klinických studiích byla incidence a závažnost trombocytopenie vyšší u asijských pacientů (viz bod 4.8).
Byly pozorovány krvácivé příhody končící úmrtím. V klinických studiích byly hlášeny případy těžkých krvácivých příhod včetně krvácení v centrálním nervovém systému; tyto příhody nebyly závislé na etnické příslušnosti. V některých pozorovaných případech byli pacienti léčeni rovněž antikoagulancii.
Doporučuje se kontrolovat počet trombocytů před každou dávkou trastuzumabu emtansinu. Pacienti s trombocytopenií (< 100 000/mm3) nebo pacienti léčení antikoagulancii (např. warfarinem, heparinem, nízkomolekulárními hepariny) mají být během léčby trastuzumabem emtansinem pečlivě sledováni. Trastuzumab emtansin nebyl hodnocen u pacientů s počtem trombocytů před zahájením léčby < 100 000/mm3. Při poklesu počtu trombocytů na stupeň 3 nebo vyšší (< 50 000/mm3) nepodávejte trastuzumab emtansin, dokud nedojde k úpravě na stupeň 1 (> 75 000/mm3) (viz bod 4.2).
Neurotoxicita
V klinických studiích s trastuzumabem emtansinem byla hlášena periferní neuropatie, zpravidla stupně 1 a především senzorická. Z klinických studií byli vyloučeni pacienti s periferní neuropatií stupně > 3 před zahájením léčby. Léčba trastuzumabem emtansinem má být dočasně přerušena u pacientů
s periferní neuropatií stupně 3 nebo 4, dokud příznaky nevymizí nebo nedojde ke zlepšení na stupeň < 2. Pacienti mají být průběžně klinicky sledováni na známky/příznaky neurotoxicity.
Obsah sodíku v pomocných látkách
Tento léčivý přípravek obsahuje v jedné dávce méně než 1 mmol (23 mg) sodíku, je v podstatě „bez sodíku“
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné formální studie interakcí.
In vitro studie metabolismu v lidských jaterních mikrozomech ukazují, že DM1 - komponenta trastuzumabu emtansinu - je metabolizován hlavně enzymem CYP3A4 a v menší míře enzymem CYP3A5.
Vzhledem k možnému zvýšení expozice DM1 a toxicity se trastuzumab emtansin nemá podávat souběžně se silnými inhibitory CYP3A4 (např. ketokonazol, itrakonazol, klaritromycin, atazanavir, indinavir, nefazodon, nelfinavir, ritonavir, sachinavir, telithromycin, vorikonazol). Zvažte jiný léčivý přípravek s nulovou nebo minimální schopností inhibovat CYP3A4. Pokud je souběžné užití silných inhibitorů CYP3A4 nevyhnutelné, zvažte pokud možno odložení léčby trastuzumabem emtansinem do doby jejich vyloučení z oběhu (přibližně 3 eliminační poločasy inhibitoru). Pokud je podáván silný inhibitor CYP3A4 a léčbu trastuzumabem emtansinem nelze odložit, má být pacient pečlivě sledován pro možné nežádoucí účinky.
4.6 Fertilita, těhotenství a kojení
Antikoncepce u mužů a žen
Ženy ve fertilním věku mají během léčby trastuzumabem emtansinem a po dobu 7 měsíců po podání poslední dávky trastuzumabu emtansinu používat účinnou antikoncepci. Muži pacienti nebo jejich partnerky mají také používat účinnou antikoncepci.
Nejsou k dispozici údaje o použití trastuzumabu emtansinu u těhotných žen. Trastuzumab - součást trastuzumabu emtansinu - může při podání těhotným ženám způsobit poškození nebo úmrtí plodu. Při postmarketingovém sledování byly u těhotných žen léčených trastuzumabem hlášeny případy oligohydramnionu, některé spojené s fatální hypoplazií plic. Studie s maytansinem - blízce příbuzná chemická látka ze stejné skupiny maytansinoidů jako DM1 - u zvířat naznačují, že lze očekávat, že DM1 - cytotoxická komponenta trastuzumabu emtansinu inhibující mikrotubuly - bude mít teratogenní a potenciálně embryotoxický efekt (viz bod 5.3).
Podání trastuzumabu emtansinu těhotným ženám se nedoporučuje a ženy mají být informovány o možném poškození plodu, než otěhotní. Pokud žena otěhotní, musí ihned kontaktovat svého lékaře. Pokud je těhotná žena léčena trastuzumabem emtansinem, doporučuje se pečlivé sledování multidisciplinárním týmem.
Kojení
Není známo, zda se trastuzumab emtansin vylučuje do lidského mateřského mléka. Vzhledem k tomu, že mnoho léčivých přípravků je vylučováno do lidského mateřského mléka, a vzhledem k možnosti závažných nežádoucích účinků u kojených dětí mají ženy ukončit kojení před zahájením léčby trastuzumabem emtansinem. Ženy mohou začít kojit 7 měsíců po ukončení léčby.
Fertilita
Nebyly provedeny studie reprodukční ani vývojové toxicity u trastuzumabu emtansinu.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Trastuzumab emtansin nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje. Vliv hlášených nežádoucích účinků, jako jsou únava, bolest hlavy, závratě nebo zastřené vidění, na schopnost řídit a obsluhovat stroje není znám. Pacienti, u kterých se vyskytly reakce na infuzi, raději nemají řídit nebo obsluhovat stroje, dokud se příznaky nezmírní.
4.8 Nežádoucí účinky
Shrnutí bezpečnostního profilu
Bezpečnost trastuzumabu emtansinu byla hodnocena u 1871 pacientů s karcinomem prsu v klinických studiích. V této populaci pacientů:
• nejčastějšími závažnými nežádoucími účinky (> 0,5 % pacientů) byly krvácení, pyrexie, dušnost, bolest svalů a kostí, trombocytopenie, bolest břicha a zvracení.
• nej častějšími nežádoucími účinky (> 25 %) trastuzumabu emtansinu byly nauzea, únava a bolest hlavy. Většina hlášených nežádoucích účinků dosáhla stupně závažnosti 1 nebo 2.
• nejčastějšími nežádoucími účinky stupně > 3 (> 2 %) dle NCI-CTCAE (National Cancer Institute
- Common Terminology Criteria for Adverse Events) byly trombocytopenie, zvýšení transamináz, anemie, neutropenie, únava, hypokalemie, bolest svalů a kostí a krvácení.
Shrnutí nežádoucích účinků do tabulky
Nežádoucí účinky u 1871 pacientů léčených trastuzumabem emtansinem jsou uvedeny v tabulce 6. Níže uvedené nežádoucí účinky jsou řazeny podle tříd orgánových systémů a absolutní četnosti databáze MedDRA. Četnosti jsou definovány jako velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1000 až < 1/100), vzácné (> 1/10 000 až < 1/1000), velmi vzácné (< 1/10 000) a není známo (z dostupných údajů nelze určit). V každé skupině četností a třídě orgánových systémů jsou nežádoucí účinky řazeny podle klesající závažnosti. Pro posouzení toxicity byly nežádoucí účinky hlášeny dle NCI-CTCAE.
Tabulka 6 Přehled nežádoucích účinků vyskytující se u pacientů léčených trastuzumabem
emtansinem
|
Třídy orgánových systémů |
Velmi časté |
Časté |
Méně časté |
|
Infekce a infestace |
Infekce močového traktu | ||
|
Poruchy krve a lymfatického systému |
Trombocytopenie, |
Neutropenie, leukopenie | |
|
Poruchy imunitního systému |
Hypersensitivita na lék | ||
|
Poruchy metabolismu a výživy |
Hypokalemie | ||
|
Psychiatrické poruchy |
Insomnie | ||
|
Poruchy nervového systému |
Periferní neuropatie, bolest hlavy |
Závratě, dysgeuzie, poruchy paměti | |
|
Poruchy oka |
Suchost oka, konjunktivitida, neostré vidění, zvýšené slzení | ||
|
Srdeční poruchy |
Dysfunkce levé srdeční komory | ||
|
Cévní poruchy |
Krvácení |
Hypertenze | |
|
Respirační, hrudní a mediastinální poruchy |
Pneumonitida (intersticiální plicní nemoc) | ||
|
Gastrointestinální poruchy |
Stomatitida, průjem, zvracení, nauzea, zácpa, sucho v ústech, bolest břicha |
Dyspepsie, krvácení dásní | |
|
Poruchy jater a žlučových cest |
Hepatotoxicita, selhávání jater, nodulární regenerační hyperplasie, portální hypertenze | ||
|
Poruchy kůže a podkožní tkáně |
Pruritus, alopecie, poruchy nehtů, syndrom palmoplantární erytrodysestezie, kopřivka | ||
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Bolest svalů a kostí, bolest kloubů, bolest svalů | ||
|
Celkové poruchy a reakce v místě aplikace |
Únava, pyrexie, astenie, třesavka |
Periferní otok |
Extravazace v místě vpichu injekce |
|
Vyšetření |
Zvýšení transamináz |
Zvýšení alkalické fosfatázy v krvi |
|
Třídy orgánových systémů |
Velmi časté |
Časté |
Méně časté |
|
Poranění, otravy a procedurální postupy |
Reakce na infuzi |
Popis vybraných nežádoucích účinků
Zvýšení transamináz (AST/ALT)
V klinických studiích bylo při léčbě trastuzumabem emtansinem pozorováno zvýšení hladiny transamináz v séru (stupeň 1-4) (viz bod 4.4). Zvýšení transamináz bylo zpravidla přechodné. Byl pozorován kumulativní vliv trastuzumabu emtansinu na transaminázy, které se po ukončení léčby normalizovaly. Zvýšení transamináz bylo v klinických studiích hlášeno u 24,2 % pacientů. Zvýšení stupně 3 nebo 4 AST a ALT bylo hlášeno u 4,2 % respektive u 2,7 % pacientů a projevilo se obvykle v časných cyklech léčby (1 až 6). Obecně nebyly jaterní příhody stupně > 3 spojeny se špatnými klinickými výsledky. Hodnoty při následných kontrolách vykazovaly trend ke zlepšení na hodnoty umožňující pacientům setrvat ve studii a nadále dostávat léčbu ve stejné nebo snížené dávce. Nebyl pozorován vztah mezi expozicí trastuzumabu emtansinu (AUC), maximální koncentrací trastuzumabu emtansinu v séru (Cmax), celkovou expozicí trastuzumabu (AUC) nebo Cmax DM1 a vzestupem transamináz. Úprava dávky v případě vzestupu transamináz viz body 4.2 a 4.4.
Dysfunkce levé srdeční komory
V klinických studiích s trastuzumabem emtansinem byla dysfunkce levé srdeční komory hlášena u 2,2 % pacientů. Většinu příhod tvořil asymptomatický pokles ejekční frakce levé komory (LVEF) stupně 1 nebo 2. Příhody stupně 3 nebo 4 byly hlášeny u 0,4 % pacientů. U pacientů s poklesem LVEF na < 45 % se doporučuje další monitorování LVEF (specifické modifikace dávky viz tabulka 5 v bodě 4.2).
Reakce související s infuzí
Reakce související s infuzí jsou charakterizovány jedním nebo více z následujících příznaků: zrudnutí, třesavka, horečka, dušnost, hypotenze, sípání, bronchospazmus a tachykardie. V klinických studiích s trastuzumabem emtansinem byly reakce související s infuzí hlášeny u 4,0 % pacientů, hlášeno bylo šest příhod stupně 3 a nebyla hlášena žádná příhoda stupně 4. Reakce související s infuzí odezněly během několika hodin až jednoho dne po ukončení infuze. V klinických studiích nebyla pozorována souvislost s dávkou. Úprava dávky v případě reakce související s infuzí viz body 4.2 a 4.4.
Reakce přecitlivělosti
V klinických studiích s trastuzumabem emtansinem byly reakce přecitlivělosti hlášeny u 2,6 % pacientů, byla hlášena jedna příhoda stupně 3 a jedna příhoda stupně 4. Většina reakcí přecitlivělosti zpravidla dosahovala mírného až středního stupně závažnosti a po léčbě odezněla. Úprava dávky
v případě reakce přecitlivělosti viz body 4.2 a 4.4.
Trombocytopenie
Trombocytopenie nebo pokles počtu trombocytů byly hlášeny u 24,9 % pacientů v klinických studiích s trastuzumabem emtansinem a byly nej častějším nežádoucím účinkem vedoucím k ukončení léčby (2,6 %). Většina pacientů měla příhodu stupně 1 nebo 2 (> 50 000/mm3) s nejnižší hodnotou do osmého dne a zpravidla se zlepšením na stupeň 0 nebo 1 (> 75 000/mm3) před další plánovanou dávkou. V klinických studiích byla incidence a závažnost trombocytopenie vyšší u asijských pacientů. Bez ohledu na rasu byla incidence příhod stupně 3 nebo 4 (< 50 000/mm3) u pacientů léčených trastuzumabem emtansinem 8,7 %. Incidence závažných krvácivých příhod (stupeň >3) byla 2,2 % všech pacientů léčených trastuzumabem emtansinem a 1,8 % asijských pacientů léčených trastuzumabem emtansinem. V některých zaznamenaných případech byli pacienti léčeni rovněž antikoagulancii. Byly zaznamenány krvácivé příhody končící úmrtím. Úprava dávky v případě trombocytopenie viz body 4.2 a 4.4.
Imunogenita
Stejně jako u všech léčebných bílkovin je možnost imunitní odpovědi na trastuzumab emtansin.
V šesti klinických studiích byla u celkem 836 pacientů v různých časových obdobích testována
přítomnost protilátkové odpovědi na trastuzumab emtansin. Po podání léku byl test na protilátky proti trastuzumabu emtansinu pozitivní v jednom nebo více časových bodech u 5,3 % (44/836) pacientů. Klinický význam protilátek proti trastuzumabu emtansinu dosud není znám.
Extravazace
V klinických studiích s trastuzumabem emtansinem byly pozorovány reakce v důsledku extravazace. Tyto reakce byly zpravidla mírné nebo středně těžké a zahrnovaly erytém, citlivost, podráždění kůže, bolest nebo zduření v místě infuze. Tyto reakce byly pozorovány nejčastěji do 24 hodin po infuzi.
V současné době není známa specifická léčba při extravazaci trastuzumabu emtansinu.
Laboratorní odchylky
V tabulce 7 jsou uvedeny laboratorní abnormality pozorované u pacientů léčených trastuzumabem emtansinem ve studii TDM4370g/BO21977.
Tabulka 7 Laboratorní odchylky zjištěné u pacientů léčených trastuzumabem emtansinem ve studii TDM4370g/BO21977
|
Parametr |
Trastuzumab emtansin | ||
|
Všechny stupně (%) |
Stupeň 3 (%) |
Stupeň 4 (%) | |
|
Jaterní | |||
|
Zvýšená hladina bilirubinu |
21 |
< 1 |
0 |
|
Zvýšení AST |
98 |
8 |
< 1 |
|
Zvýšení ALT |
82 |
5 |
< 1 |
|
Hematologické | |||
|
Snížený počet krevních destiček |
85 |
14 |
3 |
|
Snížená hladina hemoglobinu |
63 |
5 |
1 |
|
Snížený počet neutrofilů |
41 |
4 |
< 1 |
|
Draslík | |||
|
Snížená hladina draslíku |
35 |
3 |
<1 |
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Není známo žádné antidotum pro případ předávkování trastuzumabem emtansinem. V případě předávkování má být pacient pečlivě sledován pro známky a příznaky nežádoucích účinků a má být zahájena příslušná symptomatická léčba. Byly hlášeny případy předávkování při léčbě trastuzumabem emtansinem, většina byla spojena s trombocytopenií a v jednom případě došlo k úmrtí. V tomto fatálním případě dostal pacient dávku 6 mg/kg trastuzumabu emtansinu a zemřel asi za 3 týdny po předávkování. Příčinný vztah k trastuzumabu emtansinu nebyl stanoven.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: cytostatika, jiná cytostatika, monoklonální protilátky, ATC kód: L01XC14.
Mechanismus účinku
Přípravek Kadcyla, trastuzumab emtansin, je konjugát protilátky cílené na HER2 a cytostatika. Obsahuje humanizovaný IgG1 proti HER2 trastuzumab, kovalentně vázaný na mikrotubulární inhibitor DM1 (derivát maytansinu) stabilním thioéterovým vazebným můstkem MCC (4-[N-maleimidomethyl] cyklohexan-1-karboxylát). Emtansin je označení pro komplex MCC-DM1. Na každou molekulu trastuzumabu jsou navázány průměrně 3,5 molekuly DM1.
Vazba DM1 na trastuzumab zajišťuje selektivní účinek cytotoxické látky na nádorové buňky nadměrně exprimující HER2, čímž se zvyšuje intracelulární průnik DM1 přímo do nádorových buněk. Trastuzumab emtansin se po navázání na HER2 spolu s receptorem dostává do nitra buňky (internalizuje se) a následně dochází k jeho degradaci v lysozomech, při které se uvolňují cytotoxické katabolity obsahující DM1 (primárně lysin-MCC-DM1).
Trastuzumab emtansin spojuje mechanismus účinku jak trastuzumabu, tak DM1:
• Trastuzumab emtansin se podobně jako trastuzumab váže na doménu IV mimobuněčné části receptoru HER2 a rovněž na receptory Fcy a složku komplementu C1q. Kromě toho trastuzumab emtansin u lidských buněk karcinomu prsu, které nadměrně exprimují HER2, podobně jako trastuzumab inhibuje odštěpení extracelulární domény HER2, inhibuje signalizaci cestou fosfatidylinositol 3-kinázy (PI3-K) a podporuje buněčnou cytotoxicitu závislou na protilátkách.
• DM1, cytotoxická komponenta trastuzumabu emtansinu, se váže na tubulin. Inhibicí polymerizace tubulinu způsobují jak DM1, tak trastuzumab emtansin zastavení buněčného cyklu ve fázi G2/M, což v konečném důsledku vede k apoptotické smrti buňky. Výsledky in vitro hodnocení cytotoxicity ukazují, že DM1 je 20-200krát účinnější než taxany a vinca alkaloidy.
• Vazebný můstek MCC je konstruován tak, aby bylo minimalizováno uvolňování DM1 v systémovém oběhu a zvýšena cílená dodávka DM1, což je prokázáno detekcí velice nízkých hladin volného DM1 v plazmě.
Klinická účinnost
TDM4370g/BO21977
Randomizovaná multicentrická mezinárodní otevřená klinická studie fáze III byla provedena u pacientů s HER2-pozitivním neresekovatelným lokálně pokročilým nebo metastazujícím karcinomem prsu, kteří byli dříve léčeni režimem obsahujícím taxan a trastuzumab, včetně pacientů dříve léčených trastuzumabem a taxanem v adjuvantním podání, a u kterých došlo k relapsu během adjuvantní léčby nebo do šesti měsíců od ukončení adjuvantní léčby. K zařazení byli vhodní pouze pacienti se stavem tělesné výkonnosti (performance status) 0 nebo 1 dle hodnocení Eastern Cooperative Oncology Group (ECOG). Před zařazením byly vyžadovány vzorky karcinomu prsu k centrálnímu potvrzení HER2-pozitivity, která byla definována jako skóre 3+ při imunohistochemickém (IHC) stanovení nebo amplifikace genu dle in situ hybridizace (ISH). Vstupní charakteristiky pacientů a nádorů byly mezi léčebnými skupinami dobře vyváženy. Pacienty s léčenými mozkovými metastázami bylo možno zařadit, pokud u nich nebyla nutná léčba příznaků. Střední věk pacientů randomizovaných k léčbě trastuzumabem emtansinem byl 53 let, většina byly ženy (99,8 %), většinou bělošky (72 %) a v 57 % se jednalo o onemocnění s pozitivitou estrogenových a/nebo progesteronových receptorů. Studie porovnávala účinnost a bezpečnost trastuzumabu emtansinu s účinností a bezpečností lapatinibu plus kapecitabinu. Celkem 991 pacientů bylo randomizováno k léčbě buď trastuzumabem emtansinem, nebo lapatinibem plus kapecitabinem následovně:
• Rameno s trastuzumabem emtansinem: trastuzumab emtansin 3,6 mg/kg intravenózně po dobu 30-90 minut v den 1 cyklu trvajícího 21 dní. 1
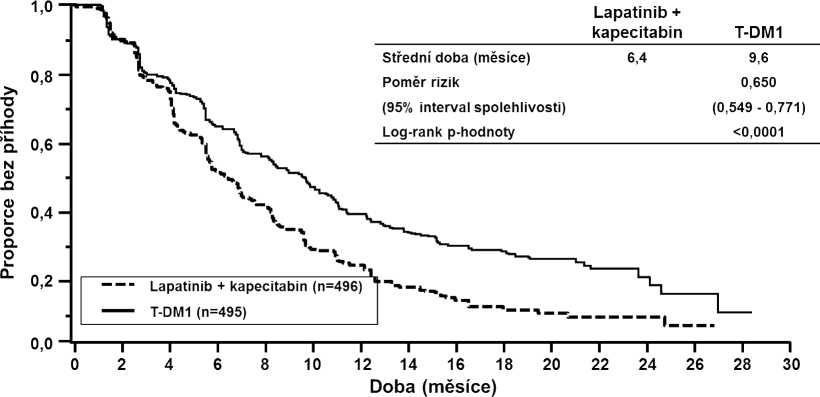
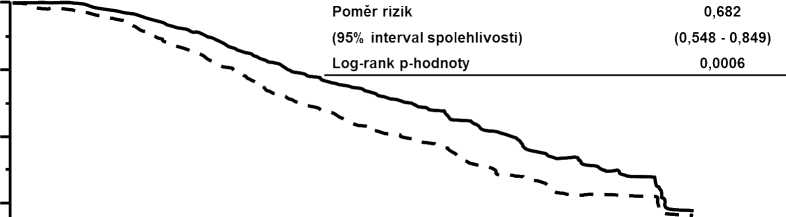
Společnými primárními cíli pro hodnocení účinnosti byly přežití bez progrese (PFS) dle hodnocení nezávislou komisí a celkové přežití (OS) (viz tabulka 8 a obrázky 1 a 2).
V klinické studii byla dále hodnocena doba do zhoršení příznaků definovaná jako pokles skóre odvozeného z podškály „outcome index-breast“ (TOI-B) dotazníku kvality života FACT-B o 5 bodů. Změna o 5 bodů v TOI-B je považována za klinicky významnou. Přípravek Kadcyla vedl k pozdějšímu zhoršení příznaků udávaných pacienty na 7,1 měsíce ve srovnání s 4,6 měsíce v kontrolním rameni (poměr rizik 0,796 [0,667; 0,951]; p-hodnota 0,0121). Údaje jsou z otevřené studie a nelze z nich vyvodit žádné pevné závěry.
Tabulka 8 Souhrn údajů o účinnosti ve studii TDM4370g/BO21977 (EMILIA)
|
Lapatinib + kapecitabin n = 496 |
Trastuzumab emtansin n = 495 | |
|
Primární cíle | ||
|
Přežití bez progrese (PFS) dle hodnocení nezávislou komisí (IRC) | ||
|
Počet (%) pacientů s příhodou |
304 (61,3 %) |
265 (53,5 %) |
|
Střední doba přežití bez progrese (měsíce) |
6,4 |
9,6 |
|
Poměr rizik (stratifikovaný1) |
0,650 | |
|
95% interval spolehlivosti pro poměr rizik |
(0,549; 0,771) | |
|
p-hodnota (Log-rank test -stratifikovaný1) |
< 0,0001 | |
|
Celkové přežití (OS) | ||
|
Počet (%) zemřelých |
182 (36,7 %) |
149 (30,1 %) |
|
Střední doba přežití (měsíce) |
25,1 |
30,9 |
|
Poměr rizik (stratifikovaný1) |
0,682 | |
|
95% interval spolehlivosti pro poměr rizik |
(0,548; 0,849) | |
|
p-hodnota (Log-rank test1) |
0,0006 | |
|
Klíčové sekundární cíle | ||
|
Přežití bez progrese (PFS) dle hodnocení řešiteli | ||
|
Počet (%) pacientů s příhodou |
335 (67,5 %) |
287 (58,0 %) |
|
Střední doba přežití bez progrese (měsíce) |
5,8 |
9,4 |
|
Poměr rizik (95% interval spolehlivosti) |
0,658 (0,560; 0,774) | |
|
p-hodnota (Log-rank test1) |
<0,0001 | |
|
Četnost objektivních odpovědí (ORR) | ||
|
Pacienti s měřitelným onemocněním |
389 |
397 |
|
Počet pacientů s objektivní odpovědí (%) |
120 (30,8 %) |
173 (43,6 %) |
|
Rozdíl (95% interval spolehlivosti) |
12,7% (6,0; 19,4) | |
|
p-hodnota (Mantel-Haenszel chí- |
0,0002 | |
|
Lapatinib + kapecitabin n = 496 |
Trastuzumab emtansin n = 495 | |
|
kvadrát test*) | ||
|
Trvání objektivní odpovědi (měsíce) | ||
|
Počet pacientů s objektivní odpovědí |
120 |
173 |
|
Medián (95% interval spolehlivosti) |
6,5 (5,5; 7,2) |
12,6 (8,4; 20,8) |
OS: celkové přežití; PFS: přežití bez progrese; ORR: četnost objektivní odpovědi; OR: objektivní odpověď; ICR: hodnocení nezávislou komisí; HR: poměr rizik; CI: interval spolehlivosti
*Stratifikace dle: oblast světa (USA, Západní Evropa, jiná), počet předchozích režimů chemoterapie v léčbě lokálně pokročilého nebo metastazujícího onemocnění (0-1 vs. > 1) a viscerální vs non-viscerální postižení.
**Průběžná analýza celkového přežití byla provedena po zaznamenání 331 příhod. Protože při této analýze byla překročena stanovená hranice účinnosti, je tato analýza považována za definitivní.
Prospěch z léčby byl pozorován v podskupině pacientů, u nichž došlo k relapsu nemoci do 6 měsíců od ukončení adjuvantní léčby a kteří předtím neměli žádnou systémovou protinádorovou léčbu pro metastatické onemocnění (n = 118). Poměr rizik pro přežití bez progrese byl 0,51 (95% interval spolehlivosti 0,30; 0,85) a poměr rizik pro celkové přežití 0,61 (95% interval spolehlivosti 0,32; 1,16). Střední doba přežití bez progrese byla ve skupině léčené trastuzumabem emtansinem 10,8 měsíce oproti 5,7 měsíce ve skupině léčené lapatinibem + kapecitabinem a střední celkové přežití nebylo ve skupině léčené trastuzumabem emtansinem dosaženo oproti 27,9 měsíce ve skupině léčené lapatinibem + kabecitabinem.
Obrázek 1 Kaplan-Meierova křivka přežití bez progrese dle hodnocení nezávislou komisí
Počet v riziku
Lap + kap 496 404 310
T-DM1 495 419 341
1 0
3 1
0
0
|
176 |
129 |
73 |
53 |
35 |
|
236 |
183 |
130 |
101 |
72 |
T-DM1: trastuzumab emtansin; Lap: lapatinib; kap: kapecítabin;
Poměr rizik je stanoven dle stratifikovaného Coxova modelu, hodnota p dle stratifikovaného log-rank testu
|
25 |
14 |
9 |
8 |
5 |
|
54 |
44 |
30 |
18 |
9 |
Obrázek 2 Kaplan-Meierova křivka celkového přežití
>
"O
o
N
Cl)
-Q
<D
O
L-
o
Q.
O
Lapatinib +
kapecitabin T-DM1
Střední doba (měsíce) 25,1 30,9
0,4
- Lapatinib + kapecitabin (n=496) — T-DM1 (n=495)
0,2'
0,0.
|
0 |
2 |
4 |
6 |
8 |
10 |
12 |
14 16 18 20 22 24 Trvání přežití (měsíce) |
26 |
28 |
30 |
32 |
34 |
36 |
|
Počet v riziku | |||||||||||||
|
Lap + Cap 496 |
471 |
453 |
435 |
403 |
368 |
297 |
240 204 159 133 110 86 |
63 |
45 |
27 |
17 |
7 |
4 |
|
T-DM1 495 |
485 |
474 |
457 |
439 |
418 |
349 |
293 242 197 164 136 111 |
86 |
62 |
38 |
28 |
13 |
5 |
T-DM1: trastuzumab emtansin; Lap: lapatinib; kap: kapecitabin;
Poměr rizik je stanoven dle stratifikovaného Coxova modelu, hodnota p dle stratifikovaného log-rank testu
Ve studii TDM4370g/BO21977 byl pozorován konzistentní prospěch při léčbě trastuzumabem emtansinem ve většině předem definovaných podskupin, což podporuje robustnost celkových výsledků. V podskupině pacientů s negativitou hormonálních receptorů (n = 426) byl poměr rizik pro přežití bez progrese 0,56 (95% interval spolehlivosti 0,44; 0,72) a poměr rizik pro celkové přežití 0,75 (95% interval spolehlivosti 0,54; 1,03). V podskupině pacientů s pozitivitou hormonálních receptorů (n = 545) byl poměr rizik pro přežití bez progrese 0,72 (95% interval spolehlivosti 0,58; 0,91) a poměr rizik pro celkové přežití 0,62 (95% interval spolehlivosti 0,46; 0,85).
V podskupině pacientů s neměřitelným onemocněním (n = 205) byl dle hodnocení nezávislou komisí poměr rizik pro přežití bez progrese 0,91 (95% interval spolehlivosti 0,59; 1,42) a poměr rizik pro celkové přežití 0,96 (95% interval spolehlivosti 0,54; 1,68). U pacientů ve věku >65 let (n=138 celkem v obou ramenech) byl poměr rizik pro přežití bez progrese 1,06 (95% interval spolehlivosti 0,68; 1,66) a poměr rizik pro celkové přežití 1,05 (95% interval spolehlivosti 0,58; 1,91). U pacientů ve věku 65 až 74 let (n = 113) byl dle hodnocení nezávislou komisí poměr rizik pro přežití bez progrese 0,88 (95% interval spolehlivosti 0,53; 1,45) a poměr rizik pro celkové přežití 0,74 (95% interval spolehlivosti 0,37; 1,47). U pacientů ve věku 75 let a více byl dle hodnocení nezávislou komisí poměr rizik pro přežití bez progrese 3,51 (95% interval spolehlivosti 1,22; 10,13) a poměr rizik pro celkové přežití 3,45 (95% interval spolehlivosti 0,94; 12,65). V podskupině pacientů ve věku 75 let a více nebyl prokázán prospěch pro přežití bez progrese ani pro celkové přežití, tato podskupina však byla příliš malá (n = 25), aby bylo možno učinit definitivní závěry.
V deskriptivní analýze sledování celkového přežití byl poměr rizik 0,75 (95% interval spolehlivostiCI 0,64; 0,88). Medián trvání celkového přežití byl 29,9 měsíce ve skupině s trastuzumab emtansinem, v porovnání s 25,9 měsíce ve skupině, kde byl podáván lapanitib plus kapecitabin. V době deskriptivní analýzy sledování celkového přežití přešlo celkem 27,4 % pacientů ze skupiny lapanitib plus kapecitabin, do skupiny s trastuzumab emtansinem. V analýze senzitivity, s cenzorovanými pacienty při přechodu ze skupiny lapanitib plus kapecitabin do skupiny s trastuzumab emtansinem, byl poměr rizik 0,69 (95% interval spolehlivostiCI 0,59; 0,82). Výsledky této deskriptivní analýzy sledování jsou v souladu s výsledky konfirmační analýzy celkového přežití.
TDM4450g
Randomizovaná otevřená multicentrická studie fáze II hodnotila účinnost trastuzumabu emtansinu proti kombinaci trastuzumab plus docetaxel u pacientů s HER2-pozitivním metastatickým karcinomem prsu, kteří neměli předchozí chemoterapii pro metastazující onemocnění. Pacienti byli randomizováni k léčbě trastuzumabem emtansinem 3,6 mg/kg intravenózně každé 3 týdny (n = 67) nebo trastuzumabem v úvodní dávce 8 mg/kg intravenózně a následně 6 mg/kg intravenózně každé 3 týdny plus docetaxelem 75 až 100 mg/m2 intravenózně každé 3 týdny (n = 70).
Primárním cílem bylo přežití bez progrese dle hodnocení řešiteli. Střední doba přežití bez progrese byla 9,2 měsíce v rameni s trastuzumabem a docetaxelem a 14,2 měsíce v rameni s trastuzumabem emtansinem (poměr rizik 0,59; p = 0,035) při střední době sledování přibližně 14 měsíců v obou ramenech. Četnost objektivních odpovědí byla 58,0 % v rameni s trastuzumabem a docetaxelem a 64,2% v rameni s trastuzumabem emtansinem. Střední doby trvání odpovědi v rameni s trastuzumabem emtansinem nebylo dosaženo a v kontrolním rameni byla 9,5 měsíce.
TDM4374g
Jednoramenná otevřená studie fáze II hodnotila účinnost trastuzumabu emtansinu u pacientů s HER2-pozitivním nevyléčitelným lokálně pokročilým nebo metastazujícím karcinomem prsu. Všichni pacienti byli dříve léčeni léky cílenými na HER2 (trastuzumab a lapatinib) a chemoterapií (antracykliny, taxany a kapecitabinem) v neoadjuvantním nebo adjuvantním podání a při léčbě lokálně pokročilého nebo metastazujícího onemocnění. Střední počet protinádorových léků, kterými byli pacienti léčeni, byl 8,5 (rozptyl 5-19) a při léčbě metastazujícího onemocnění 7,0 (3-17) při započtení všech léků užitých k léčbě karcinomu prsu.
Pacienti (n = 110) dostávali 3,6 mg/kg trastuzumabu emtansinu intravenózně každé 3 týdny do progrese nemoci nebo nepřijatelné toxicity.
Klíčovou analýzou účinnosti bylo nezávislé radiologické stanovení četnosti objektivní odpovědi a trvání objektivní odpovědi. Četnost objektivní odpovědi byla 32,7 % (95% interval spolehlivosti 24,1; 42,1), n = 36 pacientů s odpovědí dle hodnocení nezávislou komisí i dle hodnocení řešiteli. Střední doby trvání odpovědi dle hodnocení nezávislou komisí nebylo dosaženo (95% interval spolehlivosti 4,6 měsíce - nelze stanovit).
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění z povinnosti předložit výsledky studií s trastuzumabem emtansinem u všech podskupin pediatrické populace v indikaci karcinom prsu (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Absorpce
Trastuzumab emtansin je podáván intravenózně. Nebyly prováděny studie s jiným způsobem podání. Distribuce
Pacienti ve studii TDM4370g/BO21977, kteří dostávali trastuzumab emtansin intravenózně v dávce 3,6 mg/kg každé 3 týdny, měli průměrnou maximální koncentraci (Cmax) trastuzumabu emtansinu 83,4 (± 16,5) pg/ml. Podle populační analýzy farmakokinetiky je po intravenózním podání centrální distribuční objem trastuzumabu emtansinu 3,13 l, což přibližně odpovídá objemu plazmy.
Biotransformace (trastuzumab emtansin a DM1)
Očekává se, že trastuzumab emtansin projde dekonjugací a proteolytickým katabolickým procesem v buněčných lysozomech.
In vitro studie metabolismu v lidských jaterních mikrozomech ukazují, že DM1 - komponenta trastuzumabu emtansinu s malou molekulou - je metabolizován hlavně enzymem CYP3A4 a v menší míře enzymem CYP3A5. DM1 neinhiboval in vitro hlavní enzymy CYP450. V lidské plazmě byly detekovány nízké hladiny katabolitů trastuzumabu emtansinu MCC-DM1, Lys-MCC-DM1 a DM1. In
vitro byl DM1 substrátem P-glykoproteinu.
Eliminace
Podle populační analýzy farmakokinetiky po intravenózním podání trastuzumabu emtansinu pacientům s HER2-pozitivním metastatickým karcinomem prsu byla clearance trastuzumabu emtansinu 0,68 l/den a eliminační poločas (t1/2) přibližně 4 dny. Nebyla pozorována akumulace trastuzumabu emtansinu po opakovaném podávání v intravenózní infuzi každé 3 týdny.
V populační analýze farmakokinetiky byly tělesná hmotnost, albumin, součet nejdelších průměrů cílových lézí dle Kritérií pro hodnocení odpovědi solidních nádorů (RECIST), odštěpená extracelulární doména HER2, vstupní koncentrace trastuzumabu a aspartátaminotransferáza (AST) identifikovány jako statisticky významné kovariáty farmakokinetických parametrů pro trastuzumab emtansin. Míra vlivu těchto kovariátů na expozici trastuzumabu emtansinu nicméně naznačuje, že tyto kovariáty pravděpodobně nebudou mít klinicky významný vliv na expozici trastuzumabu emtansinu. Kromě toho explorativní analýzy ukázaly, že vliv kovariátů (např. funkce ledvin, rasy a věku) na farmakokinetiku celkového trastuzumabu a DM1 byl omezený a nebyl klinicky významný. V neklinických studiích jsou katabolity trastuzumabu emtansinu včetně DM1, Lys-MCC-DM1 a MCC-DM1 vylučovány zejména žlučí s minimálním vylučováním močí.
Linearita/nelinearita
Při intravenózním podání trastuzumabu emtansinu každé 3 týdny byla zjištěna lineární farmakokinetika při dávkách v rozmezí 2,4 až 4,8 mg/kg; u pacientů, kterým bylo podáno 1,2 mg/kg nebo méně, byla zaznamenána rychlejší clearance.
Starší pacienti
Populační analýza farmakokinetiky ukázala, že věk neovlivnil farmakokinetiku trastuzumabu emtansinu. Nebyly pozorovány významné rozdíly farmakokinetiky trastuzumabu emtansinu u pacientů ve věku < 65 let (n = 577), u pacientů ve věku 65-75 let (n = 78) a u pacientů ve věku > 75 let (n = 16).
Porucha funkce ledvin
U pacientů s poškozením funkce ledvin nebyly prováděny formální studie farmakokinetiky. Populační analýza farmakokinetiky ukázala, že clearance kreatininu neovlivňuje farmakokinetiku trastuzumabu emtansinu. Farmakokinetika trastuzumabu emtansinu u pacientů s mírným (clearance kreatininu 60 až 89 ml/min, n = 254) nebo středně závažným (clearance kreatininu 30 až 59 ml/min, n = 53) poškozením ledvin byla podobná jako u pacientů s normální funkcí ledvin (clearance kreatininu > 90 ml/min, n = 361). Vzhledem k minimu farmakokinetických údajů u pacientů se závažným poškozením ledvin (clearance kreatininu 15-29 ml/min, n = 1) nelze stanovit žádná doporučení pro úpravu dávky.
Porucha funkce jater
Játra jsou primárním orgánem pro eliminaci katabolitů obsahujících DM1 a DM2. Farmakokinetika trastuzumabu emtansinu a katabolitů obsahujících DM1 byla vyhodnocena po podání 3,6 mg/kg trastuzumabu emtansinu pacientům s metastazujícím HER2+ karcinomem prsu s normální funkcí jater (n=10), s lehkou poruchou funkce jater (Child-Pugh A; n=10) a se středně těžkou poruchou funkce jater (Child-Pugh B; n=8).
- Plazmatické koncentrace DM1 a katabolitů obsahujících DM1 (LYs-MCC-DM1 a MCC-DM1) byly nízké a srovnatelné mezi pacienty s poruchou funkce jater a bez poruchy funkce jater.
- Systémové expozice (AUC) trastuzumabu emtansinu v cyklu 1 byly u pacientů s lehkou poruchou funkce jater přibližně o 38 % a u pacientů se středně těžkou poruchou funkce jater přibližně o
67 % nižší než u pacientů s normální funkcí jater. Expozice trastuzumabu emtansinu (AUC) v cyklu 3 po opakovaném dávkování u pacientů s lehkou nebo středně těžkou poruchou funkce jater byla v rozmezí pozorovaných u pacientů s normální funkcí jater.
Trastuzumab emtansin nebyl studován u pacientů s těžkou poruchou funkce jater (Child-Pugh třídy C).
Další zvláštní populace
Populační analýza farmakokinetiky neukázala, že by rasa ovlivňovala farmakokinetiku trastuzumabu emtansinu. Protože většina pacientů v klinických studiích s přípravkem Kadcyla byly ženy, nebyl vliv pohlaví na farmakokinetiku trastuzumabu emtansinu formálně hodnocen.
5.3 Předklinické údaje vztahující se k bezpečnosti
Toxikologie a/nebo farmakologie u zvířat
Potkani a opice dobře snášeli podávání trastuzumabu emtansinu v dávkách až 20, respektive 10 mg/kg, což u obou druhů odpovídá 2040 pg DM1/m2, tedy dávce přibližně srovnatelné s klinickou dávkou trastuzumabu emtansinu u pacientů. Ve studiích toxicity provedených dle správné laboratorní praxe (GLP) byla u obou zvířecích modelů zjištěna částečně nebo plně reverzibilní toxicita závislá na dávce s výjimkou ireverzibilní toxicity v periferních axonech (pozorována jen u opic při dávce >10 mg/kg) a toxicity v reprodukčních orgánech (pozorována jen u potkanů při dávce 60 mg/kg). Nejdůležitější bylo toxické poškození jater (zvýšení hladiny jaterních enzymů) při dávkách > 20 mg/kg u potkanů a > 10 mg/kg u opic, kostní dřeně (snížení počtu krevních destiček a bílých krvinek)/hematologické při dávkách > 20 mg/kg u potkanů a > 10 mg/kg u opic a lymfatických orgánů při dávkách > 20 mg/kg u potkanů a > 3 mg/kg u opic.
Mutagenita
In vivo v mikrojadérkovém testu na kostní dřeni potkanů DM1 indukoval aneuploidii nebo byl klastogenní při expozicích srovnatelných s průměrnými maximálními hladinami DM1 při podání trastuzumabu emtansinu člověku. V in vitro (Amesově) testu zpětné mutace u bakterií nebyl DM1 mutagenní.
Poškození fertility a teratogenita
S trastuzumabem emtansinem nebyly prováděny studie zabývající se fertilitou. Dle výsledků obecných studií toxicity u zvířat však lze očekávat negativní účinky na fertilitu.
S trastuzumabem emtansinem nebyly prováděny studie zabývající se embryo-fetálním vývojem u zvířat. Při klinickém použití trastuzumabu byla pozorována vývojová toxicita, ač nebyla předpovězena v předklinickém programu. Kromě toho byla v neklinických studiích zjištěna vývojová toxicita maytansinu, což naznačuje, že DM1 - cytotoxický maytansinoid inhibující mikrotubuly, složka trastuzumabu emtansinu - bude podobně teratogenní a potenciálně embryotoxický.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Kyselina jantarová Hydroxid sodný Sacharóza Polysorbát 20
6.2 Inkompatibility
Tento léčivý přípravek nesmí být mísen nebo ředěn s jinými léčivými přípravky s výjimkou těch, které jsou uvedeny v bodě 6.6.
Roztok glukózy (5%) nemůže být použit k rekonstituci ani k ředění, protože způsobuje agregaci bílkoviny.
6.3 Doba použitelnosti
3 roky.
Doba použitelnosti rekonstituovaného roztoku
Byla prokázána chemická a fyzikální stabilita rekonstituovaného roztoku po dobu až 24 hodin při teplotě 2 °C až 8 °C. Z mikrobiologického hlediska má být přípravek použit okamžitě. Pokud není použit okamžitě, může být rekonstituovaná injekční lahvička uchovávána až 24 hodin při teplotě 2 °C až 8 °C za předpokladu, že rekonstituce proběhla za kontrolovaných a validovaných aseptických podmínek, a po této době musí být zlikvidována.
Doba použitelnosti zředěného roztoku
Rekonstituovaný roztok přípravku Kadcyla naředěný v infuzních vacích, obsahující 0,9% (9 mg/ml) infuzní roztok chloridu sodného nebo 0,45% (4,5 mg/ml) infuzní roztok chloridu sodného je stabilní po dobu až 24 hodin při teplotě 2 °C až 8 °C za předpokladu, že příprava proběhla za kontrolovaných a validovaných aseptických podmínek. Při rozpuštění v 0,9% roztoku chloridu sodného mohou být po uchovávání patrné částečky (viz bod 6.6).
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C).
Podmínky pro uchovávání tohoto léčivého přípravku po jeho rekonstituci a naředění jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
Přípravek Kadcyla je dodáván ve skleněné injekční lahvičce typu 1 o obsahu 15 ml (100 mg) nebo 20 ml (160 mg) uzavřené šedou butyl-pryžovou zátkou potaženou fluororesinem a zevně zabezpečené hliníkovým uzávěrem s bílým nebo purpurovým odlomitelným plastickým víčkem.
Balení obsahuje jednu injekční lahvičku.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Přípravek Kadcyla má být připravován za použití aseptických technik a postupů přiměřených pro přípravu chemoterapeutických léčivých přípravků.
Rekonstituovaný roztok přípravku Kadcyla má být ředěn v infuzním vaku z polyvinylchloridu (PVC) nebo z polyolefinu bez latexu a PVC.
Pokud je koncentrát pro infuzi ředěn v roztoku chloridu sodného pro infuzi o koncentraci 9 mg/ml (0,9%), je k infuzi nutné použít set s „in-line“ polyethersulfonovým filtrem 0,20 nebo 0,22 mikronu.
Aby se zabránilo chybám při podání léku, je nutné zkontrolovat označení na injekční lahvičce a ubezpečit se, že je připravován a podáván přípravek Kadcyla (trastuzumab emtansin) a nikoli Herceptin (trastuzumab).
Návod na rekonstituci
• 100 mg injekční lahvička trastuzumabu emtansinu: Sterilní injekční stříkačkou pomalu vstříkněte 5 ml sterilní vody na injekci do injekční lahvičky.
• 160 mg injekční lahvička trastuzumabu emtansinu: Sterilní injekční stříkačkou pomalu vstříkněte 8 ml sterilní vody na injekci do injekční lahvičky.
• Injekční lahvičkou jemně otáčejte až do úplného rozpuštění. Netřepejte!
Před použitím má být rekonstituovaný roztok vizuálně zkontrolován na přítomnost pevných částic a změnu barvy. V rekonstituovaném roztoku nemají být viditelné částice a má být čirý nebo mírně opalescentní. Barva rekonstituovaného roztoku má být bezbarvá až bledě hnědá. Nepoužívejte rekonstituovaný roztok, pokud jsou v něm viditelné částečky, je zakalený nebo jinak zabarvený.
Návod na ředění
Vypočtěte objem rekonstituovaného roztoku potřebný pro dávku 3,6 mg trastuzumabu emtansinu na kilogram tělesné hmotnosti (viz bod 4.2):
Objem (ml) = Celková dávka, která má být podána (tělesná hmotnost (kg) x dávka (mg/kg))
20 (mg/ml, koncentrace rekonstituovaného roztoku)
Natáhněte příslušné množství roztoku z injekční lahvičky a přidejte je do infuzního vaku s obsahem 250 ml infuzního roztok chloridu sodného o koncentraci 4,5 mg/ml (0,45%) nebo o koncentraci 9 mg/ml (0,9%). Nepoužívejte roztok glukózy (5%) (viz bod 6.2). Infuzní roztok chloridu sodného o koncentraci 4,5 mg/ml (0,45%) může být podán bez „in-line“ polyethersulfonového filtru 0,20 nebo 0,22 mikronu. Pokud je k infuzi použit infuzní roztok chloridu sodného o koncentraci 9 mg/ml (0,9%), je nutný „in-line“ polyethersulfonový filtr 0,20 nebo 0,22 mikronu. Připravená infuze má být podána okamžitě. Při uchovávání chraňte infuzi před mrazem a neprotřepávejte.
Likvidace
Rekonstituovaný roztok neobsahuje žádné konzervační látky a je určen pouze k jednorázovému použití. Nepoužitý zbytek zlikvidujte.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/13/885/001
EU/1/13/885/002
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 15. listopadu 2013
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY/BIOLOGICKÝCH LÉČIVÝCH LÁTEK A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY/BIOLOGICKÝCH LÉČIVÝCH LÁTEK A
VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce biologické léčivé látky
Lonza Ltd.
Lonzastrasse CH-3930 Visp Švýcarsko
Název a adresa výrobce odpovědného za propouštění šarží
Roche Pharma AG Emil-Barell-Strasse 1 D-79639 Grenzach-Wyhlen Německo
V příbalové informaci k léčivému přípravku musí být uveden název a adresa výrobce odpovědného za propouštění dané šarže.
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
• Další opatření k minimalizaci rizik
Držitel rozhodnutí o registraci odsouhlasí obsah a formát edukačních materiálů pro léčivý přípravek Kadcyla spolu s plánem komunikace s národním kompetentním úřadem členského státu před uvedením léčivého přípravku Kadcyla na trh v každém členském státě.
Držitel rozhodnutí o registraci zajistí, že současně s uvedením léčivého přípravku Kadcyla na trh
obdrží všichni zdravotničtí pracovníci, kteří mohou předepisovat, vydávat nebo podávat přípravek
Kadcyla a/nebo Herceptin, balíček edukačních materiálů pro zdravotnické pracovníky (HCP). Tento
balíček edukačních materiálů pro zdravotnické pracovníky má obsahovat následující základní součásti:
• SPC pro přípravek Kadcyla
• Informace pro zdravotnické pracovníky
Informace pro zdravotnické pracovníky má obsahovat následující klíčová sdělení:
1. Kadcyla a Herceptin jsou dva velmi odlišné produkty s odlišnými léčivými látkami, které nikdy nesmí být zaměněny. Kadcyla NENÍ generickou verzí Herceptinu a má odlišné vlastnosti, indikace a dávkování.
2. Přípravek Kadcyla je konjugát protilátky a léku, obsahuje humanizovanou IgG1 protilátku proti HER2 a DM1, maytansinoid inhibující mikrotubuly.
3. Nezaměňujte ani nekombinujte přípravek Kadcyla s přípravkem Herceptin.
4. Nepodávejte přípravek Kadcyla v kombinaci s chemoterapií.
5. Nepodávejte přípravek Kadcyla v dávkách větších než 3,6 mg/kg jednou za 3 týdny.
6. Pokud je přípravek Kadcyla předepsán elektronicky, je nezbytné zajistit, že předepsané léčivo je trastuzumab emtansin a nikoliv trastuzumab.
7. Jak obchodní název přípravku Kadcyla, tak jeho generický název (trastuzumab emtansin) mají být použity a potvrzeny, pokud je přípravek předepisován, pokud je připravován infuzní roztok a přípravek Kadcyla je podáván pacientům. Musí být ověřeno, že generický název je trastuzumab emtansin.
8. Z důvodu předcházení chybám v medikaci je důležité opakovaně číst souhrn údajů o přípravku a kontrolovat vnější obal a štítky injekčních lahviček a zajistit, že léčivý přípravek, který je připravován a podáván, je přípravek Kadcyla a nikoliv Herceptin.
9. Popsat klíčové rozdíly mezi přípravky Kadcyla a Herceptin ve vztahu k indikaci, dávce, podání a rozdílům v balení.
• Povinnost uskutečnit poregistrační opatření
Držitel rozhodnutí o registraci uskuteční v daném termínu níže uvedená opatření:
|
Popis |
Termín splnění |
|
Podání závěrečného hlášení o studii MARIANNE, jakmile bude k dispozici. |
30/04/2017 |
|
Podání závěrečného hlášení o studii TH3RESA, jakmile bude k dispozici. |
31/08/ 2016 |
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Kadcyla 100 mg prášek pro koncentrát pro infuzní roztok Trastuzumabum emtansinum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Po rekonstituci se z jedné 100 mg injekční lahvičky pro jednorázové použití obsahující prášek pro koncentrát pro infuzní roztok získá trastuzumabum emtansinum 5 ml o koncentraci 20 mg/ml.
3. SEZNAM POMOCNÝCH LÁTEK
Pomocné látky:
kyselina jantarová, hydroxid sodný, sacharóza, polysorbát 20.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Prášek pro koncentrát pro infuzní roztok 1 injekční lahvička s obsahem 100 mg
5 ZPŮSOB A CESTA/CESTY PODÁNÍ
Pro intravenózní podání po rekonstituci a naředění Před použitím si přečtěte příbalovou informaci
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do:
9 ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce (2 °C - 8 °C)
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/13/885/001
13 ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
<2D čárový kód s jedinečným identifikátorem>
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
PC:
SN:
NN:
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Kadcyla 100 mg prášek pro koncentrát pro infuzní roztok Trastuzumabum emtansinum Intravenózní podání
2. ZPŮSOB PODÁNÍ
Pro intravenózní podání po rozpuštění a naředění
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET DÁVEK
100 mg
6. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Kadcyla 160 mg prášek pro koncentrát pro infuzní roztok Trastuzumabum emtansinum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Po rekonstituci se z jedné 160 mg injekční lahvičky pro jednorázové použití obsahující prášek pro koncentrát pro infuzní roztok získá trastuzumabum emtansinum 8 ml o koncentraci 20 mg/ml.
3. SEZNAM POMOCNÝCH LÁTEK
Pomocné látky:
kyselina jantarová, hydroxid sodný, sacharóza, polysorbát 20.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Prášek pro koncentrát pro infuzní roztok 1 injekční lahvička s obsahem 160 mg
5 ZPŮSOB A CESTA/CESTY PODÁNÍ
Pro intravenózní podání po rekonstituci a naředění Před použitím si přečtěte příbalovou informaci
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do:
9 ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce (2 °C - 8 °C)
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/13/885/002
13 ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
<2D čárový kód s jedinečným identifikátorem.>
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
PC:
SN:
NN:
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Kadcyla 160 mg prášek pro koncentrát pro infuzní roztok Trastuzumabum emtansinum Intravenózní podání
2. ZPŮSOB PODÁNÍ
Pro intravenózní podání po rozpuštění a naředění
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET DÁVEK
160 mg
6. JINÉ
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro pacienta
Kadcyla 100 mg prášek pro koncentrát pro infuzní roztok Kadcyla 160 mg prášek pro koncentrát pro infuzní roztok
trastuzumabum emtansinum
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
• Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
• Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
• Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Kadcyla a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Kadcyla používat
3. Jak se přípravek Kadcyla používá
4. Možné nežádoucí účinky
5. Jak přípravek Kadcyla uchovávat
6. Obsah balení a další informace
1. Co je přípravek Kadcyla a k čemu se používá
Co je přípravek Kadcyla
Přípravek Kadcyla obsahuje léčivou látku trastuzumab emtansin, která se skládá ze dvou spojených
částí:
• trastuzumab - monoklonální protilátka, která se selektivně váže na antigen (cílovou bílkovinu) nazývanou receptor 2 pro lidský růstový epidermální faktor (označuje se anglickou zkratkou HER2). HER2 se nalézá ve velkém množství na povrchu některých nádorových buněk, jejichž růst podporuje. Když se trastuzumab naváže na HER2, může zastavit růst nádorových buněk a způsobit jejich smrt.
• DM1 - protinádorová látka, která se aktivuje až poté, co se přípravek Kadcyla dostane do nitra nádorové buňky.
Na co se přípravek Kadcyla používá
Přípravek Kadcyla se používá k léčbě nádoru prsu u dospělých, pokud:
• je na buňkách nádoru vysoké množství bílkoviny HER2 - Váš lékař provede test na přítomnost této bílkoviny na nádorových buňkách
• jste j iž byl(a) léčen(a) lékem trastuzumabem a lékem označovaným j ako taxan
• se nádor rozšířil do tkání v blízkosti prsu nebo do jiných částí Vašeho těla.
2. Čemu musíte věnovat pozornost, než začnete přípravek Kadcyla používat
Přípravek Kadcyla Vám nesmí být podán
• jestliže jste alergický(á) na trastuzumab emtansin nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Přípravek Kadcyla Vám nesmí být podán, pokud se na Vás vztahuje výše zmíněné. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo zdravotní sestrou předtím, než Vám bude přípravek Kadcyla podán.
Upozornění a opatření
Před podáním přípravku Kadcyla se poraďte se svým lékařem nebo zdravotní sestrou, pokud:
• jste někdy měl(a) závažnou reakci na infuzi po podání trastuzumabu, jejímiž známkami mohou být například zrudnutí, třesavka, horečka, dušnost, obtíže při dýchání, zrychlený srdeční tep nebo pokles krevního tlaku.
• jste léčen(a) léky na „ředění“ krve (např. warfarin, heparin).
• máte v anamnéze problémy s játry. Váš lékař Vám zkontroluje funkci jater krevním testem před léčbou a pravidelně v průběhu léčby.
Pokud se na Vás vztahuje cokoli z výše zmíněného (nebo si nejste jistý(á)), poraďte se se svým lékařem nebo lékárníkem předtím, než Vám bude přípravek Kadcyla podán.
Sledujte nežádoucí účinky
Přípravek Kadcyla může vést ke zhoršení existujících příznaků nebo způsobovat nežádoucí účinky. V bodě 4 je uvedeno více podrobností o nežádoucích účincích, na které byste měl(a) dávat pozor.
Ihned informujte svého lékaře nebo zdravotní sestru, pokud během podávání přípravku Kadcyla zaznamenáte jakékoli z následujících závažných nežádoucích účinků:
• Obtíže s dýcháním: Přípravek Kadcyla může způsobit závažné dýchací obtíže jako například dušnost (v klidu nebo při jakékoli tělesné aktivitě) a kašel. To mohou být příznaky zánětu plic, který může být závažný a dokonce vést k úmrtí. Pokud se u Vás vyvine plicní onemocnění, může Váš lékař ukončit podávání tohoto léku.
• Problémy s játry: Přípravek Kadcyla může být příčinou zánětu nebo poškození jaterních buněk, což může vést k poruše normálních funkcí jater. Zanícené nebo poškozené jaterní buňky mohou do krve uvolňovat jisté látky (jaterní enzymy) v množství větším než obvykle, což vede ke zvýšené hladině jaterních enzymů při vyšetření krve. Ve většině případů nebudete mít žádné příznaky. Příznakem může být zežloutnutí kůže a očního bělma (žloutenka). Před zahájením léčby a pravidelně v jejím průběhu bude Váš lékař pravidelně kontrolovat funkce jater pomocí vyšetření krve.
Další vzácnou odchylkou, která se může projevit v játrech, je stav označovaný jako nodulární regenerační hyperplazie. Při této odchylce dochází ke změně struktury jater, a to může vést ke změně funkce jater. Může to časem vést k příznakům, jako jsou pocit nafouknutí nebo otok břicha při nahromadění tekutiny nebo krvácení z abnormálních cév v jícnu nebo konečníku.
• Problémy se srdcem: Přípravek Kadcyla může oslabit srdeční sval. Pokud je srdeční sval oslabený, mohou se u pacienta projevit příznaky jako dušnost v klidu nebo ve spánku, bolest na hrudníku, otok nohou a paží a pocit rychlého nebo nepravidelného srdečního tepu. Před zahájením léčby a pravidelně v jejím průběhu bude Váš lékař kontrolovat funkce Vašeho srdce. Informujte ihned svého lékaře, pokud zaznamenáte jakékoli z výše uvedených příznaků.
• Reakce související s infuzí nebo alergické reakce: Přípravek Kadcyla může během infuze nebo po infuzi v průběhu prvého dne léčby způsobit zrudnutí, záchvat třesavky, horečku, dýchací obtíže, snížení krevního tlaku, zrychlení srdečního tepu, náhlý otok obličeje, jazyka nebo potíže při polykání. Váš lékař nebo zdravotní sestra budou sledovat, zda nemáte některý z těchto nežádoucích účinků. Pokud u Vás dojde k reakci, zpomalí nebo zastaví infuzi a mohou Vám podat léky působící proti těmto nežádoucím účinkům. Po úpravě příznaků může infuze pokračovat.
• Krvácení: Přípravek Kadcyla může způsobit snížení počtu krevních destiček v krvi. Krevní destičky napomáhají srážení krve, můžete tedy pozorovat neočekávaný vznik modřin nebo krvácení (například krvácení z nosu, z dásní). Váš lékař bude pravidelně provádět vyšetření krve k hodnocení sníženého počtu krevních destiček. Pokud zaznamenáte neočekávanou tvorbu modřin nebo krvácení, ihned informujte svého lékaře.
• Neurologické problémy: Přípravek Kadcyla může poškodit nervy. Můžete pociťovat brnění, bolest, necitlivost, svědění, mravenčení, bodání v rukou a nohou. Váš lékař bude sledovat známky a příznaky neurologických problémů.
Pokud zaznamenáte kterýkoli z výše uvedených nežádoucích účinků, informujte ihned svého lékaře nebo zdravotní sestru.
Děti a dospívající
Přípravek Kadcyla není určen pro nikoho ve věku do 18 let, protože nejsou informace o působení přípravku u této věkové skupiny.
Další léčivé přípravky a přípravek Kadcyla
Informujte svého lékaře nebo zdravotní sestru o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Zejména informujte svého lékaře nebo lékárníka, pokud užíváte:
• jakékoli léky na „ředění“ krve jako např. warfarin
• léky k léčbě plísňových infekcí nazývané ketokonazol, itrakonazol nebo vorikonazol
• antibiotika k léčbě infekcí nazývaná klaritromycin nebo telitromycin
• léky k léčbě HIV nazývané atazanavir, indinavir, nelfinavir, ritonavir nebo sachinavir
• lék k léčbě deprese nazývaný nefazodon
Pokud se na Vás vztahuje cokoli z výše zmíněného (nebo si nejste jistý(á)), poraďte se se svým lékařem nebo lékárníkem předtím, než Vám bude přípravek Kadcyla podán.
Těhotenství
Užívání přípravku Kadcyla se nedoporučuje, pokud jste těhotná, protože tento lék může poškodit nenarozené dítě.
• Informujte svého lékaře dříve, než začnete přípravek Kadcyla užívat, pokud jste těhotná, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět.
• Používejte účinnou metodu antikoncepce k zabránění těhotenství během léčby přípravkem Kadcyla. Informujte se u svého lékaře, jaká metoda antikoncepce je pro vás nejvhodnější.
• Účinnou antikoncepci byste měl(a) používat ještě nejméně 7 měsíců po posledním podání přípravku Kadcyla. Dříve než přestanete používat antikoncepci, poraďte se se svým lékařem.
• Mužští pacienti nebo jejich partnerky mají rovněž používat účinnou antikoncepci.
• Pokud otěhotníte během léčby přípravkem Kadcyla, okamžitě informujte svého lékaře.
Kojení
Během léčby přípravkem Kadcyla a rovněž po dobu 7 měsíců od poslední infuze přípravku Kadcyla nesmíte kojit. Není známo, zda součásti přípravku Kadcyla pronikají do mateřského mléka. Promluvte si o tom se svým lékařem.
Řízení dopravních prostředků a obsluha strojů
Nepředpokládá se, že Kadcyla ovlivňuje Vaši schopnost řídit, jezdit na kole, používat nářadí nebo stroje. Neřiďte, nejezděte na kole a nepoužívejte nářadí nebo stroje, pokud pocítíte rudnutí, záchvat třesavky, horečku, dýchací obtíže, snížení krevního tlaku, zrychlení srdečního tepu (reakce související s infuzí), zastřené vidění, únavnost, bolest hlavy nebo závratě, dokud tyto reakce neodezní.
Důležitá informace o některých látkách, které obsahuje přípravek Kadcyla
Tento léčivý přípravek obsahuje v jedné dávce méně než 1 mmol (23 mg) sodíku, je v podstatě „bez sodíku“.
Jak se přípravek Kadcyla používá
3.
Přípravek Kadcyla Vám bude podávat lékař nebo zdravotní sestra v nemocnici nebo na ambulanci:
• Podává se formou kapačky (nitrožilní infuze)
• Dostanete j ednu infuzi každé tři týdny.
Jaké množství Vám bude podáno
• Dostanete 3,6 mg přípravku Kadcyla na každý kilogram Vaší tělesné hmotnosti. Váš lékař vypočte pro Vás správnou dávku.
• První infuze bude podávána po dobu 90 minut. Během infuze a nejméně 90 minut po jejím ukončení budete sledován(a) lékařem nebo zdravotní sestrou pro případ vzniku nežádoucích účinků.
• Pokud budete první infuzi dobře snášet, mohou být další infuze podávány po dobu 30 minut. Během infuze a nejméně 30 minut po jejím ukončení budete sledován(a) lékařem nebo zdravotní sestrou pro případ vzniku nežádoucích účinků.
• Celkový počet infuzí, které dostanete, závisí na tom, jak na léčbu budete reagovat.
• Pokud se vyskytnou nežádoucí účinky, může Váš lékař rozhodnout o pokračování léčby při snížené dávce, o odložení další dávky nebo o ukončení léčby.
Jestliže jste zapomněl(a) užít přípravek Kadcyla
Jestliže zapomenete nebo zmeškáte návštěvu u lékaře a podání přípravku Kadcyla, domluvte si co
nejdříve další termín. Nečekejte až na příští plánovanou návštěvu.
Jestliže jste přestal(a) užívat přípravek Kadcyla
Nepřestávejte užívat tento přípravek bez předchozí porady se svým lékařem.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo
zdravotní sestry.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí
vyskytnout u každého.
Ihned informujte svého lékaře nebo zdravotní sestru, pokud zaznamenáte jakékoli
z následujících závažných nežádoucích účinků.
Velmi časté (mohou postihnout více než jednoho člověka z 10):
• Přípravek Kadcyla může vést k zánětu nebo poškození jaterních buněk, což vede ke zvýšení hladiny jaterních enzymů v krevních testech. Ve většině případů však během léčby přípravkem Kadcyla dochází jen k mírnému a přechodnému zvýšení hladiny jaterních enzymů, což nevede k žádným příznakům a neovlivňuje to funkci jater.
• Neočekávaná tvorba modřin nebo krvácení (jako například krvácení z nosu) může být známkou sníženého počtu krevních destiček.
• Mravenčení, bolest, necitlivost, svědění, pocit husí kůže, bodání v rukou a nohou. Tyto příznaky mohou ukazovat na poškození nervů.
Časté (mohou postihnout až jednoho člověka z 10):
• Zarudnutí, záchvaty třesavky, horečka, dýchací obtíže, snížení krevního tlaku, zrychlení srdečního tepu během infuze nebo až 24 hodin po jejím ukončení - jsou to takzvané reakce související s infuzí.
• Mohou se projevit problémy se srdcem. Většina pacientů nebude mít příznaky související
s problémy se srdcem. Pokud se příznaky projeví, můžete pozorovat kašel, dušnost v klidu nebo vleže na zádech, bolest na hrudníku, otok kotníků nebo paží nebo pocit rychlého nebo nepravidelného tepu srdce.
Méně časté (mohou postihnout až jednoho člověka ze 100):
• Zánět plic může být příčinou problémů s dýcháním, jako je například dušnost (v klidu nebo při jakékoli tělesné aktivitě), kašel nebo záchvaty kašle se suchým kašlem - jsou to známky zánětu plicní tkáně.
• Vaše kůže a oční bělmo zežloutnou (žloutenka) - mohou to být známky závažného poškození jater.
• Může se projevit alergická reakce, při níž bude mít většina pacientů mírné příznaky, jako jsou svědění nebo tíže na hrudníku. V závažnějších případech se mohou objevit otok obličeje nebo jazyka, problémy při polykání nebo dýchací obtíže.
Pokud zaznamenáte kterýkoli z výše uvedených závažných nežádoucích účinků, ihned informujte svého lékaře nebo zdravotní sestru.
Další nežádoucí účinky zahrnují
Velmi časté:
• pokles počtu červených krvinek (zjištěný při vyšetření krve)
• nevolnost, zvracení
• průjem
• suchost v ústech
• infekce močových cest
• zácpa
• bolest žaludku
• kašel
• dušnost
• zánět v ústech
• třesavka nebo příznaky podobné chřipce
• pokles hladiny draslíku (zjištěný při vyšetření krve)
• nespavost
• bolest svalů nebo kloubů
• horečka
• bolest hlavy
• kožní vyrážka
• únava
• slabost
Časté:
• pokles počtu bílých krvinek (zjištěný při vyšetření krve)
• suchost očí, zvýšené slzení, zastřené vidění
• zarudnutí očí nebo infekce očí
• zažívací obtíže
• otok nohou a/nebo paží
• krvácení z dásní
• zvýšení krevního tlaku
• závratě
• poruchy chuti
• svědění
• poruchy paměti
• vypadávání vlasů
• reakce na kůži rukou a nohou (syndrom palmoplantární erytrodysestezie)
• poruchy nehtů
Méně časté:
• další odchylkou, která může být způsobena přípravkem Kadcyla, j e stav označovaný jako nodulární regenerativní hyperplazie jater. Tato odchylka vede ke změně struktury jater. V játrech pacienta vznikají mnohočetné uzlíky, které mohou vést ke změně funkce jater. Postupem doby se mohou projevit příznaky jako pocit nafouklého nebo oteklého břicha při hromadění tekutiny nebo při krvácení z abnormálních cév v jícnu nebo konečníku.
• Pokud roztok přípravku Kadcyla prosákne do tkání v okolí místa infuze, může být kůže citlivá nebo zarudlá, nebo okolí v místě infuze je oteklé.
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků po ukončení léčby přípravkem Kadcyla, sdělte to svému lékaři nebo zdravotní sestře a řekněte jim, že jste byl(a) léčen(a) přípravkem Kadcyla.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak se přípravek Kadcyla uchovává
Přípravek Kadcyla bude uchováván zdravotnickými pracovníky v nemocnici nebo v ambulanci.
• Uchovávejte přípravek mimo dohled a dosah dětí.
• Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce za „Použitelné do:“ Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
• Uchovávejte v chladničce (2 °C - 8 °C). Chraňte před mrazem.
• Připravený roztok pro infuzi přípravku Kadcyla je stabilní po dobu až 24 hodin při teplotě 2 °C až 8 °C, po uplynutí této doby musí být zlikvidován.
• Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
Obsah balení a další informace
6.
Co přípravek Kadcyla obsahuje
• Léčivou látkou je trastuzumabum emtansinum.
• Jedna 100 mg injekční lahvička pro jedno použití obsahuje prášek pro koncentrát pro infuzní roztok, z něhož se získá trastuzumabum emtansinum 5 ml o koncentraci 20 mg/ml.
• Jedna 160 mg injekční lahvička pro jedno použití obsahuje prášek pro koncentrát pro infuzní roztok, z něhož se získá trastuzumabum emtansinum 8 ml o koncentraci 20 mg/ml.
• Dalšími složkami jsou kyselina jantarová, hydroxid sodný (viz bod 2 Důležitá informace o některých látkách, které obsahuje přípravek Kadcyla), sacharóza a polysorbát 20.
Jak přípravek Kadcyla vypadá a co obsahuje toto balení
• Přípravek Kadcyla je bílý až světle šedý lyofilizovaný prášek pro koncentrát pro infuzní roztok dodávaný ve skleněných injekčních lahvičkách.
• Přípravek Kadcyla je dostupný v balení obsahujícím jednu injekční lahvičku.
Držitel rozhodnutí o registraci
Roche Registration Limited
6 Falcon Way
Shire Park
Welwyn Garden City
AL7 1TW
Velká Británie
Výrobce
Roche Pharma AG Emil-Barell-Strasse 1 D-79639 Grenzach-Wyhlen Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgie/Belgique/Belgien Lietuva
N.V. Roche S.A. UAB “Roche Lietuva”
Tél/Tel: +32 (0) 2 525 82 11 Tel: +370 5 2546799
Luxembourg/Luxemburg
(Voir/siehe Belgique/Belgien)
Etarapnu
Pom Etarapua EOOfl
Tea: +359 2 818 44 44
|
EkXába Roche (Hellas) A.E. Tr|k: +30 210 61 66 100 |
Osterreich Roche Austria GmbH Tel: +43 (0) 1 27739 |
|
Espaňa Roche Farma S.A. Tel: +34 - 91 324 81 00 |
Polska Roche Polska Sp.z o.o. Tel: +48 - 22 345 18 88 |
|
France Roche Tél: +33 (0) 1 47 61 40 00 |
Portugal Roche Farmaceutica Química, Lda Tel: +351 - 21 425 70 00 |
|
Hrvatska Roche d.o.o Tel: +385 1 4722 333 |
Románia Roche Románia S.R.L. Tel: +40 21 206 47 01 |
|
Ireland Roche Products (Ireland) Ltd. Tel: +353 (0) 1 469 0700 |
Slovenija Roche farmacevtska družba d.o.o. Tel: +386 - 1 360 26 00 |
|
Island Roche a/s c/o Icepharma hf Sími: +354 540 8000 |
Slovenská republika Roche Slovensko, s.r.o. Tel: +421 - 2 52638201 |
|
Italia Roche S.p.A. Tel: +39 - 039 2471 |
Suomi/Finland Roche Oy Puh/Tel: +358 (0) 10 554 500 |
|
Kónpoq r.A.Exapáxn? & Eia AxS. Tr|k: +357 - 22 76 62 76 |
Sverige Roche AB Tel: +46 (0) 8 726 1200 |
|
Latvija Roche Latvija SIA Tel: +371 - 6 7 039831 |
United Kingdom Roche Products Ltd. Tel: +44 (0) 1707 366000 |
Tato příbalová informace byla naposledy revidována {měsíc RRRR}.
Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky: http://www.ema.europa.eu.
Následující informace jsou určeny pouze pro zdravotnické pracovníky
Aby se zabránilo chybám při podání léku, je nutné zkontrolovat označení na injekční lahvičce a ubezpečit se, že je připravován a podáván přípravek Kadcyla (trastuzumab emtansin) a nikoli přípravek Herceptin (trastuzumab).
Přípravek Kadcyla musí být rekonstituován a naředěn zdravotnickým pracovníkem a podán jako intravenózní infuze. Nesmí být podán jako intravenózní injekce nebo bolus.
Vždy uchovávejte tento léčivý přípravek v chladničce při teplotě 2 °C - 8 °C v původním uzavřeném obalu. Rekonstituovaný přípravek Kadcyla po rekonstituci vodou na injekci (není součástí balení) je v injekční lahvičce stabilní po dobu 24 hodin při teplotě 2 °C - 8 °C a nesmí být zmrazen.
Přípravek Kadcyla má být připravován za použití aseptických technik a postupů přiměřených pro přípravu chemoterapeutických léčivých přípravků.
Rekonstituovaný roztok přípravku Kadcyla má být ředěn v infuzním vaku z polyvinylchloridu (PVC) nebo z polyolefinu bez latexu a PVC.
Pokud je koncentrát pro infuzi ředěn v roztoku chloridu sodného pro infuzi o koncentraci 9 mg/ml (0,9%), je k infuzi nutné použít set s „in-line“ polyethersulfonovým filtrem 0,20 nebo 0,22 mikronu.
Návod na rekonstituci
• Přípravek Kadcyla 100 mg: Sterilní injekční stříkačkou pomalu vstříkněte 5 ml sterilní vody na injekci do injekční lahvičky se 100 mg trastuzumabu emtansinu.
• Přípravek Kadcyla 160 mg: Sterilní injekční stříkačkou pomalu vstříkněte 8 ml sterilní vody na injekci do injekční lahvičky se 160 mg trastuzumabu emtansinu.
• Injekční lahvičkou jemně otáčejte až do úplného rozpuštění. Netřepejte!
Před použitím má být rekonstituovaný roztok vizuálně zkontrolován na přítomnost pevných částic a změnu barvy. V rekonstituovaném roztoku nemají být viditelné částice a má být čirý nebo mírně opalescentní. Barva rekonstituovaného roztoku má být bezbarvá až bledě hnědá. Nepoužívejte rekonstituovaný roztok, pokud je zakalený nebo jinak zabarvený.
Nepoužitý zbytek zlikvidujte. Rekonstituovaný roztok neobsahuje žádné konzervační látky a je určen pouze k jednorázovému použití.
Návod na ředění
Vypočtěte objem rekonstituovaného roztoku potřebný pro dávku 3,6 mg trastuzumabu emtansinu na kilogram tělesné hmotnosti:
Objem (ml) = Celková dávka, která má být podána = (tělesná hmotnost (kg) x dávka (mg/kg))
20 (mg/ml, koncentrace rekonstituovaného roztoku)
Natáhněte příslušné množství roztoku z injekční lahvičky a přidejte je do infuzního vaku s obsahem 250 ml infuzního roztoku chloridu sodného o koncentraci 4,5 mg/ml (0,45%) nebo o koncentraci 9 mg/ml (0,9%). Nepoužívejte roztok glukózy (5%). Infuzní roztok chloridu sodného o koncentraci 4,5 mg/ml (0,45%) může být podán bez „in-line“ polyethersulfonového filtru 0,20 nebo 0,22 mikronu. Pokud je k infuzi použit infuzní roztok chloridu sodného o koncentraci 9 mg/ml (0,9%), je nutný „in-line“ polyethersulfonový filtr 0,20 nebo 0,22 mikronu. Připravená infuze má být podána okamžitě. Při uchovávání chraňte infuzi před mrazem a neprotřepávejte. Při ředění přípravku za aseptických podmínek, může být uchováván po dobu až 24 hodin při teplotě 2 °C - 8 °C.
42
Kontrolní rameno (lapatinib + kapecitabin): lapatinib 1250 mg/den perorálně jednou denně
v cyklu trvajícím 21 dní plus kapecitabin 1000 mg/m2 perorálně dvakrát denně ve dny 1-14 cyklu trvajícího 21 dní.