Jetrea 0,375 Mg/0,3 Ml
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
JETREA 0,5 mg / 0,2 ml koncentrát pro injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička obsahuje ocriplasminum* 0,5 mg v 0,2 ml roztoku.
Po naředění v 0,2 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9 %), obsahuje 0,1 ml takto naředěného roztoku ocriplasminum 0,125 mg.
*Okriplasmin je zkrácená forma lidského plazminu, která se vyrábí rekombinantní DNA technologií v expresním systému Pichia pastoris.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Koncentrát pro injekční roztok (sterilní koncentrát). Čirý bezbarvý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek JETREA je indikován k použití u dospělých pacientů k léčbě vitreomakulární trakce (VMT) včetně případů zahrnujících makulární díru o průměru menším nebo rovném 400 mikrometrů (viz bod 5.1).
4.2 Dávkování a způsob podání
Přípravu a podávání přípravku JETREA smí provádět pouze kvalifikovaný oftalmolog se zkušenostmi s intravitreálními injekcemi. Diagnóza vitreomakulární trakce (VMT) má zahrnovat kompletní klinický obraz včetně pacientovy anamnézy, klinického vyšetření a použití adekvátních diagnostických metod, jako je např. optická koherentní tomografie (OCT).
Dávkování
Doporučená dávka je 0,125 mg (0,1 ml naředěného roztoku) jednorázově aplikovaná intravitreální injekcí do postiženého oka. Jedna lahvička se smí použít pouze jednorázově a pouze k léčbě jednoho oka. Léčba druhého oka přípravkem JETREA se nedoporučuje dříve než za 7 dní od podání první injekce do iniciálně léčeného oka z důvodu monitorování postinjekční reakce včetně potenciálního rizika snížené zrakové ostrosti u léčeného oka. Opakované podávání do stejného oka se nedoporučuje (viz bod 4.4).
Pokyny ke sledování po aplikaci injekce viz bod 4.4.
Zvláštní skupiny pacientů
Porucha funkce ledvin
Nebyly provedeny žádné konkrétní studie hodnotící použití přípravku JETREA u pacientů s poruchou funkce ledvin. U pacientů s poruchou funkce ledvin se nepředpokládá nutnost úpravy dávky ani jiná zvláštní opatření (viz bod 5.2).
Porucha funkce jater
Nebyly provedeny žádné konkrétní studie hodnotící použití přípravku JETREA u pacientů s poruchou funkce jater. U pacientů s poruchou funkce jater se nepředpokládá nutnost úpravy dávky ani jiná zvláštní opatření (viz bod 5.2).
Starší pacienti
Populace starších pacientů byla hodnocena v klinických studiích. Není zapotřebí žádná úprava dávky. Pediatrická populace
Neexistuje žádné relevantní použití přípravku JETREA u dětí ve věku do 18 let v indikaci vitreomakulární trakce (VMT) včetně případů spojených s přítomností makulární díry o průměru menším nebo rovném 400 mikrometrů. V současnosti dostupné údaje o použití u dětí jsou popsány v bodě 5.1.
Způsob podání
Lahvička k jednorázové intravitreální aplikaci.
Před zákrokem mohou být dle uvážení ošetřujícího oftalmologa podány antibiotické oční kapky.
Opatření, která je nutno učinit před manipulací s léčivým přípravkem nebo před jeho podáním Intravitreální injekci je nutno provádět za kontrolovaných aseptických podmínek včetně nutnosti dezinfekce rukou operatéra, použití sterilních rukavic, sterilní roušky, sterilního rozvěrače víček (nebo ekvivalentní náhrady) a možnosti provedení sterilní paracentézy (pokud je vyžadována). Periokulární pokožka, víčko a povrch oka musí být dezinfikovány a před aplikací injekce je nutno podat adekvátní anestetikum a lokální širokospektrý mikrobiocidní přípravek dle standardní lékařské praxe.
Návod k naředění tohoto léčivého přípravku před jeho podáním je uveden v bodě 6.6.
Injekční jehla se zasune 3,5-4,0 mm posteriorně od limbu do prostoru sklivce tak, aby směřovala do centra očního bulbu, a nikoli k horizontálnímu meridiánu. Objem injekce 0,1 ml se potom aplikuje do středu sklivce.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Aktivní nebo suspektní okulární nebo periokulární infekce.
4.4 Zvláštní upozornění a opatření pro použití
Sledování po aplikaci injekce
Přípravek JETREA se podává pouze intravitreální injekcí. Intravitreální injekce mohou být spojeny s nitroočním zánětem/infekcí, s nitroočním krvácením a se zvýšeným nitroočním tlakem (NOT). Je nutno za všech okolností používat správnou aseptickou injekční techniku. Po intravitreální injekci je nutno sledovat, zda se u pacienta nerozvinou nežádoucí účinky, mimo jiné například nitrooční zánět/infekce a zvýšení NOT. Výskyt přechodného zvýšení NOT včetně přechodné slepoty a zástavy perfuze papily zrakového nervu byl pozorován do 60 minut po injekci přípravku JETREA. Sledování zvýšení NOT může sestávat z kontroly perfuze papily zrakového nervu bezprostředně po injekci a z tonometrie do 30 minut po injekci. Nitrooční zánět/infekci lze vyšetřit provedením biomikroskopie v rozmezí 2 až 7 dnů po injekci. Pacienty je nutno poučit, aby neprodleně hlásili příznaky možného nitroočního zánětu nebo infekce a jakékoli další příznaky postihující zrak nebo oko. Pokud se u pacienta vyskytne kterýkoli z výše uvedených příznaků, je nutno jej léčit podle standardní lékařské praxe.
Bilaterální léčba
Bezpečnost a účinnost přípravku JETREA při současném podávání do obou očí nebyla hodnocena. Proto se současné podávání do obou očí nedoporučuje.
Opakované podávání
Opakované podávání přípravku JETREA do stejného oka nebylo dostatečně hodnoceno, a proto se nedoporučuje.
Populace se žádnými nebo omezenými údaji
Přípravek JETREA nebyl hodnocen u pacientů s makulárními dírami o velkém průměru (> 400 mikrometrů), se silnou myopií (sférická korekce > 8 dioptrií nebo axiální délka > 28 mm), s afakií, s rhegmatogenním odchlípením sítnice v anamnéze, s nestabilitou zonuly čočky, s nedávno prodělanou oční operací nebo nitrooční injekcí (včetně laserové léčby), s proliferativní diabetickou retinopatií, s ischemickou retinopatií, s okluzí cév sítnice, s exsudativní formou věkem podmíněné makulární degenerace (VPMD) a s krvácením do sklivce. U těchto pacientů se léčba nedoporučuje.
U pacientů s neproliferativní diabetickou retinopatií, s anamnézou uveitidy (včetně závažného aktivního zánětu) nebo závažného poranění oka jsou k dispozici pouze omezené zkušenosti. Při léčbě těchto pacientů je nutno postupovat s opatrností.
Jiné
Riziko subluxace čočky nebo fakodonézy nemůže být vyloučeno. Pokud se takový případ vyskytne, je nutno jej léčit dle standardní lékařské praxe. Pacienti mají být adekvátně monitorováni (viz body 4.8 a 5.3).
U pacientů s epiretinální membránou (ERM) nebo s průměrem VMA > 1500 mikrometrů je účinnost okriplasminu (zvláště pokud jde o odstranění vitreomakulární adhese nebo docílení kompletního odloučení zadní části sklivce [PVD]) snížená (viz bod 5.1).
V důsledku možnosti nárůstu trakčních sil existuje riziko vzniku nových nebo zvětšení stávajících makulárních děr. Pacienti mají být adekvátně monitorováni (viz bod 4.8).
Během prvního týdne po aplikaci injekce existuje riziko vzniku významné, nicméně přechodné ztráty zrakové ostrosti. Pacienti mají být adekvátně monitorováni (viz bod 4.8).
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné formální studie interakcí.
Okriplasmin je proteolytický enzym s aktivitou serinové proteázy, který může být v oku přítomen po dobu několika dnů po intravitreální injekci (viz bod 5.2). Současné podání jiných léčivých přípravků v úzkém časovém rozpětí do stejného oka může ovlivnit účinek obou léčivých přípravků, a proto se nedoporučuje.
Nejsou k dispozici žádné klinické údaje ohledně současného používání okriplasminu a inhibitorů VEGF (vaskulárního endoteliálního růstového faktoru).
Nepředpokládají se žádné systémové interakce.
4.6 Fertilita, těhotenství a kojení
Údaje o podávání přípravku JETREA těhotným ženám nejsou k dispozici. Nebyly provedeny žádné studie reprodukční toxicity. Systémová expozice přípravku JETREA po intravitreální injekci je podle
předpokladů velmi nízká. Přípravek JETREA se má během těhotenství používat pouze za předpokladu, že klinické přínosy převáží potenciální rizika.
Kojení
Není známo, zda se přípravek JETREA vylučuje do lidského mateřského mléka. Přípravek JETREA se má během kojení používat pouze za předpokladu, že klinické přínosy převáží potenciální rizika.
Fertilita
O účinku přípravku JETREA na fertilitu nejsou žádné údaje.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Po intravitreální injekci přípravku JETREA může dojít k dočasným poruchám zraku (viz bod 4.8). V těchto případech pacienti nesmí řídit ani obsluhovat stroje, dokud porucha zraku neodezní.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Léčbu intravitreální injekcí přípravku JETREA obdrželo více než 800 pacientů v klinických studiích, z nichž více než 570 bylo léčeno doporučenou dávkou 0,125 mg.
Všechny nežádoucí účinky, které se vyskytly, byly oční nežádoucí účinky. Nejčastěji byly hlášeny sklivcové zákalky, bolest oka a fotopsie a také krvácení do spojivky v důsledku injekce. Většina nežádoucích účinků se objevila během prvního týdne po injekci. Většina těchto reakcí byla nezávažná, s mírnou intenzitou, a vymizela během 2 až 3 týdnů. Byly také hlášeny příznaky pozorované v kontralaterálním oku nebo bilaterálně.
Souhrnná incidence závažných nežádoucích účinků ve všech klinických studiích byla 2,2 % u pacientů léčených přípravkem JETREA a 2,4 % u kontrolní skupiny pacientů.
Tabulka s přehledem nežádoucích účinků
V následující tabulce je uveden přehled nežádoucích účinků, které byly hlášeny v klinických studiích a/nebo v postmarketingových hlášeních.
Nežádoucí účinky s reálnou možností kauzality s podáním injekce nebo s přípravkem JETREA jsou uvedeny dle systému MedDRA podle třídy orgánů a četnosti s použitím následující konvence: velmi časté (>1/10); časté (>1/100 až <1/10); méně časté (>1/1 000 až <1/100); vzácné (> 1/10 000 to <1/1 000); velmi vzácné (<1/10 000) a není známo (z dostupných údajů nelze určit). V každé skupině četnosti jsou nežádoucí účinky seřazeny podle klesající závažnosti.
Poruchy oka
Velmi časté
Sklivcové zákalky, bolest oka, krvácení do spojivky Časté
Snížená zraková ostrost*, zhoršené vidění, rozmazané vidění, krvácení do sítnice, krvácení do spojivky, roztržení sítnice , odchlípení sítnice , zvýšený nitrooční tlak, makulární díra , makulární degenerace, retinální degenerace, makulární edém , edém sítnice, retinální pigmentová epitelopatie, metamorfopsie, adheze sklivce*, edém spojivky, edém očního víčka, vitritida, přítomnost buněk v přední komoře, zarudnutí přední komory, iritida, fotopsie, hyperemie spojivky, hyperemie oka, odchlípení sklivce, abnormální retinogram*, podráždění oka, suché oko, pocit cizího tělíska v oku, svědění oka, oční diskomfort, fotofobie, chromatopsie*
Méně časté
Přechodná slepota, subluxace čočky , šeroslepost, skotom, porucha pupilárního reflexu, defekt zorného pole, diplopie, hyféma, mióza, nestejné pupily, abraze rohovky, zánět přední komory, zánět oka, podráždění spojivky
viz bod ‘Popis vybraných nežádoucích účinků‘ včetně cystoidního makulárního edému
Popis vybraných nežádoucích účinků
Snížení zrakové ostrosti
V pivotních, placebem kontrolovaných studiích fáze III došlo u 7, 7 % pacientů léčených přípravkem JETREA a 1, 6 % pacientů s placebem během prvního týdne po aplikaci injekce k akutnímu přechodnému snížení nejlépe korigované zrakové ostrosti (BCVA) o > 2 řádky (>10 ETDRS písmen) bez jiné vysvětlující příčiny. Snížení zrakové ostrosti obvykle spontánně ustoupilo do 2 týdnů bez nutnosti intervence. Doporučení ohledně monitorování viz bod 4.4.
Chromatopsie
Dyschromatopsie (obecně popisovaná jako vidění ve žlutém odstínu) byla hlášena jako častý nežádoucí účinek u pacientů, jimž byla aplikována injekce přípravku JETREA. Většina případů byla mírného, nezávazného charakteru a většina odezněla spontánně. Medián doby do odeznění byl 3 měsíce.
Abnormální retinogram
Změny elektroretinogramu (ERG) (pokles amplitudy křivek a a b) byly hlášeny jako častý nežádoucí účinek u pacientů, jimž byla aplikována injekce přípravku JETREA; ve většině případů byla také hlášena dyschromatopsie. Přibližně v polovině případů změny ERG odezněly do poslední follow-up kontroly. Medián doby do odeznění byl 6 měsíců. Změny ERG nebyly predikčním faktorem negativních výsledků ve smyslu zrakové ostrosti.
Poškození sítnice (roztržení, odchlípení)
V pivotních placebem kontrolovaných studiích fáze III, byly hlášeny případy porušení sítnice (roztržení a odchlípení) u 1,9 % pacientů, jimž byla aplikována injekce přípravku JETREA, oproti
4,3 % pacientů, jimž bylo aplikováno placebo. Většina těchto případů se u obou skupin objevila během vitrektomie nebo po ní. U skupiny léčené přípravkem JETREA se před vitrektomií vyskytlo odchlípení sítnice u 0,4 % pacientů, zatímco u skupiny s placebem k žádnému odchlípení nedošlo; oproti tomu k roztržení sítnice (bez odchlípení) došlo před vitrektomií u 0,2 % pacientů ze skupiny léčené přípravkem JETREA a u 0,5 % pacientů ve skupině s placebem.
Makulární díra
V pivotních placebem kontrolovaných studiích fáze III byly hlášeny případy vzniku nové makulární díry nebo zhoršení makulární díry u 6,7 % všech pacientů, jimž byla aplikována injekce přípravku JETREA, oproti 9,6 % pacientů, jimž byla aplikována injekce placeba. Ačkoli v pivotních placebem kontrolovaných studiích fáze III byl prokázán příznivý vliv přípravku JETREA na uzávěr makulárních děr spojených s vitreomakulární trakcí, v několika případech došlo ke zvýšení trakce s následnou progresí nebo vznikem nové makulární díry. Přestože tyto příhody jsou součástí přirozené progrese onemocnění, přispění okriplasminu v některých případech je vzhledem k mechanismu jeho působení pravděpodobné.
Vitreální adheze
V pivotních placebem kontrolovaných studiích fáze III byly hlášeny případy zhoršení vitreomakulární adheze nebo vitreomakulární trakce u 1,5 % všech pacientů, jimž byla aplikována injekce přípravku JETREA, oproti 1,1 % pacientů, jimž byla aplikována injekce placeba. Ačkoli jsou tyto příhody součástí přirozené progrese onemocnění, přispění okriplasminu v některých případech je vzhledem k mechanismu jeho působení pravděpodobné.
Subluxace čočky/fakodonéza
Subluxace čočky/ fakodonéza byla hlášena v klinických studiích s dospělými pacienty a jeví se jako možná příčina léčby přípravkem JETREA. V pediatrické studii hodnotící přípravek JETREA jako doplňkovou léčbu k vitrektomii byl hlášen jeden případ subluxace u nedonošeného dítěte, kterému byla podána jedna intravitreální injekce 0,175 mg přípravku JETREA. Subluxace čočky byla pozorována u 3 druhů zvířat při koncentracích okriplasminu vyšších, než je doporučená klinická koncentrace (viz bod 5.3).
Vzhledem k proteolytickému účinku okriplasminu a k předklinickým a klinickým výsledkům nelze možnost subluxace čočky nebo fakodonézy vyloučit. Pokud se takový případ vyskytne, je nutno jej léčit dle standardní lékařské praxe.
Doporučení ohledně sledování viz bod 4.4. Ve všech výše uvedených případech se doporučuje pravidelné sledování.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Klinické údaje o účincích přípravku JETREA při předávkování jsou omezené. Byl hlášen jeden případ náhodného předávkování 0,250 mg okriplasminu (dvojnásobkem doporučené dávky). U tohoto pacienta došlo k poklesu nejlépe korigované zrakové ostrosti BCVA o 21 písmen ETDRS vůči výchozímu stavu; na konci studie se stav vrátil k výchozímu stavu s tolerancí 9 písmen. U pacienta se také rozvinula mírná hyperemie spojivky, zánět oka a mióza, které ustoupily po podání očních kapek s obsahem kortikosteroidů.
Dojde-li k předávkování, doporučuje se pacienta pečlivě sledovat. Objeví-li se nežádoucí účinky, je nutno je léčit dle standardní lékařské praxe.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Oftalmologika, Jiná oftalmologika, ATC kód: S01XA22 Mechanismus účinku
Okriplasmin vykazuje proteolytický účinek vůči bílkovinným složkám sklivce a vitreoretinálního rozhraní (VRI) (např. lamininu, fibronektinu a kolagenu); záměrem jeho aplikace je rozpustit bílkovinnou matrix, která je zodpovědná za abnormální vitreomakulární adhezi (VMA). Těsná vazba bílkovinných složek na makulární část vitreoretinálního rozhraní přispívá k vitreomakulární trakci (VMT), což vede ke zhoršení zraku a/nebo vzniku makulárních děr.
Klinická účinnost a bezpečnost
Účinnost přípravku JETREA byla prokázána ve 2 multicentrických, randomizovaných, dvojitě zaslepených, placebem kontrolovaných 6měsíčních studiích s pacienty s VMT. V těchto 2 studiích (TG-MV-006 a TG-MV-007) bylo randomizováno celkem 652 pacientů (464 pacientů bylo léčených přípravkem JETREA, 188 placebem).
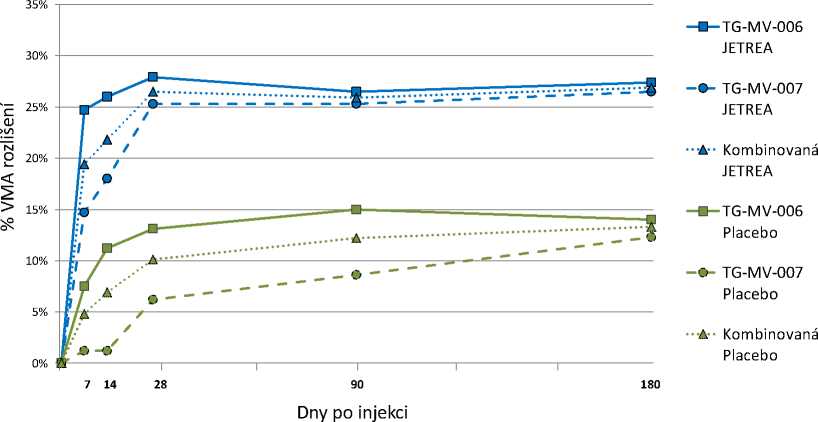
V obou pivotních studiích byl podíl pacientů, u nichž došlo k ústupu VMA ve dni 28 (primární cílový parametr) významně vyšší (p <0,003) ve skupině léčené přípravkem JETREA v porovnání se skupinou léčenou placebem. Tento rozdíl zůstal statisticky významný v obou studiích až do měsíce 6 (p < 0,024). Dle integrovaných dat bylo ve dni 28 dosaženo ústupu VMA u 26,5 % pacientů léčených přípravkem JETREA v porovnání s 10,1 % ve skupině léčené placebem (p < 0,001). Tento statisticky významný rozdíl zůstal zachován ode dne 7 až do měsíce 6 (Obrázek 1).
Obrázek 1: Podíl pacientů s ústupem VMA do dne 180 (měsíc 6) (TG-MV-006, TG-MV-007 a integrovaná data)
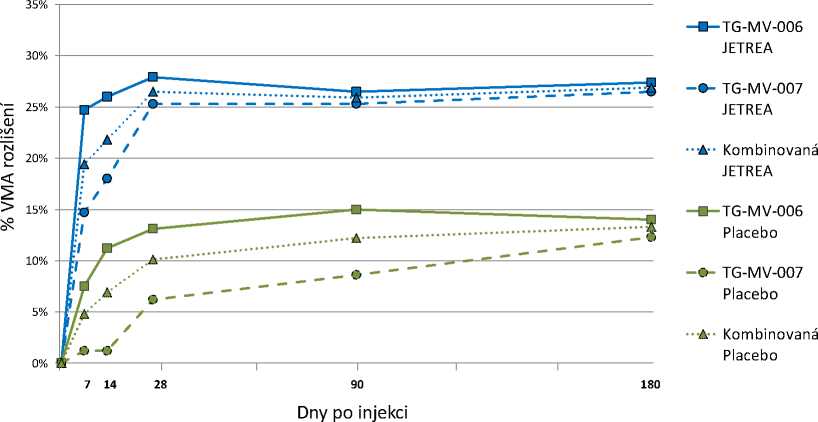
Ve všech dnech po injekci bylo p <0,024 u TG-MV-006, p <0,009 u TG-MV-007 a p <0,001 u integrovaných dat
Pacienti, kteří ve výchozím stavu neměli ERM, měli vyšší pravděpodobnost vyléčení VMA ve dni 28 ve srovnání s pacienty, kteří ve výchozím stavu měli ERM. Dle integrovaných dat byla úspěšnost léčby VMA ve dni 28 vyšší u pacientů léčených přípravkem JETREA, než u pacientů s placebem, a to jak v podskupině bez ERM (37,4 % vs. 14,3 %, p<0,001) tak s ERM (8,7% vs. 1,5%, p=0,046).
Pacienti, kteří měli ve výchozím stavu menší průměr VMA (< 1500 mikrometrů) měli větší šanci na úspěšnou léčbu VMA ve dni 28 v porovnání s pacienty s průměrem > 1500 mikrometrů.
Dle integrovaných dat byla úspěšnost léčby VMA ve dni 28 vyšší u pacientů léčených přípravkem JETREA než u pacientů s placebem, a to jak v podskupině pacientů, kteří měli ve výchozím stavu VMA < 1500 mikrometrů (34,7 % vs. 14,6 %, p<0,001), tak u pacientů s výchozím stavem VMA > 1500 mikrometrů (5,9% vs. 0%, p=0,113).
Dle integrovaných dat mělo ve výchozím stavu 106 (22,8 %) pacientů léčených přípravkem JETREA a 47 (25 %) pacientů léčených placebem makulární díru v plné tloušťce (FTMH). U těchto pacientů došlo k uzavření FTMH bez vitrektomie ve dni 28 častěji ve skupině léčené přípravkem JETREA než ve skupině léčené placebem (40,6 % oproti 10,6 %; p <0,001). Rozdíl zůstal statisticky významný až do ukončení studií (měsíc 6).
U signifikantně vyššího procenta pacientů léčených přípravkem JETREA došlo k úplnému PVD ve dni 28 v porovnání s pacienty léčenými placebem (integrovaná data: 13,4 % vs. 3,7 %; p <0,001).
Během klinických studií může být provedena vitrektomie dle uvážení zkoušejícího. Pacienti léčení přípravkem JETREA měli na konci studie (měsíc 6) nižší pravděpodobnost vitrektomie v porovnání s pacienty léčenými placebem (integrovaná data: 17,7 % oproti 26,6 %; p = 0,016).
Vyšší podíl pacientů léčených přípravkem JETREA měl v měsíci 6 nárůst BCVA o >2 nebo >3 řádky (bez ohledu na vitrektomii) (28,0 % a 12,3 %) v porovnání s pacienty léčenými placebem (17,1 % a
6,4 %) (p = 0,003 resp. p = 0,024). Stejně tak poměr pacientů s nárůstem BCVA v měsíci 6 o >2 nebo >3 řádky bez vitrektomie byl příznivější u přípravku JETREA (23,7 % vs. 11,2 %, p<0,001 pro nárůst o >2 řádky a 9,7% vs. 3,7%, p= 0,008 pro nárůst o >3 řádky).
Dle integrované analýzy výsledků dotazníku National Eye Institute Visual Function Questionnaire-25 (VFQ-25) byly výsledky jednotlivých skóre substupnic stejně jako kompozitní skóre číselně lepší u skupiny léčené přípravkem JETREA v porovnání se skupinou léčenou placebem. Tento rozdíl ve
zlepšení v celkovém sub-stupnicovém skóre zrakové ostrosti byl signifikantní (6,1 JETREA oproti
2,1 placebo, p = 0,024).
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem JETREA u všech podskupin pediatrické populace při léčbě vitreomakulární trakce (VMT) včetně současné přítomnosti makulární díry o průměru menším nebo rovném 400 mikrometrů (informace o použití u dětí viz bod 4.2).
Bezpečnost a účinnost okriplasminu u pediatrických pacientů s plánovanou vitrektomií byla zkoumána ve studii TG-MV-009. Jedna intravitreální injekce o dávce 0,175 mg (nad hranicí doporučené dávky) nebo placeba byla aplikována do středu sklivce 24 očí u dětí ve věku 0 až 16 let, 30 až 60 minut před plánovaným začátkem vitrektomie. Hlavními důvody pro podstoupení vitrektomie byli odchlípení sítnice a retinopatie nedonošených dětí. Léčba okriplasminem neprokázala vliv na míru odchlípení zadní plochy sklivce, stupeň zkapalnění sklivce, míru okamžité repozice sítnice, vývoj proliferativní vitreoretinopatie anebo stupeň retinopatie nedonošených dětí. Bezpečnostní nálezy získané ze studie TG-MV-009 byly v souladu se známým bezpečnostním profilem přípravku JETREA. Na základě výsledků dané studie se nedoporučuje použití přípravku JETREA jako doplněk vitrektomie u dětí pro ulehčení separace a odstranění sklivce.
Etnický původ
Zkušenosti s podáváním přípravku u jiné než europoidní rasy jsou omezené.
5.2 Farmakokinetické vlastnosti
Hladina okriplasminu ve sklivci po intravitreálním podání rychle klesá. V klinické studii dosahovala u pacientů s plánovanou vitrektomií, kteří obdrželi 0,125 mg přípravku JETREA (což odpovídá teoretické počáteční koncentraci ve sklivci 29 pg/ml), průměrná aktivita okriplasminu 9 % teoretické počáteční koncentrace 2-4 hodiny po injekci; po 7 dnech se nacházela pod dolní hranicí kvantifikace.
Vzhledem k malé podané dávce (0,125 mg) se po intravitreální injekci nepředpokládá detekovatelná hladina okriplasminu v systémovém oběhu.
Po intravenózním podání okriplasmin vstupuje do katabolické dráhy endogenních proteinů, skrze kterou je rychle deaktivován pomocí interakcí s inhibitorem proteázy a2-antiplazminem nebo a2-makroglobulinem. Neaktivní komplex okriplasmin/a2 antiplazmin je eliminován z oběhu s poločasem (t1/2) několika hodin.
Porucha funkce ledvin
U pacientů s poruchou funkce ledvin nebyly provedeny žádné studie hodnotící farmakokinetiku okriplasminu, protože po intravitreálním podání se předpokládá velmi nízká systémová expozice.
Porucha funkce jater
U pacientů s poruchou funkce jater nebyly provedeny žádné studie hodnotící farmakokinetiku okriplasminu, protože po intravitreálním podání se předpokládá velmi nízká systémová expozice.
5.3 Předklinické údaje vztahující se k bezpečnosti
Intravitreální toxicita okriplasminu byla hodnocena u králíků, opic a miniprasat. Okriplasmin způsoboval zánětlivou odpověď a přechodné změny ERG u králíků a opic, zatímco u laboratorních miniprasat nebyly pozorovány zánět ani změna ERG. U králíků a opic měl výskyt buněčných infiltrátů ve sklivci tendenci k ústupu v průběhu času. U opic se po podání 125 pg/oko (68 pg/ml sklivce) ERG vrátil zcela k normálu do dne 55. Subluxace čočky byla pozorována u 3 druhů při koncentracích okriplasminu ve sklivci rovných nebo vyšších než 41 pg/ml, což je koncentrace převyšující zamýšlenou klinickou koncentraci 29 pg/ml. Tento účinek se jevil jako závislý na dávce a byl pozorován u všech zvířat, kterým byl okriplasmin aplikován více než jednou. Patologické změny související s nitroočním krvácením byly pozorovány u králíků a opic. Není dosud jasné, zda je toto krvácení způsobeno injekčním výkonem samotným, nebo podáním okriplasminu. Po intravitreálním podání okriplasminu nebyla pozorována žádná systémová toxicita.
Systémová toxicita okriplasminu byla hodnocena u potkanů a psů. Intravenózní podání 10 mg/kg bylo obecně dobře tolerováno u potkanů i u psů, a to při jednorázovém i při opakovaném podání.
Nejsou dostupné žádné údaje týkající se karcinogenity a mutagenity reprodukční či vývojové toxicity.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Mannitol
Kyselina citronová
Hydroxid sodný (NaOH) (pro úpravu pH)
Voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek míchán s jinými léčivými přípravky, s výjimkou sterilního roztoku chloridu sodného 9 mg/ml (0,9 %) na injekci, bez obsahu konzervantů a pufrů.
6.3 Doba použitelnosti
3 roky při uchovávání v mrazničce (-20 °C ± 5 C).
Po rozmrazení
Přípravek se má naředit a použít okamžitě. Chemická a fyzikální stabilita neotevřeného přípravku uchovávaného v původním obale a chráněného před světlem, však byla prokázána po dobu až 8 hodin, pokud je přípravek uchováván při teplotě do 25 °C. Nezmrazujte lahvičku, pokud již byla jednou rozmrazena.
Po otevření/naředění
Z mikrobiologického hlediska je nutno přípravek použít okamžitě po otevření/naředění.
Po jednorázovém použití se musí lahvička a nepoužitá část naředěného roztoku zlikvidovat.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v mrazničce (-20°C ± 5 °C).
Podmínky uchovávání tohoto léčivého přípravku po jeho rozmrazení a otevření/naředění jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
0. 2 ml roztoku v injekční lahvičce (skleněné, třída I) uzavřené zátkou z chlorbutylové pryže a oranžovým polypropylenovým odtrhávacím víčkem. Balení obsahuje 1 lahvičku.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Lahvičky jsou určeny jen pro jedno použití.
Při přípravě přípravku JETREA pro intravitreální injekci postupujte podle následujících pokynů:
1. Vyjměte lahvičku z mrazničky a nechejte ji rozmrazit při pokojové teplotě (trvá to asi 2 minuty).
2. Po úplném rozmražení odstraňte z lahvičky oranžové ochranné polypropylenové odtrhávací víčko.
3.
4.
Dezinfikujte víčko lahvičky otřením tamponem s alkoholem.
Aseptickou technikou nařeďte přidáním 0,2 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9%) (sterilního, bez obsahu konzervantů a pufrů) do lahvičky s přípravkem JETREA a jemně promíchejte kroužením lahvičkou, dokud se roztoky nesmísí. Roztok k naředění se musí aspirovat z neotevřené nádobky, která se smí použít pouze jednou. Zbývající injekční roztok chloridu sodného 9 mg/ml (0,9%) se musí zlikvidovat. Naředěný roztok se má použít bezprostředně po naředění.
5. Vizuálně zkontrolujte lahvičku, zda neobsahuje viditelné částice. Smí se použít výhradně čirý roztok bez viditelných částic.
6. Aseptickou technikou aspirujte všechen naředěný roztok za použití vhodné sterilní jehly (lahvičku poněkud nakloňte, abyste usnadnili aspiraci) a po aspiraci obsahu lahvičky jehlu zlikvidujte. Tuto jehlu nepoužívejte k intravitreální injekci.

7. Mí sto původní j ehly nasaďte vhodnou sterilní j ehlu a pečlivě vytlačte nadbytečný roztok pomalým stlačováním pístu tak, aby se konec pístu vyrovnal s 0,1ml značkou na stříkačce (odpovídá 0,125 mg okriplasminu).
8. Injikujte 0,1 ml naředěného roztoku bezodkladně do středu sklivce.
9. Po jednorázovém použití zlikvidujte lahvičku i veškerý nepoužitý naředěný roztok.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
ThromboGenics NV Gaston Geenslaan 1 B-3001 Leuven Belgie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/13/819/001
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 13. března 2013
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
JETREA 0,375 mg / 0,3 ml injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička obsahuje ocriplasminum* 0,375 mg v 0,3 ml roztoku (1,25 mg/l). To představuje použitelné množství pro jednu dávku 0,1 ml obsahující ocriplasminum 0,125 mg.
*Okriplasmin je zkrácená forma lidského plazminu, která se vyrábí rekombinantní DNA technologií v expresním systému Pichia pastoris.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok (injekce). Čirý bezbarvý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek JETREA je indikován k použití u dospělých pacientů k léčbě vitreomakulární trakce (VMT) včetně případů zahrnujících makulární díru o průměru menším nebo rovném 400 mikrometrů (viz bod 5.1).
4.2 Dávkování a způsob podání
Podávání přípravku JETREA smí provádět pouze kvalifikovaný oftalmolog se zkušenostmi s intravitreálními injekcemi. Diagnóza vitreomakulární trakce (VMT) má zahrnovat kompletní klinický obraz včetně pacientovy anamnézy, klinického vyšetření a použití adekvátních diagnostických metod, jako je např. optická koherentní tomografie (OCT).
Dávkování
JETREA 0,375 mg/0,3 ml injekční roztok je přípravek připravený k použití a další ředění není třeba. Doporučená dávka je 0,125 mg (0,1 ml roztoku) jednorázově aplikovaná intravitreální injekcí do postiženého oka. Jedna lahvička se smí použít pouze jednorázově a pouze k léčbě jednoho oka. Léčba druhého oka přípravkem JETREA se nedoporučuje dříve než za 7 dní od podání první injekce do iniciálně léčeného oka z důvodu monitorování postinjekční reakce včetně potenciálního rizika snížené zrakové ostrosti u léčeného oka. Opakované podávání do stejného oka se nedoporučuje (viz bod 4.4).
Pokyny ke sledování po aplikaci injekce viz bod 4.4.
Zvláštní skupiny pacientů
Porucha funkce ledvin
Nebyly provedeny žádné konkrétní studie hodnotící použití přípravku JETREA u pacientů s poruchou funkce ledvin. U pacientů s poruchou funkce ledvin se nepředpokládá nutnost úpravy dávky ani jiná zvláštní opatření (viz bod 5.2).
Porucha funkce jater
Nebyly provedeny žádné konkrétní studie hodnotící použití přípravku JETREA u pacientů s poruchou funkce jater. U pacientů s poruchou funkce jater se nepředpokládá nutnost úpravy dávky ani jiná zvláštní opatření (viz bod 5.2).
Starší pacienti
Populace starších pacientů byla hodnocena v klinických studiích. Není zapotřebí žádná úprava dávky. Pediatrická populace
Neexistuje žádné relevantní použití přípravku JETREA u dětí ve věku do 18 let v indikaci vitreomakulární trakce (VMT) včetně případů spojených s přítomností makulární díry o průměru menším nebo rovném 400 mikrometrů. V současnosti dostupné údaje o použití u dětí jsou popsány v bodě 5.1.
Způsob podání
Lahvička k jednorázové intravitreální aplikaci.
Před zákrokem mohou být dle uvážení ošetřujícího oftalmologa podány antibiotické oční kapky.
Opatření, která je nutno učinit před manipulací s léčivým přípravkem nebo před jeho podáním Intravitreální injekci je nutno provádět za kontrolovaných aseptických podmínek včetně nutnosti dezinfekce rukou operatéra, použití sterilních rukavic, sterilní roušky, sterilního rozvěrače víček (nebo ekvivalentní náhrady) a možnosti provedení sterilní paracentézy (pokud je vyžadována). Periokulární pokožka, víčko a povrch oka musí být dezinfikovány a před aplikací injekce je nutno podat adekvátní anestetikum a lokální širokospektrý mikrobiocidní přípravek dle standardní lékařské praxe.
Podat se má pouze 0,1 ml z celkového objemu 0,3 ml roztoku v lahvičce. Nadbytečný objem se má před podáním injekce vytlačit tak, aby byla podána jedna dávka 0,1 ml obsahující 0,125 mg okriplasminu. Informace o zacházení s léčivým přípravkem jsou uvedeny v bodě 6.6.
Injekční jehla se zasune 3,5-4,0 mm posteriorně k limbu do centra sklivce tak, aby směřovala do centra očního bulbu, a nikoli k horizontálnímu meridiánu. Objem injekce 0,1 ml se potom aplikuje do středu sklivce.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Aktivní nebo suspektní okulární nebo periokulární infekce.
4.4 Zvláštní upozornění a opatření pro použití
Sledování po aplikaci injekce
Přípravek JETREA se podává pouze intravitreální injekcí. Intravitreální injekce mohou být spojeny s nitroočním zánětem/infekcí, s nitroočním krvácením a se zvýšeným nitroočním tlakem (NOT). Je nutno za všech okolností používat správnou aseptickou injekční techniku. Po intravitreální injekci je nutno sledovat, zda se u pacienta nerozvinou nežádoucí účinky, mimo jiné například nitrooční zánět/infekce a zvýšení NOT. Výskyt přechodného zvýšení NOT včetně přechodné slepoty a zástavy perfuze papily zrakového nervu byl pozorován do 60 minut po injekci přípravku JETREA. Sledování zvýšení NOT může sestávat z kontroly perfuze papily zrakového nervu bezprostředně po injekci a z tonometrie do 30 minut po injekci. Nitrooční zánět/infekci lze vyšetřit provedením biomikroskopie v rozmezí 2 až 7 dnů po injekci. Pacienty je nutno poučit, aby neprodleně hlásili příznaky možného nitroočního zánětu nebo infekce a jakékoli další příznaky postihující zrak nebo oko. Pokud se u pacienta vyskytne kterýkoli z výše uvedených příznaků, je nutno jej léčit podle standardní lékařské praxe.
Bilaterální léčba
Bezpečnost a účinnost přípravku JETREA při současném podávání do obou očí nebyla hodnocena. Proto se současné podávání do obou očí nedoporučuje.
Opakované podávání
Opakované podávání přípravku JETREA do stejného oka nebylo dostatečně hodnoceno, a proto se nedoporučuje.
Populace se žádnými nebo omezenými údaji
Přípravek JETREA nebyl hodnocen u pacientů s makulárními dírami o velkém průměru (> 400 mikrometrů), se silnou myopií (sférická korekce > 8 dioptrií nebo axiální délka > 28 mm), s afakií, s rhegmatogenním odchlípením sítnice v anamnéze, s nestabilitou zonuly čočky, s nedávno prodělanou oční operací nebo nitrooční injekcí (včetně laserové léčby), s proliferativní diabetickou retinopatií, s ischemickou retinopatií, s okluzí cév sítnice, s exsudativní formou věkem podmíněné makulární degenerace (VPMD) a s krvácením do sklivce. U těchto pacientů se léčba nedoporučuje.
U pacientů s neproliferativní diabetickou retinopatií, s anamnézou uveitidy (včetně závažného aktivního zánětu) nebo závažného poranění oka jsou k dispozici pouze omezené zkušenosti. Při léčbě těchto pacientů je nutno postupovat s opatrností.
Jiné
Riziko subluxace čočky nebo fakodonézy nemůže být vyloučeno. Pokud se takový případ vyskytne, je nutno jej léčit dle standardní lékařské praxe. Pacienti mají být adekvátně monitorováni (viz body 4.8 a 5.3).
U pacientů s epiretinální membránou (ERM) nebo s průměrem VMA > 1500 mikrometrů je účinnost okriplasminu (zvláště pokud jde o odstranění vitreomakulární adhese nebo docílení kompletního odloučení zadní části sklivce [PVD]) snížená (viz bod 5.1).
V důsledku možnosti nárůstu trakčních sil existuje riziko vzniku nových nebo zvětšení stávajících makulárních děr. Pacienti mají být adekvátně monitorováni (viz bod 4.8).
Během prvního týdne po aplikaci injekce existuje riziko vzniku významné, nicméně přechodné ztráty zrakové ostrosti. Pacienti mají být adekvátně monitorováni (viz bod 4.8).
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné formální studie interakcí.
Okriplasmin je proteolytický enzym s aktivitou serinové proteázy, který může být v oku přítomen po dobu několika dnů po intravitreální injekci (viz bod 5.2). Současné podání jiných léčivých přípravků v úzkém časovém rozpětí do stejného oka může ovlivnit účinek obou léčivých přípravků, a proto se nedoporučuje.
Nejsou k dispozici žádné klinické údaje ohledně současného používání okriplasminu a inhibitorů VEGF- (vaskulárního endoteliálního růstového faktoru).
Nepředpokládají se žádné systémové interakce.
4.6 Fertilita, těhotenství a kojení
Údaje o podávání přípravku JETREA těhotným ženám nejsou k dispozici. Nebyly provedeny žádné studie reprodukční toxicity. Systémová expozice přípravku JETREA po intravitreální injekci je podle předpokladů velmi nízká. Přípravek JETREA se má během těhotenství používat pouze za předpokladu, že klinické přínosy převáží potenciální rizika.
Kojení
Není známo, zda se přípravek JETREA vylučuje do lidského mateřského mléka. Přípravek JETREA se má během kojení používat pouze za předpokladu, že klinické přínosy převáží potenciální rizika.
Fertilita
O účinku přípravku JETREA na fertilitu nejsou žádné údaje.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Po intravitreální injekci přípravku JETREA může dojít k dočasným poruchám zraku (viz bod 4.8). V těchto případech pacienti nesmí řídit ani obsluhovat stroje, dokud porucha zraku neodezní.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Léčbu intravitreální injekcí přípravku JETREA obdrželo více než 800 pacientů v klinických studiích, z nichž více než 570 bylo léčeno doporučenou dávkou 0,125 mg.
Všechny nežádoucí účinky, které se vyskytly, byly oční nežádoucí účinky. Nejčastěji byly hlášeny sklivcové zákalky, bolest oka a fotopsie a také krvácení do spojivky v důsledku injekce. Většina nežádoucích účinků se objevila během prvního týdne po injekci. Většina těchto reakcí byla nezávažná, s mírnou intenzitou, a vymizela během 2 až 3 týdnů. Byly také hlášeny příznaky pozorované v kontralaterálním oku nebo bilaterálně.
Souhrnná incidence závažných nežádoucích účinků ve všech klinických studiích byla 2,2 % u pacientů léčených přípravkem JETREA a 2,4 % u kontrolní skupiny pacientů.
Tabulka s přehledem nežádoucích účinků
V následující tabulce je uveden přehled nežádoucích účinků, které byly hlášeny v klinických a/nebo v postmarketingových hlášeních.
Nežádoucí účinky s reálnou možností kauzality s podáním injekce nebo s přípravkem JETREA jsou uvedeny dle systému MedDRA podle třídy orgánů a četnosti s použitím následující konvence: velmi časté (>1/10); časté (>1/100 až <1/10); méně časté (>1/1 000 až <1/100); vzácné (> 1/10 000 to <1/1 000); velmi vzácné (<1/10 000) a není známo (z dostupných údajů nelze určit). V každé skupině četnosti jsou nežádoucí účinky seřazeny podle klesající závažnosti.
|
Poruchy oka |
Velmi časté Sklivcové zákalky, bolest oka, krvácení do spojivky |
|
Časté | |
|
Snížená zraková ostrost*, zhoršené vidění, rozmazané vidění, krvácení do sítnice, krvácení do spojivky, roztržení sítnice , odchlípení sítnice , zvýšený nitrooční tlak, makulární díra*, makulární degenerace, retinální degenerace, makulární edém**, edém sítnice, retinální pigmentová epitelopatie, metamorfopsie, adheze sklivce*, edém spojivky, edém očního víčka, vitritida, přítomnost buněk v přední komoře, zarudnutí přední komory, iritida, fotopsie, hyperemie spojivky, hyperemie oka, odchlípení sklivce, abnormální retinogram*, podráždění oka, suché oko, pocit cizího tělíska v oku, svědění oka, oční diskomfort, fotofobie, chromatopsie* | |
|
L-s—:—:---:-1 |
Méně časté Přechodná slepota, subluxace čočky , šeroslepost, skotom, porucha pupilárního reflexu, defekt zorného pole, diplopie, hyféma, mióza, nestejné pupily, abraze rohovky, zánět přední komory, zánět oka, podráždění spojivky |
včetně cystoidního makulárního edému
Popis vybraných nežádoucích účinků
Snížení zrakové ostrosti
V pivotních, placebem kontrolovaných studiích fáze III došlo u 7, 7 % pacientů léčených přípravkem JETREA a 1, 6 % pacientů s placebem během prvního týdne po aplikaci injekce k akutnímu přechodnému snížení nejlépe korigované zrakové ostrosti (BCVA) o > 2 řádky (>10 ETDRS písmen) bez jiné vysvětlující příčiny. Snížení zrakové ostrosti obvykle spontánně ustoupilo do 2 týdnů bez nutnosti intervence. Doporučení ohledně monitorování viz bod 4.4.
Chromatopsie
Dyschromatopsie (obecně popisovaná jako vidění ve žlutém odstínu) byla hlášena jako častý nežádoucí účinek u pacientů, jimž byla aplikována injekce přípravku JETREA. Většina případů byla mírného, nezávažného charakteru a většina odezněla spontánně. Medián doby do odeznění byl 3 měsíce.
Abnormální retinogram
Změny elektroretinogramu (ERG) (pokles amplitudy křivek a a b) byly hlášeny jako častý nežádoucí účinek u pacientů, jimž byla aplikována injekce přípravku JETREA; ve většině případů byla také hlášena dyschromatopsie. Přibližně v polovině případů změny ERG odezněly do poslední follow-up kontroly. Medián doby do odeznění byl 6 měsíců. Změny ERG nebyly predikčním faktorem negativních výsledků ve smyslu zrakové ostrosti.
Poškození sítnice (roztržení, odchlípení)
V pivotních placebem kontrolovaných studiích fáze III, byly hlášeny případy porušení sítnice (roztržení a odchlípení) u 1,9 % pacientů, jimž byla aplikována injekce přípravku JETREA, oproti
4,3 % pacientů, jimž bylo aplikováno placebo. Většina těchto případů se u obou skupin objevila během vitrektomie nebo po ní. U skupiny léčené přípravkem JETREA se před vitrektomií vyskytlo odchlípení sítnice u 0,4 % pacientů, zatímco u skupiny s placebem k žádnému odchlípení nedošlo; oproti tomu k roztržení sítnice (bez odchlípení) došlo před vitrektomií u 0,2 % pacientů ze skupiny léčené přípravkem JETREA a u 0,5 % pacientů ve skupině s placebem.
Makulární díra
V pivotních placebem kontrolovaných studiích fáze III byly hlášeny případy vzniku nové makulární díry nebo zhoršení makulární díry u 6,7 % všech pacientů, jimž byla aplikována injekce přípravku JETREA, oproti 9,6 % pacientů, jimž byla aplikována injekce placeba. Ačkoli v pivotních placebem kontrolovaných studiích fáze III byl prokázán příznivý vliv přípravku JETREA na uzávěr makulárních děr spojených s vitreomakulární trakcí, v několika případech došlo ke zvýšení trakce s následnou progresí nebo vznikem nové makulární díry. Přestože tyto příhody jsou součástí přirozené progrese onemocnění, přispění okriplasminu v některých případech je vzhledem k mechanismu jeho působení pravděpodobné.
Vitreální adheze
V pivotních placebem kontrolovaných studiích fáze III byly hlášeny případy zhoršení vitreomakulární adheze nebo vitreomakulární trakce u 1,5 % všech pacientů, jimž byla aplikována injekce přípravku JETREA, oproti 1,1 % pacientů, jimž byla aplikována injekce placeba. Ačkoli jsou tyto příhody součástí přirozené progrese onemocnění, přispění okriplasminu v některých případech je vzhledem k mechanismu jeho působení pravděpodobné.
Subluxace čočky/fakodonéza
Subluxace čočky/ fakodonéza byla hlášena v klinických studiích s dospělými pacienty a jeví se jako možná příčina léčby přípravkem JETREA. V pediatrické studii hodnotící přípravek JETREA jako doplňkovou léčbu k vitrektomii byl hlášen jeden případ subluxace u nedonošeného dítěte, kterému byla podána jedna intravitreální injekce 0,175 mg přípravku JETREA. Subluxace čočky byla pozorována u 3 druhů zvířat při koncentracích okriplasminu vyšších, než je doporučená klinická koncentrace (viz bod 5.3).
Vzhledem k proteolytickému účinku okriplasminu a k předklinickým a klinickým výsledkům nelze možnost subluxace čočky nebo fakodonézy vyloučit. Pokud se takový případ vyskytne, je nutno jej léčit dle standardní lékařské praxe.
Doporučení ohledně sledování viz bod 4.4. Ve všech výše uvedených případech se doporučuje pravidelné sledování.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Klinické údaje o účincích přípravku JETREA při předávkování jsou omezené. Byl hlášen jeden případ náhodného předávkování 0,250 mg okriplasminu (dvojnásobkem doporučené dávky). U tohoto pacienta došlo k poklesu nejlépe korigované zrakové ostrosti BCVA o 21 písmen ETDRS vůči výchozímu stavu; na konci studie se stav vrátil k výchozímu stavu s tolerancí 9 písmen. U pacienta se také rozvinula mírná hyperemie spojivky, zánět oka a mióza, které ustoupily po podání očních kapek s obsahem kortikosteroidů.
Dojde-li k předávkování, doporučuje se pacienta pečlivě sledovat. Objeví-li se nežádoucí účinky, je nutno je léčit dle standardní lékařské praxe.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Oftalmologika, Jiná oftalmologika, ATC kód: S01XA22 Mechanismus účinku
Okriplasmin vykazuje proteolytický účinek vůči bílkovinným složkám sklivce a vitreoretinálního rozhraní (VRI) (např. lamininu, fibronektinu a kolagenu); záměrem jeho aplikace je rozpustit bílkovinnou matrix, která je zodpovědná za abnormální vitreomakulární adhezi (VMA). Těsná vazba bílkovinných složek na makulární část vitreoretinálního rozhraní přispívá k vitreomakulární trakci (VMT), což vede ke zhoršení zraku a/nebo vzniku makulárních děr.
Klinická účinnost a bezpečnost
Účinnost přípravku JETREA byla prokázána ve 2 multicentrických, randomizovaných, dvojitě zaslepených, placebem kontrolovaných 6měsíčních studiích s pacienty s VMT. V těchto 2 studiích (TG-MV-006 a TG-MV-007) bylo randomizováno celkem 652 pacientů (464 pacientů bylo léčených přípravkem JETREA, 188 placebem).
V obou pivotních studiích byl podíl pacientů, u nichž došlo k ústupu VMA ve dni 28 (primární cílový parametr) významně vyšší (p <0,003) ve skupině léčené přípravkem JETREA v porovnání se skupinou léčenou placebem. Tento rozdíl zůstal statisticky významný v obou studiích až do měsíce 6 (p < 0,024). Dle integrovaných dat bylo ve dni 28 dosaženo ústupu VMA u 26,5 % pacientů léčených přípravkem JETREA v porovnání s 10,1 % ve skupině léčené placebem (p < 0,001). Tento statisticky významný rozdíl zůstal zachován ode dne 7 až do měsíce 6 (Obrázek 1).
Obrázek 1: Podíl pacientů s ústupem VMA do dne 180 (měsíc 6) (TG-MV-006, TG-MV-007 a integrovaná data)
Ve všech dnech po injekci bylo p <0,024 u TG-MV-006, p <0,009 u TG-MV-007 a p <0,001 u integrovaných dat
Pacienti, kteří ve výchozím stavu neměli ERM, měli vyšší pravděpodobnost vyléčení VMA ve dni 28 ve srovnání s pacienty, kteří ve výchozím stavu měli ERM. Dle integrovaných dat byla úspěšnost léčby VMA ve dni 28 vyšší u pacientů léčených přípravkem JETREA, než u pacientů s placebem, a to jak v podskupině bez ERM (37,4 % vs. 14,3 %, p<0,001) tak s ERM (8,7% vs. 1,5%, p=0,046).
Pacienti, kteří měli ve výchozím stavu menší průměr VMA (< 1500 mikrometrů) měli větší šanci na úspěšnou léčbu VMA ve dni 28 v porovnání s pacienty s průměrem > 1500 mikrometrů.
Dle integrovaných dat byla úspěšnost léčby VMA ve dni 28 vyšší u pacientů léčených přípravkem JETREA než u pacientů s placebem, a to jak v podskupině pacientů, kteří měli ve výchozím stavu VMA < 1500 mikrometrů (34,7 % vs. 14,6 %, p<0,001), tak u pacientů s výchozím stavem VMA > 1500 mikrometrů (5,9% vs. 0%, p=0,113).
Dle integrovaných dat mělo ve výchozím stavu 106 (22,8 %) pacientů léčených přípravkem JETREA a 47 (25 %) pacientů léčených placebem makulární díru v plné tloušťce (FTMH). U těchto pacientů došlo k uzavření FTMH bez vitrektomie ve dni 28 častěji ve skupině léčené přípravkem JETREA než ve skupině léčené placebem (40,6 % oproti 10,6 %; p <0,001). Rozdíl zůstal statisticky významný až do ukončení studií (měsíc 6).
U signifikantně vyššího procenta pacientů léčených přípravkem JETREA došlo k úplnému PVD ve dni 28 v porovnání s pacienty léčenými placebem (integrovaná data: 13,4 % vs. 3,7 %; p <0,001).
Během klinických studií může být provedena vitrektomie dle uvážení zkoušejícího. Pacienti léčení přípravkem JETREA měli na konci studie (měsíc 6) nižší pravděpodobnost vitrektomie v porovnání s pacienty léčenými placebem (integrovaná data: 17,7 % oproti 26,6 %; p = 0,016).
Vyšší podíl pacientů léčených přípravkem JETREA měl v měsíci 6 nárůst BCVA o >2 nebo >3 řádky (bez ohledu na vitrektomii) (28,0 % a 12,3 %) v porovnání s pacienty léčenými placebem (17,1 % a
6,4 %) (p = 0,003 resp. p = 0,024). Stejně tak poměr pacientů s nárůstem BCVA v měsíci 6 o >2 nebo >3 řádky bez vitrektomie byl příznivější u přípravku JETREA (23,7 % vs. 11,2 %, p<0,001 pro nárůst o >2 řádky a 9,7% vs. 3,7%, p= 0,008 pro nárůst o >3 řádky).
Dle integrované analýzy výsledků dotazníku National Eye Institute Visual Function Questionnaire-25 (VFQ-25) byly výsledky jednotlivých skóre substupnic stejně jako kompozitní skóre číselně lepší u skupiny léčené přípravkem JETREA v porovnání se skupinou léčenou placebem. Tento rozdíl ve
zlepšení v celkovém substupnicovém skóre zrakové ostrosti byl signifikantní (6,1 JETREA oproti
2,1 placebo, p = 0,024).
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem JETREA u všech podskupin pediatrické populace při léčbě vitreomakulární trakce (VMT) včetně současné přítomnosti makulární díry o průměru menším nebo rovném 400 mikrometrů (informace o použití u dětí viz bod 4.2).
Bezpečnost a účinnost okriplasminu u pediatrických pacientů s plánovanou vitrektomií byla zkoumána ve studii TG-MV-009. Jedna intravitreální injekce o dávce 0,175 mg (nad hranicí doporučené dávky) nebo placeba byla aplikována do středu sklivce 24 očí u dětí ve věku 0 až 16 let, 30 až 60 minut před plánovaným začátkem vitrektomie. Hlavními důvody pro podstoupení vitrektomie byli odchlípení sítnice a retinopatie nedonošených dětí. Léčba okriplasminem neprokázala vliv na míru odchlípení zadní plochy sklivce, stupeň zkapalnění sklivce, míru okamžité repozice sítnice, vývoj proliferativní vitreoretinopatie anebo stupeň retinopatie nedonošených dětí. Bezpečnostní nálezy získané ze studie TG-MV-009 byly v souladu se známým bezpečnostním profilem přípravku JETREA. Na základě výsledků dané studie se nedoporučuje použití přípravku JETREA jako doplněk vitrektomie u dětí pro ulehčení separace a odstranění sklivce.
Etnický původ
Zkušenosti s podáváním přípravku u jiné než europoidní rasy jsou omezené.
5.2 Farmakokinetické vlastnosti
Hladina okriplasminu ve sklivci po intravitreálním podání rychle klesá. V klinické studii dosahovala u pacientů s plánovanou vitrektomií, kteří obdrželi 0,125 mg přípravku JETREA (což odpovídá teoretické počáteční koncentraci ve sklivci 29 pg/ml), průměrná aktivita okriplasminu 9 % teoretické počáteční koncentrace 2-4 hodiny po injekci; po 7 dnech se nacházela pod dolní hranicí kvantifikace.
Vzhledem k malé podané dávce (0,125 mg) se po intravitreální injekci nepředpokládá detekovatelná hladina okriplasminu v systémovém oběhu.
Po intravenózním podání okriplasmin vstupuje do katabolické dráhy endogenních proteinů, skrze kterou je rychle deaktivován pomocí interakcí s inhibitorem proteázy a2_antiplazminem nebo a2_ makroglobulinem. Neaktivní komplex okriplasmin/a2 antiplazmin je eliminován z oběhu s poločasem (t1/2) několika hodin.
Porucha funkce ledvin
U pacientů s poruchou funkce ledvin nebyly provedeny žádné studie hodnotící farmakokinetiku okriplasminu, protože po intravitreálním podání se předpokládá velmi nízká systémová expozice.
Porucha funkce jater
U pacientů s poruchou funkce jater nebyly provedeny žádné studie hodnotící farmakokinetiku okriplasminu, protože po intravitreálním podání se předpokládá velmi nízká systémová expozice.
5.3 Předklinické údaje vztahující se k bezpečnosti
Intravitreální toxicita okriplasminu byla hodnocena u králíků, opic a miniprasat. Okriplasmin způsoboval zánětlivou odpověď a přechodné změny ERG u králíků a opic, zatímco u laboratorních miniprasat nebyly pozorovány zánět ani změna ERG. U králíků a opic měl výskyt buněčných infiltrátů ve sklivci tendenci k ústupu v průběhu času. U opic se po podání 125 pg/oko (68 pg/ml sklivce) ERG vrátil zcela k normálu do dne 55. Subluxace čočky byla pozorována u 3 druhů při koncentracích okriplasminu ve sklivci rovných nebo vyšších než 41 pg/ml, což je koncentrace převyšující zamýšlenou klinickou koncentraci 29 pg/ml. Tento účinek se jevil jako závislý na dávce a byl pozorován u všech zvířat, kterým byl okriplasmin aplikován více než jednou. Patologické změny související s nitroočním krvácením byly pozorovány u králíků a opic. Není dosud jasné, zda je toto krvácení způsobeno injekčním výkonem samotným, nebo podáním okriplasminu. Po intravitreálním podání okriplasminu nebyla pozorována žádná systémová toxicita.
Systémová toxicita okriplasminu byla hodnocena u potkanů a psů. Intravenózní podání 10 mg/kg bylo obecně dobře tolerováno u potkanů i u psů, a to při jednorázovém i při opakovaném podání.
Nejsou dostupné žádné údaje týkající se karcinogenity a mutagenity reprodukční či vývojové toxicity.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Chlorid sodný (NaCl)
Mannitol
Kyselina citronová
Hydroxid sodný (NaOH) (pro úpravu pH)
Kyselina chlorovodíková (HCl) (pro úpravu pH)
Voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek míchán s jinými léčivými přípravky.
6.3 Doba použitelnosti
2 roky při uchovávání v mrazničce (-20 °C ± 5 °C).
Po rozmrazení
Neotevřená lahvička v původním obale a chráněná před světlem se může uchovávat v chladničce (2 °C - 8 °C) po dobu až 1 týdne. Před umístěním do chladničky je třeba vypočítat nové datum použitelnosti a zaznamenat jej na obalu.
Po vyjmutí z mrazničky nebo chladničky smí být léčivý přípravek uchováván při teplotě do 25 °C po dobu až 8 hodin. Po uplynutí této doby se přípravek musí použít nebo zlikvidovat.
Nezmrazujte lahvičku, pokud již byla jednou rozmrazena.
Po otevření
Z mikrobiologického hlediska je nutno přípravek použít okamžitě po otevření.
Po jednorázovém použití se musí lahvička a nepoužitá část roztoku zlikvidovat.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v mrazničce (-20 °C ± 5 °C).
Podmínky uchovávání tohoto léčivého přípravku po jeho rozmrazení/otevření jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
0,3 ml roztoku v injekční lahvičce (skleněné, třída I) uzavřené zátkou z chlorbutylové pryže a modrým polypropylenovým odtrhávacím víčkem. Balení obsahuje 1 lahvičku.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Lahvičky jsou určeny jen pro jedno použití.
JETREA 0,375 mg/0,3 ml injekční roztok je přípravek připravený k použití a další ředění není třeba. Podat se má pouze 0,1 ml z celkového objemu 0,3 ml roztoku v lahvičce. Nadbytečný objem se má před podáním injekce vytlačit tak, aby byla podána jedna dávka 0,1 ml obsahující 0,125 mg okriplasminu.
Pokyny k použití
1. Vyjměte lahvičku z mrazničky a nechejte ji rozmrazit při pokojové teplotě (trvá to asi 2 minuty).
2. Po úplném rozmrazení odstraňte z lahvičky modré ochranné polypropylenové odtrhávací víčko.
3. Dezinfikujte víčko lahvičky otřením tamponem s alkoholem.
4. Vizuálně zkontrolujte lahvičku, zda neobsahuje viditelné částice. Smí se použít výhradně čirý roztok bez viditelných částic.
5. Aseptickou technikou aspirujte všechen roztok za použití vhodné sterilní jehly (lahvičku poněkud nakloňte, abyste usnadnili aspiraci) a po aspiraci obsahu lahvičky jehlu zlikvidujte. Tuto jehlu nepoužívejte k intravitreální injekci.
6. Mí sto původní j ehly nasaďte vhodnou sterilní j ehlu a pečlivě vytlačte nadbytečný roztok pomalým stlačováním pístu tak, aby se konec pístu vyrovnal se značkou s 0,1 ml na stříkačce (odpovídá 0,125 mg okriplasminu).
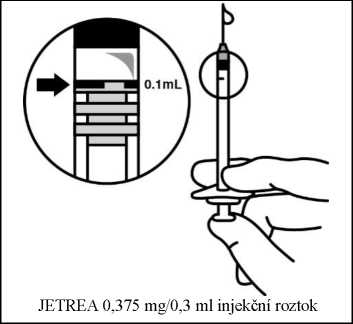
7. Injikujte 0,1 ml roztoku bezodkladně do středu sklivce.
8. Po jednorázovém použití zlikvidujte lahvičku i veškerý nepoužitý roztok.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
ThromboGenics NV Gaston Geenslaan 1 B-3001 Leuven Belgie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/13/819/002
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 24. dubna 2015
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A. VÝROBCE(I) BIOLOGICKY AKTIVNÍ(CH) LÁTKY (LÁTEK) A VYROBCE(I) ZODPOVĚDNÝ(Í) ZA PROPUŠTĚNÍ SARZE
B. PODMÍNKY NEBO OMEZENÍ V SOUVISLOSTI S DODÁVKOU A POUŽITÍM
C. OSTATNÍ PODMÍNKY A POŽADAVKY V SOUVISLOSTI S ROZHODNUTÍM O REGISTRACI
D. PODMÍNKY NEBO OMEZENÍ V SOUVISLOSTI S BEZPEČNOSTÍ A ÚČINNOSTÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY / BIOLOGICKÝCH LÉČIVÝCH LÁTEK A VÝROBCEODPOVĚDNÝ/ VÝROBCI ODPOVĚDNÍ ZA PROPUŠTĚNÍ ŠARŽÍ
Název a adresa výrobcebiologické léčivé látky/biologických léčivých látek
FUJIFILM DIOSYNTH BIOTECHNOLOGIES UK LIMITED
Belasis Avenue
Billingham, Cleveland
TS23 1LH
Velká Británie
Název a adresa výrobce odpovědného/výrobců odpovědných za propuštění šarží
ThromboGenics NV Gaston Geenslaan 1 B-3001 Leuven BELGIE
S.A. Alcon-Couvreur N.V.
Rijksweg 14 B-2870 Puurs Belgie
V tištěné příbalové informaci k léčivému přípravku musí být uveden název a adresa výrobce odpovědného za propouštění dané šarže.
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis (viz Příloha I: Souhrn údajů o přípravku, bod 4.2).
C. OSTATNÍ PODMÍNKY A POŽADAVKY V SOUVISLOSTI S ROZHODNUTÍM O REGISTRACI
• Pravidelně aktualizované zprávy o bezpečnosti (PSURs)
Držitel rozhodnutí o registraci předloží první pravidelně aktualizovanou zprávu o bezpečnosti pro tento léčivý přípravek 6 měsíců od jeho registrace. Držitel rozhodnutí o registraci dále předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
• Další opatření v souvislosti s minimalizací rizik
Před uvedením na trh v jednotlivých členských státech musí držitel rozhodnutí o registraci dohodnout s národní kompetentní autoritou příslušný edukační program.
MAH musí zajistit, po diskusi a dohodě s národní kompetentní autoritou v každém členském státě, ve kterém bude přípravek JETREA obchodován, a to v době uvedení na trh a bezprostředně po uvedení na trh, že všichni zdravotníci, u kterých se předpokládá, že budou přípravek JETREA používat budou mít k dispozici následující dokumenty:
• Souhrn údajů o přípravku (SmPC)
• Soubor informací pro pacienty
Soubor informací pro pacienta musí být k dispozici v tištěném a audio-formátu a musí obsahovat následující základní části:
• Příbalovou informaci
• Jak se připravit pro léčbu přípravkem Jetrea
• Jak je přípravek J etrea aplikován
• Jaké jsou další kroky po léčbě přípravkem Jetrea
• Hlavní známky a příznaky závažných nežádoucích účinků
• V jakých případech je nutné okamžitě vyhledat lékařskou pomoc
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
JETREA 0,5 mg/0,2 ml koncentrát pro injekční roztok ocriplasminum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna injekční lahvička obsahuje ocriplasminum 0,5 mg. Po naředění obsahuje 0,1 ml roztoku ocriplasminum 0,125 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Mannitol, kyselina citronová, hydroxid sodný, voda na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Koncentrát pro injekční roztok 1 injekční lahvička
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Pouze na jedno použití.
Před použitím si přečtěte příbalovou informaci. Intravitreální podání po naředění.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
Použijte okamžitě po naředění.
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v mrazničce.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
ThromboGenics NV Gaston Geenslaan 1 B-3001 Leuven Belgie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/13/819/001
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Nevyžaduje se - odůvodnění přijato.
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
JETREA 0,5 mg/0,2 ml sterilní koncentrát ocriplasminum
Intravitreální podání po naředění
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
JETREA 0,375 mg/0,3 ml injekční roztok ocriplasminum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna injekční lahvička obsahuje ocriplasminum 0,375 mg v 0,3 ml roztoku (1,25 mg/ml). To představuje použitelné množství pro jednu dávku 0,1 ml obsahující ocriplasminum 0,125 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Chlorid sodný, mannitol, kyselina citronová, hydroxid sodný, kyselina chlorovodíková, voda na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Injekční roztok 1 injekční lahvička
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Připraveno k použití.
Pouze na jedno použití.
Před použitím si přečtěte příbalovou informaci. Intravitreální podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v mrazničce.
Neotevřená lahvička se po rozmražení může uchovávat v chladničce po dobu až 1 týdne. Rozmražený roztok použijte do-----/-----/-----
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
ThromboGenics NV Gaston Geenslaan 1 B-3001 Leuven Belgie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/13/819/002
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Nevyžaduje se - odůvodnění přijato.
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
JETREA 0,375 mg/0,3 ml injekce
ocriplasminum
Intravitreální podání
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
B. PŘÍBALOVÁ INFORMACE
Příbalová informace:Informace pro pacienta
JETREA 0,5 mg/0,2 ml koncentrát pro injekční roztok
Ocriplasminum
'VTento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než Vám tento přípravek bude podán, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Jetrea a k čemu se používá
2. Čemu musíte věnovat pozornost, než Vám bude přípravek Jetrea podán
3. Jak se přípravek Jetrea podává
4. Možné nežádoucí účinky
5. Jak přípravek Jetrea uchovávat
6. Obsah balení a další informace
1 Co je přípravek Jetrea a k čemu se používá
Přípravek Jetrea obsahuje léčivou látku okriplasmin.
Přípravek Jetrea se používá k léčbě dospělých pacientů s onemocněním oka zvaným vitreomakulární trakce (VMT) včetně stavů, kdy je toto onemocnění spojeno s malou dírou v makule (tj. centrální části světločivné vrstvy zadní části oka).
VMT je způsobena tahem vznikajícím v důsledku pevného připojení sklivce (hmoty podobné želatině, která se nachází v zadní části oka) k makule. Makula umožňuje centrální vidění, které je zapotřebí pro běžné denní činnosti jako je řízení, čtení a rozpoznávání obličejů. VMT může vést k příznakům jako je zkreslené nebo zhoršené vidění. Když se toto onemocnění zhoršuje, v důsledku tahu může vzniknout v makule díra (tzv. makulární díra).
Přípravek Jetrea působí tak, že odděluje sklivec od makuly a pomáhá uzavřít případnou makulární díru, což může zmírnit příznaky spojené s VMT.
2 Čemu musíte věnovat pozornost, než Vám bude přípravek Jetrea podán Přípravek Jetrea Vám nesmí být podán v následujících případech
- Jestliže jste alergický(á) na okriplasmin nebo na kteroukoli jinou složku tohoto přípravku (uvedenou v bodě 6).
- Jestliže máte zánět v oku nebo jeho okolí, případně máte podezření na takový zánět. Upozornění a opatření
Než Vám bude přípravek Jetrea podán, poraďte se se svým lékařem nebo oftalmologem (očním lékařem).
Přípravek Jetrea se aplikuje injekcí do oka. Váš lékař nebo oftalmolog Vás bude sledovat pro případ, že by se u Vás po injekci rozvinula infekce nebo jakékoli jiné komplikace. Pokud se u Vás po injekci přípravku Jetrea objeví jakékoli oční příznaky popsané v bodě 4 „Možné nežádoucí účinky “, musíte ihned kontaktovat svého lékaře nebo oftalmologa.
Přípravek Jetrea Vám nebude podán současně do obou očí.
Přípravek Jetrea Vám nebude podán do stejného oka více než jedenkrát.
Máte-li nebo jste měl(a) nějaké oční onemocnění nebo léčbu, řekněte to svému lékaři nebo oftalmologovi. Váš lékař nebo oftalmolog rozhodne, zda je pro Vás léčba přípravkem Jetrea vhodná.
Děti a dospívající
Nepředpokládá se relevantní použití přípravku Jetrea u dětí a dospívajících mladších 18 let. Použití přípravku Jetrea se proto u této skupiny pacientů nedoporučuje.
Další léčivé přípravky a přípravek Jetrea
Informujte svého lékaře nebo oftalmologa o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat. Informujte svého lékaře nebo oftalmologa, pokud Vám byla v nedávné době aplikována injekce do oka. Tato informace bude zohledněna při rozhodování, zda a kdy Vám může být do stejného oka aplikována injekce přípravku Jetrea.
Těhotenství a kojení
S použitím přípravku Jetrea u těhotných nebo kojících žen nejsou žádné zkušenosti. Přípravek Jetrea se nemá používat během těhotenství nebo kojení, pokud to Váš lékař nebo oftalmolog nepovažuje za nezbytně nutné. Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo oftalmologem dříve, než Vám tento přípravek bude podán.
Řízení dopravních prostředků a obsluha strojů
Po léčbě přípravkem Jetrea můžete po omezenou dobu pociťovat určité zhoršení zraku. Dojde-li k tomu, neřiďte vozidla ani neobsluhujte stroje, dokud se Váš zrak opět nezlepší.
3 Jak se přípravek Jetrea podává
Přípravek Jetrea musí podávat kvalifikovaný oftalmolog (oční specialista), který má zkušenosti s aplikací injekcí do oka.
Přípravek Jetrea se podává jako jednorázová injekce do postiženého oka. Doporučená dávka přípravku je 0,125 mg.
Váš lékař nebo oftalmolog Vás může požádat, abyste si aplikoval(a) antibiotické oční kapky před a po injekci za účelem prevence vzniku případné oční infekce.
V den aplikace injekce Vám lékař nebo oftalmolog podá antimikrobiální oční kapky a pečlivě Vám očistí oko a víčko, aby zabránil infekci. Váš lékař nebo oftalmolog Vám také podá lokální anestetika, aby zabránil bolesti při injekci.
Po injekci bude Váš lékař nebo oftalmolog sledovat Váš zrak.
Máte-li jakékoli otázky, týkající se použití tohoto přípravku, zeptejte se svého lékaře nebo oftalmologa.
4 Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Pokud se u Vás po injekci přípravku Jetrea rozvine kterýkoli z následujících nežádoucích účinků, kontaktujte neprodleně svého lékaře nebo oftalmologa. Váš lékař nebo oftalmolog Vás bude sledovat, a bude-li třeba, provede příslušná nápravná opatření.
- U 1 z 10 pacientů bylo v období do jednoho týdne po injekci přípravku Jetrea hlášeno výrazné zhoršení zraku. Jedná se o všeobecně vratný stav, který obvykle vymizí do dvou týdnů.
- Příznaky, jako je bolest oka, zhoršující se zarudnutí oka, výrazně rozmazané nebo zhoršené vidění, zvýšená citlivost na světlo nebo zvýšený počet tmavých plovoucích skvrnek v zorném poli (zákalků) se rovněž vyskytují až u 1 z 10 pacientů a mohou být známkami infekce, krvácení, odchlípení nebo roztržení sítnice nebo zvýšení tlaku v léčeném oku
- Příznaky jako fluktuace zraku, dvojité vidění, bolest hlavy, barevné kruhy kolem zdrojů světla, pocit na zvracení nebo zvracení byly hlášeny až u 1 ze 100 pacientů a mohou být známkou vychýlení nebo nestability oční čočky oproti své normální poloze.
Pokud se u Vás objeví kterýkoli z dalších níže uvedených nežádoucích účinků, sdělte to svému lékaři nebo oftalmologovi:
Velmi časté nežádoucí účinky (mohou se vyskytnout u více než 1 pacienta z 10):
- krvácení na povrchu oka
Časté nežádoucí účinky (mohou se vyskytnout až u 1 pacienta z 10):
- zhoršení zraku
- krvácení uvnitř oka
- slepá skvrna nebo slepá ploška v zorném poli
- zkreslené vidění
- otok povrchu oka
- otok očního víčka
- zánět oka
- záblesky světla uvnitř oka
- podráždění povrchu oka
- suché oko
- pocit přítomnosti cizího tělíska v oku
- svědění oka
- nepříjemný pocit v oku
- změny vnímání barev
Méně časté nežádoucí účinky (mohou se vyskytnout až u 1 pacienta ze 100):
- obtíže při vidění v noci nebo při slabém osvětlení
- porucha reakce oka na světlo, která může zvýšit citlivost na světlo (porucha reflexu zornice)
- zhoršené vidění v části zorného pole
- hromadění krve v přední části oka
- abnormální stažlivost zornice (černé části v centru oka)
- odlišná velikost zornic
- poškrábaná nebo škrábnutá rohovka (průsvitná vrstva kryjící přední část oka)
Některé nežádoucí účinky (např. záblesky, zákalky) mohou být v některých případech pozorovány u neléčeného oka.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo oftalmologovi. Stejně postupujte i v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
Jak přípravek Jetrea uchovávat
5
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Informace o uchovávání a době, za kterou se má přípravek Jetrea použít poté, co byl rozmražen a naředěn, jsou popsány v části určené pouze pro zdravotnické pracovníky.
Váš oční specialista/lékař nebo lékárník je zodpovědný za uchovávání tohoto přípravku a za správnou likvidaci nepoužitého roztoku.
6 Obsah balení a další informace Co přípravek Jetrea obsahuje
- Léčivou látkou je ocriplasminum. Jedna injekční lahvička přípravku Jetrea obsahuje 0,5 mg ocriplasminu v 0,2 ml roztoku. Po naředění 0,2 ml injekčního roztoku chloridu sodného obsahuje 0,1 ml naředěného roztoku 0,125 mg ocriplasminu.
- Dalšími složkami jsou mannitol, kyselina citronová, hydroxid sodný (NaOH) (pro úpravu pH) a voda na injekci.
Jak přípravek Jetrea vypadá a co obsahuje toto balení
Přípravek Jetrea je koncentrát pro injekční roztok v injekční lahvičce. Koncentrát je čirý a bezbarvý. Jedno balení obsahuje jednu injekční lahvičku.
Držitel rozhodnutí o registraci
ThromboGenics NV Gaston Geenslaan 1 B-3001 Leuven Belgie
Výrobce
S.A. Alcon-Couvreur N.V. ThromboGenics NV
Rijksweg 14 Gaston Geenslaan 1
B-2870 Puurs B-3001 Leuven
Belgie Belgie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgie/Belgique/Belgien Latvija
Luxembourg/Luxemburg Alcon Pharmaceuticals Ltd
Alcon NV ® + 371 67 321 121
@ + 32 (0)2 754 32 10 (Belgiě/Belgique/Belgien)
Česká republika
Alcon Pharmaceuticals (Czech Republic) s.r.o. ® + 420 225 775 111
Danmark
Alcon Nordic A/S ® + 45 3636 4300
Magyarország
Alcon Hungária Gyógyszerkereskedelmi Kft. ® + 36-1-463-9080
Nederland
Alcon Nederland BV ® + 31 (0) 183 654321
|
Deutschland |
Norge |
|
Alcon Pharma GmbH |
Alcon Nordic A/S |
|
® + 49 (0)761 1304-0 |
® + 45 3636 4300 |
|
EkXúSa/Kúnpoq |
Osterreich |
|
AAxov Aapnopáxoprg |
Alcon Ophthalmika GmbH |
|
EAAág AEBE ® + 30 210 68 78 300 (EMáSa) |
® + 43 (0)1 596 69 70 |
|
Eesti |
Polska |
|
Alcon Pharmaceuticals Eesti filiaal |
Alcon Polska Sp. z o.o. |
|
® + 372 6 313 214 |
® + 48 22 820 3450 |
|
Espaňa |
Portugal |
|
Alcon Cusí, S.A. |
Alcon Portugal - |
|
® + 34 93 497 7000 |
Produtos e Equipamentos Oftalmológicos, Lda. ® + 351 214 400 300 |
|
France |
Románia |
|
Laboratoires Alcon |
S.C. Alcon Romania S.R.L. |
|
® + 33 (0)1 47 10 47 10 |
® + 40 21 203 93 24 |
|
Hrvatska |
Slovenija |
|
Alcon Farmaceutika d.o.o. |
Alcon d.o.o. |
|
® + 385 1 4611 988 |
® + 386 1 422 5280 |
|
Ireland |
Slovenská republika |
|
Malta |
Novartis Slovakia s.r.o. |
|
United Kingdom |
Alcon Division |
|
Alcon Laboratories (UK) Ltd ® + 44 (0) 345 266 9363 (United Kingdom) |
® + 421 2 5441 0378 |
|
Island |
Suomi/Finland |
|
Alcon Nordic A/S |
Alcon Nordic A/S |
|
® + 45 3636 4300 |
® + 45 3636 4300 |
|
Italia |
Sverige |
|
Alcon Italia S.p.A. |
Alcon Nordic A/S |
|
® + 39 02 81 80 31 |
® + 45 3636 4300 |
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu.
Následující informace jsou určeny pouze pro zdravotnické pracovníky:
Přípravu a podávání přípravku Jetrea smí provádět pouze kvalifikovaný oftalmolog se zkušenostmi s intravitreálními injekcemi. Diagnóza vitreomakulární trakce (VMT) má zahrnovat kompletní klinický obraz včetně pacientovy anamnézy, klinického vyšetření a použití adekvátních diagnostických metod, jako je např. optická koherentní tomografie (OCT).
Doporučená dávka je 0,125 mg (0,1 ml naředěného roztoku) jednorázově podaná intravitreální injekcí do postiženého oka. Jedna lahvička se smí použít pouze jednou a pouze k léčbě jednoho oka. Léčba druhého oka přípravkem JETREA není doporučena dříve než za 7 dní od podání první injekce do iniciálně léčeného oka z důvodu monitorování postinjekční reakce včetně rizika snížené zrakové ostrosti u léčeného oka. Opakované podávání do stejného oka se nedoporučuje.
Pokyny ke sledování po aplikaci injekce viz souhrn údajů o přípravku, bod 4.4.
Lahvička k jednorázovému intravitreálnímu podání.
Před zákrokem mohou být dle uvážení ošetřujícího oftalmologa podány antibiotické oční kapky.
Intravitreální injekci je nutno provádět v kontrolovaném aseptickém prostředí včetně nutnosti dezinfekce rukou operatéra, použití sterilních rukavic, sterilní roušky, sterilního rozvěrače víček (nebo ekvivalentní náhrady) a možnosti provedení sterilní paracentézy (pokud je vyžadována). Periokulární pokožka, oční víčko a povrch oka musí být dezinfikovány a před aplikací injekce je nutno podat adekvátní anestetikum a lokální širokospektrý mikrobicidní přípravek dle standardní léčebné praxe.
Injekční jehla se zasune 3,5-4,0 mm posteriorně k limbu do centra sklivce tak, aby směřovala do centra očního bulbu, a nikoli k horizontálnímu meridiánu. Objem injekce 0,1 ml se potom aplikuje do středu sklivce.
Při přípravě přípravku Jetrea pro intravitreální injekci postupujte podle následujících pokynů:
1. Vyjměte lahvičku z mrazničky a nechejte ji rozmrazit při pokojové teplotě (trvá to asi 2 minuty).
2. Po úplném rozmrazení odstraňte z lahvičky ochranné oranžové polypropylenové odtrhávací víčko (obrázek 1).
3. Dezinfikujte víčko lahvičky otřením tamponem s alkoholem (obrázek 2).
4. Aseptickou technikou nařeďte přidáním 0,2 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9 %) (sterilního, bez obsahu konzervantů a pufrů) do lahvičky s přípravkem Jetrea (obrázek 3) a jemně promíchejte kroužením lahvičkou, dokud se roztoky nesmísí (obrázek 4). Roztok k naředění se musí aspirovat z neotevřené nádobky, která se smí použít pouze jednou. Zbývající injekční roztok chloridu sodného 9 mg/ml (0,9 %) se musí zlikvidovat. Naředěný roztok má být bezprostředně po naředění použit, protože neobsahuje žádné konzervanty.
5. Vizuálně zkontrolujte lahvičku, zda neobsahuje viditelné částice. Smí se použít výhradně čirý roztok bez viditelných částic.
6. Aseptickou technikou aspirujte naředěný roztok za použití vhodné sterilní jehly (lahvičku poněkud nakloňte, abyste usnadnili aspiraci) (obrázek 5) a po aspiraci obsahu lahvičky jehlu zlikvidujte. Tuto jehlu nepoužívejte k intravitreální injekci.
7. Místo původní jehly nasaďte vhodnou sterilní jehlu a pečlivě vytlačte nadbytečný objem ze stříkačky pomalým stlačováním pístu tak, aby se konec pístu zastavil na značce 0,1 ml na stříkačce (což odpovídá 0,125 mg okriplasminu) (obrázek 6).
8. Injikujte 0,1 ml naředěného roztoku bezodkladně do středu sklivce, protože neobsahuje konzervanty.
9. Po jednorázovém použití zlikvidujte lahvičku i veškerý nepoužitý naředěný roztok.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
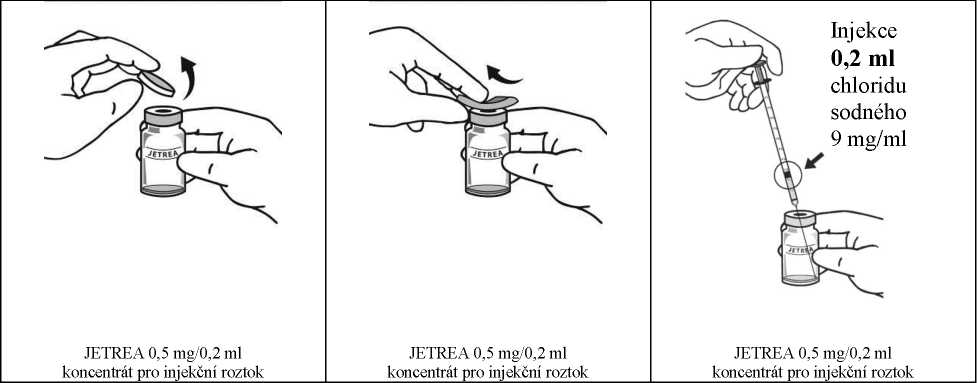
Obrázek 1
Obrázek 2
Obrázek 3
JETREA 0,5 mg/0,2 ml koncentrát pro injekční roztok
JETREA 0,5 mg/0,2 ml koncentrát pro injekční roztok
JETREA 0,5 mg/0,2 ml koncentrát pro injekční roztok
Obrázek 4
Obrázek 5
Informace o uchovávání
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na štítku a krabičce za „EXP“. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v mrazničce (-20°C±5°C).
Po rozmražení
Přípravek se má naředit a použít okamžitě. Chemická a fyzikální stabilita neotevřeného přípravku, uchovávaného v původním obale a chráněného před světlem, však byla prokázána po dobu až 8 hodin, pokud je přípravek uchováván při teplotě do 25 °C. Nezmrazujte lahvičku, pokud již byla jednou rozmrazena.
Po otevření/naředění
Z mikrobiologického hlediska je nutno přípravek použít okamžitě po otevření/naředění.
Po jednorázovém použití se musí lahvička a nepoužitá část naředěného roztoku zlikvidovat.
Příbalová informace: Informace pro pacienta
JETREA 0,375 mg/0,3 ml injekční roztok
Ocriplasminum
'VTento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než Vám tento přípravek bude podán, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Jetrea a k čemu se používá
2. Čemu musíte věnovat pozornost, než Vám bude přípravek Jetrea podán
3. Jak se přípravek Jetrea podává
4. Možné nežádoucí účinky
5. Jak přípravek Jetrea uchovávat
6. Obsah balení a další informace
1 Co je přípravek Jetrea a k čemu se používá
Přípravek Jetrea obsahuje léčivou látku okriplasmin.
Přípravek Jetrea se používá k léčbě dospělých pacientů s onemocněním oka zvaným vitreomakulární trakce (VMT) včetně stavů, kdy je toto onemocnění spojeno s malou dírou v makule (tj. centrální části světločivné vrstvy zadní části oka).
VMT je způsobena tahem vznikajícím v důsledku pevného připojení sklivce (hmoty podobné želatině, která se nachází v zadní části oka) k makule. Makula umožňuje centrální vidění, které je zapotřebí pro běžné denní činnosti jako je řízení, čtení a rozpoznávání obličejů. VMT může vést k příznakům jako je zkreslené nebo zhoršené vidění. Když se toto onemocnění zhoršuje, v důsledku tahu může vzniknout v makule díra (tzv. makulární díra).
Přípravek Jetrea působí tak, že odděluje sklivec od makuly a pomáhá uzavřít případnou makulární díru, což může zmírnit příznaky spojené s VMT.
2 Čemu musíte věnovat pozornost, než Vám bude přípravek Jetrea podán Přípravek Jetrea Vám nesmí být podán v následujících případech
- Jestliže jste alergický(á) na okriplasmin nebo na kteroukoli jinou složku tohoto přípravku (uvedenou v bodě 6).
- Jestliže máte zánět v oku nebo jeho okolí, případně máte podezření na takový zánět. Upozornění a opatření
Než Vám bude přípravek Jetrea podán, poraďte se se svým lékařem nebo oftalmologem (očním lékařem).
Přípravek Jetrea se aplikuje injekcí do oka. Váš lékař nebo oftalmolog Vás bude sledovat pro případ, že by se u Vás po injekci rozvinula infekce nebo jakékoli jiné komplikace. Pokud se u Vás po injekci přípravku Jetrea objeví jakékoli oční příznaky popsané v bodě 4 „Možné nežádoucí účinky “, musíte ihned kontaktovat svého lékaře nebo oftalmologa.
Přípravek Jetrea Vám nebude podán současně do obou očí.
Přípravek Jetrea Vám nebude podán do stejného oka více než jedenkrát.
Máte-li nebo jste měl(a) nějaké oční onemocnění nebo léčbu, řekněte to svému lékaři nebo oftalmologovi. Váš lékař nebo oftalmolog rozhodne, zda je pro Vás léčba přípravkem Jetrea vhodná.
Děti a dospívající
Nepředpokládá se relevantní použití přípravku Jetrea u dětí a dospívajících mladších 18 let. Použití přípravku Jetrea se proto u této skupiny pacientů nedoporučuje.
Další léčivé přípravky a přípravek Jetrea
Informujte svého lékaře nebo oftalmologa o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat. Informujte svého lékaře nebo oftalmologa, pokud Vám byla v nedávné době aplikována injekce do oka. Tato informace bude zohledněna při rozhodování, zda a kdy Vám může být do stejného oka aplikována injekce přípravku Jetrea.
Těhotenství a kojení
S použitím přípravku Jetrea u těhotných nebo kojících žen nejsou žádné zkušenosti. Přípravek Jetrea se nemá používat během těhotenství nebo kojení, pokud to Váš lékař nebo oftalmolog nepovažuje za nezbytně nutné. Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo oftalmologem dříve, než Vám tento přípravek bude podán.
Řízení dopravních prostředků a obsluha strojů
Po léčbě přípravkem Jetrea můžete po omezenou dobu pociťovat určité zhoršení zraku. Dojde-li k tomu, neřiďte vozidla ani neobsluhujte stroje, dokud se Váš zrak opět nezlepší.
3 Jak se přípravek Jetrea podává
Přípravek Jetrea musí podávat kvalifikovaný oftalmolog (oční specialista), který má zkušenosti s aplikací injekcí do oka.
Přípravek Jetrea se podává jako jednorázová injekce do postiženého oka. Doporučená dávka přípravku je 0,125 mg.
Váš lékař nebo oftalmolog Vás může požádat, abyste si aplikoval(a) antibiotické oční kapky před a po injekci za účelem prevence vzniku případné oční infekce.
V den aplikace injekce Vám lékař nebo oftalmolog podá antimikrobiální oční kapky a pečlivě Vám očistí oko a víčko, aby zabránil infekci. Váš lékař nebo oftalmolog Vám také podá lokální anestetika, aby zabránil bolesti při injekci.
Po injekci bude Váš lékař nebo oftalmolog sledovat Váš zrak.
Máte-li jakékoli otázky, týkající se použití tohoto přípravku, zeptejte se svého lékaře nebo oftalmologa.
4 Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Pokud se u Vás po injekci přípravku Jetrea rozvine kterýkoli z následujících nežádoucích účinků, kontaktujte neprodleně svého lékaře nebo oftalmologa. Váš lékař nebo oftalmolog Vás bude sledovat, a bude-li třeba, provede příslušná nápravná opatření.
- U 1 z 10 pacientů bylo v období do jednoho týdne po injekci přípravku Jetrea hlášeno výrazné zhoršení zraku. Jedná se o všeobecně vratný stav, který obvykle vymizí do dvou týdnů.
- Příznaky, jako je bolest oka, zhoršující se zarudnutí oka, výrazně rozmazané nebo zhoršené vidění, zvýšená citlivost na světlo nebo zvýšený počet tmavých plovoucích skvrnek v zorném poli (zákalků) se rovněž vyskytují až u 1 z 10 pacientů a mohou být známkami infekce, krvácení, odchlípení nebo roztržení sítnice nebo zvýšení tlaku v léčeném oku
- Příznaky jako fluktuace zraku, dvojité vidění, bolest hlavy, barevné kruhy kolem zdrojů světla, pocit na zvracení nebo zvracení byly hlášeny až u 1 ze 100 pacientů a mohou být známkou vychýlení nebo nestability oční čočky oproti své normální poloze.
Pokud se u Vás objeví kterýkoli z dalších níže uvedených nežádoucích účinků, sdělte to svému lékaři nebo oftalmologovi:
Velmi časté nežádoucí účinky (mohou se vyskytnout u více než 1 pacienta z 10):
- krvácení na povrchu oka
Časté nežádoucí účinky (mohou se vyskytnout až u 1 pacienta z 10):
- zhoršení zraku
- krvácení uvnitř oka
- slepá skvrna nebo slepá ploška v zorném poli
- zkreslené vidění
- otok povrchu oka
- otok očního víčka
- zánět oka
- záblesky světla uvnitř oka
- podráždění povrchu oka
- suché oko
- pocit přítomnosti cizího tělíska v oku
- svědění oka
- nepříjemný pocit v oku
- změny vnímání barev
Méně časté nežádoucí účinky (mohou se vyskytnout až u 1 pacienta ze 100):
- obtíže při vidění v noci nebo při slabém osvětlení
- porucha reakce oka na světlo, která může zvýšit citlivost na světlo (porucha reflexu zornice)
- zhoršené vidění v části zorného pole
- hromadění krve v přední části oka
- abnormální stažlivost zornice (černé části v centru oka)
- odlišná velikost zornic
- poškrábaná nebo škrábnutá rohovka (průsvitná vrstva kryjící přední část oka)
Některé nežádoucí účinky (např. záblesky, zákalky) mohou být v některých případech pozorovány u neléčeného oka.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo oftalmologovi. Stejně postupujte i v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
Jak přípravek Jetrea uchovávat
5
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Informace o uchovávání a době, za kterou se má přípravek Jetrea použít poté, co byl rozmrazen, jsou popsány v části určené pouze pro zdravotnické pracovníky.
Váš oční specialista/lékař nebo lékárník je zodpovědný za uchovávání tohoto přípravku a za správnou likvidaci nepoužitého roztoku.
6 Obsah balení a další informace Co přípravek Jetrea obsahuje
- Léčivou látkou je ocriplasminum. Jedna injekční lahvička přípravku Jetrea obsahuje ocriplasminum 0,375 mg v 0,3 ml roztoku.
- Dalšími složkami jsou chlorid sodný (NaCl), mannitol, kyselina citronová, hydroxid sodný (NaOH) (pro úpravu pH), kyselina chlorovodíková (pro úpravu pH) a voda na injekci.
Jak přípravek Jetrea vypadá a co obsahuje toto balení
Přípravek Jetrea je injekční roztok v injekční lahvičce. Roztok je čirý a bezbarvý. Jedno balení obsahuje jednu injekční lahvičku.
Držitel rozhodnutí o registraci
ThromboGenics NV Gaston Geenslaan 1 B-3001 Leuven Belgie
Výrobce
S.A. Alcon-Couvreur N.V. ThromboGenics NV
Rijksweg 14 Gaston Geenslaan 1
B-2870 Puurs B-3001 Leuven
Belgie Belgie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgie/Belgique/Belgien Latvija
Luxembourg/Luxemburg Alcon Pharmaceuticals Ltd
Alcon NV ® + 371 67 321 121
@ + 32 (0)2 754 32 10 (Belgiě/Belgique/Belgien)
Bt^rapnn
Ahkoh Etarapna EOOfl ® + 359 2 950 15 65
Danmark
Alcon Nordic A/S ® + 45 3636 4300
Nederland
Alcon Nederland BV ® + 31 (0) 183 654321
|
Deutschland |
Norge |
|
Alcon Pharma GmbH |
Alcon Nordic A/S |
|
® + 49 (0)761 1304-0 |
® + 45 3636 4300 |
|
EkXáSa/Kúrcpoq |
Osterreich |
|
AAxov Aapnopáxoprg |
Alcon Ophthalmika GmbH |
|
EAAág AEBE ® + 30 210 68 78 300 (EMáSa) |
® + 43 (0)1 596 69 70 |
|
Eesti |
Polska |
|
Alcon Pharmaceuticals Eesti filiaal |
Alcon Polska Sp. z o.o. |
|
® + 372 6 313 214 |
® + 48 22 820 3450 |
|
Espaňa |
Portugal |
|
Alcon Cusí, S.A. |
Alcon Portugal - |
|
® + 34 93 497 7000 |
Produtos e Equipamentos Oftalmológicos, Lda. ® + 351 214 400 300 |
|
France |
Románia |
|
Laboratoires Alcon |
S.C. Alcon Romania S.R.L. |
|
® + 33 (0)1 47 10 47 10 |
® + 40 21 203 93 24 |
|
Hrvatska |
Slovenija |
|
Alcon Farmaceutika d.o.o. |
Alcon d.o.o. |
|
® + 385 1 4611 988 |
® + 386 1 422 5280 |
|
Ireland |
Slovenská republika |
|
Malta |
Novartis Slovakia s.r.o. |
|
United Kingdom |
Alcon Division |
|
Alcon Laboratories (UK) Ltd ® + 44 (0) 345 266 9363 (United Kingdom) |
® + 421 2 5441 0378 |
|
Ísland |
Suomi/Finland |
|
Alcon Nordic A/S |
Alcon Nordic A/S |
|
® + 45 3636 4300 |
® + 45 3636 4300 |
|
Italia |
Sverige |
|
Alcon Italia S.p.A. |
Alcon Nordic A/S |
|
® + 39 02 81 80 31 |
® + 45 3636 4300 |
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu.
Následující informace jsou určeny pouze pro zdravotnické pracovníky:
Podávání přípravku Jetrea smí provádět pouze kvalifikovaný oftalmolog se zkušenostmi s intravitreálními injekcemi. Diagnóza vitreomakulární trakce (VMT) má zahrnovat kompletní klinický obraz včetně pacientovy anamnézy, klinického vyšetření a použití adekvátních diagnostických metod, jako je např. optická koherentní tomografie (OCT).
JETREA 0,375 mg/0,3 ml injekční roztok je přípravek připravený k použití a další ředění není třeba. Doporučená dávka je 0,125 mg v 0,1 ml roztoku jednorázově podaná intravitreální injekcí do postiženého oka. Jedna lahvička se smí použít pouze jednou a pouze k léčbě jednoho oka. Léčba druhého oka přípravkem JETREA není doporučena dříve než za 7 dní od podání první injekce do iniciálně léčeného oka z důvodu monitorování postinjekční reakce včetně rizika snížené zrakové ostrosti u léčeného oka. Opakované podávání do stejného oka se nedoporučuje.
Pokyny ke sledování po aplikaci injekce viz souhrn údajů o přípravku, bod 4.4.
Lahvička k jednorázovému intravitreálnímu podání.
Před zákrokem mohou být dle uvážení ošetřujícího oftalmologa podány antibiotické oční kapky.
Intravitreální injekci je nutno provádět v kontrolovaném aseptickém prostředí včetně nutnosti dezinfekce rukou operatéra, použití sterilních rukavic, sterilní roušky, sterilního rozvěrače víček (nebo ekvivalentní náhrady) a možnosti provedení sterilní paracentézy (pokud je vyžadována). Periokulární pokožka, oční víčko a povrch oka musí být dezinfikovány a před aplikací injekce je nutno podat adekvátní anestetikum a lokální širokospektrý mikrobicidní přípravek dle standardní léčebné praxe.
Podat se má pouze 0,1 ml z celkového objemu 0,3 ml roztoku v lahvičce. Nadbytečný objem se má před podáním injekce vytlačit tak, aby byla podána jedna dávka 0,1 ml obsahující 0,125 mg okriplasminu.
Injekční jehla se zasune 3,5-4,0 mm posteriorně k limbu do centra sklivce tak, aby směřovala do centra očního bulbu, a nikoli k horizontálnímu meridiánu. Objem injekce 0,1 ml se potom aplikuje do středu sklivce.
Pokyny k použití
1. Vyjměte lahvičku z mrazničky a nechejte ji rozmrazit při pokojové teplotě (trvá to asi 2 minuty).
2. Po úplném rozmrazení odstraňte z lahvičky modré ochranné polypropylenové odtrhávací víčko (obrázek 1).
3. Dezinfikujte víčko lahvičky otřením tamponem s alkoholem (obrázek 2).
4. Vizuálně zkontrolujte lahvičku, zda neobsahuje viditelné částice. Smí se použít výhradně čirý roztok bez viditelných částic.
5. Aseptickou technikou aspirujte roztok za použití vhodné sterilní jehly (lahvičku poněkud nakloňte, abyste usnadnili aspiraci) (obrázek 3) a po aspiraci obsahu lahvičky jehlu zlikvidujte. Tuto jehlu nepoužívejte k intravitreální injekci.
6. Místo původní jehly nasaďte vhodnou sterilní jehlu a pečlivě vytlačte nadbytečný objem ze stříkačky pomalým stlačováním pístu tak, aby se konec pístu zastavil na značce 0,1 ml na stříkačce (což odpovídá 0,125 mg okriplasminu) (obrázek 4).
7. Injikujte 0,1 ml roztoku bezodkladně do středu sklivce.
8. Po jednorázovém použití zlikvidujte lahvičku i veškerý nepoužitý roztok.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
JETREA 0,375 mg/0,3 ml injekční roztok
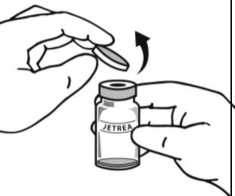
JETREA 0,375 mg/0,3 ml injekční roztok
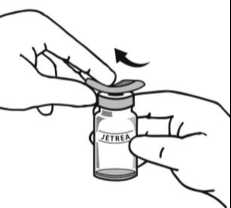
JETREA 0,375 mg/0,3 ml injekční roztok
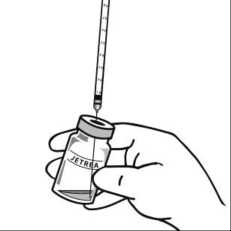
JETREA 0,375 mg/0,3 ml injekční roztok
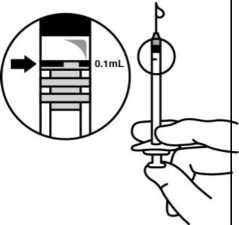
Obrázek 1 Obrázek 2 Obrázek 3 Obrázek 4
Informace o uchovávání
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na štítku a krabičce za „EXP“ Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v mrazničce (-20 °C±5 °C).
Po rozmražení
Neotevřená lahvička v původním obale a chráněná před světlem se může uchovávat v chladničce (2 °C - 8 °C) po dobu až 1 týdne. Před umístěním do chladničky je třeba vypočítat nové datum použitelnosti a zaznamenat jej na obalu.
Po vyjmutí z mrazničky nebo chladničky smí být léčivý přípravek uchováván při teplotě do 25 °C po dobu až 8 hodin. Po uplynutí této doby se přípravek musí použít nebo zlikvidovat.
Po otevření
Z mikrobiologického hlediska je nutno přípravek použít okamžitě po otevření.
Po jednorázovém použití se musí lahvička a nepoužitá část roztoku zlikvidovat.
PŘÍLOHA IV
VĚDECKÉ ZÁVĚRY A ZDŮVODNĚNÍ ZMĚNY PODMÍNEK ROZHODNUTÍ O REGISTRACI
Vědecké závěry
S ohledem na hodnotící zprávu výboru PRAC týkající se pravidelně aktualizovaných zpráv o bezpečnosti (PSUR) okriplasminu dospěl výbor CHMP k těmto vědeckým závěrům:
Celkem bylo obdrženo 29 spontánních hlášení s 48 případy „vizuálních příznaků pozorovaných v párovém oku“. Související preferované termíny (PT) zahrnovaly: oslnění, metamorfopsie, fotofobie, fotopsie, rozmazané vidění, snížená zraková ostrost, porucha zraku, sklivcové zákalky, tranzientní slepota a unilaterální slepota. U 26/48 nežádoucích účinků (54 %) byl výsledek „odeznělo“ nebo „odeznívá“, u 8 nežádoucích účinků (17 %) „neodeznělo“ a u zbývajících účinků není výsledek znám. PRAC doporučil aktualizovat informace o přípravku tak, aby zdravotničtí pracovníci a pacienti byli
0 těchto potenciálních nežádoucích účincích informováni. Celkem bylo spontánně hlášeno 29 případů „poruchy pupilárního reflexu“ (26 závažných) a 9 případů bylo pozorováno v neintervenčních studiích a klinických hodnoceních (4 závažné). Držitel rozhodnutí o registraci vysvětluje tyto nežádoucí účinky jako následek „změn fotoreceptorů“, které jsou zmíněny níže. Držitel rozhodnutí o registraci navrhuje, aby byla „porucha pupilárního reflexu“ uvedena v bodě 4.8 souhrnu údajů o přípravku, což je považováno za odpovídající. Celkem bylo obdrženo 21 spontánních hlášení s 24 případy (7 závažných a 17 nezávažných) „šerosleposti“. Pět hlášených případů bylo nezávažných a 16 závažných.
U 4 případů byla hlášena doba do nástupu nežádoucího účinku (TTO), která se pohybovala od 0 do 30 dní. U 3 nežádoucích účinků (12,5 %) byl výsledek „odeznělo“, u 1 účinku (4,2 %) „odeznívá s následky“, u 1 účinku (4,2 %) „odeznívá“, u 8 účinků (33,3 %) byl výsledek „neodeznělo“ a u 11 účinků (45,8 %) „není známo“. Kromě toho byl v neintervenční studii INJECT hlášen
1 nezávažný případ „potíží s adaptací na světlo“ (neodeznělo). Držitel rozhodnutí o registraci považuje tyto příhody za klinický projev „změny fotoreceptorů“ a navrhuje aktualizovat informace o přípravku. Je dohodnuto, že „šeroslepost“ bude uvedena v bodě 4.8 souhrn údajů o přípravku pro okriplasmin.
Z celkem 465 použivatelů okriplasminu v pivotních studiích fáze III 19 pacientů (4,1 %) nahlásilo nejméně 1 epizodu s PT „makulární edém“. Tyto epizody zahrnovaly 7 případů (1,5 %) nekomplikovaného makulárního edému a 12 případů (2,6 %) cystoidního makulárního edému.
Z 12 případů cystoidního makulárního edému bylo v 5 případech hlášeno „odeznělo“ a v 7 případech, že edém na konci studie přetrvává. Cystoidní makulární edém je považován za závažnější podtyp makulárního edému; v současné době je v souhrnu údajů o přípravku uveden pouze „makulární edém“. Z toho důvodu bylo konstatováno, že v souvislosti s makulárním edémem má být „cystoidní makulární edém“ výslovně uveden v bodě 4.8 souhrnu údajů o přípravku.
Příbalová informace bude odpovídajícím způsobem aktualizována tak, aby odrážela změny v souhrnu údajů o přípravku.
S ohledem na související dostupné údaje se tedy výbor PRAC domnívá, že změny informací o přípravku jsou oprávněné.
Výbor CHMP souhlasí s vědeckými závěry učiněnými výborem PRAC.
Zdůvodnění doporučení změnu podmínek rozhodnutí o registraci
Na základě vědeckých závěrů týkajících se okriplasminu zastává výbor CHMP stanovisko, že poměr přínosů a rizik léčivého přípravku obsahujícího léčivou látku okriplasmin je příznivý pod podmínkou, že v údajích o přípravku budou provedeny navržené změny.
Výbor CHMP doporučuje změnit podmínky rozhodnutí o registraci.
51