Jakavi 5 Mg
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Jakavi 5 mg tablety
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna tableta obsahuje ruxolitinibum 5 mg (ve formě ruxolitinibi phosphas).
Pomocné látky se známým účinkem:
Jedna tableta obsahuje 71,45 mg monohydrátu laktózy.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Tableta.
Kulaté zaoblené bílé až téměř bílé tablety o průměru přibližně 7,5 mm s vyraženým „NVR“ na jedné straně a „L5“ na straně druhé.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Myelofibróza (MF)
Přípravek Jakavi je indikován k léčbě dospělých pacientů se splenomegalií nebo s příznaky přidruženými k primární myelofibróze (chronické idiopatické myelofibróze), postpolycytemické myelofibróze nebo myelofibróze po esenciální trombocytemii.
Pravá polvcvtémie (polvcvthaemia vera - PV)
Přípravek Jakavi je indikován k léčbě dospělých pacientů s pravou polycytémií, kteří jsou rezistentní nebo intolerantní k hydroxyurei.
4.2 Dávkování a způsob podání
Léčba Jakavi má být zahajována pouze lékařem, který má zkušenosti s podáváním protinádorové terapie.
Před zahájením léčby přípravkem Jakavi musí být vyšetřen kompletní krevní obraz včetně diferenciálního rozpočtu leukocytů.
Kompletní krevní obraz včetně diferenciálního rozpočtu leukocytů má být vyšetřen každé 2-4 týdny do stabilizace dávek přípravku Jakavi, a dále pak dle klinické indikace (viz bod 4.4).
Dávkování
Počáteční dávka
Doporučená počáteční dávka přípravku Jakavi u myelofibrózy je 15 mg dvakrát denně u pacientů s počtem trombocytů 100 x 109/l až 200*109/l a 20 mg dvakrát denně u pacientů s počtem trombocytů >200x109/l. Doporučená počáteční dávka přípravku Jakavi u pravé polycytémie je 10 mg podávaných perorálně dvakrát denně.
U pacientů s počtem trombocytů 50 až <100x109/l není dostatek údajů pro stanovení přesné úvodní dávky. U těchto pacientů je maximální doporučená úvodní dávka 5 mg dvakrát denně a její další titrace má být prováděna velmi opatrně.
Úprava dávkování
Dávky jsou dále titrovány dle bezpečnosti a účinnosti. Léčba by měla být ukončena při poklesu trombocytů na méně než 50x109/l nebo při poklesu absolutního počtu neutrofilů na méně než 0,5x109/l. Při PV by měla být léčba také ukončena, pokud j e hladina hemoglobinu pod 8 g/dl. Po návratu počtu krevních elementů nad tyto hodnoty může být podávání znovu zahájeno v dávce 5 mg dvakrát denně a postupně zvyšováno za pečlivého sledování krevního obrazu včetně diferenciálního rozpočtu leukocytů.
Pokud počet trombocytů klesne pod 100x109/l, mělo by být zváženo snížení dávky, aby nebylo nutné léčbu zcela přerušit pro trombocytopenii. Při PV by se také mělo zvážit snížení dávky, pokud hladina hemoglobinu klesne pod 12 g/dl, a snížení je doporučené, pokud hladina klesne pod 10 g/dl.
Pokud není léčba účinná a počty krevních elementů jsou dostatečné, mohou být dávky zvýšeny, a to maximálně o 5 mg dvakrát denně, až na maximální dávku 25 mg dvakrát denně.
Počáteční dávka nemá být zvyšována během prvních čtyř týdnů léčby, a následně ne častěji než ve dvoutýdenních intervalech.
Maximální dávka přípravku Jakavi je 25 mg dvakrát denně.
Úprava dávkování při konkomitantní léčbě silnými inhibitory CYP3A4 nebo flukonazolem Pokud je přípravek Jakavi podáván spolu se silnými inhibitory CYP3A4 nebo duálními inhibitory enzymů CYP2C9 a CYP3A4 (např. flukonazol), má být jednotlivá dávka přípravku Jakavi, podávaná dvakrát denně, snížena o přibližně 50 % (viz bod 4.5). Je nutné se vyhnout souběžnému podávání přípravku Jakavi s flukonazolem v dávkách vyšších než 200 mg denně.
V průběhu léčby silným inhibitorem CYP3A4 nebo duálními inhibitory enzymů CYP2C9 a CYP3A4 se doporučuje častěji (např. dvakrát týdně) kontrolovat hematologické parametry a pečlivě pátrat po klinických známkách nežádoucích účinků přípravku Jakavi.
Zvláštní skupiny pacientů Pacienti s poruchou funkce ledvin
U pacientů s mírnou nebo středně závažnou poruchou funkce ledvin není nutná specifická úprava dávky.
U pacientů s MF a těžkou poruchou funkce ledvin (clearance kreatininu méně než 30 ml/min) má být doporučená počáteční dávka, podávaná dvakrát denně, stanovená podle počtu trombocytů redukovaná o přibližně 50 %. Doporučená počáteční dávka pro pacienty s PV s těžkou poruchou funkce ledvin je 5 mg dvakrát denně. U těchto pacientů je třeba pečlivě monitorovat bezpečnost a účinnost léčby.
Pro pacienty s terminálním selháním ledvin (ESRD, end-stage renal disease) léčené hemodialýzou neexistuje dostatek údajů pro stanovení optimálního dávkování. Farmakokinetické/farmakodynamické simulace založené na dostupných údajích o této populaci naznačují, že u hemodialyzovaných pacientů s MF a ESRD má být počáteční dávka podávaná pouze ve dnech hemodialýzy po jejím ukončení, a to 15-20 mg v jednotlivé dávce nebo ve dvou dávkách po 10 mg podávaných po 12 hodinách. Jednotlivá dávka 15 mg je doporučena v případě pacientů s MF a počtem trombocytů 100 * 109/l až 200* 109/l. Jednotlivá dávka 20 mg nebo dvě dávky 10 mg podávané po 12 hodinách jsou doporučeny u pacientů s MF a počtem trombocytů >200*109/l. Následující dávky (v jednotlivé dávce nebo ve dvou dávkách 10 mg podávaných po 12 hodinách) mají být podávány pouze v den hemodialýzy, a to po jejím ukončení.
Doporučená počáteční dávka pro pacienty s PV a ESRD na hemodialýze je jednorázová dávka 10 mg nebo dvě dávky 5 mg podávané po 12 hodinách po dialýze, a to pouze v den podání hemodialýzy.
Tato doporučení dávkování jsou založená na simulacích a po jakékoli úpravě dávkování u jednotlivých pacientů s ESRD by mělo následovat pečlivé sledování bezpečnosti a účinnosti léčby. Pro dávkování u pacientů podstupujících peritoneální dialýzu a kontinuální venovenózní hemofiltraci nejsou k dispozici žádná data (viz bod 5.2).
Pacienti s poruchou funkce jater
U pacientů s poruchou funkce jater má být doporučená počáteční dávka, stanovená podle počtu trombocytů a podávaná dvakrát denně, snížena o přibližně 50 %. Následující dávky mají být upraveny na základě pečlivého monitorování bezpečnosti a účinnosti léčby. Pacientům, u nichž bylo v průběhu léčby přípravkem Jakavi zjištěno poškození jater, má být vyšetřen kompletní krevní obraz včetně diferenciálního rozpočtu leukocytů alespoň jednou za jeden až dva týdny v prvních 6 týdnech po zahájení léčby a dále dle klinické potřeby, dokud nejsou jaterní funkce a krevní obraz stabilizovány. Dávka přípravku Jakavi může být dále titrována pro snížení rizika cytopenie.
Starší lidé (>65 let)
Starší lidé nevyžadují žádnou specifickou úpravu dávkování.
Pediatrická populace
Bezpečnost a účinnost přípravku Jakavi u dětí ve věku do 18 let nebyla stanovena. Nejsou dostupné žádné údaje (viz bod 5.1).
Ukončení léčby
Léčba má pokračovat, dokud přínos z léčby převažuje nad rizikem léčby. Pokud ale nedojde během 6 měsíců od započetí léčby ke zmenšení velikosti sleziny nebo zlepšení příznaků, má být léčba ukončena.
U pacientů s určitým stupněm klinického zlepšení se doporučuje léčbu ruxolitinibem přerušit v případě setrvalého zvětšování sleziny o 40 % v porovnání s velikostí ve výchozím stavu (což odpovídá zhruba 25 % zvýšení objemu sleziny), kdy zároveň nedochází k dalšímu zlepšení příznaků spojených s onemocněním.
Způsob podání
Přípravek Jakavi se užívá perorálně, s jídlem nebo bez jídla.
Při vynechání dávky nemá pacient užívat dávku navíc, ale má pokračovat další obvyklou předepsanou dávkou.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1. Těhotenství a laktace.
4.4 Zvláštní upozornění a opatření pro použití
Myelosuprese
Léčba přípravkem Jakavi může způsobit hematologické nežádoucí účinky léku, včetně trombocytopenie, anemie a neutropenie. Před zahájením léčby přípravkem Jakavi musí být vyšetřen kompletní krevní obraz včetně diferenciálního rozpočtu leukocytů. Léčba má být přerušena u pacientů, u kterých dojde k poklesu počtu trombocytů na méně než 50* 109/l nebo absolutního počtu neutrofilů na méně než 0,5*109/l (viz bod 4.2).
Bylo zjištěno, že pacienti s nižším počtem trombocytů (<200*109/l) na začátku terapie mají větší riziko vzniku trombocytopenie během léčby.
Trombocytopenie je obvykle reverzibilní a je většinou zvládnuta snížením dávky nebo přechodným vysazením přípravku Jakavi (viz body 4.2 a 4.8). Nicméně v případě klinické potřeby může být nezbytné podání transfuze trombocytů.
Vznik anemie u léčených pacientů si může vyžádat podání krevní transfuze. U těchto pacientů lze také zvážit úpravu dávky nebo přerušení léčby.
Pacienti s hladinou hemoglobinu nižší než 10,0 g/dl při započetí léčby mají vyšší riziko výskytu hladin hemoglobinu pod 8,0 g/dl během léčby v porovnání s pacienty s vyšší počáteční hladinou hemoglobinu (79,3 % oproti 30,1 %). U pacientů s počáteční hladinou hemoglobinu pod 10,0 g/dl je doporučeno častější sledování hematologických parametrů a klinických příznaků a symptomů nežádoucích účinků spojených s přípravkem Jakavi.
Neutropenie (absolutní počet neutrofilů <0,5*109/l) byla obvykle reverzibilní a bylo možné ji zvládnout přechodným vysazením přípravku Jakavi (viz body 4.2 a 4.8).
Kompletní krevní obraz by měl být sledován dle klinické indikace a dávka upravována dle doporučení (viz body 4.2 a 4.8).
Infekce
U všech pacientů má být zhodnoceno riziko vzniku závažné bakteriální, mykobakteriální, mykotické a virové infekce. U pacientů s MF léčených přípravkem Jakavi byla hlášena tuberkulóza. Před zahájením léčby by mělo být u pacientů provedeno vyšetření na aktivní a neaktivní („latentní“) tuberkulózu podle místních doporučení. Může zahrnovat anamnézu, možnost předchozího kontaktu s tuberkulózou a/nebo příslušné vyšetření, jako je rentgenové vyšetření plic, tuberkulinový test a/nebo test uvolnění interferonu gama. Lékaři si mají být vědomi rizika falešně negativních výsledků tuberkulinového kožního testu, a to zejména u pacientů, kteří jsou vážně nemocní nebo mají sníženou imunitu. Léčba přípravkem Jakavi nemá být zahajována, dokud není závažná probíhající infekce zvládnuta. Lékaři mají pečlivě sledovat pacienty léčené přípravkem Jakavi, aby rozpoznali příznaky infekce a zahájili včas adekvátní léčbu (viz bod 4.8).
Zvýšení virové zátěže hepatitidy B (HBV-DNA titru), spolu s asociovaným zvýšením hladin alaninaminotransferázy a aspartátaminotransferázy, nebo bez jejich zvýšení, bylo hlášeno u pacientů s chronickými HBV infekcemi, kteří užívali přípravek Jakavi. Účinek přípravku Jakavi na replikaci viru u pacientů s chronickou infekcí HBV není známý. Pacienti s chronickou HBV infekcí by měli být léčeni a sledováni podle klinických doporučení.
Lékaři mají poučit pacienty o časných příznacích infekce herpes zoster a doporučit jim co možná nejvčasnější vyhledání možnosti léčby v případě infekce.
Progresivní multifokální leukoencefalopatie
Při léčbě pacientů s MF přípravkem Jakavi byla hlášena progresivní multifokální leukoencefalopatie (PML). Lékaři by měli dbát zejména na příznaky, které zaznamená pacient, nasvědčující PML (např. kognitivní, neurologické nebo psychiatrické příznaky nebo známky). U pacientů musí být sledovány jakékoli z těchto nových nebo zhoršujících se příznaků nebo známek a pokud se tyto příznaky/známky objeví, má být zvažováno odeslání k neurologovi a přijetí příslušných diagnostických opatření pro PML. Pokud je podezření na PML, musí být ukončeno další podávání, dokud není PML vyloučena.
Nemelanomové nádory kůže
U pacientů užívajících ruxolitinib byly hlášeny nemelanomové nádory kůže (NMSCs) zahrnující bazocelulární karcinom, spinocelulární karcinom a karcinom z Merkelových buněk. Většina těchto pacientů byla již dříve dlouhodobě léčena hydroxyureou a vyskytovaly se u nich NMSC nebo premaligní kožní léze. Kauzální vztah k ruxolitinibu nebyl prokázán. U pacientů se zvýšeným rizikem rakoviny kůže je doporučené pravidelné vyšetření kůže.
Zvláštní skupiny pacientů
Porucha funkce ledvin
U pacientů s těžkou poruchou funkce ledvin má být snížena úvodní dávka. U hemodialyzovaných pacientů s MF a terminálním selháním ledvin má být úvodní dávka určena dle počtu trombocytů (viz bod 4.2). Následující dávky (jednorázová dávka 20 mg nebo dvě dávky 10 mg podávané po 12 hodinách u pacientů s MF; jednorázová dávka 10 mg nebo dvě dávky 5 mg podávané po 12 hodinách u pacientů s PV) mají být podávány pouze po každé hemodialýze. Další úprava dávkování má být prováděna za pečlivého monitorování bezpečnosti a účinnosti léčby (viz body 4.2 a 5.2).
Porucha funkce jater
U pacientů s poruchou funkce jater má být doporučená úvodní dávka přípravku Jakavi redukována o přibližně 50 %. Další úprava dávkování má být prováděna na základě sledování bezpečnosti a účinnosti léčby (viz body 4.2 a 5.2).
Interakce
Pokud je přípravek Jakavi podáván spolu se silnými inhibitory CYP3A4 nebo duálními inhibitory enzymů CYP3A4 a CYP2C9 (např. flukonazol), má být jednotlivá dávka přípravku Jakavi, podávaná dvakrát denně, snížena o přibližně 50 % (frekvence sledování viz body 4.2 a 4.5).
Souběžné podávání cytoredukční léčby nebo hematopoetických růstových faktorů s přípravkem Jakavi nebylo studováno. Bezpečnost a účinnost těchto souběžných podání nejsou známé (viz bod 4.5).
Následky vysazení
Po přerušení nebo ukončení léčby přípravkem Jakavi se mohou příznaky MF znovu objevit během přibližně jednoho týdne. Závažnější případy byly po přerušení léčby přípravkem Jakavi popsány zejména v souvislosti s akutním interkurentním onemocněním. Není jasné, zda k závažnosti těchto případu přispělo náhlé přerušení léčby. Pokud není náhlé přerušení léčby nutné, je vhodné zvážit postupné snižování dávky, přestože přínos tohoto postupu nebyl prokázán.
Pomocné látky
Přípravek Jakavi obsahuje laktózu. Pacienti se vzácnými dědičnými problémy s intolerancí galaktózy, vrozeným deficitem laktázy nebo malabsorpcí glukózy a galaktózy by tento přípravek neměli užívat.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Studie interakcí byly provedeny pouze u dospělých.
Ruxolitinib je eliminován metabolismem katalyzovaným CYP3A4 a CYP2C9. Léčivé přípravky inhibující tyto enzymy proto mohou zvýšit expozici ruxolitinibu.
Interakce vyžadující snížení dávky ruxolitinibu
Inhibitory CYP3A4
Silné inhibitory CYP3A4 (jako jsou např. boceprevir, klarithromycin, indinavir, itrakonazol, ketokonazol, lopinavir/ritonavir, mibefradil, nefazodon, nelfinavir, posakonazol, sachinavir, telaprevir, telithromycin, vorikonazol)
Podání přípravku Jakavi (jednorázově v dávce 10 mg) následně po podávání silného inhibitoru CYP3A4 ketokonazolu vedlo u zdravých dobrovolníků ke zvýšení Cmax ruxolitinibu o 33 % a AUC ruxolitinibu o 91 % oproti hodnotám dosaženým po podání samotného ruxolitinibu. Poločas přípravku byl při současném podání ketokonazolu prodloužen z 3,7 na 6,0 hodiny.
Pokud je přípravek Jakavi podáván se silnými inhibitory CYP3A4, má být jednotlivá dávka přípravku Jakavi, podávaná dvakrát denně, snížena přibližně o 50 %. Pacienti mají být pečlivě sledováni (např. dvakrát týdně) z důvodu možného vzniku cytopenií a dávka má být titrována na základě hodnocení bezpečnosti a účinnosti léčby (viz bod 4.2).
Duální inhibitory CYP2C9 a CYP3A4
Při společném užití s léky, které jsou duálními inhibitory enzymů CYP2C9 a CYP3A4 (např. flukonazol) má být zváženo 50 % snížení dávky. Je nutné se vyhnout souběžnému podávání přípravku Jakavi s flukonazolem v dávkách vyšších než 200 mg denně.
Induktory enzymů
Induktory CYP3A4 (jako _jsou např. avasimib, karbamazepin, _fenobarbital, _fenytoin, rifabutin, rifampin (rifampicin), třezalka tečkovaná (Hypericum perforatum))
Pacienti mají být pečlivě sledováni a dávka titrována s ohledem na bezpečnost a účinnost (viz bod 4.2).
U zdravých dobrovolníků, kterým byl ruxolitinib (jednorázová dávka 50 mg) podán po silném induktoru CYP3A4 rifampicinu (denní dávka 600 mg po dobu 10 dní), byla AUC ruxolitinibu o 70 % nižší než po podání samotného přípravku Jakavi. Expozice aktivním metabolitům ruxolitinibu se nezměnila. V souhrnu byla farmakodynamická aktivita ruxolitinibu podobná, což naznačuje, že indukce CYP3A4 má minimální vliv na farmakodynamiku. Nicméně to může být spojené s vysokou dávkou ruxolitinibu, která vede k farmakodynamickým účinkům blízkým Emax. Je možné, že je nutné u jednotlivých pacientů zvýšit dávku ruxolitinibu při zahájení léčby silným induktorem enzymů.
Další interakce, u nichž je zvažováno ovlivnění ruxolitinibu
Slabé nebo středně silné inhibitory CYP3A4 (jako _je např. ciprofloxacin, erythromycin, amprenavir, atazanavir, diltiazem, cimetidin)
Podání ruxolitinibu (jednorázově v dávce 10 mg) následně po 4denním podávání erythromycinu 500 mg dvakrát denně vedlo u zdravých dobrovolníků k zvýšení Cmax ruxolitinibu o 8 % a AUC ruxolitinibu o 27 % oproti hodnotám dosaženým po podání samotného ruxolitinibu.
Při souběžném podávání ruxolitinibu se slabými a středně silnými inhibitory CYP3A4 (např. erytromycin) není nutná úprava dávkování. Nicméně pacienti mají být po zahájení léčby středně silnými inhibitory CYP3A4 pečlivě sledováni, zda u nich nedochází k rozvoji cytopenie.
Účinky ruxolitinibu na další léčivé přípravky
Substance transportované P-glykoproteinem a dalšími transportéry
Ruxolitinib může inhibovat P-glykoprotein a protein BCRP (breast cancer resistance protein) ve střevě. To může vést ke zvýšení systémové expozice substrátů těchto transportérů, jako je dabigatran etexilát, cyklosporin, rosuvastatin a případně digoxin. Je doporučené sledování hladiny léčiva (TDM) nebo klinické sledování z důvodu možného ovlivnění takové látky.
Je možné, že potenciální inhibice P-gp a BCRP ve střevě může být minimalizována prodloužením času mezi podáním na nejdelší možnou míru.
Hemopoetické růstové _faktory
Souběžné podání hematopoetických růstových faktorů a přípravku Jakavi nebylo studováno. Není známo, zda inhibice Janus kináz (JAK) přípravkem Jakavi snižuje účinnost hematopoetických růstových faktorů nebo zda hematopoetické růstové faktory ovlivňují účinnost přípravku Jakavi (viz bod 4.4).
Cytoredukční léčba
Souběžné podávání cytoredukční léčby a přípravku Jakavi nebylo studováno. Bezpečnost a účinnost totho souběžného podávání nejsou známé (viz bod 4.4).
Studie u zdravých subjektů prokázala, že ruxolitinib neinhiboval metabolismus perorálního substrátu CYP3A4 midazolamu. Proto není očekáváno zvýšení expozice CYP3A4 substratu při kombinaci s přípravkem Jakavi. Další studie u zdravých subjektů prokázala, že přípravek Jakavi neovlivňuje farmakokinetiku perorálně podávané antikoncepce obsahující ethinylestradiol a levonorgestrel. Proto není očekáváno, že účinnost antikoncepce v této kombinaci bude oslabena souběžným podáváním ruxolitinibu.
4.6 Fertilita, těhotenství a kojení
Těhotenství a antikoncepce u žen
Údaje o podávání přípravku Jakavi těhotným ženám nejsou k dispozici.
Studie u zvířat prokázaly embryotoxický a fetotoxický účinek. Teratogenita nebyla u potkanů a králíků pozorována. Nicméně hraniční expozice byly při porovnání s nejvyšší klinickou dávkou nízké a výsledky mají proto pro člověka omezený význam (viz bod 5.3). Potenciální riziko u člověka není známo. Z hlediska bezpečnosti je podání přípravku Jakavi během těhotenství kontraindikováno (viz bod 4.3). Ženy ve fertilním věku mají během léčby přípravkem Jakavi používat účinnou antikoncepci. V případě otěhotnění v průběhu užívání přípravku Jakavi je nutné individuální zhodnocení rizika a profitu léčby a pečlivý odhad potenciálního rizika pro plod (viz bod 5.3).
Kojení
Přípravek Jakavi nesmí být podáván během kojení (viz bod 4.3) a kojení má být při zahájení léčby ukončeno. Není známo, zda se ruxolitinib a/nebo jeho metabolity vylučují do lidského mateřského mléka. Riziko pro kojené dítě nelze vyloučit. Dostupná farmakodynamická a toxikologická data prokázala vylučování ruxolitinibu a jeho metabolitů do mateřského mléka u studovaných zvířat (viz bod 5.3).
Fertilita
Nejsou k dispozici žádné klinické údaje týkající se ovlivnění fertility u lidí. Ve studiích u zvířat nebyl žádný vliv na fertilitu pozorován.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek Jakavi nemá žádný nebo jen zanedbatelný sedativní účinek. Pokud však pacient pozoruje závratě po užití přípravku Jakavi, má se vyhnout řízení a obsluze strojů.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Bezpečnost byla vyhodnocena u celkem 855 pacientů (s MF nebo PV), kteří užívali přípravek Jakavi ve studiích fáze 2 a 3.
Myelofibróza
V randomizační periodě dvou pivotních studií COMFORT-I a COMFORT-II byl medián trvání expozice přípravku Jakavi 10,8 měsíce (rozmezí 0,3 až 23,5 měsíce). Většina pacientů (68,4 %) byla léčena po dobu nejméně 9 měsíců. Z celkového počtu 301 pacientů mělo 111 (36,9 %) počet krevních destiček ve výchozím stavu mezi 100000/mm3 a 200000/mm3 a 190 (63,1 %) mělo počet krevních destiček ve výchozím stavu >200000/mm3.
V těchto klinických studiích bylo zjištěno ukončení léčby z důvodu výskytu nežádoucích účinků bez ohledu na jejich příčinu u 11,3 % pacientů.
Nejčastějšími hlášenými nežádoucími účinky byla trombocytopenie a anemie.
Hematologické nežádoucí účinky léku (všechny stupně dle CTCAE [Common Terminology Criteria for Adverse Events] klasifikace) zahrnovaly anemii (82,4 %), trombocytopenii (69,8 %) a neutropenii (16,6 %).
Výskyt anemie, trombocytopenie a neutropenie závisí na podávané dávce.
Nejčastějšími třemi nehematologickými nežádoucími účinky léku byla tvorba hematomů (21,3 %), závratě (15,3 %) a bolest hlavy (14,0 %).
Nejčastějšími třemi nehematologickými laboratorními abnormalitami bylo zvýšení ALT (27,2 %), zvýšení AST (19,9 %) a hypercholesterolemie (16,9 %). V klinických studiích fáze 3 u pacientů s MF nebyla pozorována hypercholesterolemie CTCAE stupně 3 nebo 4, zvýšená hladina aspartátaminotransferázy a ani zvýšená hladina alaninaminotransferázy CTCAE stupně 4.
Dlouhodobá bezpečnost: Jak bylo možné očekávat během prodloužené periody sledování, po vyhodnocení bezpečnostních dat 3letého sledování (medián trvání expozice 33,2 měsíců ve studích COMFORT-I a COMFORT-II u pacientů iniciálně randomizovaných k ruxolitinibu) od 457 pacientů s myelofibrózou léčených ruxolitinibem během randomizované a prodloužené periody dvou pivotních studií fáze 3 vzrostly kumulativní četnosti některých nežádoucích účinků. Toto vyhodnocení zahrnovalo data od pacientů, kteří byli iniciálně randomizovaní na ruxolitinib (N=301) a pacientů, kteří užívali ruxolitinib po přechodu z ramen s kontrolní léčbou (N=156). V těchto aktualizovaných datech bylo ukončení léčby z důvodu nežádoucích účinků zjistěno u 17,1 % pacientů léčených ruxolitinibem.
Pravá _ polycytémie
Bezpečnost přípravku Jakavi byla stanovena na základě vyhodnocení 110 pacientů s pravou polycytémií v otevřené, randomizované, kontrolované studii fáze 3 RESPONSE. Níže uvedené nežádoucí účinky reflektují počáteční periodu studie (až do týdne 32) s ekvivalentní expozicí ruxolitinibu a nejlepší dostupnou léčbou (BAT), což odpovídalo mediánu trvání expozice přípravku Jakavi 7,8 měsíce. Průměrný věk pacientů užívajících přípravek Jakavi byl přibližně 60 let.
Ukončení léčby z důvodu výskytu nežádoucích účinků bez ohledu na jejich příčinu bylo zjištěno u 3,6 % pacientů léčených přípravkem Jakavi a u 1,8 % pacientů léčených nejlepší dostupnou léčbou.
Hematologické nežádoucí účinky (jakéhokoli CTCAE stupně) zahrnovaly anemii (43,6 %) a trombocytopenii (24,5 %). Anemie nebo trombocytopenie CTCAE stupně 3 a 4 byly hlášené v 1,8 % nebo 5,5 % případů.
Tři nejčastější nehematologické nežádoucí účinky byly závrať (15,5 %), zácpa (8,2 %) a herpes zoster (6,4 %).
Tři nejčastější nehematologické laboratorní abnormality (jakéhokoli CTCAE stupně) byly hypercholesterolemie (30,0 %), zvýšená hladina alaninaminotransferázy (22,7 %) a zvýšená hladina aspartátaminotransferázy (20,9 %). Ve všech případech šlo o CTCAE stupeň 1 a 2 s výjimkou jednoho případu zvýšené hladiny alaninaminotransferázy CTCAE stupně 3.
Dlouhodobá bezpečnost: Pacienti měli medián trvání expozice přípravku Jakavi 18,6 měsíce (rozmezí 0,3 až 35,9 měsíce). S delší expozicí vzrůstala četnost nežádoucích účinků, nicméně nová bezpečnostní zjištění se neobjevila. Při přepočítání na expozici byl výskyt nežádoucích účinků obecně porovnatelný s výskytem zjištěným během počáteční periody studie.
Tabulární přehled nežádoucích účinků hlášených v klinických studiích
V klinickém studijním programu byla závažnost nežádoucích účinků hodnocena podle CTCAE klasifikace, definující stupeň 1 = lehký, stupeň 2 = středně těžký, stupeň 3 = těžký a stupeň 4 = život ohrožující.
Nežádoucí účinky hlášené v klinických studiích (Tabulka 1) jsou seřazeny podle MedDRA systémově-orgánové klasifikace. V každé systémově-orgánové třídě jsou nežádoucí účinky řazeny podle četnosti tak, že nejčastější nežádoucí účinek je na prvním místě. Četnost přiřazená ke každému nežádoucímu účinku je klasifikována podle následujících kategorií: velmi časté (>1/10); časté (>1/100 až <1/10); méně časté (>1/1 000 až <1/100); vzácné (>1/10 000 až <1/1 000); velmi vzácné (<1/10 000).
Tabulka 1 Kategorie četností nežádoucích účinků hlášených ve studiích fáze 3 (COMFORT-I, COMFORT-II, RESPONSE)
|
Nežádoucí účinky |
Kategorie četnosti pro pacienty s MF |
Kategorie četnosti pro pacienty s PV |
|
Infekce a infestace | ||
|
Infekce močových cesta,d |
Velmi časté |
Časté |
|
Časté |
Časté | |
|
Tuberkulózae |
Méně časté |
- |
|
Poruchy krve a lymfatického systémub,d | ||
|
Anemieb |
- |
- |
|
CTCAEc stupně 4 (<6,5 g/dl) |
Velmi časté |
Méně časté |
|
CTCAEc stupně 3 (<8,0 - 6,5g/dl) |
Velmi časté |
Méně časté |
|
Všechny CTCAEc stupně |
Velmi časté |
Velmi časté |
|
Trombocytopenieb | ||
|
CTCAEc stupně 4 (<25000/mm3) |
Časté |
Méně časté |
|
CTCAEc stupně 3 (50000 - 25000/mm3) |
Časté |
Časté |
|
Všechny CTCAEc stupně |
Velmi časté |
Velmi časté |
|
Neutropenieb | ||
|
CTCAEc stupně 4 (<500/mm3) |
Časté |
- |
|
CTCAEc stupně 3 (<1000 - 500/mm3) |
Časté |
- |
|
Všechny CTCAEc stupně |
Velmi časté |
- |
|
Krvácení (všechny případy krvácení zahrnující intrakraniální a gastrointestinální krvácení, podlitiny a jiné typy krvácení) |
Velmi časté |
Velmi časté |
|
Intrakraniální krvácení |
Časté |
- |
|
Gastrointestinální krvácení |
Časté |
- |
|
Podlitiny |
Velmi časté |
Velmi časté |
|
Jiné typy krvácení (zahrnují epistaxi, krvácení po zákroku a hematurii) |
Časté |
Velmi časté |
|
Poruchy metabolismu a výživy | ||
|
Nárůst tělesné hmotnosti3 |
Velmi časté |
Časté |
|
Hypercholesterolemieb CTCAEc stupně 1 a 2 |
Velmi časté |
Velmi časté |
|
Hypertriglyceridemieb CTCAEc stupně 1 |
- |
Velmi časté |
|
Poruchy nervového systému | ||
|
Závrať1 |
Velmi časté |
Velmi časté |
|
Velmi časté |
- | |
|
Gastrointestinální poruchy | ||
|
Flatulencea |
Časté |
- |
|
Zácpaa |
- |
Časté |
|
Poruchy jater a žlučových cest | ||
|
Zvýšená hladina alaninaminotransferázyb | ||
|
CTCAEc stupně 3 (>5x - 20 x ULN) |
Časté |
Méně časté |
|
Všechny CTCAEc stupně |
Velmi časté |
Velmi časté |
|
Zvýšená hladina aspartátaminotransferázyb | ||
|
Všechny CTCAEc stupně |
Velmi časté |
Velmi časté |
|
Cévní poruchy | ||
|
Hypertenze*1 |
- |
Velmi časté |
|
a Četnost podle údajů o nežádoucích účincích. - Subjekt s mnohočetným výskytem daného nežádoucího účinku (ADR) je v dané kategorii započítán pouze jednou. - Nežádoucí účinky zaznamenané při léčbě nebo 28 dní po ukončení léčby. b Četnost podle laboratorních výsledků. - Subjekt s mnohočetným výskytem daného nežádoucího účinku je v dané kategorii započítán pouze jednou. - Nežádoucí účinky zaznamenané při léčbě nebo 28 dní po ukončení léčby. c Common Terminology Criteria for Adverse Events (CTCAE) klasifikace verze 3.0; stupeň 1 = lehký, stupeň 2 = středně těžký, stupeň 3 = těžký, stupeň 4 = život ohrožující d Tyto nežádoucí účinky j sou diskutovány v textu. e Četnost je založená na údajích o všech pacientech vystavených ruxolitinibu v klinických studiích (N=4755). | ||
Po ukončení léčby se mohou u pacientů s MF znovu objevit příznaky MF jako je únava, bolesti kostí, horečka, pruritus, noční pocení, symptomatická splenomegalie a úbytek tělesné hmotnosti. Celkové symptomatické skóre pro příznaky MF se v klinických studiích s MF vrátilo k výchozí hodnotě během 7 dnů po vysazení léčby (viz bod 4.4).
Popis vybraných nežádoucích účinků
Anemie
V klinických studiích fáze 3 u pacientů s MF byla střední doba do vzniku první anemie stupně 2 dle CTCAE nebo závažnější 1,5 měsíce. U jednoho pacienta (0,3 %) byla kvůli anemii přerušena léčba.
U pacientů léčených přípravkem Jakavi klesla průměrná hladina hemoglobinu o přibližně 10 g/l oproti vstupní hodnotě s minimem po 8 až 12 týdnech léčby. Poté došlo k opětovnému vzestupu a nastavení nové rovnováhy s hodnotou hemoglobinu přibližně o 5 g/l nižší vůči vstupní hodnotě. Tento průběh byl pozorován u nemocných bez ohledu na použití transfuzní léčby.
V randomizované, placebem kontrolované studii COMFORT-I dostalo v průběhu randomizované léčby 60,6 % pacientů s MF léčených Jakavi a 37,7 % pacientů s MF léčených placebem transfuzi erytrocytů. Ve studii COMFORT-II dostalo transfuzi erytrocytů 53,4 % pacientů ve skupině léčené přípravkem Jakavi a 41,1 % pacientů ve skupině dostávající nejlepší dostupnou léčbu.
V randomizační periodě pivotních studií se anemie vyskytovala méně často u pacientů s PV než u pacientů s MF (43,6 % oproti 82,4 %). V populaci s PV byly hlášené nežádoucí účinky CTCAE stupně 3 a 4 u 1,8 % pacientů, zatímco u pacientů s MF byla četnost 42,56 %.
Trombocytopenie
V klinických studiích fáze 3 u pacientů s MF byla střední doba do vzniku trombocytopenie stupně 3 nebo 4 přibližně osm týdnů. Trombocytopenie byla obvykle po snížení dávky nebo přerušení léčby reverzibilní. Střední čas do obnovení počtu trombocytů nad 50* 109/l byl 14 dní. Během randomizované periody byla podána transfuze trombocytů 4,7 % pacientů, kteří dostávali přípravek Jakavi a 4,0 % pacientů dostávajícím kontrolní léčbu. Léčbu bylo pro trombocytopenii nutno přerušit u 0,7 % pacientů užívajících přípravek Jakavi a u 0,9 % pacientů dostávajících kontrolní léčbu.
Pacienti s počtem trombocytů 100*109/l až 200*109/l před zahájením léčby přípravkem Jakavi měli častěji trombocytopenii stupně 3 nebo 4 v porovnání s pacienty se vstupním počtem trombocytů >200*109/l (64,2 % oproti 38,5 %).
V randomizačních periodách pivotních studií byl podíl pacientů s trombocytopenií nižší u pacientů s PV (24,5 %) oproti pacientům s MF (69,8 %). Četnost vážné (tj. CTCAE stupeň 3 a 4) trombocytopenie byla nižší u pacientů s PV (5,5 %) než u pacientů s MF (11,6 %).
Neutropenie
V klinických studiích fáze 3 u pacientů s MF byla střední doba do vzniku neutropenie stupně 3 nebo 4 dvanáct týdnů. Během randomizované periody bylo zaznamenáno přerušení léčby nebo snížení dávky pro neutropenii u 1,0 % pacientů a 0,3 % pacientů muselo kvůli neutropenii léčbu ukončit.
V randomizační periodě pivotní studie u pacientů s PV byla neutropenie hlášená u dvou pacientů (1,8 %), z nichž u jednoho pacienta došlo k rozvoji neutropenie CTCAE stupně 4.
Krvácení
V pivotních klinických studiích fáze 3 u pacientů s MF byly krvácivé komplikace (zahrnující nitrolební krvácení, krvácení do zažívacího traktu, hematomy a jiné typy krvácení) zaznamenány u 32,6 % pacientů užívajících přípravek Jakavi a u 23,2 % pacientů užívajících kontrolní léčby (placebo nebo nejlepší dostupnou léčbu). Výskyt příhod krvácení stupně 3 nebo 4 byl stejný u pacientů léčených přípravkem Jakavi s pacienty léčenými kontrolními léčbami (4,7 % oproti 3,1 %). U většiny pacientů s krvácivými komplikacemi hlášenými během léčby byly hlášeny hematomy (65,3 %). Hematomy byly častěji zaznamenány u pacientů léčených přípravkem Jakavi v porovnání
s referenčními léčbami (21,3 % oproti 11,6 %). Nitrolební krvácení bylo hlášeno u 1 % pacientů léčených přípravkem Jakavi a u 0,9 % léčených kontrolními léčbami. Gastrointestinální krvácení bylo hlášeno u 5,0 % pacientů léčených přípravkem Jakavi oproti 3,1 % pacientů léčených kontrolními léčbami. Jiné typy krvácivých komplikací (zahrnující případy jako je epistaxe, krvácení po zákroku a hematurie) byly hlášeny u 13,3 % pacientů léčených přípravkem Jakavi a u 10,3 % pacientů léčených kontrolní léčbou.
V randomizační periodě pivotní studie u pacientů s PV byly krvácivé příhody (zahrnující intrakraniální a gastrointestinální krvácení, podlitiny a jiné typy krvácení) hlášeny u 20 % pacientů léčených přípravkem Jakavi a u 15,3 % pacientů s nejlepší dostupnou léčbou. Podlitiny byly hlášené s podobnými četnostmi v ramenech s přípravkem Jakavi a BAT (10,9 % vs. 8,1 %). U pacientů léčených přípravkem Jakavi nebyl hlášen ani jeden případ intrakraniálního nebo gastrointestinálního krvácení. Jeden pacient léčený přípravkem Jakavi měl krvácivou příhodu stupně 3 (krvácení po zákroku); krvácení stupně 4 nebylo hlášeno. Ostatní krvácivé příhody (zahrnující příhody jako epistaxi, krvácení po zákroku, krvácení dásní) byly hlášeny u 11,8 % pacientů léčených přípravkem Jakavi a u 6,3 % pacientů s nejlepší dostupnou léčbou.
Infekce
V klinických studiích fáze 3 u pacientů s MF byly hlášeny infekce močového traktu stupně 3 nebo 4 u 1,0 % pacientů, herpes zoster u 4,3 % a tuberkulóza u 1,0 % pacientů. V klinických studiích fáze 3 byla hlášena sepse u 3,0 % pacientů. Prodloužené sledování pacientů léčených ruxolitinibem neprokázalo žádné trendy ke zvýšení výskytu sepse v čase.
V randomizační periodě pivotní studie u pacientů s PV byl hlášen jeden případ (0,9 %) infekce močových cest CTCAE stupně 3, infekce močových cest CTCAE stupně 4 nebyla hlášena. Výskyt herpes zoster byl nepatrně vyšší u pacientů s PV (6,4 %) oproti pacientům s MF (4,0 %). U pacientů s PV se objevilo jedno hlášení postherpetické neuralgie CTCAE stupně 3.
Zvýšený systolický krevní tlak
V pivotních klinických studiích fáze 3 u pacientů s MF bylo u 31,5 % pacientů během alespoň jedné návštěvy v porovnání s 19,5 % pacientů léčených kontrolní léčbou zjištěno zvýšení systolického krevního tlaku o 20 mmHg nebo více oproti výchozímu stavu. Ve studii COMFORT-I (pacienti s MF) bylo průměrné zvýšení systolického krevního tlaku oproti výchozímu stavu o 0-2 mmHg u přípravku Jakavi v porovnání s poklesem o 2-5 mmHg v rameni s placebem. Ve studii COMFORT-II vykazovaly průměrné hodnoty u pacientů s MF léčených ruxolitinibem v porovnání s pacienty s MF léčenými kontrolní léčbou malé rozdíly.
V randomizační periodě pivotní studie u pacientů s PV se průměrný systolický krevní tlak zvýšil o 0,65 mmHg u přípravku Jakavi v porovnání s poklesem o 2 mmHg u ramene s BAT.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Není známo žádné antidotum pro předávkování přípravkem Jakavi. Jednorázové podání až 200 mg přípravku Jakavi bylo poměrně dobře snášeno. Opakované podání vyšších než doporučených dávek je spojeno s vyšším výskytem myelosuprese, zahrnující leukopenii, anemii a trombocytopenii. V těchto případech má být podávána vhodná podpůrná léčba.
Nepředpokládá se, že by hemodialýza zvyšovala vylučování ruxolitinibu.
FARMAKOLOGICKÉ VLASTNOSTI
5.
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Cytostatika, inhibitory proteinkinázy, ATC kód: L01XE18 Mechanismus účinku
Ruxolitinib je selektivní inhibitor Janus kináz (JAK) JAK1 a JAK2 (hodnoty IC50 jsou pro enzym JAK1 3,3 nM a pro JAK2 2,8 nM). Tyto kinázy jsou zapojeny do signalizace mnoha cytokinů a růstových faktorů s významnou úlohou při hematopoéze a imunitních funkcích.
Myelofibróza a pravá polycytémie jsou myeloproliferativní neoplastická onemocnění, u kterých byla popsána abnormální regulace signalizace zprostředkované JAK1 a JAK2. Předpokládá se, že příčinou poruchy regulace jsou vysoké hladiny cirkulujících cytokinů, které aktivují signální dráhu JAK-STAT, mutace zvyšující funkci enzymů jako je JAK2V617F a potlačení negativních regulačních mechanismů. U pacientů s MF nacházíme změnu regulace zprostředkované JAK bez ohledu na přítomnost mutace JAK2V617F. Aktivační mutace v JAK2 (V617F nebo exon 12) jsou zjištěné u >95 % pacientů s PV.
Ruxolitinib inhibuje signální dráhu JAK-STAT a buněčnou proliferaci u buněčných modelů hematologických malignit závislých na cytokinech, stejně jako u na cytokinech nezávislého modelu využívajícího Ba/F3 buňky exprimující JAK2V617F mutovaný protein s hodnotou IC50 v rozmezí 80-320 nM.
Farmakodynamické účinky
Ruxolitinib inhibuje cytokiny indukovanou STAT3 fosforylaci v krvi zdravých dobrovolníků i pacientů s MF a pacientů s PV. U obou skupin vedlo podávání ruxolitinibu k maximální inhibici STAT3 fosforylace 2 hodiny po podání s návratem k téměř výchozím hodnotám do 8 hodin po podání, což ukazuje na to, že nedochází k akumulaci mateřské látky ani aktivních metabolitů.
U pacientů s MF došlo při léčbě ruxolitinibem k poklesu zvýšených zánětlivých markerů jako je TNFa, IL-6 a CRP. Pacienti s MF se v průběhu času nestávali refrakterní k farmakodynamickým účinkům ruxolitinibu. Také pacienti s PV vykazovali obdobné zvýšení zánětlivých markerů ve výchozím stavu a tyto markery klesaly po léčbě ruxolitinibem.
V QT studiích u zdravých dobrovolníků nebylo zaznamenáno prodloužení QT/QTc intervalu po jednorázovém podání dávky ruxolitinibu vyšší než terapeutické (až 200 mg), což ukazuje na to, že ruxolitinib nemá vliv na srdeční repolarizaci.
Klinická účinnost a bezpečnost
Myelofibróza
U pacientů s MF (primární myelofibrózou, postpolycytemickou myelofibrózou nebo myelofibrózou po esenciální trombocytemii) byly provedeny dvě randomizované studie fáze 3 (COMFORT-I a COMFORT-II). V obou studiích měli pacienti hmatnou splenomegalii alespoň 5 cm pod žeberním obloukem a byli klasifikovaní podle kritérií International Working Group (IWG) do kategorií středního rizika 2 nebo vysokého rizika. Úvodní dávka přípravku Jakavi byla určena podle počtu trombocytů.
COMFORT-I byla dvojitě slepá, randomizovaná, placebem kontrolovaná studie zahrnující 309 pacientů, kteří byli refrakterní, nebo nebyli vhodnými kandidáty pro dostupnou léčbu. Primární cílový parametr účinnosti byl podíl nemocných, kteří dosáhnou ve 24. týdnu alespoň 35% zmenšení objemu sleziny oproti výchozí hodnotě měřeného pomocí magnetické rezonance (MRI) nebo počítačové tomografie (CT).
Sekundárními cílovými parametry bylo trvání udržení alespoň 35% redukce objemu sleziny proti výchozí hodnotě, podíl pacientů, kteří ve 24. týdnu dosáhli alespoň 50% snížení celkového symptomatického skóre, změna celkového symptomatického skóre od výchozího stavu do 24. týdne hodnocená pomocí modifikovaného MFSAF (Myelofibrosis Symptom Assessment Form) formuláře (verze 2.0, elektronický deník) a celkové přežití.
COMFORT-II byla otevřená, randomizovaná studie zahrnující 219 pacientů. Pacienti byli randomizováni v poměru 2:1 k léčbě přípravkem Jakavi oproti nejlepší dostupné léčbě. Ve skupině s nejlepší dostupnou léčbou dostávalo 47 % pacientů hydroxyureu a 16 % pacientů glukokortikoidy. Primární cílový ukazatel účinnosti léčby byl podíl nemocných, kteří dosáhnou ve 48. týdnu alespoň 35% snížení objemu sleziny oproti výchozí hodnotě měřeného pomocí MRI nebo CT.
Sekundární cílové parametry ve studii COMFORT-II zahrnovaly podíl nemocných, kteří dosáhnou ve 24. týdnu alespoň 35% snížení objemu sleziny oproti výchozí hodnotě a dobu trvání alespoň 35% snížení objemu slezinyoproti výchozí hodnotě.
Ve studiích COMFORT-I a COMFORT-II byly vstupní demografické parametry a charakteristiky onemocnění srovnatelné u obou léčených skupin.
Tabulka 2 Podíl pacientů (%) s >35% snížením objemu sleziny oproti výchozí hodnotě ve 24. týdnu ve studii COMFORT-I a ve 48. týdnu ve studii COMFORT-II (ITT)
|
COMFORT-I |
COMFORT-II | |||
|
Jakavi (N=155) |
Placebo (N=153) |
Jakavi (N=144) |
Nejlepší dostupná léčba (N=72) | |
|
Časový bod |
24. týden |
48. týden | ||
|
Počet (%) pacientů s redukcí objemu sleziny o >35 % |
65 (41,9) |
1 (0,7) |
41 (28,5) |
0 |
|
95% interval spolehlivosti |
34,1; 50,1 |
0; 3,6 |
21,3; 36,6 |
0,0; 5,0 |
|
Hodnota p |
<0,0001 |
<0,0001 | ||
Významně vyšší podíl pacientů ve skupině léčené přípravkem Jakavi dosáhl alespoň 35% snížení objemu sleziny (Tabulka 2) oproti výchozí hodnotě, a to bez ohledu na přítomnost mutace JAK2V617F nebo podtypu onemocnění (primární myelofibróza, postpolycytemická myelofibróza nebo myelofibróza po esenciální trombocytemii).
Tabulka 3 Podíl pacientů (%) s >35% snížením objemu sleziny oproti výchozí hodnotě dle mutačního stavu JAK (hodnocení bezpečnosti)
|
COMFORT-I |
COMFORT-II | |||||||
|
Jakavi |
Placebo |
Jakavi |
Nejlepší dostupná léčba | |||||
|
JAK mutační stav |
Pozitivní (N=113) n (%) |
Negativní (N=40) n (%) |
Pozitivní (N=121) n (%) |
Negativní (N=27) n (%) |
Pozitivní (N=110) n (%) |
Negativní (N=35) n (%) |
Pozitivní (N=49) n (%) |
Negativní (N=20) n (%) |
|
Počet (%) pacientů s redukcí objemu sleziny o >35 % |
54 (47,8) |
11 (27,5) |
1 (0,8) |
0 |
36 (32,7) |
5 (14,3) |
0 |
0 |
|
Časový bod |
Po 24 týdnech |
Po 48 týdnech | ||||||
Pravděpodobnost zachování odpovědi sleziny (>35% snížení) na přípravku Jakavi po dobu nejméně 24 týdnů byla 89 % ve studii COMFORT-I a 87 % ve studii COMFORT-II; 52 % udrželo odpověď sleziny po dobu nejméně 48 týdnů ve studiin COMFORT-II.
Ve studii COMFORT-I dosáhlo 45,9 % subjektů ve skupině s přípravkem Jakavi >50% zlepšení celkového symptomatického skóre od výchozího stavu ve 24. týdnu (vyhodnocovaného pomocí MFSAF deníku v2.0) v porovnání s 5,3 % pacientů ve skupině s placebem (p<0,0001 za pomoci chí-kvadrát testu). Průměrná změna celkového zdravotního stavu ve 24. týdnu měřená pomocí dotazníku EORTC QLQ C30 byla +12,3 u přípravku Jakavi a -3,4 u placeba (p<0,0001).
Ve studii COMFORT-I byl při mediánu sledování 34,3 měsíce výskyt úmrtí u pacientů randomizovaných do ramene s ruxolitinibem 27,1 % oproti 35,1 % u pacientů randomizovaných do ramene s placebem; HR 0,687; 95% CI 0,459-1,029; p=0,0668.
Ve studii COMFORT-II byl při mediánu sledování 34,7 měsíce výskyt úmrtí u pacientů randomizovaných do ramene s ruxolitinibem 19,9 % oproti 30,1 % u pacientů randomizovaných k nejlepší dostupné léčbě (BAT); HR 0,48; 95% CI 0,28-0,85; p=0,009. V obou studiích byly nižší výskyty úmrtí zjištěné v rameni s ruxolitinibem ovlivněny především výsledky získanými od pacientů z podskupin po pravé polycytémii a po esenciální trombocytémii.
Pravá _ polycytémie
Randomizovaná, otevřená, aktivně kontrolovaná studie fáze 3 (RESPONSE) byla provedena u 222 pacientů s PV, kteří byli rezistentní nebo intolerantní k hydroxyurei definované podle publikovaných kritérií mezinárodní pracovní skupiny evropské leukemické sítě (ELN). 110 pacientů bylo randomizovaných do ramene s ruxolitinibem a 112 pacientů do ramene s nejlepší dostupnou léčbou. Zahajovací dávka přípravku Jakavi byla 10 mg dvakrát denně. Poté byly u pacientů dávky individuálně upraveny na základě tolerability a účinnosti s maximální dávkou 25 mg dvakrát denně. Nejlepší dostupná léčba byla vybraná zkoušejícím individuálně pro každého pacienta a zahrnovala hydroxyureu (59,5%), interferon/pegylovaný interferon (11,7 %), anagrelid (7,2 %), pipobroman (1,8 %) a pozorování (15,3 %).
Výchozí demografické parametry a charakteristika onemocnění byly v obou ramenech léčby srovnatelné. Medián věku byl 60 let (rozmezí 33 až 90 let). Pacienti v rameni s ruxolitinibem měli medián doby od diagnózy PV 8,2 roku a dříve užívali hydroxyureu s mediánem trvání léčby přibližně 3 roky. Většina pacientů (>80 %) měla provedeny nejméně dvě flebotomie v posledních 24 týdnech před zařazením do studie. Komparativní data týkající se dlouhodobého přežívání a výskytu komplikací onemocnění chybí.
Primárním složeným cílovým parametrem byl podíl pacientů, kteří dosáhli jak nevhodnosti k flebotomii (kontrolu HCT), tak i >35% zmenšení objemu sleziny od výchozího stavu do 32. týdne. Kritérium vhodnosti k provedení flebotomie bylo definováno jako potvrzený HCT >45%, tj. minimálně o 3 procentní body vyšší oproti HCT stanovenému ve výchozím stavu nebo potvrzenému HCT >48%, dle toho toho, která hodnota je nižší. Klíčové sekundární cílové parametry zahrnovaly podíl pacientů s dosaženým primárním cílovým parametrem a bez progrese ve 48. týdnu, stejně jako podíl pacientů s dosaženou kompletní hematologickou remisí ve 32. týdnu.
Studie dosáhla svého primárního cíle, vyšší podíl pacientů ve skupině s přípravkem Jakavi dosáhl primárního složeného cílového parametru a každé z jeho individuálních komponent. Výrazně vyšší počet pacientů léčených přípravkem Jakavi (20,9 %) dosáhl primární odpovědi (p<0,0001) v porovnání s nejlepší dostupnou léčbou (0,9 %). Kontroly hematokritu bylo dosaženo u 60 % pacientů v rameni s přípravkem Jakavi v porovnání s 19,6 % v rameni s nejlepší dostupnou léčbou a >35% snížení objemu sleziny bylo dosaženo u 38,2 % pacientů v rameni s přípravkem Jakavi v porovnání s 0,9 % v rameni s nejlepší dostupnou léčbou (obrázek 1). 94 (83,9 %) pacientů randomizovaných k rameni s nejlepší dostupnou léčbou přešlo ve 32. týdnu nebo později na léčbu ruxolitinibem, což omezuje porovnání obou ramen po 32. týdnu.
Také bylo dosaženo obou klíčových sekundárních cílových parametrů. Poměr pacientů, kteří dosáhli kompletní hematologické remise, byl 23,6 % u přípravku Jakavi v porovnání s 8,9 % u nejlepší dostupné léčby (p=0,0028) a poměr pacientů, kteří dosáhli trvalé primární odpovědi ve 48. týdnu, byl
19,1 % u přípravku Jakavi a 0,9 % u nejlepší dostupné léčby (p<0,0001).
Obrázek 1 Pacienti, kteří dosáhli primárního cílového parametru a komponent primárního cílového parametru ve 32. týdnu
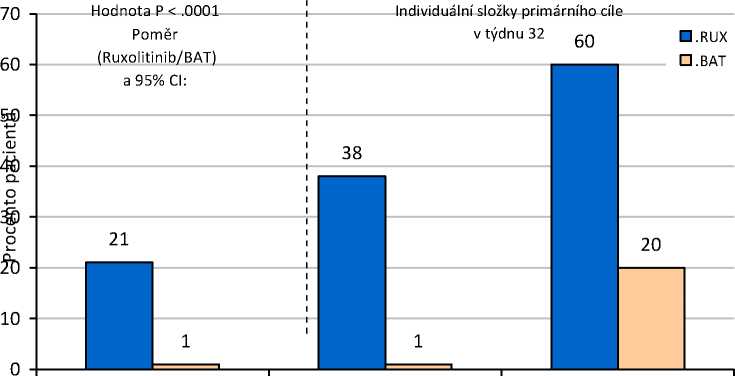
Primární složený cíl v > 35% redukce objemu Kontrola hematokritu bez týdnu 32 sleziny flebotomie
Symptomatická zátěž byla vyhodnocena pomocí elektronického pacientského dotazníku MPN-SAF celkového symptomatického skóre (TSS), který se skládá ze 14 otázek. Ve 32. týdnu dosáhlo 49 % a 64 % pacientů léčených ruxolitinibem >50% snížení TSS-14 resp. TSS-5 v porovnání s pouze 5 % a 11 % pacientů na nejlepší dostupné léčbě.
Vnímání prospěchu z léčby bylo stanoveno pomocí dotazníku Patient Global Impression of Change (PGIC). 66 % pacientů léčených ruxolitinibem hlásilo zlepšení již čtyři týdny po zahájení léčby oproti 9 % léčených nejlepší dostupnou léčbou. Zlepšení vnímání prospěchu z léčby bylo vyšší také u pacientů léčených ruxolitinibem ve 32. týdnu (78 % oproti 33 %).
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Jakavi u všech podskupin pediatrické populace pro léčbu MF (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Absorpce
Ruxolitinib patří do třídy 1 podle BCS (Biopharmaceutical Classification System), protože má vysokou prostupnost, dobrou rozpustnost a rychlý rozpad. Ruxolitinib byl v klinických studiích po perorálním podání rychle absorbován s maximální plazmatickou koncentrací (Cmax) dosaženou přibližně 1 hodinu po podání. Absorpce ruxolitinibu po perorálním podání (stanoven ruxolitinib a jeho metabolity tvořené při prvním průchodu játry) je podle farmakokinetických studií u lidí 95 % a více. Průměrné hodnoty Cmax a AUC ruxolitinibu se zvyšují úměrně s podanou jednorázovou dávkou v rozmezí 5-200 mg. Užití ruxolitinibu po tučném jídle nevedlo ke klinicky významné změně farmakokinetiky. Průměrná hodnota Cmax při požití tučného jídla mírně klesla (o 24 %) a průměrná hodnota AUC se téměř nezměnila (nárůst o 4 %).
Distribuce
Průměrný distribuční objem v ustáleném stavu je přibližně 75 litrů u pacientů s MF a PV. Vazba na plazmatické bílkoviny je in vitro při koncentracích ruxolitinibu odpovídajících klinickému využití přibližně 97 % a ruxolitinib se váže zejména na albumin. Celotělová autoradiografická studie u potkanů ukázala, že ruxolitinib neprochází hematoencefalickou bariérou.
Biotransformace
Ruxolitinib je metabolizován především CYP3A4 (>50 %) s dodatečným přispěním CYP2C9. Mateřská látka má v lidské plazmě převládající podíl a představuje 60 % látek v oběhu souvisejících s podáním přípravku. V plazmě jsou přítomné dva hlavní a zároveň aktivní metabolity, které představují 25 % a 11 % z mateřské AUC. Tyto metabolity mají polovinu až pětinu mateřské farmakologické aktivity na JAK enzymy. Celkově všechny aktivní metabolity přispívají 18 % k celkové farmakodynamické aktivitě ruxolitinibu. V klinických plazmatických koncentracích ruxolitinib podle in vitro studií neinhibuje CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 nebo CYP3A4 a není silným induktorem CYP1A2, CYP2B6 nebo CYP3A4. Na základě in vitro dat může ruxolitinib inhibovat P-gp a BCRP.
Eliminace
Ruxolitinib se eliminuje z organismu převážně metabolickou cestou. Průměrný poločas eliminace ruxolitinibu je přibližně 3 hodiny. Po jednorázovém perorálním podání [14C]-značeného ruxolitinibu zdravým dobrovolníkům bylo farmakum z převážné většiny metabolizováno a 74 % podané radioaktivity bylo vyloučeno močí a 22 % stolicí. Nezměněná mateřská látka představovala méně než 1 % celkové vyloučené radioaktivity.
Linearita/nelinearita
Ve studiích s podáním jednorázové i opakované dávky bylo prokázáno, že systémová expozice se proporcionálně zvyšuje v závislosti na dávce.
Farmakokinetika u zvláštních skupin pacientů
Vliv věku, pohlaví a rasy
Na základě studií u zdravých dobrovolníků nebyly pozorovány relevantní rozdíly ve farmakokinetice v závislosti na pohlaví a rase. Při populační farmakokinetické analýze u pacientů s MF nebyla zjištěna závislost orální clearance na věku nebo rase. Predikovaná orální clearance byla 17,7 l/h u žen a
22,1 l/h u mužů s interindividuální variabilitou 39 % u pacientů s MF. U pacientů s PV byla clearance 12,7 l/h se 42 % interindividuální variabilitou a mezi perorální clearance a pohlavím, stářím pacienta nebo rasou nebyl na základě stanovení farmakokinetiky v této populaci pacientů zřejmý žádný vztah.
Pediatrická populace
Bezpečnost a účinnost Jakavi nebyla zkoumána u pediatrické populace (viz bod 5.1 „Pediatrická populace1“).
Porucha funkce ledvin
Funkce ledvin byla stanovena pomocí MDRD (Modification of Diet in Renal Disease) a kreatininu v moči. Expozice ruxolitinibu byla po jednorázovém podání 25 mg ruxolitinibu shodná u pacientů s různým stupněm poruchy funkce ledvin a u pacientů s normálními renálními funkcemi, avšak hodnoty plazmatické AUC metabolitů ruxolitinibu měly tendenci se zvyšovat se zhoršujícím se postižením ledvin a byly nejvyšší u pacientů s těžkou poruchou funkce ledvin. Není známo, zda má zvýšená expozice metabolitům vliv na bezpečnost. Úprava dávky je doporučená u pacientů s těžkou poruchou funkce ledvin a u nemocných se selháním funkce ledvin (viz bod 4.2). Dávkování ve dnech dialýzy snižuje expozici metabolitům, ale také farmakodynamický účinek, především ve dnech mezi dialýzami.
Porucha funkce jater
Průměrná hodnota AUC ruxolitinibu byla po jednorázovém podání 25 mg ruxolitinibu pacientům s různým stupněm poškození jater zvýšená o 87 %, 28 % a 65 % u pacientů s lehkou, středně těžkou a těžkou poruchou funkce jater (v uvedeném pořadí) ve srovnání s pacienty s normální funkcí jater.
Mezi hodnotou AUC a stupněm jaterního postižení dle Child-Pugh skóre nebyl prokázán žádný vztah. Terminální poločas eliminace byl u pacientů s poškozením jater prodloužen ve srovnání se zdravými dobrovolníky (4,1-5,0 h oproti 2,8 h). U pacientů s poškozením jater je doporučeno přibližně 50% snížení dávky (viz bod 4.2).
5.3 Předklinické údaje vztahující se k bezpečnosti
S ruxolitinibem byly provedeny konvenční farmakologické studie bezpečnosti, toxicity po opakovaném podání, genotoxicity, reprodukční toxicity a hodnocení kancerogenního potenciálu.
V testech toxicity po opakovaném podání byly cílovými orgány spojenými s farmakologickým působením ruxolitinibu kostní dřeň, periferní krev a lymfatické tkáně. U psů byly zjištěny infekce, obecně asociované s imunosupresí. Při telemetrických studiích u psů byl pozorován nežádoucí pokles krevního tlaku a vzestup srdeční frekvence a v respiračních studiích u potkanů byl pozorován nežádoucí pokles minutového objemu. Hraniční dávka (podle Cmax volné látky), při které nebyly pozorovány nežádoucí účinky, byla u psů a potkanů 15,7krát respektive 10,4krát vyšší než je maximální doporučená dávka u lidí (25 mg dvakrát denně). Nebyl pozorován žádný neurofarmakologický účinek ruxolitinibu.
Ruxolitinib snižoval hmotnost plodu a zvyšoval postimplantační ztráty ve studiíích u zvířat. U potkanů a králíků nebyl zjištěn výskyt teratogeních účinků. Nicméně hraniční expozice porovnávané s nejvyšší klinickou dávkou byly nízké a výsledky proto mají pro člověka omezený význam. Nebyl pozorován žádný vliv na fertilitu. V prenatálních a postnatálních vývojových studiích bylo pozorováno mírné prodloužení gestační periody, snížení počtu implantačních míst a snížení počtu porozených mláďat.
U mláďat byla zaznamenána snížená průměrná porodní hmotnost a krátké období snížených průměrných přírůstků hmotnosti po narození. U potkanů v laktaci byly ruxolitinib a/nebo jeho metabolity vylučovány do mateřského mléka, a to v koncentracích 13krát vyšších než v mateřské plazmě. Ruxolitinib neměl mutagenní a klastogenní účinky. Ruxolitinib neměl karcinogenní účinky u Tg.rasH2 transgenních myší.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Mikrokrystalická celulóza Magnesium-stearát Koloidní bezvodý oxid křemičitý Sodná sůl karboxymethylškrobu (typ A)
Povidon Hyprolóza Monohydrát laktózy
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
3 roky
6.4 Zvláštní opatření pro uchovávání
Neuchovávejte při teplotě nad 30°C.
6.5 Druh obalu a obsah balení
Balení s PVC/PCTFE/Al blistry obsahující 14 nebo 56 tablet nebo vícečetná balení obsahující 168 (3 balení po 56) tabletách.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku
Žádné zvláštní požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/12/773/004-006
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
23.08.2012
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Jakavi 10 mg tablety
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna tableta obsahuje ruxolitinibum 10 mg (ve formě ruxolitinibi phosphas).
Pomocné látky se známým účinkem:
Jedna tableta obsahuje 142,90 mg monohydrátu laktózy.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Tableta.
Kulaté zaoblené bílé až téměř bílé tablety o průměru přibližně 9,3 mm s vyraženým „NVR“ na jedné straně a „L10“ na straně druhé.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Myelofibróza (MF)
Přípravek Jakavi je indikován k léčbě dospělých pacientů se splenomegalií nebo s příznaky přidruženými k primární myelofibróze (chronické idiopatické myelofibróze), postpolycytemické myelofibróze nebo myelofibróze po esenciální trombocytemii.
Pravá polvcvtémie (polvcvthaemia vera - PV)
Přípravek Jakavi je indikován k léčbě dospělých pacientů s pravou polycytémií, kteří jsou rezistentní nebo intolerantní k hydroxyurei.
4.2 Dávkování a způsob podání
Léčba Jakavi má být zahajována pouze lékařem, který má zkušenosti s podáváním protinádorové terapie.
Před zahájením léčby přípravkem Jakavi musí být vyšetřen kompletní krevní obraz včetně diferenciálního rozpočtu leukocytů.
Kompletní krevní obraz včetně diferenciálního rozpočtu leukocytů má být vyšetřen každé 2-4 týdny do stabilizace dávek přípravku Jakavi, a dále pak dle klinické indikace (viz bod 4.4).
Dávkování
Počáteční dávka
Doporučená počáteční dávka přípravku Jakavi u myelofibrózy je 15 mg dvakrát denně u pacientů s počtem trombocytů 100* 109/l až 200*109/l a 20 mg dvakrát denně u pacientů s počtem trombocytů >200*109/l. Doporučená počáteční dávka přípravku Jakavi u pravé polycytémie je 10 mg podávaných perorálně dvakrát denně.
U pacientů s počtem trombocytů 50 až <100*109/l není dostatek údajů pro stanovení přesné úvodní dávky. U těchto pacientů je maximální doporučená úvodní dávka 5 mg dvakrát denně a její další titrace má být prováděna velmi opatrně.
Úprava dávkování
Dávky jsou dále titrovány dle bezpečnosti a účinnosti. Léčba by měla být ukončena při poklesu trombocytů na méně než 50*109/l nebo při poklesu absolutního počtu neutrofilů na méně než 0,5*109/l. Při PV by měla být léčba také ukončena, pokud j e hladina hemoglobinu pod 8 g/dl. Po návratu počtu krevních elementů nad tyto hodnoty může být podávání znovu zahájeno v dávce 5 mg dvakrát denně a postupně zvyšováno za pečlivého sledování krevního obrazu včetně diferenciálního rozpočtu leukocytů.
Pokud počet trombocytů klesne pod 100*109/l, mělo by být zváženo snížení dávky, aby nebylo nutné léčbu zcela přerušit pro trombocytopenii. Při PV by se také mělo zvážit snížení dávky, pokud hladina hemoglobinu klesne pod 12 g/dl, a snížení je doporučené, pokud hladina klesne pod 10 g/dl.
Pokud není léčba účinná a počty krevních elementů jsou dostatečné, mohou být dávky zvýšeny, a to maximálně o 5 mg dvakrát denně, až na maximální dávku 25 mg dvakrát denně.
Počáteční dávka nemá být zvyšována během prvních čtyř týdnů léčby, a následně ne častěji než ve dvoutýdenních intervalech.
Maximální dávka přípravku Jakavi je 25 mg dvakrát denně.
Úprava dávkování při konkomitantní léčbě silnými inhibitory CYP3A4 nebo flukonazolem Pokud je přípravek Jakavi podáván spolu se silnými inhibitory CYP3A4 nebo duálními inhibitory enzymů CYP2C9 a CYP3A4 (např. flukonazol), má být jednotlivá dávka přípravku Jakavi, podávaná dvakrát denně, snížena o přibližně 50 % (viz bod 4.5). Je nutné se vyhnout souběžnému podávání přípravku Jakavi s flukonazolem v dávkách vyšších než 200 mg denně.
V průběhu léčby silným inhibitorem CYP3A4 nebo duálními inhibitory enzymů CYP2C9 a CYP3A4 se doporučuje častěji (např. dvakrát týdně) kontrolovat hematologické parametry a pečlivě pátrat po klinických známkách nežádoucích účinků přípravku Jakavi.
Zvláštní skupiny _pacientů Pacienti s poruchou funkce ledvin
U pacientů s mírnou nebo středně závažnou poruchou funkce ledvin není nutná specifická úprava dávky.
U pacientů s MF a těžkou poruchou funkce ledvin (clearance kreatininu méně než 30 ml/min) má být doporučená počáteční dávka, podávaná dvakrát denně, stanovená podle počtu trombocytů redukovaná o přibližně 50 %. Doporučená počáteční dávka pro pacienty s PV s těžkou poruchou funkce ledvin je 5 mg dvakrát denně. U těchto pacientů je třeba pečlivě monitorovat bezpečnost a účinnost léčby.
Pro pacienty s terminálním selháním ledvin (ESRD, end-stage renal disease) léčené hemodialýzou neexistuje dostatek údajů pro stanovení optimálního dávkování. Farmakokinetické/farmakodynamické simulace založené na dostupných údajích o této populaci naznačují, že u hemodialyzovaných pacientů s MF a ESRD má být počáteční dávka podávaná pouze ve dnech hemodialýzy po jejím ukončení, a to 15-20 mg v jednotlivé dávce nebo ve dvou dávkách po 10 mg podávaných po 12 hodinách. Jednotlivá dávka 15 mg je doporučena v případě pacientů s MF a počtem trombocytů 100* 109/l až 200* 109/l. Jednotlivá dávka 20 mg nebo dvě dávky 10 mg podávané po 12 hodinách jsou doporučeny u pacientů s MF a počtem trombocytů >200*109/l. Následující dávky (v jednotlivé dávce nebo ve dvou dávkách 10 mg podávaných po 12 hodinách) mají být podávány pouze v den hemodialýzy, a to po jejím ukončení.
Doporučená počáteční dávka pro pacienty s PV a ESRD na hemodialýze je jednorázová dávka 10 mg nebo dvě dávky 5 mg podávané po 12 hodinách po dialýze, a to pouze v den podání hemodialýzy.
Tato doporučení dávkování jsou založená na simulacích a po jakékoli úpravě dávkování u jednotlivých pacientů s ESRD by mělo následovat pečlivé sledování bezpečnosti a účinnosti léčby. Pro dávkování u pacientů podstupujících peritoneální dialýzu a kontinuální venovenózní hemofiltraci nejsou k dispozici žádná data (viz bod 5.2).
Pacienti s poruchou funkce jater
U pacientů s poruchou funkce jater má být doporučená počáteční dávka, stanovená podle počtu trombocytů a podávaná dvakrát denně, snížena o přibližně 50 %. Následující dávky mají být upraveny na základě pečlivého monitorování bezpečnosti a účinnosti léčby. Pacientům, u nichž bylo v průběhu léčby přípravkem Jakavi zjištěno poškození jater, má být vyšetřen kompletní krevní obraz včetně diferenciálního rozpočtu leukocytů alespoň jednou za jeden až dva týdny v prvních 6 týdnech po zahájení léčby a dále dle klinické potřeby, dokud nejsou jaterní funkce a krevní obraz stabilizovány. Dávka přípravku Jakavi může být dále titrována pro snížení rizika cytopenie.
Starší lidé (>65 let)
Starší lidé nevyžadují žádnou specifickou úpravu dávkování.
Pediatrická populace
Bezpečnost a účinnost přípravku Jakavi u dětí ve věku do 18 let nebyla stanovena. Nejsou dostupné žádné údaje (viz bod 5.1).
Ukončení léčby
Léčba má pokračovat, dokud přínos z léčby převažuje nad rizikem léčby. Pokud ale nedojde během 6 měsíců od započetí léčby ke zmenšení velikosti sleziny nebo zlepšení příznaků, má být léčba ukončena.
U pacientů s určitým stupněm klinického zlepšení se doporučuje léčbu ruxolitinibem přerušit v případě setrvalého zvětšování sleziny o 40 % v porovnání s velikostí ve výchozím stavu (což odpovídá zhruba 25 % zvýšení objemu sleziny), kdy zároveň nedochází k dalšímu zlepšení příznaků spojených s onemocněním.
Způsob podání
Přípravek Jakavi se užívá perorálně, s jídlem nebo bez jídla.
Při vynechání dávky nemá pacient užívat dávku navíc, ale má pokračovat další obvyklou předepsanou dávkou.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1. Těhotenství a laktace.
4.4 Zvláštní upozornění a opatření pro použití
Myelosuprese
Léčba přípravkem Jakavi může způsobit hematologické nežádoucí účinky léku, včetně trombocytopenie, anemie a neutropenie. Před zahájením léčby přípravkem Jakavi musí být vyšetřen kompletní krevní obraz včetně diferenciálního rozpočtu leukocytů. Léčba má být přerušena u pacientů, u kterých dojde k poklesu počtu trombocytů na méně než 50*109/l nebo absolutního počtu neutrofilů na méně než 0,5*109/l (viz bod 4.2).
Bylo zjištěno, že pacienti s nižším počtem trombocytů (<200*109/l) na začátku terapie mají větší riziko vzniku trombocytopenie během léčby.
Trombocytopenie je obvykle reverzibilní a je většinou zvládnuta snížením dávky nebo přechodným vysazením přípravku Jakavi (viz body 4.2 a 4.8). Nicméně v případě klinické potřeby může být nezbytné podání transfuze trombocytů.
Vznik anemie u léčených pacientů si může vyžádat podání krevní transfuze. U těchto pacientů lze také zvážit úpravu dávky nebo přerušení léčby.
Pacienti s hladinou hemoglobinu nižší než 10,0 g/dl při započetí léčby mají vyšší riziko výskytu hladin hemoglobinu pod 8,0 g/dl během léčby v porovnání s pacienty s vyšší počáteční hladinou hemoglobinu (79,3 % oproti 30,1 %). U pacientů s počáteční hladinou hemoglobinu pod 10,0 g/dl je doporučeno častější sledování hematologických parametrů a klinických příznaků a symptomů nežádoucích účinků spojených s přípravkem Jakavi.
Neutropenie (absolutní počet neutrofilů <0,5*109/l) byla obvykle reverzibilní a bylo možné ji zvládnout přechodným vysazením přípravku Jakavi (viz body 4.2 a 4.8).
Kompletní krevní obraz by měl být sledován dle klinické indikace a dávka upravována dle doporučení (viz body 4.2 a 4.8).
Infekce
U všech pacientů má být zhodnoceno riziko vzniku závažné bakteriální, mykobakteriální, mykotické a virové infekce. U pacientů s MF léčených přípravkem Jakavi byla hlášena tuberkulóza. Před zahájením léčby by mělo být u pacientů provedeno vyšetření na aktivní a neaktivní („latentní“) tuberkulózu podle místních doporučení. Může zahrnovat anamnézu, možnost předchozího kontaktu s tuberkulózou a/nebo příslušné vyšetření, jako je rentgenové vyšetření plic, tuberkulinový test a/nebo test uvolnění interferonu gama. Lékaři si mají být vědomi rizika falešně negativních výsledků tuberkulinového kožního testu, a to zejména u pacientů, kteří jsou vážně nemocní nebo mají sníženou imunitu. Léčba přípravkem Jakavi nemá být zahajována, dokud není závažná probíhající infekce zvládnuta. Lékaři mají pečlivě sledovat pacienty léčené přípravkem Jakavi, aby rozpoznali příznaky infekce a zahájili včas adekvátní léčbu (viz bod 4.8).
Zvýšení virové zátěže hepatitidy B (HBV-DNA titru), spolu s asociovaným zvýšením hladin alaninaminotransferázy a aspartátaminotransferázy, nebo bez jejich zvýšení, bylo hlášeno u pacientů s chronickými HBV infekcemi, kteří užívali přípravek Jakavi. Účinek přípravku Jakavi na replikaci viru u pacientů s chronickou infekcí HBV není známý. Pacienti s chronickou HBV infekcí by měli být léčeni a sledováni podle klinických doporučení.
Lékaři mají poučit pacienty o časných příznacích infekce herpes zoster a doporučit jim co možná nejvčasnější vyhledání možnosti léčby v případě infekce.
Progresivní multifokální leukoencefalopatie
Při léčbě pacientů s MF přípravkem Jakavi byla hlášena progresivní multifokální leukoencefalopatie (PML). Lékaři by měli dbát zejména na příznaky, které zaznamená pacient, nasvědčující PML (např. kognitivní, neurologické nebo psychiatrické příznaky nebo známky). U pacientů musí být sledovány jakékoli z těchto nových nebo zhoršujících se příznaků nebo známek a pokud se tyto příznaky/známky objeví, má být zvažováno odeslání k neurologovi a přijetí příslušných diagnostických opatření pro PML. Pokud je podezření na PML, musí být ukončeno další podávání, dokud není PML vyloučena.
Nemelanomové nádory kůže
U pacientů užívajících ruxolitinib byly hlášeny nemelanomové nádory kůže (NMSCs) zahrnující bazocelulární karcinom, spinocelulární karcinom a karcinom z Merkelových buněk. Většina těchto pacientů byla již dříve dlouhodobě léčena hydroxyureou a vyskytovaly se u nich NMSC nebo premaligní kožní léze. Kauzální vztah k ruxolitinibu nebyl prokázán. U pacientů se zvýšeným rizikem rakoviny kůže je doporučené pravidelné vyšetření kůže.
Zvláštní skupiny pacientů
Porucha funkce ledvin
U pacientů s těžkou poruchou funkce ledvin má být snížena úvodní dávka. U hemodialyzovaných pacientů s MF a terminálním selháním ledvin má být úvodní dávka určena dle počtu trombocytů (viz bod 4.2). Následující dávky (jednorázová dávka 20 mg nebo dvě dávky 10 mg podávané po 12 hodinách u pacientů s MF; jednorázová dávka 10 mg nebo dvě dávky 5 mg podávané po 12 hodinách u pacientů s PV) mají být podávány pouze po každé hemodialýze. Další úprava dávkování má být prováděna za pečlivého monitorování bezpečnosti a účinnosti léčby (viz body 4.2 a 5.2).
Porucha funkce jater
U pacientů s poruchou funkce jater má být doporučená úvodní dávka přípravku Jakavi redukována o přibližně 50 %. Další úprava dávkování má být prováděna na základě sledování bezpečnosti a účinnosti léčby (viz body 4.2 a 5.2).
Interakce
Pokud je přípravek Jakavi podáván spolu se silnými inhibitory CYP3A4 nebo duálními inhibitory enzymů CYP3A4 a CYP2C9 (např. flukonazol), má být jednotlivá dávka přípravku Jakavi, podávaná dvakrát denně, snížena o přibližně 50 % (frekvence sledování viz body 4.2 a 4.5).
Souběžné podávání cytoredukční léčby nebo hematopoetických růstových faktorů s přípravkem Jakavi nebylo studováno. Bezpečnost a účinnost těchto souběžných podání nejsou známé (viz bod 4.5).
Následky vysazení
Po přerušení nebo ukončení léčby přípravkem Jakavi se mohou příznaky MF znovu objevit během přibližně jednoho týdne. Závažnější případy byly po přerušení léčby přípravkem Jakavi popsány zejména v souvislosti s akutním interkurentním onemocněním. Není jasné, zda k závažnosti těchto případu přispělo náhlé přerušení léčby. Pokud není náhlé přerušení léčby nutné, je vhodné zvážit postupné snižování dávky, přestože přínos tohoto postupu nebyl prokázán.
Pomocné látky
Přípravek Jakavi obsahuje laktózu. Pacienti se vzácnými dědičnými problémy s intolerancí galaktózy, vrozeným deficitem laktázy nebo malabsorpcí glukózy a galaktózy by tento přípravek neměli užívat.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Studie interakcí byly provedeny pouze u dospělých.
Ruxolitinib je eliminován metabolismem katalyzovaným CYP3A4 a CYP2C9. Léčivé přípravky inhibující tyto enzymy proto mohou zvýšit expozici ruxolitinibu.
Interakce vyžadující snížení dávky ruxolitinibu
Inhibitory CYP3A4
Silné inhibitory CYP3A4 (jako jsou např. boceprevir, klarithromycin, indinavir, itrakonazol, ketokonazol, lopinavir/ritonavir, mibefradil, nefazodon, nefinavir, posakonazol, sachinavir, telaprevir, telithromycin, vorikonazol)
Podání přípravku Jakavi (jednorázově v dávce 10 mg) následně po podávání silného inhibitoru CYP3A4 ketokonazolu vedlo u zdravých dobrovolníků ke zvýšení Cmax ruxolitinibu o 33 % a AUC ruxolitinibu o 91 % oproti hodnotám dosaženým po podání samotného ruxolitinibu. Poločas přípravku byl při současném podání ketokonazolu prodloužen z 3,7 na 6,0 hodiny.
Pokud je přípravek Jakavi podáván se silnými inhibitory CYP3A4, má být jednotlivá dávka přípravku Jakavi, podávaná dvakrát denně, snížena přibližně o 50 %. Pacienti mají být pečlivě sledováni (např. dvakrát týdně) z důvodu možného vzniku cytopenií a dávka má být titrována na základě hodnocení bezpečnosti a účinnosti léčby (viz bod 4.2).
Duální inhibitory CYP2C9 a CYP3A4
Při společném užití s léky, které jsou duálními inhibitory enzymů CYP2C9 a CYP3A4 (např. flukonazol) má být zváženo 50 % snížení dávky. Je nutné se vyhnout souběžnému podávání přípravku Jakavi s flukonazolem v dávkách vyšších než 200 mg denně.
Induktory enzymů
Induktorv CYP3A4 (jako _jsou např. avasimib, karbamazepin, _fenobarbital, _fenvtoin, rifabutin, rifampin (rifampicin), třezalka tečkovaná (Hypericum perforatum))
Pacienti mají být pečlivě sledováni a dávka titrována s ohledem na bezpečnost a účinnost (viz bod 4.2).
U zdravých dobrovolníků, kterým byl ruxolitinib (jednorázová dávka 50 mg) podán po silném induktoru CYP3A4 rifampicinu (denní dávka 600 mg po dobu 10 dní), byla AUC ruxolitinibu o 70 % nižší než po podání samotného přípravku Jakavi. Expozice aktivním metabolitům ruxolitinibu se nezměnila. V souhrnu byla farmakodynamická aktivita ruxolitinibu podobná, což naznačuje, že indukce CYP3A4 má minimální vliv na farmakodynamiku. Nicméně to může být spojené s vysokou dávkou ruxolitinibu, která vede k farmakodynamickým účinkům blízkým Emax. Je možné, že je nutné u jednotlivých pacientů zvýšit dávku ruxolitinibu při zahájení léčby silným induktorem enzymů.
Další interakce, u nichž je zvažováno ovlivnění nixolitinihu
Slabé nebo středně silné inhibitory CYP3A4 (jako _je např. ciprofloxacin, ervthromvcin, amprenavir, atazanavir, diltiazem, cimetidin)
Podání ruxolitinibu (jednorázově v dávce 10 mg) následně po 4denním podávání erythromycinu 500 mg dvakrát denně vedlo u zdravých dobrovolníků k zvýšení Cmax ruxolitinibu o 8 % a AUC ruxolitinibu o 27 % oproti hodnotám dosaženým po podání samotného ruxolitinibu.
Při souběžném podávání ruxolitinibu se slabými a středně silnými inhibitory CYP3A4 (např. erytromycin) není nutná úprava dávkování. Nicméně pacienti mají být po zahájení léčby středně silnými inhibitory CYP3A4 pečlivě sledováni, zda u nich nedochází k rozvoji cytopenie.
Účinky ruxolitinibu na další léčivé přípravky
Substance transportované P-glykoproteinem a dalšími transportéry
Ruxolitinib může inhibovat P-glykoprotein a protein BCRP (breast cancer resistance protein) ve střevě. To může vést ke zvýšení systémové expozice substrátů těchto transportérů, jako je dabigatran etexilát, cyklosporin, rosuvastatin a případně digoxin. Je doporučené sledování hladiny léčiva (TDM) nebo klinické sledování z důvodu možného ovlivnění takové látky.
Je možné, že potenciální inhibice P-gp a BCRP ve střevě může být minimalizována prodloužením času mezi podáním na nejdelší možnou míru.
Hemopoetické růstové _faktory
Souběžné podání hematopoetických růstových faktorů a přípravku Jakavi nebylo studováno. Není známo, zda inhibice Janus kináz (JAK) přípravkem Jakavi snižuje účinnost hematopoetických růstových faktorů nebo zda hematopoetické růstové faktory ovlivňují účinnost přípravku Jakavi (viz bod 4.4).
Cytoredukční léčba
Souběžné podávání cytoredukční léčby a přípravku Jakavi nebylo studováno. Bezpečnost a účinnost totho souběžného podávání nejsou známé (viz bod 4.4).
Studie u zdravých subjektů prokázala, že ruxolitinib neinhiboval metabolismus perorálního substrátu CYP3A4 midazolamu. Proto není očekáváno zvýšení expozice CYP3A4 substratu při kombinaci s přípravkem Jakavi. Další studie u zdravých subjektů prokázala, že přípravek Jakavi neovlivňuje farmakokinetiku perorálně podávané antikoncepce obsahující ethinylestradiol a levonorgestrel. Proto není očekáváno, že účinnost antikoncepce v této kombinaci bude oslabena souběžným podáváním ruxolitinibu.
4.6 Fertilita, těhotenství a kojení
Těhotenství a antikoncepce u žen
Údaje o podávání přípravku Jakavi těhotným ženám nejsou k dispozici.
Studie u zvířat prokázaly embryotoxický a fetotoxický účinek. Teratogenita nebyla u potkanů a králíků pozorována. Nicméně hraniční expozice byly při porovnání s nejvyšší klinickou dávkou nízké a výsledky mají proto pro člověka omezený význam (viz bod 5.3). Potenciální riziko u člověka není známo. Z hlediska bezpečnosti je podání přípravku Jakavi během těhotenství kontraindikováno (viz bod 4.3). Ženy ve fertilním věku mají během léčby přípravkem Jakavi používat účinnou antikoncepci. V případě otěhotnění v průběhu užívání přípravku Jakavi je nutné individuální zhodnocení rizika a profitu léčby a pečlivý odhad potenciálního rizika pro plod (viz bod 5.3).
Kojení
Přípravek Jakavi nesmí být podáván během kojení (viz bod 4.3) a kojení má být při zahájení léčby ukončeno. Není známo, zda se ruxolitinib a/nebo jeho metabolity vylučují do lidského mateřského mléka. Riziko pro kojené dítě nelze vyloučit. Dostupná farmakodynamická a toxikologická data prokázala vylučování ruxolitinibu a jeho metabolitů do mateřského mléka u studovaných zvířat (viz bod 5.3).
Fertilita
Nejsou k dispozici žádné klinické údaje týkající se ovlivnění fertility u lidí. Ve studiích u zvířat nebyl žádný vliv na fertilitu pozorován.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek Jakavi nemá žádný nebo jen zanedbatelný sedativní účinek. Pokud však pacient pozoruje závratě po užití přípravku Jakavi, má se vyhnout řízení a obsluze strojů.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Bezpečnost byla vyhodnocena u celkem 855 pacientů (s MF nebo PV), kteří užívali přípravek Jakavi ve studiích fáze 2 a 3.
Myelofibróza
V randomizační periodě dvou pivotních studií COMFORT-I a COMFORT-II byl medián trvání expozice přípravku Jakavi 10,8 měsíce (rozmezí 0,3 až 23,5 měsíce). Většina pacientů (68,4 %) byla léčena po dobu nejméně 9 měsíců. Z celkového počtu 301 pacientů mělo 111 (36,9 %) počet krevních destiček ve výchozím stavu mezi 100000/mm3 a 200000/mm3 a 190 (63,1 %) mělo počet krevních destiček ve výchozím stavu >200000/mm3.
V těchto klinických studiích bylo zjištěno ukončení léčby z důvodu výskytu nežádoucích účinků bez ohledu na jejich příčinu u 11,3 % pacientů.
Nejčastějšími hlášenými nežádoucími účinky byla trombocytopenie a anemie.
Hematologické nežádoucí účinky léku (všechny stupně dle CTCAE [Common Terminology Criteria for Adverse Events] klasifikace) zahrnovaly anemii (82,4 %), trombocytopenii (69,8 %) a neutropenii (16,6 %).
Výskyt anemie, trombocytopenie a neutropenie závisí na podávané dávce.
Nejčastějšími třemi nehematologickými nežádoucími účinky léku byla tvorba hematomů (21,3 %), závratě (15,3 %) a bolest hlavy (14,0 %).
Nejčastějšími třemi nehematologickými laboratorními abnormalitami bylo zvýšení ALT (27,2 %), zvýšení AST (19,9 %) a hypercholesterolemie (16,9 %). V klinických studiích fáze 3 u pacientů s MF nebyla pozorována hypercholesterolemie CTCAE stupně 3 nebo 4, zvýšená hladina aspartátaminotransferázy a ani zvýšená hladina alaninaminotransferázy CTCAE stupně 4.
Dlouhodobá bezpečnost: Jak bylo možné očekávat během prodloužené periody sledování, po vyhodnocení bezpečnostních dat 3letého sledování (medián trvání expozice 33,2 měsíců ve studích COMFORT-I a COMFORT-II u pacientů iniciálně randomizovaných k ruxolitinibu) od 457 pacientů s myelofibrózou léčených ruxolitinibem během randomizované a prodloužené periody dvou pivotních studií fáze 3 vzrostly kumulativní četnosti některých nežádoucích účinků. Toto vyhodnocení zahrnovalo data od pacientů, kteří byli iniciálně randomizovaní na ruxolitinib (N=301) a pacientů, kteří užívali ruxolitinib po přechodu z ramen s kontrolní léčbou (N=156). V těchto aktualizovaných datech bylo ukončení léčby z důvodu nežádoucích účinků zjistěno u 17,1 % pacientů léčených ruxolitinibem.
Pravá _ polycytémie
Bezpečnost přípravku Jakavi byla stanovena na základě vyhodnocení 110 pacientů s pravou polycytémií v otevřené, randomizované, kontrolované studii fáze 3 RESPONSE. Níže uvedené nežádoucí účinky reflektují počáteční periodu studie (až do týdne 32) s ekvivalentní expozicí ruxolitinibu a nejlepší dostupnou léčbou (BAT), což odpovídalo mediánu trvání expozice přípravku Jakavi 7,8 měsíce. Průměrný věk pacientů užívajících přípravek Jakavi byl přibližně 60 let.
Ukončení léčby z důvodu výskytu nežádoucích účinků bez ohledu na jejich příčinu bylo zjištěno u 3,6 % pacientů léčených přípravkem Jakavi a u 1,8 % pacientů léčených nejlepší dostupnou léčbou.
Hematologické nežádoucí účinky (jakéhokoli CTCAE stupně) zahrnovaly anemii (43,6 %) a trombocytopenii (24,5 %). Anemie nebo trombocytopenie CTCAE stupně 3 a 4 byly hlášené v 1,8 % nebo 5,5 % případů.
Tři nejčastější nehematologické nežádoucí účinky byly závrať (15,5 %), zácpa (8,2 %) a herpes zoster (6,4 %).
Tři nejčastější nehematologické laboratorní abnormality (jakéhokoli CTCAE stupně) byly hypercholesterolemie (30,0 %), zvýšená hladina alaninaminotransferázy (22,7 %) a zvýšená hladina aspartátaminotransferázy (20,9 %). Ve všech případech šlo o CTCAE stupeň 1 a 2 s výjimkou jednoho případu zvýšené hladiny alaninaminotransferázy CTCAE stupně 3.
Dlouhodobá bezpečnost: Pacienti měli medián trvání expozice přípravku Jakavi 18,6 měsíce (rozmezí 0,3 až 35,9 měsíce). S delší expozicí vzrůstala četnost nežádoucích účinků, nicméně nová bezpečnostní zjištění se neobjevila. Při přepočítání na expozici byl výskyt nežádoucích účinků obecně porovnatelný s výskytem zjištěným během počáteční periody studie.
Tabulární přehled nežádoucích účinků hlášených v klinických studiích
V klinickém studijním programu byla závažnost nežádoucích účinků hodnocena podle CTCAE klasifikace, definující stupeň 1 = lehký, stupeň 2 = středně těžký, stupeň 3 = těžký a stupeň 4 = život ohrožující.
Nežádoucí účinky hlášené v klinických studiích (Tabulka 1) jsou seřazeny podle MedDRA systémově-orgánové klasifikace. V každé systémově-orgánové třídě jsou nežádoucí účinky řazeny podle četnosti tak, že nejčastější nežádoucí účinek je na prvním místě. Četnost přiřazená ke každému nežádoucímu účinku je klasifikována podle následujících kategorií: velmi časté (>1/10); časté (>1/100 až <1/10); méně časté (>1/1 000 až <1/100); vzácné (>1/10 000 až <1/1 000); velmi vzácné (<1/10 000).
Tabulka 1 Kategorie četností nežádoucích účinků hlášených ve studiích fáze 3 (COMFORT-I, COMFORT-II, RESPONSE)
|
Nežádoucí účinky |
Kategorie četnosti pro pacienty s MF |
Kategorie četnosti pro pacienty s PV |
|
Infekce a infestace | ||
|
Infekce močových cesta,d |
Velmi časté |
Časté |
|
Časté |
Časté | |
|
Tuberkulózae |
Méně časté |
- |
|
Poruchy krve a lymfatického systémub,d | ||
|
Anemieb |
- |
- |
|
CTCAEc stupně 4 (<6,5 g/dl) |
Velmi časté |
Méně časté |
|
CTCAEc stupně 3 (<8,0 - 6,5g/dl) |
Velmi časté |
Méně časté |
|
Všechny CTCAEc stupně |
Velmi časté |
Velmi časté |
|
T rombocytopenieb | ||
|
CTCAEc stupně 4 (<25000/mm3) |
Časté |
Méně časté |
|
CTCAEc stupně 3 (50000 - 25000/mm3) |
Časté |
Časté |
|
Všechny CTCAEc stupně |
Velmi časté |
Velmi časté |
|
Neutropenieb | ||
|
CTCAEc stupně 4 (<500/mm3) |
Časté |
- |
|
CTCAEc stupně 3 (<1000 - 500/mm3) |
Časté |
- |
|
Všechny CTCAEc stupně |
Velmi časté |
- |
|
Krvácení (všechny případy krvácení zahrnující intrakraniální a gastrointestinální krvácení, podlitiny a jiné typy krvácení) |
Velmi časté |
Velmi časté |
|
Intrakraniální krvácení |
Časté |
- |
|
Gastrointestinální krvácení |
Časté |
- |
|
Podlitiny |
Velmi časté |
Velmi časté |
|
Jiné typy krvácení (zahrnují epistaxi, krvácení po zákroku a hematurii) |
Časté |
Velmi časté |
|
Poruchy metabolismu a výživy | ||
|
Nárůst tělesné hmotnosti3 |
Velmi časté |
Časté |
|
Hypercholesterolemieb CTCAEc stupně 1 a 2 |
Velmi časté |
Velmi časté |
|
Hypertriglyceridemieb CTCAEc stupně 1 |
- |
Velmi časté |
|
Poruchy nervového systému | ||
|
Závrať1 |
Velmi časté |
Velmi časté |
|
Velmi časté |
- | |
|
Gastrointestinální poruchy | ||
|
Flatulencea |
Časté |
- |
|
Zácpaa |
- |
Časté |
|
Poruchy jater a žlučových cest | ||
|
Zvýšená hladina alaninaminotransferázyb | ||
|
CTCAEc stupně 3 (>5x - 20 x ULN) |
Časté |
Méně časté |
|
Všechny CTCAEc stupně |
Velmi časté |
Velmi časté |
|
Zvýšená hladina aspartátaminotransferázyb | ||
|
Všechny CTCAEc stupně |
Velmi časté |
Velmi časté |
|
Cévní poruchy | ||
|
Hypertenze*1 |
- |
Velmi časté |
|
a Četnost podle údajů o nežádoucích účincích. - Subjekt s mnohočetným výskytem daného nežádoucího účinku (ADR) je v dané kategorii započítán pouze jednou. - Nežádoucí účinky zaznamenané při léčbě nebo 28 dní po ukončení léčby. b Četnost podle laboratorních výsledků. - Subjekt s mnohočetným výskytem daného nežádoucího účinku je v dané kategorii započítán pouze jednou. - Nežádoucí účinky zaznamenané při léčbě nebo 28 dní po ukončení léčby. c Common Terminology Criteria for Adverse Events (CTCAE) klasifikace verze 3.0; stupeň 1 = lehký, stupeň 2 = středně těžký, stupeň 3 = těžký, stupeň 4 = život ohrožující d Tyto nežádoucí účinky j sou diskutovány v textu. e Četnost je založená na údajích o všech pacientech vystavených ruxolitinibu v klinických studiích (N=4755). | ||
Po ukončení léčby se mohou u pacientů s MF znovu objevit příznaky MF jako je únava, bolesti kostí, horečka, pruritus, noční pocení, symptomatická splenomegalie a úbytek tělesné hmotnosti. Celkové symptomatické skóre pro příznaky MF se v klinických studiích s MF vrátilo k výchozí hodnotě během 7 dnů po vysazení léčby (viz bod 4.4).
Popis vybraných nežádoucích účinků
Anemie
V klinických studiích fáze 3 u pacientů s MF byla střední doba do vzniku první anemie stupně 2 dle CTCAE nebo závažnější 1,5 měsíce. U jednoho pacienta (0,3 %) byla kvůli anemii přerušena léčba.
U pacientů léčených přípravkem Jakavi klesla průměrná hladina hemoglobinu o přibližně 10 g/l oproti vstupní hodnotě s minimem po 8 až 12 týdnech léčby. Poté došlo k opětovnému vzestupu a nastavení nové rovnováhy s hodnotou hemoglobinu přibližně o 5 g/l nižší vůči vstupní hodnotě. Tento průběh byl pozorován u nemocných bez ohledu na použití transfuzní léčby.
V randomizované, placebem kontrolované studii COMFORT-I dostalo v průběhu randomizované léčby 60,6 % pacientů s MF léčených Jakavi a 37,7 % pacientů s MF léčených placebem transfuzi erytrocytů. Ve studii COMFORT-II dostalo transfuzi erytrocytů 53,4 % pacientů ve skupině léčené přípravkem Jakavi a 41,1 % pacientů ve skupině dostávající nejlepší dostupnou léčbu.
V randomizační periodě pivotních studií se anemie vyskytovala méně často u pacientů s PV než u pacientů s MF (43,6 % oproti 82,4 %). V populaci s PV byly hlášené nežádoucí účinky CTCAE stupně 3 a 4 u 1,8 % pacientů, zatímco u pacientů s MF byla četnost 42,56 %.
Trombocytopenie
V klinických studiích fáze 3 u pacientů s MF byla střední doba do vzniku trombocytopenie stupně 3 nebo 4 přibližně osm týdnů. Trombocytopenie byla obvykle po snížení dávky nebo přerušení léčby reverzibilní. Střední čas do obnovení počtu trombocytů nad 50* 109/l byl 14 dní. Během randomizované periody byla podána transfuze trombocytů 4,7 % pacientů, kteří dostávali přípravek Jakavi a 4,0 % pacientů dostávajícím kontrolní léčbu. Léčbu bylo pro trombocytopenii nutno přerušit u 0,7 % pacientů užívajících přípravek Jakavi a u 0,9 % pacientů dostávajících kontrolní léčbu.
Pacienti s počtem trombocytů 100*109/l až 200*109/l před zahájením léčby přípravkem Jakavi měli častěji trombocytopenii stupně 3 nebo 4 v porovnání s pacienty se vstupním počtem trombocytů >200*109/l (64,2 % oproti 38,5 %).
V randomizačních periodách pivotních studií byl podíl pacientů s trombocytopenií nižší u pacientů s PV (24,5 %) oproti pacientům s MF (69,8 %). Četnost vážné (tj. CTCAE stupeň 3 a 4) trombocytopenie byla nižší u pacientů s PV (5,5 %) než u pacientů s MF (11,6 %).
Neutropenie
V klinických studiích fáze 3 u pacientů s MF byla střední doba do vzniku neutropenie stupně 3 nebo 4 dvanáct týdnů. Během randomizované periody bylo zaznamenáno přerušení léčby nebo snížení dávky pro neutropenii u 1,0 % pacientů a 0,3 % pacientů muselo kvůli neutropenii léčbu ukončit.
V randomizační periodě pivotní studie u pacientů s PV byla neutropenie hlášená u dvou pacientů (1,8 %), z nichž u jednoho pacienta došlo k rozvoji neutropenie CTCAE stupně 4.
Krvácení
V pivotních klinických studiích fáze 3 u pacientů s MF byly krvácivé komplikace (zahrnující nitrolební krvácení, krvácení do zažívacího traktu, hematomy a jiné typy krvácení) zaznamenány u 32,6 % pacientů užívajících přípravek Jakavi a u 23,2 % pacientů užívajících kontrolní léčby (placebo nebo nejlepší dostupnou léčbu). Výskyt příhod krvácení stupně 3 nebo 4 byl stejný u pacientů léčených přípravkem Jakavi s pacienty léčenými kontrolními léčbami (4,7 % oproti 3,1 %). U většiny pacientů s krvácivými komplikacemi hlášenými během léčby byly hlášeny hematomy (65,3 %). Hematomy byly častěji zaznamenány u pacientů léčených přípravkem Jakavi v porovnání
s referenčními léčbami (21,3 % oproti 11,6 %). Nitrolební krvácení bylo hlášeno u 1 % pacientů léčených přípravkem Jakavi a u 0,9 % léčených kontrolními léčbami. Gastrointestinální krvácení bylo hlášeno u 5,0 % pacientů léčených přípravkem Jakavi oproti 3,1 % pacientů léčených kontrolními léčbami. Jiné typy krvácivých komplikací (zahrnující případy jako je epistaxe, krvácení po zákroku a hematurie) byly hlášeny u 13,3 % pacientů léčených přípravkem Jakavi a u 10,3 % pacientů léčených kontrolní léčbou.
V randomizační periodě pivotní studie u pacientů s PV byly krvácivé příhody (zahrnující intrakraniální a gastrointestinální krvácení, podlitiny a jiné typy krvácení) hlášeny u 20 % pacientů léčených přípravkem Jakavi a u 15,3 % pacientů s nejlepší dostupnou léčbou. Podlitiny byly hlášené s podobnými četnostmi v ramenech s přípravkem Jakavi a BAT (10,9 % vs. 8,1 %). U pacientů léčených přípravkem Jakavi nebyl hlášen ani jeden případ intrakraniálního nebo gastrointestinálního krvácení. Jeden pacient léčený přípravkem Jakavi měl krvácivou příhodu stupně 3 (krvácení po zákroku); krvácení stupně 4 nebylo hlášeno. Ostatní krvácivé příhody (zahrnující příhody jako epistaxi, krvácení po zákroku, krvácení dásní) byly hlášeny u 11,8 % pacientů léčených přípravkem Jakavi a u 6,3 % pacientů s nejlepší dostupnou léčbou.
Infekce
V klinických studiích fáze 3 u pacientů s MF byly hlášeny infekce močového traktu stupně 3 nebo 4 u 1,0 % pacientů, herpes zoster u 4,3 % a tuberkulóza u 1,0 % pacientů. V klinických studiích fáze 3 byla hlášena sepse u 3,0 % pacientů. Prodloužené sledování pacientů léčených ruxolitinibem neprokázalo žádné trendy ke zvýšení výskytu sepse v čase.
V randomizační periodě pivotní studie u pacientů s PV byl hlášen jeden případ (0,9 %) infekce močových cest CTCAE stupně 3, infekce močových cest CTCAE stupně 4 nebyla hlášena. Výskyt herpes zoster byl nepatrně vyšší u pacientů s PV (6,4 %) oproti pacientům s MF (4,0 %). U pacientů s PV se objevilo jedno hlášení postherpetické neuralgie CTCAE stupně 3.
Zvýšený systolický krevní tlak
V pivotních klinických studiích fáze 3 u pacientů s MF bylo u 31,5 % pacientů během alespoň jedné návštěvy v porovnání s 19,5 % pacientů léčených kontrolní léčbou zjištěno zvýšení systolického krevního tlaku o 20 mmHg nebo více oproti výchozímu stavu. Ve studii COMFORT-I (pacienti s MF) bylo průměrné zvýšení systolického krevního tlaku oproti výchozímu stavu o 0-2 mmHg u přípravku Jakavi v porovnání s poklesem o 2-5 mmHg v rameni s placebem. Ve studii COMFORT-II vykazovaly průměrné hodnoty u pacientů s MF léčených ruxolitinibem v porovnání s pacienty s MF léčenými kontrolní léčbou malé rozdíly.
V randomizační periodě pivotní studie u pacientů s PV se průměrný systolický krevní tlak zvýšil o 0,65 mmHg u přípravku Jakavi v porovnání s poklesem o 2 mmHg u ramene s BAT.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Není známo žádné antidotum pro předávkování přípravkem Jakavi. Jednorázové podání až 200 mg přípravku Jakavi bylo poměrně dobře snášeno. Opakované podání vyšších než doporučených dávek je spojeno s vyšším výskytem myelosuprese, zahrnující leukopenii, anemii a trombocytopenii. V těchto případech má být podávána vhodná podpůrná léčba.
Nepředpokládá se, že by hemodialýza zvyšovala vylučování ruxolitinibu.
FARMAKOLOGICKÉ VLASTNOSTI
5.
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Cytostatika, inhibitory proteinkinázy, ATC kód: L01XE18 Mechanismus účinku
Ruxolitinib je selektivní inhibitor Janus kináz (JAK) JAK1 a JAK2 (hodnoty IC50 jsou pro enzym JAK1 3,3 nM a pro JAK2 2,8 nM). Tyto kinázy jsou zapojeny do signalizace mnoha cytokinů a růstových faktorů s významnou úlohou při hematopoéze a imunitních funkcích.
Myelofibróza a pravá polycytémie j sou myeloproliferativní neoplastická onemocnění, u kterých byla popsána abnormální regulace signalizace zprostředkované JAK1 a JAK2. Předpokládá se, že příčinou poruchy regulace jsou vysoké hladiny cirkulujících cytokinů, které aktivují signální dráhu JAK-STAT, mutace zvyšující funkci enzymů jako je JAK2V617F a potlačení negativních regulačních mechanismů. U pacientů s MF nacházíme změnu regulace zprostředkované JAK bez ohledu na přítomnost mutace JAK2V617F. Aktivační mutace v JAK2 (V617F nebo exon 12) jsou zjištěné u >95 % pacientů s PV.
Ruxolitinib inhibuje signální dráhu JAK-STAT a buněčnou proliferaci u buněčných modelů hematologických malignit závislých na cytokinech, stejně jako u na cytokinech nezávislého modelu využívajícího Ba/F3 buňky exprimující JAK2V617F mutovaný protein s hodnotou IC50 v rozmezí 80-320 nM.
Farmakodynamické účinky
Ruxolitinib inhibuje cytokiny indukovanou STAT3 fosforylaci v krvi zdravých dobrovolníků i pacientů s MF a pacientů s PV. U obou skupin vedlo podávání ruxolitinibu k maximální inhibici STAT3 fosforylace 2 hodiny po podání s návratem k téměř výchozím hodnotám do 8 hodin po podání, což ukazuje na to, že nedochází k akumulaci mateřské látky ani aktivních metabolitů.
U pacientů s MF došlo při léčbě ruxolitinibem k poklesu zvýšených zánětlivých markerů jako je TNFa, IL-6 a CRP. Pacienti s MF se v průběhu času nestávali refrakterní k farmakodynamickým účinkům ruxolitinibu. Také pacienti s PV vykazovali obdobné zvýšení zánětlivých markerů ve výchozím stavu a tyto markery klesaly po léčbě ruxolitinibem.
V QT studiích u zdravých dobrovolníků nebylo zaznamenáno prodloužení QT/QTc intervalu po jednorázovém podání dávky ruxolitinibu vyšší než terapeutické (až 200 mg), což ukazuje na to, že ruxolitinib nemá vliv na srdeční repolarizaci.
Klinická účinnost a bezpečnost
Myelofibróza
U pacientů s MF (primární myelofibrózou, postpolycytemickou myelofibrózou nebo myelofibrózou po esenciální trombocytemii) byly provedeny dvě randomizované studie fáze 3 (COMFORT-I a COMFORT-II). V obou studiích měli pacienti hmatnou splenomegalii alespoň 5 cm pod žeberním obloukem a byli klasifikovaní podle kritérií International Working Group (IWG) do kategorií středního rizika 2 nebo vysokého rizika. Úvodní dávka přípravku Jakavi byla určena podle počtu trombocytů.
COMFORT-I byla dvojitě slepá, randomizovaná, placebem kontrolovaná studie zahrnující 309 pacientů, kteří byli refrakterní, nebo nebyli vhodnými kandidáty pro dostupnou léčbu. Primární cílový parametr účinnosti byl podíl nemocných, kteří dosáhnou ve 24. týdnu alespoň 35% zmenšení objemu sleziny oproti výchozí hodnotě měřeného pomocí magnetické rezonance (MRI) nebo počítačové tomografie (CT).
Sekundárními cílovými parametry bylo trvání udržení alespoň 35% redukce objemu sleziny proti výchozí hodnotě, podíl pacientů, kteří ve 24. týdnu dosáhli alespoň 50% snížení celkového symptomatického skóre, změna celkového symptomatického skóre od výchozího stavu do 24. týdne hodnocená pomocí modifikovaného MFSAF (Myelofibrosis Symptom Assessment Form) formuláře (verze 2.0, elektronický deník) a celkové přežití.
COMFORT-II byla otevřená, randomizovaná studie zahrnující 219 pacientů. Pacienti byli randomizováni v poměru 2:1 k léčbě přípravkem Jakavi oproti nejlepší dostupné léčbě. Ve skupině s nejlepší dostupnou léčbou dostávalo 47 % pacientů hydroxyureu a 16 % pacientů glukokortikoidy. Primární cílový ukazatel účinnosti léčby byl podíl nemocných, kteří dosáhnou ve 48. týdnu alespoň 35% snížení objemu sleziny oproti výchozí hodnotě měřeného pomocí MRI nebo CT.
Sekundární cílové parametry ve studii COMFORT-II zahrnovaly podíl nemocných, kteří dosáhnou ve 24. týdnu alespoň 35% snížení objemu sleziny oproti výchozí hodnotě a dobu trvání alespoň 35% snížení objemu slezinyoproti výchozí hodnotě.
Ve studiích COMFORT-I a COMFORT-II byly vstupní demografické parametry a charakteristiky onemocnění srovnatelné u obou léčených skupin.
Tabulka 2 Podíl pacientů (%) s >35% snížením objemu sleziny oproti výchozí hodnotě ve 24. týdnu ve studii COMFORT-I a ve 48. týdnu ve studii COMFORT-II (ITT)
|
COMFORT-I |
COMFORT-II | |||
|
Jakavi (N=155) |
Placebo (N=153) |
Jakavi (N=144) |
Nejlepší dostupná léčba (N=72) | |
|
Časový bod |
24. týden |
48. týden | ||
|
Počet (%) pacientů s redukcí objemu sleziny o >35 % |
65 (41,9) |
1 (0,7) |
41 (28,5) |
0 |
|
95% interval spolehlivosti |
34,1; 50,1 |
0; 3,6 |
21,3; 36,6 |
0,0; 5,0 |
|
Hodnota p |
<0,0001 |
<0,0001 | ||
Významně vyšší podíl pacientů ve skupině léčené přípravkem Jakavi dosáhl alespoň 35% snížení objemu sleziny (Tabulka 2) oproti výchozí hodnotě, a to bez ohledu na přítomnost mutace JAK2V617F nebo podtypu onemocnění (primární myelofibróza, postpolycytemická myelofibróza nebo myelofibróza po esenciální trombocytemii).
Tabulka 3 Podíl pacientů (%) s >35% snížením objemu sleziny oproti výchozí hodnotě dle mutačního stavu JAK (hodnocení bezpečnosti)
|
COMFORT-I |
COMFORT-II | |||||||
|
Jakavi |
Placebo |
Jakavi |
Nejlepší dostupná léčba | |||||
|
JAK mutační stav |
Pozitivní (N=113) n (%) |
Negativní (N=40) n (%) |
Pozitivní (N=121) n (%) |
Negativní (N=27) n (%) |
Pozitivní (N=110) n (%) |
Negativní (N=35) n (%) |
Pozitivní (N=49) n (%) |
Negativní (N=20) n (%) |
|
Počet (%) pacientů s redukcí objemu sleziny o >35 % |
54 (47,8) |
11 (27,5) |
1 (0,8) |
0 |
36 (32,7) |
5 (14,3) |
0 |
0 |
|
Časový bod |
Po 24 týdnech |
Po 48 týdnech | ||||||
Pravděpodobnost zachování odpovědi sleziny (>35% snížení) na přípravku Jakavi po dobu nejméně 24 týdnů byla 89 % ve studii COMFORT-I a 87 % ve studii COMFORT-II; 52 % udrželo odpověď sleziny po dobu nejméně 48 týdnů ve studím COMFORT-II.
Ve studii COMFORT-I dosáhlo 45,9 % subjektů ve skupině s přípravkem Jakavi >50% zlepšení celkového symptomatického skóre od výchozího stavu ve 24. týdnu (vyhodnocovaného pomocí MFSAF deníku v2.0) v porovnání s 5,3 % pacientů ve skupině s placebem (p<0,0001 za pomoci chí-kvadrát testu). Průměrná změna celkového zdravotního stavu ve 24. týdnu měřená pomocí dotazníku EORTC QLQ C30 byla +12,3 u přípravku Jakavi a -3,4 u placeba (p<0,0001).
Ve studii COMFORT-I byl při mediánu sledování 34,3 měsíce výskyt úmrtí u pacientů randomizovaných do ramene s ruxolitinibem 27,1 % oproti 35,1 % u pacientů randomizovaných do ramene s placebem; HR 0,687; 95% CI 0,459-1,029; p=0,0668.
Ve studii COMFORT-II byl při mediánu sledování 34,7 měsíce výskyt úmrtí u pacientů randomizovaných do ramene s ruxolitinibem 19,9 % oproti 30,1 % u pacientů randomizovaných k nejlepší dostupné léčbě (BAT); HR 0,48; 95% CI 0,28-0,85; p=0,009. V obou studiích byly nižší výskyty úmrtí zjištěné v rameni s ruxolitinibem ovlivněny především výsledky získanými od pacientů z podskupin po pravé polycytémii a po esenciální trombocytémii.
Pravá _ polycytémie
Randomizovaná, otevřená, aktivně kontrolovaná studie fáze 3 (RESPONSE) byla provedena u 222 pacientů s PV, kteří byli rezistentní nebo intolerantní k hydroxyurei definované podle publikovaných kritérií mezinárodní pracovní skupiny evropské leukemické sítě (ELN). 110 pacientů bylo randomizovaných do ramene s ruxolitinibem a 112 pacientů do ramene s nejlepší dostupnou léčbou. Zahajovací dávka přípravku Jakavi byla 10 mg dvakrát denně. Poté byly u pacientů dávky individuálně upraveny na základě tolerability a účinnosti s maximální dávkou 25 mg dvakrát denně. Nejlepší dostupná léčba byla vybraná zkoušejícím individuálně pro každého pacienta a zahrnovala hydroxyureu (59,5%), interferon/pegylovaný interferon (11,7 %), anagrelid (7,2 %), pipobroman (1,8 %) a pozorování (15,3 %).
Výchozí demografické parametry a charakteristika onemocnění byly v obou ramenech léčby srovnatelné. Medián věku byl 60 let (rozmezí 33 až 90 let). Pacienti v rameni s ruxolitinibem měli medián doby od diagnózy PV 8,2 roku a dříve užívali hydroxyureu s mediánem trvání léčby přibližně 3 roky. Většina pacientů (>80 %) měla provedeny nejméně dvě flebotomie v posledních 24 týdnech před zařazením do studie. Komparativní data týkající se dlouhodobého přežívání a výskytu komplikací onemocnění chybí.
Primárním složeným cílovým parametrem byl podíl pacientů, kteří dosáhli jak nevhodnosti k flebotomii (kontrolu HCT), tak i >35% zmenšení objemu sleziny od výchozího stavu do 32. týdne. Kritérium vhodnosti k provedení flebotomie bylo definováno jako potvrzený HCT >45%, tj. minimálně o 3 procentní body vyšší oproti HCT stanovenému ve výchozím stavu nebo potvrzenému HCT >48%, dle toho toho, která hodnota je nižší. Klíčové sekundární cílové parametry zahrnovaly podíl pacientů s dosaženým primárním cílovým parametrem a bez progrese ve 48. týdnu, stejně jako podíl pacientů s dosaženou kompletní hematologickou remisí ve 32. týdnu.
Studie dosáhla svého primárního cíle, vyšší podíl pacientů ve skupině s přípravkem Jakavi dosáhl primárního složeného cílového parametru a každé z jeho individuálních komponent. Výrazně vyšší počet pacientů léčených přípravkem Jakavi (20,9 %) dosáhl primární odpovědi (p<0,0001) v porovnání s nejlepší dostupnou léčbou (0,9 %). Kontroly hematokritu bylo dosaženo u 60 % pacientů v rameni s přípravkem Jakavi v porovnání s 19,6 % v rameni s nejlepší dostupnou léčbou a >35% snížení objemu sleziny bylo dosaženo u 38,2 % pacientů v rameni s přípravkem Jakavi v porovnání s 0,9 % v rameni s nejlepší dostupnou léčbou (obrázek 1). 94 (83,9 %) pacientů randomizovaných k rameni s nejlepší dostupnou léčbou přešlo ve 32. týdnu nebo později na léčbu ruxolitinibem, což omezuje porovnání obou ramen po 32. týdnu.
Také bylo dosaženo obou klíčových sekundárních cílových parametrů. Poměr pacientů, kteří dosáhli kompletní hematologické remise, byl 23,6 % u přípravku Jakavi v porovnání s 8,9 % u nejlepší dostupné léčby (p=0,0028) a poměr pacientů, kteří dosáhli trvalé primární odpovědi ve 48. týdnu, byl
19,1 % u přípravku Jakavi a 0,9 % u nejlepší dostupné léčby (p<0,0001).
Obrázek 1 Pacienti, kteří dosáhli primárního cílového parametru a komponent primárního cílového parametru ve 32. týdnu
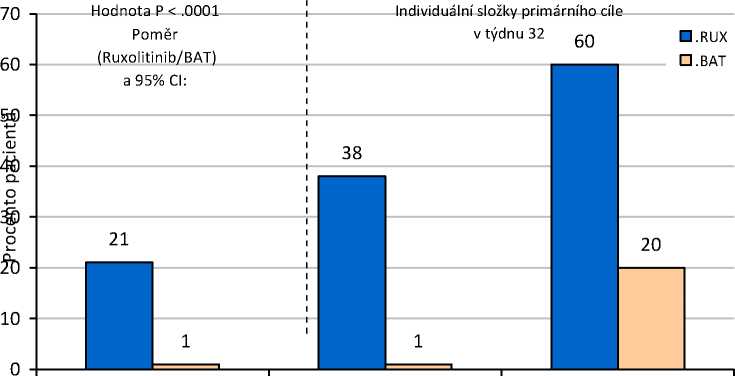
Primární složený cíl v > 35% redukce objemu Kontrola hematokritu bez týdnu 32 sleziny flebotomie
Symptomatická zátěž byla vyhodnocena pomocí elektronického pacientského dotazníku MPN-SAF celkového symptomatického skóre (TSS), který se skládá ze 14 otázek. Ve 32. týdnu dosáhlo 49 % a 64 % pacientů léčených ruxolitinibem >50% snížení TSS-14 resp. TSS-5 v porovnání s pouze 5 % a 11 % pacientů na nejlepší dostupné léčbě.
Vnímání prospěchu z léčby bylo stanoveno pomocí dotazníku Patient Global Impression of Change (PGIC). 66 % pacientů léčených ruxolitinibem hlásilo zlepšení již čtyři týdny po zahájení léčby oproti 9 % léčených nejlepší dostupnou léčbou. Zlepšení vnímání prospěchu z léčby bylo vyšší také u pacientů léčených ruxolitinibem ve 32. týdnu (78 % oproti 33 %).
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Jakavi u všech podskupin pediatrické populace pro léčbu MF (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Absorpce
Ruxolitinib patří do třídy 1 podle BCS (Biopharmaceutical Classification System), protože má vysokou prostupnost, dobrou rozpustnost a rychlý rozpad. Ruxolitinib byl v klinických studiích po perorálním podání rychle absorbován s maximální plazmatickou koncentrací (Cmax) dosaženou přibližně 1 hodinu po podání. Absorpce ruxolitinibu po perorálním podání (stanoven ruxolitinib a jeho metabolity tvořené při prvním průchodu játry) je podle farmakokinetických studií u lidí 95 % a více. Průměrné hodnoty Cmax a AUC ruxolitinibu se zvyšují úměrně s podanou jednorázovou dávkou v rozmezí 5-200 mg. Užití ruxolitinibu po tučném jídle nevedlo ke klinicky významné změně farmakokinetiky. Průměrná hodnota Cmax při požití tučného jídla mírně klesla (o 24 %) a průměrná hodnota AUC se téměř nezměnila (nárůst o 4 %).
Distribuce
Průměrný distribuční objem v ustáleném stavu je přibližně 75 litrů u pacientů s MF a PV. Vazba na plazmatické bílkoviny je in vitro při koncentracích ruxolitinibu odpovídajících klinickému využití přibližně 97 % a ruxolitinib se váže zejména na albumin. Celotělová autoradiografická studie u potkanů ukázala, že ruxolitinib neprochází hematoencefalickou bariérou.
Biotransformace
Ruxolitinib je metabolizován především CYP3A4 (>50 %) s dodatečným přispěním CYP2C9. Mateřská látka má v lidské plazmě převládající podíl a představuje 60 % látek v oběhu souvisejících s podáním přípravku. V plazmě jsou přítomné dva hlavní a zároveň aktivní metabolity, které představují 25 % a 11 % z mateřské AUC. Tyto metabolity mají polovinu až pětinu mateřské farmakologické aktivity na JAK enzymy. Celkově všechny aktivní metabolity přispívají 18 % k celkové farmakodynamické aktivitě ruxolitinibu. V klinických plazmatických koncentracích ruxolitinib podle in vitro studií neinhibuje CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 nebo CYP3A4 a není silným induktorem CYP1A2, CYP2B6 nebo CYP3A4. Na základě in vitro dat může ruxolitinib inhibovat P-gp a BCRP.
Eliminace
Ruxolitinib se eliminuje z organismu převážně metabolickou cestou. Průměrný poločas eliminace ruxolitinibu je přibližně 3 hodiny. Po jednorázovém perorálním podání [14C]-značeného ruxolitinibu zdravým dobrovolníkům bylo farmakum z převážné většiny metabolizováno a 74 % podané radioaktivity bylo vyloučeno močí a 22 % stolicí. Nezměněná mateřská látka představovala méně než 1 % celkové vyloučené radioaktivity.
Linearita/nelinearita
Ve studiích s podáním jednorázové i opakované dávky bylo prokázáno, že systémová expozice se proporcionálně zvyšuje v závislosti na dávce.
Farmakokinetika u zvláštních skupin pacientů
Vliv věku, pohlaví a rasy
Na základě studií u zdravých dobrovolníků nebyly pozorovány relevantní rozdíly ve farmakokinetice v závislosti na pohlaví a rase. Při populační farmakokinetické analýze u pacientů s MF nebyla zjištěna závislost orální clearance na věku nebo rase. Predikovaná orální clearance byla 17,7 l/h u žen a
22,1 l/h u mužů s interindividuální variabilitou 39 % u pacientů s MF. U pacientů s PV byla clearance 12,7 l/h se 42 % interindividuální variabilitou a mezi perorální clearance a pohlavím, stářím pacienta nebo rasou nebyl na základě stanovení farmakokinetiky v této populaci pacientů zřejmý žádný vztah.
Pediatrická populace
Bezpečnost a účinnost Jakavi nebyla zkoumána u pediatrické populace (viz bod 5.1 „Pediatrická populace“).
Porucha funkce ledvin
Funkce ledvin byla stanovena pomocí MDRD (Modification of Diet in Renal Disease) a kreatininu v moči. Expozice ruxolitinibu byla po jednorázovém podání 25 mg ruxolitinibu shodná u pacientů s různým stupněm poruchy funkce ledvin a u pacientů s normálními renálními funkcemi, avšak hodnoty plazmatické AUC metabolitů ruxolitinibu měly tendenci se zvyšovat se zhoršujícím se postižením ledvin a byly nejvyšší u pacientů s těžkou poruchou funkce ledvin. Není známo, zda má zvýšená expozice metabolitům vliv na bezpečnost. Úprava dávky je doporučená u pacientů s těžkou poruchou funkce ledvin a u nemocných se selháním funkce ledvin (viz bod 4.2). Dávkování ve dnech dialýzy snižuje expozici metabolitům, ale také farmakodynamický účinek, především ve dnech mezi dialýzami.
Porucha funkce jater
Průměrná hodnota AUC ruxolitinibu byla po jednorázovém podání 25 mg ruxolitinibu pacientům s různým stupněm poškození jater zvýšená o 87 %, 28 % a 65 % u pacientů s lehkou, středně těžkou a těžkou poruchou funkce jater (v uvedeném pořadí) ve srovnání s pacienty s normální funkcí jater.
Mezi hodnotou AUC a stupněm jaterního postižení dle Child-Pugh skóre nebyl prokázán žádný vztah. Terminální poločas eliminace byl u pacientů s poškozením jater prodloužen ve srovnání se zdravými dobrovolníky (4,1-5,0 h oproti 2,8 h). U pacientů s poškozením jater je doporučeno přibližně 50% snížení dávky (viz bod 4.2).
5.3 Předklinické údaje vztahující se k bezpečnosti
S ruxolitinibem byly provedeny konvenční farmakologické studie bezpečnosti, toxicity po opakovaném podání, genotoxicity, reprodukční toxicity a hodnocení kancerogenního potenciálu.
V testech toxicity po opakovaném podání byly cílovými orgány spojenými s farmakologickým působením ruxolitinibu kostní dřeň, periferní krev a lymfatické tkáně. U psů byly zjištěny infekce, obecně asociované s imunosupresí. Při telemetrických studiích u psů byl pozorován nežádoucí pokles krevního tlaku a vzestup srdeční frekvence a v respiračních studiích u potkanů byl pozorován nežádoucí pokles minutového objemu. Hraniční dávka (podle Cmax volné látky), při které nebyly pozorovány nežádoucí účinky, byla u psů a potkanů 15,7krát respektive 10,4krát vyšší než je maximální doporučená dávka u lidí (25 mg dvakrát denně). Nebyl pozorován žádný neurofarmakologický účinek ruxolitinibu.
Ruxolitinib snižoval hmotnost plodu a zvyšoval postimplantační ztráty ve studiíích u zvířat. U potkanů a králíků nebyl zjištěn výskyt teratogeních účinků. Nicméně hraniční expozice porovnávané s nejvyšší klinickou dávkou byly nízké a výsledky proto mají pro člověka omezený význam. Nebyl pozorován žádný vliv na fertilitu. V prenatálních a postnatálních vývojových studiích bylo pozorováno mírné prodloužení gestační periody, snížení počtu implantačních míst a snížení počtu porozených mláďat.
U mláďat byla zaznamenána snížená průměrná porodní hmotnost a krátké období snížených průměrných přírůstků hmotnosti po narození. U potkanů v laktaci byly ruxolitinib a/nebo jeho metabolity vylučovány do mateřského mléka, a to v koncentracích 13krát vyšších než v mateřské plazmě. Ruxolitinib neměl mutagenní a klastogenní účinky. Ruxolitinib neměl karcinogenní účinky u Tg.rasH2 transgenních myší.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Mikrokrystalická celulóza Magnesium-stearát Koloidní bezvodý oxid křemičitý Sodná sůl karboxymethylškrobu (typ A)
Povidon Hyprolóza Monohydrát laktózy
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
3 roky
6.4 Zvláštní opatření pro uchovávání
Neuchovávejte při teplotě nad 30°C.
6.5 Druh obalu a obsah balení
Balení s PVC/PCTFE/Al blistry obsahující 14 nebo 56 tablet nebo vícečetná balení obsahující 168 (3 balení po 56) tabletách.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku
Žádné zvláštní požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/12/773/014-016
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
23.08.2012
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Jakavi 15 mg tablety
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna tableta obsahuje ruxolitinibum 15 mg (ve formě ruxolitinibi phosphas).
Pomocné látky se známým účinkem:
Jedna tableta obsahuje 214,35 mg monohydrátu laktózy.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Tableta.
Oválné bílé až téměř bílé tablety o rozměrech přibližně 15,0 x 7,0 mm s vyraženým „NVR“ na jedné straně a „L15“ na straně druhé.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Myelofibróza (MF)
Přípravek Jakavi je indikován k léčbě dospělých pacientů se splenomegalií nebo s příznaky přidruženými k primární myelofibróze (chronické idiopatické myelofibróze), postpolycytemické myelofibróze nebo myelofibróze po esenciální trombocytemii.
Pravá polvcvtémie (polvcvthaemia vera - PV)
Přípravek Jakavi je indikován k léčbě dospělých pacientů s pravou polycytémií, kteří jsou rezistentní nebo intolerantní k hydroxyurei.
4.2 Dávkování a způsob podání
Léčba Jakavi má být zahajována pouze lékařem, který má zkušenosti s podáváním protinádorové terapie.
Před zahájením léčby přípravkem Jakavi musí být vyšetřen kompletní krevní obraz včetně diferenciálního rozpočtu leukocytů.
Kompletní krevní obraz včetně diferenciálního rozpočtu leukocytů má být vyšetřen každé 2-4 týdny do stabilizace dávek přípravku Jakavi, a dále pak dle klinické indikace (viz bod 4.4).
Dávkování
Počáteční dávka
Doporučená počáteční dávka přípravku Jakavi u myelofibrózy je 15 mg dvakrát denně u pacientů s počtem trombocytů 100* 109/l až 200*109/l a 20 mg dvakrát denně u pacientů s počtem trombocytů >200*109/l. Doporučená počáteční dávka přípravku Jakavi u pravé polycytémie je 10 mg podávaných perorálně dvakrát denně.
U pacientů s počtem trombocytů 50 až <100*109/l není dostatek údajů pro stanovení přesné úvodní dávky. U těchto pacientů je maximální doporučená úvodní dávka 5 mg dvakrát denně a její další titrace má být prováděna velmi opatrně.
Úprava dávkování
Dávky jsou dále titrovány dle bezpečnosti a účinnosti. Léčba by měla být ukončena při poklesu trombocytů na méně než 50*109/l nebo při poklesu absolutního počtu neutrofilů na méně než 0,5*109/l. Při PV by měla být léčba také ukončena, pokud j e hladina hemoglobinu pod 8 g/dl. Po návratu počtu krevních elementů nad tyto hodnoty může být podávání znovu zahájeno v dávce 5 mg dvakrát denně a postupně zvyšováno za pečlivého sledování krevního obrazu včetně diferenciálního rozpočtu leukocytů.
Pokud počet trombocytů klesne pod 100*109/l, mělo by být zváženo snížení dávky, aby nebylo nutné léčbu zcela přerušit pro trombocytopenii. Při PV by se také mělo zvážit snížení dávky, pokud hladina hemoglobinu klesne pod 12 g/dl, a snížení je doporučené, pokud hladina klesne pod 10 g/dl.
Pokud není léčba účinná a počty krevních elementů jsou dostatečné, mohou být dávky zvýšeny, a to maximálně o 5 mg dvakrát denně, až na maximální dávku 25 mg dvakrát denně.
Počáteční dávka nemá být zvyšována během prvních čtyř týdnů léčby, a následně ne častěji než ve dvoutýdenních intervalech.
Maximální dávka přípravku Jakavi je 25 mg dvakrát denně.
Úprava dávkování při konkomitantní léčbě silnými inhibitory CYP3A4 nebo flukonazolem Pokud je přípravek Jakavi podáván spolu se silnými inhibitory CYP3A4 nebo duálními inhibitory enzymů CYP2C9 a CYP3A4 (např. flukonazol), má být jednotlivá dávka přípravku Jakavi, podávaná dvakrát denně, snížena o přibližně 50 % (viz bod 4.5). Je nutné se vyhnout souběžnému podávání přípravku Jakavi s flukonazolem v dávkách vyšších než 200 mg denně.
V průběhu léčby silným inhibitorem CYP3A4 nebo duálními inhibitory enzymů CYP2C9 a CYP3A4 se doporučuje častěji (např. dvakrát týdně) kontrolovat hematologické parametry a pečlivě pátrat po klinických známkách nežádoucích účinků přípravku Jakavi.
Zvláštní skupiny _pacientů Pacienti s poruchou funkce ledvin
U pacientů s mírnou nebo středně závažnou poruchou funkce ledvin není nutná specifická úprava dávky.
U pacientů s MF a těžkou poruchou funkce ledvin (clearance kreatininu méně než 30 ml/min) má být doporučená počáteční dávka, podávaná dvakrát denně, stanovená podle počtu trombocytů redukovaná o přibližně 50 %. Doporučená počáteční dávka pro pacienty s PV s těžkou poruchou funkce ledvin je 5 mg dvakrát denně. U těchto pacientů je třeba pečlivě monitorovat bezpečnost a účinnost léčby.
Pro pacienty s terminálním selháním ledvin (ESRD, end-stage renal disease) léčené hemodialýzou neexistuje dostatek údajů pro stanovení optimálního dávkování. Farmakokinetické/farmakodynamické simulace založené na dostupných údajích o této populaci naznačují, že u hemodialyzovaných pacientů s MF a ESRD má být počáteční dávka podávaná pouze ve dnech hemodialýzy po jejím ukončení, a to 15-20 mg v jednotlivé dávce nebo ve dvou dávkách po 10 mg podávaných po 12 hodinách. Jednotlivá dávka 15 mg je doporučena v případě pacientů s MF a počtem trombocytů 100* 109/l až 200* 109/l. Jednotlivá dávka 20 mg nebo dvě dávky 10 mg podávané po 12 hodinách jsou doporučeny u pacientů s MF a počtem trombocytů >200*109/l. Následující dávky (v jednotlivé dávce nebo ve dvou dávkách 10 mg podávaných po 12 hodinách) mají být podávány pouze v den hemodialýzy, a to po jejím ukončení.
Doporučená počáteční dávka pro pacienty s PV a ESRD na hemodialýze je jednorázová dávka 10 mg nebo dvě dávky 5 mg podávané po 12 hodinách po dialýze, a to pouze v den podání hemodialýzy.
Tato doporučení dávkování jsou založená na simulacích a po jakékoli úpravě dávkování u jednotlivých pacientů s ESRD by mělo následovat pečlivé sledování bezpečnosti a účinnosti léčby. Pro dávkování u pacientů podstupujících peritoneální dialýzu a kontinuální venovenózní hemofiltraci nejsou k dispozici žádná data (viz bod 5.2).
Pacienti s poruchou funkce jater
U pacientů s poruchou funkce jater má být doporučená počáteční dávka, stanovená podle počtu trombocytů a podávaná dvakrát denně, snížena o přibližně 50 %. Následující dávky mají být upraveny na základě pečlivého monitorování bezpečnosti a účinnosti léčby. Pacientům, u nichž bylo v průběhu léčby přípravkem Jakavi zjištěno poškození jater, má být vyšetřen kompletní krevní obraz včetně diferenciálního rozpočtu leukocytů alespoň jednou za jeden až dva týdny v prvních 6 týdnech po zahájení léčby a dále dle klinické potřeby, dokud nejsou jaterní funkce a krevní obraz stabilizovány. Dávka přípravku Jakavi může být dále titrována pro snížení rizika cytopenie.
Starší lidé (>65 let)
Starší lidé nevyžadují žádnou specifickou úpravu dávkování.
Pediatrická populace
Bezpečnost a účinnost přípravku Jakavi u dětí ve věku do 18 let nebyla stanovena. Nejsou dostupné žádné údaje (viz bod 5.1).
Ukončení léčby
Léčba má pokračovat, dokud přínos z léčby převažuje nad rizikem léčby. Pokud ale nedojde během 6 měsíců od započetí léčby ke zmenšení velikosti sleziny nebo zlepšení příznaků, má být léčba ukončena.
U pacientů s určitým stupněm klinického zlepšení se doporučuje léčbu ruxolitinibem přerušit v případě setrvalého zvětšování sleziny o 40 % v porovnání s velikostí ve výchozím stavu (což odpovídá zhruba 25 % zvýšení objemu sleziny), kdy zároveň nedochází k dalšímu zlepšení příznaků spojených s onemocněním.
Způsob podání
Přípravek Jakavi se užívá perorálně, s jídlem nebo bez jídla.
Při vynechání dávky nemá pacient užívat dávku navíc, ale má pokračovat další obvyklou předepsanou dávkou.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1. Těhotenství a laktace.
4.4 Zvláštní upozornění a opatření pro použití
Mvelosuprese
Léčba přípravkem Jakavi může způsobit hematologické nežádoucí účinky léku, včetně trombocytopenie, anemie a neutropenie. Před zahájením léčby přípravkem Jakavi musí být vyšetřen kompletní krevní obraz včetně diferenciálního rozpočtu leukocytů. Léčba má být přerušena u pacientů, u kterých dojde k poklesu počtu trombocytů na méně než 50*109/l nebo absolutního počtu neutrofilů na méně než 0,5*109/l (viz bod 4.2).
Bylo zjištěno, že pacienti s nižším počtem trombocytů (<200*109/l) na začátku terapie mají větší riziko vzniku trombocytopenie během léčby.
Trombocytopenie je obvykle reverzibilní a je většinou zvládnuta snížením dávky nebo přechodným vysazením přípravku Jakavi (viz body 4.2 a 4.8). Nicméně v případě klinické potřeby může být nezbytné podání transfuze trombocytů.
Vznik anemie u léčených pacientů si může vyžádat podání krevní transfuze. U těchto pacientů lze také zvážit úpravu dávky nebo přerušení léčby.
Pacienti s hladinou hemoglobinu nižší než 10,0 g/dl při započetí léčby mají vyšší riziko výskytu hladin hemoglobinu pod 8,0 g/dl během léčby v porovnání s pacienty s vyšší počáteční hladinou hemoglobinu (79,3 % oproti 30,1 %). U pacientů s počáteční hladinou hemoglobinu pod 10,0 g/dl je doporučeno častější sledování hematologických parametrů a klinických příznaků a symptomů nežádoucích účinků spojených s přípravkem Jakavi.
Neutropenie (absolutní počet neutrofilů <0,5*109/l) byla obvykle reverzibilní a bylo možné ji zvládnout přechodným vysazením přípravku Jakavi (viz body 4.2 a 4.8).
Kompletní krevní obraz by měl být sledován dle klinické indikace a dávka upravována dle doporučení (viz body 4.2 a 4.8).
Infekce
U všech pacientů má být zhodnoceno riziko vzniku závažné bakteriální, mykobakteriální, mykotické a virové infekce. U pacientů s MF léčených přípravkem Jakavi byla hlášena tuberkulóza. Před zahájením léčby by mělo být u pacientů provedeno vyšetření na aktivní a neaktivní („latentní“) tuberkulózu podle místních doporučení. Může zahrnovat anamnézu, možnost předchozího kontaktu s tuberkulózou a/nebo příslušné vyšetření, jako je rentgenové vyšetření plic, tuberkulinový test a/nebo test uvolnění interferonu gama. Lékaři si mají být vědomi rizika falešně negativních výsledků tuberkulinového kožního testu, a to zejména u pacientů, kteří jsou vážně nemocní nebo mají sníženou imunitu. Léčba přípravkem Jakavi nemá být zahajována, dokud není závažná probíhající infekce zvládnuta. Lékaři mají pečlivě sledovat pacienty léčené přípravkem Jakavi, aby rozpoznali příznaky infekce a zahájili včas adekvátní léčbu (viz bod 4.8).
Zvýšení virové zátěže hepatitidy B (HBV-DNA titru), spolu s asociovaným zvýšením hladin alaninaminotransferázy a aspartátaminotransferázy, nebo bez jejich zvýšení, bylo hlášeno u pacientů s chronickými HBV infekcemi, kteří užívali přípravek Jakavi. Účinek přípravku Jakavi na replikaci viru u pacientů s chronickou infekcí HBV není známý. Pacienti s chronickou HBV infekcí by měli být léčeni a sledováni podle klinických doporučení.
Lékaři mají poučit pacienty o časných příznacích infekce herpes zoster a doporučit jim co možná nejvčasnější vyhledání možnosti léčby v případě infekce.
Progresivní multifokální leukoencefalopatie
Při léčbě pacientů s MF přípravkem Jakavi byla hlášena progresivní multifokální leukoencefalopatie (PML). Lékaři by měli dbát zejména na příznaky, které zaznamená pacient, nasvědčující PML (např. kognitivní, neurologické nebo psychiatrické příznaky nebo známky). U pacientů musí být sledovány jakékoli z těchto nových nebo zhoršujících se příznaků nebo známek a pokud se tyto příznaky/známky objeví, má být zvažováno odeslání k neurologovi a přijetí příslušných diagnostických opatření pro PML. Pokud je podezření na PML, musí být ukončeno další podávání, dokud není PML vyloučena.
Nemelanomové nádory kůže
U pacientů užívajících ruxolitinib byly hlášeny nemelanomové nádory kůže (NMSCs) zahrnující bazocelulární karcinom, spinocelulární karcinom a karcinom z Merkelových buněk. Většina těchto pacientů byla již dříve dlouhodobě léčena hydroxyureou a vyskytovaly se u nich NMSC nebo premaligní kožní léze. Kauzální vztah k ruxolitinibu nebyl prokázán. U pacientů se zvýšeným rizikem rakoviny kůže je doporučené pravidelné vyšetření kůže.
Zvláštní skupiny pacientů
Porucha funkce ledvin
U pacientů s těžkou poruchou funkce ledvin má být snížena úvodní dávka. U hemodialyzovaných pacientů s MF a terminálním selháním ledvin má být úvodní dávka určena dle počtu trombocytů (viz bod 4.2). Následující dávky (jednorázová dávka 20 mg nebo dvě dávky 10 mg podávané po 12 hodinách u pacientů s MF; jednorázová dávka 10 mg nebo dvě dávky 5 mg podávané po 12 hodinách u pacientů s PV) mají být podávány pouze po každé hemodialýze. Další úprava dávkování má být prováděna za pečlivého monitorování bezpečnosti a účinnosti léčby (viz body 4.2 a 5.2).
Porucha funkce jater
U pacientů s poruchou funkce jater má být doporučená úvodní dávka přípravku Jakavi redukována o přibližně 50 %. Další úprava dávkování má být prováděna na základě sledování bezpečnosti a účinnosti léčby (viz body 4.2 a 5.2).
Interakce
Pokud je přípravek Jakavi podáván spolu se silnými inhibitory CYP3A4 nebo duálními inhibitory enzymů CYP3A4 a CYP2C9 (např. flukonazol), má být jednotlivá dávka přípravku Jakavi, podávaná dvakrát denně, snížena o přibližně 50 % (frekvence sledování viz body 4.2 a 4.5).
Souběžné podávání cytoredukční léčby nebo hematopoetických růstových faktorů s přípravkem Jakavi nebylo studováno. Bezpečnost a účinnost těchto souběžných podání nejsou známé (viz bod 4.5).
Následky vysazení
Po přerušení nebo ukončení léčby přípravkem Jakavi se mohou příznaky MF znovu objevit během přibližně jednoho týdne. Závažnější případy byly po přerušení léčby přípravkem Jakavi popsány zejména v souvislosti s akutním interkurentním onemocněním. Není jasné, zda k závažnosti těchto případu přispělo náhlé přerušení léčby. Pokud není náhlé přerušení léčby nutné, je vhodné zvážit postupné snižování dávky, přestože přínos tohoto postupu nebyl prokázán.
Pomocné látky
Přípravek Jakavi obsahuje laktózu. Pacienti se vzácnými dědičnými problémy s intolerancí galaktózy, vrozeným deficitem laktázy nebo malabsorpcí glukózy a galaktózy by tento přípravek neměli užívat.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Studie interakcí byly provedeny pouze u dospělých.
Ruxolitinib je eliminován metabolismem katalyzovaným CYP3A4 a CYP2C9. Léčivé přípravky inhibující tyto enzymy proto mohou zvýšit expozici ruxolitinibu.
Interakce vyžadující snížení dávky ruxolitinibu
Inhibitory CYP3A4
Silné inhibitory CYP3A4 (jako jsou např. boceprevir, klarithromycin, indinavir, itrakonazol, ketokonazol, lopinavir/ritonavir, mibefradil, nefazodon, nefinavir, posakonazol, sachinavir, telaprevir, telithromycin, vorikonazol)
Podání přípravku Jakavi (jednorázově v dávce 10 mg) následně po podávání silného inhibitoru CYP3A4 ketokonazolu vedlo u zdravých dobrovolníků ke zvýšení Cmax ruxolitinibu o 33 % a AUC ruxolitinibu o 91 % oproti hodnotám dosaženým po podání samotného ruxolitinibu. Poločas přípravku byl při současném podání ketokonazolu prodloužen z 3,7 na 6,0 hodiny.
Pokud je přípravek Jakavi podáván se silnými inhibitory CYP3A4, má být jednotlivá dávka přípravku Jakavi, podávaná dvakrát denně, snížena přibližně o 50 %. Pacienti mají být pečlivě sledováni (např. dvakrát týdně) z důvodu možného vzniku cytopenií a dávka má být titrována na základě hodnocení bezpečnosti a účinnosti léčby (viz bod 4.2).
Duální inhibitory CYP2C9 a CYP3A4
Při společném užití s léky, které jsou duálními inhibitory enzymů CYP2C9 a CYP3A4 (např. flukonazol) má být zváženo 50 % snížení dávky. Je nutné se vyhnout souběžnému podávání přípravku Jakavi s flukonazolem v dávkách vyšších než 200 mg denně.
Induktory enzymů
Induktorv CYP3A4 (jako _jsou např. avasimib, karbamazepin, _fenobarbital, _fenvtoin, rifabutin, rifampin (rifampicin), třezalka tečkovaná (Hypericum perforatum))
Pacienti mají být pečlivě sledováni a dávka titrována s ohledem na bezpečnost a účinnost (viz bod 4.2).
U zdravých dobrovolníků, kterým byl ruxolitinib (jednorázová dávka 50 mg) podán po silném induktoru CYP3A4 rifampicinu (denní dávka 600 mg po dobu 10 dní), byla AUC ruxolitinibu o 70 % nižší než po podání samotného přípravku Jakavi. Expozice aktivním metabolitům ruxolitinibu se nezměnila. V souhrnu byla farmakodynamická aktivita ruxolitinibu podobná, což naznačuje, že indukce CYP3A4 má minimální vliv na farmakodynamiku. Nicméně to může být spojené s vysokou dávkou ruxolitinibu, která vede k farmakodynamickým účinkům blízkým Emax. Je možné, že je nutné u jednotlivých pacientů zvýšit dávku ruxolitinibu při zahájení léčby silným induktorem enzymů.
Další interakce, u nichž je zvažováno ovlivnění ruxolitinibu
Slabé nebo středně silné inhibitory CYP3A4 (jako _je např. ciprofloxacin, ervthromvcin, amprenavir, atazanavir, diltiazem, cimetidin)
Podání ruxolitinibu (jednorázově v dávce 10 mg) následně po 4denním podávání erythromycinu 500 mg dvakrát denně vedlo u zdravých dobrovolníků k zvýšení Cmax ruxolitinibu o 8 % a AUC ruxolitinibu o 27 % oproti hodnotám dosaženým po podání samotného ruxolitinibu.
Při souběžném podávání ruxolitinibu se slabými a středně silnými inhibitory CYP3A4 (např. erytromycin) není nutná úprava dávkování. Nicméně pacienti mají být po zahájení léčby středně silnými inhibitory CYP3A4 pečlivě sledováni, zda u nich nedochází k rozvoji cytopenie.
Účinky ruxolitinibu na další léčivé přípravky
Substance transportované P-glykoproteinem a dalšími transportéry
Ruxolitinib může inhibovat P-glykoprotein a protein BCRP (breast cancer resistance protein) ve střevě. To může vést ke zvýšení systémové expozice substrátů těchto transportérů, jako je dabigatran etexilát, cyklosporin, rosuvastatin a případně digoxin. Je doporučené sledování hladiny léčiva (TDM) nebo klinické sledování z důvodu možného ovlivnění takové látky.
Je možné, že potenciální inhibice P-gp a BCRP ve střevě může být minimalizována prodloužením času mezi podáním na nejdelší možnou míru.
Hemopoetické růstové _faktory
Souběžné podání hematopoetických růstových faktorů a přípravku Jakavi nebylo studováno. Není známo, zda inhibice Janus kináz (JAK) přípravkem Jakavi snižuje účinnost hematopoetických růstových faktorů nebo zda hematopoetické růstové faktory ovlivňují účinnost přípravku Jakavi (viz bod 4.4).
Cytoredukční léčba
Souběžné podávání cytoredukční léčby a přípravku Jakavi nebylo studováno. Bezpečnost a účinnost totho souběžného podávání nejsou známé (viz bod 4.4).
Studie u zdravých subjektů prokázala, že ruxolitinib neinhiboval metabolismus perorálního substrátu CYP3A4 midazolamu. Proto není očekáváno zvýšení expozice CYP3A4 substratu při kombinaci s přípravkem Jakavi. Další studie u zdravých subjektů prokázala, že přípravek Jakavi neovlivňuje farmakokinetiku perorálně podávané antikoncepce obsahující ethinylestradiol a levonorgestrel. Proto není očekáváno, že účinnost antikoncepce v této kombinaci bude oslabena souběžným podáváním ruxolitinibu.
4.6 Fertilita, těhotenství a kojení
Těhotenství a antikoncepce u žen
Údaje o podávání přípravku Jakavi těhotným ženám nejsou k dispozici.
Studie u zvířat prokázaly embryotoxický a fetotoxický účinek. Teratogenita nebyla u potkanů a králíků pozorována. Nicméně hraniční expozice byly při porovnání s nejvyšší klinickou dávkou nízké a výsledky mají proto pro člověka omezený význam (viz bod 5.3). Potenciální riziko u člověka není známo. Z hlediska bezpečnosti je podání přípravku Jakavi během těhotenství kontraindikováno (viz bod 4.3). Ženy ve fertilním věku mají během léčby přípravkem Jakavi používat účinnou antikoncepci. V případě otěhotnění v průběhu užívání přípravku Jakavi je nutné individuální zhodnocení rizika a profitu léčby a pečlivý odhad potenciálního rizika pro plod (viz bod 5.3).
Kojení
Přípravek Jakavi nesmí být podáván během kojení (viz bod 4.3) a kojení má být při zahájení léčby ukončeno. Není známo, zda se ruxolitinib a/nebo jeho metabolity vylučují do lidského mateřského mléka. Riziko pro kojené dítě nelze vyloučit. Dostupná farmakodynamická a toxikologická data prokázala vylučování ruxolitinibu a jeho metabolitů do mateřského mléka u studovaných zvířat (viz bod 5.3).
Fertilita
Nejsou k dispozici žádné klinické údaje týkající se ovlivnění fertility u lidí. Ve studiích u zvířat nebyl žádný vliv na fertilitu pozorován.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek Jakavi nemá žádný nebo jen zanedbatelný sedativní účinek. Pokud však pacient pozoruje závratě po užití přípravku Jakavi, má se vyhnout řízení a obsluze strojů.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Bezpečnost byla vyhodnocena u celkem 855 pacientů (s MF nebo PV), kteří užívali přípravek Jakavi ve studiích fáze 2 a 3.
Myelofibróza
V randomizační periodě dvou pivotních studií COMFORT-I a COMFORT-II byl medián trvání expozice přípravku Jakavi 10,8 měsíce (rozmezí 0,3 až 23,5 měsíce). Většina pacientů (68,4 %) byla léčena po dobu nejméně 9 měsíců. Z celkového počtu 301 pacientů mělo 111 (36,9 %) počet krevních destiček ve výchozím stavu mezi 100000/mm3 a 200000/mm3 a 190 (63,1 %) mělo počet krevních destiček ve výchozím stavu >200000/mm3.
V těchto klinických studiích bylo zjištěno ukončení léčby z důvodu výskytu nežádoucích účinků bez ohledu na jejich příčinu u 11,3 % pacientů.
Nejčastějšími hlášenými nežádoucími účinky byla trombocytopenie a anemie.
Hematologické nežádoucí účinky léku (všechny stupně dle CTCAE [Common Terminology Criteria for Adverse Events] klasifikace) zahrnovaly anemii (82,4 %), trombocytopenii (69,8 %) a neutropenii (16,6 %).
Výskyt anemie, trombocytopenie a neutropenie závisí na podávané dávce.
Nejčastějšími třemi nehematologickými nežádoucími účinky léku byla tvorba hematomů (21,3 %), závratě (15,3 %) a bolest hlavy (14,0 %).
Nejčastějšími třemi nehematologickými laboratorními abnormalitami bylo zvýšení ALT (27,2 %), zvýšení AST (19,9 %) a hypercholesterolemie (16,9 %). V klinických studiích fáze 3 u pacientů s MF nebyla pozorována hypercholesterolemie CTCAE stupně 3 nebo 4, zvýšená hladina aspartátaminotransferázy a ani zvýšená hladina alaninaminotransferázy CTCAE stupně 4.
Dlouhodobá bezpečnost: Jak bylo možné očekávat během prodloužené periody sledování, po vyhodnocení bezpečnostních dat 3letého sledování (medián trvání expozice 33,2 měsíců ve studích COMFORT-I a COMFORT-II u pacientů iniciálně randomizovaných k ruxolitinibu) od 457 pacientů s myelofibrózou léčených ruxolitinibem během randomizované a prodloužené periody dvou pivotních studií fáze 3 vzrostly kumulativní četnosti některých nežádoucích účinků. Toto vyhodnocení zahrnovalo data od pacientů, kteří byli iniciálně randomizovaní na ruxolitinib (N=301) a pacientů, kteří užívali ruxolitinib po přechodu z ramen s kontrolní léčbou (N=156). V těchto aktualizovaných datech bylo ukončení léčby z důvodu nežádoucích účinků zjistěno u 17,1 % pacientů léčených ruxolitinibem.
Pravá _ polycytémie
Bezpečnost přípravku Jakavi byla stanovena na základě vyhodnocení 110 pacientů s pravou polycytémií v otevřené, randomizované, kontrolované studii fáze 3 RESPONSE. Níže uvedené nežádoucí účinky reflektují počáteční periodu studie (až do týdne 32) s ekvivalentní expozicí ruxolitinibu a nejlepší dostupnou léčbou (BAT), což odpovídalo mediánu trvání expozice přípravku Jakavi 7,8 měsíce. Průměrný věk pacientů užívajících přípravek Jakavi byl přibližně 60 let.
Ukončení léčby z důvodu výskytu nežádoucích účinků bez ohledu na jejich příčinu bylo zjištěno u 3,6 % pacientů léčených přípravkem Jakavi a u 1,8 % pacientů léčených nejlepší dostupnou léčbou.
Hematologické nežádoucí účinky (jakéhokoli CTCAE stupně) zahrnovaly anemii (43,6 %) a trombocytopenii (24,5 %). Anemie nebo trombocytopenie CTCAE stupně 3 a 4 byly hlášené v 1,8 % nebo 5,5 % případů.
Tři nejčastější nehematologické nežádoucí účinky byly závrať (15,5 %), zácpa (8,2 %) a herpes zoster (6,4 %).
Tři nejčastější nehematologické laboratorní abnormality (jakéhokoli CTCAE stupně) byly hypercholesterolemie (30,0 %), zvýšená hladina alaninaminotransferázy (22,7 %) a zvýšená hladina aspartátaminotransferázy (20,9 %). Ve všech případech šlo o CTCAE stupeň 1 a 2 s výjimkou jednoho případu zvýšené hladiny alaninaminotransferázy CTCAE stupně 3.
Dlouhodobá bezpečnost: Pacienti měli medián trvání expozice přípravku Jakavi 18,6 měsíce (rozmezí 0,3 až 35,9 měsíce). S delší expozicí vzrůstala četnost nežádoucích účinků, nicméně nová bezpečnostní zjištění se neobjevila. Při přepočítání na expozici byl výskyt nežádoucích účinků obecně porovnatelný s výskytem zjištěným během počáteční periody studie.
Tabulární přehled nežádoucích účinků hlášených v klinických studiích
V klinickém studijním programu byla závažnost nežádoucích účinků hodnocena podle CTCAE klasifikace, definující stupeň 1 = lehký, stupeň 2 = středně těžký, stupeň 3 = těžký a stupeň 4 = život ohrožující.
Nežádoucí účinky hlášené v klinických studiích (Tabulka 1) jsou seřazeny podle MedDRA systémově-orgánové klasifikace. V každé systémově-orgánové třídě jsou nežádoucí účinky řazeny podle četnosti tak, že nejčastější nežádoucí účinek je na prvním místě. Četnost přiřazená ke každému nežádoucímu účinku je klasifikována podle následujících kategorií: velmi časté (>1/10); časté (>1/100 až <1/10); méně časté (>1/1 000 až <1/100); vzácné (>1/10 000 až <1/1 000); velmi vzácné (<1/10 000).
Tabulka 1 Kategorie četností nežádoucích účinků hlášených ve studiích fáze 3 (COMFORT-I, COMFORT-II, RESPONSE)
|
Nežádoucí účinky |
Kategorie četnosti pro pacienty s MF |
Kategorie četnosti pro pacienty s PV |
|
Infekce a infestace | ||
|
Infekce močových cesta,d |
Velmi časté |
Časté |
|
Časté |
Časté | |
|
Tuberkulózae |
Méně časté |
- |
|
Poruchy krve a lymfatického systémub,d | ||
|
Anemieb |
- |
- |
|
CTCAEc stupně 4 (<6,5 g/dl) |
Velmi časté |
Méně časté |
|
CTCAEc stupně 3 (<8,0 - 6,5g/dl) |
Velmi časté |
Méně časté |
|
Všechny CTCAEc stupně |
Velmi časté |
Velmi časté |
|
T rombocytopenieb | ||
|
CTCAEc stupně 4 (<25000/mm3) |
Časté |
Méně časté |
|
CTCAEc stupně 3 (50000 - 25000/mm3) |
Časté |
Časté |
|
Všechny CTCAEc stupně |
Velmi časté |
Velmi časté |
|
Neutropenieb | ||
|
CTCAEc stupně 4 (<500/mm3) |
Časté |
- |
|
CTCAEc stupně 3 (<1000 - 500/mm3) |
Časté |
- |
|
Všechny CTCAEc stupně |
Velmi časté |
- |
|
Krvácení (všechny případy krvácení zahrnující intrakraniální a gastrointestinální krvácení, podlitiny a jiné typy krvácení) |
Velmi časté |
Velmi časté |
|
Intrakraniální krvácení |
Časté |
- |
|
Gastrointestinální krvácení |
Časté |
- |
|
Podlitiny |
Velmi časté |
Velmi časté |
|
Jiné typy krvácení (zahrnují epistaxi, krvácení po zákroku a hematurii) |
Časté |
Velmi časté |
|
Poruchy metabolismu a výživy | ||
|
Nárůst tělesné hmotnosti3 |
Velmi časté |
Časté |
|
Hypercholesterolemieb CTCAEc stupně 1 a 2 |
Velmi časté |
Velmi časté |
|
Hypertriglyceridemieb CTCAEc stupně 1 |
- |
Velmi časté |
|
Poruchy nervového systému | ||
|
Závrať1 |
Velmi časté |
Velmi časté |
|
Velmi časté |
- | |
|
Gastrointestinální poruchy | ||
|
Flatulencea |
Časté |
- |
|
Zácpaa |
- |
Časté |
|
Poruchy jater a žlučových cest | ||
|
Zvýšená hladina alaninaminotransferázyb | ||
|
CTCAEc stupně 3 (>5x - 20 x ULN) |
Časté |
Méně časté |
|
Všechny CTCAEc stupně |
Velmi časté |
Velmi časté |
|
Zvýšená hladina aspartátaminotransferázyb | ||
|
Všechny CTCAEc stupně |
Velmi časté |
Velmi časté |
|
Cévní poruchy | ||
|
Hypertenze*1 |
- |
Velmi časté |
|
a Četnost podle údajů o nežádoucích účincích. - Subjekt s mnohočetným výskytem daného nežádoucího účinku (ADR) je v dané kategorii započítán pouze jednou. - Nežádoucí účinky zaznamenané při léčbě nebo 28 dní po ukončení léčby. b Četnost podle laboratorních výsledků. - Subjekt s mnohočetným výskytem daného nežádoucího účinku je v dané kategorii započítán pouze jednou. - Nežádoucí účinky zaznamenané při léčbě nebo 28 dní po ukončení léčby. c Common Terminology Criteria for Adverse Events (CTCAE) klasifikace verze 3.0; stupeň 1 = lehký, stupeň 2 = středně těžký, stupeň 3 = těžký, stupeň 4 = život ohrožující d Tyto nežádoucí účinky j sou diskutovány v textu. e Četnost je založená na údajích o všech pacientech vystavených ruxolitinibu v klinických studiích (N=4755). | ||
Po ukončení léčby se mohou u pacientů s MF znovu objevit příznaky MF jako je únava, bolesti kostí, horečka, pruritus, noční pocení, symptomatická splenomegalie a úbytek tělesné hmotnosti. Celkové symptomatické skóre pro příznaky MF se v klinických studiích s MF vrátilo k výchozí hodnotě během 7 dnů po vysazení léčby (viz bod 4.4).
Popis vybraných nežádoucích účinků
Anemie
V klinických studiích fáze 3 u pacientů s MF byla střední doba do vzniku první anemie stupně 2 dle CTCAE nebo závažnější 1,5 měsíce. U jednoho pacienta (0,3 %) byla kvůli anemii přerušena léčba.
U pacientů léčených přípravkem Jakavi klesla průměrná hladina hemoglobinu o přibližně 10 g/l oproti vstupní hodnotě s minimem po 8 až 12 týdnech léčby. Poté došlo k opětovnému vzestupu a nastavení nové rovnováhy s hodnotou hemoglobinu přibližně o 5 g/l nižší vůči vstupní hodnotě. Tento průběh byl pozorován u nemocných bez ohledu na použití transfuzní léčby.
V randomizované, placebem kontrolované studii COMFORT-I dostalo v průběhu randomizované léčby 60,6 % pacientů s MF léčených Jakavi a 37,7 % pacientů s MF léčených placebem transfuzi erytrocytů. Ve studii COMFORT-II dostalo transfuzi erytrocytů 53,4 % pacientů ve skupině léčené přípravkem Jakavi a 41,1 % pacientů ve skupině dostávající nejlepší dostupnou léčbu.
V randomizační periodě pivotních studií se anemie vyskytovala méně často u pacientů s PV než u pacientů s MF (43,6 % oproti 82,4 %). V populaci s PV byly hlášené nežádoucí účinky CTCAE stupně 3 a 4 u 1,8 % pacientů, zatímco u pacientů s MF byla četnost 42,56 %.
Trombocytopenie
V klinických studiích fáze 3 u pacientů s MF byla střední doba do vzniku trombocytopenie stupně 3 nebo 4 přibližně osm týdnů. Trombocytopenie byla obvykle po snížení dávky nebo přerušení léčby reverzibilní. Střední čas do obnovení počtu trombocytů nad 50* 109/l byl 14 dní. Během randomizované periody byla podána transfuze trombocytů 4,7 % pacientů, kteří dostávali přípravek Jakavi a 4,0 % pacientů dostávajícím kontrolní léčbu. Léčbu bylo pro trombocytopenii nutno přerušit u 0,7 % pacientů užívajících přípravek Jakavi a u 0,9 % pacientů dostávajících kontrolní léčbu.
Pacienti s počtem trombocytů 100*109/l až 200*109/l před zahájením léčby přípravkem Jakavi měli častěji trombocytopenii stupně 3 nebo 4 v porovnání s pacienty se vstupním počtem trombocytů >200*109/l (64,2 % oproti 38,5 %).
V randomizačních periodách pivotních studií byl podíl pacientů s trombocytopenií nižší u pacientů s PV (24,5 %) oproti pacientům s MF (69,8 %). Četnost vážné (tj. CTCAE stupeň 3 a 4) trombocytopenie byla nižší u pacientů s PV (5,5 %) než u pacientů s MF (11,6 %).
Neutropenie
V klinických studiích fáze 3 u pacientů s MF byla střední doba do vzniku neutropenie stupně 3 nebo 4 dvanáct týdnů. Během randomizované periody bylo zaznamenáno přerušení léčby nebo snížení dávky pro neutropenii u 1,0 % pacientů a 0,3 % pacientů muselo kvůli neutropenii léčbu ukončit.
V randomizační periodě pivotní studie u pacientů s PV byla neutropenie hlášená u dvou pacientů (1,8 %), z nichž u jednoho pacienta došlo k rozvoji neutropenie CTCAE stupně 4.
Krvácení
V pivotních klinických studiích fáze 3 u pacientů s MF byly krvácivé komplikace (zahrnující nitrolební krvácení, krvácení do zažívacího traktu, hematomy a jiné typy krvácení) zaznamenány u 32,6 % pacientů užívajících přípravek Jakavi a u 23,2 % pacientů užívajících kontrolní léčby (placebo nebo nejlepší dostupnou léčbu). Výskyt příhod krvácení stupně 3 nebo 4 byl stejný u pacientů léčených přípravkem Jakavi s pacienty léčenými kontrolními léčbami (4,7 % oproti 3,1 %). U většiny pacientů s krvácivými komplikacemi hlášenými během léčby byly hlášeny hematomy (65,3 %). Hematomy byly častěji zaznamenány u pacientů léčených přípravkem Jakavi v porovnání
s referenčními léčbami (21,3 % oproti 11,6 %). Nitrolební krvácení bylo hlášeno u 1 % pacientů léčených přípravkem Jakavi a u 0,9 % léčených kontrolními léčbami. Gastrointestinální krvácení bylo hlášeno u 5,0 % pacientů léčených přípravkem Jakavi oproti 3,1 % pacientů léčených kontrolními léčbami. Jiné typy krvácivých komplikací (zahrnující případy jako je epistaxe, krvácení po zákroku a hematurie) byly hlášeny u 13,3 % pacientů léčených přípravkem Jakavi a u 10,3 % pacientů léčených kontrolní léčbou.
V randomizační periodě pivotní studie u pacientů s PV byly krvácivé příhody (zahrnující intrakraniální a gastrointestinální krvácení, podlitiny a jiné typy krvácení) hlášeny u 20 % pacientů léčených přípravkem Jakavi a u 15,3 % pacientů s nejlepší dostupnou léčbou. Podlitiny byly hlášené s podobnými četnostmi v ramenech s přípravkem Jakavi a BAT (10,9 % vs. 8,1 %). U pacientů léčených přípravkem Jakavi nebyl hlášen ani jeden případ intrakraniálního nebo gastrointestinálního krvácení. Jeden pacient léčený přípravkem Jakavi měl krvácivou příhodu stupně 3 (krvácení po zákroku); krvácení stupně 4 nebylo hlášeno. Ostatní krvácivé příhody (zahrnující příhody jako epistaxi, krvácení po zákroku, krvácení dásní) byly hlášeny u 11,8 % pacientů léčených přípravkem Jakavi a u 6,3 % pacientů s nejlepší dostupnou léčbou.
Infekce
V klinických studiích fáze 3 u pacientů s MF byly hlášeny infekce močového traktu stupně 3 nebo 4 u 1,0 % pacientů, herpes zoster u 4,3 % a tuberkulóza u 1,0 % pacientů. V klinických studiích fáze 3 byla hlášena sepse u 3,0 % pacientů. Prodloužené sledování pacientů léčených ruxolitinibem neprokázalo žádné trendy ke zvýšení výskytu sepse v čase.
V randomizační periodě pivotní studie u pacientů s PV byl hlášen jeden případ (0,9 %) infekce močových cest CTCAE stupně 3, infekce močových cest CTCAE stupně 4 nebyla hlášena. Výskyt herpes zoster byl nepatrně vyšší u pacientů s PV (6,4 %) oproti pacientům s MF (4,0 %). U pacientů s PV se objevilo jedno hlášení postherpetické neuralgie CTCAE stupně 3.
Zvýšený systolický krevní tlak
V pivotních klinických studiích fáze 3 u pacientů s MF bylo u 31,5 % pacientů během alespoň jedné návštěvy v porovnání s 19,5 % pacientů léčených kontrolní léčbou zjištěno zvýšení systolického krevního tlaku o 20 mmHg nebo více oproti výchozímu stavu. Ve studii COMFORT-I (pacienti s MF) bylo průměrné zvýšení systolického krevního tlaku oproti výchozímu stavu o 0-2 mmHg u přípravku Jakavi v porovnání s poklesem o 2-5 mmHg v rameni s placebem. Ve studii COMFORT-II vykazovaly průměrné hodnoty u pacientů s MF léčených ruxolitinibem v porovnání s pacienty s MF léčenými kontrolní léčbou malé rozdíly.
V randomizační periodě pivotní studie u pacientů s PV se průměrný systolický krevní tlak zvýšil o 0,65 mmHg u přípravku Jakavi v porovnání s poklesem o 2 mmHg u ramene s BAT.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Není známo žádné antidotum pro předávkování přípravkem Jakavi. Jednorázové podání až 200 mg přípravku Jakavi bylo poměrně dobře snášeno. Opakované podání vyšších než doporučených dávek je spojeno s vyšším výskytem myelosuprese, zahrnující leukopenii, anemii a trombocytopenii. V těchto případech má být podávána vhodná podpůrná léčba.
Nepředpokládá se, že by hemodialýza zvyšovala vylučování ruxolitinibu.
FARMAKOLOGICKÉ VLASTNOSTI
5.
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Cytostatika, inhibitory proteinkinázy, ATC kód: L01XE18 Mechanismus účinku
Ruxolitinib je selektivní inhibitor Janus kináz (JAK) JAK1 a JAK2 (hodnoty IC50 jsou pro enzym JAK1 3,3 nM a pro JAK2 2,8 nM). Tyto kinázy jsou zapojeny do signalizace mnoha cytokinů a růstových faktorů s významnou úlohou při hematopoéze a imunitních funkcích.
Myelofibróza a pravá polycytémie j sou myeloproliferativní neoplastická onemocnění, u kterých byla popsána abnormální regulace signalizace zprostředkované JAK1 a JAK2. Předpokládá se, že příčinou poruchy regulace jsou vysoké hladiny cirkulujících cytokinů, které aktivují signální dráhu JAK-STAT, mutace zvyšující funkci enzymů jako je JAK2V617F a potlačení negativních regulačních mechanismů. U pacientů s MF nacházíme změnu regulace zprostředkované JAK bez ohledu na přítomnost mutace JAK2V617F. Aktivační mutace v JAK2 (V617F nebo exon 12) jsou zjištěné u >95 % pacientů s PV.
Ruxolitinib inhibuje signální dráhu JAK-STAT a buněčnou proliferaci u buněčných modelů hematologických malignit závislých na cytokinech, stejně jako u na cytokinech nezávislého modelu využívajícího Ba/F3 buňky exprimující JAK2V617F mutovaný protein s hodnotou IC50 v rozmezí 80-320 nM.
Farmakodynamické účinky
Ruxolitinib inhibuje cytokiny indukovanou STAT3 fosforylaci v krvi zdravých dobrovolníků i pacientů s MF a pacientů s PV. U obou skupin vedlo podávání ruxolitinibu k maximální inhibici STAT3 fosforylace 2 hodiny po podání s návratem k téměř výchozím hodnotám do 8 hodin po podání, což ukazuje na to, že nedochází k akumulaci mateřské látky ani aktivních metabolitů.
U pacientů s MF došlo při léčbě ruxolitinibem k poklesu zvýšených zánětlivých markerů jako je TNFa, IL-6 a CRP. Pacienti s MF se v průběhu času nestávali refrakterní k farmakodynamickým účinkům ruxolitinibu. Také pacienti s PV vykazovali obdobné zvýšení zánětlivých markerů ve výchozím stavu a tyto markery klesaly po léčbě ruxolitinibem.
V QT studiích u zdravých dobrovolníků nebylo zaznamenáno prodloužení QT/QTc intervalu po jednorázovém podání dávky ruxolitinibu vyšší než terapeutické (až 200 mg), což ukazuje na to, že ruxolitinib nemá vliv na srdeční repolarizaci.
Klinická účinnost a bezpečnost
Myelofibróza
U pacientů s MF (primární myelofibrózou, postpolycytemickou myelofibrózou nebo myelofibrózou po esenciální trombocytemii) byly provedeny dvě randomizované studie fáze 3 (COMFORT-I a COMFORT-II). V obou studiích měli pacienti hmatnou splenomegalii alespoň 5 cm pod žeberním obloukem a byli klasifikovaní podle kritérií International Working Group (IWG) do kategorií středního rizika 2 nebo vysokého rizika. Úvodní dávka přípravku Jakavi byla určena podle počtu trombocytů.
COMFORT-I byla dvojitě slepá, randomizovaná, placebem kontrolovaná studie zahrnující 309 pacientů, kteří byli refrakterní, nebo nebyli vhodnými kandidáty pro dostupnou léčbu. Primární cílový parametr účinnosti byl podíl nemocných, kteří dosáhnou ve 24. týdnu alespoň 35% zmenšení objemu sleziny oproti výchozí hodnotě měřeného pomocí magnetické rezonance (MRI) nebo počítačové tomografie (CT).
Sekundárními cílovými parametry bylo trvání udržení alespoň 35% redukce objemu sleziny proti výchozí hodnotě, podíl pacientů, kteří ve 24. týdnu dosáhli alespoň 50% snížení celkového symptomatického skóre, změna celkového symptomatického skóre od výchozího stavu do 24. týdne hodnocená pomocí modifikovaného MFSAF (Myelofibrosis Symptom Assessment Form) formuláře (verze 2.0, elektronický deník) a celkové přežití.
COMFORT-II byla otevřená, randomizovaná studie zahrnující 219 pacientů. Pacienti byli randomizováni v poměru 2:1 k léčbě přípravkem Jakavi oproti nejlepší dostupné léčbě. Ve skupině s nejlepší dostupnou léčbou dostávalo 47 % pacientů hydroxyureu a 16 % pacientů glukokortikoidy. Primární cílový ukazatel účinnosti léčby byl podíl nemocných, kteří dosáhnou ve 48. týdnu alespoň 35% snížení objemu sleziny oproti výchozí hodnotě měřeného pomocí MRI nebo CT.
Sekundární cílové parametry ve studii COMFORT-II zahrnovaly podíl nemocných, kteří dosáhnou ve 24. týdnu alespoň 35% snížení objemu sleziny oproti výchozí hodnotě a dobu trvání alespoň 35% snížení objemu slezinyoproti výchozí hodnotě.
Ve studiích COMFORT-I a COMFORT-II byly vstupní demografické parametry a charakteristiky onemocnění srovnatelné u obou léčených skupin.
Tabulka 2 Podíl pacientů (%) s >35% snížením objemu sleziny oproti výchozí hodnotě ve 24. týdnu ve studii COMFORT-I a ve 48. týdnu ve studii COMFORT-II (ITT)
|
COMFORT-I |
COMFORT-II | |||
|
Jakavi (N=155) |
Placebo (N=153) |
Jakavi (N=144) |
Nejlepší dostupná léčba (N=72) | |
|
Časový bod |
24. týden |
48. týden | ||
|
Počet (%) pacientů s redukcí objemu sleziny o >35 % |
65 (41,9) |
1 (0,7) |
41 (28,5) |
0 |
|
95% interval spolehlivosti |
34,1; 50,1 |
0; 3,6 |
21,3; 36,6 |
0,0; 5,0 |
|
Hodnota p |
<0,0001 |
<0,0001 | ||
Významně vyšší podíl pacientů ve skupině léčené přípravkem Jakavi dosáhl alespoň 35% snížení objemu sleziny (Tabulka 2) oproti výchozí hodnotě, a to bez ohledu na přítomnost mutace JAK2V617F nebo podtypu onemocnění (primární myelofibróza, postpolycytemická myelofibróza nebo myelofibróza po esenciální trombocytemii).
Tabulka 3 Podíl pacientů (%) s >35% snížením objemu sleziny oproti výchozí hodnotě dle mutačního stavu JAK (hodnocení bezpečnosti)
|
COMFORT-I |
COMFORT-II | |||||||
|
Jakavi |
Placebo |
Jakavi |
Nejlepší dostupná léčba | |||||
|
JAK mutační stav |
Pozitivní (N=113) n (%) |
Negativní (N=40) n (%) |
Pozitivní (N=121) n (%) |
Negativní (N=27) n (%) |
Pozitivní (N=110) n (%) |
Negativní (N=35) n (%) |
Pozitivní (N=49) n (%) |
Negativní (N=20) n (%) |
|
Počet (%) pacientů s redukcí objemu sleziny o >35 % |
54 (47,8) |
11 (27,5) |
1 (0,8) |
0 |
36 (32,7) |
5 (14,3) |
0 |
0 |
|
Časový bod |
Po 24 týdnech |
Po 48 týdnech | ||||||
Pravděpodobnost zachování odpovědi sleziny (>35% snížení) na přípravku Jakavi po dobu nejméně 24 týdnů byla 89 % ve studii COMFORT-I a 87 % ve studii COMFORT-II; 52 % udrželo odpověď sleziny po dobu nejméně 48 týdnů ve studím COMFORT-II.
Ve studii COMFORT-I dosáhlo 45,9 % subjektů ve skupině s přípravkem Jakavi >50% zlepšení celkového symptomatického skóre od výchozího stavu ve 24. týdnu (vyhodnocovaného pomocí MFSAF deníku v2.0) v porovnání s 5,3 % pacientů ve skupině s placebem (p<0,0001 za pomoci chí-kvadrát testu). Průměrná změna celkového zdravotního stavu ve 24. týdnu měřená pomocí dotazníku EORTC QLQ C30 byla +12,3 u přípravku Jakavi a -3,4 u placeba (p<0,0001).
Ve studii COMFORT-I byl při mediánu sledování 34,3 měsíce výskyt úmrtí u pacientů randomizovaných do ramene s ruxolitinibem 27,1 % oproti 35,1 % u pacientů randomizovaných do ramene s placebem; HR 0,687; 95% CI 0,459-1,029; p=0,0668.
Ve studii COMFORT-II byl při mediánu sledování 34,7 měsíce výskyt úmrtí u pacientů randomizovaných do ramene s ruxolitinibem 19,9 % oproti 30,1 % u pacientů randomizovaných k nejlepší dostupné léčbě (BAT); HR 0,48; 95% CI 0,28-0,85; p=0,009. V obou studiích byly nižší výskyty úmrtí zjištěné v rameni s ruxolitinibem ovlivněny především výsledky získanými od pacientů z podskupin po pravé polycytémii a po esenciální trombocytémii.
Pravá _ polycytémie
Randomizovaná, otevřená, aktivně kontrolovaná studie fáze 3 (RESPONSE) byla provedena u 222 pacientů s PV, kteří byli rezistentní nebo intolerantní k hydroxyurei definované podle publikovaných kritérií mezinárodní pracovní skupiny evropské leukemické sítě (ELN). 110 pacientů bylo randomizovaných do ramene s ruxolitinibem a 112 pacientů do ramene s nejlepší dostupnou léčbou. Zahajovací dávka přípravku Jakavi byla 10 mg dvakrát denně. Poté byly u pacientů dávky individuálně upraveny na základě tolerability a účinnosti s maximální dávkou 25 mg dvakrát denně. Nejlepší dostupná léčba byla vybraná zkoušejícím individuálně pro každého pacienta a zahrnovala hydroxyureu (59,5%), interferon/pegylovaný interferon (11,7 %), anagrelid (7,2 %), pipobroman (1,8 %) a pozorování (15,3 %).
Výchozí demografické parametry a charakteristika onemocnění byly v obou ramenech léčby srovnatelné. Medián věku byl 60 let (rozmezí 33 až 90 let). Pacienti v rameni s ruxolitinibem měli medián doby od diagnózy PV 8,2 roku a dříve užívali hydroxyureu s mediánem trvání léčby přibližně 3 roky. Většina pacientů (>80 %) měla provedeny nejméně dvě flebotomie v posledních 24 týdnech před zařazením do studie. Komparativní data týkající se dlouhodobého přežívání a výskytu komplikací onemocnění chybí.
Primárním složeným cílovým parametrem byl podíl pacientů, kteří dosáhli jak nevhodnosti k flebotomii (kontrolu HCT), tak i >35% zmenšení objemu sleziny od výchozího stavu do 32. týdne. Kritérium vhodnosti k provedení flebotomie bylo definováno jako potvrzený HCT >45%, tj. minimálně o 3 procentní body vyšší oproti HCT stanovenému ve výchozím stavu nebo potvrzenému HCT >48%, dle toho toho, která hodnota je nižší. Klíčové sekundární cílové parametry zahrnovaly podíl pacientů s dosaženým primárním cílovým parametrem a bez progrese ve 48. týdnu, stejně jako podíl pacientů s dosaženou kompletní hematologickou remisí ve 32. týdnu.
Studie dosáhla svého primárního cíle, vyšší podíl pacientů ve skupině s přípravkem Jakavi dosáhl primárního složeného cílového parametru a každé z jeho individuálních komponent. Výrazně vyšší počet pacientů léčených přípravkem Jakavi (20,9 %) dosáhl primární odpovědi (p<0,0001) v porovnání s nejlepší dostupnou léčbou (0,9 %). Kontroly hematokritu bylo dosaženo u 60 % pacientů v rameni s přípravkem Jakavi v porovnání s 19,6 % v rameni s nejlepší dostupnou léčbou a >35% snížení objemu sleziny bylo dosaženo u 38,2 % pacientů v rameni s přípravkem Jakavi v porovnání s 0,9 % v rameni s nejlepší dostupnou léčbou (obrázek 1). 94 (83,9 %) pacientů randomizovaných k rameni s nejlepší dostupnou léčbou přešlo ve 32. týdnu nebo později na léčbu ruxolitinibem, což omezuje porovnání obou ramen po 32. týdnu.
Také bylo dosaženo obou klíčových sekundárních cílových parametrů. Poměr pacientů, kteří dosáhli kompletní hematologické remise, byl 23,6 % u přípravku Jakavi v porovnání s 8,9 % u nejlepší dostupné léčby (p=0,0028) a poměr pacientů, kteří dosáhli trvalé primární odpovědi ve 48. týdnu, byl
19,1 % u přípravku Jakavi a 0,9 % u nejlepší dostupné léčby (p<0,0001).
Obrázek 1 Pacienti, kteří dosáhli primárního cílového parametru a komponent primárního cílového parametru ve 32. týdnu

Primární složený cíl v > 35% redukce objemu Kontrola hematokritu bez týdnu 32 sleziny flebotomie
Symptomatická zátěž byla vyhodnocena pomocí elektronického pacientského dotazníku MPN-SAF celkového symptomatického skóre (TSS), který se skládá ze 14 otázek. Ve 32. týdnu dosáhlo 49 % a 64 % pacientů léčených ruxolitinibem >50% snížení TSS-14 resp. TSS-5 v porovnání s pouze 5 % a 11 % pacientů na nejlepší dostupné léčbě.
Vnímání prospěchu z léčby bylo stanoveno pomocí dotazníku Patient Global Impression of Change (PGIC). 66 % pacientů léčených ruxolitinibem hlásilo zlepšení již čtyři týdny po zahájení léčby oproti 9 % léčených nejlepší dostupnou léčbou. Zlepšení vnímání prospěchu z léčby bylo vyšší také u pacientů léčených ruxolitinibem ve 32. týdnu (78 % oproti 33 %).
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Jakavi u všech podskupin pediatrické populace pro léčbu MF (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Absorpce
Ruxolitinib patří do třídy 1 podle BCS (Biopharmaceutical Classification System), protože má vysokou prostupnost, dobrou rozpustnost a rychlý rozpad. Ruxolitinib byl v klinických studiích po perorálním podání rychle absorbován s maximální plazmatickou koncentrací (Cmax) dosaženou přibližně 1 hodinu po podání. Absorpce ruxolitinibu po perorálním podání (stanoven ruxolitinib a jeho metabolity tvořené při prvním průchodu játry) je podle farmakokinetických studií u lidí 95 % a více. Průměrné hodnoty Cmax a AUC ruxolitinibu se zvyšují úměrně s podanou jednorázovou dávkou v rozmezí 5-200 mg. Užití ruxolitinibu po tučném jídle nevedlo ke klinicky významné změně farmakokinetiky. Průměrná hodnota Cmax při požití tučného jídla mírně klesla (o 24 %) a průměrná hodnota AUC se téměř nezměnila (nárůst o 4 %).
Distribuce
Průměrný distribuční objem v ustáleném stavu je přibližně 75 litrů u pacientů s MF a PV. Vazba na plazmatické bílkoviny je in vitro při koncentracích ruxolitinibu odpovídajících klinickému využití přibližně 97 % a ruxolitinib se váže zejména na albumin. Celotělová autoradiografická studie u potkanů ukázala, že ruxolitinib neprochází hematoencefalickou bariérou.
Biotransformace
Ruxolitinib je metabolizován především CYP3A4 (>50 %) s dodatečným přispěním CYP2C9. Mateřská látka má v lidské plazmě převládající podíl a představuje 60 % látek v oběhu souvisejících s podáním přípravku. V plazmě jsou přítomné dva hlavní a zároveň aktivní metabolity, které představují 25 % a 11 % z mateřské AUC. Tyto metabolity mají polovinu až pětinu mateřské farmakologické aktivity na JAK enzymy. Celkově všechny aktivní metabolity přispívají 18 % k celkové farmakodynamické aktivitě ruxolitinibu. V klinických plazmatických koncentracích ruxolitinib podle in vitro studií neinhibuje CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 nebo CYP3A4 a není silným induktorem CYP1A2, CYP2B6 nebo CYP3A4. Na základě in vitro dat může ruxolitinib inhibovat P-gp a BCRP.
Eliminace
Ruxolitinib se eliminuje z organismu převážně metabolickou cestou. Průměrný poločas eliminace ruxolitinibu je přibližně 3 hodiny. Po jednorázovém perorálním podání [14C]-značeného ruxolitinibu zdravým dobrovolníkům bylo farmakum z převážné většiny metabolizováno a 74 % podané radioaktivity bylo vyloučeno močí a 22 % stolicí. Nezměněná mateřská látka představovala méně než 1 % celkové vyloučené radioaktivity.
Linearita/nelinearita
Ve studiích s podáním jednorázové i opakované dávky bylo prokázáno, že systémová expozice se proporcionálně zvyšuje v závislosti na dávce.
Farmakokinetika u zvláštních skupin pacientů
Vliv věku, pohlaví a rasy
Na základě studií u zdravých dobrovolníků nebyly pozorovány relevantní rozdíly ve farmakokinetice v závislosti na pohlaví a rase. Při populační farmakokinetické analýze u pacientů s MF nebyla zjištěna závislost orální clearance na věku nebo rase. Predikovaná orální clearance byla 17,7 l/h u žen a
22,1 l/h u mužů s interindividuální variabilitou 39 % u pacientů s MF. U pacientů s PV byla clearance
12,7 l/h se 42 % interindividuální variabilitou a mezi perorální clearance a pohlavím, stářím pacienta nebo rasou nebyl na základě stanovení farmakokinetiky v této populaci pacientů zřejmý žádný vztah.
Pediatrická populace
Bezpečnost a účinnost Jakavi nebyla zkoumána u pediatrické populace (viz bod 5.1 „Pediatrická populace“).
Porucha funkce ledvin
Funkce ledvin byla stanovena pomocí MDRD (Modification of Diet in Renal Disease) a kreatininu v moči. Expozice ruxolitinibu byla po jednorázovém podání 25 mg ruxolitinibu shodná u pacientů s různým stupněm poruchy funkce ledvin a u pacientů s normálními renálními funkcemi, avšak hodnoty plazmatické AUC metabolitů ruxolitinibu měly tendenci se zvyšovat se zhoršujícím se postižením ledvin a byly nejvyšší u pacientů s těžkou poruchou funkce ledvin. Není známo, zda má zvýšená expozice metabolitům vliv na bezpečnost. Úprava dávky je doporučená u pacientů s těžkou poruchou funkce ledvin a u nemocných se selháním funkce ledvin (viz bod 4.2). Dávkování ve dnech dialýzy snižuje expozici metabolitům, ale také farmakodynamický účinek, především ve dnech mezi dialýzami.
Porucha funkce jater
Průměrná hodnota AUC ruxolitinibu byla po jednorázovém podání 25 mg ruxolitinibu pacientům s různým stupněm poškození jater zvýšená o 87 %, 28 % a 65 % u pacientů s lehkou, středně těžkou a těžkou poruchou funkce jater (v uvedeném pořadí) ve srovnání s pacienty s normální funkcí jater.
Mezi hodnotou AUC a stupněm jaterního postižení dle Child-Pugh skóre nebyl prokázán žádný vztah. Terminální poločas eliminace byl u pacientů s poškozením jater prodloužen ve srovnání se zdravými dobrovolníky (4,1-5,0 h oproti 2,8 h). U pacientů s poškozením jater je doporučeno přibližně 50% snížení dávky (viz bod 4.2).
5.3 Předklinické údaje vztahující se k bezpečnosti
S ruxolitinibem byly provedeny konvenční farmakologické studie bezpečnosti, toxicity po opakovaném podání, genotoxicity, reprodukční toxicity a hodnocení kancerogenního potenciálu.
V testech toxicity po opakovaném podání byly cílovými orgány spojenými s farmakologickým působením ruxolitinibu kostní dřeň, periferní krev a lymfatické tkáně. U psů byly zjištěny infekce, obecně asociované s imunosupresí. Při telemetrických studiích u psů byl pozorován nežádoucí pokles krevního tlaku a vzestup srdeční frekvence a v respiračních studiích u potkanů byl pozorován nežádoucí pokles minutového objemu. Hraniční dávka (podle Cmax volné látky), při které nebyly pozorovány nežádoucí účinky, byla u psů a potkanů 15,7krát respektive 10,4krát vyšší než je maximální doporučená dávka u lidí (25 mg dvakrát denně). Nebyl pozorován žádný neurofarmakologický účinek ruxolitinibu.
Ruxolitinib snižoval hmotnost plodu a zvyšoval postimplantační ztráty ve studiíích u zvířat. U potkanů a králíků nebyl zjištěn výskyt teratogeních účinků. Nicméně hraniční expozice porovnávané s nejvyšší klinickou dávkou byly nízké a výsledky proto mají pro člověka omezený význam. Nebyl pozorován žádný vliv na fertilitu. V prenatálních a postnatálních vývojových studiích bylo pozorováno mírné prodloužení gestační periody, snížení počtu implantačních míst a snížení počtu porozených mláďat.
U mláďat byla zaznamenána snížená průměrná porodní hmotnost a krátké období snížených průměrných přírůstků hmotnosti po narození. U potkanů v laktaci byly ruxolitinib a/nebo jeho metabolity vylučovány do mateřského mléka, a to v koncentracích 13krát vyšších než v mateřské plazmě. Ruxolitinib neměl mutagenní a klastogenní účinky. Ruxolitinib neměl karcinogenní účinky u Tg.rasH2 transgenních myší.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Mikrokrystalická celulóza Magnesium-stearát Koloidní bezvodý oxid křemičitý Sodná sůl karboxymethylškrobu (typ A)
Povidon Hyprolóza Monohydrát laktózy
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
3 roky
6.4 Zvláštní opatření pro uchovávání
Neuchovávejte při teplotě nad 30°C.
6.5 Druh obalu a obsah balení
Balení s PVC/PCTFE/Al blistry obsahující 14 nebo 56 tablet nebo vícečetná balení obsahující 168 (3 balení po 56) tabletách.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku
Žádné zvláštní požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/12/773/007-009
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
23.08.2012
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Jakavi 20 mg tablety
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna tableta obsahuje ruxolitinibum 20 mg (ve formě ruxolitinibi phosphas).
Pomocné látky se známým účinkem:
Jedna tableta obsahuje 285,80 mg monohydrátu laktózy.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Tableta.
Podlouhlé bílé až téměř bílé tablety o rozměrech přibližně 16,5 x 7,4 mm s vyraženým „NVR“ na jedné straně a „L20“ na straně druhé.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Myelofibróza (MF)
Přípravek Jakavi je indikován k léčbě dospělých pacientů se splenomegalií nebo s příznaky přidruženými k primární myelofibróze (chronické idiopatické myelofibróze), postpolycytemické myelofibróze nebo myelofibróze po esenciální trombocytemii.
Pravá polvcvtémie (polvcvthaemia vera - PV)
Přípravek Jakavi je indikován k léčbě dospělých pacientů s pravou polycytémií, kteří jsou rezistentní nebo intolerantní k hydroxyurei.
4.2 Dávkování a způsob podání
Léčba Jakavi má být zahajována pouze lékařem, který má zkušenosti s podáváním protinádorové terapie.
Před zahájením léčby přípravkem Jakavi musí být vyšetřen kompletní krevní obraz včetně diferenciálního rozpočtu leukocytů.
Kompletní krevní obraz včetně diferenciálního rozpočtu leukocytů má být vyšetřen každé 2-4 týdny do stabilizace dávek přípravku Jakavi, a dále pak dle klinické indikace (viz bod 4.4).
Dávkování
Počáteční dávka
Doporučená počáteční dávka přípravku Jakavi u myelofibrózy je 15 mg dvakrát denně u pacientů s počtem trombocytů 100* 109/l až 200*109/l a 20 mg dvakrát denně u pacientů s počtem trombocytů >200*109/l. Doporučená počáteční dávka přípravku Jakavi u pravé polycytémie je 10 mg podávaných perorálně dvakrát denně.
U pacientů s počtem trombocytů 50 až <100*109/l není dostatek údajů pro stanovení přesné úvodní dávky. U těchto pacientů je maximální doporučená úvodní dávka 5 mg dvakrát denně a její další titrace má být prováděna velmi opatrně.
Úprava dávkování
Dávky jsou dále titrovány dle bezpečnosti a účinnosti. Léčba by měla být ukončena při poklesu trombocytů na méně než 50*109/l nebo při poklesu absolutního počtu neutrofilů na méně než 0,5*109/l. Při PV by měla být léčba také ukončena, pokud j e hladina hemoglobinu pod 8 g/dl. Po návratu počtu krevních elementů nad tyto hodnoty může být podávání znovu zahájeno v dávce 5 mg dvakrát denně a postupně zvyšováno za pečlivého sledování krevního obrazu včetně diferenciálního rozpočtu leukocytů.
Pokud počet trombocytů klesne pod 100*109/l, mělo by být zváženo snížení dávky, aby nebylo nutné léčbu zcela přerušit pro trombocytopenii. Při PV by se také mělo zvážit snížení dávky, pokud hladina hemoglobinu klesne pod 12 g/dl, a snížení je doporučené, pokud hladina klesne pod 10 g/dl.
Pokud není léčba účinná a počty krevních elementů jsou dostatečné, mohou být dávky zvýšeny, a to maximálně o 5 mg dvakrát denně, až na maximální dávku 25 mg dvakrát denně.
Počáteční dávka nemá být zvyšována během prvních čtyř týdnů léčby, a následně ne častěji než ve dvoutýdenních intervalech.
Maximální dávka přípravku Jakavi je 25 mg dvakrát denně.
Úprava dávkování při konkomitantní léčbě silnými inhibitory CYP3A4 nebo flukonazolem Pokud je přípravek Jakavi podáván spolu se silnými inhibitory CYP3A4 nebo duálními inhibitory enzymů CYP2C9 a CYP3A4 (např. flukonazol), má být jednotlivá dávka přípravku Jakavi, podávaná dvakrát denně, snížena o přibližně 50 % (viz bod 4.5). Je nutné se vyhnout souběžnému podávání přípravku Jakavi s flukonazolem v dávkách vyšších než 200 mg denně.
V průběhu léčby silným inhibitorem CYP3A4 nebo duálními inhibitory enzymů CYP2C9 a CYP3A4 se doporučuje častěji (např. dvakrát týdně) kontrolovat hematologické parametry a pečlivě pátrat po klinických známkách nežádoucích účinků přípravku Jakavi.
Zvláštní skupiny _pacientů Pacienti s poruchou funkce ledvin
U pacientů s mírnou nebo středně závažnou poruchou funkce ledvin není nutná specifická úprava dávky.
U pacientů s MF a těžkou poruchou funkce ledvin (clearance kreatininu méně než 30 ml/min) má být doporučená počáteční dávka, podávaná dvakrát denně, stanovená podle počtu trombocytů redukovaná o přibližně 50 %. Doporučená počáteční dávka pro pacienty s PV s těžkou poruchou funkce ledvin je 5 mg dvakrát denně. U těchto pacientů je třeba pečlivě monitorovat bezpečnost a účinnost léčby.
Pro pacienty s terminálním selháním ledvin (ESRD, end-stage renal disease) léčené hemodialýzou neexistuje dostatek údajů pro stanovení optimálního dávkování. Farmakokinetické/farmakodynamické simulace založené na dostupných údajích o této populaci naznačují, že u hemodialyzovaných pacientů s MF a ESRD má být počáteční dávka podávaná pouze ve dnech hemodialýzy po jejím ukončení, a to 15-20 mg v jednotlivé dávce nebo ve dvou dávkách po 10 mg podávaných po 12 hodinách. Jednotlivá dávka 15 mg je doporučena v případě pacientů s MF a počtem trombocytů 100* 109/l až 200* 109/l. Jednotlivá dávka 20 mg nebo dvě dávky 10 mg podávané po 12 hodinách jsou doporučeny u pacientů s MF a počtem trombocytů >200*109/l. Následující dávky (v jednotlivé dávce nebo ve dvou dávkách 10 mg podávaných po 12 hodinách) mají být podávány pouze v den hemodialýzy, a to po jejím ukončení.
Doporučená počáteční dávka pro pacienty s PV a ESRD na hemodialýze je jednorázová dávka 10 mg nebo dvě dávky 5 mg podávané po 12 hodinách po dialýze, a to pouze v den podání hemodialýzy.
Tato doporučení dávkování jsou založená na simulacích a po jakékoli úpravě dávkování u jednotlivých pacientů s ESRD by mělo následovat pečlivé sledování bezpečnosti a účinnosti léčby. Pro dávkování u pacientů podstupujících peritoneální dialýzu a kontinuální venovenózní hemofiltraci nejsou k dispozici žádná data (viz bod 5.2).
Pacienti s poruchou funkce jater
U pacientů s poruchou funkce jater má být doporučená počáteční dávka, stanovená podle počtu trombocytů a podávaná dvakrát denně, snížena o přibližně 50 %. Následující dávky mají být upraveny na základě pečlivého monitorování bezpečnosti a účinnosti léčby. Pacientům, u nichž bylo v průběhu léčby přípravkem Jakavi zjištěno poškození jater, má být vyšetřen kompletní krevní obraz včetně diferenciálního rozpočtu leukocytů alespoň jednou za jeden až dva týdny v prvních 6 týdnech po zahájení léčby a dále dle klinické potřeby, dokud nejsou jaterní funkce a krevní obraz stabilizovány. Dávka přípravku Jakavi může být dále titrována pro snížení rizika cytopenie.
Starší lidé (>65 let)
Starší lidé nevyžadují žádnou specifickou úpravu dávkování.
Pediatrická populace
Bezpečnost a účinnost přípravku Jakavi u dětí ve věku do 18 let nebyla stanovena. Nejsou dostupné žádné údaje (viz bod 5.1).
Ukončení léčby
Léčba má pokračovat, dokud přínos z léčby převažuje nad rizikem léčby. Pokud ale nedojde během 6 měsíců od započetí léčby ke zmenšení velikosti sleziny nebo zlepšení příznaků, má být léčba ukončena.
U pacientů s určitým stupněm klinického zlepšení se doporučuje léčbu ruxolitinibem přerušit v případě setrvalého zvětšování sleziny o 40 % v porovnání s velikostí ve výchozím stavu (což odpovídá zhruba 25 % zvýšení objemu sleziny), kdy zároveň nedochází k dalšímu zlepšení příznaků spojených s onemocněním.
Způsob podání
Přípravek Jakavi se užívá perorálně, s jídlem nebo bez jídla.
Při vynechání dávky nemá pacient užívat dávku navíc, ale má pokračovat další obvyklou předepsanou dávkou.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1. Těhotenství a laktace.
4.4 Zvláštní upozornění a opatření pro použití
Myelosuprese
Léčba přípravkem Jakavi může způsobit hematologické nežádoucí účinky léku, včetně trombocytopenie, anemie a neutropenie. Před zahájením léčby přípravkem Jakavi musí být vyšetřen kompletní krevní obraz včetně diferenciálního rozpočtu leukocytů. Léčba má být přerušena u pacientů, u kterých dojde k poklesu počtu trombocytů na méně než 50*109/l nebo absolutního počtu neutrofilů na méně než 0,5*109/l (viz bod 4.2).
Bylo zjištěno, že pacienti s nižším počtem trombocytů (<200*109/l) na začátku terapie mají větší riziko vzniku trombocytopenie během léčby.
Trombocytopenie je obvykle reverzibilní a je většinou zvládnuta snížením dávky nebo přechodným vysazením přípravku Jakavi (viz body 4.2 a 4.8). Nicméně v případě klinické potřeby může být nezbytné podání transfuze trombocytů.
Vznik anemie u léčených pacientů si může vyžádat podání krevní transfuze. U těchto pacientů lze také zvážit úpravu dávky nebo přerušení léčby.
Pacienti s hladinou hemoglobinu nižší než 10,0 g/dl při započetí léčby mají vyšší riziko výskytu hladin hemoglobinu pod 8,0 g/dl během léčby v porovnání s pacienty s vyšší počáteční hladinou hemoglobinu (79,3 % oproti 30,1 %). U pacientů s počáteční hladinou hemoglobinu pod 10,0 g/dl je doporučeno častější sledování hematologických parametrů a klinických příznaků a symptomů nežádoucích účinků spojených s přípravkem Jakavi.
Neutropenie (absolutní počet neutrofilů <0,5*109/l) byla obvykle reverzibilní a bylo možné ji zvládnout přechodným vysazením přípravku Jakavi (viz body 4.2 a 4.8).
Kompletní krevní obraz by měl být sledován dle klinické indikace a dávka upravována dle doporučení (viz body 4.2 a 4.8).
Infekce
U všech pacientů má být zhodnoceno riziko vzniku závažné bakteriální, mykobakteriální, mykotické a virové infekce. U pacientů s MF léčených přípravkem Jakavi byla hlášena tuberkulóza. Před zahájením léčby by mělo být u pacientů provedeno vyšetření na aktivní a neaktivní („latentní“) tuberkulózu podle místních doporučení. Může zahrnovat anamnézu, možnost předchozího kontaktu s tuberkulózou a/nebo příslušné vyšetření, jako je rentgenové vyšetření plic, tuberkulinový test a/nebo test uvolnění interferonu gama. Lékaři si mají být vědomi rizika falešně negativních výsledků tuberkulinového kožního testu, a to zejména u pacientů, kteří jsou vážně nemocní nebo mají sníženou imunitu. Léčba přípravkem Jakavi nemá být zahajována, dokud není závažná probíhající infekce zvládnuta. Lékaři mají pečlivě sledovat pacienty léčené přípravkem Jakavi, aby rozpoznali příznaky infekce a zahájili včas adekvátní léčbu (viz bod 4.8).
Zvýšení virové zátěže hepatitidy B (HBV-DNA titru), spolu s asociovaným zvýšením hladin alaninaminotransferázy a aspartátaminotransferázy, nebo bez jejich zvýšení, bylo hlášeno u pacientů s chronickými HBV infekcemi, kteří užívali přípravek Jakavi. Účinek přípravku Jakavi na replikaci viru u pacientů s chronickou infekcí HBV není známý. Pacienti s chronickou HBV infekcí by měli být léčeni a sledováni podle klinických doporučení.
Lékaři mají poučit pacienty o časných příznacích infekce herpes zoster a doporučit jim co možná nejvčasnější vyhledání možnosti léčby v případě infekce.
Progresivní multifokální leukoencefalopatie
Při léčbě pacientů s MF přípravkem Jakavi byla hlášena progresivní multifokální leukoencefalopatie (PML). Lékaři by měli dbát zejména na příznaky, které zaznamená pacient, nasvědčující PML (např. kognitivní, neurologické nebo psychiatrické příznaky nebo známky). U pacientů musí být sledovány jakékoli z těchto nových nebo zhoršujících se příznaků nebo známek a pokud se tyto příznaky/známky objeví, má být zvažováno odeslání k neurologovi a přijetí příslušných diagnostických opatření pro PML. Pokud je podezření na PML, musí být ukončeno další podávání, dokud není PML vyloučena.
Nemelanomové nádory kůže
U pacientů užívajících ruxolitinib byly hlášeny nemelanomové nádory kůže (NMSCs) zahrnující bazocelulární karcinom, spinocelulární karcinom a karcinom z Merkelových buněk. Většina těchto pacientů byla již dříve dlouhodobě léčena hydroxyureou a vyskytovaly se u nich NMSC nebo premaligní kožní léze. Kauzální vztah k ruxolitinibu nebyl prokázán. U pacientů se zvýšeným rizikem rakoviny kůže je doporučené pravidelné vyšetření kůže.
Zvláštní skupiny pacientů
Porucha funkce ledvin
U pacientů s těžkou poruchou funkce ledvin má být snížena úvodní dávka. U hemodialyzovaných pacientů s MF a terminálním selháním ledvin má být úvodní dávka určena dle počtu trombocytů (viz bod 4.2). Následující dávky (jednorázová dávka 20 mg nebo dvě dávky 10 mg podávané po 12 hodinách u pacientů s MF; jednorázová dávka 10 mg nebo dvě dávky 5 mg podávané po 12 hodinách u pacientů s PV) mají být podávány pouze po každé hemodialýze. Další úprava dávkování má být prováděna za pečlivého monitorování bezpečnosti a účinnosti léčby (viz body 4.2 a 5.2).
Porucha funkce jater
U pacientů s poruchou funkce jater má být doporučená úvodní dávka přípravku Jakavi redukována o přibližně 50 %. Další úprava dávkování má být prováděna na základě sledování bezpečnosti a účinnosti léčby (viz body 4.2 a 5.2).
Interakce
Pokud je přípravek Jakavi podáván spolu se silnými inhibitory CYP3A4 nebo duálními inhibitory enzymů CYP3A4 a CYP2C9 (např. flukonazol), má být jednotlivá dávka přípravku Jakavi, podávaná dvakrát denně, snížena o přibližně 50 % (frekvence sledování viz body 4.2 a 4.5).
Souběžné podávání cytoredukční léčby nebo hematopoetických růstových faktorů s přípravkem Jakavi nebylo studováno. Bezpečnost a účinnost těchto souběžných podání nejsou známé (viz bod 4.5).
Následky vysazení
Po přerušení nebo ukončení léčby přípravkem Jakavi se mohou příznaky MF znovu objevit během přibližně jednoho týdne. Závažnější případy byly po přerušení léčby přípravkem Jakavi popsány zejména v souvislosti s akutním interkurentním onemocněním. Není jasné, zda k závažnosti těchto případu přispělo náhlé přerušení léčby. Pokud není náhlé přerušení léčby nutné, je vhodné zvážit postupné snižování dávky, přestože přínos tohoto postupu nebyl prokázán.
Pomocné látky
Přípravek Jakavi obsahuje laktózu. Pacienti se vzácnými dědičnými problémy s intolerancí galaktózy, vrozeným deficitem laktázy nebo malabsorpcí glukózy a galaktózy by tento přípravek neměli užívat.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Studie interakcí byly provedeny pouze u dospělých.
Ruxolitinib je eliminován metabolismem katalyzovaným CYP3A4 a CYP2C9. Léčivé přípravky inhibující tyto enzymy proto mohou zvýšit expozici ruxolitinibu.
Interakce vyžadující snížení dávky ruxolitinibu
Inhibitory CYP3A4
Silné inhibitory CYP3A4 (jako jsou např. boceprevir, klarithromycin, indinavir, itrakonazol, ketokonazol, lopinavir/ritonavir, mibefradil, nefazodon, nefinavir, posakonazol, sachinavir, telaprevir, telithromycin, vorikonazol)
Podání přípravku Jakavi (jednorázově v dávce 10 mg) následně po podávání silného inhibitoru CYP3A4 ketokonazolu vedlo u zdravých dobrovolníků ke zvýšení Cmax ruxolitinibu o 33 % a AUC ruxolitinibu o 91 % oproti hodnotám dosaženým po podání samotného ruxolitinibu. Poločas přípravku byl při současném podání ketokonazolu prodloužen z 3,7 na 6,0 hodiny.
Pokud je přípravek Jakavi podáván se silnými inhibitory CYP3A4, má být jednotlivá dávka přípravku Jakavi, podávaná dvakrát denně, snížena přibližně o 50 %. Pacienti mají být pečlivě sledováni (např. dvakrát týdně) z důvodu možného vzniku cytopenií a dávka má být titrována na základě hodnocení bezpečnosti a účinnosti léčby (viz bod 4.2).
Duální inhibitory CYP2C9 a CYP3A4
Při společném užití s léky, které jsou duálními inhibitory enzymů CYP2C9 a CYP3A4 (např. flukonazol) má být zváženo 50 % snížení dávky. Je nutné se vyhnout souběžnému podávání přípravku Jakavi s flukonazolem v dávkách vyšších než 200 mg denně.
Induktory enzymů
Induktorv CYP3A4 (jako _jsou např. avasimib, karbamazepin, _fenobarbital, _fenvtoin, rifabutin, rifampin (rifampicin), třezalka tečkovaná (Hypericum perforatum))
Pacienti mají být pečlivě sledováni a dávka titrována s ohledem na bezpečnost a účinnost (viz bod 4.2).
U zdravých dobrovolníků, kterým byl ruxolitinib (jednorázová dávka 50 mg) podán po silném induktoru CYP3A4 rifampicinu (denní dávka 600 mg po dobu 10 dní), byla AUC ruxolitinibu o 70 % nižší než po podání samotného přípravku Jakavi. Expozice aktivním metabolitům ruxolitinibu se nezměnila. V souhrnu byla farmakodynamická aktivita ruxolitinibu podobná, což naznačuje, že indukce CYP3A4 má minimální vliv na farmakodynamiku. Nicméně to může být spojené s vysokou dávkou ruxolitinibu, která vede k farmakodynamickým účinkům blízkým Emax. Je možné, že je nutné u jednotlivých pacientů zvýšit dávku ruxolitinibu při zahájení léčby silným induktorem enzymů.
Další interakce, u nichž je zvažováno ovlivnění ruxolitinibu
Slabé nebo středně silné inhibitory CYP3A4 (jako _je např. ciprofloxacin, ervthromvcin, amprenavir, atazanavir, diltiazem, cimetidin)
Podání ruxolitinibu (jednorázově v dávce 10 mg) následně po 4denním podávání erythromycinu 500 mg dvakrát denně vedlo u zdravých dobrovolníků k zvýšení Cmax ruxolitinibu o 8 % a AUC ruxolitinibu o 27 % oproti hodnotám dosaženým po podání samotného ruxolitinibu.
Při souběžném podávání ruxolitinibu se slabými a středně silnými inhibitory CYP3A4 (např. erytromycin) není nutná úprava dávkování. Nicméně pacienti mají být po zahájení léčby středně silnými inhibitory CYP3A4 pečlivě sledováni, zda u nich nedochází k rozvoji cytopenie.
Účinky ruxolitinibu na další léčivé přípravky
Substance transportované P-glykoproteinem a dalšími transportéry
Ruxolitinib může inhibovat P-glykoprotein a protein BCRP (breast cancer resistance protein) ve střevě. To může vést ke zvýšení systémové expozice substrátů těchto transportérů, jako je dabigatran etexilát, cyklosporin, rosuvastatin a případně digoxin. Je doporučené sledování hladiny léčiva (TDM) nebo klinické sledování z důvodu možného ovlivnění takové látky.
Je možné, že potenciální inhibice P-gp a BCRP ve střevě může být minimalizována prodloužením času mezi podáním na nejdelší možnou míru.
Hemopoetické růstové _faktory
Souběžné podání hematopoetických růstových faktorů a přípravku Jakavi nebylo studováno. Není známo, zda inhibice Janus kináz (JAK) přípravkem Jakavi snižuje účinnost hematopoetických růstových faktorů nebo zda hematopoetické růstové faktory ovlivňují účinnost přípravku Jakavi (viz bod 4.4).
Cytoredukční léčba
Souběžné podávání cytoredukční léčby a přípravku Jakavi nebylo studováno. Bezpečnost a účinnost totho souběžného podávání nejsou známé (viz bod 4.4).
Studie u zdravých subjektů prokázala, že ruxolitinib neinhiboval metabolismus perorálního substrátu CYP3A4 midazolamu. Proto není očekáváno zvýšení expozice CYP3A4 substratu při kombinaci s přípravkem Jakavi. Další studie u zdravých subjektů prokázala, že přípravek Jakavi neovlivňuje farmakokinetiku perorálně podávané antikoncepce obsahující ethinylestradiol a levonorgestrel. Proto není očekáváno, že účinnost antikoncepce v této kombinaci bude oslabena souběžným podáváním ruxolitinibu.
4.6 Fertilita, těhotenství a kojení
Těhotenství a antikoncepce u žen
Údaje o podávání přípravku Jakavi těhotným ženám nejsou k dispozici.
Studie u zvířat prokázaly embryotoxický a fetotoxický účinek. Teratogenita nebyla u potkanů a králíků pozorována. Nicméně hraniční expozice byly při porovnání s nejvyšší klinickou dávkou nízké a výsledky mají proto pro člověka omezený význam (viz bod 5.3). Potenciální riziko u člověka není známo. Z hlediska bezpečnosti je podání přípravku Jakavi během těhotenství kontraindikováno (viz bod 4.3). Ženy ve fertilním věku mají během léčby přípravkem Jakavi používat účinnou antikoncepci. V případě otěhotnění v průběhu užívání přípravku Jakavi je nutné individuální zhodnocení rizika a profitu léčby a pečlivý odhad potenciálního rizika pro plod (viz bod 5.3).
Kojení
Přípravek Jakavi nesmí být podáván během kojení (viz bod 4.3) a kojení má být při zahájení léčby ukončeno. Není známo, zda se ruxolitinib a/nebo jeho metabolity vylučují do lidského mateřského mléka. Riziko pro kojené dítě nelze vyloučit. Dostupná farmakodynamická a toxikologická data prokázala vylučování ruxolitinibu a jeho metabolitů do mateřského mléka u studovaných zvířat (viz bod 5.3).
Fertilita
Nejsou k dispozici žádné klinické údaje týkající se ovlivnění fertility u lidí. Ve studiích u zvířat nebyl žádný vliv na fertilitu pozorován.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek Jakavi nemá žádný nebo jen zanedbatelný sedativní účinek. Pokud však pacient pozoruje závratě po užití přípravku Jakavi, má se vyhnout řízení a obsluze strojů.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Bezpečnost byla vyhodnocena u celkem 855 pacientů (s MF nebo PV), kteří užívali přípravek Jakavi ve studiích fáze 2 a 3.
Myelofibróza
V randomizační periodě dvou pivotních studií COMFORT-I a COMFORT-II byl medián trvání expozice přípravku Jakavi 10,8 měsíce (rozmezí 0,3 až 23,5 měsíce). Většina pacientů (68,4 %) byla léčena po dobu nejméně 9 měsíců. Z celkového počtu 301 pacientů mělo 111 (36,9 %) počet krevních destiček ve výchozím stavu mezi 100000/mm3 a 200000/mm3 a 190 (63,1 %) mělo počet krevních destiček ve výchozím stavu >200000/mm3.
V těchto klinických studiích bylo zjištěno ukončení léčby z důvodu výskytu nežádoucích účinků bez ohledu na jejich příčinu u 11,3 % pacientů.
Nejčastějšími hlášenými nežádoucími účinky byla trombocytopenie a anemie.
Hematologické nežádoucí účinky léku (všechny stupně dle CTCAE [Common Terminology Criteria for Adverse Events] klasifikace) zahrnovaly anemii (82,4 %), trombocytopenii (69,8 %) a neutropenii (16,6 %).
Výskyt anemie, trombocytopenie a neutropenie závisí na podávané dávce.
Nejčastějšími třemi nehematologickými nežádoucími účinky léku byla tvorba hematomů (21,3 %), závratě (15,3 %) a bolest hlavy (14,0 %).
Nejčastějšími třemi nehematologickými laboratorními abnormalitami bylo zvýšení ALT (27,2 %), zvýšení AST (19,9 %) a hypercholesterolemie (16,9 %). V klinických studiích fáze 3 u pacientů s MF nebyla pozorována hypercholesterolemie CTCAE stupně 3 nebo 4, zvýšená hladina aspartátaminotransferázy a ani zvýšená hladina alaninaminotransferázy CTCAE stupně 4.
Dlouhodobá bezpečnost: Jak bylo možné očekávat během prodloužené periody sledování, po vyhodnocení bezpečnostních dat 3letého sledování (medián trvání expozice 33,2 měsíců ve studích COMFORT-I a COMFORT-II u pacientů iniciálně randomizovaných k ruxolitinibu) od 457 pacientů s myelofibrózou léčených ruxolitinibem během randomizované a prodloužené periody dvou pivotních studií fáze 3 vzrostly kumulativní četnosti některých nežádoucích účinků. Toto vyhodnocení zahrnovalo data od pacientů, kteří byli iniciálně randomizovaní na ruxolitinib (N=301) a pacientů, kteří užívali ruxolitinib po přechodu z ramen s kontrolní léčbou (N=156). V těchto aktualizovaných datech bylo ukončení léčby z důvodu nežádoucích účinků zjistěno u 17,1 % pacientů léčených ruxolitinibem.
Pravá _ polycytémie
Bezpečnost přípravku Jakavi byla stanovena na základě vyhodnocení 110 pacientů s pravou polycytémií v otevřené, randomizované, kontrolované studii fáze 3 RESPONSE. Níže uvedené nežádoucí účinky reflektují počáteční periodu studie (až do týdne 32) s ekvivalentní expozicí ruxolitinibu a nejlepší dostupnou léčbou (BAT), což odpovídalo mediánu trvání expozice přípravku Jakavi 7,8 měsíce. Průměrný věk pacientů užívajících přípravek Jakavi byl přibližně 60 let.
Ukončení léčby z důvodu výskytu nežádoucích účinků bez ohledu na jejich příčinu bylo zjištěno u 3,6 % pacientů léčených přípravkem Jakavi a u 1,8 % pacientů léčených nejlepší dostupnou léčbou.
Hematologické nežádoucí účinky (jakéhokoli CTCAE stupně) zahrnovaly anemii (43,6 %) a trombocytopenii (24,5 %). Anemie nebo trombocytopenie CTCAE stupně 3 a 4 byly hlášené v 1,8 % nebo 5,5 % případů.
Tři nejčastější nehematologické nežádoucí účinky byly závrať (15,5 %), zácpa (8,2 %) a herpes zoster (6,4 %).
Tři nejčastější nehematologické laboratorní abnormality (jakéhokoli CTCAE stupně) byly hypercholesterolemie (30,0 %), zvýšená hladina alaninaminotransferázy (22,7 %) a zvýšená hladina aspartátaminotransferázy (20,9 %). Ve všech případech šlo o CTCAE stupeň 1 a 2 s výjimkou jednoho případu zvýšené hladiny alaninaminotransferázy CTCAE stupně 3.
Dlouhodobá bezpečnost: Pacienti měli medián trvání expozice přípravku Jakavi 18,6 měsíce (rozmezí 0,3 až 35,9 měsíce). S delší expozicí vzrůstala četnost nežádoucích účinků, nicméně nová bezpečnostní zjištění se neobjevila. Při přepočítání na expozici byl výskyt nežádoucích účinků obecně porovnatelný s výskytem zjištěným během počáteční periody studie.
Tabulární přehled nežádoucích účinků hlášených v klinických studiích
V klinickém studijním programu byla závažnost nežádoucích účinků hodnocena podle CTCAE klasifikace, definující stupeň 1 = lehký, stupeň 2 = středně těžký, stupeň 3 = těžký a stupeň 4 = život ohrožující.
Nežádoucí účinky hlášené v klinických studiích (Tabulka 1) jsou seřazeny podle MedDRA systémově-orgánové klasifikace. V každé systémově-orgánové třídě jsou nežádoucí účinky řazeny podle četnosti tak, že nejčastější nežádoucí účinek je na prvním místě. Četnost přiřazená ke každému nežádoucímu účinku je klasifikována podle následujících kategorií: velmi časté (>1/10); časté (>1/100 až <1/10); méně časté (>1/1 000 až <1/100); vzácné (>1/10 000 až <1/1 000); velmi vzácné (<1/10 000).
Tabulka 1 Kategorie četností nežádoucích účinků hlášených ve studiích fáze 3 (COMFORT-I, COMFORT-II, RESPONSE)
|
Nežádoucí účinky |
Kategorie četnosti pro pacienty s MF |
Kategorie četnosti pro pacienty s PV |
|
Infekce a infestace | ||
|
Infekce močových cesta,d |
Velmi časté |
Časté |
|
Časté |
Časté | |
|
Tuberkulózae |
Méně časté |
- |
|
Poruchy krve a lymfatického systémub,d | ||
|
Anemieb |
- |
- |
|
CTCAEc stupně 4 (<6,5 g/dl) |
Velmi časté |
Méně časté |
|
CTCAEc stupně 3 (<8,0 - 6,5g/dl) |
Velmi časté |
Méně časté |
|
Všechny CTCAEc stupně |
Velmi časté |
Velmi časté |
|
T rombocytopenieb | ||
|
CTCAEc stupně 4 (<25000/mm3) |
Časté |
Méně časté |
|
CTCAEc stupně 3 (50000 - 25000/mm3) |
Časté |
Časté |
|
Všechny CTCAEc stupně |
Velmi časté |
Velmi časté |
|
Neutropenieb | ||
|
CTCAEc stupně 4 (<500/mm3) |
Časté |
- |
|
CTCAEc stupně 3 (<1000 - 500/mm3) |
Časté |
- |
|
Všechny CTCAEc stupně |
Velmi časté |
- |
|
Krvácení (všechny případy krvácení zahrnující intrakraniální a gastrointestinální krvácení, podlitiny a jiné typy krvácení) |
Velmi časté |
Velmi časté |
|
Intrakraniální krvácení |
Časté |
- |
|
Gastrointestinální krvácení |
Časté |
- |
|
Podlitiny |
Velmi časté |
Velmi časté |
|
Jiné typy krvácení (zahrnují epistaxi, krvácení po zákroku a hematurii) |
Časté |
Velmi časté |
|
Poruchy metabolismu a výživy | ||
|
Nárůst tělesné hmotnosti3 |
Velmi časté |
Časté |
|
Hypercholesterolemieb CTCAEc stupně 1 a 2 |
Velmi časté |
Velmi časté |
|
Hypertriglyceridemieb CTCAEc stupně 1 |
- |
Velmi časté |
|
Poruchy nervového systému | ||
|
Závrať1 |
Velmi časté |
Velmi časté |
|
Velmi časté |
- | |
|
Gastrointestinální poruchy | ||
|
Flatulencea |
Časté |
- |
|
Zácpaa |
- |
Časté |
|
Poruchy jater a žlučových cest | ||
|
Zvýšená hladina alaninaminotransferázyb | ||
|
CTCAEc stupně 3 (>5x - 20 x ULN) |
Časté |
Méně časté |
|
Všechny CTCAEc stupně |
Velmi časté |
Velmi časté |
|
Zvýšená hladina aspartátaminotransferázyb | ||
|
Všechny CTCAEc stupně |
Velmi časté |
Velmi časté |
|
Cévní poruchy | ||
|
Hypertenze*1 |
- |
Velmi časté |
|
a Četnost podle údajů o nežádoucích účincích. - Subjekt s mnohočetným výskytem daného nežádoucího účinku (ADR) je v dané kategorii započítán pouze jednou. - Nežádoucí účinky zaznamenané při léčbě nebo 28 dní po ukončení léčby. b Četnost podle laboratorních výsledků. - Subjekt s mnohočetným výskytem daného nežádoucího účinku je v dané kategorii započítán pouze jednou. - Nežádoucí účinky zaznamenané při léčbě nebo 28 dní po ukončení léčby. c Common Terminology Criteria for Adverse Events (CTCAE) klasifikace verze 3.0; stupeň 1 = lehký, stupeň 2 = středně těžký, stupeň 3 = těžký, stupeň 4 = život ohrožující d Tyto nežádoucí účinky j sou diskutovány v textu. e Četnost je založená na údajích o všech pacientech vystavených ruxolitinibu v klinických studiích (N=4755). | ||
Po ukončení léčby se mohou u pacientů s MF znovu objevit příznaky MF jako je únava, bolesti kostí, horečka, pruritus, noční pocení, symptomatická splenomegalie a úbytek tělesné hmotnosti. Celkové symptomatické skóre pro příznaky MF se v klinických studiích s MF vrátilo k výchozí hodnotě během 7 dnů po vysazení léčby (viz bod 4.4).
Popis vybraných nežádoucích účinků
Anemie
V klinických studiích fáze 3 u pacientů s MF byla střední doba do vzniku první anemie stupně 2 dle CTCAE nebo závažnější 1,5 měsíce. U jednoho pacienta (0,3 %) byla kvůli anemii přerušena léčba.
U pacientů léčených přípravkem Jakavi klesla průměrná hladina hemoglobinu o přibližně 10 g/l oproti vstupní hodnotě s minimem po 8 až 12 týdnech léčby. Poté došlo k opětovnému vzestupu a nastavení nové rovnováhy s hodnotou hemoglobinu přibližně o 5 g/l nižší vůči vstupní hodnotě. Tento průběh byl pozorován u nemocných bez ohledu na použití transfuzní léčby.
V randomizované, placebem kontrolované studii COMFORT-I dostalo v průběhu randomizované léčby 60,6 % pacientů s MF léčených Jakavi a 37,7 % pacientů s MF léčených placebem transfuzi erytrocytů. Ve studii COMFORT-II dostalo transfuzi erytrocytů 53,4 % pacientů ve skupině léčené přípravkem Jakavi a 41,1 % pacientů ve skupině dostávající nejlepší dostupnou léčbu.
V randomizační periodě pivotních studií se anemie vyskytovala méně často u pacientů s PV než u pacientů s MF (43,6 % oproti 82,4 %). V populaci s PV byly hlášené nežádoucí účinky CTCAE stupně 3 a 4 u 1,8 % pacientů, zatímco u pacientů s MF byla četnost 42,56 %.
Trombocytopenie
V klinických studiích fáze 3 u pacientů s MF byla střední doba do vzniku trombocytopenie stupně 3 nebo 4 přibližně osm týdnů. Trombocytopenie byla obvykle po snížení dávky nebo přerušení léčby reverzibilní. Střední čas do obnovení počtu trombocytů nad 50* 109/l byl 14 dní. Během randomizované periody byla podána transfuze trombocytů 4,7 % pacientů, kteří dostávali přípravek Jakavi a 4,0 % pacientů dostávajícím kontrolní léčbu. Léčbu bylo pro trombocytopenii nutno přerušit u 0,7 % pacientů užívajících přípravek Jakavi a u 0,9 % pacientů dostávajících kontrolní léčbu.
Pacienti s počtem trombocytů 100*109/l až 200*109/l před zahájením léčby přípravkem Jakavi měli častěji trombocytopenii stupně 3 nebo 4 v porovnání s pacienty se vstupním počtem trombocytů >200*109/l (64,2 % oproti 38,5 %).
V randomizačních periodách pivotních studií byl podíl pacientů s trombocytopenií nižší u pacientů s PV (24,5 %) oproti pacientům s MF (69,8 %). Četnost vážné (tj. CTCAE stupeň 3 a 4) trombocytopenie byla nižší u pacientů s PV (5,5 %) než u pacientů s MF (11,6 %).
Neutropenie
V klinických studiích fáze 3 u pacientů s MF byla střední doba do vzniku neutropenie stupně 3 nebo 4 dvanáct týdnů. Během randomizované periody bylo zaznamenáno přerušení léčby nebo snížení dávky pro neutropenii u 1,0 % pacientů a 0,3 % pacientů muselo kvůli neutropenii léčbu ukončit.
V randomizační periodě pivotní studie u pacientů s PV byla neutropenie hlášená u dvou pacientů (1,8 %), z nichž u jednoho pacienta došlo k rozvoji neutropenie CTCAE stupně 4.
Krvácení
V pivotních klinických studiích fáze 3 u pacientů s MF byly krvácivé komplikace (zahrnující nitrolební krvácení, krvácení do zažívacího traktu, hematomy a jiné typy krvácení) zaznamenány u 32,6 % pacientů užívajících přípravek Jakavi a u 23,2 % pacientů užívajících kontrolní léčby (placebo nebo nejlepší dostupnou léčbu). Výskyt příhod krvácení stupně 3 nebo 4 byl stejný u pacientů léčených přípravkem Jakavi s pacienty léčenými kontrolními léčbami (4,7 % oproti 3,1 %). U většiny pacientů s krvácivými komplikacemi hlášenými během léčby byly hlášeny hematomy (65,3 %). Hematomy byly častěji zaznamenány u pacientů léčených přípravkem Jakavi v porovnání
s referenčními léčbami (21,3 % oproti 11,6 %). Nitrolební krvácení bylo hlášeno u 1 % pacientů léčených přípravkem Jakavi a u 0,9 % léčených kontrolními léčbami. Gastrointestinální krvácení bylo hlášeno u 5,0 % pacientů léčených přípravkem Jakavi oproti 3,1 % pacientů léčených kontrolními léčbami. Jiné typy krvácivých komplikací (zahrnující případy jako je epistaxe, krvácení po zákroku a hematurie) byly hlášeny u 13,3 % pacientů léčených přípravkem Jakavi a u 10,3 % pacientů léčených kontrolní léčbou.
V randomizační periodě pivotní studie u pacientů s PV byly krvácivé příhody (zahrnující intrakraniální a gastrointestinální krvácení, podlitiny a jiné typy krvácení) hlášeny u 20 % pacientů léčených přípravkem Jakavi a u 15,3 % pacientů s nejlepší dostupnou léčbou. Podlitiny byly hlášené s podobnými četnostmi v ramenech s přípravkem Jakavi a BAT (10,9 % vs. 8,1 %). U pacientů léčených přípravkem Jakavi nebyl hlášen ani jeden případ intrakraniálního nebo gastrointestinálního krvácení. Jeden pacient léčený přípravkem Jakavi měl krvácivou příhodu stupně 3 (krvácení po zákroku); krvácení stupně 4 nebylo hlášeno. Ostatní krvácivé příhody (zahrnující příhody jako epistaxi, krvácení po zákroku, krvácení dásní) byly hlášeny u 11,8 % pacientů léčených přípravkem Jakavi a u 6,3 % pacientů s nejlepší dostupnou léčbou.
Infekce
V klinických studiích fáze 3 u pacientů s MF byly hlášeny infekce močového traktu stupně 3 nebo 4 u 1,0 % pacientů, herpes zoster u 4,3 % a tuberkulóza u 1,0 % pacientů. V klinických studiích fáze 3 byla hlášena sepse u 3,0 % pacientů. Prodloužené sledování pacientů léčených ruxolitinibem neprokázalo žádné trendy ke zvýšení výskytu sepse v čase.
V randomizační periodě pivotní studie u pacientů s PV byl hlášen jeden případ (0,9 %) infekce močových cest CTCAE stupně 3, infekce močových cest CTCAE stupně 4 nebyla hlášena. Výskyt herpes zoster byl nepatrně vyšší u pacientů s PV (6,4 %) oproti pacientům s MF (4,0 %). U pacientů s PV se objevilo jedno hlášení postherpetické neuralgie CTCAE stupně 3.
Zvýšený systolický krevní tlak
V pivotních klinických studiích fáze 3 u pacientů s MF bylo u 31,5 % pacientů během alespoň jedné návštěvy v porovnání s 19,5 % pacientů léčených kontrolní léčbou zjištěno zvýšení systolického krevního tlaku o 20 mmHg nebo více oproti výchozímu stavu. Ve studii COMFORT-I (pacienti s MF) bylo průměrné zvýšení systolického krevního tlaku oproti výchozímu stavu o 0-2 mmHg u přípravku Jakavi v porovnání s poklesem o 2-5 mmHg v rameni s placebem. Ve studii COMFORT-II vykazovaly průměrné hodnoty u pacientů s MF léčených ruxolitinibem v porovnání s pacienty s MF léčenými kontrolní léčbou malé rozdíly.
V randomizační periodě pivotní studie u pacientů s PV se průměrný systolický krevní tlak zvýšil o 0,65 mmHg u přípravku Jakavi v porovnání s poklesem o 2 mmHg u ramene s BAT.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Není známo žádné antidotum pro předávkování přípravkem Jakavi. Jednorázové podání až 200 mg přípravku Jakavi bylo poměrně dobře snášeno. Opakované podání vyšších než doporučených dávek je spojeno s vyšším výskytem myelosuprese, zahrnující leukopenii, anemii a trombocytopenii. V těchto případech má být podávána vhodná podpůrná léčba.
Nepředpokládá se, že by hemodialýza zvyšovala vylučování ruxolitinibu.
FARMAKOLOGICKÉ VLASTNOSTI
5.
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Cytostatika, inhibitory proteinkinázy, ATC kód: L01XE18 Mechanismus účinku
Ruxolitinib je selektivní inhibitor Janus kináz (JAK) JAK1 a JAK2 (hodnoty IC50 jsou pro enzym JAK1 3,3 nM a pro JAK2 2,8 nM). Tyto kinázy jsou zapojeny do signalizace mnoha cytokinů a růstových faktorů s významnou úlohou při hematopoéze a imunitních funkcích.
Myelofibróza a pravá polycytémie j sou myeloproliferativní neoplastická onemocnění, u kterých byla popsána abnormální regulace signalizace zprostředkované JAK1 a JAK2. Předpokládá se, že příčinou poruchy regulace jsou vysoké hladiny cirkulujících cytokinů, které aktivují signální dráhu JAK-STAT, mutace zvyšující funkci enzymů jako je JAK2V617F a potlačení negativních regulačních mechanismů. U pacientů s MF nacházíme změnu regulace zprostředkované JAK bez ohledu na přítomnost mutace JAK2V617F. Aktivační mutace v JAK2 (V617F nebo exon 12) jsou zjištěné u >95 % pacientů s PV.
Ruxolitinib inhibuje signální dráhu JAK-STAT a buněčnou proliferaci u buněčných modelů hematologických malignit závislých na cytokinech, stejně jako u na cytokinech nezávislého modelu využívajícího Ba/F3 buňky exprimující JAK2V617F mutovaný protein s hodnotou IC50 v rozmezí 80-320 nM.
Farmakodynamické účinky
Ruxolitinib inhibuje cytokiny indukovanou STAT3 fosforylaci v krvi zdravých dobrovolníků i pacientů s MF a pacientů s PV. U obou skupin vedlo podávání ruxolitinibu k maximální inhibici STAT3 fosforylace 2 hodiny po podání s návratem k téměř výchozím hodnotám do 8 hodin po podání, což ukazuje na to, že nedochází k akumulaci mateřské látky ani aktivních metabolitů.
U pacientů s MF došlo při léčbě ruxolitinibem k poklesu zvýšených zánětlivých markerů jako je TNFa, IL-6 a CRP. Pacienti s MF se v průběhu času nestávali refrakterní k farmakodynamickým účinkům ruxolitinibu. Také pacienti s PV vykazovali obdobné zvýšení zánětlivých markerů ve výchozím stavu a tyto markery klesaly po léčbě ruxolitinibem.
V QT studiích u zdravých dobrovolníků nebylo zaznamenáno prodloužení QT/QTc intervalu po jednorázovém podání dávky ruxolitinibu vyšší než terapeutické (až 200 mg), což ukazuje na to, že ruxolitinib nemá vliv na srdeční repolarizaci.
Klinická účinnost a bezpečnost
Myelofibróza
U pacientů s MF (primární myelofibrózou, postpolycytemickou myelofibrózou nebo myelofibrózou po esenciální trombocytemii) byly provedeny dvě randomizované studie fáze 3 (COMFORT-I a COMFORT-II). V obou studiích měli pacienti hmatnou splenomegalii alespoň 5 cm pod žeberním obloukem a byli klasifikovaní podle kritérií International Working Group (IWG) do kategorií středního rizika 2 nebo vysokého rizika. Úvodní dávka přípravku Jakavi byla určena podle počtu trombocytů.
COMFORT-I byla dvojitě slepá, randomizovaná, placebem kontrolovaná studie zahrnující 309 pacientů, kteří byli refrakterní, nebo nebyli vhodnými kandidáty pro dostupnou léčbu. Primární cílový parametr účinnosti byl podíl nemocných, kteří dosáhnou ve 24. týdnu alespoň 35% zmenšení objemu sleziny oproti výchozí hodnotě měřeného pomocí magnetické rezonance (MRI) nebo počítačové tomografie (CT).
Sekundárními cílovými parametry bylo trvání udržení alespoň 35% redukce objemu sleziny proti výchozí hodnotě, podíl pacientů, kteří ve 24. týdnu dosáhli alespoň 50% snížení celkového symptomatického skóre, změna celkového symptomatického skóre od výchozího stavu do 24. týdne hodnocená pomocí modifikovaného MFSAF (Myelofibrosis Symptom Assessment Form) formuláře (verze 2.0, elektronický deník) a celkové přežití.
COMFORT-II byla otevřená, randomizovaná studie zahrnující 219 pacientů. Pacienti byli randomizováni v poměru 2:1 k léčbě přípravkem Jakavi oproti nejlepší dostupné léčbě. Ve skupině s nejlepší dostupnou léčbou dostávalo 47 % pacientů hydroxyureu a 16 % pacientů glukokortikoidy. Primární cílový ukazatel účinnosti léčby byl podíl nemocných, kteří dosáhnou ve 48. týdnu alespoň 35% snížení objemu sleziny oproti výchozí hodnotě měřeného pomocí MRI nebo CT.
Sekundární cílové parametry ve studii COMFORT-II zahrnovaly podíl nemocných, kteří dosáhnou ve 24. týdnu alespoň 35% snížení objemu sleziny oproti výchozí hodnotě a dobu trvání alespoň 35% snížení objemu slezinyoproti výchozí hodnotě.
Ve studiích COMFORT-I a COMFORT-II byly vstupní demografické parametry a charakteristiky onemocnění srovnatelné u obou léčených skupin.
Tabulka 2 Podíl pacientů (%) s >35% snížením objemu sleziny oproti výchozí hodnotě ve 24. týdnu ve studii COMFORT-I a ve 48. týdnu ve studii COMFORT-II (ITT)
|
COMFORT-I |
COMFORT-II | |||
|
Jakavi (N=155) |
Placebo (N=153) |
Jakavi (N=144) |
Nejlepší dostupná léčba (N=72) | |
|
Časový bod |
24. týden |
48. týden | ||
|
Počet (%) pacientů s redukcí objemu sleziny o >35 % |
65 (41,9) |
1 (0,7) |
41 (28,5) |
0 |
|
95% interval spolehlivosti |
34,1; 50,1 |
0; 3,6 |
21,3; 36,6 |
0,0; 5,0 |
|
Hodnota p |
<0,0001 |
<0,0001 | ||
Významně vyšší podíl pacientů ve skupině léčené přípravkem Jakavi dosáhl alespoň 35% snížení objemu sleziny (Tabulka 2) oproti výchozí hodnotě, a to bez ohledu na přítomnost mutace JAK2V617F nebo podtypu onemocnění (primární myelofibróza, postpolycytemická myelofibróza nebo myelofibróza po esenciální trombocytemii).
Tabulka 3 Podíl pacientů (%) s >35% snížením objemu sleziny oproti výchozí hodnotě dle mutačního stavu JAK (hodnocení bezpečnosti)
|
COMFORT-I |
COMFORT-II | |||||||
|
Jakavi |
Placebo |
Jakavi |
Nejlepší dostupná léčba | |||||
|
JAK mutační stav |
Pozitivní (N=113) n (%) |
Negativní (N=40) n (%) |
Pozitivní (N=121) n (%) |
Negativní (N=27) n (%) |
Pozitivní (N=110) n (%) |
Negativní (N=35) n (%) |
Pozitivní (N=49) n (%) |
Negativní (N=20) n (%) |
|
Počet (%) pacientů s redukcí objemu sleziny o >35 % |
54 (47,8) |
11 (27,5) |
1 (0,8) |
0 |
36 (32,7) |
5 (14,3) |
0 |
0 |
|
Časový bod |
Po 24 týdnech |
Po 48 týdnech | ||||||
Pravděpodobnost zachování odpovědi sleziny (>35% snížení) na přípravku Jakavi po dobu nejméně 24 týdnů byla 89 % ve studii COMFORT-I a 87 % ve studii COMFORT-II; 52 % udrželo odpověď sleziny po dobu nejméně 48 týdnů ve studím COMFORT-II.
Ve studii COMFORT-I dosáhlo 45,9 % subjektů ve skupině s přípravkem Jakavi >50% zlepšení celkového symptomatického skóre od výchozího stavu ve 24. týdnu (vyhodnocovaného pomocí MFSAF deníku v2.0) v porovnání s 5,3 % pacientů ve skupině s placebem (p<0,0001 za pomoci chí-kvadrát testu). Průměrná změna celkového zdravotního stavu ve 24. týdnu měřená pomocí dotazníku EORTC QLQ C30 byla +12,3 u přípravku Jakavi a -3,4 u placeba (p<0,0001).
Ve studii COMFORT-I byl při mediánu sledování 34,3 měsíce výskyt úmrtí u pacientů randomizovaných do ramene s ruxolitinibem 27,1 % oproti 35,1 % u pacientů randomizovaných do ramene s placebem; HR 0,687; 95% CI 0,459-1,029; p=0,0668.
Ve studii COMFORT-II byl při mediánu sledování 34,7 měsíce výskyt úmrtí u pacientů randomizovaných do ramene s ruxolitinibem 19,9 % oproti 30,1 % u pacientů randomizovaných k nejlepší dostupné léčbě (BAT); HR 0,48; 95% CI 0,28-0,85; p=0,009. V obou studiích byly nižší výskyty úmrtí zjištěné v rameni s ruxolitinibem ovlivněny především výsledky získanými od pacientů z podskupin po pravé polycytémii a po esenciální trombocytémii.
Pravá _ polycytémie
Randomizovaná, otevřená, aktivně kontrolovaná studie fáze 3 (RESPONSE) byla provedena u 222 pacientů s PV, kteří byli rezistentní nebo intolerantní k hydroxyurei definované podle publikovaných kritérií mezinárodní pracovní skupiny evropské leukemické sítě (ELN). 110 pacientů bylo randomizovaných do ramene s ruxolitinibem a 112 pacientů do ramene s nejlepší dostupnou léčbou. Zahajovací dávka přípravku Jakavi byla 10 mg dvakrát denně. Poté byly u pacientů dávky individuálně upraveny na základě tolerability a účinnosti s maximální dávkou 25 mg dvakrát denně. Nejlepší dostupná léčba byla vybraná zkoušejícím individuálně pro každého pacienta a zahrnovala hydroxyureu (59,5%), interferon/pegylovaný interferon (11,7 %), anagrelid (7,2 %), pipobroman (1,8 %) a pozorování (15,3 %).
Výchozí demografické parametry a charakteristika onemocnění byly v obou ramenech léčby srovnatelné. Medián věku byl 60 let (rozmezí 33 až 90 let). Pacienti v rameni s ruxolitinibem měli medián doby od diagnózy PV 8,2 roku a dříve užívali hydroxyureu s mediánem trvání léčby přibližně 3 roky. Většina pacientů (>80 %) měla provedeny nejméně dvě flebotomie v posledních 24 týdnech před zařazením do studie. Komparativní data týkající se dlouhodobého přežívání a výskytu komplikací onemocnění chybí.
Primárním složeným cílovým parametrem byl podíl pacientů, kteří dosáhli jak nevhodnosti k flebotomii (kontrolu HCT), tak i >35% zmenšení objemu sleziny od výchozího stavu do 32. týdne. Kritérium vhodnosti k provedení flebotomie bylo definováno jako potvrzený HCT >45%, tj. minimálně o 3 procentní body vyšší oproti HCT stanovenému ve výchozím stavu nebo potvrzenému HCT >48%, dle toho toho, která hodnota je nižší. Klíčové sekundární cílové parametry zahrnovaly podíl pacientů s dosaženým primárním cílovým parametrem a bez progrese ve 48. týdnu, stejně jako podíl pacientů s dosaženou kompletní hematologickou remisí ve 32. týdnu.
Studie dosáhla svého primárního cíle, vyšší podíl pacientů ve skupině s přípravkem Jakavi dosáhl primárního složeného cílového parametru a každé z jeho individuálních komponent. Výrazně vyšší počet pacientů léčených přípravkem Jakavi (20,9 %) dosáhl primární odpovědi (p<0,0001) v porovnání s nejlepší dostupnou léčbou (0,9 %). Kontroly hematokritu bylo dosaženo u 60 % pacientů v rameni s přípravkem Jakavi v porovnání s 19,6 % v rameni s nejlepší dostupnou léčbou a >35% snížení objemu sleziny bylo dosaženo u 38,2 % pacientů v rameni s přípravkem Jakavi v porovnání s 0,9 % v rameni s nejlepší dostupnou léčbou (obrázek 1). 94 (83,9 %) pacientů randomizovaných k rameni s nejlepší dostupnou léčbou přešlo ve 32. týdnu nebo později na léčbu ruxolitinibem, což omezuje porovnání obou ramen po 32. týdnu.
Také bylo dosaženo obou klíčových sekundárních cílových parametrů. Poměr pacientů, kteří dosáhli kompletní hematologické remise, byl 23,6 % u přípravku Jakavi v porovnání s 8,9 % u nejlepší dostupné léčby (p=0,0028) a poměr pacientů, kteří dosáhli trvalé primární odpovědi ve 48. týdnu, byl
19,1 % u přípravku Jakavi a 0,9 % u nejlepší dostupné léčby (p<0,0001).
Obrázek 1 Pacienti, kteří dosáhli primárního cílového parametru a komponent primárního cílového parametru ve 32. týdnu

Primární složený cíl v > 35% redukce objemu Kontrola hematokritu bez týdnu 32 sleziny flebotomie
Symptomatická zátěž byla vyhodnocena pomocí elektronického pacientského dotazníku MPN-SAF celkového symptomatického skóre (TSS), který se skládá ze 14 otázek. Ve 32. týdnu dosáhlo 49 % a 64 % pacientů léčených ruxolitinibem >50% snížení TSS-14 resp. TSS-5 v porovnání s pouze 5 % a 11 % pacientů na nejlepší dostupné léčbě.
Vnímání prospěchu z léčby bylo stanoveno pomocí dotazníku Patient Global Impression of Change (PGIC). 66 % pacientů léčených ruxolitinibem hlásilo zlepšení již čtyři týdny po zahájení léčby oproti 9 % léčených nejlepší dostupnou léčbou. Zlepšení vnímání prospěchu z léčby bylo vyšší také u pacientů léčených ruxolitinibem ve 32. týdnu (78 % oproti 33 %).
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Jakavi u všech podskupin pediatrické populace pro léčbu MF (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Absorpce
Ruxolitinib patří do třídy 1 podle BCS (Biopharmaceutical Classification System), protože má vysokou prostupnost, dobrou rozpustnost a rychlý rozpad. Ruxolitinib byl v klinických studiích po perorálním podání rychle absorbován s maximální plazmatickou koncentrací (Cmax) dosaženou přibližně 1 hodinu po podání. Absorpce ruxolitinibu po perorálním podání (stanoven ruxolitinib a jeho metabolity tvořené při prvním průchodu játry) je podle farmakokinetických studií u lidí 95 % a více. Průměrné hodnoty Cmax a AUC ruxolitinibu se zvyšují úměrně s podanou jednorázovou dávkou v rozmezí 5-200 mg. Užití ruxolitinibu po tučném jídle nevedlo ke klinicky významné změně farmakokinetiky. Průměrná hodnota Cmax při požití tučného jídla mírně klesla (o 24 %) a průměrná hodnota AUC se téměř nezměnila (nárůst o 4 %).
Distribuce
Průměrný distribuční objem v ustáleném stavu je přibližně 75 litrů u pacientů s MF a PV. Vazba na plazmatické bílkoviny je in vitro při koncentracích ruxolitinibu odpovídajících klinickému využití přibližně 97 % a ruxolitinib se váže zejména na albumin. Celotělová autoradiografická studie u potkanů ukázala, že ruxolitinib neprochází hematoencefalickou bariérou.
Biotransformace
Ruxolitinib je metabolizován především CYP3A4 (>50 %) s dodatečným přispěním CYP2C9. Mateřská látka má v lidské plazmě převládající podíl a představuje 60 % látek v oběhu souvisejících s podáním přípravku. V plazmě jsou přítomné dva hlavní a zároveň aktivní metabolity, které představují 25 % a 11 % z mateřské AUC. Tyto metabolity mají polovinu až pětinu mateřské farmakologické aktivity na JAK enzymy. Celkově všechny aktivní metabolity přispívají 18 % k celkové farmakodynamické aktivitě ruxolitinibu. V klinických plazmatických koncentracích ruxolitinib podle in vitro studií neinhibuje CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 nebo CYP3A4 a není silným induktorem CYP1A2, CYP2B6 nebo CYP3A4. Na základě in vitro dat může ruxolitinib inhibovat P-gp a BCRP.
Eliminace
Ruxolitinib se eliminuje z organismu převážně metabolickou cestou. Průměrný poločas eliminace ruxolitinibu je přibližně 3 hodiny. Po jednorázovém perorálním podání [14C]-značeného ruxolitinibu zdravým dobrovolníkům bylo farmakum z převážné většiny metabolizováno a 74 % podané radioaktivity bylo vyloučeno močí a 22 % stolicí. Nezměněná mateřská látka představovala méně než 1 % celkové vyloučené radioaktivity.
Linearita/nelinearita
Ve studiích s podáním jednorázové i opakované dávky bylo prokázáno, že systémová expozice se proporcionálně zvyšuje v závislosti na dávce.
Farmakokinetika u zvláštních skupin pacientů
Vliv věku, pohlaví a rasy
Na základě studií u zdravých dobrovolníků nebyly pozorovány relevantní rozdíly ve farmakokinetice v závislosti na pohlaví a rase. Při populační farmakokinetické analýze u pacientů s MF nebyla zjištěna závislost orální clearance na věku nebo rase. Predikovaná orální clearance byla 17,7 l/h u žen a
22,1 l/h u mužů s interindividuální variabilitou 39 % u pacientů s MF. U pacientů s PV byla clearance
12,7 l/h se 42 % interindividuální variabilitou a mezi perorální clearance a pohlavím, stářím pacienta nebo rasou nebyl na základě stanovení farmakokinetiky v této populaci pacientů zřejmý žádný vztah.
Pediatrická populace
Bezpečnost a účinnost Jakavi nebyla zkoumána u pediatrické populace (viz bod 5.1 „Pediatrická populace“).
Porucha funkce ledvin
Funkce ledvin byla stanovena pomocí MDRD (Modification of Diet in Renal Disease) a kreatininu v moči. Expozice ruxolitinibu byla po jednorázovém podání 25 mg ruxolitinibu shodná u pacientů s různým stupněm poruchy funkce ledvin a u pacientů s normálními renálními funkcemi, avšak hodnoty plazmatické AUC metabolitů ruxolitinibu měly tendenci se zvyšovat se zhoršujícím se postižením ledvin a byly nejvyšší u pacientů s těžkou poruchou funkce ledvin. Není známo, zda má zvýšená expozice metabolitům vliv na bezpečnost. Úprava dávky je doporučená u pacientů s těžkou poruchou funkce ledvin a u nemocných se selháním funkce ledvin (viz bod 4.2). Dávkování ve dnech dialýzy snižuje expozici metabolitům, ale také farmakodynamický účinek, především ve dnech mezi dialýzami.
Porucha funkce jater
Průměrná hodnota AUC ruxolitinibu byla po jednorázovém podání 25 mg ruxolitinibu pacientům s různým stupněm poškození jater zvýšená o 87 %, 28 % a 65 % u pacientů s lehkou, středně těžkou a těžkou poruchou funkce jater (v uvedeném pořadí) ve srovnání s pacienty s normální funkcí jater.
Mezi hodnotou AUC a stupněm jaterního postižení dle Child-Pugh skóre nebyl prokázán žádný vztah. Terminální poločas eliminace byl u pacientů s poškozením jater prodloužen ve srovnání se zdravými dobrovolníky (4,1-5,0 h oproti 2,8 h). U pacientů s poškozením jater je doporučeno přibližně 50% snížení dávky (viz bod 4.2).
5.3 Předklinické údaje vztahující se k bezpečnosti
S ruxolitinibem byly provedeny konvenční farmakologické studie bezpečnosti, toxicity po opakovaném podání, genotoxicity, reprodukční toxicity a hodnocení kancerogenního potenciálu.
V testech toxicity po opakovaném podání byly cílovými orgány spojenými s farmakologickým působením ruxolitinibu kostní dřeň, periferní krev a lymfatické tkáně. U psů byly zjištěny infekce, obecně asociované s imunosupresí. Při telemetrických studiích u psů byl pozorován nežádoucí pokles krevního tlaku a vzestup srdeční frekvence a v respiračních studiích u potkanů byl pozorován nežádoucí pokles minutového objemu. Hraniční dávka (podle Cmax volné látky), při které nebyly pozorovány nežádoucí účinky, byla u psů a potkanů 15,7krát respektive 10,4krát vyšší než je maximální doporučená dávka u lidí (25 mg dvakrát denně). Nebyl pozorován žádný neurofarmakologický účinek ruxolitinibu.
Ruxolitinib snižoval hmotnost plodu a zvyšoval postimplantační ztráty ve studiíích u zvířat. U potkanů a králíků nebyl zjištěn výskyt teratogeních účinků. Nicméně hraniční expozice porovnávané s nejvyšší klinickou dávkou byly nízké a výsledky proto mají pro člověka omezený význam. Nebyl pozorován žádný vliv na fertilitu. V prenatálních a postnatálních vývojových studiích bylo pozorováno mírné prodloužení gestační periody, snížení počtu implantačních míst a snížení počtu porozených mláďat.
U mláďat byla zaznamenána snížená průměrná porodní hmotnost a krátké období snížených průměrných přírůstků hmotnosti po narození. U potkanů v laktaci byly ruxolitinib a/nebo jeho metabolity vylučovány do mateřského mléka, a to v koncentracích 13krát vyšších než v mateřské plazmě. Ruxolitinib neměl mutagenní a klastogenní účinky. Ruxolitinib neměl karcinogenní účinky u Tg.rasH2 transgenních myší.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Mikrokrystalická celulóza Magnesium-stearát Koloidní bezvodý oxid křemičitý Sodná sůl karboxymethylškrobu (typ A)
Povidon Hyprolóza Monohydrát laktózy
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
3 roky
6.4 Zvláštní opatření pro uchovávání
Neuchovávejte při teplotě nad 30°C.
6.5 Druh obalu a obsah balení
Balení s PVC/PCTFE/Al blistry obsahující 14 nebo 56 tablet nebo vícečetná balení obsahující 168 (3 balení po 56) tabletách.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku
Žádné zvláštní požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/12/773/010-012
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
23.08.2012
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného za propouštění šarží
Novartis Pharma GmbH
Roonstrasse 25
90429 Norimberk
Německo
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
• Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předloží první pravidelně aktualizovanou zprávu o bezpečnosti pro tento léčivý přípravek do 9měsíců od jeho registrace. Držitel rozhodnutí o registraci dále předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP j e třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
Aktualizovaný RMP se předkládá každoročně až do prodloužení registrace.
Povinnost uskutečnit poregistrační opatření
Držitel rozhodnutí o registraci uskuteční v daném termínu níže uvedená opatření
|
Popis |
Termín splnění |
|
Každoročně musí být poskytnuto sledování údajů o účinnosti a bezpečnosti z prodloužených fází studií INCB 18424-351 a INC424A2352, které v současné době zahrnují údaje podobných cílů (celkové přežití, přežití bez progrese onemocnění a přežití bez leukemie). |
Každý rok v říjnu |
|
Post-autorizační studie účinnosti k získání dlouhodobých údaj ů o účinnosti a bezpečnosti ruxolitinibu včetně (pozdního) nástupu účinku, trvání (různé) odpovědi, stejně jako incidence nežádoucích příhod zahrnujících hematologické |
CSR po týdnu 80: červen 2015 |
|
transformace a sekundární malignity ze studie B2301. |
Finální CSR: prosinec 2019 |
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Jakavi 5 mg tablety ruxolitinibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tableta obsahuje ruxolitinibum 5 mg (ve formě ruxolitinibi phosphas).
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktózu.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Tablety
14 tablet 56 tablet
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/12/773/004 14 tablet
EU/1/12/773/005 56 tablet
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Jakavi 5 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Jakavi 5 mg tablety ruxolitinibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tableta obsahuje ruxolitinibum 5 mg (ve formě ruxolitinibi phosphas).
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktózu.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Tablety
Vícečetné balení: 168 (3 balení po 56) tablet.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
POUŽITELNOST
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/12/773/006 168 tablet (3x56)
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Jakavi 5 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Jakavi 5 mg tablety ruxolitinibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tableta obsahuje ruxolitinibum 5 mg (ve formě ruxolitinibi phosphas).
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktózu.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Tablety
56 tablet. Součást vícečetného balení. Neprodávat samostatně.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA)
EU/1/12/773/006 168 tablet (3x56)
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Jakavi 5 mg
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA BLISTRECH NEBO STRIPECH BLISTRY
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Jakavi 5 mg tablety ruxolitinibum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
Pondělí
Úterý
Středa
Čtvrtek
Pátek
Sobota
Neděle
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Jakavi 10 mg tablety ruxolitinibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tableta obsahuje ruxolitinibum 10 mg (ve formě ruxolitinibi phosphas).
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktózu.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Tablety
14 tablet 56 tablet
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/12/773/014 14 tablet
EU/1/12/773/015 56 tablet
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Jakavi 10 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Jakavi 10 mg tablety ruxolitinibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tableta obsahuje ruxolitinibum 10 mg (ve formě ruxolitinibi phosphas).
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktózu.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Tablety
Vícečetné balení: 168 (3 balení po 56) tablet.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
POUŽITELNOST
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/12/773/016 168 tablet (3x56)
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Jakavi 10 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Jakavi 10 mg tablety ruxolitinibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tableta obsahuje ruxolitinibum 10 mg (ve formě ruxolitinibi phosphas).
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktózu.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Tablety
56 tablet. Součást vícečetného balení. Neprodávat samostatně.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA)
EU/1/12/773/016 168 tablet (3x56)
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Jakavi 10 mg
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA BLISTRECH NEBO STRIPECH BLISTRY
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Jakavi 10 mg tablety ruxolitinibum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
Pondělí
Úterý
Středa
Čtvrtek
Pátek
Sobota
Neděle
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Jakavi 15 mg tablety ruxolitinibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tableta obsahuje ruxolitinibum 15 mg (ve formě ruxolitinibi phosphas).
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktózu.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Tablety
14 tablet 56 tablet
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/12/773/007 14 tablet
EU/1/12/773/008 56 tablet
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Jakavi 15 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Jakavi 15 mg tablety ruxolitinibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tableta obsahuje ruxolitinibum 15 mg (ve formě ruxolitinibi phosphas).
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktózu.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Tablety
Vícečetné balení: 168 (3 balení po 56) tablet.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
POUŽITELNOST
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/12/773/009 168 tablet (3x56)
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Jakavi 15 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Jakavi 15 mg tablety ruxolitinibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tableta obsahuje ruxolitinibum 15 mg (ve formě ruxolitinibi phosphas).
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktózu.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Tablety
56 tablet. Součást vícečetného balení. Neprodávat samostatně.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA)
EU/1/12/773/009 168 tablet (3x56)
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Jakavi 15 mg
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA BLISTRECH NEBO STRIPECH BLISTRY
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Jakavi 15 mg tablety ruxolitinibum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
Pondělí
Úterý
Středa
Čtvrtek
Pátek
Sobota
Neděle
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Jakavi 20 mg tablety ruxolitinibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tableta obsahuje ruxolitinibum 20 mg (ve formě ruxolitinibi phosphas).
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktózu.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Tablety
14 tablet 56 tablet
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/12/773/010 14 tablet
EU/1/12/773/011 56 tablet
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Jakavi 20 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Jakavi 20 mg tablety ruxolitinibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tableta obsahuje ruxolitinibum 20 mg (ve formě ruxolitinibi phosphas).
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktózu.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Tablety
Vícečetné balení: 168 (3 balení po 56) tablet.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
POUŽITELNOST
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/12/773/012 168 tablet (3x56)
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Jakavi 20 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Jakavi 20 mg tablety ruxolitinibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tableta obsahuje ruxolitinibum 20 mg (ve formě ruxolitinibi phosphas).
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktózu.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Tablety
56 tablet. Součást vícečetného balení. Neprodávat samostatně.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA)
EU/1/12/773/012 168 tablet (3x56)
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Jakavi 20 mg
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA BLISTRECH NEBO STRIPECH BLISTRY
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Jakavi 20 mg tablety ruxolitinibum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
Pondělí
Úterý
Středa
Čtvrtek
Pátek
Sobota
Neděle
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro pacienta
Jakavi 5 mg tablety Jakavi 10 mg tablety Jakavi 15 mg tablety Jakavi 20 mg tablety
ruxolitinibum
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Jakavi a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Jakavi užívat
3. Jak se přípravek Jakavi užívá
4. Možné nežádoucí účinky
5. Jak přípravek Jakavi uchovávat
6. Obsah balení a další informace
1. Co je přípravek Jakavi a k čemu se používá
Přípravek Jakavi obsahuje léčivou látku ruxolitinib.
Přípravek Jakavi je léčivý přípravek užívaný k léčbě dospělých pacientů se zvětšením sleziny nebo s příznaky spojenými s myelofibrózou, vzácnou formou rakoviny krve.
Přípravek Jakavi se také užívá k léčbě pacientů s pravou polycytémií, kteří jsou rezistentní vůči hydroxyurei nebo ji netolerují.
Mechanismus účinku přípravku Jakavi
Zvětšení sleziny je jedním z příznaků myelofibrózy. Myelofibróza je onemocnění kostní dřeně, při kterém je dřeň nahrazována vazivovou tkání. Pozměněná dřeň nemůže dále tvořit dostatečné množství krvinek a následkem je zvětšování sleziny. Blokádou účinku určitých enzymů (nazývaných Janus kinázy) může přípravek Jakavi zmenšovat slezinu u pacientů s myelofibrózou a zmírňovat příznaky, jako jsou horečka, noční pocení, bolest kostí a ztráta tělesné hmotnosti u pacientů s myelofibrózou. Přípravek Jakavi může pomoci se snížením rizika závažných krevních nebo cévních komplikací.
Pravá polycytémie je onemocnění kostní dřeně, pří kterém se ve dřeni tvoří příliš vysoké množství červených krvinek. Následkem zvýšeného počtu krevních buněk stoupá hustota krve. Přípravek Jakavi může zmírnit příznaky, zmenšit velikost sleziny a množství červených krvinek tvořených u pacientů s pravou polycytémií selektivním zablokováním enzymů, které se nazývají Janusovy kinázy (JAK1 a JAK2), což potenciálně snižuje riziko závažných krevních nebo cévních komplikací.
Pokud máte jakékoli dotazy k tomu, jak přípravek Jakavi funguje nebo proč Vám byl tento přípravek předepsán, zeptejte se svého lékaře.
Pečlivě dodržujte všechny pokyny lékaře. Mohou se lišit od obecných pokynů uvedených v této příbalové informaci.
Neužívejte přípravek Jakavi
- jestliže jste alergický(á) na ruxolitinib nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
- pokud j stě těhotná nebo koj íte.
Pokud se na Vás vztahuje některý z výše uvedených bodů, informujte o tom svého lékaře, který posléze rozhodne, zda můžete zahájit léčbu přípravkem Jakavi.
Upozornění a opatření
Před užitím přípravku Jakavi se poraďte se svým lékařem nebo lékárníkem.
- Pokud se u Vás projevuje infekce. Je nezbytné vyléčit infekci před zahájením léčby přípravkem Jakavi. Je důležité, abyste sdělil(a) svému lékaři, zda jste někdy neprodělal(a) tuberkulózu, nebo zda jste nebyl(a) v blízkém kontaktu s někým, kdo tuberkulózu má nebo ji prodělal. Váš lékař může provést testy, aby zjistil, zda nemáte tuberkulózu. Je důležité, abyste informoval(a) svého lékaře, pokud jste někdy prodělal(a) hepatitidu B.
- Pokud trpíte potížemi s ledvinami. Váš lékař může předepsat odlišnou dávku přípravku Jakavi.
- Pokud trpíte, nebo jste trpěl(a) jaterními potížemi. Váš lékař může předepsat odlišnou dávku přípravku Jakavi.
- Pokud užíváte další léčivé přípravky (viz bod „Další léčivé přípravky a přípravek Jakavi “).
- Pokud j ste někdy měl(a) tuberkulózu.
- Pokud j ste někdy měl(a) rakovinu kůže.
Informujte svého lékaře nebo lékárníka během léčby přípravkem Jakavi:
- Pokud se u Vás objeví nečekané podlitiny a/nebo krvácení, neobvyklá únava, dýchavičnost během námahy nebo v klidu, neobvykle bledá barva pokožky, nebo časté infekce (jde
o příznaky poruchy funkce krve).
- Pokud se u Vás objeví horečka, zimnice nebo jiné příznaky infekcí.
- Pokud se u Vás objeví chronický kašel s hlenem obsahujícím krev, horečka, noční pocení a ztráta tělesné hmostnosti (může jít o příznaky tuberkulózy).
- Pokud se u Vás objeví následující příznaky, nebo pokud někdo z Vašeho blízkého okolí zaznamená, že máte některé z těchto příznaků: zmatenost nebo ztížená schopnost myšlení, ztráta rovnováhy nebo potíže při chůzi, nemotornost, obtíže při mluvení, omezená síla nebo slabost na jedné straně Vašeho těla, rozmazané vidění a/nebo ztráta zraku. Může jít o příznaky závažné infekce mozku a Váš lékař může navrhnout další vyšetření a sledování.
- Pokud se u Vás vyvinou bolestivé kožní vyrážky s puchýři (jde o příznaky pásového oparu).
- Pokud si všimnete kožníchzměn. Protože byly hlášeny určité typy rakoviny kůže (nejde o melanomy), může tento stav vyžadovat další sledování.
Vyšetření krve
Dříve než zahájíte léčbu přípravkem Jakavi, lékař provede vyšetření krve, aby pro Vás určil nejvhodnější zahajovací dávku. Během léčby Vám budou prováděna další vyšetření krve, aby mohl lékař sledovat množství krvinek (bílých krvinek, červených krvinek a krevních destiček) ve Vašem těle a mohl určit, jak Vaše tělo reaguje na léčbu a zda nemá přípravek Jakavi nežádoucí vliv na tyto buňky. Váš lékař může dávku upravit nebo zastavit léčbu.
Ukončení léčby přípravkem Jakavi
Pokud přerušíte užívání přípravku Jakavi, mohou se znovu objevit příznaky myelofibrózy. Váš lékař může postupně snižovat množství přípravku Jakavi užívaného každý den až do úplného ukončení léčby.
Děti a dospívající
Nepodávejte tento přípravek dětem a dospívajícím do 18 let věku, protože použití přípravku Jakavi u dětí nebylo zkoušeno.
Další léčivé přípravky a přípravek Jakavi
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Je zejména důležité, abyste uvedl(a) jakýkoli z následujících léků, které obsahují následující léčivé látky, pro možnou potřebu úpravy dávky přípravku Jakavi Vaším lékařem.
Následující přípravky mohou zvýšit riziko nežádoucí účinků spojených s přípravkem Jakavi:
- Některé léky určené k léčbě infekcí. Tato skupina zahrnuje léky určené k léčbě plísňových onemocnění (jako je ketokonazol, itrakonazol, posakonazol, flukonazol a vorikonazol), léky určené k léčbě určitých typů bakteriálních infekcí (antibiotika jako je klarithromycin, telithromycin, ciprofloxacin nebo erythromycin), léky určené k léčbě virových infekcí, včetně HIV/AIDS (jako je apranevir, atazanavir, indinavir, lopinavir/ritonavir, nelfinavir, ritonavir, sachinavir), léky určené k léčbě hepatitidy C (boceprevir, telaprevir).
- Nefazodon, lék určený k léčbě deprese.
- Mibefradil nebo diltiazem, léky určené k léčbě vysokého krevního tlaku (hypertenze) a chronické anginy pectoris (onemocnění srdce).
- Cimetidin, lék určený k léčbě pálení žáhy.
Následující léky mohou snížit účinnost přípravku Jakavi:
- Avasimib, lék užívaný k léčbě srdečních onemocnění.
- Fenytoin, karbamazepin nebo fenobarbital a další antiepileptika užívaná k zastavení záchvatů nebo křečí.
- Rifabutin nebo rifampicin, léky užívané k léčbě tuberkulózy (TBC).
- Třezalka tečkovaná (Hypericumperforatum), rostlinný přípravek určený k léčbě deprese.
Během léčby přípravkem Jakavi nikdy nezačínejte užívat nový lék bez konzultace s lékařem, který Vám předepsal přípravek Jakavi. To se týká léků na předpis, léků prodávaných bez lékařského předpisu a rostlinných nebo alternativních léků.
Těhotenství a kojení
Neužívejte přípravek Jakavi během těhotenství. Poraďte se se svým lékařem o tom, jak zabránit otěhotnění během Vaší lečby přípravkem Jakavi.
Během užívání přípravku Jakavi nekojte. Informujte svého lékaře, pokud kojíte.
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek užívat
Řízení dopravních prostředků a obsluha strojů
Pokud se u Vás při užívání přípravku objeví závratě, neřidte dopravní prostředky a neobsluhujte stroje. Přípravek Jakavi obsahuje laktózu
Přípravek Jakavi obsahuje laktózu (mléčný cukr). Pokud Vám Váš lékař řekl, že nesnášíte některé cukry, poraďte se se svým lékařem, než začnete tento léčivý přípravek užívat.
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře nebo lékárníka. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Dávka přípravku Jakavi závisí na výsledku vyšetření počtu krvinek u pacienta. Váš lékař vyhodnotí množstí krvinek ve Vašem těle a stanoví pro Vás nejvhodnější dávku, zejména pokud máte potíže s játry nebo ledvinami.
- Doporučená počáteční dávka dávka u pacientů s myelofibrózou je 15 mg dvakrát denně nebo 20 mg dvakrát denně, v závislost na množství Vašich krvinek.
- Doporučená počáteční dávka dávka u pacientů s pravou polycytémií je 10 mg dvakrát denně, v závislost na množství krvinek.
- Maximální dávka je 25 mg dvakrát denně.
Váš lékař Vám vždy sdělí přesný počet tablet přípravku Jakavi, který máte užívat.
Během léčby může Vám může lékař doporučit nižší nebo vyšší dávku, pokud výsledky krevních testů ukážou, že je to nezbytné, pokud máte potíže s játry nebo ledvinami, nebo pokud potřebujete léčbu určitými jinými léčivými přípravky.
Pokud chodíte na dialýzu, užívejte buď jednu jednotlivou dávku nebo dvě dávky přípravku Jakavi pouze ve dnech dialýzy, a to po jejím ukončení. Váš lékař Vám sdělí, zda máte užívat jednu nebo dvě dávky a kolik tablet máte v každé dávce užívat.
Měl(a) byste užívat přípravek Jakavi každý den ve stejnou dobu, s jídlem nebo bez jídla.
Měl(a) byste pokračovat s užíváním přípravku Jakavi tak dlouho, jak Vám doporučí lékař. Jde o dlouhodobou léčbu.
Váš lékař bude pravidelně sledovat Váš stav, aby zjistil, zdá má léčba požadovaný účinek.
Máte-li jakékoli další otázky, týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
Pokud se u Vás projeví nežádoucí účinky (např. poruchy krve), Váš lékař může změnit potřebné množství přípravku Jakavi, které máte užívat, nebo Vám doporučí dočasně zastavit užívání přípravku Jakavi.
Jestliže jste užil(a) více přípravku Jakavi, než jste měl(a)
Jestliže jste nedopatřením užil(a) více přípravku Jakavi než Váš lékař předepsal, okamžitě kontaktujte svého lékaře nebo lékárníka.
Jestliže jste zapomněl(a) užít přípravek Jakavi
Pokud jste zapomněl(a) užít přípravek Jakavi, užijte další dávku ve stanovenou dobou. Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku.
Jestliže jste přestal(a) užívat přípravek Jakavi
Pokud přerušíte léčbu přípravkem Jakavi, mohou se příznaky spojené s myelofibrózou znovu objevit. Proto neukončujte užívání přípravku Jakavi, aniž byste se o tom poradil(a) se svým lékařem.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
Možné nežádoucí účinky
4.
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Většina z nežádoucích účinků přípravku Jakavi jsou mírné až středně silné a obecně vymizí po několika dnech až několika týdnech léčby.
Pokud se u Vás vyskytne kterýkoli z následujících nežádoucích účinků, sdělte to svému lékaři.
Některé jsou velmi časté (mohou postihnout více než 1 člověka z 10), některé jsou časté (mohou postihnout až 1 člověka z 10):
- jakékoli příznaky krvácení do mozku, jako jsou náhlé změny vědomí, přetrvávající bolest hlavy, necitlivost, pocit brnění, slabost nebo ochrnutí (časté)
- jakékoli příznaky krvácení do žaludku nebo střeva, jako je vylučování černé nebo krvavé stolice, nebo zvracení krve (časté)
- neočekávaná tvorba podlitin a/nebo krvácení, neobvyklá únava, námahová nebo klidová dušnost, neobvykle bledá pokožka nebo časté infekce (možné příznaky poruchy funkce krve) (velmi časté)
- bolestivá kožní vyrážka s puchýři (možné příznaky pásového oparu (herpes zoster)) (časté)
- horečka, zimnice nebo jiné příznaky infekcí (velmi časté)
- nízký počet červených krvinek (anemie), nízký počet bílých krvinek (neutropenie) nebo nízký počet krevních destiček (trombocytopenie) (velmi časté)
Jiné nežádoucí účinky spojené s přípravkem Jakavi Velmi časté:
- vysoká hladina cholesterolu nebo tuku v krvi (hypertriglyceridémie)
- neobvyklé výsledky testů jaterních funkcí
- závratě
- bolest hlavy
- infekce močových cest
- přibývání na váze
Časté:
- časté nadýmání (flatulence)
- zácpa
- vysoký krevní tlak (hypertenze), který může být příčinou závrati a bolesti hlavy
Méně časté:
- tuberkulóza
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce nebo blistru za „Použitelné do:/EXP”.
Neuchovávejte při teplotě nad 30°C.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co přípravek Jakavi obsahuje
- Léčivou látkou přípravku Jakavi je ruxolitinibum.
- Jedna 5 mg Jakavi tableta obsahuje ruxolitinibum 5 mg.
- Jedna 10 mg Jakavi tableta obsahuje ruxolitinibum 10 mg.
- Jedna 15 mg Jakavi tableta obsahuje ruxolitinibu 15 mg.
- Jedna 20 mg Jakavi tableta obsahuje ruxolitinibum 20 mg.
- Dalšími složkami jsou: mikrokrystalická celulóza, magnesium-stearát, koloidní bezvodý oxid křemičitý, sodná sůl karboxymetylškrobu, povidon, hyprolóza, monohydrát laktózy.
Jak přípravek Jakavi vypadá a co obsahuje toto balení
Jakavi 5 mg tablety jsou bílé až téměř bílé kulaté tablety s vyraženým „NVR“ na jedné straně a „L5“ vyraženým na druhé straně.
Jakavi 10 mg tablety jsou bílé až téměř bílé kulaté tablety s vyraženým „NVR“ na jedné straně a „L10“ vyraženým na druhé straně.
Jakavi 15 mg tablety jsou bílé až téměř bílé oválné tablety s vyraženým „NVR“ na jedné straně a „L15“ vyraženým na druhé straně.
Jakavi 20 mg tablety jsou bílé až téměř bílé podlouhlé tablety s vyraženým „NVR“ na jedné straně a „L20“ vyraženým na druhé straně.
Jakavi tablety jsou dodávané v baleních s blistry obsahujících 14 nebo 56 tablet nebo ve vícečetných baleních obsahujících 168 (3 balení po 56) tablet.
Na trhu nemusí být všechny velkosti balení
Držitel rozhodnutí o registraci
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
Výrobce
Novartis Pharma GmbH Roonstrasse 25 90429 Norimberk Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
|
Belgie/Belgique/Belgien Novartis Pharma N.V. Tél/Tel: +32 2 246 16 11 |
Lietuva Novartis Pharma Services Inc. Tel: +370 5 269 16 50 |
|
Btarapna Novartis Pharma Services Inc. Tea.: +359 2 489 98 28 |
Luxembourg/Luxemburg Novartis Pharma N.V. Tél/Tel: +32 2 246 16 11 |
|
Česká republika Novartis s.r.o. Tel: +420 225 775 111 |
Magyarország Novartis Hungária Kft. Pharma Tel.: +36 1 457 65 00 |
|
Danmark Novartis Healthcare A/S Tlf: +45 39 16 84 00 |
Malta Novartis Pharma Services Inc. Tel: +356 2122 2872 |
|
Deutschland Novartis Pharma GmbH Tel: +49 911 273 0 |
Nederland Novartis Pharma B.V. Tel: +31 26 37 82 111 |
|
Eesti Novartis Pharma Services Inc. Tel: +372 66 30 810 |
Norge Novartis Norge AS Tlf: +47 23 05 20 00 |
|
EXXáSa Novartis (Hellas) A.E.B.E. T^: +30 210 281 17 12 |
Osterreich Novartis Pharma GmbH Tel: +43 1 86 6570 |
|
Espaňa Novartis Farmacéutica, S.A. Tel: +34 93 306 42 00 |
Polska Novartis Poland Sp. z o.o. Tel.: +48 22 375 4888 |
|
France Novartis Pharma S.A.S. Tél: +33 1 55 47 66 00 |
Portugal Novartis Farma - Produtos Farmaceuticos, S.A Tel: +351 21 000 8600 |
|
Hrvatska Novartis Hrvatska d.o.o. Tel. +385 1 6274 220 |
Románia Novartis Pharma Services Romania SRL Tel: +40 21 31299 01 |
|
Ireland Novartis Ireland Limited Tel: +353 1 260 12 55 |
Slovenija Novartis Pharma Services Inc. Tel: +386 1 300 75 50 |
Island
Vistor hf.
Sími: +354 535 7000 Italia
Novartis Farma S.p.A.
Tel: +39 02 96 54 1
Kórcpog
Novartis Pharma Services Inc. Tni: +357 22 690 690
Latvija
Novartis Pharma Services Inc. Tel: +371 67 887 070
Slovenská republika
Novartis Slovakia s.r.o.
Tel: +421 2 5542 5439
Suomi/Finland
Novartis Finland Oy Puh/Tel: +358 (0)10 6133 200
Sverige
Novartis Sverige AB Tel: +46 8 732 32 00
United Kingdom
Novartis Pharmaceuticals UK Ltd. Tel: +44 1276 698370
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu
123