Ivemend 150 Mg
SOUHRN ÚDAJŮ O PŘÍPRAVKU
PŘÍLOHA I
IVEMEND 150 mg prášek pro infuzní roztok.
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička obsahuje fosaprepitanti dimegluminum odpovídající fosaprepitantum 150 mg, což odpovídá aprepitantum 130,5 mg. Po rekonstituci a naředění obsahuje 1 ml roztoku 1 mg fosaprepitantu (1 mg/ml) (viz bod 6.6).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek pro infuzní roztok.
Bílý až téměř bílý amorfní prášek.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Prevence akutní a pozdní nauzey a zvracení u dospělých, vyvolaných vysoce emetogenní protinádorovou chemoterapií založenou na cisplatině.
Prevence nauzey a zvracení u dospělých, vyvolaných středně emetogenní protinádorovou chemoterapií.
IVEMEND 150 mg se podává jako součást kombinované terapie (viz bod 4.2).
4.2 Dávkování a způsob podání
Dávkování
Doporučená dávka je 150 mg podaná1. den ve formě infuze trvající 20 až 30 minut zahájené přibližně 30 minut před chemoterapií (viz bod 6.6). Přípravek IVEMEND se podává spolu s kortikosteroidy a antagonisty 5-HT3, jak je blíže uvedeno v tabulkách níže.
K prevenci nauzey a zvracení spojených s emetogenní protinádorovou chemoterapií se doporučují následující režimy.
Vysoce emetogenní chemoterapeutický režim
|
1. den |
2. den |
3. den |
4. den | |
|
IVEMEND |
150 mg intravenózně |
žádný |
žádný |
žádný |
|
Dexamethason |
12 mg perorálně |
8 mg perorálně |
8 mg perorálně dvakrát denně |
8 mg perorálně dvakrát denně |
|
antagonisté 5-HT3 |
Standardní dávka antagonistů 5-HT3. Ohledně příslušného dávkování zvoleného antagonisty 5-HT3 viz informace o daném přípravku |
žádný |
žádný |
žádný |
Dexamethason se podává 1. den 30 minut před chemoterapií a 2. až 4. den ráno. 3. a 4. den se dexamethason podává rovněž večer. Dávka dexamethasonu přispívá k interakcím účinných látek.
Středně emetogenní chemoterapeutický režim
|
1. den | |
|
IVEMEND |
150 mg intravenózně |
|
Dexamethason |
12 mg perorálně |
|
antagonisté 5-HT3 |
Standardní dávka antagonistů 5-HT3. Ohledně příslušného dávkování zvoleného antagonisty 5-HT3 viz informace o daném přípravku |
Dexamethason se podává 1.den 30 minut před zahájením chemoterapie . Dávka dexamethasonu přispívá k interakcím léčivých látek.
Údaje o účinnosti při kombinaci s jinými kortikosteroidy aantagonisty 5-HT3 jsou omezené. Další informace o současném podávání přípravku s kortikosteroidy jsou uvedeny v bodu 4.5.
U současně podávaných léčivých přípravků obsahujících antagonistu 5-HT3 nahlédněte do jejich souhrnu informací o přípravku.
Zvláštní _ populace Starší pacienti (>65 let)
U starších osob není nutno dávku nijak upravovat (viz bod 5.2).
Pohlaví
S ohledem na pohlaví není úprava dávkování nutná (viz bod 5.2).
Porucha funkce ledvin
U pacientů s poruchou renálních funkcí ani u pacientů v terminálním stádiu renálního onemocnění podstupujících hemodialýzu není nutno dávku nijak upravovat (viz bod 5.2).
Porucha funkce jater
U pacientů s mírnou poruchou jaterních funkcí není nutno dávku nijak upravovat. Pokud se týče pacientů se středně těžkou poruchou jaterních funkcí, je k dispozici pouze omezené množství dat a u pacientů s těžkou poruchou jaterních funkcí nejsou dostupné žádné údaje. Přípravek IVEMEND se u těchto pacientů musí používat opatrně (viz body 4.4 a 5.2).
Pediatrická populace
Bezpečnost a účinnost přípravku IVEMEND u dětí a dospívajících do 18 let věku nebyla dosud stanovena. K dispozici nejsou žádné údaje.
Způsob podání
IVEMEND 150 mg se podává intravenózně a nesmí se podávat intramuskulární nebo subkutánní cestou. Intravenózní podání je nejlepší provést trvalou intravenózní infuzí po dobu 20 až 30 minut (viz bod 6.6). Nepodávejte IVEMEND jako bolusovou injekci nebo nenaředěný roztok.
Pokyny k rekonstituci a ředění léčivého přípravku před podáním, viz bod 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku, aprepitant nebo polysorbát 80 nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1 tohoto přípravku.
Současné podávání spolu s pimozidem, terfenadinem, astemizolem nebo cisapridem (viz bod 4.5).
4.4 Zvláštní upozornění a opatření pro použití
Pacienti se středně těžkou a těžkou poruchou funkce jater
Pro pacienty se středně těžkou poruchou jaterních funkcí existuje pouze omezené množství dat a pro pacienty s těžkou poruchou jaterních funkcí nejsou k dispozici žádné údaje. U těchto pacientů je nutno IVEMEND používat s opatrností (viz bod 5.2).
Interakce na CYP3A4
IVEMEND je nutno podávat s opatrností pacientům současně užívajícím perorálně podávané léčivé látky, které se primárně metabolizují cestou CYP3A4 a které mají úzké terapeutické rozmezí, jako jsou cyklosporin, takrolimus, sirolimus, everolimus, alfentanil, deriváty námelových alkaloidů, fentanyl a chinidin (viz bod 4.5). K podávání současně s irinotekanem je navíc nutno přistupovat s obzvláštní opatrností, protože uvedená kombinace by mohla mít za následek zvýšenou toxicitu.
Současné podávání s warfarinem (substrát CYP2C9)
U pacientů dlouhodobě léčených warfarinem je nutno během 14 dnů po užití fosaprepitantu pozorně sledovat hodnotu mezinárodního normalizovaného poměru (INR) (viz bod 4.5).
Současné podávání s hormonálními kontraceptivy
Účinnost hormonální antikoncepce může být během podávání aprepitantu a 28 dní poté snížena. Během léčby fosaprepitantem a 2 měsíce po poslední dávce fosaprepitantu je nutno používat alternativní nehormonální pomocné metody antikoncepce (viz bod 4.5).
Hypersenzitivní reakce
Během podání infuze fosaprepitantu byly ojediněle hlášeny časté hypersenzitivní reakce zahrnující návaly horka, erytém a dušnost. Tyto hypersenzitivní reakce většinou po přerušení podání infuze fosaprepitantu a po příslušné léčbě ustoupily. Opětovné podání infuze přípravku pacientům, u kterých se objevila hypersenzitivní reakce, se nedoporučuje.
Podání a reakce v místě aplikace
IVEMEND se nesmí podávat jako bolusová injekce, ale vždy se musí naředit a podat jako pomalá intravenózní infuze (viz bod 4.2). IVEMEND se nesmí podávat intramuskulárně nebo subkutánně (viz bod 5.3). Při vyšších dávkách byla pozorována mírná trombóza v místě injekce. Pokud se objeví známky nebo příznaky lokálního podráždění, je nutno injekci nebo infuzi ukončit a znovu začít na jiné žíle.
Při intravenózním podání se fosaprepitant rychle přeměňuje na aprepitant.
Interakce s jinými léčivými přípravky po intravenózmm podání fosaprepitantu jsou pravděpodobné u léčivých látek, které interagují s perorálním aprepitantem. Následující informace byly odvozeny ze studií provedených s perorálním aprepitantem a ze studií provedených s intravenózním fosaprepitantem podávaným společně s dexamethasonem, midazolamem nebo diltiazemem.
Fosaprepitant v dávce 150 mg podané v jediné dávce je slabým inhibitorem CYP3A4. Nezdá se, že by fosaprepitant interagoval s P-glykoproteinovým transportérem, jak naznačuje nepřítomnost interakce perorálně podaného aprepitantu s digoxinem. Předpokládá se, že fosaprepitant by mohl způsobovat menší nebo stejnou indukci CYP2C9, CYP3A4 a glukuronidace, než jakou způsobuje perorální podání aprepitantu. Ohledně účinků na CYP2C8 a CYP2C19 údaje chybějí.
Účinek aprepitantu na farmakokinetiku dalších léčivých látek Inhibice CYP3A4
Jako slabý inhibitor CYP3A4 může fosaprepitant v jediné 150mg dávce zvýšit plazmatické koncentrace současně podávaných perorálních léčivých látek, které se metabolizují cestou CYP3A4. Celková expozice perorálně podávaným substrátům CYP3A4 se může 1. a 2. den současného podávání s jedinou 150mg dávkou fosaprepitantu zvýšit dvojnásobně. Fosaprepitant se nesmí podávat současně s pimozidem, terfenadinem, astemizolem nebo cisapridem. Inhibice CYP3A4 fosaprepitantem by mohla mít za následek zvýšené plazmatické koncentrace uvedených léčivých látek, což může vyvolat závažné nebo život ohrožující reakce (viz bod 4.3). Při současném podávání fosaprepitantu a perorálně podávaných látek, které jsou metabolizovány převážně prostřednictvím CYP3A4 a které mají úzké terapeutické rozmezí, jako je cyklosporin, takrolimus, sirolimus, everolimus, alfentanil, diergotamin, ergotamin, fentanyl a chinidin, se doporučuje opatrnost (viz bod 4.4).
Kortikosteroidy
Dexamethason: dávka perorálního dexamethasonu 1. a 2. den musí být snížena o přibližně 50 %, pokud se podává s fosaprepitantem v dávce 150 mg 1. den, aby se dosáhlo expozic dexamethasonu podobných expozicím dosahovaným při podávání bez fosaprepitantu v dávce 150 mg. Fosaprepitant v dávce 150 mg podaný 1. den jako jediná intravenózní dávka zvyšoval AUC0.24hod dexamethasonu, což je substrát CYP3A4, o 100 % 1. den, o 86 % 2. den a o 18 % 3. den, pokud se dexamethason podával jako jediná 8mg perorální dávka 1., 2. a 3. den.
Chemoterapeutické léčivé přípravky
Studie interakcí s fosaprepitantem v dávce 150 mg a chemoterapeutickými léčivými přípravky nebyly provedeny; nicméně na základě studií provedených s perorálním aprepitantem a docetaxelem a vinorelbinem se nepředpokládá, že by přípravek IVEMEND 150 mg měl klinicky významné interakce s intravenózně podávaným docetaxelem a vinorelbinem. Nelze vyloučit interakce s perorálně podávanými chemoterapeutickými léčivými přípravky, které se metabolizují převážně nebo částečně cestou CYP3A4 (např. etoposid, vinorelbin). U pacientů dostávajících léčivé přípravky, které se metabolizují především nebo i částečně CYP3A4, se doporučuje opatrnost, přičemž může být vhodné tyto pacienty dodatečně sledovat (viz bod 4.4). Po uvedení na trh byly hlášeny při současném podávání aprepitantu a ifosfamidu případy neurotoxicity, potenciální nežádoucí účinek ifosfamidu.
Imunosupresiva
Po jediné 150mg dávce fosaprepitantu se předpokládá přechodné dvoudenní střední zvýšení následované mírným poklesem expozice imunosupresivům metabolizovaným prostřednictvím CYP3A4 (např. cyklosporin, takrolimus, everolimus a sirolimus). S ohledem na krátké trvání zvýšené expozice se snížení dávky imunosupresiva založené na terapeutickém monitorování dávky v den podání přípravku IVEMEND a v den následující nedoporučuje.
Midazolam
Fosaprepitant v dávce 150 mg podaný 1. den jako jediná intravenózní dávka zvyšoval AUC0-<X) midazolamu o 77 % 1. den a 4. den neměl žádný vliv, pokud byl midazolam podáván současně jako jediná 2mg perorální dávka 1. až 4. den. Fosaprepitant v dávce 150 mg je slabým inhibitorem CYP3A4 pokud se podá v jediné dávce 1. den, přičemž 4. den se nepozorují žádné důkazy inhibice ani indukce CYP3A4.
Možné účinky zvýšených plazmatických koncentrací midazolamu nebo jiných benzodiazepinů metabolizovaných cestou CYP3A4 (alprazolam, triazolam) je nutno vzít v úvahu při současném podávání těchto léčivých přípravků spolu s přípravkem IVEMEND.
Diltiazem
Studie interakcí s fosaprepitantem v dávce 150 mg a diltiazemem nebyly provedeny, nicméně při používání přípravku IVEMEND 150 mg s diltiazemem je nutno vzít v potaz následující studii provedenou s fosaprepitantem v dávce 100 mg. U pacientů s mírnou až středně těžkou hypertenzí vedla infuze 100 mg fosaprepitantu podaná během 15 minut spolu se 120 mg diltiazemu 3krát denně k 1,4násobnému zvýšení AUC diltiazemu a k malému, nicméně klinicky významnému snížení krevního tlaku, ale nevedla ke klinicky významné změně tepové frekvence nebo intervalu PR.
Indukce
Fosaprepitant v jediné 150mg dávce ve studii interakce s midazolamem neindukoval CYP3A4 1. až 4. den. Předpokládá se, že by přípravek IVEMEND mohl způsobovat nižší nebo stejnou indukci CYP2C9, CYP3A4 a glukuronidace, než jakou způsobuje podání 3denního režimu perorálního aprepitantu, u kterého byla pozorována přechodná indukce dosahující maxima 6 až 8 dní po první dávce aprepitantu. Třídenní režim perorálního aprepitantu vedl k asi 30 až 35% snížení AUC substrátů CYP2C9 a až k 64% poklesu minimálních koncentrací ethinylestradiolu. Pokud jde o vliv na CYP2C8 a CYP2C19, nejsou údaje k dispozici. Opatrnosti je třeba, pokud se spolu s přípravkem IVEMEND podává warfarin, acenokumarol, tolbutamid, fenytoin nebo jiné léčivé látky, o nichž je známo, že jsou metabolizovány CYP2C9.
Warfarin
U pacientů chronicky léčených warfarinem je během léčby přípravkem IVEMEND k prevenci chemoterapií navozené nauzey a zvracení a 14 dnů poté nutno pečlivě sledovat protrombinový čas (INR) (viz bod 4.4).
Hormonální antikoncepce
Účinnost hormonální antikoncepce může být během podávání fosaprepitantu a po dobu 28 dní po ukončení jeho podávání snížena. Během léčby fosaprepitantem a ještě 2 měsíce po poslední dávce fosaprepitantu je třeba používat alternativních nehormonálních pomocných metod antikoncepce.
Antagonisté 5-HT3
Studie lékových interakcí s fosaprepitantem v dávce 150 mg a antagonisty 5-HT3 nebyly provedeny, nicméně v klinických studiích lékových interakcí nevykazoval aprepitant podávaný v perorálním režimu žádné klinicky významné účinky na farmakokinetiku ondansetronu, granisetronu nebo hydrodolasetronu (aktivní metabolit dolasetronu). Proto ohledně interakcí při používání přípravku IVEMEND 150 mg a antagonistů 5-HT3 neexistují žádné důkazy.
Účinky jiných léčivých přípravků na farmakokinetiku aprepitantu vyplývající z podání 150 mg fosaprepitantu
K současnému podávání fosaprepitantu s léčivými přípravky, které inhibují aktivitu CYP3A4 (např. ketokonazolem, itrakonazolem, vorikonazolem, posakonazolem, klarithromycinem, telithromycinem, nefazodonem a inhibitory proteázy), je třeba přistupovat opatrně, protože se předpokládá, že uvedené kombinace povedou k několikanásobnému zvýšení plazmatické koncentrace aprepitantu (viz bod 4.4). Ketokonazol prodlužoval terminální poločas perorálně podaného aprepitantu asi 3násobně.
Je nutno se vyvarovat současného podávání fosaprepitantu s léčivými přípravky, které silně indukují aktivitu CYP3A4 (např. rifampicin, fenytoin, karbamazepin, fenobarbital), protože tato kombinace vede ke snížení plazmatických koncentrací aprepitantu, což může vést ke snížení účinnosti. Současné podávání fosaprepitantu s třezalkou tečkovanou (Hypericum perforatum) se nedoporučuje. Rifampicin zkracoval průměrný terminální poločas perorálně podaného aprepitantu o 68 %.
Diltiazem
Studie interakcí s fosaprepitantem v dávce 150 mg a diltiazemem nebyly provedeny, nicméně při používání přípravku IVEMEND 150 mg s diltiazemem je nutno vzít v potaz následující studii provedenou s fosaprepitantem v dávce 100 mg. Infuze 100 mg fosaprepitantu podaná během 15 minut spolu se 120 mg diltiazemu 3krát denně vedla k 1,5násobnému zvýšení AUC aprepitantu. Tento účinek nebyl vyhodnocen jako klinicky významný.
4.6 Fertilita, těhotenství a kojení
Antikoncepce u mužů a žen
Účinnost hormonální antikoncepce může být během podávání fosaprepitantu a 28 dní poté snížena. Během léčby fosaprepitantem a 2 měsíce po poslední dávce fosaprepitantu je nutno používat alternativní nehormonální pomocné metody antikoncepce (viz body 4.4 a 4.5).
Nejsou k dispozici žádné klinické údaje o podávání fosaprepitantu a aprepitantu během těhotenství. Potenciální reprodukční toxicita fosaprepitantu a aprepitantu nebyla zcela stanovena, protože míra expozice vyšší než hodnoty terapeutické expozice u člověka nemohla být ve studiích se zvířaty dosažena. Tyto studie nenaznačily žádné přímé ani nepřímé škodlivé účinky na průběh těhotenství, embryonální/fetální vývoj, porod nebo postnatální vývoj (viz bod 5.3). Možné účinky na reprodukci změnami v regulaci neurokininů nejsou známy. IVEMEND se nesmí podávat během těhotenství, pokud to není nezbytně nutné.
Kojení
Aprepitant se po intravenózním podání fosaprepitantu stejně jako po perorálním podání aprepitantu vylučuje do mléka kojících potkanů. Není známo, zda se aprepitant vylučuje do lidského mléka. Proto se během léčby přípravkem IVEMEND kojení nedoporučuje.
Fertilita
Potenciál k účinkům fosaprepitantu a aprepitantu na fertilitu nebyl plně charakterizován, protože míra expozice vyšší než hodnoty terapeutické expozice u člověka nemohla být ve studiích se zvířaty dosažena. Studie fertility neukazují na přímé ani nepřímé škodlivé účinky pokud jde o páření, fertilitu, embryonální/fetální vývoj či počty a motilitu spermií (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek IVEMEND může mít mírný vliv na schopnost řídit nebo obsluhovat stroje. Po podání přípravku IVEMEND se mohou objevit závratě a únava (viz bod 4.8).
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
V klinických studiích byly různé lékové formy fosaprepitantu podány celkem 2 687 jedincům včetně 371 zdravých subjektů a 2 084 pacientů s chemoterapií navozenou nauzeou a zvracením (CINV). Jelikož se fosaprepitant přeměňuje na aprepitant, očekává se, že nežádoucí účinky spojované s aprepitantem se budou vyskytovat i u fosaprepitantu. Bezpečnostní profil aprepitantu byl hodnocen přibližně u 6 500 jedinců.
Perorální aprepitant
Nejčastějšími nežádoucími účinky u pacientů léčených vysoce emetogenní chemoterapií, které byly hlášeny s vyšší incidencí u pacientů léčených v režimu s aprepitantem než při standardní terapii, byly: škytavka (4,6 % oproti 2,9 %), zvýšení alaninaminotransferázy (ALT) (2,8 % oproti 1,1 %), dyspepsie (2,6 % oproti 2,0 %), zácpa (2,4 % oproti 2,0 %), bolesti hlavy (2,0 % 1,8 %) a pokles chuti k jídlu (2,0 % oproti 0,5 %). Nejčastějším nežádoucím účinkem hlášeným s vyšší incidencí u pacientů léčených režimem s aprepitantem než při standardní terapii u pacientů léčených středně emetogenní chemoterapií byla únava (1,4 % oproti 0,9 %).
Souhrn nežádoucích účinků v tabulce - aprepitant
Následující nežádoucí účinky byly pozorovány v souhrnné analýze studií s vysoce a středně emetogenní chemoterapií s vyšší incidencí u perorálního aprepitantu než při standardní terapii nebo po uvedení na trh:
Četnosti jsou definovány jako: velmi časté (>1/10); časté (>1/100 až <1/10); méně časté (>1/1 000 až <1/100); vzácné (>1/10 000 až <1/1 000) a velmi vzácné (<1/10 000), není známo (z dostupných údajů nelze určit).
|
Třída orgánových systémů |
Nežádoucí účinek |
Četnost |
|
Infekce a infestace |
kandidóza, stafylokokové infekce |
vzácné |
|
Poruchy krve a lymfatického systému |
febrilní neutropenie, anémie |
méně časté |
|
Poruchy imunitního systému |
hypersenzitivní reakce včetně anafylaktických reakcí |
není známo |
|
Poruchy metabolismu a výživy |
pokles chuti k jídlu |
časté |
|
polydipsie |
vzácné | |
|
Psychiatrické poruchy |
méně časté | |
|
dezorientace, euforická nálada |
vzácné | |
|
Poruchy nervového systému |
časté | |
|
závrať, somnolence |
méně časté | |
|
kognitivní porucha, letargie, dysgeuzie |
vzácné | |
|
Poruchy oka |
zánět spojivek |
vzácné |
|
Poruchy ucha a labyrintu |
tinnitus |
vzácné |
|
Srdeční poruchy |
palpitace |
méně časté |
|
bradykardie, kardiovaskulární porucha |
vzácné | |
|
Cévní poruchy |
méně časté | |
|
Respirační, hrudní a mediastinální poruchy |
škytavka |
časté |
|
bolesti v orofaryngu, kýchání, kašel, nadměrné hromadění hlenu v nasofaryngu, podráždění hrdla |
vzácné | |
|
Gastrointestinální poruchy |
zácpa, dyspepsie |
časté |
|
říhání, nauzea*, zvracení*, gastroezofageální refluxní choroba, bolesti břicha, sucho v ústech, flatulence |
méně časté | |
|
perforace duodenálního vředu, stomatitida, abdominální distenze, tuhá stolice, neutropenická kolitida |
vzácné | |
|
Poruchy kůže a podkožní tkáně |
vyrážka, akné |
méně časté |
|
Třída orgánových systémů |
Nežádoucí účinek |
Četnost |
|
fotosenzitivní reakce, hyperhidróza, seborea, kožní léze, svědivá vyrážka, Stevens-Johnsonův syndrom/toxická epidermální nekrolýza |
vzácné | |
|
svědění, kopřivka |
není známo | |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
svalová slabost, svalové spasmy |
vzácné |
|
Poruchy ledvin a močových cest |
méně časté | |
|
polakisurie |
vzácné | |
|
Celkové poruchy a reakce v místě aplikace |
časté | |
|
astenie, malátnost |
méně časté | |
|
edém, nepříjemný pocit na hrudi, poruchy chůze |
vzácné | |
|
Vyšetření |
zvýšení ALT |
časté |
|
zvýšení AST, zvýšení alkalické fosfatázy |
méně časté | |
|
pozitivní reakce na červené krvinky v moči, pokles sodíku v krvi, pokles tělesné hmotnosti, pokles počtu neutrofilů, přítomnost glukózy v moči, zvýšení výdeje moči |
vzácné |
* Nauzea a zvracení byly parametry účinnosti v prvních 5 dnech léčby po chemoterapii a potom byly hlášeny pouze jako nežádoucí účinky.
Popis vybraných nežádoucích účinků
Profily nežádoucích účinků v prodloužených studiích s vysoce nebo středně emetogenní chemoterapií s opakovanými až 6 dalšími cykly chemoterapie byly celkově podobné jako profily pozorované v 1. cyklu.
V další klinické studii kontrolované aktivním komparátorem, prováděné u 1169 pacientů, kterým byl podáván aprepitant a vysoce emetogenní chemoterapie, byl profil nežádoucích účinků celkově podobný jako profily pozorované v jiných studií vysoce emetogenní chemoterapie s aprepitantem.
U pacientů léčených aprepitantem kvůli pooperační nauzee a zvracení (PONV) byly pozorovány další nežádoucí účinky, které měly vyšší incidenci než u ondansetronu: bolesti v horní části břicha, abnormální střevní zvuky, zácpa*, dysartrie, dušnost, hypestézie, nespavost, mióza, nauzea, poruchy čití, nepříjemný pocit v oblasti břicha, subileus*, snížená ostrost vidění, sípání.
*Hlášeno u pacientů užívajících vyšší dávku aprepitantu.
Fosaprepitant
V klinické studii kontrolované aktivním komparátorem, prováděné u pacientů léčených vysoce emetogenní chemoterapií, byla bezpečnost přípravku hodnocena u 1 143 pacientů léčených v 1denním režimu s fosaprepitantem 150 mg v porovnání s 1 169 pacienty léčenými ve 3denním režimu s aprepitantem. Bezpečnost byla dále hodnocena v placebem kontrolovaném klinickém hodnocení u 504 pacientů léčených středně emetogenní chemoterapií (MEC), kteří dostali jednu dávku přípravku IVEMEND, v porovnání se 497 pacienty, kteří dostávali kontrolní režim.
Bezpečnostní profil byl celkově podobný profilu aprepitantu, který je uvedený v tabulce výše.
Souhrn nežádoucích účinků v tabulce - fosaprepitant
Níže jsou uvedeny nežádoucí účinky hlášené v klinických studiích a v poregistračních studiích u pacientů, kterým byl podáván fosaprepitant a které nebyly hlášeny při podávání aprepitantu, jak je popsáno výše.
Četnosti jsou definovány jako: velmi časté (>1/10); časté (>1/100 až <1/10); méně časté (>1/1 000 až <1/1 00); vzácné (>1/10 000 až <1/1000) a velmi vzácné (<1/10 000), není známo (z dostupných údajů nelze určit).
|
Třída orgánových systémů |
Nežádoucí účinek |
Četnost |
|
Cévní poruchy |
návaly horka, tromboflebitida (převážně tromboflebitida v místě aplikace) |
méně časté |
|
Poruchy kůže a podkožní tkáně |
erytém |
méně časté |
|
Celkové poruchy a reakce v místě aplikace |
erytém v místě aplikace, bolest v místě aplikace, svědění v místě aplikace |
méně časté |
|
indurace v místě aplikace |
vzácné | |
|
časné hypersenzitivní reakce včetně návalu horka, erytému, dušnosti |
není známo | |
|
Vyšetření |
zvýšení krevního tlaku |
méně časté |
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
V případě předávkování je nutno podávání fosaprepitantu ukončit a zajistit obecnou podpůrnou terapii a sledování pacienta. Vzhledem k antiemetogennímu účinku aprepitantu může snaha o vyvolání zvracení podáním léčivého přípravku selhat.
Aprepitant nelze odstranit hemodialýzou.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antiemetika a léčiva k terapii nauzey, ATC kód: A04AD12.
Fosaprepitant je proléčivo aprepitantu a po intravenózním podání je rychle konvertován na aprepitant (viz bod 5.2). Podíl fosaprepitantu na celkovém antiemetickém účinku nebyl zatím zcela charakterizován, ale přechodný přínos během iniciální fáze nemůže být vyloučen. Aprepitant je selektivní antagonista s vysokou afinitou k receptorům neurokinin 1 (NKi) humánní substance P. Farmakologický účinek fosaprepitantu se přičítá aprepitantu.
Vysoce emetogenní chemoterapie (HEC)
V randomizované, dvojitě zaslepené, aktivním komparátorem kontrolované studii s paralelními skupinami byl přípravek IVEMEND 150 mg (N = 1 147) porovnáván s 3denním režimem aprepitantu (N = 1 175) u pacientů léčených vysoce emetogenním režimem chemoterapie, který zahrnoval cisplatinu (> 70 mg/m2). Režim podávání fosaprepitantu sestával z fosaprepitantu v dávce 150 mg
1. den v kombinaci s ondansetronem 32 mg i.v. 1. den a dexamethasonem v dávce 12 mg 1. den, 8 mg
2. den a 8 mg dvakrát denně 3. a 4. den. Režim podávání aprepitantu sestával z aprepitantu v dávce 125 mg 1. den a 80 mg/den 2. a 3. den v kombinaci s ondansetronem v dávce 32 mg i.v. 1. den a dexamethasonem v dávce 12 mg 1. den a 8 mg denně 2. až 4. den. K zaslepení se používalo placebo fosaprepitantu, aprepitantu a dexamethasonu (večer 3. a 4. den) (viz bod 4.2). I když se v klinických hodnoceních používala 32mg intravenózní dávka ondansetronu, nejedná se již o doporučenou dávku. Ohledně příslušného dávkování zvoleného antagonisty 5-HT3 viz informace o daném přípravku.
Hodnocení účinnosti bylo založeno na vyhodnocení následujících složených kritérií hodnocení: úplná odpověď za celou dobu a v pozdní fázi a žádné zvracení za celou dobu. Prokázalo se, že přípravek IVEMEND 150 mg byl noninferiorní 3dennímu režimu aprepitantu. Souhrn primárních a sekundárních cílových parametrů je uveden v tabulce 1.
Tabulka 1
Procento pacientů léčených vysoce emetogenní terapií odpovídajících na léčbu podle léčebné skupiny
a fáze - cyklus 1
|
CÍLOVÉ PARAMETRY* |
Režim fosaprepitantu (N= 1 106) ** % |
Režim aprepitantu (N= 1 134)** % |
Rozdílf (95% interval spolehlivosti) |
|
Úplná odpověď{ | |||
|
Za celou dobu§ |
71,9 |
72,3 |
-0,4 (-4,1; 3,3) |
|
Pozdní fáze §§ |
74,3 |
74,2 |
0,1 (-3,5; 3,7) |
|
Žádné zvracení | |||
|
Za celou dobu§ |
72,9 |
74,6 |
-1,7 (-5,3; 2,0) |
*Primární cílový parametr je uveden tučně.
**N: počet pacientů zařazených do primární analýzy úplné odpovědi.
fRozdíl a interval spolehlivosti byly vypočítány za pomoci metody navržené Miettinenem a Nurminenem a upraveny podle pohlaví.
{Úplná odpověď = žádné zvracení a žádné použití záchranné terapie.
§ Celá doba = 0 až 120 hodin po zahájení chemoterapie cisplatinou.
§§Pozdní fáze = 25 až 120 hodin po zahájení chemoterapie cisplatinou.
Středně emetogenní chemoterapie (MEC)
V randomizované, dvojitě zaslepené, placebem kontrolované studii s paralelní skupinou byl u pacientů léčených režimem středně emetogenní chemoterapie přípravek IVEMEND 150 mg (N=502) v kombinaci s ondansetronem a dexamethasonem porovnáván s ondansetronem a dexamethasonem samotnými (kontrolní režim) (N=498). Režim s fosaprepitantem sestával z podání fosaprepitantu v dávce 150 mg 1. den v kombinaci se dvěma 8mg dávkami perorálního ondansetronu a perorálním dexamethasonem v dávce 12 mg. Ve 2. a 3. dni dostávali pacienti ze skupiny léčené fosaprepitantem místo ondansetronu každých 12 hodin placebo. Kontrolní režim sestával z placeba fosaprepitantu 150 mg i.v. 1. den v kombinaci se dvěma 8mg dávkami perorálního ondansetronu a perorálním dexamethasonem v dávce 20 mg. Ve 2. a 3. dni dostávali pacienti z kontrolní skupiny každých 12 hodin 8 mg perorálního ondansetronu. K uchování zaslepení se využívalo placebo fosaprepitantu a placebo dexamethasonu (1. den).
Účinnost fosaprepitantu byla hodnocena s ohledem na primární a sekundární cílové parametry uvedené v tabulce 2, přičemž se prokázalo, že s ohledem na úplnou odpověď v pozdní fázi a za celou dobu léčby je vyšší než u kontrolního režimu.
Tabulka 2
Procento pacientů léčených středně emetogenní terapií odpovídajících na léčbu podle
léčebné skupiny a fáze
|
CÍLOVÉ PARAMETRY* |
Režim fosaprepitantu (N =502)** % |
Kontrolní režim (N =498) ** % |
Hodnota p |
|
Úplná odpověďf | |||
|
Pozdní fáze} |
78,9 |
68,5 |
<0,001 |
|
Úplná odpověďf | |||
|
Za celou dobu§ |
77,1 |
66,9 |
<0,001 |
|
Akutní fáze§§ |
93,2 |
91 |
0,184 |
*Primární cílový parametr je tučně.
**N: počet pacientů zahrnutý do populace všech zařazených pacientů (intention to treat population).
f Úplná odpověď = žádné zvracení a žádné použití záchranné terapie.
} Pozdní fáze = 25 až 120 hodin po zahájení chemoterapie.
§Za celou dobu = 0 až 120 hodin po zahájení chemoterapie.
§§Akutní fáze = 0 až 24 hodin po zahájení chemoterapie.
Odhad doby do prvního zvracení je zobrazen metodou vynesení hodnot dle Kaplana-Meiera v grafu 1.
Graf 1
Procento pacientů léčených středně emetogenní chemoterapií, kteří v průběhu doby nezvraceli
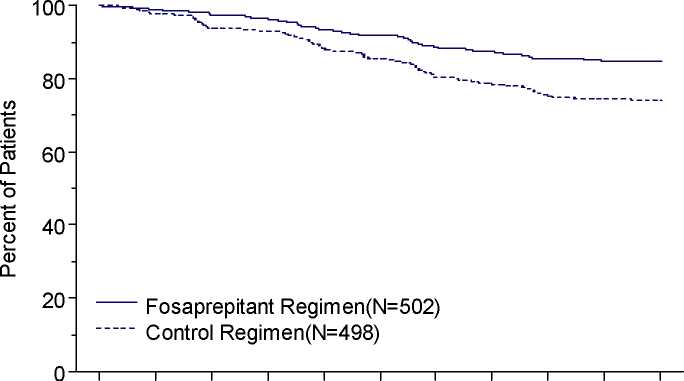
0 12 24 36 48 60 72 84 96 108 120
Time(hours) Since the First MEC Administration
Pediatrická populace
Studie hodnotící použití fosaprepitantu u pediatrických pacientů probíhají (informace o pediatrickém použití viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Fosaprepitant, proléčivo aprepitantu, se při intravenózním podání rychle přeměňuje na aprepitant. Plazmatické koncentrace fosaprepitantu jsou do 30 minut po dokončení infuze pod kvantifikovatelnými hladinami.
Aprepitant po podání fosaprepitantu
Po jedné intravenózní 150mg dávce fosaprepitantu podané ve formě 20minutové infuze zdravým dobrovolníkům byla střední hodnota AUC0-<X) aprepitantu 35,0 gg»hod/ml a střední hodnota maximální koncentrace aprepitantu byla 4,01 gg/ml.
Distribuce v organismu
Aprepitant se ve vysoké míře váže na proteiny, v průměru z 97 %. Střední geometrický zdánlivý distribuční objem za ustáleného stavu (Vdss) aprepitantu je u člověka po podání jediné 150mg intravenózní dávky fosaprepitantu přibližně 82 litrů.
Biotransformace
Fosaprepitant byl při in vitro inkubacích s lidskými jaterními preparáty rychle přeměněn na fosaprepitant. Navíc fosaprepitant podstoupil rychlou a téměř úplnou konverzi na aprepitant u preparátů S9 z jiných lidských tkání včetně ledvin, plic a ilea. Zdá se tedy, že ke konverzi fosaprepitantu na aprepitant může vedle jater docházet v rozmanitých tkáních. U člověka byl intravenózně podaný fosaprepitant rychle přeměněn na aprepitant do 30 minut po ukončení infuze.
Aprepitant prochází rozsáhlou biotransformací. Po jednorázovém intravenózním podání 100 mg [14C]-fosaprepitantu, což je proléčivo aprepitantu, zdravým mladým dospělým jedincům činil po dobu 72 hodin po aplikaci podíl aprepitantu pouze 19 % z celkové radioaktivity v plazmě, což ukazuje na značnou přítomnost metabolitů v plazmě. V lidské plazmě bylo zjištěno dvanáct metabolitů aprepitantu. Metabolismus aprepitantu probíhá ve velké míře cestou oxidace v morfolinovém kruhu a jeho postranních řetězcích a výsledné metabolity jsou pouze slabě aktivní. In vitro studie s lidskými jaterními mikrosomy ukázaly, že aprepitant se metabolizuje primárně cestou CYP3A4, případně s malým podílem CYP1A2 a CYP2C19.
Všechny metabolity zjištěné v moči, stolici a plazmě po intravenózním podání 100 mg [14C]-fosaprepitantu byly rovněž zjištěny po podání perorální dávky [14C]-aprepitantu. Po konverzi 245,3 mg dimegluminové soli fosaprepitantu (což odpovídá 150 mg fosaprepitantu) na aprepitant se uvolní 23,9 mg kyseliny fosforečné a 95,3 mg megluminu.
Eliminace z organismu
Aprepitant se nevylučuje močí v nezměněné podobě. Metabolity se vylučují močí a žlučí do stolice.
Po jednorázovém intravenózním podání dávky 100 mg [14C]-fosaprepitantu zdravým jedincům bylo 57 % radioaktivity zjištěno v moči a 45 % ve stolici.
Farmakokinetika aprepitantu je napříč rozpětím klinických dávek nelineární. Terminální biologický poločas aprepitantu po podání 150mg intravenózní dávky fosaprepitantu je přibližně 11 hodin. Střední geometrická plasmatická clearance aprepitantu po 150mg intravenózní dávce fosaprepitantu je přibližně 73 ml/min.
Farmakokinetika u zvláštních populací
Farmakokinetika fosaprepitantu nebyla u zvláštních populací hodnocena. S ohledem na věk a pohlaví se ve farmakokinetice aprepitantu neočekávají žádné klinicky významné rozdíly.
Porucha jaterních funkcí: Fosaprepitant je metabolizován v různých extrahepatálních tkáních; proto se neočekává, že by porucha jaterních funkcí měnila konverzi fosaprepitantu na aprepitant. Mírná porucha funkce jater (Child-Pughova třída A) farmakokinetiku aprepitantu v klinicky významné míře neovlivňuje. U pacientů s mírnou poruchou jaterních funkcí není třeba dávkování nijak upravovat.
Z dostupných dat nelze činit žádné závěry ohledně vlivu středně těžké poruchy jaterních funkcí
(Child-Pughova třída B) na farmakokinetiku aprepitantu. K dispozici nejsou žádné klinické ani farmakokinetické údaje od pacientů s těžkou poruchou jaterních funkcí (Child-Pughova třída C).
Porucha funkce ledvin: Pacientům s těžkou poruchou funkce ledvin (clearance kreatininu <30 ml/min) a pacientům s terminálním renálním onemocněním (end stage renal disease, ESRD) s potřebou hemodialýzy byla podána jednorázová 240mg dávka perorálního aprepitantu.
U pacientů s těžkou poruchou funkce ledvin se hodnota AUC0_<» celkového aprepitantu (nevázaného i vázaného na proteiny) ve srovnání se zdravými jedinci snížila o 21 % a hodnota Cmax se snížila o 32 %. U pacientů s ESRD podstupujících hemodialýzu se hodnota AUC0_<» celkového aprepitantu snížila o 42 % a hodnota Cmax se snížila o 32 %. Vzhledem k mírnému poklesu vazby aprepitantu na proteiny u pacientů s poruchou funkce ledvin nebyla hodnota AUC farmakologicky aktivní části nenavázaného aprepitantu u pacientů s renální nedostatečností ve srovnání se zdravými jedinci významně ovlivněna. Hemodialýza prováděná 4 nebo 48 hodin po podání dávky neměla na farmakokinetiku aprepitantu významný účinek; v dialyzátu bylo zjištěno méně než 0,2 % dávky.
U pacientů s poruchou funkce ledvin ani u pacientů s ESRD podstupujících hemodialýzu není úprava dávky nutná.
Vztah mezi koncentrací a účinkem
Zobrazovací studie využívající pozitronovou emisní tomografii (PET) s použitím vysoce specifického značení receptorů NK u zdravých mladých mužů, kterým byla podávána jednorázová intravenózní dávka 150 mg fosaprepitantu (N = 8), která se projeví obsazeností mozkových NK receptorů z > 100 % při Tmax za 24 hodin, > 97 % za 48 hodin a mezi 41 % a 75 % za 120 hodin po podání dávky. V této studii dobře koreluje obsazení mozkových NK receptorů s koncentrací aprepitantu v krevní plazmě.
5.3 Předklinické údaje vztahující se k bezpečnosti
Předklinické údaje získané při intravenózním podání fosaprepitantu a perorálním podání aprepitantu založené na konvenčních studiích toxicity po jediné dávce a po opakovaném podání, studiích genotoxicity (včetně testů in vitro) a studiích reprodukční toxicity neodhalily žádné zvláštní riziko pro člověka. U laboratorních zvířat způsobil fosaprepitant v nekomerčních lékových formách vaskulární toxicitu a hemolýzu v koncentracích pod 1 mg/ml a vyšších, v závislosti na lékové formě.
U promytých lidských krvinek se při použití nekomerčních lékových forem objevily známky hemolýzy u koncentrací fosaprepitantu 2,3 mg/ml a vyšších, avšak testy s plnou lidskou krví byly negativní. Při použití komerčních lékových forem až do koncentrace 1 mg fosaprepitantu /ml nedošlo k hemolýze ani u plné lidské krve, ani promytých lidských erytrocytů.
Kancerogenní potenciál u hlodavců byl hodnocen pouze u perorálně podávaného aprepitantu. Je však nutno podotknout, že hodnota studií toxicity provedených na hlodavcích, králících a opicích, včetně studií reprodukční toxicity, je omezená, protože systémové expozice fosaprepitantu a aprepitantu byly pouze podobné nebo dokonce nižší, než je terapeutická expozice u lidí. V provedených studiích farmakologické bezpečnosti a studiích toxicity s opakovanou dávkou u psů, byly hodnoty Cmax fosaprepitantu až 3krát a hodnoty AUC aprepitantu až 40krát vyšší než klinické hodnoty.
U králíků způsoboval přípravek IVEMEND po paravenózním, subkutánním a intramuskulárním podání počáteční přechodný akutní zánět. Na konci doby pozorování (8 dní po dávce) byl po paravenózním a intramuskulárním podání zaznamenán až lehký lokální subakutní zánět a po intramuskulárním podání až další středně silná fokální svalová degenerace/nekróza, přičemž sval se regeneroval.
FARMACEUTICKÉ ÚDAJE
6.
6.1 Seznam pomocných látek
Dinatrium-edetát (E386)
Polysorbát 80 (E433)
LaktózaHydroxid sodný (E524) (k úpravě pH) a/nebo Kyselina chlorovodíková 10% (E507) (k úpravě pH)
6.2 Inkompatibility
IVEMEND je inkompatibilní s jakýmikoli roztoky obsahujícími dvoumocné kationty (např. Ca1+, Mg1+), včetně Hartmannova a Ringerova laktátového roztoku. Tento léčivý přípravek se nesmí mísit s jinými léčivými přípravky s výjimkou těch, které jsou uvedeny v bodu 6.6.
6.3 Doba použitelnosti
2 roky.
Chemická a fyzikální stabilita po otevření před použitím (in-use stability) byla prokázána na dobu 24 hodin při 25 °C.
Z mikrobiologického hlediska má být přípravek okamžitě. Není-li použit okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou v odpovědnosti uživatele a normálně by neměla být delší než 24 hodin při teplotě 2 až 8 °C.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 až 8 °C).
Podmínky uchovávání po rekonstituci a naředění léčivého přípravku viz bod 6.3.
6.5 Druh obalu a obsah balení
10ml injekční lahvička z čirého skla typu I s chlorbutylovou nebo brombutylovou pryžovou zátkou a hliníkovým uzávěrem s šedým plastovým flip-off víčkem.
Velikosti balení: 1 nebo 10 injekčních lahviček.
Na trhu nemusí být k dispozici všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Přípravek IVEMEND musí být před podáním rekonstituován a poté naředěn.
Příprava přípravku IVEMEND 150 mg k intravenóznímu podání:
1. Vstříkněte 5 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9%) do injekční lahvičky. Zajistěte, aby byl injekční roztok chloridu sodného 9 mg/ml (0,9%) do injekční lahvičky přidáván po stěně, aby se zabránilo vzniku pěny. Obsah injekční lahvičky promíchejte mírným krouživým pohybem. Neprotřepávejte a injekční roztok chloridu sodného 9 mg/ml (0,9%)
do injekční lahvičky nevstřikujte proudem.
3. Nasajte celý objem injekční lahvičky a přeneste jej do infuzního vaku obsahujícího 145 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9%), čímž získáte celkový objem 150 ml. Infuzní vak 2-3krát opatrně obraťte.
Tento léčivý přípravek nesmí být rekonstituován nebo mísen s roztoky, jejichž fyzikální a chemická kompatibilita nebyla stanovena (viz bod 6.2).
Vzhled rekonstituovaného roztoku je stejný jako vzhled rozpouštědla.
Rekonstituovaný a naředěný přípravek se musí před podáním vizuálně zkontrolovat na výskyt částic a změnu zabarvení.
Žádné zvláštní požadavky na likvidaci.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Merck Sharp & Dohme Ltd.
Hertford Road, Hoddesdon Hertfordshire EN 11 9BU Velká Británie
8. REGISTRAČNÍ ČÍSLO
EU/1/07/437/003
EU/1/07/437/004
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 11. ledna 2008
Datum posledního prodloužení registrace: 11. ledna 2013
10. DATUM REVIZE TEXTU:
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu.
A. VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ/ VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného/výrobců odpovědných za propouštění šarží
Merck Sharp & Dohme B.V.
Waarderweg 39 2031 BN Haarlem Nizozemsko
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis.
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
IVEMEND 150 mg prášek pro infuzní roztok Fosaprepitantum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna injekční lahvička obsahuje fosaprepitanti dimegluminum odpovídající fosaprepitantum 150 mg, což odpovídá aprepitantum 130,5 mg. Po rekonstituci a naředění obsahuje 1 ml roztoku fosaprepitantum 1 mg (1 mg/ml).
3. SEZNAM POMOCNÝCH LÁTEK
Dinatrium-edetát, polysorbát 80, laktóza, NaOH a/nebo HCl 10% (k úpravě pH). Další informace viz příbalová informace.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
prášek pro infuzní roztok 1 injekční lahvička 10 injekčních lahviček
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Pouze k jednorázovému podání.
Použití perorálního aprepitantu není nutné. Před použitím si přečtěte příbalovou informaci. Intravenózní podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce.
Po rekonstituci a naředění: 24 hodin při 25°C.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Merck Sharp & Dohme Ltd. Hertford Road, Hoddesdon Hertfordshire EN11 9BU Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/07/437/003 1 x 1 injekční lahvička EU/1/07/437/004 1 x 10 injekčních lahviček
13. ČÍSLO ŠARŽE
Č.š.:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
IVEMEND 150 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
IVEMEND 150 mg prášek pro infuzní roztok
Fosaprepitantum
Intravenózní podání
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
B. PŘÍBALOVÁ INFORMACE
IVEMEND 150 mg prášek pro infuzní roztok
fosaprepitantum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je IVEMEND a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete IVEMEND používat
3. Jak se IVEMEND používá
4. Možné nežádoucí účinky
5 Jak IVEMEND uchovávat
6. Obsah balení a další informace
1. Co je IVEMEND a k čemu se používá
Přípravek IVEMEND obsahuje léčivou látku fosaprepitant, která se v těle přeměňuje na aprepitant. Patří do skupiny léčiv nazývaných "antagonisté receptoru neurokininu 1 (NK1)". Mozek má zvláštní oblast, která řídí pocit na zvracení a zvracení. Přípravek IVEMEND funguje tak, že blokuje signály do této oblasti, čímž omezuje pocit na zvracení a zvracení. Přípravek IVEMEND se používá u dospělých v kombinaci s jinými léky k zabránění vzniku pocitu na zvracení a zvracení zapříčiněných chemoterapií (léčbou rakoviny) obsahující cisplatinu (vyvolává silný pocit na zvracení a zvracení) a s chemoterapií, která vyvolává středně silný pocit na zvracení a zvracení (jako cyklofosfamid, doxorubicin nebo epirubicin).
2. Čemu musíte věnovat pozornost, než začnete IVEMEND používat Nepoužívejte IVEMEND:
• Jestliže jste alergický(á) na fosaprepitant, aprepitant nebo polysorbát 80 nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
• Spolu s léky obsahujícími pimozid (používá se k léčbě psychiatrických onemocnění), terfenadin a astemizol (používají se k léčbě senné rýmy a jiných alergických stavů), cisaprid (používá se
k léčbě trávicích potíží). Pokud tyto léky užíváte, informujte o tom svého lékaře, protože Vaše léčba musí být předtím, než začnete používat IVEMEND, upravena.
Upozornění a opatření
Před použitím tohoto přípravku se poraďte se svým lékařem, lékárníkem nebo zdravotní sestrou.
Pokud trpíte onemocněním jater, řekněte to před léčbou přípravkem IVEMEND svému lékaři, protože játra hrají důležitou roli při rozkladu léku v těle. Proto Váš lékař může potřebovat sledovat stav Vašich jater.
Děti a dospívající
Přípravek IVEMEND nepodávejte dětem a dospívajícím mladším 18 let věku, protože nebyl u této populace hodnocen.
Další léčivé přípravky a přípravek IVEMEND
IVEMEND může ovlivnit jiné léky, a to jak během léčby, tak i po léčbě přípravkem IVEMEND. Existují určité léky, které se nesmí užívat spolu s přípravkem IVEMEND (jako je pimozid, terfenadin, astemizol a cisaprid) nebo které vyžadují úpravu dávky (viz také "Nepoužívejte IVEMEND").
Účinky přípravku IVEMEND nebo jiných léčiv mohou být ovlivněny, pokud budete přípravek IVEMEND užívat spolu s dalšími léky, včetně léků uvedených dále. Prosím, informujte svého lékaře nebo lékárníka, pokud užíváte některý z níže uvedených léčivých přípravků:
- léčivé přípravky k zabránění početí, které mohou zahrnovat pilulky, kožní náplasti, implantáty a některá nitroděložní tělíska (IUD), která uvolňují hormony, nemusí odpovídajícím způsobem účinkovat, pokud se používají spolu s přípravkem IVEMEND. Během léčby přípravkem IVEMEND a po dobu až 2 měsíců po jeho použití se musí používat jiná nebo dodatečná nehormonální forma antikoncepce,
- cyklosporin, takrolimus, sirolimus, everolimus (imunosupresiva),
- alfentanil, fentanyl (používají se k léčbě bolesti),
- chinidin (používá se k léčbě nepravidelného tepu srdce),
- irinotekan, etoposid, vinorelbin, ifosfamid (léky používané k léčbě rakoviny),
- léky obsahující deriváty námelových alkaloidů, jako je ergotamin a diergotamin (používané k léčbě migrén),
- warfarin, acenokumarol (léky na ředění krve; může být požadováno provedení krevních testů),
- rifampicin, klarithromycin, telithromycin (antibiotika užívaná k léčbě infekcí),
- fenytoin (léčivý přípravek používaný k léčbě záchvatů),
- karbamazepin (léčivý přípravek používaný k léčbě depresí a epilepsie),
- midazolam, triazolam, fenobarbital (uklidňující léčivé přípravky nebo léčivé přípravky usnadňující spánek),
- třezalka tečkovaná (rostlinný přípravek používaný k léčbě deprese),
- inhibitory proteázy (používají se k léčbě infekcí HIV),
- ketokonazol, kromě šampónu (používá se k léčbě Cushingova syndromu - kdy tělo vytváří nadměrná množství kortisolu),
- nefazodon (používá se k léčbě deprese),
- diltiazem (léčivý přípravek používaný k léčbě vysokého krevního tlaku),
- kortikosteroidy (jako je dexamethason),
- léky proti úzkosti (jako je alprazolam) a
- tolbutamid (léčivý přípravek používaný k léčbě cukrovky)
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a), nebo které možná budete užívat.
Těhotenství a kojení
Neužívejte tento přípravek během těhotenství, pokud to není nezbytně nutné. Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem dříve, než začnete tento přípravek užívat.
Informace týkající se antikoncepce viz část "Další léčivé přípravky a přípravek IVEMEND".
Není známo, zda se IVEMEND u lidí vylučuje do mateřského mléka a tudíž se během léčby tímto přípravkem kojení nedoporučuje. Je důležité, abyste před použitím tohoto přípravku svého lékaře informovala, že kojíte nebo že kojení plánujete.
Řízení dopravních prostředků a obsluha strojů
Je nutné vzít v úvahu, že někteří lidé po použití přípravku IVEMEND mají závratě nebo cítí ospalost. Jestliže máte závratě nebo cítíte ospalost, měl(a) byste se po použití tohoto přípravku vyhnout řízení nebo obsluze strojů (viz "Možné nežádoucí účinky").
IVEMEND obsahuje sodík
Tento léčivý přípravek obsahuje méně než 1 mmol sodíku (23 mg) v jedné dávce, tzn. je v podstatě bez sodíku.
3. Jak se přípravek IVEMEND používá
Doporučená dávka přípravku IVEMEND je 150 mg fosaprepitantu 1. den (den chemoterapie).
Prášek se před použitím rekonstituuje a naředí. Infuzní roztok Vám podá zdravotník, jako např. lékař nebo zdravotní sestra, intravenózní infuzí (kapačkou) přibližně 30 minut před zahájením chemoterapie. Váš lékař Vás požádá, abyste užil(a) i další léky, včetně kortikosteroidů (jako dexamethason) a "antagonistů 5HT3" (jako ondansetron) k předcházení pocitu na zvracení a zvracení. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Přestaňte přípravek IVEMEND používat a ihned navštivte lékaře, pokud zaznamenáte kterýkoli z následujících nežádoucích účinků, které mohou být závažné a kvůli nimž můžete naléhavě potřebovat lékařské ošetření:
- kopřivka, vyrážka, svědění, potíže s dýcháním nebo polykáním (četnost není známa, z dostupných údajů ji nelze určit); jde o známky alergické reakce.
Další hlášené nežádoucí účinky jsou uvedeny níže.
Časté nežádoucí účinky (mohou postihovat až 1 z 10 lidí) jsou:
- zácpa, poruchy trávení,
- bolest hlavy,
- únava,
- ztráta chuti k jídlu,
- škytavka,
- zvýšené množství jaterních enzymů v krvi.
Méně časté nežádoucí účinky (mohou postihovat až 1 ze 100 lidí) jsou:
- závrať, ospalost,
- akné, vyrážka,
- úzkost,
- říhání, pocit nevolnosti, zvracení, pálení žáhy, bolest žaludku, sucho v ústech, plynatost,
- častější bolestivé nebo pálivé močení,
- slabost, celkový pocit nepohody,
- zčervenání obličeje/kůže, návaly horka,
- rychlý nebo nepravidelný srdeční tep, zvýšení krevního tlaku,
- horečka se zvýšeným rizikem infekce, snížení počtu červených krvinek,
- bolest v místě aplikace, zarudnutí v místě aplikace, svědění v místě aplikace, zánět žil v místě infuze.
Vzácné nežádoucí účinky (mohou postihovat až 1 z 1 000 lidí) jsou:
- potíže při přemýšlení, nedostatek energie, poruchy vnímání chutí,
- citlivost kůže na sluneční světlo, nadměrné pocení, mastná kůže, boláky na kůži, svědivá vyrážka, Stevens-Johnsonův syndrom/toxická epidermální nekrolýza (vzácná závažná kožní reakce),
- euforie (pocit extrémního štěstí), dezorientace,
- bakteriální infekce, plísňové infekce,
- těžká zácpa, žaludeční vřed, zánět tenkého a tlustého střeva, boláky v ústech, nadýmání,
- časté močení, větší výdej moči, než je obvyklé, přítomnost cukru nebo krve v moči,
- pocit nepohody na hrudi, otok, změny způsobu chůze,
- kašel, zahlenění zadní části hrdla, podráždění hrdla, kýchání, bolest v krku,
- výtok z očí a svědění očí,
- zvonění v uších,
- svalové křeče, svalová slabost,
- nadměrná žízeň,
- pomalý srdeční tep, onemocnění srdce a cév,
- pokles počtu bílých krvinek, nízké hladiny sodíku v krvi, pokles tělesné hmotnosti,
- zatvrdnutí v místě infuze.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek IVEMEND uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Přípravek nepoužívejte po uplynutí doby použitelnosti uvedené na krabičce a na injekční lahvičce za EXP. První 2 číslice označují měsíc; následující 4 číslice označují rok.
Uchovávejte v chladničce (2 až 8 °C).
Rekonstituovaný a naředěný roztok je stabilní po dobu 24 hodin při teplotě 25 °C.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co IVEMEND obsahuje
- Léčivou látkou je fosaprepitantum. Jedna injekční lahvička obsahuje fosaprepitanti dimegluminum odpovídající fosaprepitantum 150 mg. Po rekonstituci a naředění obsahuje 1 ml roztoku 1 mg fosaprepitantu (1 mg/ml).
- Pomocnými látkami jsou: dinatrium-edetát (E386), polysorbát 80 (E433), laktóza, hydroxid sodný (E524) (k úpravě pH) a/nebo kyselina chlorovodíková 10% (E507) (k úpravě pH).
Jak IVEMEND vypadá a co obsahuje toto balení
IVEMEND je bílý až téměř bílý prášek pro infuzní roztok.
Prášek je obsažen v čiré skleněné injekční lahvičce s pryžovou zátkou a hliníkovým uzávěrem s šedým plastovým flip-off víčkem.
Jedna injekční lahvička obsahuje fosaprepitantum 150 mg. Velikosti balení: 1 nebo 10 injekčních lahviček.
Na trhu nemusí být všechny velikosti balení.
|
Držitel rozhodnutí o registraci Merck Sharp & Dohme Ltd. Hertford Road, Hoddesdon Hertfordshire EN11 9BU Velká Británie |
Výrobce Merck Sharp & Dohme B.V. Waarderweg 39 2031 BNHaarlem Nizozemsko |
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci
|
Belgique/Belgie/Belgien MSD Belgium BVBA/SPRL Tél/Tel: 0800 38 693 (+32(0)27766211) |
Lietuva UAB Merck Sharp & Dohme Tel.:+370 5 278 02 47 msd_lietuva@merck.com |
|
BtnrapHH MepK fflapn h floyM Eturapna EOOfl Ten.:+359 2 819 3737 info-msdbg@merck.com |
Luxembourg/Luxemburg MSD Belgium BVBA/SPRL Tél/Tel: 0800 38 693 (+32(0)27766211) |
|
Česká republika Merck Sharp & Dohme s.r.o. Tel.: +420 233 010 111 dpoc_czechslovak@merck.com |
Magyarország MSD Pharma Hungary Kft. Tel.: +36 1 888 53 00 hungary_msd@merck.com |
|
Danmark MSD Danmark ApS Tlf: + 45 4482 4000 dkmail@merck.com |
Malta Merck Sharp & Dohme Cypru Limited Tel : 8007 4433 (+356 99917558) malta_info@merck.com |
|
Deutschland MSD SHARP & DOHME GMBH Tel: 0800 673 673 673 (+49 (0) 89 4561 2612) |
Nederland Merck Sharp & Dohme BV Tel: 0800 99 99 000 (+31 23 5153513) |
|
Eesti Merck Sharp & Dohme OU Tel.: +372 6144 200 msdeesti@merck.com |
Norge MSD (Norge) AS Tlf: +47 32 20 73 00 msdnorge@msd.no |
|
EXXáSa MSD A.OB.E.E. TpA,: +30 210 98 97 300 dpoc_greece@merck.com |
Osterreich Merck Sharp & Dohme Ges.m.b.H. Tel: +43 (0) 1 26 044 msd-medizin@merck.com |
|
Espaňa Merck Sharp & Dohme de Espana, S.A. Tel:+34 91 321 06 00 msd info@merck.com |
Polska MSD Polska Sp. z o.o. Tel:+48 22 549 51 00 msdpolska@merck.com |
|
France MSD France Tél: +33 (0) 1 80 46 40 40 |
Portugal Merck Sharp & Dohme, Lda Tel: +351 21 4465700 clic@merck.com |
Hrvatska
Merck Sharp & Dohme d.o.o.
Tel: + 385 1 6611 333 croatia_info@merck.com
Ireland
Merck Sharp & Dohme Ireland (Human Health) Limited
Tel: +353 (0)1 2998700 medinfo_ireland@merck.com
Ísland
Vistor hf.
Simi: +354 535 7000
Italia
MSD Italia S.r.l.
Tel:+39 06 361911 medicinalinformation.it@merck.com
Kúrcpog
Merck Sharp & Dohme Cyprus Limited TpA,: 800 00 673 (+357 22866700) cyprus_info@merck.com
Latvija
SIA Merck Sharp & Dohme Latvija Tel: +371 67364 224 msd_lv@merck.com
Románia
Merck Sharp & Dohme Romania S.R.L.
Tel: + 40 21 529 29 00 msdromania@merck.com
Slovenija
Merck Sharp & Dohme, inovativna zdravila d.o.o. Tel: + 386 1 5204201 msd_slovenia@merck.com
Slovenská republika
Merck Sharp & Dohme, s. r. o.
Tel: +421 2 58282010 dpoc_czechslovak@merck.com
Suomi/Finland
MSD Finland Oy Puh/Tel: +358 (0) 9 804650 info@msd.fi
Sverige
Merck Sharp & Dohme (Sweden) AB Tel: +46 77 5700488 medicinskinfo@merck.com
United Kingdom
Merck Sharp & Dohme Limited Tel: +44 (0) 1992 467272 medicalnformationuk@merck.com
Tato příbalová informace byla naposledy revidována:
Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese: http://www.ema.europa.eu.
Následující informace jsou určeny pouze pro zdravotnické pracovníky:
Pokyny k rekonstituci a ředění přípravku IVEMEND 150 mg
1. Vstříkněte 5 ml injekčního roztoku chloridu sodného o koncentraci 9 mg/ml (0,9%) do injekční lahvičky. Zajistěte, aby byl injekční roztok chloridu sodného 9 mg/ml (0,9%) do injekční lahvičky přidáván po stěně, aby se zabránilo vzniku pěny. Obsah injekční lahvičky promíchejte mírným krouživým pohybem. Neprotřepávejte a injekční roztok chloridu sodného 9 mg/ml (0,9%) do injekční lahvičky nevstřikujte proudem.
2. Připravte infuzní vak naplněný 145 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9%) (například odebráním 105 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9%) z infuzního vaku s 250 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9%)).
3. Odeberte celý objem injekční lahvičky a přeneste jej do infuzního vaku obsahujícího 145 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9%), čímž získáte celkový objem 150 ml. Infuzní vak 2-3krát opatrně obraťte. (viz "Jak se IVEMEND používá").
Rekonstituovaný a naředěný konečný roztok je při teplotě 25 °C stabilní po dobu 24 hodin.
Parenterální léčivé přípravky musí být před podáním vizuálně zkontrolovány na výskyt částic a změnu zbarvení kdykoli to roztok a obal umožní.
Vzhled rekonstituovaného roztoku je stejný jako vzhled rozpouštědla.
31
Připravte infuzní vak naplněný 145 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9%) (např. odebráním 105 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9%) z infuzního vaku s 250 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9%)).