Inovelon 400 Mg
SOUHRN ÚDAJŮ O PŘÍPRAVKU
PŘÍLOHA I
Inovelon 100 mg potahované tablety
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna potahovaná tableta obsahuje rufinamidum 100 mg.
Pomocná látka se známým účinkem: jedna potahovaná tableta obsahuje 20 mg monohydrátu laktosy Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Potahovaná tableta.
Růžová, „oválná“, mírně konvexní, na obou stranách s půlicí rýhou, na jedné straně s vyraženým označením „C261“ a na druhé straně bez označení.
Tabletu lze rozdělit na dvě stejné dávky.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Inovelon je indikován jako přídatná léčba při léčení záchvatů spojených s Lennox-Gastautovým syndromem u pacientů ve věku 4 let a starších.
4.2 Dávkování a způsob podání
Léčbu rufinamidem by měl předepsat lékař se specializací v oboru pediatrie nebo neurologie se zkušenostmi s léčbou epilepsie.
Inovelon perorální suspenze a Inovelon potahované tablety lze při stejných dávkách zaměnit. Pacienty je třeba v období přechodu z jedné lékové formy na druhou sledovat.
Dávkování
Podávání dětem ve věku čtyř let nebo starším o hmotnosti nižší než 30 kg Pacienti < 30 kg neužívající valproát sodný:
Léčba by měla být zahájena denní dávkou 200 mg. Podle klinické odpovědi a snášenlivosti může být ve dvoudenních intervalech denní dávka zvyšována o 200 mg, až do maximální doporučené dávky 1 000 mg/den. Dávky až 3 600 mg/den byly studovány u omezeného počtu pacientů.
Pacienti < 30 kg užívající současně valproát sodný:
Vzhledem k tomu, že valproát výrazně snižuje clearance rufinamidu, doporučuje se u pacientů < 30 kg, kteří současně užívají valproát, nižší maximální dávka Inovelonu. Léčba by měla být zahájena denní dávkou 200 mg. Podle klinické odpovědi a snášenlivosti může být minimálně po dvou dnech denní dávka zvýšena o 200 mg až do maximální doporučené dávky 600 mg/den.
Podávání dospělým, dospívajícím a dětem ve věku čtyř let nebo starším o hmotnosti 30 kg a vyšší Léčba by měla být zahájena denní dávkou 400 mg. Podle klinické odpovědi a snášenlivosti může být ve dvoudenních intervalech denní dávka zvyšována o 400 mg, až do maximální doporučené dávky, jak je uvedena v následující tabulce.
|
Rozmezí hmotnosti |
30,0 - 50,0 kg |
50,1 - 70,0 kg |
> 70,1 kg |
|
Maximální doporučená dávka |
1 800 mg/den |
2 400 mg/den |
3 200 mg/den |
Dávky až 4 000 mg/den (u hmotnosti 30-50 kg) nebo 4 800 mg/den (u hmotnosti nad 50 kg) byly studovány u omezeného počtu pacientů.
Ukončení léčby
Má-li být léčba rufinamidem ukončena, je nutné ji vysazovat postupně. V klinických studiích bylo vysazení rufinamidu dosaženo snižováním dávky o přibližně 25 % vždy po dvou dnech.
V případě jedné či více vynechaných dávek je nutné individuální posouzení klinického stavu.
Nekontrolované otevřené studie naznačují, že účinnost je dlouhodobá, přestože žádná kontrolovaná studie nebyla prováděna déle než tři měsíce.
Pediatrická populace
Bezpečnost a účinnost rufinamidu u dětí ve věku do 4 let nebyla dosud stanovena. Nejsou dostupné žádné údaje.
Starší osoby
Informace o podávání rufinamidu starším osobám jsou omezené. Protože farmakokinetika rufinamidu není u starších osob změněna (viz bod 5.2), není nutná úprava dávkování u pacientů ve věku nad 65 let.
Porucha funkce ledvin
Studie pacientů se závažnou poruchou funkce ledvin ukázala, že u těchto pacientů není nutná žádná úprava dávkování (viz bod 5.2).
Porucha funkce jater
Podávání léku u pacientů s poruchou funkce jater nebylo studováno. Při léčbě pacientů s mírnou až středně závažnou poruchou funkce jater se doporučuje opatrnost a pečlivé titrování dávky. Podávání léku pacientům s těžkou poruchou funkce jater se nedoporučuje.
Způsob podání
Rufinamid je určen k perorálnímu podání. Užívá se dvakrát denně, ráno a večer, rozdělený do dvou stejných dávek a zapíjí se vodou. Protože byl pozorován vliv jídla, měl by být Inovelon užíván s jídlem (viz bod 5.2). Má-li pacient potíže s polykáním, je možné tablety rozdrtit a podat s menším množstvím vody (půl sklenice).
4.3 Kontraindikace
Hypersenzitivita na léčivou látku, deriváty triazolu nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Status epilepticus
Při podávání rufinamidu během studií klinického vývoje byly zaznamenány případy status epilepticus, zatímco u pacientů, kteří užívali placebo, nebyly žádné takové případy zaznamenány. Tyto příhody vedly k vysazení rufinamidu ve 20 % případů. Jestliže se u pacientů objeví nové typy záchvatů a/nebo se zvýšenou frekvencí dochází ke status epilepticus, který se liší od počátečního stavu pacienta, je nutné znovu přehodnotit poměr prospěšnosti a rizik léčby.
Vysazení rufinamidu
Rufinamid je nutné vysazovat postupně, aby se snížila možnost výskytu záchvatů při vysazení léku. V klinických studiích bylo vysazení dosaženo snižováním dávky o přibližně 25 % vždy po dvou dnech. O vysazení současně podávaných antiepileptických přípravků, jakmile bylo dosaženo kontroly nad záchvaty přidáním rufinamidu, nejsou dostatečné údaje.
Reakce centrálního nervového systému
Léčba rufinamidem byla spojena se závratí, spavostí, ataxií a poruchami chůze, což mohlo zvýšit výskyt náhodných pádů v této populaci (viz bod 4.8). Je třeba, aby pacienti a ošetřující osoby dbali zvýšené opatrnosti, dokud se neseznámí s možnými účinky tohoto léčivého přípravku.
Hypersenzitivní reakce
Ve spojení s léčbou rufinamidem se vyskytly závažné syndromy přecitlivělosti na antiepileptický léčivý přípravek včetně DRESS (léková reakce s eozionfilií a systémovými příznaky) a Stevens-Johnsonova syndromu. Příznaky a symptomy tohoto onemocnění byly různorodé; avšak u pacientů se typicky vyskytovala, i když ne výlučně, horečka a vyrážka, ve spojení s postižením dalšího orgánového systému. Jiné přidružené manifestace zahrnovaly lymfadenopatii, abnormality testů jaterních funkcí a hematurii. Protože jsou projevy tohoto stavu různorodé, mohou se objevit jiné příznaky a známky postižení orgánových systémů, které v tomto textu nejsou uvedeny. Tento syndrom přecitlivělosti na antiepileptika se objevil v těsné časové souvislosti se zahájením léčby rufinamidem a u souboru pediatrických pacientů. Pokud na tuto reakci vznikne podezření, je nutné rufinamid vysadit a zahájit alternativní léčbu. Všichni pacienti, u kterých se objeví vyrážka při užívání rufinamidu, musí být pečlivě monitorováni.
Zkrácení QT
V podrobné QT studii způsobil rufinamid pokles QTc intervalu, který byl přímo úměrný jeho koncentraci. Ačkoliv mechanismus vzniku tohoto nálezu ani jeho význam s ohledem na bezpečnost přípravku nejsou známy, lékaři by na základě klinického posouzení měli zvážit, zda předepsat rufinamid pacientům, kterým hrozí další zkracování QTc intervalu (např. kongenitální syndrom krátkého QT nebo pacienti, kteří mají tento syndrom v rodinné anamnéze).
Ženy v plodném věku
Ženy v plodném věku musejí během léčby Inovelonem používat antikoncepci. Lékaři by se měli snažit zajistit, aby byla použita vhodná antikoncepce a podle klinické situace jednotlivých pacientek by měli zvážit, zda je vhodnější podávat perorální antikoncepci nebo dávky složek perorální antikoncepce (viz bod 4.5).
Laktóza
Inovelon obsahuje laktózu, proto by pacienti se vzácnými dědičnými poruchami, jako je intolerance galaktózy, vrozená deficience laktázy nebo malabsorpce glukózy a galaktózy, neměli tento léčivý přípravek užívat.
Sebevražedné myšlenky
Sebevražedné myšlenky a chování byly hlášeny u pacientů léčených antiepileptiky v několika indikacích. Metaanalýza randomizovaných placebem kontrolovaných studií antiepileptických léčivých přípravků také prokázala mírně zvýšené riziko sebevražedných myšlenek a sebevražedného chování. Mechanismus tohoto rizika není znám a dostupné údaje nevylučují možnost zvýšeného rizika u Inovelonu.
U pacientů je proto třeba sledovat známky sebevražedných myšlenek a sebevražedného chování a je třeba zvážit vhodnou léčbu. Pacienty (a ošetřovatele pacientů) je třeba poučit, aby v případě, že se objeví známky sebevražedných myšlenek nebo sebevražedného chování, vyhledali lékařskou pomoc.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Potenciál jiných léčivých přípravků k ovlivňování rufinamidu
Jiné antiepileptické přípravky
Koncentrace rufinamidu nepodléhají klinicky významným změnám při současném podávání se známými antiepileptickými léčivými přípravky indukujícími enzymy.
U pacientů léčených přípravkem Inovelon, u kterých bylo zahájeno podávání valproátu, může docházet k významnému zvýšení koncentrací rufinamidu v plazmě. Nejvýraznější zvýšení bylo pozorováno u pacientů s nízkou tělesnou hmotností (< 30 kg). Proto je třeba zvážit snížení dávky Inovelonu u pacientů < 30 kg, u kterých je zahájena léčba valproátem (viz bod 4.2).
Přidání nebo vysazení těchto léčivých přípravků nebo upravení dávky těchto léčivých přípravků během léčby rufinamidem může vyžadovat úpravu dávkování rufinamidu.
Po současném podávání lamotriginu, topiramátu nebo benzodiazepinů nebyly pozorovány žádné významné změny koncentrace rufinamidu.
Potenciál rufinamidu k ovlivňování jiných léčivých přípravků
Jiné antiepileptické léčivé přípravky
Farmakokinetické interakce mezi rufinamidem a jinými antiepileptickými léčivými přípravky byly u pacientů s epilepsií hodnoceny pomocí populačního modelování farmakokinetiky. Zdá, se že rufinamid nemá žádný klinicky relevantní účinek na koncentrace karbamazepinu, lamotriginu, fenobarbitalu, topiramátu, fenytoinu nebo valproátu v ustáleném stavu.
Perorální antikoncepce
Současné podávání rufinamidu 800 mg dvakrát denně a kombinované perorální antikoncepce (etinylestradiol 35 pg a noretindron 1 mg) po dobu 14 dní způsobilo průměrný pokles AUC0-24 etinylestradiolu o 22 % a AUC0-24 noretindronu o 14 %. Studie s jinými perorálními nebo implantovanými kontraceptivy nebyly prováděny. Ženám plodného věku, které užívají hormonální antikoncepci, se doporučuje používat další bezpečnou a účinnou antikoncepční metodu (viz body 4.4 a 4.6).
Cytochromové enzymy P450
Rufinamid je metabolizovaný hydrolýzou a cytochromovými enzymy P450 není významně metabolizován. Kromě toho rufinamid neinhibuje aktivitu cytochromových enzymů P450 (viz bod 5.2). Je tedy nepravděpodobné, aby nastaly klinicky významné interakce zprostředkované inhibicí systému cytochromů P450 rufinamidem. Bylo prokázáno, že rufinamid indukuje enzym CYP3A4 cytochromu P450 a může tudíž snížit plazmatické koncentrace látek, které jsou metabolizovány tímto enzymem. Účinek byl mírný až středně závažný. Průměrná aktivita CYP3A4, hodnocená jako clearance triazolamu, byla zvýšena o 55 % po 11 dnech léčby rufinamidem 400 mg dvakrát denně. Expozice triazolamu byla snížena o 36 %. Vyšší dávky rufinamidu mohou mít za následek výraznější indukci. Nelze vyloučit, že rufinamid může také snižovat expozici látek metabolizovaných jinými enzymy nebo dopravovaných transportními proteiny, jako je P-glykoprotein.
Doporučuje se, aby pacienti léčení látkami metabolizovanými systémem enzymu CYP3A4, byli pečlivě monitorováni po dobu dvou týdnů, a to buď na počátku nebo po skončení léčby rufinamidem, nebo po jakékoliv významné změně dávkování. Možná bude zapotřebí zvážit úpravu dávkování současně podávaného léčivého přípravku. Tato doporučení je třeba vzít v úvahu také, pokud se rufinamid používá společně s látkami s úzkým terapeutickým oknem, jako je warfarin nebo digoxin.
Studie specifické interakce u zdravých subjektů neodhalila žádný vliv rufinamidu v dávce 400 mg dvakrát denně na farmakokinetiku olanzapinu, substrátu CYP1A2.
5
Nejsou k dispozici žádné údaje o interakci rufinamidu s alkoholem.
4.6 Fertilita, těhotenství a kojení
Riziko související s epilepsií a podáváním antiepileptik všeobecně:
Ukazuje se, že v potomstvu žen s epilepsií je prevalence malformací dvakrát až třikrát větší než je četnost v celkové populaci, která je přibližně 3 %. U léčené populace byl nárůst malformací pozorován u polyterapie, avšak rozsah, za který je léčba a/nebo nemoc odpovědná, nebyl určen.
Navíc účinná antiepileptická léčba nesmí být přerušena, protože zhoršení nemoci je škodlivé jak pro matku tak pro plod.
Riziko související s rufinamidem:
Studie na zvířatech neprokázaly teratogenní účinek kromě fetotoxicity v přítomnosti maternální toxicity (viz bod 5.3). Potenciální riziko pro lidské jedince není známé.
Nejsou k dispozici klinické údaje o podávání rufinamidu během těhotenství.
Vezmeme-li v úvahu tyto údaje, rufinamid by neměl být podáván, pokud to není nezbytně nutné, během těhotenství ani u žen plodného věku, které nepoužívají kontracepční opatření.
Ženy ve fertilním věku musí během léčby rufinamidem používat antikoncepci. Lékaři by se měli snažit zajistit, aby byla použita vhodná antikoncepce a podle klinické situace jednotlivých pacientek by měli zvážit, zda je vhodnější podávat perorální antikoncepci nebo dávky složek perorální antikoncepce (viz bod 4.5).
Pokud ženy léčené rufinamidem plánují otěhotnět, je nutné pečlivě zvážit indikaci tohoto přípravku.
V průběhu těhotenství nesmí být přerušena účinná antiepileptická léčba rufinamidem, protože zhoršení nemoci je škodlivé jak pro matku, tak pro plod.
Kojení
Není známo, zda se rufinamid vylučuje do lidského mateřského mléka. Vzhledem k možným škodlivým účinkům pro kojené dítě by neměly matky při léčbě rufinamidem kojit.
Fertilita
Nejsou dostupné žádné údaje o účinku léčby rufinamidem na fertilitu.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Inovelon může způsobit závratě, spavost a rozmazané vidění. V závislosti na individuální citlivosti může mít rufinamid malý až výrazný vliv na schopnost řídit nebo obsluhovat stroje. Pacienty je třeba upozornit, aby během činností vyžadujících vysoký stupeň bdělosti, např. při řízení nebo obsluhování strojů, dbali zvýšené opatrnosti.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Klinický vývojový program zahrnoval více než 1 900 pacientů s různými typy epilepsie, kterým byl podáván rufinamid. Nejčastěji hlášené nežádoucí účinky celkově byly bolesti hlavy, závratě, únava a spavost. Nej častější nežádoucí účinky pozorované u pacientů s Lennox-Gastautovým syndromem ve vyšší incidenci než při podávání placeba byly spavost a zvracení. Nežádoucí účinky byly obvykle mírné až středně těžké. Četnost vystoupení pacientů s Lennox-Gastautovým syndromem ze studie z důvodu nežádoucích účinků byla 8,2 % pro pacienty užívající rufinamid a 0 % pro pacienty užívající placebo. Nejčastější nežádoucí účinky, které vedly k vystoupení ze studie, byly ve skupině léčené rufinamidem vyrážka a zvracení.
Tabulka nežádoucích účinků
Nežádoucí účinky hlášené s vyšší incidencí než u placeba během dvojitě zaslepených studií u pacientů s Lennox-Gastautovým syndromem nebo u celkového souboru pacientů, kterým byl podáván rufinamid, jsou uvedeny v následující tabulce podle terminologie MedDRA, třídy orgánových systémů a podle frekvence.
Frekvence jsou definovány jako: velmi časté (> 1/10), časté (> 1/100 < 1/10), méně časté (> 1/1 000 < 1/100), vzácné (> 1/10 000 až < 1/1 000).
|
Třída orgánových systémů |
Velmi časté |
Časté |
Méně časté |
Vzácné |
|
Infekce a infestace |
Chřipka Nazofaryngitida Ušní infekce Sinusitida Rinitida | |||
|
Poruchy imunitního systému |
Hypersensitivita* | |||
|
Poruchy metabolismu a výživy |
Poruchy příjmu potravy Snížená chuť k jídlu | |||
|
Psychiatrické poruchy |
Úzkost Insomnie | |||
|
Poruchy nervového systému |
Spavost* Bolesti hlavy Závratě* |
Status epilepticus* Křeče Abnormální koordinace* Nystagmus Psychomotorická hyperaktivita | ||
|
Poruchy oka |
Diplopie Rozmazané vidění | |||
|
Poruchy ucha a labyrintu | ||||
|
Respirační, hrudní a mediastinální poruchy |
Epistaxe | |||
|
Gastrointestinální poruchy |
Bolesti horní části břicha Zácpa | |||
|
Poruchy jater a žlučových cest |
Zvýšení jaterních enzymů | |||
|
Poruchy kůže a podkožní tkáně |
Vyrážka* Akné | |||
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Bolesti zad |
|
Třída orgánových systémů |
Velmi časté |
Časté |
Méně časté |
Vzácné |
|
Poruchy reprodukčního systému a prsu |
Oligomenorea | |||
|
Celkové poruchy a reakce v místě aplikace |
Únava |
Poruchy chůze* | ||
|
Vyšetření |
Pokles hmotnosti | |||
|
Poranění, otravy a procedurální komplikace |
Poranění hlavy Kontuze | |||
|
* Odkaz na bod 4/ | ||||
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.4.9 Předávkování
Po akutním předávkování může být žaludek vyprázdněn gastrickou laváží nebo vyvoláním zvracení. Pro rufinamid neexistuje žádné specifické antidotum. Léčba by měla být podpůrná a může zahrnovat hemodialýzu (viz bod 5.2).
Podávání opakovaných dávek 7 200 mg/den nebylo spojeno s žádnými závažnějšími příznaky nebo symptomy.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antiepileptika, deriváty karboxamidu; ATC kód: N03AF03. Mechanismus účinku
Rufinamid moduluje aktivitu sodíkových kanálů tak, že prodlužuje jejich inaktivní stav. Rufinamid je účinný u celé řady zvířecích modelů epilepsie.
Klinické zkušenosti
Inovelon (tablety obsahující rufinamid) byl podáván ve dvojitě zaslepené, placebem kontrolované studii v dávkách až 45 mg/kg/den po dobu 84 dnů 139 pacientům s nedostatečně kontrolovanými záchvaty spojenými s Lennox-Gastautovým syndromem (včetně atypických záchvatů typu absence a záchvatových pádů). Muži i ženy (ve věku od 4 do 30 let) byli zařazeni do studie, pokud užívali 1 až 3 současně podávané antiepileptické léčivé přípravky s pevně stanoveným dávkováním. Každý pacient musel mít alespoň 90 záchvatů během měsíce před přijetím do studie. U všech tří primárních proměnných bylo pozorováno signifikantní zlepšení: procentní změna celkové frekvence záchvatů za 28 dnů během udržovací fáze v porovnání s výchozí hodnotou (-35,8 % při podávání přípravku Inovelon oproti -1,6 % při podávání placeba, p = 0,0006), počet tonických/atonických záchvatů (-42,9 % při podávání přípravku Inovelon oproti 2,2 % při podávání placeba, p = 0,0002) a ohodnocení závažnosti záchvatu při celkovém vyhodnocení prováděném rodičem/opatrovníkem na konci dvojitě zaslepené fáze (velké nebo velmi velké zlepšení ve 32,2 % při podávání přípravku Inovelon oproti 14,5 % v rameni s placebem, p = 0,0041).
Populační modelování farmakokinetiky/farmakodynamiky ukázalo, že snížení celkové záchvatové frekvence a frekvence tonických/atonických záchvatů, zlepšení celkového vyhodnocení závažnosti záchvatů a zvýšení pravděpodobnosti snížení záchvatové frekvence bylo závislé na koncentracích rufinamidu.
5.2 Farmakokinetické vlastnosti
Absorpce
Maximální plazmatické hladiny jsou dosaženy přibližně 6 hodin po podání. Vrcholová koncentrace (Cmax) a plazmatická AUC rufinamidu se zvyšují méně než proporcionálně s dávkou u zdravých subjektů a pacientů nalačno i po jídle, což je pravděpodobně způsobeno omezenou absorpcí dávky. Po jednotlivých dávkách zvyšuje potrava biologickou dostupnost (AUC) rufinamidu přibližně o 34 % a maximální koncentraci v plazmě o 56 %.
Bylo prokázáno, že přípravky Inovelon perorální suspenze a Inovelon potahované tablety jsou biologicky ekvivalentní.
Distribuce
Ve studiích in vitro byla pouze malá frakce rufinamidu (34 %) navázána na lidské sérové proteiny, přičemž albumin odpovídal přibližně za 80 % této vazby. To ukazuje, že riziko lékových interakcí v důsledku nahrazení vazebného místa během současného podávání dalších látek je minimální. Rufinamid byl rovnoměrně rozdělen mezi erytrocyty a plazmu.
Biotransformace
Rufinamid je téměř výhradně eliminován metabolismem. Hlavní dráha metabolismu je hydrolýza karboxylamidové skupiny na farmakologicky inaktivní derivát kyseliny CGP 47292. Metabolismus zprostředkovaný cytochromem P450 se podílí v mnohem menší míře.
Nelze zcela vyloučit vznik malých množství konjugátů glutathionu.
In vitro rufinamid vykazoval malou nebo žádnou schopnost působit jako kompetitivní nebo nekompetitivní inhibitor následujících lidských enzymů P450: CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5 nebo CYP4A9/11-2.
Eliminace
Plazmatický eliminační poločas je přibližně 6-10 hodin u zdravých subjektů a pacientů s epilepsií. Rufinamid se při podávání dvakrát denně ve 12 hodinových intervalech akumuluje v rozsahu, který lze předpovídat podle jeho terminálního poločasu, což ukazuje, že farmakokinetika rufinamidu je časově nezávislá (tj. bez autoindukce metabolismu).
Ve studiích s radioaktivně značenou látkou u tří zdravých dobrovolníků byla základní sloučenina (rufinamid) hlavní radioaktivní složkou v plazmě a představovala přibližně 80 % celkové radioaktivity, metabolit CGP 47292 tvořil pouze asi 15 %. Převládající cestou eliminace látek souvisejících s léčivou látkou bylo vylučování ledvinami, které odpovídalo za 84,7 % dávky.
Linearita/nelinearita:
Biologická dostupnost rufinamidu je závislá na dávce. Se zvyšováním dávky se snižuje biologická dostupnost.
Farmakokinetika u zvláštních skupin pacientů
Pohlaví
Pro vyhodnocení vlivu pohlaví na farmakokinetiku rufinamidu bylo použito populační modelování farmakokinetiky. Tato hodnocení ukazují, že pohlaví neovlivňuje farmakokinetiku rufinamidu do klinicky významné míry.
Porucha funkce ledvin
Farmakokinetika jedné dávky 400 mg rufinamidu nebyla u pacientů s chronickým a těžkým selháním ledvin změněna, ve srovnání se zdravými dobrovolníky. Ale když byla po podání rufinamidu provedena hemodialýza, byly plazmatické hladiny sníženy přibližně o 30 %, což svědčí pro to, že by to mohl být postup použitelný v případě předávkování (viz body 4.2 a 4.9).
Porucha funkce jater
U pacientů s poruchou funkce jater nebyly prováděny žádné studie, a proto by Inovelon neměl být pacientům se těžkým poškozením jater podáván (viz bod 4.2).
Děti (2-12 let)
Děti mají obvykle nižší clearance rufinamidu než dospělé osoby a tento rozdíl souvisí s velikostí těla. Studie u novorozených dětí nebo kojenců a batolat mladších 2 let nebyly prováděny.
Starší osoby
Farmakokinetická studie u starších zdravých dobrovolníků neukázala významný rozdíl ve farmakokinetických parametrech ve srovnání s mladšími dospělými osobami.
5.3 Předklinické údaje vztahující se k bezpečnosti
Konvenční farmakologické studie bezpečnosti v klinicky relevantních dávkách neodhalily žádná zvláštní rizika.
Toxické účinky pozorované u psů v hladinách podobných expozici u člověka při maximální doporučené dávce byly změny na játrech, včetně žlučových trombů, cholestázy a zvýšení hodnot jaterních enzymů, o kterých se předpokládá, že souvisejí se zvýšenou sekrecí žluče u tohoto živočišného druhu. Ve studiích toxicity po opakovaném podávání u laboratorních potkanů a opic nebyl zjištěn žádný důkaz svědčící pro přidružené riziko.
Ve studiích reprodukční a vývojové toxicity se vyskytlo zpomalení růstu plodu a snížení přežití a několik mrtvě narozených plodů v důsledku maternální toxicity. Ale u potomstva nebyly pozorovány žádné účinky na morfologii a funkce, včetně učení nebo paměti. Rufinamid nebyl teratogenní u myší, laboratorních potkanů či králíků.
Rufinamid nebyl genotoxický a neměl žádný kancerogenní potenciál. Nežádoucím účinkem, který nebyl pozorován během klinických studií, ale byl zaznamenán u zvířat při expozici podobné hodnotám klinické expozice, a který by mohl být relevantní z hlediska použití u lidských jedinců, byla ve studii kancerogenity u myší myelofibróza kostní dřeně. Výskyt nezhoubných kostních novotvarů (osteomů) a hyperostóza zaznamenané u myší byly považovány za výsledek aktivace viru specifického pro myši ionty flóru uvolněnými během oxidačního metabolismu rufinamidu.
Pokud jde o imunotoxický potenciál, byl u psů během třináctitýdenní studie pozorován malý brzlík a involuce brzlíku s výraznou reakcí při vysokých dávkách u samců. Změny kostní dřeně a lymfatické změny u samic byly při podávání vysokých dávek během třináctitýdenní studie hlášeny s malou incidencí. U potkanů byla snížená buněčnost kostní dřeně a atrofie brzlíku pozorována pouze u studie kancerogenity.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Jádro
Monohydrát laktosy Mikrokrystalická celulosa Kukuřičný škrob Sodná sůl kroskarmelosy Hypromelosa Magnesium-stearát
Natrium-lauryl-sulfát Koloidní bezvodý oxid křemičitý
Potah tablety Hypromelosa Makrogoly (8000)
Oxid titaničitý (E171)
Mastek
Červený oxid železitý (E172)
6.2 Inkompatibility Neuplatňuje se.
6.3 Doba použitelnosti
4 roky.
6.4 Zvláštní opatření pro uchovávání
Neuchovávejte při teplotě nad 30 °C.
6.5 Druh obalu a obsah balení
Blistry z hliníku/hliníku (Al/Al blistry), balení po 10, 30, 50, 60 a 100 potahovaných tabletách. Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku
Žádné zvláštní požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Eisai Limited, European Knowledge Centre, Mosquito Way, Hatfield, Hertfordshire, AL10 9SN, Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/06/378/001-005
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 16. ledna 2007
Datum posledního prodloužení registrace: 16. ledna 2012
10. DATUM REVIZE TEXTU
{MM/RRRR}
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu.
NÁZEV PŘÍPRAVKU
1.
Inovelon 200 mg potahované tablety
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna potahovaná tableta obsahuje rufinamidum 200 mg.
Pomocná látka se známým účinkem: jedna potahovaná tableta obsahuje 40 mg monohydrátu laktosy Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Potahovaná tableta.
Růžová, „oválná“, mírně konvexní, na obou stranách s půlicí rýhou, na jedné straně s vyraženým označením „C262“ a na druhé straně bez označení.
Tabletu lze rozdělit na dvě stejné dávky.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Inovelon je indikován jako přídatná léčba při léčení záchvatů spojených s Lennox-Gastautovým syndromem u pacientů ve věku 4 let a starších.
4.2 Dávkování a způsob podání
Léčbu rufinamidem by měl předepsat lékař se specializací v oboru pediatrie nebo neurologie se zkušenostmi s léčbou epilepsie.
Inovelon perorální suspenze a Inovelon potahované tablety lze při stejných dávkách zaměnit. Pacienty je třeba v období přechodu z jedné lékové formy na druhou sledovat.
Dávkování
Podávání dětem ve věku čtyř let nebo starším o hmotnosti nižší než 30 kg Pacienti < 30 kg neužívající valproát sodný:
Léčba by měla být zahájena denní dávkou 200 mg. Podle klinické odpovědi a snášenlivosti může být ve dvoudenních intervalech denní dávka zvyšována o 200 mg, až do maximální doporučené dávky 1 000 mg/den. Dávky až 3 600 mg/den byly studovány u omezeného počtu pacientů.
Pacienti < 30 kg užívající současně valproát sodný:
Vzhledem k tomu, že valproát výrazně snižuje clearance rufinamidu, doporučuje se u pacientů < 30 kg, kteří současně užívají valproát, nižší maximální dávka Inovelonu. Léčba by měla být zahájena denní dávkou 200 mg. Podle klinické odpovědi a snášenlivosti může být minimálně po dvou dnech denní dávka zvýšena o 200 mg až do maximální doporučené dávky 600 mg/den.
Podávání dospělým, dospívajícím a dětem ve věku čtyř let nebo starším o hmotnosti 30 kg a vyšší Léčba by měla být zahájena denní dávkou 400 mg. Podle klinické odpovědi a snášenlivosti může být ve dvoudenních intervalech denní dávka zvyšována o 400 mg, až do maximální doporučené dávky, jak je uvedena v následující tabulce.
|
Rozmezí hmotnosti |
30,0 - 50,0 kg |
50,1 - 70,0 kg |
> 70,1 kg |
|
Maximální doporučená dávka |
1 800 mg/den |
2 400 mg/den |
3 200 mg/den |
Dávky až 4 000 mg/den (u hmotnosti 30-50 kg) nebo 4 800 mg/den (v kategorii nad 50 kg) byly studovány u omezeného počtu pacientů.
Vysazení rufinamidu
Má-li být léčba rufinamidem ukončena, je nutné ji vysazovat postupně. V klinických studiích bylo vysazení rufinamidu dosaženo snižováním dávky o přibližně 25 % vždy po dvou dnech.
V případě jedné či více vynechaných dávek je nutné individuální posouzení klinického stavu.
Nekontrolované otevřené studie naznačují, že účinnost je dlouhodobá, přestože žádná kontrolovaná studie nebyla prováděna déle než tři měsíce.
Pediatrická populace
Bezpečnost a účinnost rufinamidu u dětí ve věku do 4 let nebyla dosud stanovena. Nejsou dostupné žádné údaje.
Starší osoby
Informace o podávání rufinamidu starším osobám jsou omezené. Protože farmakokinetika rufinamidu není u starších osob změněna (viz bod 5.2), není nutná úprava dávkování u pacientů ve věku nad 65 let.
Porucha funkce ledvin
Studie pacientů se závažnou poruchou funkce ledvin ukázala, že u těchto pacientů není nutná žádná úprava dávkování (viz bod 5.2).
Porucha funkce jater
Podávání léku u pacientů s poruchou funkce jater nebylo studováno. Při léčbě pacientů s mírnou až středně závažnou poruchou funkce jater se doporučuje opatrnost a pečlivé titrování dávky. Podávání léku pacientům s těžkou poruchou funkce jater se nedoporučuje.
Způsob podání
Rufinamid je určen k perorálnímu podání. Užívá se dvakrát denně, ráno a večer, rozdělený do dvou stejných dávek a zapíjí se vodou. Protože byl pozorován vliv jídla, měl by být Inovelon užíván s jídlem (viz bod 5.2). Má-li pacient potíže s polykáním, je možné tablety rozdrtit a podat s menším množstvím vody (půl sklenice).
4.3 Kontraindikace
Hypersenzitivita na léčivou látku, deriváty triazolu nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Status epilepticus
Při podávání rufinamidu během studií klinického vývoje byly zaznamenány případy status epilepticus, zatímco u pacientů, kteří užívali placebo, nebyly žádné takové případy zaznamenány. Tyto příhody vedly k vysazení rufinamidu ve 20 % případů. Jestliže se u pacientů objeví nové typy záchvatů a/nebo se zvýšenou frekvencí dochází ke status epilepticus, který se liší od počátečního stavu pacienta, je nutné znovu přehodnotit poměr prospěšnosti a rizik léčby.
Vysazení rufinamidu
Rufinamid je nutné vysazovat postupně, aby se snížila možnost výskytu záchvatů při vysazení léku. V klinických studiích bylo vysazení dosaženo snižováním dávky o přibližně 25 % vždy po dvou dnech.
O vysazení současně podávaných antiepileptických přípravků, jakmile bylo dosaženo kontroly nad záchvaty přidáním rufinamidu, nejsou dostatečné údaje.
Reakce centrálního nervového systému
Léčba rufinamidem byla spojena se závratí, spavostí, ataxií a poruchami chůze, což mohlo zvýšit výskyt náhodných pádů v této populaci (viz bod 4.8). Je třeba, aby pacienti a ošetřující osoby dbali zvýšené opatrnosti, dokud se neseznámí s možnými účinky tohoto léčivého přípravku.
Hypersenzitivní reakce
Ve spojení s léčbou rufinamidem se vyskytly závažné syndromy přecitlivělosti na antiepileptický léčivý přípravek včetně DRESS (Léková reakce s eozionfilií a systémovými příznaky) a Stevens-Johnosonova syndromu. Příznaky a symptomy tohoto onemocnění byly různorodé; avšak u pacientů se typicky vyskytovala, i když ne výlučně, horečka a vyrážka, ve spojení s postižením dalšího orgánového systému. Jiné přidružené manifestace zahrnovaly lymfadenopatii, abnormality testů jaterních funkcí a hematurii. Protože jsou projevy tohoto stavu různorodé, mohou se objevit jiné příznaky a známky postižení orgánových systémů, které v tomto textu nejsou uvedeny. Tento syndrom přecitlivělosti na antiepileptika se objevil v těsné časové souvislosti se zahájením léčby rufinamidem a u souboru pediatrických pacientů. Pokud na tuto reakci vznikne podezření, je nutné rufinamid vysadit a zahájit alternativní léčbu. Všichni pacienti, u kterých se objeví vyrážka při užívání rufinamidu, musí být pečlivě monitorováni.
Zkrácení QT
V podrobné QT studii způsobil rufinamid pokles QTc intervalu, který byl přímo úměrný jeho koncentraci. Ačkoliv mechanismus vzniku tohoto nálezu ani jeho význam s ohledem na bezpečnost přípravku nejsou známy, lékaři by na základě klinického posouzení měli zvážit, zda předepsat rufinamid pacientům, kterým hrozí další zkracování QTc intervalu (např. kongenitální syndrom krátkého QT nebo pacienti, kteří mají tento syndrom v rodinné anamnéze).
Ženy v plodném věku
Ženy v plodném věku musejí během léčby Inovelonem používat antikoncepci. Lékaři by se měli snažit zajistit, aby byla použita vhodná antikoncepce a podle klinické situace jednotlivých pacientek by měli zvážit, zda je vhodnější podávat perorální antikoncepci nebo dávky složek perorální antikoncepce (viz bod 4.5).
Laktóza
Inovelon obsahuje laktózu, proto by pacienti se vzácnými dědičnými poruchami, jako je intolerance galaktózy, Lappova deficience laktázy nebo malabsorpce glukózy a galaktózy, neměli tento léčivý přípravek užívat.
Sebevražedné myšlenky
Sebevražedné myšlenky a chování byly hlášeny u pacientů léčených antiepileptiky v několika indikacích. Metaanalýza randomizovaných placebem kontrolovaných studií antiepileptických léčivých přípravků také prokázala mírně zvýšené riziko sebevražedných myšlenek a sebevražedného chování. Mechanismus tohoto rizika není znám a dostupné údaje nevylučují možnost zvýšeného rizika u Inovelonu.
U pacientů je proto třeba sledovat známky sebevražedných myšlenek a sebevražedného chování a je třeba zvážit vhodnou léčbu. Pacienty (a ošetřovatele pacientů) je třeba poučit, aby v případě, že se objeví známky sebevražedných myšlenek nebo sebevražedného chování, vyhledali lékařskou pomoc.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Potenciál jiných léčivých přípravků k ovlivňování rufinamidu
Jiné antiepileptické přípravky
Koncentrace rufinamidu nepodléhají klinicky významným změnám při současném podávání se známými antiepileptickými léčivými přípravky indukujícími enzymy.
U pacientů léčených přípravkem Inovelon, u kterých bylo zahájeno podávání valproátu, může docházet k významnému zvýšení koncentrací rufinamidu v plazmě. Nejvýraznější zvýšení bylo pozorováno u pacientů s nízkou tělesnou hmotností (< 30 kg). Proto je třeba zvážit snížení dávky Inovelonu u pacientů < 30 kg, u kterých je zahájena léčba valproátem (viz bod 4.2).
Přidání nebo vysazení těchto léčivých přípravků nebo upravení dávky těchto léčivých přípravků během léčby rufinamidem může vyžadovat úpravu dávkování rufinamidu.
Po současném podávání lamotriginu, topiramátu nebo benzodiazepinů nebyly pozorovány žádné významné změny koncentrace rufinamidu.
Potenciál rufinamidu k ovlivňování jiných léčivých přípravků
Jiné antiepileptické léčivé přípravky
Farmakokinetické interakce mezi rufinamidem a jinými antiepileptickými léčivými přípravky byly u pacientů s epilepsií hodnoceny pomocí populačního modelování farmakokinetiky. Zdá, se že rufinamid nemá žádný klinicky relevantní účinek na koncentrace karbamazepinu, lamotriginu, fenobarbitalu, topiramátu, fenytoinu nebo valproátu v ustáleném stavu
Perorální antikoncepce
Současné podávání rufinamidu 800 mg dvakrát denně a kombinované perorální antikoncepce (etinylestradiol 35 pg a noretindron 1 mg) po dobu 14 dní způsobilo průměrný pokles AUC0-24 etinylestradiolu o 22 % a AUC0-24 noretindronu o 14 %. Studie s jinými perorálními nebo implantovanými kontraceptivy nebyly prováděny. Ženám plodného věku, které užívají hormonální antikoncepci, se doporučuje používat další bezpečnou a účinnou antikoncepční metodu (viz body 4.4 a 4.6).
Cytochromové enzymy P450
Rufinamid je metabolizovaný hydrolýzou a cytochromovými enzymy P450 není významně metabolizován. Kromě toho rufinamid neinhibuje aktivitu cytochromových enzymů P450 (viz bod 5.2). Je tedy nepravděpodobné, aby nastaly klinicky významné interakce zprostředkované inhibicí systému cytochromů P450 rufinamidem. Bylo prokázáno, že rufinamid indukuje enzym CYP3A4 cytochromu P450 a může tudíž snížit plazmatické koncentrace látek, které jsou metabolizovány tímto enzymem. Účinek byl mírný až středně závažný. Průměrná aktivita CYP3A4, hodnocená jako clearance triazolamu, byla zvýšena o 55 % po 11 dnech léčby rufinamidem 400 mg dvakrát denně. Expozice triazolamu byla snížena o 36 %. Vyšší dávky rufinamidu mohou mít za následek výraznější indukci. Nelze vyloučit, že rufinamid může také snižovat expozici látek metabolizovaných jinými enzymy nebo dopravovaných transportními proteiny, jako je P-glykoprotein.
Doporučuje se, aby pacienti léčení látkami metabolizovanými systémem enzymu CYP3A4, byli pečlivě monitorováni po dobu dvou týdnů, a to buď na počátku nebo po skončení léčby rufinamidem, nebo po jakékoliv významné změně dávkování. Možná bude zapotřebí zvážit úpravu dávkování současně podávaného léčivého přípravku. Tato doporučení je třeba vzít v úvahu také, pokud se rufinamid používá společně s látkami s úzkým terapeutickým oknem, jako je warfarin nebo digoxin.
Studie specifické interakce u zdravých subjektů neodhalila žádný vliv rufinamidu v dávce 400 mg dvakrát denně na farmakokinetiku olanzapinu, substrátu CYP1A2.
15
Nejsou k dispozici žádné údaje o interakci rufinamidu s alkoholem.
4.6 Fertilita, těhotenství a kojení
Riziko související s epilepsií a podáváním antiepileptik všeobecně:
Ukazuje se, že v potomstvu žen s epilepsií je prevalence malformací dvakrát až třikrát větší než je četnost v celkové populaci, která je přibližně 3 %. U léčené populace byl nárůst malformací pozorován u polyterapie, avšak rozsah, za který je léčba a/nebo nemoc odpovědná, nebyl určen.
Navíc účinná antiepileptická léčba nesmí být přerušena, protože zhoršení nemoci je škodlivé jak pro matku tak pro plod.
Riziko související s rufinamidem:
Studie na zvířatech neprokázaly teratogenní účinek kromě fetotoxicity v přítomnosti maternální toxicity (viz bod 5.3). Potenciální riziko pro lidské jedince není známé.
Nejsou k dispozici klinické údaje o podávání rufinamidu během těhotenství.
Vezmeme-li v úvahu tyto údaje, rufinamid by neměl být podáván, pokud to není nezbytně nutné, během těhotenství ani u žen plodného věku, které nepoužívají kontracepční opatření.
Ženy ve fertilním věku musí během léčby rufinamidem používat antikoncepci. Lékaři by se měli snažit zajistit, aby byla použita vhodná antikoncepce a podle klinické situace jednotlivých pacientek by měli zvážit, zda je vhodnější podávat perorální antikoncepci nebo dávky složek perorální antikoncepce (viz bod 4.5).
Pokud ženy léčené rufinamidem plánují otěhotnět, je nutné pečlivě zvážit indikaci tohoto přípravku.
V průběhu těhotenství nesmí být přerušena účinná antiepileptická léčba rufinamidem, protože zhoršení nemoci je škodlivé jak pro matku, tak pro plod.
Kojení
Není známo, zda se rufinamid vylučuje do lidského mateřského mléka. Vzhledem k možným škodlivým účinkům pro kojené dítě by neměly matky při léčbě rufinamidem kojit.
Fertilita
Nejsou dostupné žádné údaje o účinku léčby rufinamidem na fertilitu.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Inovelon může způsobit závratě, spavost a rozmazané vidění. V závislosti na individuální citlivosti může mít rufinamid malý až výrazný vliv na schopnost řídit nebo obsluhovat stroje. Pacienty je třeba upozornit, aby během činností vyžadujících vysoký stupeň bdělosti, např. při řízení nebo obsluhování strojů, dbali zvýšené opatrnosti.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Klinický vývojový program zahrnoval více než 1 900 pacientů s různými typy epilepsie, kterým byl podáván rufinamid. Nejčastěji hlášené nežádoucí účinky celkově byly bolesti hlavy, závratě, únava a spavost. Nej častější nežádoucí účinky pozorované u pacientů s Lennox-Gastautovým syndromem ve vyšší incidenci než při podávání placeba byly spavost a zvracení. Nežádoucí účinky byly obvykle mírné až středně těžké. Četnost vystoupení pacientů s Lennox-Gastautovým syndromem ze studie z důvodu nežádoucích účinků byla 8,2 % pro pacienty užívající rufinamid a 0 % pro pacienty užívající placebo. Nejčastější nežádoucí účinky, které vedly k vystoupení ze studie, byly ve skupině léčené rufinamidem vyrážka a zvracení.
Tabulka nežádoucích účinků
Nežádoucí účinky hlášené s vyšší incidencí než u placeba během dvojitě zaslepených studií u pacientů s Lennox-Gastautovým syndromem nebo u celkového souboru pacientů, kterým byl podáván rufinamid, jsou uvedeny v následující tabulce podle terminologie MedDRA, třídy orgánových systémů a podle frekvence.
Frekvence jsou definovány jako: velmi časté (> 1/10), časté (> 1/100 < 1/10), méně časté (> 1/1 000 < 1/100), vzácné (> 1/10 000 až < 1/1 000).
|
Třída orgánových systémů |
Velmi časté |
Časté |
Méně časté |
Vzácné |
|
Infekce a infestace |
Chřipka Nazofaryngitida Ušní infekce Sinusitida Rinitida | |||
|
Poruchy imunitního systému |
Hypersensitivita* | |||
|
Poruchy metabolismu a výživy |
Poruchy příjmu potravy Snížená chuť k jídlu | |||
|
Psychiatrické poruchy |
Úzkost Insomnie | |||
|
Poruchy nervového systému |
Spavost* Bolesti hlavy Závratě* |
Status epilepticus* Křeče Abnormální koordinace* Nystagmus Psychomotorická hyperaktivita | ||
|
Poruchy oka |
Diplopie Rozmazané vidění | |||
|
Poruchy ucha a labyrintu | ||||
|
Respirační, hrudní a mediastinální poruchy |
Epistaxe | |||
|
Gastrointestinální poruchy |
Bolesti horní části břicha Zácpa | |||
|
Poruchy jater a žlučových cest |
Zvýšení jaterních enzymů | |||
|
Poruchy kůže a podkožní tkáně |
Vyrážka* Akné | |||
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Bolesti zad |
|
Třída orgánových systémů |
Velmi časté |
Časté |
Méně časté |
Vzácné |
|
Poruchy reprodukčního systému a prsu |
Oligomenorea | |||
|
Celkové poruchy a reakce v místě aplikace |
Únava |
Poruchy chůze* | ||
|
Vyšetření e |
Pokles hmotnosti | |||
|
Poranění, otravy a procedurálních komplikací |
Poranění hlavy Kontuze |
* Odkaz na bod 4.4
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Po akutním předávkování může být žaludek vyprázdněn gastrickou laváží nebo vyvoláním zvracení. Pro rufinamid neexistuje žádné specifické antidotum. Léčba by měla být podpůrná a může zahrnovat hemodialýzu (viz bod 5.2).
Podávání opakovaných dávek 7 200 mg/den nebylo spojeno s žádnými závažnějšími příznaky nebo symptomy.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antiepileptika, deriváty karboxamidu; ATC kód: N03AF03. Mechanismus účinku
Rufinamid moduluje aktivitu sodíkových kanálů tak, že prodlužuje jejich inaktivní stav. Rufinamid je účinný u celé řady zvířecích modelů epilepsie.
Klinické zkušenosti
Inovelon (tablety obsahující rufinamid) byl podáván ve dvojitě zaslepené, placebem kontrolované studii v dávkách až 45 mg/kg/den po dobu 84 dnů 139 pacientům s nedostatečně kontrolovanými záchvaty spojenými s Lennox-Gastautovým syndromem (včetně atypických záchvatů typu absence a záchvatových pádů). Muži i ženy (ve věku od 4 do 30 let) byli zařazeni do studie, pokud užívali 1 až 3 současně podávané antiepileptické léčivé přípravky s pevně stanoveným dávkováním. Každý pacient musel mít alespoň 90 záchvatů během měsíce před přijetím do studie. U všech tří primárních proměnných bylo pozorováno signifikantní zlepšení: procentní změna celkové frekvence záchvatů za 28 dnů během udržovací fáze v porovnání s výchozí hodnotou (-35,8 % při podávání přípravku Inovelon oproti -1,6 % při podávání placeba, p = 0,0006), počet tonických/atonických záchvatů (-42,9 % při podávání přípravku Inovelon oproti 2,2 % při podávání placeba, p = 0,0002) a ohodnocení závažnosti záchvatu při celkovém vyhodnocení prováděném rodičem/opatrovníkem na konci dvojitě zaslepené fáze (velké nebo velmi velké zlepšení ve 32,2 % při podávání přípravku Inovelon oproti 14,5 % v rameni s placebem, p = 0,0041).
Populační modelování farmakokinetiky/farmakodynamiky ukázalo, že snížení celkové záchvatové frekvence a frekvence tonických/atonických záchvatů, zlepšení celkového vyhodnocení závažnosti záchvatů a zvýšení pravděpodobnosti snížení záchvatové frekvence bylo závislé na koncentracích rufinamidu.
5.2 Farmakokinetické vlastnosti
Absorpce
Maximální plazmatické hladiny jsou dosaženy přibližně 6 hodin po podání. Vrcholová koncentrace (Cmax) a plazmatická AUC rufinamidu se zvyšují méně než proporcionálně s dávkou u zdravých subjektů a pacientů nalačno i po jídle, což je pravděpodobně způsobeno omezenou absorpcí dávky. Po jednotlivých dávkách zvyšuje potrava biologickou dostupnost (AUC) rufinamidu přibližně o 34 % a maximální koncentraci v plazmě o 56 %.
Bylo prokázáno, že přípravky Inovelon perorální suspenze a Inovelon potahované tablety jsou biologicky ekvivalentní.
Distribuce
Ve studiích in vitro byla pouze malá frakce rufinamidu (34 %) navázána na lidské sérové proteiny, přičemž albumin odpovídal přibližně za 80 % této vazby. To ukazuje, že riziko lékových interakcí v důsledku nahrazení vazebného místa během současného podávání dalších látek je minimální. Rufinamid byl rovnoměrně rozdělen mezi erytrocyty a plazmu.
Biotransformace
Rufinamid je téměř výhradně eliminován metabolismem. Hlavní dráha metabolismu je hydrolýza karboxylamidové skupiny na farmakologicky inaktivní derivát kyseliny CGP 47292. Metabolismus zprostředkovaný cytochromem P450 se podílí v mnohem menší míře.
Nelze zcela vyloučit vznik malých množství konjugátů glutathionu.
In vitro rufinamid vykazoval malou nebo žádnou schopnost působit jako kompetitivní nebo nekompetitivní inhibitor následujících lidských enzymů P450: CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5 nebo CYP4A9/11-2.
Eliminace Plazmatický eliminační poločas je přibližně 6-10 hodin u zdravých subjektů a pacientů s epilepsií. Rufinamid se při podávání dvakrát denně ve 12 hodinových intervalech akumuluje v rozsahu, který lze předpovídat podle jeho terminálního poločasu, což ukazuje, že farmakokinetika rufinamidu je časově nezávislá (tj. bez autoindukce metabolismu).
Ve studiích s radioaktivně značenou látkou u tří zdravých dobrovolníků byla základní sloučenina (rufinamid) hlavní radioaktivní složkou v plazmě a představovala přibližně 80 % celkové radioaktivity, metabolit CGP 47292 tvořil pouze asi 15 %. Převládající cestou eliminace látek souvisejících s léčivou látkou bylo vylučování ledvinami, které odpovídalo za 84,7 % dávky.
Linearita/nelinearita:
Biologická dostupnost rufinamidu je závislá na dávce. Se zvyšováním dávky se snižuje biologická dostupnost.
Farmakokinetika u zvláštních skupin pacientů
Pohlaví
Pro vyhodnocení vlivu pohlaví na farmakokinetiku rufinamidu bylo použito populační modelování farmakokinetiky. Tato hodnocení ukazují, že pohlaví neovlivňuje farmakokinetiku rufinamidu do klinicky významné míry.
Porucha funkce ledvin
Farmakokinetika jedné dávky 400 mg rufinamidu nebyla u pacientů s chronickým a těžkým selháním ledvin změněna, ve srovnání se zdravými dobrovolníky. Ale když byla po podání rufinamidu
provedena hemodialýza, byly plazmatické hladiny sníženy přibližně o 30 %, což svědčí pro to, že by to mohl být postup použitelný v případě předávkování (viz body 4.2 a 4.9).
Porucha funkce jater
U pacientů s poruchou funkce jater nebyly prováděny žádné studie, a proto by Inovelon neměl být pacientům se těžkým poškozením jater podáván (viz bod 4.2).
Děti (2-12 let)
Děti mají obvykle nižší clearance rufinamidu než dospělé osoby a tento rozdíl souvisí s velikostí těla. Studie u novorozených dětí nebo kojenců a batolat mladších 2 let nebyly prováděny.
Starší osoby
Farmakokinetická studie u starších zdravých dobrovolníků neukázala významný rozdíl ve farmakokinetických parametrech ve srovnání s mladšími dospělými osobami.
5.3 Předklinické údaje vztahující se k bezpečnosti
Konvenční farmakologické studie bezpečnosti v klinicky relevantních dávkách neodhalily žádná zvláštní rizika.
Toxické účinky pozorované u psů v hladinách podobných expozici u člověka při maximální doporučené dávce byly změny na játrech, včetně žlučových trombů, cholestázy a zvýšení hodnot jaterních enzymů, o kterých se předpokládá, že souvisejí se zvýšenou sekrecí žluče u tohoto živočišného druhu. Ve studiích toxicity po opakovaném podávání u laboratorních potkanů a opic nebyl zjištěn žádný důkaz svědčící pro přidružené riziko.
Ve studiích reprodukční a vývojové toxicity se vyskytlo zpomalení růstu plodu a snížení přežití a několik mrtvě narozených plodů v důsledku maternální toxicity. Ale u potomstva nebyly pozorovány žádné účinky na morfologii a funkce, včetně učení nebo paměti. Rufinamid nebyl teratogenní u myší, laboratorních potkanů či králíků.
Rufinamid nebyl genotoxický a neměl žádný kancerogenní potenciál. Nežádoucím účinkem, který nebyl pozorován během klinických studií, ale byl zaznamenán u zvířat při expozici podobné hodnotám klinické expozice, a který by mohl být relevantní z hlediska použití u lidských jedinců, byla ve studii kancerogenity u myší myelofibróza kostní dřeně. Výskyt nezhoubných kostních novotvarů (osteomů) a hyperostóza zaznamenané u myší byly považovány za výsledek aktivace viru specifického pro myši ionty flóru uvolněnými během oxidačního metabolismu rufinamidu.
Pokud jde o imunotoxický potenciál, byl u psů během třináctitýdenní studie pozorován malý brzlík a involuce brzlíku s výraznou reakcí při vysokých dávkách u samců. Změny kostní dřeně a lymfatické změny u samic byly při podávání vysokých dávek během třináctitýdenní studie hlášeny s malou incidencí. U potkanů byla snížená buněčnost kostní dřeně a atrofie brzlíku pozorována pouze u studie kancerogenity.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Jádro:
Monohydrát laktosy Mikrokrystalická celulosa Kukuřičný škrob Sodná sůl kroskarmelosy Hypromelosa Magnesium-stearát
Natrium-lauryl-sulfát Koloidní bezvodý oxid křemičitý
Potah tablety:
Hypromelosa Makrogoly (8000)
Oxid titaničitý (E171)
Mastek
Červený oxid železitý (E172)
6.2 Inkompatibility Neuplatňuje se.
6.3 Doba použitelnosti
4 roky.
6.4 Zvláštní opatření pro uchovávání
Neuchovávejte při teplotě nad 30 °C.
6.5 Druh obalu a obsah balení
Blistry z hliníku/hliníku (Al/Al blistry), balení po 10, 30, 50, 60 a 100 potahovaných tabletách. Na trhu nemusí být všechny velikosti balení
6.6 Zvláštní opatření pro likvidaci přípravku
Žádné zvláštní požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Eisai Limited, European Knowledge Centre, Mosquito Way, Hatfield, Hertfordshire, AL10 9SN, Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/06/378/006-010
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 16. ledna 2007
Datum posledního prodloužení registrace: 16. ledna 2012
10. DATUM REVIZE TEXTU
{MM/RRRR}
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu.
NÁZEV PŘÍPRAVKU
1.
Inovelon 400 mg potahované tablety
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna potahovaná tableta obsahuje rufinamidum 400 mg.
Pomocná látka se známým účinkem: jedna potahovaná tableta obsahuje 80 mg monohydrátu laktosy Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Potahovaná tableta.
Růžová, „oválná“, mírně konvexní, na obou stranách s půlicí rýhou, na jedné straně s vyraženým označením „C263“ a na druhé straně bez označení.
Tabletu lze rozdělit na dvě stejné dávky.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Inovelon je indikován jako přídatná léčba při léčení záchvatů spojených s Lennox-Gastautovým syndromem u pacientů ve věku 4 let a starších.
4.2 Dávkování a způsob podání
Léčbu rufinamidem by měl předepsat lékař se specializací v oboru pediatrie nebo neurologie se zkušenostmi s léčbou epilepsie.
Inovelon perorální suspenze a Inovelon potahované tablety lze při stejných dávkách zaměnit. Pacienty je třeba v období přechodu z jedné lékové formy na druhou sledovat.
Dávkování
Podávání dětem ve věku čtyř let nebo starším o hmotnosti nižší než 30 kg Pacienti < 30 kg neužívající valproát sodný:
Léčba by měla být zahájena denní dávkou 200 mg. Podle klinické odpovědi a snášenlivosti může být ve dvoudenních intervalech denní dávka zvyšována o 200 mg, až do maximální doporučené dávky 1 000 mg/den. Dávky až 3 600 mg/den byly studovány u omezeného počtu pacientů.
Pacienti < 30 kg užívající současně valproát sodný:
Vzhledem k tomu, že valproát výrazně snižuje clearance rufinamidu, doporučuje se u pacientů < 30 kg, kteří současně užívají valproát, nižší maximální dávka Inovelonu. Léčba by měla být zahájena denní dávkou 200 mg. Podle klinické odpovědi a snášenlivosti může být minimálně po dvou dnech denní dávka zvýšena o 200 mg až do maximální doporučené dávky 600 mg/den.
Podávání dospělým, dospívajícím a dětem ve věku čtyř let nebo starším o hmotnosti 30 kg a vyšší Léčba by měla být zahájena denní dávkou 400 mg. Podle klinické odpovědi a snášenlivosti může být ve dvoudenních intervalech denní dávka zvyšována o 400 mg, až do maximální doporučené dávky, jak je uvedena v následující tabulce.
|
Rozmezí hmotnosti |
30,0 - 50,0 kg |
50,1 - 70,0 kg |
> 70,1 kg |
|
Maximální doporučená dávka |
1 800 mg/den |
2 400 mg/den |
3 200 mg/den |
Dávky až 4 000 mg/den (u hmotnosti 30-50 kg) nebo 4 800 mg/den (v kategorii nad 50 kg) byly studovány u omezeného počtu pacientů.
Vysazení rufinamidu
Má-li být léčba rufinamidem ukončena, je nutné ji vysazovat postupně. V klinických studiích bylo vysazení rufinamidu dosaženo snižováním dávky o přibližně 25 % vždy po dvou dnech.
V případě jedné či více vynechaných dávek je nutné individuální posouzení klinického stavu.
Nekontrolované otevřené studie naznačují, že účinnost je dlouhodobá, přestože žádná kontrolovaná studie nebyla prováděna déle než tři měsíce.
Pediatrická populace
Bezpečnost a účinnost rufinamidu u dětí ve věku do 4 let nebyla dosud stanovena. Nejsou dostupné žádné údaje.
Starší osoby
Informace o podávání rufinamidu starším osobám jsou omezené. Protože farmakokinetika rufinamidu není u starších osob změněna (viz bod 5.2), není nutná úprava dávkování u pacientů ve věku nad 65 let.
Porucha funkce ledvin
Studie pacientů se závažnou poruchou funkce ledvin ukázala, že u těchto pacientů není nutná žádná úprava dávkování (viz bod 5.2).
Porucha funkce jater
Podávání léku u pacientů s poruchou funkce jater nebylo studováno. Při léčbě pacientů s mírnou až středně závažnou poruchou funkce jater se doporučuje opatrnost a pečlivé titrování dávky. Podávání léku pacientům s těžkou poruchou funkce jater se nedoporučuje.
Způsob podání
Rufinamid je určen k perorálnímu podání. Užívá se s vodou dvakrát denně, ráno a večer, rozdělený do dvou stejných dávek Protože byl pozorován vliv jídla, měl by být Inovelon užíván s jídlem (viz bod 5.2). Má-li pacient potíže s polykáním, je možné tablety rozdrtit a podat s menším množstvím vody (půl sklenice).
4.3 Kontraindikace
Hypersenzitivita na léčivou látku, deriváty triazolu nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Status epilepticus
Při podávání rufinamidu během studií klinického vývoje byly zaznamenány případy status epilepticus, zatímco u pacientů, kteří užívali placebo, nebyly žádné takové případy zaznamenány. Tyto příhody vedly k vysazení rufinamidu ve 20 % případů. Jestliže se u pacientů objeví nové typy záchvatů a/nebo se zvýšenou frekvencí dochází ke status epilepticus, který se liší od počátečního stavu pacienta, je nutné znovu přehodnotit poměr prospěšnosti a rizik léčby.
Vysazení rufinamidu
Rufinamid je nutné vysazovat postupně, aby se snížila možnost výskytu záchvatů při vysazení léku. V klinických studiích bylo vysazení dosaženo snižováním dávky o přibližně 25 % vždy po dvou dnech.
O vysazení současně podávaných antiepileptických přípravků, jakmile bylo dosaženo kontroly nad záchvaty přidáním rufinamidu, nejsou dostatečné údaje.
Reakce centrálního nervového systému
Léčba rufinamidem byla spojena se závratí, spavostí, ataxií a poruchami chůze, což mohlo zvýšit výskyt náhodných pádů v této populaci (viz bod 4.8). Je třeba, aby pacienti a ošetřující osoby dbali zvýšené opatrnosti, dokud se neseznámí s možnými účinky tohoto léčivého přípravku.
Hypersenzitivní reakce
Ve spojení s léčbou rufinamidem se vyskytly závažné syndromy přecitlivělosti na antiepileptický léčivý přípravek včetně DRESS (Léková reakce s eozionfilií a systémovými příznaky) a Stevens-Johnosonova syndromu. Příznaky a symptomy tohoto onemocnění byly různorodé; avšak u pacientů se typicky vyskytovala, i když ne výlučně, horečka a vyrážka, ve spojení s postižením dalšího orgánového systému. Jiné přidružené manifestace zahrnovaly lymfadenopatii, abnormality testů jaterních funkcí a hematurii. Protože jsou projevy tohoto stavu různorodé, mohou se objevit jiné příznaky a známky postižení orgánových systémů, které v tomto textu nejsou uvedeny. Tento syndrom přecitlivělosti na antiepileptika se objevil v těsné časové souvislosti se zahájením léčby rufinamidem a u souboru pediatrických pacientů. Pokud na tuto reakci vznikne podezření, je nutné rufinamid vysadit a zahájit alternativní léčbu. Všichni pacienti, u kterých se objeví vyrážka při užívání rufinamidu, musí být pečlivě monitorováni.
Zkrácení QT
V podrobné QT studii způsobil rufinamid pokles QTc intervalu, který byl přímo úměrný jeho koncentraci.
Ačkoliv mechanismus vzniku tohoto nálezu ani jeho význam s ohledem na bezpečnost přípravku nejsou známy, lékaři by na základě klinického posouzení měli zvážit, zda předepsat rufinamid pacientům, kterým hrozí další zkracování QTc intervalu (např. kongenitální syndrom krátkého QT nebo pacienti, kteří mají tento syndrom v rodinné anamnéze).
Ženy v plodném věku
Ženy v plodném věku musejí během léčby Inovelonem používat antikoncepci. Lékaři by se měli snažit zajistit, aby byla použita vhodná antikoncepce a podle klinické situace jednotlivých pacientek by měli zvážit, zda je vhodnější podávat perorální antikoncepci nebo dávky složek perorální antikoncepce (viz bod 4.5).
Laktóza
Inovelon obsahuje laktózu, proto by pacienti se vzácnými dědičnými poruchami, jako je intolerance galaktózy, Lappova deficience laktázy nebo malabsorpce glukózy a galaktózy, neměli tento léčivý přípravek užívat.
Sebevražedné myšlenky
Sebevražedné myšlenky a chování byly hlášeny u pacientů léčených antiepileptiky v několika indikacích. Metaanalýza randomizovaných placebem kontrolovaných studií antiepileptických léčivých přípravků také prokázala mírně zvýšené riziko sebevražedných myšlenek a sebevražedného chování. Mechanismus tohoto rizika není znám a dostupné údaje nevylučují možnost zvýšeného rizika u Inovelonu.
U pacientů je proto třeba sledovat známky sebevražedných myšlenek a sebevražedného chování a je třeba zvážit vhodnou léčbu. Pacienty (a ošetřovatele pacientů) je třeba poučit, aby v případě, že se objeví známky sebevražedných myšlenek nebo sebevražedného chování, vyhledali lékařskou pomoc.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Potenciál jiných léčivých přípravků k ovlivňování rufinamidu
Jiné antiepileptické přípravky
Koncentrace rufinamidu nepodléhají klinicky významným změnám při současném podávání se známými antiepileptickými léčivými přípravky indukujícími enzymy.
U pacientů léčených přípravkem Inovelon, u kterých bylo zahájeno podávání valproátu, může docházet k významnému zvýšení koncentrací rufinamidu v plazmě. Nejvýraznější zvýšení bylo pozorováno u pacientů s nízkou tělesnou hmotností (< 30 kg). Proto je třeba zvážit snížení dávky Inovelonu u pacientů < 30 kg, u kterých je zahájena léčba valproátem (viz bod 4.2).
Přidání nebo vysazení těchto léčivých přípravků nebo upravení dávky těchto léčivých přípravků během léčby rufinamidem může vyžadovat úpravu dávkování rufinamidu.
Po současném podávání lamotriginu, topiramátu nebo benzodiazepinů nebyly pozorovány žádné významné změny koncentrace rufinamidu.
Potenciál rufinamidu k ovlivňování jiných léčivých přípravků
Jiné antiepileptické léčivé přípravky
Farmakokinetické interakce mezi rufinamidem a jinými antiepileptickými léčivými přípravky byly u pacientů s epilepsií hodnoceny pomocí populačního modelování farmakokinetiky. Zdá, se že rufinamid nemá žádný klinicky relevantní účinek na koncentrace karbamazepinu, lamotriginu, fenobarbitalu, topiramátu, phenytoinu nebo valproátu v ustáleném stavu.
Perorální antikoncepce
Současné podávání rufinamidu 800 mg dvakrát denně a kombinované perorální antikoncepce (etinylestradiol 35 pg a noretindron 1 mg) po dobu 14 dní způsobilo průměrný pokles AUC0-24 etinylestradiolu o 22 % a AUC0-24 noretindronu o 14 %. Studie s jinými perorálními nebo implantovanými kontraceptivy nebyly prováděny. Ženám plodného věku, které užívají hormonální antikoncepci, se doporučuje používat další bezpečnou a účinnou antikoncepční metodu (viz body 4.4 a 4.6).
Cytochromové enzymy P450
Rufinamid je metabolizovaný hydrolýzou a cytochromovými enzymy P450 není významně metabolizován. Kromě toho rufinamid neinhibuje aktivitu cytochromových enzymů P450 (viz bod 5.2). Je tedy nepravděpodobné, aby nastaly klinicky významné interakce zprostředkované inhibicí systému cytochromů P450 rufinamidem. Bylo prokázáno, že rufinamid indukuje enzym CYP3A4 cytochromu P450 a může tudíž snížit plazmatické koncentrace látek, které jsou metabolizovány tímto enzymem. Účinek byl mírný až středně závažný. Průměrná aktivita CYP3A4, hodnocená jako clearance triazolamu, byla zvýšena o 55 % po 11 dnech léčby rufinamidem 400 mg dvakrát denně. Expozice triazolamu byla snížena o 36 %. Vyšší dávky rufinamidu mohou mít za následek výraznější indukci. Nelze vyloučit, že rufinamid může také snižovat expozici látek metabolizovaných jinými enzymy nebo dopravovaných transportními proteiny, jako je P-glykoprotein.
Doporučuje se, aby pacienti léčení látkami metabolizovanými systémem enzymu CYP3A4, byli pečlivě monitorováni po dobu dvou týdnů, a to buď na počátku nebo po skončení léčby rufinamidem, nebo po jakékoliv významné změně dávkování. Možná bude zapotřebí zvážit úpravu dávkování současně podávaného léčivého přípravku. Tato doporučení je třeba vzít v úvahu také, pokud se rufinamid používá společně s látkami s úzkým terapeutickým oknem, jako je warfarin nebo digoxin.
Studie specifické interakce u zdravých subjektů neodhalila žádný vliv rufinamidu v dávce 400 mg dvakrát denně na farmakokinetiku olanzapinu, substrátu CYP1A2.
25
Nejsou k dispozici žádné údaje o interakci rufinamidu s alkoholem.
4.6 Fertilita, těhotenství a kojení
Riziko související s epilepsií a podáváním antiepileptik všeobecně:
Ukazuje se, že v potomstvu žen s epilepsií je prevalence malformací dvakrát až třikrát větší než je četnost v celkové populaci, která je přibližně 3 %. U léčené populace byl nárůst malformací pozorován u polyterapie, avšak rozsah, za který je léčba a/nebo nemoc odpovědná, nebyl určen.
Navíc účinná antiepileptická léčba nesmí být přerušena, protože zhoršení nemoci je škodlivé jak pro matku tak pro plod.
Riziko související s rufinamidem:
Studie na zvířatech neprokázaly teratogenní účinek kromě fetotoxicity v přítomnosti maternální toxicity (viz bod 5.3). Potenciální riziko pro lidské jedince není známé.
Nejsou k dispozici klinické údaje o podávání rufinamidu během těhotenství.
Vezmeme-li v úvahu tyto údaje, rufinamid by neměl být podáván, pokud to není nezbytně nutné, během těhotenství ani u žen plodného věku, které nepoužívají kontracepční opatření.
Ženy ve fertilním věku musí během léčby rufinamidem používat antikoncepci. Lékaři by se měli snažit zajistit, aby byla použita vhodná antikoncepce a podle klinické situace jednotlivých pacientek by měli zvážit, zda je vhodnější podávat perorální antikoncepci nebo dávky složek perorální antikoncepce (viz bod 4.5).
Pokud ženy léčené rufinamidem plánují otěhotnět, je nutné pečlivě zvážit indikaci tohoto přípravku.
V průběhu těhotenství nesmí být přerušena účinná antiepileptická léčba rufinamidem, protože zhoršení nemoci je škodlivé jak pro matku, tak pro plod.
Kojení
Není známo, zda se rufinamid vylučuje do lidského mateřského mléka. Vzhledem k možným škodlivým účinkům pro kojené dítě by neměly matky při léčbě rufinamidem kojit.
Fertilita
Nejsou dostupné žádné údaje o účinku léčby rufinamidem na fertilitu.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Inovelon může způsobit závratě, spavost a rozmazané vidění. V závislosti na individuální citlivosti může mít rufinamid malý až výrazný vliv na schopnost řídit nebo obsluhovat stroje. Pacienty je třeba upozornit, aby během činností vyžadujících vysoký stupeň bdělosti, např. při řízení nebo obsluhování strojů, dbali zvýšené opatrnosti.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Klinický vývojový program zahrnoval více než 1 900 pacientů s různými typy epilepsie, kterým byl podáván rufinamid. Nejčastěji hlášené nežádoucí účinky celkově byly bolesti hlavy, závratě, únava a spavost. Nej častější nežádoucí účinky pozorované u pacientů s Lennox-Gastautovým syndromem ve vyšší incidenci než při podávání placeba byly spavost a zvracení. Nežádoucí účinky byly obvykle mírné až středně těžké. Četnost vystoupení pacientů s Lennox-Gastautovým syndromem ze studie z důvodu nežádoucích účinků byla 8,2 % pro pacienty užívající rufinamid a 0 % pro pacienty užívající placebo. Nejčastější nežádoucí účinky, které vedly k vystoupení ze studie, byly ve skupině léčené rufinamidem vyrážka a zvracení.
Tabulka nežádoucích účinků
Nežádoucí účinky hlášené s vyšší incidencí než u placeba během dvojitě zaslepených studií u pacientů s Lennox-Gastautovým syndromem nebo u celkového souboru pacientů, kterým byl podáván rufinamid, jsou uvedeny v následující tabulce podle terminologie MedDRA, třídy orgánových systémů a podle frekvence.
Frekvence jsou definovány jako: velmi časté (> 1/10), časté (> 1/100 < 1/10), méně časté (> 1/1 000 < 1/100), vzácné (> 1/10 000 až < 1/1 000).
|
Třída orgánových systémů |
Velmi časté |
Časté |
Méně časté |
Vzácné |
|
Infekce a infestace |
Chřipka Nazofaryngitida Ušní infekce Sinusitida Rinitida | |||
|
Poruchy imunitního systému |
Hypersensitivita* | |||
|
Poruchy metabolismu a výživy |
Poruchy příjmu potravy Snížená chuť k jídlu | |||
|
Psychiatrické poruchy |
Úzkost Insomnie | |||
|
Poruchy nervového systému |
Spavost* Bolesti hlavy Závratě* |
Status epilepticus* Křeče Abnormální koordinace* Nystagmus Psychomotorická hyperaktivita | ||
|
Poruchy oka |
Diplopie Rozmazané vidění | |||
|
Poruchy ucha a labyrintu | ||||
|
Respirační, hrudní a mediastinální poruchy |
Epistaxe | |||
|
Gastrointestinální poruchy |
Bolesti horní části břicha Zácpa | |||
|
Poruchy jater a žlučových cest |
Zvýšení jaterních enzymů | |||
|
Poruchy kůže a podkožní tkáně |
Vyrážka* Akné | |||
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Bolesti zad |
|
Třída orgánových systémů |
Velmi časté |
Časté |
Méně časté |
Vzácné |
|
Poruchy reprodukčního systému a prsu |
Oligomenorea | |||
|
Celkové poruchy a reakce v místě aplikace |
Únava |
Poruchy chůze* | ||
|
Vyšetření |
Pokles hmotnosti | |||
|
Poranění, otravy a procedurální komplikace |
Poranění hlavy Kontuze |
* Odkaz na bod 4.4
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Po akutním předávkování může být žaludek vyprázdněn gastrickou laváží nebo vyvoláním zvracení. Pro rufinamid neexistuje žádné specifické antidotum. Léčba by měla být podpůrná a může zahrnovat hemodialýzu (viz bod 5.2).
Podávání opakovaných dávek 7 200 mg/den nebylo spojeno s žádnými závažnějšími příznaky nebo symptomy.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antiepileptika, deriváty karboxamidu; ATC kód: N03AF03. Mechanismus účinku
Rufinamid moduluje aktivitu sodíkových kanálů tak, že prodlužuje jejich inaktivní stav. Rufinamid je účinný u celé řady zvířecích modelů epilepsie.
Klinické zkušenosti
Inovelon (tablety obsahující rufinamid) byl podáván ve dvojitě zaslepené, placebem kontrolované studii v dávkách až 45 mg/kg/den po dobu 84 dnů 139 pacientům s nedostatečně kontrolovanými záchvaty spojenými s Lennox-Gastautovým syndromem (včetně atypických záchvatů typu absence a záchvatových pádů). Muži i ženy (ve věku od 4 do 30 let) byli zařazeni do studie, pokud užívali 1 až 3 současně podávané antiepileptické léčivé přípravky s pevně stanoveným dávkováním. Každý pacient musel mít alespoň 90 záchvatů během měsíce před přijetím do studie. U všech tří primárních proměnných bylo pozorováno signifikantní zlepšení: procentní změna celkové frekvence záchvatů za 28 dnů během udržovací fáze v porovnání s výchozí hodnotou (-35,8 % při podávání přípravku Inovelon oproti -1,6 % při podávání placeba, p = 0,0006), počet tonických/atonických záchvatů (-42,9 % při podávání přípravku Inovelon oproti 2,2 % při podávání placeba, p = 0,0002) a ohodnocení závažnosti záchvatu při celkovém vyhodnocení prováděném rodičem/opatrovníkem na konci dvojitě zaslepené fáze (velké nebo velmi velké zlepšení ve 32,2 % při podávání přípravku Inovelon oproti 14,5 % v rameni s placebem, p = 0,0041).
Populační modelování farmakokinetiky/farmakodynamiky ukázalo, že snížení celkové záchvatové frekvence a frekvence tonických/atonických záchvatů, zlepšení celkového vyhodnocení závažnosti záchvatů a zvýšení pravděpodobnosti snížení záchvatové frekvence bylo závislé na koncentracích rufinamidu.
5.2 Farmakokinetické vlastnosti
Absorpce
Maximální plazmatické hladiny jsou dosaženy přibližně 6 hodin po podání. Vrcholová koncentrace (Cmax) a plazmatická AUC rufinamidu se zvyšují méně než proporcionálně s dávkou u zdravých subjektů a pacientů nalačno i po jídle, což je pravděpodobně způsobeno omezenou absorpcí dávky. Po jednotlivých dávkách zvyšuje potrava biologickou dostupnost (AUC) rufinamidu přibližně o 34 % a maximální koncentraci v plazmě o 56 %.
Bylo prokázáno, že přípravky Inovelon perorální suspenze a Inovelon potahované tablety jsou biologicky ekvivalentní.
Distribuce
Ve studiích in vitro byla pouze malá frakce rufinamidu (34 %) navázána na lidské sérové proteiny, přičemž albumin odpovídal přibližně za 80 % této vazby. To ukazuje, že riziko lékových interakcí v důsledku nahrazení vazebného místa během současného podávání dalších látek je minimální. Rufinamid byl rovnoměrně rozdělen mezi erytrocyty a plazmu.
Biotransformace
Rufinamid je téměř výhradně eliminován metabolismem. Hlavní dráha metabolismu je hydrolýza karboxylamidové skupiny na farmakologicky inaktivní derivát kyseliny CGP 47292. Metabolismus zprostředkovaný cytochromem P450 se podílí v mnohem menší míře.
Nelze zcela vyloučit vznik malých množství konjugátů glutathionu.
In vitro rufinamid vykazoval malou nebo žádnou schopnost působit jako kompetitivní nebo nekompetitivní inhibitor následujících lidských enzymů P450: CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5 nebo CYP4A9/11-2.
Eliminace
Plazmatický eliminační poločas je přibližně 6-10 hodin u zdravých subjektů a pacientů s epilepsií. Rufinamid se při podávání dvakrát denně ve 12 hodinových intervalech akumuluje v rozsahu, který lze předpovídat podle jeho terminálního poločasu, což ukazuje, že farmakokinetika rufinamidu je časově nezávislá (tj. bez autoindukce metabolismu).
Ve studiích s radioaktivně značenou látkou u tří zdravých dobrovolníků byla základní sloučenina (rufinamid) hlavní radioaktivní složkou v plazmě a představovala přibližně 80 % celkové radioaktivity, metabolit CGP 47292 tvořil pouze asi 15 %. Převládající cestou eliminace látek souvisejících s léčivou látkou bylo vylučování ledvinami, které odpovídalo za 84,7 % dávky.
Linearita/nelinearita:
Biologická dostupnost rufinamidu je závislá na dávce. Se zvyšováním dávky se snižuje biologická dostupnost.
Farmakokinetika u zvláštních skupin pacientů
Pohlaví
Pro vyhodnocení vlivu pohlaví na farmakokinetiku rufinamidu bylo použito populační modelování farmakokinetiky. Tato hodnocení ukazují, že pohlaví neovlivňuje farmakokinetiku rufinamidu do klinicky významné míry.
Porucha funkce ledvin
Farmakokinetika jedné dávky 400 mg rufinamidu nebyla u pacientů s chronickým a těžkým selháním ledvin změněna, ve srovnání se zdravými dobrovolníky. Ale když byla po podání rufinamidu provedena hemodialýza, byly plazmatické hladiny sníženy přibližně o 30 %, což svědčí pro to, že by to mohl být postup použitelný v případě předávkování (viz body 4.2 a 4.9).
Porucha funkce jater
U pacientů s poruchou funkce jater nebyly prováděny žádné studie, a proto by Inovelon neměl být pacientům se těžkým poškozením jater podáván (viz bod 4.2).
Děti (2-12 let)
Děti mají obvykle nižší clearance rufinamidu než dospělé osoby a tento rozdíl souvisí s velikostí těla. Studie u novorozených dětí nebo kojenců a batolat mladších 2 let nebyly prováděny.
Starší osoby
Farmakokinetická studie u starších zdravých dobrovolníků neukázala významný rozdíl ve farmakokinetických parametrech ve srovnání s mladšími dospělými osobami.
5.3 Předklinické údaje vztahující se k bezpečnosti
Konvenční farmakologické studie bezpečnosti v klinicky relevantních dávkách neodhalily žádná zvláštní rizika.
Toxické účinky pozorované u psů v hladinách podobných expozici u člověka při maximální doporučené dávce byly změny na játrech, včetně žlučových trombů, cholestázy a zvýšení hodnot jaterních enzymů, o kterých se předpokládá, že souvisejí se zvýšenou sekrecí žluče u tohoto živočišného druhu. Ve studiích toxicity po opakovaném podávání u laboratorních potkanů a opic nebyl zjištěn žádný důkaz svědčící pro přidružené riziko.
Ve studiích reprodukční a vývojové toxicity se vyskytlo zpomalení růstu plodu a snížení přežití a několik mrtvě narozených plodů v důsledku maternální toxicity. Ale u potomstva nebyly pozorovány žádné účinky na morfologii a funkce, včetně učení nebo paměti. Rufinamid nebyl teratogenní u myší, laboratorních potkanů či králíků.
Rufinamid nebyl genotoxický a neměl žádný kancerogenní potenciál. Nežádoucím účinkem, který nebyl pozorován během klinických studií, ale byl zaznamenán u zvířat při expozici podobné hodnotám klinické expozice, a který by mohl být relevantní z hlediska použití u lidských jedinců, byla ve studii kancerogenity u myší myelofibróza kostní dřeně. Výskyt nezhoubných kostních novotvarů (osteomů) a hyperostóza zaznamenané u myší byly považovány za výsledek aktivace viru specifického pro myši ionty flóru uvolněnými během oxidačního metabolismu rufinamidu.
Pokud jde o imunotoxický potenciál, byl u psů během třináctitýdenní studie pozorován malý brzlík a involuce brzlíku s výraznou reakcí při vysokých dávkách u samců. Změny kostní dřeně a lymfatické změny u samic byly při podávání vysokých dávek během třináctitýdenní studie hlášeny s malou incidencí. U potkanů byla snížená buněčnost kostní dřeně a atrofie brzlíku pozorována pouze u studie kancerogenity.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Jádro
Monohydrát laktosy Mikrokrystalická celulosa Kukuřičný škrob
Sodná sůl kroskarmelosy
Hypromelosa
Magnesium-stearát
Natrium-lauryl-sulfát
Koloidní bezvodý oxid křemičitý
Potah tablety-Hypromelosa Makrogoly (8000)
Oxid titaničitý (E171)
Mastek
Červený oxid železitý (E172)
6.2 Inkompatibility Neuplatňuje se.
6.3 Doba použitelnosti
4 roky.
6.4 Zvláštní opatření pro uchovávání
Neuchovávejte při teplotě nad 30 °C.
6.5 Druh obalu a obsah balení
Blistry z hliníku/hliníku (Al/Al blistry), balení po 10, 30, 50, 60, 100 a 200 potahovaných tabletách. Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku Žádné zvláštní požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Eisai Limited, European Knowledge Centre, Mosquito Way, Hatfield, Hertfordshire, AL10 9SN, Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/06/378/011-016
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 16. ledna 2007
Datum posledního prodloužení registrace: 16. ledna 2012
10. DATUM REVIZE TEXTU
{MM/RRRR}
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu.
NÁZEV PŘÍPRAVKU
1.
Inovelon 40 mg/ml perorální suspenze
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden ml perorální suspenze obsahuje rufinamidum 40 mg.
Jedna 460ml lahev obsahuje 18 400 mg rufinamidum.
Pomocné látky se známým účinkem:
Jeden ml perorální suspenze obsahuje 1,2 mg methylparabenu (E218), 0,3 mg propylparabenu (E216) a 250 mg sorbitolu (E420).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Perorální suspenze.
Bílá, mírně viskózní suspenze.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Inovelon je indikován jako přídatná léčba při léčení záchvatů spojených s Lennox-Gastautovým syndromem u pacientů ve věku 4 let a starších.
4.2 Dávkování a způsob podání
Léčbu rufinamidem by měl předepsat lékař se specializací v oboru pediatrie nebo neurologie se zkušenostmi s léčbou epilepsie.
Inovelon perorální suspenze a Inovelon potahované tablety lze při stejných dávkách zaměnit. Pacienty je třeba v období přechodu z jedné lékové formy na druhou sledovat.
Dávkování
Podávání dětem ve věku čtyř let nebo starším o hmotnosti nižší než 30 kg Pacienti < 30 kg neužívající valproát sodný:
Léčba by měla být zahájena denní dávkou 200 mg (dávka 5 ml suspenze podaná jako dvě 2,5 ml dávky, jedna ráno a jedna večer). Podle klinické odpovědi a snášenlivosti může být ve dvoudenních intervalech denní dávka zvyšována o 200 mg, až do maximální doporučené dávky 1 000 mg/den (25 ml/den). Dávky až 3 600 mg/den (90 ml/den) byly studovány u omezeného počtu pacientů.
Pacienti < 30 kg užívající současně valproát sodný:
Vzhledem k tomu, že valproát výrazně snižuje clearance rufinamidu, doporučuje se u pacientů < 30 kg, kteří současně užívají valproát, nižší maximální dávka Inovelonu. Léčba by měla být zahájena denní dávkou 200 mg. Podle klinické odpovědi a snášenlivosti může být minimálně po dvou dnech denní dávka zvýšena o 200 mg až do maximální doporučené dávky 600 mg/den (15 ml/den).
Podávání dospělým, dospívajícím a dětem ve věku čtyř let nebo starším o hmotnosti 30 kg a vyšší Léčba by měla být zahájena denní dávkou 400 mg (dávka 10 ml suspenze podaná jako dvě 5 ml dávky). Podle klinické odpovědi a snášenlivosti může být ve dvoudenních intervalech denní dávka zvyšována o 400 mg, až do maximální doporučené dávky, jak je uvedena v následující tabulce.
|
Rozmezí hmotnosti |
30,0 - 50,0 kg |
50,1 - 70,0 kg |
> 70,1 kg |
|
Maximální doporučená dávka |
1 800 mg/den nebo 45 ml/den |
2 400 mg/den nebo 60 ml/den |
3 200 mg/den nebo 80 ml/den |
Dávky až 4 000 mg/den (100 ml/den) u hmotnosti 30-50 kg nebo 4 800 mg/den (120 ml/den) v kategorii nad 50 kg byly studovány u omezeného počtu pacientů.
Ukončení léčby
Má-li být léčba rufinamidem ukončena, je nutné ji vysazovat postupně. V klinických studiích bylo vysazení rufinamidu dosaženo snižováním dávky o přibližně 25 % vždy po dvou dnech.
V případě jedné či více vynechaných dávek je nutné individuální posouzení klinického stavu.
Nekontrolované otevřené studie naznačují, že účinnost je dlouhodobá, přestože žádná kontrolovaná studie nebyla prováděna déle než tři měsíce.
Pediatrická populace
Bezpečnost a účinnost rufinamidu u dětí ve věku do 4 let nebyla dosud stanovena. Nejsou dostupné žádné údaje.
Starší osoby
Informace o podávání rufinamidu starším osobám jsou omezené. Protože farmakokinetika rufinamidu není u starších osob změněna (viz bod 5.2), není nutná úprava dávkování u pacientů ve věku nad 65 let.
Porucha funkce ledvin
Studie pacientů se závažnou poruchou funkce ledvin ukázala, že u těchto pacientů není nutná žádná úprava dávkování (viz bod 5.2).
Porucha funkce jater
Podávání léku u pacientů s poruchou funkce jater nebylo studováno. Při léčbě pacientů s mírnou až středně závažnou poruchou funkce jater se doporučuje opatrnost a pečlivé titrování dávky. Podávání léku pacientům s těžkou poruchou funkce jater se nedoporučuje.
Způsob podání
Rufinamid je určen k perorálnímu podání. Má se užívat dvakrát denně, ráno a večer, rozdělený do dvou stejných dávek. Protože byl pozorován účinek jídla, má se přípravek podávat s jídlem (viz bod 5.2).
Perorální suspenzi je nutné před každým podáním pořádně protřepat. Další podrobnosti jsou uvedeny v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku, deriváty triazolu nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Status epilepticus
Při podávání rufinamidu během studií klinického vývoje byly zaznamenány případy status epilepticus, zatímco u pacientů, kteří užívali placebo, nebyly žádné takové případy zaznamenány. Tyto příhody vedly k vysazení rufinamidu ve 20 % případů. Jestliže se u pacientů objeví nové typy záchvatů a/nebo se zvýšenou frekvencí dochází ke status epilepticus, který se liší od počátečního stavu pacienta, je nutné znovu přehodnotit poměr prospěšnosti a rizik léčby.
Vysazení rufinamidu
Rufinamid je nutné vysazovat postupně, aby se snížila možnost výskytu záchvatů při vysazení léku. V klinických studiích bylo vysazení dosaženo snižováním dávky o přibližně 25 % vždy po dvou dnech. O vysazení současně podávaných antiepileptických přípravků, jakmile bylo dosaženo kontroly nad záchvaty přidáním rufinamidu, nejsou dostatečné údaje.
Reakce centrálního nervového systému
Léčba rufinamidem byla spojena se závratí, spavostí, ataxií a poruchami chůze, což mohlo zvýšit výskyt náhodných pádů v této populaci (viz bod 4.8). Je třeba, aby pacienti a ošetřující osoby dbali zvýšené opatrnosti, dokud se neseznámí s možnými účinky tohoto léčivého přípravku.
Hypersenzitivní reakce
Ve spojení s léčbou rufinamidem se vyskytly závažné syndromy přecitlivělosti na antiepileptický léčivý přípravek včetně DRESS (Léková reakce s eozionfilií a systémovými příznaky) a Stevens-Johnosonova syndromu. Příznaky a symptomy tohoto onemocnění byly různorodé; avšak u pacientů se typicky, i když ne výlučně, vyskytovala horečka a vyrážka, ve spojení s postižením dalšího orgánového systému. Jiné přidružené manifestace zahrnovaly lymfadenopatii, abnormality testů jaterních funkcí a hematurii. Protože jsou projevy tohoto stavu různorodé, mohou se objevit jiné příznaky a známky postižení orgánových systémů, které v tomto textu nejsou uvedeny. Tento syndrom přecitlivělosti na antiepileptika se objevil v těsné časové souvislosti se zahájením léčby rufinamidem a u souboru pediatrických pacientů. Pokud na tuto reakci vznikne podezření, je nutné rufinamid vysadit a zahájit alternativní léčbu. Všichni pacienti, u kterých se objeví vyrážka při užívání rufinamidu, musí být pečlivě monitorováni.
Zkrácení QT
V podrobné QT studii způsobil rufinamid pokles QTc intervalu, který byl přímo úměrný jeho koncentraci. Ačkoliv mechanismus vzniku tohoto nálezu ani jeho význam s ohledem na bezpečnost přípravku nejsou známy, lékaři by na základě klinického posouzení měli zvážit, zda předepsat rufinamid pacientům, kterým hrozí další zkracování QTc intervalu (např. kongenitální syndrom krátkého QT nebo pacienti, kteří mají tento syndrom v rodinné anamnéze).
Ženy v plodném věku
Ženy v plodném věku musejí během léčby Inovelonem používat antikoncepci. Lékaři by se měli snažit zajistit, aby byla použita vhodná antikoncepce, a podle klinické situace jednotlivých pacientek by měli zvážit, zda je vhodnější podávat perorální antikoncepci nebo dávky složek perorální antikoncepce (viz bod 4.5).
Parabeny, sorbitol
Inovelon perorální suspenze obsahuje parabeny, které mohou vyvolávat alergické reakce (též opožděné). Obsahuje také sorbitol, a proto se nemá podávat pacientům se vzácnými dědičnými poruchami intolerance fruktózy.
Sebevražedné myšlenky
Sebevražedné myšlenky a chování byly hlášeny u pacientů léčených antiepileptiky v několika indikacích. Metaanalýza randomizovaných placebem kontrolovaných studií antiepileptických léčivých přípravků také prokázala mírně zvýšené riziko sebevražedných myšlenek a sebevražedného chování.
Mechanismus tohoto rizika není znám a dostupné údaje nevylučují možnost zvýšeného rizika u Inovelonu.
U pacientů je proto třeba sledovat možné známky sebevražedných myšlenek a sebevražednéhochování a je třeba zvážit vhodnou léčbu. Pacienty (a ošetřovatele pacientů) je třeba poučit, aby v případě, že se objeví známky sebevražedných myšlenek nebo sebevražedného chování, vyhledali lékařskou pomoc.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Potenciál jiných léčivých přípravků k ovlivňování rufinamidu
Jiné antiepileptické léčivé přípravky
Koncentrace rufinamidu nepodléhají klinicky významným změnám při současném podávání se známými antiepileptickými léčivými přípravky indukujícími enzymy.
U pacientů léčených přípravkem Inovelon, u kterých bylo zahájeno podávání valproátu, může docházet k významnému zvýšení koncentrací rufinamidu v plazmě. Nejvýraznější zvýšení bylo pozorováno u pacientů s nízkou tělesnou hmotností (< 30 kg). Proto je třeba zvážit snížení dávky Inovelonu u pacientů < 30 kg, u kterých je zahájena léčba valproátem (viz bod 4.2).
Přidání nebo vysazení těchto léčivých přípravků nebo upravení dávky těchto léčivých přípravků během léčby rufinamidem může vyžadovat úpravu dávkování rufinamidu.
Po současném podávání lamotriginu, topiramátu nebo benzodiazepinů nebyly pozorovány žádné významné změny koncentrace rufinamidu.
Potenciál rufinamidu k ovlivňování jiných léčivých přípravků
Jiné antiepileptické léčivé přípravky
Farmakokinetické interakce mezi rufinamidem a jinými antiepileptickými léčivými přípravky byly u pacientů s epilepsií hodnoceny pomocí populačního modelování farmakokinetiky. Zdá, se že rufinamid nemá žádný klinicky relevantní účinek na koncentrace karbamazepinu, lamotriginu, fenobarbitalu, topiramátu phenytoinu nebo valproátu v ustáleném stavu.
Perorální antikoncepce
Současné podávání rufinamidu 800 mg dvakrát denně a kombinované perorální antikoncepce (ethinylestradiol 35 pg a norethindron 1 mg) po dobu 14 dní způsobilo průměrný pokles AUC0-24 ethinylestradiolu o 22 % a AUC0-24 norethindronu o 14 %. Studie s jinými perorálními nebo implantovanými kontraceptivy nebyly prováděny. Ženám plodného věku, které užívají hormonální antikoncepci, se doporučuje používat další bezpečnou a účinnou antikoncepční metodu (viz body 4.4 a 4.6).
Cytochromové enzymy P450
Rufinamid je metabolizovaný hydrolýzou a cytochromovými enzymy P450 není významně metabolizován. Kromě toho rufinamid neinhibuje aktivitu cytochromových enzymů P450 (viz bod 5.2). Je tedy nepravděpodobné, aby nastaly klinicky významné interakce zprostředkované inhibicí systému cytochromů P450 rufinamidem. Bylo prokázáno, že rufinamid indukuje enzym CYP3A4 cytochromu P450 a může tudíž snížit plazmatické koncentrace látek, které jsou metabolizovány tímto enzymem. Účinek byl mírný až středně závažný. Průměrná aktivita CYP3A4, hodnocená jako clearance triazolamu, se zvýšila o 55 % po 11 dnech léčby rufinamidem 400 mg dvakrát denně. Expozice triazolamu se snížila o 36 %. Vyšší dávky rufinamidu mohou mít za následek výraznější indukci. Nelze vyloučit, že rufinamid může také snižovat expozici látek metabolizovaných jinými enzymy nebo dopravovaných transportními proteiny, jako je P-glykoprotein.
Doporučuje se, aby pacienti léčení látkami metabolizovanými systémem enzymu CYP3A4, byli pečlivě monitorováni po dobu dvou týdnů, a to buď na počátku nebo po skončení léčby rufinamidem, nebo po jakékoliv významné změně dávkování. Možná bude zapotřebí zvážit úpravu dávkování
36
současně podávaného léčivého přípravku. Tato doporučení je třeba vzít v úvahu také, pokud se rufinamid používá společně s látkami s úzkým terapeutickým oknem, jako je warfarin nebo digoxin.
Studie specifické interakce u zdravých subjektů neodhalila žádný vliv rufinamidu v dávce 400 mg dvakrát denně na farmakokinetiku olanzapinu, substrátu CYP1A2.
Nejsou k dispozici žádné údaje o interakci rufinamidu s alkoholem.
4.6 Fertilita, těhotenství a kojení
Riziko související s epilepsií a podáváním antiepileptik všeobecně:
Ukazuje se, že v potomstvu žen s epilepsií je prevalence malformací dvakrát až třikrát větší než je četnost v celkové populaci, která je přibližně 3 %. U léčené populace byl nárůst malformací pozorován u polyterapie, avšak rozsah, za který je léčba a/nebo nemoc odpovědná, nebyl určen.
Navíc účinná antiepileptická léčba nesmí být přerušena, protože zhoršení nemoci je škodlivé jak pro matku, tak pro plod.
Riziko související s rufinamidem:
Studie na zvířatech neprokázaly teratogenní účinek kromě fetotoxicity v přítomnosti maternální toxicity (viz bod 5.3). Potenciální riziko pro člověka není známé.
Nejsou k dispozici klinické údaje o podávání rufinamidu během těhotenství.
Vezmeme-li v úvahu tyto údaje, rufinamid by se neměl podávat, pokud to není nezbytně nutné, během těhotenství ani u žen plodného věku, které nepoužívají antikoncepci.
Ženy ve fertilním věku musí během léčby rufinamidem používat antikoncepci. Lékaři by se měli snažit zajistit, aby byla použita vhodná antikoncepce, a podle klinické situace jednotlivých pacientek by měli zvážit, zda je vhodnější podávat perorální antikoncepci nebo dávky složek perorální antikoncepce (viz bod 4.5).
Pokud ženy léčené rufinamidem plánují otěhotnět, je nutné pečlivě zvážit indikaci tohoto přípravku.
V průběhu těhotenství nesmí být účinná antiepileptická léčba rufinamidem přerušena, protože zhoršení nemoci je škodlivé jak pro matku, tak pro plod.
Kojení
Není známo, zda se rufinamid vylučuje do lidského mateřského mléka. Vzhledem k možným škodlivým účinkům pro kojené dítě by neměly matky při léčbě rufinamidem kojit.
Fertilita
Nejsou dostupné žádné údaje o účinku léčby rufinamidem na fertilitu.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Inovelon může způsobit závratě, spavost a rozmazané vidění. V závislosti na individuální citlivosti může mít rufinamid malý až výrazný vliv na schopnost řídit nebo obsluhovat stroje. Pacienty je třeba upozornit, aby během činností vyžadujících vysoký stupeň bdělosti, např. při řízení nebo obsluhování strojů, dbali zvýšené opatrnosti.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Klinický vývojový program zahrnoval více než 1 900 pacientů s různými typy epilepsie, kterým byl podáván rufinamid. Nejčastěji hlášené nežádoucí účinky celkově byly bolesti hlavy, závratě, únava a
37
spavost. Nej častější nežádoucí účinky pozorované u pacientů s Lennox-Gastautovým syndromem ve vyšší incidenci než při podávání placeba byly spavost a zvracení. Nežádoucí účinky byly obvykle mírné až středně těžké. Četnost vystoupení pacientů s Lennox-Gastautovým syndromem ze studie z důvodu nežádoucích účinků byla 8,2 % u pacientů užívajících rufinamid a 0 % u pacientů užívajících placebo. Nejčastější nežádoucí účinky, které vedly k vystoupení ze studie, byly ve skupině léčené rufinamidem vyrážka a zvracení.
Tabulka nežádoucích účinků
Nežádoucí účinky hlášené s vyšší incidencí než u placeba během dvojitě zaslepených studií u pacientů s Lennox-Gastautovým syndromem nebo u celkového souboru pacientů, kterým byl podáván rufinamid, jsou uvedeny v následující tabulce podle terminologie MedDRA, třídy orgánových systémů a podle frekvence.
Frekvence jsou definovány jako: velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté
(> 1/1 000 až < 1/100), vzácné (> 1/10 000 až < 1/1 000).
|
Třída orgánových systémů |
Velmi časté |
Časté |
Méně časté |
Vzácné |
|
Infekce a infestace |
Chřipka Nazofaryngitida Ušní infekce Sinusitida Rinitida | |||
|
Poruchy imunitního systému |
Hypersenzitivita* | |||
|
Poruchy metabolismu a výživy |
Poruchy příjmu potravy Snížená chuť k jídlu | |||
|
Psychiatrické poruchy |
Úzkost Insomnie | |||
|
Poruchy nervového systému |
Spavost* Bolesti hlavy Závratě* |
Status epilepticus* Křeče Abnormální koordinace* Nystagmus Psychomotorická hyperaktivita | ||
|
Poruchy oka |
Diplopie Rozmazané vidění | |||
|
Poruchy ucha a labyrintu | ||||
|
Respirační, hrudní a mediastinální poruchy |
Epistaxe | |||
|
Gastrointestinální poruchy |
Bolesti horní části břicha Zácpa | |||
|
Poruchy jater a žlučových cest |
° |
Zvýšení jaterních enzymů | ||
|
Poruchy kůže a |
Vyrážka* |
|
Třída orgánových systémů |
Velmi časté |
Časté |
Méně časté |
Vzácné |
|
podkožní tkáně |
Akné | |||
|
Poruchy svalové a kosterní soustavy a pojivové tkáně a poruchy kostí |
Bolesti zad | |||
|
Poruchy reprodukčního systému a prsu |
Oligomenorea | |||
|
Celkové poruchy a reakce v místě aplikace |
Únava |
Poruchy chůze* | ||
|
Vyšetření |
Pokles hmotnosti | |||
|
Poranění, otravy a procedurální komplikace |
Poranění hlavy Kontuze |
*Odkaz na bod 4.4.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Po akutním předávkování může být žaludek vyprázdněn gastrickou laváží nebo vyvoláním zvracení. Pro rufinamid neexistuje žádné specifické antidotum. Léčba by měla být podpůrná a může zahrnovat hemodialýzu (viz bod 5.2).
Podávání opakovaných dávek 7 200 mg/den nebylo spojeno s žádnými závažnějšími příznaky nebo symptomy.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antiepileptika, deriváty karboxamidu; ATC kód: N03AF03. Mechanismus účinku
Rufinamid moduluje aktivitu sodíkových kanálů tak, že prodlužuje jejich inaktivní stav. Rufinamid je účinný u celé řady zvířecích modelů epilepsie.
Klinické zkušenosti
Inovelon (tablety obsahující rufinamid) byl podáván ve dvojitě zaslepené, placebem kontrolované studii v dávkách až 45 mg/kg/den po dobu 84 dnů 139 pacientům s nedostatečně kontrolovanými záchvaty spojenými s Lennox-Gastautovým syndromem (včetně atypických záchvatů typu absence a záchvatových pádů). Muži i ženy (ve věku od 4 do 30 let) byli do studie zařazeni, pokud užívali 1 až 3 současně podávané antiepileptické léčivé přípravky s pevně stanoveným dávkováním. Každý pacient musel mít alespoň 90 záchvatů během měsíce před přijetím do studie. U všech tří primárních proměnných bylo pozorováno signifikantní zlepšení: procentuální změna celkové frekvence záchvatů za 28 dnů během udržovací fáze v porovnání s výchozí hodnotou (-35,8 % při podávání přípravku
Inovelon oproti -1,6 % při podávání placeba, p = 0,0006), počet tonických/atonických záchvatů (-42,9 % při podávání přípravku Inovelon oproti 2,2 % při podávání placeba, p = 0,0002) a ohodnocení závažnosti záchvatu při celkovém vyhodnocení prováděném rodičem/opatrovníkem na konci dvojitě zaslepené fáze (velké nebo velmi velké zlepšení ve 32,2 % při podávání přípravku Inovelon oproti 14,5 % v rameni s placebem, p = 0,0041).
Populační modelování farmakokinetiky/farmakodynamiky ukázalo, že snížení celkové záchvatové frekvence a frekvence tonických/atonických záchvatů, zlepšení celkového vyhodnocení závažnosti záchvatů a zvýšení pravděpodobnosti snížení záchvatové frekvence bylo závislé na koncentracích rufinamidu.
5.2 Farmakokinetické vlastnosti
Absorpce
Maximální plazmatické hladiny jsou dosaženy přibližně 6 hodin po podání. Vrcholová koncentrace (Cmax) a plazmatická AUC rufinamidu se zvyšují méně než proporcionálně s dávkou u zdravých subjektů a pacientů nalačno i po jídle, což je pravděpodobně způsobeno omezenou absorpcí dávky. Po jednotlivých dávkách zvyšuje potrava biologickou dostupnost (AUC) rufinamidu přibližně o 34 % a maximální koncentraci v plazmě o 56 %.
Bylo prokázáno, že přípravky Inovelon perorální suspenze a Inovelon potahované tablety jsou biologicky ekvivalentní.
Distribuce
Ve studiích in vitro byla pouze malá frakce rufinamidu (34 %) navázána na lidské sérové proteiny, přičemž albumin odpovídal přibližně za 80 % této vazby. To ukazuje, že riziko lékových interakcí v důsledku nahrazení vazebného místa během současného podávání dalších látek je minimální. Rufinamid byl rovnoměrně rozdělen mezi erytrocyty a plazmu.
Biotransformace
Rufinamid je téměř výhradně eliminován metabolismem. Hlavní drahou metabolismu je hydrolýza karboxylamidové skupiny na farmakologicky inaktivní derivát kyseliny CGP 47292. Metabolismus zprostředkovaný cytochromem P450 se podílí v mnohem menší míře. Nelze zcela vyloučit vznik malých množství konjugátů glutathionu.
In vitro rufinamid vykazoval malou nebo žádnou schopnost působit jako kompetitivní nebo nekompetitivní inhibitor následujících lidských enzymů P450: CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5 nebo CYP4A9/11-2.
Eliminace
Plazmatický eliminační poločas je přibližně 6-10 hodin u zdravých subjektů a pacientů s epilepsií. Rufinamid se při podávání dvakrát denně ve 12 hodinových intervalech akumuluje v rozsahu, který lze předpovídat podle jeho terminálního poločasu, což ukazuje, že farmakokinetika rufinamidu je časově nezávislá (tj. bez autoindukce metabolismu).
Ve studiích s radioaktivně značenou látkou u tří zdravých dobrovolníků byla základní sloučenina (rufinamid) hlavní radioaktivní složkou v plazmě a představovala přibližně 80 % celkové radioaktivity, metabolit CGP 47292 tvořil pouze asi 15 %. Převládající cestou eliminace látek souvisejících s léčivou látkou bylo vylučování ledvinami, které odpovídalo za 84,7 % dávky.
Linearita/nelinearita:
Biologická dostupnost rufinamidu je závislá na dávce. Se zvyšováním dávky se snižuje biologická dostupnost.
Farmakokinetika u zvláštních skupin pacientů
Pohlaví
Pro vyhodnocení vlivu pohlaví na farmakokinetiku rufinamidu bylo použito populační modelování farmakokinetiky. Tato hodnocení ukazují, že pohlaví neovlivňuje farmakokinetiku rufinamidu do klinicky významné míry.
Porucha funkce ledvin
Farmakokinetika jedné dávky 400 mg rufinamidu nebyla u pacientů s chronickým a těžkým selháním ledvin změněna, ve srovnání se zdravými dobrovolníky. Ale když byla po podání rufinamidu provedena hemodialýza, snížily se plazmatické hladiny přibližně o 30 %, což naznačuje, že by mohl být postup použitelný v případě předávkování (viz body 4.2 a 4.9).
Porucha funkce jater
U pacientů s poruchou funkce jater nebyly prováděny žádné studie, a proto by Inovelon neměl být pacientům se těžkým poškozením jater podáván (viz bod 4.2).
Děti (2-12 let)
Děti mají obvykle nižší clearance rufinamidu než dospělé osoby a tento rozdíl souvisí s velikostí těla. Studie u novorozených dětí nebo kojenců a batolat mladších 2 let nebyly prováděny.
Starší osoby
Farmakokinetická studie u starších zdravých dobrovolníků neukázala významný rozdíl ve farmakokinetických parametrech ve srovnání s mladšími dospělými osobami.
5.3 Předklinické údaje vztahující se k bezpečnosti
Konvenční farmakologické studie bezpečnosti v klinicky relevantních dávkách neodhalily žádná zvláštní rizika.
Toxické účinky pozorované u psů v hladinách podobných expozici u člověka při maximální doporučené dávce zahrnovaly změny na játrech, včetně žlučových trombů, cholestázy a zvýšení hodnot jaterních enzymů, o kterých se předpokládá, že souvisejí se zvýšenou sekrecí žluče u tohoto živočišného druhu. Ve studiích toxicity po opakovaném podávání u potkanů a opic nebyl zjištěn žádný důkaz svědčící pro přidružené riziko.
Ve studiích reprodukční a vývojové toxicity se vyskytlo zpomalení růstu plodu a snížení přežití a několik mrtvě narozených plodů v důsledku maternální toxicity. Ale u potomstva nebyly pozorovány žádné účinky na morfologii a funkce, včetně učení nebo paměti. Rufinamid nebyl teratogenní u myší, potkanů ani králíků.
Rufinamid nebyl genotoxický a neměl žádný kancerogenní potenciál. Nežádoucím účinkem, který nebyl pozorován během klinických studií, ale byl zaznamenán u zvířat při expozici podobné hodnotám klinické expozice, a který by mohl být relevantní z hlediska použití u lidských jedinců, byla ve studii karcinogenity u myší myelofibróza kostní dřeně. Výskyt nezhoubných kostních novotvarů (osteomů) a hyperostóza zaznamenané u myší byly považovány za výsledek aktivace viru specifického pro myši ionty fluoru uvolněnými během oxidačního metabolismu rufinamidu.
Pokud jde o imunotoxický potenciál, byl u psů během třináctitýdenní studie pozorován malý brzlík a involuce brzlíku s výraznou reakcí při vysokých dávkách u samců. Změny kostní dřeně a lymfatické změny u samic byly při podávání vysokých dávek během třináctitýdenní studie hlášeny s malou incidencí. U potkanů byla snížená buněčnost kostní dřeně a atrofie brzlíku pozorována pouze u studie karcinogenity.
FARMACEUTICKÉ ÚDAJE
6.
6.1 Seznam pomocných látek
Mikrokrystalická celulosa (E460)
Sodná sůl karmelosy (E466)
Hyetelosa
Kyselina citronová (E330)
Simetikonová emulze 30% obsahující kyselinu benzoovou, cyklotetrasiloxan, dimetikon, ethylenglykol-distearát a glycerol-distearát, methylcelulosu, PEG-40 stearát (makrogol-stearát), polysorbát 65, silikagel, kyselinu sorbovou, kyselinu sírovou a vodu Poloxamer 188 Methylparaben (E218)
Propylparaben (E216)
Propylenglykol (E1520)
Kalium-sorbát (E202)
Sorbitol (E420) tekutý (nekrystalizující)
Pomerančová příchuť Voda
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
3 roky.
Po prvním otevření: 90 dní.
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání. Podmínky uchovávání tohoto léčivého přípravku po prvním otevření jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
Lahev z orientovaného polyethylentereftalátu (o-PET) s polypropylenovým (PP) uzávěrem s dětskou pojistkou; jedna lahev obsahuje 460 ml suspenze; krabička.
Jedna krabička obsahuje jednu lahev, dvě stejné kalibrované stříkačky na perorální dávkování a zatlačovací adaptér lahve (PIBA). Stříkačky na perorální dávkování jsou opatřené stupnicí po 0,5 ml.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Příprava: Zatlačovací adaptér lahve (PIBA), který se dodává v krabičce s přípravkem, je nutné před použitím pevně zasunout do hrdla lahve, kde zůstane po celou dobu používání lahve. Dávkovací stříkačku je nutné zasunout do PIBA a dávku natáhnout z převrácené lahve. Víčko je nutné po každém použití opět uzavřít. Víčko správně pasuje, když je PIBA v lahvi.
Žádné zvláštní požadavky na likvidaci.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Eisai Limited, European Knowledge Centre, Mosquito Way, Hatfield, Hertfordshire, AL10 9SN, Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 16. ledna 2007
Datum posledního prodloužení registrace: 16. ledna 2012
10. DATUM REVIZE TEXTU
{MM/RRRR}
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky: http://www.ema.europa.eu.
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
E. ZVLÁŠTNÍ POVINNOST USKUTEČNIT POREGISTRAČNÍ OPATŘENÍ PRO REGISTRACI PŘÍPRAVKU ZA VÝJIMEČNÝCH OKOLNOSTÍ
Název a adresa výrobce odpovědného za propouštění šarží
Eisai Manufacturing Limited
Mosquito Way
Hatfield
Hertfordshire
AL10 9SN
Velká Británie
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz Příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
• Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci bude předkládat pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ TOHOTO LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
E. ZVLÁŠTNÍ POVINNOST USKUTEČNIT POREGISTRAČNÍ
OPATŘENÍ PRO REGISTRACI PŘÍPRAVKU ZA VÝJIMEČNÝCH OKOLNOSTÍ
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Inovelon 100 mg potahované tablety Rufinamidum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tableta obsahuje 100 mg rufinamidu.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktosu. Pro další informace si přečtěte příbalovou informaci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
10
10 potahovaných tablet 30
30 potahovaných tablet 50
50 potahovaných tablet 60
60 potahovaných tablet 100
100 potahovaných tablet
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP: (MM/RRRR)
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Neuchovávejte při teplotě nad 30 °C.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH
PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Eisai Ltd., European Knowledge Centre, Mosquito Way, Hatfield, Hertfordshire, AL10 9SN, Velká Británie.
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/06/378/001-005
13. ČÍSLO ŠARŽE
č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Inovelon 100 mg tablety

Inovelon 100 mg potahované tablety Rufinamidum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Eisai Ltd.
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Inovelon 200 mg potahované tablety Rufinamidum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tableta obsahuje 200 mg rufinamidu.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktosu. Pro další informace si přečtěte příbalovou informaci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
10
10 potahovaných tablet 30
30 potahovaných tablet 50
50 potahovaných tablet 60
60 potahovaných tablet 100
100 potahovaných tablet
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP: (MM/RRRR)
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Neuchovávejte při teplotě nad 30 °C.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Eisai Ltd., European Knowledge Centre, Mosquito Way, Hatfield, Hertfordshire, AL10 9SN, Velká Británie.
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/06/378/006-010
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Inovelon 200 mg tablety

Inovelon 200 mg potahované tablety Rufinamidum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Eisai Ltd.
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Inovelon 400 mg potahované tablety Rufinamidum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tableta obsahuje 400 mg rufinamidu.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktosu. Pro další informace si přečtěte příbalovou informaci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
10
10 potahovaných tablet 30
30 potahovaných tablet 50
50 potahovaných tablet 60
60 potahovaných tablet 100
100 potahovaných tablet 200
200 potahovaných tablet
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP: (MM/RRRR)
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Neuchovávejte při teplotě nad 30 °C.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH
PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Eisai Ltd., European Knowledge Centre, Mosquito Way, Hatfield, Hertfordshire, AL10 9SN, Velká Británie.
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/06/378/011-016
13. ČÍSLO ŠARŽE
č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Inovelon 400 mg tablety

Inovelon 400 mg potahované tablety Rufinamidum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Eisai Ltd.
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Inovelon 40 mg/ml perorální suspenze Rufinamidum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
1 ml přípravku Inovelon perorální suspenze obsahuje 40 mg rufinamidu. 1 lahev obsahuje 18 400 mg rufinamidu.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje také methylparaben (E218) propylparaben (E216) sorbitol (E420)
Pro další informace si přečtěte příbalovou informaci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Perorální suspenze 460 ml.
Jedna krabička obsahuje 1 lahev, 2 stříkačky a 1 adaptér PIBA.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Před použitím dobře protřepejte.
Perorální podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP:
Po prvním otevření: spotřebujte do 90 dní.
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Eisai Ltd., European Knowledge Centre, Mosquito Way, Hatfield, Hertfordshire, AL10 9SN, Velká Británie.
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/06/378/017
13. ČÍSLO ŠARŽE
č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Inovelon 40 mg/ml
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro uživatele
Inovelon 100 mg potahované tablety Inovelon 200 mg potahované tablety Inovelon 400 mg potahované tablety
Rufinamidum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí
ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je Inovelon a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete Inovelon užívat
3. Jak se Inovelon používá
4. Možné nežádoucí účinky
5. Jak Inovelon uchovávat
6. Obsah balení a další informace
1. Co je Inovelon a k čemu se používá
Inovelon obsahuje lék rufinamid. Patří do skupiny léků zvaných antiepileptika, která se používají k léčbě epilepsie (stavu, kdy může docházet k záchvatům).
Inovelon se používá v kombinaci s dalšími léky k léčbě záchvatů spojených s Lennox-Gastautovým syndromem u dospělých, dospívajících a dětí starších 4 let. Lennox-Gastautův syndrom je označení pro skupinu závažných epilepsií, u nichž dochází k opakovaným záchvatům různého typu.
Inovelon Vám lékař předepsal ke snížení počtu záchvatů.
2. Čemu musíte věnovat pozornost, než začnete Inovelon užívat
Neužívejte přípravek Inovelon:
- jestliže jste alergický(á) na rufinamid, deriváty triazolu nebo na kteroukoli další složku přípravku Inovelon (uvedenou v bodě 6).
Upozornění a opatření
Informujte prosím lékaře nebo lékárníka:
- jestliže trpíte vrozeným syndromem krátkého QT intervalu nebo máte takový syndrom (poruchu elektrické aktivity srdce) v rodinné anamnéze, protože užívání rufinamidu by mohlo tento stav zhoršit.
- jestliže máte potíže s játry. O užívání rufinamidu jsou v této skupině pacientů k dispozici omezené informace, takže možná bude nutné dávku Vašeho léku zvyšovat pomaleji. Pokud trpíte těžkým onemocněním jater, může lékař rozhodnout, že pro Vás není přípravek Inovelon vhodný.
- jestliže dostanete kožní vyrážku nebo horečku. Může jít o známky alergické reakce. Okamžitě vyhledejte lékaře, protože ve velmi vzácných případech to může být závažné.
- jestliže se počet záchvatů, či jejich závažnost nebo trvání zvýší, měli byste okamžitě kontaktovat lékaře.
- jestliže se u Vás objeví potíže s chůzí, abnormální pohyby, závrať nebo ospalost, informujte lékaře.
Poraďte se prosím s lékařem, i když výše uvedené stavy nastaly kdykoliv v minulosti.
Děti
Přípravek Inovelon se nesmí podávat dětem do 4 let věku, protože o použití u této věkové skupiny nejsou k dispozici dostatečné informace.
Další léčivé přípravky a Inovelon
Informujte lékaře nebo lékárníka o všech lécích, které užíváte nebo jste užíval(a) v nedávné době, a to i o lécích, které jsou dostupné bez lékařského předpisu. Jestliže užíváte jiné léky, které se z těla odstraňují enzymovým systémem CYP3A4, může být nutné, aby vás lékař sledoval po dobu dvou týdnů na začátku nebo po ukončení léčby rufinamidem, nebo po jakékoliv výrazné změně dávky. Může být nutná úprava dávkování jiných léků, které užíváte, protože při podávání s rufinamidem se může mírně snížit účinnost těchto léků.
Informujte lékaře, jestliže užíváte hormonální/perorální antikoncepci. Přípravek Inovelon může snižovat účinek hormonální/perorální antikoncepce, například antikoncepčních pilulek. Proto se při užívání Inovelonu doporučuje používat ještě další bezpečnou a účinnou antikoncepční metodu.
Informujte lékaře, jestliže užíváte lék na ředění krve - warfarin. Může být nutné, aby lékař upravil dávkování.
Informujte lékaře, jestliže užíváte digoxin (lék používaný k léčbě srdečních onemocnění). Může být nutné, aby lékař upravil dávkování.
Pokud Vám lékař předepíše nebo doporučí doplňující léčbu epilepsie (např. valproát), musíte informovat lékaře, že užíváte Inovelon, protože možná bude nutné upravit dávkování.
Přípravek Inovelon s jídlem a pitím
Pokyny pro užívání Inovelonu s jídlem a pitím viz bod 3 „Jak se Inovelon používá“.
Těhotenství a kojení
Pokud jste žena v plodném věku, musíte během užívání přípravku Inovelon používat antikoncepci.
Pokud jste těhotná nebo se domníváte, že těhotná můžete být, či plánujete-li otěhotnět, poraďte se s lékařem nebo lékárníkem dříve, než začnete Inovelon užívat. Inovelon smíte užívat během těhotenství pouze v případě, že tak rozhodne lékař.
Během užívání přípravku Inovelon se nedoporučuje kojit, protože není známo, zda se rufinamid vylučuje do mateřského mléka.
Poraďte se s lékařem nebo lékárníkem dříve, než začnete užívat jakýkoliv lék.
Řízení dopravních prostředků a obsluha strojů
Užívání Inovelonu může způsobovat závratě a ospalost a může mít vliv na Vaše vidění, především na začátku léčby nebo po zvýšení dávky. Pokud k tomu dojde, neřiďte dopravní prostředek ani neobsluhujte žádné stroje.
Inovelon obsahuje laktózu
Pokud Vám v minulosti lékař sdělil, že trpíte nesnášenlivostí některých cukrů, řekněte to lékaři, než začnete tento přípravek užívat.
Jak se Inovelon používá
3.
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý/á, poraďte se s lékařem nebo lékárníkem.
Děti ve věku 4 let a starší o hmotnosti menší než 30 kg (neužívající valproát)
Doporučená počáteční dávka přípravku je 200 mg denně podávaných ve dvou dávkách. Dávku Vám upraví lékař a může ji obden zvyšovat až o 200 mg až na denní dávku nepřesahující 1 000 mg.
Děti ve věku 4 let a starší o hmotnosti menší než 30 kg (užívající valproát)
U dětí o hmotnosti menší než 30 kg, které užívají valproát (lék na epilepsii), je maximální doporučená denní dávka Inovelonu 600 mg denně.
Doporučená počáteční dávka přípravku je 200 mg denně ve dvou dávkách. Lékař Vám dávku upraví a může ji zvyšovat o 200 mg každé dva dny až na maximální doporučenou dávku 600 mg denně.
Dospělí a děti o hmotnosti 30 kg nebo více
Obvyklá počáteční dávka je 400 mg denně podávaných ve dvou dávkách. Dávku Vám upraví lékař a může ji zvyšovat až o 400 mg ob den na denní dávku nepřesahující 3 200 mg, v závislosti na Vaší hmotnosti.
Někteří pacienti mohou reagovat na nižší dávky a lékař Vám může dávku upravit v závislosti na Vaší odpovědi na léčbu.
Pokud zaznamenáte nežádoucí účinky, lékař Vám může dávku zvyšovat pomaleji.
Tablety Inovelon se musí užívat dvakrát denně, ráno a večer, s vodou. Inovelon se má užívat s jídlem. Pokud máte obtíže při polykání, můžete tabletu rozdrtit. Pak smíchejte prášek přibližně s polovinou sklenice (100 ml) vody a ihned vypijte.
Jestliže jste užil(a) více přípravku Inovelon, než jste měl(a)
Jestliže jste snad užil(a) více přípravku Inovelon než jste měl(a), oznamte to okamžitě lékaři nebo lékárníkovi, nebo se obraťte na oddělení pohotovosti nejbližší nemocnice a vezměte si tento lék s sebou.
Jestliže jste zapomněl(a) užít Inovelon
Jestliže jste zapomněl(a) užít jednu dávku, pokračujte v užívání léku jako obvykle. Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku. Jestliže jste vynechal(a) více než jednu dávku, poraďte se s lékařem.
Jestliže jste přestal(a) užívat Inovelon
Jestliže Vám lékař řekne, abyste léčbu ukončil(a), řiďte se jeho pokyny ohledně postupného snižování dávky přípravku Inovelon, aby se zmenšilo riziko zvýšení výskytu záchvatů.
Máte-li jakékoli další otázky, týkající se užívání tohoto přípravku, zeptejte se lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i Inovelon nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Následující vedlejší účinky mohou být velmi závažné:
Vyrážka a/nebo horečka. Může jít o známky alergické reakce. Pokud se u Vás tyto účinky objeví, okamžitě informujte svého lékaře nebo navštivte nemocnici.
Změna typu záchvatů / častější status epilepticus (záchvaty, které trvají delší dobu, opakované záchvaty). Okamžitě informujte svého lékaře.
Malý počet lidí léčených antiepileptiky, například Inovelonem, měli myšlenky na sebepoškozování nebo sebevraždu. Jakmile se u Vás tyto myšlenky vyskytnou, okamžitě se obraťte na svého lékaře.
Při užívání tohoto léku se u Vás mohou objevit následující nežádoucí účinky. Pokud pozorujete některý z níže uvedených účinků, sdělte to lékaři:
Velmi časté (více než u 1 z 10 pacientů) nežádoucí účinky přípravku Inovelon jsou:
Závratě, bolesti hlavy, nauzea, zvracení, ospalost, únavu.
Časté (více než u 1 ze 100 pacientů) nežádoucí účinky přípravku Inovelon jsou:
Problémy spojené s nervovým systémem zahrnující.: potíže při chůzi, abnormální pohyby, křeče/záchvaty, neobvyklé pohyby očí, rozmazané vidění, třes.
Problémy spojené se zažíváním zahrnující.: bolesti břicha, zácpa, zažívací potíže, řídká stolice (průjem), ztráta nebo změna chuti k jídlu, ztráta hmotnosti.
Infekce: ušní infekce, chřipka, ucpaný nos, hrudní infekce.
Pacienti dále mohou zaznamenat: úzkost, nespavost, krvácení z nosu, trudovitost, vyrážku, bolesti zad, méně častou menstruaci, vznik modřin, poranění hlavy (v důsledku náhodného poranění během záchvatu).
Méně časté (mezi 1 ze 100 a 1 z 1 000 pacientů) nežádoucí účinky přípravku Inovelon jsou:
Alergické reakce a zvýšení hladin jaterních enzymů (ukazatelů funkce jater).
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak Inovelon uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na blistru a papírové krabičce. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Neuchovávejte při teplotě nad 30°C.
Nepoužívejte tento přípravek, pokud si všimnete změny vzhledu přípravku.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co Inovelon obsahuje
- Léčivou látkou je rufinamid.
Jedna potahovaná tableta 100 mg obsahuje 100 mg rufinamidu.
Jedna potahovaná tableta 200 mg obsahuje 200 mg rufinamidu.
Jedna potahovaná tableta 400 mg obsahuje 400 mg rufinamidu.
- Pomocnými látkami jsou monohydrát laktosy, mikrokrystalická celulosa, kukuřičný škrob, sodná sůl kroskarmelosy, hypromelosa, magnesium-stearát, natrium-lauryl-sulfát a koloidní bezvodý oxid křemičitý. Potah tablety obsahuje hypromelosu, makrogoly (8000), oxid titaničitý (E171), talek a červený oxid železitý (E172).
Jak Inovelon vypadá a co obsahuje toto balení
• Inovelon 100 mg tablety jsou růžové, oválné, mírně vypouklé potahované tablety, na obou stranách s půlicí rýhou, na jedné straně s vyraženým označením „C261“ a na druhé straně bez označení.
Jsou dispozici v balení po 10, 30, 50, 60 a 100 potahovaných tabletách.
• Inovelon 200 mg tablety jsou růžové, oválné, mírně vypouklé potahované tablety, na obou stranách s půlicí rýhou, na jedné straně s vyraženým označením „C262“ a na druhé straně bez označení.
Jsou dispozici v balení po 10, 30, 50, 60 a 100 potahovaných tabletách.
• Inovelon 400 mg tablety jsou růžové, oválné, mírně vypouklé potahované tablety, na obou stranách s půlicí rýhou, na jedné straně s vyraženým označením „C263“ a na druhé straně bez označení.
Jsou dispozici v balení po 10, 30, 50, 60, 100 a 200 potahovaných tabletách.
Držitel rozhodnutí o registraci a výrobce
Držitel rozhodnutí o registraci:
Eisai Ltd, European Knowledge Centre, Mosquito Way, Hatfield, Hertfordshire, AL10 9SN, Velká Británie.
Výrobce:
Eisai Manufacturing Ltd, Mosquito Way, Hatfield, Hertfordshire, AL10 9SN, Velká Británie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgie/Belgique/Belgien
Eisai Europe Ltd.
Tél/Tel: + 32 (0) 2 502 58 04
Et.urapHH
Eisai Ltd.
Tel: + 44 (0) 20 8600 1400 (OdeguHeHOTO KpaacTBo)
Česká republika
Eisai GesmbH organizační složka Tel: + 420 242 485 839
Lietuva
Eisai Ltd.
Tel: + 44 (0) 20 8600 1400 (Jungtiné Karalysté)
Luxembourg/Luxemburg
Eisai Europe Ltd.
Tél/Tel: + 32 (0) 2 502 58 04 (Belgique/Belgien)
Magyarország Eisai Ltd.
Tel.: + 44 (0) 20 8600 1400 Egyesult Királyság (Nagy-Britannia)
|
Danmark Eisai AB Tlf: +46 (0) 8 501 01 600 (Sverige) |
Malta Associated Drug Company Ltd Tel: + 356 (0) 2277 8000 |
|
Deutschland Eisai GmbH Tel: + 49 (0) 696 65 85-0 |
Nederland Eisai BV. Tel: + 31 (0) 900 575 3340 |
|
Eesti (Eisai Ltd. Tel: + 44 (0) 20 8600 1400 (Uhendkuningriik) |
Norge Eisai AB Tlf: + 46 (0) 8 501 01 600 (Sverige) |
|
EXláSa Arriani Pharmaceuticals S.A. Tr(k: + 30 210 668 3000 |
Osterreich Eisai GesmbH Tel: + 43 (0) 1 535 1980-0 |
|
Espaňa Eisai Farmacéutica, S.A. Tel: +(34) 91 455 94 55 |
Polska Eisai Ltd. Tel.: + 44 (0) 20 8600 1400 (Wielka Brytania) |
|
France Eisai SAS Tél: + (33) 1 47 67 00 05 |
Portugal Eisai Farmacéutica, Unipessoal Lda Tel: + 351 21 487 55 40 |
|
Hrvatska Eisai Ltd. Tel: + 44 (0) 20 8600 1400 (Velika Britanija) |
Románia Eisai Ltd. Tel: + 44 (0) 208 600 1400 (Marea Britanie) |
|
Ireland Eisai Ltd. Tel: + 44 (0) 20 8600 1400 (United Kingdom) |
Slovenská republika Eisai GesmbH organizační složka Tel: + 420 242 485 839 (Česká republika) |
|
Italia Eisai S.r.l. Tel: + 39 02 5181401 |
Slovenija Eisai Ltd. Tel: + 44 (0) 20 8600 1400 (Velika Britanija) |
|
Ísland Eisai AB Sími: + 46 (0) 8 501 01 600 (Svífrjóó) |
Slovenská republika Eisai GesmbH organizační složka Tel: + 420 242 485 839 (Česká republika) |
|
Italia Eisai S.r.l. Tel: + 39 02 5181401 |
Suomi/Finland Eisai AB Puh/Tel: + 46 (0) 8 501 01 600 (Ruotsi/Sverige) |
|
Kónpoq Arriani Pharmaceuticals S.A. Tqk +30 210 668 3000 (EAMSa) |
Sverige Eisai AB Tel: + 46 (0) 8 501 01 600 |
Tato příbalová informace byla naposledy revidována {MM/RRRR}.
Podrobné informace o tomto přípravku jsou uveřejněny na webových stránkách Evropské agentury pro léčivé přípravky: http://www.ema.europa.eu
Příbalová informace: informace pro uživatele Inovelon 40 mg/ml perorální suspenze
Rufinamidum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků sdělte to lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci:
1. Co je Inovelon a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Inovelon užívat
3. Jak se Inovelon používá
4. Možné nežádoucí účinky
5. Jak přípravek Inovelon uchovávat
6. Obsah balení a další informace
1. Co je Inovelon a k čemu se používá
Inovelon obsahuje lék rufinamid. Patří do skupiny léků zvaných antiepileptika, která se používají k léčbě epilepsie (stavu, kdy může docházek k záchvatům).
Inovelon se používá v kombinaci s dalšími léky k léčbě záchvatů spojených s Lennox-Gastautovým syndromem u dospělých, dospívajících a dětí starších 4 let. Lennox-Gastautův syndrom je označení pro skupinu závažných epilepsií, u nichž dochází k opakovaným záchvatům různého typu.
Inovelon Vám lékař předepsal ke snížení počtu záchvatů.
2. Čemu musíte věnovat pozornost, než začnete přípravek Inovelon užívat
Neužívejte přípravek Inovelon:
- jestliže jste alergický(á) na rufinamid, deriváty triazolu nebo na kteroukoli další složku přípravku Inovelon (uvedenou v bodě 6)
Upozornění a opatření
Informujte prosím lékaře nebo lékárníka:
- jestliže trpíte vrozeným syndromem krátkého QT intervalu nebo máte takový syndrom (poruchu elektrické aktivity srdce) v rodinné anamnéze, protože užívání rufinamidu by mohlo tento stav zhoršit.
- jestliže máte potíže s játry. O užívání rufinamidu jsou v této skupině pacientů k dispozici omezené informace, takže možná bude nutné dávku Vašeho léku zvyšovat pomaleji. Pokud trpíte těžkým onemocněním jater, může lékař rozhodnout, že pro Vás není přípravek Inovelon vhodný.
- jestliže dostanete kožní vyrážku nebo horečku. Může jít o známky alergické reakce. Okamžitě vyhledejte lékaře, protože ve velmi vzácných případech to může být závažné.
- jestliže se počet záchvatů, či jejich závažnost nebo trvání zvýší, měl(a) byste okamžitě kontaktovat lékaře.
- jestliže se u Vás objeví potíže s chůzí, abnormální pohyby, závrať nebo ospalost, informujte lékaře.
Poraďte se prosím s lékařem, i když výše uvedené stavy nastaly kdykoliv v minulosti.
Děti
Přípravek Inovelon se nesmí podávat dětem do 4 let věku, protože o použití u této věkové skupiny nejsou k dispozici dostatečné informace.
Další léčivé přípravky a Inovelon
Informujte lékaře nebo lékárníka o všech lécích, které užíváte nebo jste užíval(a) v nedávné době, a to i o lécích, které jsou dostupné bez lékařského předpisu. Jestliže užíváte jiné léky, které se z těla odstraňují enzymovým systémem CYP3A4, můžete vyžadovat důkladné sledování po dobu dvou týdnů na začátku nebo po ukončení léčby rufinamidem, nebo po jakékoliv výrazné změně dávky. Může být nutná změna dávky dalšího léku, který užíváte, protože při podávání s rufinamidem se může mírně snížit účinnost tohoto dalšího léku.
Informujte lékaře, jestliže užíváte hormonální/perorální antikoncepci. Přípravek Inovelon může snižovat účinek hormonální/perorální antikoncepce, například antikoncepčních pilulek. Proto se při užívání Inovelonu doporučuje používat ještě další bezpečnou a účinnou antikoncepční metodu.
Informujte lékaře, jestliže užíváte lék na ředění krve - warfarin. Může být nutné, aby lékař upravil dávkování.
Informujte lékaře, jestliže užíváte digoxin (lék používaný k léčbě srdečních onemocnění). Může být nutné, aby lékař upravil dávkování.
Pokud Vám lékař předepíše nebo doporučí doplňující léčbu epilepsie (např. valproát), musíte informovat lékaře, že užíváte Inovelon, protože možná bude nutné upravit dávkování.
Přípravek Inovelon s jídlem a pitím
Pokyny pro užívání Inovelonu s jídlem a pitím viz bod 3 „Jak se Inovelon používá“.
Těhotenství a kojení
Pokud jste žena v plodném věku, musíte během užívání přípravku Inovelon používat antikoncepci.
Pokud jste těhotná nebo se domníváte, že těhotná můžete být, či plánujete-li otěhotnět, poraďte se s lékařem nebo lékárníkem dříve, než začnete Inovelon užívat. Inovelon smíte užívat během těhotenství pouze v případě, že tak rozhodne lékař.
Během užívání přípravku Inovelon se nedoporučuje kojit, protože není známo, zda se rufinamid vylučuje do mateřského mléka.
Poraďte se s lékařem nebo lékárníkem dříve, než začnete užívat jakýkoliv lék.
Řízení dopravních prostředků a obsluha strojů
Užívání Inovelonu může způsobovat závratě a ospalost a může mít vliv na Vaše vidění, především na začátku léčby nebo po zvýšení dávky. Pokud k tomu dojde, neřiďte dopravní prostředek ani neobsluhujte žádné stroje.
Inovelon obsahuje methylparaben (E218) a propylparaben (E216)
Tyto složky mohou vyvolat alergické reakce (též opožděné).
3. Jak se Inovelon používá
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se s lékařem nebo lékárníkem.
Děti ve věku 4 let a starší o hmotnosti menší než 30 kg (neužívající valproát)
Doporučená počáteční dávka přípravku je 200 mg denně podávaných ve dvou dávkách. To je 5 ml suspenze podaných jako jedna 2,5ml dávka ráno a druhá 2,5ml dávka večer.
Lékař Vám dávku upraví a může ji zvyšovat o 200 mg každé dva dny až na maximální doporučenou dávku nepřesahující 1 000 mg denně (25 ml).
Děti ve věku 4 let a starší o hmotnosti menší než 30 kg (užívající valproát)
U dětí o hmotnosti menší než 30 kg, které užívají valproát (lék na epilepsii), je maximální doporučená denní dávka Inovelonu 600 mg denně.
Doporučená počáteční dávka přípravku je 200 mg denně ve dvou dávkách. To je 5 ml suspenze podaných jako jedna 2,5ml dávka ráno a druhá 2,5ml dávka večer.
Lékař Vám dávku upraví a může ji zvyšovat o 200 mg každé dva dny až na maximální doporučenou dávku 600 mg denně (15 ml).
Dospělí a děti o hmotnosti 30 kg nebo více
Obvyklá počáteční dávka je 400 mg denně podávaných ve dvou dávkách. To je 10 ml suspenze podaných jako jedna 5 ml dávka ráno a druhá 5 ml dávka večer.
Dávku Vám upraví lékař a může ji obden zvyšovat až o 400 mg až na denní dávku nepřesahující 3 200 mg, v závislosti na Vaší hmotnosti.
Někteří pacienti mohou reagovat na nižší dávky a lékař Vám může dávku upravit v závislosti na Vaší odpovědi na léčbu.
Pokud zaznamenáte nežádoucí účinky, lékař Vám může dávku zvyšovat pomaleji.
Inovelon se musí užívat dvakrát denně, jednou ráno a jednou večer, a má se užívat s jídlem.
Způsob podání
Na dávkování prosím používejte přiloženou stříkačku a adaptér.
Pokyny k použití stříkačky a adaptéru jsou uvedeny níže:
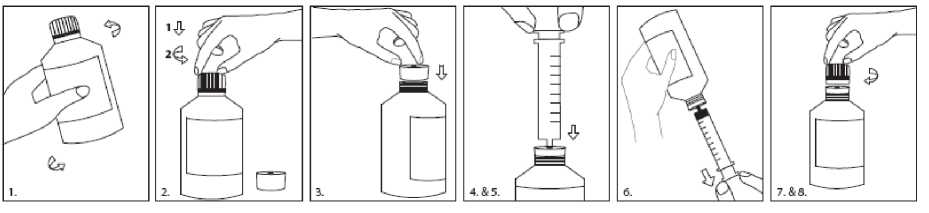
1. Před použitím dobře protřepejte.
2. Zamáčkněte víčko a pootočte jím. Lahev se otevře.
3. Zamáčkněte adaptér do hrdla lahve, až pevně zapadne.
4. Zatlačte píst stříkačky úplně dolů.
5. Zasuňte stříkačku co nejdál do otvoru adaptéru.
6. Otočte lahev dnem vzhůru a natáhněte předepsané množství přípravku Inovelon.
7. Otočte lahev zpět a vyjměte stříkačku.
8. Nechte adaptér na místě a uzavřete lahev víčkem. Umyjte stříkačku čistou vodou a pořádně ji osušte.
Nesnižujte si dávku ani nepřestávejte tento lék užívat, pokud Vám k tomu lékař nedá pokyn.
Jestliže jste užil(a) více přípravku Inovelon, než jste měl(a)
Jestliže jste snad užil(a) více přípravku Inovelon než jste měl(a), oznamte to okamžitě lékaři nebo lékárníkovi, nebo se obraťte na oddělení pohotovosti nejbližší nemocnice a vezměte si tento lék s sebou.
Jestliže jste zapomněl(a) užít Inovelon
Jestliže jste zapomněl(a) užít jednu dávku, pokračujte v užívání léku jako obvykle. Nezdvojujte následující dávku, abyste nahradil(a) vynechanou dávku. Jestliže jste vynechal(a) více než jednu dávku, poraďte se s lékařem.
Jestliže jste přestal(a) užívat Inovelon
Jestliže Vám lékař řekne, abyste léčbu ukončil(a), řiďte se jeho pokyny ohledně postupného snižování dávky přípravku Inovelon, aby se zmenšilo riziko zvýšení výskytu záchvatů.
Máte-li jakékoli další otázky, týkající se užívání tohoto přípravku, zeptejte se lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i Inovelon nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Následující vedlejší účinky mohou být velmi závažné:
Vyrážka a/nebo horečka. Může jít o známky alergické reakce. Pokud se u Vás tyto účinky objeví, okamžitě informujte svého lékaře nebo navštivte nemocnici.
Změna typu záchvatů / častější status epilepticus (záchvaty, které trvají delší dobu, opakované záchvaty). Okamžitě informujte svého lékaře.
Malý počet lidí léčených antiepileptiky, například Inovelonem, měl myšlenky na sebepoškozování nebo sebevraždu. Jakmile se u Vás tyto myšlenky vyskytnou, okamžitě se obraťte na svého lékaře.
Při užívání tohoto léku se u Vás mohou objevit následující nežádoucí účinky. Pokud pozorujete některý z níže uvedených účinků, sdělte to lékaři:
Velmi časté (více než u 1 z 10 pacientů) nežádoucí účinky přípravku Inovelon jsou:
Závratě, bolesti hlavy, nevolnost, zvracení, ospalost, únava.
Časté (více než u 1 ze 100 pacientů) nežádoucí účinky přípravku Inovelon jsou:
Problémy spojené s nervovým systémem zahrnující: potíže při chůzi, abnormální pohyby, křeče/záchvaty, neobvyklé pohyby očí, rozmazané vidění, třes.
Problémy spojené se zažíváním zahrnující: bolesti břicha, zácpu, zažívací potíže, řídkou stolici (průjem), ztrátu nebo změnu chuti k jídlu, ztrátu hmotnosti.
Infekce: ušní infekce, chřipka, ucpaný nos, hrudní infekce.
Pacienti dále mohou zaznamenat: úzkost, nespavost, krvácení z nosu, akné, vyrážku, bolesti zad, méně častou menstruaci, vznik modřin, poranění hlavy (v důsledku náhodného poranění během záchvatu).
Méně časté (mezi 1 ze 100 a 1 z 1 000 pacientů) nežádoucí účinky přípravku Inovelon jsou:
Alergické reakce a zvýšení hladin jaterních enzymů (ukazatelů funkce jater).
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v . Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Inovelon uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti, uvedené na štítku lahve a na krabičce. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Pokud Vám více než 90 dní od prvního otevření zbude v lahvi suspenze, nepoužívejte ji.
Nepoužívejte suspenzi, pokud si všimnete, že se vzhled nebo zápach Vašeho léku změnil. Vraťte lahev lékárníkovi.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co Inovelon obsahuje
- Léčivou látkou je rufinamid. Jeden mililitr obsahuje 40 mg rufinamidu. 5 ml obsahuje 200 mg rufinamidu.
- Pomocnými látkami jsou mikrokrystalická celulosa a karmelosa sodná sůl, kyselina citronová bezvodá, simetikonová emulze 30% (obsahující kyselinu benzoovou, cyklotetrasiloxan, dimetikon, glykol-stearát a glyceryl-distearát, methylcelulosu, PEG-40 stearát [polyethylenglykol-stearát], polysorbát 65, oxid křemičitý gel, kyselinu sorbovou, kyselinu sírovou a vodu), poloxamer 188, hydroxyethylcelulosa, methylparaben (E218), propylparaben (E216), kalium-sorbát, propylenglykol (E1520), sorbitol tekutý (nekrystalizující), pomerančová příchuť a voda.
Jak Inovelon vypadá a co obsahuje toto balení
• Inovelon je bílá, mírně viskózní suspenze. Dodává se v lahvi o objemu 460 ml se dvěma stejnými stříkačkami a zatlačovacím adaptérem lahve (PIBA). Stříkačky jsou opatřené stupnicí po 0,5 ml.
Držitel rozhodnutí o registraci a výrobce
Držitel rozhodnutí o registraci:
Eisai Ltd, European Knowledge Centre, Mosquito Way, Hatfield, Hertfordshire AL10 9SN, Velká Británie.
Výrobce:
Eisai Manufacturing Ltd, Mosquito Way, Hatfield, Hertfordshire AL10 9SN, Velká Británie.
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci
|
Belgie/Belgique/Belgien Eisai Europe Ltd. Tél/Tel: + 32 (0) 2 502 58 04 |
Lietuva Eisai Ltd. Tel: + 44 (0) 20 8600 1400 (Jungtiné Karalysté) |
|
EfcnrapHH Eisai Ltd. Tel: + 44 (0) 20 8600 1400 (OdegHHeHOTO KpaacTBo) |
Luxembourg/Luxemburg Eisai Europe Ltd. Tél/Tel: + 32 (0) 2 502 58 04 (Belgique/Belgien) |
|
Česká republika Eisai GesmbH organizační složka Tel: + 420 242 485 839 |
Magyarország Eisai Ltd. Tel.: + 44 (0) 20 8600 1400 Egyesult Királyság (Nagy-Britannia) |
|
Danmark Eisai AB Tlf: +46 (0) 8 501 01 600 (Sverige) |
Malta Associated Drug Company Ltd Tel: + 356 (0) 2277 8000 |
|
Deutschland Eisai GmbH Tel: + 49 (0) 696 65 85-0 |
Nederland Eisai BV. Tel: + 31 (0) 900 575 3340 |
|
Eesti (Eisai Ltd. Tel: + 44 (0) 20 8600 1400 (Ůhendkuningriik) |
Norge Eisai AB Tlf: + 46 (0) 8 501 01 600 (Sverige) |
|
EXlába Arriani Pharmaceuticals S.A. T qk: + 30 210 668 3000 |
Osterreich Eisai GesmbH Tel: + 43 (0) 1 535 1980-0 |
|
Espaňa Eisai Farmacéutica, S.A. Tel: +(34) 91 455 94 55 |
Polska Eisai Ltd. Tel.: + 44 (0) 20 8600 1400 (Wielka Brytania) |
|
France Eisai SAS Tél: + (33) 1 47 67 00 05 |
Portugal Eisai Farmacéutica, Unipessoal Lda Tel: + 351 21 487 55 40 |
|
Hrvatska Eisai Ltd. Tel: + 44 (0) 20 8600 1400 (Velika Britanija) |
Románia Eisai Ltd. Tel: + 44 (0) 208 600 1400 (Marea Britanie) |
|
Ireland Eisai Ltd. Tel: + 44 (0) 20 8600 1400 (United Kingdom) |
Slovenija Eisai Ltd. Tel: + 44 (0) 20 8600 1400 (Velika Britanija) |
|
Ísland Eisai AB Sími: + 46 (0) 8 501 01 600 (Svífejóó) |
Slovenská republika Eisai GesmbH organizační složka Tel: + 420 242 485 839 (Česká republika) |
|
Italia Eisai S.r.l. Tel: + 39 02 5181401 |
Suomi/Finland Eisai AB Puh/Tel: + 46 (0) 8 501 01 600 (Ruotsi/Sverige) |
|
Kónpoq Arriani Pharmaceuticals S.A. Tnk: +30 210 668 3000 (EkkáSa) |
Sverige Eisai AB Tel: + 46 (0) 8 501 01 600 |
|
Latvija Eisai Ltd. Tel: + 44 (0) 20 8600 1400 (Liebritánija) |
United Kingdom Eisai Ltd. Tel: + 44 (0) 20 8600 1400 |
Tato příbalová informace byla naposledy revidována {MM/RRRR}.
Podrobné informace o tomto přípravku jsou uveřejněny na webových stránkách Evropské agentury pro léčivé přípravky: http://www.ema.europa.eu
73