Inlyta 5 Mg
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Inlyta 1 mg potahované tablety Inlyta 3 mg potahované tablety Inlyta 5 mg potahované tablety Inlyta 7 mg potahované tablety
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Inlyta 1 mg potahované tablety
Jedna potahovaná tableta obsahuje axitinibum 1 mg.
Inlyta 3 mg potahované tablety
Jedna potahovaná tableta obsahuje axitinibum 3 mg.
Inlyta 5 mg potahované tablety
Jedna potahovaná tableta obsahuje axitinibum 5 mg.
Inlyta 7 mg potahované tablety
Jedna potahovaná tableta obsahuje axitinibum 7 mg.
Pomocné látky se známým účinkem Inlyta 1 mg potahované tablety
Jedna potahovaná tableta obsahuje 33,6 mg monohydrátu laktosy
Inlyta 3 mg potahované tablety
Jedna potahovaná tableta obsahuje 35,3 mg monohydrátu laktosy
Inlyta 5 mg potahované tablety
Jedna potahovaná tableta obsahuje 58,8 mg monohydrátu laktosy
Inlyta 7 mg potahované tablety
Jedna potahovaná tableta obsahuje 82,3 mg monohydrátu laktosy
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Potahovaná tableta (tableta).
Inlyta 1 mg potahované tablety
Červené potahované tablety oválného tvaru s vyraženým nápisem “Pfizer” na jedné straně a “1 XNB” na druhé straně.
Inlyta 3 mg potahované tablety
Červené potahované tablety kulatého tvaru s vyraženým nápisem “Pfizer” na jedné straně a “3 XNB” na druhé straně.
Inlyta 5 mg potahované tablety
Červené potahované tablety trojúhelníkového tvaru s vyraženým nápisem “Pfizer” na jedné straně a “5 XNB” na druhé straně.
Inlyta 7 mg potahované tablety
Červené potahované tablety čtyřúhelníkového tvaru s vyraženým nápisem “Pfizer” na jedné straně a “7 XNB” na druhé straně.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Inlyta je indikován k léčbě dospělých pacientů s pokročilým renálním karcinomem (RCC) po selhání předchozí léčby sunitinibem nebo cytokiny.
4.2 Dávkování a způsob podání
Léčba přípravkem Inlyta má být vedena lékařem se zkušenostmi s podáváním protinádorových léčivých přípravků.
Dávkování
Doporučená dávka axitinibu je 5 mg dvakrát denně.
Léčba by měla pokračovat tak dlouho, dokud je pozorován klinický přínos nebo dokud se nevyskytne netolerovatelná toxicita, kterou nelze zvládnout souběžně podávanými léčivými přípravky nebo úpravou dávky.
Pokud pacient zvrací nebo vynechá dávku, nemá se podávat dodatečná dávka. Další předepsaná dávka se má vzít v obvyklou dobu.
Úprava dávky
Zvýšení nebo snížení dávky se doporučuje na základě individuální bezpečnosti a snášenlivosti.
Pacientům, kteří snáší zahajovací dávku axitinibu 5 mg dvakrát denně bez nežádoucích účinků > stupně 2 (t.j. bez závažných nežádoucích účinků podle obecných terminologických kritérií pro nežádoucí účinky [CTCAE] verze 3.0) dva po sobě následující týdny, lze zvýšit dávku na 7 mg dvakrát denně, pokud není krevní tlak pacienta vyšší než 150/90 mmHg nebo pokud není pacient léčen antihypertenzními léky. Dále může být za použití stejných kritérií pacientům, kteří snášejí dávku 7 mg dvakrát denně, zvýšena dávka na maximální dávku 10 mg dvakrát denně.
Léčba některých nežádoucích účinků může vyžadovat dočasné nebo trvalé vysazení axitinibu a/nebo snížení dávky axitinibu (viz bod 4.4). Pokud je nutné snížení dávky, může být dávka axitinibu snížena na 3 mg dvakrát denně a dále na 2 mg dvakrát denně.
Úprava dávky není nutná z důvodu věku, rasy, pohlaví nebo tělesné hmotnosti pacienta.
Souběžně podávané silné inhibitory CYP3A4/5
Souběžné podávání axitinibu se silnými inhibitory CYP3A4/5 může zvýšit plazmatickou koncentraci axitinibu (viz bod 4.5). Doporučuje se, aby byl souběžně podáván alternativní léčivý přípravek s žádným nebo minimálním potenciálem pro inhibici CYP3A4/5.
Přestože úprava dávky axitinibu nebyla u pacientů léčených silnými inhibitory CYP3A4/5 sledována, pokud musí být silný inhibitor CYP3A4/5 souběžně podáván, doporučuje se snížení dávky axitinibu přibližně na polovinu (např. zahajovací dávka má být snížena z 5 mg dvakrát denně na 2 mg dvakrát denně). Léčba některých nežádoucích účinků může vyžadovat dočasné nebo trvalé vysazení axitinibu (viz bod 4.4). Pokud je souběžné podávání silného inhibitoru ukončeno, je nutno zvážit návrat k dávce axitinibu používané před zahájením podávání silného inhibitoru CYP3A4/5 (viz bod 4.5).
Souběžně podávané silné induktory CYP3A4/5
Souběžné podávání axitinibu se silnými induktory CYP3A4/5 může snížit plazmatickou koncentraci axitinibu (viz bod 4.5). Doporučuje se, aby byl souběžně podáván alternativní léčivý přípravek s žádným nebo minimálním potenciálem pro indukci CYP3A4/5.
Přestože úprava dávky axitinibu nebyla u pacientů léčených silnými induktory CYP3A4/5 sledována, pokud musí být silný induktor CYP3A4/5 souběžně podáván, doporučuje se postupné zvyšování dávky axitinibu. Bylo hlášeno, že k maximální indukci při podávání vysokých dávek silných induktorů CYP3A4/5 dochází v průběhu jednoho týdne léčby induktory. Pokud je dávka axitinibu zvýšena, má být pacient pečlivě monitorován z hlediska toxicity. Léčba některých nežádoucích účinků může vyžadovat dočasné nebo trvalé vysazení a/nebo snížení dávky axitinibu (viz bod 4.4). Pokud je souběžné podávání silného induktoru ukončeno, je nutno se okamžitě vrátit k dávce axitinibu používané před zahájením podávání silného induktoru CYP3A4/5 (viz bod 4.5).
Zvláštní _ populace
Starší osoby (> 65 let): Není nutná úprava dávky (viz body 4.4 a 5.2).
Porucha funkce ledvin: Není nutná úprava dávky (viz bod 5.2). Nejsou k dispozici prakticky žádné údaje týkající se léčby axitinibem u pacientů s clearance kreatininu <15 ml/min.
Porucha funkce jater: Při podávání axitinibu pacientům s mírnou poruchou jater (Child-Pugh třída A) není nutná úprava dávky. Pokud je axitinib podáván pacientům se středně závažnou poruchou jater (Child-Pugh třída B), doporučuje se snížení dávky (např. zahajovací dávka by měla být snížena z 5 mg dvakrát denně na 2 mg dvakrát denně). Axitinib nebyl zkoumán u pacientů se závažným poškozením jater (Child-Pugh třída C) a u této populace se nemá používat (viz body 4.4 a 5.2).
Pediatrická populace
Bezpečnost a účinnost axitinibu u dětí a dospívajících ve věku <18 let nebyly stanoveny. Nejsou dostupné žádné údaje.
Způsob podání
Axitinib se užívá perorálně dvakrát denně v přibližně 12hodinovém odstupu, s jídlem nebo bez jídla (viz bod 5.2). Tablety axitinibu se mají spolknout celé, zapít sklenicí vody.
4.3 Kontraindikace
Hypersenzitivita na axitinib nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Specifické bezpečnostní příhody je nutno sledovat před zahájením podávání axitinibu a pravidelně v průběhu jeho podávání, jak je uvedeno níže.
Příhody srdečního selhání
V klinických studiích s axitinibem při léčbě pacientů s RCC byly hlášeny příhody srdečního selhání (včetně srdečního selhání, městnavého srdečního selhání, kardiopulmonálního selhání, dysfunkce levé komory, snížené ejekční frakce a selhání pravé komory) (viz bod 4.8).
Známky a příznaky srdečního selhání je třeba během léčby axinitibem pravidelně sledovat. Léčba příhod srdečního selhání může vyžadovat dočasné nebo trvalé vysazení a/nebo snížení dávky axitinibu.
Hypertenze
V klinických studiích s axitinibem při léčbě pacientů s RCC byla velmi často hlášena hypertenze (viz bod 4.8).
V kontrolované klinické studii byla střední doba nástupu hypertenze (systolický krevní tlak
> 150 mmHg nebo diastolický krevní tlak > 100 mmHg) v průběhu prvního měsíce po zahájení léčby axitinibem a zvýšení krevního tlaku bylo zjištěno dokonce již po 4 dnech po zahájení léčby axitinibem.
Před zahájením podávání axitinibu by měl být dobře kontrolován krevní tlak. U pacienta by měla být monitorována hypertenze a léčena standardními antihypertenzními léky. V případě hypertenze přetrvávající navzdory podávání antihypertenzních léčivých přípravků by měla být dávka axitinibu snížena. U pacientů, u kterých se vyvine závažná hypertenze, přerušte dočasně podávání axitinibu a znovu zahajte léčbu s nižší dávkou, až bude pacient normotenzní. Při přerušení podávání axitinibu mají být pacienti léčení antihypertenzními přípravky monitorováni pro možnou hypotenzi (viz bod 4.2).
V případě závažné nebo přetrvávající arteriální hypertenze a symptomů ukazujících na syndrom posteriorní reverzibilní encefalopatie (PRES) (viz níže), je nutno zvážit diagnostické zobrazení mozku magnetickou rezonancí (MRI).
Dysfunkce štítné žlázy
V klinických studiích s axitinibem při léčbě pacientů s RCC byly hlášeny případy hypotyreózy a v menší míře hypertyreózy (viz bod 4.8).
Funkci štítné žlázy je nutno monitorovat před zahájením podávání axitinibu a pravidelně v průběhu jeho podávání. Hypotyreóza nebo hypertyreóza by měly být léčeny podle standardní léčebné praxe, aby byl zachován eutyreoidní stav.
Arteriální tromboembolické příhody
V klinických studiích s axitinibem byly hlášeny arteriální tromboembolické příhody (včetně tranzitorní ischemické ataky, infarktu myokardu, cerebrovaskulární příhody a okluze retinální arterie), (viz bod 4.8).
S opatrností má být axitinib používán u pacientů s rizikem těchto příhod a u pacientů, kteří je prodělali. Axitinib nebyl zkoušen u pacientů, kteří prodělali arteriální tromboembolickou příhodu v uplynulých 12 měsících.
Venózní tromboembolické příhody
V klinických studiích s axitinibem byly hlášeny venózní tromboembolické příhody (včetně plicní embolie, hluboké žilní trombózy a okluze/trombózy retinální vény), (viz bod 4.8).
S opatrností má být axitinib používán u pacientů s rizikem těchto příhod a u pacientů, kteří je prodělali. Axitinib nebyl zkoušen u pacientů, kteří prodělali venózní tromboembolickou příhodu v uplynulých 6 měsících.
Zvýšení hodnot hemoglobinu nebo hematokritu
Během léčby axitinibem může dojít ke zvýšení hodnot hemoglobinu nebo hematokritu, jako důsledek zvýšení celkového počtu červených krvinek (viz bod 4.8, polycytemie). Zvýšení celkového počtu červených krvinek může zvýšit riziko tromboembolických příhod.
Hodnoty hemoglobinu nebo hematokritu je nutno monitorovat před zahájením podávání axitinibu a pravidelně v průběhu jeho podávání. Pokud se hodnoty hemoglobinu nebo hematokritu zvýší nad normální hodnotu, mají být pacienti léčeni podle standardní léčebné praxe ke snížení hodnoty hemoglobinu nebo hematokritu na přijatelnou úroveň.
Krvácení
V klinických studiích s axitinibem byly hlášeny krvácivé příhody (viz bod 4.8).
Axitinib nebyl zkoušen u pacientů s prokázanými neléčenými mozkovými metastázami nebo s recentním aktivním gastrointestinálním krvácením, a u těchto pacientů by neměl být podáván. Pokud jakékoli krvácení vyžaduje léčebný zásah, má být podávání axitinibu přechodně přerušeno.
Gastrointestinální _perforace a tvorba _píštělí
V klinických studiích s axitinibem byly hlášeny příhody gastrointestinální perforace a píštělí (viz bod 4.8).
Příznaky gastrointestinální perforace a píštěle musí být pravidelně monitorovány po celou dobu léčby axitinibem.
Komplikace hojení ran
Nebyly provedeny žádné oficiální studie vlivu axitinibu na hojení ran.
Léčba axitinibem má být vysazena nejméně 24 hodin před plánovaným operačním výkonem. Rozhodnutí o opětovném nasazení axitinibu po operačním výkonu má být učiněno podle klinického posouzení dostatečného hojení rány.
Syndrom _posteriorní reverzibilní encefalopatie (PRES)
V klinických studiích s axitinibem byly hlášeny případy PRES, (viz bod 4.8).
PRES je neurologická porucha, která se může projevit bolestí hlavy, křečemi, letargií, zmateností, oslepnutím a dalšími poruchami vidění a neurologickými poruchami. Může být přítomna mírná až závažná hypertenze. Zobrazení magnetickou resonancí je nezbytné pro potvrzení diagnózy PRES. U pacientů s příznaky PRES je třeba přechodně přerušit léčbu axitinibem nebo ji trvale vysadit. Bezpečnost opětovného zahájení léčby axitinibem u pacientů, u kterých se dříve vyskytl PRES, není známa.
Proteinurie
V klinických studiích s axitinibem byla hlášena proteinurie včetně proteinurie závažnosti stupně 3 a 4 (viz bod 4.8).
Proteinurii je nutno monitorovat před zahájením podávání axitinibu a pravidelně v průběhu jeho podávání. U pacientů, u nichž se vyvine středně závažná až závažná proteinurie, je třeba snížit dávku axitinibu nebo přechodně axitinib vysadit (viz bod 4.2). Léčba axitinibem má být přerušena, pokud u pacienta dojde k rozvoji nefrotického syndromu.
Nežádoucí účinky související s játry
V kontrolované klinické studii s axitinibem při léčbě pacientů s RCC byly hlášeny nežádoucí účinky související s játry. Mezi nejčastěji hlášené nežádoucí účinky související s játry patří zvýšení alaninaminotransferázy (ALT), aspartátaminotransferázy (AST) a bilirubinu v krvi (viz bod 4.8). Nebylo pozorováno současné zvýšení ALT (více než 3násobné překročení horní hranice normálu [ULN]) a bilirubinu (více než 2násobek ULN).
V klinické studii zjišťující dávku bylo pozorováno u 1 pacienta, který dostával axitinib v zahajovací dávce 20 mg dvakrát denně (4násobek doporučené zahajovací dávky) současné zvýšení ALT (12násobek ULN) a bilirubinu (2,3násobek ULN), posouzené jako hepatotoxicita související s léčbou.
Jaterní testy je nutno monitorovat před zahájením podávání axitinibu a pravidelně v průběhu jeho podávání.
Porucha funkce jater
V klinických studiích s axitinibem byla systémová expozice axitinibu přibližně dvakrát vyšší u subjektů se středně závažnou poruchou jater (Child-Pugh třída B) v porovnání se subjekty s normální jatemí funkcí. Když je axitinib podáván pacientům se středně závažnou poruchou jater (Child-Pugh třída B), doporučuje se snížení dávky (viz bod 4.2).
Axitinib nebyl zkoumán u pacientů se závažným poškozením jater (Child-Pugh třída C) a nemá být používán pro tyto pacienty.
Starší osoby (> 65 let) a rasa
V kontrolované klinické studii s axitinibem při léčbě pacientů s RCC bylo 34 % pacientů léčených axitinibem ve věku > 65 let. Většina pacientů byli běloši (77 %) nebo Asiaté (21 %). Přestože u starších pacientů a Asiatů nelze vyloučit vyšší náchylnost k vývoji nežádoucích účinků, nebyly vcelku pozorovány větší rozdíly v bezpečnosti a účinnosti axitinibu mezi pacienty ve věku > 65 let a mladšími, ani mezi bělochy a pacienty jiných ras.
Není nutná úprava dávky podle věku či rasy pacienta (viz body 4.2 a 5.2).
Laktóza
Tento léčivý přípravek obsahuje laktózu. Pacienti se vzácnými dědičnými problémy s intolerancí galaktózy, vrozeným nedostatkem laktázy nebo malabsorpcí glukózy a galaktózy by tento léčivý přípravek neměli užívat.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Data in vitro ukazují, že axitinib je metabolizován hlavně cytochromem CYP3A4/5 a v menší míře cytochromy CYP1A2, CYP2C19 a uridin difosfát-glukuronosyltransferázou (UGT) 1A1.
Inhibitory CYP3A4/5
Ketokonazol, silný inhibitor CYP3A4/5, podávaný v dávce 400 mg jednou denně po 7 dnů zvyšoval po jedné perorální dávce 5 mg axitinibu u zdravých dobrovolníků střední plochu pod křivkou (AUC) 2násobně a Cmax 1,5násobně. Souběžné podávání axitinibu a silných inhibitorů CYP3A4/5 (např. ketokonazolu, itrakonazolu, klarithromycinu, erythromycinu, atazanaviru, indinaviru, nefazodonu, nelfinaviru, ritonaviru, sachinaviru a telithromycinu) může zvyšovat plasmatickou koncentraci axitinibu. Grapefruit může také zvýšit plasmatickou koncentraci axitinibu. Doporučuje se, aby byly souběžně podávány léčivé přípravky s žádným nebo minimálním potenciálem pro inhibici CYP3A4/5. Pokud musí být silný inhibitor CYP3A4/5 souběžně podáván, doporučuje se úprava dávky axitinibu (viz bod 4.2).
Inhibitory CYP1A2 a CYP2C19
CYP1A2 a CYP2C19 představují méně významné cesty (< 10 %) v metabolismu axitinibu. Účinek silných inhibitorů těchto isoenzymů na farmakokinetiku axitinibu nebyl zkoumán. Je nutná opatrnost vzhledem k riziku zvýšení plasmatické koncentrace axitinibu u pacientů užívajících silné inhibitory těchto isoenzymů.
Induktory CYP3A4/5
Rifampicin, silný induktor CYP3A4/5, podávaný v dávce 600 mg jednou denně po 9 dnů, snižoval po jedné dávce 5 mg axitinibu u zdravých dobrovolníků střední AUC o 79 % a Cmax o 71 %.
Souběžné podávání axitinibu se silnými induktory CYP3A4/5 (např. rifampicinem, dexamethasonem, fenytoinem, karbamazepinem, rifabutinem, rifapentinem, fenobarbitalem a Hypericum perforatum [třezalka tečkovaná]) může snížit plasmatickou koncentraci axitinibu. Doporučuje se, aby byly souběžně podávány léčivé přípravky s žádným nebo minimálním potenciálem pro indukci CYP3A4/5. Pokud musí být silný induktor CYP3A4/5 souběžně podáván, doporučuje se úprava dávky axitinibu (viz bod 4.2).
Studie inhibice a indukce CYP a UGT in vitro
Studie in vitro ukázaly, že axitinib v terapeutické plasmatické koncentraci neinhibuje CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5 ani UGT1A1.
Studie in vitro ukázaly, že axitinib má potenciál pro inhibici CYP1A2. Proto může souběžné podávání axitinibu se substráty CYP1A2 vést ke zvýšení plasmatické koncentrace substrátů CYP1A2 (např. theofylinu).
Studie in vitro také ukázaly, že axitinib má potenciál pro inhibici CYP2C8. Souběžné podávání axitinibu s paklitaxelem, známým substrátem CYP2C8, však nevedlo ke zvýšení plasmatické koncentrace paklitaxelu u pacientů s pokročilou malignitou, což ukazuje na chybění klinické inhibice CYP2C8.
Studie in vitro na lidských hepatocytech také ukázaly, že axitinib neindukuje CYP1A1, CYP1A2 ani CYP3A4/5. Proto se neočekává, že by souběžné podávání axitinibu snižovalo plasmatickou koncentraci souběžně podávaných substrátů CYP1A1, CYP1A2 či CYP3A4/5 in vivo.
Studie in vitro s P-glykoproteinem
Studie in vitro ukázaly, že axitinib inhibuje P-glykoprotein. Neočekává se však, že by axitinib v terapeutické plasmatické koncentraci inhiboval P-glykoprotein. Proto se neočekává, že by souběžné podávání axitinibu zvyšovalo plasmatickou koncentraci digoxinu či jiných substrátů P-glykoproteinu
in vivo.
4.6 Fertilita, těhotenství a kojení
O použití axitinibu u těhotných žen neexistují žádné údaje. Na základě farmakologických vlastností axitinibu může při podání těhotným ženám dojít k poškození plodu. Studie na zvířatech ukázaly reprodukční toxicitu včetně vzniku malformací (viz bod 5.3). Axitinib nemá být používán v těhotenství, pokud léčbu tímto léčivým přípravkem nevyžaduje klinický stav ženy.
Ženy schopné otěhotnění musí během léčby a 1 týden po jejím skončení používat účinnou antikoncepční metodu.
Kojení
Není známo, zda se axitinib vylučuje mateřským mlékem. Riziko pro kojence nelze vyloučit. Axitinib nemá být u kojících žen používán.
Fertilita
Na základě neklinických zjištění má axitinib u lidí potenciál pro narušení reprodukčních funkcí a fertility (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Axitinib má malý vliv na schopnost řídit nebo obsluhovat stroje. Pacienty je nutno upozornit, že se u nich během léčby axitinibem mohou vyskytnout příhody, jako jsou např. závratě a/nebo únava.
4.8 Nežádoucí účinky
Souhrn bezpečnostního _profilu
Následující rizika a příslušná opatření jsou podrobněji probrána v bodu 4.4: příhody srdečního selhání, hypertenze, dysfunkce štítné žlázy, arteriální tromboembolické příhody, venózní tromboembolické příhody, zvýšení hodnot hemoglobinu nebo hematokritu, krvácení, gastrointestinální perforace a tvorba píštělí, komplikace hojení ran, PRES, proteinurie a zvýšení hodnot jaterních enzymů.
Nejčastější (> 20 %) nežádoucí účinky pozorované po léčbě axitinibem byl průjem, hypertenze, únava, snížená chuť k jídlu, nauzea, úbytek tělesné hmotnosti, dysfonie, syndrom palmární-plantární erytrodysestézie (syndrom ruka-noha), krvácení, hypotyreóza, zvracení, proteinurie, kašel a zácpa.
Tabulkový seznam nežádoucích účinků
V tabulce 1 jsou uvedeny nežádoucí účinky hlášené v souhrnném souboru dat od 672 pacientů léčených axitinibem v klinických studiích hodnotících léčbu pacientů s RCC (viz bod 5.1).
Nežádoucí účinky jsou uvedeny podle orgánových systémů, četnosti a stupně závažnosti. V každé skupině četnosti jsou nežádoucí účinky definovány jako: velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1 000 až < 1/100), vzácné (> 1/10 000 až < 1/1 000), velmi vzácné (< 1/10 000) a není známo (z dostupných údajů nelze určit). Současná bezpečnostní databáze pro axitinib je příliš malá a nelze zjistit vzácné a velmi vzácné nežádoucí účinky.
Kategorie byly vytvořeny na základě absolutní četnosti v údajích ze souhrnných klinických studií.
V každé skupině orgánového systému jsou nežádoucí účinky se stejnou četností seřazeny podle klesající závažnosti.
Tabulka 1. Nežádoucí účinky hlášené
ve studiích s RCC u pacientů léčených axitinibem
N= 672)
|
Orgánový systém |
Četnost |
Nežádoucí účinkya |
Všechny . vb stupně % |
Stupeň 3b % |
Stupeň 4b % |
|
Poruchy krevního a lymfatického systému |
Časté |
6,3 |
1,2 |
0,4 | |
|
Trombocytopenie |
1,6 |
0,1 |
0 | ||
|
Polycytémiec |
1,5 |
0,1 |
0 | ||
|
Méně časté |
Neutropenie |
0,3 |
0,1 |
0 | |
|
Leukopenie |
0,4 |
0 |
0 | ||
|
Endokrinní poruchy |
Velmi časté |
Hypotyreózac |
24,6 |
0,3 |
0 |
|
Časté |
Hypertyreózac |
1,6 |
0,1 |
0,1 | |
|
Poruchy metabolismu a výživy |
Velmi časté |
Snížená chuť k jídlu |
39,0 |
3,6 |
0,3 |
|
Časté |
Dehydratace |
6,7 |
3,1 |
0,3 | |
|
Hyperkalémie |
2,7 |
1,2 |
0,1 | ||
|
Hyperkalcémie |
2,2 |
0,1 |
0,3 | ||
|
Poruchy nervového systému |
Velmi časté |
16,2 |
0,7 |
0 | |
|
Poruchy chuti |
11,5 |
0 |
0 | ||
|
Časté |
Závratě |
9,1 |
0,6 |
0 | |
|
Méně časté |
Syndrom posteriorní reverzibilní encefalopatiee |
0,3 |
0,1 |
0 | |
|
Poruchy ucha a labyrintu |
Časté |
Tinnitus |
3,1 |
0 |
0 |
|
Srdeční poruchy |
Časté |
Příhody srdečního selhání0,4 f |
1,8 |
0,3 |
0,7 |
|
Cévní poruchy |
Velmi časté |
Hypertenze8 |
51,2 |
22,0 |
1,0 |
|
1/ ^ ,c, d, h Krvácení |
25,7 |
3,0 |
1,0 | ||
|
Časté |
Venózní tromboembolické příhody4 d i |
2,8 |
0,9 |
1,2 | |
|
Arteriální tromboembolické příhody4 d j |
2,8 |
1,2 |
1,3 | ||
|
Respirační, hrudní a mediastinální poruchy |
Velmi časté |
17,1 |
3,6 |
0,6 | |
|
20,4 |
0,6 |
0 | |||
|
Dysfonie |
32,7 |
0 |
0,1 | ||
|
Časté |
Orofaryngeální bolest |
7,4 |
0 |
0 |
|
Orgánový systém |
Četnost |
Nežádoucí účinkya |
Všechny stupněb % |
Stupeň 3b % |
Stupeň 4b % |
|
Gastrointestinální poruchy |
Velmi časté |
55,4 |
10,1 |
0,1 | |
|
23,7 |
2,7 |
0,1 | |||
|
33,0 |
2,2 |
0,1 | |||
|
14,7 |
2,5 |
0,3 | |||
|
Zácpa |
20,2 |
1,0 |
0 | ||
|
Stomatitida |
15,5 |
1,8 |
0 | ||
|
11,2 |
0,1 |
0 | |||
|
Časté |
Bolest v epigastriu |
9,4 |
0,9 |
0 | |
|
Glosodynie |
2,8 |
0 |
0 | ||
|
Flatulence |
4,5 |
0 |
0 | ||
|
Hemoroidy |
3,3 |
0 |
0 | ||
|
Gastrointestinální perforace a píštěl c k |
1,9 |
0,9 |
0,3 | ||
|
Poruchy jater a žlučových cest |
Časté |
Hyperbilirubinémie |
1,3 |
0,1 |
0,1 |
|
Poruchy kůže a podkožní tkáně |
Velmi časté |
Syndrom palmární-plantární erytrodysestézie (syndrom ruka-noha) |
32,1 |
7,6 |
0 |
|
14,3 |
0,1 |
0 | |||
|
Suchá kůže |
10,1 |
0,1 |
0 | ||
|
Časté |
6,0 |
0 |
0 | ||
|
Erytém |
3,7 |
0 |
0 | ||
|
Alopecie |
5,7 |
0 |
0 | ||
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Velmi časté |
Bolest končetin |
14,1 |
1,0 |
0,3 |
|
Artralgie |
17,7 |
1,9 |
0,3 | ||
|
Časté |
Myalgie |
8,2 |
0,6 |
0,1 | |
|
Poruchy ledvin a močových cest |
Velmi časté |
| Proteinurie |
21,1 |
4,8 |
0,1 |
|
Časté |
Renální selháním |
1,6 |
0,9 |
0,1 | |
|
Celkové poruchy a reakce v místě aplikace |
Velmi časté |
Únava |
45,1 |
10,6 |
0,3 |
|
Astenied |
13,8 |
2,8 |
0,3 | ||
|
Zánět sliznic |
13,7 |
1,0 |
0 | ||
|
Vyšetření |
Velmi časté |
Úbytek tělesné hmotnosti |
32,7 |
4,9 |
0 |
|
Časté |
Zvýšení lipázy |
3,7 |
0,7 |
0,7 | |
|
Zvýšení alaninaminotransferázy |
6,5 |
1,2 |
0 | ||
|
Zvýšení amylázy |
3,4 |
0,6 |
0,4 | ||
|
Zvýšení aspartátaminotransferázy |
6,1 |
1,0 |
0 | ||
|
Zvýšení alkalické fosfatázy |
4,8 |
0,3 |
0 | ||
|
Zvýšení kreatininu |
5,7 |
0,4 |
0 | ||
|
Zvýšení TSH |
7,9 |
0 |
0 |
a Nežádoucí účinky podle frekvence všech událostí během léčby
bObecná terminologická kritéria National Cancer Institute Common Terminology Criteria pro nežádoucí příhody, verze 3.0 cViz bod Popis vybraných nežádoucích účinků d Byly hlášeny případy úmrtí (stupeň 5) e Včetně leukoencefalopatie
f
Včetně srdečního selhání, městnavého srdečního selhání, kardiopulmonálního selhání, snížení ejekční frakce, dysfunkce levé komory a selhání pravé komory g Včetně akcelerované hypertenze, zvýšeného krevního tlaku, hypertenze ahypertenzní krize h Včetně prodlouženého aktivovaného parciálního tromboplastinového času, análního krvácení, arteriálního krvácení, krve přítomné v moči, krvácení do centrálního nervového systému, krvácení do mozku, prodlouženého koagulačního času, krvácení spojivky, kontuze, hemoragického průjmu, dysfunkčního děložního krvácení, epistaxe, krvácení do žaludku, gastrointestinálního krvácení, krvácení z dásní, hematemeze, hematochezie, sníženého hematokritu, hematomu, hematurie, sníženého hemoglobinu, hemoptýzy, krvácení, krvácení z koronární arterie, krvácení z močového traktu, hemoroidálního krvácení, hemostázy, zvýšené náchylnosti k tvoření modřin, zvýšeného INR (international normalized ratio), krvácení z dolní části gastrointestinálního traktu, meleny, petechií, krvácení z faryngu, prodlouženého protrombinového času, plicního krvácení, purpury, krvácení z rekta, sníženého počtu erytrocytů, renálního krvácení, krvácení do skléry, skrotální hematokély, hematomu sleziny, třískovité hemorhagie, subarachnoidálního krvácení, krvácení jazyka, krvácení v horní části gastrointestinálního traktu a vaginálního krvácení i Včetně Buddova-Chiariho syndromu, hluboké žilní trombózy, trombózy vena jugularis, žilní trombózy pánve, plicní embolie, okluze retinální vény, trombózy retinální vény, trombózy vena subclavia, žilní trombózy a žilní trombózy končetiny j Včetně akutního infarktu myokardu, embolizace, infarktu myokardu, okluze retinální arterie a tranzitorní ischemické ataky
k Gastrointestinální perforace a píštěl zahrnuje následující preferované termíny: abdominální absces, anální absces, anální píštěl, píštěl, gastrointestinální anastomotické prosakování, gastrointestinální perforace, perforace tračníku, ezofagobronchiální píštěl a peritonitida l Proteinurie zahrnuje následující preferované termíny: bílkovina v moči, přítomná bílkovina v moči a proteinurie
m Včetně akutního renálního selhání
Popis vybraných nežádoucích účinků Příhody srdečního selhání (viz bod 4.4)
V kontrolované klinické studii s axitinibem (N = 359) při léčbě pacientů s RCC byly u 1,7 % pacientů užívajících axitinib hlášeny příhody srdečního selhání, včetně srdečního selhání (0,6 %), kardiopulmonálního selhání (0,6 %), dysfunkce levé komory (0,3 %) a selhání pravé komory (0,3 %). Nežádoucí účinky srdečního selhání stupně 4 byly hlášeny u 0,6 % pacientů užívajících axitinib. Srdeční selhání vedoucí k úmrtí bylo hlášeno u 0,6 % pacientů užívajících axitinib.
Ve studiích s monoterapií axitinibem (N = 672) při léčbě pacientů s RCC byly u 1,8 % pacientů užívajících axitinib hlášeny příhody srdečního selhání (včetně srdečního selhání, městnavého srdečního selhání, kardiopulmonálního selhání, dysfunkce levé komory, snížené ejekční frakce a selhání pravé komory). Příhody srdečního selhání stupně 3/4 byly hlášeny u 1,0 % pacientů a příhody srdečního selhání vedoucího k úmrtí byly hlášeny u 0,3 % pacientů užívajících axitinib.
Dysfunkce štítné žlázy (viz bod 4.4)
V kontrolované klinické studii s axitinibem při léčbě pacientů s RCC byla hlášena hypotyreóza v 20,9 % případů a hypertyreóza v 1,1 % případů. Zvýšení tyreotropního hormonu (TSH) bylo hlášeno jako nežádoucí účinek u 5,3 % pacientů léčených axitinibem. Při rutinním laboratorním vyšetření u pacientů, kteří měli před léčbou TSH < 5 ^U/ml, došlo ke zvýšení TSH na > 10 ^U/ml u 32,2 % pacientů léčených axitinibem.
V souhrnných klinických studiích s axitinibem (N = 672) při léčbě pacientů s RCC byla hlášena hypotyreóza u 24,6 % pacientů léčených axitinibem. Hypertyreóza byla hlášena u 1,6 % pacientů léčených axitinibem.
Venózní tromboembolicképříhody (viz bod 4.4)
V kontrolované klinické studii s axitinibem při léčbě pacientů s RCC byly hlášeny venózní tromboembolické nežádoucí účinky u 3,9 % pacientů léčených axitinibem, včetně plicní embolie (2,2 %), okluze/trombózy retinální vény (0,6 %) a hluboké žilní trombózy (0,6 %). Venózní tromboembolické nežádoucí účinky stupně 3/4 byly hlášeny u 3,1 % pacientů léčených axitinibem. Fatální plicní embolie byla hlášena u jednoho pacienta (0,3 %) léčeného axitinibem.
V souhrnných klinických studiích s axitinibem (N = 672) při léčbě pacientů s RCC byly hlášeny venózní tromboembolické příhody u 2,8 % pacientů léčených axitinibem. Venózní tromboembolické příhody stupně 3 byly hlášeny u 0,9 % pacientů. Venózní tromboembolické příhody stupně 4 byly hlášeny u 1,2 % pacientů. Fatální venózní tromboembolické příhody byly hlášeny u 0,1 % pacientů léčených axitinibem.
Arteriální tromboembolické příhody (viz bod 4.4)
V kontrolované klinické studii s axitinibem při léčbě pacientů s RCC byly hlášeny arteriální tromboembolické nežádoucí účinky u 4,7 % pacientů léčených axitinibem, včetně infarktu myokardu (1,4 %), tranzitorní ischemické ataky (0,8 %) a cerebrovaskulární příhody (0,6 %). Arteriální tromboembolické nežádoucí účinky stupně 3/4 byly hlášeny u 3,3 % pacientů léčených axitinibem. Fatální akutní infarkt myokardu a mozková cévní příhoda byly hlášeny každá u jednoho pacienta (0,3 %) léčeného axitinibem. Ve studiích s monoterapií axitinibem (N=850) byly hlášeny arteriální tromboembolické nežádoucí účinky u 5,3 % pacientů léčených axitinibem.
V souhrnných klinických studiích s axitinibem (N = 672) při léčbě pacientů s RCC byly hlášeny arteriální tromboembolické příhody u 2,8 % pacientů léčených axitinibem. Arteriální tromboembolické příhody stupně 3 byly hlášeny u 1,2 % pacientů. Arteriální tromboembolické příhody stupně 4 byly hlášeny u 1,3 % pacientů. Fatální arteriální tromboembolické příhody byly hlášeny u 0,3 % pacientů léčených axitinibem.
Polycytémie (viz Zvýšení hodnot hemoglobinu nebo hematokritu v bodu 4.4)
V kontrolované klinické studii s axitinibem při léčbě pacientů s RCC byla hlášena polycytemie jako nežádoucí účinek u 1,4 % pacientů léčených axitinibem. Při rutinním laboratorním vyšetření byla zjištěna hodnota hemoglobinu zvýšená nad ULN u 9,7 % pacientů léčených axitinibem. Ve čtyřech klinických studiích s axitinibem při léčbě pacientů s RCC (N=537) byla pozorována hodnota hemoglobinu zvýšená nad ULN u 13,6 % pacientů léčených axitinibem.
V souhrnných klinických studiích s axitinibem (N = 672) při léčbě pacientů s RCC byla hlášena polycytemie u 1,5 % pacientů léčených axitinibem.
Krvácení (viz bod 4.4)
V kontrolované klinické studii s axitinibem při léčbě pacientů s RCC s vyloučením pacientů
s neléčenými mozkovými metastázami bylo hlášeno krvácení jako nežádoucí účinek u 21,4 % pacientů léčených axitinibem. Krvácivé nežádoucí účinky u pacientů léčených axitinibem byly epistaxe (7,8 %), hematurie (3,6 %), hemoptýza (2,5 %), krvácení z rekta (2,2 %), krvácení z dásní (1,1 %), krvácení do žaludku (0,6 %), krvácení do mozku (0,3 %) a krvácení z dolní části gastrointestinálního traktu (0,3 %). Krvácivé nežádoucí účinky stupně > 3 byly hlášeny u 3,1 % pacientů léčených axitinibem (včetně krvácení do mozku, krvácení do žaludku, krvácení z dolní části gastrointestinálního traktu a hemoptýzy). Fatální krvácení bylo hlášeno u jednoho pacienta (0,3 %) léčeného axitinibem (krvácení do žaludku). Ve studiích s monoterapií axitinibem (N=850) byla hemoptýza jako nežádoucí účinek hlášena u 3,9 % pacientů. Hemoptýza stupně > 3 byla hlášena u 0,5 % pacientů.
V souhrnných klinických studiích s axitinibem (N = 672) při léčbě pacientů s RCC byly hlášeny krvácivé příhody u 25,7 % pacientů léčených axitinibem. Krvácivé nežádoucí účinky stupně 3 byly hlášeny u 3 % pacientů. Krvácivé nežádoucí účinky stupně 4 byly hlášeny u 1 % pacientů a fatální krvácení bylo hlášeno u 0,4 % pacientů léčených axitinibem.
Gastrointestinálníperforace a tvorbapíštěle (viz bod 4.4)
V kontrolované klinické studii s axitinibem při léčbě pacientů s RCC byly u 1,7 % pacientů léčených axitinibem hlášeny události typu gastrointestinální perforace, včetně análních píštělí (0,6 %), píštělí (0,3 %) a gastrointestinální perforace (0,3 %). Ve studiích s monoterapií axitinibem (N=850) byly
hlášeny události typu gastrointestinální perforace u 1,9 % pacientů a fatální gastrointestinální perforace byla hlášena u jednoho pacienta (0,1 %).
V souhrnných klinických studiích s axitinibem (N = 672) při léčbě pacientů s RCC byly hlášeny gastrointestinální perforace a píštěle u 1,9 % pacientů léčených axitinibem.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Na předávkování axitinibem neexistuje specifická léčba.
V kontrolované klinické studii s axitinibem při léčbě pacientů s RCC užíval jeden pacient nedopatřením dávku 20 mg dvakrát denně po 4 dny a vyskytly se u něj závratě (stupeň 1).
V klinické studii zjišťující dávku axitinibu došlo u subjektů, které užívaly zahajovací dávku 10 mg dvakrát denně nebo 20 mg dvakrát denně k nežádoucím účinkům, mezi něž patřila hypertenze, křeče související s hypertenzí a fatální hemoptýza.
V případech podezření na předávkování by mělo být podávání axitinibu pozastaveno a zavedena podpůrná léčba.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: cytostatika, inhibitory proteinkináz, ATC kód: L01XE17
Mechanismus účinku
Axitinib je silný a selektivní inhibitor tyrosinkinázových receptorů růstového faktoru cévního endotelu (VEGFR)-1, VEGFR-2 a VEGFR-3. Tyto receptory se účastní na patologické angiogenezi, růstu tumoru a progresi metastatického procesu u malignit. Bylo prokázáno, že axitinib je silný inhibitor proliferace a přežívání endoteliálních buněk zprostředkovaných VEGF. Axitinib inhiboval fosforylaci VEGFR-2 v cévách xenogenních nádorových štěpů, které exprimovaly receptory in vivo a vedl v mnoha experimentálních modelech malignity ke zpomalení růstu a regresi nádoru a inhibici metastáz.
Účinek na interval QTc
V randomizované studii typu dvoucestně zkřížené byla 35 zdravým dobrovolníkům podána jedna perorální dávka axitinibu (5 mg) bez současného podávání ketokonazolu nebo po 7 dnech podávání 400 mg ketokonazolu. Výsledky této studie ukázaly, že ke klinicky významnému prodloužení intervalu QT nevedla plasmatická expozice axitinibu až dvojnásobně vyšší, než je terapeutická úroveň koncentrace předpokládaná po dávce 5 mg.
Klinická účinnost
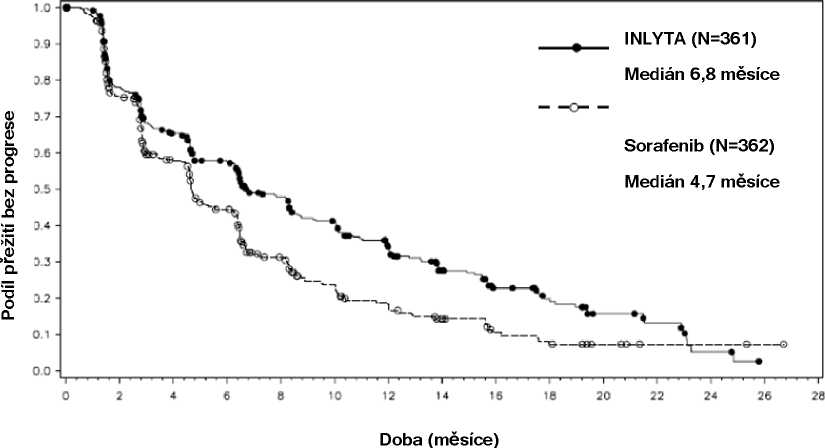
Bezpečnost a účinnost axitinibu byla hodnocena v randomizované otevřené, multicentrické studii fáze 3. Pacienti (N=723) s pokročilým RCC, jejichž onemocnění progredovalo během či po skončení jedné předchozí systémové léčby zahrnující režimy obsahující sunitinib, bevacizumab, temsirolimus nebo cytokin, byli randomizováni (1:1) k léčbě axitinibem (N=361) nebo sorafenibem (N=362). Primární cílový ukazatel, doba přežití bez progrese (PFS), byl hodnocen pomocí centrálního zaslepeného nezávislého hodnocení. Sekundární cílové ukazatele zahrnovaly výskyt objektivní odpovědi (ORR) a celkové přežití (OS).
Z pacientů zařazených do této studie absolvovalo 389 pacientů (53,8 %) jednu předchozí léčbu se sunitinibem, 251 pacientů (34,7 %) jednu předchozí léčbu s cytokiny (interleukin-2 nebo interferon-alfa), 59 pacientů (8,2 %) jednu předchozí léčbu s bevacizumabem a 24 pacientů (3,3 %) jednu předchozí léčbu s temsirolimem. Výchozí demografické údaje a charakteristiky onemocnění byly u skupiny s axitinibem a u skupiny se sorafenibem podobné, co se týče věku, pohlaví, rasy, stavu výkonnosti podle Eastern Cooperative Oncology Group (ECOG), geografické oblasti a předchozí léčby.
V celkové populaci pacientů a ve dvou hlavních podskupinách (předchozí léčba sunitinibem a předchozí léčba cytokinem) měl v prvním cílovém ukazateli PFS významnou převahu axitinib v porovnání se sorafenibem (viz tabulku 2 a obrázky 1, 2 a 3). Významnost výsledku mediánu PFS byla různá/odlišná ve skupinách podle předchozí léčby. Dvě z podskupin byly příliš malé pro poskytnutí spolehlivých výsledků (předchozí léčba temsirolimusem nebo předchozí léčba bevacizumabem). Nebyly statisticky významné rozdíly mezi rameny v OS v celkové populaci nebo v podskupinách podle předchozí léčby.
Tabulka 2. Výsledky účinnosti
|
Cílový ukazatel / hodnocená populace |
Axitinib |
Sorafenib |
HR (95% IS) |
hodnota p |
|
Celkový ITT |
N = 361 |
N = 362 | ||
|
Medián PFSa,b v měsících |
6,8 (6,4, 8,3) |
4,7 (4,6, 6,3) |
0,67 (0,56, 0,81) |
<0,0001c |
|
(95% IS) | ||||
|
Medián OS d v měsících |
20,1 (16,7, 23,4) |
19,2 (17,5, 22,3) |
0,97 (0,80, 1,17) |
NS |
|
(95% IS) | ||||
|
ORRb,e % (95% IS) |
19,4 (15,4, 23,9) |
9,4 (6,6, 12,9) |
2,06f (1,41, 3,00) |
0,0001g |
|
Předcházejícíc léčba sunitinibem |
N = 194 |
N = 195 | ||
|
Medián PFSa,b v měsících |
4,8 (4,5, 6,5) |
3,4 (2,8, 4,7) |
0,74 (0,58, 0,94) |
0,0063h |
|
(95% IS) | ||||
|
Medián OS d v měsících |
15,2 (12,8, 18,3) |
16,5 (13,7, 19,2) |
1,00 (0,78, 1,27) |
NS |
|
(95% IS) |
1,48f (0,79, 2,75) | |||
|
ORRb,e % (95% IS) |
11,3 (7,2, 16,7) |
7,7 (4,4, 12,4) |
NS | |
|
Předcházejícíc léčba cytokinem |
N = 126 |
N = 125 | ||
|
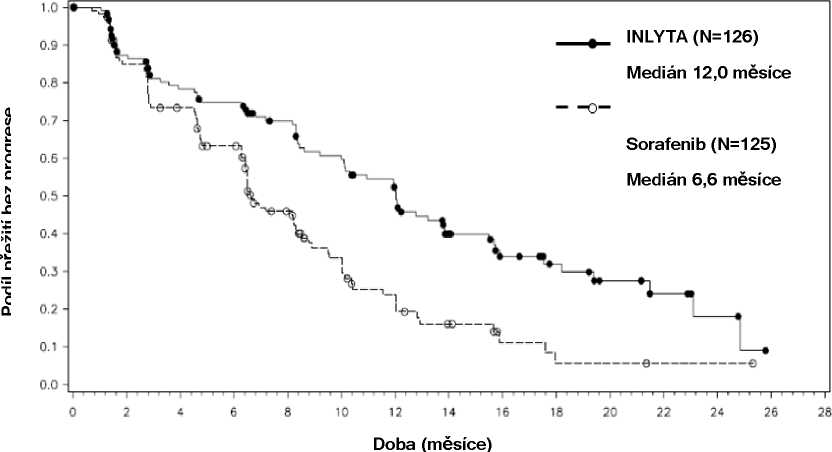
Medián PFSa,b v měsících |
12,0 (10,1, 13,9) |
6,6 (6,4, 8,3) |
0,52 (0,38, 0,72) |
<0,0001h |
|
(95% IS) | ||||
|
Medián OS d v měsících |
29,4 (24,5, NE) |
27,8 (23,1, 34,5) |
0,81 (0,56, 1,19) |
NS |
|
(95% IS) |
2,39f (1,43-3,99) | |||
|
ORRb,e % (95% IS) |
32,5 (24,5, 41,5) |
13,6 (8,1, 20,9) |
0,0002i |
IS=interval spolehlivosti, HR=relativní riziko (axitinib/sorafenib); ITT: záměr léčit; NE: nehodnotitelné; NS: statisticky nevýznamné ORR: míra objektivní odpovědi; OS: celkové přežití, PFS: doba přežití bez progrese a Doba od randomizace do progrese nebo úmrtí z jakékoli příčiny, k čemu dojde dříve. Přerušeno k datu:
3. června2011.
b Hodnoceno nezávislou radiologickou kontrolou podle RECIST (Response Evaluation Criteria in Solid Tumours).
c Jednostranná p-hodnota podle log-rank testu léčby stratifikovaného podle stavu výkonnosti ECOG a před
léčbou.
d Přerušeno k datu 1. listopadu 2011.
e Přerušeno k datu: 31. srpna 2010.
f Relativní riziko se používá pro ORR. Relativní riziko > 1 ukazovalo na vyšší pravděpodobnost odpovědi v rameni s axitinibem; relativní riziko < 1 ukazovalo na vyšší pravděpodobnost odpovědi v rameni se sorafenibem.
g Jednostranná p-hodnota podle Cochran-Mantel-Haenszelova testu léčby stratifikovaného podle stavu
výkonnosti ECOG a předchozí léčby.
h Jednostranna p-hodnota podle log-rank testu léčby stratifikovaného podle stavu výkonnosti ECOG.
i Jednostranná p-hodnota podle Cochran-Mantel-Haenszelova testu léčby stratifikovaného podle stavu výkonnosti
Obrázek 1. Kaplan-Meierova křivka doby přežití bez progrese podle nezávislého hodnocení u celkové populace
Obrázek 2. Kaplan-Meierova křivka doby přežití bez progrese podle nezávislého hodnocení u podskupiny s předcházející léčbou sunitibem
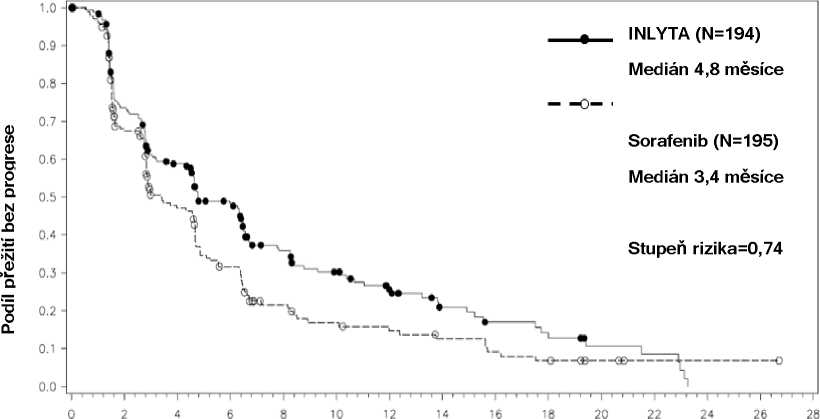
Doba (měsíce)
Obrázek 3. Kaplan-Meierova křivka doby přežití bez progrese podle nezávislého hodnocení u podskupiny s předcházející léčbou cytokiny
Pediatrická _ populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s axitinibem u všech podskupin pediatrické populace při léčbě renálních karcinomů a karcinomů ledvinné pánvičky (vyjma nefroblastomu, nefroblastomatózy, sarkomu z jasných buněk, mesoblastického nefromu, medulárního karcinomu ledviny a rabdoidního tumoru ledviny), (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Po perorálním podání tablet axitinibu je průměrná absolutní biologická dostupnost v porovnání s intravenózním podáním 58 %. Rozmezí plasmatického poločasu axitinibu je 2,5 až 6,1 hodin. Dávka axitinibu 5 mg dvakrát denně způsobovala méně než dvojnásobnou kumulaci v porovnání s podáním jedné dávky. Vzhledem ke krátkému poločasu axitinibu se dosažení rovnovážného stavu předpokládá během 2 až 3 dnů po podání první dávky.
Absorpce a distribuce
Maximální koncentrace axitinibu v plasmě je obvykle dosaženo během 4 hodin po perorálním podání axitinibu, přičemž medián Tmax je v rozmezí od 2,5 do 4,1 hodin. Podání axitinibu se středně tučným jídlem vedlo k expozici nižší o 10 % v porovnání s nočním lačněním. Vysoce tučné jídlo s vysokým obsahem kalorií vedlo k expozici o 19 % vyšší než noční lačnění. Axitinib lze podávat s jídlem nebo bez jídla (viz bod 4.2).
Průměrná Cmax a AUC rovnoměrně rostou v rozsahu dávky od 5 do 10 mg axitinibu. Vazba na proteiny lidské plasmy in vitro je > 99 % s preferenční vazbou na albumin a s mírnou vazbou na ai-kyselý glykoprotein. Při dávkování 5 mg dvakrát denně v sytém stavu byl u pacientů s pokročilým RCC geometrický průměr vrcholu plasmatické koncentrace 27,8 ng/ml a 24hodinová AUC 265 ng.h/ml. Geometrický průměr perorální clearance byl 38 l/h a geometrický průměr zjevného distribučního objemu 160 l.
Biotransformace a eliminace
Axitinib je metabolizován hlavně v játrech cytochromem CYP3A4/5 a v menší míře cytochromy CYP1A2, CYP2C19 a UGT1A1.
Po perorálním podání 5 mg radioaktivně značeného axitinibu se 30-60 % radioaktivity objevilo ve stolici a 23 % radioaktivity v moči. Nezměněný axitinib, odpovídající 12 % dávky, byla hlavní složka zjištěná ve stolici. Nezměněný axitinib nebyl zjištěn v moči; za většinu radioaktivity v moči odpovídá kyselina karboxylová a sulfoxidové metabolity. V plasmě představuje N-glukuronidový metabolit hlavní radioaktivní složku (50 % radioaktivity v oběhu) a jak nezměněný axitinib, tak i sulfoxidový metabolit odpovídaly za přibližně 20 % radioaktivity v oběhu.
Sulfoxidové a N-glukuronidové metabolity vykazují in vitro v porovnání s axitinibem přibližně 400krát, respektive až 8000krát nižší účinnost proti VEGFR-2.
Zvláštní populace
Starší osoby, pohlaví a rasa
Populační farmakokinetické analýzy u pacientů s pokročilou malignitou (včetně pokročilého RCC) a u zdravých dobrovolníků ukazují, že neexistuje klinicky relevantní vliv věku, pohlaví, tělesné hmotnosti, rasy, ledvinné funkce, genotypu UGT1A1 či genotypu CYP2C19.
Pediatrická populace
Axitinib nebyl zkoumán u pacientů ve věku <18 let.
Porucha jater
Údaje in vitro a in vivo ukazují, že axitinib je metabolizován hlavně v játrech.
V porovnání se subjekty s normální jaterní funkcí byla u subjektů s lehkým poškozením jater (Child-Pugh třída A) systémová expozice po jedné dávce axitinibu podobná a u subjektů se středně závažným poškozením jater (Child-Pugh třída B) vyšší (přibližně dvojnásobná). Axitinib nebyl zkoušen u subjektů se závažným poškozením jater (Child-Pugh třída C) a u této populace by se neměl používat (viz bod 4.2, doporučení pro úpravu dávky).
Porucha funkce ledvin
Nezměněný axitinib nebyl zjištěn v moči.
Axitinib nebyl zkoumán u pacientů s poruchou funkce ledvin. V klinických studiích s axitinibem při léčbě pacientů s RCC byli pacienti s kreatininem v séru > 1,5krát vyšším než ULN nebo s vypočtenou clearance kreatininu < 60 ml/min vyloučeni. Populační farmakokinetické analýzy ukázaly, že clearance axitinibu nebyla u subjektů s poruchou funkce ledvin změněna a úprava dávky není nutná.
5.3 Předklinické údaje vztahující se k bezpečnosti
Toxicita u opakovaných dávek
Hlavní zjištění toxicity u myší a psů po opakovaných dávkách po dobu až 9 měsíců se týkalo gastrointestinálního, hematopoetického, reprodukčního, kosterního a zubního systému, s koncentrací bez zjistitelných nežádoucích účinků (NOAEL) přibližně ekvivalentní nebo nižší, než je předpokládaná expozice u lidí při doporučené zahajovací dávce (na základě hodnot AUC).
Kancerogenita
Studie kancerogenity nebyly s axitinibem provedeny.
Genotoxicita
V klasických testech genotoxicity in vitro nebyl axitinib mutagenní ani klastogenní. Významně zvýšený počet polyploidie byl pozorován in vitro při koncentraci > 0,22 ^g/ml a zvýšený počet mikrojaderných polychromatických erytrocytů byl pozorován in vivo přičemž koncentrace bez zjistitelných nežádoucích účinků (NOAEL) byla 69krát vyšší, než je předpokládaná expozice u lidí. Zjištění genotoxicity není pokládáno za klinicky relevantní při hodnotách expozice pozorovaných u lidí.
Reprodukční toxicita
K nálezům na varlatech a nadvarleti souvisejícím s axitinibem patří snížená hmotnost, atrofie či degenerace orgánu, snížený počet zárodečných buněk, hypospermie nebo abnormální tvar spermií a snížená denzita a počet spermií. Tyto nálezy byly pozorovány u myší při hladině expozice přibližně 12krát vyšší, než je předpokládaná expozice u lidí, a u psů při hladině expozice nižší, než je předpokládaná expozice u lidí. Při hladině expozice přibližně 57krát vyšší, než je předpokládaná expozice u lidí, nedošlo k žádnému účinku na páření či fertilitu u myších samců. K nálezům u samic při expozici přibližně ekvivalentní předpokládané expozici u lidí patřily známky opožděné pohlavní zralosti, snížený počet nebo chybění žlutého tělíska, snížená hmotnost dělohy a atrofie dělohy. Snížení fertility a životaschopnosti embryí bylo u myších samic pozorováno při všech testovaných dávkách, s nejnižší hladinou expozice přibližně 10krát vyšší, než je předpokládaná expozice u lidí.
Březí myši s expozicí axitinibu vykazovaly při hladině expozice nižší, než je předpokládaná expozice u lidí, zvýšený výskyt rozštěpu patra a změn na kostře včetně opožděné osifikace. Vývojové studie perinatální a postnatální toxicity nebyly provedeny.
Zjištění toxicity u nezralých zvířat
Reverzibilní dysplazie dlouhých kostí byla pozorována u myší a psů, kterým byl podáván axitinib po dobu nejméně 1 měsíce při hladině expozice přibližně šestkrát vyšší, než je předpokládaná expozice u lidí. Částečně reverzibilní zubní kazy byly pozorovány u myší léčených po dobu delší než 1 měsíc při hladině expozice podobné předpokládané expozici u lidí. U mláďat nebyly hodnoceny jiné projevy toxicity potenciálně se týkající pediatrických pacientů.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Jádro tablet:
Mikrokrystalická celulosa Monohydrát laktosy Sodná sůl kroskarmelosy Magnesium-stearát
Potahová vrstva tablet:
Hypromelosa Oxid titaničitý (E171)
Monohydrát laktosy Triacetin (E1518)
Červený oxid železitý (E172)
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
3 roky.
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
6.5 Druh obalu a obsah balení
Inlyta 1 mg potahované tablety
Al/Al blistr obsahující 14 potahovaných tablet. Jedno balení obsahuje 28 nebo 56 potahovaných tablet.
HDPE lahvičky s vysoušedlem (silikagel) a polypropylenovým uzávěrem obsahují 180 potahovaných tablet.
Inlyta 3 mg potahované tablety
Al/Al blistr obsahující 14 potahovaných tablet. Jedno balení obsahuje 28 nebo 56 potahovaných tablet.
HDPE lahvičky s vysoušedlem (silikagel) a polypropylenovým uzávěrem obsahují 60 potahovaných tablet.
Inlyta 5 mg potahované tablety
Al/Al blistr obsahující 14 potahovaných tablet. Jedno balení obsahuje 28 nebo 56 potahovaných tablet.
HDPE lahvičky s vysoušedlem (silikagel) a polypropylenovým uzávěrem obsahují 60 potahovaných tablet.
Inlyta 7 mg potahované tablety
Al/Al blistr obsahující 14 potahovaných tablet. Jedno balení obsahuje 28 nebo 56 potahovaných tablet.
HDPE lahvičky s vysoušedlem (silikagel) a polypropylenovým uzávěrem obsahují 60 potahovaných tablet.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Pfizer Ltd Ramsgate Road Sandwich, Kent CT13 9NJ Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
Inlyta 1 mg potahované tablety EU/1/12/777/001 EU/1/12/777/002 EU/1/12/777/003
Inlyta 3 mg potahované tablety EU/1/12/777/007 EU/1/12/777/008 EU/1/12/777/009
Inlyta 5 mg potahované tablety EU/1/12/777/004
EU/1/12/777/005
EU/1/12/777/006
Inlyta 7 mg potahované tablety EU/1/12/777/010 EU/1/12/777/011 EU/1/12/777/012
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 3. září 2012
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného za propouštění šarží
Pfizer Manufacturing Deutschland GmbH Betriebsstatte Freiburg Mooswaldallee 1 D-79090 Freiburg Německo
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace ave veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Inlyta 1 mg potahované tablety axitinibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna potahovaná tableta obsahuje axitinibum 1 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktosu. Viz příbalová informace pro další podrobnosti.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
28 tablet 56 tablet
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
|
11. |
NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI | |
|
Pfizer Limited | ||
|
Ramsgate Road | ||
|
Sandwich, Kent, CT13 9NJ | ||
|
Velká Británie | ||
|
12. |
REGISTRAČNÍ ČÍSLO/ČÍSLA | |
|
EU/1/12/777/001 |
28 tablet | |
|
EU/1/12/777/002 |
56 tablet | |
|
13. |
ČÍSLO ŠARŽE | |
|
Číslo šarže: | ||
|
14. |
KLASIFIKACE PRO VÝDEJ | |
|
Výdej léčivého přípravku |
vázán na lékařský předpis. | |
|
15. |
NÁVOD K POUŽITÍ | |
|
16. |
INFORMACE V BRAILLOVĚ PÍSMU | |
Inlyta 1 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Inlyta 1 mg potahované tablety axitinibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna potahovaná tableta obsahuje axitinibum 1 mg
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktosu. Viz příbalová informace pro další podrobnosti.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
180 tablet
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Pfizer Limited Ramsgate Road Sandwich, Kent, CT13 9NJ Velká Británie
12. REGISTRAČNÍ CÍSLO/CÍSLA
EU/1/12/777/003 13. ČÍSLO ŠARŽE
Číslo šarže:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ 16. INFORMACE V BRAILLOVE PÍSMU
Inlyta 1 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Inlyta 1 mg potahované tablety axitinibum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Pfizer
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Inlyta 3 mg potahované tablety axitinibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna potahovaná tableta obsahuje axitinibum 3 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktosu. Viz příbalová informace pro další podrobnosti.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
28 tablet 56 tablet
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
|
11. |
NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI | |
|
Pfizer Limited | ||
|
Ramsgate Road | ||
|
Sandwich, Kent, CT13 9NJ | ||
|
Velká Británie | ||
|
12. |
REGISTRAČNÍ ČÍSLO/ČÍSLA | |
|
EU/1/12/777/007 |
28 tablet | |
|
EU/1/12/777/008 |
56 tablet | |
|
13. |
ČÍSLO ŠARŽE | |
|
Číslo šarže: | ||
|
14. |
KLASIFIKACE PRO VÝDEJ | |
|
Výdej léčivého přípravku |
vázán na lékařský předpis. | |
|
15. |
NÁVOD K POUŽITÍ | |
|
16. |
INFORMACE V BRAILLOVĚ PÍSMU | |
Inlyta 3 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Inlyta 3 mg potahované tablety axitinibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna potahovaná tableta obsahuje axitinibum 3 mg
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktosu. Viz příbalová informace pro další podrobnosti.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
60 tablet
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Pfizer Limited Ramsgate Road Sandwich, Kent, CT13 9NJ Velká Británie
12. REGISTRAČNÍ CÍSLO/CÍSLA
EU/1/12/777/009 13. ČÍSLO ŠARŽE
Číslo šarže:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ 16. INFORMACE V BRAILLOVE PÍSMU
Inlyta 3 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Inlyta 3 mg potahované tablety axitinibum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Pfizer
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Inlyta 5 mg potahované tablety axitinibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna potahovaná tableta obsahuje axitinibum 5 mg
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktosu. Viz příbalová informace pro další podrobnosti.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
28 tablet 56 tablet
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
|
11. |
NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI | |
|
Pfizer Limited | ||
|
Ramsgate Road | ||
|
Sandwich, Kent, CT13 9NJ | ||
|
Velká Británie | ||
|
12. |
REGISTRAČNÍ ČÍSLO/ČÍSLA | |
|
EU/1/12/777/004 |
28 tablet | |
|
EU/1/12/777/005 |
56 tablet | |
|
13. |
ČÍSLO ŠARŽE | |
|
Číslo šarže: | ||
|
14. |
KLASIFIKACE PRO VÝDEJ | |
|
Výdej léčivého přípravku |
vázán na lékařský předpis. | |
|
15. |
NÁVOD K POUŽITÍ | |
|
16. |
INFORMACE V BRAILLOVĚ PÍSMU | |
Inlyta 5 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Inlyta 5 mg potahované tablety axitinibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna potahovaná tableta obsahuje axitinibum 5 mg
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktosu. Viz příbalová informace pro další podrobnosti.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
60 tablet
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Pfizer Limited Ramsgate Road Sandwich, Kent, CT13 9NJ Velká Británie
12. REGISTRAČNÍ CÍSLO/CÍSLA
EU/1/12/777/006
13. ČÍSLO ŠARŽE <, KÓD DÁRCE A KÓD LÉČIVÉHO PRÍPRAVKU>
Číslo šarže:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ 16. INFORMACE V BRAILLOVE PÍSMU
Inlyta 5 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Inlyta 5 mg potahované tablety axitinibum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Pfizer
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE <, KÓD DÁRCE A KÓD LÉČIVÉHO PŘÍPRAVKU>
Lot
5. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Inlyta 7 mg potahované tablety axitinibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna potahovaná tableta obsahuje axitinibum 7 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktosu. Viz příbalová informace pro další podrobnosti.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
28 tablet 56 tablet
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
|
11. |
NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI | |
|
Pfizer Limited | ||
|
Ramsgate Road | ||
|
Sandwich, Kent, CT13 9NJ | ||
|
Velká Británie | ||
|
12. |
REGISTRAČNÍ ČÍSLO/ČÍSLA | |
|
EU/1/12/777/010 |
28 tablet | |
|
EU/1/12/777/011 |
56 tablet | |
|
13. |
ČÍSLO ŠARŽE | |
|
Číslo šarže: | ||
|
14. |
KLASIFIKACE PRO VÝDEJ | |
|
Výdej léčivého přípravku |
vázán na lékařský předpis. | |
|
15. |
NÁVOD K POUŽITÍ | |
|
16. |
INFORMACE V BRAILLOVĚ PÍSMU | |
Inlyta 7 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Inlyta 7 mg potahované tablety axitinibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna potahovaná tableta obsahuje axitinibum 7 mg
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktosu. Viz příbalová informace pro další podrobnosti.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
60 tablet
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Pfizer Limited Ramsgate Road Sandwich, Kent, CT13 9NJ Velká Británie
12. REGISTRAČNÍ CÍSLO/CÍSLA
EU/1/12/777/012 13. ČÍSLO ŠARŽE
Číslo šarže:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ 16. INFORMACE V BRAILLOVE PÍSMU
Inlyta 7 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Inlyta 7 mg potahované tablety axitinibum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Pfizer
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro pacienta
Inlyta 1 mg potahované tablety Inlyta 3 mg potahované tablety Inlyta 5 mg potahované tablety Inlyta 7 mg potahované tablety
axitinibum
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka nebo zdravotní sestry.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Inlyta a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Inlyta užívat
3. Jak se přípravek Inlyta užívá
4. Možné nežádoucí účinky
5. Jak přípravek Inlyta uchovávat
6. Obsah balení a další informace
1. Co je přípravek Inlyta a k čemu se používá
Inlyta je léčivý přípravek, který obsahuje léčivou látku axitinib. Axitinib snižuje krevní zásobování tumoru a zpomaluje růst nádoru.
Přípravek Inlyta je určen k léčbě pokročilé rakoviny ledviny (pokročilý renální karcinom) u dospělých, když jiné léčivé látky (zvané sunitinib nebo cytokiny) už dále nezastavují rozvoj choroby.
Pokud máte jakékoliv dotazy ohledně toho, jak léčivý přípravek účinkuje, nebo proč Vám byl tento lék předepsán, zeptejte se svého lékaře.
2. Čemu musíte věnovat pozornost, než začnete přípravek Inlyta užívat Neužívejte přípravek Inlyta
jestliže jste alergický(á) na axitinib nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Pokud si myslíte, že byste mohl(a) být alergický(á), požádejte o radu lékaře.
Upozornění a opatření
Před použitím přípravku Inlyta se poraďte se svým lékařem:
• Pokud máte vysoký krevní tlak.
Inlyta může zvýšit krevní tlak. Je důležité kontrolovat Váš krevní tlak předtím, než začnete tento přípravek užívat a dále pravidelně během užívání přípravku. Pokud máte vysoký krevní tlak (hypertenzi) je možné, že budete léčeni přípravky na snížení krevního tlaku. Váš lékař zajistí, aby byl Váš krevní tlak pod kontrolou před zahájením léčby přípravkem Inlyta, a v průběhu léčby tímto přípravkem.
• Pokud máte problémy se štítnou žlázou.
Inlyta může způsobit problémy se štítnou žlázou. Sdělte svému lékaři, pokud během užívání tohoto přípravku pozorujete, že se snadno unavíte, vnímáte chlad více než ostatní lidé nebo máte hlubší hlas. Před zahájením léčby přípravkem Inlyta a pravidelně během léčby Vám má být provedena kontrola funkce štítné žlázy. Pokud Vaše štítná žláza před zahájením nebo v průběhu léčby tímto přípravkem neprodukuje dostatečné množství hormonu, máte být léčeni náhradním hormonem štítné žlázy.
• Jestliže jste měl(a) v nedávné době problém s tvorbou krevních sraženin v žilách a/nebo tepnách (typ krevních cév), včetně mrtvice, srdečního infarktu, ucpání cév krevními sraženinami.
Sežeňte okamžitě pohotovost a informujte lékaře, pokud máte během léčby tímto přípravkem příznaky, jako jsou bolest na hrudi nebo tlak, bolest v rukou, zádech, krku nebo čelisti, zkrácení dechu, sníženou citlivosti nebo slabost na jedné straně těla, obtíže s mluvením, bolest hlavy, změny vidění nebo závratě.
• Pokud trpíte problémy s krvácením.
Inlyta může zvýšit riziko krvácení. Sdělte svému lékaři, pokud během užívání tohoto přípravku se u Vás vyskytne jakékoliv krvácení, vykašlávání krve nebo krvavého hlenu.
• Pokud se u Vás během léčby tímto přípravkem objeví vážné bolesti žaludku (břicha) nebo přetrvávající bolest žaludku.
Inlyta může zvýšit riziko rozvoje proděravění žaludku nebo střeva nebo vznikem píštěle (abnormální kanálek spojující jednu tělní dutinu s jinou tělní dutinou či s povrchem kůže). Informujte svého lékaře, jestliže během léčby tímto přípravkem máte silné bolesti břicha.
• Pokud podstupujete nebo jste nedávno podstoupil(a) chirurgický zákrok nebo pokud máte nezahojenou ránu.
Inlyta může mít vliv na hojení, proto by Váš lékař měl přerušit léčbu tímto přípravkem alespoň 24 hodin před operací. Léčba tímto přípravkem by měla být znovu zahájena, až když je rána dostatečně zahojena.
• Pokud se během léčby tímto přípravkem u Vás objeví příznaky, jako jsou bolest hlavy, zmatenost, křeče (záchvaty) nebo změny vidění s nebo bez vysokého tlaku krve.
Vyhledejte okamžitě pohotovost a informujte svého lékaře. Mohlo by se jednat o vzácný neurologický nežádoucí účinek nazývaný syndrom posteriorní reverzibilní encefalopatie.
• Pokud máte problémy s játry.
Váš lékař Vám má provést krevní testy kvůli kontrole funkce jater před zahájením a v průběhu léčby přípravkem Inlyta.
• Pokud se u Vás během léčby tímto přípravkem objeví příznaky, jako jsou nadměrná únava, otoky břicha, nohou nebo kotníků, dýchavičnost nebo vystupující žíly na krku.
Přípravek Inlyta může zvyšovat riziko vzniku příhod srdečního selhání. Váš lékař by měl během léčby axitinibem pravidelně sledovat, zda se u Vás neobjevují známky nebo příznaky příhod srdečního selhání.
Použití u dětí a dospívajících
Inlyta není doporučena osobám mladším než 18 let. Tento lék nebyl u dětí a dospívajících hodnocen. Další léčivé přípravky a přípravek Inlyta
Některé léky mohou mít vliv na přípravek Inlyta nebo mohou být tímto přípravkem ovlivněny. Informujte svého lékaře, lékárníka nebo zdravotní sestru o všech lécích, které užíváte nebo jste v nedávné době užíval(a), užíváte nyní nebo plánujete /hodláte užívat, a to i o lécích, které jsou dostupné bez lékařského předpisu, vitamíny a rostlinné přípravky. Léky uvedené v této příbalové informaci nemusí být jediné, které se mohou s přípravkem Inlyta ovlivňovat.
Následující léky mohou při současném podávání s přípravkem Inlyta zvyšovat riziko nežádoucích účinků:
• ketokonazol nebo itrakonazol, používané k léčbě plísňových infekcí;
• klarithromycin, erythromycin nebo telithromycin, antibiotika užívaná k léčbě bakteriálních infekcí;
• atazanavir, indinavir, nelfinavir, ritonavir nebo sachinavir, užívané k léčbě HIV infekcí/AIDS;
• nefazodon, užívaný k léčbě deprese.
Následující léky mohou snižovat účinnost přípravku Inlyta:
• rifampicin, rifabutin nebo rifapentin, užívaný k léčbě tuberkulózy (TBC);
• dexamethason, steroidní lék předepisovaný pro mnoho různých stavů včetně závažných onemocnění;
• fenytoin, karbamazepin nebo fenobarbital, antiepileptika užívaná k zastavení záchvatů nebo křečí;
• třezalka tečkovaná (Hypericumperforatum), rostlinný přípravek užívaný k léčbě depresí.
Neměl(a) byste tyto léky užívat během léčby přípravkem Inlyta. Pokud užíváte kterýkoliv z nich, informujte svého lékaře nebo lékárníka. Váš lékař může změnit dávku léků, změnit dávku přípravku Inlyta nebo Vás převést na jiný lék.
Inlyta může zvyšovat nežádoucí účinky spojené s theofylinem, užívaným k léčbě astmatu nebo jiných plicních chorob.
Přípravek Inlyta s jídlem a pitím
Tento lék můžete užívat s jídlem nebo bez jídla.
Tento lék neužívejte s grapefruitem nebo grapefruitovou šťávou, protože se tak může zvýšit riziko nežádoucích účinků.
Těhotenství a kojení
• Inlyta by mohla poškodit nenarozené dítě nebo kojené dítě.
• Tento lék neužívejte během těhotenství. Jestliže jste těhotná nebo byste mohla být těhotná, informujte svého lékaře ještě před zahájením léčby.
• Během užívání přípravku Inlyta a 1 týden po poslední dávce tohoto přípravku, používejte spolehlivou metodu antikoncepce, abyste předešla otěhotnění.
• Během léčby přípravkem Inlyta nekojte. Pokud jste kojící, proberte s Vaším lékařem, zda přerušit kojení nebo ukončit léčbu přípravkem Inlyta.
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem, lékárníkem nebo zdravotní sestrou dříve, než začnete tento přípravek užívat.
Řízení dopravních prostředků a obsluha strojů
Pokud se u Vás objeví závratě nebo pokud se cítíte unavený(á), věnujte zvláštní pozornost při řízení vozidel a obsluze strojů.
Inlyta obsahuje laktózu (mléčný cukr)
Pokud Vám Váš lékař řekl, že nesnášíte některé cukry, poraďte se se svým lékařem, než začnete tento přípravek užívat..
3. Jak se přípravek Inlyta užívá
Přípravek užívejte vždy přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo zdravotní sestrou.
Doporučená dávka je 5 mg dvakrát denně. Váš lékař Vám následně může zvýšit nebo snížit Vaši dávku v závislosti na tom, jak léčbu přípravkem Inlyta snášíte.
Tablety polykejte celé s vodou, s jídlem nebo bez jídla. Dávky přípravku Inlyta užívejte v rozmezí přibližně 12 hodin.
Jestliže jste užil(a) více přípravku Inlyta, než jste měl(a)
Pokud náhodně užijete příliš mnoho tablet nebo vyšší dávku než potřebujete, informujte o tom ihned svého lékaře. Je-li to možné, ukažte lékaři balení nebo tuto příbalovou informaci. Budete možná potřebovat lékařskou péči.
Jestliže jste zapomněl(a) užít přípravek Inlyta
Vezměte si další dávku ve Vaší pravidelné době. Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou tabletu.
Jestliže jste přestal(a) užívat přípravek Inlyta
Jestliže si nemůžete vzít tento přípravek tak, jak Vám předepsal lékař nebo máte pocit, že ho již dále nepotřebujete, okamžitě kontaktujte svého lékaře.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Některé nežádoucí účinky by mohly být závažné. Musíte okamžitě informovat svého lékaře, pokud proděláte kterýkoliv z následujících závažných nežádoucích účinků (viz rovněž bod 2 Čemu musíte věnovat pozornost, než začnete přípravek Inlyta užívat):
• Příhody srdečního selhání. Informujte svého lékaře, pokud se u Vás objeví nadměrná únava, otoky břicha, nohou nebo kotníků, dýchavičnost nebo vystupující žíly na krku.
• Srážení krve v žilách a/nebo tepnách (typ krevních cév), včetně cévní mozkové příhody, srdečního infarktu, tvorby krevních sraženin, ucpání cévy krevní sraženinou.
Sežeňte okamžitě pohotovost a informujte lékaře, pokud máte příznaky, jako jsou bolest nebo tlak na hrudi, bolest v rukou, zádech, krku nebo čelisti, pocit zkrácení dechu, sníženou citlivost nebo slabost na jedné straně těla, obtíže s mluvením, bolest hlavy, změny vidění nebo závratě.
• Krvácení
Sdělte svému lékaři okamžitě, pokud máte následující příznaky nebo vážné problémy s krvácením během užívání přípravku Inlyta: černá dehtovitá stolice, vykašlávání krve nebo krvavého hlenu nebo změny Vašeho mentálního stavu.
• Proděravění žaludku nebo střev nebo tvorba píštěle (abnormální kanálek spojující jednu tělní dutinu s jinou tělní dutinou či s povrchem kůže)
Sdělte svému lékaři, pokud se u Vás objeví silné bolesti břicha.
• Vysoký krevní tlak (hypertenzní krize)
Sdělte svému lékaři, pokud máte velmi vysoký krevní tlak, silné bolesti hlavy nebo silnou bolest
na hrudi.
• Reverzibilní (zvratný)otok mozku (syndrom posteriorní reverzibilní encefalopatie)
Vyhledejte okamžitě pohotovost a informujte lékaře, pokud máte příznaky, jako jsou bolest hlavy, zmatenost, křeče (záchvaty) nebo změny vidění s nebo bez vysokého tlaku krve.
Další nežádoucí účinky přípravku Inlyta mohou zahrnovat:
Velmi časté nežádoucí účinky (mohou postihnout více než 1 z 10 pacientů)
• Velmi vysoký tlak krve nebo zvýšení krevního tlaku
• Průjem, pocit nevolnosti nebo zvracení, bolesti břicha, zažívací potíže, sucho v ústech, jazyka nebo krku, zácpa
• Nedostatek energie, pocit slabosti nebo únavy
• Snížení činnosti štítné žlázy (může se projevit v krevních testech)
• Zarudnutí a otok dlaní a chodidel (syndrom ruce-nohy), kožní vyrážka, suchost kůže
• Bolest kloubů, bolest v rukou nebo nohou
• Ztráta chuti k jídlu
• Bílkovina v moči (může se projevit v testech moči)
• Úbytek tělesné hmotnosti
• Bolest hlavy, poruchy chuti, ztráta chuti
Časté nežádoucí účinky (mohou postihnout až 1 z 10 pacientů)
• Dehydratace (ztráta tělesných tekutin)
• Selhání ledvin
• Nadýmání, hemoroidy, krvácení z dásní, krvácení z konečníku, pálivé nebo bodavé pocity v ústech
• Zvýšená funkce štítné žlázy (může se projevit v krevních testech)
• Vředy v krku nebo nose a podráždění v krku
• Bolest svalů
• Krvácení z nosu
• Svědění kůže, zarudnutí kůže, ztráta vlasů
• Ušní šelest (tinitus)
• Snížení počtu červených krvinek (může se projevit v krevních testech)
• Snížení počtu krevních destiček (buněk, které napomáhají srážení krve), (může se projevit v krevních testech)
• Přítomnost červených krvinek/krve v moči (může se projevit v testech moči)
• Změny hladin různých látek/enzymů v krvi (může se projevit v krevních testech)
• Zvýšení počtu červených krvinek (může se projevit v krevních testech)
• Otok břicha, nohou nebo kotníků, vystupující žíly na krku, nadměrná únava, dýchavičnost (příznak srdečního selhání)
• Píštěl (abnormální kanálek spojující jednu tělní dutinu s jinou tělní dutinou či s povrchem kůže)
• Závrať
Méně časté nežádoucí účinky (mohou postihnout až 1 ze 100 pacientů)
• Snížení počtu bílých krvinek (může se projevit v krevních testech)
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Inlyta uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a na fólii blistru nebo na lahvičce za EXP (zkratka používaná pro dobu použitelnosti). Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Tento přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
Nepoužívejte tento přípravek, jestliže si všimnete, že obal je poškozený nebo vykazuje známky porušení.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co přípravek Inlyta obsahuje
• Léčivou látkou je axitinibum. Inlyta potahované tablety se dodává v různých silách.
Inlyta 1 mg: jedna tableta obsahuje axitinibum 1 mg.
Inlyta 3 mg: jedna tableta obsahuje axitinibum 3 mg.
Inlyta 5 mg: jedna tableta obsahuje axitinibum 5 mg.
Inlyta 7 mg: jedna tableta obsahuje axitinibum 7 mg.
• Dalšími složkami jsou mikrokrystalická celulosa, monohydrát laktosy, sodná sůl kroskarmelosy, magnesium-stearát, hypromelosa, oxid titaničitý (E171), triacetin (E1518), červený oxid železitý (E172).
Jak přípravek Inlyta vypadá a co obsahuje toto balení
Inlyta 1 mg potahované tablety jsou červené potahované tablety oválného tvaru s vyraženým nápisem “Pfizer” na jedné straně a “1 XNB” na druhé straně. Inlyta 1 mg tablety jsou dostupné v lahvičkách po 180 tabletách a balení v blistrech po 28 tabletách a 56 tabletách.
Inlyta 3 mg potahované tablety jsou červené potahované tablety kulatého tvaru s vyraženým nápisem “Pfizer” na jedné straně a “3 XNB” na druhé straně. Inlyta 3 mg tablety jsou dostupné v lahvičkách po 60 tabletách a balení v blistrech po 28 tabletách a 56 tabletách.
Inlyta 5 mg potahované tablety jsou červené potahované tablety ve tvaru trojúhelníku s vyraženým nápisem “Pfizer” na jedné straně a “5 XNB” na druhé straně. Inlyta 5 mg tablety jsou dostupné v lahvičkách po 60 tabletách a balení v blistrech po 28 tabletách a 56 tabletách.
Inlyta 7 mg potahované tablety jsou červené potahované tablety ve tvaru kosočtverce s vyraženým nápisem “Pfizer” na jedné straně a “7 XNB” na druhé straně. Inlyta 7 mg tablety jsou dostupné v lahvičkách po 60 tabletách a balení v blistrech po 28 tabletách a 56 tabletách.
Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci a výrobce
Pfizer Limited Ramsgate Road Sandwich, Kent CT13 9NJ Velká Británie
Výrobce
Pfizer Manufacturing Deutschland GmbH Betriebsstátte Freiburg Mooswaldallee 1 79090 Freiburg Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Tel. + 370 52 51 4000
Bt^rapuu Luxembourg/Luxemburg
n$aň3ep HroKceMÓypr CAPH, KnoH Btarapua Pfizer S.A.
Ten.: +359 2 970 4333 Tél/Tel: +32 (0)2 554 62 11
Danmark
Pfizer ApS
Tlf: +45 44 20 11 00
Malta
V.J. Salomone Pharma Ltd. Tel. +356 21220174
Deutschland
Nederland
Pfizer BV
Tel: +31 (0)10 406 43 01
Pfizer Pharma GmbH Tel: +49 (0)30 550055 51000
|
Eesti Pfizer Luxembourg SARL Eesti filiaal Tel.: +372 666 7500 |
Norge Pfizer Norge AS Tlf: +47 67 52 61 00 |
|
EXláSa Pfizer EALág A.E. T^L +30 210 6785 800 |
Osterreich Pfizer Corporation Austria Ges.m.b.H. Tel: +43 (0)1 521 15-0 |
|
Espaňa Pfizer S.L. Tél: +34 91 490 99 00 |
Polska Pfizer Polska Sp.z.o.o Tel.:+48 22 335 61 00 |
|
France Pfizer Tél: +33 (0)1 58 07 34 40 |
Portugal Pfizer Biofarmaceutica, Sociedade Unipessoal Lda Tel: +351 21 423 5500 |
|
Hrvatska Pfizer Croatia d.o.o. Tel: + 385 1 3908 777 |
Románia Pfizer Romania S.R.L. Tel: +40 (0) 21 207 28 00 |
|
Ireland Pfizer Healthcare Ireland Tel: 1800 633 363 (toll free) +44 (0)1304 616161 |
Slovenija Pfizer Luxembourg SARL Pfizer, podružnica za svetovanje s področja farmacevtske dejavnosti, Ljubljana Tel.: + 386 (0)1 52 11 400 |
|
Ísland Icepharma hf. Sími: +354 540 8000 |
Slovenská republika Pfizer Luxembourg SARL, organizačná zložka Tel.: + 421 2 3355 5500 |
|
Italia Pfizer S.r.l. Tel: +39 06 33 18 21 |
Suomi/Finland Pfizer Oy Puh./Tel: +358 (0)9 43 00 40 |
|
Kúnpoq Pfizer EALág A.E. (Cyprus Branch) TpL +357 22 817690 |
Sverige Pfizer Innovations AB Tel: +46 (0)8 550-52000 |
|
Latvija Pfizer Luxembourg SARL filiale Latvija Tel.: + 371 670 35 775 |
United Kingdom Pfizer Limited Tel: +44 (0) 1304 616161 |
Tato příbalová informace byla naposledy revidována
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu
53