Increlex 10 Mg/Ml
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
INCRELEX 10 mg/ml injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden ml obsahuje mecaserminum 10 mg*.
Jedna injekční lahvička obsahuje mecaserminum 40 mg.
*Mecasermin je lidskému inzulinu podobný růstový faktor 1(IGF-1), odvozený z rekombinantní DNA, produkovaný v Escherichia coli.
Pomocné látky se známým účinkem:
Jeden ml obsahuje 9 mg benzylalkoholu.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok (injekce). Vodný roztok, čirý, bezbarvý.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
K dlouhodobé léčbě růstových poruch dětí a dospívajících od 2 do 18 let se závažnou primární deficiencí inzulinu podobného růstového faktoru 1 (primární IGFD).
Závažná primární IGFD je definována jako:
• skóre standardní odchylky od růstové normy < -3,0 a
• bazální hladina IGF-1 pod 2,5. percentil pro daný věk a pohlaví a
• dostatek GH.
• Vyloučení sekundárních forem deficience IGF-1, například podvýživy, hypothyroidismu nebo dlouhodobé léčby farmakologickými dávkami protizánětlivých steroidů.
Mezi pacienty se závažnou primární IGFD patří pacienti s mutacemi receptoru GH (GHR) a signální dráhy post-GHR a s poruchami genu IGF-1; tito pacienti nemají nedostatek růstového hormonu a proto se u nich nedá očekávat adekvátní odpověď na léčbu exogenním růstovým hormonem. Diagnózu se doporučuje potvrdit provedením testu tvorby IGF-1.
4.2 Dávkování a způsob podání
Léčbu přípravkem INCRELEX smějí předepisovat pouze lékaři s praxí v diagnostice a léčbě pacientů s poruchami růstu.
Dávkování
Dávka má být přizpůsobena konkrétnímu pacientovi. Doporučená počáteční dávka mecaserminu je 0,04 mg/kg tělesné hmotnosti dvakrát denně subkutánní injekcí. Pokud se po dobu nejméně jednoho
týdne neobjeví žádné nežádoucí reakce, dávku lze zvyšovat po krocích po 0,04 mg/kg na maximální dávku 0,12 mg/kg podávanou dvakrát denně. Nejsou žádné zkušenosti s podáváním dávky vyšší než 0,12 mg/kg dvakrát denně u dětí se závažnou primární IGFD.
Jestliže pacient netoleruje doporučenou dávku, může být zvážena léčba nižší dávkou. Úspěch léčby by měl být hodnocen na základě růstové rychlosti. Nejnižší dávka spojená s významným zvýšením růstu na individuální bázi je 0,04 mg/kg 2x denně.
Pediatrická populace
Bezpečnost a účinnost přípravku INCRELEX u dětí mladších 2 let nebyla stanovena. Nejsou dostupné žádné údaje.
Proto není doporučeno používat přípravek INCRELEX u dětí mladších 2 let.
Způsob podání
INCRELEX se má podávat subkutánní injekcí krátce před nebo po jídle nebo svačině. Pokud se při doporučené dávce vyskytne hypoglykémie i přes adekvátní příjem potravy, dávku je třeba snížit. Pokud pacient není schopen z jakéhokoli důvodu jíst, přípravek INCRELEX se nesmí podávat. Dávka mecaserminu se nikdy nesmí zvyšovat jako náhrada za jednu nebo více vynechaných dávek.
Místa aplikace se musí měnit tak, aby se každá injekce aplikovala na jiné místo.
INCRELEX se nesmí podávat intravenózně.
Opatření, která je nutno učinit před zacházením s léčivým přípravkem nebo před jeho podáním Roztok by měl být čirý ihned po vyjmutí z chladničky. Pokud je roztok zakalený nebo obsahuje částice, nesmí být aplikován (viz bod 6.6).
INCRELEX se má podávat pomocí sterilních stříkaček a injekčních jehel na jedno použití. Stříkačky musí mít vhodnou velikost tak, aby bylo možno předepsanou dávku natáhnout z injekční lahvičky s přiměřenou přesností.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Aktivní nebo suspektní nádorové onemocnění. Pokud se rozvinou příznaky nádorového onemocnění, léčbu je třeba přerušit.
Jelikož přípravek INCRELEX obsahuje benzylalkohol, nesmí se podávat nedonošeným ani donošeným novorozencům.
4.4 Zvláštní upozornění a opatření pro použití
Před zahájením léčby přípravkem INCRELEX je nutno korigovat funkci štítné žlázy a nutriční deficience.
INCRELEX není náhrada za léčbu GH.
INCRELEX se nesmí používat k podpoře růstu u pacientů s uzavřenými epifýzami.
INCRELEX se má podávat krátce před jídlem nebo po jídle, protože může způsobovat hypoglykemické stavy podobně jako inzulín. Zvláštní pozornost je třeba věnovat malým dětem, dětem s hypoglykémií v anamnéze a dětem s nestálým příjmem potravy. Pacienti se musí vyhýbat jakýmkoli rizikovým činnostem 2-3 hodiny po aplikaci dávky, a to zejména na začátku léčby přípravkem
INCRELEX, dokud nebude zjištěna dobře tolerovaná dávka přípravku INCRELEX. Pokud je pacient se závažnou hypoglykémií v bezvědomí nebo není z jakéhokoli jiného důvodu schopen normálně požít potravu, může být zapotřebí podat injekčně glukagon. Pacienti se závažnou hypoglykémií v anamnéze musí mít glukagon k dispozici. Na počátku léčby musí lékař poučit rodiče o známkách, příznacích a léčbě hypoglykémie, mimo jiné i o injekční aplikaci glukagonu.
U diabetiků, jimž je podáván INCRELEX, může být zapotřebí snížit dávky inzulínu a/nebo jiných hypoglykemických léčivých přípravků.
Před zahájením léčby přípravkem INCRELEX se doporučuje u všech pacientů provést echokardiografické vyšetření. Po ukončení léčby se má provést kontrolní echokardiografie. U pacientů s abnormálním nálezem v echokardiogramu nebo s příznaky kardiovaskulárního onemocnění se musí pravidelně provádět kontrolní echokardiografické vyšetření.
V souvislosti s použitím přípravku INCRELEX byla hlášena hypertrofie lymfoidní tkáně (např. tonzil), spojená s komplikacemi, jako je chrápání, spánkové apnoe a chronické efuze ze středouší. Pacienty je třeba pravidelně sledovat, zda se u nich neobjevují klinické příznaky, a případně vyloučit tyto potenciální komplikace nebo zahájit vhodnou léčbu.
U pacientů léčených přípravkem INCRELEX byly hlášeny intrakraniální hypertenze (IH) s papiloedémem, změnami vidění, bolestí hlavy, nevolností a/nebo zvracením, stejně jako u pacientů léčených terapeutickými dávkami GH. Známky a příznaky související s IH vymizely po přerušení podávání tohoto přípravku. Na počátku léčby přípravkem INCRELEX, pravidelně v jejím průběhu a při výskytu klinických příznaků se doporučuje provést fundoskopické vyšetření.
U pacientů s rychlým růstem se může objevit sklouznutí hlavice femorální epifýzy (v potenciálem vést k avaskulární nehroze') a progrese skoliózy. Tyto stavy a další známky a příznaky, o nichž je známo, že obecně souvisejí s léčbou GH, je nutno sledovat i během léčby přípravkem INCRELEX. Je nutno vyšetřit všechny pacienty, u nichž se objeví kulhání nebo kteří si stěžují na bolest v koleni nebo kyčli.
V postmarketingových zkušenostech u pacientů léčených přípravkem INCRELEX jsou hlášeny případy přecitlivělosti, kopřivky, svědění a zarudnutí. Tyto byly pozorovány jako systémové a/nebo lokální v místě injekce. Byl hlášen malý počet případů svědčících pro anafylaxi vyžadující hospitalizaci. Rodiče i pacienty je nutno poučit o tom, že se tyto reakce mohou vyskytnout a že
v případě, že se vyskytnou systémové alergické reakce, je nutno přerušit léčbu a vyhledat okamžitou lékařskou pomoc.
Léčba by se měla znovu zvážit, pokud po roce zůstává pacient bez odpovědi.
Pacienti s alergickou reakcí na injekci IGF-1, pacienti s neobvykle vysokou hladinou IGF-1 v krvi po injekci nebo pacienti bez růstové reakce mohou mít reakci protilátek na injikovaný IGF-1. V takových případech by se mělo postupovat podle instrukcí pro testování protilátek.
Pomocné látky
INCRELEX obsahuje 9 mg/ml benzylalkoholu jako konzervační látku.
Benzylalkohol může způsobit toxické reakce a anafylaktoidní reakce u kojenců a dětí do tří let.
Tento léčivý přípravek obsahuje méně než 1 mmol sodíku (23 mg) v jedné lahvičce, tj. v podstatě "bez sodíku".
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Žádné studie interakcí nebyly provedeny.
Možná bude nutné snížit dávky inzulínu a/nebo jiných hypoglykemických léčivých přípravků (viz bod 4.4).
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku / Antikoncepce u mužů a žen
U všech žen v plodném věku je před zahájením léčby přípravkem INCRELEX vhodné ujistit se o negativitě těhotenského testu. U všech žen v plodném věku je také během léčby doporučeno používat vhodnou antikoncepci.
O podávání mecaserminu těhotným ženám nejsou žádné informace nebo jen omezené množství informací.
Studie na zvířatech jsou nedostačující s ohledem na reprodukční toxicitu (viz bod 5.3). Potenciální riziko pro lidské pacienty není známé.
INCRELEX se nesmí používat během těhotenství, pokud to není z jasných důvodů nezbytné.
Kojení
Podávání přípravku INCRELEX kojícím ženám se nedoporučuje.
Fertilita
U potkanů byla provedena s přípravkem INCRELEX teratologická studie bez účinku na plod až do dávky 16 mg/kg (20 násobek MRHD založený na Body surface Area) a teratologická studie u králíků bez účinku na plod při dávce 0,5 mg/kg (2 násobek MRHD založený na Bodu surface Area). U potkanů nemá INCRELEX žádný vliv na fertilitu při použití intravenózních dávek 0,25, 1 a 4 mg/den (až do 4 násobku klinické expozice s MRHD založené na AUC).
Účinky přípravku INCRELEX na nenarozené děti nebyly studovány. Proto nejsou dostatečná medicínská data ke stanovení, zda existují závažná rizika pro plod. Studie s přípravkem INCRELEX nebyly provedeny u kojících matek. INCRELEX se nesmí podávat těhotným a kojícím ženám. Negativní těhotenský test a používání vhodné antikoncepce je nutné u všech premenopauzálních žen užívajících INCRELEX.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Hypoglykémie je velmi častým nežádoučím účinkem. V případě hypoglykemické epizody může mít INCRELEX výrazný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky
Shrnutí bezpečnostního profilu
Údaje o nežádoucích účincích byly převzaté celkem od 413 pacientů klinické studie s těžkou primární IGFD. Údaje byly získávány také z post-marketingových zdrojů.
Nejčastěji hlášené nežádoucí účinky v rámci klinických studií byly bolest hlavy (44%), hypoglykémie (28%), zvracení (26%), hypertrofie v místě vpichu injekce (17%) a zánět středouší (17%). Intrakraniální hypertenze/zvýšený intrakraniální tlak se objevily u 0,96% pacientů z klinických studií a u nově léčených osob ve věku 7 - 9 let.
Během klinických studií v jiných indikacích čítajících přibližně 300 pacientů bylo obdrženo hlášení lokální a/nebo systémové hypersenzitivity u 8 % pacientů. Systémová hypersenzitivita byla také hlášena z post-marketingového použití, některé z těchto případů svědčily o anafylaxi. V rámci post-marketingového použití byly hlášeny také místní alergické reakce.
U některých pacientů se mohou vyvinout protilátky na INCRELEX. Žádný útlum růstu nebyl pozorován jako následek rozvoje protilátek.
Tabulkový seznam nežádoucích účinků
Tabulka 1 uvádí velmi časté (> 1/10), časté (> 1/100 až < 1/10) a méně časté (> 1/1000, < 1/100) nežádoucí reakce, ke kterým došlo při klinických zkouškách. V každé skupině četností jsou nežádoucí reakce seřazeny podle klesající závažnosti. Další nežádoucí účinky byly zjištěny při poregistračním použití přípravku INCRELEX. Jelikož jsou tyto reakce hlášeny dobrovolně z populace o neurčité velikosti, není možné spolehlivě určit jejich frekvenci.
Tabulka 1: Nežádoucí účinky
|
Klasifikace orgánového systému |
Reakce hlášené v klinických studiích |
Reakce hlášené z post-marketingového prostředí |
|
Poruchy krve a lymfatického systému |
Časté: hypertrofie thymu | |
|
Poruchy imunitního systému |
Neznámé: systémová hypersensitivita (anafylaxe, generalizovaná kopřivka, angioedém, dyspnoe) Lokální alergická reakce v místě vpichu (svědění, kopřivka). | |
|
Poruchy metabolismu a výživy |
Velmi časté: hypoglykémie Časté: hypoglykemický záchvat, hyperglykémie, | |
|
Psychiatrické poruchy | ||
|
Poruchy nervového systému |
Velmi časté: bolest hlavy Časté: křeče, závrať, třes Méně časté: Benigní intrakraniální hypertenze | |
|
Poruchy zraku |
Časté: papiloedém | |
|
Poruchy ucha a labyrintu |
Časté: Bolest ucha, tekutina ve středním uchu Méně časté: nedoslýchavost, abnormální tympanometrie | |
|
Srdeční poruchy |
Časté: srdeční šelest, tachykardie Méně časté: kardiomegalie, ventrikulární hypertrofie, mitrální insuficience, trikuspidální insuficience | |
|
Respirační, hrudní a mediastinální poruchy |
Časté: syndrom spánkového apnoe, adenoidní hypertrofie, tonsilární hypertrofie, chrápání, syndrom spánkové apnoe | |
|
Gastrointestinální poruchy |
Velmi časté: zvracení, bolest břicha, bolest v horní části břicha | |
|
Poruchy kůže a podkožní tkáně |
Časté: hypertrofie kůže, abnormální struktura vlasu |
Neznámé: alopecie |
|
Poruchy pohybového systému a pojivové tkáně |
Časté: artralgie, bolest končetin, skolióza | |
|
Novotvary benigní, maligní a blíže neurčené (zahrnující cysty a polypy) |
Časté: Melanocytární névus | |
|
Poruchy reprodukčního |
Časté: Gynekomastie |
|
systému a choroby prsů | ||
|
Celkové poruchy a reakce v místě podání |
Velmi časté: Hypertrofie v místě vpichu Časté: Reakce v místě injekce (např. erytém, bolest, podráždění, hematom, krvácení, zatvrdnutí, podlitina) Méně časté: Reakce v místě injekce (např. vyrážka, otok), lipohypertrofie |
Neznámé: Lokální alergické reakce v místě injekce (pruritus, vyrážka) |
|
Laboratorní a funkční vyšetření |
Méně časté: Zvýšení hmotnosti | |
|
Chirurgické a léčebné postupy |
Časté: Zavedení ušní trubice |
Popis vybraných nežádoucích reakcí Systémová/lokální hypersenzitivita
Klinické studie: Během klinických studíí v jiných indikacích (čítajících přibližně 300 pacientů) bylo obdrženo hlášení lokální a/nebo systémové hypersenzitivity u 8% pacientů. Všechny případy byly mírné nebo středně závažné a žádný nebyl závažný.
Post-marketingové hlášení: systémová hypersenzitivita zahrnovala příznaky jako anafylaxe, generalizovaná kopřivka, angioedém a dušnost. Tyto příznaky v těchto případech poukazovaly na anafylaxi včetně kopřivky, angioedému a dyspnoe. Někteří pacienti vyžadovali hospitalizaci. Při opakovaném podání se příznaky neobjevily znovu u všech pacientů. Byly hlášeny také lokální alergické reakce v místě vpichu. Obvykle se jednalo o svědění a kopřivku.
Hypoglykémie
Ze 115 subjektů (28%), kteří měli jednu nebo více epizod hypoglykémie, 6 subjektů prodělalo hypoglykemický záchvat, který se vyskytl jednou nebo vícekrát. Symptomatické hypoglykémii se zpravidla zabránilo, pokud pacient krátce před nebo po podání přípravku INCRELEX snědl jídlo nebo svačinu.
Hypertrofie místa vpichu
Tato reakce se objevila u 71 (17%) subjektů z klinických studií a byla obecně spojena s nesprávným způsobem měnění místa vpichu. Pokud se injekce správně rozptýlily, tento stav vymizel.
Hypertrofie tonzil
Toto bylo zaznamenáno u 38 (9%) subjektů, zejména v prvním nebo druhém roce léčby, s menším nárůstem tonziliární tkáně v následujících letech.
Chrápání
Chrápání se většinou objevilo obvykle v prvním roce léčby, bylo hlášeno u 30 subjektů (7%). Intrakraniální hypertenze/zvýšený intrakraniální tlak
U čtyř subjektů (0,96%) se objevila intrakraniální hypertenze; u dvou subjektů byla léčba přípravkem INCRELEX přerušena a již nebyla obnovena; u dvou subjektů se po opětovném nasazení přípravku INCRELEX ve snížené dávce příhoda neobjevila. Všechny čtyři subjekty se z nežádoucího účinku zotavily bez následků.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím prostřednictvím národního systému hlášení nežádoucích účinků uvedeného Dodatku V.
4.9 Předávkování
Akutní předávkování může způsobit hypoglykémii. Při dlouhodobém předávkování se mohou objevit známky a příznaky akromegalie a gigantismu.
Léčba akutního předávkování mecaserminem má být zaměřena na zmírnění vlivu hypoglykémie. Má se podat orálně glukóza nebo jídlo. Pokud při předávkování dojde ke ztrátě vědomí, pravděpodobně bude zapotřebí podat intravenózně glukózu nebo parenterálně glukagon, aby se odstranily účinky hypoglykémie.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina:Hormony hypofýzy a hypotalamu a jejich analoga, somatropin a agonisté somatropinu, ATC kód: H01AC03
Mecasermin je lidskému inzulínu podobný růstový faktor 1 (rhIGF-1), vyrobený technologií rekombinantní DNA. IGF-1 se skládá ze 70 aminokyselin v jednom řetězci se třemi intramolekulárními disulfidovými můstky a jeho molekulová hmotnost je 7649 daltonů. Pořadí aminokyselin v produktu je identické s pořadím aminokyselin v endogenním lidském IGF-1. Protein rhIGF-1 je syntetizován v bakteriích (E. coli), které byly modifikovány přidáním genu lidského IGF-1.
Mechanismus účinku
Inzulínu podobný růstový faktor-1 (IGF-1) je hlavním hormonálním mediátorem tělesného růstu. Za normálních okolností se růstový hormon (GH) váže na svůj receptor v játrech a v dalších tkáních a stimuluje syntézu nebo sekreci IGF-1. V cílové tkáni je působením IGF-1 aktivován receptor IGF-1 typu 1, který je homologní s receptorem inzulínu, což vede k intracelulární signalizaci, která stimuluje více procesů vedoucích k tělesnému růstu. Metabolické účinky IGF-1 jsou zčásti směrovány na stimulaci absorpce glukózy, mastných kyselin a aminokyselin tak, aby metabolismus podporoval rostoucí tkáně.
Farmakodynamické účinky
U endogenního lidského IGF-1 byly demonstrovány následující účinky:
Růst tkání
K růstu skeletu dochází v epifyzárních ploténkách na koncích rostoucí kosti. Růst a metabolismus buněk epifyzárních plotének je přímo stimulován GH a IGF-1.
Růst orgánů: Léčba potkanů s deficitem IGF-1 pomocí rhIGF-1 způsobuje růst celého těla i orgánů. Růst buněk: Receptory IGF-1 jsou přítomny ve většině typů buněk a tkání. IGF-1 má mitogenní aktivitu vedoucí ke zvýšení počtu tělesných buněk.
Metabolismus sacharidů
IGF-1 potlačuje produkci glukózy v játrech, stimuluje využití periferní glukózy a může snižovat hladinu glukózy v krvi a způsobovat hypoglykémii.
IGF-1 potlačuje sekreci inzulínu.
Metabolismus kostí a minerálů
Cirkulující IGF-1 hraje důležitou roli při získávání a udržování kostní hmoty. IGF-1 zvyšuje hustotu kostní tkáně.
Přípravek INCRELEX byl testován v pěti klinických studiích (4 otevřených a 1 dvojitě zaslepené, kontrolované placebem). 92 pediatrickým subjektům se závažnou primární IGFD byly podkožně aplikovány dávky mecaserminu, všeobecně v rozsahu od 60 do 120 pg/kg dvakrát denně (BID). Pacienti byli zahrnuti do studií na základě extrémně malé tělesné výšky, pomalého růstu, nízké koncentrace IGF-1 v séru a normální sekrece GH. Osmdesáttři (83) z 92 pacientů bylo při vstupu do studie přípravkem INCRELEX léčeno nově a 81 pacientů dokončilo alespoň jeden rok léčby přípravkem INCRELEX. Vstupní hodnoty 81 pacientů vyhodnocovaných v analýzách primární a sekundární účinnosti v kombinovaných studiích byly následující (průměr ± SD): chronologický věk (let): 6,8 ± 3,8; výška (cm): 84,1 ± 15,8; skóre odchylky od standardní výšky (SDS): -6,9 ± 1,8; rychlost růstu (cm/rok): 2,6 ± 1,7; SDS rychlosti růstu: -3,4 ± 1,6; IGF-1 (ng/ml): 24,5 ± 27,9; SDS IGF-1: -4,2 ± 2,0; a kostní věk (roky): 3,8 ± 2,8. Z těchto mělo 72 (85 %) fenotyp podobný Laronovu syndromu; 7 (9 %) mělo deleci genu GH a 1 (1 %) měl neutralizující protilátky proti GH a 1 (1%) měl izolovanou genetickou nedostatečnost GH. Čtyřicetšest (57%) těchto subjektů bylo pohlaví mužského; 66 (81 %) subjektů byli běloši. Sedmdesátčtyři (91%) subjektů bylo při vstupu do studie v prepubertálním věku.
Roční výsledky rychlosti růstu, SDS rychlosti růstu a SDS výšky až do 8 let uvádí Tabulka 2. Údaje o rychlosti růstu před léčbou byly k dispozici u 75 subjektů. Rychlost růstu v daném roce léčby byla po dokončení daného roku léčby porovnávána párovanými t-testy s rychlostí růstu stejného subjektu před léčbou. Rychlosti růstu v letech 2 až 8 zůstaly statisticky vyšší než výchozí hodnoty. U 21 nově léčených subjektů s výškou blízkou dospělé osobě byl průměr (±SD) rozdílu mezi dosaženým nárustem výšky a očekávaným dle Laron přibližně 13 cm (±8 cm) po průměrně 11 letech léčby.
|
Pre-Tx |
1. rok |
2. rok |
3. rok |
4. rok |
5. rok |
6. rok |
7. rok |
8. rok | |
|
Rychlost růstu (cm/rok) | |||||||||
|
N Průměr (SD) Průměr (SD) změny oproti Pre-Tx P-hodnota pro změnu oproti Pre-Tx [1] |
75 2,6 (1,7) |
75 8,0 (2,3) +5,4 (2,6) <0,0001 |
63 5,9 (1,7) +3,2 (2,6) <0,0001 |
62 5,5 (1,8) +2,8 (2,4) <0,0001 |
60 5,2 (1.5) +2,5 (2.5) <0,0001 |
53 4,9 (1,5) +2,1 (2,1) <0,0001 |
39 4,8 (1,4) +1,9 (2,1) <0,0001 |
25 4,3 (1,5) +1,4 (2,2) 0,0042 |
19 4,4 (1,5) +1,3 (2,8) 0,0486 |
|
SDS rychlosti růstu | |||||||||
|
N Průměr (SD) Průměr (SD) změny oproti Pre-Tx P-hodnota pro změnu oproti Pre-Tx [1] |
75 2,6 (1,7) |
75 8,0 (2,3) +5,4 (2,6) <0,0001 |
63 5,9 (1,7) +3,2 (2,6) <0,0001 |
62 5,5 (1,8) +2,8 (2,4) <0,0001 |
60 5,2 (1.5) +2,5 (2.5) <0,0001 |
53 4,9 (1,5) +2,1 (2,1) <0,0001 |
39 4,8 (1,4) +1,9 (2,1) <0,0001 |
25 4,3 (1,5) +1,4 (2,2) 0,0042 |
19 4,4 (1,5) +1,3 (2,8) 0,0486 |
|
SDS výšky | |||||||||
|
N Průměr (SD) Průměr (SD) změny oproti Pre-Tx |
81 -6,9 (1,8) |
81 -6,1 (1,8) +0,8 (0,6) |
67 -5,6 (1,7) +1,2 (0,9) |
66 -5,3 (1,7) +1,4 (1,1) |
64 -5,1 (1,7) +1,6 (1,2) |
57 -5,0 (1,7) +1,7 (1,3) |
41 -4,9 (1,6) +1,8 (1,1) |
26 -4,9 (1,7) +1,7 (1,0) |
19 -5,1 (1,7) +1,7 (1,0) |
|
P-hodnota pro změnu oproti Pre-Tx [1] |
<0,0001 |
<0,0001 |
<0,0001 |
<0,0001 |
<0,0001 |
<0,0001 |
0,0001 |
<0,0001 |
Pre-Tx = před léčbou; SD = standardní odchylka; SDS = skóre standardní odchylky
[1] P-hodnoty pro srovnání s hodnotami Pre-Tx byly vypočítány pomocí párovaných t-testů.
U subjektů s dostupnými údaji o kostním věku po dobu nejméně 6 let od zahájení léčby bylo průměrné zvýšení v kostním věku srovnatelné s průměrným zvýšením v chronologickém věku; u těchto subjektů se kostní věk blízký chronologickému věku nejevil jako klinicky signifikantní výhoda.
Účinnost je závislá na dávce. Dávka 120 pg/kg podávaná subkutánně a 2x denně byla spojována s největší odpovědí růstu.
Ze všech subjektů zahrnutých do hodnocení bezpečnosti (n=92), byl u 83% subjektů hlášen alespoň jeden nežádoucí účinek v průběhu trvání studií. V průběhu studií se nevyskytlo žádné úmrtí. Žádný ze subjektů nepřerušil účast ve studii z důvodu nežádoucích účinků.
Nejčastěji hlášeným nežádoucím účinkem byla hypoglykémie a je třeba klást dostatečnou pozornost na jídlo ve vztahu k podávání.
Tento léčivý přípravek byl registrován postupem tzv. „podmíněného schválení“
Znamená to, že vzhledem k vzácnosti onemocnění nebylo možné získat úplné informace o tomto léčivém přípravku.
Evropská léková agentura každoročně vyhodnotí nové informace o tomto přípravku a tento souhrn údajů o přípravku bude podle potřeby aktualizován.
5.2 Farmakokinetické vlastnosti
Všeobecné vlastnosti
Absorpce
Absolutní subkutánní biologická dostupnost mecaserminu nebyla u subjektů se závažnou primární IGFD stanovena. Biologická dostupnost mecaserminu po subkutánním podání u zdravých subjektů byla hlášena jako přibližně 100 %.
Distribuce
V krvi se IGF-1 váže na šest vazebných bílkovin IGF (IGFBP), s přibližně 80 % vazbou jako komplex s IGFBP-3 a podjednotkou IGF labilní na kyseliny. Koncentrace IGFBP-3 je u subjektů se závažnou primární IGFD snížená, což má za následek zvýšenou clearanci IGF-1 u těchto subjektů v porovnání se zdravými subjekty. Celkový distribuční objem IGF-1 (průměr ± SD) po podkožním podání přípravku INCRELEX u 12 subjektů se závažnou primární IGFD je odhadován na 0,257 (± 0,073) l/kg při dávce mecaserminu 0,045 mg/kg a předpokládá se, že při vyšší dávce mecaserminu bude vyšší. O koncentraci volného IGF-1 po podání přípravku INCRELEX jsou k dispozici pouze omezené údaje.
Biotransformace
Bylo prokázáno, že IGF-1 je metabolizován v játrech i v ledvinách.
Eliminace z organismu
Průměrná odhadovaná hodnota eliminačního poločasu tm celkového IGF-1 po jednorázovém podkožním podání 0,12 mg/kg u tří pediatrických subjektů se závažnou primární IGFD je 5,8 hodin. Clearance celkového IGF-1 je nepřímo úměrná sérové koncentraci IGFBP-3 a odhadovaná systémová clearance celkového IGF-1 (CL/F) je 0,04 l/h/kg při 3 mg/l IGFBP-3 u 12 subjektů.
Zvláštní skupiny pacientů
Starší pacienti
Farmakokinetické vlastnosti přípravku INCRELEX u subjektů starších 65 let nebyly zjišťovány.
Děti
Farmakokinetické vlastnosti přípravku INCRELEX u subjektů mladších 12 let nebyly zjišťovány. Pohlaví
U dětí starších 12 let s primární IGFD a u zdravých dospělých nebyly ve farmakokinetice přípravku INCRELEX žádné zjevné rozdíly mezi muži a ženami.
Rasa
K dispozici nejsou žádné údaje. Renální _ poškození
Nebyly provedeny žádné klinické studie s dětmi se zhoršenou funkcí ledvin.
Hepatická _ poškození
Nebyly provedeny žádné studie ke zjištění vlivu zhoršené funkce jater na farmakokinetiku mecaserminu.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti, toxicity po opakovaném podávání a genotoxicity neodhalily žádné zvláštní riziko pro člověka.
Nežádoucí účinky, které nebyly zaznamenány v klinických studiích, avšak vyskytly se v pokusech na zvířatech po expozici podobné klinické expozici, a pravděpodobně důležité pro klinické použití, byly následující:
Reprodukční toxicita
Byla studována reprodukční toxicita u potkanů a králíků po intravenózním podávání, nikoli po subkutánním (normální klinický způsob podání). Tyto studie neprokázaly přímý ani nepřímý škodlivý vliv na plodnost a březost, ale v důsledku odlišného způsobu podání je relevance těchto nálezů nejasná. Placentální přenos mecaserminu nebyl studován.
Karcinogenní potenciál
Mecasermin byl podáván subkutánně u potkanů Sprague Dawley v dávkách 0; 0,25; 1; 4 a 10 mg/kg/den po dobu 2 let. U potkaních samců při dávkách 1 mg/kg/den a vyšších (> 1násobek klinické expozice s maximální doporučenou humánní dávkou [MRHD] podle AUC) a u potkaních samic při všech úrovních dávek (> 0,3násobek klinické expozice MRHD podle AUC) byl pozorován zvýšený výskyt adrenální medulární hyperplazie a feochromocytomu.
Zvýšený výskyt keratoakantomu pokožky byl pozorován u potkanich samců při dávkách 4 a 10 mg/kg/den (> čtyřnásobek expozice MRHD podle AUC). Zvýšený výskyt karcinomu mléčné žlázy u potkaních samců i samic byl pozorován u zvířat, jimž byly podávány dávky 10 mg/kg/den (sedminásobek expozice MRHD podle AUC). Ve studiích karcinogeneze byla pozorována mimořádná mortalita v důsledku hypoglykémie navozené IGF-1.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Benzylalkohol Chlorid sodný Polysorbát 20 Kyselina octová 99%
Octan sodný Voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto tento léčivý přípravek nesmí být mísen s žádnými dalšími léčivými přípravky.
6.3 Doba použitelnosti
3 roky
Po otevření:
Byla prokázána chemická a fyzikální stabilita přípravku po dobu 30 dnů při teplotě od 2 °C do 8 °C.
Z mikrobiologického hlediska se přípravek po otevření může uchovávat maximálně 30 dnů při teplotě od 2 °C do 8 °C. Za použití jiné doby a podmínek uchovávání otevřeného výrobku odpovídá uživatel.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Injekční lahvičku uchovávejte v krabičce, aby byla chráněna před světlem.
Podmínky uchovávání tohoto léčivého přípravku po jeho prvním otevření jsou uvedeny v bodě 6.3.
6.5 Druh obalu a velikost balení
4 ml roztoku v 5 ml injekční lahvičce (sklo typ I) uzavřené zátkou ( bromobutyl/izopren) a těsnicím uzávěrem (lakovaný plast).
Velikost balení: 1 injekční lahvička.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
INCRELEX se dodává jako vícedávkový roztok.
Roztok by měl být ihned po vyjmutí z chladničky čirý. Je-li roztok zkalený nebo obsahuje-li částice, nesmí být podáván (viz bod 4.2).
Všechen nepoužitý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Ipsen Pharma 65, quai Georges Gorse 92100 Boulogne-Billancourt Francie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/07/402/001
9. DATUM PRVNÍ REGISTRACE / PRODLOUŽENÍ REGISTRACE
Datum první registrace: 03/08/2007
Datum posledního prodloužení registrace: 03/08/2012
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu.
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
E. ZVLÁŠTNÍ POVINNOST USKUTEČNIT POREGISTRAČNÍ OPATŘENÍ PRO REGISTRACI PŘÍPRAVKU ZA VÝJIMEČNÝCH OKOLNOSTÍ
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce biologické léčivé látky
Lonza Biologics, Inc.
97 South Street
Hopkinton, Massachusetts 01748 USA
Název a adresa výrobce odpovědného/výrobců odpovědných za propouštění šarží
Beaufour Ipsen Industrie Rue ďEthe Virton 28100 Dreux Francie
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
• Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
Další opatření k minimalizaci rizik
Držitel rozhodnutí o registraci musí při uvedení na trh zajistit, aby všichni lékaři, u nichž se předpokládá, že mohou předepisovat INCRELEX, dostali „informační balíček pro lékaře“, obsahující následující:
• Informace o přípravku
• Informace o přípravku INCRELEX pro lékaře (informační karta, návod k dávkování a kalkulačka pro výpočet dávkování)
• Informační balíček pro pacienty
Informace o přípravku INCRELEX pro lékaře mají obsahovat následující klíčové pokyny:
• Poučení pro rodiče o známkách, příznacích a léčbě hypoglykémie včetně injekční aplikace glukagonu.
• Pacienty je třeba pravidelně otorhinolaryngologicky vyšetřovat, zda se u nich neobjevují klinické příznaky, a případně vyloučit potenciální komplikace nebo zahájit vhodnou léčbu.
• Je nutno provést běžné funduskopické vyšetření před zahájením léčby a pravidelně během ní nebo při výskytu klinických příznaků.
• INCRELEX je kontraindikován při výskytu aktivních nebo suspektních novotvarů, a pokud se prokáže vývoj novotvaru, je třeba léčbu přerušit.
• U pacientů s rychlým růstem se může objevit sklouznutí hlavice femorální epifýzy a progrese skoliózy. Tyto stavy je třeba v průběhu léčby přípravkem INCRELEX sledovat.
• Rodiče i pacienty je nutno poučit o tom, že se systémové alergické reakce mohou vyskytnout a že v případě jejich výskytu je nutno přerušit léčbu a vyhledat okamžitou lékařskou pomoc.
• Informace o odběru vzorků pro vyšetření imunogenicity.
Informace o přípravku INCRELEX pro pacienty mají obsahovat následující informace:
• INCRELEX se má podávat krátce po jídle nebo před jídlem, protože může způsobovat hypoglykemické stavy podobně jako inzulin.
• Známky a příznaky hypoglykémie. Instrukce k léčbě hypoglykémie. Rodiče a pečující osoby musí dítěti trvale zajistit možnost podání cukru. Instrukce k podávání glukagonu v případě výskytu závažné hypoglykémie.
• INCRELEX se nesmí podávat, pokud pacient není schopen z jakéhokoli důvodu jíst. Dávka přípravku INCRELEX se nikdy nesmí zdvojnásobit jako náhrada za jednu nebo dvě vynechané dávky.
• Pacienti se musí vyhýbat jakýmkoli rizikovým činnostem (tj. namáhavé tělesné aktivitě) 2-3 hodiny po aplikaci dávky, a to zejména na začátku léčby přípravkem INCRELEX, dokud nebude zjištěna dobře tolerovaná dávka přípravku INCRELEX.
• Instrukce ohledně změn místa vpichu při každé injekci, aby se zamezilo vzniku lipohypertrofie.
• Instrukce k ohlášení vzniku nebo zhoršení chrápání, které může indikovat zvětšení mandlí a/nebo adenoidů po zahájení léčby přípravkem INCRELEX.
• Instrukce k ohlášení výskytu závažné bolesti hlavy, rozostřeného vidění a související nevolnosti a zvracení svému lékaři.
• Instrukce k ohlášení výskytu kulhání nebo bolesti v kyčli nebo koleni, aby bylo možno provést klasifikační vyšetření.
Kromě toho bude balíček obsahovat návod k dávkování a kalkulačku pro výpočet dávkování k použití pro lékaře a pacienty; tyto pomůcky budou obsahovat informace o individuální eskalaci dávky se současnou minimalizací rizika medikačních chyb a vzniku hypoglykémie.
E. ZVLÁŠTNÍ POVINNOST USKUTEČNIT POREGISTRAČNÍ OPATŘENÍ PRO REGISTRACI PŘÍPRAVKU ZA VÝJIMEČNÝCH OKOLNOSTÍ
Tato registrace byla schválena za „výjimečných okolností“, a proto podle článku 14(8) nařízení (ES) č 726/2004 držitel rozhodnutí o registraci uskuteční v daném termínu následující opatření:
|
Provést dlouhodobou bezpečnostní studii, v níž bude léčba mecaserminem zahájena v rané fázi dětství s pokračováním až do dospělosti za účelem vyšetření: • Dlouhodobé toxicity u pacientů procházejících vývojovými změnami • Možného výskytu malignit a jiných rizik Předběžné zprávy budou předkládány každé dva roku a to až do doby, kdy bude ukončeno 5-ti leté sledování posledního pacienta. |
Finální zpráva v roce 2023 |
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU KARTÓNOVÁ KRABIČKA
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
INCRELEX 10 mg/ml injekční roztok mecaserminum
2. OBSAH LÉČIVÉ LÁTKY / LÉČIVÝCH LÁTEK
Jeden ml obsahuje 10 mg mecaserminum.
Jedna injekční lahvička obsahuje 40 mg mecaserminum.
3. SEZNAM POMOCNÝCH LÁTEK
Pomocnými látkami jsou: benzylalkohol, chlorid sodný, polysorbát 20, kyselina octová 99%, octan sodný a voda na injekci.
Viz příbalová informace.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Injekční roztok.
Jedna injekční lahvička na více použití, 4 ml.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Subkutánní podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
Použijte do 30 dnů po prvním otevření.
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce.
Nezmrazujte.
Injekční lahvičku uchovávejte v krabičce, aby byla chráněna před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Všechen nepoužitý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Ipsen Pharma 65, quai Georges Gorse 92100 Boulogne-Billancourt Francie
12. REGISTRAČNÍ ČÍSLO(A)
EU/1/07/402/001
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku je vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
INCRELEX
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA JEDNOTKÁCH BALENÍ K BEZPROSTŘEDNÍMU POUŽITÍ
INJEKČNÍ LAHVIČKA
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
INCRELEX 10 mg/ml injekce
mecaserminum
SC
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
4 ml
6. JINÉ
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro uživatele
INCRELEX 10 mg/ml injekční roztok
mecaserminum
'VTento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité informace.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu. Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci:
1. Co je INCRELEX a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete INCRELEX používat
3. Jak se INCRELEX používá
4. Možné nežádoucí účinky
5. Jak INCRELEX uchovávat
6. Obsah balení a další informace
1. Co je INCRELEX a k čemu se používá
- INCRELEX je kapalina obsahující růstový faktor-1 podobný lidskému inzulinu (IGF-1), který je podobný IGF-1 produkovanému v lidském těle.
- INCRELEX se používá k léčbě dětí nebo dospívajících, kteří jsou na svůj věk velmi malí, protože jejich tělo nedokáže vyrobit dostatek IGF-1. Tento stav se nazývá primární deficience IGF-1.
2. Čemu musíte věnovat pozornost, než začnete INCRELEX používat Nepoužívejte INCRELEX
- jestliže jste alergický(á)/ na mecasermin nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
- jestliže máte rakovinu.
- u nedonošených ani donošených novorozenců, protože obsahuje benzylalkohol. Upozornění a opatření
Před použitím přípravku INCRELEX se poraďte se svým lékařem nebo lékarníkem
- jestliže máte křivou páteř (skoliózu). Vývoj skoliózy musí být u Vás monitorován.
- jestliže se u Vás objeví kulhání nebo bolest kyčelního nebo kolenního kloubu.
- jestliže máte zvětšené mandle (tonsilární hypertrofii). Pak byste měl chodit na pravidelná vyšetření.
- jestliže máte příznaky zvýšeného tlaku v mozku (intrakraniální hypertenze), například změny vidění, bolesti hlavy, nevolnost a/nebo zvracení, je třeba vyhledat lékaře, který poradí, co dělat.
- jestliže máte lokalizovanou reakci v místě injekce nebo generalizovanou alergickou reakci na přípravek INCRELEX. Vyhledejte co nejrychleji lékaře, pokud máte lokalizovanou vyrážku. Okamžitou lékařskou pomoc vyhledejte, pokud máte generalizovanou alergickou reakci (kopřivku, ztížené dýchání, mdloby nebo kolaps a pocit pacienta, že se celkově necídí dobře).
- jestliže u Vás došlo k ukončení tělesného růstu (růstové ploténky jsou uzavřeny).
V takovém případě Vám přípravek INCRELEX nepomůže růst a neměl by být používán.
Děti mladší 2 let
Použití tohoto přípravku u dětí mladších 2 let nebylo studováno, a proto se nedoporučuje.
Další léčivé přípravky a přípravek INCRELEX
Prosím, informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, v nedávné době jste užíval(a) nebo jste mohl(a) užívat.
Lékaře informujte zejména, jestliže berete inzulin nebo jiné léky proti diabetu. U těchto léků bude možná nutné upravit dávkování.
Těhotenství, kojení a fertilita
U všech žen v plodném věku je před zahájením léčby přípravkem INCRELEX nutný negativní těhotenský test. U všech žen v plodném věku je také během léčby doporučeno používat vhodnou antikoncepci.
Léčbu přípravkem INCRELEX je třeba přerušit, pokud pacientka otěhotní.
INCRELEX se nesmí podávat kojícím matkám.
Řízení dopravních prostředků a obsluha strojů
INCRELEX může způsobit hypoglykémii (velmi častý nežádoucí účinek), která může zhoršit Vaši schopnost řídit a obsluhovat stroje, protože Vaše schopnost soustředit se nebo reagovat může být tímto snížena.
Měl(a) byste se vyhýbat jakýmkoli rizikovým činnostem (tj. řízení apod.) 2-3 hodiny po aplikaci dávky, a to zejména na začátku léčby přípravkem INCRELEX, dokud nebude zjištěna příslušná dávka přípravku INCRELEX, při které nedochází k výskytu nežádoucích účinků, které dělají tyto činnosti rizikovými.
INCRELEX obsahuje benzylalkohol
INCRELEX obsahuje 9 mg/ml benzylalkoholu jako konzervační látku. Benzylalkohol může způsobit toxické reakce a alergické reakce u kojenců a dětí do tří let.
Tento léčivý přípravek obsahuje méně než 1 mmol sodíku (23 mg) v jedné lahvičce, tj. v podstatě "bez sodíku".
3. Jak se INCRELEX používá
Vždy používejte tento přípravek přesně podle pokynů, které Vám sdělil lékař. Pokud si nejste jistý(á), poraďte se s lékařem nebo s lékárníkem.
Obvyklá dávka je 0,04 až 0,12 mg/kg hmotnosti pacienta podávaná dvakrát denně. Viz „Návod k použití“ na konci této příbalové informace.
INCRELEX aplikujte injekcí těsně pod kůži pacienta krátce před nebo po jídle nebo svačině, protože může mít podobně jako inzulin hypoglykemický účinek a tedy může snižovat hladinu cukru v krvi (viz část hypoglykémie v bodě 4). Pokud z nějakého důvodu pacient nemůže jíst, dávku přípravku INCRELEX nepodávejte. Nezdvojujte následující dávku, abyste nahradil(a) vynechanou dávku. Následující dávka by měla být podána jako obvykle s jídlem nebo svačinou.
INCRELEX aplikujte injekcí těsně pod kůži pacienta na paži, stehně, břiše nebo hýždích. Přípravek se nikdy nesmí aplikovat přímo do žíly ani do svalu. Při každé injekci změňte místo vpichu.
Přípravek INCRELEX používejte pouze tehdy, je-li čirý a bezbarvý.
Léčba přípravkem INCRELEX je dlouhodobá. Další informace žádejte u lékaře.
Jestliže jste použil(a) více přípravku INCRELEX, než jste měl(a)
INCRELEX, podobně jako inzulin, může snižovat hladinu cukru v krvi (viz hypoglykémie v bodě 4).
Pokud jste injekčně aplikoval(a) více přípravku INCRELEX, než jste měl(a), okamžitě kontaktujte Vašeho lékaře.
Akutní předávkování může způsobit hypoglykémii (nízký krevní cukr). Při dlouhodobém předávkování může dojít ke zvětšení některých částí těla (např. rukou, chodidel, částí obličeje) nebo k nadměrnému růstu celého těla.
Léčba akutního předávkování přípravkem INCRELEX má být zaměřena na odstranění hypoglykémie. Mají se konzumovat tekutiny nebo potraviny obsahující cukr. Pokud pacient není při vědomí nebo není dostatečně bdělý, aby mohl vypít tekutinu obsahující cukr, může být k odstranění nízké hladiny krevního cukru zapotřebí aplikovat nitrosvalovou injekci glukagonu. Váš lékař nebo zdravotní sestra Vám ukáže, jak se podává injekce glukagonu.
Jestliže jste zapomněl(a) použít INCRELEX
Nezdvojujte následující dávku, abyste nahradil(a) vynechanou dávku.
Jestliže dojde k vynechání dávky, nezvyšujte následující dávku jako kompenzaci. Další dávka by měla být přijata jako obvykle, s jídlem nebo svačinou.
Jestliže jste přestal/a používat INCRELEX
Předčasné ukončení léčby přípravkem INCRELEX může zhoršit úspěšnost růstové terapie. Před ukončením léčby se prosím poraďte s lékařem.
Máte-li jakékoli další otázky, týkající se užívání tohoto léku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento lék nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci.
Nejčastější nežádoucí účinky přípravku INCRELEX jsou: nízká hladina krevního cukru (hypoglykémie), zvracení, reakce v místě vpichu, bolest hlavy a infekce středního ucha. Byly hlášeny
26
také závažné alergické reakce při léčbě přípravkem INCRELEX. Pokud se u Vás projeví některá z těchto událostí, řiďte se, prosím, pokyny uvedenými v následujících odstavcích pro danou událost.
Frekvence není známa (četnost nelze z dostupných dat stanovit)
Závažné alergické reakce (anafylaxe)
Generalizovaná kopřivka, dýchací potíže, závrať, byl hlášen otok tváře a / nebo hrdla po podání přípravku INCRELEX. Objeví-li se u Vás závažná alergická reakce, ihned přestaňte přípravek INCRELEX používat a vyhledejte okamžitou lékařskou pomoc.
Byly také hlášeny místní alergické reakce v místě vpichu (svědění, kopřivka).
Vypadávání vlasů (alopecie)
Vypadávání vlasů bylo také hlášeno po použití léčivého přípravku INCRELEX.
Velmi časté (mohou postihnout více než 1 pacienta z 10)
Nízká hladina krevního cukru (hypoglykémie)
INCRELEX může snižovat hladinu cukru v krvi. Známky nízké hladiny krevního cukru jsou: závratě, únava, neklid, hlad, podrážděnost, poruchy soustředění, pocení, nevolnost a rychlý nebo nepravidelný puls.
Závažná hypoglykémie může způsobit bezvědomí, záchvaty / křeče nebo smrt. Objeví-li se u Vás záchvaty/křeče nebo bezvědomí, ihned přestaňte přípravek INCRELEX používat a vyhledejte okamžitou lékařskou pomoc.
Pokud používáte INCRELEX, musíte vyloučit účast na vysoce rizikových činnostech (jako je namáhavá tělesná činnost) 2 až 3 hodiny po aplikaci injekce přípravku INCRELEX, a to zejména na začátku léčby přípravkem INCRELEX.
Před zahájením léčby přípravkem INCRELEX Vám lékař nebo zdravotní sestra vysvětlí, jak se léčí hypoglykémie. Musíte mít neustále k dispozici zdroj cukru, jako je pomerančový džus, glukózové želé, bonbón nebo mléko pro případ, že se objeví příznaky hypoglykémie. Při závažné hypoglykémii, když nereagujete a nemůžete vypít tekutinu obsahující cukr, je nutno podat injekci glukagonu. Lékař nebo zdravotní sestra Vám ukáže, jak se injekce aplikuje. Glukagon po injekční aplikaci zvyšuje hladinu krevního cukru. Je důležité, abyste měl(a) dobře vyváženou stravu obsahující kromě potravin s obsahem cukru také potraviny s bílkovinami a tuky (např. v mase a sýrech).
Hypertrofie v místě injekce (zvětšení tkáně v místě injekce) a podlitina Tomuto lze předejít střídáním místa vpichu při každé injekci (rotace místa vpichu).
Zažívací systém
Během léčby přípravkem INCRELEX se vyskytlo zvracení a bolest v horní části břicha.
Infekce
U dětí léčených přípravkem INCRELEX byly pozorovány infekce středního ucha.
Svalová a kosterní soustava
Během léčby přípravkem INCRELEX se vyskytly bolesti kloubů a končetin.
Nervový systém
Během léčby přípravkem INCRELEX se vyskytly bolesti hlavy.
Časté (mohou se objevit u 1 z 10 osob)
Při léčbě lékem INCRELEX byly pozorovány záchvaty/křeče.
Závratě a třes byly také hlášeny během léčby přípravkem INCRELEX.
Srdeční abnormality
Rychlý srdeční puls a abnormální srdeční ozvy byly hlášeny během léčby přípravkem INCRELEX. Zvýšený cukr v krvi (hyperglykémie)
Při léčbě lékem INCRELEX bylo také pozorováno zvýšení cukru v krvi.
Zvětšené mandle/nosní mandle
Přípravek INCRELEX může způsobit zvětšení mandlí/nosních mandlí. Některé příznaky zvětšených mandlí/nosních mandlí zahrnují: chrápání, obtížné dýchání nebo polykání, spánkové apnoe (stav, kdy se ve spánku nakrátko zastaví dech) nebo tekutina ve středním uchu a také infekce ucha. Spánkové apnoe mohou způsobovat nepřiměřenou denní ospalost. Pokud Vám tyto příznaky vadí, vyhledejte lékaře. Lékař by Vám měl pravidelně vyšetřovat mandle / nosní mandle.
Zvětšený brzlík
Během léčby přípravkem INCRELEX bylo pozorováno zvětšení brzlíku (specializovaný orgán imunitního systému).
Papiloedem
Lékař nebo optik může v průběhu léčby přípravkem INCRELEX zaznamenat otok v zadní části oka (z důvodu zvýšeného tlaku v mozku).
Nedoslýchavost (ztráta sluchu)
Nedoslýchavost (ztráta sluchu), bolesti uší a tekutina ve středním uchu byly pozorovány v průběhu léčby s přípravkem INCRELEX. Řekněte svému lékaři, pokud se u Vás objeví potíže se sluchem.
Zhoršení skoliózy (způsobené rychlým růstem)
Máte-li skoliózu, bude Vás potřeba často kontrolovat na zvýšení zakřivení páteře. Při léčbě lékem INCRELEX byla pozorována bolest svalů .
Reprodukční systém
Během léčby přípravkem INCRELEX bylo pozorováno zvětšení prsů.
Zažívací systém
V souvislosti s léčbou přípravkem INCRELEX se vyskytly bolesti břicha.
Změny kůže a vlasů
V souvislosti s léčbou přípravkem INCRELEX bylo pozorováno ztluštění kůže a abnormální struktura vlasů.
Reakce v místě vpichu
Při léčbě přípravkem INCRELEX byly hlášeny reakce zahrnující bolest, podráždění, krvácení, modřiny, červenání a zatvrdnutí. Reakci v místě vpichu lze předejít střídáním místa vpichu při každé injekci (rotace místa vpichu).
Méně časté (mohou se objevit až u 1 ze 100 osob)
Zvýšený tlak v mozku (intrakraniální hypertenze)
INCRELEX může někdy způsobit dočasné zvýšení tlaku v mozku. Mezi příznaky nitrolební hypertenze mohou patřit změny vidění, bolest hlavy, nevolnost a / nebo zvracení. Okamžitě informujte lékaře, pokud máte některý z těchto příznaků. Váš lékař dokáže zkontrolovat, zda se jedná o intrakraniální hypertenzi. Pokud se opravdu jedná o tento stav, může lékař rozhodnout o dočasném omezení nebo přerušení léčby přípravkem INCRELEX. Po odeznění příhody je možné opět začít užívat přípravek INCRELEX.
Srdeční abnormality
U některých pacientů léčených přípravkem INCRELEX ukázalo ultrazvukové vyšetření srdce (echokardiogram) zvětšení srdečního svalu. Váš lékař může před léčbou přípravkem INCRELEX, během ní a po ní provést echokardiografické vyšetření.
Reakce v místě vpichu
Při léčbě přípravkem INCRELEX byly hlášeny reakce zahrnující vyrážku, otok a tukové boule. Reakci v místě vpichu se lze vyhnout změnou místa injekce při každém podání (rotace místa vpichu).
Zvýšení tělesné váhy
Při léčbě přípravkem INCRELEX bylo pozorováno zvýšení tělesné váhy.
Další méně časté nežádoucí účinky pozorované během léčby přípravkem INCRELEX zahrnovaly deprese, nervozitu.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného Dodatku VNahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5.
Jak INCRELEX uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento lék po uplynutí doby použitelnosti, uvedené na štítku vedle značky EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v chladničce (2 °C - 8 °C). Chraňte před mrazem.
Injekční lahvičku uchovávejte v krabičce, aby byla chráněna před světlem.
Po prvním použití lze injekční lahvičku uchovávat maximálně 30 dnů při 2 až 8 °C.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka. jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co INCRELEX obsahuje
• Léčivou látkou je mecaserminum. Jeden ml obsahuje mecaserminum 10 mg. Jedna injekční lahvička obsahuje mecaserminum 40 mg.
• Pomocnými látkami jsou benzylalkohol, chlorid sodný. polysorbát 20. kyselina octová 99%. octan sodný a voda na injekci.
Jak INCRELEX vypadá a co obsahuje toto balení
INCRELEX je čirý a bezbarvý roztok k injekcím, dodávaný ve skleněné lahvičce uzavřené zátkou a těsnicím uzávěrem. Jedna injekční lahvička obsahuje 4 ml kapaliny.
INCRELEX se dodává v balení obsahujícím jednu skleněnou lahvičku.
Velikost balení je jedna lahvička.
Držitel rozhodnutí o registraci a výrobce
Držitel rozhodnutí o registraci:
Ipsen Pharma 65. quai Georges Gorse 92100 Boulogne-Billancourt Francie
Výrobce:
Beaufour Ipsen Industrie Rue d'Ethe Virton 28100 Dreux Francie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
|
Belgie/Belgique/Belgien, Luxembourg/Luxemburg Ipsen NV Guldensporenpark 87 B-9820 Merelbeke Belgie /Belgique/Belgien Tél/Tel: + 32 9 243 96 00 |
Italia Ipsen SpA Via del Rinnovato n. 6 Milanofiori Nord Palazzo U7 20090 Assago (Mi) Tel: + 39 - 02 - 39 22 41 |
|
Románia, Et.irapírn Ipsen Pharma Str. Grigore Alexandrescu nr. 59, cladirea HQ Sector 1, 010623, Bucure§ti, Románia Tel/Tea.: + 40 (021) 231 27 20 |
Latvija Ipsen Pharma Kalnciema iela 33-5Riga LV 1046 Tel: +371 67622233 |
|
Česká Republika Ipsen Pharma, o.s. Evropská 136/810 CZ-160 00 Praha 6 Tel: + 420 242 481 821 |
Lietuva, Hrvatska Ipsen Pharma Lietuvos filialas 9-ojo Forto 47 LT -48100 Kaunas Tel. + 370 37 337854 |
|
Danmark, Norge, Suomi/Finland, Sverige, Ísland Institut Produits Synthese (IPSEN) AB Kista Science Tower Fárogatan 33 SE - 164 51 Kista Sverige/Ruotsi/Svíkjóó Tlf/Puh/Tel/Sími: +46 8 451 60 00 |
Magyarország Ipsen Pharma SAS Magyarországi Kereskedelmi Képviselet Árbóc utca 6. H-1133 Budapest Tel: +36 1 555 5930 |
|
Deutschland, Osterreich Ipsen Pharma GmbH Willy-Brandt-Str. 3 D-76275 Ettlingen Tel: + 49 7243 184-80 |
Nederland Ipsen Farmaceutica B.V. Taurusavenue 33 B NL-2132 LS Hoofddorp Tel: + 31 23 55 41 600 |
|
Eesti ESTOBIIN OU Udeselja 4-4 EE-11913 Tallinn Tel: +372 51 55 810 |
Polska Ipsen Poland Sp. z o.o. Al. Jana Pawla II 29 PL-00-867 Warszawa Tel.: + 48 (0) 22 653 68 00 |
|
EkXába, Kúnpoq, Malta Ipsen EnE Ay. App^rpíou 63 ATapog GR-17456 A0pva ETAáSa TpT,: + 30 - 210 - 984 3324 |
Portugal Ipsen Portugal - Produtos Farmaceuticos S.A. Alameda Fernáo Lopes, no 16-11°, Miraflores P-1495 - 190 Algés Tel: + 351 - 21 - 412 3550 |
|
Espaňa Ipsen Pharma S.A. Torre Realia, Plaza de Europa, 41-43 08908 LHospitalet de Llobregat Barcelona Tel: + 34 - 936 - 858 100 |
Slovenija Pharmaswiss d.o.o Brodišče 32 SI-1236 TrzinTel: + 386 1 236 47 00 |
Ireland
Ipsen Pharmaceuticals Ltd. Blanchardstown Industrial Park Blanchardstown IRL-Dublin 15
Tel: + 353 - 1 - 809 8200
United Kingdom
Ipsen Ltd.
190 Bath Road Slough, Berkshire SL13XE
Tel: + 44 - (0)1753 - 62 77 00
Slovenská republika
Liek s.r.o.
Hviezdoslavova 19 SK-90301 Senec Tel: + 421 245 646 322
Tato příbalová informace byla naposledy schválena
Tento léčivý přípravek byl registrován za „výjimečných okolností“
Znamená to, že vzhledem k vzácné povaze tohoto onemocnění nebylo možné získat o tomto léčivém přípravku úplné informace.
Evropská agentura pro léčivé přípravky každoročně vyhodnotí jakékoli nové informace týkající se tohoto léčivého přípravku a tato příbalová informace bude podle potřeby aktualizována.
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu. Na těchto stránkách naleznete též odkazy na další webové stránky, týkající se vzácných onemocnění a jejich léčby.
Na webových stránkách Evropské agentury pro léčivé přípravky je tato příbalová informace k dispozici ve všech úředních jazycích EU/EHP.
<------------------------------------------------------------------------------------------------------------------------
NÁVOD K POUŽITÍ
INCRELEX se má podávat pomocí sterilních stříkaček a injekčních jehel na jedno použití, které
mohou být poskytnuty lékařem/lékárníkem/sestrou. Stříkačky musí mít vhodnou velikost tak, aby bylo
možno předepsanou dávku natáhnout z injekční lahvičky s přiměřenou přesností.
Příprava dávky:
1. Předtím, než přípravek INCRELEX začnete připravovat k injekci, si umyjte ruce.
2. Pro každou dávku použijte novou jehlu a stříkačku na jedno použití. Stříkačky a jehly používejte pouze jednou. Správným způsobem je zlikvidujte do kontejneru na ostré předměty (jakým je kontejner na biologicky nebezpečný odpad), do kontejneru z tvrdého plastu (jakým je láhev na saponát) nebo do kovového kontejneru (jakým je prázdná kovová nádoba). Nikdy nepoužívejte stříkačky ani jehly použité jinou osobou.
3. Zkontrolujte kapalinu, zda je čirá a bezbarvá. Přípravek nepoužívejte po datu použitelnosti (které je uvedeno na obalu za EXP a odpovídá poslednímu dni uvedeného měsíce) ani pokud je zakalený nebo pokud jsou v něm viditelné částice. Pokud lahvička zmrzne, zlikvidujte ji. Zeptejte se svého lékárníka, jak zlikvidovat léky, které již nepoužíváte.
4. Jestliže používáte novou lahvičku, sejměte ochranné víčko. Neodstraňujte gumovou zátku.
5. Gumovou zátku v lahvičce otřete tamponem namočeným v alkoholu, abyste zabránili kontaminaci lahvičky choroboplodnými zárodky, které se mohou zavléci při jejím opakovaném používání (viz Obrázek 1).
Obrázek 1: Otřete zátku alkoholem
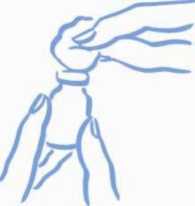
6. Před zasunutím jehly do lahvičky vytáhněte píst a natáhněte do stříkačky vzduch odpovídající
předepsané dávce léku. Propíchněte jehlou gumovou zátku lahvičky a zasunutím pístu vytlačte do lahvičky vzduch (viz Obrázek 2).
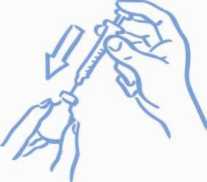
Obrázek 2: Vytlačte vzduch do stříkačky
7.
Ponechte stříkačku v lahvičce a stříkačku i lahvičku obraťte dnem vzhůru. Stříkačku i lahvičku pevně držte (viz Obrázek 3).
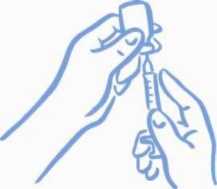
Obrázek 3: Příprava k nasátí
8. Ujistěte se, že je hrot jehly ponořený v kapalině (viz Obrázek 4). Zatáhnutím za píst natáhněte do stříkačky správnou dávku (viz Obrázek 5).
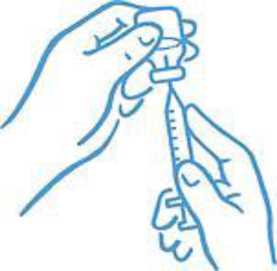
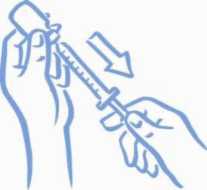
Obrázek 4: Hrot v tekutině
Obrázek 5: Natáhněte správnou dávku
9. Před vytažením jehly z lahvičky zkontrolujte, zda ve stříkačce nejsou vzduchové bublinky. Jsou-li ve stříkačce vzduchové bublinky, podržte stříkačku s lahvičkou dnem vzhůru a poklepejte na stěnu stříkačky tak, aby bublinky vystoupaly nahoru. Bublinky vytlačte pomocí pístu a natáhněte správné množství kapaliny tak, abyste ve stříkačce měli správnou dávku (viz Obrázek 6).

Obrázek 6: Odstraňte bubliny a doplňte správnou dávku
10. Vytáhněte jehlu z lahvičky a vyměňte ochranný kryt. Jehla se nesmí ničeho dotknout. Nyní jste připraveni k aplikaci injekce (viz Obrázek 7).

Injekce dávky:
Injekci přípravku INCRELEX aplikujte podle pokynů lékaře.
Injekci nepodávejte, pokud nejste schopen/schopna přijmout jídlo krátce před injekcí nebo po ní.
1. Vyberte si místo aplikace injekce - na paži, stehně, hýždích nebo břichu (viz níže). Místo vpichu se musí při každé injekci měnit.

Horní část paže Stehno
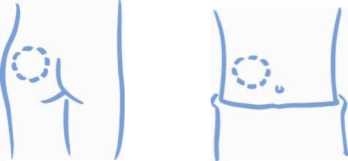
Hýždě Břicho
2. Na místě, kde se má provést vpich injekce, očistěte pokožku alkoholem nebo mýdlem a vodou. Před injekcí musí být místo vpichu suché.
Pokožku lehce stiskněte. Zaveďte jehlu ve směru, který Vám ukázal lékař. Uvolněte pokožku (viz Obrázek A).
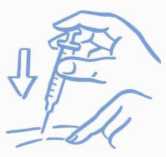
Obrázek A: Zlehka stiskněte kůži a aplikujte podle návodu
4. Pomalu stlačte píst stříkačky úplně dolů a ujistěte se, že j ste aplikovali všechnu kapalinu. Vytáhněte jehlu přímo nahoru a místo vpichu jemně na několik sekund stlačte gázou nebo vatovým tamponem. Místo vpichu netřete (viz Obrázek B).

Přitiskněte gázu nebo tampon (netřete)
5. Při likvidaci jehly a stříkačky postupujte podle pokynů lékaře. Stříkačku neuzavírejte. Použitá jehla a stříkačka se musí odložit do speciálního kontejneru na ostrý odpad (např. do kontejneru na biologicky nebezpečný odpad), do obalu z tvrdého plastu (např. do lahve od čisticího prostředku) nebo do kovové nádoby (např. do prázdné plechovky od kávy). Tyto nádoby se musí uzavřít a správně zlikvidovat způsobem, jaký Vám lékař popsal.
36