Incivo 375 Mg
&
O
O
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘípra/KU
JŮ O PŘÍPRAV
\>
A
í;
<b
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
INCIVO 375 mg potahované tablety
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna tableta obsahuje telaprevirum 375 mg.
Pomocné látky: 2,3 mg sodíku v potahované tabletě.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
O4
fd
Potahovaná tableta
Žlutá tableta ve tvaru tobolky o délce přibližně 20 mm označená na jedné straně „T375“.
V
INCIVO v kombinaci s peginterferonem alfa a ribavirinem je určeno k léčbě chronické hepatitidy C genotypu 1 u dospělých pacientů s kompenzovanou poruchou jater (včetně cirhózy):
- kteří dosud nebyli léčeni;
- kteří byli dříve léčeni interferonem alfa (pegylovaným nebo nepegylovaným) samostatně nebo v kombinaci s ribavirinem, vče+n* relabujících pacientů, pacientů, kteří na léčbu odpovídali částečně a těch, kteří na léčb'' neodpovídali (viz body 4.4 a 5.1).
4.
4.1
KLINICKÉ ÚDAJE Terapeutické indikace

4.2 Dávkování a způsob podán
Léčba přípravkem INCIVO musí být zahájena a monitorována lékařem, který má zkušenosti s léčbou chronické hepatitidy C.
/V
Dávkování
Užívá se 1 125 mg přípravku INCIVO (tři 375mg potahované tablety) perorálně dvakrát denně spolu s jídlem. Přípa'ně lze užívat 750 mg (dvě 375 mg tablety) perorálně každých 8 hodin spolu s jídlem. Celková denní dávka je 6 tablet (2 250 mg). Užívání přípravku INCIVO bez jídla nebo bez ohledu na interval dávkování může vést ke snížení koncentrací telapreviru v plazmě, což může snížit jeho ^terapeutický účinek.
terap
INC
mez
CIVO je nutno podávat společně s ribavirinem a buď peginterferonem alfa-2a nebo 2b. Pro výběr mezi peginterferonem alfa-2a nebo 2b si přečtěte body 4.4 a 5.1. Pro zvláštní pokyny týkající se dávkování peginterferonu alfa a ribavirinu je nutno nahlédnout do souhrnu údajů o přípravku těchto léčivých přípravků.
Doba trvání léčby - Léčba dosud neléčených pacientů a pacientů relabujících po předchozí léčbě Léčba přípravkem INCIVO musí být zahájena v kombinaci s peginterferonem alfa a ribavirinem a podávaná 12 týdnů (viz obrázek 1).
Pacienti s nedetekovatelnou kyselinou ribonukleovou viru hepatitidy C (HCV RNA) (hladiny detekčního limitu nebylo dosaženo) v týdnech 4 a 12 dostávají dalších 12 týdnů samotný peginterferon alfa a ribavirin, takže celková doba léčby je 24 týdnů.
Pacienti s detekovatelnou HCV RNA buď v týdnu 4 nebo 12 dostávají dalších 36 týdnů samotný peginterferon alfa a ribavirin, takže celková doba léčby je 48 týdnů.
Všichni pacienti s cirhózou bez ohledu na detekovatelnost HCV RNA (hladiny detekčního limitu nebylo dosaženo) v týdnech 4 a 12 dostávají dalších 36 týdnů samotný peginterferon alfa a ribavirin, takže celková doporučená doba léčby je 48 týdnů (viz bod 5.1).
Obrázek 1: Trvání léčby u dosud neléčených pacientů a pacientů relabujících po předchozí léčbě
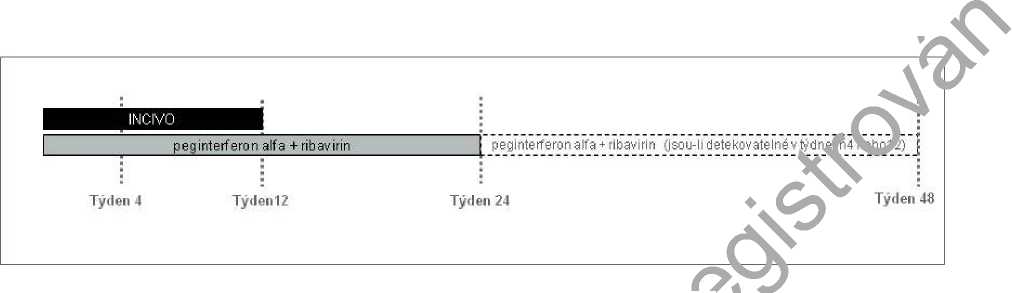
Pro stanovení délky léčby je nutno v týdnech 4 a 12 monitorovat hladiny HC'V RNA. V hodnoceních fáze III bylo pro stanovení nedetekovatelnosti hladin HCV RNA (hladiny detekčního limitu nebylo dosaženo) použito citlivé stanovení PCR v reálném čase s limitem kvantifikace 25 IU/ml a limitem detekce 10-15 IU/ml (viz bod 5.1). Pro rozhodnutí o době trvání léčby nelze zaměňovat detekovatelnou HCV RNA pod dolní hranicí stanovitelnosti za nedetekovatelnou“ (hladiny detekčního limitu nebylo dosaženo), protože to by mohlo vést k nedostatečné době trvání léčby a vyššímu výskytu relapsů. Pokyny pro ukončení léčby kombinací přípravku INCIVO, peginterferonu alfa a ribavirinu je uveden v tabulce 1.
Doba trvání léčby - Dříve léčení dospělí pacienti s částečnou nebo žádnou předchozí odpovědí Léčba přípravkem INCIVO musí být zahájena v kombinaci s peginterferonem alfa a ribavirinem a podávaná 12 týdnů, poté následuje podávání samotného peginterferonu alfa a ribavirinu (bez přípravku INCIVO) tak, aby celková doba léčby byla 48 týdnů (viz obrázek 2).
Obrázek 2: Trvání léčby u dřr. ^ léct-iých pacientů s částečnou nebo žádnou předchozí odpovědí
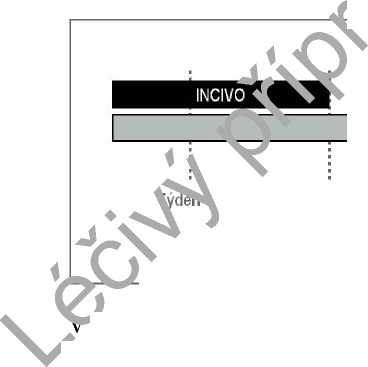
Týden 12
peginterferon alfa + ribavirin
Týden 48
týdnech 4 a 12 je nutno monitorovat hladiny HCV RNA. Viz tabulka Pokyny pro ukončení léčby přípravkem INCIVO, peginterferonem alfa a ribavirinem.
Všichni pacienti
Vzhledem k tomu, že je velmi nepravděpodobné, že pacienti s neadekvátní virovou odpovědí dosáhnou trvalé virologické odpovědi (sustained virological response = SVR), doporučuje se, aby u pacientů s HCV RNA > 1 000 IU/ml v týdnu 4 nebo 12 byla léčba přípravkem INCIVO ukončena (viz tabulka 1).
|
Tabulka 1: Pokyny pro ukončení léčby přípravkem INCIVO, peginterferonem alfa a ribavirinem | ||
|
Léčivý přípravek |
HCV RNA > 1 000 IU/ml v týdnu 4 léčbya |
HCV RNA > 1 000 IU/ml v týdnu 12 léčbya |
|
INCIVO |
Trvale ukončit |
Léčba přípravkem INCIVO trvale ukončena |
|
Peginterferon alfa a ribavirin |
Trvale ukončit | |
léčba přípravkem INCIVO, peginterferonem alfa a ribavirinem. Tyto pokyny nemusejí být stejné, pokud před zahájením léčby přípravkem INCIVO byla již použita léčba s peginterferonem alfa a ribavirinem (viz bod 5.1)
V hodnoceních fáze III žádný z pacientů s HCV RNA > 1 000 IU/ml buď v týdnu 4 nebo v týdnu 12 nedosáhl při pokračování léčby peginterferonem alfa a ribavirinem SVR. U dosud neléčených paci'ntů v hodnoceních fáze III dosáhli SVR 4/16 (25 %) pacientů s hladinami HCV RNA mezi 100 IU/m1 a 1 000 IU/ml v týdnu 4. Z pacientů s HCV RNA mezi 100 IU/ml a 1 000 IU/ml v týdnu 12 dosáhli SVR 2/8 (25 %).

RNA ravku
U pacientů, kteří dříve vůbec neodpovídali, je nutno zvážit provedení dodatečnéh mezi týdnem 4 a 12. Je-li koncentrace HCV RNA > 1 000 IU/ml, ukončí se podá INCIVO, peginterferonu alfa a ribavirinu.
U pacientů, kteří podstupují celých 48 týdnů léčby, se podávání pegintefTOn" alfa a ribavirinu ukončí, pokud HCV RNA je detekovatelná buď v týdnu 24 nebo v týdnu 36
Aby se zabránilo selhání léčby, musí být INCIVO užíváno v kombinaci s peginterferonem alfa a ribavirinem.
Aby se zabránilo selhání léčby, dávka přípravku INCIVO x nesmí snižovat nebo přerušovat.
Dojde-li k ukončení léčby přípravkem INCIVO kvůli íPžádoucím účinkům nebo kvůli nedostatečné virologické odpovědi, nesmí být léčba tímto přípravkem znovu zahájena.
Návod k úpravě dávky, přerušení, ukončení le^o znovu zahájení podávání peginterferonu alfa a ribavirinu je uveden v jejich souhrnech úda'ů o přípravku (viz bod 4.4).
Podává-li se dvakrát denně a dojde -li k vynechání dávky přípravku INCIVO o méně než 6 hodin, je pacienty nutno poučit, aby si vzali další dávku přípravku INCIVO spolu s jídlem, jakmile to bude možné. Dojde-li k vynechání dávky přípravku INCIVO o více než 6 hodin, dávka přípravku INCIVO se vynechá a pacient dále pokračuje v normálním dávkovacím režimu.
~ >
Podává-li se každých 8 hodin a dojde-li k vynechání dávky přípravku INCIVO o méně než 4 hodiny, je pacienty nutno poučit, aby si vzali další dávku přípravku INCIVO spolu s jídlem, jakmile to bude možné. Dojde -li k vynechání dávky přípravku INCIVO o více než 4 hodiny, dávka přípravku INCIVO se vynechá a pacient dále pokračuje v normálním dávkovacím režimu.
ZJáštik populace Porucha funkce ledvin
Pro pacienty se středně těžkou nebo těžkou poruchou funkce ledvin (CrCl <50 ml/min) nejsou k dispozici žádné klinické údaje o podávání přípravku INCIVO (viz bod 4.4). U HCV-negativních pacientů s těžkou poruchou funkce ledvin nebyla v expozici telapreviru pozorována klinicky relevantní změna (viz bod 5.2). Proto se pro HCV pacienty s poruchou funkce ledvin nedoporučuje úprava dávky přípravku INCIVO.
Pro pacienty na hemodialýze nejsou k dispozici žádné klinické údaje.
U pacientů s CrCl <50 ml/min je nutné se řídit také souhrnem údajů o přípravku ribavirinu.
Porucha funkce jater
INCIVO se nedoporučuje u pacientů se středně těžkou nebo těžkou poruchou funkce jater (Child-Pugh B nebo C, skóre > 7) nebo s dekompenzovanou poruchou funkce jater (ascites, krvácení v důsledku portální hypertenze, encefalopatie, a/nebo žloutenka jiného charakteru než Gilbertův syndrom, viz bod 4.4). Při podávání přípravku INCIVO pacientům s hepatitidou C s mírnou poruchou funkce jater (Child-Pugh A, skóre 5-6) není nutná úprava dávky.
Je nutno se řídit také souhrny údajů o přípravku peginterferonu alfa a ribavirinu, které jsou kontraindikovány u Child-Pugh skóre > 6.
nutno
Koinfekce HCV/virem lidské imunodeficience (HIV) typu 1 Pacienti koinfikovaní HCV/HIV-1 se musí léčit stejně jako pacienti infikovaní pouze HCV. Je nutí pečlivě zvážit lékové interakce, viz body 4.4 a 4.5. Pacienti léčení režimem založeným na efavi musí dostávat přípravek INCIVO v dávce 1 125 mg každých 8 hodin. Výsledky získané u pacie současně infikovaných HIV, viz bod 5.1.
Pacienti po transplantaci jater bez cirhózy ♦
irinem a
Léčba přípravkem INCIVO musí být zahájena v kombinaci s peginterferonem alfa a podávána po dobu 12 týdnů. Následujících 36 týdnů se podává peginterferon al a a :bavirin samostatně do celkové doby léčby 48 týdnů. U stabilizovaných pacientů s transplantovanými játry není nutná úprava dávkování přípravku INCIVO (viz body 4.8 a 5.1). Při zahojení léčby přípravkem INCIVO se doporučuje nižší dávka ribavirinu (600 mg/den) (viz bod 5.1). Při záhajování a ukončování léčby přípravkem INCIVO se musí výrazně upravit dávkování současí^ podávaného takrolimu či cyklosporinu A (viz body 4.4 a 4.5, Imunosupresiva).
> 65 le
Cř
Starší pacienti
:t jsou k dispozici jen omezené údaje.
Pro použití přípravku INCIVO u HCV pacientů ve věku >
Pediatrická populace
Nejsou dostupné žádné údaje o bezpečnosti a účinnosti přípravku INCIVO u dětí mladších 18 let.
Způsob podání
Pacienty je nutno poučit, aby tablety polykali celé (pacienti např. nesmějí tablety kousat, drtit nebo rozpouštět).
4.3 Kontraindikace
Hypersenzitivita na léčivo

ty pol
;ku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Současné podávání s léčivými látkami, jejichž vylučování je vysoce závislé na CYP3A a jejichž zvýšené hladiny v plazmě jsou spojeny se závažnými a/nebo život ohrožujícími nežádoucími účinky. K těmto léčivvm látkám patří alfuzosin, amiodaron, bepridil, chinidin, astemizol, terfenadin, cisaprid, pimozid, námelové alkaloidy (dihydroergotamin, ergometrin, ergotamin, methylergometrin), lovastakn, simvistatin, atorvastatin, sildenafil nebo tadalafil (pouze jsou-li užity k léčbě plicní arteriální hypertenze) kvetiapin a perorálně podávané midazolam nebo triazolam (viz bod 4.5).
i
Současné podávání s antiarytmiky třídy Ia nebo III s výjimkou intravenózního lidokainu (viz bod 4.5).
oučasné podávání přípravku INCIVO s léčivými látkami, které silně indukují CYP3A4, např. rifampicinem, třezalkou tečkovanou (Hypericum perforatum), karbamazepinem, fenytoinem a fenobarbitalem, může vést k nižší expozici a ztrátě účinnosti přípravku INCIVO.
Závažná vyrážka
Při kombinované léčbě s přípravkem INCIVO byly hlášeny závažné potenciálně život ohrožující a fatální kožní reakce. Po uvedení na trh byla pozorována toxická epidermální nekrolýza (TEN) včetně TEN s fatálním koncem (viz bod 4.8). Fatální případy byly hlášeny u pacientů s progresivní vyrážkou a systémovými příznaky, kteří pokračovali v kombinované léčbě s přípravkem INCIVO po identifikaci závažné kožní rekace.
a a
V placebem kontrolovaných hodnoceních fází II a III byla závažná vyrážka (převážně ekzematická, svědivá a zahrnující více než 50 % tělesného povrchu) hlášena u 4,8 % pacientů, kteří dostávali kombinovanou léčbu s přípravkem INCIVO, oproti 0,4 % pacientů, kteří dostávali peginterferon alfa i ribavirin. Dostupné údaje ukazují, že peginterferon alfa, a možná také ribavirin, mohou přispívat k frekvenci a závažnosti vyrážky spojené s kombinovanou léčbou s přípravkem INCIVO. <o
ezření n
Kvůli vyrážce byla ukončena léčba samotným přípravkem INCIVO u 5,8 % pacientů , léčba ko mbinací s přípravkem INCIVO byla ukončena u 2,6 % pacientů, zatímco z pacientů dostávajících peginterferon alfa a ribavirin neukončil léčbu nikdo.
V placebem kontrolovaných hodnoceních fází II a III mělo 0,4 % pacientů podezření na lékovou vyrážku spojenou s eosinofilií a systémovými příznaky (DRESS). Podle klinické zkušenosti s přípravkem INCIVO mělo Stevens-Johnsonův syndrom (SJS) méně než 0,1 % pacientů. Všechny tyto reakce vymizely po ukončení léčby.
DRESS se jeví jako vyrážka s eosinofilií spojená s jedním z msmdujících příznaků: horečka, lymfadenopatie, otok obličeje a příznaky na interních org?n.Xh (jaterní, ledvinové, plicní). Může se objevit kdykoli po začátku léčby, ačkoli většina případů s. objevila mezi šesti a deseti týdny od začátku léčby přípravkem INCIVO.
Předepisující lékař se musí ujistit, že pacienti jsou úplně informováni o riziku závažné vyrážky a nutnosti konzultovat se svým lékařem okamžitě, jakmile se objeví nová vyrážka nebo se existující vyrážka zhorší. Všechny vyrážky je nutno monitorovat na progresi, dokud vyrážka nevymizí. Vymizení vyrážky může trvat několik týdnů. Další léčivé přípravky spojené se závažnými kožními reakcemi je nutno během podávání kombinované léčby s přípravkem INCIVO užívat s opatrností, aby se zabránilo potenciálním dohadům, který léčivý přípravek vedl k závažné kožní reakci. V případě závažné kožní reakce je nutno zvážit ukončení podávání dalších léčivých přípravků, o kterých je známo, že jsou spojeny se závažnými kožními reakcemi.
' <J
Další informace o mírných až středně závažných vyrážkách viz bod 4.8.
Doporučení pro monitorování kožních reakcí a pro ukončení léčby přípravkem INCIVO, ribavirinem a peginterferonem alfa jsou shrnuty v tabulce níže:
|
\ |
Rozsah a chaakter kožních reakcí &° |
Doporučení pro monitorování kožních reakcí a ukončení léčby přípravkem INCIVO, ribavirinem a peginterferonem alfa u závažné vyrážky |
|
v |
Mírná vyrážka: lokalizované výsevy na kůži a/nebo výsev na kůži s omezenou distribucí (až několik izolovaných míst na těle) |
Monitorování na progresi nebo systémové příznaky, dokud vyrážka nevymizí. |
Středně závažná vyrážka: Difuzní vyrážka < 50 % povrchu těla
Monitorování na progresi nebo systémové příznaky, dokud vyrážka nevymizí. Zvážit konzultaci s dermatologem.
U středně závažné vyrážky, která progreduje, je nutno zvážit trvalé vysazení přípravku INCIVO. Nezlepší-li se vyrážka během 7 dnů po vysazení přípravku INCIVO, je nutno přerušit podávání ribavirinu. Přerušení podávání ribavirinu může být nutné dříve, pokud se vyrážka i přes vysazení telapreviru zhoršuje. Podávání peginterferonu alfa může pokračovat, pokud není vysazení indikován léčebně.
U středně závažné vyrážky, která progre duje na závažnou (> 50 % povrchu těla), je nutno trvale ukončit podávání přípravku INCIVO +'..'1e (viz níže).
áno
le (viz
Závažná vyrážka: Rozsah vyrážky > 50 % povrchu těla nebo spojení s puchýřky, bulami, ulceracemi jinými než SJS
1
Okamžitě ukončit podávání přípravku INCIVO natrvalo. Doporučuje se koizuhace s dermatologem. Monitorování na progresi nebo systémové příznaky, dokud vyrážka nevymizí.
Podávání peginterferonu alfa a ribavirinu může pokračovat. Nezlepší-li se vyrážka během 7 dnů po vysazení př pra ku INCIVO, je nutno zvážit postupné nebo současné přerušení nebo ukončení podávání ribavirinu a/nebo peginterferonu alfa. Je-li to léčebně indikováno, může být nutné dřívější přerušení nebo ukončení podávání peginterferonu alfa a ribavirinu.
Závažné kožní reakce včetně vyrážky se systémovými příznaky, progresivní závažná vyrážka, podezření nebo diagnóza generalizovaného bulózního v^so^u, DRESS, SJS/TEN, akutní geneializované exantematické pustulosis, erythema multiforme
stulosiř, ery;
<ř- -
Trvalé a okamžité ukončení podávání přípravku INCIVO, peginterferonu alfa a ribavirinu. Konzultace s dermatologem.
Je-li léčba přípravkem INCIVO ukončena kvůli kožní rekaci, nesmí být znovu zahájena. Další informace o závažných kožních reakcích spojených s podáváním peginterferonu alfa a ribavirinu jsou uvedeny v jejich souhrnech udajů o přípravku.
Anei _
kontrolovaných klinických hodnoceních fází II a III se celková incidence a závažnost zvyšovaly u kombinované léčby s přípravkem INCIVO ve srovnání s podáváním samotných interferonu alfa a ribavirinu. Hodnoty hemoglobinu <10 g/dl (6,2 mmol/l) byly pozorovány 4 % pacientů, kteří dostávali kombinovanou léčbu s přípravkem INCIVO, a u 14 % pacientů, kteří dostávali peginterferon alfa a ribavirin. Hodnoty hemoglobinu < 8,5 g/dl (5,3 mmol/l) byly pozorovány u 8 % pacientů, kteří dostávali kombinovanou léčbu s přípravkem INCIVO, a u 2 % pacientů, kteří dostávali peginterferon alfa a ribavirin. Snížení hladin hemoglobinu se vyskytlo během prvních 4 týdnů léčby s nejnižšími hodnotami dosaženými na konci podávání přípravku INCIVO. Po dokončení podávání přípravku INCIVO se hodnoty hemoglobinu pomalu zlepšovaly.
Pro zvládnutí akutní anemie vyvolané léčbou je preferováno snížit dávky ribavirinu. Pokyny pro snížení dávky a/nebo ukončení léčby ribavirinem jsou uvedeny v souhrnu údajů o přípravku obsahujícího ribavirin. Je-li permanentně ukončeno podávání ribavirinu, aby byla zvládnuta anemie, musí být také permanentně ukončeno podávání přípravku INCIVO. Je-li ukončeno podávání přípravku INCIVO z důvodu anemie, pacienti mohou pokračovat v léčbě peginterferonem alfa a ribavirinem. Podávání ribavirinu lze obnovit podle pokynů pro změnu jeho dávky v příslušném souhrnu údajů o přípravku. Dávku přípravku INCIVO není možno snižovat a podávání přípravku nelze po jeho vysazení obnovit.
o
Těhotenství a požadavky na antikoncepci Vzhledem k tomu, že přípravek INCIVO je nutno užívat v kombinaci s peginterferonem alfa a ribavirinem, jsou pro kombinovanou léčbu aplikovatelné kontraindikace a upozornění pro tyto léčivé přípravky.
U všech zvířecích druhů vystavených ribavirinu byly prokázány významné teratogenní a/n b embryocidní účinky; u pacientek a partnerek pacientů mužů je tedy nutno věnovat zvýšenou péči zabránění těhotenství.
vV
Pacientky ve fertilním věku a jejich partneři a také pacienti muži a jejich partnerky musejí užívat 2 účinné antikoncepční metody během léčby přípravkem INCIVO a dále dle doporučení v souhrnu údajů o přípravku pro ribavirin a jak je popsáno níže.
Během podávání přípravku INCIVO a až do dvou měsíců po ukončení jeho podávání lze pokračovat v užívání hormonální antikoncepce, ale ta nemusí být spolehlivá (viz bod 4.5). Během tohoto období musejí proto pacientky ve fertilním věku užívat dvě účinné nfhCrmonální metody antikoncepce. Dva měsíce po ukončení léčby přípravkem INCIVO je hormonální antikoncepce opět vhodná jako jedna ze dvou požadovaných účinných metod kontroly početí.
Další informace viz body 4.5 a 4.6.
Kardiovaskulární systém
Výsledky hodnocení u zdravých dobrovolnou prokázaly mírný vliv telapreviru v dávce 1 875 mg každých 8 hodin na QTcF interval; středn íodnota prodloužení, po úpravě hodnot naměřených pro placebo, byla 8,0 msec (90 % CI: 5,1 - - i0,9) (viz bod 5.1). Expozice při této dávce byla srovnatelná s expozicí u HCV infikovaných pacientů při dávce 750 mg přípravku INCIVO každých 8 hodin s peginterferonem alfa a ribavinrem. Potenciální klinický význam těchto nálezů není zřejmý.
INCIVO je nutno užívat opatrně v kombinaci s antiarytmiky třídy Ic propafenonem a flekainidem, včetně příslušného klinickém monitorování EKG.
Opatrnost se doporučuje, pokud je přípravek INCIVO předepsán současně s léčivými přípravky, o nichž je známo, že prodlužují QT interval a které jsou substráty CYP3A, jako je erythromycin, klarithromycin, telithromycin, posakonazol, vorikonazol, ketokonazol, takrolimus, salmeterol (viz bod 4.5) Je nutio se vyvarovat podávání přípravku INCIVO spolu s domperidonem (viz bod 4.5). INCIVO může zvyšovat koncentrace současně podávaných léčivých přípravků, což může vést ke zvýšenému riziku s nimi spojených kardiovaskulárních nežádoucích účinků. Je-li současné podání ovýchto léčivých přípravků s přípravkem INCIVO opravdu nutné, doporučuje se klinické jnitorování včetně hodnocení EKG. Léčivé přípravky, které jsou současně s přípravkem INCIVO ontraindikovány, jsou uvedeny také v bodu 4.3.
Použití přípravku INCIVO je nutno se vyvarovat u pacientů s vrozeným prodloužením QT intervalu nebo s rodinnou anamnézou vrozeného prodloužení QT nebo náhlého úmrtí. Je-li léčba přípravkem INCIVO v takovém případě opravdu nutná, je pacienty nutno důkladně monitorovat, včetně hodnocení
EKG.
INCIVO je nutno užívat opatrně u pacientů s: - anamnézou získaného prodloužení QT;
- klinicky relevantní bradykardií (přetrvávající srdeční frekvence < 50 tepů/min);
- anamnézou srdečního selhání se sníženou ejekční frakcí levé komory;
- nutností užívání léčivých přípravků, o nichž je známo, že prodlužují QT interval, ale jejichž metabolismus není primárně závislý na CYP3A4 (např. methadon, viz bod 4.5).
Tyto pacienty je nutno důkladně monitorovat, včetně hodnocení EKG.
Před zahájením léčby přípravkem INCIVO a během ní je nutno monitorovat a v případě nutnosti upravovat poruchy elektrolytů (např. hypokalemii, hypomagnesemii a hypokalcemii).
Použití u pacientů s pokročilým stádiem onemocnění jater Hypalbuminemie a nízký počet trombocytů byly identifikovány jako faktory závažných komplikací onemocnění jater a také léčebných postupů založených na použití interferonu (např. dekompenzac jater, závažná bakteriální infekce). Navíc byl pozorován vysoký stupeň anémie v případě užívání přípravku INCIVO s peginterferonem a ribavirinem u pacientů s těmito charakteristikami. Přpraek INCIVO není v kombinaci s peginterferonem a ribavirinem doporučen u pacientů s počtem trombocytů < 90 000/mm3 a/nebo albuminem < 3,3 g/dl. V případě užívání přípravku INCIVO u pacientů s pokročilým stádiem onemocnění jater je doporučen přísný dohled a včasné zvládnutí nežádoucích účinků.
eí u
Laboratorní testy
V týdnech 4 a 12 a podle klinické indikace je nutno monitorovat hladiny HC pro ukončení léčby přípravkem INCIVO, bod 4.2).
CVRN.
A (viz také pokyny
.
Před zahájením kombinované léčby s přípravkem INCIVO je nutno u všech pacientů provést následující laboratorní vyšetření - krevní obraz s diferenciálů m počtem bílých krvinek, elektrolyty, kreatinin v séru, jaterní testy, TSH, kyselina močová.
Doporučené výchozí hodnoty pro zahájení kombi:
- Hemoglobin: >12 g/dl (ženy); >13 g/dl (:
- Počet krevních destiček > 90 000/mm
- Absolutní počet neutrofilů > 1 500/
- Adekvátně kontrolovaná funkce
- Vypočítaná clearance kreatininu
- Draslík >3,5 mmol/l.
- Albumin > 3,3g/dl
é léčby s přípravkem INCIVO jsou:
četně difer
Hematologická vyšetření (včetně diferenciálního počtu bílých krvinek) jsou doporučena v týdnech 2, 4, 8 a 12 a poté dle klinického obrazu.
Biochemická vyšetřen' (elektrolyty, kreatinin v séru, kyselina močová, jaterní enzymy, bilirubin a TSH) jsou doporučeny ve stejných intervalech jako hematologická vyšetření nebo dle klinické indikace (viz bod 4.8).
Další informace, včetně požadavků na testování těhotenství, jsou uvedeny v souhrnech údajů o přípravku peginterferonu alfa a ribavirinu (viz bod 4.6).
\TVvam přípravku INCIVO v kombinaci s peginterferonem alfa-2b
Hodnocení fáze III byly provedeny s peginterferonem alfa-2a v kombinaci s přípravkem INCIVO a ribavirinem. Pro užívání přípravku INCIVO v kombinaci s peginterferonem alfa-2b nejsou pro již léčené pacienty k dispozici žádné údaje a pro dosud neléčené pacienty pouze omezené údaje. Dosud neléčení pacienti léčení buď peginterferonem alfa-2a/ribavirinem (n = 80) nebo peginterferonem alfa-2b/ribavirinem (n = 81) v kombinaci s přípravkem INCIVO v otevřeném hodnocení měli srovnatelné výskyty SVR. U pacientů léčených peginterferonem alfa-2b však došlo častěji k virovému průlomu (breakthrough) a méně často dosahovali kritérií pro zkrácenou celkovou dobu léčby (viz bod 5.1).
Všeobecně
INCIVO se nesmí podávat jako monoterapie a musí se předpisovat pouze v kombinaci jak s peginterferonem alfa tak i ribavirinem. Před zahájením léčby přípravkem INCIVO je nutno proto prostudovat souhrny údajů o přípravku peginterferonu alfa a ribavirinu.
K dispozici nejsou žádné klinické údaje pro opakovanou léčbu pacientů, u kterých selhala terapie založená na inhibitorech HCV NS3-4A proteázy (viz bod 5.1).
Nedostatečná virologická odpověď
U pacientů, u kterých se projeví nedostatečná virologická odpověď, je nutno léčbu ukončit (viz body 4.2 a 4.4, Laboratorní testy).
,,
Použití přípravku INCIVO v léčbě jiných HCV genotypů
Není dostatek klinických údajů podporujících léčbu pacientů s HCV genotypy jinými než genotyp Proto se použití přípravku INCIVO u jiných genotypů než HCV 1 nedoporučuje.
Porucha funkce ledvin
♦
U pacientů s mírnou nebo středně těžkou poruchou funkce ledvin (CrCl <50 ml/min) nebo u pacientů na hemodialýze nebyly bezpečnost a účinnost stanoveny. Viz bod 4.4, Laboratorní vyšetření.
U pacientů s CrCl < 50 ml/min je nutno se řídit také souhrnem údajů o přípravku liOavirinu (viz také body 4.2 a 5.2).
V N
Porucha funkce jater
INCIVO nebylo hodnoceno u pacientů s těžkou poruchou funkce jater (Child-Pugh C, skóre >10) nebo s dekompenzovaným onemocněním jater (ascites, krvácení v důsledku portální hypertenze, encefalopatie, a/nebo žloutenka jiného charakteru než Gilbertů' syndrom )a u této populace se nedoporučuje.
INCIVO nebylo hodnoceno u HCV-infikovaných pacientů se středně těžkou poruchou funkce jater (Child-Pugh B, skóre 7-9). U HCV negativních pacientů se středně těžkou poruchou funkce jater byla pozorována snížená expozice telapreviu. Příslušná dávka přípravku INCIVO u hepatitidou C infikovaných pacientů se středně těžkou poruchou funkce jater nebyla stanovena. U těchto pacientů se tedy INCIVO nedoporučuje (viz body 4.2 a 5.2).
Další informace v souhrnech ú společně s přípravkem INCI
Pacienti po transplantaci -rgaiu
Přípravek INCIVO v kombinaci s peginterferonem alfa a ribavirinem byl hodnocen u 74 pacientů infikovaných HCV 1, keří po transplantaci jater bez cirhózy dostávali buď takrolimus nebo cyklosporin A. Při zahájení léčby přípravkem INCIVO je třeba dávky současně podávaného takrolimu nebo cyklosporinu A výrazně snížit, včetně prodloužení dávkovacího intervalu takrolimu pro udržení terapeutické plasmatické koncentrace imunosupresiva. Po vysazení přípravku INCIVO je třeba dávky takrolimu nebo cyklosporinu A zvýšit a dávkovací interval takrolimu zkrátit. Někteří pacienti mohou io at vyšší dávky takrolimu nebo cyklosporinu A než na počátku léčby. Tyto změny v dávkování být založeny na častém sledování plasmatických koncentrací takrolimu nebo cyklosporinu A v ěhu léčby přípravkem INCIVO. Informace o používání přípravku INCIVO v kombinaci s interferonem alfa a ribavirinem u dosud neléčených a již léčených pacientů infikovaných HCV-1 po transplantaci jater, kteří byli na stabilním režimu imunosupresiv takrolimu nebo cyklosporinu A, viz body 4.2, 4.5, Imunosupresiva, 4.8 a 5.1.
pravku peginterferonu alfa a ribavirinu, které je nutno podávat
Pro léčbu přípravkem INCIVO v kombinaci s peginterferonem alfa a ribavirinem u pre nebo peri transplantovaných pacientů (transplantace jater nebo jiných orgánů) nejsou dostupné žádné údaje.
Koinfekce HCV/HIV
Interakce mezi telaprevirem a antiretrovirovými léky proti HIV jsou časté, přičemž je nutno pečlivě dodržovat doporučení uvedená v tabulce 2 v bodě 4.5.
Mezi režimy léčby HIV, které lze použít (neomezuje se na režimy níže uvedené), je nutno vzít v úvahu následující:
Atazanavir/ritonavir: tato kombinace je spojena s vysokou frekvencí hyperbilirubinemie/ikteru.
V klinickém hodnocení HPC3008 (viz body 4.8 a 5.1), bylo během léčby přípravkem INCIVO u 39 %, respektive u 22 % z 59 pacientů léčených kombinací atazanavir/ritonavir, pozorováno přechodné zvýšení bilirubinu stupně 3 (2,5 až < 5násobek horního limitu normálu), respektive stupně 4 (> 5násobek horního limitu normálu).
Efavirenz: při této kombinaci se dávka telapreviru musí zvýšit na 1 125 mg třikrát denně (každých 8 hodin).
Koinfekce HCV/HBV Pro léčbu přípravkem INCIVO u pacientů infikovaných HCV/HBV nejsou dostupné ž;
A
ádné úd
u této
Pediatrická populace
INCIVO se nedoporučuje k používání u dětí a dospívajících do 18 let věku, protone u té nebyly stanoveny bezpečnost a účinnost.
Porucha štítné žlázy
Při kombinované léčbě s přípravkem INCIVO může dojít k vzestupu thyree+ropního hormonu (TSH), což může ukázat na zhoršení nebo rekurenci preexistující nebo předchozí hypothyreózy nebo na nově vzniklou hypothyreózu (viz bod 4.8). Před zahájením podávání a během kombinované léčby s přípravkem INCIVO je nutno stanovit hladiny TSH a léčit tik jak je klinicky vhodné, včetně potenciální úpravy hormonální substituční léčby u pacientů ' preexistující hypothyreózou (viz bod 4.4, Laboratorní vyšetření).
populace
Interakce s léčivými přípravky Telaprevir je silným inhibitorem důležitého enzymu CYP3A4 metabolizujícího léčivé přípravky. Je-li telaprevir kombinován s léčivými přípravky vysoce metabolizovanými tímto enzymem, očekávají se zvýšené systémové expozice. Seznam léčivých přípravků, které jsou kontraindikovány v kombinaci s přípravkem INCIVO vzhledem k život ohrožujícím nežádoucím účinkům nebo potenciální ztrátě terapeutického účinku přípravku INCiVo, je uveden v bodě 4.3. V bodě 4.5 jsou uvedeny zjištěné nebo jiné potenciálně významné lékové interakce.
ých
é lékov
Důležité informace o některých složkách přípravku INCIVO
Tento léčivý přípravek obsahuje 2,3 mg sodíku v jedné tabletě, což je nutno vzít v úvahu u pacientů na kontrolované sodíkové detě
4.5 Interakc
ir je čá <
e ■’ jiný
mi léčivými přípravky a jiné formy interakce
Telaprevi" je částečně metabolizován v játrech CYP3A a je substrátem P-glykoproteinu (P-gp). Na metábo,ismu se podílejí i další enzymy (viz bod 5.2). Současné podání přípravku INCIVO spolu s léčivými přípravky, které indukují CYP3A a/nebo P-gp, může významně snižovat koncentrace tel.previru v plazmě. Současné podání přípravku INCIVO spolu s léčivými přípravky, které inhibují CYP3A a/nebo P-gp, může zvyšovat koncentrace telapreviru v plazmě.
I NCIVO je silným na čase závislým inhibitorem CYP3A4 a také významně inhibuje P-gp. Časová závislost ukazuje, že inhibice CYP3A4 může být zvýšena během prvních 2 týdnů léčby. Po ukončení léčby může být pro úplné vymizení inhibice nutný přibližně jeden týden. Podání přípravku INCIVO může zvyšovat systémovou expozici léčivým přípravkům, které jsou substtráty CYP3A nebo P-gp, což může zvýšit nebo prodloužit jejich terapeutický účinek a nežádoucí účinky. Na základě výsledků hodnocení interakcí léčivo - léčivo (např. escitalopram, zolpidem, ethinylestradiol) nelze vyloučit indukci metabolických enzymů telaprevirem.
Telaprevir inhibuje transportní polypeptidy organických aniontů (OATP) OATP1B1 a OATP2B1. Současné podávání přípravku INCIVO a léčivých přípravků transportovaných těmito transportéry, jako fluvastatin, pravastatin, rosuvastatin, pitavastatin, bosentan a repaglinid, je nutno provádět s opatrností (viz tabulka 2). Simvastatin je kontraindikován vzhledem k předpokládanému významnému zvýšení expozice způsobenému vícerými mechanismy.

Na základě studií in vitro může telaprevir potenciálně zvýšit plazmatickou koncentraci léčivých přípravků, u nichž je vylučování závislé na vícelékové a toxinové extruzi (MATE)-1 a MATE2-K (viz tabulka 2).
Studie interakcí byly provedeny pouze u dospělých.
Kontraindikace při současném podávání (viz bod 4.3)
v v
dy Ia nebo III s výjimkou
(?\
třídy Ic
Přípravek INCIVO se nesmí podávat současně s léčivými látkami, jejichž clearance je vysoce závi na CYP3A a u nichž jsou zvýšené koncentrace v plazmě spojeny se závažnými a/nebo živ t ohrožujícími stavy jako je srdeční arytmie (např. amiodaron, astemizol, bepridil, cisaprid, pimozid, chinidin, terfenadin), nebo periferní vasospasmus nebo ischemie (např. dihydroergotamin, ergometrin, ergotamin, methylergometrin), nebo myopatie včetně rhabdomyolýzy (např. lovastatin, simvastatin, atorvastatin), nebo prodloužená nebo zesílená sedace nebo respirační deprese (např. kvetiapin, perorálně podaný midazolam nebo triazolam), nebo hypotenze nebo srdeční arytuue (např. alfuzosin a sildenafil při plicní arteriální hypertenzi).
Přípravek INCIVO se nesmí podávat současně s antiarytmiky tříd intravenózního lidokainu.
Ic propafenonem a flekainidem,
INCIVO je nutno užívat opatrně v kombinaci s antiarytmiky t včetně příslušného klinického monitorování EKG (viz bod 4.4).
Rifampicin
.....
Rifampicin snižuje AUC telapreviru v plazmě o p^ižně 92 %. INCIVO se tedy nesmí podávat současně s rifampicinem. VÍ >
Třezalka tečkovaná (Hypericum perforatum)
Koncentrace telapreviru v plazmě mohou být při současném užívání rostlinných přípravků obsahujících třezalku tečkovanou 'Hypericum perforatum) sníženy. Rostlinné přípravky obsahující třezalku tečkovanou se tedy nesměj spřípravkem INCIVO kombinovat.
Karbamazepin, fenytoin a fenobarbital
Současné podáván í s induktory ■eviru s rizikem nižší účinnosti. Silné
induktory CYP3Ajako karbamazepin, fenytoin a fenobarbital jsou kontraindikovány (viz bod 4.3).

i
Slabé a středně silné induktory CYP3A
Slabým a středně silným induktorům CYP3A je nutno se vyhnout zejména u pacientů, kteří dříve neodpovídali nf léčbu (částeční nebo nuloví respondeři na peginterferon alfa/ribavirin), pokud neexistují speciální doporučení pro dávkování (viz tabulka 2).
\
Pďši kombinace
IV
tabulce 2 jsou uvedena doporučení pro dávkování vycházející z lékových interakcí přípravku NCIVO. Tato doporučení jsou založena buď na hodnoceních interakcí (označeno *) nebo interakcích předpokládaných na základě významu interakce a potenciálu pro závažné nežádoucí účinky nebo ztrátu účinnosti. Většina studií interakcí léčivo - léčivo byla provedena s dávkou telapreviru 750 mg každých 8 hodin. Vzhledem k tomu, že režim 1 125 mg dvakrát denně vede k téže denní dávce s podobnou expozicí telapreviru, očekává se, že relativní lékové interakce budou podobné.
Směr šipky (| = zvýšení, [ = snížení, = beze změny) u každého farmakokinetického parametru je
založen na 90 % intervalu spolehlivosti středního geometrického poměru uvnitř (^) rozmezí 80 -125 %, pod (4) rozmezím 80 - 125 % nebo nad (|) rozmezím 80 - 125 %.
|
Tabulka 2: INTERAKCE A DOPORUČENÍ PRO DÁVKOVÁNÍ S DALŠÍMI LÉČIVÝMI PŘÍPRAVKY | ||
|
Léčivé přípravky podle terapeutické oblasti |
Vliv na koncentraci přípravku INCIVO nebo současně podaného léčivého přípravku a možný mechanismus |
Klinický komentář |
|
ANALGETIKA | ||
|
alfentanil fentanyl |
t alfentanil t fentanyl |
Při současném podání telapreviru s alfentanilem nebo fentanylem, včetně perorálních, bukálních, nosních a transdermálních nebo transmukosálních forem fentanylu s prodlouženým uvolňováním, se zejména na počátku léčby doporučuje důkladné monitorování terapeutických i nežádoucích účinků (včetně respirační deprese) Může být nutná úprava dávky fen^n^u nebo alfentanilu. Nevýznamnější účinky se očekávají u perorálních, nosních a bukálníh/sublingválních forem fentanylu. |
|
ANTIARYTMIKA | ||
|
lidokain (intravenózní) |
t lidokain inhibice CYP3A OC- |
Při podávání intravenózního lidokainu k léčbě akutní ventrikulární arytmie je třeba opatrnost a doporučuje se klinické monitorování. |
|
digoxin* |
t digoxin AUC 1,85 (1,7" - 2,00) Cmax 1,50 (1,36 1,65) účinek na P gp transport ve stsvě |
Na úvod je třeba předepsat nejnižší dávku digoxinu. Je třeba monitorovat hladinu digoxinu v séru a použít ji pro titraci dávky digoxinu, aby bylo dosaženo požadovaného klinického účinku. |
|
ANTIBIOTIKA | ||
|
klarithromycin erythromycin telithromycin troleandomycin _,Ov |
t telaprevir t antibiotikum inhibice CYP3A |
Při současném podávání s přípravkem INCIVO je třeba opatrnost a doporučuje se klinické monitorování. U klarithromycinu a erytromycinu byly hlášeny prodloužení QT intervalu a Torsade de Pointes. S telithromycinem bylo hlášeno prodloužení QT intervalu (viz bod 4.4). |
|
ANTIKOAGULANCIA | ||
|
warfarin |
t nebo ^ warfarin modulace metabolických enzymů |
Při současném podávání s telaprevirem se doporučuje monitorovat INR. |
|
t dabigatran ^ telaprevir účinek na P-gp transport ve střevě |
Je nutná opatrnost, doporučuje se laboratorní a klinické monitorování. | |

|
ANTIEPILEPTIKA | ||
|
karbamazepin* |
i telaprevir AUC 0,68 (0,58-0,79) Cmax 0,79 (0,70-0,90) Cmin 0,53 (0,44-0,65) ^ karbamazepin AUC 1,10 (0,99-1,23) Cmax 1,09 (0,98-1,21) Cmin 1,10 (0,97-1,24) indukce CYP3A karbamazepinem a inhibice CYP3A telaprevirem |
Současné podávání s karbamazepinem je kontraindikováno. X |
|
fenytoin* |
i telaprevir AUC 0,53 (0,47-0,60) Cmax 0,68 (0,60-0,77) Cmin 0,32 (0,25-0,42) t fenytoin AUC 1,31 (1,15-1,49) Cmax 1,27 (1,09-1,47) Cmin 1,36 (1,21-1,53) indukce CYP3A fenytoinem a inhibice CYP3A telaprevirem |
Současné podávání s fenytoinem je kontraindikováno. . ^ |
|
fenobarbital* |
i telaprevir t nebo i fenobarbital < indukce CYP3A fenobarbitalem a inhibice CYP3A telaprevirem |
Současné podávání s fenobarbitalem je kontraindikováno. r |
|
ANTIDEPRESIVA | ||
|
escitalopram* |
^ telaprevir ^ escitalopiam AUC 0,65 (0,60 - 0,70) Cm.x 0,70 (0,( 5 - 0,76) Cin ',58 (0,52-0,64) mechanismus není znám |
Klinický význam není znám. Při kombinaci s telaprevirem může být nutno zvýšit dávky. |
|
trazodon |
l trazodon inhibice CYP3A |
Současné podávání může vést k nežádoucím účinkům jako nauzea, závrať, hypotenze a synkopa. Užívá-li se trazodon s telaprevirem, je třeba postupovat opatrně a zvážit podání nižší dávky trazodonu. |
|
ANTIDIABETIKA | ||
|
metformin |
t metformin inhibice MATE-1 a MATE2-K |
U pacientů léčených metforminem, kteří začínají nebo ukončují léčbu přípravkem INCIVO, je doporučeno pečlivě sledovat účinnost a bezpečnost metforminu. Může být nutná úprava dávkování metforminu. |
|
AVTliMETIKA | ||
|
acmperidon ► |
l domperidon inhibice CYP3A |
Současného podávání přípravku INCIVO s domperidonem je třeba se vyvarovat (viz bod 4.4). |
|
ANTIMYKOTIKA | ||
|
ketokonazol* itrakoknazol posakonazol vorikonazol |
t ketokonazol (200 mg) AUC 2,25 (1,93 -2,61) Cmax 1,75 (1,51 -2,03) t ketokonazol (400 mg) AUC 1,46 (1,35 - 1,58) Cmax 1,23 (1,14 - 1,33) t telaprevir (s ketokonazolem 400 mg) AUC 1,62 (1,45- 1,81) Cmax 1,24 (1,10-1,41) t itrakonazol t posakonazol t nebo ^ vorikonazol Inhibice CYP3A. Vzhledem k zapojení různých enzymů do metabolismu vorikonazolu je těžké předpovědět interakci s telaprevirem. |
Je-li nutné současné podávání, nedoporučují se vysoké dávky itrakonazolu (> 200 mg/den) nebo ketokonazolu (> 200 mg/den). U itrakonazolu, posakonazolu a vorikonazolu je nutná opatrnost a doporučuje se klinické monitorování. U vorikonazolu a posakonazolu byly hlášeny prodloužení QT intervalu a Torsade de Pointes. U ketokonazolu bylo hlášeno prodloužení QT intervalu (viz bod 4.4). Pacientům léčeným telaprevirem by e vorikonazol neměl podávat, pokud to nezdůvodní posouzení poměru prospěch/riziko. cv |
|
ANTIURATIKA | ||
|
kolchicin |
t kolchicin inhibice CYP3A |
Pacientům s poruchou funkce ledvin nebo jater se INCIVO nesmí podávat vzhledem k riziku kolchicinové toxicity. U pacientů s normální funkcí ledvin a jater se doporučuje přerušení léčby kolchicinem nebo pouze omezená léčba, při které se použijí snížené dávky kolchicinu. |
|
ANTITUBERKULOTIKA | ||
|
rifabutin |
^ telaprevir t rifabutin indukce CYP3A rifabutinem, inhibice CYP3A telaprevirem |
Telaprevir může být vzhledem ke sníženým koncentracím méně účinný. Současné podávání rifabutinu a telapreviru se nedoporučuje. |
|
""" |
^ telaprevir AUC 0,08 (0,07-0,11) Cmax 0,14 (0,11-0,18) t rifampicin indukce CYP3A rifampicinem, inhibice CYP3A telaprevirem |
Současné podávání rifampicinu a telapreviru je kontraindikováno. |
|
-ANTIPSYCHOTIKA | ||
|
kvetiapin |
Protože telaprevir inhibuje CYP3 A, očekává se zvýšení koncentrace kvetiapinu. |
Souběžné podávání přípravku INCIVO a kvetiapinu je kontraindikováno, protože by to mohlo zvýšit toxicitu související s kvetiapinem. Zvýšená koncentrace kvetiapinu může vést ke komatu. |
|
BENZODIAZEPINY | ||
|
alprazolam* |
t alprazolam AUC 1,35 (1,23 - 1,49) Cmax 0,97 (0,92- 1,03) |
Klinický význam není znám. |
|
parenterálně podaný midazolam* perorální midazolam* perorální triazolam |
t midazolam (intravenózní) AUC 3,40 (3,04 - 3,79) Cmax 1,02 (0,80- 1,31) t midazolam (p.o.) AUC 8,96 (7,75 - 10,35) Cmax 2,86 (2,52-3,25) t triazolam inhibice CYP3A |
Současné podávání je možné pouze v prostředí, které zaručí klinické monitorování a příslušnou lékařskou péči v případě respirační deprese a/nebo prodloužené sedace. Je třeba zvážit snížení dávky u parenterálně podaného midazolamu, zejména při podání více než jedné dávky. Současné podávání perorálního midazolamu nebo triazolamu a teplapreviru je kontraindikováno. |
|
zolpidem (nebenzodiazepinové sedativum)* |
^ zolpidem AUC 0,53 (0,45 - 0,64) Cmax 0,58 (0,52 - 0,66) mechanismus není znám |
Klinický význam není znám. K udržení účinnosti může být uu.ná zvýšená dávka zolpidemu. |
|
BLOKÁTORY KALCIOVÝCH KANÁLŮ | ||
|
amlodipin* |
t amlodipin AUC 2,79 (2,58-3,01) Cmax 1,27 (1,21 - 1,33) inhibice CYP3A |
Je třeba opatrnosti a je nutno zvážit snížení dávky am-'od,pinu. Doporučuje se klinické monito^vání. QfV |
|
diltiazem felodipin nikardipin nifedipin nisoldipin verapamil |
t blokátory kalciových kanálů inhibice CYP3A a/nebo účinek na P-gp transport ve střevě |
Je nutn opatrnost a doporučuje se klinické monitorování pacientů. |
|
ANTAGONISTÉ CCR5 | ||
|
maravirok* |
t AUCJ2 ma^aviroku 9,49 (7,94-11,3 4) Cmax maravirok’ 7,81 (5,92-10,32) C12 naraviroiu 10,17 Souasné podání maraviroku nemá pravděpodobně vliv na koncentrace telapreviru (dle historických údajů a způsobu eliminace telapreviru) |
Dávkování maraviroku je 150 mg dvakrát denně, pokud je současně podáván s telaprevirem. |
|
KORTIKOSTEROiDY | ||
|
Systémový dexamethason ■ iV |
^ telaprevir indukce CYP3A |
Současné užívání může vést ke ztrátě terapeutického účinku telapreviru. Tuto kombinaci je tedy nutno užívat s opatrností nebo zvážit jinou alternativu. |
|
iuhalacní/nosní flutikason budesonid |
t flutikason t budesonid inhibice CYP3A |
Současné podávání flutikasonu nebo budesonidu a telapreviru se nedoporučuje, pokud potenciální přínos nepřeváží riziko systémových nežádoucích účinků steroidů. |
|
ANTAGONISTÉ ENDOTELINOVÝCH RECEPTORŮ | ||
|
Bosentan |
t bosentan ^ telaprevir indukce CYP3A bosentanem, inhibice CYP3A a transportních polypeptidů organických aniontů (OATP) telaprevirem |
Je nutná opatrnost a doporučuje se klinické monitorování. |
|
HIV-ANTIVIROVÉ PŘÍPRAVKY: INHIBITORY HIV-PROTEÁZY (PI) | ||
|
atazanavir/ritonavir* |
i telaprevir AUC 0,80 (0,76 - 0,85) Cmax 0,79 (0,74-0,84) Cmin 0,85 (0,75 - 0,98) t atazanavir AUC 1,17 (0,97- 1,43) Cmax 0,85 (0,73 - 0,98) Cmin 1,85 (1,40 - 2,44) inhibice CYP3A telaprevirem |
U této kombinace je častá hyperbilirubinemie. Doporučuje se klinické a laboratorní monitorování hyperbilirubinemie (viz body 4.4 a 4.8). A |
|
darunavir/ritonavir* |
i telaprevir AUC 0,65 (0,61-0,69) Cmax 0,64 (0,61-0,67) Cmin 0,68 (0,63 - 0,74) i darunavir AUC 0,60 (0,57 - 0,63) Cmax 0,60 (0,56 - 0,64) Cmin 0,58 (0,52 - 0,63) mechanismus není znám |
Současné podávání kombinace darunavir/ritonavir a telapreviru se nedoporučuje (viz bod 4.4). j£° |
|
fosamprenavir/ritonavir* |
i telaprevir AUC 0,68 (0,63 - 0,72) Cmax 0,67 (0,63 -0,71) Cmin 0,70 (0,64 - 0,77) i amprenavir AUC 0,53 (0,49 - 0,58) Cmax 0,65 (0,59-0,70) Cmin 0,44 (0,40-0,50) mechanismus není ^nám |
Současné podávání kombinace fosamprenavir/ritonavir a telapreviru se nedoporučuje (viz bod 4.4). |
|
lopinavir/ritonavir* /V |
i telaprevir AUC 0,46 (0,4 -0,52) Cmax 0,47 (0,4-1 -0,52) Cmin 0,48 (0,40-0,56) ^ V'iriqvir aUC \06 (0,96- 1,17) Cm, 0,96 (0,87- 1,05) Cmin 1,14 (0,96- 1,36) mechanismus není znám |
Současné podávání kombinace lopinavir/ritonavir a telapreviru se nedoporučuje (viz bod 4.4). |
|
HIV-ANTIVIROVÉ PŘÍPRAVKY- INHIBITORY REVERZNÍ TRANSKRIPTÁZY | ||
|
efavirenz* O |
i telaprevir 1 125 mg každých 8 hodin (odpovídá 750 mg každých 8 hodin) AUC 0,82 (0,73 - 0,92) Cmax 0,86 (0,76 - 0,97) Cmin 0,75 (0,66 - 0,86) i efavirenz (+ TVR 1 125 mg každých 8 hodin AUC 0,82 (0,74 - 0,90) Cmax 0,76 (0,68 - 0,85) Cmin 0,90 (0,81-1,01) indukce CYP3A efavirenzem |
Při společném podání se užije telaprevir 1 125 mg každých 8 hodin (viz bod 4.4). |

|
tenofovir-disoproxyl-fumarát* |
^ telaprevir AUC 1,00 (0,94 - 1,07) Cmax 1,01 (0,96- 1,05) Cmin 1,03 (0,93 - 1,14) t tenofovir AUC 1,30 (1,22 - 1,39) Cmax 1,30 (1,16- 1,45) Cmin 1,41 (1,29- 1,54) účinek na P-gp transport ve střevě |
Je nutné zvýšené klinické a laboratorní monitorování (viz bod 4.4). |
|
abakavir zidovudin |
Nebylo studováno. |
Účinek telapreviru na UDP-glukuronyltransferázu nelze vyloučit a může ovlivnit koncentrace abakaviru nebo zidovudinu v plazme |
|
etravirin* |
i telaprevir 750 mg každých 8 hodin AUC 0,84 (0,71-0,98) Cmax 0,90 (0,79- 1,02) Cmin 0,75 (0,61-0,92) ^ etravirin (+ TVR 750 mg každých 8 hodin) AUC 0,94 (0,85 - 1,04) Cmax 0,93 (0,84 - 1,03) Cmin 0,97 (0,86- 1,10) |
Při současném podávání není nutná úprava dávky. . & |
|
rilpivirin* |
i telaprevir 750 mg každých 8 hodin AUC 0,95 (0,76- 1,18) Cmax 0,97 (0,79- 1,2D Cmin 0,89 (0,67 - 1,18) t rilpivirin (+ TVR 750 mg každých 8 hodin) AUC 1,78 (\4‘ -2,20) Cmax 1,49 (1 20 - 1,84) Cm n ,93 (1,5 5-2,41) |
Při současném podávání není nutná úpiava dávky. r |
|
INHIBITORY INTEGRÁZY | ||
|
raltegravir* |
< ? tlaprevir auC 1,07 (1,00- 1,15) Cmax 1,07 (0,98- 1,16) Cmin 1,14 (1,04- 1,26) t raltegravir AUC 1,31 (1,03 - 1,67) Cmax 1,26 (0,97 - 1,62) Cmin 1,78 (1,26-253) |
Při současném podávání není nutná úprava dávky. |
|
INHIBITORY HMG-CoA REDUKTÁZY | ||
|
atorvastatin* |
t atorvastatin AUC 7,88 (6,82 - 9,07) Cmax 10,6 (8,74 - 12,85) inhibice CYP3A a OATP telaprevirem |
Současné podávání atorvastatinu a telapreviru je kontraindikováno (viz bod 4.3). |
|
pitavastatin pravastatin ^ rosuvastatin |
t statin inhibice CYP3A a OATP telaprevirem |
Je nutná opatrnost a doporučuje se klinické monitorování. Spolu s přípravkem INCIVO kontraindikované inhibitory HMG-CoA reduktázy viz bod 4.3. |
|
HORMONÁLNÍ ANTIKONCEPCE/ESTROGEN | ||
|
ethinylestradiol* |
i ethinylestradiol |
Je-li hormonální antikoncepce |
|
norethisteron* |
AUC 0,72 (0,69 - 0,75) Cmax 0,74 (0,68-0,80) Cmin 0,67 (0,63-0,71) ^ norethisteron AUC 0,89 (0,86 - 0,93) Cmax 0,85 (0,81 -0,89) Cmin 0,94 (0,87 - 1,00) mechanismus není znám |
podávána spolu s telaprevirem, je nutno užít ještě další nehormonální metody antikoncepce. Pacientky užívající estrogeny v rámci hormonální substituční léčby je nutno klinicky monitorovat na příznaky estrogenové deficience. Viz body 4.4 a 4.6. |
|
IMUNOSUPRESIVA | ||
|
cyklosporin* |
t cyklosporin |
Je nutné výrazné snížení dávky |
|
takrolimus* |
AUC 4,64 (3,90-5,51) |
imunosupresiva s prodloužením |
|
sirolimus |
Cmax 1,32 (1,08 - 1,60) t takrolimus AUC 70,3 (52,9 - 93,4)** Cmax 9,35 (6,73 - 13,0)** t sirolimus t telaprevir ** počítáno na základě údajů se sníženou dávkou inhibice CYP3A inhibice transportních bílkovin |
intervalu dávkování nebo bez něj. Pii současném podávání s telaprrvirtu se doporučuje důsledné monitorování hladin imunosupresiva v krvi a funkce ledvin a nežádoucích účinků spojených s imunosupresivy. Takrolimus může prodlužovat QT interval (viz bod 4 + |
|
INHALAČNÍ BETAMIMETKA |
aV |
} |
|
salmeterol |
t salmeterol inhibice CYP^A \L> />> |
Současné podávání salmeterolu a telapreviru se nedoporučuje. Kombinace může vést ke zvýšenému riziku kardiovaskulárních nežádoucích účinků spojených se salmeterolem včetně prodloužení QT, palpitací a sinusové tachykardii (viz bod 4.4). |
|
SEKRETAGOGA INSULINU |
\Q | |
|
repaglinid |
t repaglinid inhibice OATP telaprevirem |
Je nutná opatrnost a doporučuje se klinické monitorování. |
|
OPIOIDNÍ ANALGETIKA | ||
|
methadon* |
i R-methadon |
Při zahájení podávání telapreviru není |
|
AUC 0,71 (0,66-0,76) Cmax 0,71 (0,66-0,76) Cmin 0,69 (0,64 - 0,75) Žádný účinek na koncentraci nevázaného |
nutná úprava dávky methadonu. Doporučuje se však klinické monitorování, protože u některých pacientů může být nutno během udržovací terapie upravit dávku methadonu. | |
|
;cf O |
R-methadonu. Vytěsnění methadonu z vazby na bílkoviny v plazmě. |
U methadonu byly hlášeny prodloužení QT intervalu a Torsade de Pointes (viz bod 4.4). Na počátku léčby a pravidelně i během léčby telaprevirem je nutno monitorovat EKG. |
|
buprenorfin* |
^ buprenorfin AUC 0,96 (0,84- 1,10) Cmax 0,80 (0,69 - 0,93) Cmin 1,06 (0,87 - 1,30) |
Při současném podávání s telaprevirem není nutná úprava dávky buprenorfinu. |
|
INHIBITORY PDE-5 | ||
|
sildenafil |
T inhibitory PDE-5 |
Současné podávání sildenafilu nebo |
|
tadalafil |
inhibice CYP3A |
vardenafilu a telapreviru se |
|
vardenafil |
nedoporučuje. Tadalafil v léčbě erektilní dysfunkce lze užívat s opatrností v jednotlivé dávce nepřesahující 10 mg za 72 hodin a se zvýšeným monitorováním s tadalafilem spojených nežádoucích účinků. Současné podávání sildenafilu nebo tadalafilu a telapreviru v léčbě plicní arteriální hypertenze je kontraindikováno (viz bod 4.3). | |
|
INHIBITORY PROTONOVÉ PUMPY | ||
|
esomeprazol* |
^ telaprevir |
Inhibitory protonové pumpy lze uívat |
|
AUC 0,98 (0,91 - 1,05) Cmax 0,95 (0,86- 1,06) |
bez úpravy dávkování. -:—♦ — | |
4.6 Fertilita, těhotenství a kojení

Údaje o podávání přípravku INCIVO těhotným ženám nejsou k dispozici. Studie reprodukční toxicity na zvířatech jsou nedostatečné (viz bod 5.3). Podávání přípravku INCWO se v těhotenství a u žen ve fertilním věku, které nepoužívají antikoncepci, nedoporučuje.
Antikoncepce u mužů a žen
Vzhledem k tomu, že přípravek INCIVO je nutno užívat ^ kombinaci s peginterferonem alfa a ribavirinem, jsou pro kombinovanou léčbu aplikovatelné kontraindikace a upozornění pro tyto léčivé přípravky.
Kvůli kombinované léčbě s peginterferonem alfa a ribavirinem musejí pacientky ve fertilním věku a jejich partneři a také pacienti - muži a jejich partneiky během léčby přípravkem INCIVO užívat 2 účinné antikoncepční metody. Po ukončení ’éčby přípravkem INCIVO je nutno pro antikoncepční doporučení sledovat souhrn údajů o přípravku pro ribavirin a pokyny níže.

Během podávání přípravku INCFO a až do dvou měsíců po ukončení jeho podávání lze pokračovat v hormonální antikoncepci, ta ale nemusí být spolehlivá (viz bod 4.5). Během tohoto období musejí proto pacientky ve fekáln m věku užívat dvě účinné nehormonální metody antikoncepce. Dva měsíce po ukončení léčby přípravkem INCIVO je hormonální antikoncepce opět vhodná jako jedna ze dvou požadovaných účinných metod kontroly početí.
Další informace jsoi uvedeny v souhrnech údajů o přípravku pro ribavirin a peginterferon alfa. Kojení
Telaprevir a jeho hlavní metabolit jsou vylučovány do mléka potkanů (viz bod 5.3). Není známo, zda je telaprevir vylučován do lidského mateřského mléka. Vzhledem k potenciálu možnosti nežádoucích účinků u kojených dětí kvůli kombinované léčbě přípravkem INCIVO s peginterferonem alfa a ribavirinem musí být kojení ukončeno před zahájením léčby. Viz také souhrn údajů o přípravku ribavirinu.
Fertilita
Ve studiích na potkanech neměl telaprevir vliv na plodnost nebo reprodukční schopnost.
4.7 Účinky na schopnost řídit a obsluhovat stroje
INCIVO nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje. Nebyly provedeny žádné studie vlivu přípravku INCIVO na schopnost řídit a obsluhovat stroje. U některých
pacientů užívajících INCIVO byly hlášeny synkopa a retinopatie, což je nutno vzít v úvahu při posouzení schopnosti pacienta řídit a obsluhovat stroje. Další informace jsou uvedeny v souhrnech údajů o přípravku peginterferonu alfa a ribavirinu.
4.8 Nežádoucí účinky
Shrnutí bezpečnostního profilu
Celkový bezpečnostní profil přípravku INCIVO je založen na údajích z klinických hodnocení fází II a III (jak kontrolovaných tak i nekontrolovaných), kterých se účastnilo 3 441 pacientů léčených přípravkem INCIVO v kombinované terapii a ze spontánních hlášení po uvedení na trh.
Ca
INCIVO je nutno podávat s peginterferonem alfa a ribavirinem. S nimi spojené nežádoucí účinky jsou uvedeny v jejich souhrnech údajů o přípravku. .<0
Četnost nežádoucích účinků alespoň střední intenzity (> stupeň 2) byla vyšší ve skupin ě s pňp ravkem INCIVO než ve skupině s placebem.
Během fáze léčby s přípravkem INCIVO/placebem byly nejčastěji hlášenými nižáa /ucími účinky stupně závažnosti alespoň 2 ve skupině s přípravkem INCIVO (s incidencí > 5,0 %> anemie, vyrážka, pruritus, nauzea a průjem.
Během fáze léčby s přípravkem INCIVO/placebem byly nejčastěji hlášenými nežádoucími účinky stupně alespoň 3 ve skupině s přípravkem INCIVO (s incidencí >10%) anemie, vyrážka, trombocytopenie, lymfopenie, pruritus a nauzea.
ČT
Tabelovanv přehled nežádoucích účinků
Nežádoucí účinky přípravku INCIVO jsou uvedeny v tabulce 3.
Nežádoucí účinky seřazené dle tříd orgánových systémů jsou uvedeny s následujícími frekvencemi: velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1 000 až < 1/100) a vzácné (> 1/10 000 až < 1/1 000). V každé skupině četností jsou nežádoucí účinky uvedeny s klesající závažností.
|
Tabulka 3: Nežádoucí účinky přípravku INCIVO (užívaného v kombinaci s peginterferonem alfa a ribavirinem) u pacientů s HCV infekcí v klinických hodnoceních3 a po uvedení na trh | ||
|
Třída orgánových systémů |
Skupiny četnosti |
Nežádoucí účinek kombinované léčby přípravkem INCIVO, peginterferonem alfa a ribavirinem |
|
Infekce a infesta^e |
časté |
kandidóza úst |
|
Poruchy krve a lymatického systému |
velmi časté |
anemie |
|
časté |
trombocytopenieb, lymfopenieb | |
|
Endokrinní pruchy |
časté |
hypothyreóza |
|
Poruchy metabolismu a výživy VN |
časté |
hyperurikemieb, hypokalemieb |
|
méně časté |
dna | |
|
Potichy nervového systému |
časté |
dysgeuzie, synkopa |
|
Poruchy oka |
méně časté |
retinopatie |
|
Gastrointestinální poruchy |
velmi časté | |
|
časté |
svědění konečníku, krvácení z konečníku, anální fisura | |
|
méně časté |
proktitida, pankreatitida | |
|
Poruchy jater a žlučových cest |
časté |
hyperbilirubinemieb |
|
Poruchy kůže a podkožní tkáně |
velmi časté |
pruritus, vyrážka |
|
časté |
ekzém, otok obličeje, exfoliativní vyrážka | |
|
méně časté |
léková vyrážka s eosinofilií a systémovými příznaky (DRESS), kopřivka | |
|
vzácné |
SJS, TEN, erythema multiforme | |
|
Poruchy ledvin a močových cest |
méně časté |
zvýšení kreatininu v krvib, prerenální azotemie s nebo bez akutního selhání ledvin |
|
Celkové poruchy a reakce v místě aplikace |
časté |
periferní otok, abnormální chuť přípravku |
placebem kontrolovaná hodnocení fáze II a fáze III (souhrnné údaje) zahrnovala 1 346 pacientů nakažených HCV četnost výskytu je založena na četnosti hlášení nežádoucích účinků (dále viz Laboratorní abnormality níže)
b
V analýze dalšího hodnocení, hodnocení C211, byl bezpečnostní profil u kombinované léčby s přípravkem INCIVO v dávce 1 125 mg dvakrát denně podobný bezpečnostnímu profilu u pacientů léčených kombinovanou léčbou s přípravkem INCIVO v dávce 750 mg každých 8 hodin. Ne’ identifikovány žádné nové bezpečnostní nálezy.
Laboratorní abnormality Vybrané laboratorní abnormality alespoň střední intenzity (> stupeň 2), které pře-úaovaly zhoršení oproti výchozímu stavu a byly považovány za nežádoucí účinky pozorované u pacientů s HCV infekcí léčených přípravkem INCIVO v kombinované terapii podle souhrnných údiiů z pla^bem kontrolovaných hodnocení fází II a III jsou uvedeny v tabulce níže:
|
Tabulka 4: Vybrané laboratorní abnormality (DAIDSa stupeň > 2) které představovaly zhoršení oproti výchozímu stavu a byly považovány za nežádoucí účinky pozorované u pacientů s HCV infekcí léčených přípravkem INCIVO v kombinované terapii podle souhrnných údajů z placebem kontrolovaných hodnocení fází II a III | ||||
|
Stupeň 2 |
Stupeň 3 |
Stupeň 4 | ||
|
Zvýšeníb | ||||
|
- i ^ |
kyselina močová |
17,9 % (10,1- N0 mg/dl) |
4,6 % (12,1 - 15,0 mg/dl) |
1,1 % (> 15,0 mg/dl) |
|
bilirubin |
13,6% (1,6 - 2,5 x ULN) |
3,6 % (2,6 - 5,0 x ULN) |
0,3 % (> 5,0 x ULN) | |
|
celkový cholesterol |
15,4 % (6,20 -7,77 mmol/l 240 - 300 mg/dl) |
2,0 % (> 7,77 mmol/l > 300 mg/dl) |
NA | |
|
LDL |
6,9 % (4,134,90 mmol/l 160- 190 mg/dl) |
2,5 % (> 4,91 mmol/l >191 mg/dl) |
NA | |
|
kreatinin |
0,9 % (1,4- 1,8 x ULN) |
0,2 % (1,9-3,4 x ULN) |
0% (> 3,4 x ULN) | |
|
Sníženíb | ||||
|
£ |
hemoglobin |
27,0 % (9,0 - 9,9 g/dl nebo jakékoli snížení 3,5 -4,4 g/dl) |
51,1 % (7,0 - 8,9 g/dl nebo jakékoli snížení > 4,5 g/dl) |
1,1 % (< 7,0 g/dl) |
|
počet krevních destiček |
24,4 % (50 00099 999/mm3) |
2,8 % (25 000 -49 999/mm3) |
0,2 % (< 25 000/mm3) | |
|
celkový počet lymfocytů |
13,1 % (500 - 599/mm3) |
11,8 % (350 - 499/mm3) |
4,8 % (< 350/mm3) | |
|
draslík |
1,6 % (2,5 - 2,9 mEq/l) |
0% (2,0 - 2,4 mEq/l) |
0% (< 2,0 mEq/l) | |
NA = není aplikovatelné
a The Division of AIDS Table for Grading the Severity of Adult and Paediatric Adverse Events (DAIDS, verze 1.0, prosinec 2004) bylo užito v celkovém hodnocení laboratorních výsledků. b Výskyt byl vypočítán podle počtu pacientů pro každý parametr.
Většina laboratorních hodnot se vrátila na hladiny pozorované u peginterferonu alfa a ribavirinu v týdnu 24, s výjimkou počtu krevních destiček, který zůstal na nižších hodnotách než u peginterferonu alfa a ribavirinu až do týdnu 48 (viz bod 4.4).
Během kombinované léčby s přípravkem INCIVO, peginterferonem alfa a ribavirinem docházelo velmi často ke zvýšení hladin kyseliny močové v séru. Po ukončení léčby přípravkem INCIVO se hladiny kyseliny močové většinou snížily během následujících 8 týdnů a byly srovnatelné s hladinami pozorovanými u pacientů dostávajících pouze peginterferon alfa a ribavirin.
Popis vybraných nežádoucích účinků >.<o
anou léčb
Při kombinované léčbě s přípravkem INCIVO byly hlášeny závažné potenciálně život ohrožující a fatální kožní reakce včetně DRESS, SJS a TEN (viz bod 4.4). V placebem kontrolo aných hodnoceních fází II a III se celková četnost a závažnost vyrážky při podání přípravku INCIVO s peginterferonem alfa a ribavirinem zvýšila. Během léčby přípravkem INCIVO byly případy vyrážky (všechny stupně) hlášeny u 55 % pacientů, kteří dostávali kombinovanou léčbu s přípravkem INCIVO, a u 33 % pacientů, kteří dostávali peginterferon alfa a ribavirin.
Více než 90 % případů vyrážky bylo slabé nebo střední závažnosti. Vyrážka hlášená během kombinované léčby s přípravkem INCIVO byla posuzována jak typická svědivá ekzematózní vyrážka, která zahrnovala méně než 30 % tělesného povrchu. Polovina případů vyrážky nastala během 4 počátečních týdnů, vyrážka se však může objevit kdykoi během kombinované léčby s přípravkem INCIVO. U mírné a středně závažné vyrážky není Tutné ukončení léčby přípravkem INCIVO.
Doporučení pro monitorování vyrážky a ukončení podávání přípravku INCIVO, ribavirinu a peginterferonu alfa jsou uvedena v bodu 4.4 Pacienty s mírnou až středně závažnou vyrážkou je nutno monitorovat na známky progrese; progrese nebyla častá (méně než 10 %). V klinických hodnoceních se většině pacientů podávala antihistaminika a lokální kortikoidy. Po ukončení podávání přípravku INCIVO nebo po přerušení léčby došlo ke zlepšení vyrážky; k vymizení vyrážky však může dojít až po několika týdnech.
Aneme
lsé
V placebem kontrolovaných hodnoceních fází II a III byla anemie (všech stupňů) hlášena u 32,1 % pacientů, kteří dostávali kombinovanou léčbu s přípravkem INCIVO, a u 14,8 %, kteří dostávali peginterferon alfa a libavirin. Pro zvládnutí anemie se užilo snížení dávek ribavirinu. Z pacientů léčených kombinovanou léčbou s přípravkem INCIVO vyžadovalo snížení dávek ribavirinu 21,6 %, zatímco z pacientů dostávajících pouze peginterferon alfa a ribavirin to bylo 9,4 %. Látky stimulující erytropoézu (ESA) většinou nebyly povoleny a v hodnoceních fází II a III byly použity pouze u 1 % pacientů. 1 placebem kontrolovaných hodnoceních fází II a III byly transfuze hlášeny během fáze léčby přípravkem INCIVO/placebem u 2,5 % pacientů dostávajících INCIVO v kombinované léčbě a u 0,7 % pacientů dostávajících pouze peginterferon alfa a ribavirin. Podíly transfuzí během celé doby hodnocení byly 4,6 %, resp. 1,6 %. V placebem kontrolovaných hodnoceních fází II a III ukončilo ■čju pouze přípravkem INCIVO kvůli anemii 1,9 %, 0,9 % pacientů ukončilo kombinovanou léčbu přípravkem INCIVO, zatímco z pacientů dostávající peginterferon alfa a ribavirin, ukončilo léčbu 0,5 % pacientů (viz bod 4.4).
Anorektální subjektivní a objektivní příznaky
V klinických hodnoceních byla většina těchto účinků (např. hemoroidy, anorektální diskomfort, anální pruritus a pálení konečníku) slabé nebo střední intenzity, velmi málo z nich vedlo k ukončení léčby a po dokončení podávání přípravku INCIVO došlo k jejich zahojení.
Pacienti současně nakažení HIV-1
Celkový bezpečnostní profil přípravku INCIVO u pacientů infikovaných současně HCV/HIV-1 (buď bez antiretrovirové léčby nebo s antiretrovirovou léčbou) byl podobný bezpečnostnímu profilu u pacientů nakažených pouze HCV, s výjimkou pacientů léčených atazanavirem/ritonavirem, u kterých se často během 2. týdne vyskytl přechodný vzestup hladin nepřímého bilirubinu (včetně stupně 3 až 4), který se do 12. týdne vrátil téměř k normálu (viz bod 4.4).
Pacienti po transplantaci jater bez cirhózy
Celkový bezpečnostní profil přípravku INCIVO při léčbě dosud neléčených a již léčených pacientů infikovaných HCV-1 po transplantaci jater, kteří byli na stabilním režimu imunosupresiv takrolimu nebo cyklosporinu A, byl obecně podobný bezpečnostnímu profilu přípravku INCIVO u pacientů, kterým játra transplantována nebyla, i když častěji byla během fáze léčby přípravkem INCIVO anémie (55,4 % versus 32,1 % v souhrnném hodnocení bezpečnosti ve fázi II až III). Ke zvládni anémie se při zahájení léčby přípravkem INCIVO použila nižší zahajovací dávka ribavirinu (600 mg/den); během celé léčebné fáze byla dávka ribavirinu dále snížena u 36,5 % pacien ů, 4,9 dostávalo látky stimulující erytropoézu a 21,6 % dostávalo krevní transfuze (viz body 4.4 ■ Imunosupresiva).
Pediatrická populace
Bezpečnost a účinnost přípravku INCIVO u dětí ve věku < 18 let nebyly stáno'
K dispozici nejsou žádné údaje.
ý\
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého příp avku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím náio^ního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
itů,
hlášena
0/
%
eny.
4.9 Předávkování
aná zdravým
Nejvyšší dávka přípravku INCIVO podávaná, zdravým dobrovolníkům je 1 875 mg každých 8 hodin po dobu 4 dnů. V tomto hodnocení při dávkvání 1 875 mg každých 8 hodin byly hlášeny následující obvyklé nežádoucí účinky častěji než pa dávkování 750 mg každých 8 hodin: nauzea, bolest hlavy, průjem, snížení chuti k jídlu, dysgeuzie a zvracení.

Na předávkování telaprevirem není dostupné specifické antidotum. Léčba předávkování přípravkem INCIVO zahrnuje celková podpůrná opatření včetně monitorování vitálních známek a sledování klinického stavu pacienta Je- i to indikováno, může být neabsorbovaná léčivá látka odstraněna vyvoláním zvracení rebo výplachem žaludku. Výplach žaludku se provede pouze, pokud to lze stihnout do jedné hodiny po požití. K odstranění neabsorbované léčivé látky lze použít také aktivní
uhlí.
Není zná
Iv
námo, zd
X
a je telaprevir odstranitelný dialýzou nebo peritoneální dialýzou.
ARMAKOLOGICKE VLASTNOSTI
Farmakodynamické vlastnosti
Farmakoterapeutická skupina: přímo účinkující antivirotikum, ATC kód: J05AE11.
Mechanismus účinku
Telaprevir je inhibitor HCV NS3^4A serinové protézy, která je důležitá pro virovou replikaci.
Studie in vitro
Účinek telapreviru proti HCV
Při analýze replikonu HCV subtypu 1b byla IC50 telapreviru proti divokému typu HCV 0,354 ^M podobně jako u infekčního viru subtypu 1a, kde IC50 byla 0,28 ^M.
Rezistence
A
Varianty HCV spojené s virologickým selháním při léčbě nebo s relapsem byly hodnoceny místně vedenou mutagenezí při analýze replikonu. Varianty V36A/M, T54A/S, R155K/T a A156S vykazovaly in vitro nižší hladiny rezistence k telapreviru (3 až 25násobné zvýšení IC50 telapreviru); varianty A156V/T a V36M+R155K vykazovaly in vitro vyšší hladiny rezistence k telapreviru (> 25násobné zvýšení IC50 telapreviru). Varianty replikonu získané ze sekvencí odvozených od pacientů vykazovaly podobné výsledky.
In vitro replikační kapacita variant rezistentních k telapreviru byla nižší než u divokých typů viru Zkřížená rezistence
Varianty rezistentní k telapreviru byly testovány na zkříženou rezistenci k reprezenta hmím inhibitorům proteázy v systému replikonů HCV. Replikony s jednou substitucí na pozici 155 nebo 156 a dvojité varianty se substitucí na reziduech 36 a 155 vykázaly zkříženou rezistnci 'x všem testovaným inhibitorům proteázy v širokém rozsahu citlivostí. Všechny studované varianty rezistentní k telapreviru zůstaly plně citlivé k interferonu alfa, ribavirinu a reprezentativním HCV nukleosidovým a nenukleosidovým inhibitorům polymerázy v systému replikonů. K dispoz:ci nejsou žádné klinické údaje týkající se nového zahájení léčby u pacientů, u kterých selhaH léčba založená na HCV NS3-4A inhibitorech proteázy jako je telaprevir, ani neexistují údaje o opakovaných cyklech léčby telaprevirem.
Klinická virologická hodnocení
V hodnoceních fází II a III s telaprevirem se pacienti dříve neléčení nebo pacienti, u nichž selhala předchozí léčba, s predominantně rezistentním’ variantami k telapreviru na počátku (V36M, T54A a R155K < 1 % a T54S 2,7 %) vyskytovali zřídka. Predominantní rezistence k telapreviru na počátku nevylučuje úspěšnou léčbu telaprevirem, peginterferonem alfa a ribavirinem. Vliv predominantně rezistentních variant k telapreviru na počátku je pravděpodobně větší u pacientů se špatnou odpovědí na interferon, např. u pacientů nereagujících na předchozí léčbu.
Ve fázi III klinického hodnocení s T12,PR režimem došlo u215 z 1169 pacientů k virologickému selhání během léčby (n = 125' nebo k relapsu (n = 90). Na základě populačních sekvenčních analýz HCV u těchto 215 pacientů by1 výskyt HCV variant rezistentních k telapreviru detekován u 105 (84 %) virologických selhání i u 55 (61 %) pacientů s relapsem; divoký typ viru byl detekován u 15 (12 %) virologických selhán a u 24 (27 %) pacientů s relapsem. HCV sekvenční údaje nebyly dostupné pro 16 (7 0% pacientů. Sekvenční analýzy variant rezistentních k telapreviru identifikovaly substituce na 4 pozic ch v oblasti NS3-4A proteázy, což odpovídá mechanismu účinku telapreviru (V36A/M, T54A/S, R155K/T a A156S/T/V). Ve fázi III hodnocení C211 nebyl rozdíl v typu objevujících se variant mezi pacienty léčenými 1 125 mg telapreviru dvakrát denně a pacienty léčeným' 7 50 mg telapreviru každých 8 hodin. V době selhání měly varianty rezistentní k telapreviru bné podíly pacientů v obou léčebných skupinách. Virologické selhání během léčby telaprevirem spojeno především s vysoce rezistentními variantami a relaps byl zejména spojen orezistentními variantami nebo viry divokého typu.
acienti s 1a genotypem HCV mají především jednotlivé nebo kombinované varianty V36M a R155K, zatímco pacienti s 1b genotypem HCV mají především varianty V36A, T54A/S a A156S/T/V. Tento rozdíl je pravděpodobně způsoben vyšší genetickou bariérou substituce pro V36M a R155K u genotypu 1b oproti genotypu 1a. U pacientů léčených telaprevirem bylo virologické selhání během léčby častější u genotypu 1a než u genotypu 1b a bylo také častější u dříve neodpovídající populace než u dalších populací (dosud neléčení, dříve relabující, dříve částečně reagující; viz bod 5.1 Klinická zkušenost, Účinnost u dříve léčených dospělých).
Profil rezistence pozorovaný v hodnocení HPC3008 u pacientů koinfikovaných HCV/HIV-1 byl podobný profilu rezistence u pacientů infikovaných pouze HCV.
Profil rezistence pozorovaný v hodnocení HPC3006 při léčbě dosud neléčených a již léčených pacientů infikovaných HCV-1 po transplantaci jater, kteří byli na stabilním režimu imunosupresiv takrolimu nebo cyklosporinu A, byl podobný profilu rezistence u pacientů infikovaných HCV bez transplantace jater.
ých

Analýza následného sledování hodnocení u pacientů léčených přípravkem INCIVO, kteří nedosáhli SVR, ukázala, že po ukončení léčby telaprevirem se během času populace divokého viru zvyšovala a populace variant rezistentních k telapreviru se stala nedetekovatelnou. Z celkem 255 dosud neléčen a dříve léčených pacientů z hodnocení fáze III 108, 111 a C216, u kterých se během léčby objevily varianty rezistentní k telapreviru, nebyly u 152 pacientů (60 %) populačním sekvencováním detekovány rezistentní varianty (medián následného sledování 10 měsíců). Z 393 rezistentních variant detekovaných u 255 pacientů nebylo již detekováno 68 % z NS3-36, 84 % z NS3-54, 59 % z Nc3-155 86 % z NS3-156 a 52 % z NS3-36M+NS3-155K varianty.
V hodnocení následného sledování nebyly z 98 dosud neléčených pacientů a pacientů, u kterých léčba dříve selhala a kteří byli v hodnocení fáze II nebo fáze III léčeni režimem s přípcv^m INCIVO a nedosáhli SVR, varianty rezistentní k telapreviru již detekovány u 85 % (83 98) pacientů (medián doby sledování 27,5 měsíce). Klonální sekvenční analýza u podskupiny prcK^ů, kteří měli divoký typ HCV při populačním sekvencování (n = 20), která porovnávala frekvenci rezistentních variant před zahájením léčby telaprevirem a během následného sledování, prokázala, že se populace HCV variant
u všech pacientů vrátila na úrovně před léčbou. U variant rezistencích ktelapreviru byl delší medián doby dosažení nedetekovatelnosti podle sekvencované populace u variant NS3-36 (6 měsíců),
NS3-155 (9 měsíců) a NS3-36M+NS3-155K (12 měsíců) pozorovaný především u pacientů s genotypem 1a než u variant NS3-54 (2 měsíce) a NS3-156 (3 měsíce) pozorovaný především u pacientů s genotypem 1b.
♦A/
Klinická účinnost a bezpečnost
Účinnost a bezpečnost přípravku INCIVO u pacientů s chronickou hepatitidou C genotypu 1 byly sledovány ve čtyřech hodnoceních fáze III: ' u dosud neléčených pacientů a 1 u již dříve léčených pacientů (relabující, částečně odpovídaící a nereagující). Pacienti v těchto hodnoceních, 108, 111 a C216, měli kompenzované jaterní onemocnění, detekovatelnou HCV RNA a histopatologii jater konzistentní s chronickou hepatitidou C. Není-li uvedeno jinak, podával se přípravek INCIVO v dávce 750 mg každých 8 hodin; dávka pegmterferonu alfa-2a byla 180 ^g/týden a dávka ribavirinu 1 000 mg/den (u pacientů c hmotností < 75 kg) nebo 1 200 mg/den (u pacientů s hmotností > 75 kg). Hodnoty HCV RNA v pkzmt byly měřeny pomocí COBAS TaqMan HCV testu (verze 2.0) k použití s vysoce čištěným systémem. Analýza měla nižší limit kvantifikace 25 IU/ml.
V popisu výstupu z hodnocení fáze III u hodnocení 108, 111 a C216, byla SVR považovaná za virologické vyléčení definována na základě posouzení HCV RNA v okně při návštěvě v týdnu 72, s použitím pos edního měření v okně. V případě chybějících údajů z okna v týdnu 72 byl použit poslední údaj o HCV RNA od týdne 12 výše. Navíc byl pro stanovení SVR použit limit kvantifikace 25 IU/ml.
pisu výstupů z hodnocení fáze III u hodnocení C211, HPC3008 a HPC3006 byla SVR12 ažovaná za virologické vyléčení definována na základě HCV RNA pod limitem kvantifikace 25 IU/ml) za použití posledního měření v okně při návštěvě ve 12. týdnu po plánovaném ukončení léčby.
Účinnost u dosud neléčených _pacientů Hodnocení C211
Hodnocení C211 bylo randomizované otevřené hodnocení fáze III provedené u dosud neléčených pacientů, kteří byli randomizováni do jedné ze dvou skupin: INCIVO 750 mg každých 8 hodin [T12(každých 8 hodin - dále q8h)/PR] nebo INCIVO 1 125 mg dvakrát denně [T12(2x denně - dále b.i.d.)/PR] v kombinaci s peginterferonem alfa-2a a ribavirinem. Primárním účelem hodnocení bylo prokázat noninferioritu T12(b.i.d.)/PRnad režimem T12(q8h)/PR. Všichni pacienti podstoupili 12 týdnů léčby přípravkem INCIVO v kombinaci s peginterferonem alfa-2a a ribavirinem. Ve 12. týdnu léčba přípravkem INCIVO skončila a pacienti pokračovali s léčbou peginterferonem alfa-2a a ribavirinem. Celkové trvání léčby bylo založeno na individuální pacientově virové odpovědi. Dosáhl-li pacient nedetekovatelné HCV RNA (hladiny detekčního limitu nebylo dosaženo) ve 4. týdnu, celková doba trvání léčby byla 24 týdnů. Jinak byla celková doba trvání léčby 48 týdnů.
Medián věku zahrnutých 740 pacientů byl 51 let (rozpětí: 18 až 70); 60 % pacientů byli muži; 21 % mělo body mass index > 30 kg/m2; 5 % byli černoši; 2 % byli asiati; 85 % mělo počáteční hladiny HCV RNA > 800 000 IU/ml; 15 % mělo přemosťující fibrózu; 14 % mělo cirhózu; 57 % mělo HCV genotyp 1a a 43 % mělo HCV genotyp 1b.
71)
Výskyt podílu SVR12 u skupiny T12(b.i.d.)/PR byl 74 % (274/369) ve srovnání se 73 % (270/3 u skupiny T12(q8h)/PR s 95 % intervalem spolehlivosti rozdílu: -4,9 %, 12,0 %. Spodní hranice 95 % intervalu spolehlivosti (-4,9 %) byla vyšší než předem stanovená hranice noninferiority -11 %, " proto
povědí
byla prokázána noninferiorita T12(b.i.d.)/PR oproti T12(q8h)/PR. Tabulka 5 ukazuje výs u skupin T12(b.i.d.)/PR a T12(q8h)/PR.
|
Tabulka 5: Výskyt odpovědí: Hodnocení C211 | ||
|
Výsledek léčby |
T12(b.i.d.)/PR N = 369 % (n/N) |
T12(q8h)/PR N = 371 % (n/N) |
|
SVR12 |
74 % (274/3 69) |
73 % (270/371) |
|
Nedetekovatelná HCV RNA (hladiny detekčního limitu nebylo dosaženo) v týdnu 4a |
69% 2rc'369) |
67% (250/371) |
|
Nedetekovatelná HCV RNA (hladiny detekčního limitu nebylo dosaženo) v týdnech 4 a 12 |
. <n 66 % (244/369) |
63 % (234/371) |
|
SVR u pacientů s nedetekovatelnou HCV RNA (hladiny detekčního limitu nebylo dosaženo) v týdnech 4 a 12 |
89 % (218/244) |
89 % (209/234) |
|
SVR u pacientů, kteří neměli nedetekova^nou HCV RNA (hladiny detekčního limitu nebylo dosaženo) v týdnech 4 a 12 |
45 % (56/125) |
45 % (61/137) |
|
Pacienti bez SVR |
26 % (95/369) |
27% (101/371) |
|
Virologické selhání během léčby6 |
10 % (38/369) |
10% (36/371) |
|
Relapsc |
8 % (23/300) |
6 % (19/293) |
|
Dalšíd |
9 % (34/369) |
12% (46/371) |
T12(b.i.d.)/PR: INCIVO 1 125 mg dvakrát denně po 12 týdnů s peginterferonem alfa-2a a ribavirinem po 24 nebo 48 týdnů; T12(q8h) PR: INCIVO 750 mg každých 8 hodin po 12 týdnů s peginterferonem alfa-2a a ribavirinem po 24 nebo 48 týdnů
Pacienti s pánovaným celkovým trváním léčby 24 týdnů.
Virolcnck' elhání během léčby zahrnuje pacienty, kteří dosáhli protokolem definovaného pravidla virologického selh ání '/neb' měli virový průlom (breakthrough).

Reops byl definován jako méně než 25 IU/ml při plánovaném ukončení léčby a následně HCV RNA > 25 IU/ml při poUedním pozorování během následného návštěvního okna. Denominátor při počítání podílu relapsů reprezentuje počet pacientů s odpovědí na konci léčby (HCV RNA < 25 IU/ml).
Další zahrnuje pacienty s detekovatelnou HCV RNA při plánovaném ukončení léčby, kteří ale neměli virový průlom (breakthrough), a pacienty s chybějícím zhodnocením SVR během dalšího sledování.
Tabulka 6 shrnuje výskyt SVR u genotypu IL28B při fibróze jater na počátku léčby.
|
Tabulka 6: Výskyt SVR u podskupin pacientů: Hodnocení C211 | ||
|
Podskupina |
T12(b.i.d.)/PR N = 369 % (n/N) |
T12(q8h)/PR N = 371 % (n/N) |
|
Genotyp IL28B | ||
|
CC |
92 % (97/105) |
87 % (92/106) |
|
CT |
67 % (139/206) |
68 % (141/208) |
|
TT |
66 % (38/58) |
65 % (37/57) |
|
Fibróza jater na počátku léčby | ||
|
Žádná nebo minimální fibróza |
80 % (138/172) |
79 % (140/177) |
|
Portální fibróza |
79 % (75/95) |
80 % (68/85) |
|
Přemosťující fibróza |
67 % (32/48) |
64 % (38/59) |
|
54 % (29/54) |
49 % (24/49) |
T12(b.i.d.)/PR: INCIVO 1 125 mg dvakrát denně po 12 týdnů s peginterferonem alfa-2a a ribavirinem po 24 nebo 48 týdnů;
T12(q8h)/PR: INCIVO 750 mg každých 8 hodin po 12 týdnů s peginterferonem alfa-2a a ribavirinem po 24 nebo 48 týdi
v paralelním uspořádání u dosud neléčených pacientů. INCIVO se podávalo prvních 8 týd~ů kxby (režim T8/PR) nebo prvních 12 týdnů léčby (režim T12/PR) v kombinaci s peginterferonem J.fa-2a a ribavirinem buď 24 nebo 48 týdnů. Pacienti, kteří měli nedetekovatelnou HCV RNA (hVdiny detekčního limitu nebylo dosaženo) v týdnech 4 a 12, byli 24 týdnů léčeni peginterferonem alfa-2a a ribavirinem; pacienti, kteří v týdnech 4 a 12 neměli nedetekovatelnou HCV RNA (ťadiny detekčního limitu nebylo dosaženo), byli léčeni peginterferonem alfa-2a a ribavirinem 4 8 týdnů. Kontrolní režim (Pbo/PR) měl fixní trvání léčby 48 týdnů a podávaly se v něm placebo napodobující telaprevir počátečních 12 týdnů a peginterferon alfa-2a a ribavirin 48 týdnů.
Hodnocení 108 (ADVANCE)
Hodnocení 108 bylo randomizované dvojitě zaslepené placebem kontrolované hodnocení fáze IIi
U 1 088 zahrnutých pacientů byl medián věku 49 let (rozmezí 18 až 6°), 58 % pacientů bylo mužů,
23 % mělo body mass index >30 kg/m2, 9 % byli černoši, 11 % uyli hispánci nebo latinoameričané, 77 % mělo hladiny HCV RNA na počátku > 800 000 IU/ml, k % mělo přemosťující fibrózu, 6 % mělo cirhózu, 59 % mělo genotyp 1a a 40 % mělo genotyp 1b.
Výskyt SVR u skupiny T8/PR byl 72 % (261/364) (P < 0,0001 ve srovnání se skupinou Pbo/PR48). Tabulka 7 shrnuje výskyt odpovědí pro skupiny nktR a Pbo/PR.
|
Tabulka 7: Výskyt odpovědí: Hodnocen 108 | ||
|
Výsledek léčby |
T12/PR N = 363 n/N (%) |
Pbo/PR48 N = 361 n/N (%) |
|
SVR- |
79 % (285/363) |
46 % (166/361) |
|
(74 %, 83 %)b |
(41 %, 51 %)b | |
|
NedetekovatelnáHCV RNA (hladiny detekčního limitu nebylo dosr.enn) v týdnech 4 a 12 (eRVR) |
58 % (212/363) |
8 % (29/361) |
|
SVR u eRVR pacientů |
92% (195/212) |
93 % (27/29) |
|
Žádné eRVR |
42% (151/363) |
92 % (332/361) |
|
SVR u pacientů bez eRVR |
60% (90/151) |
42 % (139/332) |
|
HCV RNA < 25 IU/ml na konci léčby |
82 % (299/363) |
62 % (225/361) |
|
Pelans |
4 % (13/299) |
26 % (58/225) |
T12/PR:
T 2/PR: iNCIVO po dobu 12 týdnů s peginterferonem alfa-2a a ribavirinem po dobu 24 nebo 48 týdnů; placebo po dobu 12 týdnů s peginterferonem alfa-2a a ribavirinem po dobu 48 týdnů;
0,0001; T12/PR ve srovnání s Pbo/PR48. Rozdíl ve výskytu SVR (95 % CI) mezi T12/PR a Pbo/PR skupinami byl 33 (26, 39).
95 % CI
pv;r:
Výskyt SVR byl vyšší (s absolutním rozdílem alespoň 28 %) u skupiny T12/PR než u skupiny Pbo/PR48 pro všechny podskupiny dle pohlaví, věku, rasy, etnicity, body mass indexu, subtypu genotypu HCV, počáteční HCV RNA (< 800 000, > 800 000 IU/ml) a rozsahu jaterní fibrózy. Tabulka 8 ukazuje podíly SVR u těchto podskupin pacientů.
|
Tabulka 8: Výskyt SVR u podskupin pacientů v hodnocení 108 | ||
|
Podskupiny |
T12/PR |
Pbo/PR |
|
Muži |
78 % (166/214) |
46 % (97/211) |
|
Věk 45 až <65 let |
73 % (157/214) |
39 % (85/216) |
|
Černoši |
62 % (16/26) |
29 % (8/28) |
|
Hispánci a latinoameričané |
77 % (27/35) |
39 % (15/38) |
|
BMI >30 kg/m2 |
73 % (56/77) |
44 % (38/87) |
|
Počáteční HCV RNA > 800 000 IU/ml |
77% (215/281) |
39 % (109/279) |
|
HCV genotyp 1a |
75 % (162/217) |
43 % (90/210) |
|
HCV genotyp 1b |
84% (119/142) |
51 % (76/149) |
|
Počáteční jaterní fibróza |
X | |
|
Bez fibrózy, lehká fibróza nebo portální fibróza |
82 % (237/290) |
49 % (140/288) |
|
Přemosťující fibróza |
63 % (33/52) |
35 % (18 52) |
|
71 % (15/21) |
38 % (8/21) | |
T12/PR: INCIVO 12 týdnů s peginterferonem alfa-2a a ribavirinem po dobu 24 nebo 48 týdnů; Pbo/PR: placebo 12 týdnů s peginterferonem alfa-2a a ribavirinem po dobu 48 týdnů
Hodnocením (ILLUMINATE)
Hodnocení 111 bylo randomizované otevřené hodnocení fáze III u dosud le.Včeaých pacientů. Hodnocení bylo navrženo tak, aby srovnávalo výskyt SVR u pacienJů s nedetekovatelnou HCV RNA (hladiny detekčního limitu nebylo dosaženo) v týdnech 4 a 12, kteří byli léčeni přípravkem INCIVO 12 týdnů v kombinaci s peginterferonem alfa-2a a ribavirinem buď po dobu 24 týdnů (režim T12/PR24) nebo po dobu 48 týdnů (režim T12/PR48). Pacienti s nedetekovatelnou HCV RNA (hladiny detekčního limitu nebylo dosaženo) v týdnech 4 a 12 byl. ve 20. týdnu randomizováni tak, aby dostávali buď 24týdenní nebo 48týdenní léčbu peginterferonem alfa-2a a ribavirinem. Primárním výstupem bylo hodnocení neinferiority s použitím ^dílu -10,5 % mezi 24týdenním a 48týdenním režimem u pacientů s nedetekovatelnou HCV RNA (hladiny detekčního limitu nebylo dosaženo) v týdnech 4 a 12.
U 540 zahrnutých pacientů byl medián věk’ 51 let (rozmezí 19 až 70), 60 % pacientů bylo mužů,
32 % mělo body mass index > 30 kg/m2, '4 % byli černoši, 10 % byli hispánci nebo latinoameričané, 82 % mělo hladiny HCV RNA na poeáu.” > 800 000 IU/ml, 16 % mělo přemosťující fibrózu, 11 % mělo cirhózu, 72 % mělo genotyp 1a a 27 % mělo genotyp 1b.
Celkem 352 (65 %) pacientů mělo v týdnech 4 a 12 nedetekovatelnou HCV RNA (hladiny detekčního limitu nebylo dosaženo). Tabulka 9 shrnuje výskyt odpovědí. U pacientů, kteří měli nedetekovatelnou HCV RNA (hladiny detekčního limitu nebylo dosaženo) v týdnech 4 a 12, nepřineslo pokračování léčby peginterferonem alfa-2a a ribavirinem do 48. týdnu žádný další přínos (rozdíl ve výskytu SVR byl 2 %; 95 % in erv.l spolehlivosti: -4 %, 8 %).
i 4
|
Tabulka 9 Výskyt odpovědí v hodnocení 111 | |||
|
r |
Pacienti s nedetekovatelnou HCV RNA (hladiny detekčního limitu nebylo dosaženo) v týdnech 4 a 12 |
T12/PR Všichni pacienti3 N = 540 | |
|
Výsledek léčby |
T12/PR24 N = 162 |
T12/PR48 N = 160 | |
|
SVR |
92 % (149/162) (87 %, 96 %)b |
90 % (144/160) (84 %, 94 %)b |
74 % (398/540) (70 %, 77 %)b |
|
HCV RNA < 25 IU/ml na konci léčby |
98 % (159/162) |
93 % (149/160) |
79 % (424/540) |
|
Relaps |
6 % (10/159) |
1 % (2/149) |
4 % (19/424) |
T12/PR24: INCIVO 12 týdnů s peginterferonem alfa-2a a ribavirinem po dobu 24 týdnů;
T12/PR48: INCIVO 12 týdnů s peginterferonem alfa-2a a ribavirinem po dobu 48 týdnů
a Všichni pacienti zahrnují 322 pacientů s nedetekovatelnou HCV RNA (hladiny detekčního limitu nebylo dosaženo) v týdnech 4 a12 a 218 dalších pacientů léčených v hodnocení (118, kteří neměli nedetekovatelnou HCV RNA (hladiny detekčního limitu nebylo dosaženo) v týdnech 4 a 12 a 100, kteří ukončili léčbu před týdnem 20, kdy došlo k randomizaci).
b
95 % CI
Výskyt SVR u černochů byl 62 % (45/73). Tabulka 10 ukazuje vyskyt SVR v závislosti na rozsahu fibrózy jater na počátku léčby.
|
Tabulka 10: Výskyt SVR podle rozsahu fibrózy jater na počátku: Hodnocení 111 | |||
|
Podskupiny |
Pacienti s nedetekovatelnou HCV RNA (hladiny detekčního limitu nebylo dosaženo) v týdnech 4 a 12 |
T12.PR Všichni pacienti3 | |
|
T12/PR24 |
T12/PR48 | ||
|
Bez fibrózy, lehká fibróza nebo portální fibróza |
96 % (119/124) |
91 % (115/127) |
7/ % (302/391) |
|
Přemosťující fibróza |
95 % (19/20) |
86% (18/21) i ’4% (65/88) | |
|
61 % (11/18) |
92% (11/12) , 51% (31/61) | ||
T12/PR24: INCIVO 12 týdnů s peginterferonem alfa-2a a ribavirinem po dobu 24 týdnů;
T12/PR48: INCIVO 12 týdnů s peginterferonem alfa-2a a ribavirinem po dobu 48 týdnů
a Všichni pacienti zahrnují 322 pacientů s nedetekovatelnou HCV RNA (hladiny detekčního limitu nebylo dosaženo) v týdnech 4 a 12 a 218 dalších pacientů léčených v hodnocení (118, kteří neměli nedetekovatelnou HCV RNA (hladiny detekčního limitu nebylo dosaženo) v týdnech 4 a 12 a 100, kteří ukončili léčbu před týdnem 20, kdy došlo k randomizaci).
zaslepené h
Ví
Účinnost u již dříve léčených _pacientů Hodnocení C216 (REALIZE)
Hodnocení C216 bylo randomizované dvojitě zas’epmé hodnocení fáze III v paralelním uspořádání u pacientů, kteří nedosáhli SVR před léčbou peginteferonem alfa-2a a ribavirinem nebo peginterferonem alfa-2b a ribavirinem. Do hodnocení byli zahrnuti dříve relabující pacienti (pacienti s nedetekovatelnou HCV RNA na konci 'éčby režimem na bázi pegylovaného interferonu, ale s detekovatelnou HCV RNA během 24 týdnů následného sledování) a dříve nereagující pacienti (pacienti, kteří neměli nedekovatelné hladiny HCV RNA během předchozí alespoň 12týdenní léčby nebo na jejím konci). Nereagujíc' populace byla rozdělena do 2 podskupin: pacienti dříve odpovídající částečně (se snížením HCV Rt tA v týdnu 12 rovným nebo větším než 2 logio, ale bez dosažení nedetekovatelné HCV RNA na konci léčby peginterferonem alfa a ribavirinem) a dříve nereagující (se snížením HCV RNa v týdnu 12 léčby peginterferonem a ribavirinem menším než 2 log10).
Pacienti byli random'zováni v poměru 2:2:1 do jedné ze tří skupin: simultánní start (T12/PR48): INCIVO ode dne 1 do týdnu 12; odložený start (T12(DS)/PR48); INCIVO od týdnu 5 do týdnu 16; Pbo/PR48: pNcebo do týdnu 16. U všech režimů se léčba peginterferonem alfa-2a a ribavirinem podávala 48 týdnů.
U 662 zahrnutých pacientů byl medián věku 51 let (rozmezí 21 až 70), 70 % pacientů bylo mužů,
9o % mělo body mass index >30 kg/m2, 5 % byli černoši, 11 % byli hispánci nebo latinoameričané,
V89 % mělo hladiny HCV RNA na počátku > 800 000 IU/ml, 22 % mělo přemosťující fibrózu, 26 % mělo cirhózu, 54 % mělo genotyp 1a a 46 % mělo genotyp 1b.
Podíly SVR u skupiny T12(DS)/PR byly 88 % (124/141) z dříve relabujících, 56 % (27/48) z dříve částečně odpovídajících a 33 % (25/75) z dříve nereagujících pacientů. V tabulce 11 jsou uvedeny odpovědi u skupin s doporučeným simultánním startem (T12/PR48) a s Pbo/PR48.
|
Tabulka 11: Výskyt odpovědí v hodnocení C216 | ||
|
T12/PR48 |
Pbo/PR48 | |
|
Výsledek léčby |
% (n/N) |
% (n/N) |
|
SVR | ||
|
Dříve relabující3 |
84 % (122/145) (77 %, 90 %)b |
22 % (15/68) (13 %, 34 %)b |
|
Dříve částečně odpovídající*1 |
61 % (30/49) (46 %, 75 %)b |
15 % (4/27) (4 %, 34 %)b |
|
Dříve nereagující*1 |
31 % (22/72) (20 %, 43 %)b |
5 % (2/37) (1 %, 18 %)b |
|
HCV RNA < 25 IU/ml na konci léčby | ||
|
Dříve relabující |
87 % (126/145) |
63 % (43/68) |
|
Dříve částečně odpovídající |
73 % (36/49) |
15 % (4/27) |
|
Dříve nereagující |
39 % (28/72) |
11 % (4'37) |
|
Relaps | ||
|
Dříve relabující |
3 % (4/126) |
63 % (27/43) |
|
Dříve částečně odpovídající |
17 % (6/36) |
0% (0/4) |
|
Dříve nereagující |
21 % (6/28) |
50 % (2/4) |
T12/PR48: INCIVO 12 týdnů následováno placebem po dobu 4 týdnů v kombinaci s peginJerteionem alfa-2a a ribavirinem po dobu 48 týdnů;
Pbo/PR48: placebo 16 týdnů v kombinaci s peginterferonem alfa-2a a ribavirinem po dou' 48 týdnů a P < 0,001, T12/PR ve srovnání sPbo/PR48. Rozdíly ve výskytu SVR (95 % inmrval spolehlivosti) mezi skupinami T12/PR a Pbo/PR byly 63 (51, 74) u dříve relabujících, 46 (27, 66) u dříve částečně ( u dříve nereagujících pacientů.
U všech populací v hodnocení (dříve relabující, dříve částečně odpovídající a dříve nereagující pacienti) byl výskyt SVR vyšší u skupiny T12/PR než u skupiny Pbo/PR48 u všech podskupin dle pohlaví, rasy, etnicity, body mass indexu, subtypu geiotypu HCV, počátečních hladin HCV RNA a rozsahu jaterní fibrózy. Tabulka 12 ukazuje výskyt sVR v závislosti na rozsahu fibrózy jater.
Tabulka 12:_Výskyt SVR podle rozsahu fibrózy jater na počátku hodnocení C216
|
Rozsah fibrózy jater |
T12/PR |
Pbo/PR48 |
|
Dříve relabující | ||
|
Bez fibrózy, lehká fibróza nebj rortální fibróza |
84 % (68/81) |
32 % (12/38) |
|
Přemosťující fibróza |
86 % (31/36) |
13 % (2/15) |
|
82 % (23/28) |
7 % (1/15) | |
|
Dříve částečně odpovídající | ||
|
Bez fibrózy, lehká fbióza nebo portální fibróza |
79 % (19/24) |
18% (3/17) |
|
Přemosťující fibróza |
71 % (5/7) |
0 (0/5) |
|
33 % (6/18) |
20 % (1/5) | |
|
Tn'vc nereagující | ||
|
Eez fibrózy, lehká fibróza nebo portální fibróza |
31 % (9/29) |
6% (1/18) |
|
Přemosťující fibróza |
47 % (8/17) |
0 (0/9) |
|
19 % (5/26) |
10 % (1/10) |
T12/PR48: INCIVO 12 týdnů následováno placebem po dobu 4 týdnů v kombinaci s peginterferonem alfa-2a a ribavirinem po 48 týdnů;
Pbo/PR48: placebo 16 týdnů v kombinaci s peginterferonem alfa-2a a ribavirinem po dobu 48 týdnů
Tabulka 13 ukazuje podíly SVR u odpovědí v týdnu 4 (snížení HCV RNA < 1 logi0 nebo > 1 logi0) u dříve částečně odpovídajících a u dříve neodpovídajících ve skupině T12(DS)/PR.
|
Tabulka 13: Výskyt SVR v týd T12(DS)/PR48: Hodnocení C21 |
nu 4 (snížení HCV RNA < 1 logi0 nebo > 1 logi0) u skupiny 6 | |
|
Dřívější odpověď na léčbu |
T12(DS)/PR % (n/N)a | |
|
snížení HCV RNA < 1 log10 v týdnu 4 |
snížení HCV RNA > 1 log10 v týdnu 4 | |
|
Dříve částečně odpovídající |
56% (10/18) |
63 % (17/27) |
|
Dříve neodpovídající |
15 % (6/41) |
54 % (15/28) |
zahrnuje pouze údaje o pacientech, u kterých byla dostupná HCV RNA v týdnu 4
Hodnocení 106 a hodnocení 107 Hodnocení 106 bylo randomizované dvojitě zaslepené placebem kontrolované hodnocení fáze II u pacientů, u kterých selhala dřívější léčba peginterferonem alfa-2a a ribavirinem nebo peginterferonem alfa-2b a ribavirinem. Udříve relabujících pacientů ve skupině T12/PR24, kteří něli nedetekovatelnou HCV RNA (hladiny detekčního limitu nebylo dosaženo) v týdnech 4 a 12 léčby, byl výskyt SVR 89 % (25/28) a výskyt relapsů 7 %.
v kontroln
Hodnocení 107 bylo otevřené převrácené hodnocení u pacientů, kteří byli léčeni kontrolní skupině (placebo, peginterferon alfa-2a a ribavirin) fáze II telaprevirem a kteří v hodnocení ■ áze II nedosáhli SVR. U dříve relabujících pacientů ve skupině T12/PR24, kteří měli nedetekovatelnou HCV RNA (hladiny detekčního limitu nebylo dosaženo) v týdnech 4 a 12 léčby, byl ýsky SVR 100 % (24/24).

tevřeného randomizovaného
CIVO v kombinaci
Používání peginterferonu alfa-2a nebo 2b Oba typy peginterferonu alfa (2a a 2b) byly studovány ve fázi IIa hodnocení C208 u dosud neléčených pacientů.
Všichni pacienti dostávali 12 týdnů standardní léčbu přípravkem s peginterferonem alfa/ribavirinem. Pacienti byli randomizováni do 1 ze 4 skupin léčby:
- INCIVO 750 mg každých 8 hodin s peginterfeonem alfa-2a 180 pg/týden a ribavirinem 1 000 nebo 1 200 mg/den
- INCIVO 750 mg každých 8 hodin s peginteferonem alfa-2b 1,5 pg/kg/týden a ribavirinem 800 nebo 1 200 mg/den
- INCIVO 1 125 mg každých 12 ho^n s peginterferonem alfa-2a 180 pg/týden a ribavirinem 1 000 nebo 1 200 mg/den
- INCIVO 1 125 mg každých rodin speginterferonem alfa-2b 1,5 pg/kg/týden a ribavirinem 800 nebo 1 200 mg/den.
Peginterferon alfa-2a/peginte fer>n alfa-2b a ribavirin byly užity dle jejich souhrnů údajů o přípravku. Ve 12. týdnu skončilo podáván1 přípravku INCIVO a pacienti pokračovali pouze na standardní léčbě. V souhrnné skupině s peginte feronem alfa-2a dosáhlo kritérií [nedetekovatelná HCV RNA (hladiny detekčního limitu nebylo dosaženo) mezi týdny 4 a 20] pro zkrácené 24týdenní trvání léčby peginterferonem/ ioWirinem 73,8 % (59/80) pacientů oproti 61,7 % (50/81) pacientů v souhrnné skupině s peginterferonem alfa-2b.
|
Tabulka 14: Souhrnné výskyty odpovědí v hodnocení C208 | ||
|
^ ^4 * Výsledek léčby |
T12P(2a)R48 N = 80 (%) n/N |
T12P(2b)R48 N = 81 (%) n/N |
|
^SVRa |
83,8 (67/80) |
81,5 (66/81) |
|
Virový průlom (breakthrough) |
5 (4/80) |
12,3 (10/81) |
|
Relaps |
8,1 (6/74b) |
4,2 (3/71b) |
T12/P(2a)R48: INCIVO po dobu 12 týdnů v kombinaci s peginterferonem alfa-2a a ribavirinem po dobu 24 nebo 48 týdnů T12/P(2b)R48: INCIVO po dobu 12 týdnů v kombinaci s peginterferonem alfa-2b a ribavirinem po dobu 24 nebo 48 týdnů
95 % interval spolehlivosti pro rozdíl byl (-10,8; 12,1)
Jmenovatelem byl počet pacientů s nedetekovatelnou HCV RNA (hladiny detekčního limitu nebylo dosaženo) na konci léčby
Údaje o dlouhodobé účinnosti Hodnocení H2 (EXTEND)
Tříleté hodnocení následného sledování u pacientů, kteří dosáhli SVR při léčbě založené na přípravku INCIVO, ukázalo, že > 99 % (122/123) si udrželo SVR stav během doby následného sledování (medián trvání 22 měsíců).
Účinnost u dospělých infikovaných HCV/HIV-1 Hodnocení 110
Hodnocení 110 bylo randomizované dvojitě zaslepené placebem kontrolované hodnocení fáze II u pacientů s chronickou koinfekcí genotypem 1 HCV/HIV, u nichž dosud hepatitida C nebyla léčen; Pacienti buď nedostávali antiretrovirovou léčbu (počet CD4 buněk >500 buněk/mm3) nebo je u nic] HIV onemocnění stabilní (HIV RNA <50 kopií/ml, počet CD4 buněk > 300 buněk/mm3) léčbou efavirenzem nebo atazanavirem/ritonavirem v kombinaci s tenofovir-disoproxyl-fumarátem a emtricitabinem nebo lamivudinem. Pacienti byli randomizováni na 12 týdnů do skupiny léčené přípravkem INCIVO (750 mg každých 8 hodin při použití v kombinaci s atazanavirem/ritciavirem, tenofovir-disoproxyl-fumarátem a emtricitabinem nebo lamivudinem NEBO 1 125 mg každých 8 hodin při použití v kombinaci s efavirenzem, tenofovir-disoproxyl-fumarátem a smrr>itaDinem) nebo do skupiny s placebem. Všichni pacienti dostávali peginterferon alfa-2a a ribavrin po 48 týdnů. Padesát pět ze šedesáti pacientů dostávalo ribavirin ve fixní dávce 800 mg/den . zbývajících 5 pacientů dostávalo dávku ribavirinu závislou na tělesné hmotnosti. Ve skupině ii? PR48 měli na počátku léčby 3 pacienti (8 %) přemosťující fibrózu a 2 pacienti (5 %) cřióz-' Ve skupině Pbo/PR měli na počátku léčby přemosťující fibrózu 2 pacienti (9 % ) a žádný pacient neměl na počátku cirhózu. Tabulka 15 ukazuje výskyt odpovědí pro skupiny T12/PR48 a Pbo/PR48. Podíly odpovědí ve skupině Pbo/PR byly vyšší než podíly pozorované v jiných klinických hodnoceních s biterapií peginterferonem (historický podíl SVR < 36 %).
|
Tabulka 15: Výskyt odpovědí: Hodnocení 110 | ||
|
Výsledek léčby |
T12/PR48 % (n/N) |
Pbo/PR % (n/N) |
|
Celkový podíl SVR12a |
74 % (28/38) |
45 % (10/22) |
|
Pacienti s režimem založeným na 69 % (11/16) efavirenzu |
50 % (4/8) | |
|
Pacienti s režimem založeným na atazanaviru/ritonaviru |
80 % (12/15) |
50 % (4/8) |
|
Pacienti nedostávající antiretrovrovou léčbu |
71 % (5/7) |
33 % (2/6) |
T12//PR48: INCIVO po 12 týdnů s p’ginterferonem alfa-2a a ribavirinem po 48 týdnů; Pbo/PR: placebo po 12 týdnů s peginterferonem alfa-2a a n, avir nem po 48 týdnů a HCV RNA< 25 Iu/ml př kontrole ve 12. týdnu
Hodnocení HPC.008
Hodnocení HDC30b8 bylo otevřené hodnocení fáze IIIb u pacientů s chronickou koinfekcí HCV genotypu 1/H'V-1, kteří dosud na hepatitidu C nebyli léčeni nebo kteří při předchozí léčbě peginterferonem alfa (2a nebo 2b) a ribavirinem nedosáhli SVR (včetně pacientů s předchozím relapsem, pacientů s předchozí částečnou odpovědí a pacientů s předchozí nulovou odpovědí). Bylo požadováno, aby pacienti při screeningu měli HIV-1 RNA <50 kopií/ml a počty r,'
sv
IN
CD4 > 300 buněk/mm3. Pacienti dostávali přípravek INCIVO v dávkách 750 mg každých 8 hodin, ýjimkou pacientů, kteří byli léčeni režimem založeným na efavirenzu, kteří dostávali přípravek CIVO v dávkách 1 125 mg každých 8 hodin. Dosud neléčení pacienti nebo pacienti s předchozím relapsem, kteří netrpěli cirhózou a dosáhli rozšířené rychlé virologické odpovědi (eRVR), dostávali po 12 týdnů přípravek INCIVO plus peginterferon alfa-2a a ribavirin, po které následovalo 12 týdnů léčby peginterferonem alfa-2a a ribavirinem (celková doba léčby 24 týdnů). Dosud neléčení pacienti a pacienti s předchozím relapsem, kteří nedosáhli eRVR, pacienti s předchozí částečnou odpovědí a pacienti s předchozí nulovou odpovědí a všichni pacienti s cirhózou dostali po 12 týdnů přípravek INCIVO plus peginterferon alfa-2a a ribavirin, následovano 36 týdny léčby peginterferonem alfa-2a a ribavirinem (celková doba léčby 48 týdnů). Všichni pacienti dostávali ribavirin ve fixní dávce 800 mg/den. Režimy antiretrovirové léčby zahrnovaly efavirenz, atazanavir/ritonavir, raltegravir,
etravirin nebo darunavir/ritonavir v kombinaci s tenofovirem nebo abakavirem a buď lamivudinem nebo emtricitabinem.
Primárním cílem studie bylo vyhodnocení antivirové účinnosti přípravku INCIVO, peginterferonu alfa-2a a ribavirinu u pacientů koinfikovaných HCV/HIV-1, měřeno pomocí SVR12.
162 zařazených pacientů mělo medián věku 46 let (rozmezí: 20 až 67 let); 78,4 % pacientů byli muži;
6,8 % mělo index tělesné hmotnosti > 30 kg/m2; 4,3 % byli černosši; 1,9 % byli Asiaté; 87,0 % mělo výchozí hladiny HCV RNA > 800 000 IU/ml; 17,3 % mělo přemosťující fibrózu; 13,0 % mělo cirhózu; 65,6 % mělo HCV genotypu 1a; 33,8 % mělo HCV genotypu 1b; 39,5 % (n = 64) pacientů na HCV dosud neléčených; 17,9 % (n = 29) mělo předchozí relaps; 11,1 % (n = 18) byli pacienti s předchozí částečnou odpovědí; 31,5 % (n = 51) byli pacienti s předchozí nulovou odpovědí. Medán (rozmezí) výchozích hodnot počtu buněk CD4 byl 651 (277 až 1551 buněk/mm3).
Tabulka 16 ukazuje míry odpovědí u dosud neléčených pacientů a u pacientů se zkušen iosť s léčbou podle podskupin (dosud neléčení pacienti, pacienti s předchozím relapsem a pacienti s předchozí nulovou odpovědí na léčbu).
|
Tabulka 16: léčebné výsh koinfikovaných HIV-1 v |
;dky u dospělých pacientů s infekcí HCV genotypu 1 a íodnocení HPC3008 | ||
|
Pacienti se zkušeností s léčbou podle podskupin | |||
|
Léčebný výsledek |
Dosud neléčení pacienti N = 64 % (n/N) |
Pacienti s předchozím relapsem N =29 % (n/N) |
Pacienti bez předchozí odpovědi3 N = 69 % (n/N) |
|
SVR12 |
64,1 % (41/64) |
62,1 % (18/29) |
49,3 % (34/69) |
|
Nedetekovatelná HCV RNA (hladiny detekčního limitu nebylo dosaženo) ve 4. a 12. týdnu |
57,8 % (37/64) |
48,3 % (14/29) V |
42,0 % (29/69) |
|
SVR u pacientů s nedetekovatelnou HCV RNA (hladiny detekčního limitu nebylo dosaženo) v týdnech 4 a 12 |
83,8 % (31/37) |
92,9 % (13/14) |
89,7 % (26/29) |
|
SVR u pacientů, kteří neměli nedetekovatelnou HCV RNA (hladiny detekčního limitu nebylo dosaženo) v týdnech 4 a 12 |
37,0 % (10/27) |
33,3 % (5/15) |
20,0 % (8/40) |
|
pStu^eň SVR u pacientů s cirhózou nebo bez ní | |||
|
P; cienti bez cirhózy |
65,5 % (38/58) |
61,5 % (16/26) |
52,6 % (30/57) |
|
Pacienti s cirhózou |
50,0 % (3/6) |
66,7 % (2/3) |
33,3 % (4/12) |
|
Výsledky u pacientů bez SVR12 | |||
|
Virologické selhání během léčbyb |
21,9% (14/64) |
3,4 % (1/29) |
37,7 % (26/69) |
|
Relapsc |
8,9 % (4/45) |
5,3 % (1/19) |
8,1 % (3/37) |
|
Další3 |
7,8 % (5/64) |
31,0% (9/29) |
8,7 % (6/69) |
Příjemci transplantovaných jater
v<>?
VO, peginterferonu řeno pomocí
1,9 pacie
Hodnocení HPC3006 bylo otevřené hodnocení fáze IlIb u dosud neléčených a již léčených pacientů chronicky infikovaných genotypem HCV-1, kterým byla poprvé transplantována játra a kteří byli na stabilním režimu imunosupresiv takrolimu nebo cyklosporinu A. Žádný pacient netrpěl cirhózou transplantovaných jater. Pacienti dostávali přípravek INCIVO v dávce 750 mg každých 8 hodin Všichni pacienti zahajovali dávkou 600 mg/den ribavirinu a 180 pg/týden peginterferonu alfa -2a. Všichni pacienti dostávali po dobu 12 týdnů přípravek INCIVO plus peginterferon alfa 9a a ribavirin, následovalo 36 týdnů léčby peginterferonem alfa-2a a ribavirinem (celková doba léčby 48 +ýdnů).
mcí SVR12.
a
b
c
d
Podskupina pacientů bez předchozí odpovědi zahrnuje pacienty s předchozí částečnou odpovědí a pacienty s předchozí nulovou odpovědí.
Virologické selhání během léčby bylo definováno jako splnění pravidla pro ukončení léčby a/nebo pacienti, u kterých došlo k náhlému relapsu (viral breakthrough).
Relaps byl definován jako HCV RNA >25 IU/ml během následného pozorování po předchozím HCV RNA < 25 IU/ml na plánovaném konci léčby a nedosažení SVR.
Další zahrnuje pacienty s detekovatelnou HCV RNA při ukončení léčby, u kterých nedošlo k náhlému relapsu (viral breakthrough), a pacienty s chybějícím zhodnocením SVR během plánovaného následného sledování.
Primárním cílem studie bylo vyhodnocení antivirové účinnosti přípravku INCI alfa-2a a ribavirinu u pacientů infikovaných HCV po transplantaci jater, měř
74 zařazených pacientů mělo medián věku 56 let (rozmezí: 43 až 68 let); 91,9 % pacientů byli muži;
24,3 % mělo index tělesné hmotnosti > 30 kg/m2; 1,4 % byli černoši; 95,9 % mělo výchozí hladinu HCV RNA > 800 000 IU/ml; 10,8 % mělo přemosťující fibrózu; žádny neměl cirhózu; 38,9 % mělo HCV genotypu 1a; 58,3 % mělo HCV genotypu 1b; 2,8 % mělo HCV genotypu 1d; 21,6 % mělo IL28B genotypu CC; 54,1 % mělo IL28B genotypu CT; 24,3 % mělo IL28B genotypu TT; 28,4 %
(n = 21) bylo na HCV dosud neléčeno; 71,6 % (n = 53) již lepeno [14,9 % (n = 11) byli pacienti s relapsem; 40,5 % (n = 30) byli pacienti s předchozí nulovo' odpovědí; 16,2 % (n = 12) nemohlo být klasifikováno]; medián doby od transplantace jater byl 2,5 roku (rozmezí: 0,6 až 9,5 roku); 67,6 %
(n = 50) dostávalo takrolimus; 32,4 % (n = 24) doAávťo cyklosporin A.
Tabulka 17 ukazuje celkové míry odpovědí u dosud neléčených a již léčených pacientů s chronickou infekcí HCV genotypu 1 po transplantaci jater a podle podskupin (pacienti léčení takrolimem nebo cyklosporinem A).
b
c
Tabulka 17: léčebné výsled k
acientů infikovaných HCV genotypu 1 po transplantaci
|
Léčebný výsledek |
t Pacienti léčení takrolimem N = 50 % (n/N) |
Pacienti léčení cyklosporinem A N = 24 % (n/N) |
Všichni pacienti N = 74 % (n/N) |
|
SVR12 |
66 % (33/50) |
83 % (20/24) |
72 % (53/74) |
|
Výsledky u pacientů bez SVR |
12 | ||
|
Všichni pacienti | |||
|
V:rologické selhání během léčbya |
12 % (6/50) |
8 % (2/24) |
11 % (8/74) |
|
Relap? |
11 % (4/37) |
0 |
7 % (4/56) |
|
Dalšíc |
14 % (7/50) |
8 % (2/24) |
12 % (9/74) |
Virologické selhání během léčby bylo definováno jako splnění pravidla pro ukončení léčby nebo pacienti, u kterých došlo k náhlému relapsu (viral breakthrough). Virologická pravidla pro ukončení léčby z této analýzy léčby jsou skutečnými pravidly pro ukončení léčby, tj. pravidly odvozenými z dat o dispozici a expozici, oproti matematickým pravidlům pro ukončení, tj. odvozeným z údajů o HCV RNA.
Relaps byl definován jako detekovatelná HCV RNA v plasmě na plánovaném konci léčby po předchozích hodnotách HCV RNA < 25 IU/ml při plánovaném konci léčby HCV a nedosažení SVR12. Jmenovatelem je počet pacientů s HCV RNA < 25 IU/ml při plánovaném konci léčby nebo chybějící vyhodnocení HCV RNA při plánovaném konci léčby a HCV RNA < 25 IU/ml během následného pozorování na plánovaném konci léčby.
Zahrnuje pacienty s detekovatelnou HCV RNA při skutečném ukončení léčby, kteří však nesplňovali definici virologického selhání během léčby, a pacienty s chybějícím vyhodnocením HCV RNA během plánovaného následného sledování.
Klinická hodnocení sledující QT interval
Ve dvou dvojitě zaslepených randomizovaných placebem a aktivně kontrolovaných hodnoceních ke sledování vlivu na QT interval nebyla monoterapie telaprevirem v dávce 750 mg každých 8 hodin spojena s klinicky relevantním účinkem na QTcF interval. V jednom z těchto hodnocení byl posuzován režim podávání telapreviru 1 875 mg každých 8 hodin a největší nárůst QTcF, po korekci hodnot pro placebo, byl 8,0 ms (90 % CI: 5,1 - 10,9). Plazmatické koncentrace telapreviru podávaného v dávce 1 875 mg každých 8 hodin byly srovnatelné s koncentracemi pozorovanými v hodnocení u HCV infikovaných pacientů, kteří dostávali 750 mg každých 8 hodin v kombinaci s peginterferonem alfa-2a a ribavirinem.
Pediatrická populace
U pediatrických pacientů nebyla provedena žádná klinická hodnocení.

Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s přípravkem INCIVO u jedné nebo více podskupin pediatrické populace s chronickou heritiudoa (informace o použití u dětí viz bod 4.2).
h dobrov
5.2 Farmakokinetické vlastnosti
'&>
Farmakokinetické vlastnosti telapreviru byly hodnoceny u zdravých dospělých dobrovolníků a u pacientů s chronickou HCV infekcí. Telaprevir lze podávat perorálně s ídlC^. jako 375mg tablety,
1 125 mg dvakrát denně po dobu 12 týdnů v kombinaci s peginterferonem Jfa a ribavirinem. Případně lze telaprevir podávat perorálně sjídlem jako 375mg tablety, 750mg každých 8 hodin po dobu 12 týdnů v kombinaci s peginterferonem alfa a ribavirinem. Po podání s peginterferonem alfa a ribavirinem je expozice telapreviru vyšší než po podání jeho 'amotného.
ravděpo
Expozice telapreviru jsou srovantelné jak při podání pegin+erferonu alfa-2a a ribavirinu, tak i peginterferonu alfa-2b a ribavirinu.
Absorpce
Telaprevir je dostupný po perorálním po pro vstřebávání v tlustém střevě nejsou důka

pravděpodobně se absorbuje v tenkém střevě, zatímco aximálních koncentrací v plazmě po podání jednotlivé dávky telapreviru je většinou dosaženo po 4-5 hodinách. In vitro studie provedené s lidskými Caco-2 buňkami ukazují, že telaprevir je substrátem P-glykoproteinu (P-gp).
Expopzice telapreviru byly prdobm bez ohledu na to, zda byla podána celková denní dávka 2 250 mg jako 750 mg každých 8 ho^n nebo 1 125 mg dvakrát denně.Na základě populačního farmakokinetického modelu expozic telapreviru v rovnovážném stavu byly poměry geometrických průměrů nejmenších čtverců (90 % interval spolehlivosti) u 1 125 mg dvakrát denně oproti 750 mg každých 8 hodin 1,08 (1,02; 1,13) pro AUC24,ss; 0,878 (0,827; 0,930) pro Qrough,ss a 1,18 (1,12; 1,24)
pro Cmax,ss
Při podán' po kalorickém jídle s vysokým obsahem tuků (56 g tuku, 928 kcal) byla zvýšena expozice telaprev;ru o 20 % ve srovnání s podáním spolu se standardně kalorickým jídlem (21 g tuku,
:0a1). Ve srovnání s podáním po standardně kalorickém jídle byla expozice (AUC) telapreviru při alačno snížena o 73 %, při podání s nízkokalorickým jídlem bohatým na bílkoviny (9 g tuku, cal) o 26 % a při podání s nízkokalorickým nízkotučným jídlem (3,6 g tuku, 249 kcal) o 39 %. laprevir je tedy nutno užívat sjídlem.
Distribuce
Telaprevir se váže na bílkoviny krevní plazmy přibližně z 59 % - 76 %. Telaprevir se primárně váže na kyselý alfa-1 protein a albumin.
Po perorálním podání byl typický zdánlivý distribuční objem (Vd) odhadnutý na 252 l s interindividuální variabilitou 72,2 %.
Biotransformace
Telaprevir je metabolizován převážně v játrech, včetně hydrolýzy, oxidace a redukce. Ve stolici, plazmě a moči byly nalezeny různé metabolity. Po opakovaném perorálním podání byly nalezeny tyto hlavní metabolity: R-diastereomer telaprenaviru (30x méně účinný), kyselina pyrazinová a metabolit vzniklý redukcí na a-ketoamidové vazbě (neaktivní).
CYP3A4 je částečně odpovědný za metabolismus telapreviru. Metabolismu se účastní i další enzymy jako aldo-ketoreduktázy a další proteolytické enzymy. Studie s rekombinantními lidskými supresory CYP ukázaly, že telaprevir je inhibitor CYP3A4 a v lidských jaterních mikrozomech byla pozorována na čase a koncentraci závislá inhibice CYP3A4. In vitro nebyly pozorovány relevantní inhibice CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 a CYP2E1. In vitro nebyly pozorovány u telapreviru žádné relevantní indukce CYP1A2, CYP2B6, CYP2C a CYP3A isozomr Na základě klinických hodnocení interakcí léčivo - léčivo (např. escitalopram, zolpidem, ethinylestradiol) nelze vyloučit indukci metabolických enzymů telaprevirem. „<0
nu. In vitro nebyla dovány
In vitro studie prokázaly, že telaprevir není inhibitorem UGT1A9 nebo UGT2B7. In vitro studie s rekombinantním UGT1A3 ukazují, že telaprevir může inhibovat tento enzym. Klin>i-.á "elevance tohoto zjištění není jistá, protože podání telapreviru s jednotlivou dávkou buprenorfinu, částečného substrátu UGT1A3, zdravým dobrovolníkům nevedlo ke zvýšení expozic bupr pozorována relevantní inhibice alkoholdehydrogenázy telaprevirem. Nebyl dostatečně vysoké koncentrace pro vyloučení inhibice ve střevě.
torem O
V lidských hepatocytech in vitro byla porozována suprese telaprevirem a VRT-127394 enzymů CYP které jsou regulované přes nukleární receptory CAR, PXR a Ah. Klinické studie interakcí lék-lék se substráty CYP2B6, CYP2C8, CYP2D6, CYP2C19 a UGTlAi, UGT2B7 a UGT1A3 neukázaly na klinicky relevantní vliv suprese in vitro. Klinické působen ostatních enzymů a přenašečů (např. CYP1A1, CYP1A3, BCRP, OATPs), které jsou regulovaiK stejnými nukleárními receptory, není známo.
Transportéry
In vitro studie prokázaly, že telaprevir je nh bitorem OATP1B1 a OATP2B1.
In vitro nebyla pozorována inhibice tel^eim organickými kationtovými přenašeči (OCT) OCT2.
,'<9
Telaprevir je slabý in vitro inhibitor pienašečů vícelékové a toxinové extruze (MATE) MATE-1 a MATE2-K s IC50 28,3 pM, respektive 32,5 pM. Klinický význam tohoto zjištění není v současné době znám.
Eliminace
Po podání jednorázové perorální dávky 750 mg 14C-telapreviru zdravým dobrovolníkům se 90 % celkové radioaktivity objevilo ve stolici a moči a vymizelo během 96 hodin po podání. Medián zachycené radioaktivity byl 82 % ve stolici, 9 % ve vydechovaném vzduchu a 1 % v moči. Podíl nezměněného 4C-telapreviru z celkové radioaktivity byl 31,8 % a u VRT-127394 18,7 %.

Po perorálním podání byla celková clearance (Cl/F) stanovena na 32,4 l/hod s interindividuální variabilitou 27,2 %. Průměrný eliminační poločas po jednorázovém perorálním podání 750 mg reviru byl většinou v rozmezí 4,0 až 4,7 hodiny. V rovnovážném stavu je efektivní poločas 9 -hodin.
Linearita/nelinearita
Expozice (AUC) telapreviru se po jednorázovém podání 375 - 1 875 mg telapreviru spolu s jídlem zvyšovala o něco více, než by bylo úměrné dávce, pravděpodobně vzhledem k saturaci metabolických cest nebo efluxních transportérů.
Zvláštní populace
Pediatrická populace
Údaje pro pediatrickou populaci nejsou zatím dostupné.
Porucha funkce ledvin
Farmakokinetika telapreviru byla hodnocena po podání jednotlivé dávky 750 mg HCV-negativním subjektům s těžkou poruchou funkce ledvin (CrCl <30 ml/min). Průměrná Cmax byla o 10 % vyšší a průměrná AUC byla o 21 % větší než u zdravých subjektů (viz bod 4.2).
Porucha funkce jater
Telaprevir je primárně metabolizován v játrech. Expozice telapreviru v rovnovážném stavu byla u subjektů s mírnou poruchou funkce jater (Child-Pugh třídy A, skóre 5-6) o 15 % nižší ve srovn; se zdravými subjekty. Expozice telapreviru v rovnovážném stavu byla u subjektů se středně těž poruchou funkce jater (Child-Pugh třídy B, skóre 7-9) o 46 % nižší ve srovnání se zdravým' subjekty. Účinek na koncentrace nevázaného telapreviru není znám (viz body 4.2 a 4.4).
a
ovnání
kou
$>
Pohlaví
).
opulač
Vliv pohlaví na farmakokinetiku telapreviru byl hodnocen prostřednictvím údajů o p farmakokinetice z hodnocení fází II a III přípravku INCIVO. Nebyl identifikov.n vznamný vliv pohlaví.
i o populační
erJ
Rasa
Analýza populační farmakokinetiky přípravku INCIVO u HCV-infikovaných subjektů naznačuje, že expozice telapreviru byla podobná u černochů/afroameričanů a bělochů.
Starší osoby
Pro užití přípravku INCIVO u pacientů ve věku > 65 let je omezené množství farmakokinetických údajů a pro pacienty ve věku > 70 let nejsou žádné údaje.
1/
5.3 Předklinické údaje vztahující se k bezpexnoS(i
Toxicita a/nebo farmakologie u zvířat
U potkanů a psů byl telaprevir spojen s reverzibilním snížením parametrů červených krvinek doprovázeným regenerační odpovědí. Tak u potkanů tak i psů byla ve většině studií pozorována zvýšení AST/ALT, zvýšená ALT u potkanů se po zotavení nenormalizovala. Histopatologické nálezy v játrech byly podobné jak ve studiích u potkanů tak i psů; po zotavení se nenormalizovaly všechny nálezy. U potkanů (ne však u psů) způsoboval telaprevir degenerativní změny na varlatech, které byly reverzibilní a neovlivňovaly plodnost. Obecně byly hodnoty expozice ve vztahu k hodnotám u člověka ve studiích farmakologie a toxikologie u zvířat nízké.
Kancerogeneze a muagneze
Kancerogenn' potenciál telapreviru nebyl testován. Ani telaprevir ani jeho hlavní metabolity nezpůsobovaly poškození DNA ve standardním souboru testů na stanovení mutageneze v přítomnosti i nepřítomnosti aktivace metabolismu.

plodnosti
oumání na potkanech neměl telaprevir vliv na plodnost nebo rozmnožovací schopnost.
mbryofetální vývoj
Telaprevir přestupuje placentu jak u potkanů tak i myší s poměrem fetální/maternální expozice 19 -50 %. Telaprevir neměl u potkanů a myší teratogenní potenciál. Ve studiích fertility a časného embryonálního vývoje u potkanů bylo pozorováno zvýšení počtu neživotaschopných zárodků. Dávkování u zvířat nevedlo ke stanovení rozpětí expozice při srovnání s expozicí u lidí.
normální porodní hmotnost. Při krmení mlékem samic potkanů léčených telaprevirem byly však přírůstky na hmotnosti mláďat nižší než normálně (vzhledem k odporné chuti). Po odstavení se hmotnost mláďat vrátila k normálu.
6. FARMACEUTICKÉ ÚDAJE 6.1 Seznam pomocných látek

Jádro tablety
acetátosukcinát hypromelosy hydrogenfosforečnan vápenatý mikrokrystalická celulosa koloidní bezvodý oxid křemičitý natrium-lauryl-sulfát sodná sůl kroskarmelosy natrium-stearyl-fumarát
Potah tablety polyvinylalkohol makrogol mastek
oxid titaničitý (E171) žlutý oxid železitý (E172)
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
2 roky
6.4 Zvláštní opatření pro uch
Uchovávejte v původní lahvičce. Lfnvičku uchovávejte dobře uzavřenou, aby byl přípravek chráněn před vlhkostí. Neodstraňme vysoušedlo.
6.5 Druh obal u a obsah ba lení
Lahvička z polyethylenu o vysoké hustotě (HDPE) obsahující 42 potahovaných tablet uzavřená dětským bezpečnostním uzávěrem z polypropylenu (PP) s indikátorem neporušenosti. Přibaleno je vysoušedlo (jeden nebo dva sáčky).
Přípravek INCIVO je dostupný v baleních obsahujících 1 lahvičku (celkem 42 potahovaných tablet) ne^o 4 lahvičky (celkem 168 tablet).
N
trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku
Žádné zvláštní požadavky.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Janssen Cilag International NV Tumhoutseweg 30 B-2340 Beerse Belgie
8. REGISTRAČNÍ CÍSLO(A)
EU/1/11/720/001 balení se 4 lahvičkami EU/1/11/720/002 balení s 1 lahvičkou
9. DATUM PRVNÍ REGISTRACE/PRODLOUZENÍ REGISTRACE
&
&
Datum první registrace: 19. září 2011
10. DATUM REVIZE TEXTU
.v®
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A
95 % CI
Jd
: odpovídajících a 26 (13, 39)
&
£>
A.
B.
C.
D.
<0
VÝROBCE ODPOVĚDNÝ ZA pr°POUŠTĚNÍ ŠARŽÍ PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
v,
další podmínky a požadavky registrace
PŘÍLOHA II
PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU

A. VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného/výrobců odpovědných za propouštění šarží
Janssen-Cilag SpA Via C. Janssen IT-04100 Borgo San Michele Latina, Itálie
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ *
v
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2). O
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
o oezpe
s>
• Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci dále předkládá pravidelně aktualizované zprávy o Bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dV Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D.
BEZP
PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A UCINNE POUŽÍVANÍ LÉČIVÉHO PŘÍPRAVKU

• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP upečeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích

Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik),
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
• Další opatření k minimalizaci rizik
Držitel rozhodnutí o registraci odsouhlasí s členským státem před uvedením přípravku na trh formát a obsah vzdělávacího balíčku pro zdravotnické pracovníky.
•n
Držitel rozhodnutí o registraci zajistí, aby všichni lékaři, u kterých se očekává, že budou předpisovat ebo používat INCIVO, obdrželi vzdělávací balíček pro zdravotnické pracovníky, který obsahuje ásledující:
Souhrn údajů o přípravku;
Příbalovou informaci pro pacienta;
Informaci pro lékaře.
Informace pro lékaře bude obsahovat dále uvedené základní prvky:
• Údaje o bezpečnosti z fází II a III vztahující se k vyrážce a závažným kožním nežádoucím účinkům;
• Výskyt vyrážky a závažných kožních nežádoucích účinků;
Klasifikaci a postupy pro zvládnutí vyrážky a závažných kožních rekací zejména s ohledem na kritéria pro pokračovaní léčby telaprevirem a dalšími složkami, nebo její ukončení;
Obrázky vyrážky podle jednotlivých stupňů závažnosti.



1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
INCIVO 375 mg potahované tablety telaprevirum
|
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK |
-A'- | |
|
Jedna potahovaná tableta obsahuje telaprevirum 375 mg |
<£° | |
|
3. SEZNAM POMOCNÝCH LÁTEK |
<? | |
|
Obsahuje sodík. Další podrobnosti viz příbalová informace. |
<<£ ■o |
!> |
|
4. LÉKOVÁ FORMA A OBSAH BALENÍ | ||
|
42 potahovaných tablet | ||
|
5. ZPŮSOB A CESTA/CESTY PODÁNÍ | ||
Před použitím si přečtěte příbalovou info: Perorální podání.
Tablety polykejte celé.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7
DALŠÍ ZVLÁŠ
VLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
r8. POUŽITELNOST
P oužitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Janssen Cilag International NV Tumhoutseweg 30 B-2340 Beerse Belgie
|
12. |
REGISTRAČNÍ ČÍSLO/ČÍSLA | ||
|
EU/1/11/720/002 | |||
|
13. |
ČÍSLO ŠARŽE |
<V | |
|
č.š.: |
<&> | ||
|
14. |
KLASIFIKACE PRO VÝDEJ | ||
|
Výdej léčivého přípravku vázán na lékařský předpis. JA |
~č& ^ | ||
|
15. |
NÁVOD K POUŽITÍ |
nnJ |
/ |
|
16. |
INFORMACE V BRAILLOVĚ pÍSM |
U | |

|
ÚDAJE UVÁDĚNÉ NA VNITŘNÍM OBALU | |
|
ŠTÍTEK (balení s 1 lahvičkou) | |
|
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU | |
|
INCIVO 375 mg potahované tablety telaprevirum |
v> |
|
2. OBSAH LÉČIVÉ LÁTKY/lÉČIVÝCH LÁTEK | |
|
Jedna potahovaná tableta obsahuje telaprevirum 375 mg |
■é° |
|
3. SEZNAM POMOCNÝCH LÁTEK |
■sp |
|
Obsahuje sodík. | |
|
4. LÉKOVÁ FORMA A OBSAH BALENÍ |
A |
|
42 potahovaných tablet __ | |
|
5. ZPŮSOB A CESTA/CESTY PODÁNÍ | |
Před použitím si přečtěte příbalovou informaci Perorální podání.
Tablety polykejte celé. .GV
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVY PŘÍPRAVEK MUSÍ BYT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo
.1
dohled a dosa
osah dětí.
:vl-Á
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
DALŠÍ ZV
ř“dn necd;
straňujte.
r
8. POUŽITELNOST
E XP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Janssen Cilag International NV Tumhoutseweg 30 B-2340 Beerse Belgie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
|
EU/1/11/720/002 |
•A | |
|
13. ČÍSLO ŠARŽE |
<V | |
|
Lot |
<&> JO | |
|
14. KLASIFIKACE PRO VÝDEJ | ||
|
Výdej léčivého přípravku vázán na lékařský předpis. JA |
~č& ^ | |
|
15. NÁVOD K POUŽITÍ |
nnJ |
/ |
|
^ 4 | ||
|
16. INFORMACE V BRAILLOVĚ pÍSM |
U | |

INCIVO 375 mg potahované tablety telaprevirum
|
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK | ||
|
Jedna potahovaná tableta obsahuje telaprevirum 375 mg |
r$° | |
|
3. SEZNAM POMOCNÝCH LÁTEK |
< |
<? |
|
Obsahuje sodík. Další podrobnosti viz příbalová informace. |
<& JO |
!> |
|
4. LÉKOVÁ FORMA A OBSAH BALENÍ |
<N | |
|
Vy 168 potahovaných tablet (4 lahvičky, každá obsahuje 42 tablet) Jednotlivé lahvičky nelze distribuovat sA/ | ||
|
5. ZPŮSOB A CESTA/CESTY PODÁNÍ | ||
|
Před použitím si přečtěte příbalovou mf^mah. Perorální podání. Tablety polykejte celé. r V r V ř V r ss r V r r r r r | ||
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DAŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, pokud je POTŘEBNÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
L-
k;
POUŽITELNOST
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
A
|
12. |
REGISTRAČNÍ ČÍSLO/ČÍSLA |
/O |
|
EU/1/11/720/001 | ||
|
13. |
ČÍSLO ŠARŽE | |
|
č.š.: |
<y | |
|
14. |
KLASIFIKACE PRO VÝDEJ |
__ |
|
Výdej léčivého přípravku vázán na lékařský předpis. |
<r / | |
|
15. |
NÁVOD K POUŽITÍ | |
|
16. |
INFORMACE V BRAILLO^Ě PÍSMU | |
incivo 375 mg
í;
Janssen Cilag International NV Tumhoutseweg 30 B-2340 Beerse Belgie
v v*'
|
ÚDAJE UVÁDĚNÉ NA VNITŘNÍM OBALU | |
|
KRABIČKA (balení se 4 lahvičkami) | |
|
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU | |
|
INCIVO 375 mg potahované tablety telaprevirum |
v> |
|
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK | |
|
Jedna potahovaná tableta obsahuje telaprevirum 375 mg |
■é° |
|
3. SEZNAM POMOCNÝCH LÁTEK |
■sp |
|
Obsahuje sodík. | |
|
4. LÉKOVÁ FORMA A OBSAH BALENÍ |
A |
|
42 potahovaných tablet __ | |
|
5. ZPŮSOB A CESTA/CESTY PODÁNÍ | |
Před použitím si přečtěte příbalovou informaci Perorální podání.
Tablety polykejte celé. .GV
|
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ | |
|
A Uchovávejte mimo doh.ed . dosah dětí. <Y | |
|
7. DALŠÍ ZVláŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ | |
|
' 8. POUŽITELNOST | |
|
\/XP | |
|
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ | |
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Janssen Cilag International NV Tumhoutseweg 30 B-2340 Beerse Belgie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
|
EU/1/11/720/001 |
•A | |
|
13. ČÍSLO ŠARŽE |
<V | |
|
Lot |
<&> JO | |
|
14. KLASIFIKACE PRO VÝDEJ | ||
|
Výdej léčivého přípravku vázán na lékařský předpis. JA |
~č& ^ | |
|
15. NÁVOD K POUŽITÍ |
nnJ |
/ |
|
^ 4 | ||
|
16. INFORMACE V BRAILLOVĚ pÍSM |
U | |


Příbalová informace: informace pro uživatele
INCIVO 375 mg potahované tablety
telaprevirum
'V Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl ly jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1.
2.
3.
4.
5.
6.
1.
t
Co je INCIVO a k čemu se používá
Čemu musíte věnovat pozornost, než začnete INCIVO užívat Jak se INCIVO užívá Možné nežádoucí účinky Jak INCIVO uchovávat Obsah balení a další informace
Co je INCIVO a k čemu se používá
INCIVO účinkuje proti viru, který způso infekce hepatitidou C u dospělých paci

patitid
hepatródu C (zánět jater), a používá se k léčbě chronické 'e věku 18-65 let) v kombinaci s peginterferonem alfa a ribavirinem. INCIVO obsahuje látku zvanoi telaprevir a patřící do skupiny léčiv zvaných „inhibitory NS3-4A proteázy“. Inhibitory NS3 4a proteázy snižují množství viru hepatitidy C ve Vašem těle. INCIVO nelze užívat samotné, musí se užívat v kombinaci s peginterferonem alfa a ribavirinem, aby se zajistil účinek léčby. INCrVO mohou užívat pacienti s infekcí chronickou hepatitidou C, kteří dosud nebyli léčeni, nebo jej mohou užívat pacienti s infekcí chronickou hepatitidou C, kteří byli dříve léčeni režimem založeným na interferonu.
2.
Čemu musíte věnovat pozornost, než začnete INCIVO užívat
Neužívejte iN^IVO, jestliže jste alergický(á) na telaprevir nebo na kteroukoli další složku tohoto přípravku 1 uvedenou v bodě 6).

m k tomu, že INCIVO je nutno užívat v kombinaci s peginterferonem alfa a ribavirinem, tudujte také jejich kontraindikace (např. opatření pro zamezení těhotenství pro muže a ženy) příbalových informacích peginterferonu alfa a ribavirinu. Nejste-li si jist(a) jakoukoli kontraindikací zmíněnou v příbalových informacích, poraďte se s lékařem.
Neužívejte INCIVO v kombinaci s žádným z dále uvedených léčivých přípravků, protože to může zvýšit riziko závažných nežádoucích účinků a/nebo ovlivnit způsob, jakým INCIVO nebo další léčivý přípravek účinkuje:
|
Léčivý přípravek (název léčivé látky) |
Účinek přípravku |
|
alfuzosin |
k léčbě příznaků zvětšené prostaty (antagonista alfa-1-adrenergních receptorů) |
|
amiodaron, bepridil, chinidin, další antiarytmika třídy Ia nebo III |
k léčbě určitých onemocnění srdce jako je nepravidelný tlukot (antiarytmika) |
|
astemizol, terfenadin |
k léčbě příznaků alergie (antihistaminika) |
|
rifampicin |
k léčbě některých infekcí jako je tuberkulóza (antituberkulotika) V |
|
dihydroergotamin, ergometrin, ergotamin, methylergometrin |
k léčbě migrény a bolesti hlavy (námelové alkaloidy) |
|
cisaprid |
k léčbě některých žaludečních obtíží (k ovlivnění pohybu v trávicích cestách) |
|
třezalka tečkovaná (Hypericum perforatum) |
rostlinný přípravek k úlevě od úzkosti t\£) ^ |
|
atorvastatin, lovastatin, simvastatin |
ke snížení hladin cholesterolu (inhibitory HMG CoA reduktázy) |
|
pimozid |
k léčbě psychiatrických obtíží (neuroleptika) |
|
sildenafil, tadalafil |
Sildeiaíi1 nebo tadalafil se nesmějí užívat k léčbě onemocnění srdce a plic zvaného plicní ateriální hypertenze. Existují i jiné stavy léčené sldenafilem a tadalafilem. Viz bod Další léčivé přípravky a INCIVO. |
|
kvetiapin |
k léčbě schizofrenie, bipolární poruchy a závažné depresivní poruchy |
|
midazolam (užitý ústy), triazolam (už tý ústy) |
k usnadnění spánku a/nebo úlevě od úzkosti (sedativa/hypnotika) |
|
karbamazepin, fenobarb tal, penytoin _JAV_ |
k léčbě epileptických záchvatů (antiepileptika) |
INCIVO je nutno užívat v kombinaci s peginterferonem alfa a ribavirinem. Proto je velmi důležité, abyste si také přečetl(a) příbalové informace dodávané s těmito léčivými přípravky. Máte-li jakékoli tázky k Vašim léčivým přípravkům, zeptejte se svého lékaře nebo lékárníka.
Užíváte-li cokoli z výše uvedeného, domluvte se s lékařem na změně na jiný léčivý přípravek.
hX
Upozornění a opatření
Před už'tím příiravku INCIVO se poraďte se svým lékařem nebo lékárníkem.
o
Ujistěte se, že jste zkontroloval(a) všechny dále uvedené body a týká-li se Vás cokoli z dále uvedeného, oznamte to lékaři, který Vám léčí virus hepatitidy C (HCV).
Kožní vyrážka
U pacientů užívajících INCIVO se může objevit kožní vyrážka. Vyrážka může svědit. Vyrážka je většinou slabá/mírná nebo středně závažná, ale může být také závažná a/nebo život ohrožující nebo se může v závažnou a/nebo život ohrožující změnit. Pokud se u Vás nově objeví vyrážka nebo se vyrážka zhorší, kontaktujte okamžitě svého lékaře. Nesmíte začít znovu užívat
přípravek INCIVO, je-li Vaším lékařem vysazen. Musíte si pečlivě přečíst informace o vyrážce v bodu 4 Možné nežádoucí účinky.
Anemie (snížení počtu červených krvinek)
Oznamte svému lékaři, pokud se u Vás objeví únava, slabost, dušnost, točení hlavy a/nebo pocit rychlého tlukotu srdce. Může se jednat o příznaky anemie.
Srdeční problémy
Oznamte lékaři, pokud trpíte srdečním selháním, nepravidelným srdečním rytmem, pomalým srdečním rytmem, abnormalitou ukazující se na EKG (vyšetření srdce) zvanou „syndrom prodlouženého QT“ nebo máte v rodině onemocnění zvané „vrozený QT syndrom“.
Lékař může během léčby přípravkem INCIVO požadovat další vyšetření.
.P~znaky
Porucha jater
Oznamte svému lékaři, pokud jste kdykoli měl(a) problémy s játry jako jaterní selhání. mohou zahrnovat zežloutnutí kůže nebo očí (žloutenka), hromadění tekutiny v břiše (ascites) nebo otoky dolních končetin a krvácení ze zduřených žil (varixy) v jícnu. Psed rozhodnutím o zahájení léčby přípravkem INCIVO lékař vyhodnotí, jak jsou Vaše problémy s játry závažné.
Infekce
Oznamte lékaři, pokud máte infekci hepatitidou B, aby mohl lékai INCIVO vhodné.

out, zda je pro Vás
Transplantace orgánů
Oznamte lékaři, pokud jste proděl(a) transplantaci jate protože INCIVO v této situaci nemusí být pro Vás
idelně

ř rozh
bo jiného orgánu nebo na ni čekáte,
Krevní testy
Před zahájením léčby a během ní bude lékař pravideliě provádět krevní testy:
- aby viděl, kolik je ve Vaší krvi viru a určil, zd^ máte typ viru (genotyp 1), který lze léčit přípravkem INCIVO. Na základě těch to testů lze provést rozhodnutí o léčbě. Lékař bude sledovat Vaši časnou odpověď na léču a kolik viru máte v krvi. Pokud léčba neúčinkuje, může lékař léčivý přípravek vysadit. Vysadí-li lékař INCIVO, nesmí se začít užívat znovu.
- aby zkontroloval, zda máte anemii (snížení počtu červených krvinek).
- aby zkontroloval změny, ke kte^m dochází v počtu krvinek nebo složení krve. To lze poznat z výsledků krevních term. Lékař Vám to vysvětlí. Příklady jsou: krevní obraz, hladiny hormonu
štítné žlázy (žláza INCIVO bylo užito pouze u
ciku, která kontroluje metabolismus), jaterní a ledvinové testy.
ezeného množství pacientů starších 65 let. Patříte-li do této věkové přípravku INCIVO se svým lékařem.
d
skupiny, poraďte se o u Děti a dospívající
INCIVO není u dětí a dospívajících, protože u pacientů do 18 let věku nebylo
dostatečně studováno.
Jry
Da'ší léčivé přípravky a INCIVO
INCIVO může ovlivňovat jiné léčivé přípravky nebo jiné léčivé přípravky mohou ovlivňovat CIVO. Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Informujte svého lékaře, pokud užíváte kterýkoli z dále uvedených léčivých přípravků:
|
Léčivý přípravek (název léčivé látky) |
Účinek přípravku |
|
flekainid, propafenon |
k léčbě některých poruch srdce jako je nepravidlený tlukot srdce (antiarytmika) |
|
alfentanil, fentanyl |
k léčbě bolesti (analgetika) nebo během chirurgického zákroku k navození spánku |
|
digoxin, intravenózní lidokain |
k léčbě některých poruch srdce jako je nenormální tlukot srdce (antiarytmika) |
|
klarithromycin, erythromycin, telithromycin, troleandomycin |
k léčbě bakteriálních infekcí (antibiotika) |
|
warfarin, dabigatran |
k prevenci srážení krve (antikoagulancia) |
|
escitalopram, trazodon |
k léčbě poruch nálady (antidepresiva) |
|
metformin |
k léčbě cukrovky (antidiabetika) |
|
domperidon |
k léčbě zvracení a nevolnosti (antiemetika) |
|
itrakonazol, ketokonazol, posakonazol, vorikonazol |
k léčbě plísňových infekcí (antimykotika) 4 |
|
kolchicin |
k léčbě zánětlivé artritidy (antiuratika) |
|
rifabutin |
k léčbě některých infekcí (antituberkulotika) |
|
alprazolam, midazolam podávaný injekčně |
k podpoře spánku a/nebo úlevě od úzkost’ (benzodiazepiny) |
|
zolpidem |
k podpoře spánku a/nebo úlevě od ^zk^sti (ne-benzodiazepinová sedativa) |
|
amlodipin, diltiazem, felodipin, nikardipin, nifedipin, nisoldipin, verapamil |
ke snížení tlaku krve (blokátory kalmových kanálů) |
|
maravirok |
k léčbě HIV infekce (ar+"gouíst. CCR5) |
|
budesonid, inhalační/nasální flutikason, dexamethason užívaný ústy nebo injekčně |
k léčbě astmatu nebo z'nět ivych a autoimunitních onemocnění (kortikosteroidy) |
|
bosentan |
k léčbě onemocnění srdce a plic zvaného plicní arteriální hypeiťnze (antagonisté endoteliro.-vch receptorů) |
|
atazanavir/ritonavir, darunavir/ritonavir, fosamprenavir/ritonavir, lopinavir/ritonavir |
k léčbě HIV nfekce (inhibitory HIV-proteázy) |
|
abakavir, efavirenz, ^ tenofovir-disoproxyl-fumarát, zidovudin |
k léčbě HIV infekce (inhibitory reverzní t anskriptázy) |
|
fluvastatin, pitavastatin, pravastatin, rosuvastatn |
ke snížení hladin cholesterolu (inhibitory HMG CoA reduktázy) |
|
všechny typy hormonální antikoncepce |
hormonální kontraceptiva |
|
léčivé přípravky na základě estrogenů |
hormonální substituční léčba |
|
cyklosporin, sirolimus, takrolimus < Ty |
k potlačení imunitního systému (imunosupresiva), léčivé přípravky užívané u některých revmatických chorob nebo k zabránění problémům s transplantovanými orgány |
|
salmeterol |
ke zlepšení dýchání u astmatu (inhalační beta mimetika) |
|
repaglinid |
k léčbě cukrovky typu II (antidiabetikum) |
|
methadon |
k léčbě závislosti na opiátech |
|
sildenafil, taoalafil, vardenafil _ |
k léčbě erektilní dysfunkce nebo k léčbě onemocnění srdce a plic zvaného plicní arteriální hypertenze (inhibitory PDE-5) |
i
s>
INCIVO s jídlem a pitím
CIVO je nutno vždy užívat spolu s jídlem. Jídlo je důležité, aby se do Vašeho těla dostaly správné ladiny léku.
Těhotenství a kojení
Jste-li těhotná, nesmíte INCIVO užívat. INCIVO je nutno užívat v kombinaci s peginterferonem alfa a ribavirinem. Ribavirin může poškodit Vaše nenarozené dítě. Je proto nezbytně nutné, abyste během této léčby dodržovala veškerá opatření k zabránění početí.
Pokud otěhotníte Vy nebo Vaše partnerka během užívání přípravku INCIVO nebo měsíc po jeho ukočení, je nutno okamžitě navštívit lékaře (viz bod „Opatření k zabránění těhotenství pro muže a ženy“ níže).
Pokud kojíte, musíte před zahájením užívání přípravku INCIVO přestat s kojením. Není známo, zda se telaprevir, léčivá látka přípravku INCIVO, nachází v mateřském mléce.
Poraďte se se svým lékařem nebo lékárníkem dříve, než začnete užívat jakýkoliv lék.
&
em
Opatření k zabránění těhotenství pro muže a ženy
Protože INCIVO je nutno užívat v kombinaci s ribavirinem a ribavirin může být velmi škodlivý pro nenarozené dítě, musejí pacientky i pacienti dbát zvláštních opatření k zabránění těhotenství.
Jakákoli metoda kontroly početí může selhat, proto musíte Vy i Váš partner během léčby přípravk INCIVO a po ní užívat alespoň dvě účinné metody k zabránění početí. Po ukončení léčby přípravkem INCIVO se při pokračování antikoncepčních opatření řiďte příbalovou informací ribavirin-.
/V
Pacientky v plodném věku a leiich partneři
Hormonální antikoncepce nemusí být během užívání přípravku INCIVO spolehli vá. Proto musíte Vy a Váš partner během užívání přípravku INCIVO a 2 měsíce po něm užívat dvě jiié mžody kontroly početí.
Přečtěte si příbalové informace peginterferonu alfa a ribavirinu, kde naleznete další informace.
Řízení dopravních prostředků a obsluha strojů
Někteří pacienti mohou omdlévat nebo mít během léčby přípravkem INCIVO problémy se zrakem. Neřiďte nebo neobsluhujte stroje, pokud je Vám během užíván1 přípravku INCIVO na omdlení nebo máte problémy se zrakem. Další informace naleznete v piíbalových informacích peginterferonu alfa a ribavirinu.
INCIVO obsahuje sodík
Tento léčivý přípravek obsahuje 2,3 mg sodíku v jedné tabletě, což je nutno zohlednit u pacientů na dietě s kontrolovaným obsahem sodíku. Oznamte svému lékaři, pokud musíte kontrolovat svůj příjem sodíku a dodržujte dietu s nízkým obsahem sodíku.
á
3. Jak se INCIVO užívá
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře nebo lékárníka. Pokud si nejste jistý(á), poraďte se se svým 'ékařem nebo lékárníkem.
Návod ke správnému užívání
Lékař rozhodne, který dávkovací režim je pro Vás vhodný.
Doporučený dávkovací režim je:
- 3 tablety přípravku INCIVO dvakrát denně (ráno a večer) spolu s jídlem. Celková dávka je 6 tablet za den.
- 2 tablety přípravku INCIVO každých 8 hodin spolu s jídlem. Celková dávka je 6 tablet za den.
Pokud trpíte současně infekcí virem hepatitidy C a virem lidské imunodeficience, a užíváte efavirenz, je doporučený dávkovací režim přípravku INCIVO 3 tablety každých 8 hodin s jídlem.
INCIVO musíte vždy užívat s jídlem; což je důležité, aby se do těla dostalo náležité množství léčivého přípravku. Dávku přípravku INCIVO nesmíte snižovat. Tablety polykejte celé. Před spolknutím tablety nekousejte, nerozlamujte nebo nerozpouštějte. Máte-li problém s jejich polykáním, poraďte se se svým lékařem.
Vzhledem k tomu, že se INCIVO vždy užívá spolu s peginterferonem alfa a ribavirinem, seznamte se, prosím, s dávkováním těchto léčivých přípravků v jejich příbalových informacích. Potřebujete-li pomoci, poraďte se se svým lékařem nebo lékárníkem.
INCIVO užívejte spolu s peginterferonem alfa a ribavirinem po 12 týdnů. Celková doba léčby peginterferonem alfa a ribavirinem kolísá mezi 24 a 48 týdny v závislosti na odpovědi na léčbu a na tom, zda jste už byl(a) léčen(a) dříve. Lékař Vám bude kontrolovat hladiny viru v krvi ve 4. a 12. týdnu, aby stanovil délku trvání léčby. Celková doporučená doba léčby u pacientů po transplantaci jater je 48 týdnů. Poraďte se se svým lékařem a dodržujte doporučenou délku léčby.
Pokud lékař ukončí léčbu přípravkem INCIVO kvůli nežádoucím účinkům nebo protože léčba neúčinkuje, nelze léčbu znovu zahájit.
ěla by být
Otevření dětského bezpečnostního uzávěru
Plastová lahvička se dodává s dětským bezpečnostním uzávěrem otevřena následovně:
- Zatlačte plastový šroubovací uzávěr směrem dolů a souča otáčejte proti směru hodinových ručiček.
- Sejměte odšroubovaný uzávěr.
Jestliže jste užil(a) více přípravku INCIVO, než jste měl(a)
Okamžitě se obraťte na svého lékaře nebo lékárníka s žádostí o radu.
V případě předávkování se může vyskytnout nevolnost, bolest hlavy, průjem, snížení chuti k jídlu,


uzávěrem
abnormální chuť a zvracení.
Jestliže jste zapomněl(a) užít INCIVO
Užíváte-li INCIVO dvakrát denně (ráno a večer)
Pokud si vzpomenete do 6 hodin, okamžitě užijte vzpomenete po více než 6 hodinách, vynechejte jednu dávku a další si vezměte v obvyklou dobu. Nezdvojnásobujte následující dávku, abyste íahradil(a) vynechanou dávku.
tři table
lety. Tablety vždy užívejte s jídlem. Pokud si
Užíváte-li INCIVO každých 8 hodin)
Pokud si vzpomenete do 4 hodin, okamžitě užijte dvě tablety. Tablety vždy užívejte s jídlem. Pokud si vzpomenete po více než 4 hodinách, vynechejte jednu dávku a další si vezměte v obvyklou dobu. Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku.
Jestliže jste přestal(a) užívat INCIVO
Dokud Vám lékař netkne, abyste léčbu ukončil(a), pokračujte v užívání přípravku INCIVO, aby se zajistil stálý účinek leku proti viru. Pokud lékař léčbu ukončil, nesmí se léčba přípravkem INCIVO znovu zahajovat.
Máte-li jakékol' další otázky týkající se užívání tohoto léčivého přípravku, zeptejte se svého lékaře nebo lékárníka.
4 . Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
U pacientů užívajících INCIVO se často objevila svědivá kožní vyrážka. Vyrážka je většinou mírná nebo středně závažná, ale může být také závažná a/nebo život ohrožující nebo se na závažnou a/nebo život ohrožující může zhoršit. Vzácně mohou pacienti mít spolu s vyrážkou i jiné příznaky, které mohou být známkou závažné kožní reakce.
Objeví-li se u Vás kožní vyrážka, okamžitě kontaktujte lékaře.
Okamžitě vyhledejte lékaře také:
- pokud se vyrážka zhorší NEBO
- pokud se spolu s vyrážkou objeví další příznaky jako:
- horečka
- únava
- otok obličeje
- otok lymfatických uzlin NEBO
- pokud máte rozsáhlou vyrážku s olupující se kůží, která může být doprovázena horečkou, příznaky podobnými chřipce, bolestivými puchýři na kůži a puchýřky v ústech, očích a/nebc na genitáliích.
Lékař musí vyrážku prohlédnout, aby mohl určit, jak ji léčit. Lékař může ukončit Vaši léčba lékařem ukončena, nesmí se INCIVO začít znovu užívat.
\
Okamžitě vyhledejte lékaře také, pokud se u Vás objeví kterýkoli z následujících příznaků:
- únava, slabost, dušnost, točení hlavy a/nebo pocit rychlého bušení srdce. Mm' se jednat o příznaky anemie (sníženého počtu červených krvinek);
- mdloba;
- bolestivý zánět kloubů většinou na noze (dna);
- problémy se zrakem;
- krvácení z konečníku;
- otok obličeje.
Četnosti nežádoucích účinků spojených s přípravkem INcIVO jsou uvedeny dále.
j sob):
1 z 0 os ím otvoru (he
Velmi časté nežádoucí účinky (postihují více než
- nízký počet červených krvinek (anemie);
- nevolnost, průjem, zvracení;
- zduření cév v konečníku nebo řitním otvoru (hemoroidy), bolest konečníku nebo řiti;
- kožní vyrážka a svědění kůže.
Časté nežádoucí účinky (postihují méně než 1 z 10 osob):
- plísňová infekce úst;
- nízký počet krevních destiček, snížení počtu lymfocytů (druh bílých krvinek), snížení činnosti
štítné žlázy, zvýšení hladiny kyseliny močové v krvi, snížení hladiny draslíku v krvi, zvýšení hladiny bilirubinu v krvi;
- změna chutý
- mdloba;
- svěděn' řitního otvoru nebo jeho okolí, krvácení z řitního otvoru nebo jeho okolí, malé trhlinky v kůži, která obklopuje řitní otvor, které mohou způsobovat bolest a/nebo krvácení při stolici
- červená rozpraskaná suchá šupinatá kůže (ekzém), vyrážka doprovázená červenou popraskanou suchou olupující se kůží (exfoliativní vyrážka);
- otok obličeje, otok paží a/nebo nohou (edém);
abnormální chuť přípravku.
-
Méně časté nežádoucí účinky (postihují méně než 1 ze 100 osob):
- zvýšení kreatininu v krvi;
- bolestivý zánět kloubů většinou na noze (dna);
- poškození očního pozadí (sítnice);
- zánět konečníku a řiti;
- zánět slinivky břišní
závažná vyrážka, která může být provázena horečkou, únavou, otokem obličeje nebo mízních uzlin, zvýšením počtu eosinofilů (druh bílých krvinek), účinky na játra, ledviny nebo plíce (reakce zvaná DRESS); puchýřky (kopřivka).
dehydratace. Známky a příznaky dehydratace zahrnující žízeň, sucho v ústech, sníženou četnost močení nebo objem moči a tmavě zbarvenou moč. V průběhu užívání kombinované léčby přípravkem INCIVO je důležité přijímat dostatek tekutin.
Vzácné nežádoucí účinky (postihují méně než 1 z 1 000 osob):
A
- hodně rozšířená závažná vyrážka s loupající se pokožkou, která může být provázena horečkou příznaky podobnými chřipce, puchýřky v ústech, očích a/nebo na genitáliích (Stevens-Johnsonův syndrom).
Hlášení nežádoucích účinků
cích pe
můžete
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo léká'n: Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního syitémi hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků m^ž^e přispět k získání více informací o bezpečnosti tohoto přípravku.
Seznamte se také s nežádoucími účinky uvedenými v příbalových informacích peginterferonu alfa a ribavirinu.
5. Jak INCIVO uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
...
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na štítku za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
INCIVO je nutno uchovávat v původní lahvčce. Lahvičku uchovávejte dobře uzavřenou, aby byl přípravek chráněn před vlhkostí. Lahvička obsahuje jeden nebo dva sáčky s vysoušedlem, které uchovávají tablety suché. Toto vysoušedlo z lahvičky neodstraňujte. Vysoušedlo se nepolyká.
s příprav.-
6. Obsah ba^níi další informace CIVO obsahuje
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
Co INCI ♦

u látkou je
Dalš
je telaprevirum. Jedna tableta přípravku INCIVO obsahuje telaprevirum 375 mg. ími pomocnými látkami jsou:
Jádro tablety
acetátosukcinát hypromelosy, hydrogenfosforečnan vápenatý, mikrokrystalická celulosa, koloidní bezvodý oxid křemičitý, natrium-lauryl-sulfát, sodná sůl kroskarmelosy, natrium-stearyl-fumarát
Potah tablety
polyvinylalkohol, makrogol, mastek, oxid titaničitý (E171), žlutý oxid železitý (E172)
INCIVO je dostupné v baleních obsahujících jednu lahvičku nebo 4 lahvičky v krabičce. Každá lahvička obsahuje jeden nebo dva sáčky, které udržují tablety suché (vysoušedlo).
Na trhu nemusí být k dispozici všechny velikosti balení.
Držitel rozhodnutí o registraci
Janssen Cilag International NV Turnhoutseweg 30 B-2340 Beerse Belgie
Výrobce
Janssen-Cilag SpA,
Via C. Janssen,
041 00 Borgo San Michele, Latina, Itálie

utí o re
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnut-' o registraci:
Belgie/Belgique/Belgien
Janssen-Cilag NV Antwerpseweg 15-17 B-2340 Beerse Tel/Tél: +32 14 64 94 11
EtnrapHH
„fl^OHCtH & fl^OHCtH E-barapna” EOOfl ^.k. Mnagocr 4 En3Hec napK Co$na, crpaga 4 Co^na 1766 Ten.: +359 2 489 94 00
Lietuva
UAB „Johnson & Joln: Geležinio Vilko g. 18a LT-08104 Vilnius Tel: +370 5 278 68 88
so-
S
e msourg/]
JJaonh
D-4
Česká republika
Janssen-Cilag s.r.o.
Karla Engliše 3201/06 CZ-150 00 Praha 5 - Smíchov Tel: +420 227 012 227
Danmark
Janssen-Cilag Bregneredvej DK-3460 Bi-kera Tlf: +45 45 94 82 82
Deutschland
anssm-Cilag GmbH ’ nson & Johnson Platz 1 -41470 Neuss Tel: +49 2137 955-955

Antwe T
Luxembo^rg/Luxemburg
Janssen-Cilag NV twerpseweg 15-17 0 Beerse gique/Belgien él/Tel: +32 14 64 94 11

Magyarország
Janssen-Cilag Kft.
Nagyenyed u. 8-14 H-Budapest, 1123 Tel: +36 1 884 2858
Malta
AM MANGION LTD.
Mangion Building, Triq Gdida fi Triq Valletta MT-Hal-Luqa LQA 6000 Tel: +356 2397 6000
Nederland
Janssen-Cilag B.V.
Dr. Paul Janssenweg 150 NL-5026 RH Tilburg Tel: +31 13 583 73 73
Eesti
Janssen-Cilag Polska Sp. z o.o. Eesti filiaal LSStsa 2
EE-11415 Tallinn Tel: +372 617 7410
Norge
Janssen-Cilag AS Postboks 144 NO-1325-Lysaker Tlf: +47 24 12 65 00
EXXáSa
Janssen-Cilag OappaKenxiK^ A.E.B.E. Asro^ópog Eip^vn? 56 GR-151 21 neÚKn, A0^va Tn^: +30 210 80 90 000
Osterreich
Janssen-Cilag Pharma GmbH VorgartenstraBe 206B A-1020 Wien Tel: +43 1 610 300
Espaňa
Janssen-Cilag, S.A.
Paseo de las Doce Estrellas, 5-7 E-28042 Madrid Tel: +34 91 722 81 00
France
Janssen-Cilag
1, rue Camille Desmoulins, TSA 91003 F-92787 Issy Les Moulineaux, Cedex 9 Tél: 0 800 25 50 75 / +33 1 55 00 40 03
Polska
Janssen-Cilag Polska Sp. z o.o. ul. Uzecka 24 PL-02-135 Warszawa Tel: +48 22 237 60 00
Portugal
Janssen-Cilag Farmaceutica, Lda. Estrada Consiglieri Pedroso, 69 A Queluz de Baixo PT-2734-503 Barcarena Tel: +351 21 43 68 835 -X*'
Hrvatska
Johnson & Johnson S.E. d.o.o. Oreškoviceva 6h 10010 Zagreb Tel: +385 1 6610 700
Románia
Johnson & Johnson Rom,r ia SRL Str. Tipografilor nr. 11 15 Cládirea S-Park Corp A2, Etaj 5 013714 Bucu-e§ti, ROMÁNIA
Ireland
Janssen-Cilag Ltd.
50-100 Holmers Farm Way High Wycombe Buckinghamshire HP12 4EG United Kingdom Tel: +44 1 494 567 444
Ísland
Janssen-Cilag AB c/o Vistor hf. Horgatúni 2
IS-210 Garóabs ',0
Sími: +354 535
5 7000
Tel: +40 21 207 1800
Slovenija
Tohnson
Šmartinska cesta 53 Si-1000 Ljubljana Tel: +386 1 401 18 30
Slovenská republika
Johnson & Johnson s.r.o. CBC III, Karadžičova 12 SK-821 08 Bratislava Tel: +421 232 408 400
Italia
Janssen-Cilag SpA Via M.Buonarroti, 23 I-20093 Cologno Monzese MI
Suomi/Finland
Janssen-Cilag Oy Vaisalantie/Vaisalavagen 2 FI-02130 Espoo/Esbo
T'l: +39 02 2510 1
v
Kúnpoq
BapváPag Xax^nnavay^g AxS, Asro^ópog riávvou KpaviSiróxn 226 Aaxaiá
CY-2234 AeuKroaía Tn^: +357 22 207 700
Puh/Tel: +358 207 531 300 Sverige
Janssen-Cilag AB Box 7073
SE-192 07 Sollentuna Tel: +46 8 626 50 00
Latvija
Janssen-Cilag Polska Sp. z o.o. filiale Latvija Mukusalas iela 101 Riga, LV-1004 Tel: +371 678 93561
United Kingdom
Janssen-Cilag Ltd.
50-100 Holmers Farm Way High Wycombe
Buckinghamshire HP12 4EG - UK Tel: +44 1 494 567 444
Tato příbalová informace byla naposledy revidována {MM/RRRR}.
Ostatní zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
ké
0
V
0^
0
&
A
65