Imnovid 2 Mg
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Imnovid 1 mg tvrdé tobolky
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna tvrdá tobolka obsahuje pomalidomidum 1 mg.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Tvrdá tobolka.
Tvrdá tobolka Imnovid 1 mg: tmavě modré neprůhledné víčko a žluté neprůhledné tělo s nápisem „POML“ v bílé barvě a „1 mg“ v černé barvě, velikost 4, tvrdá želatinová tobolka.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Imnovid je v kombinaci s dexamethasonem indikován k léčbě dospělých pacientů s relabujícím a refrakterním mnohočetným myelomem, kteří absolvovali alespoň dvě předchozí léčebná schémata, zahrnující jak lenalidomid, tak i bortezomib, a při poslední terapii vykazovali progresi onemocnění.
4.2 Dávkování a způsob podání
Léčba musí být zahájena a vedena pod dohledem lékaře se zkušenostmi v léčbě mnohočetného myelomu.
Dávkování
Doporučená počáteční dávka přípravku Imnovid je 4 mg jednou denně perorálně v 1. až 21. den opakovaných 28denních cyklů. Doporučená dávka dexamethasonu je 40 mg perorálně jednou denně v 1., 8., 15., a 22. den každého 28denního léčebného cyklu.
Dávkování je třeba udržovat a upravovat na základě klinických a laboratorních nálezů.
Při progresi onemocnění je nutné léčbu přerušit.
Úprava dávky _pomalidomidu nebo _přerušení léčby
Pokyny pro přerušení léčby nebo snížení dávky pomalidomidu v souvislosti s hematologickými nežádoucími účinky jsou uvedeny v tabulce níže.
• Pokyny pro úpravu dávky pomalidomidu
|
Toxicita |
Úprava dávky |
|
Neutropenie • ANC* < 0,5 x 109/l nebo febrilní neutropenie (horečka > 38,5 °C a ANC < 1 x 109/l) |
Přerušení léčby pomalidomidem, sledování kompletního KO** v týdenních intervalech. |
|
• Návrat ANC na > 1 x 109/l |
Pokračování v léčbě pomalidomidem v dávce 3 mg denně. |
|
• Každý následný pokles na < 0,5 x 109/l |
Přerušení léčby pomalidomidem. |
|
• Návrat ANC na > 1 x 109/l |
Pokračování v léčbě pomalidomidem v dávce o 1 mg nižší než předchozí dávka. |
|
Trombocvtopenie • Počet trombocytů < 25 x 109/l |
Přerušení léčby pomalidomidem, sledování kompletního KO** v týdenních intervalech. |
|
• Návrat počtu trombocytů na > 50 x 109/l |
Pokračování v léčbě pomalidomidem v dávce 3 mg denně. |
|
• Každý následný pokles na < 25 x 109/l |
Přerušení léčby pomalidomidem. |
|
• Návrat počtu trombocytů na > 50 x 109/l |
Pokračování v léčbě pomalidomidem v dávce o 1 mg nižší než předchozí dávka. |
*ANC - absolutní počet neutrofilů; **KO - krevní obraz;
Nový cyklus léčby pomalidomidem lze zahájit, pouze pokud je počet neutrofilů > 1 x 109/l a počet trombocytů > 50 x 109/l.
V případě neutropenie má lékař zvážit použití růstových faktorů.
V případě jiných nežádoucích účinků 3. a 4. stupně, u nichž se usuzuje na spojitost s léčbou pomalidomidem, ukončete léčbu. Po zmírnění nežádoucího účinku na < 2. stupeň, dle uvážení lékaře, obnovte léčbu s dávkou o 1 mg nižší, než byla předchozí dávka.
Pokud se nežádoucí účinky vyskytnou po snížení dávky na 1 mg, je nutné léčivý přípravek vysadit.
Při výskytu kožní vyrážky 2. až 3. stupně je třeba zvážit přerušení nebo ukončení léčby pomalidomidem. Léčba pomalidomidem musí být ukončena při výskytu angioedému, kožní vyrážky 4. stupně a exfoliativní nebo bulózní vyrážky; po ukončení léčby z důvodu těchto reakcí se nemá léčba znovu zahajovat.
Jestliže jsou s pomalidomidem současně podávány silné inhibitory CYP1A2 (např. ciprofloxacin, enoxacin a fluvoxamin), snižte dávku pomalidomidu o 50 %.
• Pokyny pro úpravu dávky dexamethasonu
|
Toxicita |
Úprava dávky |
|
Dyspepsie = 1.-2. stupeň |
Udržování dávky a léčba blokátory histaminu (H2) nebo ekvivalenty. V případě přetrvávání příznaků snížení dávky o jednu úroveň. |
|
Dyspepsie > 3. stupeň |
Přerušení podávání dávky až do zvládnutí příznaků. Přidání blokátoru H2 nebo ekvivalentu a podávání snížené dávky o jednu úroveň po obnovení podávání. |
|
Edém > 3. stupeň |
Použití diuretik podle potřeby a snížení dávky o jednu úroveň. |
|
Zmatenost nebo změny nálady > 2. stupeň |
Přerušení podávání dávky do vymizení příznaků. Po obnovení podávání snížit dávku o jednu úroveň. |
|
Toxicita |
Úprava dávky |
|
Svalová slabost > 2. stupeň |
Přerušení podávání dávky do fáze svalové slabosti < 1. stupeň. Obnovení podávání s dávkou sníženou o jednu úroveň. |
|
Hyperglykemie > 3. stupeň |
Snížení dávky o jednu úroveň. Léčba insulinem nebo perorálními hypoglykemickými přípravky dle potřeby. |
|
Akutní pankreatitida |
Ukončení podávání dexamethasonu pacientovi. |
|
Další nežádoucí účinky související s dexamethasonem > 3. stupeň |
Přerušení podávání dexamethasonu do vymizení nežádoucích účinků do < 2. stupeň. Pokračování podávání s dávkou sníženou o jednu úroveň. |
Úrovně snížení dávky dexamethasonu:
Úrovně snížení dávky (< 75 let): počáteční dávka 40 mg; úroveň 1 snížení dávky - 20 mg; úroveň 2 snížení dávky- 10 mg v 1., 8., 15., a 22. den každého 28denního léčebného cyklu.
Úrovně snížení dávky (> 75 let): počáteční dávka 20 mg; úroveň 1 snížení dávky - 12 mg; úroveň 2 snížení dávky - 8 mg v 1., 8., 15., a 22. den každého 28denního léčebného cyklu.
Pokud zotavování z toxicit trvá déle než 14 dní, pak se dávka dexamethasonu sníží o jednu úroveň dávky.
Zvláštní populace
Pediatrická _ populace
Použití přípravku Imnovid v indikaci mnohočetného myelomu u dětí ve věku 0-17 let není relevantní.
Starší _ pacienti
Úprava dávky pomalidomidu není nutná. U pacientů ve věku > 75 let je počáteční dávka dexamethasonu 20 mg jednou denně v 1., 8., 15. a 22. den každého 28denního léčebného cyklu.
Porucha _ funkce ledvin
U pacientů s poruchou funkce ledvin není nutná žádná úprava dávky pomalidomidu. Ve dnech, kdy pacienti podstupují hemodialýzu, má být dávka pomalidomidu užita po hemodialýze..
Porucha _ funkce _ jater
Pacienti s celkovým sérovým bilirubinem > 2,0 mg/dl byli z klinických studií vyřazeni. Porucha funkce jater má mírný vliv na farmakokinetiku pomalidomidu (viz bod 5.2). U pacientů s poruchou funkce jater definovanou za použití kritérií podle Childa a Pugha není nutná žádná úprava počáteční dávky pomalidomidu. Je však nutné pacienty s poruchou funkce jater důkladně sledovat pro případ výskytu nežádoucích účinků a podle potřeby snížit dávku nebo přerušit podávání pomalidomidu.
Způsob podání Perorální podání.
Přípravek Imnovid se má užívat každý den ve stejnou dobu. Tobolky se nesmí otevírat, lámat ani žvýkat (viz bod 6.6). Tento léčivý přípravek je třeba polykat vcelku, nejlépe zapít vodou. Tobolky se mohou užívat s jídlem nebo bez něho. Pokud pacient zapomene jeden den užít dávku přípravku Imnovid, může užít normální předepsanou dávku v plánovaný čas následujícího dne. Pacienti nesmí upravovat dávku, aby nahradili vynechanou dávku z předchozího dne.
Pro vyjmutí tobolky z blistru se doporučuje zatlačit pouze na jedné straně, aby se minimalizovalo riziko deformace či rozlomení tobolky.
4.3 Kontraindikace
- Těhotenství.
- Ženy, které mohou otěhotnět, pokud nejsou splněny všechny podmínky Programu prevence početí (viz body 4.4 a 4.6).
- Pacienti muži, kteří nejsou schopni dodržovat požadovaná antikoncepční opatření (viz bod 4.4).
- Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Teratogenita
Pomalidomid se nesmí užívat v průběhu těhotenství, neboť se očekávají teratogenní účinky. Pomalidomid je strukturálně podobný thalidomidu. Thalidomid je známý lidský teratogen, který způsobuje těžké a život ohrožující vrozené vady. Pomalidomid vykazoval teratogenní účinky jak u potkanů, tak i u králíků, pokud byl podáván v období hlavní organogeneze (viz bod 5.3).
Všechny pacientky musí splňovat podmínky Programu prevence početí (PPP), pokud není spolehlivý důkaz o tom, že u pacientky je možnost otěhotnění vyloučena.
Kritéria pro ženu, která nemůže otěhotnět
Pacientka nebo partnerka pacienta-muže je považována za neschopnou otěhotnět, pokud splňuje alespoň jedno z následujících kritérií:
• věk > 50 let a přirozená amenorea po dobu > 1 rok*
• předčasné selhání vaječníků potvrzené gynekologem
• předchozí oboustranná adnexektomie nebo hysterektomie
• genotyp XY, Turnerův syndrom, ageneze dělohy.
*Amenorea po protinádorové terapii nebo během kojení nevylučuje možnost otěhotnění pacientky.
Poradenství
U žen, které mohou otěhotnět, je pomalidomid kontraindikován, pokud nejsou splněna všechna následující kritéria:
• Žena si je vědoma očekávaného teratogenního rizika pro nenarozené dítě.
• Žena chápe nutnost účinné antikoncepce praktikované bez přerušení po 4 týdny před začátkem léčby, po celou dobu během ní a 4 týdny po jejím ukončení.
• I když má fertilní žena amenoreu, musí používat účinnou antikoncepci.
• Žena musí být schopna dodržovat účinná antikoncepční opatření.
• Žena je informována a je si vědoma potenciálních následků těhotenství a nutnosti rychle informovat lékaře, pokud hrozí riziko těhotenství.
• Žena chápe nutnost zahájení léčby hned po vydání pomalidomidu, kterému předchází negativní těhotenský test.
• Žena chápe nutnost a je ochotna absolvovat těhotenské testy každé 4 týdny, kromě případů potvrzené sterilizace podvazem vejcovodů.
• Žena potvrdí, že si je vědoma rizik a nutných bezpečnostních opatření spojených s užíváním pomalidomidu.
Předepisující lékař musí u žen, které mohou otěhotnět, zajistit, že:
• Pacientka splňuje podmínky Programu prevence početí (PPP), včetně ujištění, že těmto podmínkám patřičně porozuměla.
• Pacientka výše uvedené podmínky potvrdila.
U mužů užívajících pomalidomid farmakokinetická data ukázala, že pomalidomid je během léčby přítomen v semeni. Z preventivních důvodů musí všichni muži užívající pomalidomid splňovat následující podmínky:
• Muž si je vědom očekávaného teratogenního rizika při pohlavním styku s těhotnou ženou nebo se ženou, která může otěhotnět.
• Muž chápe nutnost používání kondomu, pokud má pohlavní styk s těhotnou ženou nebo se ženou, která může otěhotnět a nepoužívá účinnou antikoncepci, během léčby a po dobu 7 dní
po přerušení a/nebo ukončení léčby. Muži, kteří podstoupili vasektomii, mají při pohlavním styku s těhotnou ženou nebo se ženou ve fertilním věku, používat kondom, jelikož semenná tekutina může i při absenci spermií obsahovat pomalidomid.
• Muž si je vědom, že musí ihned informovat svého ošetřujícího lékaře, pokud jeho partnerka otěhotní v průběhu jeho léčby pomalidomidem nebo 7 dní po ukončení léčby, a že se doporučuje partnerku předat specialistovi nebo zkušenému teratologovi, aby posoudil riziko a poskytl doporučení.
Antikoncepce
Ženy, které mohou otěhotnět, musí používat jednu účinnou metodu antikoncepce po 4 týdny před léčbou, během ní a 4 týdny po léčbě pomalidomidem, a také po dobu případného přerušení léčby, pokud se nezavážou k absolutní a trvalé pohlavní abstinenci, kterou musí každý měsíc potvrdit. Pokud pacientka nepoužívá účinnou antikoncepci, musí být odeslána k vyškolenému zdravotníkovi, který jí s výběrem antikoncepční metody poradí, aby antikoncepce mohla být nasazena.
Vhodné metody antikoncepce například jsou:
• implantát
• nitroděložní tělísko uvolňující levonorgestrel
• depotní medroxyprogesteron-acetát
• sterilizace podvazem vejcovodů
• pohlavní styk pouze s mužem po vasektomii; vasektomie musí být potvrzena dvěma negativními testy semene
• antikoncepční tablety inhibující ovulaci obsahující pouze progesteron (tj. desogestrel)
Vzhledem ke zvýšenému riziku žilní tromboembolie u pacientek s mnohočetným myelomem užívajících pomalidomid a dexamethason se kombinovaná perorální antikoncepce nedoporučuje (viz také bod 4.5). Pokud pacientka v současnosti používá kombinovanou perorální antikoncepci, je třeba přejít na některou z účinných antikoncepčních metod uvedených výše. Riziko žilní tromboembolie trvá po dobu 4-6 týdnů po vysazení kombinované perorální antikoncepce. Účinnost steroidních antikoncepčních přípravků může být během současného podávání dexamethasonu snížena (viz bod 4.5).
Implantáty a nitroděložní tělíska uvolňující levonorgestrel jsou spojeny se zvýšeným rizikem infekce v době zavedení a nepravidelného vaginálního krvácení. Je třeba zvážit profylaktické podávání antibiotik, zvláště u pacientek s neutropenií.
Zavádění nitroděložních tělísek uvolňujících měď se nedoporučuje vzhledem k potenciálnímu riziku infekce v době zavedení a ztrátě menstruační krve, která může způsobit komplikace u pacientek trpících těžkou neutropenií nebo těžkou trombocytopenií.
Těhotenské testy
V souladu s místní praxí je třeba zajistit provádění těhotenských testů s minimální citlivostí 25 mIU/ml pod dohledem lékaře u žen, které mohou otěhotnět, jak je uvedeno níže. Tento požadavek se týká i žen, které mohou otěhotnět a praktikují absolutní a trvalou pohlavní abstinenci. Je ideální, aby byl ve stejný den proveden těhotenský test a lék předepsán i vydán. Vydání pomalidomidu ženám, které mohou otěhotnět, se má provést během 7 dnů od předepsání.
Před začátkem léčby
Je třeba provést těhotenský test pod lékařským dohledem při návštěvě lékaře, kdy je pomalidomid předepsán, nebo ve 3 dnech předcházejících návštěvě u předepisujícího lékaře - poté, co pacientka používá účinnou antikoncepci přinejmenším 4 týdny. Test musí potvrdit, že pacientka není při zahájení léčby pomalidomidem těhotná.
Ukončení léčby a další sledování
Těhotenský test pod lékařským dohledem musí být opakován každé 4 týdny včetně 4 týdnů po ukončení léčby, kromě případů potvrzené sterilizace podvazem vejcovodů. Tyto testy je třeba provést v den předepsání přípravku nebo během 3 dnů před návštěvou předepisujícího lékaře.
Muži
Pomalidomid je během léčby přítomen v semeni. Z preventivních důvodů, a s ohledem na zvláštní populace s potenciálně prodlouženou dobou eliminace, např. s poruchou funkce ledvin, musí všichni pacienti muži užívající pomalidomid, včetně těch, co podstoupili vasektomii, používat kondom po celou dobu léčby, během jejího přerušení a 7 dní po ukončení léčby, pokud je jejich partnerka těhotná nebo pokud u ní nelze možnost otěhotnění vyloučit, a pokud ta nepoužívá jinou antikoncepci.
Pacienti muži nesmí darovat semeno nebo sperma během léčby (včetně období přerušení léčby) a 7 dní po vysazení pomalidomidu.
Další opatření
Pacienti musí být poučeni, aby nikdy tento léčivý přípravek nedávali jiným osobám a nepoužité tobolky vrátili na konci léčby do lékárny.
Pacienti nesmí darovat krev, semeno nebo sperma během léčby (včetně období přerušení léčby) a 7 dní po vysazení pomalidomidu.
Vzdělávací materiály, omezení týkající se předepisování a výdeje
Držitel rozhodnutí o registraci bude vydávat vzdělávací materiály pro zdravotníky, aby byli schopni odborně poradit pacientům, jak zabránit vlivu pomalidomidu na plod. Materiály obsahují varování před očekávanými teratogenními účinky pomalidomidu, rady ohledně antikoncepce před začátkem terapie a poučení o nutnosti těhotenských testů. Předepisující lékař musí pacienta informovat o očekávaném teratogenním riziku a přísných antikoncepčních opatřeních tak, jak jsou uvedena v Programu prevence početí (PPP) a poskytnout pacientům odpovídající informační brožurku, kartu pacienta a/nebo ekvivalentní pomůcky v souladu se zavedeným národním systémem evidence karet pacientů. Ve spolupráci s jednotlivými příslušnými národními orgány byl zaveden systém regulované národní distribuce. Tento systém regulované distribuce zahrnuje využívání karet pacientů a/nebo ekvivalentního nástroje k regulaci předepisování a/nebo výdeje a shromažďování podrobných údajů týkajících se indikace za účelem monitorování použití mimo schválenou indikaci na území daného státu. Ideálně se mají procesy předepsání a výdeje léku uskutečnit ve stejný den jako těhotenský test. Pomalidomid je nutné ženám, které mohou otěhotnět, vydat do 7 dní od předpisu a po provedení těhotenského testu pod lékařským dohledem s negativním výsledkem. Ženám, které mohou otěhotnět, může být předepsán na maximální dobu 4 týdnů a všem ostatním pacientům na maximální dobu 12 týdnů.
Hematologické příhody
Neutropenie byla nejčastěji hlášeným hematologickým nežádoucím účinkem 3. nebo 4. stupně u pacientů s relabovaným /s refrakterním mnohočetným myelomem, dále následovaly anémie a trombocytopenie. U pacientů je třeba sledovat výskyt hematologických nežádoucích účinků, především neutropenie. Pacienty je nutné informovat, aby febrilní epizody neprodleně hlásili. Lékaři musí u pacientů sledovat známky krvácení, jako jsou epistaxe, zejména při současném užívání léčivých přípravků, o nichž je známo, že zvyšují riziko krvácení (viz bod 4.8). Celkový krevní obraz je nutné sledovat na počátku, jednou týdně po dobu prvních 8 týdnů a dále jednou za měsíc. Může být nutné upravit dávky (viz bod 4.2). U pacientů může být potřeba použít podpůrné krevní produkty a/nebo růstové faktory.
Tromboembolické příhody
U pacientů užívajících pomalidomid v kombinaci s dexamethasonem se vyskytují žilní tromboembolické příhody (především hluboká žilní trombóza a plicní embolie) a tepenné trombotické příhody (infarkt myokardu a cerebrovaskulární příhoda). Pacienty se známými rizikovými faktory pro tromboembolismus - včetně trombózy v anamnéze - je nutné důkladně sledovat. Je nutné přijmout opatření k minimalizaci veškerých modifikovatelných rizikových faktorů (např. kouření, hypertenze a hyperlipidemie). Pacientům a lékařům se doporučuje sledovat možný výskyt známek a příznaků tromboembolismu. Pacienty je nutné poučit, aby vyhledali lékařskou péči, pokud se u nich objeví příznaky, jako je dechová nedostatečnost, bolest na hrudi a otoky paží nebo nohou. Doporučuje se antikoagulační terapie (pokud není kontraindikována) (například kyselina acetylsalicylová, warfarin, heparin, nebo klopidogrel), především u pacientů s dalšími trombotickými rizikovými faktory. O profylaktických opatřeních má být rozhodnuto po pečlivém zhodnocení základních rizikových faktorů u jednotlivých pacientů. V klinických studiích pacienti dostávali v rámci profylaxe kyselinu acetylsalicylovou nebo alternativní antitrombotickou terapii. Použití erytropoetických látek s sebou nese riziko tromboembolických příhod včetně tromboembolismu. Proto je nutné erytropoetické látky i jiné látky, které mohou zvyšovat riziko tromboembolických příhod, používat s opatrností.
Periferní neuropatie
Pacienti s probíhající periferní neuropatií > 2. stupně byli z klinických studií s pomalidomidem vyloučeni. Při zvažování léčby těchto pacientů pomalidomidem je třeba postupovat s patřičnou opatrností.
Významná srdeční dysfunkce
Pacienti s významnou srdeční dysfunkcí (městnavým srdečním selháním [třídy III nebo IV podle NYHA], infarktem myokardu v průběhu 12 měsíců od zahájení studie, nestabilní nebo nedostatečně kontrolovanou anginou pectoris) byli z klinických studií s pomalidomidem vyloučeni. Byly hlášeny případy srdečního selhání, včetně městnavého srdečního selhání a plicního edému (viz bod 4.8), zejména u pacientů s již existujícím srdečním onemocněním nebo s kardiálními rizikovými faktory.
Při zvažování léčby těchto pacientů pomalidomidem je třeba postupovat s patřičnou opatrností, včetně pravidelného sledování známek a příznaků srdečního selhání.
Syndrom nádorového rozpadu
Může se vyskytnout syndrom nádorového rozpadu. Mezi pacienty s největším rizikem syndromu nádorového rozpadu patří pacienti s vysokou nádorovou zátěží před léčbou. Tyto pacienty je nutné důkladně sledovat a přijmout vhodná opatření.
Další primární malignity
U pacientů léčených pomalidomidem byly hlášeny další primární malignity, např. nemelanomové kožní nádory (viz bod 4.8). Lékaři musí pacienty důkladně vyšetřit před léčbou i během léčby pomocí standardních metod pro zjišťování karcinomu pro případ výskytu dalších primárních malignit a zahájit léčbu podle indikace.
Alergická reakce
Byly hlášeny případy angioedému a závažných kožních reakcí (viz bod 4.8). Pacienti s předchozí závažnou alergickou reakcí spojenou s léčbou thalidomidem nebo lenalidomidem v anamnéze byli z klinických studií vyloučeni. Tito pacienti mohou být vystaveni zvýšenému riziku hypersenzitivitních reakcí a pomalidomid jim nemá být podáván. Při výskytu kožní vyrážky stupně 2 až 3 je třeba zvážit přerušení nebo ukončení léčby pomalidomidem. Léčba pomalidomidem musí být trvale ukončena při výskytu angioedému, kožní vyrážky stupně 4, exfoliativní nebo bulózní vyrážky.
Závratě a zmatenost
Při užívání pomalidomidu byly hlášeny závratě a zmatenost. Pacienti se musí vyvarovat situací, kdy pro ně závratě nebo zmatenost mohou představovat problém, a nesmí bez předchozí konzultace s lékařem užívat jiné léčivé přípravky, které mohou způsobovat závratě nebo zmatenost.
Intersticiální plicní nemoc (Interstitial lung disease - ILD)
Při léčbě pomalidomidem byly pozorovány případy ILD a souvisejících onemocnění, včetně pneumonitidy. Pacienti s akutním nástupem nebo s nevysvětleným náhlým zhoršením plicních příznaků mají být pečlivě vyšetřeni za účelem vyloučení ILD. Pomalidomid by měl být do doby vyšetření těchto příznaků vysazen a pokud se ILD potvrdí, má být zahájena vhodná terapie. Podávání pomalidomidu lze obnovit pouze po důkladném vyhodnocení přínosů a rizik.
Poruchy jaterních funkcí
U pacientů léčených pomalidomidem bylo pozorováno významné zvýšení hladin alaninaminotransferázy a bilirubinu (viz bod 4.8). Vyskytly se také případy hepatitidy, které vedly k ukončení léčby pomalidomidem. Po dobu prvních 6 měsíců léčby pomalidomidem a dále podle klinické indikace se doporučuje pravidelné sledování jaterních funkcí.
Infekce
U pacientů, kteří byli v minulosti infikováni virem hepatitidy B (HBV), léčených pomalidomidem v kombinaci s dexamethasonem, byla vzácně hlášena reaktivace hepatitidy B. Některé z těchto případů progredovaly do akutního selhání jater, což vedlo k ukončení léčby pomalidomidem. Před zahájením léčby pomalidomidem se mají provést testy na stanovení viru hepatitidy B. U pacientů pozitivních na infekci HBV se doporučuje konzultace s lékařem specializovaným na léčbu hepatitidy B. Při použití kombinace pomalidomidu s dexamethasonem u pacientů, kteří byli v minulosti infikováni HBV, včetně pacientů, kteří jsou anti-HBc pozitivní, ale HBsAg negativní, je třeba postupovat s opatrností. Tyto pacienty je nutné v průběhu celé léčby pečlivě sledovat kvůli výskytu známek a příznaků aktivní infekce HBV.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Účinek přípravku Imnovid na jiné léčivé přípravky
Při současném podávání pomalidomidu a substrátů enzymů nebo transportérů se nepředpokládají klinicky relevantní farmakokinetické lékové interakce v důsledku inhibice nebo indukce izoenzymu P450 nebo inhibice transportérů. Potenciál k takovým lékovým interakcím, včetně potenciálního dopadu pomalidomidu na farmakokinetiku kombinovaných perorálních kontraceptiv, nebyl klinicky hodnocen (viz bod 4.4 Teratogenita).
Účinek jiných léčivých přípravků na přípravek Imnovid
Pomalidomid je částečně metabolizován enzymy CYP1A2 a CYP3A4/5. Je také substrát P-glykoproteinu. Současné podávání pomalidomidu s ketokonazolem, silným inhibitorem CYP3A4/5, a P-glykoproteinem, nebo karbamazepinem, silným induktorem CYP3A4/5, nemá žádný klinicky relevantní účinek na expozici pomalidomidu. Současné podávání pomalidomidu s fluvoxaminem, silným inhibitorem CYP1A2, za přítomnosti ketokonazolu zvýšilo střední expozici pomalidomidu o 107 % při 90% intervalu spolehlivosti [91 % až 124 %] v porovnání s kombinací pomalidomidu a ketokonazolu. Ve druhé studii, která hodnotila přispění samotného inhibitoru CYP1A2 ke změnám metabolismu, zvýšilo současné podávání samotného fluvoxaminu s pomalidomidem střední expozici pomalidomidu o 125 % při 90% intervalu spolehlivosti [98 % až 157 %] v porovnání se samotným pomalidomidem. Jestliže jsou s pomalidomidem současně podávány silné inhibitory CYP1A2 (např. ciprofloxacin, enoxacin a fluvoxamin), snižte dávku pomalidomidu o 50 %.
Dexamethason
Současné podávání opakovaných dávek až 4 mg pomalidomidu s 20 mg až 40 mg dexamethasonu (slabý až středně silný induktor několika enzymů CYP, včetně CYP3A) pacientům s mnohočetným myelomem nemělo žádný účinek na farmakokinetiku pomalidomidu v porovnání s pomalidomidem podávaným v monoterapii.
Účinky dexamethasonu na warfarin nejsou známy. Během léčby se doporučuje pečlivě sledovat hladinu warfarinu.
4.6 Fertilita, těhotenství a kojení
Ženy, které mohou otěhotnět / antikoncepce u mužů a žen
Ženy, které mohou otěhotnět, musí používat účinnou metodu antikoncepce. Pokud žena léčená pomalidomidem otěhotní, musí být léčba zastavena a pacientka odeslána ke specializovanému lékaři nebo zkušenému teratologovi, aby posoudil riziko a navrhl doporučení. Pokud otěhotní partnerka pacienta-muže léčeného pomalidomidem, doporučuje se tuto partnerku odeslat ke specializovanému lékaři nebo zkušenému teratologovi, aby posoudil riziko a poskytl doporučení. Pomalidomid je přítomen v lidském semeni. Z preventivních důvodů musí všichni pacienti muži užívající pomalidomid používat kondom po celou dobu léčby, během jejího přerušení a 7 dní po ukončení léčby, pokud je jejich partnerka těhotná nebo pokud u ní nelze možnost otěhotnění vyloučit, a pokud ta nepoužívá jinou antikoncepci (viz body 4.3 a 4.4).
U člověka lze očekávat teratogenní účinek pomalidomidu. Pomalidomid je v těhotenství a u žen ve fertilním věku kontraindikován, vyjma případů, kdy jsou splněny všechny podmínky prevence početí, viz body 4.3 a 4.4.
Kojení
Není známo, zda se pomalidomid vylučuje do lidského mateřského mléka. Pomalidomid byl zjištěn v mateřském mléce laktujících samic potkanů po podání matkám. Vzhledem k potenciálu k nežádoucím účinkům pomalidomidu u kojených dětí je třeba posoudit nezbytnost léčivého přípravku pro matku a rozhodnout, zda bude přerušeno kojení nebo zda ukončit podávání léčivého přípravku matce.
Fertilita
Bylo zjištěno, že u zvířat má pomalidomid negativní dopad na fertilitu a vykazuje teratogenní účinky. Pomalidomid po podání březím samicím králíka prostupoval placentou a byl přítomen v krvi plodu. Viz bod 5.3.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek Imnovid má malý nebo mírný vliv na schopnost řídit nebo obsluhovat stroje.
Při užívání pomalidomidu byly hlášeny únava, snížený stupeň vědomí, zmatenost a závratě. Při výskytu těchto příznaků mají být pacienti poučeni, aby po dobu léčby pomalidomidem neřídili vozidla, nepoužívali stroje a nevykonávali nebezpečné činnosti.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Nejčastěji hlášenými nežádoucími účinky v klinických studiích byly poruchy krve a lymfatického systému včetně anémie (45,7 %), neutropenie (45,3 %) a trombocytopenie (27 %); celkové poruchy a reakce v místě aplikace včetně únavy (28,3 %), pyrexie (21 %) a periferního edému (13 %); a infekce a infestace včetně pneumonie (10,7 %). Nežádoucí účinky periferní neuropatie byly hlášeny u 12,3 % pacientů a nežádoucí účinky žilní embolické nebo trombotické příhody (VTE) byly hlášeny u 3,3 % pacientů. Nejčastěji hlášenými nežádoucími účinky 3. a 4. stupně byly poruchy krve a lymfatického systému včetně neutropenie (41,7 %), anémie (27 %) a trombocytopenie (20,7 %); infekce a infestace včetně pneumonie (9 %); a celkové poruchy a reakce v místě aplikace včetně únavy (4,7 %), pyrexie (3 %) a periferního edému (1,3 %). Nejčastěji hlášeným závažným nežádoucím účinkem byla pneumonie (9,3 %). Další hlášené závažné nežádoucí účinky zahrnovaly febrilní neutropenii (4,0 %), neutropenii (2,0 %), trombocytopenii (1,7 %) a nežádoucí účinky VTE (1,7 %).
Nežádoucí účinky se vyskytovaly častěji spíše v prvních 2 cyklech léčby pomalidomidem.
Přehled nežádoucích účinků v tabulce
V randomizované studii (CC-4047-MM-003), byly 302 pacientům s relabovaným a refrakterním mnohočetným myelomem podávány 4 mg pomalidomidu jednou denně po dobu 21 dní v každém 28denním cyklu v kombinaci s nízkou dávkou dexamethasonu jednou týdně.
Nežádoucí účinky pozorované u pacientů léčených kombinací pomalidomidu a dexamethasonu jsou uvedeny níže a seřazeny podle tříd orgánových systémů a četnosti veškerých nežádoucích účinků a nežádoucích účinků 3. a 4. stupně.
Četnosti nežádoucích účinků j sou četnosti hlášené ve skupině léčené kombinací pomalidomidu a dexamethasonu ve studii CC-4047-MM-003 (n = 302) a z údajů po uvedení přípravku na trh. V každé třídě orgánových systémů a skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti. Četnosti jsou definovány v souladu s platnými předpisy takto: velmi časté (> 1/10), časté (> 1/100 až < 1/10) a méně časté (> 1/1000 až < 1/100).
|
Třídy orgánových systémů / Preferovaný termín |
Veškeré nežádoucí účinky / Frekvence |
Nežádoucí účinky 3.4. stupně / F rekvence |
|
Infekce a infestace |
Velmi časté |
Časté |
|
Pneumonie (bakteriální, virové |
Neutropenická sepse | |
|
a mykotické infekce, včetně |
Pneumonie (bakteriální, virové | |
|
oportunních infekcí) |
a mykotické infekce, včetně oportunních infekcí) | |
|
Časté |
Bronchopneumonie | |
|
Neutropenická sepse |
Infekce dýchacích cest | |
|
Bronchopneumonie Bronchitida |
Infekce horních cest dýchacích | |
|
Infekce dýchacích cest |
Méně časté | |
|
Infekce horních cest dýchacích |
Bronchitida | |
|
Nazofaryngitida Herpes zoster |
Herpes zoster Není známo | |
|
Není známo Reaktivace hepatitidy B |
Reaktivace hepatitidy B | |
|
Novotvary benigní, maligní |
Méně časté |
Méně časté |
|
a blíže neurčené (zahrnující |
Bazocelulámí karcinom kůže |
Bazocelulámí karcinom kůže |
|
cysty a polypy) |
Spinocelulámí karcinom kůže |
Spinocelulámí karcinom kůže |
|
Poruchy krve a lymfatického |
Velmi časté |
Velmi časté |
|
systému |
Neutropenie |
Neutropenie |
|
Trombocytopenie |
T rombocytopenie | |
|
Leukopenie |
Časté | |
|
Časté |
Febrilní neutropenie | |
|
Febrilní neutropenie |
Leukopenie | |
|
Pancytopenie* |
Pancytopenie15 | |
|
Poruchy metabolismu a |
Velmi časté |
Časté |
|
výživy |
Snížená chuť k jídlu |
Hyperkalemie Hyponatremie |
|
Časté Hyperkalemie |
Hyperurikemie* | |
|
Hyponatremie |
Méně časté | |
|
Hyperurikemie* Méně časté Syndrom nádorového rozpadu* |
Snížená chuť k jídlu Syndrom nádorového rozpadu* | |
|
Psychiatrické poruchy |
Časté |
Časté |
|
Stav zmatenosti |
Stav zmatenosti | |
|
Poruchy nervového systému |
Časté |
Časté |
|
Snížený stupeň vědomí Periferní senzorická neuropatie |
Snížený stupeň vědomí | |
|
Závratě |
Méně časté | |
|
Periferní senzorická neuropatie | ||
|
Intrakraniální krvácení* |
Závratě | |
|
Méně časté |
Cerebrovaskulární příhoda* | |
|
Cerebrovaskulární příhoda* |
Intrakraniální krvácení* |
|
Třídy orgánových systémů / |
Veškeré nežádoucí účinky / |
Nežádoucí účinky 3.- |
|
Preferovaný termín |
Frekvence |
4. stupně / F rekvence |
|
Poruchy ucha a labyrintu |
Časté |
Časté |
|
Cévní poruchy |
Časté |
Méně časté |
|
Hluboká žilní trombóza |
Hluboká žilní trombóza | |
|
Srdeční poruchy |
Časté |
Časté |
|
Srdeční selháni* |
Srdeční selhání* | |
|
Fibrilace síní* Infarkt myokardu* |
Fibrilace síní* | |
|
Méně časté Infarkt myokardu* | ||
|
Poruchy imunitního systému |
Časté |
Méně časté |
|
Angioedém* |
Angioedém* | |
|
Urtikárie* |
Urtikárie* | |
|
Respirační, hrudní a |
Velmi časté |
Časté |
|
mediastinální poruchy | ||
|
Méně časté | ||
|
Časté | ||
|
Epistaxe* |
Epistaxe* | |
|
Intersticiálni plicní nemoc* |
Intersticiální plicní nemoc* | |
|
Gastrointestinální poruchy |
Velmi časté |
V Časté |
|
Zácpa |
Zácpa | |
|
Časté |
Méně časté | |
|
Gastrointestinální krvácení |
Gastrointestinální krvácení | |
|
Poruchy jater a žlučových |
Méně časté |
Méně časté |
|
cest |
Hyperbilirubinemie Hepatitida5 |
Hyperbilirubinemie |
|
Poruchy kůže a podkožní |
Časté |
Časté |
|
tkáně | ||
|
Poruchy svalové a kosterní |
Velmi časté |
Časté |
|
soustavy a pojivové tkáně |
Bolest kostí |
Bolest kostí |
|
Svalové křeče |
Méně časté Svalové křeče | |
|
Poruchy ledvin a močových |
Časté |
Časté |
|
cest |
Renální selhání |
Renální selhání |
|
Retence moči |
Méně časté Retence moči | |
|
Poruchy reprodukčního |
v Časté |
v Časté |
|
systému a prsu |
Pánevní bolest |
Pánevní bolest |
|
Třídy orgánových systémů / Preferovaný termín |
Veškeré nežádoucí účinky / Frekvence |
Nežádoucí účinky 3.4. stupně / F rekvence |
|
Celkové poruchy a reakce |
Velmi časté |
Časté |
|
v místě aplikace |
Únava |
Únava |
|
Pyrexie |
Pyrexie | |
|
Periferní edém |
Periferní edém | |
|
Vyšetření |
Časté |
Časté |
|
Snížený počet neutrofilů |
Snížený počet neutrofilů | |
|
Snížený počet leukocytů |
Snížený počet leukocytů | |
|
Snížený počet trombocytů |
Snížený počet trombocytů | |
|
Zvýšená hladina |
Zvýšená hladina | |
|
alaninaminotransferázy Zvýšená hladina kyseliny močové |
alaninaminotransferázy | |
|
v krvi* |
Méně časté Zvýšená hladina kyseliny močové v krvf |
^Zjištěno z údajů po uvedení přípravku na trh, s frekvencemi vycházejícími z údajů klinických studií.
Popis vybraných nežádoucích účinků
Teratogenita
Pomalidomid je strukturálně podobný thalidomidu. Thalidomid je známá lidská teratogenní léčivá látka, která způsobuje těžké a život ohrožující vrozené vady. Pomalidomid vykazoval teratogenní účinky jak u potkanů, tak i u králíků, pokud byl podáván v období hlavní organogeneze (viz body 4.6 a 5.3). Pokud je pomalidomid užíván během těhotenství, očekávají se u lidí teratogenní účinky pomalidomidu (viz bod 4.4).
Neutropenie a trombocytopenie
Neutropenie se vyskytla u 45,3 % pacientů, kterým byl podáván pomalidomid s nízkou dávkou dexamethasonu (Pom + LD-Dex) a u 19,5 % pacientů, kterým byla podávána vysoká dávka dexamethasonu (HD-Dex). Neutropenie byla 3. nebo 4. stupně u 41,7 % pacientů, kterým byl podáván Pom + LD-Dex, v porovnání se 14,8 %, kterým byl podáván HD-Dex. Neutropenie u pacientů ve skupině Pom + LD-Dex byla zřídka závažná (2,0 % pacientů), nevedla k ukončení léčby a byla spojena s přerušením léčby u 21 % pacientů a se snížením dávky u 7,7 % pacientů.
Febrilní neutropenie (FN) byla pozorována u 6,7 % pacientů, kterým byl podáván Pom + LD-Dex a u žádného z pacientů, kterým byl podáván HD-Dex. Veškeré FN byly hlášeny se závažností 3. a 4. stupně. FN byla hlášena jako závažná u 4,0 % pacientů. FN byla spojena s přerušením léčby u 3,7 % pacientů a se snížením dávky u 1,3 % a nebyla spojena s žádným ukončením léčby.
Trombocytopenie se vyskytla u 27,0 % pacientů, kterým byl podáván Pom + LD-Dex a u 26,8 % pacientů, kterým byl podáván HD-Dex. Trombocytopenie byla 3. a 4. stupně u 20,7 % pacientů, kterým byl podáván Pom + LD-Dex a u 24,2 % pacientů, kterým byl podáván HD-Dex. U pacientů léčených Pom + LD-Dex byla trombocytopenie závažná u 1,7 % pacientů, vedla ke snížení dávky u 6,3 % pacientů, k přerušení léčby u 8 % pacientů a k ukončení léčby u 0,7 % pacientů (viz body 4.2 a 4.4).
Infekce
Infekce byla nejčastější nehematologickou toxicitou a vyskytla se u 55,0 % pacientů, kterým byl podávánPom + LD-Dex a u 48,3 % pacientů, kterým byl podáván HD-Dex. Přibližně polovina z těchto infekcí dosahovala 3. a 4. stupně, 24,0 % u pacientů léčených Pom + LD-Dex a 22,8 % u pacientů, kterým byl podáván HD-Dex.
U pacientů léčených Pom + LD-Dex byly nejčastěji hlášenými infekcemi pneumonie a infekce horních cest dýchacích (10,7 %, resp. 9,3 % pacientů), přičemž 24,3 % hlášených infekcí bylo závažných a fatálních (5. stupeň) a vyskytovaly se u 2,7 % léčených pacientů. U pacientů léčených Pom + LD-Dex vedly infekce k ukončení léčby u 2,0 % pacientů, k přerušení léčby u 14,3 % pacientů a ke snížení dávky u 1,3 % pacientů.
Tromboembolické příhody
Žilní embolickou nebo trombotickou příhodu (VTE) mělo 3,3 % pacientů, kterým byl podáván Pom + LD-Dex a 2,0 % pacientů, kterým byl podáván HD-Dex. Reakce 3. a 4. stupně se vyskytly u 1,3 % pacientů, kterým byl podáván Pom + LD-Dex a u žádného z pacientů, kterým byl podáván HD-Dex. U pacientů léčených Pom + LD-Dex byla VTE hlášena jako závažná u 1,7 % pacientů, nebyly hlášeny žádné fatální reakce v klinických studiích a VTE nebyla spojena s ukončením léčby.
Profylaktické podávání kyseliny acetylsalicylové (a dalších antikoagulancií u pacientů s vysokým rizikem) bylo povinné pro všechny pacienty v klinických studiích. Doporučuje se antikoagulační léčba, pokud není kontraindikována (viz bod 4.4).
Periferní neuropatie
Pacienti s probíhající periferní neuropatií > 2. stupně byli z klinických studií vyloučeni. Periferní neuropatie byla většinou 1. nebo 2. stupně a vyskytla se u 12,3 % pacientů, kterým byl podáván Pom + LD-Dex a u 10,7 % pacientů, kterým byl podáván HD-Dex. Reakce 3. a 4. stupně se vyskytly u
1,0 % pacientů, kterým byl podáván Pom + LD-Dex a u 1,3 % pacientů, kterým byl podáván HD-Dex. U pacientů léčených Pom + LD-Dex nebyla v klinických zkouškách žádná periferní neuropatie hlášena jako závažná; periferní neuropatie vedla k ukončení léčby u 0,3 % pacientů (viz bod 4.4).
Střední doba do nástupu neuropatie činila 2,1 týdnů, přičemž se pohybovala od 0,1 do 48,3 týdnů. Střední doba do nástupu byla ve studii kratší u pacientů, kterým byl podáván HD-Dex ve srovnání s Pom + LD-Dex (1,3 týdnů oproti 2,1 týdnům).
Střední doba do vyřešení činila ve studii 22,4 týdnů u pacientů, kterým byl podáván Pom + LD-Dex a 13,6 týdnů u pacientů, kterým byl podáván HD-Dex. Spodní limit 95% intervalu spolehlivosti činil 5,3 týdnů u pacientů léčených Pom +LD-Dex a 2,0 týdny u pacientů, kterým byl podáván HD-Dex.
Krvácení
Při léčbě pomalidomidem byly zejména u pacientů s rizikovými faktory, např. se současně podávanými léčivými přípravky, které zvyšují náchylnost ke krvácení, hlášeny hemoragické poruchy. Hemoragické příhody zahrnovaly epistaxi, intrakraniální krvácení a gastrointestinální krvácení.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Pomalidomid byl hodnocen v dávkách 50 mg podávaných v jediné dávce zdravým dobrovolníkům a 10 mg podávaných v opakovaných dávkách jednou denně pacientům s mnohočetným myelomem; nebyly hlášeny žádné závažné nežádoucí příhody v důsledku předávkování. Pomalidomid byl odstraněn hemodialýzou.
V případě předávkování se doporučuje poskytnutí symptomatické péče.
FARMAKOLOGICKÉ VLASTNOSTI
5.
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Imunosupresiva. ATC kód: L04AX06 Mechanismus účinku
Pomalidomid má přímý tumoricidní účinek na myelomy, imunomodulační účinky a inhibuje podporu stromálních buněk pro růst nádorových buněk mnohočetného myelomu. Konkrétně pomalidomid inhibuje proliferaci a indukuje apoptózu hematopoetických nádorových buněk. Pomalidomid rovněž inhibuje proliferaci buněčných linií mnohočetného myelomu rezistentních vůči lenalidomidu a v kombinaci s dexamethasonem působí jak na buněčné linie odpovídající na lenalidomid, tak i na ty, které jsou vůči lenalidomidu rezistentní, a indukuje apoptózu nádorových buněk. Pomalidomid zvyšuje imunitu zprostředkovanou T buňkami a přirozenými zabíječi (NK (Natural Killer) buňkami) a inhibuje tvorbu prozánětlivých cytokinů (např. TNF-a a IL-6) monocyty. Pomalidomid také inhibuje angiogenezi blokováním migrace a adheze endotelových buněk.
Klinická účinnost a bezpečnost
Účinnost a bezpečnost pomalidomidu v kombinaci s dexamethasonem byla hodnocena v multicentrické, randomizované, otevřené studii fáze III (CC-4047-MM-03), v níž byla léčba pomalidomidem v kombinaci s nízkými dávkami dexamethasonu (Pom + LD-Dex) porovnávána s vysokými dávkami dexamethasonu samotného (HD-Dex). Do studie byli zařazeni dříve léčení dospělí pacienti s relabovaným a refrakterním mnohočetným myelomem, kteří absolvovali nejméně dvě předchozí léčebná schémata, včetně lenalidomidu i bortezomibu, a u nichž při poslední léčbě došlo k progresi onemocnění. Do studie bylo zařazeno celkem 455 pacientů: 302 pacientů ve skupině Pom + LD-Dex a 153 pacientů ve skupině HD-Dex. Většina pacientů byla mužského pohlaví (59 %) a bílé pleti (79 %); střední věk v celkové populaci činil 64 let (min. 35 let, max. 87 let).
Pacienti ve skupině Pom + LD-Dex dostávali 4 mg pomalidomidu perorálně v 1.-21. den každého 28denního cyklu. LD-Dex (40 mg) byl podáván jednou denně v 1., 8., 15. a 22. den 28denního cyklu. Ve skupině HD-Dex byl dexamethason (40 mg) podáván jednou denně v 1.-4., 9.-12. a 17.-20. den každého 28denního cyklu. Pacienti ve věku > 75 let zahajovali léčbu 20 mg dexamethasonu. Léčba pacientů pokračovala až do progrese nemoci.
Primárním cílovým parametrem účinnosti bylo přežití bez progrese (progression free survival, PFS) podle kritérií IMWG (International Myeloma Working Group - Mezinárodní pracovní skupina zabývající se mnohočetným myelomem). V populaci ITT činil medián doby PFS po revizi IRAC (Nezávislá revizní a posudková komise Independent Review Adjudication Committee) na základě kritérií IMWG 15,7 týdne (95% IS: 13,0; 20,1) ve skupině Pom + LD-Dex a vypočtená četnost přežití bez příhod po dobu 26 týdnů byla 35,99 % (± 3,46 %). Ve skupině HD-Dex byl medián doby PFS 8,0 týdne (95% IS: 7,0; 9,0) a vypočtená četnost přežití bez příhod po dobu 26 týdnů byla 12,15 % (± 3,63 %).
Parametr přežití bez progrese byl hodnocen v několika relevantních podskupinách: podle pohlaví, rasy, stavu ECOG,stratifikačních faktorů (věk, populace s onemocněním, předchozí léčby myelomu [2, > 2]), vybraných prognosticky významných parametrů (vstupní hladina beta-2 mikroglobulinu, vstupní hladiny albuminu, vstupní poruchy funkce ledvin a cytogenetické riziko) a expozice a refrakterita k předchozím léčbám myelomu. Bez ohledu na hodnocené podskupiny byla hodnota PFS celkově konzistentní s hodnotami pozorovanými v populaci ITT v obou léčebných skupinách.
Výsledky přežití bez progrese v populaci ITT jsou shrnuty v tabulce 1. Kaplan-Meierova křivka parametru PFS v populaci ITT je znázorněna na obrázku 1.
|
(stratifikovaný test log-rank' |
(populace ITT) | |
|
Pom + LD- Dex (N=302) |
HD-Dex (N=153) | |
|
Přežití bez progrese, N |
302 (100,0) |
153 (100,0) |
|
S cenzurou, n (%) |
138 (45,7) |
50 (32,7) |
|
S progresí/zemřelo, n (%) |
164 (54,3) |
103 (67,3) |
|
Doba přežití bez progrese (týdny) | ||
|
Medián3 |
15,7 |
8,0 |
|
Oboustranný 95% ISb |
[13,0; 20,11 |
[7,0; 9,01 |
|
Poměr rizik (Pom + LD-Dex : HD-Dex) oboustranný 95% ISc |
0,45 [0,35; 0,59] | |
|
P hodnota oboustranného log-rank testud |
< 0,001 | |
Poznámka: IS = interval spolehlivosti; IRAC = Nezávislá revizní a posudková komise (Independent Review Adjudication Commitee);
NE = nelze určit.
a Medián podle Kaplan-Meierova odhadu. b 95% interval spolehlivosti mediánu doby přežití bez progrese.
c Vychází z Coxova modelu poměrných rizik porovnávajícího rizikové funkce spojené s léčebnými skupinami, stratifikovaných podle věku (< 75 let vs. > 75 let), populace s onemocněním (refrakterita jak k lenalidomidu, tak i bortezomibu vs. bez refrakterity k oběma lékům) a počet předchozích léčeb myelomu (= 2 vs. > 2).
d p hodnota vychází ze stratifikovaného log-rank testu se stejnými stratifikačními faktory jako u výše uvedeného Coxova modelu.
Údaje ke dni: 7. září 2012
Obrázek 1:
Přežití bez progrese po revizi odezvy IRAC podle kritérií IMWG (stratifikovaný log-rank test) (populace ITT)
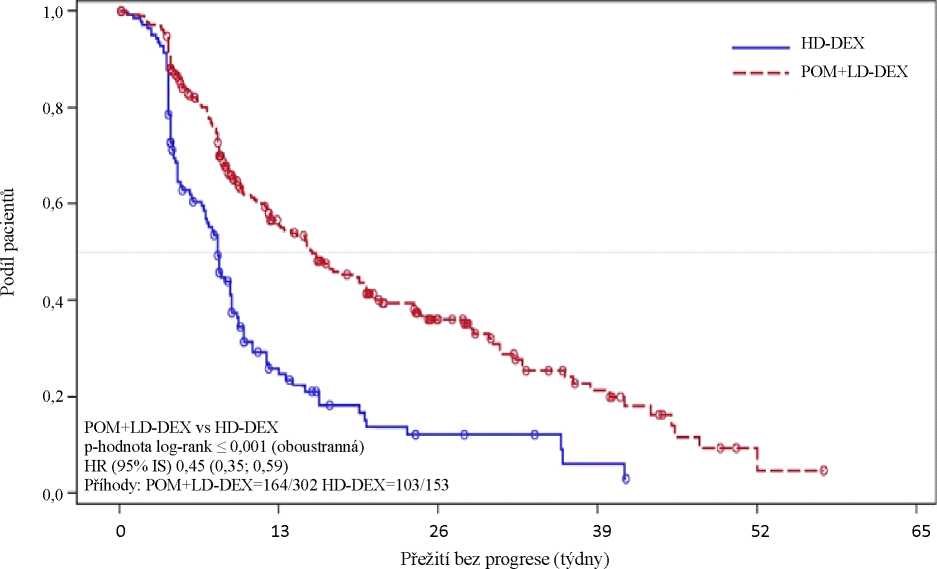
Údaje ke dni: 7. září 2012
Hlavním sekundárním cílovým parametrem studie bylo celkové přežití. Celkem 226 (74,8 %) pacientů ve skupině Pom + LD-Dex a 95 (62,1 %) pacientů ve skupině HD-Dex přežilo ke dni uzávěrky (7. září 2012). Medián doby celkového přežití podle Kaplan-Meierova odhadu nebyl ve skupině Pom + LD-Dex dosažen, lze však očekávat, že bude činit nejméně 48 týdnů, což je spodní hranice
95% IS. Medián doby celkového přežití ve skupině HD-Dex činil 34 týdnů (95% IS: 23,4; 39,9). Četnost přežití po dobu 1 roku bez příhody byla 52,6 % (± 5,72 %) ve skupině Pom + LD-Dex a 28,4 % (± 7,51 %) ve skupině HD-Dex. Rozdíl mezi celkovým přežitím v obou léčebných skupinách byl statisticky významný (p < 0,001).
Výsledky celkového přežití v populaci ITT jsou shrnuty v tabulce 2. Kaplan-Meierova křivka celkového přežití v populaci ITT je znázorněna na obrázku 2.
Na základě výsledků obou cílových parametrů, přežití bez progrese a celkového přežití, doporučila komise monitorující údaje, která byla pro tuto studii ustavena, studii dokončit a pacienty ve skupině HD-Dex převést do skupiny Pom + LD-Dex.
Tabulka 2: Celkové přežití: populace ITT
|
Statistika |
Pom + LD-Dex (N=302) |
HD-Dex (N=153) | |
|
N |
302 (100,0) |
153 (100,0) | |
|
S cenzurou |
n (%) |
226 (74,8) |
95 (62,1) |
|
Zemřelo |
n (%) |
76 (25,2) |
58 (37,9) |
|
Doba přežití (týdny) |
Medián3 |
NE |
34,0 |
|
Oboustranný 95% ISb |
[48,1; NE] |
[23,4; 39,9] | |
|
Poměr rizik (Pom + LD-Dex : HD-Dex) [oboustranný 95% ISc] |
0,53[0,37; 0,74] | ||
|
P hodnota oboustranného log-rank testud |
< 0,001 | ||
Poznámka: IS = interval spolehlivosti; NE = nelze určit. a Medián podle Kaplan-Meierova odhadu. b 95% interval spolehlivosti mediánu doby celkového přežití.
c Vychází z Coxova modelu poměrných rizik porovnávajícího rizikové funkce spojené s léčebnými skupinami. d p hodnota vychází z nestratifikovaného log-rank testu.
Údaje ke dni: 7. září 2012
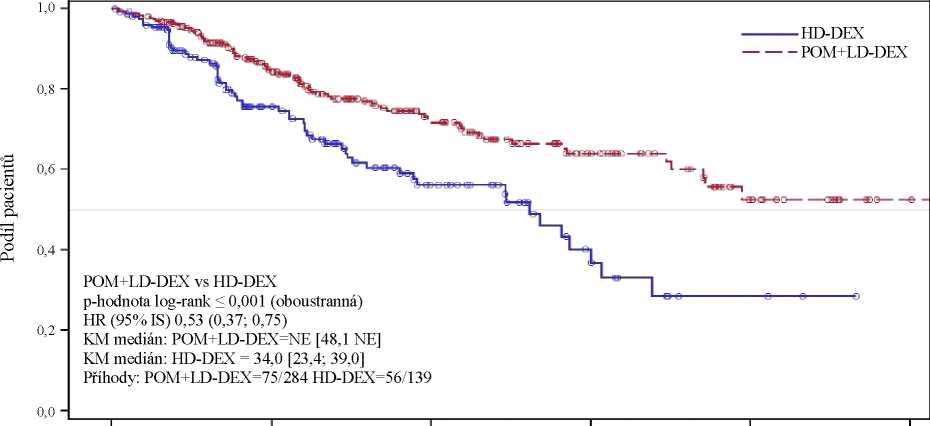
0 13 26 39 52 65
Celkové přežití (týdny)
Údaje ke dni: 7. září 2012
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Imnovid u všech podskupin pediatrické populace u mnohočetného myelomu (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Absorpce
Pomalidomid dosahuje maximální plazmatické koncentrace (Cmax) za 2 až 3 hodiny a po perorálním podání jediné dávky je absorbováno nejméně 73 %. Systémová expozice (AUC) pomalidomidu se zvyšuje přibližně lineárně a úměrně dávce. Po podání několika dávek má pomalidomid akumulační poměr 27 až 31 % AUC.
Při současném podávání stravy s vysokým obsahem tuku a vysoce kalorické stravy dochází ke zpomalení absorpce a snížení střední plazmatické Cmax přibližně o 27 %, ale účinek na celkovou míru absorpce je s 8% poklesem střední AUC minimální. Pomalidomid lze tedy podávat bez ohledu na příjem potravy.
Distribuce
Střední zdánlivý distribuční objem (Vd/F) pomalidomidu je 62 až 138 l v ustáleném stavu. Pomalidomid je distribuován do semene zdravých jedinců v množství přibližně 67 % plazmatické koncentrace 4 hodiny po podání dávky (přibližně Tmax) po 4 dnech dávkování 2 mg jednou denně. Vazba enantiomerů pomalidomidu na lidské plazmatické proteiny in vitro je v rozmezí 12 % až 44 % a není závislá na koncentraci.
Biotransformace
Pomalidomid je hlavní cirkulující složkou (přibližně 70 % plazmatické radioaktivity) in vivo u zdravých jedinců, kteří dostali jedinou perorální dávku [14C]-pomalidomidu (2 mg). V plazmě nebyly přítomny žádné metabolity při > 10% poměru k základní nebo celkové radioaktivitě v plazmě.
Převládající metabolickou cestou eliminace radioaktivity jsou hydroxylace s následnou glukuronidací nebo hydrolýza. In vitro bylo zjištěno, že CYP1A2 a CYP3A4 jsou primárními enzymy, které se podílejí na CYP zprostředkované hydroxylaci pomalidomidu, k níž v menší míře také přispívají CYP2C19 a CYP2D6. Pomalidomid je rovněž substrátem P-glykoproteinu in vitro. Současné podávání pomalidomidu s ketokonazolem, silným inhibitorem CYP3A4/5 a P-glykoproteinu, nebo karbamazepinem, silným induktorem CYP3A4/5, nemělo žádný klinicky relevantní účinek na expozici pomalidomidu. Současné podávání pomalidomidu s fluvoxaminem, silným inhibitorem CYP1A2, za přítomnosti ketokonazolu zvýšilo střední expozici pomalidomidu o 107 % při 90% intervalu spolehlivosti [91 %; 124 %] v porovnání s kombinací pomalidomidu a ketokonazolu. Ve druhé studii, která hodnotila přispění samotného inhibitoru CYP1A2 ke změnám metabolismu, zvýšilo současné podávání samotného fluvoxaminu s pomalidomidem střední expozici pomalidomidu o 125 % při 90% intervalu spolehlivosti [98 % až 157 %] v porovnání se samotným pomalidomidem. Jestliže jsou s pomalidomidem současně podávány silné inhibitory CYP1A2 (např. ciprofloxacin, enoxacin a fluvoxamin), snižte dávku pomalidomidu na 50 %. Podávání pomalidomidu u kuřáků nemělo klinicky relevantní účinek na expozici pomalidomidu v porovnání s expozicí pomalidomidu pozorovanou u nekuřáků, ačkoli je známo, že kouření tabáku indukuje izoformu CYP1A2.
Dle údajů in vitro pomalidomid není inhibitorem ani induktorem izoenzymů cytochromu P-450 a neinhibuje žádné transportéry léčiv, které byly studovány. Klinicky relevantní lékové interakce se při současném podávání pomalidomidu a substrátů těchto cest neočekávají.
Eliminace
Pomalidomid je vylučován se středním plazmatickým poločasem přibližně 9,5 hodiny u zdravých jedinců a přibližně 7,5 hodiny u pacientů s mnohočetným myelomem. Pomalidomid má střední celkovou tělesnou clearance (CL/F) přibližně 7-10 l/h.
Po jediném perorálním podání [14C]-pomalidomidu (2 mg) zdravým jedincům bylo přibližně 73 % radioaktivní dávky vyloučeno močí a 15 % stolicí, s přibližně 2 %, resp. 8 % podaného radioaktivního uhlíku vyloučenými ve formě pomalidomidu v moči a stolici.
Pomalidomid je ve značné míře metabolizován dříve, než dojde k vyloučení, s výslednými metabolity vylučovanými primárně v moči. Tři převládající metabolity v moči (tvořené prostřednictvím hydrolýzy nebo hydroxylace s následnou glukuronidací) odpovídají za přibližně 23 %, 17 % a 12 % dávky v moči.
Metabolity závislé na CYP odpovídají přibližně za 43 % celkové vyloučené radioaktivity, zatímco hydrolytické metabolity nezávislé na CYP odpovídají za 25 % a vyloučení nezměněného pomalidomidu odpovídalo za 10 % (2 % v moči a 8 % ve stolici).
Farmakokinetika populace
Na základě farmakokinetických analýz populace za použití dvojkompartmentového modelu měli zdraví a pacienti s mnohočetným myelomem srovnatelnou zdánlivou clearance (CL/F) a zdánlivý centrální distribuční objem (V2/F). V periferních tkáních byl pomalidomid přednostně vstřebán nádory, se zdánlivou periferní distribuční clearance (Q/F) 3,7krát vyšší než u zdravých jedinců a zdánlivým distribučním objemem (V3/F) 8krát vyšším než u zdravých jedinců.
Pediatrická populace
Nejsou dostupné žádné údaje o podávání pomalidomidu pediatrickým nebo dospívajícím pacientům (< 18 let věku).
Starší lidé
Na základě farmakokinetických analýz populace u zdravých jedinců a u pacientů s mnohočetným myelomem nebyl pozorován významný vliv věku (19-83 let) na perorální clearance pomalidomidu.
V klinických studiích nebyla u starších pacientů (> 65 let) vystavených pomalidomidu vyžadována žádná úprava dávkování. Viz bod 4.2.
Pacienti s poruchou funkce ledvin
Ve farmakokinetických analýzách populace bylo prokázáno, že farmakokinetické parametry pomalidomidu nejsou výrazně ovlivněny u pacientů s poruchou funkce ledvin (definovanou pomocí clearance kreatininu nebo odhadu glomerulární filtrace [estimatedglomerularfiltration rate, eGFR]) v porovnání s pacienty s normální funkcí ledvin (CrCl > 60 ml/min). Střední normalizovaná AUC expozice pomalidomidu byla 98,2 % při 90% intervalu spolehlivosti [77,4 % až 120,6 %] u pacientů se středně těžkou poruchou funkce ledvin (eGFR > 30 až < 45 ml/min/1,73 m2) v porovnání s pacienty s normální funkcí ledvin.Střední normalizovaná AUC expozice pomalidomidu byla 100,2 % při 90% intervalu spolehlivosti [79,7 % až 127,0 %] u pacientů s těžkou poruchou funkce ledvin, jejichž stav nevyžadoval dialýzu (CrCl < 30 nebo eGFR < 30 ml/min/1,73 m2) v porovnání s pacienty s normální funkcí ledvin. Střední normalizovaná AUC expozice pomalidomidu se zvýšila o 35,8 % při 90% intervalu spolehlivosti [7,5 % až 70,0 %] u pacientů s těžkou poruchou funkce ledvin, jejichž stav vyžadoval dialýzu (CrCl < 30 ml/min, stav vyžadující dialýzu) v porovnání s pacienty s normální funkcí ledvin. Střední změny expozice pomalidomidu v každé z těchto skupin poruch funkce ledvin nejsou tak významné, aby bylo nutné upravovat dávkování.
Pacienti s poruchou funkce jater
U pacientů s poruchou funkce jater (definovanou za použití kritérií podle Childa a Pugha) byly mírně změněné farmakokinetické parametry v porovnání se zdravými jedinci. Střední expozice pomalidomidu se zvýšila o 51 % při 90% intervalu spolehlivosti [9 % až 110 %] u pacientů s lehkou poruchou funkce jater v porovnání se zdravými jedinci. Střední expozice pomalidomidu se zvýšila
0 58 % při 90% intervalu spolehlivosti [13 % až 119 %] u pacientů se středně těžkou poruchou funkce jater v porovnání se zdravými jedinci. Střední expozice pomalidomidu se zvýšila o 72 % při 90% intervalu spolehlivosti [24 % až 138 %] u pacientů s těžkou poruchou funkce jater v porovnání se zdravými jedinci. Střední zvýšení expozice pomalidomidu v každé z těchto skupin poruch funkce jater nejsou tak významná, aby bylo nutné upravovat harmonogram dávek nebo dávku (viz bod 4.2).
5.3 Předklinické údaje vztahující se k bezpečnosti
Toxikologické studie s opakovanými dávkami
U potkanů bylo dlouhodobé podávání pomalidomidu v dávkách 50, 250 a 1000 mg/kg/den po dobu 6 měsíců dobře tolerováno. Žádné nepříznivé nálezy nebyly zaznamenány až do 1000 mg/kg/den (175násobný poměr expozice pro 4mg klinickou dávku).
Pomalidomid byl hodnocen ve studiích s opakovanými dávkami podávanými opicím po dobu až 9 měsíců. V těchto studiích vykazovaly opice větší vnímavost k účinkům pomalidomidu než potkani. Primární toxicity pozorované u opic byly spojeny s hematopoetickými/lymforetikulárními systémy.
V 9měsíční studii, ve které byly opicím podávány dávky 0,05, 0,1 a 1 mg/kg/den, byla u 6 zvířat pozorována morbidita a předčasná eutanázie při dávkách 1 mg/kg/den a tyto účinky byly připisovány imunosupresivním účinkům (stafylokokové infekce, snížené lymfocyty v periferní krvi, chronický zánět tlustého střeva, histologická lymfoidní deplece a hypocelularita kostní dřeně) při vysokých expozicích pomalidomidu (15násobný poměr expozice pro 4mg klinickou dávku). Tyto imunosupresivní účinky vedly u 4 opic k předčasné eutanázii v důsledku špatného zdravotního stavu (vodnatá stolice, nechuť k příjmu potravy, snížený příjem potravy a váhový úbytek). Histopatologické vyšetření těchto zvířat prokázalo chronický zánět tlustého střeva a vilózní atrofii tenkého střeva. Stafylokoková infekce byla pozorována u 4 opic, z nichž 3 zvířata odpověděla na antibiotickou léčbu a
1 zvíře uhynulo bez léčby. Nálezy konzistentní s akutní myelogenní leukemií také vedly u 1 opice k předčasné eutanázii. Klinická pozorování a klinické patologické stavy a/nebo změny kostní dřeně pozorované u tohoto zvířete byly konzistentní s imunosupresí. Při dávkách 1 mg/kg/den byla rovněž pozorována minimální nebo mírná proliferace žlučových cest a související zvýšení hladin ALP a GGT. Vyhodnocení zotavených zvířat prokázala, že všechny nálezy v souvislosti s léčbou byly reverzibilní po 8 týdnech vysazení dávky, vyjma proliferace intrahepatálních žlučových cest, která byla pozorována u 1 zvířete ve skupině užívající 1 mg/kg/den. Hladina bez jakýchkoli nežádoucích účinků (No Observed Adverse Effect Level, NOAEL) byla 0,1 mg/kg/den (0,5násobný poměr expozice pro 4mg klinickou dávku).
Genotoxicita/karcinogenicita
Pomalidomid nebyl mutagenní v testech mutací na bakteriálních a savčích buňkách a nepřivodil chromozomální aberace v lidských lymfocytech z periferní krve nebo mikronukleární formaci v polychromatických erytrocytech v kostní dřeni potkanů, kteří dostávali dávky až 2000 mg/kg/den. Studie karcinogenicity nebyly provedeny.
Fertilita a časný embryonální vývoj
Ve studiích fertility a časného embryonálního vývoje u potkanů byl pomalidomid podáván samcům i samicím v dávkách 25, 250 a 1000 mg/kg/den. Vyšetření dělohy ve 13. den gestace prokázalo snížení středního počtu životaschopných embryí a zvýšení postimplantační ztráty při všech dávkových hladinách. Hladina NOAEL pro tyto pozorované účinky byla tedy < 25 mg/kg/den (AUC24h byla 39 960 ng»h/ml [nanogram»hodina/mililitr] při této nejnižší hodnocené dávce a poměr expozice byl 99násobný pro 4mg klinickou dávku). Při spáření samců léčených v této studii s neléčenými samicemi byly všechny parametry týkající se dělohy srovnatelné s kontrolní skupinou. Na základě těchto výsledků byly pozorované účinky připsány léčbě samic.
Embryofetální vývoj
Pomalidomid vykazoval teratogenní účinky jak u potkanů, tak i u králíků, pokud byl podáván v období hlavní organogeneze. V toxikologické studii embryofetálního vývoje u potkanů byly při všech dávkových hladinách (25, 250 a 1000 mg/kg/den) pozorovány malformace v podobě nepřítomnosti močového měchýře, nepřítomnosti štítné žlázy a fúze a vychýlení bederních a hrudních obratlů (středové a/nebo neurální oblouky).
V této studii nebyla pozorována žádná maternální toxicita. Hladina NOAEL pro maternální toxicitu byla tedy 1000 mg/kg/den a hladina NOAEL pro vývojovou toxicitu byla < 25 mg/kg/den (AUC24h byla 34 340 ng»h/ml v 17. den gestace při této nejnižší hodnocené dávce a poměr expozice byl 85násobný pro 4mg klinickou dávku). Pomalidomid v dávkách 10 až 250 mg/kg u králíků způsoboval embryofetální vývojové malformace. Zvýšené srdeční anomálie byly pozorovány při všech dávkách, významný nárůst byl zaznamenán při dávce 250 mg/kg/den. Při dávkách 100 a 250 mg/kg/den byl pozorován mírný nárůst postimplantační ztráty a mírný pokles tělesné hmotnosti plodu. Fetální malformace při dávce 250 mg/kg/den zahrnovaly anomálie končetin (ohnuté a/nebo přetočené přední a/nebo zadní končetiny, nepřirostlý nebo chybějící prst) a související kosterní malformace (nezkostnatělý metakarpus, vychýlená falanga nebo metakarpus, chybějící prst, nezkostnatělá falanga a krátká nezkostnatělá nebo ohnutá tibie), mírnou dilataci laterálního ventrikulu v mozku, abnormální umístění pravé podklíčkové tepny, nepřítomnost středního plicního laloku, nízko posazené ledviny, změněnou morfologii jater, nekompletně nebo zcela nezkostnatělou pánev, zvýšený průměr nadpočetných hrudních žeber a snížený průměr zkostnatělých tarsů. Při dávkách 100 a 250 mg/kg/den bylo pozorováno mírné snížení maternálního váhového přírůstku, významné snížení triglyceridů a významné snížení absolutních a relativních hmotností sleziny. Maternální hladina NOAEL byla 10 mg/kg/den a hladina NOAEL pro vývojovou toxicitu byla < 10 mg/kg/den (AUC24h byla 418 ng»h/ml v 19. den gestace při této nejnižší hodnocené dávce, což je obdobná hodnota, jaká byla získána při 4mg klinické dávce).
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Obsah tobolky:
Mannitol
Předbobtnalý škrob Natrium-stearyl-fumarát
Obal tobolky:
Obal 1 mg tobolky obsahuje: želatinu, oxid titaničitý (E171), indigokarmín (E132), žlutý oxid železitý (E172) a bílý a černý inkoust.
Potisková barva:
Obal 1 mg tobolky obsahuje: bílý inkoust - šelak, oxid titaničitý (E171), simetikon, propylenglykol (E1520) a koncentrovaný roztok amoniaku (E527); černý inkoust - šelak, černý oxid železitý (E172), propylenglykol (E1520) a koncentrovaný roztok amoniaku (E527).
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
4 roky.
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
6.5 Druh obalu a obsah balení
Tobolky jsou baleny v blistrech z polyvinylchloridu (PVC) / polychlortrifluorethylenu (PCTFE), s protlačovací hliníkovou fólií.
Velikost balení: 21 tobolek.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Tobolky se nesmí otvírat ani drtit. Pokud se prášek pomalidomidu dostane do kontaktu s pokožkou, je nutné pokožku okamžitě a důkladně umýt mýdlem a vodou. Pokud se pomalidomid dostane do kontaktu se sliznicí, je nutné postižené místo důkladně opláchnout vodou.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky. Nepoužitý léčivý přípravek se musí na konci léčby vrátit lékárníkovi.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Celgene Europe Ltd.
1 Longwalk Road Stockley Park Uxbridge UB11 1DB Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/13/850/001
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 5. srpna 2013
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Imnovid 2 mg tvrdé tobolky
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna tvrdá tobolka obsahuje pomalidomidum 2 mg.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Tvrdá tobolka.
Tvrdá tobolka Imnovid 2 mg: tmavě modré neprůhledné víčko a oranžové neprůhledné tělo s nápisem „POML 2 mg“ v bílé barvě, velikost 2, tvrdá želatinová tobolka.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Imnovid je v kombinaci s dexamethasonem indikován k léčbě dospělých pacientů s relabujícím a refrakterním mnohočetným myelomem, kteří absolvovali alespoň dvě předchozí léčebná schémata, zahrnující jak lenalidomid, tak i bortezomib, a při poslední terapii vykazovali progresi onemocnění.
4.2 Dávkování a způsob podání
Léčba musí být zahájena a vedena pod dohledem lékaře se zkušenostmi v léčbě mnohočetného myelomu.
Dávkování
Doporučená počáteční dávka přípravku Imnovid je 4 mg jednou denně perorálně v 1. až 21. den opakovaných 28denních cyklů. Doporučená dávka dexamethasonu je 40 mg perorálně jednou denně v 1., 8., 15., a 22. den každého 28denního léčebného cyklu.
Dávkování je třeba udržovat a upravovat na základě klinických a laboratorních nálezů.
Při progresi onemocnění je nutné léčbu přerušit.
Úprava dávky _pomalidomidu nebo _přerušení léčby
Pokyny pro přerušení léčby nebo snížení dávky pomalidomidu v souvislosti s hematologickými nežádoucími účinky jsou uvedeny v tabulce níže.
• Pokyny pro úpravu dávky pomalidomidu
|
Toxicita |
Úprava dávky |
|
Neutropenie • ANC* < 0,5 x 109/l nebo febrilní neutropenie (horečka > 38,5 °C a ANC < 1 x 109/l) |
Přerušení léčby pomalidomidem, sledování kompletního KO** v týdenních intervalech. |
|
• Návrat ANC na > 1 x 109/l |
Pokračování v léčbě pomalidomidem v dávce 3 mg denně. |
|
• Každý následný pokles na < 0,5 x 109/l |
Přerušení léčby pomalidomidem. |
|
• Návrat ANC na > 1 x 109/l |
Pokračování v léčbě pomalidomidem v dávce o 1 mg nižší než předchozí dávka. |
|
Trombocvtopenie • Počet trombocytů < 25 x 109/l |
Přerušení léčby pomalidomidem, sledování kompletního KO** v týdenních intervalech. |
|
• Návrat počtu trombocytů na > 50 x 109/l |
Pokračování v léčbě pomalidomidem v dávce 3 mg denně. |
|
• Každý následný pokles na < 25 x 109/l |
Přerušení léčby pomalidomidem. |
|
• Návrat počtu trombocytů na > 50 x 109/l |
Pokračování v léčbě pomalidomidem v dávce o 1 mg nižší než předchozí dávka. |
*ANC - absolutní počet neutrofilů; **KO - krevní obraz;
Nový cyklus léčby pomalidomidem lze zahájit, pouze pokud je počet neutrofilů > 1 x 109/l a počet trombocytů > 50 x 109/l.
V případě neutropenie má lékař zvážit použití růstových faktorů.
V případě jiných nežádoucích účinků 3. a 4. stupně, u nichž se usuzuje na spojitost s léčbou pomalidomidem, ukončete léčbu. Po zmírnění nežádoucího účinku na < 2. stupeň, dle uvážení lékaře, obnovte léčbu s dávkou o 1 mg nižší, než byla předchozí dávka.
Pokud se nežádoucí účinky vyskytnou po snížení dávky na 1 mg, je nutné léčivý přípravek vysadit.
Při výskytu kožní vyrážky 2. až 3. stupně je třeba zvážit přerušení nebo ukončení léčby pomalidomidem. Léčba pomalidomidem musí být ukončena při výskytu angioedému, kožní vyrážky 4. stupně a exfoliativní nebo bulózní vyrážky; po ukončení léčby z důvodu těchto reakcí se nemá léčba znovu zahajovat.
Jestliže jsou s pomalidomidem současně podávány silné inhibitory CYP1A2 (např. ciprofloxacin, enoxacin a fluvoxamin), snižte dávku pomalidomidu o 50 %.
• Pokyny pro úpravu dávky dexamethasonu
|
Toxicita |
Úprava dávky |
|
Dyspepsie = 1.-2. stupeň |
Udržování dávky a léčba blokátory histaminu (H2) nebo ekvivalenty. V případě přetrvávání příznaků snížení dávky o jednu úroveň. |
|
Dyspepsie > 3. stupeň |
Přerušení podávání dávky až do zvládnutí příznaků. Přidání blokátoru H2 nebo ekvivalentu a podávání snížené dávky o jednu úroveň po obnovení podávání. |
|
Edém > 3. stupeň |
Použití diuretik podle potřeby a snížení dávky o jednu úroveň. |
|
Zmatenost nebo změny nálady > 2. stupeň |
Přerušení podávání dávky do vymizení příznaků. Po obnovení podávání snížit dávku o jednu úroveň. |
|
Toxicita |
Úprava dávky |
|
Svalová slabost > 2. stupeň |
Přerušení podávání dávky do fáze svalové slabosti < 1. stupeň. Obnovení podávání s dávkou sníženou o jednu úroveň. |
|
Hyperglykemie > 3. stupeň |
Snížení dávky o jednu úroveň. Léčba insulinem nebo perorálními hypoglykemickými přípravky dle potřeby. |
|
Akutní pankreatitida |
Ukončení podávání dexamethasonu pacientovi. |
|
Další nežádoucí účinky související s dexamethasonem > 3. stupeň |
Přerušení podávání dexamethasonu do vymizení nežádoucích účinků do < 2. stupeň. Pokračování podávání s dávkou sníženou o jednu úroveň. |
Úrovně snížení dávky dexamethasonu:
Úrovně snížení dávky (< 75 let): počáteční dávka 40 mg; úroveň 1 snížení dávky - 20 mg; úroveň 2 snížení dávky- 10 mg v 1., 8., 15., a 22. den každého 28denního léčebného cyklu.
Úrovně snížení dávky (> 75 let): počáteční dávka 20 mg; úroveň 1 snížení dávky - 12 mg; úroveň 2 snížení dávky - 8 mg v 1., 8., 15., a 22. den každého 28denního léčebného cyklu.
Pokud zotavování z toxicit trvá déle než 14 dní, pak se dávka dexamethasonu sníží o jednu úroveň dávky.
Zvláštní populace
Pediatrická _ populace
Použití přípravku Imnovid v indikaci mnohočetného myelomu u dětí ve věku 0-17 let není relevantní.
Starší _ pacienti
Úprava dávky pomalidomidu není nutná. U pacientů ve věku > 75 let je počáteční dávka dexamethasonu 20 mg jednou denně v 1., 8., 15. a 22. den každého 28denního léčebného cyklu.
Porucha _ funkce ledvin
U pacientů s poruchou funkce ledvin není nutná žádná úprava dávky pomalidomidu. Ve dnech, kdy pacienti podstupují hemodialýzu, má být dávka pomalidomidu užita po hemodialýze..
Porucha _ funkce _ jater
Pacienti s celkovým sérovým bilirubinem > 2,0 mg/dl byli z klinických studií vyřazeni. Porucha funkce jater má mírný vliv na farmakokinetiku pomalidomidu (viz bod 5.2). U pacientů s poruchou funkce jater definovanou za použití kritérií podle Childa a Pugha není nutná žádná úprava počáteční dávky pomalidomidu. Je však nutné pacienty s poruchou funkce jater důkladně sledovat pro případ výskytu nežádoucích účinků a podle potřeby snížit dávku nebo přerušit podávání pomalidomidu.
Způsob podání Perorální podání.
Přípravek Imnovid se má užívat každý den ve stejnou dobu. Tobolky se nesmí otevírat, lámat ani žvýkat (viz bod 6.6). Tento léčivý přípravek je třeba polykat vcelku, nejlépe zapít vodou. Tobolky se mohou užívat s jídlem nebo bez něho. Pokud pacient zapomene jeden den užít dávku přípravku Imnovid, může užít normální předepsanou dávku v plánovaný čas následujícího dne. Pacienti nesmí upravovat dávku, aby nahradili vynechanou dávku z předchozího dne.
Pro vyjmutí tobolky z blistru se doporučuje zatlačit pouze na jedné straně, aby se minimalizovalo riziko deformace či rozlomení tobolky.
4.3 Kontraindikace
- Těhotenství.
- Ženy, které mohou otěhotnět, pokud nejsou splněny všechny podmínky Programu prevence početí (viz body 4.4 a 4.6).
- Pacienti muži, kteří nejsou schopni dodržovat požadovaná antikoncepční opatření (viz bod 4.4).
- Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Teratogenita
Pomalidomid se nesmí užívat v průběhu těhotenství, neboť se očekávají teratogenní účinky. Pomalidomid je strukturálně podobný thalidomidu. Thalidomid je známý lidský teratogen, který způsobuje těžké a život ohrožující vrozené vady. Pomalidomid vykazoval teratogenní účinky jak u potkanů, tak i u králíků, pokud byl podáván v období hlavní organogeneze (viz bod 5.3).
Všechny pacientky musí splňovat podmínky Programu prevence početí (PPP), pokud není spolehlivý důkaz o tom, že u pacientky je možnost otěhotnění vyloučena.
Kritéria pro ženu, která nemůže otěhotnět
Pacientka nebo partnerka pacienta-muže je považována za neschopnou otěhotnět, pokud splňuje alespoň jedno z následujících kritérií:
• věk > 50 let a přirozená amenorea po dobu > 1 rok*
• předčasné selhání vaječníků potvrzené gynekologem
• předchozí oboustranná adnexektomie nebo hysterektomie
• genotyp XY, Turnerův syndrom, ageneze dělohy.
*Amenorea po protinádorové terapii nebo během kojení nevylučuje možnost otěhotnění pacientky.
Poradenství
U žen, které mohou otěhotnět, je pomalidomid kontraindikován, pokud nejsou splněna všechna následující kritéria:
• Žena si je vědoma očekávaného teratogenního rizika pro nenarozené dítě.
• Žena chápe nutnost účinné antikoncepce praktikované bez přerušení po 4 týdny před začátkem léčby, po celou dobu během ní a 4 týdny po jejím ukončení.
• I když má fertilní žena amenoreu, musí používat účinnou antikoncepci.
• Žena musí být schopna dodržovat účinná antikoncepční opatření.
• Žena je informována a je si vědoma potenciálních následků těhotenství a nutnosti rychle informovat lékaře, pokud hrozí riziko těhotenství.
• Žena chápe nutnost zahájení léčby hned po vydání pomalidomidu, kterému předchází negativní těhotenský test.
• Žena chápe nutnost a je ochotna absolvovat těhotenské testy každé 4 týdny, kromě případů potvrzené sterilizace podvazem vejcovodů.
• Žena potvrdí, že si je vědoma rizik a nutných bezpečnostních opatření spojených s užíváním pomalidomidu.
Předepisující lékař musí u žen, které mohou otěhotnět, zajistit, že:
• Pacientka splňuje podmínky Programu prevence početí (PPP), včetně ujištění, že těmto podmínkám patřičně porozuměla.
• Pacientka výše uvedené podmínky potvrdila.
U mužů užívajících pomalidomid farmakokinetická data ukázala, že pomalidomid je během léčby přítomen v semeni. Z preventivních důvodů musí všichni muži užívající pomalidomid splňovat následující podmínky:
• Muž si je vědom očekávaného teratogenního rizika při pohlavním styku s těhotnou ženou nebo se ženou, která může otěhotnět.
• Muž chápe nutnost používání kondomu, pokud má pohlavní styk s těhotnou ženou nebo se ženou, která může otěhotnět a nepoužívá účinnou antikoncepci, během léčby a po dobu 7 dní
po přerušení a/nebo ukončení léčby. Muži, kteří podstoupili vasektomii, mají při pohlavním styku s těhotnou ženou nebo se ženou ve fertilním věku, používat kondom, jelikož semenná tekutina může i při absenci spermií obsahovat pomalidomid.
• Muž si je vědom, že musí ihned informovat svého ošetřujícího lékaře, pokud jeho partnerka otěhotní v průběhu jeho léčby pomalidomidem nebo 7 dní po ukončení léčby, a že se doporučuje partnerku předat specialistovi nebo zkušenému teratologovi, aby posoudil riziko a poskytl doporučení.
Antikoncepce
Ženy, které mohou otěhotnět, musí používat jednu účinnou metodu antikoncepce po 4 týdny před léčbou, během ní a 4 týdny po léčbě pomalidomidem, a také po dobu případného přerušení léčby, pokud se nezavážou k absolutní a trvalé pohlavní abstinenci, kterou musí každý měsíc potvrdit. Pokud pacientka nepoužívá účinnou antikoncepci, musí být odeslána k vyškolenému zdravotníkovi, který jí s výběrem antikoncepční metody poradí, aby antikoncepce mohla být nasazena.
Vhodné metody antikoncepce například jsou:
• implantát
• nitroděložní tělísko uvolňující levonorgestrel
• depotní medroxyprogesteron-acetát
• sterilizace podvazem vejcovodů
• pohlavní styk pouze s mužem po vasektomii; vasektomie musí být potvrzena dvěma negativními testy semene
• antikoncepční tablety inhibující ovulaci obsahující pouze progesteron (tj. desogestrel)
Vzhledem ke zvýšenému riziku žilní tromboembolie u pacientek s mnohočetným myelomem užívajících pomalidomid a dexamethason se kombinovaná perorální antikoncepce nedoporučuje (viz také bod 4.5). Pokud pacientka v současnosti používá kombinovanou perorální antikoncepci, je třeba přejít na některou z účinných antikoncepčních metod uvedených výše. Riziko žilní tromboembolie trvá po dobu 4-6 týdnů po vysazení kombinované perorální antikoncepce. Účinnost steroidních antikoncepčních přípravků může být během současného podávání dexamethasonu snížena (viz bod 4.5).
Implantáty a nitroděložní tělíska uvolňující levonorgestrel jsou spojeny se zvýšeným rizikem infekce v době zavedení a nepravidelného vaginálního krvácení. Je třeba zvážit profylaktické podávání antibiotik, zvláště u pacientek s neutropenií.
Zavádění nitroděložních tělísek uvolňujících měď se nedoporučuje vzhledem k potenciálnímu riziku infekce v době zavedení a ztrátě menstruační krve, která může způsobit komplikace u pacientek trpících těžkou neutropenií nebo těžkou trombocytopenií.
Těhotenské testy
V souladu s místní praxí je třeba zajistit provádění těhotenských testů s minimální citlivostí 25 mIU/ml pod dohledem lékaře u žen, které mohou otěhotnět, jak je uvedeno níže. Tento požadavek se týká i žen, které mohou otěhotnět a praktikují absolutní a trvalou pohlavní abstinenci. Je ideální, aby byl ve stejný den proveden těhotenský test a lék předepsán i vydán. Vydání pomalidomidu ženám, které mohou otěhotnět, se má provést během 7 dnů od předepsání.
Před začátkem léčby
Je třeba provést těhotenský test pod lékařským dohledem při návštěvě lékaře, kdy je pomalidomid předepsán, nebo ve 3 dnech předcházejících návštěvě u předepisujícího lékaře - poté, co pacientka používá účinnou antikoncepci přinejmenším 4 týdny. Test musí potvrdit, že pacientka není při zahájení léčby pomalidomidem těhotná.
Ukončení léčby a další sledování
Těhotenský test pod lékařským dohledem musí být opakován každé 4 týdny včetně 4 týdnů po ukončení léčby, kromě případů potvrzené sterilizace podvazem vejcovodů. Tyto testy je třeba provést v den předepsání přípravku nebo během 3 dnů před návštěvou předepisujícího lékaře.
Muži
Pomalidomid je během léčby přítomen v semeni. Z preventivních důvodů, a s ohledem na zvláštní populace s potenciálně prodlouženou dobou eliminace, např. s poruchou funkce ledvin, musí všichni pacienti muži užívající pomalidomid, včetně těch, co podstoupili vasektomii, používat kondom po celou dobu léčby, během jejího přerušení a 7 dní po ukončení léčby, pokud je jejich partnerka těhotná nebo pokud u ní nelze možnost otěhotnění vyloučit, a pokud ta nepoužívá jinou antikoncepci.
Pacienti muži nesmí darovat semeno nebo sperma během léčby (včetně období přerušení léčby) a 7 dní po vysazení pomalidomidu.
Další opatření
Pacienti musí být poučeni, aby nikdy tento léčivý přípravek nedávali jiným osobám a nepoužité tobolky vrátili na konci léčby do lékárny.
Pacienti nesmí darovat krev, semeno nebo sperma během léčby (včetně období přerušení léčby) a 7 dní po vysazení pomalidomidu.
Vzdělávací materiály, omezení týkající se předepisování a výdeje
Držitel rozhodnutí o registraci bude vydávat vzdělávací materiály pro zdravotníky, aby byli schopni odborně poradit pacientům, jak zabránit vlivu pomalidomidu na plod. Materiály obsahují varování před očekávanými teratogenními účinky pomalidomidu, rady ohledně antikoncepce před začátkem terapie a poučení o nutnosti těhotenských testů. Předepisující lékař musí pacienta informovat o očekávaném teratogenním riziku a přísných antikoncepčních opatřeních tak, jak jsou uvedena v Programu prevence početí (PPP) a poskytnout pacientům odpovídající informační brožurku, kartu pacienta a/nebo ekvivalentní pomůcky v souladu se zavedeným národním systémem evidence karet pacientů. Ve spolupráci s jednotlivými příslušnými národními orgány byl zaveden systém regulované národní distribuce. Tento systém regulované distribuce zahrnuje využívání karet pacientů a/nebo ekvivalentního nástroje k regulaci předepisování a/nebo výdeje a shromažďování podrobných údajů týkajících se indikace za účelem monitorování použití mimo schválenou indikaci na území daného státu. Ideálně se mají procesy předepsání a výdeje léku uskutečnit ve stejný den jako těhotenský test. Pomalidomid je nutné ženám, které mohou otěhotnět, vydat do 7 dní od předpisu a po provedení těhotenského testu pod lékařským dohledem s negativním výsledkem. Ženám, které mohou otěhotnět, může být předepsán na maximální dobu 4 týdnů a všem ostatním pacientům na maximální dobu 12 týdnů.
Hematologické příhody
Neutropenie byla nejčastěji hlášeným hematologickým nežádoucím účinkem 3. nebo 4. stupně u pacientů s relabovaným /s refrakterním mnohočetným myelomem, dále následovaly anémie a trombocytopenie. U pacientů je třeba sledovat výskyt hematologických nežádoucích účinků, především neutropenie. Pacienty je nutné informovat, aby febrilní epizody neprodleně hlásili. Lékaři musí u pacientů sledovat známky krvácení, jako jsou epistaxe, zejména při současném užívání léčivých přípravků, o nichž je známo, že zvyšují riziko krvácení (viz bod 4.8). Celkový krevní obraz je nutné sledovat na počátku, jednou týdně po dobu prvních 8 týdnů a dále jednou za měsíc. Může být nutné upravit dávky (viz bod 4.2). U pacientů může být potřeba použít podpůrné krevní produkty a/nebo růstové faktory.
Tromboembolické příhody
U pacientů užívajících pomalidomid v kombinaci s dexamethasonem se vyskytují žilní tromboembolické příhody (především hluboká žilní trombóza a plicní embolie) a tepenné trombotické příhody (infarkt myokardu a cerebrovaskulární příhoda). Pacienty se známými rizikovými faktory pro tromboembolismus - včetně trombózy v anamnéze - je nutné důkladně sledovat. Je nutné přijmout opatření k minimalizaci veškerých modifikovatelných rizikových faktorů (např. kouření, hypertenze a hyperlipidemie). Pacientům a lékařům se doporučuje sledovat možný výskyt známek a příznaků tromboembolismu. Pacienty je nutné poučit, aby vyhledali lékařskou péči, pokud se u nich objeví příznaky, jako je dechová nedostatečnost, bolest na hrudi a otoky paží nebo nohou. Doporučuje se antikoagulační terapie (pokud není kontraindikována) (například kyselina acetylsalicylová, warfarin, heparin, nebo klopidogrel), především u pacientů s dalšími trombotickými rizikovými faktory. O profylaktických opatřeních má být rozhodnuto po pečlivém zhodnocení základních rizikových faktorů u jednotlivých pacientů. V klinických studiích pacienti dostávali v rámci profylaxe kyselinu acetylsalicylovou nebo alternativní antitrombotickou terapii. Použití erytropoetických látek s sebou nese riziko tromboembolických příhod včetně tromboembolismu. Proto je nutné erytropoetické látky i jiné látky, které mohou zvyšovat riziko tromboembolických příhod, používat s opatrností.
Periferní neuropatie
Pacienti s probíhající periferní neuropatií > 2. stupně byli z klinických studií s pomalidomidem vyloučeni. Při zvažování léčby těchto pacientů pomalidomidem je třeba postupovat s patřičnou opatrností.
Významná srdeční dysfunkce
Pacienti s významnou srdeční dysfunkcí (městnavým srdečním selháním [třídy III nebo IV podle NYHA], infarktem myokardu v průběhu 12 měsíců od zahájení studie, nestabilní nebo nedostatečně kontrolovanou anginou pectoris) byli z klinických studií s pomalidomidem vyloučeni. Byly hlášeny případy srdečního selhání, včetně městnavého srdečního selhání a plicního edému (viz bod 4.8), zejména u pacientů s již existujícím srdečním onemocněním nebo s kardiálními rizikovými faktory.
Při zvažování léčby těchto pacientů pomalidomidem je třeba postupovat s patřičnou opatrností, včetně pravidelného sledování známek a příznaků srdečního selhání.
Syndrom nádorového rozpadu
Může se vyskytnout syndrom nádorového rozpadu. Mezi pacienty s největším rizikem syndromu nádorového rozpadu patří pacienti s vysokou nádorovou zátěží před léčbou. Tyto pacienty je nutné důkladně sledovat a přijmout vhodná opatření.
Další primární malignity
U pacientů léčených pomalidomidem byly hlášeny další primární malignity, např. nemelanomové kožní nádory (viz bod 4.8). Lékaři musí pacienty důkladně vyšetřit před léčbou i během léčby pomocí standardních metod pro zjišťování karcinomu pro případ výskytu dalších primárních malignit a zahájit léčbu podle indikace.
Alergická reakce
Byly hlášeny případy angioedému a závažných kožních reakcí (viz bod 4.8). Pacienti s předchozí závažnou alergickou reakcí spojenou s léčbou thalidomidem nebo lenalidomidem v anamnéze byli z klinických studií vyloučeni. Tito pacienti mohou být vystaveni zvýšenému riziku hypersenzitivitních reakcí a pomalidomid jim nemá být podáván. Při výskytu kožní vyrážky stupně 2 až 3 je třeba zvážit přerušení nebo ukončení léčby pomalidomidem. Léčba pomalidomidem musí být trvale ukončena při výskytu angioedému, kožní vyrážky stupně 4, exfoliativní nebo bulózní vyrážky.
Závratě a zmatenost
Při užívání pomalidomidu byly hlášeny závratě a zmatenost. Pacienti se musí vyvarovat situací, kdy pro ně závratě nebo zmatenost mohou představovat problém, a nesmí bez předchozí konzultace s lékařem užívat jiné léčivé přípravky, které mohou způsobovat závratě nebo zmatenost.
Intersticiální plicní nemoc (Interstitial lung disease - ILD)
Při léčbě pomalidomidem byly pozorovány případy ILD a souvisejících onemocnění, včetně pneumonitidy. Pacienti s akutním nástupem nebo s nevysvětleným náhlým zhoršením plicních příznaků mají být pečlivě vyšetřeni za účelem vyloučení ILD. Pomalidomid by měl být do doby vyšetření těchto příznaků vysazen a pokud se ILD potvrdí, má být zahájena vhodná terapie. Podávání pomalidomidu lze obnovit pouze po důkladném vyhodnocení přínosů a rizik.
Poruchy jaterních funkcí
U pacientů léčených pomalidomidem bylo pozorováno významné zvýšení hladin alaninaminotransferázy a bilirubinu (viz bod 4.8). Vyskytly se také případy hepatitidy, které vedly k ukončení léčby pomalidomidem. Po dobu prvních 6 měsíců léčby pomalidomidem a dále podle klinické indikace se doporučuje pravidelné sledování jaterních funkcí.
Infekce
U pacientů, kteří byli v minulosti infikováni virem hepatitidy B (HBV), léčených pomalidomidem v kombinaci s dexamethasonem, byla vzácně hlášena reaktivace hepatitidy B. Některé z těchto případů progredovaly do akutního selhání jater, což vedlo k ukončení léčby pomalidomidem. Před zahájením léčby pomalidomidem se mají provést testy na stanovení viru hepatitidy B. U pacientů pozitivních na infekci HBV se doporučuje konzultace s lékařem specializovaným na léčbu hepatitidy B. Při použití kombinace pomalidomidu s dexamethasonem u pacientů, kteří byli v minulosti infikováni HBV, včetně pacientů, kteří jsou anti-HBc pozitivní, ale HBsAg negativní, je třeba postupovat s opatrností. Tyto pacienty je nutné v průběhu celé léčby pečlivě sledovat kvůli výskytu známek a příznaků aktivní infekce HBV.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Účinek přípravku Imnovid na jiné léčivé přípravky
Při současném podávání pomalidomidu a substrátů enzymů nebo transportérů se nepředpokládají klinicky relevantní farmakokinetické lékové interakce v důsledku inhibice nebo indukce izoenzymu P450 nebo inhibice transportérů. Potenciál k takovým lékovým interakcím, včetně potenciálního dopadu pomalidomidu na farmakokinetiku kombinovaných perorálních kontraceptiv, nebyl klinicky hodnocen (viz bod 4.4 Teratogenita).
Účinek jiných léčivých přípravků na přípravek Imnovid
Pomalidomid je částečně metabolizován enzymy CYP1A2 a CYP3A4/5. Je také substrát P-glykoproteinu. Současné podávání pomalidomidu s ketokonazolem, silným inhibitorem CYP3A4/5, a P-glykoproteinem, nebo karbamazepinem, silným induktorem CYP3A4/5, nemá žádný klinicky relevantní účinek na expozici pomalidomidu. Současné podávání pomalidomidu s fluvoxaminem, silným inhibitorem CYP1A2, za přítomnosti ketokonazolu zvýšilo střední expozici pomalidomidu o 107 % při 90% intervalu spolehlivosti [91 % až 124 %] v porovnání s kombinací pomalidomidu a ketokonazolu. Ve druhé studii, která hodnotila přispění samotného inhibitoru CYP1A2 ke změnám metabolismu, zvýšilo současné podávání samotného fluvoxaminu s pomalidomidem střední expozici pomalidomidu o 125 % při 90% intervalu spolehlivosti [98 % až 157 %] v porovnání se samotným pomalidomidem. Jestliže jsou s pomalidomidem současně podávány silné inhibitory CYP1A2 (např. ciprofloxacin, enoxacin a fluvoxamin), snižte dávku pomalidomidu o 50 %.
Dexamethason
Současné podávání opakovaných dávek až 4 mg pomalidomidu s 20 mg až 40 mg dexamethasonu (slabý až středně silný induktor několika enzymů CYP, včetně CYP3A) pacientům s mnohočetným myelomem nemělo žádný účinek na farmakokinetiku pomalidomidu v porovnání s pomalidomidem podávaným v monoterapii.
Účinky dexamethasonu na warfarin nejsou známy. Během léčby se doporučuje pečlivě sledovat hladinu warfarinu.
4.6 Fertilita, těhotenství a kojení
Ženy, které mohou otěhotnět / antikoncepce u mužů a žen
Ženy, které mohou otěhotnět, musí používat účinnou metodu antikoncepce. Pokud žena léčená pomalidomidem otěhotní, musí být léčba zastavena a pacientka odeslána ke specializovanému lékaři nebo zkušenému teratologovi, aby posoudil riziko a navrhl doporučení. Pokud otěhotní partnerka pacienta-muže léčeného pomalidomidem, doporučuje se tuto partnerku odeslat ke specializovanému lékaři nebo zkušenému teratologovi, aby posoudil riziko a poskytl doporučení. Pomalidomid je přítomen v lidském semeni. Z preventivních důvodů musí všichni pacienti muži užívající pomalidomid používat kondom po celou dobu léčby, během jejího přerušení a 7 dní po ukončení léčby, pokud je jejich partnerka těhotná nebo pokud u ní nelze možnost otěhotnění vyloučit, a pokud ta nepoužívá jinou antikoncepci (viz body 4.3 a 4.4).
U člověka lze očekávat teratogenní účinek pomalidomidu. Pomalidomid je v těhotenství a u žen ve fertilním věku kontraindikován, vyjma případů, kdy jsou splněny všechny podmínky prevence početí, viz body 4.3 a 4.4.
Kojení
Není známo, zda se pomalidomid vylučuje do lidského mateřského mléka. Pomalidomid byl zjištěn v mateřském mléce laktujících samic potkanů po podání matkám. Vzhledem k potenciálu k nežádoucím účinkům pomalidomidu u kojených dětí je třeba posoudit nezbytnost léčivého přípravku pro matku a rozhodnout, zda bude přerušeno kojení nebo zda ukončit podávání léčivého přípravku matce.
Fertilita
Bylo zjištěno, že u zvířat má pomalidomid negativní dopad na fertilitu a vykazuje teratogenní účinky. Pomalidomid po podání březím samicím králíka prostupoval placentou a byl přítomen v krvi plodu. Viz bod 5.3.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek Imnovid má malý nebo mírný vliv na schopnost řídit nebo obsluhovat stroje.
Při užívání pomalidomidu byly hlášeny únava, snížený stupeň vědomí, zmatenost a závratě. Při výskytu těchto příznaků mají být pacienti poučeni, aby po dobu léčby pomalidomidem neřídili vozidla, nepoužívali stroje a nevykonávali nebezpečné činnosti.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Nejčastěji hlášenými nežádoucími účinky v klinických studiích byly poruchy krve a lymfatického systému včetně anémie (45,7 %), neutropenie (45,3 %) a trombocytopenie (27 %); celkové poruchy a reakce v místě aplikace včetně únavy (28,3 %), pyrexie (21 %) a periferního edému (13 %); a infekce a infestace včetně pneumonie (10,7 %). Nežádoucí účinky periferní neuropatie byly hlášeny u 12,3 % pacientů a nežádoucí účinky žilní embolické nebo trombotické příhody (VTE) byly hlášeny u 3,3 % pacientů. Nejčastěji hlášenými nežádoucími účinky 3. a 4. stupně byly poruchy krve a lymfatického systému včetně neutropenie (41,7 %), anémie (27 %) a trombocytopenie (20,7 %); infekce a infestace včetně pneumonie (9 %); a celkové poruchy a reakce v místě aplikace včetně únavy (4,7 %), pyrexie (3 %) a periferního edému (1,3 %). Nejčastěji hlášeným závažným nežádoucím účinkem byla pneumonie (9,3 %). Další hlášené závažné nežádoucí účinky zahrnovaly febrilní neutropenii (4,0 %), neutropenii (2,0 %), trombocytopenii (1,7 %) a nežádoucí účinky VTE (1,7 %).
Nežádoucí účinky se vyskytovaly častěji spíše v prvních 2 cyklech léčby pomalidomidem.
Přehled nežádoucích účinků v tabulce
V randomizované studii (CC-4047-MM-003), byly 302 pacientům s relabovaným a refrakterním mnohočetným myelomem podávány 4 mg pomalidomidu jednou denně po dobu 21 dní v každém 28denním cyklu v kombinaci s nízkou dávkou dexamethasonu jednou týdně.
Nežádoucí účinky pozorované u pacientů léčených kombinací pomalidomidu a dexamethasonu jsou uvedeny níže a seřazeny podle tříd orgánových systémů a četnosti veškerých nežádoucích účinků a nežádoucích účinků 3. a 4. stupně.
Četnosti nežádoucích účinků j sou četnosti hlášené ve skupině léčené kombinací pomalidomidu a dexamethasonu ve studii CC-4047-MM-003 (n = 302) a z údajů po uvedení přípravku na trh. V každé třídě orgánových systémů a skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti. Četnosti jsou definovány v souladu s platnými předpisy takto: velmi časté (> 1/10), časté (> 1/100 až < 1/10) a méně časté (> 1/1000 až < 1/100).
|
Třídy orgánových systémů / Preferovaný termín |
Veškeré nežádoucí účinky / Frekvence |
Nežádoucí účinky 3.4. stupně / F rekvence |
|
Infekce a infestace |
Velmi časté |
Časté |
|
Pneumonie (bakteriální, virové |
Neutropenická sepse | |
|
a mykotické infekce, včetně |
Pneumonie (bakteriální, virové | |
|
oportunních infekcí) |
a mykotické infekce, včetně oportunních infekcí) | |
|
Časté |
Bronchopneumonie | |
|
Neutropenická sepse |
Infekce dýchacích cest | |
|
Bronchopneumonie Bronchitida |
Infekce horních cest dýchacích | |
|
Infekce dýchacích cest |
Méně časté | |
|
Infekce horních cest dýchacích |
Bronchitida | |
|
Nazofaryngitida Herpes zoster |
Herpes zoster Není známo | |
|
Není známo Reaktivace hepatitidy B |
Reaktivace hepatitidy B | |
|
Novotvary benigní, maligní |
Méně časté |
Méně časté |
|
a blíže neurčené (zahrnující |
Bazocelulámí karcinom kůže |
Bazocelulámí karcinom kůže |
|
cysty a polypy) |
Spinocelulámí karcinom kůže |
Spinocelulámí karcinom kůže |
|
Poruchy krve a lymfatického |
Velmi časté |
Velmi časté |
|
systému |
Neutropenie |
Neutropenie |
|
Trombocytopenie |
T rombocytopenie | |
|
Leukopenie |
Časté | |
|
Časté |
Febrilní neutropenie | |
|
Febrilní neutropenie |
Leukopenie | |
|
Pancytopenie* |
Pancytopenie15 | |
|
Poruchy metabolismu a |
Velmi časté |
Časté |
|
výživy |
Snížená chuť k jídlu |
Hyperkalemie Hyponatremie |
|
Časté Hyperkalemie |
Hyperurikémie* | |
|
Hyponatremie |
Méně časté | |
|
Hyperurikémie* Méně časté Syndrom nádorového rozpadu* |
Snížená chuť k jídlu Syndrom nádorového rozpadu* | |
|
Psychiatrické poruchy |
Časté |
Časté |
|
Stav zmatenosti |
Stav zmatenosti | |
|
Poruchy nervového systému |
Časté |
Časté |
|
Snížený stupeň vědomí Periferní senzorická neuropatie |
Snížený stupeň vědomí | |
|
Závratě |
Méně časté | |
|
Periferní senzorická neuropatie | ||
|
Intrakraniální krvácení* |
Závratě | |
|
Méně časté |
Cerebrovaskulární příhoda* | |
|
Cerebrovaskulární příhoda* |
Intrakraniální krvácení* |
|
Třídy orgánových systémů / |
Veškeré nežádoucí účinky / |
Nežádoucí účinky 3.- |
|
Preferovaný termín |
Frekvence |
4. stupně / F rekvence |
|
Poruchy ucha a labyrintu |
Časté |
Časté |
|
Cévní poruchy |
Časté |
Méně časté |
|
Hluboká žilní trombóza |
Hluboká žilní trombóza | |
|
Srdeční poruchy |
Časté |
Časté |
|
Srdeční selháni* |
Srdeční selhání* | |
|
Fibrilace síní* Infarkt myokardu* |
Fibrilace síní* | |
|
Méně časté Infarkt myokardu* | ||
|
Poruchy imunitního systému |
Časté |
Méně časté |
|
Angioedém* |
Angioedém* | |
|
Urtikárie* |
Urtikárie* | |
|
Respirační, hrudní a |
Velmi časté |
Časté |
|
mediastinální poruchy | ||
|
Méně časté | ||
|
Časté | ||
|
Epistaxe* |
Epistaxe* | |
|
Intersticiálni plicní nemoc* |
Intersticiální plicní nemoc* | |
|
Gastrointestinální poruchy |
Velmi časté |
V Časté |
|
Zácpa |
Zácpa | |
|
Časté |
Méně časté | |
|
Gastrointestinální krvácení |
Gastrointestinální krvácení | |
|
Poruchy jater a žlučových |
Méně časté |
Méně časté |
|
cest |
Hyperbilirubinemie Hepatitida5 |
Hyperbilirubinemie |
|
Poruchy kůže a podkožní |
Časté |
Časté |
|
tkáně | ||
|
Poruchy svalové a kosterní |
Velmi časté |
Časté |
|
soustavy a pojivové tkáně |
Bolest kostí |
Bolest kostí |
|
Svalové křeče |
Méně časté Svalové křeče | |
|
Poruchy ledvin a močových |
Časté |
Časté |
|
cest |
Renální selhání |
Renální selhání |
|
Retence moči |
Méně časté Retence moči | |
|
Poruchy reprodukčního |
v Časté |
v Časté |
|
systému a prsu |
Pánevní bolest |
Pánevní bolest |
|
Třídy orgánových systémů / Preferovaný termín |
Veškeré nežádoucí účinky / Frekvence |
Nežádoucí účinky 3.4. stupně / F rekvence |
|
Celkové poruchy a reakce |
Velmi časté |
Časté |
|
v místě aplikace |
Únava |
Únava |
|
Pyrexie |
Pyrexie | |
|
Periferní edém |
Periferní edém | |
|
Vyšetření |
Časté |
Časté |
|
Snížený počet neutrofilů |
Snížený počet neutrofilů | |
|
Snížený počet leukocytů |
Snížený počet leukocytů | |
|
Snížený počet trombocytů |
Snížený počet trombocytů | |
|
Zvýšená hladina |
Zvýšená hladina | |
|
alaninaminotransferázy Zvýšená hladina kyseliny močové |
alaninaminotransferázy | |
|
v krvi* |
Méně časté Zvýšená hladina kyseliny močové v krvf |
^Zjištěno z údajů po uvedení přípravku na trh, s frekvencemi vycházejícími z údajů klinických studií.
Popis vybraných nežádoucích účinků
Teratogenita
Pomalidomid je strukturálně podobný thalidomidu. Thalidomid je známá lidská teratogenní léčivá látka, která způsobuje těžké a život ohrožující vrozené vady. Pomalidomid vykazoval teratogenní účinky jak u potkanů, tak i u králíků, pokud byl podáván v období hlavní organogeneze (viz body 4.6 a 5.3). Pokud je pomalidomid užíván během těhotenství, očekávají se u lidí teratogenní účinky pomalidomidu (viz bod 4.4).
Neutropenie a trombocytopenie
Neutropenie se vyskytla u 45,3 % pacientů, kterým byl podáván pomalidomid s nízkou dávkou dexamethasonu (Pom + LD-Dex) a u 19,5 % pacientů, kterým byla podávána vysoká dávka dexamethasonu (HD-Dex). Neutropenie byla 3. nebo 4. stupně u 41,7 % pacientů, kterým byl podáván Pom + LD-Dex, v porovnání se 14,8 %, kterým byl podáván HD-Dex. Neutropenie u pacientů ve skupině Pom + LD-Dex byla zřídka závažná (2,0 % pacientů), nevedla k ukončení léčby a byla spojena s přerušením léčby u 21 % pacientů a se snížením dávky u 7,7 % pacientů.
Febrilní neutropenie (FN) byla pozorována u 6,7 % pacientů, kterým byl podáván Pom + LD-Dex a u žádného z pacientů, kterým byl podáván HD-Dex. Veškeré FN byly hlášeny se závažností 3. a 4. stupně. FN byla hlášena jako závažná u 4,0 % pacientů. FN byla spojena s přerušením léčby u 3,7 % pacientů a se snížením dávky u 1,3 % a nebyla spojena s žádným ukončením léčby.
Trombocytopenie se vyskytla u 27,0 % pacientů, kterým byl podáván Pom + LD-Dex a u 26,8 % pacientů, kterým byl podáván HD-Dex. Trombocytopenie byla 3. a 4. stupně u 20,7 % pacientů, kterým byl podáván Pom + LD-Dex a u 24,2 % pacientů, kterým byl podáván HD-Dex. U pacientů léčených Pom + LD-Dex byla trombocytopenie závažná u 1,7 % pacientů, vedla ke snížení dávky u 6,3 % pacientů, k přerušení léčby u 8 % pacientů a k ukončení léčby u 0,7 % pacientů (viz body 4.2 a 4.4).
Infekce
Infekce byla nejčastější nehematologickou toxicitou a vyskytla se u 55,0 % pacientů, kterým byl podávánPom + LD-Dex a u 48,3 % pacientů, kterým byl podáván HD-Dex. Přibližně polovina z těchto infekcí dosahovala 3. a 4. stupně, 24,0 % u pacientů léčených Pom + LD-Dex a 22,8 % u pacientů, kterým byl podáván HD-Dex.
U pacientů léčených Pom + LD-Dex byly nejčastěji hlášenými infekcemi pneumonie a infekce horních cest dýchacích (10,7 %, resp. 9,3 % pacientů), přičemž 24,3 % hlášených infekcí bylo závažných a fatálních (5. stupeň) a vyskytovaly se u 2,7 % léčených pacientů. U pacientů léčených Pom + LD-Dex vedly infekce k ukončení léčby u 2,0 % pacientů, k přerušení léčby u 14,3 % pacientů a ke snížení dávky u 1,3 % pacientů.
Tromboembolické příhody
Žilní embolickou nebo trombotickou příhodu (VTE) mělo 3,3 % pacientů, kterým byl podáván Pom + LD-Dex a 2,0 % pacientů, kterým byl podáván HD-Dex. Reakce 3. a 4. stupně se vyskytly u 1,3 % pacientů, kterým byl podáván Pom + LD-Dex a u žádného z pacientů, kterým byl podáván HD-Dex. U pacientů léčených Pom + LD-Dex byla VTE hlášena jako závažná u 1,7 % pacientů, nebyly hlášeny žádné fatální reakce v klinických studiích a VTE nebyla spojena s ukončením léčby.
Profylaktické podávání kyseliny acetylsalicylové (a dalších antikoagulancií u pacientů s vysokým rizikem) bylo povinné pro všechny pacienty v klinických studiích. Doporučuje se antikoagulační léčba, pokud není kontraindikována (viz bod 4.4).
Periferní neuropatie
Pacienti s probíhající periferní neuropatií > 2. stupně byli z klinických studií vyloučeni. Periferní neuropatie byla většinou 1. nebo 2. stupně a vyskytla se u 12,3 % pacientů, kterým byl podáván Pom + LD-Dex a u 10,7 % pacientů, kterým byl podáván HD-Dex. Reakce 3. a 4. stupně se vyskytly u 1,0 % pacientů, kterým byl podáván Pom + LD-Dex a u 1,3 % pacientů, kterým byl podáván HD-Dex. U pacientů léčených Pom + LD-Dex nebyla v klinických zkouškách žádná periferní neuropatie hlášena jako závažná; periferní neuropatie vedla k ukončení léčby u 0,3 % pacientů (viz bod 4.4).
Střední doba do nástupu neuropatie činila 2,1 týdnů, přičemž se pohybovala od 0,1 do 48,3 týdnů. Střední doba do nástupu byla ve studii kratší u pacientů, kterým byl podáván HD-Dex ve srovnání s Pom + LD-Dex (1,3 týdnů oproti 2,1 týdnům).
Střední doba do vyřešení činila ve studii 22,4 týdnů u pacientů, kterým byl podáván Pom + LD-Dex a 13,6 týdnů u pacientů, kterým byl podáván HD-Dex. Spodní limit 95% intervalu spolehlivosti činil 5,3 týdnů u pacientů léčených Pom +LD-Dex a 2,0 týdny u pacientů, kterým byl podáván HD-Dex.
Krvácení
Při léčbě pomalidomidem byly zejména u pacientů s rizikovými faktory, např. se současně podávanými léčivými přípravky, které zvyšují náchylnost ke krvácení, hlášeny hemoragické poruchy. Hemoragické příhody zahrnovaly epistaxi, intrakraniální krvácení a gastrointestinální krvácení.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Pomalidomid byl hodnocen v dávkách 50 mg podávaných v jediné dávce zdravým dobrovolníkům a 10 mg podávaných v opakovaných dávkách jednou denně pacientům s mnohočetným myelomem; nebyly hlášeny žádné závažné nežádoucí příhody v důsledku předávkování. Pomalidomid byl odstraněn hemodialýzou.
V případě předávkování se doporučuje poskytnutí symptomatické péče.
FARMAKOLOGICKÉ VLASTNOSTI
5.
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Imunosupresiva. ATC kód: L04AX06 Mechanismus účinku
Pomalidomid má přímý tumoricidní účinek na myelomy, imunomodulační účinky a inhibuje podporu stromálních buněk pro růst nádorových buněk mnohočetného myelomu. Konkrétně pomalidomid inhibuje proliferaci a indukuje apoptózu hematopoetických nádorových buněk. Pomalidomid rovněž inhibuje proliferaci buněčných linií mnohočetného myelomu rezistentních vůči lenalidomidu a v kombinaci s dexamethasonem působí jak na buněčné linie odpovídající na lenalidomid, tak i na ty, které jsou vůči lenalidomidu rezistentní, a indukuje apoptózu nádorových buněk. Pomalidomid zvyšuje imunitu zprostředkovanou T buňkami a přirozenými zabíječi (NK (Natural Killer) buňkami) a inhibuje tvorbu prozánětlivých cytokinů (např. TNF-a a IL-6) monocyty. Pomalidomid také inhibuje angiogenezi blokováním migrace a adheze endotelových buněk.
Klinická účinnost a bezpečnost
Účinnost a bezpečnost pomalidomidu v kombinaci s dexamethasonem byla hodnocena v multicentrické, randomizované, otevřené studii fáze III (CC-4047-MM-03), v níž byla léčba pomalidomidem v kombinaci s nízkými dávkami dexamethasonu (Pom + LD-Dex) porovnávána s vysokými dávkami dexamethasonu samotného (HD-Dex). Do studie byli zařazeni dříve léčení dospělí pacienti s relabovaným a refrakterním mnohočetným myelomem, kteří absolvovali nejméně dvě předchozí léčebná schémata, včetně lenalidomidu i bortezomibu, a u nichž při poslední léčbě došlo k progresi onemocnění. Do studie bylo zařazeno celkem 455 pacientů: 302 pacientů ve skupině Pom + LD-Dex a 153 pacientů ve skupině HD-Dex. Většina pacientů byla mužského pohlaví (59 %) a bílé pleti (79 %); střední věk v celkové populaci činil 64 let (min. 35 let, max. 87 let).
Pacienti ve skupině Pom + LD-Dex dostávali 4 mg pomalidomidu perorálně v 1.-21. den každého 28denního cyklu. LD-Dex (40 mg) byl podáván jednou denně v 1., 8., 15. a 22. den 28denního cyklu. Ve skupině HD-Dex byl dexamethason (40 mg) podáván jednou denně v 1.-4., 9.-12. a 17.-20. den každého 28denního cyklu. Pacienti ve věku > 75 let zahajovali léčbu 20 mg dexamethasonu. Léčba pacientů pokračovala až do progrese nemoci.
Primárním cílovým parametrem účinnosti bylo přežití bez progrese (progression free survival, PFS) podle kritérií IMWG (International Myeloma Working Group - Mezinárodní pracovní skupina zabývající se mnohočetným myelomem). V populaci ITT činil medián doby PFS po revizi IRAC (Nezávislá revizní a posudková komise Independent Review Adjudication Committee) na základě kritérií IMWG 15,7 týdne (95% IS: 13,0; 20,1) ve skupině Pom + LD-Dex a vypočtená četnost přežití bez příhod po dobu 26 týdnů byla 35,99 % (± 3,46 %). Ve skupině HD-Dex byl medián doby PFS 8,0 týdne (95% IS: 7,0; 9,0) a vypočtená četnost přežití bez příhod po dobu 26 týdnů byla 12,15 % (± 3,63 %).
Parametr přežití bez progrese byl hodnocen v několika relevantních podskupinách: podle pohlaví, rasy, stavu ECOG,stratifikačních faktorů (věk, populace s onemocněním, předchozí léčby myelomu [2, > 2]), vybraných prognosticky významných parametrů (vstupní hladina beta-2 mikroglobulinu, vstupní hladiny albuminu, vstupní poruchy funkce ledvin a cytogenetické riziko) a expozice a refrakterita k předchozím léčbám myelomu. Bez ohledu na hodnocené podskupiny byla hodnota PFS celkově konzistentní s hodnotami pozorovanými v populaci ITT v obou léčebných skupinách.
Výsledky přežití bez progrese v populaci ITT jsou shrnuty v tabulce 1. Kaplan-Meierova křivka parametru PFS v populaci ITT je znázorněna na obrázku 1.
|
(stratifikovaný test log-rank' |
(populace ITT) | |
|
Pom + LD- Dex (N=302) |
HD-Dex (N=153) | |
|
Přežití bez progrese, N |
302 (100,0) |
153 (100,0) |
|
S cenzurou, n (%) |
138 (45,7) |
50 (32,7) |
|
S progresí/zemřelo, n (%) |
164 (54,3) |
103 (67,3) |
|
Doba přežití bez progrese (týdny) | ||
|
Medián3 |
15,7 |
8,0 |
|
Oboustranný 95% ISb |
[13,0; 20,11 |
[7,0; 9,01 |
|
Poměr rizik (Pom + LD-Dex : HD-Dex) oboustranný 95% ISc |
0,45 [0,35; 0,59] | |
|
P hodnota oboustranného log-rank testud |
< 0,001 | |
Poznámka: IS = interval spolehlivosti; IRAC = Nezávislá revizní a posudková komise (Independent Review Adjudication Commitee);
NE = nelze určit.
a Medián podle Kaplan-Meierova odhadu. b 95% interval spolehlivosti mediánu doby přežití bez progrese.
c Vychází z Coxova modelu poměrných rizik porovnávajícího rizikové funkce spojené s léčebnými skupinami, stratifikovaných podle věku (< 75 let vs. > 75 let), populace s onemocněním (refrakterita jak k lenalidomidu, tak i bortezomibu vs. bez refrakterity k oběma lékům) a počet předchozích léčeb myelomu (= 2 vs. > 2).
d p hodnota vychází ze stratifikovaného log-rank testu se stejnými stratifikačními faktory jako u výše uvedeného Coxova modelu.
Údaje ke dni: 7. září 2012
Obrázek 1:
Přežití bez progrese po revizi odezvy IRAC podle kritérií IMWG (stratifikovaný log-rank test) (populace ITT)
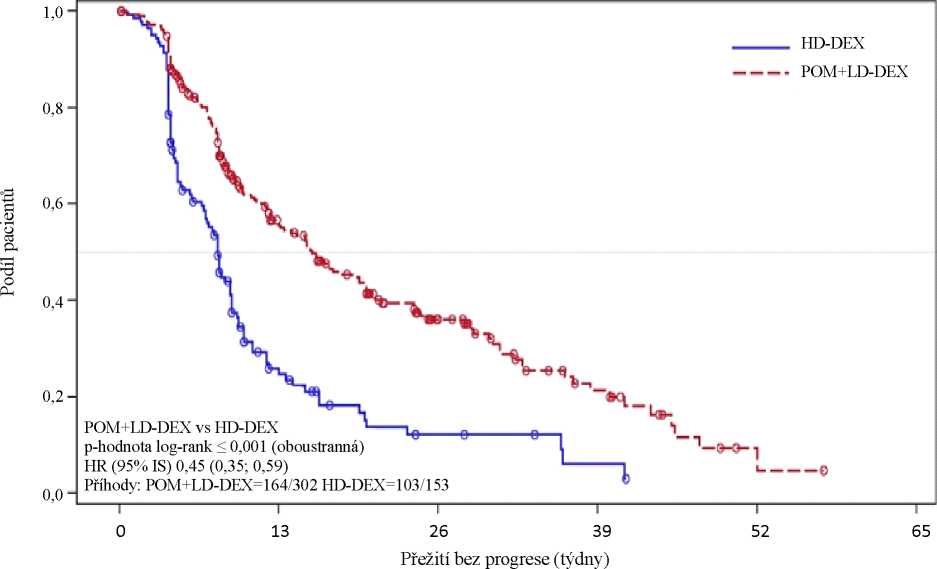
Údaje ke dni: 7. září 2012
Hlavním sekundárním cílovým parametrem studie bylo celkové přežití. Celkem 226 (74,8 %) pacientů ve skupině Pom + LD-Dex a 95 (62,1 %) pacientů ve skupině HD-Dex přežilo ke dni uzávěrky (7. září 2012). Medián doby celkového přežití podle Kaplan-Meierova odhadu nebyl ve skupině Pom + LD-Dex dosažen, lze však očekávat, že bude činit nejméně 48 týdnů, což je spodní hranice
95% IS. Medián doby celkového přežití ve skupině HD-Dex činil 34 týdnů (95% IS: 23,4; 39,9). Četnost přežití po dobu 1 roku bez příhody byla 52,6 % (± 5,72 %) ve skupině Pom + LD-Dex a 28,4 % (± 7,51 %) ve skupině HD-Dex. Rozdíl mezi celkovým přežitím v obou léčebných skupinách byl statisticky významný (p < 0,001).
Výsledky celkového přežití v populaci ITT jsou shrnuty v tabulce 2. Kaplan-Meierova křivka celkového přežití v populaci ITT je znázorněna na obrázku 2.
Na základě výsledků obou cílových parametrů, přežití bez progrese a celkového přežití, doporučila komise monitorující údaje, která byla pro tuto studii ustavena, studii dokončit a pacienty ve skupině HD-Dex převést do skupiny Pom + LD-Dex.
Tabulka 2: Celkové přežití: populace ITT
|
Statistika |
Pom + LD-Dex (N=302) |
HD-Dex (N=153) | |
|
N |
302 (100,0) |
153 (100,0) | |
|
S cenzurou |
n (%) |
226 (74,8) |
95 (62,1) |
|
Zemřelo |
n (%) |
76 (25,2) |
58 (37,9) |
|
Doba přežití (týdny) |
Medián3 |
NE |
34,0 |
|
Oboustranný 95% ISb |
[48,1; NE] |
[23,4; 39,9] | |
|
Poměr rizik (Pom + LD-Dex : HD-Dex) [oboustranný 95% ISc] |
0,53[0,37; 0,74] | ||
|
P hodnota oboustranného log-rank testud |
< 0,001 | ||
Poznámka: IS = interval spolehlivosti; NE = nelze určit. a Medián podle Kaplan-Meierova odhadu. b 95% interval spolehlivosti mediánu doby celkového přežití.
c Vychází z Coxova modelu poměrných rizik porovnávajícího rizikové funkce spojené s léčebnými skupinami. d p hodnota vychází z nestratifikovaného log-rank testu.
Údaje ke dni: 7. září 2012
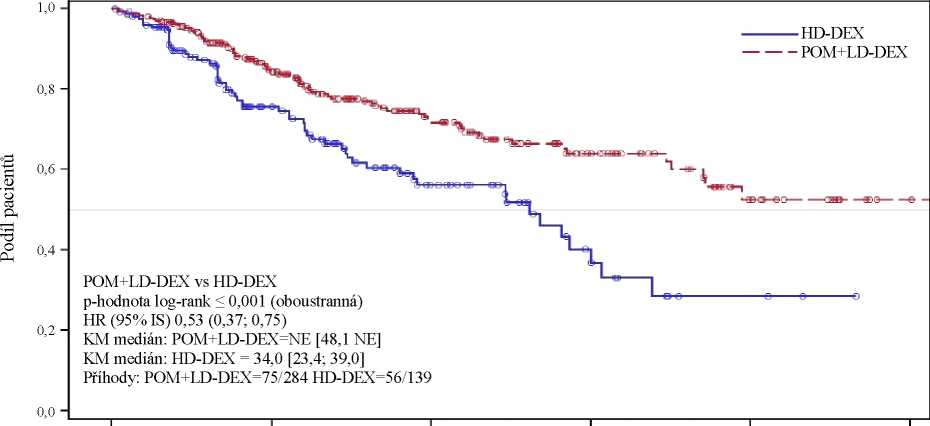
0 13 26 39 52 65
Celkové přežití (týdny)
Údaje ke dni: 7. září 2012
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Imnovid u všech podskupin pediatrické populace u mnohočetného myelomu (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Absorpce
Pomalidomid dosahuje maximální plazmatické koncentrace (Cmax) za 2 až 3 hodiny a po perorálním podání jediné dávky je absorbováno nejméně 73 %. Systémová expozice (AUC) pomalidomidu se zvyšuje přibližně lineárně a úměrně dávce. Po podání několika dávek má pomalidomid akumulační poměr 27 až 31 % AUC.
Při současném podávání stravy s vysokým obsahem tuku a vysoce kalorické stravy dochází ke zpomalení absorpce a snížení střední plazmatické Cmax přibližně o 27 %, ale účinek na celkovou míru absorpce je s 8% poklesem střední AUC minimální. Pomalidomid lze tedy podávat bez ohledu na příjem potravy.
Distribuce
Střední zdánlivý distribuční objem (Vd/F) pomalidomidu je 62 až 138 l v ustáleném stavu. Pomalidomid je distribuován do semene zdravých jedinců v množství přibližně 67 % plazmatické koncentrace 4 hodiny po podání dávky (přibližně Tmax) po 4 dnech dávkování 2 mg jednou denně. Vazba enantiomerů pomalidomidu na lidské plazmatické proteiny in vitro je v rozmezí 12 % až 44 % a není závislá na koncentraci.
Biotransformace
Pomalidomid je hlavní cirkulující složkou (přibližně 70 % plazmatické radioaktivity) in vivo u zdravých jedinců, kteří dostali jedinou perorální dávku [14C]-pomalidomidu (2 mg). V plazmě nebyly přítomny žádné metabolity při > 10% poměru k základní nebo celkové radioaktivitě v plazmě.
Převládající metabolickou cestou eliminace radioaktivity jsou hydroxylace s následnou glukuronidací nebo hydrolýza. In vitro bylo zjištěno, že CYP1A2 a CYP3A4 jsou primárními enzymy, které se podílejí na CYP zprostředkované hydroxylaci pomalidomidu, k níž v menší míře také přispívají CYP2C19 a CYP2D6. Pomalidomid je rovněž substrátem P-glykoproteinu in vitro. Současné podávání pomalidomidu s ketokonazolem, silným inhibitorem CYP3A4/5 a P-glykoproteinu, nebo karbamazepinem, silným induktorem CYP3A4/5, nemělo žádný klinicky relevantní účinek na expozici pomalidomidu. Současné podávání pomalidomidu s fluvoxaminem, silným inhibitorem CYP1A2, za přítomnosti ketokonazolu zvýšilo střední expozici pomalidomidu o 107 % při 90% intervalu spolehlivosti [91 %; 124 %] v porovnání s kombinací pomalidomidu a ketokonazolu. Ve druhé studii, která hodnotila přispění samotného inhibitoru CYP1A2 ke změnám metabolismu, zvýšilo současné podávání samotného fluvoxaminu s pomalidomidem střední expozici pomalidomidu o 125 % při 90% intervalu spolehlivosti [98 % až 157 %] v porovnání se samotným pomalidomidem. Jestliže jsou s pomalidomidem současně podávány silné inhibitory CYP1A2 (např. ciprofloxacin, enoxacin a fluvoxamin), snižte dávku pomalidomidu na 50 %. Podávání pomalidomidu u kuřáků nemělo klinicky relevantní účinek na expozici pomalidomidu v porovnání s expozicí pomalidomidu pozorovanou u nekuřáků, ačkoli je známo, že kouření tabáku indukuje izoformu CYP1A2.
Dle údajů in vitro pomalidomid není inhibitorem ani induktorem izoenzymů cytochromu P-450 a neinhibuje žádné transportéry léčiv, které byly studovány. Klinicky relevantní lékové interakce se při současném podávání pomalidomidu a substrátů těchto cest neočekávají.
Eliminace
Pomalidomid je vylučován se středním plazmatickým poločasem přibližně 9,5 hodiny u zdravých jedinců a přibližně 7,5 hodiny u pacientů s mnohočetným myelomem. Pomalidomid má střední celkovou tělesnou clearance (CL/F) přibližně 7-10 l/h.
Po jediném perorálním podání [14C]-pomalidomidu (2 mg) zdravým jedincům bylo přibližně 73 % radioaktivní dávky vyloučeno močí a 15 % stolicí, s přibližně 2 %, resp. 8 % podaného radioaktivního uhlíku vyloučenými ve formě pomalidomidu v moči a stolici.
Pomalidomid je ve značné míře metabolizován dříve, než dojde k vyloučení, s výslednými metabolity vylučovanými primárně v moči. Tři převládající metabolity v moči (tvořené prostřednictvím hydrolýzy nebo hydroxylace s následnou glukuronidací) odpovídají za přibližně 23 %, 17 % a 12 % dávky v moči.
Metabolity závislé na CYP odpovídají přibližně za 43 % celkové vyloučené radioaktivity, zatímco hydrolytické metabolity nezávislé na CYP odpovídají za 25 % a vyloučení nezměněného pomalidomidu odpovídalo za 10 % (2 % v moči a 8 % ve stolici).
Farmakokinetika populace
Na základě farmakokinetických analýz populace za použití dvojkompartmentového modelu měli zdraví a pacienti s mnohočetným myelomem srovnatelnou zdánlivou clearance (CL/F) a zdánlivý centrální distribuční objem (V2/F). V periferních tkáních byl pomalidomid přednostně vstřebán nádory, se zdánlivou periferní distribuční clearance (Q/F) 3,7krát vyšší než u zdravých jedinců a zdánlivým distribučním objemem (V3/F) 8krát vyšším než u zdravých jedinců.
Pediatrická populace
Nejsou dostupné žádné údaje o podávání pomalidomidu pediatrickým nebo dospívajícím pacientům (< 18 let věku).
Starší lidé
Na základě farmakokinetických analýz populace u zdravých jedinců a u pacientů s mnohočetným myelomem nebyl pozorován významný vliv věku (19-83 let) na perorální clearance pomalidomidu. V klinických studiích nebyla u starších pacientů (> 65 let) vystavených pomalidomidu vyžadována žádná úprava dávkování. Viz bod 4.2.
Pacienti s poruchou funkce ledvin
Ve farmakokinetických analýzác