Immunate Stim Plus 1000 Iu Fviii/750 Iu Vwf
sp.zn. sukls132850/2016
SOUHRN ÚDAJŮ O PŘÍPRAVKU
1. NÁZEV PŘÍPRAVKU
Immunate Stim Plus 1000 IU FVIII/750 IU VWF
Prášek a rozpouštědlo pro injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Léčivé látky: Factor VIII coagulationis humanus/ Factor von Willebrand humanus
Jedna lahvička obsahuje nominálně 1000 IU lidského koagulačního faktoru VIII1 a 750 IU lidského plazmatického von Willebrandova faktoru2 (vWF:RCo).
Přípravek Immunate Stim Plus 1000 IU FVIII/750 IU VWF obsahuje přibližně 100 IU/ml lidského plazmatického koagulačního faktoru VIII a 75 IU/ml lidského plazmatického von Willebrandova faktoru.
Účinnost faktoru VIII (IU) se určuje chromogenní analýzou podle evropského lékopisu (European Pharmacopoeia, EP). Specifická aktivita přípravku Immunate je 70 (± 30) IU FVIII/mg proteinu3. Účinnost von Willebrandova faktoru (IU) se určuje analýzou ristocetin kofaktoru podle Evropského lékopisu (vWF:RCo).
Vyrobeno z lidské plazmy.
Pomocné látky se známým účinkem:
1 lahvička obsahuje přibližně 19,6 mg sodíku.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční roztok. Bílý nebo světle žlutý prášek či drolivá hmota.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba a profylaxe krvácení u pacientů s vrozeným (hemofilie A) nebo získaným nedostatkem faktoru VIII.
Léčba krvácení u pacientů s von Willebrandovou chorobou s nedostatkem faktoru VIII, pokud není dostupný jiný přípravek účinný proti von Willebrandově chorobě a pokud léčba samotným desmopresinem (DDAVP) je neúčinná nebo kontraindikovaná.
4.2 Dávkování a způsob podání
Léčba by měla být pod dozorem lékaře, který má zkušenosti s léčbou hemostatických poruch. Dávkování
Dávkování u hemofilie A
Dávkování a délka substituční léčby závisí na závažnosti deficitu faktoru VIII, místě a rozsahu krvácení a na klinickém stavu pacienta.
Podávaná dávka faktoru VIII se vyjadřuje v mezinárodních jednotkách (International Units, IU), které se vztahují k aktuálnímu standardu WHO pro přípravky s faktorem VIII. Aktivita faktoru VIII v plazmě se vyjadřuje buď v procentech (ve vztahu k normální lidské plazmě) nebo v IU (podle mezinárodního standardu pro koncentráty faktoru VIII v plazmě).
Jedna mezinárodní jednotka (IU) aktivity faktoru VIII se rovná množství faktoru VIII v jednom ml normální lidské plazmy.
Výpočet požadované dávky faktoru VIIIje založen na empirickém zjištění, že 1 mezinárodní jednotka (IU) faktoru VIII na 1 kg tělesné hmotnosti zvyšuje aktivitu plazmatického faktoru VIII přibližně o 2 % normální aktivity.
Potřebná dávka se určuje podle následuj ícího vzorce:
Potřebné jednotky = tělesná hmotnost (kg) x žádaný vzestup faktoru VIII (%) x 0,5
Dávka a četnost podávání by měly být vždy vztaženy ke klinické účinnosti v individuálním případě.
Krvácení a operace
V případě následujících krvácivých příhod by v daném období neměla poklesnout aktivita faktoru VIII pod danou plazmatickou aktivitu (v % z normálu nebo IU/dl).
Následující tabulka může být vodítkem pro určení dávkování při krvácivých příhodách a chirurgických výkonech:
|
Stupeň krvácení / Typ chirurgického výkonu |
Požadovaná hodnota faktoru VIII (% normálu) (IU/dl) |
Frekvence dávkování (hodiny) / Délka trvání léčby (dny) |
|
Krvácení | ||
|
Časná hemartróza, krvácení do svalů nebo ústní dutiny |
20-40 |
Opakujte infuze každých 12 až 24 hodin, nejméně jeden den, dokud se krvácení, které se vyznačuje bolestí, nezastaví nebo se nedosáhne vyléčení. |
|
Intenzivnější hemartróza, krvácení do svalů nebo hematom |
30-60 |
Opakujte infuze každých 12 až 24 hodin po 3-4 dny či více, dokud bolest a akutní nemohoucnost nepřejdou. |
|
Život ohrožující krvácení |
60-100 |
Opakujte infuze každých 8 až 24, dokud není nebezpečí zažehnáno. |
|
Operace | ||
|
Menší Včetně extrakce zubu |
30-60 |
Každých 24 hodin, nejméně 1 den, dokud se nedosáhne vyléčení. |
|
Větší |
80-100 (před operací a po operaci) |
Opakujte infuze každých 8 až 24 hodin až do adekvátního zahojení rány, pak pokračujte v terapii nejméně dalších 7 dní, abyste udrželi aktivitu faktoru VIII na 30 % až 60 % (IU/dl). |
Dávkování a četnost podávání je třeba upravit podle klinické odpovědi v individuálním případě. Za jistých okolností (např. přítomnost nízkého titru inhibitoru) mohou být nezbytné dávky vyšší než dávky vypočítané podle vzorce.
Jako vodítko k určení potřebné dávky a frekvence opakovaných infuzí se doporučuje sledování hladin faktoru VIII v průběhu léčby. Zejména v případě větších chirurgických výkonů je přesné sledování substituční terapie pomocí koagulačních vyšetření (aktivity plazmatického faktoru VIII) nezbytné. Jednotliví pacienti se mohou lišit v odpovědi na faktor VIII vykazováním různého poločasu a obnovením.
Pediatrická populace
U dětí mladších 6 let, které byly jen omezeně vystaveny působení přípravků obsahujících faktor VIII, používejte přípravek obezřetně, jelikož pro tuto skupinu pacientů jsou k dispozici jen omezené klinické údaje.
Dlouhodobá profylaxe
Pro dlouhodobou profylaxi krvácení u pacientů s těžkou hemofilií A jsou obvyklé dávky 20 až 40 IU faktoru VIII na kg tělesné hmotnosti v intervalu dvou až tří dnů. V některých případech, zejména u mladších pacientů, však mohou být nezbytné kratší intervaly mezi dávkami nebo vyšší dávky.
Dávkování při von Willebrandově chorobě
Substituční terapie s použitím přípravku Immunate pro regulaci krvácení se řídí doporučením pro hemofilii A.
Vzhledem k tomu, že přípravek Immunate obsahuje poměrně vysoké množství faktoru VIII ve vztahu k vWF, měl by si být ošetřující lékař vědom, že trvalá léčba může způsobit nadměrné zvýšení faktoru VIII:C, což může vést ke zvýšenému riziku trombózy.
Způsob podání
Intravenózní podání.
Přípravek Immunate by se měl podávat pomalu intravenózně. Maximální rychlost infuze by neměla překročit 2 ml za minutu.
Návod pro rekonstituci tohoto přípravku před podáním, viz bod 6.6
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Hypersenzitivita
U přípravku Immunate jsou možné hypersenzitivní reakce alergického typu. Jestliže se u pacienta objeví příznaky hypersenzitivity, měli by být pacienti poučeni, aby ihned přerušili používání přípravku a kontaktovali svého lékaře. Pacienti by měli být informováni o časných příznacích hypersenzitivních reakcí, mezi něž patří vyrážka, generalizovaná kopřivka, exantém, návaly horka, svědění, otok (včetně otoku tváře a očních víček), tlak na hrudi, sípání, dyspnoe, bolest na hrudi, tachykardie, hypotenze a anafylaxe až alergický šok. V případě šoku je nutno dodržovat standardní lékařské postupy pro léčbu šoku.
Pacienti s hemofílií A
Inhibitory
Tvorba neutralizujících protilátek (inhibitorů) proti faktoru VIII je známou komplikací léčby pacientů s hemofilií A. Tyto inhibitory jsou obvykle IgG imunoglobuliny zaměřené proti prokoagulační aktivitě faktoru VIII, které jsou kvantifikovány v Bethesda jednotkách (Bethesda Units, BU) na 1 ml plazmy při použití modifikovaného testu. Riziko vzniku inhibitorů je úměrné rozsahu expozice faktoru VIII a je nejvyšší během prvních 20 dnů expozice. Vzácně mohou inhibitory vzniknout po prvních 100 dnech expozice.
Byly pozorovány případy rekurentního inhibitoru (nízký titr) po přechodu z jednoho faktoru VIII na jiný u pacientů, kteří se léčili po dobu delší než 100 dnů a kteří mají v předchozí anamnéze rozvinutí inhibitorů. Proto se doporučuje u všech pacientů, kteří přešli na jiný přípravek, pečlivě sledovat výskyt inhibitorů.
Všeobecně pacienti léčení přípravky s koagulačním faktorem VIII musí být pečlivě sledováni vzhledem k vzniku inhibitorů příslušným klinickým pozorováním a laboratorními testy.
Pokud očekávané hladiny faktoru VIII v plazmě nejsou dosaženy nebo pokud není krvácení zvládnuto odpovídající dávkou, měl by být proveden test na přítomnost inhibitoru faktoru VIII.
U pacientů s vysokými hladinami inhibitoru nemusí být léčba faktorem VIII účinná a měla by se zvážit jiná možnost léčby. Léčba takových pacientů by měla být vedena lékaři se zkušenostmi s péčí o hemofílii a inhibitorů faktoru VIII.
Inhibitory byly hlášeny převážně u pacientů dříve neléčených.
Pacienti s von Willebrandovou chorobou
Inhibitory
U pacientů s von Willebrandovou chorobou, zvláště u pacientů s typem 3, se mohou vyvinout neutralizující protilátky (inhibitory) na von Willebrandův faktor. Pokud není dosažena očekávaná VWR: RCo hladina aktivity plazmy nebo krvácení není kontrolováno pomocí odpovídající dávky, má být proveden příslušný test ke stanovení přítomnosti inhibitoru von Willebrandova faktoru. U pacientů s vysokou hladinou inhibitoru léčba von Willebrandovým faktorem nemusí být účinná a měly by být zváženy jiné terapeutické možnosti.
Trombotické příhody
Existuje riziko výskytu trombotických příhod, zejména u pacientů se známými klinickými nebo laboratorními rizikovými faktory. Proto musí být pacienti sledováni na výskyt časných příznaků trombózy. Podle současných doporučení by měla být zahájena profylaxe žilního tromboembolismu. Vzhledem k tomu, přípravek Immunate obsahuje poměrně vysoké množství faktoru VIII ve vztahu k VWF, měl by si ošetřující lékař být vědom, že pokračující léčba může způsobit nadměrné zvýšení FVIII: C. U pacientů užívajících přípravek Immunate, by se měly sledovat plazmatické hladiny FVIII: C, aby se zabránilo trvalým nadměrným hladinám FVIII: C v plazmě, které mohou zvýšit riziko trombotických příhod.
Jelikož množství sodíku v maximální denní dávce může překročit hodnotu 200 mg, je nutno u osob s dietou s nízkým obsahem sodíku tuto skutečnost zohlednit.
U dětí mladších 6 let, jejichž expozice přípravkům s faktorem VIII je omezená, používejte přípravek obezřetně, jelikož pro tuto skupinu pacientů jsou k dispozici jen omezené klinické údaje.
Standardní opatření zabraňující přenosu infekce v souvislosti s používáním léčivých přípravků vyrobených z lidské krve nebo plazmy zahrnují pečlivý výběr dárců, testování jednotlivých odběrů krve a plazmatických poolů na specifické ukazatele infekce a zařazení účinných výrobních kroků, při nichž jsou deaktivovány nebo odstraněny viry. Přesto nemůže být při přípravě léků vyráběných z lidské krve nebo plazmy zcela vyloučena možnost přenosu infekce. To platí i pro jakékoli neznámé nebo vznikající viry a jiné patogeny.
Přijatá opatření jsou považována za účinná u tzv. obalených virů, například viru lidské imunodeficience (HIV), viru hepatitidy B (HBV) a viru hepatitidy C (HCV), a u neobaleného viru hepatitidy A (HAV). Omezený účinek mají tato opatření u neobalených virů, jako je parvovirus B19. Infekce parvovirem B19 může být velmi závažná u těhotných žen (fetální infekce) a u imunodeficientních jedinců nebo jedinců se zvýšenou erytropoézou (například hemolytická anemie).
U pacientů, kteří pravidelně nebo opakovaně dostávají přípravky s faktorem VIII izolovaným z lidské plazmy, je třeba zvážit vhodné očkování (hepatitida A a B).
Při každé aplikaci přípravku Immunate doporučujeme zaznamenat název a číslo šarže přípravku, aby bylo možné zpětně přiřadit k pacientovi číslo použité šarže.
Přípravek Immunate obsahuje iso-agglutininy krevních skupin (anti-A a anti-B). U pacientů s krevní skupinou A, B nebo AB se může objevit hemolýza po opakovaném podání v krátkých časových intervalech nebo po podání velmi vysokých dávek.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
S přípravkem Immunate se neprováděly žádné studie interakcí.
Nebyly popsány žádné interakce lidského koagulačního faktoru VIII s jinými léčivými přípravky.
4.6 Fertilita, těhotenství a kojení
S faktorem VIII nebyly prováděny zvířecí reprodukční studie. Vzhledem ke vzácnému výskytu hemofilie A u žen nejsou k dispozici zkušenosti s podáváním faktoru VIII během těhotenství a kojení. Proto by se přípravek Immunate měl během těhotenství nebo kojení používat pouze tehdy, je-li to nezbytně nutné.
Informace k parvoviru B19, viz bod 4.4
Účinky přípravku Immunate na fertilitu nebyly stanoveny.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Neexistují žádné informace o účincích přípravku Immunate na schopnost řídit a obsluhovat stroje.
4.8 Nežádoucí účinky
Možné nežádoucí účinky přípravků obsahujících lidský plazmatický faktor VIII: Shrnutí bezpečnostního profilu
Hypersenzitivita nebo alergické reakce, k nimž patří angioedém, pálení a podráždění v místě vpichu, mrazení, návaly, generalizovaná kopřivka, vyrážka, bolest hlavy, kopřivka, pruritus, hypotenze, letargie, nevolnost, neklid, tachykardie, tlak na hrudi, dyspnoe, mravenčení, zvracení,sípání. Tyto reakce byly pozorovány vzácně a mohou v některých případech vyústit v těžkou anafylaxi (včetně šoku). Pacienti by měli být poučeni, aby v případě vzniku těchto příznaků kontaktovali svého lékaře (viz bod 4.4).
U pacientů s hemofilií A se mohou tvořit neutralizující protilátky (inhibitory) proti faktoru VIII. Pokud se tyto inhibitory vyskytnou, projeví se tento stav nedostatečnou klinickou odpovědí na léčbu. V takových případech doporučujeme kontaktovat specializované hemofilické centrum.
U pacientů s von Willebrandovou chorobou, zejména pacienti s typem 3, se vzácně mohou objevit neutralizující protilátky (inhibitory) na von Willebrandův faktor. Pokud se tyto inhibitory objeví, budou se manifestovat jako nepřiměřená klinická odpověď. Tyto protilátky mohou být v úzkém vztahu s anafylaktickými reakcemi. Z tohoto důvodu by pacienti, kteří prodělali anafylaktickou reakci, měli být vyšetřeni na přítomnost inhibitoru. Ve všech těchto případech se doporučuje vyhledat specializované hemofilické centrum.
Po podání vysokých dávek pacientům krevních skupin A, B nebo AB se může objevit hemolýza.
Informace o bezpečnosti z hlediska přenosných agens viz bod 4.4.
Nežádoucí účinky z hlášení z klinických studií a z post-marketingových zkušeností s přípravkem Immunate:
Přehled nežádoucích účinků v tabulce
Tabulka uvedená níže je podle MedRA databáze třídy orgánových systémů (TOS a preferovaný termín)
Četnost byla posouzena podle následující konvence:
velmi časté (> 1/10), časté (> 1/100, < 1/10), méně časté (> 1/1 000, < 1/100), vzácné (>
1/10 000, < 1/1 000), velmi vzácné (< 1/10 000), není známo (z dostupných údajů nelze určit). Z nežádoucích účinků uvedených níže v tabulce, byla hypersenzitivita hlášena z klinické studie a všechny ostatní nežádoucí účinky byly hlášeny z post-marketingové zkušenosti.
|
MedDRA standardní třída orgánového systému |
Nežádoucí účinek |
Frekvence |
|
Poruchy imunitního systému |
Hypersenzitivita |
Méně časté * |
|
Poruchy krve a lymfatického systému |
Inhibice faktoru VIII |
Není známo |
|
Koagulopatie |
Není známo | |
|
Psychiatrické poruchy |
Neklid |
Není známo |
|
Poruchy nervového systému |
Parestézie |
Není známo |
|
Závrať |
Není známo | |
|
Není známo | ||
|
Poruchy oka |
Zánět spojivek |
Není známo |
|
Srdeční poruchy |
Není známo | |
|
Palpitace |
Není známo | |
|
Cévní poruchy |
Není známo | |
|
Zrudnutí |
Není známo | |
|
Bledost |
Není známo | |
|
Respirační, hrudní a mediastinální poruchy |
Není známo | |
|
Není známo | ||
|
Gastrointestinální poruchy |
Není známo | |
|
Není známo | ||
|
Poruchy kůže a podkožní tkáně |
Urtikaria |
Není známo |
|
Vyrážka (včetně erytematózní a papulární vyrážky) |
Není známo | |
|
Pruritus |
Není známo | |
|
Erytém |
Není známo | |
|
Zvýšené pocení |
Není známo | |
|
Neurodermatitida |
Není známo |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Myalgie |
Není známo |
|
Celkové poruchy a reakce v místě aplikace |
Není známo | |
|
Není známo | ||
|
Edém (včetně periferního otoku , otoku očních víček a tváře) |
Není známo | |
|
Pyrexie |
Není známo | |
|
Není známo | ||
|
Podráždění v místě vpichu (včetně pálení) |
Není známo | |
|
Bolest |
Není známo |
*jedna hypersenzitivní reakce u 5 pacientů z 329 infuzí v jedné klinické studii
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na adresu: Státní ústav pro kontrolu léčiv, Šrobárova 48,
100 41 Praha 10, webové stránky: www.sukl.cz/nahlasit-nezadouci-ucinek.
4.9 Předávkování
Nebyl zaznamenán žádný případ předávkování.
Mohou se vyskytnout tromboembolické příhody, viz bod 4.4
U pacientů s krevní skupinou A, B nebo AB se může vyskytnout hemolýza, viz bod 4.4
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antihemoragika: von Willebrandův faktor a koagulační faktor VIII v kombinaci. ATC kód: B02BD06.
Komplex faktoru VIII/von Willebrandova faktoru se skládá ze dvou molekul (faktor VIII a von Willebrandův faktor) s různými fyziologickými funkcemi.
Při infuzi pacientovi s hemofilií se faktor VIII váže na von Willebrandův faktor v krevním oběhu.
Aktivovaný faktor VIII působí jako kofaktor aktivovaného faktoru IX, který akceleruje konverzi faktoru X na aktivovaný faktor X. Aktivovaný faktor X konvertuje protrombin na trombin. Trombin potom konvertuje fibrinogen na fibrin a může se vytvořit sraženina. Hemofilie A je na pohlaví závislá dědičná porucha srážení krve způsobená sníženými hladinami faktoru VIII a má za následek silné krvácení do kloubů, svalů a vnitřních orgánů, ať už spontánní nebo jako důsledek úrazu nebo chirurgického traumatu. Hladiny plazmatického faktoru VIII se zvyšují substituční terapií, která umožňuje dočasnou úpravu nedostatku faktoru a úpravu sklonu ke krvácení.
Kromě toho, že von Willebrandův faktor funguje jako ochranný protein faktoru VIII, zprostředkovává rovněž přilnutí destiček k místům cévních poranění a je důležitým prvkem při shlukování destiček.
5.2 Farmakokinetické vlastnosti
Veškeré farmakokinetické parametry přípravku Immunate byly měřeny u subjektů s těžkou hemofilií A (hladina faktoru VIII <1%). Vyšetření vzorků plazmy bylo provedeno v centrální laboratoři s použitím chromogenního testu FVIII. Farmakokinetické parametry pocházející ze zkřížené studie s přípravkem Immunate u 18 dříve léčených pacientů starších 12 let jsou uvedeny v tabulce níže.
Shrnutí farmakokinetických vlastností přípravku Immunate u 18 pacientů s těžkou hemofilií A (dávka = 50 IU/kg):
|
Parametr |
Průměr |
SD |
Medián |
90% Cl |
|
AUC0-* ([IUxh]/ml) |
12,2 |
3,1 |
12,4 |
11,1 až 13,2 |
|
Cmax (IU/ml) |
1,0 |
0,3 |
0,9 |
0,8 až 1,0 |
|
Tmax (h) |
0,3 |
0,1 |
0,3 |
0,3 až 0,3 |
|
Konečný poločas rozpadu (v hodinách) |
12,7 |
3,2 |
12,2 |
10,8 až 15,3 |
|
Clearance (ml/h) |
283 |
146 |
232 |
199 až 254 |
|
Střední doba prodlení (h) |
15,3 |
3,6 |
15,3 |
12,1 až 17,2 |
|
Vss (ml) |
4166 |
2021 |
3613 |
2815 až 4034 |
|
Přírůstek recovery ([IU/ml]/[IU/kg]) |
0,020 |
0,006 |
0,019 |
0,016 až 0,020 |
5.3 Předklinické údaje vztahující se k bezpečnosti
Lidský krevní koagulační faktor VIII obsažený v přípravku Immunate je běžnou složkou lidské plazmy a působí jako endogenní faktor VIII.
Předklinické údaje získané na základě konvenčních studií bezpečnostní farmakologie, toxicity opakované dávky, lokální tolerance a imunogennosti neodhalují žádné zvláštní riziko pro člověka.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek:
Lidský albumin Glycin
Chlorid sodný Citronan sodný Lysin-hydrochlorid Chlorid vápenatý
Rozpouštědlo:
Voda na injekci
6.2 Inkompatibility
Tento léčivý přípravek nesmí být mísen s jinými přípravky s výjimkou těch, které jsou uvedeny v bodě 6.6.
Měla by být použita pouze infuzní souprava dodávaná s přípravkem, protože v důsledku adsorpce koagulačního faktoru VIII na vnitřní povrchy některých infuzních zařízení může dojít k selhání léčby.
6.3 Doba použitelnosti
2 roky.
Chemická a fyzikální stabilita při použití byla prokázána na dobu 3 hodin při pokojové teplotě. Z mikrobiologického hlediska by měl být přípravek použít ihned po rekonstituci. To ovšem neplatí v případě, že způsob rekonstituce vylučuje riziko mikrobiální kontaminace (kontrolované a validované aseptické podmínky). Není-li přípravek ihned použit, odpovídá za dobu a podmínky uchovávání během použití uživatel. Rekonstituovaný přípravek nevracejte zpět do chladničky.
Během doby použitelnosti může být přípravek uchováván při pokojové teplotě (do 25 °C) maximálně po jedno období nepřesahující 6 měsíců. Dobu uchovávání při pokojové teplotě vyznačte na obalu. Po uplynutí této doby neukládejte přípravek zpět do chladničky, ale spotřebujte jej nebo jej zlikvidujte.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte a převážejte chlazené (2°C- 8°C).
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Podmínky uchovávání léčivého přípravku po rekonstituci, viz bod 6.3
6.5 Druh obalu a velikost balení
Prášek i rozpouštědlo jsou dodávány ve skleněných lahvičkách na jednorázovou dávku, EP, (prášek: hydrolytický typ II, rozpouštědlo: hydrolytický typ I), uzavřených zátkami z butylové pryže, EP.
Každé balení obsahuje:
1 lahvičku přípravku Immunate Stim Plus 1000 IU FVIII/750 IU VWF 1 lahvičku s vodou na injekci (10 ml)
1 převodní a filtrační soupravu 1 jednorázovou injekční stříkačku (10 ml)
1 jednorázovou jehlu 1 infuzní soupravu (motýlek)
Velikost balení: 1 x 1000 IU FVIII/750 IU VWF
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
K rekonstituci použijte pouze infuzní soupravu, která je součástí balení. Přípravek Immunate je určen k rekonstituci bezprostředně před podáním, jelikož neobsahuje konzervační přísady. Léčivý přípravek po rekonstituci by měl být před podáním vizuálně zkontrolován na výskyt částic a změny zabarvení. Roztok má být čirý nebo lehce opalescentní. Roztoky rekonstituovaného přípravku, které jsou zakalené nebo obsahují usazeniny, se nesmí používat.
Před infuzí přípravku Immunate a po ní se doporučuje propláchnout žilní přístup izotonickým fyziologickým roztokem.
Rekonstituce prášku:
Použijte aseptický postup!
1. Zahřejte neotevřenou lahvičku obsahující rozpouštědlo (sterilizovanou vodu na injekci) na pokojovou teplotu (maximálně 37°C).
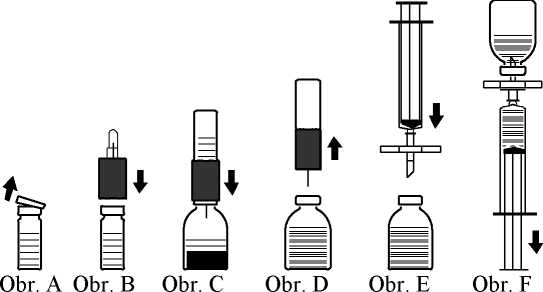
2. Odstraňte ochranná víčka z lahvičky s práškem a z lahvičky s rozpouštědlem (obr. A) a očistěte pryžové zátky obou lahviček.
3. Umístěte zvlněný okraj převodní soupravy na lahvičku s rozpouštědlem a přitlačte (obr. B).
4. Odstraňte ochranný kryt z druhého konce převodní soupravy; nedotýkejte se obnaženého konce.
5. Převraťte převodní soupravu s připojenou lahvičkou s rozpouštědlem nad lahvičku s práškem a propíchněte volnou jehlou pryžovou zátku lahvičky s práškem (obr. C). Rozpouštědlo se pod tlakem samo natáhne do lahvičky s práškem.
6. Asi po jedné minutě oddělte obě lahvičky od sebe odpojením převodní soupravy
s připojenou lahvičkou s rozpouštědlem od lahvičky s práškem (obr. D). Jelikož se přípravek snadno rozpouští, pohybujte lahvičkou s koncentrátem pouze jemně - pokud je to vůbec nutné. OBSAH LAHVIČKY NEPROTŘEPÁVEJTE. NEPŘEVRACEJTE LAHVIČKU S PRÁŠKEM, DOKUD NEBUDETE PŘIPRAVENI NATÁHNOUT OBSAH.
7. Po rekonstituci je nutno pohledem zkontrolovat, zda připravený roztok neobsahuje pevné částice nebo zda roztok neztratil svou barvu. I přes přísné dodržování postupu rekonstituce se může občas objevit několik drobných částic. Přiložená filtrační souprava částice odstraní a účinnost vyznačená na obalu nebude snížena.
Podávání:
Použijte aseptický postup!
1. Aby nedošlo k podání částí pryže ze zátky spolu s léčivým přípravkem (riziko mikroembolie), použijte přiloženou filtrační soupravu. Chcete-li natáhnout rozpuštěný přípravek, nasaďte filtrační soupravu na přiloženou jednorázovou injekční stříkačku a propíchněte jí pryžovou zátku (obr. E).
2. Na okamžik odpojte injekční stříkačku od filtrační soupravy. Do lahvičky s práškem se dostane vzduch a případná pěna opadne. Poté přes filtrační soupravu natáhněte roztok do injekční stříkačky (obr. F).
3. Odpojte injekční stříkačku od filtrační soupravy a pomalu roztok intravenózně vstříkněte (maximální rychlost vstřikování: 2 ml za minutu) pomocí přiložené infuzní soupravy (motýlek), případně přiložené jednorázové jehly.
Všechen nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Do 30.11.2016:
Baxter AG Industriestrasse 67 A-1221 Vídeň Rakousko
Od 1.12.2016:
Baxalta Innovations GmbH
Industriestrasse 67 A-1221 Vídeň Rakousko
8. REGISTRAČNÍ ČÍSLO
75/618/97-C
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
30.7.1997/9.12.2012
10. DATUM REVIZE TEXTU
10.8.2016
11/11
Účinnost FVIII byla určena podle mezinárodní normy Světové zdravotnické organizace pro koncentráty FVIII.
Aktivita ristocetin kofaktoru lidského von Willebrandova faktoru byla určena podle mezinárodní normy Světové zdravotnické organizace pro koncentrát von Willebrandova faktoru.
Bez stabilizátoru (albuminu); maximální specifická aktivita při poměru aktivity faktoru VIII k antigenu von Willebrandova faktoru 1:1 je 100 IU faktoru VIII na mg proteinu.