Imlygic 10^8 Jednotek Tvořících Plaky (Pfu)/Ml
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Imlygic 106 jednotek tvořících plaky (PFU)/ml injekční roztok Imlygic 108 jednotek tvořících plaky (PFU)/ml injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
2.1 Obecný popis
Talimogenum laherparepvecum je oslabený virus herpes simplex typu 1 (HSV-1) získaný funkční delecí dvou genů (ICP34.5 a ICP47) a vložením kódující sekvence pro lidský faktor stimulující granulocyto-makrofágové kolonie (granulocyte macrophage colony-stimulating factor - GM-CSF; viz bod 5.1).
Talimogen laherparepvek je produkován ve Vero buňkách rekombinantní DNA technologií.
2.2 Kvalitativní a kvantitativní složení
Imlygic 106 jednotek tvořících plaky (PFU)/ml injekční roztok
Jedna injekční lahvička obsahuje 1 ml připraveného objemu přípravku Imlygic o nominální koncentraci 1 x 106 (1 milion) jednotek tvořících plaky (plaque forming units - PFU)/ml.
Imlygic 108 jednotek tvořících plaky (PFU)/ml injekční roztok
Jedna injekční lahvička obsahuje 1 ml připraveného objemu přípravku Imlygic o nominální koncentraci 1 x 108 (100 milionů) jednotek tvořících plaky (PFU)/ml.
Pomocná látka se známým účinkem
Každé 4 ml dávky obsahují přibližně 30 mg (1,3 mmol) sodíku a 80 mg sorbitolu. Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok.
Imlygic 106 jednotek tvořících plaky (PFU)/ml injekční roztok Čirá až poloprůsvitná tekutina po rozmrazení ze zmrzlého stavu.
Imlygic 108 jednotek tvořících plaky (PFU)/ml injekční roztok Poloprůsvitná až neprůsvitná tekutina po rozmrazení ze zmrzlého stavu. Přípravek může obsahovat bílé, viditelné částice různého tvaru obsahující virus.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Imlygic je indikován k léčbě dospělých s neresekovatelným melanomem s regionálními nebo vzdálenými metastázami (stadium IIIB, IIIC a IVM1a) bez postižení kostí, mozku, plic nebo jiného viscerálního postižení (viz body 4.4 a 5.1).
4.2 Dávkování a způsob podání
Léčbu talimogen laherparepvekem má zahájit a dohlížet kvalifikovaný lékař se zkušenostmi s léčbou nádorových onemocnění.
Pacienti léčení přípravkem Imlygic musí dostat Kartu pacienta a musí být informováni o rizicích přípravku Imlygic (viz též příbalová informace).
Dávkování
Imlygic se dodává v injekčních lahvičkách na jedno použití o objemu 1 ml ve dvou různých koncentracích:
• 106 (1 milion) PFU/ml - pouze pro úvodní dávku.
• 108 (100 milionů) PFU/ml - pro všechny další dávky.
Celkový injekční objem pro každou léčebnou návštěvu má být maximálně 4 ml. Úvodní doporučená dávka je maximálně 4 ml přípravku Imlygic o koncentraci 106 (1 milion) PFU/ml. Další dávky se podávají v objemu maximálně 4 ml přípravku Imlygic o koncentraci 108 (100 milionů) PFU/ml.
Doporučené dávkovací schéma pro přípravek Imlygic je uvedeno v tabulce 1.
Tabulka 1: Doporučené dávkovací schéma přípravku Imlygic
|
Léčebná návštěva |
Léčebný interval |
Maximální celkový injekční objem |
Dávky o koncentraci |
Stanovení priorit lézí, do nichž se podá injekce |
|
Úvodní |
- |
Až 4 ml |
106 (1 milion) PFU/ml |
• Nejdřív podejte injekci do největší léze/největších lézí. • Seřaďte zbylé léze podle velikosti a pokračujte v aplikaci až do dosažení maximálního injekčního objemu. |
|
Druhá |
3 týdny po úvodní léčbě |
Až 4 ml |
108 (100 milionů) PFU/ml |
• Nejdřív podejte injekci do nových lézí (léze, které se mohly vytvořit od úvodní léčby). • Zbylé léze seřaďte podle velikosti a pokračujte v aplikaci až do dosažení maximálního injekčního objemu. |
|
Léčebná návštěva |
Léčebný interval |
Maximální celkový injekční objem |
Dávky o koncentraci |
Stanovení priorit lézí, do nichž se podá injekce |
|
Všechny další léčebné návštěvy (včetně reiniciace) |
2 týdny po předchozí léčbě |
Až 4 ml |
108 (100 milionů) PFU/ml |
• Nejdřív podejte injekci do nových lézí (léze, které se mohly vytvořit od úvodní léčby). • Zbylé léze seřaďte podle velikosti a pokračujte v aplikaci až do dosažení maximálního injekčního objemu. |
Stanovení objemu dávky přípravku Imlygic (na lézi)
Objem přípravku Imlygic, který se má aplikovat do každé léze, závisí na velikosti léze a stanovuje se podle tabulky 2. Celkový injekční objem pro každé léčebné sezení má být maximálně 4 ml.
Tabulka 2: Výběr injekčního objemu přípravku Imlygic na základě velikosti léze
|
Velikost léze (největší rozměr) |
Injekční objem přípravku Imlygic |
|
> 5 cm |
až 4 ml |
|
> 2,5 cm až 5 cm |
až 2 ml |
|
> 1,5 cm až 2,5 cm |
až 1 ml |
|
> 0,5 cm až 1,5 cm |
až 0,5 ml |
|
< 0,5 cm |
až 0,1 ml |
Před dosažením odpovědi na léčbu může u pacientů dojít ke zvětšení stávající léze/lézí nebo objevení se nové léze. Dokud zůstávají léze, do kterých lze přípravek aplikovat, v léčbě přípravkem Imlygic se má pokračovat po dobu nejméně 6 měsíců, dokud lékař nerozhodne, že další léčba přípravkem Imlygic již není pro pacienta přínosná, nebo že je nutná jiná léčba.
Léčbu přípravkem Imlygic lze opět zahájit, pokud se objeví nové léze po úplné odpovědi na léčbu a lékař rozhodne, že léčba bude pro pacienta přínosná.
Zvláštní populace
Pediatrická populace
Bezpečnost a účinnost přípravku Imlygic u pediatrických pacientů nebyla stanovena. Nejsou dostupné žádné údaje.
Starší pacienti
Úprava dávky u pacientů > 65 let není nutná (viz bod 5.1).
Porucha funkce jater a ledvin
Nebyly provedené žádné klinické studie hodnotící vliv poruchy funkce jater nebo ledvin na farmakokinetiku talimogen laherparepveku. Úprava dávky u pacientů s poruchou funkce jater nebo ledvin ale není nutná.
Způsob podání
Imlygic se podává intralezionální injekcí do kožních, podkožních a/nebo uzlinových lézí, které jsou viditelné, hmatné nebo jsou dostupné pod sonografickou kontrolou.
Jestliže jsou zdravotničtí pracovníci náhodně vystaveni přípravku Imlygic, postupujte podle bodů 4.4 a 6.6.
Zdravotničtí pracovníci, kteří jsou imunokompromitovaní, nebo těhotné ženy nesmějí podávat
přípravek Imlygic a nesmějí přijít do přímého kontaktu s místem/místy injekčního vpichu přípravku
Imlygic nebo s tělesnými tekutinami léčených pacientů (viz body 4.3 a 4.4).
Při přípravě a podání přípravku Imlygic pacientům dodržujte tyto pokyny:
Před injekcí
• Nechte injekční lahvičku/lahvičky s přípravkem Imlygic rozmrznout při pokojové teplotě. Rozmražený Imlygic může být před podáním uchováván (viz bod 6.3).
• Odeberte požadované množství přípravku Imlygic z injekční lahvičky do stříkačky za použití aseptické techniky. Doporučují se injekční jehly 22 - 26 gauge.
• Místo injekce lze ošetřit lokálním anestetikem. Injekční anestetikum lze aplikovat kolem periférie léze, ale nesmí se aplikovat přímo do léze.
• Očistěte lézi a okolní oblasti tampónem s alkoholem a nechte místo oschnout.
Injekce
• Podejte přípravek Imlygic intralezionální injekcí do kožních, podkožních a/nebo uzlinových lézí, které jsou viditelné, hmatné nebo dostupné pod sonografickou kontrolou.
• Pomocí tabulky 2 stanovte injekční objem pro každou lézi.
• Pomocí jednoho injekčního vpichu podejte přípravek Imlygic do různých míst léze tak, aby při paprskovitém pohybu jehlou v lézi bylo dosaženo stejnoměrného a úplného rozptýlení podaného přípravku. Více vpichů je možné použít tehdy, pokud je léze větší než paprskovitý dosah jehly.
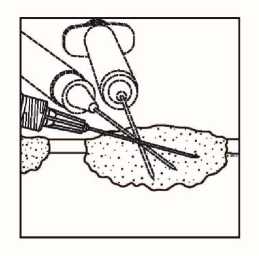
Podkožní léze
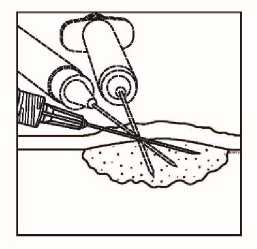
Uzlinové léze
Obrázek 2.
Injekční podání do podkožních lézí
Obrázek 3.
Injekční podání do uzlinových lézí
• Rozptylte přípravek Imlygic rovnoměrně a úplně v lézi posunováním jehly, aniž byste ji z léze zcela vytáhli. Měňte směr jehly tolikrát, kolikrát je to nutné a přitom aplikujte zbylou dávku přípravku Imlygic. V této činnosti pokračujte až do rovnoměrného a úplného rozptýlení dávky.
• Při vytahování jehlu z léze vytahujte pomalu, aby nedošlo k úniku nebo vystříknutí přípravku Imlygic z místa injekčního vpichu.
• Tento postup opakujte i u dalších lézí, do kterých se má přípravek aplikovat. Použijte novou jehlu vždy po úplném vytáhnutí jehly z léze a taktéž vždy, když se přípravek aplikuje do jiné
léze.
Po injekci
• Stlačujte místo injekčního vpichu sterilní gázou nejméně 30 sekund.
• Otřete místo vpichu a jeho okolí alkoholem a přikryjte injikovanou lézi absorpčním tampónem a suchým okluzivním obvazem.
Likvidace
Veškerý materiál, který přišel do kontaktu s přípravkem Imlygic (např. injekční lahvička, stříkačka, jehla, obvazový materiál nebo gáza), zlikvidujte v souladu s místními požadavky zdravotnického zařízení (viz bod 6.6).
4.3 Kontraindikace
• Pacienti s anamnézou hypersenzitivity na talimogen laherparepvek nebo na kteroukoli pomocnou látku.
• Těžce imunokompromitovaní pacienti (např. pacienti s těžkou vrozenou nebo získanou buněčnou a/anebo humorální imunodeficiencí) (viz bod 4.4).
4.4 Zvláštní upozornění a opatření pro použití
Dříve léčení pacienti
Údaje o účinnosti přípravku Imlygic v druhé nebo další linii léčby jsou omezené.
Imunokopromitovaní pacienti
Imlygic nebyl hodnocen u imunokompromitovaných pacientů. Na základě údajů ze studií na zvířatech mohou mít těžce imunokompromitovaní pacienti zvýšené riziko diseminované herpetické infekce a nesmějí být léčeni přípravkem Imlygic (viz body 4.3 a 5.3). Diseminovaná herpetická infekce se může rovněž vyskytnout u imunokompromitovaných pacientů (jako jsou pacienti s HIV/AIDS, leukemií, lymfomem, běžnou variabilní imunodeficiencí nebo pacienti, kteří vyžadují chronickou léčbu vysokými dávkami steroidů nebo jinými imunosupresivy). Před podáním přípravku Imlygic těmto pacientům se musí zvážit rizika a přínosy léčby.
Náhodná expozice přípravku Imlygic
Náhodná expozice může vést k přenosu přípravku Imlygic a herpetické infekci. Zdravotničtí pracovníci a osoby, které jsou s pacienty v blízkém kontaktu (např. členové domácnosti, pečovatelé, sexuální partneři nebo osoby sdílející stejnou postel), se mají vyvarovat přímého kontaktu s injikovanými lézemi nebo s tělesnými tekutinami léčených pacientů během celého léčebného období a až 30 dní po posledním podání přípravku (viz bod 6.6). U zdravotnických pracovníků byly zaznamenány případy náhodného píchnutí jehlou a vystříknutí přípravku Imlygic během jeho přípravy a podávání.
Těhotné ženy nebo imunokompromitované osoby, které jsou v blízkém kontaktu s pacientem, nesmí měnit pacientovi obvazy nebo čistit místo injekčního vpichu. Těhotné ženy, novorozenci a imunokompromitovaní jedinci nesmí být vystaveni potenciálně kontaminovaným materiálům.
Zdravotničtí pracovníci mají zajistit, aby pacienti byli schopni dostát požadavku na přikrytí injekčních míst okluzivním obvazem (viz bod 6.6). Pacientům se má taktéž doporučit, aby se nedotýkali nebo neškrábali místa injekčního vpichu, jelikož to může vést k nechtěnému přenosu přípravku Imlygic na jiné části těla nebo na osoby, které jsou s nimi v blízkém kontaktu.
I když není známo, zda se Imlygic může přenášet pohlavním stykem, je známo, že divoký typ viru HSV-1 lze přenést sexuálním kontaktem. Pacienti mají být poučeni, aby při pohlavním styku používali latexový kondom, aby zabránili možnému přenosu přípravku Imlygic. Ženy ve fertilním věku mají být poučeny, aby během léčby přípravkem Imlygic používaly účinnou antikoncepční metodu k zabránění otěhotnění (viz bod 4.6).
Pečovatelé mají být poučeni, aby používali ochranné rukavice, když pomáhají pacientům při přikládání nebo výměně okluzivního obvazu a aby dodržovali bezpečnostní opatření při likvidaci použitých obvazů a čistícího materiálu (viz body 4.2 a 6.6).
Při náhodné expozici přípravku Imlygic mají být zasažené osoby poučeny, aby si důkladně očistily postiženou oblast mýdlem a vodou a/nebo dezinfekčním činidlem. Jestliže se rozvinou známky nebo příznaky herpetické infekce, musí tyto osoby kontaktovat svého zdravotnického pracovníka.
Talimogen laherparepvek je citlivý na acyklovir.
Herpetická infekce u pacientů léčených přípravkem Imlygic
V klinických studiích byly u pacientů léčených přípravkem Imlygic hlášeny případy herpetické infekce (včetně oparů a herpetické keratitidy). Předpokládá se, že symptomy lokální nebo systémové infekce v možné souvislosti s přípravkem Imlygic budou podobné symptomům vyvolaným infekcí divokým typem viru HSV-1.
Je známo, že osoby s infekcí divokým typem viru HSV-1 mohou mít celoživotní riziko symptomatické herpetické infekce vzhledem k reaktivaci latentního divokého typu viru HSV-1. Je nutné brát v úvahu symptomatickou herpetickou infekci způsobenou možnou reaktivací přípravku Imlygic.
Pacienti, u nichž se rozvine herpetická infekce, mají být poučeni, aby dodržovali standardní hygienická opatření k zabránění přenosu viru.
Talimogen laherparepvek je citlivý na acyklovir. Rizika a přínosy léčby přípravkem Imlygic se musí zvážit před podáním acykloviru nebo jiných antivirotik indikovaných k léčbě herpetické infekce. Tato léčiva mohou mít vliv na účinnost přípravku Imlygic, jsou-li podávána systémově nebo lokálně přímo do místa injekčního vpichu.
Celulitida v místě injekčního vpichu
Po léčbě přípravkem Imlygic se může objevit nekróza nebo ulcerace nádorové tkáně. Byly popsány případy celulitidy a systémové bakteriální infekce. Doporučuje se důkladná péče o ránu a bezpečnostní opatření k prevenci infekce, a to zejména pokud nekróza tkáně vyústí v otevřené rány.
Zhoršené hojení v místě injekčního vpichu
V klinických studiích byly hlášeny případy zhoršeného hojení v místě injekčního vpichu. Imlygic může zvýšit riziko zhoršeného hojení u pacientů s výchozími rizikovými faktory (např. předchozí ozařování v místě injekčního vpichu nebo léze v místech se špatnou vaskularizací).
Jestliže se rozvine perzistující infekce nebo opožděné hojení, je třeba před pokračováním léčby zvážit rizika a přínosy léčby přípravkem Imlygic.
Imunitně zprostředkované příhody
V klinických studiích byly u pacientů léčených přípravkem Imlygic hlášeny imunitně zprostředkované příhody jako glomerulonefritida, vaskulitida, pneumonitida, zhoršení psoriázy a vitiligo.
Je třeba zvážit rizika a přínosy léčby přípravkem Imlygic u pacientů s výchozím autoimunitním onemocněním nebo před pokračováním léčby u pacientů, u kterých se rozvinuly imunitně zprostředkované příhody.
Plazmocytom v místě injekčního vpichu
Po podání přípravku Imlygic byly popsány případy plazmocytomu v blízkosti místa injekčního vpichu. Je třeba zvážit rizika a přínosy léčby přípravkem Imlygic u pacientů s mnohočetným myelomem nebo u pacientů, u nichž se během léčby objevil plazmocytom.
Obstrukční porucha dýchacích cest
Po léčbě přípravkem Imlygic byly popsány případy obstrukční poruchy dýchacích cest. Při aplikaci do lézí v blízkosti velkých dýchacích cest je třeba postupovat s opatrností.
HSV-1 séronegativní pacienti
U HSV-1 séronegativních pacientů před zahájením léčby byl hlášen vyšší výskyt pyrexie, zimnice a onemocnění podobných chřipce v porovnání s HSV-1 séropozitivními pacienty před zahájením léčby, a to zejména v období prvních 6 léčebných cyklů (viz bod 4.8).
Všichni pacienti
Imlygic obsahuje sorbitol (E420). Pacienti se vzácnými dědičnými problémy s intolerancí fruktózy by tento přípravek neměli používat.
Přípravek Imlygic obsahuje přibližně 30 mg (1,3 mmol) sodíku v jedné dávce (4 ml). Nutno vzít v úvahu u pacientů na dietě s nízkým obsahem sodíku.
Sledovatelnost přípravku Imlygic
Aby se zlepšila sledovatelnost biologických léčivých přípravků, je třeba do zdravotní dokumentace pacienta zřetelně zapsat (nebo uvést) obchodní název a číslo šarže podaného přípravku.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
S přípravkem Imlygic nebyly provedeny žádné studie interakcí. Acyklovir a další antivirotika mohou mít vliv na účinnost přípravku Imlygic, pokud se podávají systémově nebo lokálně přímo do místa injekčního vpichu. Před podáním acykloviru nebo jiných antivirotik indikovaných k léčbě herpetické infekce zvažte rizika a přínosy léčby přípravkem Imlygic.
4.6 Fertilita, těhotenství a koj ení
Ženy ve fertilním věku/antikoncepce
Ženy ve fertilním věku mají být poučeny, aby během léčby přípravkem Imlygic používaly účinnou antikoncepční metodu k zabránění otěhotnění.
Všichni pacienti mají být poučeni, aby při pohlavním styku používali latexový kondom k zabránění možného přenosu přípravku Imlygic (viz bod 4.4).
U těhotných žen nebyly provedeny adekvátní a dobře kontrolované studie s talimogen laherparepvekem.
Jestliže těhotná žena má infekci divokým typem viru HSV-1 (primární infekce nebo její reaktivace), existuje možné riziko přestupu viru placentární bariérou a rovněž riziko přenosu během porodu z důvodu uvolňování viru. Infekce divokým typem viru HSV-1 jsou spojovány se závažnými nežádoucími účinky včetně multiorgánového selhání a úmrtí, pokud se plod nebo novorozenec nakazí herpetickou infekcí divokým typem viru. I když dosud neexistují žádné klinické údaje o infekcích talimogenem laherparepvekem u těhotných žen, mohlo by existovat riziko pro plod nebo novorozence, pokud by měl talimogen laherparepvek působit stejně. Ve studiích na zvířatech nebyly pozorovány žádné účinky na embryo-fetální vývoj (viz bod 5.3). Jako bezpečnostní opatření je vhodné se vyvarovat použití talimogen laherparepveku během těhotenství.
Mohou se vyskytnout transplacentární metastázy maligního melanomu. Vzhledem k tomu, že talimogen laherparepvek je určen k tomu, aby vstupoval a replikoval se v nádorové tkáni, může existovat riziko expozice plodu talimogen laherparepveku z nádorové tkáně, která prošla placentou.
Pokud se Imlygic používá v těhotenství, nebo když pacientka během léčby přípravkem Imlygic otěhotní, musí být seznámena s potenciálním nebezpečím pro plod a/nebo novorozence.
Kojení
Není známo, zda se talimogen laherparepvek vylučuje do lidského mateřského mléka. Je třeba se rozhodnout, zda přerušit kojení nebo přerušit/ukončit léčbu přípravkem Imlygic, přičemž se má vzít v úvahu přínos kojení pro dítě a přínos léčby pro ženu.
Fertilita
Nebyly provedeny žádné klinické studie hodnotící účinky talimogen laherparepveku na fertilitu (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Talimogen laherparepvek má malý vliv na schopnost řídit a obsluhovat stroje. Vzhledem k možným nežádoucím účinkům, jako jsou závratě a stav zmatenosti (viz bod 4.8), je třeba pacienty poučit, aby byli při řízení a obsluze strojů opatrní, dokud není jisté, že na ně talimogen laherparepvek v tomto smyslu nepůsobí negativně.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Bezpečnost přípravku Imlygic byla hodnocena v pivotní studii, kde 292 pacientů dostalo nejméně 1 dávku přípravku Imlygic (viz bod 5.1). Medián doby expozice přípravku Imlygic byl 23 týdnů (5,3 měsíců). Celkem 26 pacientů bylo vystaveno přípravku Imlygic po dobu nejméně 1 roku.
Nejčastěji hlášené nežádoucí účinky (> 25 %) u pacientů léčených přípravkem Imlygic byly únava (50,3 %), zimnice (48,6 %), pyrexie (42,8 %), nauzea (35,6 %), onemocnění podobné chřipce (30,5 %) a bolest v místě injekčního vpichu (27,7 %). Celkem devadesát osm procent (98 %) těchto hlášených nežádoucích účinků bylo lehkých nebo středně závažných. Nejčastějším nežádoucím účinkem stupně 3 nebo vyššího byla celulitida (2,1 %) (viz bod 4.4).
Tabulkový seznam nežádoucích účinků
V tabulce 3 jsou uvedené nežádoucí účinky pozorované v klinických studiích u pacientů s melanomem léčených přípravkem Imlygic v porovnání s GM-CSF. Nežádoucí účinky jsou uvedeny podle třídy orgánových systémů a frekvence. Frekvence jsou definované jako velmi časté (> 1/10), časté (> 1/100 až < 1/10) a méně časté (> 1/1000 až < 1/100). V každé skupině četnosti jsou nežádoucí účinky seřazeny podle klesající závažnosti.
Tabulka 3: Nežádoucí účinky pozorované v klinických studiích u pacientů s melanomem léčených přípravkem Imlygic
|
Infekce a infestace | |
|
Časté |
Celulitida, herpetická infekce v ústech |
|
Méně časté |
Infekce v místě vpichu |
|
Novotvary benigní |
, maligní a blíže neurčené (zahrnující cysty a polypy) |
|
Časté |
Bolest v nádoru, infekce novotvaru |
|
Méně časté |
Plazmocytom v místě injekčního vpichu |
|
Poruchy krve a lymfatického systému | |
|
Velmi časté |
Periferní otoky |
|
Časté | |
|
Poruchy imunitního systému | |
|
Časté |
Imunitně zprostředkované příhody^ |
|
Poruchy metabolismu a výživy | |
|
Časté |
Dehydratace |
|
Poruchy nervového systému | |
|
Velmi časté | |
|
Časté |
Stav zmatenosti, anxieta, deprese, závratě, insomnie |
|
Poruchy oka | |
|
Méně časté | |
|
Poruchy ucha a labyrintu | |
|
Časté |
Bolest ucha |
|
Srdeční poruchy | |
|
Časté | |
|
Cévní poruchy | |
|
Časté |
Hluboká žilní trombóza, hypertenze, zrudnutí kůže |
|
Respirační, hrudní a mediastinální poruchy | |
|
Velmi časté | |
|
Časté |
Námahová dušnost, bolest orofaryngu, infekce horních cest dýchacích |
|
Méně časté |
Obstrukční porucha dýchacích cest |
|
Gastrointestinální |
poruchy |
|
Velmi časté | |
|
Časté |
Bolest břicha, břišní diskomfort |
|
Poruchy kůže a podkožní tkáně | |
|
Časté |
Vitiligo, vyrážka, dermatitida |
|
Poruchy svalové a |
kosterní soustavy a pojivové tkáně |
|
Velmi časté |
Myalgie, artralgie, bolest končetiny |
|
Časté |
Bolest zad, bolest v tříslech |
|
Celkové poruchy a reakce v místě aplikace | |
|
Velmi časté |
Onemocnění podobné chřipce, pyrexie, zimnice, únava, bolest, reakce v místě |
§ Reakce v místě injekčního vpichu zahrnují: velmi častý termín bolest v místě injekčního vpichu, časté termíny erytém v místě injekčního vpichu, krvácení v místě injekčního vpichu, otok v místě injekčního vpichu, reakce v místě injekčního vpichu, zánět v místě injekčního vpichu, sekrece, výtok v místě injekčního vpichu a méně častý termín horko v místě injekčního vpichu.
|
injekčního vpichu§ | |
|
Časté |
Malátnost, bolest v axile |
|
Vyšetření | |
|
Časté |
Pokles tělesné hmotnosti |
|
Poranění, otravy a procedurální komplikace | |
|
Časté |
Komplikace v ráně, sekrece z rány, kontuze, bolest při výkonu |
Ť Imunitně zprostředkované příhody zahrnují: méně časté termíny vaskulitida, pneumonitida, zhoršení psoriázy a glomerulonefritida.
Popis vybraných nežádoucích účinků
Imunitně zprostředkované příhody
Imunitně zprostředkované příhody hlášené v pivotní klinické studii zahrnovaly případ zhoršení psoriázy u pacienta s anamnézou psoriázy, jeden případ pneumonitidy u pacienta s anamnézou autoimunitního onemocnění, jeden případ vaskulitidy a dva případy glomerulonefritidy, z nichž jeden se projevil jako akutní selhání ledvin.
Plazmocytom
V klinických studiích byl pozorován jeden případ plazmocytomu v místě injekčního vpichu u pacienta, u kterého byl zjištěn mnohočetný myelom.
Celulitida
V pivotní klinické studii (studie 005/05) byly zaznamenané případy celulitidy, z nichž některé byly hodnocené jako závažné nežádoucí příhody. Žádný z nich však nevedl k trvalému vysazení léčby přípravkem Imlygic. Doporučuje se důkladná péče o rány a dodržování opatření k zabránění vzniku infekce, zejména pokud nekróza tkání vyústí v otevřené rány.
Symptomy podobné chřipce
U 90 % pacientů léčených přípravkem Imlygic se vyskytly symptomy podobné chřipce. Pyrexie, zimnice a onemocnění podobné chřipce, které se mohou vyskytnout kdykoliv během léčby přípravkem Imlygic, obvykle odezněly během 72 hodin. Tyto příhody byly hlášené častěji během prvních 6 léčebných cyklů, a to hlavně u pacientů, kteří byli před léčbou HSV-1 negativní.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Klinické zkušenosti s předávkováním přípravkem Imlygic nejsou. V klinických studiích se podávaly dávky až do 4 ml o koncentraci 108 PFU/ml každé 2 týdny bez průkazu dávku limitující toxicity. Maximální dávka přípravku Imlygic, kterou lze bezpečně podávat, nebyla stanovena. V případě podezření na předávkování nebo při neúmyslném intravenózním podání se má pacient léčit symptomaticky, např. acyklovirem nebo jinými antivirotiky (viz bod 4.4), a podle potřeby se mají zavést podpůrná opatření.
FARMAKOLOGICKÉ VLASTNOSTI
5.
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antineoplastika a imunomodulující léčiva, ATC kód: L01XX51. Mechanismus účinku
Talimogen laherparepvek je onkolytické imunoterapeutikum, které je odvozeno od HSV-1. Talimogen laherparepvek je modifikovaný tak, aby se replikoval v nádorech a vytvářel lidský imunitu stimulující protein GM-CSF. Talimogen laherparepvek způsobuje smrt nádorových buněk a uvolňování nádorových antigenů. Předpokládá se, že talimogen laherparepvek spolu s GM-CSF podporuje systémovou protinádorovou imunitní odpověď a efektorovou odpověď T-buněk. Myši, u kterých došlo po léčbě k úplné regresi primárních nádorů, byly rezistentní na následnou opakovanou expozici nádoru.
Modifikace vedoucí od HSV-1 k talimogen laherparepveku zahrnují deleci genů ICP34.5 a ICP47. Zatímco antivirová imunitní odpověď chrání normální buňky před infekcí vyvolanou talimogen laherparepvekem, bylo prokázáno, že nádory jsou citlivé na poškození a buněčnou smrt způsobenou viry HSV-1 s deficitem genu ICP34.5, včetně talimogen laherparepveku. Delece genu ICP47 zabraňuje downregulaci molekul prezentujících antigen a zvyšuje expresi genu HSV US11, čímž zvyšuje replikaci viru v nádorových buňkách.
Klinická účinnost a bezpečnost
Studie 005/05
Bezpečnost a účinnost monoterapie přípravkem Imlygic v porovnání se subkutánně podávaným GM-CSF byla hodnocena v mezinárodní, otevřené a randomizované klinické studie fáze III u pacientů s melanomem stupně IIIB, IIIC, a IV, který nebyl indikovaný k chirurgické resekci. Předchozí systémová léčba melanomu byla povolená, ale nebyla vyžadována. Ze studie byli vyloučeni pacienti s aktivními mozkovými metastázami, metastázami v kostech, rozsáhlým viscerálním onemocněním, primárním očním nebo slizničním melanomem a pacienti na systémové léčbě antiherpetickým přípravkem.
Pacienti byli randomizováni v poměru 2:1 k léčbě přípravkem Imlygic nebo GM-CSF (n = 436; 295 pacientů dostávalo Imlygic, 141 pacientů dostávalo GM-CSF). Imlygic byl podáván intralezionální injekcí v úvodní koncentraci 106 (1 milion) PFU/ml 1. den a dále v koncentraci 108 (100 milionů) PFU/ml 21. den a poté každé 2 týdny v dávce až 4 ml. GM-CSF byl podáván subkutánně v dávce 125 pg/m2 denně po dobu 14 dní a poté následovalo 14denní období bez léčby v opakujících se intervalech.
Aby se mohl projevit opožděný imunitně zprostředkovaný protinádorový účinek, pacienti byli léčeni minimálně 6 měsíců nebo do doby, kdy již nebyly přítomné žádné léze vhodné k injekční léčbě.
Během této doby měla léčba pokračovat i při zvětšování již přítomné léze/přítomných lézí a/nebo rozvoji nové léze/nových lézí, pokud se u pacienta nevyvinula netolerovatelná toxicita nebo pokud zkoušející neusoudil, že v nejlepším zájmu pacienta bylo léčbu ukončit nebo léčit melanom jinou terapií. Po 6 měsících léčby pacienti pokračovali v léčbě až do klinicky významné progrese onemocnění (tj. progrese nemoci spojená s poklesem performance statusu a/nebo potřeba alternativní léčby na základě posouzení zkoušejícícho). Pacienti, u nichž byla dosažena odpověď po 12 měsících léčby, mohli pokračovat v léčbě dalších až 6 měsíců. Průměrné (SD) trvání léčby u ITT populace (ITT - intent to treat) bylo 15,76 týdnů (15,79) v rameni s GM-CSF a 26,83 týdnů (18,39) v rameni s přípravkem Imlygic. Primárním cílovým parametrem byl výskyt trvající odpovědi (DRR - durable response rate) [definovaný jako procento pacientů s kompletní odpovědí (CR) nebo částečnou odpovědí (PR) přetrvávající nepřetržitě minimálně 6 měsíců] podle zaslepeného centrálního vyhodnocení. Sekundárními cílovými parametry byly celkové přežití (OS), celkový výskyt objektivní odpovědi (ORR - overall response rate) [PR+CR], doba do odpovědi, trvání odpovědi a doba do selhání léčby (doba od randomizace do první epizody klinicky významné progrese onemocnění, kdy není dosažená odpověď po příhodě progrese nebo až do úmrtí pacienta).
Průměrný věk byl 63 let (rozptyl: 22 až 94 let); 26,5 % pacientů bylo starších 65 let a 23,3 % pacientů bylo starších 74 let. Většina pacientů (98 %) byli běloši. Muži tvořili 57 % studijní populace a 70 % pacientů mělo výchozí ECOG performance status 0. Ze zařazených pacientů mělo 22 % stadium IV M1c nemoci a 53 % pacientů podstoupilo v minulosti léčbu melanomu jako je chemoterapie a imunoterapie založená na cytokinu navíc k chirurgickému výkonu, adjuvantní léčbě nebo ozařování. Celkem 58 % všech pacientů zařazených do studie bylo séropozitivních na divoký typ viru HSV-1 před zahájením léčby a 32,6 % pacientů bylo séronegativních. Sérologický stav pro HSV-1 u zbylých 9,4 % pacientů byl neznámý.
Rozdíl v DRR mezi přípravkem Imlygic a GM-CSF v populaci ITT byl statisticky významný (viz tabulka 4) ve prospěch přípravku Imlygic.
Tabulka 4: Souhrn výsledků u populace ITT ve studii 005/05 s přípravkem Imlygic
|
Cílový parametr studie |
Imlygic N = 295 |
GM-CSF N = 141 | |
|
Výskyt trvající odpovědi |
Primární |
16,3 % (n = 48) (95% CI: 12,1; 20,5) |
2,1 % (n = 3) (95% CI: 0,0; 4,5) |
|
Poměr šancí 8,9; (95% CI: 2,7; 29,2) P < 0,0001 | |||
|
Výskyt objektivní odpovědi (% CR, % PR) |
Sekundární |
26,4 % (n = 78) (95% CI: 21,4 %, 31,5 %) (10,8% CR, 15,6% PR) |
5,7 % (n = 8) (95% CI: 1,9%, 9,5%) (0,7% CR, 5% PR) |
|
Celkové přežití |
Sekundární |
Medián 23,3 (95% CI: 19,5;29,6) měsíců |
Medián 18,9 (95% CI: 16,0; 23,7) měsíců |
|
HR: 0,79; (95% CI: 0,62; 1,00) p = 0,051 | |||
|
Trvání odpovědi (přetrvávající odpověď při posledním vyšetření nádoru) |
Sekundární |
Nedosažen (rozptyl: > 0,0 až > 16,8 měsíců) |
Medián 2,8 měsíců (rozptyl: 1,2 to > 14,9 měsíců) |
|
HR: 0,46; (95% CI: 0,35; 0,60) | |||
|
Doba do odpovědi (medián) |
Sekundární |
4,1 měsíce |
3,7 měsíců |
|
Doba do selhání léčby (medián) |
Sekundární |
8,2 měsíců (95% CI: 6,5; 9,9) |
2,9 měsíců (95% CI: 2,8; 4,0) |
|
HR: 0,42; (95% CI: 0,32; 0,54) | |||
U pacientů odpovídajících na léčbu přípravkem Imlygic přetrvávalo v době primární analýzy ještě 56 (72 %) odpovědí. Z pacientů odpovídajících na léčbu mělo 42 (54 %) o > 25 % větší celkovou velikost existující léze/existujících lézí a/nebo se u nich vytvořila nová léze/nové léze před konečným dosažením odpovědi na léčbu.
V analýze hodnotící systémové působení přípravku Imlygic mělo 27 ze 79 pacientů (34,2 %) > 50 % celkové zmenšení mimoviscerálních lézí, které nebyly léčené injekcemi přípravku Imlygic a 8 ze 71 pacientů (11,3 %) mělo > 50 % celkové zmenšení viscerálních lézí, které nebyly léčené injekcemi přípravku Imlygic.
Obrázek 4: Kaplan-Meierova křivka - celkové přežití (ITT populace)
100% -90 % -80 % -70 % -60 % -50 % -40% -30% -20% -10% -0 % -
Imlygic
GM-CSF
Medián [95% Cl] 23,3 [19,5; 29,6] měsíců
Medián [95% Cl] 18,9 [16,0; 23,7] měsíců
log-rank hodnota p = 0,0511
Poměr rizika 95% Cl = 0,79 0,62; 1,00
■JLIl.. ...........lili I
:-LUil____ili
Počet měsíců ve studii
Počet subjektu v riziku
Imlygic
GM-CSF
Cenzorovane subjekty jsou vyjádřeny svislou čarou I.
Mezi staršími (> 65 let věku) a mladšími dospělými pacienty nebyly pozorovány žádné rozdíly v bezpečnosti nebo účinnosti.
Exploratorní podskupiny
Byly rovněž provedeny exploratorní analýzy podskupin pro DRR a celkové přežití podle stadia nemoci (viz obrázek 5 a tabulku 5). I když pivotní studie neměla statistickou sílu na hodnocení účinnosti u těchto jednotlivých podskupin, pacienti bez viscerálního postižení měli větší prospěch z léčby přípravkem Imlygic než pacienti s pokročilejším onemocněním.
Tabulka 5: Souhrn výsledků z exploratorní analýzy podskupin ze studie 005/05 s přípravkem Imlygic
|
DRR, (%) |
ORR, (%) |
OS (poměr rizika) | |||
|
Imlygic |
GM-CSF |
Imlygic |
GM-CSF |
Imlygic versus GM-CSF | |
|
Stadium5 IIIb/IIIc/ stadium IVMla (Imlygic, n = 163; GM-CSF, n = 86) |
25,2 |
1,2 |
40,5 |
2,3 |
0,57, (95% CI: 0,40; 0,80); |
|
Stadium5 IVMlb/ IVMlc (Imlygic, n = 131; GM-CSF, n = 55) |
5,3 |
3,6 |
9,2 |
10,9 |
1,07, (95% CI: 0,75; 1,52); |
§ American Joint Committee on Cancer (AJCC) 6. vydání.
Obrázek 5: Kaplan-Meierův odhad celkového přežití podle randomizovaného léčebného ramene u onemocnění stadia IIIb/IIIc/ IVMla (exploratorní analýza podskupin)
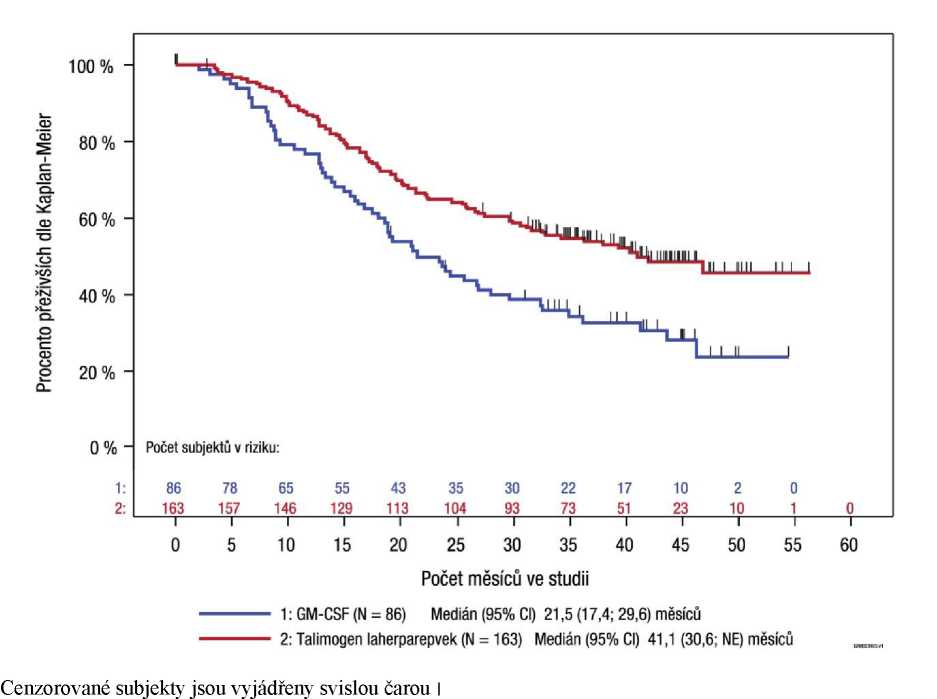
NE = neodhadnutelný
Vzhledem k exploratorní povaze analýzy a na základě současných poznatků nebylo prokázáno, že přípravek Imlygic je spojen s účinkem na celkové přežití.
Pediatrická populace
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s přípravkem Imlygic u jedné nebo více podskupin pediatrické populace s melanomem (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Talimogen laherparepvek je geneticky modifikovaný virus HSV-1, který je schopný replikace. Farmakokinetika a biodistribuce jsou proto dány místem intralezionální injekce, nádorově selektivní replikací a uvolňováním z nádorové tkáně.
Absorpce
K buněčnému vychytávání talimogen laherparepveku po lokální injekci do nádorů dochází přes HSV-1 receptory na nádorech a nenádorových buňkách. Protože talimogen laherparepvek se injikuje a replikuje v nádoru, biologická dostupnost a systémová koncentrace talimogen laherparepveku nejsou prediktivní pro aktivitu léčivé látky, a proto nebyly hodnoceny.
Metabolismus/eliminace
Talimogen laherparepvek se odstraňuje pomocí obecných obranných mechanismů hostitele (např. autofagií, adaptivní imunitní odpovědí). Talimogen laherparepvek se odbourává typickými endogenními katabolickými metabolickými dráhami pro proteiny a DNA. Stejně jako u ostatních infekcí divokým typem HSV-1, latentní pool DNA talimogen laherparepveku může přetrvávat v tělech neuronů inervujících místa vpichu; proto nelze vyloučit výskyt latentní infekce způsobené talimogen laherparepvekem.
Biodistribuce (v těle) a vylučování viru (exkrece/sekrece)
DNA talimogen laherparepveku byla kvantifikována pomocí vysoce senzitivní a specifické kvantitativní polymerázové řetězové reakce (quantitative Polymerase Chain Reaction - qPCR), která nemusí korelovat s rizikem virové infekčnosti. Talimogen laherparepvek byl rovněž kvantifikován u vybraných vzorků pacientů v klinických studiích pomocí analýzy virové infekčnosti v místě injekčního vpichu a v některých případech potenciálních herpetických lézí.
Klinická biodistribuce, eliminace a vylučování
Biodistribuce a vylučování intralezionálně podaného talimogen laherparepveku jsou zkoumány ve studii s melanomem. Průběžné výsledky od 30 pacientů prokazují, že DNA talimogen laherparepveku byla detekována v přechodných a nízkých koncentracích v krvi u 90 % pacientů a v moči u 20 % pacientů ve studii. Podíl pacientů s detekovatelnou DNA talimogen laherparepveku v krvi a moči byl nejvyšší v druhém léčebném cyklu. Ve vzorcích z injikovaných lézí byla DNA talimogen laherparepveku detekovaná přibližně u 90 % pacientů, avšak pouze 14 % pacientů mělo pozitivní testy na infekční virus pomocí analýzy TCID50 (50% Tissue Culture Infectious Dose), u všech to bylo během 8 dní po podání léku. Celkem sedmnáct procent vyšetřených vzorků z vnější části okluzivního obvazu bylo pozitivních na DNA talimogen laherparepveku, ale žádný testovaný vzorek nebyl pozitivní na přítomnost infekčního viru. Ze vzorků ze sliznice úst měl během studie pouze 1 vzorek detekovatelnou DNA talimogen laherparepveku, avšak vzorek nebyl pozitivně testován na přítomnost infekčního viru.
Farmakokinetika u zvláštních populací
U zvláštních populací pacientů nebyly prováděny farmakokinetické studie s talimogen laherparepvekem.
5.3 Předklinické údaje vztahující se k bezpečnosti
Imunokompetentní myši, potkani a psi dobře tolerovali dávky až do 4 x 108 PFU/kg nebo 107 PFU/dávka (60násobek nejvyšší navrhované klinické dávky) talimogen laherparepveku podávaného v jedné nebo opakovaných dávkách subkutánní, intravenózní nebo intratumorální injekcí. Nebyla pozorována žádná neurologická patologie nebo nežádoucí neurologické účinky. Ve studii in vivo s intracerebrální injekcí byl talimogen laherparepvek 10 000x méně neurovirulentní než dávka divokého typu HSV-1, která vede k úmrtí 50 % myší v čase.
Talimogen laherparepvek byl injikován do různých xenograftových nádorů v dávkách až do 2 x 108 PFU/kg (30násobek nejvyšší navrhované klinické dávky) imunodeficitním myším (holé a s těžkou kombinovanou imunodeficiencí - SCID). Letální systémová virová infekce byla pozorovaná až u 20 % holých myší (především s deficitem funkce T-lymfocytů) a u 100 % SCID myší (postrádající T- i B-lymfocyty).
Ve všech studiích byla fatální diseminovaná virová infekce pozorovaná u 14 % holých myší po léčbě talimogen laherparepvekem v dávkách, které jsou 10 až 100x vyšší než dávky, které vedou k 100% letalitě u divokého typu HSV-1.
Mutagenita
Genotoxický potenciál talimogen laherparepveku nebyl hodnocen v dlouhodobých studiích na zvířatech nebo u člověka. Vzhledem k tomu, že divoký typ HSV-1 se neintegruje do hostitelského genomu, je riziko inzerční mutageneze talimogen laherparepvekem zanedbatelné.
Kancerogenita
Kancerogenní potenciál talimogen laherparepveku nebyl hodnocen v dlouhodobých studiích na zvířatech nebo u člověka. Dostupné údaje o talimogen laherparepveku a divokém typu HSV-1 ale neukazují na kancerogenní riziko u člověka.
Reprodukční a vývojová toxicita
Po léčbě dospělých myší dávkami až do 4 x 108 PFU/kg (60x vyššími na základě PFU/kg v porovnání s maximální klinickou dávkou) nebylo pozorováno žádné působení na samčí nebo samičí reprodukční tkáně. Nebyl pozorován žádný vliv na embryofetální vývoj při podávání talimogen laherparepveku březím myším během organogeneze v dávkách až do 4 x 108 (400 milionů) PFU/kg (60x vyššími na základě PFU/kg v porovnání s maximální klinickou dávkou). V krvi plodů bylo zjištěno zanedbatelné množství (< 0,001 % hladiny v krvi matek) DNA talimogen laherparepveku.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Dihydrát hydrogenfosforečnanu sodného Dihydrát dihydrogenfosforečnanu sodného Chlorid sodný Inositol
Sorbitol (E420)
Voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti
Neotevřená injekční lahvička 4 roky.
Rozmrazování injekčních lahviček
• Před použitím nechte při pokojové teplotě (20 °C až 25 °C) rozmrznout zmrazené injekční lahvičky s přípravkem Imlygic, dokud nebude přípravek Imlygic tekutý (přibližně 30 minut). Injekční lahvičkou jemně otáčejte. NETREPEJTE.
• Injekční lahvičky nechte rozmrznout a uchovávejte je v původním obalu až do podání, aby byly chráněny před světlem.
Po rozmražení
• Po rozmražení podejte Imlygic jakmile je to prakticky možné.
• Rozmražený Imlygic je stabilní při uchovávání za teploty 2 °C až 25 °C chráněn před světlem v původní injekční lahvičce, v injekční stříkačce nebo v původní injekční lahvičce a následně v injekční stříkačce. Nepřekračujte dobu uchovávání uvedenou v tabulce 6 a 7.
• Pokud je rozmražený Imlygic uchováván v původní injekční lahvičce a následně v injekční stříkačce:
o po celou dobu uchovávání až do podání má být dodrženo stejné rozmezí teplot.
o doba uchovávání v injekční stříkačce za pokojové teploty do 25 °C nesmí přesáhnout
2,5 hodiny pro 106 (1 milion) PFU/ml a 4 hodiny pro 108 (100 milionů) PFU/ml (viz tabulka 6).
o maximální kumulativní doba uchovávání (doba uchovávání v injekční lahvičce plus doba uchovávání v injekční stříkačce) nesmí přesáhnout dobu uvedenou v tabulce 7.
• Po rozmrazení se Imlygic nesmí nechat znovu zmrazit. Zlikvidujte rozmražený Imlygic v injekční lahvičce nebo v injekční stříkačce, který byl uchováván déle, než je níže uvedená doba.
Tabulka 6: Maximální doba uchovávání pro rozmražený Imlygic v injekční stříkačce
|
106 (1 milion) PFU/ml |
108 (100 milionů) PFU/ml | |
|
2 °C až 8 °C |
8 hodin |
16 hodin |
|
do 25 °C |
2,5 hodiny |
4 hodiny |
Tabulka 7: Maximální kumulativní doba uchovávání (doba uchovávání v injekční lahvičce plus doba uchovávání v injekční stříkačce) pro rozmražený Imlygic
|
106 (1 milion) PFU/ml |
108 (100 milionů) PFU/ml | |
|
2 °C až 8 °C |
31 hodin |
6 týdnů (42 dnů) |
|
do 25 °C |
17 hodin |
85 hodin |
6.4 Zvláštní opatření pro uchovávání
Uchovávejte a převážejte zmrazené (-90 °C až -70 °C).
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
6.5 Druh obalu a obsah balení
1 ml roztoku bez konzervačních látek v injekční lahvičce na jedno použití (pryskyřice z cyklického olefinového polymerového plastu) se zátkou (chlorobutylový elastomer) a závěrem (hliník) s odtrhovacím víčkem (polypropylen).
Víčko injekční lahvičky je barevně odlišeno: 106 (1 milion) PFU/ml je světle zelené a 108 (100 milionů) PFU/ml je tmavě modré.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Dodržujte místní předpisy zdravotnického zařízení pro zacházení a podávání přípravku, osobní ochranné pomůcky, opatření při náhodném rozlití a pro nakládání s odpadem.
• Při přípravě nebo podávání přípravku Imlygic noste ochranný oblek nebo laboratorní plášť, ochranné brýle nebo obličejový štít a rukavice. Před podáním přípravku přikryjte všechny odkryté rány. Vyvarujte se kontaktu s pokožkou, očima nebo sliznicemi.
• Po podání přípravku si vyměňte rukavice před přiložením okluzivního obvazu na injikované léze. Otřete vnější plochu okluzivního obvazu ubrouskem s alkoholem. Doporučuje se, aby pacienti měli místa injekčního vpichu pokryta pokud možno po celou dobu vzduchotěsným a vodotěsným obvazem. Aby se minimalizovalo riziko virového přenosu, pacienti mají mít
přikryté místo injekčního vpichu nejméně po dobu 8 dní od poslední léčby nebo déle, jestliže místo injekčního vpichu mokvá anebo prosakuje. Doporučte pacientům, aby si přikládali obvazy podle pokynů zdravotnických pracovníků, a aby obvaz vyměnili, pokud jim spadne.
• Zlikviduje všechen materiál, který přišel do kontaktu s přípravkem Imlygic (např. injekční lahvička, stříkačka, jehla, obvazový materiál) v souladu s místními postupy zdravotnického zařízení.
Náhodná expozice
• V případě náhodné expozice přípravku Imlygic při práci (např. při stříknutí do očí nebo na sliznice) během přípravy nebo podávání oplachujte postižená místa čistou vodou nejméně 15 minut. V případě expozice porušené pokožky nebo při píchnutí jehlou umyjte pečlivě postiženou oblast mýdlem a vodou a/nebo dezinfekční látkou.
• Ošetřete všechna místa potřísněná přípravkem Imlygic viricidní látkou a savým materiálem.
• Doporučte pacientům, aby použitý obvazový a čistící materiál umístili do neprodyšně uzavřeného plastového pytle, protože tento materiál může být kontaminovaný, a pytel vyhodili do domovního odpadu.
Tento přípravek obsahuje geneticky modifikované organismy. Veškerý nepoužitý léčivý přípravek musí být zlikvidován v souladu s požadavky poskytovatele zdravotních služeb pro nakládání s geneticky upravenými organismy nebo s nebezpečným biologickým odpadem, podle toho, co je vhodné.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Amgen Europe B.V.
Minervum 7061 NL-4817 ZK Breda Nizozemsko
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/15/1064/001 EU/1/15/1064/002
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 16.prosince 2015
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu
PŘÍLOHA II
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa vvrobce/vvrobců biologické léčivé látky/biologických léčivých látek BioVex Inc. - Subsidiary of Amgen, Inc.
34 Commerce Way Woburn Massachusetts 01801
Spojené státy americké
Název a adresa výrobce odpovědného/výrobců odpovědných za propouštění šarží Amgen Europe B.V.
Minervum 7061 4817 ZK Breda Nizozemsko
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl.
107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
Držitel rozhodnutí o registraci předloží první pravidelně aktualizovanou zprávu o bezpečnosti pro tento léčivý přípravek do 6 měsíců od jeho registrace.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
• Další opatření k minimalizaci rizik
Před uvedením přípravku IMLYGIC na trh v každém členském státu se držitel rozhodnutí o registraci (MAH) musí s příslušnými národními orgány dohodnout na obsahu a formě edukačního a kontrolovaného distribučního programu včetně komunikačních médií, způsobů distribuce a všech dalších aspektů programu.
Záměrem edukačního programu je informovat o důležitých rizicích spojených s přípravkem IMLYGIC:
• Herpetická infekce vyskytující se v celém těle (disseminovaná herpetická infekce) u imunokompromitovaných osob (osoby s jakoukoli vrozenou nebo získanou buněčnou a/nebo humorální imunitní nedostatečností, tj, HIV/AIDS, leukémie, lymfom, běžná variabilní imunodeficience, nebo osoby vyžadující vysoké dávky steroidů nebo jiných imunosupresiv)
• Náhodná expozice zdravotnických pracovníků přípravku IMLYGIC
• Přenos přípravku IMLYGIC na blízké kontakty nebo poskytovatele zdravotní péče po přímém kontaktu s injikovanou lézí nebo s tělními tekutinami
• Symptomatická herpetická infekce v důsledku latence a reaktivace přípravku IMLYGIC nebo herpesu (divoký typ HSV-1) u pacientů
• Pacienti s oslabeným imunitním systémem (imunokompromitovaní pacienti) léčení přípravkem IMLYGIC a trpící konkomitatní infekcí
• Kombinace s jinými terapiemi, jako je chemoterapie nebo imunosupresivní látky
• Těhotné a kojící ženy
Držitel rozhodnutí o registraci zabezpečí, aby v každém členském státu, kde je přípravek IMLYGIC obchodovaný, všichni zdravotničtí pracovníci a pacienti/pečovatelé, u nichž lze předpokládat, že budou přípravek IMLYGIC předepisovat, vydávat a používat, mají přístup k/obdrží následující edukační sadu:
• Edukační materiál pro lékaře
• Edukační materiál pro pacienty
Edukační materiál pro lékaře musí obsahovat:
• Souhrn údajů o přípravku
• Pokyny pro zdravotnické pracovníky
• Kartu pacienta
• Pokyny pro zdravotnické pracovníky musí obsahovat tyto klíčové prvky:
o Informaci o riziku herpetické infekce u pacientů léčených přípravkem IMLY GIC o Informaci o riziku disseminované herpetické infekce u imunokompromitovaných pacientů léčených přípravkem IMLYGIC
o Doporučení při náhodném vystavení zdravotnických pracovníků přípravku IMLYGIC: o Požadavek používat vždy ochranný oděv/laboratorní plášť, bezpečnostní brýle a rukavice při přípravě nebo aplikaci přípravku IMLYGIC; o Zabránit kontaktu s pokožkou, očima, sliznicemi a vyvarovat se přímého kontaktu bez rukavic s injikovanou lézí nebo tělesnými tekutinami léčených pacientů; o Pokyny o první pomoci po náhodném vystavení;
o Zdravotníci s oslabeným imunitním systémem nebo těhotné zdravotnické pracovnice
nesmí připravovat a podávat IMLYGIC.
o Doporučení týkající se náhodného přenosu přípravku IMLYGIC z pacienta na blízký kontakt nebo zdravotnické pracovníky:
o Poučení o tom, jak se chovat po podání/náhodném přenosu a jakým způsobem a jak často
měnit obvaz a kdo to nesmí provádět;
o Instrukce k minimalizaci rizika vystavení blízkých kontaktů krvi a tělním tekutinám po dobu léčby přípravkem IMLYGIC až do 30 dní po posledním podání přípravku IMLYGIC. Je třeba se vyvarovat následujících činností:
■ Pohlavní styk bez latexového kondomu
■ Líbání, pokud má některá ze stran otevřenou ránu v ústech
■ Společné používání příborů, nádobí a nádob na pití
■ Společné používání injekčních jehel, holicích čepelek a zubních kartáčků;
o Odpovídající likvidace a dekontaminace odpadu v souladu s doporučeními pro likvidaci biologicky nebezpečného odpadu.
o Informace o použití přípravku IMLYGIC v těhotenství
o Pokyny, jak hlásit případné nežádoucí příhody, včetně uvádění čísla šarže při hlášení nežádoucích účinků
• Karta pacienta musí obsahovat následující klíčová sdělení:
o Upozornění pro zdravotnické pracovníky kdykoli ošetřující pacienta, včetně naléhavých
situací, že pacient užívá IMLYGIC o Kontaktní údaje lékaře předepisujícího IMLYGIC
o Podrobnosti o datu zahájení léčby přípravkem IMLYGIC, číslu šarže, datu podání, výrobci přípravku a držiteli rozhodnutí o registraci o Informace o herpetických lézích
• Sada informací pro pacienta musí obsahovat:
o Příbalovou informaci
o Pokyny pro pacienta/pečovatele a blízké kontakty
• Pokyny pro pacienta/pečovatele a blízké kontakty musí obsahovat následující klíčová sdělení:
o Popis důležitých rizik spojených s používáním přípravku IMLYGIC;
o Poučení o tom, jak se chovat po podání a jakým způsobem a jak často měnit obvaz, a kdo
to nesmí provádět.
o Informace o známkách a příznacích rizika herpetické infekce; o Informace o použití přípravku IMLYGIC v těhotenství;
o Doporučení týkající se náhodného přenosu přípravku IMLYGIC z pacienta na blízký
kontakt nebo zdravotnické pracovníky:
o Instrukce k minimalizaci rizika vystavení blízkých kontaktů krvi a tělním tekutinám po dobu léčby přípravkem IMLYGIC až do 30 dní po posledním podání přípravku IMLYGIC. Je třeba se vyvarovat následujících činností:
■ Pohlavní styk bez latexového kondomu
■ Líbání, pokud některá ze stran má otevřenou ránu v ústech
■ Společné používání příborů, nádobí a nádob na pití
■ Společné používání injekčních jehel, holicích čepelek a zubních kartáčků;
o Odpovídající likvidace a dekontaminace odpadu v souladu s doporučeními pro likvidaci biologicky nebezpečného odpadu. o Poučení o tom, jak se chovat po náhodném přenosu.
Program kontrolované distribuce je zaměřen na řízení dodavatelského řetězce přípravku, aby bylo zabezpečeno, že jsou dodržovány požadavky na uchovávání v chladu, a na kontrolu distribuce přípravku IMLYGIC kvalifikovaným centrům až po dodání pacientům.
MAH musí zajistit, aby v každém členském státě, kde je IMLYGIC obchodován, byl systém zaměřený na kontrolovanou distribuci přípravku IMLYGIC nad rámec kontroly vyplývající z rutinních opatření k minimalizaci rizik. Před zahájením vydávání přípravku musí být splněny následující požadavky:
• Dostatečně vyškolení a zkušení zdravotničtí pracovníci, aby se minimalizovalo riziko výskytu určitých nežádoucích účinků u pacientů, zdravotnických pracovníků a blízkých kontaktů pacientů;
• Vyškolení zdravotnických pracovníků a pomocných pracovníků s ohledem na bezpečné a náležité uchovávání, zacházení a podávání přípravku IMLYGIC a klinické sledování pacientů léčených přípravkem IMLYGIC;
• Poskytnutí přesně stanovených bezpečnostních informací pacientům a informování pacientů, že je důležité sdělit tyto informace rodině a pečovatelům;
• Vyškolit zdravotnické pracovníky, aby u všech podaných injekcí zaznamenali číslo šarže v dokumentaci pacienta a na kartě pacienta s upozorněním, aby číslo šarže uvedli při hlášení nežádoucích účinků.
• Povinnost uskutečnit poregistrační opatření
Držitel rozhodnutí o registraci uskuteční v daném termínu níže uvedená opatření:
|
Popis |
Termín splnění |
|
Držitel rozhodnutí o registraci musí předložit předběžné výsledky studie 20120325 (multicentrická, otevřená jednoramenná studie fáze 2 hodnotící souvislost mezi výskytem objektivní odpovědi a výchozí intratumorální hustotou CD8+ T-lymfocytů u pacientů s neresekovaným melanomem stadia IIIB až IVM1c léčených talimogen laherparepvekem) |
31. prosinec 2018 |
|
Předložit předběžné výsledky studie 20110266 (multicentrická, randomizovaná, otevřená studie fáze 2 hodnotící účinnost a bezpečnost talimogen laherparepveku v neoadjuvantní léčbě s chirurgickým výkonem oproti samotnému chirurgickému výkonu u resekabilního melanomu stadia IIIB až IVM1a) |
31. prosinec 2019 |
|
Poskytnout předběžné výsledky účinnosti ze studie 20110265 z části fáze III (multicentrická studie hodnotící kombinaci talimogen laherparepveku s pembrolizumabem) |
30. červen 2019 |
PŘÍLOHA III
Imlygic 106 jednotek tvořících plaky (PFU)/ml injekční roztok talimogenum laherparepvecum
2. OBSAH LÉČIVÉ LÁTKY/LÉCIVÝCH LÁTEK
Jedna injekční lahvička obsahuje 1 ml talimogenum laherparepvecum 1 x 106 (1 milion) jednotek tvořících plaky (PFU).
3. SEZNAM POMOCNÝCH LÁTEK
Dihydrát hydrogenfosforečnanu sodného, dihydrát dihydrogenfosforečnanu sodného, chlorid sodný, inositol, sorbitol (E420), voda na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Injekční roztok 1 injekční lahvička.
5 ZPŮSOB A CESTA/CESTY PODÁNÍ
Intralezionální podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9 ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte a převážejte zmrazené od -90 °C do - 70 °C.
Tento léčivý přípravek obsahuje geneticky modifikované organismy.
Nepoužitý léčivý přípravek musí být zlikvidován v souladu se směrnicemi zdravotnického zařízení pro geneticky modifikované organismy nebo biologicky nebezpečné odpady.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Amgen Europe B.V. Minervum 7061 NL-4817 ZK Breda Nizozemsko
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/15/1064/001
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15 NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
Nevyžaduje se - odůvodnění přijato
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Imlygic 106 PFU/ml injekce talimogenum laherparepvecum Intralezionální podání
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO SARZE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
1 ml
6. JINÉ
Imlygic 108 jednotek tvořících plaky (PFU)/ml injekční roztok talimogenum laherparepvecum
2. OBSAH LÉČIVÉ LÁTKY/LÉCIVÝCH LÁTEK
Jedna injekční lahvička obsahuje 1 ml talimogenum laherparepvecum 1 x 108 (100 milionů) jednotek tvořících plaky (PFU).
3. SEZNAM POMOCNÝCH LÁTEK
Dihydrát hydrogenfosforečnanu sodného, dihydrát dihydrogenfosforečnanu sodného, chlorid sodný, inositol, sorbitol (E420), voda na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Injekční roztok 1 injekční lahvička.
5 ZPŮSOB A CESTA/CESTY PODÁNÍ
Intralezionální podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8 POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte a převážejte zmrazené od -90 °C do - 70 °C.
Tento léčivý přípravek obsahuje geneticky modifikované organismy.
Nepoužitý léčivý přípravek musí být zlikvidován v souladu se směrnicemi zdravotnického zařízení pro geneticky modifikované organismy nebo biologicky nebezpečné odpady.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Amgen Europe B.V. Minervum 7061 NL-4817 ZK Breda Nizozemsko 12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/15/1064/002 13 ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Nevyžaduje se - odůvodnění přijato
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Imlygic 108 PFU/ml injekce talimogenum laherparepvecum Intralezionální podání
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO SARZE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
1 ml
6. JINÉ
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro pacienta
Imlygic 101 2 3 4 5 6 jednotek tvořících plaky (PFU)/ml injekční roztok Imlygic 107 jednotek tvořících plaky (PFU)/ml injekční roztok
talimogenum laherparepvecum
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo zdravotní sestry.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je Imlygic a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete Imlygic používat
3. Jak se Imlygic používá
4. Možné nežádoucí účinky
5 Jak Imlygic uchovávat
6. Obsah balení a další informace
1. Co je Imlygic a k čemu se používá
Imlygic se používá k léčbě dospělých pacientů s určitým typem kožního nádoru zvaným melanom, který se rozšířil v kůži nebo do mízních uzlin a který se nedá léčit chirurgicky.
Léčivou látkou přípravku Imlygic je talimogen laherparepvek. Je to oslabená forma viru herpes simplex typu 1 (HSV-1), který se běžně nazývá virus oparu. Pro získání přípravku Imlygic z HSV-1 byl virus změněn tak, že se množí efektivněji v nádorech než v normálních buňkách. To vede ke zničení infikovaných nádorových buněk. Imlygic účinkuje rovněž tak, že napomáhá imunitnímu systému rozeznat a ničit nádory v těle.
onemocnění krve nebo kostní dřeně, nebo pokud užíváte steroidy či jiné léky potlačující imunitní systém, oznamte to svému zdravotnickému pracovníkovi.
Náhodné potřísnění přípravkem Imlygic sebe nebo druhých
Imlygic může potřísnit jiné části Vašeho těla nebo jiné osoby přímým kontaktem s Vašimi tělesnými tekutinami nebo s místy injekčního vpichu.
Abyste zabránil(a) potřísnění přípravkem Imlygic jiných oblastí Vašeho těla nebo osob, se kterými přicházíte do blízkého kontaktu (blízké kontakty zahrnují členy domácnosti, pečovatele, sexuální partnery nebo osoby se kterými sdílíte stejnou postel), musíte učinit tato opatření:
• Zabraňte přímému kontaktu s místy injekčního vpichu nebo tělesnými tekutinami (např. krví a močí) a těsnému kontaktu (např. používejte latexový kondom při pohlavním styku, vyhněte se líbání s blízkými osobami, pokud Vy nebo dotyčná osoba máte otevřený vřed v ústech), zatímco jste léčen(a) přípravkem Imlygic a až 30 dní po poslední dávce.
• Nedotýkejte se ani se neškrábejte v místech injekčního vpichu.
• Mějte místa injekčního vpichu po celou dobu přikrytá vzduchotěsným a vodotěsným obvazem. Obvaz si přikládejte podle pokynů svého zdravotnického pracovníka. Jestliže se obvaz uvolní nebo spadne, ihned ho nahraďte čistým obvazem.
• Všechen použitý obvazový a čistící materiál umístěte do neprodyšně uzavřeného plastového pytle a vyhoďte do domovního odpadu.
Osobám, se kterými jste v blízkém kontaktu, musíte říct, aby:
• se vyvarovaly přímého kontaktu s Vašimi tělesnými tekutinami nebo místy injekčního vpichu.
• při výměně Vašich obvazů nosily rukavice.
Jestliže jsou osoby, se kterými jste v blízkém kontaktu, náhodně vystaveny přípravku Imlygic, musí si omýt postiženou oblast těla mýdlem a vodou a/nebo dezinfekční látkou. Jestliže se u nich rozvinou příznaky nebo projevy herpetické infekce, požádejte je, aby kontaktovaly svého zdravotnického pracovníka.
Těhotné ženy nebo osoby s oslabeným imunitním systémem, se kterými jste v blízkém kontaktu, a novorozenci
Zajistěte, aby se osoby, s nimiž jste v blízkém kontaktu a jsou těhotné nebo mají oslabený imunitní systém, nedotýkaly míst injekčního vpichu, použitých obvazů a čistícího materiálu. Použité obvazy a čistící materiál uchovávejte z dosahu novorozenců.
Herpetická infekce
Během léčby nebo po léčbě přípravkem Imlygic se může rozvinout opar nebo závažnější herpetická infekce. Známky a příznaky související s léčbou přípravkem Imlygic mohou být stejné jako u herpetické infekce a zahrnují bolest, pálení nebo brnění v puchýři na ústech nebo genitáliích, či na prstech nebo uších, dále bolest oka, citlivost na světlo, výtok z očí nebo rozmazané vidění, slabost v rukou či nohou, výrazná únava (pocit ospalosti) a zmatenost. Jestliže se u Vás vyskytují tyto příznaky, měl(a) byste dodržovat standardní hygienická opatření k zabránění přenosu viru na jiné osoby.
Infekce a opožděné hojení v místě injekčního vpichu přípravku Imlygic
Imlygic může způsobit infekci v místě injekčního vpichu. Příznaky a projevy infekce zahrnují bolest, zarudnutí, horkost, otok, výtok nebo vznik vředu, horečku a zimnici. Místo injekčního vpichu se může hojit déle než za normálních okolností. Jestliže zpozorujete některý z těchto příznaků, sdělte to svému zdravotnickému pracovníkovi.
Autoimunitní reakce
Imlygic může vyvolat autoimunitní reakce (nadměrná reakce imunitního systému). U některých osob léčených přípravkem Imlygic se rozvinul zánět ledvin (glomerulonefritida), zúžení nebo ucpání krevních cév (vaskulitida), otok plic (pneumonitida), zhoršení šupinatění kůže (lupénka) a oblasti kůže bez zbarvení (vitiligo). V případě, že máte nebo jste v minulosti měl(a) autoimunitní onemocnění, sdělte to svému lékaři.
Plazmocytom
Imlygic může způsobit, že se nádorové bílé krvinky hromadí v místě injekčního vpichu nebo jeho blízkosti (plazmocytom). Informujte svého zdravotnického pracovníka, pokud máte nebo jste v minulosti měl(a) nádorové onemocnění krve včetně mnohočetného myelomu.
Dýchací potíže
Jestliže máte nádor v krku, může Vás zdravotnický pracovník upozornit, že během léčby přípravkem Imlygic můžete mít pocit tlaku v dýchacích cestách.
Pacienti, kteří neměli v minulosti herpetickou infekci
Jestliže jste v minulosti nikdy neměl(a) herpetickou infekci, s větší pravděpodobností můžete mít během prvních 6 léčebných cyklů přípravkem Imlygic horečku, zimnici a onemocnění podobné chřipce.
Děti a dospívající
Imlygic se nedoporučuje u dětí a dospívajících, protože účinky přípravku Imlygic u osob mladších 18 let nejsou známé.
Další léčivé přípravky a Imlygic
Informujte svého zdravotnického pracovníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat, včetně léků, jako je např. acyklovir, které se používají k léčbě nebo prevenci herpetických infekcí. Acyklovir a další protivirové léky mohou snížit účinnost přípravku Imlygic.
Těhotenství a kojení
Poraďte se se svým zdravotnickým pracovníkem:
• pokud se domníváte, že byste mohla být těhotná
• nebo plánujete otěhotnět.
Váš lékař určí, zda je Imlygic pro Vás vhodný.
Jestliže jste těhotná nebo kojíte, poraďte se se svým zdravotnickým pracovníkem dříve, než začnete tento přípravek používat. Imlygic může poškodit nenarozené dítě.
Ženy v plodném věku mají používat účinnou antikoncepci, aby se zabránilo otěhotnění během léčby přípravkem Imlygic. Informujte se u svého zdravotnického pracovníka o vhodné antikoncepční metodě.
Není známo, zda se Imlygic vylučuje do mateřského mléka. Jestliže kojíte nebo plánujete kojit, oznamte to svému zdravotnickému pracovníkovi. Pomůže Vám při rozhodování, zda ukončit kojení, nebo přestat používat Imlygic při zvážení přínosu kojení pro Vaše dítě a přínosu přípravku Imlygic pro Vás.
Řízení dopravních prostředků a obsluha strojů
Během léčby přípravkem Imlygic můžete pozorovat příznaky, jako jsou závratě nebo zmatenost. To může zhoršit Vaši schopnost řídit nebo obsluhovat stroje. Při řízení nebo obsluze strojů buďte opatrní, dokud si nebudete jistý(á), že na Vás Imlygic nepůsobí nepříznivě.
Imlygic obsahuje sodík a sorbitol
Imlygic obsahuje přibližně 30 mg (1,3 mmol) sodíku v jedné dávce (4 ml). Nutno vzít v úvahu u pacientů na dietě s nízkým obsahem sodíku.
Imlygic obsahuje sorbitol (E420). Pokud Vám zdravotnický pracovník řekl, že nesnášíte některé cukry, poraďte se se svým zdravotnickým pracovníkem dříve, než začnete tento přípravek používat.
3. Jak se Imlygic používá
Imlygic se podává ve zdravotnickém zařízení pod dohledem zdravotnického pracovníka. Doporučená úvodní dávka je až 4 ml přípravku Imlygic o koncentraci 106 (1 milion) PFU/ml. Další dávky budou v množství až 4 ml přípravku Imlygic o koncentraci 108 (100 milionů) PFU/ml.
Váš zdravotnický pracovník Vám podá přípravek Imlygic přímo do nádoru/ů pomocí injekční jehly a stříkačky. Druhou injekci dostanete za 3 týdny po první injekci. Poté budete dostávat injekce každé 2 týdny, dokud budete mít nádor/y.
Váš zdravotnický pracovník rozhodne, do kterého nádoru/ů se injekce podá a nemusí ji podat do všech nádorů. Během léčby přípravkem Imlygic se existující nádor/y může/mohou zvětšit a může/mohou se objevit i nový/é nádor/y.
Očekávejte, že přípravkem Imlygic budete léčen(a) nejméně 6 měsíců nebo déle.
Jestliže jste zapomněl(a) použít dávku přípravku Imlygic
Je důležité, abyste dodržoval(a) termíny návštěv, při kterých budete dostávat přípravek Imlygic. Jestliže zapomenete přijít na návštěvu, zeptejte se svého zdravotnického pracovníka, kdy se máte dostavit na příští dávku.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
U pacientů léčených přípravkem Imlygic byly často pozorované infekce v místě injekčního vpichu způsobené bakteriemi (celulitida). V prevenci těchto typů infekcí napomáhá udržování ran v čistotě a jejich převazování.
U pacientů léčených přípravkem Imlygic byla velmi často pozorována onemocnění podobná chřipce, horečka a zimnice. Tyto projevy obvykle odezní během prvních 72 hodin po léčbě.
U pacientů léčených přípravkem Imlygic byly hlášeny níže uvedené nežádoucí účinky:
Velmi časté nežádoucí účinky (mohou se vyskytnout u více než 1 z 10 léčených osob):
• Otok tkání (periferní otok)
• Bolest hlavy
• Kašel
• Zvracení, průjem, zácpa, pocit na zvracení
• Bolest svalů (myalgie), bolestivé/oteklé klouby (artralgie), bolest končetin
• Onemocnění podobné chřipce, horečka (pyrexie), zimnice, únava, bolest
• Bolest, zarudnutí, krvácení, otok, zánět, sekrece, výtok a pocit horka v místě injekčního vpichu
Časté nežádoucí účinky (mohou se vyskytnout až u 1 z 10 léčených osob):
• Infekce způsobená bakteriemi (celulitida), opar v ústech
• Bolest nádoru, infekce nádoru
• Nižší počet červených krvinek (anémie)
• Nežádoucí účinky týkající se imunitního systému jako je zúžení nebo uzávěr krevních cév (vaskulitida), zánět plic (pneumonitida), zhoršení olupování kůže (zhoršení lupénky) a zánět ledvin (glomerulonefritida)
• Dehydratace (nedostatek vody v organismu)
• Zmatenost, úzkost, deprese, závratě, nespavost (insomnie)
• Bolest ucha, hrdla, břicha, v tříslech, zad a podpaží
• Rychlejší srdeční akce v klidu (tachykardie)
• Tvorba sraženin v hlubokých žilách (hluboká žilní trombóza), vysoký krevní tlak (hypertenze), zarudnutí v obličeji
• Dušnost (námahová dušnost), infekce horních cest dýchacích
• Nepříjemný pocit v oblasti břicha
• Nezbarvené oblasti kůže (vitiligo), vyrážka, zánět kůže (dermatitida)
• Celkový pocit nemoci
• Pokles tělesné hmotnosti
• Komplikace ran, výtok z rány, modřiny (zhmožděniny), bolest po výkonu
Méně časté nežádoucí účinky (mohou se se vyskytnout až u 1 ze 100 léčených osob):
• Infekce v místě vpichu
• Nádorové bílé krvinky v místě injekčního vpichu nebo v jeho okolí (plazmocytom)
• Oční infekce způsobené herpetickým virem (herpetická keratitida)
• Stlačené dýchací cesty (obstrukční porucha dýchacích cest)
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému zdravotnickému pracovníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak Imlygic uchovávat
Imlygic budou uchovávat zdravotničtí pracovníci ve Vašem zdravotnickém zařízení.
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na štítku a na krabičce za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte a převážejte zmrazené při teplotě -90 °C až -70 °C.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Obsah balení a další informace
6.
Co Imlygic obsahuje
- Léčivou látkou je talimogenum laherparepvecum.
Jedna injekční lahvička obsahuje 1 extrahovatelný ml roztoku o nominální koncentraci 1 x 106 (1 milion) jednotek tvořících plaky (PFU)/ml nebo 1 x 108 (100 milionů) PFU/ml.
- Dalšími složkami jsou dihydrát hydrogenfosforečnanu sodného, dihydrát dihydrogenfosforečnanu sodného, chlorid sodný, inositol, sorbitol (E420), voda na injekci (viz bod 2).
Jak Imlygic vypadá a co obsahuje toto balení
Imlygic je čirá až poloprůsvitná (106 PFU/ml) nebo poloprůsvitná až neprůsvitná (108 PFU/ml) tekutina. Dodává se jako 1 ml roztoku bez konzervačních látek v injekční lahvičce na jedno použití (pryskyřice z cyklického olefinového polymerového plastu) se zátkou (chlorobutylový elastomer) a závěrem (hliník) s odtrhovacím víčkem (polypropylen).
Víčko injekční lahvičky je barevně odlišeno: 106 PFU/ml je světle zelené a 108 PFU/ml je tmavě modré.
Držitel rozhodnutí o registraci a výrobce
Amgen Europe B.V.
Minervum 7061 NL-4817 ZK Breda Nizozemsko
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci
Deutschland
AMGEN GmbH Tel.: +49 89 1490960
Eesti
Amgen Switzerland AG Vilniaus filialas Tel: +372 586 09553
Belgie/Belgique/Belgien
s.a. Amgen n.v.
Tel/Tél: +32 (0)2 7752711
Etarapnu
AMg^eH Etarapna EOOfl Tea.: +359 (0)2 424 7440
Česká republika
Amgen s.r.o.
Tel: +420 221 773 500
Danmark
Amgen, filial af Amgen AB, Sverige Tlf: +45 39617500
Lietuva
Amgen Switzerland AG Vilniaus filialas Tel: +370 5 219 7474
Luxembourg/Luxemburg
s.a. Amgen Belgique/Belgien Tel/Tél: +32 (0)2 7752711
Magyarország
Amgen Kft.
Tel.: +36 1 35 44 700
Malta
Amgen B.V.
The Netherlands Tel: +31 (0)76 5732500
Nederland
Amgen B.V.
Tel: +31 (0)76 5732500
Norge
Amgen AB Tel: +47 23308000
Suomi/Finland
Italia
Amgen S.r.l.
Tel: +39 02 6241121
EiiúSa
Amgen Eiiág OappaKenuKá E.n.E. Tni.: +30 210 3447000
Espaňa
Amgen S.A.
Tel: +34 93 600 18 60
France
Amgen S.A.S.
Tél: +33 (0)9 69 363 363
Hrvatska
Amgen d.o.o.
Tel: +385 (0)1 562 57 20
Ireland
Amgen Limited United Kingdom Tel: +44 (0)1223 420305
Island
Vistor hf.
Sími: +354 535 7000
Osterreich
Amgen GmbH Tel: +43 (0)1 50 217
Polska
Amgen Biotechnologia Sp. z o.o. Tel.: +48 22 581 3000
Portugal
Amgen Biofarmaceutica, Lda. Tel: +351 21 4220550
Románia
Amgen Románia SRL Tel: +4021 527 3000
Slovenija
AMGEN zdravila d.o.o.
Tel: +386 (0)1 585 1767
Slovenská republika
Amgen Slovakia s.r.o.
Tel: +421 33 321 13 22
Amgen AB, sivuliike Suomessa/Amgen AB, filial i Finland
Puh/Tel: +358 (0)9 54900500
Latvija United Kingdom
Amgen Switzerland AG Rlgas filiale Amgen Limited
Tel: +371257 25888 Tel: +44 (0)1223 420305
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Následující informace jsou určeny pouze pro zdravotnické pracovníky:
Rozmrazování injekčních lahviček přípravku Imlygic
• Před použitím nechte zmražené injekční lahvičky s přípravkem Imlygic rozmrznout při pokojové teplotě (20 °C až 25 °C), dokud nebude Imlygic tekutý (přibližně 30 minut). Injekční lahvičkou jemně otáčejte. NETŘEPEJTE.
• Injekční lahvičky nechte rozmrznout a uchovávejte v původním obalu až do podání, aby byly chráněny před světlem.
Po rozmražení
• Po rozmražení podejte Imlygic jakmile je to prakticky možné.
• Rozmražený Imlygic je stabilní při uchovávání za teploty 2 °C až 25 °C chráněn před světlem v původní injekční lahvičce, v injekční stříkačce nebo v původní injekční lahvičce a následně v injekční stříkačce. Nepřekračujte dobu uchovávání uvedenou v tabulce 1 a 2.
• Pokud je rozmražený Imlygic uchováván v původní injekční lahvičce a následně v injekční stříkačce:
o po celou dobu uchovávání až do podání má být dodrženo stejné rozmezí teplot.
o doba uchovávání v injekční stříkačce za pokojové teploty do 25 °C nesmí přesáhnout
2,5 hodiny pro 106 (1 milion) PFU/ml a 4 hodiny pro 108 (100 milionů) PFU/ml (viz tabulka 1).
o maximální kumulativní doba uchovávání (doba uchovávání v injekční lahvičce plus doba uchovávání v injekční stříkačce) nesmí přesáhnout dobu uvedenou v tabulce 2.
• Po rozmrazení se Imlygic nesmí nechat znovu zmrazit. Zlikvidujte rozmražený Imlygic v injekční lahvičce nebo v injekční stříkačce, který byl uchováván déle, než je níže uvedená doba.
Tabulka 1: Maximální doba uchovávání pro rozmražený Imlygic v injekční stříkačce
|
106 (1 milion) PFU/ml |
108 (100 milionů) PFU/ml | |
|
2 °C až 8 °C |
8 hodin |
16 hodin |
|
do 25 °C |
2,5 hodiny |
4 hodiny |
Tabulka 2: Maximální kumulativní doba uchovávání (doba uchovávání v injekční lahvičce plus doba uchovávání v injekční stříkačce) pro rozmražený Imlygic
|
106 (1 milion) PFU/ml |
108 (100 milionů) PFU/ml | |
|
2 °C až 8 °C |
31 hodin |
6 týdnů (42 dnů) |
|
do 25 °C |
17 hodin |
85 hodin |
Tento přípravek obsahuje geneticky modifikované organismy. Nepoužitý léčivý přípravek musí být zlikvidován v souladu s požadavky poskytovatele zdravotních služeb pro nakládání s geneticky upravenými organismy nebo nebezpečným biologickým odpadem, podle toho, co je vhodné.
41
Čemu musíte věnovat pozornost, než začnete Imlygic používat
Imlygic nebudete dostávat v těchto případech:
- jestliže jste alergický(á) na talimogen laherparepvek nebo na kteroukoliv další složku tohoto přípravku (uvedenou v bodě 6).
- jestliže Vám Váš zdravotnický pracovník řekl, že máte výrazně oslabený imunitní systém. Upozornění a opatření
Před použitím přípravku Imlygic se poraďte se svým zdravotnickým pracovníkem.
Pacienti s oslabeným imunitním systémem
U pacientů s oslabeným imunitním systémem se může vyskytnout život ohrožující herpetická infekce. Jestliže máte nebo jste někdy měl(a) oslabený imunitní systém, jestliže máte HIV/AIDS, nádorové