Imbruvica 140 Mg
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
IMBRUVICA 140 mg tvrdé tobolky.
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna tvrdá tobolka obsahuje ibrutinibum 140 mg.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Tvrdá tobolka (tobolka).
Bílá neprůhledná, tvrdá tobolka o délce 22 mm, označená „ibr 140 mg“ černým inkoustem.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek IMBRUVICA je v monoterapii indikován k léčbě dospělých pacientů s relabujícím nebo refrakterním lymfomem z plášťových buněk (mantle cell lymphoma, MCL).
Přípravek IMBRUVICA je v monoterapii indikován k léčbě dospělých pacientů s dosud neléčenou chronickou lymfocytární leukemií (CLL) (viz. bod 5.1)
Přípravek IMBRUVICA je v monoterapii nebo v kominaci s bendamustinem a rituximabem (BR) indikován k léčbě dospělých pacientů s CLL, kteří podstoupili alespoň jednu předchozí terapii.
Přípravek IMBRUVICA je v monoterapii indikován k léčbě dospělých pacientů s Waldenstromovou makroglobulinemií (WM), kteří již podstoupili alespoň jednu předchozí terapii, nebo v první linii u pacientů, u nichž není vhodná chemo-imunoterapie.
4.2 Dávkování a způsob podání
Léčba tímto léčivým přípravkem musí být zahájena a monitorována lékařem, který má zkušenosti s používáním protinádorových léčivých přípravků.
Dávkování
Lymfom z plášťových buněk
Doporučená dávka pro léčbu MCL je 560 mg (čtyři tobolky) jednou denně.
Chronická lymfocytární leukemie a Waldenstromova makroglobulinemie
Doporučená dávka pro léčbu CLL buď v monoterapii nebo v kombinaci je 420 mg (tři tobolky) jednou denně (viz bod 5.1 pro podrobnosti o kombinované léčbě).
Doporučená dávka pro léčbu WM je 420 mg (tři tobolky) jednou denně.
Léčba by měla pokračovat až do progrese onemocnění nebo dokud nepřestane být tolerována pacientem.
Úpravy dávkování
Středně silné a silné inhibitory CYP3A4 zvyšují expozici ibrutinibu (viz body 4.4 a 4.5).
Dávku přípravku IMBRUVICA je nutné snížit na 140 mg jednou denně (1 tobolka) v případě, že je přípravek užíván společně se středně silnými inhibitory CYP3A4.
Dávku přípravku IMBRUVICA je nutné omezit na 140 mg jednou denně (1 tobolka) nebo vysadit až na 7 dní v případě, že je přípravek užíván společně se silnými inhibitory CYP3A4.
Léčba přípravkem IMBRUVICA by měla být přerušena při každém novém výskytu nebo zhoršení nehematologické toxicity na stupeň > 3, neutropenie s infekcí nebo horečkou na stupeň 3 nebo vyšší nebo hematologických toxicit stupně 4. Jakmile projevy toxicity ustoupí na 1. stupeň nebo k výchozímu stavu (uzdravení), může být léčba přípravkem IMBRUVICA obnovena v počáteční dávce. Pokud dojde k recidivě toxicity, denní dávku je třeba snížit o jednu tobolku (140 mg). V případě potřeby lze zvážit druhé snížení dávky o 140 mg. Pokud budou toxické projevy přetrvávat nebo recidivovat i po dvou redukcích dávky, léčivý přípravek je nutné vysadit.
Doporučené úpravy dávky jsou popsány níže:
|
Výskyt toxicity |
Úprava dávky u MCL po odeznění toxicity |
Úprava dávky u CLL/WM po odeznění toxicity |
|
První |
Obnovte léčbu dávkou 560 mg denně |
Obnovte léčbu dávkou 420 mg denně |
|
Druhý |
Obnovte léčbu dávkou 420 mg denně |
Obnovte léčbu dávkou 280 mg denně |
|
Třetí |
Obnovte léčbu dávkou 280 mg denně |
Obnovte léčbu dávkou 140 mg denně |
|
Čtvrtý |
Přípravek IMBRUVICA vysaďte |
Přípravek IMBRUVICA vysaďte |
Vynechaná dávka
V případě, že není dávka užita v plánovaném čase, lze ji užít co nejdříve tentýž den a následující den pokračovat v normálním rozvrhu dávkování. Pacient však nesmí užít tobolky navíc, aby nahradil vynechanou dávku.
Zvláštní populace
Starší osoby
U starších pacientů (> 65 let) není nutná úprava dávkování.
Porucha funkce ledvin
U pacientů s poruchou funkce ledvin nebyly provedeny žádné specifické klinické studie. V klinických studiích hodnotících přípravek IMBRUVICA byli léčeni pacienti s mírnou nebo středně závažnou poruchou funkce ledvin. U pacientů s mírnou až středně závažnou poruchou funkce ledvin (clearance kreatininu vyšší než 30 ml/min) není nutná úprava dávky. Je nutné zajistit hydrataci a pravidelně monitorovat hladinu kreatininu v plazmě. Pacientům se závažnou poruchou funkce ledvin (clearance kreatininu <30 ml/min) lze podávat přípravek IMBRUVICA pouze tehdy, pokud přínos léčby převýší její rizika, a pacient je pečlivě sledován na známky toxicity. U pacientů se závažnou poruchou funkce ledvin nebo u pacientů na dialýze nejsou k dispozici žádné údaje (viz bod 5.2).
Porucha funkce jater
Ibrutinib je metabolizován v játrech. Údaje ze studie zahrnující neonkologické pacienty s poruchou funkce jater prokázaly nárůst expozice ibrutinibu (viz bod 5.2). U pacientů s mírnou poruchou funkce jater (Child-Pugh třída A) je doporučená dávka 280 mg denně (dvě tobolky). U pacientů se středně závažnou poruchou funkce jater (Child-Pugh třídy B) je doporučená dávka 140 mg denně (jedna tobolka). Pacienty je třeba sledovat na příznaky toxicity přípravku IMBRUVICA a podle potřeby postupovat podle pokynů pro úpravu dávkování. Podávání přípravku IMBRUVICA pacientům se závažnou poruchou funkce jater (Child-Pugh třída C) se nedoporučuje.
Závažné onemocnění srdce
Pacienti se závažným kardiovaskulárním onemocněním byli vyloučeni z klinických studií přípravku IMBRUVICA.
Pediatrická populace
Bezpečnost a účinnost přípravku IMBRUVICA u dětí ve věku 0 až 18 let nebyla dosud stanovena. Nejsou k dispozici žádné údaje.
Způsob podání
Přípravek IMBRUVICA se užívá perorálně jednou denně a zapíjí se sklenicí vody, každý den přibližně ve stejnou dobu. Tobolky se polykají celé s vodou a nesmí se otevírat, lámat nebo kousat. Přípravek IMBRUVICA se nesmí užívat s grapefruitovou šťávou nebo plody pomerančovníku hořkého (viz bod 4.5).
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Použití přípravků s obsahem třezalky tečkované (Hypericumperforatum) je kontraindikováno u pacientů léčených přípravkem IMBRUVICA.
4.4 Zvláštní upozornění a opatření pro použití
Příhody související s krvácením
U pacientů léčených přípravkem IMBRUVICA byly hlášeny hemoragické příhody s trombocytopenií i bez trombocytopenie. Mezi tyto příhody patří drobné krvácivé příhody, jako jsou kontuze, epistaxe a petechie; a velké krvácivé příhody, některé fatální, včetně gastrointestinálního krvácení, intrakraniálního krvácení a hematurie.
Pacienti byli vyloučeni z účasti ve studiích přípravku IMBRUVICA fáze 2 a 3, pokud vyžadovali warfarin nebo jiné antagonisty vitaminu K. Warfarin a další antagonisté vitaminu K se nemají podávat současně s přípravkem IMBRUVICA. Je třeba vyhnout se některým doplňkům stravy, jako jsou rybí olej a přípravky obsahující vitamin E. Použití přípravku IMBRUVICA u pacientů vyžadujících jiná antikoagulancia nebo léčivé přípravky, které inhibují funkci krevních destiček, může zvýšit riziko krvácení a zvláštní péče je nutná v případě, že je použita antikoagulační léčba. Pacienti s vrozeným sklonem ke krvácení nebyli studováni.
Podávání přípravku IMBRUVICA je třeba přerušit na dobu alespoň 3-7 dnů před operací a po operaci v závislosti na druhu chirurgického zákroku a riziku krvácení.
Leukostáza
U pacientů léčených přípravkem IMBRUVICA byly hlášeny případy leukostázy. Vysoký počet cirkulujících lymfocytů (> 400 000/^l) může vést ke zvýšení rizika. Zvažte dočasné přerušení léčby přípravku IMBRUVICA. Pacienty je třeba pečlivě sledovat. Zajistěte podpůrnou péči, včetně hydratace a/nebo cytoredukce, pokud je indikována.
Infekce
U pacientů léčených přípravkem IMBRUVICA byly pozorovány infekce (včetně sepse, neutropenické sepse, bakteriálních, virových nebo mykotických infekcí). Některé z těchto infekcí byly spojeny s hospitalizací a úmrtím pacienta. Většina pacientů s fatální infekcí měla taktéž neutropenii. Pacienti by měli být sledováni na výskyt horečky, neutropenie a infekcí, a dle indikace by měla být zahájena odpovídající protiinfekční terapie.
Cytopenie
U pacientů léčených přípravkem IMBRUVICA byly hlášeny cytopenie stupně 3 nebo 4 vzniklé při léčbě (neutropenie, trombocytopenie a anemie). Jednou měsíčně je nutné kontrolovat krevní obraz.
Intersticiální plicní nemoc (ILD)
U pacientů léčených přípravkem IMBRUVICA byly hlášeny případy ILD. U pacientů je třeba sledovat rozvoj plicních příznaků svědčících o ILD. Pokud se příznaky rozvinou, je třeba přerušit léčbu přípravkem IMBRUVICA a odpovídajícím způsobem léčit ILD. Pokud příznaky přetrvávají, je třeba zvážit poměr rizik a přínosů léčby přípravkem IMBRUVICA a dodržovat pokyny pro úpravu dávkování.
Fibrilace síní/síňový flutter
U pacientů léčených přípravkem IMBRUVICA byly hlášeny případy fibrilace síní a flutteru síní, a to zejména u pacientů s kardiovaskulárními rizikovými faktory, akutní infekcí a fibrilací síní v anamnéze. Pacienty je třeba pravidelně klinicky sledovat na fibrilaci síní. Pacienty, u nichž se vyvinou příznaky arytmie nebo nově vzniklé dušnosti, je třeba klinicky vyšetřit a v případě potřeby provést EKG vyšetření (elektrokardiogram).
U pacientů s fibrilací síní v anamnéze, kteří vyžadují antikoagulační léčbu, je nutné zvážit možnost alternativní léčby k přípravku IMBRUVICA. U pacientů, u nichž došlo k rozvoji fibrilace síní na základě léčby přípravkem IMBRUVICA, je třeba provést pečlivé zhodnocení rizika tromboembolického onemocnění. U pacientů s vysokým rizikem, a u nichž není možné použít alternativní léčbu k přípravku IMBRUVICA, je nutné zvážit přísně kontrolovanou léčbu antikoagulancii.
Syndrom nádorového rozpadu
V souvislosti s léčbou přípravkem IMBRUVICA byl hlášen syndrom nádorového rozpadu. Riziko vzniku syndromu nádorového rozpadu se vyskytuje u pacientů, kteří mají velkou masu nádoru před léčbou. Pacienty je třeba pečlivě sledovat a přijmout náležitá preventivní opatření.
Nemelanomový karcinom kůže
U pacientů léčených přípravkem IMBRUVICA byly častěji hlášeny nemelanomové karcinomy kůže ve srovnání s pacienty léčenými srovnávacím přípravkem ve sdružených, randomizovaných, srovnávacích klinických studiích fáze 3. U pacientů je třeba sledovat výskyt nemelanomového karcinomu kůže.
Vliv na QT interval
Ve studii fáze 2 bylo na základě EKG vyšetření prokázáno, že přípravek IMBRUVICA vyvolává mírný pokles QTcF intervalu (průměr 7,5 ms). Související mechanismus a význam tohoto nálezu pro bezpečnost nejsou známy, lékař by proto měl rozhodovat na základě vlastního klinického úsudku, zda předepsat ibrutinib pacientům, u nichž je přítomno riziko dalšího zkrácení QTc intervalu (např. vrozený syndrom krátkého intervalu QT nebo pacienti s výskytem tohoto syndromu v rodinné anamnéze).
Lékové interakce
Souběžné podání silných nebo středně silných inhibitorů CYP3A4 s přípravkem IMBRUVICA může vést ke zvýšené expozici ibrutinibu a současně k vyššímu riziku toxicity. Naopak, souběžné podání induktorů CYP3A4 může vést ke snížené expozici přípravku IMBRUVICA a současně k riziku ztráty účinnosti. Proto je třeba vyhnout se současnému užívání přípravku IMBRUVICA se silnými nebo středně silnými inhibitory CYP3A4. Současné podání lze zvážit pouze v případě, že prospěch z léčby zcela převáží možná rizika. V případě, že je nezbytné použít středně silný nebo silný inhibitor CYP3A (viz body 4.2 a 4.5), je nutné pacienty pečlivě sledovat na příznaky toxicity přípravku IMBRUVICA. Jestliže je nutné použít induktor CYP3A4, je třeba pacienty pečlivě sledovat na příznaky nedostatečné účinnosti.
Ženy ve fertilním věku
Ženy ve fertilním věku musí během léčby přípravkem IMBRUVICA používat vysoce účinnou metodu antikoncepce (viz bod 4.6).
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Ibrutinib je primárně metabolizován cytochromem P450 3A4 (CYP3A4).
Léky, které mohou zvyšovat plazmatické koncentrace ibrutinibu
Současné užívání přípravku IMBRUVICA a léků, které silně nebo středně silně inhibují CYP3A4, může zvýšit expozici ibrutinibu, proto je třeba se této kombinaci vyhnout.
Silné inhibitory CYP3A4
Současné podávání silného inhibitoru CYP3A4 ketokonazolu u 18 lačných zdravých subjektů zvýšilo expozici ibrutinibu (Cmax a AUC) 29krát resp. 24krát. Simulace za použití podání nalačno naznačila, že silný inhibitor CYP3A4 klarithromycin může zvýšit AUC ibrutinibu 14krát. Je třeba se vyhnout silným inhibitorům CYP3A4 (např. ketokonazol, indinavir, nelfinavir, ritonavir, sachinavir, klarithromycin, telithromycin, itrakonazol, nefazodon a kobicistat). Pokud přínos převažuje riziko a silný inhibitor CYP3A4 je nezbytné použít, je nutné snížit dávku přípravku IMBRUVICA na 140 mg (jedna tobolka) nebo léčbu dočasně vysadit (po dobu 7 dnů nebo méně). Pacienty je třeba pečlivě sledovat na známky toxicity a pokud je třeba, postupovat dle pokynů k úpravě dávkování (viz body 4.2 a 4.4).
Středně silné inhibitory CYP3A4
Simulace za použití podání nalačno naznačila, že středně silné inhibitory CYP3A4 diltiazem, erythromycin a vorikonazol mohou zvýšit AUC ibrutinibu 5-9krát. Je třeba se vyhnout středně silným inhibitorům CYP3A (např. vorikonazol, erythromycin, amprenavir, aprepitant, atazanavir, ciprofloxacin, krizotinib, darunavir/ritonavir, diltiazem, flukonazol, fosamprenavir, imatinib, verapamil, amiodaron, dronedaron). Pokud je nezbytné použít středně silný inhibitor CYP3A, je nutné dávku přípravku IMBRUVICA snížit na 140 mg (jedna tobolka) po dobu použití inhibitoru. Je třeba pečlivě sledovat pacienty na příznaky toxicity a podle potřeby postupovat podle pokynů pro úpravu dávkování (viz body 4.2 a 4.4).
Slabé inhibitory CYP3A4
Simulace za použití klinicky relevantního podání nalačno naznačila, že slabé inhibitory CYP3A4 azythromycin a fluvoxamin mohou zvýšit AUC ibrutinibu < 2 krát. Není třeba žádná úprava dávkování v kombinaci se slabými inhibitory. Je třeba pečlivě sledovat pacienty na příznaky toxicity a podle potřeby postupovat podle pokynů pro úpravu dávkování.
Současné podávání grapefruitové šťávy, která obsahuje inhibitory CYP3A4, osmi zdravým dobrovolníkům zvýšilo expozici (Cmax a AUC) ibrutinibu přibližně 4krát, resp. 2krát.
Během léčby přípravkem IMBRUVICA se nesmí užívat grapefruitová šťáva a plody pomerančovníku hořkého, protože obsahují středně silné inhibitory CYP3A4 (viz bod 4.2).
Léky, které mohou snižovat plazmatické koncentrace ibrutinibu
Podání přípravku IMBRUVICA s induktory CYP3A4 může snížit plazmatické koncentrace ibrutinibu.
Současné podávání silného induktoru CYP3A4 rifampicinu u 18 lačných zdravých subjektů snížilo expozici ibrutinibu (Cmax a AUC) o 92 % resp. 90 %. Je nutné vyvarovat se současného užívání silných nebo středně silných induktorů CYP3A4 (např. karbamazepin, rifampicin, fenytoin). V průběhu léčby přípravkem IMBRUVICA je kontraindikováno používat přípravky s obsahem třezalky tečkované (Hypericum perforatum), protože by mohla být snížena účinnost. Je třeba zvážit alternativní léky se slabší indukcí CYP3A4. V případě, že musí být použit silný nebo středně silný induktor CYP3A4 a prospěch převáží nad rizikem, je nutné u pacienta pečlivě sledovat ztrátu účinnosti (viz body 4.3 a 4.4). Použití středně silných induktorů lze zvážit při současném podávání přípravku IMBRUVICA, avšak pacienty je nutné sledovat na možnou nedostatečnou účinnost přípravku.
Vzhledem k tomu, že rozpustnost ibrutinibu je závislá na pH, je zde teoretické riziko, že léčivé přípravky zvyšující pH žaludku (např. inhibitory protonové pumpy) mohou snížit expozici ibrutinibu. Tato interakce nebyla studována in vivo.
Látky, jejichž plazmatické koncentrace mohou být ovlivněny ibrutinibem
Ibrutinib je in vitro inhibitorem glykoproteinu P (P-gp) a proteinu BCRP (breast cancer resistance protein). Vzhledem ktomu, že nejsou dostupné žádné klinické údaje o této interakci, nelze vyloučit, že by ibrutinib po podání terapeutické dávky mohl inhibovat intestinální P-gp a BCRP. Pro minimalizaci potenciálních interakcí v GI traktu je třeba užívat substráty P-gp nebo BCRP s úzkým perorálním terapeutickým rozmezím, jako například digoxin nebo methotrexát, nejméně 6 hodin před podáním nebo 6 hodin po podání přípravku IMBRUVICA. Ibrutinib může také inhibovat BCRP v játrech a zvýšit tak expozici látkám, které jsou substrátem hepatálního efluxního transportéru BCRP, například rosuvastatinu.
Na základě údajů in vitro je ibrutinib slabým reverzibilním inhibitorem střevní CYP3A4, a tudíž může zvyšovat expozici substrátům CYP3A4 citlivých na střevní metabolismus zprostředkovaný tímto cytochromem. O této interakci nejsou dostupné klinické údaje. Pozornosti je třeba při současném podávání ibrutinibu s perorálně podávanými substráty CYP3A4 s úzkým terapeutickým rozmezím (jako je dihydroergotamin, ergotamin, fentanyl, cyklosporin, sirolimus a takrolimus).
Na základě údajů in vitro je ibrutinib slabým induktorem CYP2B6 a potenciálně tak může mít vliv na expresi jiných enzymů a transportérů regulovaných prostřednictvím konstitutivního androstanového receptoru (CAR), např. CYP2C9, CYP2C19, UGT1A1 a MRP2. Klinický význam není znám, ale expozice substrátům CYP2B6 (jako je efavirenz a bupropion) a koregulačním enzymům může být při společném užívání s ibrutinibem snížena.
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku/antikoncepce u žen
Z nálezů u zvířat vyplývá, že přípravek IMBRUVICA může způsobit poškození plodu, pokud by byl podán těhotné ženě. Ženy by se měly vyvarovat těhotenství v průběhu užívání přípravku IMBRUVICA a další 3 měsíce po ukončení léčby. Ženy ve fertilním věku proto musí během léčby přípravkem IMBRUVICA a další 3 měsíce po ukončení léčby používat vysoce účinnou antikoncepci. V současné době není známo, zda ibrutinib může snižovat účinnost hormonální kontracepce, proto u žen užívajících hormonální antikoncepci je třeba přidat bariérovou metodu.
Přípravek IMBRUVICA se nesmí používat během těhotenství. Údaje o použití přípravku IMBRUVICA u těhotných žen nejsou k dispozici. Studie na zvířatech prokázaly reprodukční toxicitu (viz bod 5.3).
Kojení
Není známo, zda se ibrutinib nebo jeho metabolity vylučují do lidského mateřského mléka. Riziko pro kojené novorozence/děti nelze vyloučit. Kojení má být během léčby přípravkem IMBRUVICA přerušeno.
Fertilita
Nebyly pozorovány žádné účinky na fertilitu nebo reprodukční schopnost u samců nebo samic potkanů, kteří byli testováni maximální dávkou 100 mg/kg/den (Human Equivalent Dose [HED]
16 mg/kg/den) viz bod 5.3). Nejsou dostupné žádné údaje o účincích ibrutinibu na fertilitu u člověka.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Někteří pacienti užívající přípravek IMBRUVICA si stěžovali na únavu, závratě a slabost a tuto možnost je třeba zohlednit při posuzování schopnosti pacienta řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Bezpečnostní profil je založen na údajích získaných od 981 pacientů léčených přípravkem IMBRUVICA ve třech klinických studiích fáze 2 a čtyřech randomizovaných klinických studiích fáze 3 a z postmarketingového sledování. Pacienti s MCL užívali v klinickém hodnocení 560 mg přípravku IMBRUVICA jednou denně a pacienti s CLL nebo WM užívali v klinickém hodnocení 420 mg přípravku IMBRUVICA jednou denně. Všichni pacienti užívali v klinickém hodnocení přípravek IMBRUVICA do progrese onemocnění nebo do doby, kdy již přípravek IMBRUVICA nebyl tolerován.
Nejčastěji se vyskytující nežádoucí účinky (> 20 %) byly průjem, neutropenie, krvácení (např. tvorba modřin), muskuloskeletální bolest, nauzea, vyrážka a pyrexie. Nejčastější nežádoucí účinky stupně 3/4 (> 5 %) byly neutropenie, pneumonie, trombocytopenie a febrilní neutropenie.
Nežádoucí účinky
Nežádoucí účinky u pacientů s malignitami B-buněk léčených ibrutinibem a nežádoucí účinky hlášené z postmarketingového sledování jsou uvedeny níže podle tříd orgánových systémů a četností. Četnosti jsou definovány jako: velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1 000 až < 1/100), není známo (z dostupných údajů nelze určit). V každé skupině četností jsou nežádoucí účinky uvedeny v pořadí podle klesající závažnosti.
Tabulka 1: Nežádoucí účinky hlášené v klinických studiích nebo z postmarketingového sledování u _pacientů s malignitami B-buněk_
|
Třída orgánových systémů |
Frekvence (Všechny stupně) |
Nežádoucí účinky |
Všechny stupně (%) |
Stupeň >3 (%) |
|
Infekce a infestace |
Velmi časté |
Pneumonie*# |
16 |
10 |
|
Infekce horních cest dýchacích |
19 |
1 | ||
|
Sinusitida* |
11 |
1 | ||
|
Kožní infekce* |
10 |
3 | ||
|
Časté |
Sepse*# |
4 |
4 | |
|
Infekce močových cest |
9 |
2 | ||
|
Novotvary maligní, |
Časté |
Nemelanomový karcinom kůže* |
6 |
1 |
|
benigní a blíže neurčené |
Bazocelulární karcinom |
3 |
< 1 | |
|
(zahrnující cysty a polypy) |
Spinocelulární karcinom |
2 |
< 1 | |
|
Poruchy krve a |
Velmi časté |
Neutropenie |
30 |
26 |
|
lymfatického systému |
Trombocytopenie |
20 |
10 | |
|
Časté |
Febrilní neutropenie |
5 |
5 | |
|
Leukocytóza |
2 |
1 | ||
|
Lymfocytóza |
2 |
1 | ||
|
Méně časté |
Syndrom leukostázy |
< 1 |
< 1 | |
|
Poruchy imunitního |
Časté |
Intersticiální plicní nemoc*#,a |
2 |
< 1 |
|
systému | ||||
|
Poruchy metabolismu a |
Časté |
Syndrom nádorového rozpadu |
1 |
1 |
|
výživy |
Hyperurikemie |
7 |
2 | |
|
Poruchy nervového |
Velmi časté |
13 |
1 | |
|
systému |
Časté |
Závratě |
9 |
0 |
|
Poruchy oka |
Časté |
Rozmazané vidění |
7 |
0 |
|
Srdeční poruchy |
Časté |
Fibrilace síní |
6 |
3 |
|
Cévní poruchy |
Velmi časté |
Krvácení *# |
30 |
1 |
|
Tvorba modřin* |
22 |
< 1 | ||
|
Časté |
Subdurální hematom# |
1 |
1 | |
|
Epistaxe |
8 |
< 1 | ||
|
Petechie |
7 |
0 | ||
|
Hypertenze* |
10 |
4 |
|
Gastrointestinální poruchy |
Velmi časté |
Stomatitida* Zácpa |
41 14 13 27 16 |
3 < 1 1 1 < 1 |
|
Poruchy jater a žlučových |
Není známo |
Jaterní selhání*,a |
Není |
Není známo |
|
cest |
známo | |||
|
Poruchy kůže a podkožní |
Velmi časté |
Vyrážka* |
22 |
2 |
|
tkáně |
Časté |
Kopřivka |
1 |
< 1 |
|
Erytém |
2 |
0 | ||
|
Méně časté |
Angioedém |
< 1 |
< 1 | |
|
Poruchy svalové a kosterní |
Velmi časté |
Artralgie |
12 |
1 |
|
soustavy a pojivové tkáně |
Svalové křeče |
14 |
< 1 | |
|
Muskuloskeletální bolest* |
28 |
3 | ||
|
Celkové poruchy a reakce |
Velmi časté |
Pyrexie |
20 |
2 |
|
v místě aplikace |
Periferní otok |
14 |
1 |
^ Frekvence jsou zaokrouhleny na nejbližsí celé číslo. * Zahrnuje více termínů pro nežádoucí účinek
Zahrnuje příhody s fatálními následky. a Spontánní hlášení z postmarketingového sledování
Ukončení léčby a úprava dávkování z důvodu nežádoucích účinků
Z 981 pacientů s s B-buněčnými malignitami léčených přípravkem IMBRUVICA ukončilo 5 % léčbu primárně v důsledku výskytu nežádoucích účinků. Ty zahrnovaly pneumonii, fibrilaci síní a krvácení. Nežádoucí účinky, které vedly ke snížení dávky, se vyskytly přibližně u 5 % pacientů.
Starší pacienti
Z 981 pacientů léčených přípravkem IMBRUVICA bylo 62 % ve věku 65 let nebo starších. Pneumonie stupně 3 a vyššího se vyskytovala častěji u starších pacientů léčených přípravkem IMBRUVICA (13 % u pacientů starších > 65 let oproti 7 % u pacientů mladších < 65 let).
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
K dispozici jsou pouze omezené údaje o účincích předávkování přípravkem IMBRUVICA. Ve studii fáze 1, ve které pacienti užívali až 12,5 mg/kg/den (1400 mg/den), nebylo dosaženo maximální tolerované dávky. V jiné studii, u jednoho zdravého subjektu, který užíval dávku 1680 mg, došlo k reverzibilnímu zvýšení jaterních enzymů stupně 4 [aspartátaminotransferáza (AST) a alaninaminotransferáza (ALT)]. Neexistuje žádné specifické antidotum pro přípravek IMBRUVICA. Pacienty, kteří užijí větší množství, než je doporučená dávka, je třeba pečlivě sledovat a poskytovat jim vhodnou podpůrnou léčbu.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Cytostatika, inhibitory proteinkinázy, ATC kód: L01XE27. Mechanismus účinku
Ibrutinib je silný inhibitor Brutonovy tyrosinkinázy (BTK) o malé molekule. Ibrutinib tvoří kovalentní vazbu s cysteinovým zbytkem (Cys-481) v aktivním místě BTK, která vede k trvalé inhibici
enzymatické aktivity BTK. BTK je člen rodiny Tec kináz a představuje důležitou signální molekulu drah B-buněčného receptoru (BCR) a cytokinového receptoru. BCR dráha se podílí na patogenezi některých B-buněčných malignit, včetně MCL, difúzního velkobuněčného B-lymfomu (DLBCL), folikulárního lymfomu a chronické lymfocytární leukemie (CLL). BTK zastává ústřední roli v signalizaci přes povrchové receptory B-buněk, která vede k aktivaci drah nezbytných pro prostup (trafficking), chemotaxi a adhezi B-buněk. Preklinické studie ukazují, že ibrutinib účinně inhibuje maligní proliferaci a přežívání B-buněk in vivo, stejně jako migraci buněk a adhezi k substrátu in vitro.
Lvmfocvtóza
Po zahájení léčby bylo přibližně u tří čtvrtin pacientů s CLL léčených přípravkem IMBRUVICA pozorováno reverzibilní zvýšení počtu lymfocytů (tj. nárůst o > 50 % oproti výchozí hodnotě a absolutní počet > 5 000/p.l), často spojené s redukcí lymfadenopatie. Tento efekt byl rovněž pozorován asi u jedné třetiny pacientů s relabující nebo refrakterní MCL, kteří byli léčeni přípravkem IMBRUVICA. Pozorovaná lymfocytóza s farmakodynamickým účinkem a při absenci jiných klinických nálezů by neměla být považována za progresivní onemocnění. U obou typů onemocnění se lymfocytóza typicky objevuje během prvního měsíce léčby přípravkem IMBRUVICA a obvykle vymizí během mediánu doby 8,0 týdnů u pacientů s MCL a 14 týdnů u pacientů s CLL. U některých pacientů bylo pozorováno velké zvýšení počtu cirkulujících lymfocytů (např. > 400 000/^l). Lymfocytóza nebyla pozorována u pacientů s WM léčených přípravkem IMBRUVICA.
Klinická účinnost a bezpečnost
Lymfom z plášťových buněk
Bezpečnost a účinnost přípravku IMBRUVICA u pacientů s relabujícím nebo refrakterním MCL byly hodnoceny v jediné otevřené, multicentrické studii fáze 2 (PCYC-1104-CA) u 111 pacientů. Medián věku byl 68 let (rozsah: 40-84 let), 77 % účastníků byli muži a 92 % účastníků bylo bílé (europoidní) rasy. Pacienti s hodnotou ECOG (Eastern Cooperative Oncology Group) performance status 3 nebo vyšší byli ze studie vyloučeni. Medián doby od diagnózy činil 42 měsíců a medián počtu předchozích terapií činil 3 terapie (rozsah: 1 až 5 terapií), včetně 35 % pacientů s předchozí vysokodávkovanou chemoterapií, 43 % s předchozí léčbou bortezomibem, 24 % s předchozí léčbou lenalidomidem a 11 % s předchozí transplantací autologních nebo allogenních kmenových buněk. Při screeningu mělo 39 % pacientů uzlinové postižení (tzv. bulky disease) (> 5 cm), 49 % pacientů mělo vysoce rizikové skóre podle zjednodušeného mezinárodního prognostického indexu MCL (MIPI) a 72 % pacientů bylo v pokročilém stadiu nemoci (extranodální postižení a/nebo postižení kostní dřeně).
Přípravek IMBRUVICA byl podáván perorálně v dávce 560 mg jednou denně až do progrese onemocnění nebo nepřijatelné toxicity. Byla hodnocena odpověď nádoru podle kritérií revidované Mezinárodní pracovní skupiny (IWG) pro non-Hodgkinské lymfomy (NHL). Primárním cílovým parametrem v této studii byl celkový výskyt léčebné odpovědi (ORR) hodnocený zkoušejícím. Odpovědi na léčbu přípravkem IMBRUVICA jsou uvedeny v tabulce 2.
Tabulka 2: Celkový výskyt léčebné odpovědi (ORR) a doba trvání odpovědi (DOR) u
pacientů s relabujícím nebo refrakterním MCL (studie PCYC-1104-CA)
|
Celkem N = 111 | |
|
ORR (%) |
67,6 |
|
95 % CI (%) |
(58,0;76,1) |
|
CR (%) |
20,7 |
|
PR (%) |
46,8 |
|
Medián DOR (CR+PR) (měsíce) |
17,5 (15,8; NR) |
|
Medián doby do úvodní odpovědi, měsíce (rozsah) |
1,9 (1,4-13,7) |
|
Medián doby do CR, měsíce (rozsah) |
5,5 (1,7; 11,5) |
CI = interval spolehlivosti; CR = kompletní odpověď; PR = částečná odpověď; NR = nebylo dosaženo
Údaje o účinnosti byly dále hodnoceny nezávislou hodnotící komisí (IRC), která prokázala hodnotu ORR 69 %, výskyt kompletní odpovědi (CR) 21 % a výskyt částečné odpovědi (PR) 48 %. Medián DOR podle odhadu komise IRC činil 19,6 měsíců.
Celková odpověď na přípravek IMBRUVICA byla nezávislá na předchozí léčbě, včetně bortezomibu a lenalidomidu, nebo na základních rizikových/prognostických faktorech, bulky disease, pohlaví nebo věku.
Bezpečnost a účinnost přípravku IMBRUVICA byla prokázána v randomizované otevřené multicentrické studii fáze 3, které se účastnilo 280 pacientů s MCL, kteří předtím absolvovali alespoň jednu předchozí léčbu (studie MCL3001). Pacienti byli randomizováni v poměru 1:1 do skupiny užívající perorálně přípravek IMBRUVICA v dávce 560 mg jednou denně po dobu 21 dnů nebo temsirolimus intravenózně v dávce 175 mg v den 1, 8, 15 prvního cyklu následované 75 mg v den 1, 8, 15 každého následujícího 21denního cyklu. Léčba v obou ramenech pokračovala až do progrese onemocnění nebo nepřijatelné toxicity. Medián věku činil 68 let (rozmezí 34 až 88 let), 74 % pacientů byli muži, 87 % účastníků bylo bílé (europoidní) rasy. Medián doby od diagnózy činil 43 měsíců, medián počtu předchozích terapií činil 2 terapie (rozmezí: 1 až 9 terapií): 51 % s předchozí vysokodávkovou chemoterapií, 18 % s předchozí léčbou bortezomibem, 5 % s předchozí léčbou lenalidomidem a 24 % po předchozí transplantaci kmenových buněk. V úvodu mělo 53 % pacientů lymfomové masy (> 5 cm), 21 % mělo simplifikované skóre MIPI odpovídající vysokému riziku,
60 % mělo extranodální onemocnění a 54 % trpělo postižením kostní dřeně při screeningu.
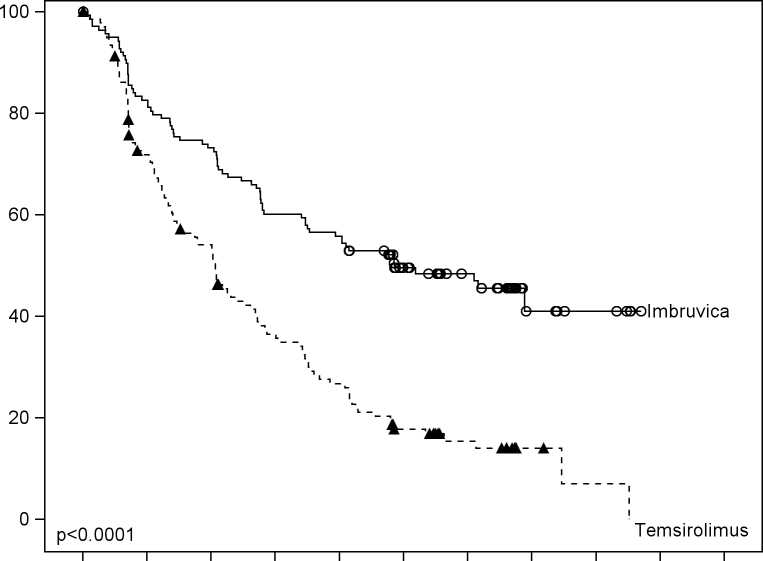
Přežívání bez progrese (PFS) hodnotila nezávislá hodnotící komise (IRC) podle revidovaných kritérií mezinárodní pracovní skupiny (IWG) pro non-Hodgkinské lymfomy (NHL). Výsledky účinnosti ve studii MCL3001 jsou uvedeny v tabulce 3 a Kaplan-Meierova křivka pro PFS je na obrázku 1.
Tabulka 3: Výsledky účinnosti u pacientů s relabujícím nebo refrakterním MCL (studie
MCL3001)
|
Cílový parametr |
IMBRUVICA N = 139 |
Temsirolimus N = 141 |
|
Přežívání bez progrese3 | ||
|
Medián přežití bez progrese (95% CI), (měsíce) |
14,6 (10,4, NE) |
6,2 (4,2, 7,9) |
|
HR = 0,43 [95% CI: 0,32, 0,581 | ||
|
Celková léčebná odpověď (%) |
71,9 40,4 | |
|
P-hodnota |
p< 0,0001 | |
Hodnoceno IRC;
Menší procento pacientů léčených ibrutinibem mělo klinicky významné zhoršení příznaků lymfomu oproti skupině užívající temsirolimus (27 % versus 52 %) a doba do zhoršení příznaků byla pomalejší u ibrutinibu než u temsirolimu (HR 0,27, p < 0,0001).
|
0 |
3 |
6 |
9 |
12 |
15 |
18 21 |
24 |
27 |
30 |
|
Počet pacientů v riziku |
Měsíce | ||||||||
|
Imbruvica 139 |
114 |
101 |
83 |
77 |
45 |
34 8 |
5 |
0 |
0 |
|
Temsirolimus 141 |
93 |
69 -e— |
45 Imbruvica |
33 ---A- |
19 |
11 3 Temsirolimus |
1 |
0 |
0 |
Chronická lymfocytární leukemie Dosud neléčení pacienti s CLL
U pacientů ve věku 65 let a starších dosud neléčených s CLL byla provedena randomizovaná, multicentrická, otevřená studie fáze 3 hodnotící přípravek IMBRUVICA oproti chlorambucilu (PCYC-1115-CA). Pacienti ve věku mezi 65 a 70 lety museli mít nejméně jednu komorbiditu, která předem vyloučila v první linii použití chemo-imunoterapie fludarabinem, cyklofosfamidem a rituximabem. Pacienti (n = 269) byli randomizováni v poměru 1:1 k léčbě buď přípravkem IMBRUVICA 420 mg denně až do progrese onemocnění nebo nepřijatelné toxicity nebo chlorambucilem s počáteční dávkou 0,5 mg/kg ve dnech 1a 15 každého 28denního cyklu po dobu maximálně 12 cyklů, s povoleným zvýšením dávky až na 0,8 mg/kg v závislosti na snášenlivosti. Pacienti léčení chlorambucilem mohli být převedeni na ibrutinib po potvrzení progrese onemocnění.
Medián věku činil 73 let (rozsah: 65 až 90 let) 63 % účastníků byli muži a 91 % účastníků bylo bílé (europoidní) rasy. Devadesát jedna procent pacientů mělo vstupní hodnotu ECOG skóre 0 nebo 1 a 9 % mělo vstupní hodnotu ECOG skóre 2. Do studie bylo zařazeno 269 pacientů s CLL. Na počátku studie mělo 45 % pokročilé klinické stadium (Rai Stage III nebo IV), 35 % pacientů mělo minimálně jeden tumor > 5 cm, 39 % se vstupní anemií, 23 % se vstupní trombocytopenií, 65 % mělo zvýšený P2 mikroglobulin > 3500 mcg/l, 47 % mělo CrCl < 60 ml/min a 20 % pacientů mělo del11q.
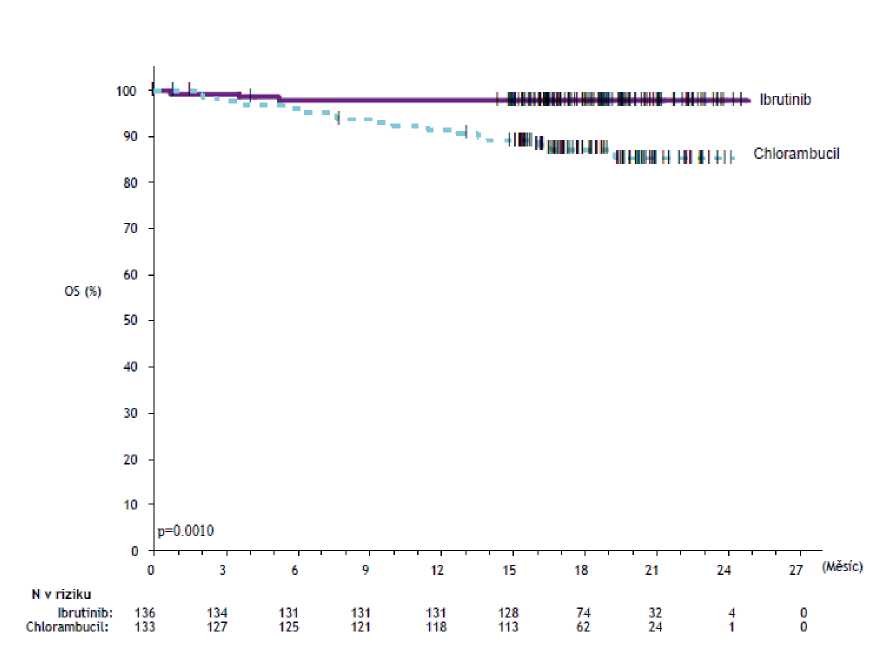
Přežití bez progrese (PFS) hodnocené nezávislou hodnotící komisí (IRC) podle kritérií International Workshop on CLL (IWCLL) prokázalo statisticky významné 84% snížení rizika úmrtí nebo progrese u pacientů v rameni s přípravkem IMBRUVICA. Výsledky účinnosti ve studii PCYC-1115-CA jsou uvedeny v tabulce 4 a Kaplan-Meierova křivka pro PFS a OS je jednotlivě uvedena na obrázku 2 a 3.
U ITT populace došlo ke statisticky významnému trvalému zlepšení krevních destiček nebo hemoglobinu ve prospěch ibrutinibu oproti chlorambucilu. U pacientů s výchozí cytopenií bylo trvalé hematologické zlepšení: krevní destičky 77,1 % oproti 42,9 %; hemoglobin 84,3 % oproti 45,5 % pro inbrutinib a chlorambucil, v uvedeném pořadí.
Tabulka 4: Výsledky účinnosti ve studii PCYC-1115-CA
|
Cílový parametr |
IMBRUVICA N = 136 |
Chlorambucil N = 133 |
|
Přežití bez progrese3 | ||
|
Počet případů (%) |
15 (11,0) |
64 (48,1) |
|
Medián (95% CI), měsíce |
nedosaženo |
18,9 (14,1; 22,0) |
|
HRb (95% CI) |
0,161 (0,091; 0,283) | |
|
Celková léčebná odpověď11 (CR + PR) |
82,4% |
35,3% |
|
P-hodnota |
<0,0001 | |
|
Celkové přežití' | ||
|
Počet úmrtí (%) |
3 (2,2) |
17 (12,8) |
|
HR (95% CI) |
0,163 (0,048; 0,558) | |
a
b
hodnocení IRC, medián sledování 18,4 měsíců; HR = poměr rizika;
c
Medián OS nebyl dosažen u obou ramen. p < 0,005 pro OS.
Obrázek 2: Kaplan-Meierova křivka přežití bez progrese (populace ITT) ve studii
PCYC-1115-CA
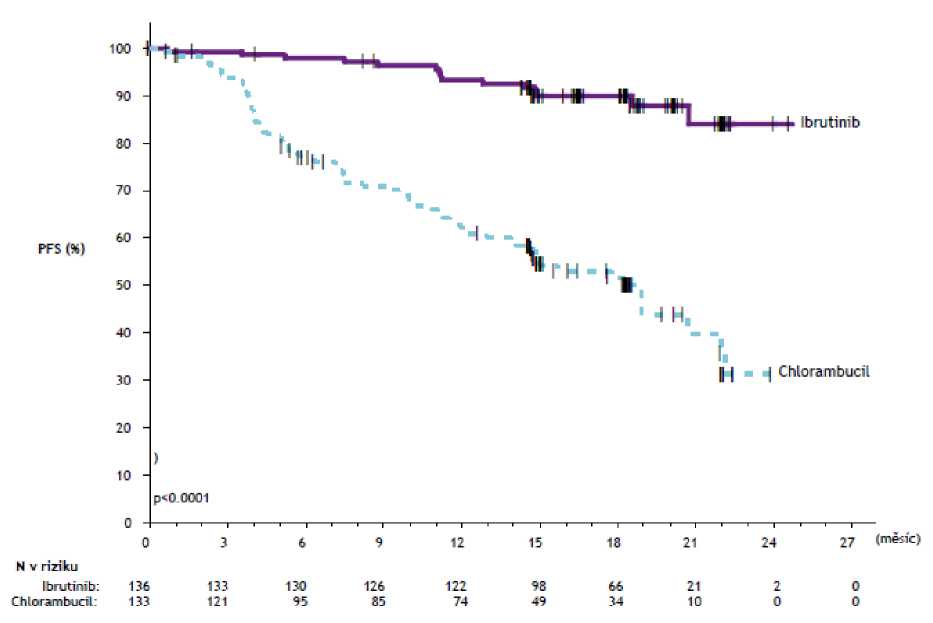
Obrázek 3: Kaplan-Meierova křivka celkového přežití (populace ITT) ve studii PCYC-1115-CA
Pacienti s CLL, kteří dostali minimálně jednu předchozí léčbu
Bezpečnost a účinnost přípravku IMBRUVICA u pacientů s CLL byla prokázána v jedné nekontrolované studii a v jedné randomizované kontrolované studii. Otevřená multicentrická studie (PCYC-1102-CA) zahrnovala 51 pacientů s relabujícím nebo refrakterním CLL, kterým byla podávána dávka 420 mg jednou denně. Přípravek IMBRUVICA byl podáván až do progrese onemocnění nebo nepřijatelné toxicity. Medián věku činil 68 let (rozsah: 37 až 82 let), medián doby od diagnózy činil 80 měsíců a medián počtu předchozích terapií činil 4 terapie (rozsah: 1 až 12 terapií), včetně 92,2 % pacientů s předchozí léčbou nukleosidovým analogem, 98,0 % s předchozí léčbou rituximabem, 86,3 % s předchozí léčbou alkylačním činidlem, 39,2 % s předchozí léčbou bendamustinem a 19,6 % s předchozí léčbou ofatumumabem. Na počátku mělo 39,2 % pacientů klinické stadium Rai IV; 45,1 % pacientů mělo bulky disease (> 5 cm), 35,3 % mělo del17p a 31,4% mělo del11q.
Celkový výskyt léčebné odpovědi (ORR) byl hodnocen podle kritérií 2008 IWCLL zkoušejícími a nezávislou hodnotící komisí (IRC). Celkový výskyt léčebné odpovědi (ORR) hodnocený pomocí nezávislé hodnotící komise (IRC) při následném sledování s mediánem doby 16,4 měsíců u 51 pacientů s relabujícím nebo refrakterním onemocněním byl 64,7 % (95 % CI: 50,1 %, 77,6 %), ve všech případech se jednalo o parciální odpověď. Celkový výskyt léčebné odpovědi (ORR) zahrnující výskyt částečných odpovědí (PR) s lymfocytózou byl 70,6 %. Medián doby do odpovědi činil 1,9 měsíce. Hodnota DOR se pohybovala v rozmezí 3,9 až 24,2 měsíce. Mediánu DOR nebylo dosaženo.
U pacientů s relabujícím nebo refrakterním CLL byla provedena randomizovaná, multicentrická, otevřená studie fáze 3 hodnotící přípravek IMBRUVICA oproti ofatumumabu (PCYC-1112-CA). Pacienti (n = 391) byli randomizováni v poměru 1:1 k léčbě buď přípravkem IMBRUVICA 420 mg denně až do progrese onemocnění nebo nepřijatelné toxicity nebo ofatumumabem po dobu až 12 dávek (300/2 000 mg). Padesát sedm (57) pacientů randomizovaných k léčbě ofatumumabem bylo po progresi převedeno na přípravek IMBRUVICA. Medián věku činil 67 let (rozsah: 30-88 let), 68 % účastníků byli muži a 90 % účastníků bylo bílé (europoidní) rasy. Všichni pacienti měli vstupní hodnotu ECOG performance status 0 nebo 1. Medián doby od diagnózy činil 91 měsíců a medián počtu předchozích terapií činil 2 terapie (rozsah: 1 až 13 terapií). Na počátku studie mělo 58 % pacientů minimálně jeden tumor > 5 cm. Třicet dva procent (32 %) pacientů mělo deleci 17p a 31 % mělo deleci 11q.
Přežití bez progrese (PFS) hodnocené nezávislou hodnotící komisi (IRC) podle kritérii IWCLL prokázalo statisticky významné 78 % snížení rizika úmrtí nebo progrese u pacientů v rameni užívajícím přípravek IMBRUVICA. Analýza celkového přežití (OS) prokázala statisticky významné 57 % snížení rizika úmrtí u pacientů v rameni léčeném přípravkem IMBRUVICA. Výsledky účinnosti ve studii PCYC-1112-CA jsou uvedeny v tabulce 5.
Tabulka 5: Výsledky účinnosti u pacientů s chronickou lymfocytární leukemií (studie _PCYC-1112-CA)_
|
Cílový parametr |
IMBRUVICA N = 195 |
Ofatumumab N = 196 |
|
Medián doby přežití bez progrese |
Nedosaženo |
8,1 měsíců |
|
HR = 0,215 [95 %CI: 0,146; 0,3171 | ||
|
Celkové přežití3 |
HR= 0,434 [95 % CI: 0,238; 0,789]b HR= 0,387 [95 % CI: 0,216; 0,6951c | |
|
Celkový výskyt léčebné odpovědi4 e (%) |
42,6 |
4,1 |
|
Celkový výskyt léčebné odpovědi včetně PR s lymfocytózoud (%) |
62,6 |
4,1 |
a
b
Medián OS nebyl dosažen u obou ramen. p < 0,005 pro OS.
Pacienti randomizovaní k léčbě ofatumumabem byli v případě potřeby při zahájení léčby přípravkem IMBRUVICA vyloučeni.
Analýza senzitivity, ve které nebyli pacienti převedení z ramene s ofatumumabem vyloučeni k datu první dávky přípravku IMBRUVICA.
Podle IRC. Ke konfirmaci odpovědi je zapotřebí opakované CT vyšetření.
Všechny dosažené PR; p < 0,0001 pro ORR.
c d
e
Účinnost byla obdobná v rámci všech zkoumaných podskupin, včetně pacientů s delecí a bez delece 17p, předem stanoveným stratifikačním faktorem (tabulka 6).
Tabulka 6: Analýza podskupiny s ohledem na přežití bez progrese (Studie PCYC-1112-CA)
|
N |
Poměr rizika |
95 % CI | |
|
Všichni jedinci |
391 |
0,210 |
(0,143; 0,308) |
|
Delece17P Ano |
127 |
0,247 |
(0,136; 0,450) |
|
Ne |
264 |
0,194 |
(0,117; 0,323) |
|
Refrakterní onemocnění na léčbu na bázi purinových analogů Ano |
175 |
0,178 |
(0,100; 0,320) |
|
Ne |
216 |
0,242 |
(0,145; 0,404) |
|
Věk <65 |
152 |
0,166 |
(0,088; 0,315) |
|
>65 |
239 |
0,243 |
(0,149; 0,395) |
|
Počet předchozích léčení <3 |
198 |
0,189 |
(0,100; 0,358) |
|
>3 |
193 |
0,212 |
(0,130; 0,344) |
|
Bulky disease < 5 cm |
163 |
0,237 |
(0,127; 0,442) |
|
> 5 cm |
225 |
0,191 |
(0,117; 0,311) |
Na obrázku 4 jsou znázorněny Kaplan-Meierovy křivky pro PFS.
Obrázek 4: Kaplan-Meierova křivka přežití bez progrese (populace ITT) ve studii PCYC-1112-CA
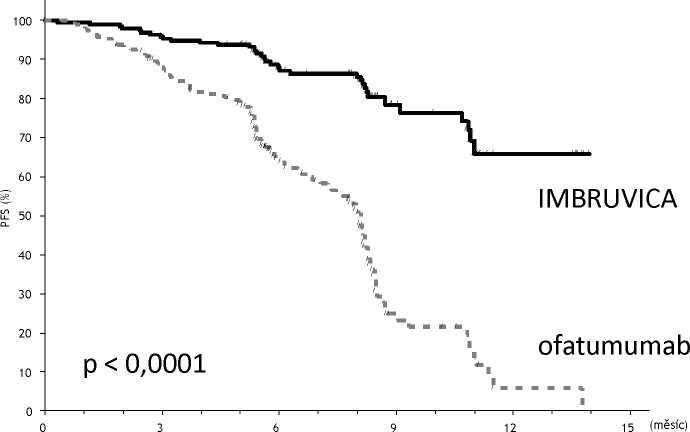
Počet pacientů v ohrožení
Ibrutinib: 195 183 116 38
Ofatumumab: 196 161 83 15
7
1 0
Kombinovaná léčba
Bezpečnost a účinnost přípravku IMBRUVICA u pacientů, po předchozí léčbě CLL, byly dále hodnoceny v randomizované multicentrické dvojitě zaslepené studii fáze 3 s přípravkem IMBRUVICA v kombinaci s BR ve srovnání s kombinací placebo + BR (studie CLL3001). Pacienti (n = 578) byli randomizováni v poměru 1:1 do skupiny k léčbě přípravkem IMBRUVICA 420 mg užívaným denně nebo placebem v kombinaci s BR až do progrese onemocnění nebo rozvoje nepřijatelné toxicity. Všichni pacienti dostávali BR v maximálně šesti 28denních cyklech.
Bendamustin byl podáván v dávce 70 mg/m2 intravenózně po dobu 30 minut v cyklu 1, v dnech 2 a 3 a v cyklech 2 až 6 v dnech 1 a 2. Celkem bylo podáno maximálně 6 cyklů. Rituximab byl podáván v dávce 375 mg/m2 první den prvního cyklu a v dávce 500 mg/m2 vždy první den v cyklech 2 až 6. 90 pacientů randomizovaných do skupiny placebo + BR přešlo do skupiny léčené přípravkem IMBRUVICA v návaznosti na progresi potvrzenou IRC. Medián věku byl 64 let (rozmezí 31 až 86 let), 66 % pacientů byli muži, 91 % bylo bílé (europoidní) rasy. Všichni pacienti měli vstupní hodnotu ECOG performance status 0 nebo 1. Medián doby od diagnózy činil 6 let, medián počtu předchozích terapií činil 2 terapie (rozmezí: 1 až 11). Při vstupu mělo 56 % pacientů minimálně jeden tumor > 5 cm, 26 % mělo deleci 11q.
Přežití bez progrese (PFS) hodnotila nezávislá hodnotící komise (IRC) dle kritérií IWCLL. Výsledky účinnosti ve studii CLL3001 jsou uvedenyv tabulce 7.
Tabulka 7: Výsledky účinnosti u pacientů s chronickou lymfocytární leukemií (studie
CLL3001)
|
Cílový parametr |
IMBRUVICA + BR N = 289 |
Placebo + BR N = 289 |
|
Přežití bez progrese | ||
|
Medián (95% CI), měsíce |
Nedosaženo |
13,3 (11,3, 13,9) |
|
HR= 0,203 [95% CI: 0,150, 0,2761 | ||
|
Celkový výskyt léčebné odpovědi a % |
82,7 |
67,8 |
|
Celkové přežití (OS)b |
HR= 0,628 [95% CI: 0,385, 1,0241 | |
a
b
Hodnoceno IRC, ORR (CR, Cri, nPR, PR) Medián OS nebyl dosažen u obou ramen
WaldenstrOmova makroglobulinemie (WM)
Bezpečnost a účinnost přípravku IMBRUVICA u WM (lymfoplazmocytámí lymfom vylučující IgM) byla vyhodnocena v otevřené, multicentrické, jednoramenné studii u 63 již léčených pacientů_Medián věku činil 63 let (rozsah: 44 až 86 let), 76 % účastníků byli muži a 95 % účastníků bylo bílé (europoidní) rasy. Všichni pacienti měli vstupní ECOG performance status 0 nebo 1. Medián doby od diagnózy činil 74 měsíců a medián počtu předchozích terapií činil 2 terapie (rozsah: 1 až 11 terapií). Na počátku studie byl medián hodnoty IgM v séru 3,5 g/dl a 60 % pacientů bylo anemických (hodnota hemoglobinu <11 g/dl nebo 6,8 mmol/l).
Přípravek IMBRUVICA byl podáván perorálně v dávce 420 mg jednou denně až do progrese onemocnění nebo nepřijatelné toxicity. Primárním cílovým parametrem v této studii byl celkový výskyt léčebné odpovědi (ORR) hodnocený zkoušejícím. ORR a DOR byly hodnoceny podle kritérií z Third International Workshop of Waldenstrom’s macroglobulinaemia. Odpovědi na přípravek IMBRUVICA jsou uvedeny v tabulce 8.
Tabulka 8: Celkový výskyt léčebné odpovědi (ORR) a doba trvání odpovědi (DOR) u
pacientů s WM
|
Total (N = 63) | |
|
ORR (%) |
87,3 |
|
95 % CI (%) |
(76,5; 94,4) |
|
VGPR (%) |
14,3 |
|
PR (%) |
55,6 |
|
MR (%) |
17,5 |
|
Medián DOR v měsících (rozsah) |
NR (0,03+, 18,8+) |
CI = interval spolehlivosti; NR = nebylo dosaženo ; MR = malá léčebná odpověď; PR = částečná odpověď; VGPR = velmi dobrá částečná odpověď; ORR = MR + PR + VGPR
Medián doby do odpovědi byl 1,0 měsíce (rozsah: 0,7 až 13,4 měsíce).
Výsledky účinnosti léčby byly taktéž posouzenynezávislou hodnotící komisí (IRC) a ukázaly 83 % ORR, přičemž velmi dobrá částečná odpověď (VGPR) byla zaznamenána u 11 % a částečná odpověď (PR) u 51 % pacientů.
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem IMBRUVICA u všech podskupin pediatrické populace s MCL, CLL a lymfoplazmacytickým lymfomem (LPL) (informace o pediatrickém použití viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Absorpce
Ibrutinib je po perorálním podání rychle absorbován s mediánem hodnoty Tmax 1 až 2 hodiny. Absolutní biologická dostupnost na lačno (n = 8) byla 2,9 % (90 %CI = 2,1 - 3,9) a dvojnásobná v případě kombinace s jídlem. Farmakokinetika ibrutinibu se významně neliší u pacientů s různými B-buněčnými malignitami. Expozice ibrutinibu se zvyšuje s dávkou až do 840 mg. Hodnota AUC v rovnovážném stavu pozorovaná u pacientů při dávce 560 mg činila 953 ± 705 ng h/ml (průměr ± směrodatná odchylka). Podání ibrutinibu v lačném stavu vedlo přibližně k 60 % expozici (AUClast) v porovnání se stavem 30 minut před, 30 minut po (v sytém stavu) nebo 2 hodiny po snídani s vysokým obsahem tuků.
Distribuce
Reverzibilní vazba ibrutinibu na lidské plazmatické proteiny in vitro činila 97,3 %, bez závislosti na koncentraci v rozmezí od 50 do 1 000 ng/ml. Zdánlivý distribuční objem v rovnovážném stavu (Vd,ss/F) byl přibližně 10 000 l.
Metabolismus
Ibrutinib je primárně metabolizován enzymem CYP3A4 za vzniku dihydrodiolového metabolitu s inhibiční aktivitou vůči BTK přibližně 15krát nižší než je aktivita ibrutinibu. Podíl CYP2D6 na metabolizmu ibrutinibu je minimální.
Z tohoto důvodu nejsou nutná žádná bezpečnostní opatření u pacientů s různými genotypy CYP2D6. Eliminace
Zdánlivá clearance (CL/F) je přibližně 1 000 l/h. Poločas ibrutinibu je 4 až 13 hodin.
Po jednorázovém perorálním podání radioaktivně značeného [14C]-ibrutinibu zdravým jedincům bylo přibližně 90 % radioaktivity vyloučeno do 168 hodin, přičemž většina (80 %) se vylučuje stolicí a < 10 % močí. Nezměněný ibrutinib tvořil přibližně 1 % radioaktivně značeného exkrečního produktu ve stolici a v moči nebyl přítomen.
Zvláštní populace
Starší osoby
Populační farmakokinetika ukázala, že věk nemá významný vliv na clearance ibrutinibu z cirkulace.
Pediatrická populace
U pacientů mladších 18 let nebyly provedeny žádné farmakokinetické studie s přípravkem IMBRUVICA.
Pohlaví
Údaje z populační farmakokinetiky ukázaly, že pohlaví nemá významný vliv na clearance ibrutinibu z cirkulace.
Rasa
Nejsou k dispozici dostatečná data k vyhodnocení vlivu rasy na farmakokinetiku ibrutinibu.
Tělesná hmotnost
Údaje z populační farmakokinetiky ukazují, že tělesná hmotnost (rozsah: 41 až 146 kg; průměr [SD]): 83 [19 kg]) měla zanedbatelný vliv na clearance ibrutinibu.
Porucha funkce ledvin
Ibrutinib má minimální renální clearance; vylučování metabolitů močí činí < 10 % dávky. U pacientů s poruchou funkce ledvin nebyly provedeny žádné specifické studie. U pacientů se závažnou poruchou funkce ledvin nebo u pacientů na dialýze nejsou k dispozici žádné údaje (viz bod 4.2).
Porucha funkce jater
Ibrutinib je metabolizován v játrech. Byla provedena studie u neonkologických pacientů s poruchou funkce jater, kterým byla nalačno podána jednorázová dávka 140 mg léčivého přípravku. Vliv poruchy funkce jater se podstatně lišil mezi jednotlivci, ale v průměru bylo pozorováno 2,7násobné,
8,2násobné, respektive 9,8násobné zvýšení expozice (AUClast) ibrutinibu u subjektů s mírnou (n = 6, Child-Pugh třída A), středně závažnou (n = 10, Child-Pugh třída B) a závažnou (n = 8, Child-Pugh třída C) poruchou funkce jater. Volná frakce ibrutinibu se zvyšovala s mírou poruchy funkce, a dosáhla hodnoty 3,0, 3,8, resp. 4,8 % u pacientů s mírnou, středně závažnou a závažnou poruchou funkce jater oproti 3,3 % v plazmě odpovídajících zdravých kontrol v rámci této studie. Odpovídající zvýšení expozice (AUCunbound, last) nevázanému ibrutinibu u pacientů s mírnou, středně závažnou a závažnou poruchou funkce jater se odhaduje na 4,1násobek, 9,8násobek, resp. 13násobek původní hodnoty (viz bod 4.2).
Společné podání se substráty CYP
In vitro studie prokázaly, že ibrutinib je slabým reverzibilním inhibitorem CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 a intestinální (nikoli jaterní) CYP3A4 a neukazuje klinicky relevantní časově závislou inhibici CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19 a CYP2D6. Dihydrodiolový metabolit ibrutinibu je slabým inhibitorem CYP2B6, CYP2C8, CYP2C9 a CYP2D6. Dihydrodiolový metabolit je slabým induktorem izoenzymů CYP450 in vitro. Přestože je ibrutinib citlivým substrátem CYP3A4, nemá klinicky relevantní účinek na vlastní expozici.
Společné podání s transportními substráty/inhibitory
In vitro studie ukázaly, že ibrutinib není substrátem P-gp ani jiných významných transportérů vyjma OCT2. Dihydrodiolový metabolit a jiné metabolity jsou substráty P-gp. Ibrutinib je in vitro inhibitorem P-gp a BCRP (viz bod 4.5).
5.3 Předklinické údaje vztahující se k bezpečnosti
Níže uvedené nežádoucí účinky byly pozorovány ve studiích s délkou trvání 13 týdnů u potkanů a psů. Bylo zjištěno, že ibrutinib vyvolává gastrointestinální účinky (měkká stolice/průjem a/nebo zánět) a lymfoidní deplece u potkanů a psů léčených dávkou, při které ještě nebyl pozorován škodlivý účinek (NOAEL), odpovídající 30 mg/kg/den u obou těchto zvířecích druhů. Na základě průměrné expozice (AUC) při klinické dávce 560 mg/den činil poměr AUC 2,6 resp. 21 při podání dávky NOAEL (No Observed Adverse Effect Level) u samců a samic potkanů a 0,4 resp. 1,8 při podání dávky NOAEL u samců a samic psů. Nejnižší dávka s pozorovatelným negativním účinkem LOEL (Lowest Observed Effect Level) (60 mg/kg/den) u psa odpovídala 3,6násobku (samci) resp. 2,3násobku (samice) klinické dávky. U potkanů byla pozorována středně závažná atrofie acinárních buněk pankreatu (považována za nežádoucí účinek) při dávkách >100 mg/kg u potkaních samců (2,6x AUC expozice), zatímco u samic nebyla tato atrofie pozorována při dávkách až do 300 mg/kg/den (21,3násobné rozpětí AUC expozice). U samic potkanů, kterým byla podána dávka >100 mg/kg/den (20,3násobné rozpětí AUC expozice), byl pozorován mírný úbytek trabekulární a kortikální kosti. Všechny gastrointestinální, lymfatické a kostní potíže se upravily po rekonvalescenci v délce 6-13 týdnů. Nálezy na pankreatu se částečně upravily po srovnatelné rekonvalescenční periodě.
Studie juvenilní toxicity nebyly provedeny.
Karcinogenicita/genotoxicita
Studie karcinogenity u ibrutinibu nebyly provedeny.
Ibrutinib neměl genotoxické vlastnosti při experimentech na bakteriích, savčích buňkách nebo myších.
Reprodukční toxicita
U březích samic potkanů byl ibrutinib v dávce 80 mg/kg/den spojen se zvýšeným výskytem postimplantačních ztrát, viscerálních malformací (na úrovni srdce a velkých cév) a změn na skeletu při expozici odpovídající 14násobku AUC u pacientů s denní dávkou 560 mg. Při dávce > 40 mg/kg/den byl ibrutinib spojen se sníženou fetální tělesnou hmotností (poměr AUC > 5,6 v porovnání s denní dávkou 560 mg u pacientů). Prahová dávka bez nežádoucích účinků (NOAEL) pro plod tak činila 10 mg/kg/den (přibližně 1,3násobek AUC ibrutinibu při dávce 560 mg denně (viz bod 4.6).
U březích králíků byl ibrutinib v dávce 15 mg/kg/den nebo vyšší spojován se skeletálními malformacemi (fúze sterna) a ibrutinib v dávce 45 mg/kg/den byl spojován se zvýšenou post-implantační ztrátou. Ibrutinib v dávce 15 mg/kg/den způsobil malforamce u králíků (přibližně 2násobek AUC ibrutinibu u pacientů s MCL s denní dávkou 560 mg a 2,8násobek expozice u pacientů s CLL nebo WM užívající ibrutinib v denní dávce 420 mg). Prahová dávka bez nežádoucích účinků (NOAEL) pro plod tak činila 5 mg/kg/den (přibližně 0,7násobek AUC ibrutinibu při dávce 560 mg denně) (viz bod 4.6).
Fertilita
Nebyly pozorovány žádné účinky na fertilitu nebo reprodukční schopnost samců nebo samic potkanů při maximálních testovaných dávkách 100 mg/kg/den (Human Equivalent Dose [HED]16 mg/kg/den).
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Obsah tobolky
sodná sůl kroskarmelózy
magnesium-stearát
mikrokrystalická celulóza
natrium-lauryl-sulfát
Obal tobolky želatina
oxid titaničitý (E171)
Černý inkoust: šelak
černý oxid železitý (E172) propylenglykol
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
2 roky.
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
6.5 Druh obalu a obsah balení
HDPE lahvičky s polypropylenovým s dětským bezpečnostním uzávěrem.
Každá krabička obsahuje jednu lahvičku obsahující buď 90 nebo 120 tvrdých tobolek.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Janssen-Cilag International NV Turnhoutseweg 30 B-2340 Beerse Belgie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/14/945/001 (90 tvrdých tobolek)
EU/1/14/945/002 (120 tvrdých tobolek)
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 21. října 2014
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
PŘÍLOHA II
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného za propouštění šarží
Janssen Pharmaceutica NV Turnhoutseweg 30 B-2340 Beerse Belgie
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
• Povinnost uskutečnit poregistrační opatření
Držitel rozhodnutí o registraci uskuteční v daném termínu níže uvedená opatření:
|
Popis |
Termín splnění |
|
Předložení ročních aktualizovaných výsledků studie 1112 s ohledem na progresi a úmrtí - bude poskytováno až do dosažení zralosti (dat) v rameni s ibrutinibem, např. 70 %, a mělo by nejlépe zahrnovat rovněž PFS2, nebo alespoň dobu trvání následující léčby. |
4Q 2017 |
PŘÍLOHA III
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU KRABIČKA
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
IMBRUVICA 140 mg tvrdé tobolky ibrutinibum
2. OBSAH LÉČIVÉ LÁTKY / LÉČIVÝCH LÁTEK
Jedna tvrdá tobolka obsahuje ibrutinibum 140 mg.
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH BALENÍ
90 tvrdých tobolek 120 tvrdých tobolek
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Janssen-Cilag International NV Tumhoutseweg 30 B-2340 Beerse Belgie
12. REGISTRAČNÍ ČÍSLO(A)
EU/1/14/945/001 (90 tvrdých tobolek) EU/1/14/945/002 (120 tvrdých tobolek)
13. ČÍSLO ŠARŽE
Č. šarže
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
imbruvica
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
2D čárový kód s jedinečným identifikátorem.
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
PC:
SN:
NN:
ÚDAJE UVÁDĚNÉ NA VNITŘNÍM OBALU ŠTÍTEK NA LAHVIČCE
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
IMBRUVICA 140 mg tobolky ibrutinibum
2. OBSAH LÉČIVÉ LÁTKY / LÉČIVÝCH LÁTEK
Jedna tobolka obsahuje ibrutinibum 140 mg
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH BALENÍ
90 tobolek 120 tobolek
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Janssen-Cilag International NV Tumhoutseweg 30 B-2340 Beerse Belgie
12. REGISTRAČNÍ ČÍSLO(A)
EU/1/14/945/001 (90 tvrdých tobolek) EU/1/14/945/002 (120 tvrdých tobolek)
13. ČÍSLO ŠARŽE
Č. šarže
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
B. PŘÍBALOVÁ INFORMACE
IMBRUVICA 140 mg tvrdé tobolky
ibrutinibum
V Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
• Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
• Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka nebo zdravotní sestry.
• Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
• Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou v této příbalové informaci uvedeny. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek IMBRUVICA a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek IMBRUVICA užívat
3. Jak se přípravek IMBRUVICA užívá
4. Možné nežádoucí účinky
5. Jak přípravek IMBRUVICA uchovávat
6. Obsah balení a další informace
1. Co je přípravek IMBRUVICA a k čemu se používá Co je přípravek IMBRUVICA
IMBRUVICA je protinádorový lék, který obsahuje léčivou látku ibrutinib. Patří do skupiny léků nazývaných inhibitory proteinkinázy.
K čemu se přípravek IMBRUVICA používá
Používá se k léčbě následujících typů rakoviny krve u dospělých.
• lymfom z plášťových buněk (MCL), typ rakoviny postihující lymfatické uzliny, pokud se onemocnění vrátilo nebo nereagovalo na léčbu.
• chronická lymfocytární leukemie (CLL), typ rakoviny, který postihuje bílé krvinky označované jako lymfocyty, a zároveň i lymfatické uzliny. Přípravek IMBRUVICA se používá u pacientů
s dosud neléčenou CLL nebo v případě, že se onemocnění vrátilo nebo nereagovalo na léčbu.
• Waldenstromova makroglobulinemie (WM), typ rakoviny postihující bílé krvinky označované jako lymfocyty. Přípravek je používán v případě, že se onemocnění vrátilo nebo pokud nereagovalo na léčbu nebo u pacientů, u kterých není chemoterapie se současně podávanými protilátkami vhodnou léčbou.
Jak přípravek IMBRUVICA působí
U MCL, CLL a WM přípravek IMBRUVICA působí tak, že blokuje Brutonovu tyrokinázu, protein v lidském těle, který pomáhá těmto nádorovým buňkám přežívat a růst. Blokací tohoto proteinu pomáhá přípravek IMBRUVICA usmrcovat rakovinné buňky a snižovat jejich počet. Také zpomaluje zhoršování rakoviny.
2. Čemu musíte věnovat pozornost, než začnete přípravek IMBRUVICA užívat Neužívejte přípravek IMBRUVICA
• jestliže jste alergický(á) na ibrutinib nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
• jestliže užíváte rostlinný přípravek s obsahem třezalky tečkované, používaný k léčbě deprese. Pokud si nejste jistý(á), poraďte se před užitím tohoto přípravku se svým lékařem, lékárníkem nebo zdravotní sestrou.
Upozornění a opatření
Před použitím přípravku IMBRUVICA se poraďte se svým lékařem, lékárníkem nebo zdravotní sestrou:
• pokud se u Vás v minulosti vyskytly neobvyklé modřiny nebo krvácení, nebo pokud užíváte jakékoli léky nebo doplňky, které zvyšují riziko krvácení (viz bod „Další léčivé přípravky a přípravek IMBRUVICA“)
• pokud se u Vás v minulosti vyskytl nepravidelný srdeční rytmus (fibrilaci síní) nebo závažné srdeční selhání, které u Vás vyvolává dušnost nebo může vést k otokům nohou.
• pokud máte problémy s ledvinami nebo játry
• pokud jste v nedávné době podstoupil(a) operaci, zejména pokud tato operace mohla ovlivnit vstřebávání jídla nebo léků ze žaludku nebo střeva.
• pokud je u vás operace plánována, může Vás lékař požádat, abyste krátkodobě přerušil(a) užívání přípravku IMBRUVICA.
Pokud se Vás cokoli z výše uvedeného týká (nebo si nejste jistý/á), poraďte se před užitím tohoto léku se svým lékařem, lékárníkem nebo zdravotní sestrou.
Testy a kontrolní vyšetření před zahájením léčby a v jejím průběhu
Syndrom nádorového rozpadu (TLS): V průběhu léčby rakoviny, a v některých případech i mimo období léčby, může dojít k výskytu neobvyklých hladin chemických látek v krvi, které jsou způsobené rychlým rozpadem nádorových buněk. To může vést ke změnám funkce ledvin, abnormálnímu srdečnímu tepu, nebo křečím. Váš lékař nebo jiný zdravotnický pracovník Vám může udělat krevní testy pro kontrolu TLS.
Lymfocytóza: Laboratorní testy během prvních několika týdnů léčby mohou ukazovat zvýšený počet bílých krvinek (tzv. „lymfocytů“) v krvi. Tento stav se očekává a může trvat několik měsíců. Nemusí však nutně znamenat, že se rakovina krve zhoršuje. Váš lékař zkontroluje Váš krevní obraz před léčbou nebo v průběhu léčby a ve vzácných případech Vám podá jiný lék. Promluvte si se svým lékařem o tom, co vaše výsledky testů znamenají.
Děti a dospívající
Přípravek IMBRUVICA se nemá používat u dětí a dospívajících. Důvodem je, že v těchto věkových skupinách nebyl hodnocen.
Další léčivé přípravky a přípravek IMBRUVICA
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat. To platí i pro volně prodejné léky, rostlinné přípravky a doplňky stravy. Důvodem je, že přípravek IMBRUVICA může ovlivnit způsob, jakým účinkují některé další léky. Také některé jiné léky mohou ovlivnit účinek přípravku IMBRUVICA.
Přípravek IMBRUVICA může zvýšit riziko krvácení. To znamená, že byste měl(a) informovat svého lékaře, pokud užíváte jiné léky, které zvyšují riziko krvácení. Mezi tyto léky patří:
• kyselina acetylsalicylová a nesteroidní protizánětlivé látky (NSAID), jako je ibuprofen nebo naproxen
• léky na ředění krve, jako je warfarin, heparin nebo jiné léky k léčbě krevních sraženin
• doplňky, které mohou zvyšovat riziko krvácení, jako jsou rybí olej, vitamin E nebo lněné semínko.
Pokud se Vás cokoli z výše uvedeného týká (nebo si nejste jistý/á), poraďte se před užitím přípravku IMBRUVICA se svým lékařem, lékárníkem nebo zdravotní sestrou.
Informujte rovněž svého lékaře, pokud užíváte kterýkoli z následujících léků - účinky přípravku IMBRUVICA nebo jiných přípravků mohou být ovlivněny, pokud užíváte přípravek IMBRUVICA spolu s některým z následujících přípravků:
• léky označované jako antibiotika používané k léčbě bakteriálních infekcí - klarithromycin, telithromycin, ciprofloxacin, erythromycin nebo rifampicin
• léky k léčbě plísňových infekcí - ketokonazol, itrakonazol, flukonazol nebo vorikonazol
• léky k léčbě HIV infekce - ritonavir, kobicistat, indinavir, nelfinavir, sachinavir, amprenavir, atazanavir, darunavir/ritonavir nebo fosamprenavir.
• léky k prevenci pocitu na zvracení a zvracení v souvislosti s chemoterapií - aprepitant.
• léky k léčbě deprese - nefazodon.
• léky označované jako inhibitory kináz používané k léčbě jiných zhoubných nádorů - krizotinib, nebo imatinib
• léky označované jako blokátory kalciového kanálu používané k léčbě vysokého krevního tlaku nebo bolesti na hrudi - diltiazem nebo verapamil
• léky k léčbě vysoké hladiny cholesterolu nazývané statiny - rosuvastatin
• léky k léčbě onemocnění srdce/antiarytmika - amiodaron nebo dronedaron
• léky používané k prevenci křečí, k léčbě epilepsie nebo léky k léčbě bolestivých stavů obličeje označovaných jako neuralgie trojklanného nervu - karbamazepin nebo fenytoin
Pokud se Vás cokoli z výše uvedeného týká (nebo si nejste jistý/á), poraďte se před užitím přípravku IMBRUVICA se svým lékařem, lékárníkem nebo zdravotní sestrou.
Jestliže užíváte digoxin, lék užívaný k léčbě problémů se srdcem, nebo methotrexát k léčbě jiných nádorů a ke snížení aktivity imunitního systému (např. na revmatoidní artritidu nebo lupénku), je nutné ho užívat minimálně 6 hodin před nebo po přípravku IMBRUVICA.
Přípravek IMBRUVICA s jídlem
Neužívejte přípravek IMBRUVICA s grapefruitem nebo plody pomerančovníku hořkého - tzn. vyvarujte se jejich konzumace, pití šťáv nebo užívání doplňků, které by je mohly obsahovat. Tyto produkty totiž mohou zvýšit množství přípravku IMBRUVICA v krvi.
Těhotenství, kojení a plodnost
Během léčby tímto přípravkem nesmíte otěhotnět. Pokud jste těhotná, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem, lékárníkem nebo zdravotní sestrou dříve, než začnete tento přípravek užívat.
Přípravek IMBRUVICA se nemá používat během těhotenství.
Neexistují žádné informace o bezpečnosti přípravku IMBRUVICA u těhotných žen.
Ženy v plodném věku musí používat vysoce účinnou metodu antikoncepce během léčby a tři měsíce po léčbě přípravku IMBRUVICA, aby nedošlo k otěhotnění během léčby přípravkem IMBRUVICA. Pokud užíváte hormonální antikoncepci, například antikoncepční pilulky nebo antikoncepční prostředky, je nezbytné používat také i bariérovou metodu antikoncepce (např. kondom).
• Pokud otěhotníte, okamžitě informujte svého lékaře.
• Během užívání tohoto léku nekojte.
Řízení dopravních prostředků a obsluha strojů
Po užití přípravku IMBRUVICA můžete pocit únavu nebo závratě, což může ovlivnit Vaši schopnost řídit dopravní prostředky nebo obsluhovat přístroje nebo strojní zařízení.
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře nebo lékárníka. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Jaké množství se užívá
Lymfom z plášťových buněk (MCL)
Doporučená dávka přípravku IMBRUVICA je čtyři tobolky (560 mg) jednou denně.
Chronická lymfocytární leukémie (CLL)/Waldenstromova makroglobulinemie (WM)
Doporučená dávka přípravku IMBRUVICA je tři tobolky (420 mg) jednou denně.
Váš lékař může dávku upravit.
Jak se přípravek užívá
• Tobolky se užívají perorálně (ústy) a zapíjí se sklenicí vody.
• Užívejte tobolky přibližně ve stejnou dobu každý den.
• Tobolky se polykají celé. Tobolky neotevírejte, nedrťte a nekousejte.
Jestliže jste užil(a) více přípravku IMBRUVICA, než jste měl(a)
Jestliže užijete více přípravku IMBRUVICA, než jste měl(a), poraďte se s lékařem nebo neprodleně navštivte nemocnici. Tobolky a tuto příbalovou informaci vezměte s sebou.
Jestliže jste zapomněl(a) užít přípravek IMBRUVICA
• Pokud vynecháte dávku, užijte ji co nejdříve tentýž den, a počínaje následujícím dnem se vraťte k normálnímu rozvrhu užívání.
• Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku.
• Pokud si nejste jistý(á), poraďte se s lékařem, lékárníkem nebo zdravotní sestrou, kdy máte užít následující dávku.
Jestliže jste přestal(a) užívat přípravek IMBRUVICA
Nepřestávejte užívat tento přípravek, pokud Vás k tomu nevyzve Váš lékař.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
U tohoto přípravku se mohou vyskytnout následující nežádoucí účinky:
Jestliže zaznamenáte některý z následujících nežádoucích účinků, přestaňte užívat přípravek IMBRUVICA a neprodleně informujte lékaře:
svědivá vystouplá vyrážka, dýchací obtíže, otok obličeje, rtů, jazyka nebo hrdla - můžete mít alergickou reakci na tento lék.
Jestliže zaznamenáte některý z následujících nežádoucích účinků, informujte neprodleně svého lékaře.
Velmi časté (mohou postihnout více než 1 osobu z 10)
• horečka, zimnice, bolest těla, pocit únavy, nachlazení nebo příznaky podobné chřipce, dušnost -může se jednat o příznaky infekce (virové, bakteriální nebo plísňové). Ty by mohly zahrnovat infekce nosu, vedlejších nosních dutin nebo krku (infekce horních cest dýchacích), nebo plic, nebo kůže.
• tvorba modřin nebo zvýšený sklon k tvorbě modřin.
Časté (mohou postihnout více než 1 osobu ze 100)
• závažné infekce v celém těle (sepse)
• infekce močových cest
• krvácení z nosu, malé červené nebo fialové skvrny způsobené krvácením pod kůži
• krev ve stolici nebo v moči, silnější menstruační krvácení, krvácení ze zranění, které nelze zastavit, zmatenost, bolest hlavy s poruchou řečí nebo pocit na omdlení - může se jednat o příznaky závažného vnitřního krvácení do žaludku, střev nebo mozku.
• zrychlený srdeční tep, vynechávání srdeční činnosti, slabý nebo nerovnoměrný puls (příznaky fibrilace síní)
• zvýšený počet nebo podíl bílých krvinek prokázaný v krevních testech
• nízký počet bílých krvinek s horečkou (febrilní neutropenie)
• v průběhu léčby rakoviny, a v některých případech i mimo období léčby, může dojít k výskytu neobvyklých hladin chemických látek v krvi, které jsou způsobené rychlým rozpadem nádorových buněk (syndrom nádorového rozpadu)
• nemelanomová rakovina kůže, nejčastěji rakovina skvamózních buněk a rakovina bazálních kožních buněk
• pocit závratě
• rozmazané vidění
• krvácení z nosu
• malé červené nebo fialové skrvny způsobené krvácením pod kůži
• vysoký krevní tlak
• zčervenání kůže
• vysoká hladina „kyseliny močové“ v krvi, která může způsobit dnu - prokázaná v krevních testech
• zánět v plicích, který může vést až k trvalému poškození plic.
Méně časté (mohou postihnout více než 1 osobu z 1 000)
• závažné zvýšení počtu bílých krvinek, které může způsobit jejich shlukování
• alergická reakce, v některých případech závažná, která může zahrnovat otok obličeje, rtů, úst, jazyka nebo krku, obtíže s polykáním nebo dýcháním, svědivá vyrážka (kopřivka).
Jiné velmi časté nežádoucí účinky
• afty
• bolest hlavy
• zácpa
• nevolnost (pocit na zvracení nebo zvracení)
• průjem - může být nezbytné, aby Vám lékař doplnil tekutiny nebo soli nebo podal jiný lék
• kožní vyrážka
• bolest rukou nebo nohou
• bolest zad nebo kloubů
• svalové křeče, bolesti nebo spasmy
• nízký počet buněk, které se podílejí na tvorbě krevní sraženiny (krevní destičky), velmi nízký počet bílých krvinek - prokázané v krevních testech
• otok rukou, kotníků nebo nohou.
Není známo (z dostupných údajů nelze určit)
• jaterní selhání
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na štítku, krabičce, lahvičce za označením „Použitelné do:“. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Tento přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co přípravek IMBRUVICA obsahuje
• Léčivou látkou je ibrutinibum. Jedna tvrdá tobolka obsahuje 140 mg ibrutinibum.
• Dalšími složkami jsou:
- Obsah tobolky: sodná sůl kroskarmelózy, magnesium-stearát, mikrokrystalická celulóza a natrium-lauryl-sulfát.
- Obal tobolky: želatina a oxid titaničitý
- Potiskový inkoust: šelak, černý oxid železitý (E172) a propylenglykol.
Jak přípravek IMBRUVICA vypadá a co obsahuje toto balení
Přípravek IMBRUVICA jsou bílé neprůhledné tvrdé tobolky označené na jedné straně černým potiskem „ibr 140 mg“.
Tobolky se dodávají v plastové lahvičce s polypropylenovým dětským bezpečnostním uzávěrem. Každá lahvička obsahuje buď 90 nebo 120 tobolek. Každé balení obsahuje po jedné lahvičce.
Držitel rozhodnutí o registraci
Janssen-Cilag International NV Turnhoutseweg 30 B-2340 Beerse Belgie
Výrobce
Janssen Pharmaceutica NV Turnhoutseweg 30 2340 Beerse Belgie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgie/Belgique/Belgien
Janssen-Cilag NV Antwerpseweg 15-17 B-2340 Beerse Tel/Tél: +32 14 64 94 11
Lietuva
UAB „JOHNSON & JOHNSON Geležinio Vilko g. 18A LT-08104 Vilnius Tel: +370 5 278 68 88
Luxembourg/Luxemburg
Janssen-Cilag Antwerpseweg 15-17 B-2340 Beerse Belgique/Belgien Tél/Tel: +32 14 649 411
Eunrapnu
„fl^OHCbH & fl^OHCtH Etnrapna” EOOfl
^.k. Mnagocr 4
En3Hec napK Co$na, crpaga 4
Co^na 1766
Ten.: +359 2 489 94 00
|
Česká republika Janssen-Cilag s.r.o. Karla Engliše 3201/06 CZ-150 00 Praha 5 - Smíchov Tel. +420 227 012 227 |
Magyarország Janssen-Cilag Kft. Nagyenyed u. 8-14 H-Budapest, 1123 Tel.: +36 1 884 2858 |
|
Danmark Janssen-Cilag A/S Bregneredvej 133 DK-3460 Birkered Tlf: +45 45 94 82 82 |
Malta Am Mangion Ltd. Mangion Building, Triq Gdida fi Triq Valletta MT-Hal-Luqa LQA 6000 Tel: +356 2397 6000 |
|
Deutschland Janssen-Cilag GmbH Johnson & Johnson Platz 1 D-41470 Neuss Tel: +49 2137 955 955 |
Nederland Janssen-Cilag B.V. Dr. Paul Janssenweg 150 NL-5026 RH Tilburg Tel: +31 13 583 73 73 |
|
Eesti UAB „JOHNSON & JOHNSON“ Eesti filiaal LSStsa 2 EE-11415 Tallinn Tel: +372 617 7410 |
Norge Janssen-Cilag AS Postboks 144 NO-1325-Lysaker Tlf: +47 24 12 65 00 |
|
EXláSa Janssen-Cilag Oap^aKerniK^ A.E.B.E. Aero^ópog Erp^vn? 56 GR-151 21 neÚKn, A0^va T^: +30 210 80 90 000 |
Osterreich Janssen-Cilag Pharma GmbH VorgartenstraBe 206B A-1020 Wien Tel: +43 1 610 300 |
|
Espaňa Janssen-Cilag, S.A. Paseo de las Doce Estrellas, 5-7 E-28042 Madrid Tel: +34 91 722 81 00 |
Polska Janssen-Cilag Polska Sp. z o.o. ul. Uzecka 24 PL-02-135 Warszawa Tel.+48 22 237 60 00 |
|
France Janssen-Cilag 1, rue Camille Desmoulins, TSA 91003 F-92787 Issy Les Moulineaux, Cedex 9 Tél: 0 800 25 50 75 / +33 1 55 00 40 03 |
Portugal Janssen-Cilag Farmaceutica, Lda. Estrada Consiglieri Pedroso, 69 A Queluz de Baixo PT-2734-503 Barcarena Tel: +351 21 43 68 835 |
|
Hrvatska Johnson & Johnson S.E. d.o.o. Oreškoviceva 6H HR-10010 Zagreb Tel: + 385 1 66 10 700 |
Románia Johnson & Johnson Románia SRL Strada Tipografilor Nr. 11-15 Cládirea S-Park, Corp B3-B4, Etaj 3 013714 Bucure§ti, ROMANIA Tel: +40 21 207 18 00 |
|
Ireland Janssen-Cilag Ltd. 50-100 Holmers Farm Way High Wycombe Buckinghamshire HP12 4EG United Kingdom Tel: +44 1494 567 567 |
Slovenija Johnson & Johnson d.o.o. Šmartinska cesta 53 SI-1000 Ljubljana Tel: +386 1 401 18 30 |
|
Ísland Janssen-Cilag AB c/o Vistor hf. Horgatúni 2 IS-210 Garóab^r Sími: +354 535 7000 |
Slovenská republika Johnson & Johnson, s.r.o. CBC III, Karadžičova 12 SK-821 08 Bratislava Tel: +421 232 408 400 |
|
Italia Janssen-Cilag SpA Via M.Buonarroti, 23 I-20093 Cologno Monzese MI Tel: +39 02 2510 1 |
Suomi/Finland Janssen-Cilag Oy Vaisalantie/Vaisalavágen 2 FI-02130 Espoo/Esbo Puh/Tel: +358 207 531 300 |
|
Kúnpoq BapváPag XaxZnnavay^g AxS Asro^ópog riávvou KpavrSrróxn 226 Aaxorá CY-2234 AsuK^oía T^: ++357 22 207 700 |
Sverige Janssen-Cilag AB Box 4042 SE-16904 Solna Tel: +46 8 626 50 00 |
|
Latvija UAB „JOHNSON & JOHNSON filiale Latvija Mukusalas iela 101 Riga, LV-1004 Tel: +371 678 93561 |
United Kingdom Janssen-Cilag Ltd. 50-100 Holmers Farm Way High Wycombe Buckinghamshire HP12 4EG - UK Tel: +44 1494 567 444. |
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
38