Ikervis 1 Mg/Ml
SOUHRN ÚDAJŮ O PŘÍPRAVKU
PŘÍLOHA I
IKERVIS 1 mg/ml oční kapky, emulze
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden ml emulze obsahuje ciclosporinum 1 mg (ciclosporin).
Pomocná látka se známým účinkem:
Jeden ml emulze obsahuje 0,05 mg cetalkonium-chloridu (viz bod 4.4). Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Oční kapky, emulze. Mléčně bílá emulze.
4. KLINICKÉ ÚDAJE
4.1 Terapeutická indikace
Léčba závažné keratitidy u dospělých pacientů se syndromem suchého oka, který se nezlepšil navzdory léčbě umělými slzami (viz bod 5.1).
4.2 Dávkování a způsob podání
Léčbu přípravkem IKERVIS musí zahájit oftalmolog nebo zdravotník s odbornou kvalifikací v očním lékařství.
Dávkování
Dospělí
Doporučená dávka je jedna kapka přípravku IKERVIS do postiženého oka(očí) jednou denně večer před spaním.
Odpověď na léčbu je nutné přehodnotit nejméně jednou za 6 měsíců.
Pokud je dávka vynechána, léčba pokračuje následující den jako obvykle. Pacienty je zapotřebí poučit, aby nepodávali více než jednu kapku do postiženého oka(očí).
Starší pacienti
Populace starších pacientů byla v klinických studiích hodnocena. Není zapotřebí žádná úprava dávek.
Pacienti s poruchou funkce ledvin nebo jater
Účinek přípravku IKERVIS nebyl u pacientů s poruchou funkce jater nebo ledvin studován. U těchto populací však nejsou třeba žádná zvláštní opatření.
Pediatrická populace
Použití přípravku IKERVIS u dětí a dospívajících ve věku do 18 let není relevantní při léčbě závažné keratitidy u pacientů se syndromem suchého oka, který se nezlepšil navzdory léčbě umělými slzami.
Způsob podání Oční podání.
Opatření, která je nutno učinit před podáním léčivého přípravku Pacienty je nutné upozornit, aby si nejprve omyli ruce.
Před podáním je zapotřebí jednodávkovým obalem jemně zatřepat.
Pouze pro jednorázové použití. Jedna jednodávková nádobka dostačuje na léčbu obou očí. Jakoukoliv nepoužitou emulzi je nutno ihned zlikvidovat.
Pacienty je nutné upozornit, aby používali nasolakrimální okluzi a zavřeli víčka na 2 minuty po vkápnutí do oka, aby se snížila systémová absorpce. Omezí se tak rovněž celkové nežádoucí účinky a zvýší se lokální účinnost přípravku (viz bod 4.4).
Jestliže se používá více než jeden lokálně aplikovaný oftalmologický léčivý přípravek, léčivé přípravky se musí podávat jednotlivě v rozpětí nejméně 15 minut po sobě. Přípravek IKERVIS je zapotřebí podávat poslední (viz bod 4.4).
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Aktivní okulární či periokulární infekce či podezření na ni.
4.4 Zvláštní upozornění a opatření pro použití
Přípravek IKERVIS nebyl studován u pacientů s herpes zoster oftalmicus v anamnéze, a proto by se těchto pacientů měl používat s opatrností.
Kontaktní čočky
Pacienti s kontaktními čočkami nebyli studováni. Doporučuje se pečlivě sledovat pacienty se závažnou keratitidou. Kontaktní čočky je třeba před instilací očních kapek večer před usnutím vyjmout a po probuzení je možné je opět nasadit.
Souběžná terapie
Zkušenosti s použitím přípravku IKERVIS při léčbě pacientů s glaukomem jsou omezené. Při souběžné léčbě těchto pacientů přípravkem IKERVIS je nutné postupovat opatrně zvláště u beta blokátorů, o nichž je známo, že snižují sekreci slz.
Účinky na. imunitní systém
Léčivé přípravky ovlivňující imunitní systém, včetně cyklosporinu, mohou ovlivňovat obranyschopnost hostitele proti infekcím a malignitám.
Společné podávání přípravku IKERVIS s očními kapkami obsahujícími kortikosteroidy by mohlo zesilovat účinky přípravku IKERVIS na imunitní systém (viz bod 4.5).
Pomocná látka
Přípravek IKERVIS obsahuje cetalkonium-chlorid, který může způsobit podráždění očí.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné studie interakcí s přípravkem IKERVIS.
Kombinace s jinými léčivými přípravky, které ovlivňují imunitní systém
Společné podávání přípravku IKERVIS s očními kapkami obsahujícími kortikosteroidy by mohlo zesilovat účinky cyklosporinu na imunitní systém (viz bod 4.4).
4.6 Fertilita, těhotenství a kojení
Ženy v reprodukčním věku/ženy používající antikoncepci
Podávání přípravku IKERVIS se u žen v reprodukčním věku, které nepoužívají účinnou antikoncepci, nedoporučuje.
Údaje o podávání přípravku IKERVIS těhotným ženám nejsou k dispozici.
Studie na zvířatech prokázaly reprodukční toxicitu po systémovém podání cyklosporinu při expozici dostatečně převyšující maximální expozici u člověka. To svědčí o malém významu při klinickém použití přípravku IKERVIS.
Podávání přípravku IKERVIS se v těhotenství nedoporučuje, pokud potenciální výhody pro matku nepřeváží nad riziky pro plod.
Kojení
Po perorálním podání se cyklosporin vylučuje do mateřského mléka. Informace o účincích cyklosporinu na novorozence/kojence jsou nedostatečné. Při terapeutických dávkách cyklosporinu v očních kapkách však není pravděpodobné, že by bylo v mateřském mléce obsaženo dostatečné množství. Na základě posouzení prospěšnosti kojení pro dítě a prospěšnosti léčby pro matku je nutno rozhodnout, zda přerušit kojení nebo ukončit/přerušit podávání přípravku IKERVIS.
Fertilita
Údaje o účincích přípravku IKERVIS na lidskou fertilitu nejsou k dispozici.
U zvířat, která dostávala cyklosporin intravenózně, nebylo hlášeno žádné zhoršení fertility (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek IKERVIS má mírný vliv na schopnost řídit nebo obsluhovat stroje.
Tento léčivý přípravek může vyvolat dočasně rozmazané vidění nebo jiné poruchy vidění, které mohou ovlivňovat schopnost řídit nebo obsluhovat stroje (viz bod 4.8). Pacienty je proto zapotřebí upozornit, aby neřídili ani neobsluhovali stroje, dokud se jim nevrátí ostré vidění.
4.8 Nežádoucí účinky
Shrnutí bezpečnostního profilu
V pěti klinických studiích zahrnujících 532 pacientů, kteří dostávali přípravek IKERVIS, a 398 pacientů, kteří dostávali vehikulum přípravku IKERVIS (kontrola), byl přípravek IKERVIS podáván nejméně jednou denně do obou očí až po dobu jednoho roku. Nejčastějšími nežádoucími účinky byly bolest oka (19,2 %), podráždění oka (17,8 %), slzení (6,4 %), oční hyperémie (5,5 %) a erytém očního víčka (1,7 %), které byly obvykle přechodné a vyskytovaly se během instilace.
Většina nežádoucích účinků hlášených v klinických studiích s použitím přípravku IKERVIS byla očních a měla slabou až mírnou závažnost.
Tabulkový seznam nežádoucích účinků
V klinických studiích byly pozorovány následující nežádoucí účinky uvedené níže. Nežádoucí účinky jsou seřazeny podle orgánových systémů a četnosti při použití následujících kategorií: velmi časté (>1/10); časté (>1/100 až <1/10); méně časté (>1/1 000 až <1/100); vzácné (>1/10 000 až <1/1 000); velmi vzácné (<1/10 000), není známo (z dostupných údajů nelze určit).
|
Infekce a infestace |
Méně časté |
Bakteriální keratitida, herpes zoster ophthalmicus, |
|
Poruchy oka |
Časté |
Erytém očního víčka, zvýšené slzení, oční hyperémie, rozmazané vidění, edém očních víček, hyperémie spojivek, podráždění oka, bolest oka. |
|
Méně časté |
Edém spojivek, porucha slzení, výtok z očí, oční pruritus, podráždění spojivek, konjunktivitida, pocit cizího tělesa v oku, oční depozitum, keratitida, blefaritida, dekompenzace rohovky, chalazion, infiltráty rohovky, zjizvení rohovky, pruritus očního víčka, iridocyklitida. |
|
Celkové poruchy a |
Velmi časté |
Bolest v místě instilace |
|
reakce v místě aplikace |
Časté |
Iritace v místě instilace, erytém v místě instilace, slzení v místě instilace. |
|
Méně časté |
Reakce v místě instilace, diskomfort v místě instilace, pruritus v místě instilace, pocit přítomnosti cizího tělesa v místě instilace. |
Popis vybraných nežádoucích účinků
Bolest v místě instilace byla často hlášenými lokálním nežádoucím účinkem spojeným s používáním přípravku IKERVIS během klinických studií. Pravděpodobně ji lze připsat na vrub cyklosporinu.
V jednom případě byla hlášena závažná eroze rohovkového epitelu, kterou zkoušející označil jako dekompenzaci rohovky a která se vyléčila bez následků.
Pacienti, kteří podstupují imunosupresivní terapie včetně podávání cyklosporinu, jsou ve zvýšené míře ohroženi infekcemi. Mohou se vyskytovat jak generalizované, tak lokalizované infekce. Mohou se také zhoršit již dříve existující infekce (viz bod 4.3). Případy infekcí byly méně často hlášeny ve spojení s používáním přípravku IKERVIS. Snížení systémové absorpce viz bod 4.2.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměrů přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Po očním podání je nepravděpodobné, že by došlo k lokálnímu předávkování. Jestliže dojde k předávkování přípravkem IKERVIS, léčba má být symptomatická a podpůrná.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Oftalmologika, jiná oftalmologika, ATC kód: S01XA18.
Mechanismus účinku a farmakodynamické účinky
Cyklosporin (rovněž známý jako cyklosporin A) je imunomodulátor tvořený cyklickým polypeptidem s imunosupresivními vlastnostmi. Prokázalo se, že prodlužuje přežití alogenních transplantátů u zvířat a významně zlepšil přežití všech typů štěpů při transplantaci solidních orgánů u člověka.
Rovněž se prokázalo, že cyklosporin má protizánětlivý účinek. Studie se zvířaty naznačují, že cyklosporin inhibuje rozvoj reakcí mediovaných buňkou. Bylo prokázáno, že cyklosporin inhibuje tvorbu a/nebo uvolňování prozánětlivých cytokinů, včetně interleukinu 2 (IL-2) nebo T-buněčného růstového faktoru (TCGF) Je rovněž známo, že reguluje uvolňování protizánětlivých cytokinů směrem ke zvýšení. Ukazuje se, že cyklosporin blokuje lymfocyty spočívající ve fázi G0 nebo G1 buněčného cyklu. Veškeré dostupné důkazy naznačují, že cyklosporin specificky a reverzibilně působí na lymfocyty a nepotlačuje hematopoézu ani nemá žádný účinek na funkci fagocytárních buněk.
U pacientů se syndromem suchého oka možná bude zapotřebí vzít v úvahu onemocnění se zánětlivým imunologickým mechanismem, kdy je po očním podání cyklosporin pasivně absorbován do infiltrátů T-lymfocytů v rohovce a spojivce a inaktivuje kalcineurinovou fosfatázu. Cyklosporinem indukovaná inaktivace kalcineurinu inhibuje defosforylaci transkripčního faktoru NF-AT a brání translokaci NF-AT do nukleu, čímž blokuje uvolnění prozánětlivých cytokinů, jako je IL-2.
Klinická účinnost a bezpečnost
Účinnost a bezpečnost přípravku IKERVIS byly hodnoceny ve dvou randomizovaných dvojitě zaslepených, vehikulem kontrolovaných, klinických studiích u dospělých pacientů se syndromem suchého oka (suchou keratokonjunktivitidou), kteří splňovali kritéria Mezinárodního pracovního semináře pro suché oko (International Dry Eye Workshop).
Ve 12měsíčním, klíčovém, dvojitě zaslepených, vehikulem kontrolované klinické studii (studie SANSIKA), 246 pacientů se syndromem suchého oka (DED) se závažnou keratitidou (definovanou jako skóre barvení rohovky fluoresceinem (corneal fluorescein staining, CFS) 4 na modifikované Oxfordské stupnici), tito pacienti byly randomizováni pro jednu kapku přípravku IKERVIS, nebo vehikula denně před usnutím po 6 měsíců. Pacienti randomizovaní do skupiny s vehikulem byli převedeni na přípravek IKERVIS po 6 měsících. Primárním cílovým parametrem byl podíl pacientů, kteří do 6. měsíce dosáhli nejméně dvoustupňového zlepšení keratitidy (CFS) a 30% zlepšení symptomů měřeného pomocí indexu poškození povrchu oka (Ocular Surface Disease Index, OSDI). Podíl respondérů ve skupině s přípravkem IKERVIS byl 28,6 % v porovnání s 23,1 % ve skupině s vehikulem. Rozdíl nebyl statisticky signifikantní (p=0,326).
Závažnost keratitidy hodnocená barvením CFS se významně zvýšila od výchozího stavu do 6. měsíce přípravkem IKERVIS v porovnání s vehikulem (průměrná změna vůči výchozímu stavu byla -1,764 u přípravku IKERVIS vs. -1,418 s vehikulem, p=0,037). Podíl pacientů léčených přípravkem IKERVIS s 3stupňovým zlepšením skóre CFS v 6. měsíci (ze 4 na 1) byl 28,8 % v porovnání s 9,6 % u subjektů hodnocení léčených vehikulem, ale jednalo se o analýzu post-hoc, která omezuje robustnost tohoto výsledku. Prospěšný účinek na keratitidu se udržovat v otevřené fázi studie od 6. do 12. měsíce. Průměrná změna vůči výchozímu stavu na 100bodové škále OSDI byla -13,6 u přípravku IKERVIS a -
14,1 u vehikula v 6. měsíci (p=0,858). Navíc nebylo u přípravku IKERVIS pozorováno žádné zlepšení v porovnání s vehikulem v 6. měsíci u jiných sekundárních cílových parametrů včetně skóre očních obtíží, Schirmerova testu, souběžného používání umělých slz, globálního hodnocení účinnosti zkoušejícím, doby rozložení slzné vrstvy, barvení lisaminovou zelení, skóre kvality života a osmolarity slzné vrstvy.
Snížení zánětu povrchu oka hodnocené expresí humánního leukocitního antigenu-DR (HLA-DR) (výzkumný cílový parametr) bylo pozorováno v 6. měsíci ve prospěch přípravku IKERVIS (p=0,021).
V 6měsíčním, dvojitě zaslepeném, vehikulem kontrolovaném, podpůrném klinickém hodnocení (studie SICCANOVE), 492 DED pacientů se středně až vysoce závažnou keratitidou (definovanou jako skóre CFS od 2 do 4) bylo rovněž randomizováno na přípravek IKERVIS nebo vehikulum denně před usnutím po 6 měsíců. Společné primární cílové parametry zahrnovaly změnu skóre CFS a změnu globálního skóre očních obtíží nesouvisejících s instilací hodnoceného léku a byly oba měřeny v 6. měsíci. Byl pozorován malý, ale statisticky významný rozdíl ve zlepšení CFS mezi léčebnými skupinami v 6. měsíci ve prospěch přípravku IKERVIS (a to u průměrné změny vůči výchozímu stavu v CFS -1,05 u přípravku IKERVIS a -0,82 u vehikula, p=0,009). Průměrná změna vůči výchozímu stavu u skóre očních obtíží (hodnocená pomocí vizuální analogové škály) byl -12,82 u přípravku IKERVIS a -11,21 u vehikula (p=0.808).
V obou studiích nebylo pozorováno významné zlepšení příznaků po 6 měsících léčby u přípravku IKERVIS v porovnání s vehikulem, ať již se použila vizuálních analogová škála, nebo OSDI.
V obou studiích měla Sjorgrenův syndrom průměrně jedna třetina pacientů; pokud jde o celkovou populaci, bylo v této podskupině pacientů pozorováno statistické zlepšení CFS ve prospěch přípravku IKERVIS.
Při dokončení studie SANSIKA (12měsíční studie) byli pacienti požádáni o vstup do studie následující po SANSIKA. Tato studie byla otevřená, nerandomizovaná, 24měsíční prodloužená studie původní studie Sansika s jedním ramenem. Ve studii následující po SANSIKA pacienti alternativně dostávali léčbu přípravkem IKERVIS nebo byli bez léčby v závislosti na skóre CFS (pacienti dostávali IKERVIS, když došlo ke zhoršení keratitidy).
Tato studie byla navržena ke sledování dlouhodobé účinnosti a míry relapsu u pacientů, kteří předtím dostávali přípravek IKERVIS.
Primárním cílem studie bylo vyhodnotit dobu trvání zlepšení po přerušení léčby přípravkem IKERVIS, jakmile se pacient zlepšil vůči výchozímu stavu ve studii SANSIKA (tj. zlepšení nejméně o 2 stupně na upravené Oxfordské stupnici).
Bylo zařazeno 67 pacientů (37,9 % ze 177 pacientů studii Sansika ukončilo). Po 24měsíčním období nezaznamenalo 61,3 % z 62 pacientů zahrnutých do populace primární účinnosti relaps na základě skóre CFS. Procento pacientů, u nichž došlo k rekurenci závažné keratitidy bylo 35 % u pacientů léčených přípravkem IKERVIS 12 měsíců a 48 % pacientů léčených 6 měsíců ve studii SANSIKA.
Na základě prvního kvartilu (medián nebylo možné odhadnout kvůli malému počtu relapsů) doba do relapsu (zpět na stupeň CFS 4) byla < 224 dnů u pacientů dříve léčených 12 měsíců přípravkem IKERVIS a < 175 dnů u pacientů léčených 6 měsíců. Pacienti strávili více času na stupni 2 CFS (medián doby 12,7 týdne/rok) a stupni 1 (medián doby 6,6 týdnů/rok) než na stupni 3 CFS (medián doby 2,4 týdne/rok) stupních 4 a 5 CFS (medián doby 0 týdne/rok).
Hodnocení příznaků DED pomocí VAS ukázalo zhoršení diskomfortu pacienta od doby prvního zastavení léčby do doby jejího opětovného zahájení s výjimkou bolesti, která zůstala relativně nízká a stabilní. Medián globálního skóre VAS vzrostl od doby, kdy byla léčba poprvé zastavena (23,3 %) do doby obnovení léčby (45,1 %).
U jiných sekundárních cílových parametrů (TBUT, barvení lisaminovou zelení a Schirmerův test, NEI,-VFQ a EQ-5d) nebyly v průběhu prodloužené studie pozorovány žádné změny.
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem IKERVIS u všech podskupin pediatrické populace pro syndrom suchého oka (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Formální farmakokinetické studie s přípravkem IKERVIS u lidí nebyly provedeny.
Koncentrace přípravku IKERVIS v krvi byly měřeny pomocí specifické analýzy vysokotlakou kapalinovou chromatografií a hmotnostní spektrometrií. Koncentrace cyklosporinu v plazmě byly měřeny u 374 pacientů ve dvou studiích účinnosti před podáním a za 6 měsíců (studie SICCANOVE a studie SANSIKA) a za 12 měsíců léčby (studie SANSIKA). Po 6 měsících instilace do očí přípravku IKERVIS jednou za den mělo 327 pacientů hodnoty pod dolní mezí detekce (0,050 ng/ml) a 35 pacientů bylo pod dolní mezí kvantifikace (0,100 ng/ml). Měřitelné hodnoty nepřekračující 0,206 ng/ml byly naměřeny u osmi pacientů a hodnoty byly považovány za zanedbatelné. Tři pacienti měli hodnoty nad horní mezí kvantifikace (5 ng/ml), ovšem pacienti již užívali cyklosporin perorálně ve stabilní dávce, což dovoloval protokol studie. Po 12 měsících léčby byly tyto hodnoty pod dolní mezí detekce u 56 pacientů a pod dolní mezí kvantifikace u 19 pacientů. Sedm pacientů mělo měřitelné hodnoty (od 0,105 do 1,27 ng/ml), ale všechny hodnoty byly považovány za zanedbatelné. Dva pacienti měli hodnoty nad horní mezí kvantifikace, ovšem pacienti již užívali cyklosporin perorálně ve stabilní dávce při zařazení studie.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti, toxicity po opakovaném podávání, fototoxicity a fotoalergie, genotoxicity, hodnocení kancerogenního potenciálu, reprodukční a vývojové toxicity neodhalily žádné zvláštní riziko pro člověka.
Účinky v neklinických studiích byly pozorovány pouze u systémového podání nebo po expozicích dostatečně převyšujících maximální expozici u člověka, což svědčí o malém významu při klinickém použití.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Střední nasycené triacylglyceroly
Cetalkonium-chlorid
Glycerol
Tyloxapol
Poloxamer 188
Hydroxid sodný (k úpravě pH)
Voda na injekci
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
3 roky.
6.4 Zvláštní opatření pro uchovávání
Chraňte před mrazem.
Po otevření hliníkových váčků je třeba uchovávat jednodávkový obal ve váčcích, aby byl chráněn před světlem a nedocházelo k odpařování. Zlikvidujte jakýkoliv jednotlivý otevřený jednodávkový obal se zbývající emulzí bezprostředně po použití.
6.5 Druh obalu a obsah balení
Přípravek IKERVIS je dodáván v 0,3ml jednodávkových obalech z nízkohustotního polyethylenu (LDPE) vložených do uzavřeného lamináto-hliníkového váčku.
Jeden váček obsahuje pět jednodávkových obalů.
Velikost balení: 30 a 90 jednodávkových obalů.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
SANTEN Oy Niittyhaankatu 20 33720 Tampere Finsko
8. REGISTRAČNÍ ČÍSLA
EU/1/15/990/001
EU/1/15/990/002
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 19. března 2015
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
PŘÍLOHA II
A. VÝROBCE ODPOVĚDNÝ /VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného za propouštění šarží EXCELVISION
27 RUE DE LA LOMBARDIERE, ZI LA LOMBARDIERE
07100 ANNONAY
Francie
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předloží první pravidelně aktualizovanou zprávu o bezpečnosti pro tento léčivý přípravek do 6 měsíců od jeho registrace. Držitel rozhodnutí o registraci dále předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
• Podmínky nebo omezení s ohledem na bezpečné a účinné používání léčivého přípravku, které mají realizovat členské státy
Neuplatňuje se.
PŘÍLOHA III
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU VNĚJŠÍ OBAL
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
IKERVIS 1 mg/ml oční kapky, emulze ciclosporinum
2. OBSAH LÉČIVÉ LÁTKY
Jeden ml emulze obsahuje ciclosporinum 1 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Pomocné látky: střední nasycené triacylglyceroly, cetalkonium-chlorid, glycerol, tyloxapol, poloxamer 188, hydroxid sodný (k úpravě pH) a vodu na injekci.
Další údaje naleznete v příbalové informaci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Oční kapky, emulze.
30jednodávkových obalů 90jednodávkových obalů
5. ZPŮSOB A CESTA PODÁNÍ
Pouze k jednorázovému použití.
Před použitím si přečtěte příbalovou informaci. Oční podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
Před použitím vyjměte kontaktní čočky.
8. POUŽITELNOST
EXP
Bezprostředně po použití zlikvidujte jakýkoliv jednotlivý otevřený jednodávkový obal se zbývající emulzí.
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Chraňte před mrazem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH,POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
SANTEN Oy Niittyhaankatu 20 33720 Tampere Finsko
12. REGISTRAČNÍ ČÍSLA
EU/1/15/990/001
EU/1/15/990/002
13. ČÍSLO ŠARŽE
č.š.:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
ikervis
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
IKERVIS 1 mg/ml oční kapky, emulze ciclosporinum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
SANTEN Oy
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
Oční podání.
5 jednodávkových obalů Pouze k jednorázovému použití.
Chraňte před mrazem.
Další údaje naleznete v příbalové informaci.
Po otevření hliníkových váčků je třeba uchovávat jednodávkový obal ve váčcích, aby byl chráněn před světlem a nedocházelo k odpařování.
Bezprostředně po použití zlikvidujte jakýkoliv jednotlivý otevřený jednodávkový obal se zbývající emulzí.
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
IKERVIS 1 mg/ml oční kapky, emulze
ciclosporinum
ciclosporin
2. ZPŮSOB PODÁNÍ
Oční podání
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
0,3 ml
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro pacienta
IKERVIS 1 mg/ml oční kapky, emulze
ciclosporinum (ciclosporin)
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoliv další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoliv nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek IKERVIS a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek IKERVIS používat
3. Jak se přípravek IKERVIS používá
4. Možné nežádoucí účinky
5. Jak přípravek IKERVIS uchovávat
6. Obsah balení a další informace
1. Co je přípravek IKERVIS a k čemu se používá
Přípravek IKERVIS obsahuje léčivou látku cyklosporin. Cyklosporin patří do skupiny léčiv známých jako imunosupresiva, která se používají k potlačení zánětu.
Přípravek IKERVIS se používá k léčbě dospělých se závažnou keratitidou (zánět rohovky, průhledné vrstvy v přední části oka). Používá se u pacientů se syndromem suchého oka, který se nezlepšil navzdory léčbě náhradami slzami (umělé slzy).
Pokud se nebudete cítit lépe, nebo se Vám přitíží, musíte se poradit s lékařem.
Měl(a) byste navštívit lékaře nejméně jednou za 6 měsíců, aby vyhodnotil účinek přípravku IKERVIS.
2. Čemu musíte věnovat pozornost, než začnete přípravek IKERVIS používat NEPOUŽÍVEJTE přípravek IKERVIS
- jestliže jste alergický(á) na cyklosporin nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
- jestliže máte oční infekci.
Upozornění a opatření
Přípravek IKERVIS používejte pouze ke vkapávání do oka(očí).
Před použitím přípravku IKERVIS se poraďte se svým lékařem nebo lékárníkem,
- jestliže jste měl(a) oční infekci vyvolanou virem herpes, která by mohla poškodit průhlednou přední část oka (rohovka),
- jestliže užíváte jakékoliv přípravky obsahující steroidy,
- jestliže užíváte jakékoliv přípravky k léčbě glaukomu.
Kontaktní čočky mohou dále poškodit průhlednou přední část oka (rohovka). Proto je nutné, abyste si vyndal(a) kontaktní čočky před ulehnutím před použitím přípravku IKERVIS; po probuzení si je můžete znovu nasadit.
Děti a dospívající
Přípravek IKERVIS by neměl být používán u dětí a dospívajících mladších 18 let.
Další léčivé přípravky a přípravek IKERVIS
Informujte svého lékaře nebo lékárníka o všech lécích, které používáte, které jste v nedávné době používal(a) nebo které možná budete používat.
Informujte svého lékaře, pokut používáte oční kapky obsahující steroidy s přípravkem IKERVIS, protože ty by mohly zvýšit riziko nežádoucích účinků.
Oční kapky IKERVIS je nutno používat nejméně za 15 minut po použití jakýchkoliv jiných očních kapek.
Těhotenství a kojení
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek používat.
Přípravek IKERVIS byste neměla používat, pokud jste těhotná.
Jestliže byste mohla otěhotnět, musíte během používání tohoto přípravku používat antikoncepci.
Je pravděpodobné, že přípravek IKERVIS bude přítomen v mateřském mléce ve velmi malých množstvích. Pokud kojíte, informujte o tom před používáním tohoto léčivého přípravku svého lékaře.
Řízení dopravních prostředků a obsluha strojů
Bezprostředně po použití očních kapek IKERVIS můžete vidět rozmazaně. Pokud k tomu dojde, neřiďte a neobsluhujte žádné žádné stroje, dokud nebudete opět jasně vidět.
3. Jak se přípravek IKERVIS používá
Vždy používejte tento přípravek přesně podle pokynů svého lékaře nebo lékárníka. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Doporučená dávka přípravku je jedna kapka do každého postiženého oka jednou denně večer před spaním.
Návod k použití
Pečlivě dodržujte tyto pokyny, a pokud něčemu nebudete rozumět, požádejte svého lékaře nebo lékárníka o vysvětlení.
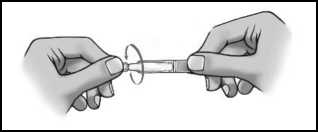
1
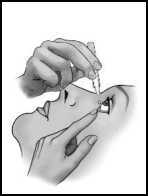
2
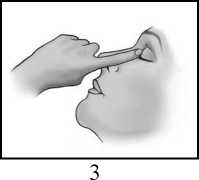
• Umyjte si ruce.
• Pokud nosíte kontaktní čočky, vyndejte si je před spaním před použitím očních kapek; po probuzení si je můžete znovu nasadit.
• Otevřete hliníkový váček, který obsahuje pět jednodávkových obalů.
• Vyjměte jednodávkový obal z hliníkového váčku.
• Před použitím j emně protřepejte j ednodávkový obal.
• Odlomte uzávěr (obrázek 1).
• Stáhněte dolů dolní víčko oka (obrázek 2).
• Zakloňte hlavu dozadu a podívejte se nahoru ke stropu.
• Jemně vymáčkněte jednu kapku přípravku do oka. Nesmíte se dotknout oka hrotem jednodávkového obalu.
• Několikrát mrkněte, aby přípravek pokryl vaše oko.
• Po použití přípravku IKERVIS zatlačte prstem do koutku oka v blízkosti nosu a zavřete víčko na 2 minuty (obrázek 3). To pomůže zastavit přípravek IKERVIS v pronikání do zbytku těla.
• Jestliže kapky používáte do obou očí, opakujte kroky u druhého oka.
• Jednodávkový obal zlikvidujte ihned po použití, a to i v případě, že v něm stále zůstává nějaká tekutina.
• Zbývající jednodávkové obaly je nutné uchovávat v hliníkovém váčku.
Jestliže kapka oko mine, zkuste to znovu.
Jestliže jste použil(a) více přípravku IKERVIS, než jste měl(a), vypláchněte si oko vodou. Nevkapávejte si žádné další kapky, dokud nenastane doba na další pravidelnou dávku.
Jestliže jste zapomněl(a) použít přípravek IKERVIS, pokračujte následující plánovanou dávkou. Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku. Nepoužívejte více než jednu kapku každý den do postiženého oka(očí).
Jestliže jste přestal(a) používat přípravek IKERVIS, aniž byste o tom informoval(a) svého lékaře, zánět průhledné přední části oka (známý jako keratitida) nebude pod kontrolou a mohl by vést ke zhoršenému vidění.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Byly hlášeny následující nežádoucí účinky:
Nejčastější nežádoucí účinky se vyskytují v očích a jejich okolí.
Velmi časté (mohou postihnout více než 1 osobu z 10)
Bolest v místě, kde se kapky vkapávají do oka.
Časté (mohou postihnout až 1 osobu z 10)
Podráždění, zarudnutí a zvýšená tvorba slz při vkapávání kapek do oka, zarudnutí víčka, vodnaté oči, zarudnutí oka, rozmazané vidění. Otok očního víčka, zarudnutí spojivky (tenká membrána zakrývající přední část oka), podráždění oka, bolest oka.
Méně časté (mohou postihnout až 1 osobu ze 100)
Méně časté nežádoucí účinky týkající se oka:
Nepříjemné pocity v oku, svědění či podráždění v oku nebo okolo něj včetně pocitu, že máte něco v oku.
Podráždění nebo otok spojivky (tenká membrána pokrývající přední část oka), oční alergie, porucha slzení, výtok z oka, zánět duhovky (barevná část oka) nebo očního víčka, depozita (usazeniny) v oku, bakteriální infekce nebo zánět rohovky (průhledná přední část oka), abraze (poškrabání) vnější vrstvy rohovky, bělavé skvrny na rohovce, cysta v očním víčku, svědění v očním víčku, bolestivá vyrážka okolo oka způsobená virem herpes zoster.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek IKERVIS uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na vnějším obalu, hliníkovém váčku a na jednodávkových obalech za „EXP“. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Chraňte před mrazem.
Po otevření hliníkových váčků je třeba uchovávat jednodávkový obal ve váčcích, aby byl chráněn před světlem a nedocházelo k odpařování. Zlikvidujte jakýkoliv jednotlivý otevřený jednodávkový obal se zbývající emulzí bezprostředně po použití.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co přípravek IKERVIS obsahuje
- Léčivou látkou je ciclosporinum. Jeden mililitr přípravku IKERVIS obsahuje ciclosporinum 1 mg.
- Dalšími složkami jsou střední nasycené triacylglyceroly, cetalkonium-chlorid, glycerol, tyloxapol, poloxamer 188, hydroxid sodný (k úpravě pH) a vodu na injekci.
Jak přípravek IKERVIS vypadá a co obsahuje toto balení
Přípravek IKERVIS jsou oční kapky v podobě mléčné bílé emulze.
Dodávají se v jednodávkových obalech vyrobených z polyethylenu o nízké hustotě (LDPE).
Jeden jednodávkový obal obsahuje 0,3 ml očních kapek, emulze.
Jednodávkové obaly jsou zabaleny v uzavřeném hliníkovém váčku.
Velikost balení: 30 a 90 jednodávkových obalů.
Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci
SANTEN Oy Niittyhaankatu 20 33720 Tampere Finsko
Výrobce
EXCELVISION Rue de la Lombardiére ZI la Lombardiére F-07100 Annonay Francie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci
|
Belgie/Belgique/Belgien Santen Oy Tél/Tel : + 358 (0) 3 284 8111 |
Lietuva Santen Oy Tel: + 358 (0) 3 284 8111 |
|
Btarapna Santen Oy Tea.: + 358 (0) 3 284 8111 |
Luxembourg/Luxemburg Santen Oy Tél/Tel: + 358 (0) 3 284 8111 |
|
Česká republika Santen Oy Tel: + 358 (0) 3 284 8111 |
Magyarország Santen Oy Tel.: + 358 (0) 3 284 8111 |
|
Danmark SantenPharma AB Tlf: + 46 (0) 8 444 75 60 |
Malta Santen Oy Tel: + 358 (0) 3 284 8111 |
|
Deutschland Santen GmbH Tel: + 49 (0) 89 84 80 78-0 |
Nederland S anten Oy Tel: + 358 (0) 3 284 8111 |
|
Eesti Santen Oy Tel: + 358 (0) 3 284 8111 |
Norge SantenPharma AB Tlf: + 46 (0) 8 444 75 60 |
|
EXXáda Santen Oy T^: + 358 (0) 3 284 8111 |
Osterreich Santen Oy Tel: + 358 (0) 3 284 8111 |
|
Espaňa Santen Pharmaceutical Spain S.L. Tel: + 34 914 142 485 |
Polska Santen Oy Tel.: + 358 (0) 3 284 8111 |
|
France Santen S.A.S. Tél: + 33 (0)1 69 87 40 20 |
Portugal Santen Oy Tel: + 351 308 805 912 |
|
Hrvatska Santen Oy Tel: + 358 (0) 3 284 8111 |
Románia Santen Oy Tel: + 358 (0) 3 284 8111 |
|
Ireland Santen UK Limited Tel: + 353 (0) 16950008 |
Slovenija Santen Oy Tel: + 358 (0) 3 284 8111 |
|
Ísland Santen Oy Sími: + 358 (0) 3 284 8111 |
Slovenská republika Santen Oy Tel: + 358 (0) 3 284 8111 |
Italia
Santen Italy S.r.l.
Tel: + 39 02 620019.1
Kúrcpog
Santen Oy
Tni: + 358 (0) 3 284 8111
Latvija
Santen Oy
Tel: + 358 (0) 3 284 8111
Suomi/Finland
Santen Oy
Puh/Tel: + 358 (0) 3 284 8111 Sverige
SantenPharma AB Tel: + 46 (0) 8 444 75 60
United Kingdom
Santen UK Limited Tel: + 44 (0) 845 075 4863
Tato příbalová informace byla naposledy revidována
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
24