Idelvion 250 Iu
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
IDELVION 250 IU prášek a rozpouštědlo pro injekční roztok IDELVION 500 IU prášek a rozpouštědlo pro injekční roztok IDELVION 1000 IU prášek a rozpouštědlo pro injekční roztok IDELVION 2000 IU prášek a rozpouštědlo pro injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
IDELVION 250 IU prášek a rozpouštědlo pro injekční roztok
Jedna injekční lahvička obsahuje jmenovitě 250 IU rekombinantního fúzního proteinu spojujícího koagulační faktor IX s albuminem (rIX-FP), (INN = albutrepenonacogum alfa). Po rekonstituci s 2,5 ml vody na injekci roztok obsahuje albutrepenonacogum alfa 100 IU/ml.
IDELVION 500 IU prášek a rozpouštědlo pro injekční roztok
Jedna injekční lahvička obsahuje jmenovitě 500 IU rekombinantního fúzního proteinu spojujícího koagulační faktor IX s albuminem (rIX-FP), (INN = albutrepenonacogum alfa). Po rekonstituci s 2,5 ml vody na injekci roztok obsahuje albutrepenonacogum alfa 200 IU/ml.
IDELVION 1000 IU prášek a rozpouštědlo pro injekční roztok
Jedna injekční lahvička obsahuje jmenovitě 1000 IU rekombinantního fúzního proteinu spojujícího koagulační faktor IX s albuminem (rIX-FP), (INN = albutrepenonacogum alfa). Po rekonstituci s 2,5 ml vody na injekci roztok obsahuje albutrepenonacogum alfa 400 IU/ml.
IDELVION 2000 IU prášek a rozpouštědlo pro injekční roztok
Jedna injekční lahvička obsahuje jmenovitě 2000 IU rekombinantního fúzního proteinu spojujícího koagulační faktor IX s albuminem (rIX-FP), (INN = albutrepenonacogum alfa). Po rekonstituci s 5 ml vody na injekci roztok obsahuje albutrepenonacogum alfa 400 IU/ml.
Účinnost (v mezinárodních jednotkách [IU]) se stanoví in-vitro pomocí jednostupňového testu srážení krve, založeného na aktivovaném parciálním tromboplastinovém času (aPTT), kalibrovaného proti mezinárodnímu standardu Světové zdravotnické organizace (WHO) pro koncentrát faktoru IX.
Albutrepenonacogum alfa je purifikovaný protein produkovaný rekombinantní DNA technologií, vytvořený genetickou fúzí rekombinantního albuminu s rekombinantním koagulačním faktorem IX. Genetická fúze cDNA lidského albuminu s cDNA lidského koagulačního faktoru IX umožňuje protein, který byl vyroben jako jeden rekombinantní protein a zabezpečuje homogennost výrobku tím, že zabraňuje chemické konjugaci. Část rekombinantního faktoru IX je totožná s Thr148 alelickou formou faktoru IX z plazmy. Štěpný linker mezi molekulami rekombinantního faktoru IX a albuminu je odvozen z endogenního "aktivačního peptidu" v nativním faktoru IX.
Pomocná látka se známým účinkem:
Až 25,8 mg (1,13 mmol) sodíku na dávku (tělesná hmotnost 70 kg).
Úplný seznam pomocných látek viz bod 6.1.
LÉKOVÁ FORMA
3.
Prášek a rozpouštědlo pro injekční roztok.
Světle žlutý až bílý prášek a čiré, bezbarvé rozpouštědlo pro injekční roztok.
pH: 6,6 - 7,2
Osmolalita:
IDELVION 250 IU prášek a rozpouštědlo pro injekční roztok 175 - 215 mOsm/kg
IDELVION 500 IU prášek a rozpouštědlo pro injekční roztok 260 - 300 mOsm/kg
IDELVION 1000 IU prášek a rozpouštědlo pro injekční roztok 260 - 300 mOsm/kg
IDELVION 2000 IU prášek a rozpouštědlo pro injekční roztok 260 - 300 mOsm/kg
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba a profylaxe krvácení u pacientů s hemofilií B (vrozený nedostatek faktoru IX).
Přípravek IDELVION je možné použít pro všechny věkové skupiny.
4.2 Dávkování a způsob podání
Léčba má být pod dohledem lékaře, který má zkušenosti s léčbou hemofilie B.
Dosud neléčení pacienti
Bezpečnost a účinnost přípravku IDELVION nebyla stanovena u dříve neléčených pacientů. Monitoring léčby
Během léčby se doporučuje vhodné stanovení hladin faktoru IX jako vodítko pro dávky, které mají být podávány a frekvence opakovaných infuzí. Jednotliví pacienti se mohou lišit v odpovědích na faktor IX, což demonstruje různé poločasy a recovery. Dávka vychází z tělesné hmotnosti a může vyžadovat úpravu s podvýživou nebo nadváhou pacientů. V případě velkých chirurgických zákroků je nezbytné přesné monitorování průběhu substituční terapie pomocí koagulační analýzy (aktivita plazmatického faktoru IX).
Při použití in vitro tromboplastinového času (aPTT) založeného na jednostupňovém testu srážení krve pro stanovení aktivity faktoru IX ve vzorcích krve pacientů, výsledky aktivity faktoru IX v plazmě mohou být významně ovlivněny typem aPTT činidla i referenčním standardem používaným v testu. Měření jednofázovým testem srážení s použitím na kaolinu založeném aPTT činidlu nebo aktin FS aPTT činidlu, bude pravděpodobně mít za následek podcenění úrovně aktivity. To je důležité zejména při změně laboratoře a/nebo činidel použitých v testu.
Dávkování
Dávka a délka substituční léčby závisí na závažnosti nedostatku faktoru IX, na místě a rozsahu krvácení a na klinickém stavu pacienta.
Počet jednotek podaného faktoru IX se vyjadřuje v mezinárodních jednotkách (IU), které jsou vztaženy k aktuálnímu standardu WHO pro přípravky s faktorem IX. Aktivita faktoru IX v plazmě se vyjadřuje buď v procentech (vzhledem k normální lidské plazmě) nebo v mezinárodních jednotkách (vzhledem k mezinárodnímu standardu pro faktor IX v plazmě).
Jedna mezinárodní jednotka (IU) aktivity faktoru IX je ekvivalentní množství faktoru IX v jednom ml normální lidské plazmy.
Požadovaná léčba
Výpočet požadované dávky faktoru IX je založen na empirickém zjištění, že při podání 1 mezinárodní jednotky (IU) faktoru IX na kg tělesné hmotnosti se předpokládá zvýšení cirkulující hladiny faktoru IX v průměru o 1,3 IU/dl (1,3% normálu) u pacientů ve věku > 12 let a o 1,0 IU / dl (1,0% normálu) u pacientů ve věku <12 let. Požadovaná dávka se určuje pomocí následujícího vzorce:
Požadovaná dávka (IU) = tělesná hmotnost (kg) x požadovaný vzestup faktoru IX (% normálu nebo IU/dl) x {převrácená hodnota pozorované recovery (IU/kg na IU/dl)}
Očekávané zvýšení hladiny faktoru IX (IU/dl nebo% normální hladiny) = dávka (IU) x Recovery (IU/dl na IU/kg)/tělesná hmotnost (kg)
Množství přípravku, které má být podáno a frekvence podávání má být vždy orientována na klinickou účinnost v konkrétním případě.
Pacienti ve věku <12 let
Pro kumulativní recovery 1 IU/dl na 1 IU/kg se dávka vypočte takto:
Dávka (IU) = tělesná hmotnost (kg) x požadované zvýšení faktoru IX (IU/dl) x 1 dl/kg
Příklad
1. U pacienta s hmotností 20 kg s těžkou hemofilií B se požaduje maximální hladina 50% normálu. Vhodná dávka bude 20 kg x 50 IU/dl x 1 dl/kg = 1000 IU.
2. Dávka 1000 IU přípravku IDELVION podaná 25 kg pacientovi bude mít očekávaný výsledek zvýšení maximální hladiny po injekci faktoru IX 1000 IU/25kg, x 1,0 (IU/dl na IU/kg)
= 40 IU/dl (40% běžné hodnoty).
Pacienti ve věku > 12 let
Pro kumulativní recovery 1,3 IU/dl na 1 IU/kg se dávka vypočte takto:
Dávka (IU) = tělesná hmotnost (kg) x požadované zvýšení faktoru IX (IU/dl) x 0,77 dl/kg
Příklad
3. U pacienta s hmotností 80 kg s těžkou hemofilií B se požaduje maximální hladina 50% normálu. Vhodná dávka bude 80 kg x 50 IU/dl x 0,77 dl/kg = 3080 IU.
4. Dávka 2000 IU přípravku IDELVION podaná 80 kg pacientovi bude mít očekávaný výsledek zvýšení maximální hladiny po injekci faktoru IX 2000 IU x 1,3 (IU/dl na IU/kg)/80 kg = 32,5 IU/dl (32,5% normálu).
V případě následujících krvácivých příhod by aktivita faktoru IX neměla v daném období klesnout pod určenou hladinu plazmatické aktivity (v % normálu nebo IU/dl). Tuto tabulku lze použít jako ukazatel v případě krvácení nebo operace:
|
Stupeň krvácení/typ chirurgického výkonu |
Požadovaná hladina faktoru IX (%) (IU/dl) |
Frekvence dávkování (hodiny)/délka trvání léčby (dny) |
|
Krvácení Začínající nebo mírná hemartróza, krvácení do svalů (s výjimkou iliopsoas) nebo ústní dutiny |
30-60 |
Jedna dávka je dostatečná pro většinu krvácení. Udržovací dávka po 24 - 72 hodinách, v případě, že krvácení pokračuje. |
|
Intenzivnější krvácení Život ohrožující krvácení, hluboké svalové krvácení včetně iliopsoas |
O O 1 O kO |
Opakovat každých 24-72 hodin, během prvního týdne a poté udržovací dávka každý týden až do zastavení krvácení a zahojení. |
|
Menší chirurgický výkon Včetně nekomplikovaného vytržení zubu |
50-80 (počáteční hladina) |
Jedna dávka je dostačující pro většinu menších operací. V případě potřeby udržovací dávka může být za 24 - 72 hodin, až do zastavení krvácení a zahojení. |
|
Velké chirurgické výkony |
60 - 100 (počáteční hladina) |
Opakovat každých 24 - 72 hodin během prvního týdne a poté udržovací dávka 1-2 krát týdně, dokud se krvácení nezastaví a do zahojení. |
Profylaxe
Pro dlouhodobou profylaxi krvácení u pacientů s těžkou hemofilií B jsou obvyklé dávky 35 až 50 IU/kgjednou týdně.
Někteří pacienti, kteří jsou dobře kontrolováni v jednotýdenním režimu, mohou být léčeni dávkou až 75 IU/kg v intervalu 10 až 14 dnů (viz bod 5.1).
V některých případech, zejména u mladších pacientů, mohou být nutné kratší intervaly nebo vyšší dávky.
Po výskytu krvácivé příhody během profylaxe mají pacienti co nejpřísněji dodržovat svůj profylaktický režim s 2 dávkami přípravku IDELVION podanými s odstupem alespoň 24 hodin, nebo déle, pokud se to považuje za vhodné pro pacienta.
Pediatrická populace
Pro rutinní profylaxi pediatrických pacientů je doporučený režim dávkování 35 až 50 IU/kg jednou týdně (viz body 5.1 a 5.2).
Způsob podání Intravenózní podání.
Pokyny pro rekonstituci přípravku před podáním, viz bod 6.6. Rekonstituovaný přípravek má být aplikován pomalu intravenózně rychlostí pohodlnou pro pacienta až do maximálně 5 ml/min.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku (rekombinantní fúzní protein spojující koagulační faktor IX s albuminem (rIX-FP)) nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Známá alergická reakce na křeččí bílkovinu.
4.4 Zvláštní upozornění a opatření pro použití
Hypersenzitivita
U přípravku IDELVION jsou možné hypersenzitivní reakce alergického typu. Přípravek obsahuje stopy křeččích proteinů. Objeví-li se příznaky hypersenzitivity, pacienti mají být poučeni, že musí okamžitě přerušit používání přípravku a kontaktovat svého lékaře. Pacienti mají být informováni o časných příznacích alergických reakcí včetně vyrážky, generalizované kopřivky, tlaku na hrudi, sípání, hypotenze a anafylaxe. Doporučuje se, aby první podání faktoru IX bylo podle požadavků ošetřujícího lékaře prováděno pod lékařským dohledem, kde je poskytnuta řádná lékařská péče při alergické reakci. V případě šoku mají být provedeny standardní lékařské postupy pro léčbu šoku.
Inhibitory
Po opakované léčbě lidským koagulačním faktorem IX mají být pacienti sledováni s ohledem na vývoj neutralizujících protilátek (inhibitorů), které mají být kvantifikovány v jednotkách Bethesda (BU) pomocí vhodného biologického testování.
Zprávy z literatury ukazují korelaci mezi výskytem inhibitoru faktoru IX a alergickými reakcemi. Proto pacienti trpící alergickými reakcemi musí být vyšetřeni na přítomnost inhibitoru.
Je třeba poznamenat, že u pacientů s inhibitory faktoru IX může být ve zvýšené riziko anafylaxe s následnou stimulací faktorem IX.
Vzhledem k riziku alergických reakcí u přípravků s faktorem IX, počáteční podání faktoru IX má být podle požadavku ošetřujícího lékaře prováděno pod lékařským dohledem, kde může být poskytnuta řádná lékařská péče při alergické reakci.
Tromboembolismus
Vzhledem k potenciálnímu riziku trombotických komplikací, klinický dohled pro časné příznaky trombotické a konsumpční koagulopatie má být zahájen vhodným biologickým testováním při podávání tohoto přípravku pacientům s onemocněním jater, pacientům po operaci, novorozencům nebo pacientům s rizikem trombotických jevů nebo DIC. V každé z těchto situací přínos léčby s přípravkem IDELVION má být zvážen oproti riziku těchto komplikací.
Kardiovaskulární příhody
U pacientů s existujícími kardiovaskulárními rizikovými faktory může substituční terapie s FIX zvyšovat kardiovaskulární riziko.
Komplikace související s katetrem
Jestliže je třeba použít zařízení pro centrální žilní přístup (CVAD), musí se uvážit riziko vzniku komplikací souvisejících s CVAD, včetně lokální infekce, bakteriémie a trombózy v místě vstupu katetru.
Pediatrická populace
Uvedené upozornění a opatření platí jak pro dospělé tak i pro děti.
Starší pacienti
Klinické studie přípravku IDELVION nezahrnovaly pacienty ve věku 65 a více let. Není známo, zda reagují odlišně než mladší subjekty.
Indukce imunitní tolerance
Bezpečnost a účinnost použití přípravku IDELVION při indukci imunitní tolerance nebyla dosud stanovena.
Obsah sodíku
Tento přípravek obsahuje až 25,8 mg (1,13 mmol) sodíku v jedné dávce (tělesná hmotnost 70 kg) jestliže je aplikována maximální dávka (15 ml = 6000 IU). Nutno vzít v úvahu u pacientů na dietě s nízkým obsahem sodíku.
Záznam o použití
Důrazně se doporučuje, aby při každém podání přípravku IDELVION pacientovi byl zaznamenáván název a číslo šarže přípravku tak, aby se zachovala souvislost mezi pacientem a šarží léčivého přípravku.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly hlášeny žádné interakce lidského koagulačního faktoru IX s jinými léčivými přípravky.
4.6 Fertilita, těhotenství a kojení
Reprodukční studie s faktorem IX na zvířatech nebyly provedeny. Vzhledem ke vzácnému výskytu hemofilie B u žen, zkušenosti s použitím faktoru IX během těhotenství a kojení nejsou k dispozici.
Proto má faktor IX být během těhotenství a kojení podáván pouze tehdy, pokud je to nezbytně nutné.
K dispozici nejsou žádné informace o účincích faktoru IX na plodnost.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek IDELVION nemá žádný vliv na schopnost řídit a obsluhovat stroje.
4.8 Nežádoucí účinky Souhrn bezpečnostního profilu
Hypersenzitivita nebo alergické reakce (které mohou zahrnovat angioedém, pálení a bodání v místě infuze, zimnice, zarudnutí, generalizovanou kopřivku, bolest hlavy, kopřivku, hypotenzi, letargii, nauzeu, neklid, tachykardii, tlak na hrudi, mravenčení, zvracení, sípání) byly pozorovány vzácně a mohou se v některých případech rozvinout do závažné anafylaxe (včetně šoku). V některých případech tyto reakce vyústily až v závažnou anafylaxi a došlo k ní v těsné časové souvislosti s rozvojem inhibitorů faktoru IX (viz také bod 4.4). Nefrotický syndrom byl hlášen při pokusech o navození imunitní tolerance u pacientů s hemofilií B s inhibitorem faktoru IX a anamnézou alergických reakcí.
Při použití přípravků s faktorem IX získaných z buněk CHO byl pozorován velmi zřídka vývoj protilátek na křeččí protein.
Pacienti s hemofilií B si mohou vyvinout neutralizující protilátky (inhibitory) proti faktoru IX. Pokud se takové inhibitory vyskytnou, tento stav se projeví jako nedostatečná klinická odpověď. V takových případech se doporučuje kontaktovat specializované centrum pro léčbu hemofilie.
Existuje potenciální riziko tromboembolických příhod po podání faktoru IX, s vyšším rizikem u přípravků s nižší čistotou. Použití faktoru IX nízké čistoty bylo spojeno s případy infarktu myokardu, diseminované intravaskulární koagulace, žilní trombózy a plicní embolie. Použití vysoce čistého faktoru IX je zřídka spojeno s těmito nežádoucími účinky.
Tabulkový seznam nežádoucích účinků
Bylo hlášeno 13 nežádoucích reakcí u 7 subjektů ze čtyř otevřených klinických studiích, které zahrnovaly 107 subjektů s nejméně jednou expozicí přípravkem IDELVION.
Tabulka uvedená níže je podle klasifikace orgánových systémů MedDRA (SOC a frekvence výskytu).
Frekvence byly hodnoceny podle následující konvence: velmi časté (>1/10); časté (>1/100 až <1/10); méně časté (>1/1000 až <1/100); vzácné (>1/10000 až <1/1000); velmi vzácné (<1/10000), není známo (z dostupných údajů nelze určit).
V každé skupině četností jsou nežádoucí účinky prezentovány v pořadí klesající závažnosti.
|
Třída orgánových systémů MedDRA |
Nežádoucí účinky |
Frekvence na pacienta |
|
Celkové poruchy a reakce v místě aplikace |
Reakce v místě vpichu |
Časté |
|
Poruchy nervového systému |
Časté | |
|
Závrať |
Méně časté | |
|
Poruchy imunitního systému |
Hypersenzitivita |
Méně časté |
|
Poruchy kůže a podkožní tkáně |
Méně časté | |
|
Ekzém |
Méně časté |
Popis vybraných nežádoucích účinků
U jednoho dříve neléčeného pacienta (PNP) byl hlášen nízký titr inhibitoru proti faktoru IX u probíhající klinické studie. Nejsou k dispozici dostatečné údaje, které poskytují informace o výskytu inhibitoru u PNP.
Pediatrická populace
Očekává se, že frekvence, typ a závažnost nežádoucích účinků u dětí budou podobné jako u dospělých.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Nebyly hlášeny žádné příznaky předávkování s přípravkem IDELVION.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antihemoragika: krevní koagulační faktor IX.
ATC kód: B02BD04
Mechanismus účinku
Přípravek IDELVION (INN: albutrepenonacogum alfa) je rekombinantní koagulační faktor IX. Prodloužení poločasu přípravku IDELVION a lepší systémová expozice je dosaženo fúzí s rekombinantním albuminem. Albumin je přírodní, inertní proteinový nosič v plazmě s poločasem přibližně 20 dnů. Genetická fuze rekombinantního koagulačního faktoru IX s albuminem prodlužuje poločas faktoru IX (viz bod 5.2).
Přípravek IDELVION zůstává neporušený v oběhu, dokud nedojde k aktivaci faktoru IX, načež se albumin štěpí, uvolňuje aktivovaný faktor IX (FIXa), když je to potřeba pro koagulaci.
Farmakodynamické účinky
Hemofilie B je pohlavně vázaná dědičná porucha srážení krve způsobená sníženou hladinou faktoru IX a výsledkem je silné krvácení do kloubů, svalů nebo vnitřních orgánů, a to buď spontánně nebo v důsledku náhodného nebo chirurgického traumatu. Substituční terapií se zvýší plazmatické hladiny faktoru IX a tím umožní dočasnou korekci nedostatku faktoru a úpravu sklonů ke krvácení.
Faktor IX je aktivován faktorem VII/komplexem tkáňového faktoru vnější cestou a také faktorem XIa ve vnitřním koagulačním systému. Aktivovaný faktor IX v kombinaci s aktivovaným faktorem VIII aktivuje faktor X. To vede v konečném důsledku ke konverzi protrombinu na trombin. Trombin pak přeměňuje fibrinogen na fibrin a může se vytvořit sraženina. Aktivita faktoru IX chybí nebo je značně snížena u pacientů s hemofilií B a může požadovaná substituční terapie.
Klinická účinnost a bezpečnost
Studie fáze 1/2 hodnotí účinnost léčby a prevence krvácivých příhod pro rIX-FP u 17 subjektů (ve věku 13 - 46 let), 13 subjektů v profylaktické skupině dostávalo týdenní profylaxi s přípravkem IDELVION po dobu asi 11 měsíců a 4 subjekty ve skupině použití přípravku podle potřeby dostávaly přípravek IDELVION po výskytu krvácivých příhod. Všech 85 krvácivých příhod bylo úspěšně léčeno s 1 nebo 2 dávkami přípravku IDELVION.
Účinnost přípravku IDELVION byla hodnocena v otevřené nekontrolované části studie fáze 2/3, ve které dostávalo celkem 63 dříve léčených pacientů (PLP) mužského pohlaví ve věku od 12 do 61 let přípravek IDELVION jednou za 7, 10 a/nebo 14 dní v rámci profylaxe a/nebo podle potřeby v rámci léčby krvácivých příhod. Všechny subjekty měly těžkou (hladina FIX <1%) nebo středně těžkou (hladina FIX < 2%) hemofilii B. Čtyřicet PLP dostávalo přípravek IDELVION v rámci profylaxe.
Pacienti, kteří dostávali profylaktickou léčbu, začínali s 35-50 IU/kg jednou týdně. Podskupiny pacientů, kteří přešli na prodloužené intervaly léčby (každých 10 nebo 14 dnů) s doporučenou dávkou 75 IU/kg a individuálním nastavením. 21 PLP zůstalo na prodlouženém intervalu 14 dnů profylaxe po celou dobu léčby 98 až 575 (medián 386) dnů. Z těchto subjektů, u 8 (38%) se vyskytlo alespoň jedno krvácení během 14ti denní profylaxe, zatímco ostatní neměli krvácivou příhodou při profylaxi jednou týdně. Průměrná celková roční míra krvácení (ABR) 7. den profylaxe s přípravkem IDELVION byl 0,0 (rozmezí 0-6) pro všechna krvácení a při 14ti denní profylaxi byla 1,08 (rozmezí 0 - 9,1).
V současné době jsou k dispozici dostupné informace k podpoře prodlouženého intervalu léčby, u některých pacientů však mohou být spojeny se zvýšeným rizikem krvácení ve srovnání s jednotýdenním režimem.
Za zmínku stojí, že ABR není kompatibilní mezi různými koncentráty faktoru a mezi různými klinickými studiemi.
Profylaxe a kontrola krvácení u PLP do 12 let
Účinnost přípravku IDELVION byla hodnocena ve studii fáze 3, ve které dostávalo celkem 27 PLP mužského pohlaví ve věku od 1 do 10 let (střední věk 6,0 let) s 12 pacienty <6 let přípravek IDELVION v rámci profylaxe a kontroly krvácivých příhod. Všech 27 subjektů dostávalo týdenní profylaktickou léčbu s přípravkem IDELVION po průměrnou dobu studie 13,1 měsíců (9, 18 měsíců).
Ze 106 krvácivých příhod byla většina (94; 88,7%) léčena jednou injekcí, 103; 97,2% bylo léčeno 1-2 injekcemi. Hemostatická účinnost při řešení krvácení byla hodnocena jako výborná nebo dobrá u 96% všech léčených krvácivých příhod.
Nadále probíhají klinické studie zkoumající bezpečnost a účinnost léčby v delších intervalech než jednou týdně.
Perioperační řízení
Bezpečnost a účinnost v perioperačním období byla hodnocena ve dvou hlavních klinických studiích fáze 3 (Studie 3001 a 3002) a v probíhající prodloužené bezpečnostní studii fáze 3 (Studie 3003). "Per-protocol" analýza účinnosti zahrnuje 15 operací provedených u 12 pacientů ve věku od 8 do 51 let, kteří podstoupili větší či menší chirurgické, stomatologické nebo jiné chirurgické invazivní zákroky.
Přípravek IDELVION byl podáván bolusovou injekcí.
Hemostáza byla udržována po celou dobu trvání studie.
Pediatrická populace
Evropská léková agentura udělila odklad povinnosti předložit výsledky studií s přípravkem IDELVION při léčbě a profylaxi krvácení u dříve neléčených pacientů s hemofilií B (viz bod 4.2 informace o použití u dětí).
5.2 Farmakokinetické vlastnosti
Dospělá populace
Farmakokinetiky (PK) přípravku IDELVION byly hodnoceny po intravenózní injekci jedné dávky 25, 50 a 75 IU/kg. Parametry PK po jednorázové injekci 50 a 75 IU/kg přípravku IDELVION (viz tabulka níže) byly založeny na aktivitě faktoru IXv plazmě, měřeno jednostupňovým testem srážlivosti. Průměrná aktivita faktoru IX v den 7 a den 14 byla 13,76 % a 6,10 %, v tomto pořadí, po jednorázové dávce 50 IU/kg přípravku IDELVION. Opakované hodnocení PK po dobu až 30 týdnů prokázala stabilní farmakokinetický profil a inkrementální recovery bylo v průběhu času stálé.
V cílených klinických studiích pro dosažení kontroly nad krvácením při profylaxi byly pozorovány průměrné hladiny 5-10 %. PK simulace naznačují, že čas k dosažení 5% aktivity FIX v plazmě po jednorázové injekci 50 IU/kg přípravku IDELVION byl 12,5 dnů pro dospělé.
Farmakokinetické parametry u pacientů s těžkou hemofilií (medián (min, max)) po jedné injekci 50 IU/kg přípravku IDELVION
|
PK Parametry |
IDELVION (50 (IU/kg)) (N=22) |
|
IR (IU/dl)/(IU/kg) |
1,18 (0,86; 1,86) |
|
C '-/max (IU/dl) |
62,7 (40,5; 87,0) |
|
AUC0-inf (h*IU/dl) |
6638 (2810; 9921) |
|
Eliminace tJ/2 (h) |
95,3 (51,5; 135,7) |
|
CL (ml/h/kg) |
0,875 (0,748; 1,294) |
IR = přírůstková recovery; AUC = plocha pod křivkou aktivity faktoru IX; CL = clearance nastavená podle tělesné hmotnosti; Eliminace t1/2 = Eliminační poločas
Pediatrická populace
Farmakokinetické (PK) parametry přípravku IDELVION byly hodnoceny u dospívajících (ve věku 12 až <18 let) a dětí (ve věku 1 až <12 let) po intravenózní injekci jednorázové dávky 50 IU/kg. PK parametry (uvedené níže) byly odhadnuty na základě aktivity faktoru IX v plazmě v průběhu času profilu, měřená jednostupňovým testem srážlivosti.
Srovnání farmakokinetických parametrů přípravku IDELVION podle věkové kategorie (medián (min, max)) po jedné injekci 50 IU/kg přípravku IDELVION _
|
PK Paramety |
1 až <6 let (N=12) |
6 až <12 let (N=15) |
12 až <18 let (N=8) |
|
IR (IU/dl)/(IU/kg) |
0,968 (0,660; 1,280) |
1,07 (0,70; 1,47) |
1,11 (0,84; 1,61) |
|
C v-/max (IU/dl) |
48,2 (33,0; 64,0) |
50,5 (34,9; 73,6) |
55,3 (40,5; 80,3) |
|
AUC0-inf (h*IU/dl) |
4301 (2900; 8263) |
4663 (3212; 7720) |
4804 (2810;9595) |
|
Eliminace tJ/2 (h) |
86,2 (72,6; 105,8) |
89,0 (62,1; 123,0) |
88,8 (51,5; 130,0) |
|
CLa (ml/h/kg) |
1,16 (0,61; 1,72) |
1,07 (0,65; 1,56) |
1,04 (0,52; 1,67) |
a = odhad doby mediánu aktivity faktoru IX výše předem stanovených %
IR = přírůstková recovery; AUC = plocha pod křivkou aktivity faktoru IX; CL = clearance nastavená podle tělesné hmotnosti; Eliminace t1/2 = Eliminační poločas
V cílených klinických studiích pro dosažení kontroly nad krvácením při profylaxi byly pozorovány průměrné hladiny 5-10 %.
PK simulace naznačuje, že čas k dosažení 5% aktivity FIX v plazmě po jednorázové injekci 50 IU/kg přípravku IDELVION je 7 dnů pro 1- <6 let, 9 dnů pro 6- <12 let a 11 dnů pro 12- <18 let věku).
5.3 Předklinické údaje vztahující se k bezpečnosti
Předklinické údaje neodhalily žádné zvláštní riziko pro člověka na základě konvenčních farmakologických studií bezpečnosti, toxicity po jednorázovém a opakovaném podání, genotoxicity, trombogenicity a lokální tolerance.
Studie karcinogenity a reprodukční toxicity nebyly provedeny.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek:
dihydrát natrium-citrátu, polysorbát 80, mannitol, sacharóza, kyselina chlorovodíková (na úpravu pH).
Rozpouštědlo:
Voda na injekci
6.2 Inkompatibility
Vzhledem k absenci studií kompatibility, tento přípravek nesmí být smíchán s jinými léky, ředidly nebo rozpouštědly, kromě těch, které jsou uvedeny v bodě 6.1.
6.3 Doba použitelnosti
IDELVION 250 IU prášek a rozpouštědlo pro injekční roztok
2 roky
IDELVION 500 IU prášek a rozpouštědlo pro injekční roztok
2 roky
IDELVION 1000 IU prášek a rozpouštědlo pro injekční roztok
3 roky
IDELVION 2000 IU prášek a rozpouštědlo pro injekční roztok
3 roky
Po rekonstituci byla prokázána chemická a fyzikální stabilita pro použití během 8 hodin při 2-25 °C). Z mikrobiologického hlediska má být přípravek použit okamžitě. Není-li použit okamžitě, doba a podmínky uchovávání před použitím jsou v odpovědnosti uživatele.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte při teplotě do 25 °C.
Chraňte před mrazem. Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
Podmínky uchovávání tohoto léčivého přípravku po jeho rekonstituci jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
IDELVION 250 IU prášek a rozpouštědlo pro injekční roztok
Prášek (250 IU) v 6 ml injekční lahvičce (sklo třídy I) se zátkou (pryž) víčkem (plast) a uzávěrem (hliník).
2.5 ml rozpouštědla v injekční lahvičce (sklo třídy I) se zátkou (pryž) víčkem (plast) a uzávěrem (hliník).
IDELVION 500 IU prášek a rozpouštědlo pro injekční roztok
Prášek (500 IU) v 6 ml injekční lahvičce (sklo třídy I) se zátkou (pryž) víčkem (plast) a uzávěrem (hliník).
2.5 ml rozpouštědla v injekční lahvičce (sklo třídy I) se zátkou (pryž) víčkem (plast) a uzávěrem (hliník).
IDELVION 1000 IU prášek a rozpouštědlo pro injekční roztok
Prášek (1000 IU) v 6 ml injekční lahvičce (sklo třídy I) se zátkou (pryž) víčkem (plast) a uzávěrem (hliník).
2.5 ml rozpouštědla v injekční lahvičce (sklo třídy I) se zátkou (pryž) víčkem (plast) a uzávěrem (hliník).
IDELVION 2000 IU prášek a rozpouštědlo pro injekční roztok
Prášek (2000 IU) v 10 ml injekční lahvičce (sklo třídy I) se zátkou (pryž) víčkem (plast) a uzávěrem (hliník).
5 ml rozpouštědla v injekční lahvičce (sklo třídy I) se zátkou (pryž) víčkem (plast) a uzávěrem (hliník).
Velikost balení Každé balení obsahuje:
IDELVION 250 IU prášek a rozpouštědlo pro injekční roztok 1 injekční lahvičku s práškem 1 injekční lahvičku s 2,5 ml vody na injekci Jedna aplikační souprava obsahuje:
1 přepouštěcí adaptér s filtrem 20/20 1 injekční stříkačka 5 ml k jednorázovému použití
1 venepunkční set
2 tampony s alkoholem 1 nesterilní náplast
IDELVION 500 IU prášek a rozpouštědlo pro injekční roztok 1 injekční lahvičku s práškem 1 injekční lahvičku s 2,5 ml vody na injekci Jedna aplikační souprava obsahuje:
1 přepouštěcí adaptér s filtrem 20/20 1 injekční stříkačka 5 ml k jednorázovému použití
1 venepunkční set
2 tampony s alkoholem 1 nesterilní náplast
IDELVION 1000 IU prášek a rozpouštědlo pro injekční roztok 1 injekční lahvičku s práškem 1 injekční lahvičku s 2,5 ml vody na injekci Jedna aplikační souprava obsahuje:
1 přepouštěcí adaptér s filtrem 20/20 1 injekční stříkačka 5 ml k jednorázovému použití
1 venepunkční set
2 tampony s alkoholem 1 nesterilní náplast
IDELVION 2000 IU prášek a rozpouštědlo pro injekční roztok 1 injekční lahvičku s práškem 1 injekční lahvičku s 5 ml vody na injekci Jedna aplikační souprava obsahuje:
1 přepouštěcí adaptér s filtrem 20/20 1 injekční stříkačka 10 ml k jednorázovému použití
1 venepunkční set
2 tampony s alkoholem 1 nesterilní náplast
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Všeobecné pokyny
- Rekonstituovaný roztok má být čirý nebo slabě opalizující. Po filtraci/natáhnutí (viz dále) má být rekonstituovaný přípravek před aplikací vizuálně zkontrolován, zda neobsahuje částice nebo nezměnil zbarvení.
- Nepoužívejte roztoky, které jsou zakalené nebo obsahují usazeniny.
- Rekonstituce a natáhnutí roztoku se musí provádět za aseptických podmínek.
Rekonstituce
Ohřejte rozpouštědlo na pokojovou teplotu (pod 25 °C). Ujistěte se, že vyklápěcí uzávěry injekčních lahviček s produktem a rozpouštědlem byly odstraněny a zátky byly ošetřeny antiseptickým roztokem a nechaly se vysušit před otevřením Mix2Vial (zařízení pro rekonstituci).
1
2
3
4
5
6
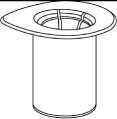
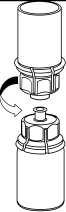
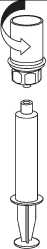
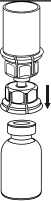
1. Otevřete Mix2Vial sloupnutím víčka. Nevytahujte Mix2Vial z blistru!
2. Postavte injekční lahvičku s rozpouštědlem na rovný a čistý povrch a pevně ji držte. Uchopte Mix2Vial společně s blistrem a zatlačte hrot konce modrého adaptéru přímo dolů přes zátku injekční lahvičky s rozpouštědlem.
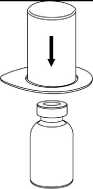
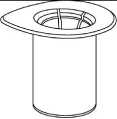
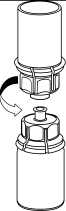
3. Opatrně odstraňte blistr ze soupravy Mix2Vial tak, že ho držíte za okraj a táhnete svisle nahoru. Přesvědčte se, že jste vytáhli jen blistrový obal, nikoli soupravu Mix2Vial.
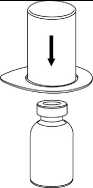
4. Postavte injekční lahvičku s práškem na
rovný a pevný povrch. Otočte injekční lahvičku s rozpouštědlem a připojeným setem Mix2Vial dnem nahoru a zatlačte hrot průhledného konce adaptéru rovně dolů přes zátku injekční lahvičky s přípravkem. Rozpouštědlo automaticky přeteče do injekční lahvičky s přípravkem.
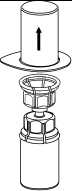
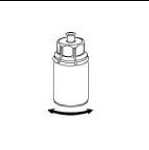
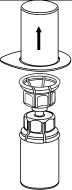
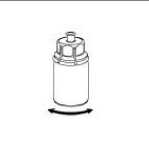
5. Uchopte jednou rukou část setu Mix2Vial uchycenou na injekční lahvičce s práškem a druhou rukou uchopte část setu uchycenou na injekční lahvičce s rozpouštědlem a odšroubováním proti směru hodinových ručiček rozdělte opatrně set na dvě části. Injekční lahvičku s rozpouštědlem a připojeným modrým adaptérem setu Mix2Vial zlikvidujte.
6. Injekční lahvičku s připojeným průhledným adaptérem jemně otáčejte, dokud se přípravek úplně nerozpustí. Injekční lahvičkou netřepejte.
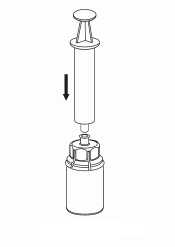
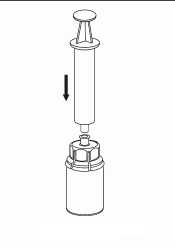
7. Nasajte vzduch do prázdné sterilní injekční stříkačky. Zatímco je injekční lahvička s přípravkem dnem dolů, spojte injekční stříkačku s koncovkou Luer Lock soupravy Mix2Vial šroubováním ve směru hodinových ručiček. Vstříkněte vzduch do injekční lahvičky s přípravkem
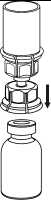
Natáhnutí a aplikace
8
8. Otočte systém dnem vzhůru a současně držte píst injekční stříkačky stlačený. Natáhněte roztok do injekční stříkačky pomalým vytahováním pístu.
9
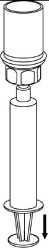
9. Po natáhnutí roztoku do injekční stříkačky uchopte pevně válec stříkačky (píst směřuje stále dolů) a odpojte průhledný adaptér setu Mix2Vial od injekční stříkačky odšroubováním proti směru hodinových ručiček.
K injekci přípravku IDELVION je nutné používat pouze dodávanou aplikační sadu, protože může dojít k selhání léčby v důsledku adsorpce faktoru IX na vnitřní povrch některého injekčního zařízení.
Je třeba dbát na to, aby se žádná krev nedostala do injekční stříkačky s produktem, protože je zde nebezpečí, že by se mohla srážet krev v injekční stříkačce a sraženina fibrinu by mohla být podána pacientovi.
Roztok přípravku IDELVION se nesmí ředit.
Rekonstituovaný roztok má být podáván pomalou intravenózní injekcí. Rychlost podání má být určena komfortem pacienta, a to maximálně do 5 ml/min.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
CSL Behring GmbH Emil-von-Behring-Str. 76 35041 Marburg Německo
8.
REGISTRAČNÍ ČÍSLO(A)
EU/1/16/1095/001
EU/1/16/1095/002
EU/1/16/1095/003
EU/1/16/1095/004
DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
9.
Datum první registrace:
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce biologické léčivé látky
CSL Behring GmbH Emil-von-Behring Strasse 76 35041 Marburg Německo
Název a adresa výrobce odpovědného za propouštění šarží
CSL Behring GmbH Emil-von-Behring Strasse 76 35041 Marburg Německo
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl.
107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
Držitel rozhodnutí o registraci předloží první pravidelně aktualizovanou zprávu o bezpečnosti pro tento léčivý přípravek do 6 měsíců od jeho registrace.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU Krabička 250IU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
IDELVION 250 IU
prášek a rozpouštědlo pro injekční roztok Albutrepenonacogum alfa
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Albutrepenonacogum alfa 250 IU
3. SEZNAM POMOCNÝCH LÁTEK
Pomocné látky: dihydrát natrium-citrátu, polysorbát 80, mannitol, sacharóza, kyselina chlorovodíková
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Prášek a rozpouštědlo pro injekční roztok
1 injekční lahvička s práškem: albutrepenonacogum alfa 250 IU (100 IU/ml po rekonstituci) 1 injekční lahvička s 2,5 ml vody na injekci Jedna aplikační souprava obsahuje:
1 přepouštěcí adaptér s filtrem 20/20 1 injekční stříkačka 5 ml k jednorázovému použití
1 venepunkční set
2 tampony s alkoholem 1 nesterilní náplast
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci.
Intravenózní podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte při teplotě do 25 °C. Chraňte před mrazem.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
CSL Behring GmbH, 35041 Marburg, Německo 12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/16/1095/001
13. ČÍSLO ŠARŽE
Č. šarže:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
IDELVION 250 IU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
IDELVION 250 IU prášek pro injekční roztok Albutrepenonacogum alfa Intravenózní podání
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Voda na injekci 2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
2,5 ml
6. JINÉ
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU Krabička 500IU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
IDELVION 500 IU
prášek a rozpouštědlo pro injekční roztok Albutrepenonacogum alfa
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Albutrepenonacogum alfa 500 IU
3. SEZNAM POMOCNÝCH LÁTEK
Pomocné látky: dihydrát natrium-citrátu, polysorbát 80, mannitol, sacharóza, kyselina chlorovodíková
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Prášek a rozpouštědlo pro injekční roztok
1 injekční lahvička s práškem: albutrepenonacogum alfa 500 IU (200 IU/ml po rekonstituci) 1 injekční lahvička s 2,5 ml vody na injekci Jedna aplikační souprava obsahuje:
1 přepouštěcí adaptér s filtrem 20/20 1 injekční stříkačka 5 ml k jednorázovému použití
1 venepunkční set
2 tampony s alkoholem 1 nesterilní náplast
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci.
Intravenózní podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte při teplotě do 25 °C. Chraňte před mrazem.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
CSL Behring GmbH, 35041 Marburg, Německo 12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/16/1095/002
13. ČÍSLO ŠARŽE
Č. šarže:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
IDELVION 500 IU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
IDELVION 500 IU prášek pro injekční roztok Albutrepenonacogum alfa Intravenózní podání
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Voda na injekci 2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
2,5 ml
6. JINÉ
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU Krabička 1000IU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
IDELVION 1000 IU
prášek a rozpouštědlo pro injekční roztok
Albutrepenonacogum alfa
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Albutrepenonacogum alfa 1000 IU
3. SEZNAM POMOCNÝCH LÁTEK
Pomocné látky: dihydrát natrium-citrátu, polysorbát 80, mannitol, sacharóza, kyselina chlorovodíková
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Prášek a rozpouštědlo pro injekční roztok
1 injekční lahvička s práškem: albutrepenonacogum alfa 1000 IU (400 IU/ml po rekonstituci) 1 injekční lahvička s 2,5 ml vody na injekci Jedna aplikační souprava obsahuje:
1 přepouštěcí adaptér s filtrem 20/20 1 injekční stříkačka 5 ml k jednorázovému použití
1 venepunkční set
2 tampony s alkoholem 1 nesterilní náplast
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci.
Intravenózní podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte při teplotě do 25 °C. Chraňte před mrazem.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
CSL Behring GmbH, 35041 Marburg, Německo 12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/16/1095/003
13. ČÍSLO ŠARŽE
Č. šarže:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
IDELVION 1000 IU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
IDELVION 1000 IU prášek pro injekční roztok Albutrepenonacogum alfa Intravenózní podání
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Voda na injekci 2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
2,5 ml
6. JINÉ
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU Krabička 2000IU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
IDELVION 2000 IU
prášek a rozpouštědlo pro injekční roztok
Albutrepenonacogum alfa
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Albutrepenonacogum alfa 2000 IU
3. SEZNAM POMOCNÝCH LÁTEK
Pomocné látky: dihydrát natrium-citrátu, polysorbát 80, mannitol, sacharóza, kyselina chlorovodíková
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Prášek a rozpouštědlo pro injekční roztok
1 injekční lahvička s práškem: albutrepenonacogum alfa 2000 IU (400 IU/ml po rekonstituci) 1 injekční lahvička s 5 ml vody na injekci Jedna aplikační souprava obsahuje:
1 přepouštěcí adaptér s filtrem 20/20 1 injekční stříkačka 10 ml k jednorázovému použití
1 venepunkční set
2 tampony s alkoholem 1 nesterilní náplast
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Intravenózní podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte při teplotě do 25 °C. Chraňte před mrazem.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
CSL Behring GmbH, 35041 Marburg, Německo 12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/16/1095/004
13. ČÍSLO ŠARŽE
Č. šarže:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
IDELVION 2000 IU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
IDELVION 2000 IU prášek pro injekční roztok Albutrepenonacogum alfa Intravenózní podání
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Voda na injekci 2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
5 ml
6. JINÉ
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro uživatele
IDELVION 250 IU prášek a rozpouštědlo pro injekční roztok IDELVION 500 IU prášek a rozpouštědlo pro injekční roztok IDELVION 1000 IU prášek a rozpouštědlo pro injekční roztok IDELVION 2000 IU prášek a rozpouštědlo pro injekční roztok
Albutrepenonacogum alfa
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
• Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
• Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
• Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy
• Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek IDELVION a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek IDELVION používat
3. Jak se přípravek IDELVION používá
4. Možné nežádoucí účinky
5 Jak přípravek IDELVION uchovávat
6. Obsah balení a další informace
1. Co je přípravek IDELVION a k čemu se používá Co je přípravek IDELVION?
Přípravek IDELVION je lék k léčbě hemofilie, který nahrazuje přirozený (koagulační) faktor IX na srážení krve. Léčivá látka přípravku IDELVION je albutrepenonakog alfa (rekombinantní fúzní protein spojující koagulační faktor IX s albuminem (rIX-FP)).
Faktor IX se podílí na srážení krve. Pacienti s hemofilii B mají nedostatek tohoto faktoru, který znamená, že jejich krev není srážená tak rychle, jak je třeba a zvyšuje se sklon ke krvácení. Přípravek IDELVION funguje jako náhrada faktoru IX u pacientů s hemofilií B, která umožní jejich srážení krve.
K čemu se přípravek IDELVION používá?
IDELVION se používá k prevenci nebo k zastavení krvácení způsobené nedostatkem faktoru IX u pacientů všech věkových skupin s hemofilií B (také nazývaný vrozený nedostatek faktoru IX nebo Christmasova nemoc).
2. Čemu musíte věnovat pozornost, než začnete přípravek IDELVION používat Nepoužívejte přípravek IDELVION
• Jestliže jste alergický(á) na léčivou látku (albutrepenonakog alfa) nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
• Jestliže j ste alergický(á) na křeččí proteiny.
Upozornění a opatření
Před použitím přípravku IDELVION se poraďte se svým lékařem, lékárníkem nebo zdravotní sestrou.
• Jsou možné alergické reakce (přecitlivělost). Přípravek obsahuje stopy křeččích proteinů (viz také "Nepoužívejte IDELVION"). Objeví-li se příznaky alergických reakcí, okamžitě přestaňte používat tento přípravek a kontaktujte svého lékaře. Váš lékař by Vás měl informovat o časných příznacích hypersenzitivních reakcí. Patří mezi ně vyrážku, generalizovaná kožní vyrážka, pocit tíže na hrudi, dušnost, nízký krevní tlak (hypotenze) a anafylaxe (závažná alergická reakce, která způsobuje závažné potíže s dýcháním, nebo závratě).
• Vzhledem k riziku alergických reakcí s faktorem IX, Vaše první podání přípravku IDELVION má být provedeno pod lékařským dohledem, kde je možno poskytnout řádnou lékařskou péči pro alergické reakce.
• Tvorba inhibitorů (neutralizačních protilátek) je známou komplikací, která může nastat během léčby, a která zastaví správně fungující léčbu. Pokud krvácení s přípravkem IDELVION není kontrolováno, ihned informujte svého lékaře. Měl(a) byste být pečlivě monitorován(a) na rozvoj inhibitorů.
• Pokud trpíte onemocněním jater nebo srdečním onemocněním nebo pokud jste v nedávné době měl(a) velký chirurgický zákrok, informujte svého lékaře, protože je zde zvýšené riziko komplikací srážení krve (koagulace).
• Pokud potřebujete centrální žilní přístup (CVAD) pro injekci přípravku IDELVION, má být lékařem zváženo riziko komplikací včetně lokálních infekcí, bakterií v krvi (bakteriémie) a vytváření krevních sraženin v cévách (trombóza) v místě zavedení katetru.
Záznam o použití
Důrazně se doporučuje, že pokaždé, když je Vám podán přípravek IDELVION, datum podání, číslo šarže a aplikovaný objem je zaznamenán ve Vašem deníku léčby.
Další léčivé přípravky a přípravek IDELVION
• Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Těhotenství, kojení a plodnost
• Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek používat.
• Během těhotenství a kojení, má být přípravek IDELVION podán pouze tehdy, pokud je to nezbytně nutné.
Řízení dopravních prostředků a obsluha strojů
Přípravek IDELVION nemá vliv na schopnost řídit motorová vozidla a obsluhovat stroje.
Přípravek IDELVION obsahuje sodík
Přípravek IDELVION obsahuje až 25,8 mg (1,13 mmol) sodíku na dávku (tělesná hmotnost 70 kg) jestliže se aplikuje maximální dávka (15 ml = 6000 IU). Prosím, vezměte toto v úvahu, pokud jste na dietě s nízkým obsahem sodíku.
Vaše léčba má být zahájena a sledována lékařem, který má zkušenosti s léčbou poruch krevní srážlivosti.
Dávkování
Váš doktor vypočte dávku přípravku IDELVION, kterou potřebujete. Množství přípravku IDELVION které musíte dostat a délka léčby závisí na:
• závažnosti onemocnění
• místě a intenzitě krvácení
• Vašem klinickém stavu a reakci
• Vaší tělesné hmotnosti
Postupujte podle pokynů, které Vám dal Váš lékař.
Jestliže jste použil(a) více přípravku IDELVION, než jste měl(a)
Prosím, kontaktujte okamžitě svého lékaře, pokud jste si aplikoval(a) více přípravku IDELVION než Vám doporučil lékař.
Jestliže jste přestal(a) používat přípravek IDELVION
Nepřestávejte používat přípravek IDELVION bez konzultace s lékařem.
Rekonstituce a aplikace
Všeobecné pokyny:
• Prášek se musí rozpustit v rozpouštědle (tekutině) a natáhnout z injekční lahvičky za aseptických podmínek.
• Přípravek IDELVION nesmí být mísen s jinými léčivými přípravky nebo rozpouštědly, s výjimkou těch, které jsou uvedeny v bodě 6.
• Roztok má být čirý nebo slabě opalizující, žlutý až bezbarvý, to znamená, že může drobnými částečkami rozptylovat světlo, ale nesmí obsahovat žádné viditelné částice. Po filtraci nebo natažení (viz dále) se roztok před použitím vizuálně zkontroluje. Nepoužívejte zakalený roztok, nebo roztok obsahující vločky nebo částice.
• Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky a podle pokynů vašeho lékaře.
Rekonstituce
Ohřejte neotevřené injekční lahvičky s práškem přípravku IDELVION a rozpouštědlem na pokojovou nebo tělesnou teplotu. Toto můžete udělat buď ponecháním injekčních lahviček při pokojové teplotě přibližně 1 hodinu, nebo ohřátím injekčních lahviček v rukách po dobu několika minut. Injekční lahvičky NESMÍTE vystavit přímému zdroji tepla. Injekční lahvičky nesmí být ohřáté na teplotu převyšující teplotu lidského těla (37 °C).
Opatrně odstraňte ochranná víčka z injekčních lahviček, očistěte odkryté gumové zátky tampónem napuštěným alkoholem. Před otevřením Mix2Vial ( zařízení pro rekonstituci) nechte injekční lahvičky oschnout, potom postupujte podle pokynů níže.
1
2
3
4
5
6
1. Otevřete Mix2Vial sloupnutím víčka. Nevytahujte Mix2Vial z blistru!
2. Postavte injekční lahvičku s rozpouštědlem
na rovný a čistý povrch a pevně ji držte. Uchopte Mix2Vial společně s blistrem a zatlačte hrot konce modrého adaptéru přímo dolů přes zátku injekční lahvičky s rozpouštědlem.
3. Opatrně odstraňte blistr ze soupravy Mix2Vial tak, že ho držíte za okraj a táhnete svisle nahoru. Přesvědčte se, že jste vytáhli jen blistrový obal, nikoli soupravu Mix2Vial.
4. Postavte injekční lahvičku s práškem na
rovný a pevný povrch. Otočte injekční lahvičku s rozpouštědlem a připojeným setem Mix2Vial dnem nahoru a zatlačte hrot průhledného konce adaptéru rovně dolů přes zátku injekční lahvičky s přípravkem. Rozpouštědlo automaticky přeteče do injekční lahvičky s přípravkem.
5. Uchopte jednou rukou část setu Mix2Vial uchycenou na injekční lahvičce s práškem a druhou rukou uchopte část setu uchycenou na injekční lahvičce s rozpouštědlem a odšroubováním proti směru hodinových ručiček rozdělte opatrně set na dvě části.
Injekční lahvičku s rozpouštědlem a připojeným modrým adaptérem setu Mix2Vial zlikvidujte.
6. Injekční lahvičku s připojeným průhledným adaptérem jemně otáčejte, dokud se přípravek úplně nerozpustí. Injekční lahvičkou netřepejte.
7. Nasajte vzduch do prázdné sterilní injekční stříkačky. Zatímco je injekční lahvička s přípravkem dnem dolů, spojte injekční stříkačku s koncovkou Luer Lock soupravy Mix2Vial šroubováním ve směru hodinových ručiček. Vstříkněte vzduch do injekční lahvičky s přípravkem.
Natáhnutí a aplikace
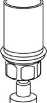
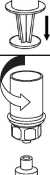
8
9
8. Otočte systém dnem vzhůru a současně držte píst injekční stříkačky stlačený. Natáhněte roztok do injekční stříkačky pomalým vytahováním pístu.
9. Po natažení roztoku do injekční stříkačky uchopte pevně válec stříkačky (píst směřuje stále dolů) a odpojte průhledný adaptér setu Mix2Vial od injekční stříkačky odšroubováním proti směru hodinových ručiček.
Použijte venepunkční set dodávaný spolu s přípravkem, zaveďte jehlu do žíly. Nechte protékat krev zpět na konec hadičky. Připojte stříkačku se závitem na uzavřený konec venepunkční soupravy. Aplikujte rekonstituovaný roztok pomalu (jak je to pohodlné pro Vás, maximálně do 5 ml/min) do žíly podle pokynů získaných od Vašeho lékaře. Dávejte pozor, aby se do naplněné injekční stříkačky nedostala žádná krev.
Pozorujte se, zda se u vás nevyskytnou jakékoli nežádoucí účinky. Pokud se vyskytnou nežádoucí účinky, které mohou souviset s podáním přípravku IDELVION, injekci musíte přerušit (viz též body 2 a 4).
Máte-li jakékoli další otázky, týkající se užívání tohoto přípravku, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí
vyskytnout u každého.
Kontaktujte prosím ihned svého lékaře:
• pokud si všimnete příznaků alergických reakcí (viz níže uvedených)
• pokud si všimnete, že lék přestane správně účinkovat
Následující nežádoucí účinky byly pozorovány u léků s faktorem IX:
• Hypersenzitivní reakce alergického typu jsou možné a mohou zahrnovat následující příznaky: kopřivka, kožní vyrážky (generalizovaná kopřivka), tlak na hrudi, sípání, nízký krevní tlak (hypotenze) a anafylaxe (Závažná reakce, která způsobuje vážné potíže s dýcháním a závratě). Pokud k tomu dojde, měli byste okamžitě přestat používat tento lék a kontaktovat svého lékaře.
• Inhibitory: lék přestane správně fungovat (pokračující krvácení). Může se vyvinout inhibitor (neutralizační protilátka) proti faktoru IX, v takovém případě nebude faktor IX správně fungovat. Pokud se toto stane, přestaňte používat tento lék a kontaktujte svého lékaře.
Následující nežádoucí účinky byly pozorovány u přípravku IDELVION často (mohou postihnout až 1 z 10 pacientů):
• Bolest hlavy
• Reakce v místě vpichu
Následující nežádoucí účinky se objevily méně často (mohou postihnout až 1 ze 100 pacientů):
• Závratě
• Alergické reakce (přecitlivělost)
• Vyrážka
• Ekzém
• Nežádoucí účinky u dětí a dospívajících
Očekávané nežádoucí účinky u dětí budou stejné jako u dospělých.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, zdravotní sestře nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek IDELVION uchovávat
• Uchovávejte tento přípravek mimo dohled a dosah dětí.
• Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na štítku a na krabičce.
• Uchovávejte při teplotě do 25 °C
• Chraňte před mrazem.
• Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
• Rekonstituovaný přípravek je nutné přednostně použít okamžitě.
• Pokud rekonstituovaný přípravek není podán okamžitě, doba a podmínky uchovávání před použitím jsou v odpovědnosti uživatele.
6. Obsah balení a další informace Co přípravek IDELVION obsahuje
Léčivou látkou je:
na injekci roztok obsahuje na injekci roztok obsahuje
250 IU v injekční lahvičce, po rekonstituci s 2,5 ml vody albutrepenonacogum alfa 100 IU/ml.
500 IU v injekční lahvičce, po rekonstituci s 2,5 ml vody albutrepenonacogum alfa 200 IU/ml.
1000 IU v injekční lahvičce, po rekonstituci s 2,5 ml vody na injekci roztok obsahuje albutrepenonacogum alfa 400 IU/ml.
2000 IU v injekční lahvičce, po rekonstituci s 5 ml vody na injekci roztok obsahuje albutrepenonacogum alfa 400 IU/ml.
Dalšími pomocnými látkami jsou:
dihydrát trinatrium-citrátu, polysorbát 80, mannitol, sacharóza a kyselina chlorovodíková (na úpravu
pH)
Viz poslední odstavec bodu 2.
Rozpouštědlo: voda na injekci
Přípravek IDELVION je jako světle žlutý až bílý prášek a je dodáván s vodou pro injekci jako rozpouštědlem.
Rekonstituovaný roztok má být čirý až lehce opalizující, žlutý až bezbarvý to znamená, že může jiskřit, když ho držíte proti světlu, ale nesmí obsahovat žádné viditelné částice.
Velikost balení
Jedno balení s 250 IU, 500 IU nebo 1000 IU obsahuje:
1 injekční lahvičku s práškem 1 injekční lahvičku s 2,5 ml vody na injekci Jedna aplikační souprava obsahuje:
1 přepouštěcí adaptér s filtrem 20/20 1 injekční stříkačka 5 ml k jednorázovému použití
1 venepunkční set
2 tampony s alkoholem 1 nesterilní náplast
Jedno balení s 2000 IU obsahuje:
1 injekční lahvičku s práškem 1 injekční lahvičku s 5 ml vody na injekci Jedna aplikační souprava obsahuje:
1 přepouštěcí adaptér s filtrem 20/20 1 injekční stříkačka 10 ml k jednorázovému použití
1 venepunkční set
2 tampony s alkoholem 1 nesterilní náplast
Držitel rozhodnutí o registraci a výrobce
CSL Behring GmbH Emil-von-Behring-StraBe 76 35041 Marburg Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Eunrapnu
HoBHMeg OOfl
Ten: +359 2 850 8617
Luxembourg/Luxemburg
CSL Behring NV Tél/Tel: +32 16 38 80 80
|
Deutschland CSL Behring GmbH Tel: +49 69 30584437 |
Nederland CSL Behring BV Tel: + 31 85 111 96 00 |
|
Eesti CSL Behring GmbH Tel: +49 69 30584437 |
Norge CSL Behring AB Tlf: +46 8 544 966 70 |
|
EXXáSa CSL Behring MEnE Tn^: +30 210 7255 660 |
Osterreich CSL Behring GmbH Tel: +43 1 80101 2463 |
|
Espaňa CSL Behring S.A. Tel: +34 933 67 1870 |
Polska Imed Poland Sp.z o.o. Tel: +48 22 663 43 10 CSL Behring Sp.z o.o. Tel: +48 22 213 22 65 |
|
France CSL Behring S.A. Tél: + 33 -(0)-1 53 58 54 00 |
Portugal CSL Behring Lda Tel: +351 21 782 62 30 |
|
Hrvatska PharmaSwiss d.o.o. Tel: +385 (1) 631-1833 |
Románia Prisum International Trading srl Tel: +40 21 322 0171 |
|
Ireland CSL Behring UK Ltd. Tel: +44 1444 447405 |
Slovenija MediSanus d.o.o. Tel: +386 1 25 71 496 |
|
Ísland CSL Behring AB Sími: +46 8 544 966 70 |
Slovenská republika CSL Behring s.r.o. Tel: +421 911 653 862 |
|
Italia CSL Behring S.p.A. Tel: +39 02 34964 200 |
Suomi/Finland CSL Behring AB Puh/Tel: +46 8 544 966 70 |
|
Kúnpoq AKH! nANAOQTOY & YIO! ATA Tn^: +357 22677038 |
Sverige CSL Behring AB Tel: +46 8 544 966 70 |
|
Latvija CSL Behring GmbH Tel: +49 69 30584437 |
United Kingdom CSL Behring UK Ltd. Tel: +44 1444 447405 |
Tato příbalová informace byla naposledy revidována {MM.RRRR}.
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu a na webových stránkách Státního ústavu pro kontrolu léčiv http://www.sukl.cz
Následující informace jsou určeny pouze pro zdravotnické pracovníky:
Dávkování
Dávka a délka substituční léčby závisí na závažnosti nedostatku faktoru IX, na místě a rozsahu krvácení a na klinickém stavu pacienta.
Počet jednotek podaného faktoru IX se vyjadřuje v mezinárodních jednotkách (IU), které jsou vztaženy k aktuálnímu standardu WHO pro přípravky s faktorem IX. Aktivita faktoru IX v plazmě se vyjadřuje buď v procentech (vzhledem k normální lidské plazmě) nebo v mezinárodních jednotkách (vzhledem k mezinárodnímu standardu pro faktor IX v plazmě).
Jedna mezinárodní jednotka (IU) aktivity faktoru IX je ekvivalentní množství faktoru IX v jednom ml normální lidské plazmy.
Požadovaná léčba
Výpočet požadované dávky faktoru IX je založen na empirickém zjištění, že při podání 1 mezinárodní jednotky (IU) faktoru IX na kg tělesné hmotnosti se předpokládá zvýšení cirkulující hladiny faktoru IX v průměru o 1,3 IU/dl (1,3% normálu) u pacientů ve věku > 12 let a o 1,0 IU / dl (1,0% normálu) u pacientů ve věku <12 let. Požadovaná dávka se určuje pomocí následujícího vzorce:
Požadovaná dávka (IU) = tělesná hmotnost (kg) x požadovaný vzestup faktoru IX (% normálu nebo IU/dl) x {převrácená hodnota pozorované recovery (IU/kg na IU/dl)}
Očekávané zvýšení hladiny faktoru IX (IU/dl nebo% normální hladiny) = dávka (IU) x Recovery (IU/dl na IU/kg)/tělesná hmotnost (kg)
Množství, které má být podáno a frekvence podávání má být vždy orientována na klinickou účinnost v konkrétním případě.
Pacienti ve věku <12 let
Pro kumulativní recovery 1 IU/dl na 1 IU/kg se dávka vypočte takto:
Dávka (IU) = tělesná hmotnost (kg) x požadované zvýšení faktoru IX (IU/dl) x 1 dl/kg
Příklad
1. U pacienta s hmotností 20 kg s těžkou hemofilií B se požaduje maximální hladina 50% normálu. Vhodná dávka bude 20 kg x 50 IU/dl x 1 dl/kg = 1000 IU.
2. Dávka 1000 IU přípravku IDELVION podaná 25 kg pacientovi bude mít očekávaný výsledek zvýšení maximální hladiny po injekci faktoru IX 1000 IU/25kg, x 1,0 (IU/dl na IU/kg)
= 40 IU/dl (40% běžné hodnoty).
Pacienti ve věku > 12 let
Pro kumulativní recovery 1,3 IU/dl na 1 IU/kg se dávka vypočte takto:
Dávka (IU) = tělesná hmotnost (kg) x požadované zvýšení faktoru IX (IU/dl) x 0,77 dl/kg
Příklad
3. U pacienta s hmotností 80 kg s těžkou hemofilií B se požaduje maximální hladina 50% normálu. Vhodná dávka bude 80 kg x 50 IU/dl x 0,77 dl/kg = 3080 IU.
4. Dávka 2000 IU přípravku IDELVION podaná 80 kg pacientovi bude mít očekávaný výsledek zvýšení maximální hladiny po injekci faktoru IX 2000 IU x 1,3 (IU/dl na IU/kg)/80 kg = 32,5 IU/dl (32,5% normálu).
V případě následujících krvácivých příhod by aktivita faktoru IX neměla v daném období klesnout pod určenou hladinu plazmatické aktivity (v % normálu nebo IU/dl). Tuto tabulku lze použít jako ukazatel v případě krvácení nebo operace:
|
Stupeň krvácení/typ chirurgického výkonu |
Požadovaná hladina faktoru IX (%) (IU/dl) |
Frekvence dávkování (hodiny)/délka trvání léčby (dny) |
|
Krvácení Začínající nebo mírná hemartróza, krvácení do svalů (s výjimkou iliopsoas) nebo ústní dutiny |
30-60 |
Jedna dávka je dostatečná pro většinu krvácení. Udržovací dávka po 24 - 72 hodinách, v případě, že krvácení pokračuje. |
|
Intenzivnější krvácení Život ohrožující krvácení, hluboké svalové krvácení včetně iliopsoas |
O O 1 O kO |
Opakovat každých 24-72 hodin, během prvního týdne a poté udržovací dávka každý týden až do zastavení krvácení a zahojení. |
|
Menší chirurgický výkon včetně nekomplikovaného vytržení zubu |
50-80 (počáteční hladina) |
Jedna dávka je dostačující pro většinu menších operací. V případě potřeby udržovací dávka může být za 24 - 72 hodin, až do zastavení krvácení a zahojení. |
|
Velké chirurgické výkony |
60-100 (počáteční hladina) |
Opakovat každých 24-72 hodin během prvního týdne a poté udržovací dávka 1-2 krát týdně, dokud se krvácení nezastaví a do zahojení. |
Profylaxe
Pro dlouhodobou profylaxi krvácení u pacientů s těžkou hemofilií B jsou obvyklé dávky 35 až 50 IU/kgjednou týdně.
Někteří pacienti, kteří jsou dobře kontrolováni v jednotýdenním režimu, mohou být léčeni dávkou až 75 IU/kg v intervalu 10 až 14 dnů (viz bod 5.1).
V některých případech, zejména u mladších pacientů, , mohou být nutné kratší intervaly nebo vyšší dávky.
Po výskytu krvácivé příhody během profylaxe mají pacienti co nejpřísněji dodržovat svůj profylaktický režim s 2 dávkami přípravku IDELVION podanými s odstupem alespoň 24 hodin, nebo déle, pokud se to považuje za vhodné pro pacienta.
Pediatrická populace
Pro rutinní profylaxi pediatrických pacientů je doporučený režim dávkování 35 až 50 IU/kg jednou týdně.
Zvláštní upozornění a opatření pro použití
Inhibitory
Po opakované léčbě lidským koagulačním faktorem IX mají být pacienti sledováni s ohledem na vývoj neutralizujících protilátek (inhibitorů), které mají být kvantifikovány v jednotkách Bethesda (BU) pomocí vhodného biologického testování.
Zprávy z literatury ukazují korelaci mezi výskytem inhibitoru faktoru IX a alergickými reakcemi. Proto pacienti trpící alergickými reakcemi musí být vyšetřeni na přítomnost inhibitoru. Je třeba poznamenat, že u pacientů s inhibitory faktoru IX může být ve zvýšené riziko anafylaxe s následnou stimulací faktorem IX.
Monitoring léčby
Během léčby se doporučuje vhodné stanovení hladin faktoru IX jako vodítko pro dávky, které mají být podávány a frekvence opakovaných infuzí. Jednotliví pacienti se mohou lišit v odpovědích na faktor IX, což demonstruje různé poločasy a recovery. Dávka vychází z tělesné hmotnosti a může vyžadovat úpravu s podvýživou nebo nadváhou pacientů. V případě velkých chirurgických zákroků je nezbytné přesné monitorování průběhu substituční terapie pomocí koagulační analýzy (aktivity plazmového faktoru IX).
Při použití in vitro tromboplastinového času (aPTT) založeného na jednostupňovém testu srážení krve pro stanovení aktivity faktoru IX ve vzorcích krve pacientů, výsledky aktivity faktoru IX v plazmě mohou být významně ovlivněny typem aPTT činidla i referenčním standardem používaným v testu. Měření jednofázovým testem srážení s použitím na kaolinu založeném aPTT činidlu nebo aktin FS aPTT činidlu, bude pravděpodobně mít za následek podcenění úrovně aktivity. To je důležité zejména při změně laboratoře a/nebo činidel použitých v testu.
48