Ibandronic Acid Accord 3 Mg
SOUHRN ÚDAJŮ O PŘÍPRAVKU
PŘÍLOHA I
Ibandronic Acid Accord 2 mg koncentrát pro infuzní roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička s 2 ml koncentrátu pro přípravu infuzního roztoku obsahuje acidum ibandronicum 2 mg (jako natrii monohydricus).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Koncentrát pro přípravu infuzního roztoku Čirý, bezbarvý roztok
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Kyselina ibandronová je indikována u dospělých pacientů k
- prevenci kostních příhod (patologických zlomenin, kostních komplikací, které vyžadují radiologickou nebo chirurgickou léčbu) u pacientů s karcinomem prsu a kostními metastázami
- léčbě hyperkalcemie vyvolané nádorem s výskytem nebo bez výskytu metastáz
4.2 Dávkování a způsob podání
Pacientům, kteří budou léčeni kyselinou ibandronovou, musí být předána příbalová informace a karta s připomínkami.
Léčba kyselinou ibandronovou má být zahájena pouze lékařem se zkušenostmi s léčbou onkologického onemocnění.
Dávkování
Prevence kostních _příhod u _pacientů s karcinomem _prsu a kostními metastázami
Doporučené dávkování v prevenci kostních příhod u pacientů s karcinomem prsu a kostními metastázami je 6 mg ve formě intravenózní injekce podávané každé 3-4 týdny. Jednotlivá dávka má být podána infuzí trvající alespoň 15 minut.
Kratší doba (tj. 15 min) infuze má být použita pouze u pacientů s normální funkcí ledvin nebo s mírně zhoršenou funkcí ledvin. Nejsou k dispozici žádné údaje charakterizující užití kratší doby infuze u pacientů s clearance kreatininu pod 50 ml/min. Předepisující lékař má věnovat pozornost doporučení o dávkování a podávání přípravku této skupině pacientů, které je uvedeno v bodu Pacienti s onemocněním ledvin (viz bod 4.2).
Léčba hyperkalcemie vyvolané nádorem
Před léčbou kyselinou ibandronovou musí být pacienti adekvátně rehydratováni 0,9% roztokem chloridu sodného. Je třeba brát v úvahu stupeň závažnosti hyperkalcemie stejně jako typ nádoru. Pacienti s osteolytickými metastázami kostí potřebují obecně nižší dávky než pacienti s humorálním typem hyperkalcemie. U většiny pacientů s těžkou hyperkalcemií (albuminem korigovaný sérový vápník* > 3 mmol/l nebo > 12 mg/dl) postačuje jednotlivá dávka 4 mg. U pacientů s mírnou hyperkalcemií (albuminem korigovaný sérový vápník < 3 mmol/l nebo < 12 mg/dl) je účinná dávka 2 mg. Nejvyšší dávka použitá v klinických studiích byla 6 mg, ale tato dávka se neprojevila vyšším účinkem.
* Poznámka: koncentrace albuminem korigovaného sérového vápníku se vypočte následujícím způsobem:
|
Koncentrace albuminem korigovaného sérového vápníku (mmol/l) |
vápník v séru (mmol/l) - [0,02 x albumin (g/l)] + 0,8 | |
|
Nebo | ||
|
Koncentrace albuminem korigovaného sérového vápníku (mg/dl) |
vápník v séru (mg/dl) + 0,8 x [4 - albumin (g/dl)] | |
|
Hodnoty albuminem korigované |
io sérového vápníku v mmol/l lze převést na mg/dl vynásobením 4. | |
Ve většině případů může být zvýšená hladina sérového vápníku snížena na normální hladinu během 7 dnů. Střední doba do relapsu (znovuzvýšení albuminem korigovaného sérového vápníku nad 3 mmol/l) byla 18 - 19 dnů při dávkách 2 mg a 4 mg. Střední doba do relapsu při dávce 6 mg byla 26 dnů.
Omezený počet pacientů (50 pacientů) dostal druhou infuzi z důvodů hyperkalcemie. Léčbu je třeba opakovat v případě vracející se hyperkalcemie nebo při nedostatečné účinnosti.
Koncentrát kyseliny ibandronové pro infuzní roztok má být podáván jako intravenózní infuze po dobu minimálně 2 hodin.
Zvláštní skupiny _pacientů
Pacienti s poruchou jaterních funkcí
Úprava dávky není nutná (viz bod 5.2).
Pacienti s poruchou renálních funkcí
U pacientů s mírnou poruchou funkce ledvin (CLcr >50 a <80 ml/min) není úprava dávky nutná. U pacientů s karcinomem prsu a kostními metastázami se středně těžkou poruchou funkce ledvin (CLcr > 30 a < 50 ml/min) nebo těžkou poruchou funkce ledvin (CLcr < 30 ml/min), kteří jsou léčeni z důvodu prevence kostních příhod, by se měla dodržovat následující dávkovací doporučení (viz bod 5.2):
|
Clearance kreatininu (ml/min) |
Dávka |
Objem 1 a doba 2 infuze |
|
>50 CLcr <80 >30_CLcr <50 < 30 |
6 mg (6 ml koncentrátu pro infuzní roztok) 4 mg (4 ml koncentrátu pro infuzní roztok) 2 mg (2 ml koncentrátu pro infuzní roztok) |
100 ml po dobu 15 min 500 ml po dobu 1 hodiny 500 ml po dobu 1 hodiny |
1 0,9% roztok chloridu sodného nebo 5% roztok glukózy
2 Při podávání každé 3 až 4 týdny
U pacientů s nádorovým onemocněním a CLCr < 50 ml/min nebyla doba infuze trvající 15 minut dosud studována.
Starší pacienti (>65 let)
Úprava dávky není nutná (viz bod 5.2).
Pediatrická populace
U dětí a dospívajících mladších než 18 let nebyla bezpečnost a účinnost kyseliny ibandronové stanovena. Žádné údaje nejsou k dispozici (viz bod 5.1 a 5.2).
Způsob podání K intravenóznímu podání.
Obsah injekční lahvičky má být použit následujícím způsobem:
- Prevence kostních příhod - obsah přidat ke 100 ml izotonického roztoku chloridu sodného nebo ke 100 ml 5% roztoku glukózy a aplikovat infuzi po dobu nejméně 15 minut. Viz také bod výše týkající se dávkování u pacientů s poruchou renálních funkcí
- Léčba hyperkalcemie vyvolané nádorem - obsah přidat k 500 ml izotonického roztoku chloridu sodného nebo k 500 ml 5% roztoku glukózy a aplikovat infuzi po dobu minimálně 2 hodin
Pouze k jednorázovému použití. Měl by být použit pouze čirý roztok bez jakýchkoli částic.
Koncentrát kyseliny ibandronové pro přípravu infuzního roztoku se aplikuje jako intravenózní infuze. Je nutno ve zvýšené míře dbát na to, aby nebyl koncentrát kyseliny ibandronové pro přípravu infuzního roztoku podán intraarteriálně nebo paravenózně, neboť by mohlo dojít k poškození tkáně.
4.3 Kontraindikace
- Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku tohoto přípravku uvedenou v bodě 6.1.
- Hypokalcemie.
4.4 Zvláštní upozornění a opatření pro použití
Pacienti s poruchami kostního a minerálového metabolismu
Před zahájením léčby kyselinou ibandronovou u metastatického postižení kostí je vhodné účinně léčit hypokalcemii a další poruchy kostního a minerálového metabolismu.
U všech pacientů je důležitý dostatečný přísun vápníku a vitaminu D. V případě jejich nedostatečného přísunu ve stravě mají pacienti dostávat doplňky vápníku a/nebo vitaminu D.
Anafylaktická reakce/šok
U pacientů léčených kyselinou ibandronovou v intravenózní formě byly hlášeny případy anafylaktické reakce/šoku včetně příhod končících úmrtím.
Pokud je podávána intravenózní injekce přípravku Ibandronic Acid Accord, je třeba, aby byla
dostupná vhodná lékařská péče a zajištěno odpovídající sledování pacienta. Jestliže se objeví anafylaktickénebo jiné závažné hypersenzitivní/alergické reakce, okamžitě ukončete podávání injekce a zahajte příslušnou léčbu.
Osteonekróza čelisti
Velmi vzácně byla u pacientů léčených kyselinou ibandronovou z důvodu onkologických indikací (viz odstavec 4.8) hlášena v poprodejním sledování osteonekróza čelisti (ONČ).
U pacientů s nezhojenými lézemi měkkých tkání v ústech je třeba odložit zahájení léčby nebo přechod na novou léčbu.
U pacientů s průvodními rizikovými faktory se před zahájením léčby kyselinou ibandronovou doporučuje provést preventivní zubní prohlídku a individuální posouzení přínosů a rizik.
Při vyhodnocování rizika rozvoje ONČ u pacienta je třeba zvažovat následující rizikové faktory:
- Účinnost léčivého přípravku, který inhibuje resorpci kostí (vyšší riziko u vysoce účinných přípravků), způsob podávání (vyšší riziko při parenterálním podávání) a kumulovaná dávka při léčbě resorpce kostí
- Onkologické onemocnění, další chorobné stavy (například anémie, koagulopatie, infekce), kouření
- Doprovodná léčba: kortikosteroidy, chemoterapie, angiogenní inhibitory, radioterapie hlavy a krku
Nedostatečná ústní hygiena, periodontální onemocnění, nedostatečně sedící umělý chrup, dentální onemocnění v anamnéze, invazivní dentální postupy léčby například extrakce zubů
Všem pacientům je třeba doporučit, aby během léčby kyselinou ibandronovou udržovali dobrou ústní hygienu, absolvovali rutinní zubní prohlídky a ihned ohlásili všechny příznaky onemocnění ústní dutiny jako pohyblivost zubů, bolesti nebo otoky, nehojící se vředy nebo výtoky. V průběhu léčby je třeba provádět invazivní zubní zákroky a postupy jedině po pečlivém zvážení a vyvarovat se jim v bezprostřední blízkosti podávání kyseliny ibandronové.
Plán léčby pacientů, u kterých se rozvine ONČ, musí být stanoven ve spolupráci ošetřujícího lékaře a zubaře nebo ústního chirurga s odbornými znalostmi o ONČ. Dokud se příslušný stav nevyřeší a nebude případně možné zmírnit rizikové faktory, je třeba zvažovat dočasné přerušení léčby kyselinou ibandronovou.
Osteonekróza vnějšího zvukovodu
V případě léčby bisfosfonáty, zejména ve spojení s dlouhodobou léčbou, byla hlášena osteonekróza vnějšího zvukovodu. Možné rizikové faktory osteonekrózy vnějšího zvukovodu zahrnují užívání steroidů a chemoterapii a/nebo lokální rizikové faktory, jakým je infekce nebo trauma. Možnost osteonekrózy vnějšího zvukovodu je třeba zvažovat u pacientů léčených bisfosfonáty, u kterých se projevují příznaky ušních onemocnění včetně chronických ušních infekcí.
Atypické zlomeniny femuru
V souvislosti s léčbou bisfosfonáty byly hlášeny atypické subtrochanterické a diafyzární zlomeniny femuru, zejména u pacientů dlouhodobě léčených pro osteoporózu. Tyto příčné nebo krátké šikmé zlomeniny se mohou objevit kdekoli v celé délce femuru od oblasti těsně pod malým trochanterem až do části těsně nad suprakondylickým rozšířením. Tyto zlomeniny se objevují po minimálním traumatu nebo bez souvislosti s traumatem a u některých pacientů se mohou projevovat bolestí ve stehně nebo třísle, často sdružené na zobrazovacích vyšetřeních s obrazem typickým pro stresové zlomeniny (neobvyklé nízkotraumatické zlomeniny, v angličtině známé jako „insufficiency fractures“)týdny až měsíce před manifestací kompletní zlomeniny femuru. Zlomeniny jsou často oboustranné, proto je nutné u pacientů léčených bisfosfonáty, kteří utrpěli zlomeninu diafýzy femuru, vyšetřit i kontralaterální femur. Rovněž bylo zaznamenáno špatné hojení těchto zlomenin. U pacientů, u kterých je podezření na atypickou zlomeninu femuru, je třeba při hodnocení jejich stavu zvážit i přerušení léčby bisfosfonáty, a to na základě zhodnocení prospěchu a rizika léčby u jednotlivého pacienta. Pacienty je třeba poučit, aby během léčby bisfosfonáty hlásili jakoukoli bolest v oblasti stehna, boku nebo třísla, a všechny pacienty, u kterých se tyto příznaky objeví, je třeba vyšetřit s ohledem na možnou inkompletní zlomeninu femuru.
Pacienti s poruchou renálních funkcí
Klinické studie neprokázaly, že by při dlouhodobé léčbě kyselinou ibandronovou docházelo ke zhoršení renálních funkcí. Nicméně se doporučuje na základě zhodnocení klinického stavu každého pacienta sledovat při léčbě kyselinou ibandronovou renální funkce, hladinu vápníku, fosfátu a hořčíku v séru (viz bod 4.2).
Pacienti s poruchou jaterních funkcí
Z důvodu nedostačujících klinických údajů není možné doporučit dávky u pacientů s těžkou jaterní dysfunkcí (viz bod 4.2).
Pacienti se srdeční poruchou
U pacientů ohrožených rozvojem srdečního selhání je třeba zabránit převodnění.
Pacienti se známou hvpersenzitivitou na jiné bisfosfonáty
Opatrnosti je třeba u pacientů se známou hypersenzitivitou na jiné bisfosfonáty.
Pomocné látky se známým účinkem
Tento léčivý přípravek obsahuje méně než 1 mmol sodíku (23 mg) v jedné injekční lahvičce, tzn., že je v podstatě “bez sodíku”.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Metabolické interakce se nepředpokládají vzhledem k tomu, že kyselina ibandronová neinhibuje hlavní lidské hepatické izoenzymy P450, a bylo prokázáno, že neindukuje systém hepatického cytochromu P 450 u potkanů (viz bod 5.2). Kyselina ibandronová se vylučuje pouze renální sekrecí a neprochází žádnou metabolickou přeměnou.
Zvýšená opatrnost se doporučuje při podávání bisfosfonátů souběžně s aminoglykosidy, neboť obě látky snižují koncentraci vápníku v séru na delší dobu. Je třeba také věnovat pozornost možnému rozvoji souběžné hypomagnesemie.
4.6 Fertilita, těhotenství a kojení
O podávání kyseliny ibandronové těhotným ženám nejsou k dispozici potřebné údaje. Studie u potkanů prokázaly reprodukční toxicitu (viz bod 5.3). Možné riziko u lidí není známo. Proto nemá být kyselina ibandronová těhotným ženám podávána.
Kojení
Není známo, zda se kyselina ibandronová vylučuje do lidského mléka. Studie u kojících potkanů prokázaly po intravenózním podání kyseliny ibandronové přítomnost její nízké koncentrace v mléce. Kojícím ženám nemá být kyselina ibandronová podávána.
Fertilita
Údaje týkající se účinků kyseliny ibandronové u člověka nejsou k dispozici. V reprodukčních studiích s perorálním podáním u potkanů kyselina ibandronová snižovala fertilitu. Ve studiích s intravenózním podáním u potkanů kyselina ibandronová snižovala fertilitu při vysokých denních dávkách (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Na základě farmakodynamického a farmakokinetického profilu a hlášených nežádoucích účinků lze předpokládat, že kyselina ibandronová nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky
Shrnutí bezpečnostního profilu
Nejzávažnější hlášené nežádoucí účinky jsou anafylaktická reakce/šok, atypické zlomeniny femuru, osteonekróza čelisti a oční záněty (viz odstavec „Popis vybraných nežádoucích účinků“ a bod 4.4).
Při hyperkalcemii vyvolané nádorem je léčba nejčastěji doprovázena zvýšením tělesné teploty. Méně často je hlášen pokles sérového vápníku pod normální hodnotu (hypokalcemie). Ve většině případů není zapotřebí zvláštní léčby a příznaky za pár hodin/dnů odezní.
V prevenci kostních příhod u pacientů s karcinomem prsu a kostními metastázami je léčba nejčastěji spojena s astenií doprovázenou zvýšením tělesné teploty a bolestí hlavy.
Shrnutí nežádoucích účinků do tabulky
V tabulce 1 jsou uvedeny nežádoucí účinky z klíčových studií fáze III (Léčba hyperkalcemie vyvolané nádorem: 311 pacientů léčených kyselinou ibandronovou 2 mg nebo 4 mg;
Prevence kostních příhod u pacientů s karcinomem prsu a kostními metastázami: 152 pacientů léčených kyselinou ibandronovou 6 mg) a po uvedení přípravku na trh.
Nežádoucí účinky jsou uvedeny podle tříd orgánových systémů dle databáze MedDRA a četnosti výskytu. Četnosti výskytu jsou definovány podle následujících konvencí: velmi časté (>1/10), časté (>1/100 až <1/10), méně časté (>1/1000 až <1/100), vzácné (>1/10000 až <1/1000), velmi vzácné (<1/10000), není známo (z dostupných údajů nelze určit). V rámci každé skupiny četností jsou nežádoucí účinky uvedeny v pořadí dle klesající závažnosti.
Tabulka 1 Nežádoucí účinky hlášené po intravenózním podání kyseliny ibandronové
|
Třída orgánových systémů |
Velmi časté |
Časté |
Méně časté |
Vzácné |
Velmi vzácné |
Není známo |
|
Infekce a infestace |
Infekce |
Cystitida, vaginitida, orální kandidóza | ||||
|
Novotvary benigní, maligní a blíže neurčené |
Benigní kožní novotvar | |||||
|
Poruchy krve a lymfatického systému |
Anémie, krevní dyskrazie | |||||
|
Poruchy imunitního systému |
Hypersenz itivitaf, bronchosp asmusf, angioedé mf, anafylakti cká reakce/šok f** |
Exacerbace astmatuf | ||||
|
Endokrinní poruchy |
Poruchy příštitných tělísek | |||||
|
Poruchy metabolismu a výživy |
Hypokalce mie** |
Hypofosfatemie | ||||
|
Psychiatrické poruchy |
Poruchy spánku, úzkost, afektivní labilita | |||||
|
Poruchy nervového systému |
Bolest hlavy, závratě, dysgeuzie (změny chuti) |
Cerebrovaskulární onemocnění, léze nervového kořene, amnezie, migréna, neuralgie, hypertonie, hyperestezie, cirkumorální parestezie, parosmie | ||||
|
Poruchy oka |
Katarakta |
Zánět okaf** | ||||
|
Poruchy ucha a labyrintu |
Hluchota |
|
Třída orgánových systémů |
Velmi časté |
Časté |
Méně časté |
Vzácné |
Velmi vzácné |
Není známo |
|
Srdeční poruchy |
Raménkov ý blok |
Ischemie myokardu, kardiovaskulární onemocnění, palpitace | ||||
|
Respirační, hrudní a mediastinální poruchy |
Faryngitida |
Plicní edém, stridor | ||||
|
Gastrointestinál ní poruchy |
gastrointest inální bolesti, zubní potíže |
Gastroenteritida, gastritida, ulcerace v ústech, dysfagie, cheilitida | ||||
|
Poruchy jater a žlučových cest |
Žlučové kameny | |||||
|
Poruchy kůže a podkožní tkáně |
Kožní onemocněn í, ekchymóza |
Vyrážka, alopecie |
Stevens- Johnsonův syndromf, erythema multiform ef, bulózní dermatitid af | |||
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Osteoartró za, myalgie, artralgie, onemocněn í kloubů |
Atypické subtrochante rické a diafyzární zlomeniny femuruf (skupinový nežádoucí účinek bisfosfonátů ) |
Osteonekr óza čelistif** Osteonekr óza vněj šího zvukovod u (nežádouc í účinek bisfosfoná tů) f | |||
|
Poruchy ledvin a močových cest |
Močová retence, ledvinná cysta | |||||
|
Poruchy reprodukčního systému a prsu |
Bolesti v pánvi | |||||
|
Celkové poruchy a reakce v místě aplikace |
Pyrexie, onemocněn í podobné chřipce**, periferní edém, astenie, žízeň |
Hypotermie | ||||
|
Vyšetření |
Zvýšení |
Zvýšení alkalické |
|
Třída orgánových systémů |
Velmi časté |
Časté |
Méně časté |
Vzácné |
Velmi vzácné |
Není známo |
|
gama-GT, zvýšení kreatininu |
fosfatázy v krvi, snížení tělesné hmotnosti | |||||
|
Poranění, otravy a procedurální komplikace |
Poranění, bolest v místě vpichu |
**Viz další informace níže fldentifikovány po uvedení přípravku na trh
Popis vybraných nežádoucích účinků
Hypokalcemie
Snížené vylučování kalcia ledvinami může být doprovázeno poklesem hladiny fosfátů v séru, které nevyžaduje terapeutický zásah. Hladina vápníku v séru může klesnout na hypokalcemické hodnoty.
Onemocnění _podobné chřipce
Objevily se příznaky podobné chřipce zahrnující horečku, třesavku, bolesti kostí a/nebo svalů. Ve většině případů nebylo zapotřebí zvláštní léčby a příznaky za pár hodin/dnů odezněly.
Osteonekróza čelisti
Případy osteonekrózy čelisti byly hlášeny převážně u pacientů s onkologickým onemocněním, léčených léčivými přípravky, které inhibují resorpci kostí, například kyselinou ibandronovou (viz odstavec 4.4.). Případy ONČ byly hlášeny v poprodejním sledování kyseliny ibandronové.
Zánět oka
Při podávání kyseliny ibandronové byly hlášeny oční zánětlivé reakce, jako jsou uveitida, episkleritida, skleritida. V některých případech tyto nežádoucí reakce neustoupily, dokud podávání kyseliny ibandronové nebylo ukončeno.
Anafylaktická reakce/šok
U pacientů léčených kyselinou ibandronovou v intravenózní formě byly hlášeny případy anafylaktické reakce/šoku včetně příhod končících úmrtím.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Zkušenosti s akutní otravou koncentrátem kyseliny ibandronové pro přípravu infuzního roztoku dosud nejsou. Vzhledem k tomu, že v preklinických studiích s vysokými dávkami přípravku byly toxicitou nejvíce postiženy játra a ledviny, je nutné jejich činnost během léčby monitorovat. Klinicky závažnou hypokalcemii lze upravit intravenózním podáním kalcium-glukonátu.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Léčivé přípravky ovlivňující stavbu a mineralizaci kostí, bisfosfonáty, ATC kód: M05BA06.
Kyselina ibandronová patří do skupiny bisfosfonátových sloučenin, které mají specifický účinek na kosti. Její selektivní působení na kostní tkáň je založeno na vysoké afinitě bisfosfonátů ke kostním minerálům. Bisfosfonáty inhibují aktivitu osteoklastů, ale přesný mechanizmus není dosud znám.
In vivo kyselina ibandronová zabraňuje pokusně způsobené destrukci kostí, která byla navozena přerušením funkcí gonád, retinoidy, nádory nebo extrakty z nádorů. Potlačení endogenní resorpce kostí bylo dokumentováno studiemi kinetiky 45Ca a uvolněním radioaktivního tetracyklinu předtím zavedeného do kostry.
V dávkách, které byly zřetelně vyšší než je farmakologicky účinná dávka, neměla kyselina ibandronová žádný účinek na mineralizaci kostí.
Pro resorpci kosti při malignitě je typická nadměrná resorpce, která není vyrovnána odpovídající novotvorbou kosti. Kyselina ibandronová selektivně inhibuje aktivitu osteoklastů, což vede ke zpomalení kostní resorpce a tím k sníženému výskytu kostních komplikací u maligních onemocnění.
Klinické studie léčby nádorem vyvolané hyperkalcemie
Klinické studie hyperkalcemie u malignit ukázaly, že inhibiční efekt kyseliny ibandronové na tumory indukovanou osteolýzu, a zvláště na nádorem vyvolanou hyperkalcemii, je charakterizován poklesem hladiny sérového vápníku a poklesem vylučování vápníku močí.
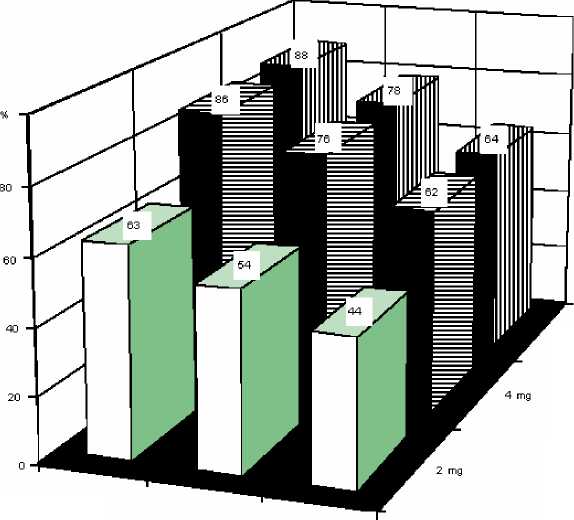
V dávkování doporučovaném pro léčbu byly zjištěny následující četnosti odezvy s odpovídajícím intervalem spolehlivosti na základě výsledků klinických studií u pacientů se vstupní koncentrací albuminem korigovaného sérového vápníku > 3,0 mmol/l po náležité rehydrataci.
6 mg
Dávky
kyseliny
ibandronové
Vysoký 90%
interval
spolehlivosti
Poměr léčebné odpovědi
Nízký 90%
interval
spolehlivosti
|
Dávka kyseliny ibandronové |
% pacientů s odpovědí |
90% interval spolehlivosti |
|
2 mg |
54 |
44-63 |
|
4 mg |
76 |
62-86 |
|
6 mg |
78 |
64-88 |
U těchto pacientů dostávajících výše uvedené dávky byl průměrný čas pro dosažení normokalcemie 4 -7 dnů. Průměrná doba recidivy (návrat albuminem korigovaného sérového vápníku nad 3,0 mmol/l) byla 18 až 26 dnů.
Klinické studie _prevence kostních _příhod u _pacientů s karcinomem _prsu a kostními metastázami Klinické studie u pacientů s karcinomem prsu a kostními metastázami ukázaly, že zde existuje na dávce závislý inhibiční účinek na osteolýzu kosti, který lze hodnotit markery kostní resorpce, a na dávce závislý účinek na kostní příhody.
Prevence kostních komplikací u pacientů s karcinomem prsu a kostními metastázami při intravenózním podání 6 mg kyseliny ibandronové byla hodnocena v jedné randomizované placebem kontrolované klinické studii fáze III, která trvala 96 týdnů. Pacientky s karcinomem prsu a radiologicky potvrzenými kostními metastázami byly randomizovány do skupin léčených placebem (158 pacientek) nebo 6 mg kyseliny ibandronové (154 pacientek). Výsledky těchto studií jsou shrnuty níže.
Primární výsledný ukazatel účinnosti
Primárním výsledným ukazatelem účinnosti v klinické studii byla doba do objevení se kostní komplikace (SMPR). Jednalo se o sloučený výsledný ukazatel, do kterého patřily následující kostní komplikace (SREs):
- ozařování kosti z indikace léčby zlomeniny nebo hrozící zlomeniny
- chirurgická léčba zlomenin
- zlomeniny obratlů
- nevertebrální zlomeniny
Analýza SMPR byla korigovaná na čas a zohledňovala skutečnost, že jedna nebo více příhod, které se staly v odstupu 12 týdnů, mohou navzájem souviset. Vícečetné příhody jsou tedy pro účely analýzy započítány pouze jednou. Údaje z této studie prokázaly významný přínos léčby intravenózně podávanou kyselinou ibandronovou 6 mg oproti placebu z hlediska snížení počtu SRE při hodnocení parametru SMPR (p = 0,004). Počet SRE byl také významně nižší při léčbě kyselinou ibandronovou 6 mg a oproti placebu došlo k 40% snížení rizika SRE (relativní riziko 0,6, p = 0,003). Výsledky hodnocení účinnosti jsou shrnuty v tabulce 2.
Tabulka 2 Výsledky hodnocení účinnosti (pacienti s karcinomem prsu a s kostními metastázami)
|
Všechny kostní komplikace (SREs) | |||
|
Placebo n=158 |
Kyselina ibandronová 6 mg n=154 |
Hodnota P | |
|
SMPR (na jednoho pacienta a rok) |
1,48 |
1,19 |
p=0,004 |
|
Počet příhod (na jednu pacientku) |
3,64 |
2,65 |
p=0,025 |
|
Relativní riziko SRE |
- |
0,60 |
p=0,003 |
Sekundární výsledné ukazatele účinnosti
Při léčbě intravenózně podávanou kyselinou ibandronovou v dávce 6 mg bylo ve srovnání s placebem zjištěno statisticky významné zlepšení skóre bolesti kostí. Stupeň zmírnění bolesti byl po celou dobu trvání studie setrvale pod úrovní, která byla při zahájení studie, a při srovnání s placebem byl provázen významným snížením spotřeby analgetik. Zhoršení při hodnocení kvality života bylo ve skupině léčené kyselinou ibandronovou menší než ve skupině léčené placebem. Shrnutí sekundárních výsledných parametrů je uvedeno v tabulce 3.
Tabulka 3 Sekundární výsledné parametry účinnosti (pacienti s karcinomem prsu s kostními metastázami)
|
Placebo n=158 |
Kyselina ibandronová 6 mg n=154 |
Hodnota P | |
|
Bolesti kostí * |
0,21 |
-0,28 |
p<0,001 |
|
Potřeba analgetik * |
0,90 |
0,51 |
p=0,083 |
|
Kvalita života * |
-45,4 |
-10,3 |
p=0,004 |
|
* Průměrná změna od vstupního vyšetření do posledního |
hodnocení | ||
U pacientek léčených kyselinou ibandronovou byl také zjištěn významný pokles markerů kostní resorpce v moči (pyridinolin a deoxypyridinolin), který byl ve srovnání s placebem statisticky významný.
Ve studii zahrnující 130 pacientů s metastazujícím karcinomem prsu byla srovnávána bezpečnost kyselinou ibandronovou podávanou v infuzi trvající 1 hodinu oproti infuzi trvající 15 minut.
Z hlediska indikátorů funkce ledvin nebyl pozorován žádný rozdíl. Celkový profil nežádoucích příhod vyskytujících se po 15minutové infuzi kyseliny ibandronové byl ve shodě se známým bezpečnostním profilem po delší době infuze a v souvislosti s infuzí trvající 15 minut nebyly zaznamenány žádné nové skutečnosti týkající se bezpečnosti přípravku.
U pacientů s nádory a s clearance kreatininu < 50 ml/min nebyla infuze trvající 15 minut dosud studována.
Pediatrická populace (viz bod 4.2 a 5.2)
U dětí a dospívajících mladších než 18 let nebyla bezpečnost a účinnost kyseliny ibandronové stanovena. Žádné údaje nejsou k dispozici.
5.2 Farmakokinetické vlastnosti
Po dvouhodinové infuzi 2, 4 a 6 mg kyseliny ibandronové jsou farmakokinetické parametry přímo úměrné výši podané dávky.
Distribuce
Po prvotním systémovém rozptýlení se kyselina ibandronová rychle váže na kostní tkáň nebo se vylučuje do moči. U lidí je účinný terminální distribuční objem nejméně 90 litrů a množství léku, které se dostane do kosti, se odhaduje na 40-50 % množství, které se dostane do krevního oběhu. Vazba na bílkoviny lidské plazmy je v terapeutických koncentracích přibližně 87 % a tak nejsou interakce s jinými léčivými přípravky v důsledku vytěsňování z této vazby pravděpodobné.
Biotransformace
Žádné důkazy nenasvědčují tomu, že by u zvířat nebo lidí podléhala kyselina ibandronová metabolické přeměně.
Eliminace
Rozpětí zjištěních poločasů je široké a závisí na dávce a citlivosti zvolené vyšetřovací metody, ale zjevný terminální poločas se pohybuje v rozpětí 10 - 60 hodin. Časné plazmatické koncentrace však klesají rychle a dosahují 10 % maximální koncentrace za 3 a 8 hodin po intravenózním, respektive perorálním podání. Při intravenózním podání kyseliny ibandronové jednou za 4 týdny po dobu 48 týdnů nebyly zjištěny projevy systémové kumulace.
Celková clearance kyseliny ibandronové je nízká s průměrnou hodnotou v rozpětí 84 -160 ml/min. Renální clearance (přibližně 60 ml/min u zdravých žen po menopause) tvoří 50 - 60 % celkové clearance a souvisí s clearance kreatininu. Rozdíl mezi zjevnou celkovou a renální clearance odpovídá stupni vychytání látky v kosti.
Sekreční cesta se nepřekrývá s žádným ze známých kyselých nebo zásaditých transportních systémů, které se podílejí na vylučování jiných léčivých látek. Kyselina ibandronová dále neinhibuje hlavní lidské hepatické izoenzymy P450 a neindukuje systém hepatického cytochromu P 450 u potkanů.
Farmakokinetika u zvláštních skupin _pacientů
Pohlaví
Biologická dostupnost a farmakokinetika kyseliny ibandronové jsou u mužů i žen podobné.
Rasa
Žádné důkazy nenasvědčují tomu, že by chování kyseliny ibandronové v organismu bylo odlišné u různých etnických skupin bělochů a Asiatů. Od pacientů afroamerického původu je k dispozici pouze velmi malé množství údajů.
Pacienti s onemocněním ledvin
U pacientů s různým stupněm poškození funkce ledvin souvisí expozice kyselině ibandronové s clearance kreatininu (CLcr). U jedinců s těžkou poruchou funkce ledvin (střední hodnota CLcr =
21,2 ml/min) byla střední hodnota AUC0.24h ,korigovaná na dávku, o 110 % vyšší v porovnání se zdravými dobrovolníky. V klinické farmakologické studii WP18551 bylo zjištěno, že po jediné intravenózní dávce 6 mg (15minutová infuze) se průměrná AUC0-24 u subjektů s mírným stupněm poškození ledvin zvýšila o 14 % (průměrná odhadovaná CLcr = 68,1 ml/min) a u subjektů se středním stupněm poškození ledvin o 86 % (průměrná odhadovaná CLcr= 41,2 ml/min) ve srovnání se zdravými dobrovolníky (průměrná odhadovaná CLcr = 120 ml/min). Průměrná hodnota Cmax se u pacientů s mírným stupněm poškození ledvin nezvýšila a u pacientů se středním stupněm poškození ledvin byla zvýšena o 12 %. U pacientů s mírnou poruchou funkce ledvin (CLcr >50 a <80 ml/min) není úprava dávkování nutná. U pacientů s karcinomem prsu a kostními metastázami se středně těžkou poruchou funkce ledvin (CLcr > 30 a < 50 ml/min) nebo těžkou poruchou funkce ledvin (CLcr < 30 ml/min), kteří jsou léčeni z důvodu prevence kostních příhod, je úprava dávkování doporučena (viz bod 4.2).
Pacienti s onemocněním jater (viz bod 4.2)
U pacientů s poškozením funkce jater nejsou pro kyselinu ibandronovou k dispozici žádné farmakokinetické údaje. Játra se na clearance kyseliny ibandronové zásadním způsobem nepodílejí, neboť tato kyselina se nemetabolizuje, nýbrž se vylučuje ledvinami a vychytává se v kosti. Proto u nemocných s porušenou funkcí j ater není úprava dávkování nutná. Kromě toho j e vazba kyseliny ibandronové v terapeutických koncentracích na plazmatické bílkoviny přibližně 87 %, takže hypoproteinemie v případě těžkého poškození funkce jater pravděpodobně nepovede k významnému zvýšení volné frakce kyseliny ibandronové v plazmě.
Starší pacienti (viz bod 4.2)
Ve víceproměnné analýze nebyl věk mezi nezávislými faktory, které ovlivňovaly hodnocené farmakokinetické parametry. Vzhledem k tomu, že s narůstajícím věkem dochází ke zhoršování renálních funkcí, je tento faktor třeba vzít v úvahu (viz bod poškození ledvin).
Pediatrická populace (viz bod 4.2 a 5.1)
U pacientů mladších 18 let nejsou pro použití kyseliny ibandronové k dispozici žádné údaje.
5.3 Předklinické údaje vztahující se k bezpečnosti
Účinky v neklinických studiích byly pozorovány pouze po expozicích dostatečně převyšujících maximální expozice u člověka, což svědčí o malém významu při použití. Podobně jako u ostatních bisfosfonátů bylo prokázáno, že primárním orgánem systémové toxicity jsou ledviny.
Mutagenita/Karcinogenita:
Karcinogenní potenciál nebyl pozorován. Testy genotoxicity nepřinesly žádné důkazy o genetickém účinku kyseliny ibandronové.
Reprodukční toxicita:
U potkanů a králíků, kterým byla podávána kyselina ibandronová intravenózně, nebyly zjištěny žádné důkazy o přímé fetální toxicitě nebo teratogenních účincích. V reprodukčních studiích s perorálním podáním u potkanů se účinky na fertilitu sestávaly ze zvýšených preimplantačních ztrát při dávkách 1 mg/kg/den a vyšších. V reprodukčních studiích s intravenózním podáním u potkanů kyselina ibandronová snižovala počet spermií při dávkách 0,3 a 1 mg/kg/den a snižovala fertilitu u samců při dávkách 1 mg/kg/den a u samic při dávkách 1,2 mg/kg/den. Nežádoucí účinky kyseliny ibandronové pozorované ve studiích reprodukční toxicity u potkanů odpovídaly očekávanému spektru pro tuto skupinu léčivých látek (bisfosfonáty). Patří mezi ně snížení počtu implantačních míst, narušení přirozené porodní činnosti (dystokie) a zvýšení výskytu viscerálních variací (syndrom ledvinné pánvičky a ureteru) a abnormality zubů u první filiální generace potkanů.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Chlorid sodný Trihydrát octanu sodného Kyselina octová 98%
Voda na injekci
6.2 Inkompatibility
Aby bylo možno vyloučit inkompatibilitu, může být koncentrát kyseliny ibandronové pro přípravu infuzního roztoku ředěn pouze izotonickým roztokem chloridu sodného nebo 5% roztokem glukózy.
Koncentrát kyseliny ibandronové pro přípravu infuzního roztoku nesmí být mísen s roztoky obsahujícími vápník.
6.3 Doba použitelnosti
2 roky
Po naředění:
Chemická a fyzikální stabilita při použití po naředění v 0,9% roztoku chloridu sodného nebo 5% roztoku glukózy byla prokázána po dobu 36 hodin při teplotě 25 °C a 2 °C až 8 °C.
Z mikrobiologického hlediska má být roztok spotřebován okamžitě. Pokud není spotřebován okamžitě, jsou doba a podmínky uchovávání plně v odpovědnosti uživatele a neměly by přesáhnout dobu 24 hodin při teplotě 2 °C až 8 °C, jestliže naředění proběhlo za kontrolovaných a validovaných aseptických podmínek.
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
Podmínky pro uchovávání naředěného přípravku naleznete v bodě 6.3.
6.5 Druh obalu a obsah balení
6ml skleněná injekční lahvička (typ I) s fluorotec/pryžovou zátkou a hliníkovým uzávěrem s levandulovým flip off víčkem. Je dodáván v baleních obsahujících 1 injekční lahvičku.
6.6 Zvláštní opatření pro likvidaci přípravku
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky. Léčivé přípravky se nesmí vyhazovat do odpadních vod nebo domácího odpadu.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Accord Healthcare Limited Sage House 319, Pinner Road North Harrow Middlesex HA1 4HF Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/12/798/001
9. DATUM PRVNÍ REGISTRACE
Datum první registrace: 19 listopad 2012
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
NÁZEV PŘÍPRAVKU
1.
Ibandronic Acid Accord 6 mg koncentrát pro infuzní roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna lahvička se 6 ml koncentrátu pro přípravu infuzního roztoku obsahuje acidum ibandronicum 6 mg (jako natrii monohydricus).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Koncentrát pro přípravu infuzního roztoku Čirý, bezbarvý roztok
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Kyselina ibandronová je indikována u dospělých pacientů k
- prevenci kostních příhod (patologických zlomenin, kostních komplikací, které vyžadují radiologickou nebo chirurgickou léčbu) u pacientů s karcinomem prsu a kostními metastázami
- léčbě hyperkalcemie vyvolané nádorem s výskytem nebo bez výskytu metastáz
4.2 Dávkování a způsob podání
Pacientům, kteří budou léčeni kyselinou ibandronovou, musí být předána příbalová informace a karta s připomínkami.
Léčba kyselinou ibandronovou má být zahájena pouze lékařem se zkušenostmi s léčbou onkologického onemocnění.
Dávkování
Prevence kostních _příhod u _pacientů s karcinomem _prsu a kostními metastázami
Doporučené dávkování v prevenci kostních příhod u pacientů s karcinomem prsu a kostními metastázami je 6 mg ve formě intravenózní injekce podávané každé 3-4 týdny. Jednotlivá dávka má být podána infuzí trvající alespoň 15 minut.
Kratší doba (tj. 15 min) infuze má být použita pouze u pacientů s normální funkcí ledvin nebo s mírně zhoršenou funkcí ledvin. Nejsou k dispozici žádné údaje charakterizující užití kratší doby infuze u pacientů s clearance kreatininu pod 50 ml/min. Předepisující lékař má věnovat pozornost doporučení o dávkování a podávání přípravku této skupině pacientů, které je uvedeno v bodu Pacienti s onemocněním ledvin (viz bod 4.2).
Léčba hyperkalcemie vyvolané nádorem
Před léčbou kyselinou ibandronovou musí být pacienti adekvátně rehydratováni 0,9% roztokem chloridu sodného. Je třeba brát v úvahu stupeň závažnosti hyperkalcemie stejně jako typ nádoru. Pacienti s osteolytickými metastázami kostí potřebují obecně nižší dávky než pacienti s humorálním typem hyperkalcemie. U většiny pacientů s těžkou hyperkalcemií (albuminem korigovaný sérový vápník* > 3 mmol/l nebo > 12 mg/dl) postačuje jednotlivá dávka 4 mg. U pacientů s mírnou hyperkalcemií (albuminem korigovaný sérový vápník < 3 mmol/l nebo < 12 mg/dl) je účinná dávka 2 mg. Nejvyšší dávka použitá v klinických studiích byla 6 mg, ale tato dávka se neprojevila vyšším účinkem.
* Poznámka: koncentrace albuminem korigovaného sérového vápníku se vypočte následujícím způsobem:
|
Koncentrace albuminem korigovaného sérového vápníku (mmol/l) |
vápník v séru (mmol/l) - [0,02 x albumin (g/l)] + 0,8 | |
|
Nebo | ||
|
Koncentrace albuminem korigovaného sérového vápníku (mg/dl) |
vápník v séru (mg/dl) + 0,8 x [4 - albumin (g/dl)] | |
|
Hodnoty albuminem korigované |
io sérového vápníku v mmol/l lze převést na mg/dl vynásobením 4. | |
Ve většině případů může být zvýšená hladina sérového vápníku snížena na normální hladinu během 7 dnů. Střední doba do relapsu (znovuzvýšení albuminem korigovaného sérového vápníku nad 3 mmol/l) byla 18 - 19 dnů při dávkách 2 mg a 4 mg. Střední doba do relapsu při dávce 6 mg byla 26 dnů.
Omezený počet pacientů (50 pacientů) dostal druhou infuzi z důvodů hyperkalcemie. Léčbu je třeba opakovat v případě vracející se hyperkalcemie nebo při nedostatečné účinnosti.
Koncentrát kyseliny ibandronové pro infuzní roztok má být podáván jako intravenózní infuze po dobu minimálně 2 hodin.
Zvláštní skupiny _pacientů
Pacienti s poruchou jaterních funkcí
Úprava dávky není nutná (viz bod 5.2).
Pacienti s poruchou renálních funkcí
U pacientů s mírnou poruchou funkce ledvin (CLcr >50 a <80 ml/min) není úprava dávky nutná. U pacientů s karcinomem prsu a kostními metastázami se středně těžkou poruchou funkce ledvin (CLcr > 30 a < 50 ml/min) nebo těžkou poruchou funkce ledvin (CLcr < 30 ml/min), kteří jsou léčeni z důvodu prevence kostních příhod, by se měla dodržovat následující dávkovací doporučení (viz bod 5.2):
|
Clearance kreatininu (ml/min) |
Dávka |
Objem 1 a doba 2 infuze | |
|
>50 CLcr <80 >30 CLcr <50 <30 |
6 mg 4 mg 2 mg |
(6 ml koncentrátu pro infuzní roztok) (4 ml koncentrátu pro infuzní roztok) (2 ml koncentrátu pro infuzní roztok) |
100 ml po dobu 15 min 500 ml po dobu 1 hodiny 500 ml po dobu 1 hodiny |
1 0,9% roztok chloridu sodného nebo 5% roztok glukózy
2 Při podávání každé 3 až 4 týdny
U pacientů s nádorovým onemocněním a CLcr < 50 ml/min nebyla doba infuze trvající 15 minut dosud studována.
Starší pacienti (>65 let)
Úprava dávky není nutná (viz bod 5.1 a 5.2).
Pediatrická populace
U dětí a dospívajících mladších než 18 let nebyla bezpečnost a účinnost kyseliny ibandronové stanovena. Žádné údaje nejsou k dispozici (viz bod 5.1 a 5.2).
Způsob podání K intravenóznímu podání.
Obsah injekční lahvičky má být použit následujícím způsobem:
- Prevence kostních příhod - obsah přidat ke 100 ml izotonického roztoku chloridu sodného nebo ke 100 ml 5% roztoku glukózy a aplikovat infuzi po dobu nejméně 15 minut. Viz také bod výše týkající se dávkování u pacientů s poruchou renálních funkcí
- Léčba hyperkalcemie vyvolané nádorem - obsah přidat k 500 ml izotonického roztoku chloridu sodného nebo k 500 ml 5% roztoku glukózy a aplikovat infuzi po dobu minimálně 2 hodin
Pouze k jednorázovému použití. Měl by být použit pouze čirý roztok bez jakýchkoli částic.
Koncentrát kyseliny ibandronové pro přípravu infuzního roztoku se aplikuje jako intravenózní infuze. Je nutno ve zvýšené míře dbát na to, aby nebyl koncentrát kyseliny ibandronové pro přípravu infuzního roztoku podán intraarteriálně nebo paravevenózně, neboť by mohlo dojít k poškození tkáně.
4.3 Kontraindikace
- Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku tohoto přípravku uvedenou v bodě 6.1.
- Hypokalcemie.
4.4 Zvláštní upozornění a opatření pro použití
Pacienti s poruchami kostního a minerálového metabolismu
Před zahájením léčby kyselinou ibandronovou u metastatického postižení kostí je vhodné účinně léčit hypokalcemii a další poruchy kostního a minerálového metabolismu.
U všech pacientů je důležitý dostatečný přísun vápníku a vitaminu D. V případě jejich nedostatečného přísunu ve stravě mají pacienti dostávat doplňky vápníku a/nebo vitaminu D.
Anafylaktická reakce/šok
U pacientů léčených kyselinou ibandronovou v intravenózní formě byly hlášeny případy anafylaktické reakce/šoku včetně příhod končících úmrtím.
Pokud je podávána intravenózní injekce přípravku Ibandronic Acid Accord, je třeba, aby byla dostupná vhodná lékařská péče a zajištěno odpovídající sledování pacienta. Jestliže se objeví anafylaktické nebo jiné závažné hypersenzitivní/alergické reakce, okamžitě ukončete podávání injekce a zahajte příslušnou léčbu.
Osteonekróza čelisti
Velmi vzácně byla u pacientů léčených kyselinou ibandronovou z důvodu onkologických indikací (viz odstavec 4.8) hlášena v poprodejním sledování osteonekróza čelisti (ONČ).
U pacientů s nezhojenými lézemi měkkých tkání v ústech je třeba odložit zahájení léčby nebo přechod na novou léčbu.
U pacientů s průvodními rizikovými faktory se před zahájením léčby kyselinou ibandronovou doporučuje provést preventivní zubní prohlídku a individuální posouzení přínosů a rizik.
Při vyhodnocování rizika rozvoje ONČ u pacienta je třeba zvažovat následující rizikové faktory:
- Účinnost léčivého přípravku, který inhibuje resorpci kostí (vyšší riziko u vysoce účinných přípravků), způsob podávání (vyšší riziko při parenterálním podávání) a kumulovaná dávka při léčbě resorpce kostí
- Onkologické onemocnění, další chorobné stavy (například anémie, koagulopatie, infekce), kouření
- Doprovodná léčba: kortikosteroidy, chemoterapie, angiogenní inhibitory, radioterapie hlavy a krku
Nedostatečná ústní hygiena, periodontální onemocnění, nedostatečně sedící umělý chrup, dentální
onemocnění v anamnéze, invazivní dentální postupy léčby například extrakce zubů
Všem pacientům je třeba doporučit, aby během léčby kyselinou ibandronovou udržovali dobrou ústní hygienu, absolvovali rutinní zubní prohlídky a ihned ohlásili všechny příznaky onemocnění ústní dutiny jako pohyblivost zubů, bolesti nebo otoky, nehojící se vředy nebo výtoky. V průběhu léčby je třeba provádět invazivní zubní zákroky a postupy jedině po pečlivém zvážení a vyvarovat se jim v bezprostřední blízkosti podávání kyseliny ibandronové.
Plán léčby pacientů, u kterých se rozvine ONČ, musí být stanoven ve spolupráci ošetřujícího lékaře a zubaře nebo ústního chirurga s odbornými znalostmi o ONČ. Dokud se příslušný stav nevyřeší a nebude případně možné zmírnit rizikové faktory, je třeba zvažovat dočasné přerušení léčby kyselinou ibandronovou.
Osteonekróza vnějšího zvukovodu
V případě léčby bisfosfonáty, zejména ve spojení s dlouhodobou léčbou, byla hlášena osteonekróza vnějšího zvukovodu. Možné rizikové faktory osteonekrózy vnějšího zvukovodu zahrnují užívání steroidů a chemoterapii a/nebo lokální rizikové faktory, jakým je infekce nebo trauma. Možnost osteonekrózy vnějšího zvukovodu je třeba zvažovat u pacientů léčených bisfosfonáty, u kterých se projevují příznaky ušních onemocnění včetně chronických ušních infekcí.
Atypické zlomeniny femuru
V souvislosti s léčbou bisfosfonáty byly hlášeny atypické subtrochanterické a diafyzární zlomeniny femuru, zejména u pacientů dlouhodobě léčených pro osteoporózu. Tyto příčné nebo krátké šikmé zlomeniny se mohou objevit kdekoli v celé délce femuru od oblasti těsně pod malým trochanterem až do části těsně nad suprakondylickým rozšířením. Tyto zlomeniny se objevují po minimálním traumatu nebo bez souvislosti s traumatem a u některých pacientů se mohou projevovat bolestí ve stehně nebo třísle, často sdružené na zobrazovacích vyšetřeních s obrazem typickým pro stresové zlomeniny (neobvyklé nízkotraumatické zlomeniny, v angličtině známé jako „insufficiency fractures“)týdny až měsíce před manifestací kompletní zlomeniny femuru. Zlomeniny jsou často oboustranné, proto je nutné u pacientů léčených bisfosfonáty, kteří utrpěli zlomeninu diafýzy femuru, vyšetřit i kontralaterální femur. Rovněž bylo zaznamenáno špatné hojení těchto zlomenin. U pacientů, u kterých je podezření na atypickou zlomeninu femuru, je třeba při hodnocení jejich stavu zvážit i přerušení léčby bisfosfonáty, a to na základě zhodnocení prospěchu a rizika léčby u jednotlivého pacienta. Pacienty je třeba poučit, aby během léčby bisfosfonáty hlásili jakoukoli bolest v oblasti stehna, boku nebo třísla, a všechny pacienty, u kterých se tyto příznaky objeví, je třeba vyšetřit s ohledem na možnou inkompletní zlomeninu femuru.
Pacienti s poruchou renálních funkcí
Klinické studie neprokázaly, že by při dlouhodobé léčbě kyselinou ibandronovou docházelo ke zhoršení renálních funkcí. Nicméně se doporučuje na základě zhodnocení klinického stavu každého pacienta sledovat při léčbě kyselinou ibandronovou renální funkce, hladinu vápníku, fosfátu a hořčíku v séru (viz bod 4.2).
Pacienti s poruchou jaterních funkcí
Z důvodu nedostačujících klinických údajů není možné doporučit dávkování u pacientů s těžkou jaterní dysfunkcí (viz bod 4.2).
Pacienti se srdeční poruchou
U pacientů ohrožených rozvojem srdečního selhání je třeba zabránit převodnění.
Pacienti se známou hypersenzitivitou na jiné bisfosfonáty
Opatrnosti je třeba u pacientů se známou hypersenzitivitou na jiné bisfosfonáty.
Pomocné látky se známým účinkem
Tento léčivý přípravek obsahuje méně než 1 mmol sodíku (23 mg) v jedné injekční lahvičce, tzn., že je v podstatě “bez sodíku”.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Metabolické interakce se nepředpokládají vzhledem k tomu, že kyselina ibandronová neinhibuje hlavní lidské hepatické izoenzymy P450, a bylo prokázáno, že neindukuje systém hepatického cytochromu P 450 u potkanů (viz bod 5.2). Kyselina ibandronová se vylučuje pouze renální sekrecí a neprochází žádnou metabolickou přeměnou.
V klinických studiích byla kyselina ibandronová podávána souběžně s běžně užívanými antineoplastiky, diuretiky, antibiotiky a analgetiky bez klinických projevů interakce.
4.6 Fertilita, těhotenství a kojení
O podávání kyseliny ibandronové těhotným ženám nejsou k dispozici potřebné údaje. Studie u potkanů prokázaly reprodukční toxicitu (viz bod 5.3). Možné riziko u lidí není známo. Proto nemá být kyselina ibandronová těhotným ženám podávána.
Kojení
Není známo, zda se kyselina ibandronová vylučuje do lidského mléka. Studie u kojících potkanů prokázaly po intravenózním podání kyseliny ibandronové přítomnost její nízké koncentrace v mléce. Kojícím ženám nemá být kyselina ibandronová podávána.
Fertilita
Údaje týkající se účinků kyseliny ibandronové u člověka nejsou k dispozici. V reprodukčních studiích s perorálním podáním u potkanů kyselina ibandronová snižovala fertilitu. Ve studiích s intravenózním podáním u potkanů kyselina ibandronová snižovala fertilitu při vysokých denních dávkách (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Na základě farmakodynamického a farmakokinetického profilu a hlášených nežádoucích účinků lze předpokládat, že kyselina ibandronová nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky
Shrnutí bezpečnostního profilu
Nejzávažnější hlášené nežádoucí účinky jsou anafylaktická reakce/šok, atypické zlomeniny femuru, osteonekróza čelisti a oční záněty (viz odstavec „Popis vybraných nežádoucích účinků“ a bod 4.4).Při hyperkalcemii vyvolané nádorem je léčba nejčastěji doprovázena zvýšením tělesné teploty. Méně často je hlášen pokles sérového vápníku pod normální hodnotu (hypokalcemie).
Ve většině případů není zapotřebí zvláštní léčby a příznaky za pár hodin/dnů odezní.
V prevenci kostních příhod u pacientů s karcinomem prsu a kostními metastázami je léčba nejčastěji spojena s astenií doprovázenou zvýšením tělesné teploty a bolestí hlavy.
Shrnutí nežádoucích účinků do tabulky
V tabulce 1 jsou uvedeny nežádoucí účinky z klíčových studií fáze III (Léčba hyperkalcemie vyvolané nádorem: 311 pacientů léčených kyselinou ibandronovou 2 mg nebo 4 mg; Prevence kostních příhod u pacientů s karcinomem prsu a kostními metastázami: 152 pacientů léčených kyselinou ibandronovou 6 mg) a po uvedení přípravku na trh.
Nežádoucí účinky jsou uvedeny podle tříd orgánových systémů dle databáze MedDRA a četnosti výskytu. Četnosti výskytu jsou definovány podle následujících konvencí: velmi časté (>1/10), časté (>1/100 až <1/10), méně časté (>1/1000 až <1/100), vzácné (>1/10000 až <1/1000), velmi vzácné (<1/10000), není známo (z dostupných údajů nelze určit). V rámci každé skupiny četností jsou nežádoucí účinky uvedeny v pořadí dle klesající závažnosti.
Tabulka 1
Nežádoucí účinky hlášené po intravenózním podáníkyseliny ibandronové
|
Třída orgánových systémů |
Velmi časté |
Časté |
Méně časté |
Vzácné |
Velmi vzácné |
Není známo |
|
Infekce a infestace |
Infekce |
Cystitida, vaginitida, orální kandidóza | ||||
|
Novotvary benigní, maligní a blíže neurčené |
Benigní kožní novotvar | |||||
|
Poruchy krve a lymfatického systému |
Anémie, krevní dyskrazie | |||||
|
Poruchy imunitního systému |
Hypersenzitivitaf, bronchospasmusf, angioedémf, anafylaktická reakce/šokf** |
Exacerbace astmatuf | ||||
|
Endokrinní poruchy |
Poruchy příštitných tělísek | |||||
|
Poruchy metabolismu a výživy |
Hypokalce mie** |
Hypofosfatemie | ||||
|
Psychiatrické poruchy |
Poruchy spánku, úzkost, afektivní labilita | |||||
|
Poruchy nervového systému |
Bolest hlavy, závratě, dysgeuzie (změny chuti) |
Cerebrovaskulární onemocnění, léze nervového kořene, amnezie, migréna, neuralgie, hypertonie, hyperestezie, cirkumorální parestezie, parosmie | ||||
|
Poruchy oka |
Katarakta |
Zánět okaf** | ||||
|
Poruchy ucha a labyrintu |
Hluchota | |||||
|
Srdeční poruchy |
Raménkov ý blok |
Ischemie myokardu, kardiovaskulární onemocnění, palpitace | ||||
|
Respirační, hrudní a mediastinální poruchy |
Faryngitid a |
Plicní edém, stridor | ||||
|
Gastrointestin ální poruchy |
Gastroenteritida, gastritida, ulcerace |
|
Třída orgánových systémů |
Velmi časté |
Časté |
Méně časté |
Vzácné |
Velmi vzácné |
Není známo |
|
gastrointes tinální bolesti, zubní potíže |
v ústech, dysfagie, cheilitida | |||||
|
Poruchy jater a žlučových cest |
Žlučové kameny | |||||
|
Poruchy kůže a podkožní tkáně |
Kožní onemocně ní, ekchymóz a |
Vyrážka, alopecie |
Stevens- Johnsonův syndromf, erythema multiformef, bulózní dermatitidaf | |||
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Osteoartró za, myalgie, artralgie, onemocně ní kloubů |
Atypické subtrocha nterické a diafyzárn í zlomenin y femuruf (skupino vý nežádouc í účinek bisfosfon átů) |
Osteonekróza čelistif** Osteonekróza vněj šího zvukovodu (nežádoucí účinek bisfosfonátů) f | |||
|
Poruchy ledvin a močových cest |
Močová retence, ledvinná cysta | |||||
|
Poruchy reprodukčního systému a prsu |
Bolesti v pánvi | |||||
|
Celkové poruchy a reakce v místě aplikace |
Pyrexie, onemocně ní podobné chřipce**, periferní edém, astenie, žízeň |
Hypotermie | ||||
|
Vyšetření |
Zvýšení gama-GT, zvýšení kreatininu |
Zvýšení alkalické fosfatázy v krvi, snížení tělesné hmotnosti | ||||
|
Poranění, otravy a procedurální komplikace |
Poranění, bolest v místě vpichu |
**Viz další informace níže
fldentifikovány po uvedení přípravku na trh Popis vybraných nežádoucích účinků
Hypokalcemie
Snížené vylučování kalcia ledvinami může být doprovázeno poklesem hladiny fosfátů v séru, které nevyžaduje terapeutický zásah. Hladina vápníku v séru může klesnout na hypokalcemické hodnoty.
Onemocnění podobné chřipce
Objevily se příznaky podobné chřipce zahrnující horečku, třesavku, bolesti kostí a/nebo svalů. Ve většině případů nebylo zapotřebí zvláštní léčby a příznaky za pár hodin/dnů odezněly.
Osteonekróza čelisti
Případy osteonekrózy čelisti byly hlášeny převážně u pacientů s onkologickým onemocněním, léčených léčivými přípravky, které inhibují resorpci kostí, například kyselinou ibandronovou (viz odstavec 4.4.). Případy ONČ byly hlášeny v poprodejním sledování kyseliny ibandronové.
Zánět oka
Při podávání kyseliny ibandronové byly hlášeny oční zánětlivé reakce, jako jsou uveitida, episkleritida, skleritida. V některých případech tyto nežádoucí reakce neustoupily, dokud podávání kyseliny ibandronové nebylo ukončeno.
Anafylaktická reakce/šok
U pacientů léčených kyselinou ibandronovou v intravenózní formě byly hlášeny případy anafylaktické reakce/šoku včetně příhod končících úmrtím.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Zkušenosti s akutní otravou koncentrátem kyseliny ibandronové pro přípravu infuzního roztoku dosud nejsou. Vzhledem k tomu, že v preklinických studiích s vysokými dávkami přípravku byly toxicitou nejvíce postiženy játra a ledviny, je nutné jejich činnost během léčby monitorovat. Klinicky závažnou hypokalcemii lze upravit intravenózním podáním kalcium-glukonátu.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Léčivé přípravky ovlivňující stavbu a mineralizaci kostí, bisfosfonáty, ATC kód: M05BA06.
Kyselina ibandronová patří do skupiny bisfosfonátových sloučenin, které mají specifický účinek na kosti. Její selektivní působení na kostní tkáň je založeno na vysoké afinitě bisfosfonátů ke kostním minerálům. Bisfosfonáty inhibují aktivitu osteoklastů, ale přesný mechanizmus není dosud znám.
In vivo kyselina ibandronová zabraňuje pokusně způsobené destrukci kostí, která byla navozena přerušením funkcí gonád, retinoidy, nádory nebo extrakty z nádorů. Potlačení endogenní resorpce kostí bylo dokumentováno studiemi kinetiky 45Ca a uvolněním radioaktivního tetracyklinu předtím zavedeného do kostry.
V dávkách, které byly zřetelně vyšší než je farmakologicky účinná dávka, neměla kyselina ibandronová žádný účinek na mineralizaci kostí.
Pro resorpci kosti při malignitě je typická nadměrná resorpce, která není vyrovnána odpovídající novotvorbou kosti. Kyselina ibandronová selektivně inhibuje aktivitu osteoklastů, což vede ke zpomalení kostní resorpce a tím k sníženému výskytu kostních komplikací u maligních onemocnění.
Klinické studie léčby nádorem vyvolané hyperkalcemie
Klinické studie hyperkalcemie u malignit ukázaly, že inhibiční efekt kyseliny ibandronové na tumory indukovanou osteolýzu, a zvláště na nádorem vyvolanou hyperkalcemii, je charakterizován poklesem hladiny sérového vápníku a poklesem vylučování vápníku močí.
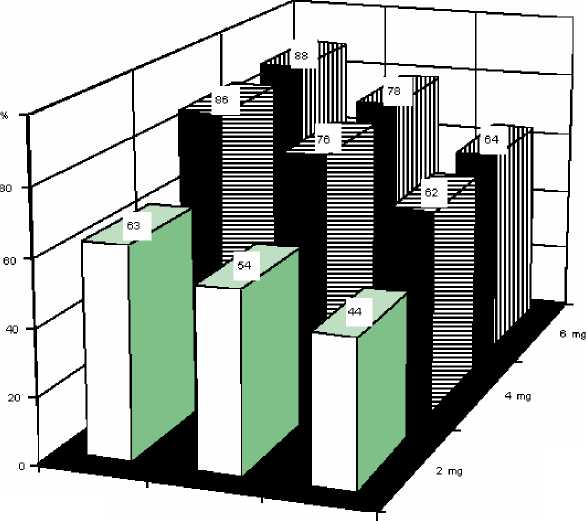
V dávkování doporučovaném pro léčbu byly zjištěny následující četnosti odezvy s odpovídajícím intervalem spolehlivosti na základě výsledků klinických studií u pacientů se vstupní koncentrací albuminem korigovaného sérového vápníku > 3,0 mmol/l po náležité rehydrataci.
Vysoký 90%
interval
spolehlivosti
Poměr léčebné odpovědi
Dávky
kyseliny
ibandronové
Nízký 90%
interval
spolehlivosti
|
Dávka kyseliny ibandronové |
% pacientů s odpovědí |
90% interval spolehlivosti |
|
2 mg |
54 |
44-63 |
|
4 mg |
76 |
62-86 |
|
6 mg |
78 |
64-88 |
U těchto pacientů dostávajících výše uvedené dávky byl průměrný čas pro dosažení normokalcemie 4 -7 dnů. Průměrná doba recidivy (návrat albuminem korigovaného sérového vápníku nad 3,0 mmol/l) byla 18 až 26 dnů.
Klinické studie _prevence kostních _příhod u _pacientů s karcinomem prsu a kostními metastázami
Klinické studie u pacientů s karcinomem prsu a kostními metastázami ukázaly, že zde existuje na dávce závislý inhibiční účinek na osteolýzu kosti, který lze hodnotit markery kostní resorpce, a na dávce závislý účinek na kostní příhody.
Prevence kostních komplikací u pacientů s karcinomem prsu a kostními metastázami při intravenózním podání 6 mg kyseliny ibandronové byla hodnocena v jedné randomizované placebem kontrolované klinické studii fáze III, která trvala 96 týdnů. Pacientky s karcinomem prsu a radiologicky potvrzenými kostními metastázami byly randomizovány do skupin léčených placebem (158 pacientek) nebo 6 mg kyseliny ibandronové (154 pacientek). Výsledky těchto studií jsou shrnuty níže.
Primární výsledný ukazatel účinnosti
Primárním výsledným ukazatelem účinnosti v klinické studii byla doba do objevení se kostní komplikace (SMPR). Jednalo se o sloučený výsledný ukazatel, do kterého patřily následující kostní komplikace (SREs):
- ozařování kosti z indikace léčby zlomeniny nebo hrozící zlomeniny
- chirurgická léčba zlomenin
- zlomeniny obratlů
- nevertebrální zlomeniny
Analýza SMPR byla korigovaná na čas a zohledňovala skutečnost, že jedna nebo více příhod, které se staly v odstupu 12 týdnů, mohou navzájem souviset. Vícečetné příhody jsou tedy pro účely analýzy započítány pouze jednou. Údaje z této studie prokázaly významný přínos léčby intravenózně podávanou kyselinou ibandronovou 6 mg oproti placebu z hlediska snížení počtu SRE při hodnocení parametru SMPR (p = 0,004). Počet SRE byl také významně nižší při léčbě kyselinou ibandronovou 6 mg a oproti placebu došlo k 40% snížení rizika SRE (relativní riziko 0,6, p = 0,003). Výsledky hodnocení účinnosti jsou shrnuty v tabulce 2.
Tabulka 2 Výsledky hodnocení účinnosti (pacienti s karcinomem prsu a s kostními metastázami)
|
Všechny kostní komplikace (SREs) | |||
|
Placebo n=158 |
Kyselina ibandronová 6 mg n=154 |
Hodnota P | |
|
SMPR (na jednoho pacienta a rok) |
1,48 |
1,19 |
p=0,004 |
|
Počet příhod (na jednu pacientku) |
3,64 |
2,65 |
p=0,025 |
|
Relativní riziko SRE |
- |
0,60 |
p=0,003 |
Sekundární výsledné ukazatele účinnosti
Při léčbě intravenózně podávanou kyselinou ibandronovou v dávce 6 mg bylo ve srovnání s placebem zjištěno statisticky významné zlepšení skóre bolesti kostí. Stupeň zmírnění bolesti byl po celou dobu trvání studie setrvale pod úrovní, která byla při zahájení studie, a při srovnání s placebem byl provázen významným snížením spotřeby analgetik. Zhoršení při hodnocení kvality života bylo ve skupině léčené kyselinou ibandronovou menší než ve skupině léčené placebem. Shrnutí sekundárních výsledných parametrů je uvedeno v tabulce 3.
Tabulka 3 Sekundární výsledné parametry účinnosti (pacienti s karcinomem prsu s kostními metastázami)
|
Placebo n=158 |
Kyselina ibandronová 6 mg n=154 |
Hodnota P | |
|
Bolesti kostí * |
0,21 |
-0,28 |
p<0,001 |
|
Potřeba analgetik * |
0,90 |
0,51 |
p=0,083 |
|
Kvalita života * |
-45,4 |
-10,3 |
p=0,004 |
|
* Průměrná změna od vstupního vyšetření do posledního |
hodnocení | ||
U pacientek léčených kyselinou ibandronovou byl také zjištěn významný pokles markérů kostní resorpce v moči (pyridinolin a deoxypyridinolin), který byl ve srovnání s placebem statisticky významný.
Ve studii zahrnující 130 pacientů s metastazujícím karcinomem prsu byla srovnávána bezpečnost kyselinou ibandronovou podávanou v infuzi trvající 1 hodinu oproti infuzi trvající 15 minut.
Z hlediska indikátorů funkce ledvin nebyl pozorován žádný rozdíl. Celkový profil nežádoucích příhod vyskytujících se po 15minutové infuzi kyseliny ibandronové byl ve shodě se známým bezpečnostním profilem po delší době infuze a v souvislosti s infuzí trvající 15 minut nebyly zaznamenány žádné nové skutečnosti týkající se bezpečnosti přípravku.
U pacientů s nádory a s clearance kreatininu < 50 ml/min nebyla infuze trvající 15 minut dosud studována.
Pediatrická populace (viz bod 4.2 a 5.2)
U dětí a dospívajících mladších než 18 let nebyla bezpečnost a účinnost kyseliny ibandronové stanovena. Žádné údaje nejsou k dispozici.
5.2 Farmakokinetické vlastnosti
Po dvouhodinové infuzi 2, 4 a 6 mg kyseliny ibandronové jsou farmakokinetické parametry přímo úměrné výši podané dávky.
Distribuce
Po prvotním systémovém rozptýlení se kyselina ibandronová rychle váže na kostní tkáň nebo se vylučuje do moči. U lidí je účinný terminální distribuční objem nejméně 90 litrů a množství léku, které se dostane do kosti, se odhaduje na 40-50 % množství, které se dostane do krevního oběhu. Vazba na bílkoviny lidské plazmy je v terapeutických koncentracích přibližně 87 % a tak nejsou interakce s jinými léčivými přípravky v důsledku vytěsňování z této vazby pravděpodobné.
Biotransformace
Žádné důkazy nenasvědčují tomu, že by u zvířat nebo lidí podléhala kyselina ibandronová metabolické přeměně.
Vylučování
Rozpětí zjištěných poločasů je široké a závisí na dávce a citlivosti zvolené vyšetřovací metody, ale zjevný terminální poločas se pohybuje v rozpětí 10 - 60 hodin. Časné plazmatické koncentrace však klesají rychle a dosahují 10 % maximální koncentrace za 3 a 8 hodin po intravenózním, respektive perorálním podání. Při intravenózním podání kyseliny ibandronové jednou za 4 týdny po dobu 48 týdnů nebyly zjištěny projevy systémové kumulace.
Celková clearance kyseliny ibandronové je nízká s průměrnou hodnotou v rozpětí 84 -160 ml/min. Renální clearance (přibližně 60 ml/min u zdravých žen po menopause) tvoří 50 - 60 % celkové clearance a souvisí s clearance kreatininu. Rozdíl mezi zjevnou celkovou a renální clearance odpovídá stupni vychytání látky v kosti.
Sekreční cesta se nepřekrývá s žádným ze známých kyselých nebo zásaditých transportních systémů, které se podílejí na vylučování jiných léčivých látek. Kyselina ibandronová dále neinhibuje hlavní lidské hepatické izoenzymy P450 a neindukuje systém hepatického cytochromu P 450 u potkanů.
Farmakokinetika u zvláštních skupin _pacientů
Pohlaví
Biologická dostupnost a farmakokinetika kyseliny ibandronové jsou u mužů i žen podobné.
Rasa
Žádné důkazy nenasvědčují tomu, že by chování kyseliny ibandronové v organismu bylo odlišné u různých etnických skupin bělochů a Asiatů. Od pacientů afroamerického původu je k dispozici pouze velmi malé množství údajů.
Pacienti s onemocněním ledvin
U pacientů s různým stupněm poškození funkce ledvin souvisí expozice kyselině ibandronové s clearance kreatininu (CLcr). U jedinců s těžkou poruchou funkce ledvin (střední hodnota CLcr =
21,2 ml/min) byla střední hodnota AUC0-24h ,korigovaná na dávku, o 110 % vyšší v porovnání se zdravými dobrovolníky. V klinické farmakologické studii WP18551 bylo zjištěno, že po jediné intravenózní dávce 6 mg (15minutová infuze) se průměrná AUC0-24 u subjektů s mírným stupněm poškození ledvin zvýšila o 14 % (průměrná odhadovaná CLcr = 68,1 ml/min) a u subjektů se středním stupněm poškození ledvin o 86 % (průměrná odhadovaná CLcr= 41,2 ml/min) ve srovnání se zdravými dobrovolníky (průměrná odhadovaná CLcr = 120 ml/min). Průměrná hodnota Cmax se u pacientů s mírným stupněm poškození ledvin nezvýšila a u pacientů se středním stupněm poškození ledvin byla zvýšena o 12 %. U pacientů s mírnou poruchou funkce ledvin (CLcr >50 a <80 ml/min) není úprava dávkování nutná. U pacientů s karcinomem prsu a kostními metastázami se středně těžkou poruchou funkce ledvin (CLcr > 30 a < 50 ml/min) nebo těžkou poruchou funkce ledvin (CLcr < 30 ml/min), kteří jsou léčeni z důvodu prevence kostních příhod, je úprava dávkování doporučena (viz bod 4.2).
Pacienti s onemocněním jater (viz bod 4.2)
U pacientů s poškozením funkce jater nejsou pro kyselinu ibandronovou k dispozici žádné farmakokinetické údaje. Játra se na clearance kyseliny ibandronové zásadním způsobem nepodílejí, neboť tato kyselina se nemetabolizuje, nýbrž se vylučuje ledvinami a vychytává se v kosti. Proto u nemocných s porušenou funkcí jater není úprava dávkování nutná. Kromě toho je vazba kyseliny ibandronové v terapeutických koncentracích na plazmatické bílkoviny přibližně 87 %, takže hypoproteinemie v případě těžkého poškození funkce jater pravděpodobně nepovede k významnému zvýšení volné frakce kyseliny ibandronové v plazmě.
Starší pacienti (viz bod 4.2)
Ve víceproměnné analýze nebyl věk mezi nezávislými faktory, které ovlivňovaly hodnocené farmakokinetické parametry. Vzhledem k tomu, že s narůstajícím věkem dochází ke zhoršování renálních funkcí, je tento faktor třeba vzít v úvahu (viz bod poškození ledvin).
Pediatrická populace (viz bod 4.2 a 5.1)
U pacientů mladších 18 let nejsou pro použití kyseliny ibandronové k dispozici žádné údaje.
5.3 Předklinické údaje vztahující se k bezpečnosti
Účinky v neklinických studiích byly pozorovány pouze po expozicích dostatečně převyšujících maximální expozice u člověka, což svědčí o malém významu při použití. Podobně jako u ostatních bisfosfonátů bylo prokázáno, že primárním orgánem systémové toxicity jsou ledviny.
Mutagenita/Karcinogenita:
Karcinogenní potenciál nebyl pozorován. Testy genotoxicity nepřinesly žádné důkazy o genetickém účinku kyseliny ibandronové.
Reprodukční toxicita:
U potkanů a králíků, kterým byla podávána kyselina ibandronová intravenózně, nebyly zjištěny žádné důkazy o přímé fetální toxicitě nebo teratogenních účincích. V reprodukčních studiích s perorálním podáním u potkanů se účinky na fertilitu sestávaly ze zvýšených preimplantačních ztrát při dávkách 1 mg/kg/den a vyšších. V reprodukčních studiích s intravenózním podáním u potkanů kyselina ibandronová snižovala počet spermií při dávkách 0,3 a 1 mg/kg/den a snižovala fertilitu u samců při dávkách 1 mg/kg/den a u samic při dávkách 1,2 mg/kg/den. Nežádoucí účinky kyseliny ibandronové pozorované ve studiích reprodukční toxicity u potkanů odpovídaly očekávanému spektru pro tuto skupinu léčivých látek (bisfosfonáty). Patří mezi ně snížení počtu implantačních míst, narušení přirozené porodní činnosti (dystokie) a zvýšení výskytu viscerálních variací (syndrom ledvinné pánvičky a ureteru) a abnormality zubů u první filiální generace potkanů.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Chlorid sodný Trihydrát octanu sodného Kyselina octová 98%
Voda na injekci
6.2 Inkompatibility
Aby bylo možno vyloučit inkompatibilitu, může být koncentrát kyseliny ibandronové pro přípravu infuzního roztoku ředěn pouze izotonickým roztokem chloridu sodného nebo 5% roztokem glukózy.
Koncentrát kyseliny ibandronové pro přípravu infuzního roztoku nesmí být mísen s roztoky obsahujícími vápník.
6.3 Doba použitelnosti
2 roky
Po naředění:
Chemická a fyzikální stabilita při použití po naředění v 0,9% roztoku chloridu sodného nebo 5% roztoku glukózy byla prokázána po dobu 36 hodin při teplotě 25 °C a 2 °C až 8 °C.
Z mikrobiologického hlediska má být roztok spotřebován okamžitě. Pokud není spotřebován okamžitě, jsou doba a podmínky uchovávání plně v odpovědnosti uživatele a neměly by přesáhnout dobu 24 hodin při teplotě 2 °C až 8 °C, jestliže naředění proběhlo za kontrolovaných a validovaných aseptických podmínek.
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
Podmínky pro uchovávání naředěného přípravku naleznete v bodě 6.3.
6.5 Druh obalu a obsah balení
6ml skleněná lahvička (typ I) s fluorotec/pryžovou zátkou a hliníkovým uzávěrem s růžovým flip off víčkem. Je dodáván v baleních obsahujících 1, 5 a 10 injekčních lahviček.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky. Léčivé přípravky se nesmí vyhazovat do odpadních vod nebo domácího odpadu.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Accord Healthcare Limited Sage House 319, Pinner Road North Harrow Middlesex HA1 4HF Velká Británie
8. REGISTRAČNÍ ČÍSLO (A)
EU/1/12/798/002
EU/1/12/798/003
EU/1/12/798/004
9. DATUM PRVNÍ REGISTRACE
Datum první registrace :19 listopad 2012
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
1. NÁZEV PŘÍPRAVKU
Ibandronic Acid Accord 3 mg injekční roztok v předplněné injekční stříkačce
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna předplněná injekční stříkačka obsahuje acidum ibandronicum 3mg (jako natrii ibandronas monohydricus) ve 3ml roztoku.
Koncentrace kyseliny ibandronové v injekčním roztoku je 1 mg/ml.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok (injekce) Čirý, bezbarvý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba osteoporózy u postmenopauzálních žen se zvýšeným rizikem zlomenin (viz bod 5.1).
Bylo prokázáno snížení rizika zlomenin obratlů, účinnost na zlomeniny krčku proximálního femuru nebyla stanovena.
4.2 Dávkování a způsob podání
Pacientům, kteří budou léčeni kyselinou ibandronovou, musí být předána příbalová informace a karta s připomínkami.
Dávkování
Doporučená dávka kyseliny ibandronové je 3 mg podávaná formou intravenózní injekce během 15-30 vteřin, jedenkrát za tři měsíce.
Pacienti musí dostávat doplňky vápníku a vitamínu D (viz bod 4.4 a bod 4.5).
Pokud dojde k vynechání dávky, měla by být injekce aplikována co možná nejdříve. Poté je třeba naplánovat další aplikaci injekcí v intervalu 3 měsíců od data poslední injekce.
Optimální délka léčby bisfosfonáty u osteoporózy nebyla zatím stanovena. Nutnost pokračování v léčbě je třeba pravidelně opakovaně hodnotit s ohledem na prospěch a možná rizika kyseliny ibandronové u jednotlivého pacienta, a to zvláště po 5 letech léčby a později.
Zvláštní skupiny _pacientů Pacienti s poruchou funkce ledvin
Injekční podávání kyseliny ibandronové u pacientů s hodnotou kreatininu v séru nad 200 ^mol/l (2,3 mg/dl) nebo s clearance kreatininu (zjištěnou nebo předpokládanou) pod 30 ml/min se pro omezené údaje z klinických studií u těchto pacientů nedoporučuje (viz body 4.4 a 5.2).
U pacientů s mírnou až středně těžkou poruchou funkce ledvin, kdy hodnota kreatininu v séru je rovna nebo menší než 200 ^.mol/l (2,3 mg/dl) nebo u nichž je clearance kreatininu (zjištěná nebo předpokládaná) rovna 30 ml/min nebo vyšší, není úprava dávky nutná.
Pacienti s poruchou funkce jater
Úprava dávky není nutná (viz bod 5.2).
Starší populace (>65 let)
Úprava dávky není nutná (viz bod 5.2).
Pediatrická populace
U dětí mladších 18 let nemá kyselina ibandronová příslušné použití, proto nebyla kyselina ibandronová u této populace studována (viz body 5.1 a 5.2).
Způsob podání
Intravenózní podání po dobu 15 - 30 vteřin, každé 3 měsíce.
Je nutné přísně dodržovat intravenózní způsob podání (viz bod 4.4).
4.3 Kontraindikace
- Hypersenzitivita na na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1
- Hypokalcemie
4.4 Zvláštní upozornění a opatření pro použití
Chybné podání
Musí být zabezpečeno, aby nedošlo k aplikaci injekce kyseliny ibandronové intraarteriálně nebo paravenózně, což by mohlo vést k poškození tkáně.
Hypokalcemie
Kyselina ibandronová, stejně jako jiné bisfosfonáty podávané intravenózně, může zapříčinit přechodné snížení hladiny vápníku v séru.
Před zahájením injekční léčby kyselinou ibandronovou musí být upravena přítomná hypokalcemie. Stejně účinně by měly být před zahájením injekční terapie kyselinou ibandronovou léčeny i jiné poruchy kostního a minerálního metabolismu.
Všichni pacienti musí dostávat přiměřené množství doplňků obsahujících vápník a vitamín D. AnafVlaktická reakce/šok
U pacientů léčených kyselinou ibandronovou v intravenózní formě byly hlášeny případy anafylaktické reakce/šoku včetně příhod končících úmrtím.
Pokud je podávána intravenózní injekce kyseliny ibandronové, je třeba, aby byla dostupná vhodná lékařská péče a zajištěno odpovídající sledování pacienta. Jestliže se objeví anafylaktické nebo jiné závažné hypersenzitivní/alergické reakce, okamžitě ukončete podávání injekce a zahajte příslušnou léčbu.
Porucha funkce ledvin
Pacienti trpící současně jinými chorobami nebo pacienti užívající léčivé přípravky, které mohou mít nežádoucí účinky na ledviny, by měli být během léčby pravidelně kontrolováni v souladu se zásadami správné lékařské praxe.
Pro nedostatek klinických zkušeností není kyselina ibandronová v injekční formě doporučována u pacientů s hodnotami kreatininu v séru vyššími než 200 ^.mol/l (2,3 mg/dl) nebo s hodnotami clearance kreatininu pod 30 ml/min (viz bod 4.2 a bod 5.2).
Pacienti s poruchou srdečních funkcí
U pacientů, u kterých je riziko srdečního selhání, je třeba se vyvarovat převodnění.
Osteonekróza čelisti
Velmi vzácně byla u pacientů léčených kyselinou ibandronovou z důvodu onkologických indikací (viz
odstavec 4.8) hlášena v poprodejním sledování osteonekróza čelisti (ONČ).
U pacientů s nezhojenými lézemi měkkých tkání v ústech je třeba odložit zahájení léčby nebo přechod na novou léčbu.
U pacientů s průvodními rizikovými faktory se před zahájením léčby kyselinou ibandronovou doporučuje provést preventivní zubní prohlídku a individuální posouzení přínosů a rizik.
Při vyhodnocování rizika rozvoje ONČ u pacienta je třeba zvažovat následující rizikové faktory:
- Účinnost léčivého přípravku, který inhibuje resorpci kostí (vyšší riziko u vysoce účinných přípravků), způsob podávání (vyšší riziko při parenterálním podávání) a kumulovaná dávka při léčbě resorpce kostí
- Onkologické onemocnění, další chorobné stavy (například anémie, koagulopatie, infekce), kouření
- Doprovodná léčba: kortikosteroidy, chemoterapie, angiogenní inhibitory, radioterapie hlavy a krku
- Nedostatečná ústní hygiena, periodontální onemocnění, nedostatečně sedící umělý chrup, dentální onemocnění v anamnéze, invazivní dentální postupy léčby například extrakce zubů
Všem pacientům je třeba doporučit, aby během léčby kyselinou ibandronovou udržovali dobrou ústní hygienu, absolvovali rutinní zubní prohlídky a ihned ohlásili všechny příznaky onemocnění ústní dutiny jako pohyblivost zubů, bolesti nebo otoky, nehojící se vředy nebo výtoky. V průběhu léčby je třeba provádět invazivní zubní zákroky a postupy jedině po pečlivém zvážení a vyvarovat se jim v bezprostřední blízkosti podávání kyseliny ibandronové.
Plán léčby pacientů, u kterých se rozvine ONČ, musí být stanoven ve spolupráci ošetřujícího lékaře a zubaře nebo ústního chirurga s odbornými znalostmi o ONČ. Dokud se příslušný stav nevyřeší a nebude případně možné zmírnit rizikové faktory, je třeba zvažovat dočasné přerušení léčby kyselinou ibandronovou.
Osteonekróza vnějšího zvukovodu
V případě léčby bisfosfonáty, zejména ve spojení s dlouhodobou léčbou, byla hlášena osteonekróza vnějšího zvukovodu. Možné rizikové faktory osteonekrózy vnějšího zvukovodu zahrnují užívání steroidů a chemoterapii a/nebo lokální rizikové faktory, jakým je infekce nebo trauma. Možnost osteonekrózy vnějšího zvukovodu je třeba zvažovat u pacientů léčených bisfosfonáty, u kterých se projevují příznaky ušních onemocnění včetně chronických ušních infekcí.
Atypické zlomeniny femuru
V souvislosti s léčbou bisfosfonáty byly hlášeny atypické subtrochanterické a diafyzární zlomeniny femuru, zejména u pacientů dlouhodobě léčených pro osteoporózu. Tyto příčné nebo krátké šikmé zlomeniny se mohou objevit kdekoli v celé délce femuru od oblasti těsně pod malým trochanterem až do části těsně nad suprakondylickým rozšířením. Tyto zlomeniny se objevují po minimálním traumatu nebo bez souvislosti s ním a u některých pacientů se mohou projevovat bolestí ve stehně nebo třísle, často sdružené na zobrazovacích vyšetřeních s obrazem typickým pro stresové zlomeniny (neobvyklé nízkotraumatické zlomeniny, v angličtině známé jako „insufficiency fractures“), a to týdny až měsíce před manifestací kompletní zlomeniny kosti stehenní. Zlomeniny jsou často oboustranné, proto je nutné u pacientů léčených bisfosfonáty, kteří utrpěli zlomeninu diafýzy femuru, vyšetřit i kontralaterální femur. Rovněž bylo zaznamenáno špatné hojení těchto zlomenin. U pacientů, u kterých je podezření na atypickou zlomeninu femuru, je třeba při hodnocení jejich stavu zvážit i přerušení léčby bisfosfonáty, a to na základě zhodnocení prospěchu a rizika léčby u jednotlivého pacienta. Pacienty je třeba poučit, aby během léčby bisfosfonáty hlásili jakoukoli bolest v oblasti stehna, kyčle nebo třísla, a všechny pacienty, u kterých se tyto příznaky objeví, je třeba vyšetřit z hlediska možné neúplné zlomeniny femuru.
Injekce kyseliny ibandronové v podstatě neobsahuje sodík.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Metabolické interakce nejsou pravděpodobné, protože kyselina ibandronová neinhibuje hlavní lidské jatemí izoenzymy P450 a bylo prokázáno, že neindukuje jaterní systém cytochromu P450 u potkanů (viz bod 5.2). Kyselina ibandronová se vylučuje pouze ledvinami a nepodléhá žádné biotransformaci.
4.6 Fertilita, těhotenství a kojení
Kyselina ibandronová je určena pouze pro ženy v postmenopauze a nesmí ji používat ženy ve fertilním věku.
Neexistují dostatečné údaje o používání kyseliny ibandronové u těhotných žen. Studie na potkanech prokázaly určitou reprodukční toxicitu (viz bod 5.3). Potenciální riziko pro člověka není známo. Kyselina ibandronová nemá být podávána během těhotenství.
Kojení
Není známo, zda se kyselina ibandronová vylučuje do lidského mléka. Studie na potkanech v laktaci prokázaly po intravenózním podání přítomnost nízkých hladin kyseliny ibandronové v mléce. Kyselina ibandronová nemá být podávána během kojení.
Fertilita
Údaje týkající se účinků kyseliny ibandronové u člověka nejsou k dispozici. V reprodukčních studiích s perorálním podáním u potkanů kyselina ibandronová snižovala fertilitu. Ve studiích s intravenózním podáním u potkanů kyselina ibandronová snižovala fertilitu při vysokých denních dávkách (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Na základě farmakodynamického a farmakokinetického profilu a hlášených nežádoucích účinků lze předpokládat, že kyselina ibandronová nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky
Shrnutí bezpečnostního profilu
Nejzávažnější hlášené nežádoucí účinky jsou anafylaktická reakce/šok, atypické zlomeniny femuru, osteonekróza čelisti, zánět oka (viz bod „Popis vybraných nežádoucích účinků“ a bod 4.4).
Nejčastěji hlášené nežádoucí účinky jsou artralgie a příznaky podobné chřipce. Tyto příznaky jsou obvyklé ve spojitosti s podáním první dávky, obecně mají krátké trvání, jsou mírné nebo středně závažné intenzity a obvykle odezní během pokračující léčby bez potřeby zvláštních opatření (viz bod “Onemocnění podobné chřipce“).
Shrnutí nežádoucích účinků do tabulky
V tabulce 1 je uveden úplný seznam známých nežádoucích účinků.
Bezpečnost perorální léčby kyselinou ibandronovou v dávce 2,5 mg denně byla hodnocena u 1251 pacientů léčených ve 4 placebem kontrolovaných klinických studiích, z nichž většina pacientů pocházela z pivotní tříleté studie se zlomeninami (MF4411).
V pivotní dvouleté studii u postmenopauzálních žen s osteoporózou (BM16550) byla celková bezpečnost intravenózní injekce kyseliny ibandronové 3mg podávané každé tři měsíce podobná jako u perorálního podání kyseliny ibandronové v dávce 2,5mg denně. Celkový podíl pacientů, u kterých se vyskytl nežádoucí účinek, byl 26,0% pro injekce kyseliny ibandronové 3mg podávané každé tři měsíce po jednom roce a 28,6% po dvou letech. Většina případů nežádoucích účinků nevedla k ukončení léčby.
Nežádoucí účinky jsou uvedeny podle tříd orgánových systémů dle databáze MedDRA a četnosti výskytu. Četnosti výskytu jsou definovány podle následujících konvencí: velmi časté (>1/10), časté (>1/100 až <1/10), méně časté (>1/1000 až <1/100), vzácné (>1/10000 až <1/1000), velmi vzácné (<1/10000), není známo (z dostupných údajů nelze určit). V rámci každé skupiny četností jsou nežádoucí účinky uvedeny v pořadí dle klesající závažnosti.
Tabulka 1: Nežádoucí účinky, které se objevily u postmenopauzálních žen léčených kyselinou
ibandronovou 3 mg jednou za 3 měsíce nebo kyselinou ibandronovou 2,5 mg denně ve fázi III studií BM16550 a MF4411 a po uvedení přípravku na trh.
|
Třída orgánových systémů |
Časté |
Méně časté |
Vzácné |
Velmi vzácné |
|
Poruchy imunitního systému |
Exacerbace astmatu |
Hypersenzitivní reakce |
Anafylaktická reakce/šok*f | |
|
Poruchy nervového systému | ||||
|
Poruchy oka |
Zánět oka*f | |||
|
Cévní poruchy |
Flebitida/thrombofle- bitida | |||
|
Gastrointestinální poruchy | ||||
|
Poruchy kůže a podkožní tkáně |
Angioedém, otok obličeje/edém, kopřivka |
Stevens- Johnsonův syndromf, erythema multiformef, bulózní dermatitidaf | ||
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Artralgie, myalgie, muskuloskeletální bolest, bolest zad |
Bolest kostí |
Atypické subtrochanterické a diafyzární zlomeniny femuruf |
Osteonekróza čelisti*f Osteonekróza vnějšího zvukovodu (nežádoucí účinek bisfosfonátů) |
|
Celkové poruchy a reakce v místě aplikace |
Onemocnění podobné chřipce*, únava |
Reakce v místě aplikace, astenie |
*Viz další informace níže fIdentifikovány po uvedení přípravku na trh
Popis vybraných nežádoucích účinků
Onemocnění _podobné chřipce
Příznaky a reakce včetně příznaků chřipkového typu během akutní fáze zahrnovaly bolest svalů, bolest kloubů, horečku, třesavku, únavu, nevolnost, ztrátu chuti k jídlu a bolest kostí.
Osteonekróza čelisti
Případy osteonekrózy čelisti byly hlášeny převážně u pacientů s onkologickým onemocněním, léčených léčivými přípravky, které inhibují resorpci kostí, například kyselinou ibandronovou (viz odstavec 4.4.). Případy ONČ byly hlášeny v poprodejním sledování kyseliny ibandronové.
Při podávání kyseliny ibandronové byly hlášeny oční zánětlivé reakce, jako jsou uveitida, episkleritida, skleritida. V některých případech tyto nežádoucí reakce neustoupily, dokud podávání kyseliny ibandronové nebylo ukončeno.
Anafylaktická reakce/šok
U pacientů léčených kyselinou ibandronovou v intravenózní formě byly hlášeny případy anafylaktické reakce/šoku včetně příhod končících úmrtím.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Neexistují žádné specifické informace o léčbě v případě předávkování injekčně podávanou kyselinou ibandronovou.
Na základě známých vlastností této terapeutické skupiny může intravenózní předávkování vyvolat hypokalcemii, hypofosfatemii a hypomagnezemii. Klinicky významný pokles hladin vápníku, fosforu a hořčíku v séru by měl být upraven podáním glukonátu vápenatého, fosforečnanu draselného nebo sodného resp. síranu hořečnatého.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Léčivé přípravky ovlivňující stavbu a mineralizaci kostí, bisfosfonáty, ATC kód: M05BA06
Mechanismus účinku
Kyselina ibandronová je vysoce účinný bisfosfonát, který patří do skupiny aminobisfosfonátů, působí selektivně na kostní tkáň a specificky inhibuje aktivitu osteoklastů bez přímého ovlivnění kostní novotvorby. Neinterferuje s aktivací osteoklastů. Podávání kyseliny ibandronové u žen po menopauze vede k postupnému přibývání kostní hmoty a snížení výskytu zlomenin v důsledku potlačení zvýšeného kostního obratu až na premenopauzální úroveň.
Farmakodynamické účinky
Farmakodynamickým účinkem kyseliny ibandronové je inhibice kostní resorpce. V pokusech provedených in vivo zabraňuje podávání kyseliny ibandronové destrukci kostí experimentálně navozené při poruše funkce gonád, podání retinoidů, přítomnosti nádorů nebo extraktů z nádorů. Endogenní kostní resorpce je inhibována rovněž u mladých (rychle rostoucích) potkanů, což vede ke zvýšení normální kostní hmoty ve srovnání s neléčenými zvířaty.
Zvířecí modely potvrzují, že kyselina ibandronová je vysoce účinný inhibitor aktivity osteoklastů. U rostoucích potkanů nebyly prokázány žádné známky porušené mineralizace, a to dokonce ani v dávkách 5000krát vyšších než je dávka nezbytná pro léčbu osteoporózy.
Jak denní, tak intermitentní dlouhodobé podávání (s prodlouženými intervaly bez aplikace dávky) vedlo u potkanů, psů a opic k tvorbě nové kostní hmoty se zachovanou normální kvalitou a udrželo nebo zvýšilo mechanickou pevnost dokonce v dávkách v rozmezí toxicity. V klinické studii (MF 4411), která byla zaměřena na účinnost kyseliny ibandronové proti vzniku zlomenin, byla potvrzena účinnost kyseliny ibandronové při každodenním i intermitentním podávání s intervalem bez aplikace dávky v rozmezí 9 - 10 týdnů.
Při studiu na živočišných modelech navodilo podávání kyseliny ibandronové biochemické změny vypovídající o dávkově závislé inhibici kostní resorpce, včetně suprese močových biochemických markerů degradace kostního kolagenu (jako jsou deoxypyridinolin a příčně vázané N- telopeptidy kolagenu typu I (NTX)).
U postmenopauzálních žen došlo při každodenním i při intermitentním podávání (s intervalem bez podávání dávky trvajícím 9 - 10 týdnů ve čtvrtletí) kyseliny ibandronové perorálně i intravenózně k biochemickým změnám značícím inhibici kostní resorpce v závislosti na dávce přípravku.
Po intravenózní injekci kyseliny ibandronové došlo k poklesu hladiny alfa řetězce C-telopeptidu kolagenu typu I v séru (CTX) během 3 - 7 dnů od zahájení léčby a ke snížení hladin osteokalcinu během 3 měsíců.
Po zastavení léčby dochází k opětovnému obnovení patologických hodnot zvýšené kostní přeměny, které se vyskytovaly před léčbou v souvislosti s postmenopauzální osteoporózou.
Histologická analýza biopsií kosti u postmenopauzálních pacientek, jimž byla podávána kyselina ibandronová perorálně v dávce 2,5 mg jednou denně a intermitentně intravenózní dávky až do 1 mg vždy jednou za 3 měsíce, prokázala po dvou a třech letech léčby normální kvalitu kostí a žádné známky poruchy mineralizace kostí. Po dvou letech léčby kyselinou ibandronovou při injekčním podávání dávky 3 mg bylo rovněž pozorováno očekávané snížení kostního obratu ("bone turnover"), normální kvalita kosti a nepřítomnost poruch mineralizace.
Klinická, účinnost
Užití nezávislých rizikových faktorů, jako například nízký BMD, věk, přítomnost již prodělaných zlomenin, zlomeniny v rodinné anamnéze, rychlý úbytek kostní hmoty a nízký body mass index, by mělo být zváženo z důvodu identifikace žen, u kterých je zvýšené riziko výskytu osteoporotických zlomenin.
Kyselina ibandronová 3 mg injekčně jedenkrát za 3 měsíce Denzita kostní hmoty ("bone minerál density", BMD)
Ve dvouleté, randomizované, dvojitě zaslepené multicentrické, non-inferioritní klinické studii (BM16550) prováděné u postmenopauzálních žen (1386 žen ve věku 55 - 80 let) s osteoporózou (vstupní hodnota BMD T-skóre v bederní páteři pod -2,5SD) bylo prokázáno, že podávání kyseliny ibandronové v dávce 3 mg jedenkrát za 3 měsíce je přinejmenším stejně účinné, jako podávání dávky 2,5mg kyseliny ibandronové jednou denně. Tato skutečnost byla potvrzena jak primární analýzou po prvním roce léčby, tak i konfirmační analýzou po dvou letech léčby (tabulka 2).
Primární analýzou údajů získaných ve studii BM16550 po prvním roce a konfirmační analýzou po 2 letech léčby nebyla prokázána nižší účinnost dávkovacího režimu 3mg intravenózně jedenkrát za 3 měsíce v porovnání s dávkovacím režimem 2,5mg perorálně jedenkrát denně, z hlediska průměrného zvýšení BMD v bederní páteři, v celkovém proximálním femuru ("total hip"), v krčku femuru a trochanteru (tabulka 2).
Tabulka 2: Průměrná relativní změna BMD od výchozích hodnot v bederní páteři, celkovém
proximálním femuru, v krčku femuru a trochanteru po jednom roce léčby (primární analýza) a po dvou letech léčby (populace per-protokol) zjištěná ve studii BM16550
|
Údaje ze studie BM16550 po prvním roce léčby |
Údaje ze studie BM16550 po dvou letech léčby | |||
|
Průměrná relativní změna od výchozích hodnot v % [95% CI] |
Kyselina ibandronová 2,5 mg jednou denně (N=377) |
Kyselina ibandronová 3 mg injekce jednou za 3 měsíce (N=365) |
Kyselina ibandronová 2,5 mg jednou denně (N=334) |
Kyselina ibandronová 3 mg injekce jednou za 3 měsíce (N=334) |
|
BMD v bederní páteři L2-L4 |
3,8 [3,4, 4,2] |
4,8 [4,5, 5,2] |
4,8 [4,3, 5,4] |
6,3 [5,7, 6,8] |
|
BMD v celkovém proximálním femuru |
1,8 [1,5,2,1] |
2,4 [2,0, 2,7] |
2,2 [1,8, 2,6] |
3,1 [2,6, 3,6] |
|
BMD v krčku femuru |
1,6 [1,2, 2,0] |
2,3 [1,9, 2,7] |
2,2 [1,8, 2,7] |
2,8 [2,3, 3,3] |
|
BMD v trochanteru |
3,0 [2,6, 3,4] |
3,8 [3,2, 4,4] |
3,5 [3,0, 4,0] |
4,9 [4,1, 5,7] |
Bylo prokázáno, že injekce kyseliny ibandronové 3mg aplikované jedenkrát za 3 měsíce měly lepší účinek než perorálně podávaná kyselina ibandronová v dávce 2,5mg jednou denně, z hlediska zvýšení BMD v bederní páteři v prospektivně plánované analýze po prvním roce bylo p<0,001 a po dvou letech p< 0,001.
V případě BMD v bederní páteři došlo po jednom roce léčby u 92,1 % pacientek, které obdržely 3mg injekce jedenkrát za 3 měsíce, ke zvýšení BMD nebo tato hodnota zůstala beze změn (jednalo se tedy o pacientky odpovídající na léčbu), v porovnání s 84,9% pacientek, kterým byla perorálně podávána dávka 2,5mg jedenkrát denně (p=0,002). Po dvou letech léčby byla u 92,8 % pacientek, jimž byly aplikovány 3 mg injekce jedenkrát za 3 měsíce a u 84,7% pacientek léčených dávkou 2,5mg perorálně jednou denně, hodnota BMD v bederní páteři vyšší nebo se nezměnila (p=0,001).
Z hlediska BMD v celkovém proximálním femuru odpovídalo na léčbu po prvním roce 82,3% pacientek, kterým byly aplikovány injekce 3mg jedenkrát za 3 měsíce v porovnání se 75,1% pacientek, které dostávaly 2,5mg perorálně jedenkrát denně (p=0,02). Po dvou letech léčby se hodnota BMD v celkovém proximálním femuru zvýšila nebo nezměnila u 85,6% pacientek, jimž byly aplikovány 3mg injekce jedenkrát za 3 měsíce a u 77,0% pacientek léčených dávkou 2,5mg perorálně jednou denně (p=0,004).
Podíl pacientek, u nichž došlo ke zvýšení BMD v bederní páteři i v celkovém proximálním femuru nebo se tato hodnota nezměnila po prvním roce léčby, byl 76,2% ve skupině, jíž byly aplikovány 3mg injekce jedenkrát za 3 měsíce a 67,2% ve skupině léčené dávkou 2,5mg perorálně jednou denně (p=0,007). Po dvou letech bylo toto kritérium splněno u 80,1 % pacientek ve skupině léčené 3 mg injekcemi jedenkrát za 3 měsíce a u 68,8% pacientek ve skupině léčené dávkou 2,5mg perorálně jednou denně (p=0,001).
Biochemické markéry kostního obratu
Klinicky významné snížení hladin CTX v séru bylo pozorováno ve všech časových bodech studie.
V 12. měsíci byl u léčebného režimu s podáváním intravenózních injekcí 3mg jedenkrát za 3 měsíce medián vyjadřující relativní změnu od výchozích hodnot -58,6% a u režimu s podáváním dávky 2,5mg perorálně jednou denně -62,6%. Na léčbu odpovídalo 64,8% pacientů, jimž byla intravenózně aplikována dávka 3mg jedenkrát za tři měsíce a 64,9% pacientů léčených perorálně podávanou dávkou 2,5mg jednou denně (odpovídavost byla definována jako snížení o >50% od výchozích hodnot). Snížení CTX v séru přetrvávalo více než 2 roky u více než poloviny pacientů, kteří odpovídali na léčbu v obou léčebných skupinách.
Na základě výsledků studie BM16550 lze očekávat, že kyselina ibandronová podávaná formou intravenózních injekcí v dávce 3mg jedenkrát za tři měsíce bude v prevenci zlomenin přinejmenším stejně účinná jako podávání kyseliny ibandronové perorálně v dávce 2,5mg jednou denně.
Tablety kyseliny ibandronové 2,5mg jedenkrát denně
V úvodní tříleté, randomizované, dvojitě zaslepené, placebem kontrolované studii sledující výskyt zlomenin (MF 4411) bylo prokázáno statisticky významné a léčebně relevantní snížení incidence nových radiograficky prokázaných, morfometrických a klinických zlomenin obratlů (tabulka 3). V této studii bylo posuzováno perorální podávání kyseliny ibandronové v dávce 2,5mg denně a 20mg periodicky v rámci výzkumného režimu. Kyselina ibandronová byla užívána 60minut před prvním denním jídlem nebo nápojem (období lačnění po požití přípravku). Do studie byly zařazeny ženy ve věku od 55 do 80 let, alespoň 5 let po menopauze, s BMD v bederní páteři v rozmezí od 2 do 5 SD pod průměr premenopauzálních žen (T-skóre) a to alespoň v jednom obratli [L1 - L4] a s jednou až čtyřmi předchozími zlomeninami obratlů. Všechny pacientky dostávaly 500mg vápníku a 400m.j. vitaminu D denně. Účinnost byla posuzována u 2928 pacientek. Užívání kyseliny ibandronové v dávce 2,5mg denně vedlo ke statisticky významnému a léčebně relevantnímu snížení incidence nových zlomenin obratlů. Za tři roky trvání studie byl snížen výskyt nových radiograficky potvrzených zlomenin obratlů o 62% (p=0,0001). Po 2 letech bylo pozorováno snížení relativního rizika o 61% (p=0,0006). Po jednom roce léčby nebyl statisticky významný rozdíl zaznamenán (p=0,056). Účinek na zlomeniny byl během trvání celé studie konzistentní. Nebyl žádný náznak svědčící o oslabení tohoto účinku v čase.
Po třech letech léčby byla také signifikantně snížena incidence klinických zlomenin obratlů o 49% (p=0,011). Výrazný účinek na vertebrální zlomeniny se dále odrazil ve statisticky významném snížení ztráty tělesné výšky ve srovnání s placebem (p<0,0001).
Tabulka 3: Výsledky 3-leté studie MF 4411 sledující účinek na zlomeniny (%, 95% CI)
|
Placebo (N=974) |
Kyselina ibandronová 2,5 mg jednou denně (N=977) | |
|
Snížení relativního rizika výskytu nových morfometrických vertebrálních fraktur |
62 % (40,9, 75,1) | |
|
Incidence nových morfometrických vertebrálních fraktur |
9,56% (7,5, 11,7) |
4,68% (3,2,6,2) |
|
Snížení relativního rizika klinických vertebrálních fraktur |
49% (14,03, 69,49) | |
|
Incidence klinických vertebrálních |
5,33% |
2,75 % |
|
fraktur |
(3,73, 6,92) |
(1,61, 3,89) |
|
BMD - průměrná změna oproti vstupní hodnotě v bederní páteři ve 3. roce |
1,26 % (0,8, 1,7) |
6,54 % (6,1, 7,0) |
|
BMD - průměrná změna oproti vstupní hodnotě v celkovém proximálním femuru ve 3. roce |
-0,69 % (-1,0, -0,4) |
3,36% (3,0, 3,7) |
Terapeutický efekt kyseliny ibandronové byl dále posuzován v analýze subpopulace pacientek se vstupní hodnotou BMD T-skóre v bederní páteři pod -2,5 (tabulka 4). Snížení rizika zlomeniny obratle bylo velmi obdobné jako u celé sledované populace.
Tabulka 4: Výsledky 3-leté studie MF 4411 (%, 95 % CI) sledující účinek na zlomeniny u pacientek se vstupní hodnotou BMD T-skóre pod -2,5
|
Placebo (N=587) |
Kyselina ibandronová 2,5 mg jednou denně (N=575) | |
|
Snížení relativního rizika výskytu nových morfometrických vertebrálních fraktur |
59% (34,5, 74,3) | |
|
Incidence nových morfometrických vertebrálních fraktur |
12,54% (9,53, 15,55) |
5,36% (3,31, 7,41) |
|
Snížení relativního rizika klinických vertebrálních fraktur |
50% (9,49, 71,91) | |
|
Incidence klinických vertebrálních fraktur |
6,97% (4,67, 9,27) |
3,57% (1,89, 5,24) |
|
BMD - průměrná změna oproti vstupní hodnotě v bederní páteři ve 3. roce |
1,13% (0,6, 1,7) |
7,01% (6,5, 7,6) |
|
BMD - průměrná změna oproti vstupní hodnotě v celkovém proximálním femuru ve 3. roce |
-0,70% (-1,1, -0,2) |
3,59% (3,1, 4,1) |
V celkové populaci pacientů zařazených do studie MF4411 nebylo zaznamenáno snížení počtu nevertebrálních zlomenin, avšak při denním podávání kyseliny ibandronové byla prokázána účinnost u vysoce rizikové subpopulace (s hodnotami BMD T-skóre < -3.0 v krčku proximálního femuru), kde bylo zaznamenáno snížení rizika nevertebrálních zlomenin o 69%.
Denní perorální podávání tablet kyseliny ibandronové v dávce 2,5mg vedlo k postupnému zvyšování BMD jak v oblasti obratlů, tak na ostatních místech skeletu.
Za tři roky léčby bylo zvýšení BMD v bederní páteři ve srovnání s placebem 5,3% a 6,5% oproti vstupní hodnotě. V oblasti proximálního femuru byla zaznamenána zvýšení oproti vstupním hodnotám 2,8% v krčku femuru, 3,4% v celkovém proximálním femuru a 5,5% v trochanteru.
Biochemické markery kostního obratu (jako jsou močový CTX a sérový osteokalcin) vykázaly očekávanou supresi na premenopauzální úroveň a maximální suprese bylo dosaženo během období 3 -6 měsíců užívání 2,5mg kyseliny ibandronové jednou denně.
Klinicky významný pokles biochemických markerů kostní resorpce o 50% byl pozorován již jeden měsíc po zahájení léčby kyselinou ibandronovou 2,5mg.
Pediatrická populace (viz body 4.2 a 5.2)
Podávání kyseliny ibandronové nebylo studováno v pediatrické populaci, proto nejsou k dispozici žádné údaje o účinnosti nebo bezpečnosti pro tuto skupinu pacientů.
5.2 Farmakokinetické vlastnosti
Různé studie u zvířat i lidí prokázaly, že primární farmakologické účinky kyseliny ibandronové na kost nejsou v přímém vztahu k aktuálním plazmatickým koncentracím.
Po intravenózním podání 0,5mg - 6mg kyseliny ibandronové se úměrně k dávce zvýšila koncentrace této látky v plazmě.
Absorpce Není relevantní
Po počáteční systémové expozici se kyselina ibandronová rychle váže v kosti nebo je vyloučena do moči. Konečný distribuční objem u člověka je alespoň 90 l a velikost dávky, která se dostane do kosti je odhadována na 40 - 50% cirkulující dávky. Vazba na plazmatické bílkoviny u člověka je přibližně 85% - 87% (zjištěno in vitro při terapeutických koncentracích kyseliny ibandronové) a tudíž potenciál pro interakce s jinými léčivými přípravky způsobené vytěsněním z vazby je nízký.
Biotransformace
Metabolizace kyseliny ibandronové nebyla prokázána u zvířat ani lidí.
Eliminace
Kyselina ibandronová je z krevního oběhu odstraněna absorpcí do kostí (u postmenopauzálních žen jde zhruba o 40 - 50%), zbývající množství je v nezměněné formě vyloučeno ledvinami.
Rozmezí pozorovaných poločasů je široké, ale terminální poločas se obecně pohybuje v rozmezí 10 -72 hodin. Vypočtené hodnoty jsou z velké části závislé na trvání studie, použité dávce a citlivosti stanovení, proto je skutečný terminální poločas stejně jako u jiných bisfosfonátů pravděpodobně mnohem delší. Časně dosažené plazmatické koncentrace se rychle snižují a dosahují 10 % vrcholových hodnot během 3 a 8 hodin po intravenózním, resp. perorálním podání.
Celková clearance kyseliny ibandronové je nízká, s průměrnými hodnotami v rozmezí 84 -160 ml/min. Renální clearance (zhruba 60 ml/min u zdravých postmenopauzálních žen) odpovídá za 50 - 60 % celkové clearance a je v relaci s clearancí kreatininu. Rozdíl mezi celkovou a renální clearancí odráží vychytávání přípravku kostí.
Sekreční cesta se nepřekrývá s žádným ze známých kyselých nebo zásaditých transportních systémů, které se podílejí na vylučování jiných léčivých látek (viz bod 4.5). Kyselina ibandronová dále neinhibuje hlavní lidské hepatické izoenzymy P450 a neindukuje systém hepatického cytochromu P 450 u potkanů.
Farmakokinetika. ve zvláštních klinických situacích
Pohlaví
Farmakokinetika kyseliny ibandronové je u mužů i žen obdobná.
Rasa
Neexistují žádné důkazy pro klinicky významné rozdíly v chování kyseliny ibandronové u příslušníků jednotlivých etnických skupin bělošského a asijského původu. U pacientů afro-amerického původu jsou k dispozici pouze omezená data.
Pacienti s poruchou funkce ledvin
Renální clearance kyseliny ibandronové u pacientů s různým stupněm renální insuficience je přímo úměrná clearanci kreatininu (CLcr).
U pacientů s mírnou nebo středně těžkou renální insuficiencí (CLcr vyšší nebo rovna 30 ml/min) není nutná žádná úprava dávky.
Pacienti s těžkou renální insuficiencí (CLcr nižší než 30 ml/min), kteří denně dostávali 10 mg kyseliny ibandronové v perorální formě po dobu 21 dnů, měli 2 - 3krát vyšší plazmatické koncentrace než osoby s normální funkcí ledvin a celková clearance kyseliny ibandronové činila 44 ml/min. Po intravenózním podání dávky 0,5 mg kyseliny ibandronové se u osob s těžkou renální insuficiencí snížila celková, renální a nerenální clearance o 67 %, 77 %, resp. 50 %, nebyl však pozorován pokles snášenlivosti spojený se zvýšenou expozicí. Pro nedostatečné klinické zkušenosti není kyselina ibandronová doporučována u pacientů s těžkou renální insuficiencí (viz bod 4.2 a bod 4.4). Farmakokinetika kyseliny ibandronové byla posuzována u nízkého počtu pacientů v konečné fázi onemocnění ledvin léčených hemodialýzou, u pacientů, kteří nepodstupují dialýzu, není farmakokinetika kyseliny ibandronové známa. U pacientů v konečné fázi onemocnění ledvin jsou k
dispozici pouze omezené údaje, proto by kyselina ibandronová neměla být v těchto případech podávána.
Pacienti s poruchou funkce jater (viz bod 4.2)
Neexistují žádné údaje o farmakokinetice při podávání kyseliny ibandronové u pacientů s jaterní insuficiencí. Játra nemají významnou úlohu v clearanci kyseliny ibandronové, která není metabolizována, ale je vylučována ledvinami a absorbována v kostech. U pacientů s jaterní insuficiencí není tedy úprava dávky nutná.
Starší populace (viz bod 4.2)
V multivariační analýze nebyl prokázán věk jako nezávislý faktor žádného ze sledovaných farmakokinetických parametrů. Jediným faktorem, který je nutné vzít v úvahu, je pokles funkce ledvin s věkem (viz bod Pacienti s poruchou funkce ledvin).
Pediatrická populace (viz body 4.2 a 5.1)
Neexistují žádné údaje o užívání kyseliny ibandronové v těchto věkových skupinách.
5.3 Předklinické údaje vztahující se k bezpečnosti
Toxické účinky, např. známky poškození ledvin, byly pozorovány u psů pouze po expozicích dostatečně převyšujících maximální expozici u člověka, což svědčí pro malý význam při klinickém použití.
Mutagenita/Kancerogenita:
Nebyly pozorovány žádné známky kancerogenního potenciálu. Studie na genotoxicitu neodhalily žádný důkaz genetické aktivity kyseliny ibandronové.
Reprodukční toxicita:
Specifické studie zaměřené na tříměsíční dávkovací režim nebyly prováděny. Ve studiích dávkovacího režimu založeného na denní i.v. aplikaci nebyl nalezen žádný důkaz přímé fetální toxicity nebo teratogenního účinku kyseliny ibandronové u potkanů ani králíků. U potkanů v generaci F1 došlo k poklesu tělesné hmotnosti. V reprodukčních studiích s perorálním podáním u potkanů se účinky na fertilitu sestávaly ze zvýšených preimplantačních ztrát při dávkách 1 mg/kg/den a vyšších.
V reprodukčních studiích s intravenózním podáním u potkanů kyselina ibandronová snižovala počet spermií při dávkách 0,3 a 1 mg/kg/den a snižovala fertilitu u samců při dávkách 1 mg/kg/den a u samic při dávkách 1,2 mg/kg/den. Další nežádoucí účinky kyseliny ibandronové ve studiích reprodukční toxicity u potkanů se nelišily od nežádoucích účinků pozorovaných u bisfosfonátů jako celé skupiny. Zahrnují snížený počet implantačních míst, interferenci s přirozeným způsobem porodu (dystocie) a zvýšení výskytu viscerálních variací (tzv. "renal pelvis ureter syndrom").
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Chlorid sodný Kyselina octová, ledová Trihydrát octanu sodného Voda na injekci
6.2 Inkompatibility
Injekční roztok kyseliny ibandronové nesmí být mísen s roztoky obsahujícími kalcium ani s jinými intravenózně podávanými léčivými přípravky.
6.3 Doba použitelnosti
2 roky.
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
6.5 Druh obalu a obsah balení
Předplněné injekční stříkačky vyrobené z bezbarvého skla, opatřené šedou pryžovou plunžrovou zátkou a víčkem, obsahují 3 ml injekčního roztoku.
Balení po 1 předplněné injekční stříkačce s 1 injekční jehlou nebo po 4 předplněných injekčních stříkačkách se 4 injekčními jehlami.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku
V případě, že léčivý přípravek je podán za využití již zavedené i.v. infuzní sestavy, může být k infuzi použit pouze fyziologický roztok nebo roztok glukózy o koncentraci 50mg/ml (5%). Toto platí to také pro roztoky používané k promytí křídelkového adaptéru a dalších zařízení.
Veškerý nepoužitý injekční roztok, stříkačky i injekční jehly musí být zlikvidovány v souladu s místními požadavky. Léčivé přípravky se nesmí vyhazovat do odpadních vod nebo domácího odpadu.
Je třeba přesně dodržovat následující body týkající se použití a likvidace stříkaček a jiných ostrých předmětů:
• Jehly a stříkačky se nikdy nesmí použít znovu.
• Odložte všechny použité jehly a stříkačky do nádoby na ostré předměty (nepropíchnutelné nádoby, kterou máte k dispozici).
• Uchovávejte tuto nádobu mimo dosah dětí.
• Použité nádoby na ostré předměty se nesmí vyhazovat do domácího odpadu.
• Naplněnou nádobu zlikvidujte v souladu s místními požadavky nebo dle instrukcí zdravotnického personálu.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Accord Healthcare Limited Sage House 319, Pinner Road North Harrow Middlesex HA1 4HF Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/12/798/005
EU/1/12/798/006
9. DATUM PRVNÍ REGISTRACE
Datum první registrace : 23.března 2015
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A. VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného/výrobců odpovědných za propouštění šarží
Accord Healthcare Ltd.
Sage House 319 Pinner road
North Harrow, Middlesex HA1 4HF Velká Británie
CEMELOG- BRS Ltd.
H-2040 Budaors, Vasut u.13.
Maďarsko
Tištěný příbalový leták léčivého přípravku musí uvádět název a adresu výrobce odpovědného za výrobu předmětné dávky.
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Ibandronic Acid Accord 2 mg a 6 mg koncentrát pro infuzní roztok (onkologické indikace):
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
Ibandronic Acid Accord 3 mg roztok pro injekční roztok (indikace při osteoporóze): Výdej léčivého přípravku vázán na lékařský předpis.
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky na předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou stanoveny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a v dalších aktualizacích zveřejněných na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné předložit současně.
Další opatření k minimalizace rizik
Držitel registrace musí zajistit, aby byla implementována karta připomínek pro pacienta týkající osteonekrózy.
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Ibandronic Acid Accord 2 mg koncentrát pro infuzní roztok acidum ibandronicum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna injekční lahvička obsahuje acidum ibandronicum 2 mg (jako natrii ibandronas monohydricus 2,25 mg).
3. SEZNAM POMOCNÝCH LÁTEK
Chlorid sodný, trihydrát octanu sodného, kyselina octová 98% a voda na injekci. Další údaje naleznete v příbalové informaci
4. LÉKOVÁ FORMA A VELIKOST BALENÍ
Koncentrát pro infuzní roztok 1 injekční lahvička (2 mg/2 ml)
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci Intravenózní podání, pro infuzi po naředění
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP Přečtěte si příbalovou informaci, kde najdete dobu použitelnosti po naředění.
9 ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z TAKOVÝCH LÉČIVÝCH PŘÍPRAVKŮ, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Accord Healthcare Limited Sage House 319, Pinner Road North Harrow Middlesex HA1 4 HF Velká Británie
12. REGISTRAČNÍ CÍSLO/CÍSLA
1 injekční lahvička: EU/1/12/798/001
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
Nevyžaduje se - odůvodnění přijato
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Ibandronic Acid Accord 2 mg sterilní koncentrát acidum ibandronicum i.v. podání
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP:
4 ČÍSLO ŠARŽE<KÓDY ODBĚRU A LÉKŮ>
Lot:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET DÁVEK
2 mg/2 ml
6. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Ibandronic Acid Accord 6 mg koncentrát pro infuzní roztok acidum ibandronicum
2. OBSAH LÉČIVÉ LÁTKY/ LÉČIVÝCH LÁTEK
Jedna injekční lahvička obsahuje acidum ibandronicum 6 mg (jako natrii ibandronas monohydricus 6,75 mg).
3. SEZNAM POMOCNÝCH LÁTEK
Chlorid sodný, trihydrát octanu sodného kyselina octová 98% a voda na injekci. Další údaje naleznete v příbalové informaci.
4. LÉKOVÁ FORMA A VELIKOST BALENÍ
Koncentrát pro infuzní roztok 1 injekční lahvička (6 mg/6 ml)
5 injekčních lahviček (6 mg/6 ml) 10 injekčních lahviček (6 mg/6 ml
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci Intravenózní podání, pro infuzi po naředění
6 ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
Přečtěte si příbalovou informaci, kde najdete dobu použitelnosti po naředění.
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
11 NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Accord Healthcare Limited Sage House 319, Pinner Road North Harrow Middlesex HA1 4 HF Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA 1 injekční lahvička: EU/1/12/798/002 5 injekční lahvička: EU/1/12/798/003 10 injekční lahvička: EU/1/12/798/004 13. ČÍSLO ŠARŽE
Lot:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis
15. NÁVOD K POUŽITÍ 16. INFORMACE V BRAILLOVE PÍSMU
Nevyžaduje se - odůvodnění přijato
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Ibandronic Acid Accord 6 mg sterilní koncentrát acidum ibandronicum i.v. podání
2 ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4 ČÍSLO ŠARŽE<KÓDY odběru a léků>
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET DÁVEK
6 mg/6 ml
6. JINÉ
Ibandronic Acid Accord 3 mg injekční roztok v předplněné injekční stříkačce Acidum ibandronicum
2. OBSAH LÉČIVÉ LÁTKY/LÁTEK
Jedna předplněná injekční stříkačka obsahuje acidum ibandronicum 3 mg (jako natrii ibandronas monohydricus) ve 3 ml roztoku.
3. SEZNAM POMOCNÝCH LÁTEK
Pomocné látky: chlorid sodný, kyselina octová, ledová, trihydrát octanu sodného a voda na injekci. Další údaje naleznete v příbalové informaci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Injekční roztok
1 předplněná injekční stříkačka + 1 injekční jehla 4 předplněné injekční stříkačky + 4 injekční jehly
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci Pouze k intravenóznímu podání
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do:
9 ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Accord Healthcare Limited Sage House 319, Pinner Road North Harrow Middlesex HA1 4HF Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
1 předplněná injekční stříkačka: EU/1/12/798/005 4 předplněné injekční stříkačky: EU/1/12/798/006
13. ČÍSLO ŠARŽE
č.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
[Neuplatňuje se - odůvodnění přijato.]
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Ibandronic Acid Accord 3 mg injekční roztok Acidum ibandronicum Pouze pro i.v. podání
2 ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro pacienta
Ibandronic Acid Accord 2 mg koncentrát pro infuzní roztok Ibandronic Acid Accord 6 mg koncentrát pro infuzní roztok
acidum ibandronicum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
• Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
• Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
• Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři,lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Ibandronic Acid Accord a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Ibandronic Acid Accord používat
3. Jak se přípravek Ibandronic Acid Accord používá
4. Možné nežádoucí účinky
5. Jak přípravek Ibandronic Acid Accord uchovávat
6. Obsah balení a další informace
1. Co je přípravek Ibandronic Acid Accord a k čemu se používá
Přípravek Ibandronic Acid Accord obsahuje léčivou látku kyselinu ibandronovou. Ta patří do skupiny léků nazývaných bisfosfonáty.
Přípravek Ibandronic Acid Accord je používán u dospělých pacientů a předepisuje se při rakovině prsu, která se šíří do kostí („kostní metastázy“).
• Napomáhá předcházet vzniku kostních zlomenin (fraktur).
• Napomáhá předcházet dalším kostním komplikacím, které by mohly vyžadovat chirurgický zákrok nebo radioterapii.
Přípravek Ibandronic Acid Accord lze rovněž předepsat v případě zvýšené hladiny vápníku v krvi v důsledku nádorového onemocnění.
Přípravek Ibandronic Acid Accord snižuje ztráty vápníku z kostí a tím zamezuje oslabování kostí.
2. Čemu musíte věnovat pozornost, než začnete přípravek Ibandronic Acid Accord používat Nepoužívejte přípravek Ibandronic Acid Accord
• jestliže jste alergický/á na kyselinu ibandronovou nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
• jestliže máte nebo jste někdy měl(a) nízkou hladinu vápníku v krvi.
Pokud se Vás cokoli z výše zmíněného týká, nepoužívejte tento lék. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem dříve, než začnete přípravek Ibandronic Acid Accord používat.
Upozornění a opatření
Velmi vzácně byla u pacientů léčených kyselinou ibandronovou z důvodu onkologických indikací hlášena v poprodejním sledování osteonekróza čelisti (ONČ). ONČ se může projevit i po ukončení léčby.
Je velmi důležité pokusit se předcházet rozvoji ONČ, protože jde o bolestivý stav, jehož léčba může být obtížná. Ke snížení rizika rozvoje osteonekrózy čelisti je třeba přijmout určitá preventivní opatření.
Před zahájením léčby informujte svého lékaře/zdravotní sestru (zdravotnický personál) o případných následujících stavech:
- Máte jakékoli problémy se svými ústy nebo zuby, například nedostatečný zdravotní stav ústní dutiny, onemocnění dásní nebo plánovaná extrakce zubu.
- Nemáte pravidelnou zubní péči nebo jste dlouho nebyl(a) na preventivní zubní prohlídce.
- Jste kuřák/kuřačka (protože to může zvyšovat riziko zubních problémů).
- Byl(a) jste v minulosti léčen(a) bisfosfonáty (používanými k léčbě onemocnění kostí).
- Užíváte léky ze skupiny kortikosteroidů (jako prednisolon nebo dexametazon).
- Máte onkologické onemocnění.
Než bude u vás zahájena léčba kyselinou ibandronovou, může vás lékař požádat, abyste absolvoval(a) preventivní zubní prohlídku.
Během léčby kyselinou ibandronovou udržujte dobrou ústní hygienu (včetně pravidelného čištění zubů), absolvujte rutinní zubní prohlídky. Jestliže máte umělý chrup, zajistěte, aby správně seděl. Jestliže jste léčeni stomatologem nebo se podrobíte zubnímu chirurgickému zákroku (například extrakce zubů), informujte svého lékaře o stomatologické léčbě a svého zubního lékaře informujte, že jste léčeni kyselinou ibandronovou.
Pokud se u vás projeví problémy s ústní dutinou nebo se zuby, jako jsou uvolněné zuby, bolest nebo otoky, nehojící se vředy nebo výtoky, ihned informujte svého lékaře a zubního lékaře, protože může jít o příznaky osteonekrózy čelisti.
Před použitím přípravku Ibandronic Acid Accord se poraďte se svým lékařem, lékárníkem nebo zdravotní sestrou:
• jestliže j ste alergický(á) na jakékoli jiné bisfosfonáty
• jestliže máte vysokou nebo nízkou hladinu vitamínu D, vápník nebo jakýchkoli jiných minerálů
• jestliže máte problémy s ledvinami
• jestliže máte srdeční obtíže a Váš lékař Vám doporučil omezit denní příjem tekutin
U pacientů léčených kyselinou ibandronovou v intravenózní formě byly hlášeny případy závažné alergické reakce, někdy končící úmrtím.
Okamžitě oznamte svému lékaři nebo zdravotní sestře, pokud zaznamenáte některý z následujících příznaků, jako je zkrácení dechu/obtížné dýchání, pocit tísně v krku, otok jazyka, závrať, pocit ztráty vědomí, zčervenání nebo otok obličeje, vyrážka na těle, nevolnost a zvracení (viz bod 4).
Děti a dospívající
Přípravek Ibandronic Acid Accord se nesmí používat u dětí a dospívajících ve věku nižším než 18 let. Další léčivé přípravky a přípravek Ibandronic Acid Accord
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat. To je proto, že přípravek Ibandronic Acid Accord může ovlivňovat způsob, jakým některé další léky účinkují a naopak některé jiné léky mohou ovlivňovat způsob, jakým účinkuje přípravek Ibandronic Acid Accord.
Zejména sdělte svému lékaři nebo lékárníkovi, pokud dostáváte určitý typ injekčního antibiotika ze skupiny aminoglykosidů, jako je např. gentamicin. To je proto, že aminoglykosidy i přípravek Ibandronic Acid Accord mohou snižovat množství vápníku v krvi.
Těhotenství a kojení
Pokud jste těhotná, těhotenství plánujete nebo pokud kojíte, neměla byste přípravek Ibandronic Acid Accord používat.
Poraďte se se svým lékařem nebo lékárníkem dříve, než začnete užívat jakýkoliv lék.
Řízení dopravních prostředků a obsluha strojů
Není známo, zda přípravek Ibandronic Acid Accord ovlivňuje schopnost řídit dopravní prostředky, obsluhovat stroje nebo přístroje. Pokud chcete řídit, obsluhovat stroje nebo přístroje, poraďte se nejprve se svým lékařem.
Přípravek Ibandronic Acid Accord obsahuje méně než 1 mmol sodíku (23 mg) v jedné injekční lahvičce, tzn., že je v podstatě “bez sodíku”.
3. Jak se přípravek Ibandronic Acid Accord používá Používání tohoto léku
• Přípravek Ibandronic Acid Accord obvykle podává lékař nebo zdravotnický personál, který má zkušenosti s léčbou nádorových onemocnění
• Obvykle se podává ve formě infuze do žíly
Při podávání přípravku Ibandronic Acid Accord Vám lékař může pravidelně provádět krevní testy.
Tím kontroluje, zda Vám podává správné množství léku.
Kolik přípravku budete dostávat
Podle Vašeho onemocnění lékař rozhodne, jaké množství přípravku Ibandronic Acid Accord budete dostávat.
Pokud máte rakovinu prsu, která se rozšířila do kostí, doporučená dávka je 6 mg každé 3-4 týdny podaná ve formě infuze do žíly po dobu alespoň 15 minut.
Pokud máte zvýšené hladiny vápníku v krvi v důsledku nádorového onemocnění, doporučená dávka je jednorázové podání 2 mg nebo 4 mg v závislosti na závažnosti Vašeho onemocnění. Lék má být podán ve formě infuze do žíly po dobu alespoň dvou hodin.
V případě nedostatečné odpovědi nebo při opětném propuknutí Vašeho onemocnění může být zváženo opakované podání dávky.
Pokud máte problémy s ledvinami, může lékař dávku a trvání nitrožilní infuze upravit.
Máte-li jakékoli další otázky, týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí
vyskytnout u každého.
Pokud zaznamenáte jakýkoli z následujících závažných nežádoucích účinků, sdělte to ihned
zdravotní sestře nebo lékaři - je možné, že budete potřebovat akutní lékařské ošetření:
Vzácné (mohou postihnout až 1 osobu z 1000)
• přetrvávající bolest a zánět oka
• nová bolest, slabost nebo nepříjemné pocity v oblasti stehna, kyčle nebo třísla. Můžete mít časné známky možné neobvyklé zlomeniny stehenní kosti
Velmi vzácné (mohou postihnout až 1 osobu z 10000)
• bolest nebo bolavé místo v ústech nebo čelisti. Můžete mít časné známky závažných potíží s čelistí (nekrózy (mrtvé kostní tkáně) v čelistní kosti)
• jestliže trpíte bolestmi uší, výtoky z ucha a/nebo ušní infekcí, informujte svého lékaře. Tyto
příznaky mohou být symptomy poškození kostí v uchu.^ svědění, otok obličeje, rtů, jazyka a
hrdla, s obtížným dýcháním. Můžete mít závažnou, možno život ohrožující, alergickou reakci (viz bod 2)
závažné nežádoucí kožní reakce
Není známo (četnost z dostupných údajů nelze určit)
• astmatický záchvat
Další možné nežádoucí účinky
Velmi časté (mohou postihnou až 1 osobu z 10)
• příznaky podobné chřipce, včetně horečky, třesu a zimnice, člověk se celkově necítí dobře, únavy, bolesti kostí a bolesti svalů a kloubů. Tyto příznaky obvykle vymizí během několika hodin nebo dnů. Pokud Vás kterýkoli z těchto účinků začne obtěžovat nebo trvá více než několik dní, sdělte to zdravotní sestře nebo lékaři
• zvýšení tělesné teploty.
• bolest žaludku a břicha, porucha trávení, nevolnost, zvracení nebo průjem (řídká stolice)
• nízká hladina vápníku nebo fosfátů v krvi
• změny výsledků krevních testů, např. gama-glutamyltransferázy nebo kreatininu
• problém se srdečním rytmem nazývaný „blokáda Tawarova raménka“
• bolest kostí nebo svalů
• bolest hlavy, závratě nebo pocit slabosti
• pocit žízně, bolest v krku, změny chuti
• otoky nohou
• bolesti kloubů, zánět kloubů nebo jiné kloubní problémy
• problémy s příštítnými tělísky
• tvorba modřin
• infekce
• problémy s očima nazývané šedý zákal
• kožní problémy
• problémy se zuby.
Méně časté (mohou postihnout méně než 1 osobu ze 100)
• třes nebo chvění
• příliš nízká tělesná teplota („hypotermie“)
• postižení krevních cév v mozku nazývané „cerebrovaskulární onemocnění“ (cévní mozková příhoda nebo krvácení do mozku)
• problémy se srdcem nebo krevním oběhem (včetně bušení srdce, srdečního infarktu, vysokého krevního tlaku a křečových žil)
• změny počtu krvinek („anémie“)
• vysoká hladina alkalické fosfatázy v krvi
• zadržování tekutin a otoky („lymfedém“)
• tekutina na plicích
• žaludeční problémy, např. „gastroenteritida“ nebo „gastritida“
• žlučové kameny
• neschopnost močení, zánět močového měchýře
• migréna
• bolest v průběhu nervů, poškození nervových kořenů
• hluchota
• zvýšená citlivost ke sluchovým, chuťovým nebo dotykovým podnětům, nebo změny čichu
• obtíže s polykáním
• vředy v ústech, otoky rtů (zánět rtů), ústní moučnivka
• svědění nebo brnění v okolí úst
• pánevní bolest, výtok, svědění nebo bolest v pochvě
• kožní výrůstky (nezhoubné kožní útvary)
• ztráta paměti
• problémy se spaním, pocit úzkosti, emoční labilita, nebo výkyvy nálady
• kožní vyrážka
• vypadávání vlasů
• poranění nebo bolest v místě vpichu injekce
• ztráta tělesné hmotnosti
• cysty na ledvinách (tekutinou naplněný útvar v ledvinách).
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Ibandronic Acid Accord uchovávat
• Uchovávejte tento přípravek mimo dohled a dosah dětí.
• Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce za „Použitelné do:“ a nálepce injekční lahvičky za „EXP“. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
• Tento léčivý přípravek nevyžaduje před naředěním žádné zvláštní podmínky uchovávání.
Po naředění:
Chemická a fyzikální stabilita při použití po naředění v 0,9% roztoku chloridu sodného nebo 5% roztoku glukózy byla prokázána po dobu 36 hodin při teplotě 25 °C a 2 °C až 8 °C. Z mikrobiologického hlediska má být roztok spotřebován okamžitě. Pokud není spotřebován okamžitě, jsou doba a podmínky uchovávání plně v odpovědnosti uživatele a neměly by přesáhnout dobu 24 hodin při teplotě 2 až 8 °C, jestliže naředění proběhlo za kontrolovaných a validovaných aseptických podmínek.
Nepoužívejte tento přípravek, pokud si všimnete, že roztok není čirý nebo obsahuje částice.
6. Obsah balení a další informace Co přípravek Ibandronic Acid Accord obsahuje
• Léčivou látkou je acidum ibandronicum.
Ibandronic Acid Accord 2 mg koncentrát pro infuzní roztok
Jedna injekční lahvička s 2 ml koncentrátu pro infuzní roztok obsahuje acidum ibandronicum 2 mg (natrii ibandronas monohydricus 2,25 mg).
Ibandronic Acid Accord 6 mg koncentrát pro infuzní roztok
Jedna injekční lahvička s 6 ml koncentrátu pro infuzní roztok obsahuje acidum ibandronicum 6 mg (natrii ibandronas monohydricus 6,75 mg).
• Pomocnými látkami jsou chlorid sodný, trihydrát octanu sodného, kyselina octová 98%,a voda na injekci.
Jak přípravek Ibandronic Acid Accord vypadá a co obsahuje toto balení
Ibandronic Acid Accord je bezbarvý, čirý roztok.
Je dodáván v:
Ibandronic Acid Accord 2 mg koncentrát pro infuzní roztok
6ml skleněná injekční lahvička (typ I) s fluorotec/pryžovou zátkou a hliníkovým uzávěrem s levandulovým flip- off uzávěrem.
Ibandronic Acid Accord 6 mg koncentrát pro infuzní roztok
6ml skleněná injekční lahvička (typ I) s fluorotec/pryžovou zátkou a hliníkovým uzávěrem s růžovým flip- off uzávěrem.
Velikost balení:
Ibandronic Acid Accord 2 mg koncentrát pro infuzní roztok
Je dodáván v baleních obsahujících 1 injekční lahvičku.
Ibandronic Acid Accord 6 mg koncentrát pro infuzní roztok
Je dodáván v baleních obsahujících 1, 5 a 10 injekčních lahviček. Na trhu nemusí být k dispozici všechny velikosti balení.
Držitel rozhodnutí o registraci a výrobce
Accord Healthcare Limited Sage House 319, Pinner Road North Harrow Middlesex HA1 4 HF Velká Británie
Výrobce
Accord Healthcare Limited Sage House 319, Pinner Road North Harrow Middlesex HA1 4 HF Velká Británie
CEMELOG- BRS Ltd.
H-2040 Budaors, Vasut u.13.
Maďarsko
Tato příbalová informace byla naposledy revidována v květnu 2014:
Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/.
Tato příbalová informace je k dispozici ve všech jazycích EU/EHP na webových stránkách Evropské agentury pro léčivé přípravky.
Následující informace jsou určeny pouze pro zdravotnické pracovníky:
Dávkování: Léčba kostních příhod u pacientů s rakovinou prsu a kostními metastázami
Doporučené dávkování k prevenci kostních příhod u pacientů s karcinomem prsu a kostními metastázami je 6 mg nitrožilně každé 3-4 týdny. Jednotlivá dávka by měla být podána infuzí v průběhu alespoň 15 minut.
Pacienti s onemocněním ledvin
|
Clearance kreatininu (ml/min) |
Dávka |
Objem 1 a doba 2 infuze |
|
> 50 CLcr < 80 > 30 CLcr < 50 <30 |
6 mg (6 ml koncentrátu pro infuzní roztok) 4 mg (4 ml koncentrátu pro infuzní roztok) 2 mg (2 ml koncentrátu pro infuzní roztok) |
100 ml po dobu 15 min 500 ml po dobu 1 hodiny 500 ml po dobu 1 hodiny |
1 0,9% roztok chloridu sodného nebo 5% roztok glukózy
2 Při podávání každé 3 až 4 týdny
U pacientů s mírnou formou poškození funkce ledvin (CLcr >50 a <80 ml/min) není úprava dávkování nutná. U pacientů s rakovinou prsu a kostními metastázami se středně závažnou formou poškození funkce ledvin (CLcr > 30 a < 50 ml/min) nebo závažnou formou poškození funkce ledvin (CLcr < 30 ml/min), kteří jsou léčeni z důvodu prevence kostních příhod, by se měla dodržovat následující doporučení:_
U pacientů s nádorovým onemocněním a CLcr < 50 ml/min nebyla doba infuze trvající 15 minut dosud studována.
Dávkování: Léčba hyperkalcemie vyvolané nádorem
Ibandronic Acid Accord koncentrát pro přípravu infuzního roztoku se obvykle podává během hospitalizace. Dávku určí lékař po posouzení následujících faktorů:
Před léčbou přípravkem Ibandronic Acid Accord musí být pacienti adekvátně rehydratováni 0,9% roztokem chloridu sodného. Je třeba vzít v úvahu stupeň závažnosti hyperkalcemie stejně jako typ nádoru. U většiny pacientů s těžkou hyperkalcemií (albuminem korigovaný sérový vápník* > 3 mmol/l nebo > 12 mg/dl) postačuje jednotlivá dávka 4 mg. U pacientů s mírnou hyperkalcemií (albuminem korigovaný sérový vápník < 3 mmol/l nebo < 12 mg/dl) je účinná dávka 2 mg. Nejvyšší dávka použitá v klinických studiích byla 6 mg, ale tato dávka se neprojevila vyšším účinkem.
* Poznámka: koncentrace albuminem korigovaného sérového vápníku se vypočte následujícím způsobem:
|
Koncentrace albuminem korigovaného sérového vápníku (mmol/l) |
vápník v séru (mmol/l) - [0,02 x albumin (g/l)] + 0,8 | |
|
N |
ebo | |
|
Koncentrace albuminem korigovaného sérového vápníku (mg/dl) |
vápník v séru (mg/dl) + 0,8 x [4 - albumin (g/dl)] | |
|
Hodnoty albuminem korigovaného sérového vápníku v mmol/l lze převést na mg/dl vynásobením 4. | ||
Ve většině případů může zvýšená hladina sérového vápníku poklesnout na normální hladinu během 7 dnů. Střední doba recidivy (znovuzvýšení albuminem korigovaného sérového vápníku nad 3 mmol/l) byla 1819 dnů při dávkách 2 mg a 4 mg. Střední doba recidivy při dávce 6 mg byla 26 dnů.
Způsob a cesta podání
Ibandronic Acid Accord koncentrát pro přípravu infuzního roztoku se aplikuje jako intravenózní infuze.
K tomuto účelu se obsah injekční lahvičky používá následujícím způsobem:
- Prevence kostních příhod u pacientů s karcinomem prsu a kostními metastázami - obsah
lahvičky se přidá ke 100 ml izotonického roztoku chloridu sodného nebo 100 ml 5% roztoku glukózy a infuze se podává po dobu nejméně 15 minut. Viz také výše uvedený odstavec o dávkování u pacientů s poruchou funkce ledvin
- Léčba hyperkalcemie vyvolané nádorem - obsah lahvičky se přidá k 500 ml izotonického
roztoku chloridu sodného nebo 500 ml 5% roztoku glukózy a infuze se podává po dobu minimálně 2 hodin
Poznámka:
Aby byl vyloučen možný vznik inkompatibility, musí být Ibandronic Acid Accord koncentrát pro přípravu infuzního roztoku mísen pouze s izotonickým roztokem chloridu sodného nebo s 5% roztokem dextrózy. Ibandronic Acid Accord koncentrát pro přípravu infuzního roztoku nesmí být mísen s roztoky obsahujícími vápník.
Naředěný roztok je určen k jednorázovému podání. Použit může být pouze čirý roztok bez částic.
Doporučuje se použít roztok okamžitě po naředění (viz bod 5 této příbalové informace - „Jak přípravek Ibandronic Acid Accord uchovávat“).
Přípravek Ibandronic Acid Accord koncentrát pro infuzní roztok se aplikuje jako intravenózní infuze. Je nutno ve zvýšené míře dbát na to, aby nebyl Ibandronic Acid Accord koncentrát pro přípravu infuzního roztoku podán intraarteriálně nebo paravenózně, neboť by mohlo dojít k poškození tkáně.
Četnost podání
Při léčbě nádorové hyperkalcemie se Ibandronic Acid Accord koncentrát pro přípravu infuzního roztoku obvykle podává jednorázovou infuzí.
Doporučený interval opakování infuze v prevenci kostních příhod u pacientů s karcinomem prsu a kostními metastázami je 3 - 4 týdny.
Délka léčby
Omezený počet pacientů (50 pacientů) dostal druhou infuzi z důvodu hyperkalcemie. Léčbu je třeba opakovat v případě vracející se hyperkalcemie nebo při nedostatečné účinnosti.
U pacientů s karcinomem prsu a kostními metastázami se doporučuje podávat infuzi přípravku Ibandronic Acid Accord každé 3-4 týdny. V klinických studiích trvala léčba až 96 týdnů.
Předávkování
S akutní otravou přípravkem Ibandronic acid Accord koncentrátem pro přípravu infuzního roztoku dosud nejsou zkušenosti. Vzhledem k tomu, že v preklinických studiích s vysokými dávkami přípravku byly toxicitou nejvíce postiženy játra a ledviny, je nutné jejich činnost během léčby monitorovat.
Klinicky závažnou hypokalcemii (velmi nízká koncentrace vápníku v séru) lze upravit intravenózním podáním glukonanu vápenatého.
Příbalová informace: informace pro uživatele
Ibandronic Acid Accord 3 mg injekční roztok v předplněné injekční stříkačce
Acidum ibandronicum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
• Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
• Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
• Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci:
1. Co je Ibandronic Acid Accord a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete Ibandronic Acid Accord používat
3. Jak se Ibandronic Acid Accord používá
4. Možné nežádoucí účinky
5. Jak Ibandronic Acid Accord uchovávat
6. Obsah balení a další informace
1. Co je Ibandronic Acid Accord a k čemu se používá
Ibandronic Acid Accord patří do skupiny léčivých přípravků zvaných bisfosfonáty. Obsahuje léčivou látku kyselinu ibandronovou.
Ibandronic Acid Accord může zvrátit kostní ztráty zastavením dalších ztrát kosti a zvýšením kostní masy u většiny žen, které jej užívají, a to i přesto, že ženy nemusí vidět nebo pociťovat rozdíl. Ibandronic Acid Accord může pomoci snížit riziko zlomenin kostí (fraktur). Toto snížení zlomenin bylo prokázáno u páteře (obratlů), ale ne u kyčelní kosti.
Ibandronic Acid Accord Vám byl předepsán k léčbě postmenopauzální osteoporózy, neboť je u Vás zvýšené riziko výskytu zlomenin. Osteoporóza znamená řídnutí a slábnutí kostí, běžně se vyskytuje u žen po menopauze. V období menopauzy přestávají vaječníky tvořit ženský pohlavní hormon, estrogen, který pomáhá udržovat kostní skelet v dobrém stavu. Čím dříve žena dosáhne menopauzy, tím vyšší je u ní riziko zlomenin z důvodu osteoporózy.
Další faktory, které mohou zvyšovat riziko zlomenin, zahrnují:
• nedostatečný přívod vápníku a vitamínu D v potravě
• kouření cigaret nebo nadměrné pití alkoholu
• nedostatek pohybu nebo jiné fyzické aktivity zatěžující skelet
• výskyt osteoporózy v rodině
Zdravý životní styl Vám rovněž pomůže, aby pro Vás byla léčba co možná nejpřínosnější. Znamená to:
• příjem vyvážené potravy bohaté na vápník a vitamín D
• pravidelnou chůzi nebo cvičení zatěžující skelet
• nekouřit a nepít nadměrné množství alkoholu.
2. Čemu musíte věnovat pozornost, než začnete Ibandronic Acid Accord používat Nepoužívejte Ibandronic Acid Accord
• pokud máte, nebo jste měl(a) sníženou hladinu vápníku v krvi. Prosím, konzultujte tuto skutečnost s Vaším lékařem
• jestliže j ste alergický(á) na kyselinu ibandronovou nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6)
Upozornění a opatření
Velmi vzácně byla u pacientů léčených kyselinou ibandronovou z důvodu onkologických indikací hlášena v poprodejním sledování osteonekróza čelisti (ONČ). ONČ se může projevit i po ukončení léčby.
Je velmi důležité pokusit se předcházet rozvoji ONČ, protože jde o bolestivý stav, jehož léčba může být obtížná. Ke snížení rizika rozvoje osteonekrózy čelisti je třeba přijmout určitá preventivní opatření.
Před zahájením léčby informujte svého lékaře/zdravotní sestru (zdravotnický personál) o případných následujících stavech:
- Máte jakékoli problémy se svými ústy nebo zuby, například nedostatečný zdravotní stav ústní dutiny, onemocnění dásní nebo plánovaná extrakce zubu.
- Nemáte pravidelnou zubní péči nebo jste dlouho nebyl(a) na preventivní zubní prohlídce.
- Jste kuřák/kuřačka (protože to může zvyšovat riziko zubních problémů).
- Byl(a) jste v minulosti léčen(a) bisfosfonáty (používanými k léčbě onemocnění kostí).
- Užíváte léky ze skupiny kortikosteroidů (jako prednisolon nebo dexametazon).
- Máte onkologické onemocnění.
Než bude u vás zahájena léčba kyselinou ibandronovou, může vás lékař požádat, abyste absolvoval(a) preventivní zubní prohlídku.
Během léčby kyselinou ibandronovou udržujte dobrou ústní hygienu (včetně pravidelného čištění zubů), absolvujte rutinní zubní prohlídky. Jestliže máte umělý chrup, zajistěte, aby správně seděl. Jestliže jste léčeni stomatologem nebo se podrobíte zubnímu chirurgickému zákroku (například extrakce zubů), informujte svého lékaře o stomatologické léčbě a svého zubního lékaře informujte, že jste léčeni kyselinou ibandronovou.
Pokud se u vás projeví problémy s ústní dutinou nebo se zuby, jako jsou uvolněné zuby, bolest nebo otoky, nehojící se vředy nebo výtoky, ihned informujte svého lékaře a zubního lékaře, protože může jít o příznaky osteonekrózy čelisti.
Některé osoby musí být při používání přípravku Ibandronic Acid Accord obzvlášť opatrné. Před použitím přípravku Ibandronic Acid Accord se poraďte se svým lékařem:
• Pokud máte nebo jste někdy měl(a) potíže s ledvinami, selhání ledvin, pokud jste někdy podstoupil(a) dialýzu nebo trpíte jiným onemocněním, které může ovlivňovat Vaše ledviny
• Jestliže Vám byla prokázána porucha metabolismu minerálů (jako je nedostatek vitamínu D)
• Pokud dostáváte Ibandronic Acid Accord, měl(a) byste užívat doplňky obsahující vápník a vitamín D.
Pokud je nemůžete užívat, měl(a) byste informovat svého lékaře.
• Pokud máte srdeční obtíže a Váš lékař Vám doporučil omezit denní příjem tekutin.
U pacientů léčených kyselinou ibandronovou v intravenózní formě byly hlášeny případy závažné alergické reakce, někdy končící úmrtím.
Okamžitě oznamte svému lékaři nebo zdravotní sestře, pokud zaznamenáte některý z následujících příznaků, jako je zkrácení dechu/obtížné dýchání, pocit tísně v krku, otok jazyka, závrať, pocit ztráty vědomí, zčervenání nebo otok obličeje, vyrážka na těle, nevolnost a zvracení (viz bod 4).
Děti a dospívající
Ibandronic Acid Accord nesmí být používán u dětí nebo dospívajících mladších 18 let.
Další léčivé přípravky a Ibandronic Acid Accord
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Těhotenství a kojení
Ibandronic Acid Accord je určen pouze pro ženy v postmenopauze a nesmí ho užívat ženy, které stále mohou otěhotnět.
Nepoužívejte Ibandronic Acid Accord, jestliže jste těhotná nebo kojíte.
Poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek používat.
Řízení dopravních prostředků a obsluha strojů
Můžete řídit dopravní prostředky a obsluhovat stroje, protože lze předpokládat, že Ibandronic Acid Accord nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje.
Ibandronic Acid Accord obsahuje méně než 1 mmol sodíku (23 mg) v jedné dávce (3 ml), tzn. že je v podstatě “bez sodíku”.
3. Jak se Ibandronic Acid Accord používá
Doporučené dávkování přípravku Ibandronic Acid Accord ve formě intravenózních injekcí je 3 mg (1 předplněná injekční stříkačka) jedenkrát za 3 měsíce.
Injekce podává lékař nebo vyškolený zdravotnický personál do žíly. Injekce si neaplikujte sami. Injekční roztok musí být podán výhradně do žíly, nikoli do jiné části těla.
Další používání přípravku Ibandronic Acid Accord
Pro dosažení maximálního účinku léčby je důležité pokračovat v aplikaci injekcí každé tři měsíce tak dlouho, jak Vám je Váš lékař bude předepisovat. Ibandronic Acid Accord může léčit osteoporózu pouze po dobu, po kterou budete podstupovat léčbu, přestože nemusíte zaznamenat či pociťovat rozdíl. Po 5 letech užívání přípravku Ibandronic Acid Accord se, prosím, zeptejte svého lékaře, zda máte pokračovat v užívání přípravku Ibandronic Acid Accord.
Rovněž byste měl(a) užívat potravinové doplňky obsahující vápník a vitamín D, tak jak Vám doporučí lékař.
Jestliže jste použil(a) více přípravku Ibandronic Acid Accord, než jste měl(a)
Může se stát, že budete mít nízkou hladinu vápníku, fosforu nebo hořčíku v krvi. Váš lékař podnikne odpovídající kroky k nápravě změn a může Vám dát injekci obsahující zmíněné minerály.
Jestliže jste zapomněla(a) použít Ibandronic Acid Accord
Měl(a) byste si co nejdříve sjednat novou návštěvu lékaře, který Vám dá další injekci. Poté budete opět dostávat injekce vždy po 3 měsících od data poslední injekce, kterou jste dostal(a).
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Pokud zaznamenáte jakýkoli z následujících závažných nežádoucích účinků, sdělte to ihned zdravotní sestře nebo lékaři - je možné, že budete potřebovat akutní lékařské ošetření:
Vzácné (mohou postihnout až 1 osobu z 1000):
• svědění, otok obličeje, rtů, jazyka a hrdla, s obtížným dýcháním
• přetrvávaj ící bolest a zánět oka (pokud j e dlouhotrvající)
• nová bolest, slabost nebo nepříjemné pocity v oblasti stehna, kyčle nebo třísla. Můžete mít časné známky možné neobvyklé zlomeniny stehenní kosti.
Velmi vzácné (mohou postihnout až 1 osobu z 10000):
• bolest nebo bolavé místo v ústech nebo čelisti. Můžete mít časné známky závažných potíží s čelistí (nekrózy (mrtvé kostní tkáně) v čelistní kosti).
• jestliže trpíte bolestmi uší, výtoky z ucha a/nebo ušní infekcí, informujte svého lékaře. Tyto příznaky mohou být symptomy poškození kostí v uchu.
• závažná, možno život ohrožující, alergická reakce (viz bod 2)
• závažné odmítavé reakce kůže
Další možné nežádoucí účinky
Časté (mohou postihnout až 1 osobu z 10):
• bolest hlavy
• bolest žaludku (jako je gastritida) nebo bolest břicha, porucha trávení, nevolnost, průjem (řídká stolice) nebo zácpa
• bolest svalů, kloubů nebo zad
• pocit únavy a vyčerpanosti
• příznaky podobné chřipce, včetně horečky, třesu a chvění, člověk se celkově necítí dobře, bolesti kostí a bolesti svalů a kloubů. Pokud Vás kterýkoli z těchto účinků začne obtěžovat nebo trvá více než několik dní, sdělte to zdravotní sestře nebo lékaři.
• vyrážka
Méně časté (mohou postihnout až 1 osobu ze 100):
• zánět žil
• bolest nebo poranění v místě vpichu
• bolest kostí
• pocit slabosti
• záchvaty astmatu
Vzácné (mohou postihnout až 1 osobu z 1000):
• kopřivka
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak Ibandronic Acid Accord uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce za „Použitelné do:“ a na injekční stříkačce za „EXP“. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Zdravotník, který aplikuje injekci, musí zlikvidovat nevyužitý zbývající roztok a odložit injekční stříkačku a injekční jehlu do příslušné nádoby na odpad.
6. Obsah balení a další informace Co Ibandronic Acid Accord obsahuje
• Léčivou látkou je acidum ibandronicum. Jedna předplněná injekční stříkačka obsahuje 3 mg kyseliny ibandronové ve 3ml roztoku (ve formě monohydrátu natrium-ibandronátu).
• Dalšími složkami jsou chlorid sodný, kyselina octová, trihydrát kyseliny octové a voda na injekci.
Jak Ibandronic Acid Accord vypadá a co obsahuje toto balení
Ibandronic Acid Accord 3 mg injekční roztok je čirý bezbarvý roztok, který je dodáván v předplněných injekčních stříkačkách. Jedna předplněná injekční stříkačka obsahuje 3ml roztoku. Ibandronic Acid Accord je dodáván v balení po 1 předplněné injekční stříkačce s 1 injekční jehlou nebo v balení po 4 předplněných injekčních stříkačkách se 4 injekčními jehlami.
Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci a výrobce
Držitel rozhodnutí o registraci
Accord Healthcare Limited Sage House
319, Pinner Road. North Harrow Middlesex HA1 4HF Velká Británie
Výrobce
Accord Healthcare Limited Sage House
319, Pinner Road, North Harrow Middlesex HA1 4HF Velká Británie
CEMELOG- BRS Ltd.
H-2040 Budaors, Vasut u.13.
Maďarsko
Tato příbalová informace byla naposledy revidována: {datum}.
Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky: http://www.ema.europa.eu.
Následující informace jsou určeny pouze pro zdravotnické pracovníky:
INFORMACE PRO ZDRAVOTNÍKY
Další informace jsou uvedeny v souhrnu údajů o přípravku.
Aplikace přípravku Ibandronic Acid Accord 3mg injekční roztok v předplněné injekční stříkačce:
Ibandronic Acid Accord 3 mg injekční roztok v předplněné injekční stříkačce musí být aplikován intravenózně během 15 - 30 vteřin.
Roztok je dráždivý, proto je důležité přísně dodržovat intravenózní způsob podání. V případě neúmyslné aplikace roztoku do tkáně v okolí žíly může u pacienta dojít k lokálnímu podráždění, bolestivosti a zánětu místa vpichu.
Ibandronic Acid Accord 3 mg injekční roztok v předplněné injekční stříkačce nesmí být mísen s jinými roztoky s obsahem vápníku (např. s Ringer laktátovým roztokem, kalcium heparinem) ani s jinými intravenózně podávanými léčivými přípravky. V případě, že je Ibandronic Acid Accord podán za využití již zavedené i.v. infuzní sestavy, může být k infuzi použit pouze fyziologický roztok nebo roztok glukózy o koncentraci 50mg/ml (5%).
Vynechání dávky:
Při vynechání dávky by injekce měla být aplikována co možná nejdříve. Další injekce by měly být podávány každé 3 měsíce od data poslední injekce.
Předávkování:
K dispozici nejsou žádné specifické údaje o léčbě předávkování přípravkem Ibandronic Acid Accord .
Podle dostupných údajů může intravenózní předávkování touto třídou sloučenin vést k hypokalcemii, hypofosfatemii a hypomagnezemii, které jsou příčinou parestézie. V závažných případech je možné podat intravenózní infuzi odpovídajících dávek kalcium glukonátu, fosfátu draselného nebo sodného a magnézium sulfátu.
Obecné údaje:
Ibandronic Acid Accord 3 mg injekční roztok v předplněné injekční stříkačce, stejně jako další bisfosfonáty podávané intravenózně, může vyvolat přechodné snížení hladin vápníku v krevním séru.
Je třeba provést vyšetření na hypokalcemii a další poruchy kostního a minerálního metabolismu, které by měly být léčeny před zahájením injekčního podávání přípravku Ibandronic Acid Accord . U všech pacientů je důležitý příjem vápníku a vitamínu D v přiměřeném množství. Všichni pacienti musí dostávat potravinové doplňky obsahující vápník a vitamín D.
Pacienti současně trpící onemocněním ledvin nebo užívající léčivé přípravky, které potenciálně mohou mít nežádoucí účinky na ledviny, by měli být během léčby pravidelně kontrolováni v souladu s postupy správné lékařské praxe.
Veškerý nepoužitý injekční roztok, injekční stříkačka a injekční jehla musí být zlikvidovány v souladu s místními požadavky.
71