Hypnomidate
sp.zn. sukls129638/2010 a sukls113515/2014
SOUHRN ÚDAJŮ O PŘÍPRAVKU
1. NÁZEV PŘÍPRAVKU
HYPN OMIDATE etomidatum 2 mg/ml inj ekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna 10ml ampulka obsahuje etomidatum 20 mg (2 mg/ml).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok.
Čirý bezbarvý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
HYPNOMIDATE je určen k úvodu do celkové anestezie.
HYPNOMIDATE je určen ke krátkodobým diagnostickým výkonům nebo k výkonům v ambulantních podmínkách, kdy je žádoucí rychlé zotavení pacienta bez reziduálních projevů.
Vzhledem k tomu, že při doporučeném dávkování ovlivňuje etomidát hemodynamické parametry minimálně (viz body 4.4 Zvláštní upozornění a opatření pro použití a bod 4.8 Nežádoucí účinky), je jeho použití vhodné zejména v kardiochirurgii nebo u kardiaků.
4.2 Dávkování a způsob podání
Dávkování
Dospělí
Jedna ampulka přípravku HYPNOMIDATE obsahuje 20 mg etomidátu v 10 ml roztoku určeného k okamžité aplikaci, tj. 2 mg etomidátu v 1 ml roztoku. Účinná hypnotická dávka je 0,3 mg/kg tělesné hmotnosti. Pro dospělého pacienta je proto obvykle k navození 4 - 5minutového spánku dostačující aplikovat obsah jedné ampulky. Dávku lze upravovat v závislosti na tělesné hmotnosti.
Přípravek mohou používat pouze lékaři školení v endotracheální intubaci. Musí být dostupné zařízení pro umělou plicní ventilaci.
Hypnotický stav může být prodloužen dalšími injekcemi přípravku HYPNOMIDATE. Nepřekračujte celkové množství 3 ampulek (30 ml).
Vzhledem k absenci analgetického účinku přípravku HYPNOMIDATE se 1 - 2 minuty před injekcí přípravku HYPNOMIDATE doporučuje podat vhodný opioid, např. 1 - 2 ml fentanylu intravenózně.
Dávka by měla být upravena podle individuální odpovědi pacienta a klinických
účinkůPediatrická populace
U dětí do 15 let může být dávka navýšena: k dosažení spánku stejné hloubky a stejného trvání jako u dospělého je zapotřebí podat doplňující dávku až do 30 % obvyklé dávky pro dospělé (viz bod 5.2 Zvláštní populace: Pediatrická populace).
Starší pacienti
U starších osob by měla být aplikována jednorázová dávka 0,15 - 0,2 mg/kg tělesné hmotnosti, kterou lze posléze upravit dle účinku (viz body 4.4 Zvláštní upozornění a opatření pro použití a
5.2 Zvláštní populace: Starší pacienti).
Způsob podání
HYPNOMIDATE se podává pomalu intravenózně.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1 tohoto přípravku.
4.4 Zvláštní upozornění a opatření pro použití
HYPNOMIDATE je určen výhradně k intravenózní aplikaci.
Indukce narkózy pomocí přípravku HYPNOMIDATE může být doprovázena mírným a přechodným poklesem krevního tlaku, který je způsoben snížením periferní vaskulární rezistence. U zesláblých pacientů, u nichž může být hypotenze škodlivá, by měla být provedena tato opatření:
1. Indukci narkózy provádějte u pacienta ležícího naznak.
2. Zajistěte dobrý žilní přístup, aby bylo možné ovlivňovat objem cirkulující krve.
3. Aplikujte HYPNOMIDATE pomalu intravenózně (tj. 10 ml za 1 minutu).
Pokud možno, nepoužívejte k indukci narkózy jiné látky.
Pokud je použit HYPNOMIDATE, je nutné mít k dispozici vybavení pro resuscitaci, aby bylo možné zvládnout respirační depresi a případné apnoe.
Indukční dávky etomidátu byly provázeny poklesem plazmatických koncentrací kortizolu a aldosteronu (viz bod 5.1 Farmakodynamické vlastnosti). Pokud jde o pacienty, kteří prodělali těžký stres, zejména pacienty s adrenokortikální dysfunkcí, mělo by být zváženo podání exogenního kortizolu. Prodloužená suprese endogenního kortizolu a aldosteronu se může objevit jako přímý následek podávání etomidátu kontinuální infuzí nebo po jeho opakovaném podání, a proto by měl být tento způsob podávání vyloučen. V těchto případech není stimulace nadledvin ACTH účinná.
Etomidát je nutno používat s opatrností u pacientů s kortikoadrenální insuficiencí jako jsou pacienti se sepsí.
U pacientů s jaterní cirhózou nebo u těch pacientů, kterým byla již dříve podávána neuroleptika, opioidy nebo sedativa, by měla být dávka etomidátu snížena.
U jedné nebo více svalových skupin se mohou objevit spontánní stahy, zejména pokud nebyla podána premedikace. Je to připisováno nedostatečné subkortikální inhibici. Tomuto jevu je možné z větší míry zamezit intravenózní aplikací malých dávek fentanylu s diazepamem podaným 1 - 2 minuty před indukcí anestezie přípravkem HYPNOMIDATE.
Myoklonii a bolestem při aplikaci, včetně žilní bolestivosti, která byla pozorována během aplikace přípravku HYPNOMIDATE, zejména pokud je aplikován do malé žíly, lze většinou zamezit nitrožilním podáním nízké dávky vhodných opioidů, např.fentanylu 1 - 2 minuty před úvodem do anestezie.
HYPNOMIDATE má být používán opatrně u starších pacientů vzhledem k jeho schopnosti snižovat srdeční výkon, což bylo pozorováno při vyšších než doporučených dávkách (viz bod 4.2 Dávkování a způsob podání, Starší pacienti).
S ohledem na absenci analgetického účinku přípravku HYPNOMIDATE je nutné během chirurgických výkonů současně podávat vhodná analgetika.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Hypnotický účinek etomidátu mohou zvýšit neuroleptika, opioidy a sedativa a také alkohol.
Indukce anestezie etomidátem může být provázena slabým a přechodným snížením periferního odporu, což může zvyšovat účinek jiných léčivých přípravků snižujících krevní tlak.
Účinek jiných léčivých přípravků na etomidát
Bylo hlášeno, že současná aplikace etomidátu s alfentanilem snižuje výsledný biologický poločas etomidátu přibližně na 29 minut. Při současné aplikaci etomidátu a alfentanilu je nutná opatrnost, protože koncentrace etomidátu může klesnout pod hypnotický práh.
Při současném podání s intravenózním (i.v.) fentanylem jsou sníženy celková plazmatická clearance a distribuční objem etomidátu 2 až 3krát beze změny poločasu. Při současném podání etomidátu s i.v. fentanylem může dojít za určitých okolností k nutnosti snížit dávku etomidátu.
Účinek etomidátu na jiné léčivé přípravky
Zdá se, že současná aplikace etomidátu a ketaminu nemá významný vliv na plazmatické koncentrace nebo farmakokinetické parametry ketaminu nebo jeho hlavního metabolitu norketaminu.
4.6 Fertilita, těhotenství a kojení
U zvířat nebyly prokázány přímé embryotoxické nebo teratogenní účinky etomidátu (viz Předklinické údaje). HYPNOMIDATE může být aplikován v těhotenství pouze tehdy, když potenciální přínos léčby převyšuje riziko pro plod.
Během anestezie v porodnictví etomidát prochází placentou. Apgar score novorozenců, jejichž matky obdržely etomidát, je srovnatelné s Apgar score novorozenců narozených po podání jiných hypnotik. U novorozenců, jejichž matkám byl aplikován HYPNOMIDATE, byl zjištěn přechodný pokles hladiny kortizolu v trvání přibližně 6 hodin, snížené hladiny však zůstaly v normálním rozmezí.
Kojení
Etomidát se vylučuje do mateřského mléka. Účinek etomidátu na novorozence není znám. Kojení by mělo být přerušeno během léčby přípravkem HYPNOMIDATE a zahájeno 24 hodin po ukončení léčby.
Fertilita
Výsledky reprodukčních studií u zvířat ukázaly, že přípravek HYPNOMIDATE nemá v doporučených dávkách vliv na fertilitu.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Etomidát má výrazný vliv na schopnost řídit a obsluhovat stroje. Nedoporučuje se používat potenciálně nebezpečné stroje nebo řídit auto během 24 hodin po podání. Návrat normální pozornosti se může lišit podle trvání operace, celkové podané dávky etomidátu a současně použitých léčivých přípravků. Proto rozhodnutí o povolení řízení nebo obsluze strojů musí být provedeno na základě posouzení ošetřujícím lékařem.
4.8 Nežádoucí účinky
V tomto bodě jsou prezentovány nežádoucí účinky, které jsou považovány za důvodně spojené s užíváním etomidátu na základě komplexního posouzení dostupných informací o každém nežádoucím účinku. V jednotlivých případech nemůže být spolehlivě stanoven kauzální vztah s etomidátem. Protože klinické studie jsou prováděny za velmi proměnlivých podmínek, výskyt nežádoucích účinků pozorovaných v klinickém hodnocení s jedním léčivým přípravkem nelze přímo srovnávat s výskytem nežádoucích účinků v klinickém hodnocení s jiným léčivým přípravkem a nemusí odrážet výskyt pozorovaný v klinické praxi.
Údaje z klinických hodnocení
Bezpečnost přípravku HYPNOMIDATE byla hodnocena u 812 pacientů, kteří se účastnili 4 otevřených klinických studií s přípravkem HYPNOMIDATE užitým k navození celkové anestezie. Těmto pacientům byla podána nejméně jedna dávka přípravku HYPNOMIDATE a byly vyhodnoceny údaje o bezpečnosti. Na základě souhrnných údajů o bezpečnosti z těchto klinických studií byly nejčastěji hlášenými nežádoucími účinky (> 5% incidencí) dyskineze (10,3 %) a bolest žil (7,6 %). Následující tabulka zahrnuje jak nežádoucí účinky uvedené výše hlášené při použití přípravku HYPNOMIDATE v klinických studiích, tak i z postmarketingového sledování.
Četnosti nežádoucích účinků jsou uvedeny podle následující konvence: Velmi časté (> 1/10); časté (> 1/100 až < 1/10); méně časté (> 1/1 000 až < 1/100); vzácné (> 1/10 000 až < 1/1 000); velmi vzácné (< 1/10 000), není známo (z dostupných údajů nelze určit).
|
Třídy orgánových systémů |
Nežádoucí účinek | |||
|
Kategorie četnosti | ||||
|
Velmi časté (> 1/10) |
Časté (> 1/100 až < 1/10) |
Méně časté (> 1/1 000 až < 1/100) |
Není známo | |
|
Poruchy imunitního systému |
Hypersenzitivita (jako anafylaktický šok, anafylaktická reakce, anafylaktoidní reakce) | |||
|
Endokrinní poruchy |
Adrenální insuficience | |||
|
Poruchy nervového systému |
Dyskineze |
Myoklonus |
Hypertonie, mimovolní svalové kontrakce, nystagmus |
Konvulze (včetně záchvatů typu grand mal) |
|
Srdeční poruchy |
Bradykardie, |
Srdeční zástava, | ||
|
Třídy orgánových systémů |
Nežádoucí účinek | |||
|
Kategorie četnosti | ||||
|
Velmi časté (> 1/10) |
Časté (> 1/100 až < 1/10) |
Méně časté (> 1/1 000 až < 1/100) |
Není známo | |
|
extrasystoly, ventrikulární extrasystoly |
kompletní atrioventrikulární blokáda | |||
|
Cévní poruchy |
Bolest žil, hypotenze |
Flebitida, hypertenze |
Šok, tromboflebitida (včetně superficiální tromboflebitidy a hluboké žilní trombózy) | |
|
Respirační, hrudní a mediastinální poruchy |
Apnoe, hyperventilace, stridor |
Hypoventilace, škytavka, kašel |
Respirační bronchospasmus (včetně bronchospasmu s fatálním koncem) | |
|
Gastrointestinální poruchy |
Hypersekrece slin | |||
|
Poruchy kůže a podkožní tkáně |
Erytém |
Stevens-Johnsonův syndrom, kopřivka | ||
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Svalová ztuhlost |
Trismus | ||
|
Celkové poruchy a reakce v místě aplikace |
Bolest v místě injekce | |||
|
Poranění, otravy a procedurální komplikace |
Komplikace anestezie, prodloužené zotavení z anestezie, neadekvátní analgesie, nauzea spojená s anestezií | |||
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na adresu:
Státní ústav pro kontrolu léčiv Šrobárova 48 100 41 Praha 10
Webové stránky: www.sukl.cz/nahlasit-nezadouci-ucinek
4.9 Předávkování
Známky a příznaky
Předávkování etomidátem aplikovaným ve formě bolusu navozuje hlubší spánek a může způsobit hypotenzi, sníženou sekreci kůry nadledvin, respirační depresi a dokonce zástavu dechu. Pokud
zástava dechu nastane, je nutné mít k dispozici vybavení pro adekvátní podporu dechových funkcí. Také se může vyskytnout dezorientace a opožděné probuzení.
Léčba
Kromě podpůrných opatření (např. respiračních) může být nutná aplikace 50 - 100 mg hydrokortizonu (nikoli adrenokortikotropního hormonu ACTH).
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: anestetika celková ATC kód: N01AX07, etomidát
Mechanismus účinku
Etomidát je krátkodobě působící intravenózní hypnotikum užívané k anestezii. U dospělých vyvolá dávka 0,2 - 0,3 mg/kg tělesné hmotnosti (přibližně 1 ampulka s 10 ml přípravku HYPNOMIDATE) během 10 vteřin hypnotický stav, který trvá přibližně 5 minut (nebo při premedikaci sedativy zpravidla déle).
Při dostatečné úrovni centrálního útlumu vykazuje etomidát i vlastnosti antikonvulzivní a ochranné pro mozkovou tkáň před hypoxickým poškozením nervových buněk. Nevykazuje však účinek analgetický, což vylučuje jeho použití jako samostatného anestetika. Etomidát je rychle hydrolyzován, především v játrech. Zotavení je proto rychlé a jen vzácně provázené ospalostí a závratěmi. Srdeční a oběhové funkce jsou ovlivněny etomidátem pouze minimálně. Nedochází k uvolňování histaminu ani ke změnám jaterních funkcí.
Adrenální suprese
Při použití etomidátu k navození anestezie dochází k poklesu plazmatických hladin kortizolu a aldosteronu, které zůstávají sníženy po 6 až 8 hodin. Tyto hladiny se vrátí k výchozím hodnotám obvykle během 24 hodin. Etomidát se zdá být specifickým a reverzibilním inhibitorem 11-beta-hydroxylace při syntéze nadledvinových steroidů.
5.2 Farmakokinetické vlastnosti Plazmatický profil
Po intravenózní aplikaci může být časový průběh plazmatických hladin etomidátu definován tříkompartmentovým modelem odrážejícím proces distribuce, metabolismu a vylučování.
Distribuce
Etomidát je vázán přibližně z 76,5 % na plazmatické bílkoviny. Etomidát je rychle distribuován do mozku a ostatních tkání. Jeho distribuční objem činí přibližně 4,5 l/kg.
Biotransformace a eliminace
Etomidát je metabolizován v játrech. Za 24 hodin je 75 % dávky vyloučeno do moči, převážně ve formě metabolitů. Pouze 2 % etomidátu jsou vyloučena močí v nezměněné formě. Výsledný biologický poločas přibližně 3 - 5 hodin je projevem pomalé distribuce etomidátu z hlubokého periferního kompartmentu.
Vztah koncentrace - účinek
Minimální plazmatická koncentrace navozující hypnotický účinek činí přibližně 0,3 gg/ml. Zvláštní populace
Pediatrická populace: V hodnocení provedeném u 12 dětí (věk 7 - 13 let, tělesná hmotnost 22 -48 kg) byl počáteční distribuční objem vztažený k tělesné hmotnosti 2,4krát vyšší než u dospělých (0,66 vs 0,27 l/kg) a clearance léčiva byla u dětí přibližně o 58 % vyšší než u dospělých. Tyto údaje naznačují potřebu aplikace vyšší dávky u dětí ve srovnání s dospělými.
Zhoršená funkce jater. U pacientů s cirhózou jater, kteří obdrželi etomidát v kombinaci s fentanylem, byl hlášen prodloužený poločas vylučování. U těchto pacientů je nutné posoudit snížení rychlosti infúze.
Starší pacienti: Ve srovnání s mladými jedinci je u starších jedinců (> 65 let) snížena clearance etomidátu. Ve srovnání s mladými jedinci jsou u starších jedinců vyšší počáteční plazmatické koncentrace z důvodu menšího počátečního distribučního objemu. U starších jedinců může být proto nutné snížit dávku.
5.3 Předklinické údaje vztahující se k bezpečnosti
Předklinické účinky byly pozorovány pouze po expozicích dostatečně převyšujících maximální expozici u člověka, což svědčí pro malý význam při klinickém použití. Bylo pozorováno zkrácené přežívání mláďat laboratorních potkanů, jejichž matky obdržely toxické dávky.
Etomidát byl testován kompletními studiemi bezpečnosti zahrnujícími toxicitu po jednorázovém podání; toxicitu po opakovaném podávání po dobu až 8 týdnů; reprodukční studie testování fertility a celkovou reprodukci, teratogenitu a embryotoxicitu, perinatální a postnatální reprodukci. Mutagenita byla hodnocena sérií in vitro testů a/nebo studiemi genové mutace.
Akutní letální dávky (LD50) činily pro hlodavce 19 mg/kg nebo přibližně 63násobek účinné hypnotické dávky pro člověka (EHHD - Effective Human Hypnotic Dose) po jednorázovém intravenózním podání. Všeobecně byla hypnóza vyvolána okamžitě u všech zvířat po všech testovaných dávkách. Při srovnání s klinicky účinnou hypnotickou dávkou 0,3 mg/kg představují tyto hodnoty LD50 dostatečně široké bezpečnostní rozmezí. Výsledky studií toxicity po opakovaném intravenózním podání prokázaly ataxii závislou na dávce a/nebo spánek/hypnózu s odpovídajícím snížením příjmu potravy a tělesné hmotnosti. Při nejvyšších testovaných dávkách (5 mg/kg nebo ~ 17x EHHD) se vyskytla nepříliš četná úmrtí. Snížení tělesné hmotnosti se ukázalo v průběhu zotavení jako dobře reverzibilní.
Výsledky reprodukční studie fertility neprokázaly vliv na fertilitu nebo celkové parametry březosti a žádné známky embryotoxicity nebo teratogenity. Ve standardních studiích embryotoxicity a teratogenity se ve skupině s vysokou dávkou (5 mg/kg nebo ~ 17x EHHD) vyskytla úmrtí, avšak žádné embryotoxické nebo teratogenní účinky nebyly výslovně přisouzeny testované látce. Podávání etomidátu během peri a postnatálního období mělo za následek mateřskou mortalitu a toxicitu závislou na dávce. Tomu byl přisuzován mírný pokles v přežívání mláďat při vysokých dávkách (5 mg/kg nebo ~ 17x EHHD). Žádné nežádoucí účinky nebyly pozorovány na průběh březosti, velikost vrhu zvířat, hmotnost při narození nebo přírůstek tělesné hmotnosti a nebyly zaznamenány žádné abnormality u mláďat. Hodnocení genové toxicity neodhalilo důkaz pro mutagenní potenciál. Hodnocení intravenózního a subplantárního podráždění neprokázalo žádné intravenózní podráždění v místě vpichu a po subplantární injekci byl pozorován pouze mírný reverzibilní otok.
Souhrnně lze konstatovat, že popsané výsledky toxicity po jednorázovém a opakovaném podání, reprodukční studie a studie mutagenity ukazují, že pozorovaná toxicita u těchto zvířecích modelů souvisí obecně s vysokými toxickými dávkami, které jsou zcela mimo předpokládané rozmezí klinických terapeutických dávek a sekundárně mimo farmakologický účinek tohoto léku.
Závěrem lze shrnout, že získané předklinické výsledky svědčí o celkové bezpečnosti etomidátu.
FARMACEUTICKÉ ÚDAJE
6.
6.1 Seznam pomocných látek
Propylenglykol, voda na injekci.
6.2 Inkompatibility
Tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti
3 roky
6.4 Zvláštní opatření pro uchovávání
Uchovávejte při teplotě do 25 °C.
Z mikrobiologického hlediska, pokud způsob otevření nevyloučí riziko mikrobiologické kontaminace, má být přípravek použit okamžitě.
Pokud není použit okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou v odpovědnosti uživatele.
6.5 Druh obalu a obsah balení
HYPNOMLDATE je dodáván v 10ml skleněných ampulkách (2 mg/1 ml).
Obal: bezbarvá skleněná ampulka, kartonová vložka, krabička.
Velikost balení: 5x 10 ml
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Výdej přípravku vázán na lékařský předpis.
Při otevírání ampulky používejte ochranné rukavice.
Návod k otevření ampulky:
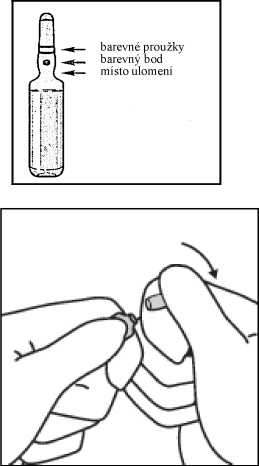
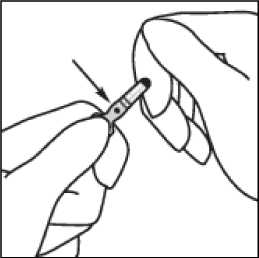
1. Podržte ampulku mezi palcem a ukazováčkem tak, aby zúžená část zůstala volná.
2. Druhou rukou uchopte zúženou část ampulky tak, že opřete krček ampulky o ukazováček a palec přitiskněte na barevně označený bod souběžně
s barevnými proužky.
3. Přidržujte palec na označeném místě a zprudka ulomte zúženou část ampulky. Držte ampulku pevně v ruce.
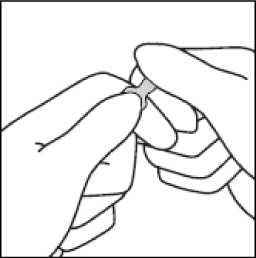
V případě náhodné dermální expozice zasažené místo opláchněte vodou. Vyhněte se použití mýdla, alkoholu a jiných čisticích látek, které mohou způsobit chemické nebo fyzické oděrky na kůži.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Janssen-Cilag s.r.o., Karla Engliše 3201/6, 150 00 Praha 5, Česká republika
8. REGISTRAČNÍ ČÍSLO(A)
05/160/80-C
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace|: 30. 12. 1980
Datum posledního prodloužení registrace: 10.9.2014
10. DATUM REVIZE TEXTU
10.9.2014
9/9