Holoclar 79 000 - 316 000 Buněk/Cm2
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
▼ Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Holoclar 79 000 - 316 000 buněk/cm2 náhrada živé tkáně
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
2.1 Obecný popis
Autologní lidský rohovkový epitel expandovaný ex vivo obsahující kmenové buňky.
2.2 Kvalitativní a kvantitativní složení
Holoclar je transparentní kruhová membrána s 300 000 až 1 200 000 životaschopných autologních buněk lidského rohovkového epitelu (79 000 - 316 000 buněk/cm2) včetně průměrně 3,5 % (0,4 až 10 %) limbálních kmenových buněk a přechodně amplifikujících a terminálně diferencovaných buněk odvozených z kmenových buněk, které jsou fixovány na podkladní fibrinové matrici o průměru 2,2 cm a uloženy v transportním médiu.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Náhrada živé tkáně.
Transparentní kruhová membrána
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba dospělých pacientů se středním až závažným deficitem limbálních kmenových buněk (definovaným jako přítomnost superficiální korneální neovaskularizace v nejméně dvou kvadrantech rohovky se zahrnutím centrální části rohovky a se závažně zhoršenou ostrostí zraku), jednostranným nebo oboustranným, způsobeným fyzikálním nebo chemickým popálením očí. Pro biopsii se vyžaduje minimálně 1 -2 mm2 nepoškozeného limbu.
4.2 Dávkování a způsob podání
Tento léčivý přípravek je určen pouze k autolognímu použití.
Holoclar musí aplikovat pouze příslušně vyškolený a kvalifikovaný chirurg a jeho použití je omezeno na prostředí zdravotnického zařízení.
Dávkování
Počet buněk, které se mají aplikovat, záleží na velikosti povrchu rohovky (povrchu v cm2).
Každý přípravek Holoclar obsahuje individuální léčebnou dávku s počtem buněk dostačujícím k pokrytí celého povrchu rohovky. Doporučená dávka přípravku Holoclar je 79 000 - 316 000 buněk/cm2, což odpovídá 1 cm2 přípravku na cm2 defektu. Každý přípravek Holoclar je určen k jednomu ošetření. Ošetření se může opakovat, pokud to ošetřující lékař považuje za indikované.
Po aplikaci musí následovat vhodný plán antibiotické a protizánětlivé léčby podle doporučení lékaře (viz bod 4.4).
Zvláštní populace Starší pacienti
Údaje o použití přípravku Holoclar u starších pacientů jsou omezené. Nelze učinit žádná doporučení ohledně dávkování (viz body 4.8 a 5.1).
Porucha funkce jater a ledvin
Údaje o použití přípravku Holoclar u pacientů s poruchou funkce jater a ledvin nejsou k dispozici.
Pediatrická populace
Bezpečnost a účinnost přípravku Holoclar u dětí a dospívajících ve věku od 0 do 18 let nebyla dosud stanovena. V současnosti dostupné údaje jsou popsány v bodě 4.8 a 5.1, ale na jejich základě nelze učinit žádná doporučení ohledně dávkování.
Způsob podání Určeno k implantaci.
Úplné technické informace o postupech spojených s použitím přípravku Holoclar jsou uvedeny ve školicí příručce.
Biopsie
Pro výrobu přípravku Holoclar je nutné provedení biopsie 1-2 mm2 nepoškozené tkáně limbu. Biopsie se provádí při lokální anestézii. Provede se povrchová laváž oka balancovaným fyziologickým roztokem pro irigaci oka, po níž následuje oddělení spojivky od limbu pro obnažení místa odběru vzorku z rohovky. K odebrání biopsie se provede incize 2x2 mm.
Bioptický vzorek se umístí do dodané sterilní zkumavky s transportním médiem. Bioptický vzorek musí výrobce obdržet do 24 hodin po jeho opatření.
Léčba po biopsii
Po biopsii se musí aplikovat vhodný režim profylaktické léčby antibiotiky.
V některých případech se může stát, že zdrojové limbální kmenové buňky pacienta nelze pomnožit nebo že nejsou splněna kritéria pro propuštění šarže, a to z důvodu nedostatečné kvality biopsie, kvůli charakteristice pacienta nebo kvůli selhání při výrobě. Proto se může stát, že Holoclar nebude možno dodat. Operující lékař bude informován, jakmile to bude během výroby možné, a v tom případě musí zvolit pro léčbu pacienta alternativní metodu.
Implantace
Holoclar je určen výhradně k použití pro autologní regeneraci limbálních kmenových buněk v souladu se schválenou terapeutickou indikací a musí se aplikovat za aseptických podmínek cestou limbální peritomie, podpovrchového řezu spojivky a excize fibrovaskulární tkáně rohovky při přípravě lože defektu. Následuje uložení implantátu pod podpovrchově incidovanou spojivku. Přebytek implantátu se odstřihne a okraj se pokryje spojivkou při založení 2 nebo 3 stehů z vicrylu nebo hedvábí 8/0 pro mechanické utěsnění léze a pro zajištění implantátu. Oční víčka jsou fixována uzavřená přes implantát a zajištěna náplastí Steri-strip.
Přípravek Holoclar se obecně implantuje při lokální retrobulbární nebo parabulbární anestézii. Podle rozhodnutí chirurga mohou následovat další anesteziologické postupy.
Pooperační léčba
Po implantaci je nutno aplikovat vhodný režim lokální a systémové protizánětlivé a profylaktické antibiotické léčby.
Doporučuje se následující režim: Doxycyklin 100 mg tablety dvakrát denně (nebo amoxicillin 500 mg dvakrát denně) a prednison perorálně při denní dávce 0,5 mg/kg (do maximální dávky 25 mg) za den se podává ode dne operace po dobu 2 týdnů. Po 2 týdnech se má systémové podávání antibiotik vysadit a denní dávka prednisonu se sníží na 0,25 mg/kg (max. 12,5 mg) za den po dobu 1 týdne, na 0,125 mg/kg (max. 5,0 mg) za den po dobu následujícího týdne a pak se vysadí.
Dva týdny po operaci se zahájí topická léčba kortikosteroidy za použití dexamethasonu bez konzervantů 0,1% očních kapek, 1 kapka třikrát denně po dobu 2 týdnů; dále se sníží na 1 kapku dvakrát denně po dobu 1 týdne a na 1 kapku jednou denně následující týden. V případě přetrvávajícího zánětu oka lze topickou léčbu kortikosteroidy udržovat.
Po implantaci musí následovat vhodný režim sledování.
Informace o přípravě přípravku Holocar a pro zacházení s ním viz bod 6.6.
4.3 Kontraindikace
Hypersenzitivita na kteroukoli pomocnou látku uvedenou v bodě 6.1 nebo na hovězí sérum a myší buňky 3T3-J2.
4.4 Zvláštní upozornění a opatření pro použití
Obecně
Holoclar je autologní výrobek a za žádných okolností se nesmí aplikovat jiné osobě než dárcovskému pacientovi.
Holoclar obsahuje letálně ozářené myší fibroblasty 3T3 a může obsahovat stopy fetálního hovězího séra. Pacienti se známou hypersenzitivitou na myši nebo na fetální hovězí sérum nesmí být léčeni (viz bod 4.3).
Holoclar může obsahovat potenciálně infekční biologický materiál. Nicméně riziko se považuje za nízké a kontrolované při výrobě.
Opatření pro použití
Potenciálními komplikujícími faktory jsou současná nesprávná pozice očních víček, zjizvení spojivky se zkrácením fornixu, anestézie rohovky a/nebo anestézie nebo závažná hypoestézie spojivky, pterygium a závažná suchost oka. Pokud je to možné, je nutno současné oční potíže korigovat před implantací přípravku Holoclar.
U pacientů s akutním zánětem nebo infekcí oka musí být výkon odložen až do zdokumentovaného zotavení, protože zánět může ohrozit úspěšnost zákroku.
Postup aplikace přípravku Holoclar zahrnuje použití antibiotik a kortikosteroidů (viz bod 4.2). Lékaři mají zjistit příslušné informace o bezpečnosti v souhrnech údajů o přípravku pro tyto uvedené přípravky.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné studie interakcí.
Oční kapky obsahující benzalkonium-chlorid a/nebo jiné konzervanty je nutno vyloučit. Benzalkonium-chlorid (stejně jako jiné kvartérní amoniové sloučeniny) je cytotoxický a oční kapky obsahující tento konzervant mohou poškozovat nově regenerovaný epitel rohovky. Je nutno vyloučit i jiné cytotoxické přípravky.
Nebyly hlášeny žádné interakce mezi přípravkem Holoclar a léčbou po biopsii/operaci doporučovanou v bodě 4.2.
4.6 Fertilita, těhotenství a kojení
Údaje o použití přípravku Holoclar u těhotných žen nejsou k dispozici.
Studie reprodukční toxicity na zvířatech nejsou k dispozici (viz bod 5.3).
Použití přípravku Holoclar v těhotenství se vzhledem k požadavku pooperační farmakologické léčby z preventivních důvodů nedoporučuje.
Kojení
Použití přípravku Holoclar k implantaci během kojení se z preventivních důvodů nedoporučuje. Fertilita
Nejsou k dispozici žádné klinické údaje o účincích přípravku Holoclar na fertilitu.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Vzhledem k chirurgické povaze doprovodného postupu implantace má přípravek Holoclar výrazný vliv na schopnost řídit nebo obsluhovat stroje. Proto má být po léčbě přípravkem Holoclar řízení a obsluha strojů omezeno a pacienti musí dodržovat pokyny svého ošetřujícího lékaře.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Nejzávažnějšími nežádoucími účinky jsou perforace rohovky a ulcerativní keratitida, které mohou nastat do 3 měsíců od implantace přípravku Holoclar a které jsou spojeny s nestabilitou rohovkového epitelu, a vazovagální synkopa s výskytem první den po operaci, způsobená bolestí oka. Nejčastějšími nežádoucími účinky jsou poruchy oka. Nejčastěji hlášeným nežádoucím účinkem souvisejícím s chirurgickým výkonem bylo krvácení spojivky (5 %), které se objevuje většinou první den po operaci a má sklon k mírné intenzitě a odezní bez léčby do několika dnů.
Tabulka s přehledem nežádoucích účinků
V tabulce jsou uvedeny nežádoucí účinky u pacientů s implantovaným přípravkem Holoclar.
K řazení nežádoucích účinků podle četnosti výskytu se používají následující kategorie: velmi časté (>1/10), časté (>1/100 až <1/10), méně časté (>1/1000 až <1/100), vzácné (>1/10000 až <1/1000), velmi vzácné (<1/10000) a není známo (z dostupných údajů nelze určit).
|
Třída orgánových systémů |
Nežádoucí účinek |
Četnost |
|
Infekce a infestace |
Infekce rohovky |
Méně časté |
|
Poruchy nervového systému |
Vazovagální synkopa |
Méně časté |
|
Poruchy oka |
Blefaritida |
Velmi časté |
|
Krvácení spojivky, krvácení oka, defekt rohovkového epitelu, bolest oka, glaukom/zvýšený nitrooční tlak, |
Časté |
|
ulcerativní keratitida | ||
|
Adheze spojivky, hyperémie spojivky, edém rohovky, perforace rohovky, podráždění oka, fotofobie |
Méně časté | |
|
Poruchy kůže a podkožní tkáně |
Podkožní krvácení |
Méně časté |
|
Celkové poruchy a reakce v místě aplikace |
Metaplazie implantátu |
Méně časté |
|
Poranění, otravy a procedurální komplikace |
Prasknutí stehů |
Méně časté |
Popis vybraných nežádoucích účinků
Nejčastějšími individuálními nežádoucími účinky nesouvisejícími s chirurgickým výkonem byly blefaritida (10,5 %) a defekt rohovkového epitelu (3,5 %). Nejčastějším nežádoucím účinkem považovaným za související s léčbou kortikosteroidy byl glaukom (3,5 %) (viz body 4.2 a 4.4). Hlášení glaukomu zahrnovala nežádoucí účinky na nitrooční tlak.
Pediatrická populace
O bezpečnosti přípravku Holoclar u dětí do 7 let nejsou k dispozici žádné údaje; o jeho bezpečnosti u dětí ve věku 8-17 let jsou pouze omezené údaje. U pediatrických pacientů zařazených do studií HLSTM01 (věk 13, 14 a 16 let) a HLSTM02 (věk 8 a 14 let) se profil nežádoucích účinků nelišil od dospělé populace.
Starší pacienti
Existují pouze omezené údaje o starších pacientech (n = 12; věk >65 let) a o velmi starých pacientech (n = 2; věk 75-84 let).
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Nebyl hlášen žádný případ předávkování.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Oftalmologika, jiná oftalmologika, ATC kód: S01XA19 Mechanismus účinku a farmakodynamické účinky
Mechanismus účinku přípravku Holoclar je náhrada rohovkového epitelu a ztracených limbálních kmenových buněk u pacientů, u kterých byl limbus zničen v důsledku popálení oka. Během procesu reparace rohovky mají implantované buňky cestou částečného pomnožení, diferenciace a migrace regenerovat rohovkový epitel a také zachovat rezervoár kmenových buněk, který může kontinuálně regenerovat rohovkový epitel.
Konvenční farmakodynamické studie přípravku Holoclar nebyly provedeny.
Klinická účinnost a bezpečnost
Účinnost léčivého přípravku byla vyhodnocena v multicentrické, nekontrolované, retrospektivní kohortové studii se sériemi případů u 106 pacientů (studie HLSTM01) obou pohlaví, léčených pro přítomnost středního nebo závažného deficitu limbálních kmenových buněk (LSCD). Střední až závažný LSCD byl definován podle zasažení nejméně dvou kvadrantů povrchu rohovky superficiální novotvorbou cév. Do primární analýzy účinnosti byli zahrnuti 104 pacienti ve věku od 13 do 79 let (průměr 46,8 let). V době implantace přípravku bylo průměrné trvání stavu od úrazu 18 let (medián 10 let); 99 % pacientů mělo zakalení rohovky a 90 % z nich mělo závažné zhoršení zraku (1/10 nebo méně na Snellenově tabuli). Úspěšnost postupu byla vyhodnocena na základě přítomnosti stabilního rohovkového epitelu (tj. nepřítomnosti defektů epitelu) bez signifikantní rekurence neovaskularizace (ne více než jeden kvadrant, bez zasažení centrální rohovky) 12 měsíců po intervenci. Bylo hlášeno celkem 75 (72,1 %) případů léčby s úspěšným výsledkem. Tyto výsledky byly potvrzeny analýzou senzitivity, v níž povrchová neovaskularizace byla také vyhodnocena nezávislým hodnotitelem ze zaslepených fotografií očí pacientů, pořízených před a po implantaci přípravku Holoclar.
Byly vyhodnoceny další klinicky relevantní parametry jako sekundární hodnocení účinnosti.
Podíl pacientů se symptomy (bolest, pálení nebo fotofobie) se významně snížil oproti stavu před operací (40 pacientů s nejméně jedním příznakem; 38,5 %) jeden rok po operaci (12 pacientů;
11,5 %).
U padesáti jednoho pacienta (49,0 %) bylo dosaženo zlepšení zrakové ostrosti nejméně o jeden úplný řádek na Snellenově tabuli (nebo o jednu kategorii u případů se závažným zhoršením). Podíl pacientů se zlepšenou zrakovou ostrostí byl vyšší u případů bez zjizvení stromatu rohovky (15/18 pacientů;
83,3 %) než u pacientů se zjizvením (36/81 pacientů; 44,4 %). Po převedení kategorických hodnot zrakové ostrosti do logaritmu minimálního úhlu rozlišení (LogMAR) došlo u 47 % případů (40 z 85 s nechybějícími hodnotami) ke zlepšení rovnajícímu se nebo vyššímu než 3 ekvivalenty řádku Snellenovy tabule.
Padesát sedm pacientů podstoupilo po použití přípravku keratoplastiku s úspěšností 42,1 % (N = 24) jeden rok po transplantaci rohovky (tj. se stabilním rohovkovým epitelem bez signifikantní rekurence neovaskularizace).
Starší pacienti
Do studie HLSTM01 bylo zařazeno celkem sedm pacientů (6,7 % studijní populace) ve věku při vstupu 65 let nebo více; sedm (24,1 %) dalších pacientů bylo zařazeno do studie HLSTM02. I když byly tyto studie omezené co do počtu subjektů, data z obou ukázala úspěšnost kolem 70 % léčených případů u starší populace. Tato úroveň účinnosti je podobná úrovni pozorované u celkové populace léčených pacientů.
Pediatrická populace
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s přípravkem Holoclar u jedné nebo více podskupin pediatrické populace ohledně léčby deficitu limbálních kmenových buněk způsobených popálením očí (informace o použití u dětí viz bod 4.2).
Tento léčivý přípravek byl registrován postupem tzv. podmíněného schválení. Znamená to, že j sou očekávány další důkazy o jeho přínosech.
Evropská agentura pro léčivé přípravky nejméně jednou za rok vyhodnotí nové informace o tomto léčivém přípravku a tento souhrn údajů o přípravku bude podle potřeby aktualizován.
5.2 Farmakokinetické vlastnosti
Přípravek je implantován lokálně.
Vzhledem k povaze a k určenému klinickému použití přípravku Holoclar nejsou konvenční farmakokinetické studie vyhodnocující absorpci, biotransformaci a eliminaci relevantní. Imunohistochemická analýza rohovky odebrané pacientům s keratoplastikou po léčbě přípravkem Holoclar prokázala, že transplantované kmenové buňky vytvořily normální vrstvu stratifikovaného rohovkového epitelu, bez migrace nebo invaze do bazálních očních struktur.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje o bezpečnosti byly omezeny na in vitro testování tumorigenicity lidských autologních buněčných kultur. Tyto testy zahrnovaly buněčný karyotyp, buněčný růst v měkkém agaru a proliferaci závislou na růstovém faktoru. Studie in vitro neprokázaly žádný důkaz růstu nezávislého na substrátu, indikujícího tumorigenní potenciál.
Bezpečnost přípravku Holoclar je prokázána ve výsledcích získaných ze dvou retrospektivních klinických studií.
Konvenční neklinické studie reprodukce a vývojové toxicity se nepovažují za relevantní, vezmeme-li v úvahu charakter a určené klinické použití tohoto autologního přípravku.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Transportní médium (Dulbecco's Modified Eagle's Medium)
Fibrinová membrána.
6.2 Inkompatibility
U přípravku Holoclar nebyly provedeny žádné formální studie kompatibility, a proto se tento léčivý přípravek nesmí používat s jinými léčivými přípravky během pooperačního období, dokud nebude zcela obnovena integrita rohovkového epitelu. Výjimkou jsou systémově podaná antibiotika pro profylaxi a kortikosteroidy během bezprostředního pooperačního období.
6.3 Doba použitelnosti
36 hodin.
Holoclar se musí aplikovat nejpozději do 15 minut po otevření primární nádobky.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte při teplotě od 15 °C do 25 °C.
Chraňte před chladem nebo mrazem Neozařujte (např. rentgenovými paprsky).
Nesterilizujte.
Ocelovou primární nádobku uchovávejte dobře uzavřenou, aby byl přípravek chráněn před kontaminací bakteriemi, plísněmi a viry.
6.5 Druh obalu a obsah balení a zvláštní vybavení pro použití, podání nebo implantaci
Holoclar se dodává jako jedna individuální léčebná dávka obsažená v nádobce se šroubovacím uzávěrem. Jedna nádobka obsahuje 3,8 cm2 autologního lidského rohovkového epitelu nasazeného na fibrinovou membránu a pokrytého transportním médiem.
Nádobka je uložena v sekundární plastové nádobě, která je následně vložena v zataveném sterilním plastovém sáčku. Zatavený sáček je uložený v nesterilní tepelně izolované krabici pro transplantaci orgánů, opatřené monitorem teploty. Nakonec je tepelně izolovaná krabice uložena do uzavíratelné transportní tašky opatřené zipem.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Holoclar je určen výhradně pro autologní použití. Před implantací je nutno pečlivě překontrolovat jméno pacienta a porovnat je s identifikací pacienta/dárce na dokumentaci k zásilce a na nádobě s přípravkem.
Je nutno se vyvarovat jakéhokoli potřásání, převracení nebo jiného mechanického namáhání nádoby s přípravkem Holoclar.
Další údaje jsou uvedeny v edukačním materiálu.
Holoclar se nesmí sterilizovat. Nádoba a uzávěr se musí pečlivě vizuálně zkontrolovat, zda nejsou jakkoli poškozené. Je-li primární nádoba přípravku Holoclar poškozená, je ovlivněn vizuální vzhled přípravku nebo jsou identifikovány viditelné částice, přípravek se nesmí používat a musí se vrátit výrobci. Pokud se teplota monitorovaná na izolované krabici odlišuje od podmínek uchovávání, kontaktujte výrobce.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být navrácen výrobci.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Chiesi Farmaceutici SPA,
Via Palermo 26/A,
43122, Parma,
Itálie
Telefon: +3905212791 Fax: +390521 774468 E-Mail: info@chiesigroup.com
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/14/987/001
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 17/02/2015 Datum posledního prodloužení registrace:
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
E. ZVLÁŠTNÍ POVINNOST USKUTEČNIT POREGISTRAČNÍ OPATŘENÍ PRO PODMÍNĚNOU REGISTRACI PŘÍPRAVKU
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce biologické léčivé látky
Holostem Terapie Avanzate S.R.L.
Via Glauco Gottardi,100, Modena, 41100, Italy
Název a adresa výrobce odpovědného za propouštění šarží
Holostem Terapie Avanzate S.R.L.
Via Glauco Gottardi,100, Modena, 41100, Italy
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
• Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoliv následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
Držitel rozhodnutí o registraci předloží první pravidelně aktualizovanou zprávu o bezpečnosti pro tento léčivý přípravek do 6 měsíců od jeho registrace.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky;
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
• Další opatření k minimalizaci rizik
Pro bezpečné a účinné užívání přípravku jsou nezbytná následující doplňková opatření pro minimalizaci rizika:
Edukační materiál pro zdravotnické pracovníky, který slouží ke školení správného použití přípravku a minimalizaci rizik a obsahuje následující klíčová témata:
- Výběr pacienta
- Sledovatelnost pacientů a použití identifikátorů
- Biopsie, implantace a následná péče
- Kontraindikace použití očních kapek obsahujících benzalkoniumchlorid
- Riziko glaukomu a blefaritidy
- Podpora zařazení do registru
- Hlášení podezření na nežádoucí účinky
Součástí edukačního materiálu by také měly být jak Edukační příručka, tak školicí program, jehož součástí bude kontrola osvojení školené tématiky lékařem.
Edukační materiál pro pacienty a/nebo pečovatele, který obsahuje následující klíčová témata:
- Kontraindikace použití očních kapek obsahujících benzalkoniumchlorid
- Nežádoucí účinky léčby antibiotiky a kortikosteroidy po transplantaci
- Informace o registru pro pacienty
- Hlášení podezření na nežádoucí účinky
E. ZVLÁŠTNÍ POVINNOST USKUTEČNIT POREGISTRAČNÍ OPATŘENÍ PRO PODMÍNĚNOU REGISTRACI PŘÍPRAVKU
Tato registrace byla schválena postupem tzv. podmíněného schválení, a proto podle článku 14(7) nařízení (ES) č. 726/2004 držitel rozhodnutí o registraci uskuteční v daném termínu následující opatření:
|
Popis |
Termín splnění |
|
Mnohonárodnostní, multicentrická, prospektivní, otevřená, nekontrolovaná intervenční studie (HLSTM03) hodnotící účinnost a bezpečnost štěpu z autologních kultivovaných limbálních kmenových buněk při obnově rohovkového epitelu u pacientů s deficitem limbálních kmenových buněk způsobeným popálením očí |
Definitivní uzavření v prosinci 2020 |
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
Holoclar 79 000 - 316 000 buněk/cm2 náhrada živé tkáně.
Autologní lidský rohovkový epitel expandovaný ex-vivo obsahující kmenové buňky
Tento přípravek obsahuje buňky lidského původu.
Holoclar je transparentní kruhová membrána s 300 000 až 1 200 000 životaschopných autologních buněk lidského rohovkového epitelu (79 000 - 316 000 buněk/cm2) včetně průměrně 3,5 % (0,4 až 10 %) limbálních kmenových buněk a přechodně amplifikujících a terminálně diferencovaných buněk odvozených z kmenových buněk, které jsou fixovány na podkladní fibrinové vrstvě o průměru 2,2 cm a uloženy v transportním prostředí.
Transportní médium (Dulbecco's Modified Eagle's Medium) Fibrinová membrána.
Náhrada živé tkáně.
Jedna nádobka obsahuje 3,8 cm2 autologního lidského rohovkového epitelu nasazeného na fibrinovou membránu a ponořeného v transportním médiu.
Pouze k jednorázovému použití.
Před použitím si přečtěte příbalovou informaci. Určeno k implantaci.
Uchovávejte mimo dohled a dosah dětí.
Potenciálně infekční biologický materiál.
Zacházejte s ním opatrně; neprotřepávejte, nepřevracejte a nevystavujte žádnému mechanickému namáhání.
EXP : Den / měsíc / rok Čas: Hodiny / minuty (CET)
Uchovávejte při teplotě od 15°C do 25°C.
Ocelovou primární nádobku uchovávejte dobře uzavřenou, aby byl přípravek chráněn před kontaminací bakteriemi, plísněmi a viry.
Nezmrazujte.
Nesterilizujte.
Neozařujte (např. rentgenovými paprsky).
Každá šarže se dodává v teplotně kontrolované tepelně izolované krabici pro transplantaci orgánů.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Veškerý nepoužitý léčivý přípravek nebo odpad musí být navrácen výrobci.
Chiesi Farmaceutici S.P.A, Via Palermo 26/A 43122 Parma, Itálie
EU/1/14/987/001
Č. ŠARŽE:
Výdej léčivého přípravku vázán na lékařský předpis.
Nevyžaduje se - odůvodnění přijato
Holoclar 79 000 - 316 000 buněk/cm2 náhrada živé tkáně.
Autologní lidský rohovkový epitel expandovaný ex-vivo obsahující kmenové buňky
Tento přípravek obsahuje buňky lidského původu.
Holoclar je transparentní kruhová membrána s 300 000 až 1 200 000 životaschopných autologních buněk lidského rohovkového epitelu (79 000 - 316 000 buněk/cm2) včetně průměrně 3,5 % (0,4 až 10 %) limbálních kmenových buněk a přechodně amplifikujících a terminálně diferencovaných buněk odvozených z kmenových buněk, které jsou fixovány na podkladní fibrinové vrstvě o průměru 2,2 cm a uloženy v transportním prostředí.
Transportní médium (Dulbecco's Modified Eagle's Medium) Fibrinová membrána.
Náhrada živé tkáně.
Jedna nádobka obsahuje 3,8 cm2 autologního lidského rohovkového epitelu nasazeného na fibrinovou membránu a ponořeného v transportním médiu.
Pouze k jednorázovému použití.
Před použitím si přečtěte příbalovou informaci. Určeno k implantaci.
Uchovávejte mimo dohled a dosah dětí.
Potenciálně infekční biologický materiál.
Zacházejte s ním opatrně; neprotřepávejte, nepřevracejte a nevystavujte žádnému mechanickému namáhání.
EXP: Den / měsíc / rok Čas: Hodiny / minuty (CET)
Uchovávejte při teplotě od 15 °C do 25 °C.
Ocelovou primární nádobku uchovávejte dobře uzavřenou, aby byl přípravek chráněn před kontaminací bakteriemi, plísněmi a viry.
Nezmrazujte.
Nesterilizujte.
Neozařujte (např. rentgenovými paprsky).
Každá šarže se dodává v teplotně kontrolované tepelně izolované krabici pro transplantaci orgánů.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Veškerý nepoužitý léčivý přípravek nebo odpad musí být navrácen výrobci.
Chiesi Farmaceutici S.P.A, Via Palermo 26/A 43122 Parma, Itálie
EU/1/14/987/001
Č. ŠARŽE :
Jméno a příjmení pacienta: Datum narození pacienta:
Výdej léčivého přípravku vázán na lékařský předpis.
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITŘNÍM OBALU NÁDOBKA SE ŠROUBOVACÍM UZÁVĚREM_
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ Holoclar
2. ZPŮSOB PODÁNÍ_
3. POUŽITELNOST_
EXP :
DATUM:
HODINA: (časové pásmo)
4. ČÍSLO ŠARŽE<, KÓD DÁRCE A KÓD LÉČIVÉHO PŘÍPRAVKU>
Č. ŠARŽE:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ_
Držitel rozhodnutí o registraci: CHIESI FARMACEUTICI S.P.A
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro pacienta
Holoclar 79 000 - 316 000 buněk/cm2 náhrada živé tkáně.
Autologní lidský rohovkový epitel expandovaný ex-vivo obsahující kmenové buňky.
▼ Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než Vám bude tento přípravek aplikován, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého chirurga.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému chirurgovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Holoclar a k čemu se používá
2. Čemu musíte věnovat pozornost, než Vám bude přípravek Holoclar aplikován
3. Jak se Holoclar aplikuje
4. Možné nežádoucí účinky
5. Jak uchovávat Holoclar
6. Obsah balení a další informace
1. Co je přípravek Holoclar a k čemu se používá
Holoclar je léčivý přípravek používaný k náhradě poškozených buněk rohovky (čiré vrstvy, která pokrývá barevnou duhovku v přední části oka), který obsahuje limbální buňky, jež normálně pomáhají udržovat Vaše oko zdravé.
Holoclar se skládá z vrstvy Vašich vlastních buněk, které byly pomnoženy (expandovány ex vivo) ze vzorku limbálních buněk odebraného z Vašeho oka během drobného chirurgického výkonu zvaného biopsie. Každá výroba přípravku Holoclar se provádí individuálně a je určena pouze pro jedno ošetření; ošetření se však mohou opakovat. Buňky použité k výrobě přípravku Holoclar jsou známy jako limbální buňky.
• Autologní znamená, že se jedná o Vaše vlastní buňky.
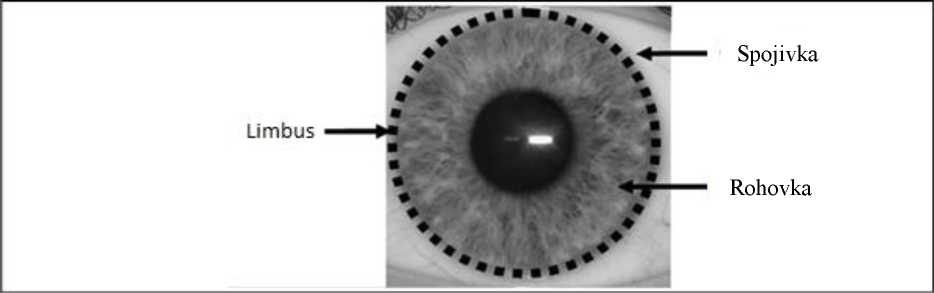
• Limbus je část Vašeho oka. Je to okraj obklopující barevný střed oka (duhovku).
Obrázek ukazuje, kde se ve Vašem oku nachází limbus.
• Limbus obsahuje limbální buňky, které za normálních okolností pomáhají udržovat oko zdravé, a některé z nich jsou kmenové buňky, které mohou produkovat nové buňky. Tyto nové buňky mohou nahradit poškozené buňky ve Vašem oku.
Holoclar se implantuje s cílem opravit poškozený povrch oka u dospělých. Pokud je oko těžce poškozeno popálením ohněm nebo poleptáním chemikálií, může dojít k velkému zjizvení a limbus se může poškodit. Při poškození limbu se zastaví proces hojení, což znamená, že poškození oka se nikdy řádně nezhojí.
Po odebrání několika zdravých limbálních buněk se v laboratoři vypěstuje nová vrstva zdravé tkáně na podkladní vrstvě vyrobené z fibrinu, což je bílkovinná mřížka. Tuto vrstvu tkáně chirurg následně implantuje do poškozené rohovky, což pomůže tomu, aby se Vaše oko normálně zhojilo.
2. Čemu musíte věnovat pozornost, než Vám bude přípravek Holoclar aplikován
Přípravek Holoclar Vám nesmí být aplikován v následujících případech:
- jestliže jste alergický(á) na kteroukoli složku tohoto přípravku (uvedenou v bodě 6) nebo na hovězí sérum a myší buňky.
Upozornění a opatření
Předtím, než Vám bude aplikován přípravek Holoclar, se poraďte se svým chirurgem.
Holoclar se připravuje individuálně z Vašich vlastních buněk tak, aby Vám vyhovoval, a nesmí se použít u jiné osoby než u Vás.
Máte-li akutní infekci oka nebo oteklé, zčervenalé (zanícené) oči, Vaši léčbu je nutno odložit do doby, než se uzdravíte.
Při výrobě přípravku Holoclar se používají dvě složky zvířecího původu. Jednou z nich je hovězí fetální sérum, které pochází od krav a používá se jako pomocný prostředek k vypěstování buněk.
Další složkou je speciální druh inaktivovaných myších buněk, který se používá při pěstování Vašich limbálních buněk. Pokud jste alergický(á) na kteroukoli z těchto složek, může se stát, že Vám tento léčivý přípravek nebude moci být aplikován (viz výše bod „Přípravek Holoclar Vám nesmí být aplikován v následujících případech").
Máte-li kteroukoli z následujících očních potíží, musí se tyto potíže vyléčit před použitím tohoto léčivého přípravku:
• Nerovnoměrná víčka.
• Zjizvení spojivky (ochranné vrstvy přes oční bělmo) s poškozením v místě jejího připojení
na vnitřní část víčka (zkrácený fomix).
• Oko není schopno vnímat bolest (anestézie nebo hypoestézie rohovky nebo spojivky).
• Přerůstání spojivky přes rohovku (pterygium).
• Závažně suché oko.
Další případy, kdy se nesmí Holoclar použít
I když již chirurg odebral malý vzorek limbálních buněk (biopsii) potřebný k výrobě léčivého přípravku, je možné, že u Vás nebude možné použít léčbu přípravkem Holoclar. Je to v případě, kdy biopsie nedostačuje k výrobě přípravku Holoclar; buňky nemohou být v laboratoři pomnoženy nebo vypěstované buňky nesplňují všechna kritéria kvality. Váš chirurg Vás v tomto případě bude informovat.
Děti a dospívající
Dosud byl léčen velmi malý počet dětí, takže není známo, zda je tento přípravek bezpečný při použití u dětí a jak účinný může být.
Problémy s ledvinami a játry
Jestliže trpíte onemocněním jater nebo s ledvin, sdělte to svému lékaři před zahájením léčby.
Další léčivé přípravky a Holoclar
Některé oční kapky obsahují konzervační látku zvanou „benzalkoniumchlorid“. Tato složka může poškodit buňky, ze kterých je přípravek Holoclar vyroben. Nepoužívejte oční kapky obsahující benzalkoniumchlorid a/nebo jiné konzervační látky. Poraďte se se svým lékařem nebo lékárníkem.
Těhotenství a kojení
Pokud jste těhotná, domníváte se, že můžete být těhotná, nebo pokud kojíte, léčba tímto léčivým přípravkem musí být odložena.
Řízení dopravních prostředků a obsluha strojů
Holoclar se aplikuje chirurgicky do oka, a tím bude ovlivněna Vaše schopnost řídit a obsluhovat stroje. Proto po aplikaci přípravku Holoclar neřiďte ani neobsluhujte stroje, dokud Vám chirurg nesdělí, že je to bezpečné. Pečlivě dodržujte jeho pokyny.
3. Jak se Holoclar aplikuje
Holoclar může předepsat a aplikovat výhradně oční chirurg v nemocnici.
Léčba přípravkem Holoclar je dvoustupňový proces.
Návštěva 1: Odběr biopsie
Na první návštěvě chirurg provede biopsii, což znamená, že (Vám z oka) odebere velmi malé množství tkáně obsahující limbální buňky. Před biopsií Vám chirurg aplikuje oční kapky, aby Vám znecitlivěl oko, a potom chirurgicky odebere biopsii. Tato biopsie se použije při výrobě přípravku Holoclar. Po odběru biopsie Vám chirurg předepíše léčebnou dávku antibiotik, aby se snížilo riziko infekce.
Výroba přípravku Holoclar trvá několik týdnů.
Návštěva 2: Implantace přípravku Holoclar:
Na druhé návštěvě chirurg provede následující:
• Provede anestézii (znecitlivění) oka.
• Odstraní zjizvenou tkáň z rohovky.
• Nahradí ji přípravkem Holoclar.
V den operace Vám chirurg znecitliví oko a potom připevní okraj Vaší nové rohovky pomocí stehů, aby zajistil že přípravek Holoclar zůstane na svém místě. Oční víčko budete mít zavřené a přelepené náplastí na tři dny a oko bude zakryté obvazem po dobu 10 až 15 dnů po implantaci.
Po operaci Vám bude předepsána dávka léků pro zajištění úplného zhojení: antibiotika ke snížení rizika infekce a steroidy ke zmírnění otoku a podráždění. Je velmi důležité, abyste užíval(a) všechny léky, které Vám předepsal Váš chirurg, protože jinak se může stát, že Holoclar nebude fungovat. Prosím, prostudujte příbalové informace jednotlivých léčivých přípravků, které máte užívat, protože obsahují další informace o těchto léčivých přípravcích.
Máte-li jakékoli další otázky týkající se léčby přípravkem Holoclar, zeptejte se svého chirurga.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Většina nežádoucích účinků postihuje oko a některé z nich jsou způsobeny operací. Většina nežádoucích účinků je mírné povahy a odezní v průběhu několika týdnů po operaci.
Nejzávažnějšími nežádoucími účinky jsou obtíže s rohovou (eroze) a perforace rohovky, které mohou nastat během 3 měsíců od nasazení přípravku Holoclar. V takovém případě se spojte se svým očním lékařem.
Velmi časté: mohou se vyskytnout u více než 1 osoby z 10
• Zánět očních víček (blefaritida)
Časté: mohou se vyskytnout až u 1 osoby z 10
• Krvácení kolem místa operace, kde byl implantován přípravek Holoclar.
• Potíže s rohovkou (eroze).
• Zvýšený nitrooční tlak (glaukom).
• Bolest oka.
• Zánět rohovky.
Méně časté: mohou se vyskytnout až u 1 osoby ze 100
• Poruchy oka - lepivost očního víčka, krví podlité oči, otok oka, perforace rohovky a podráždění oka.
• Citlivost na světlo.
• Přerůstání kolem implantátu (metaplazie).
• Infekce rohovky.
• Prasknutí stehů.
• Mdloby.
• Krvácení z kůže očního víčka.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete také hlásit přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
Nežádoucí účinky můžete hlásit také přímo na adresu:
Státní ústav pro kontrolu léčiv Šrobárova 48 100 41 Praha 10
webové stránky: www.sukl.cz/nahlasit-nezadouci-ucinek.
Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Holocar uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na štítku.
Neuchovávejte při teplotě nad 25 °C nebo do 15 °C.
Chraňte před chladem nebo mrazem.
Holoclar uchovávejte v ocelové nádobce v plastovém sáčku až do operace. Je to z důvodu ochrany před bakteriální kontaminací.
Holoclar se nesmí ozařovat ani sterilizovat.
Vzhledem k tomu, že se má tento léčivý přípravek použít při Vaší operaci, je za jeho správné uchovávání před jeho použitím a během něj zodpovědný personál zdravotnického zařízení, stejně jako za správnou likvidaci.
6. Obsah balení a další informace Co Holoclar obsahuje
- Léčivou látkou je 300 000 až 1 200 000 Vašich živých očních buněk, z nichž průměrně
3,5 % jsou kmenové buňky. Každý čtvereční centimetr přípravku Holoclar obsahuje 79 000 až 316 000 buněk.
- Přípravek obsahuje dvě pomocné látky: jednou z nich je fibrin - čirá podkladová vrstva používaná k zachování neporušenosti přípravku Holoclar, a druhou je kapalina obsahující aminokyseliny, vitaminy, soli a karbohydráty, která slouží k uchovávání buněk v nádobce a nazývá se Dulbecco's Modified Eagle's Medium.
Jak Holoclar vypadá a co obsahuje toto balení
Holoclar je vrstva buněk určená k implantaci do oka. Buňky jsou udržovány naživu v malé sterilní nádobce. Léčivý přípravek je uložen v několika vrstvách obalů, které jej chrání před bakteriemi a zajišťují uchovávání léčivého přípravku Holoclar při stabilní teplotě po dobu 36 hodin, je-li uložen v prostředí s pokojovou teplotou.
Jedno balení obsahuje individuální léčebnou dávku, která je dostatečně velká k tomu, aby pokryla Vaši rohovku.
Držitel rozhodnutí o registraci
Chiesi Farmaceutici S.p.A.
Via Palermo, 26/A, Parma, 43122, Itálie Telefon: +39 0521 2791 Fax: +39 0521 774468 E-mail: info@chiesi.com
Výrobce
Holostem Terapie Avanzate S.R.L, presso Centro di Medicina Rigenerativa “Stefano Ferrari”,
Via Glauco Gottardi 100, 41100 Modena, Itálie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgie/Belgique/Belgien Lietuva
Chiesi sa/nv Chiesi Pharmaceuticals GmbH
Tél/Tel: + 32 (0)2 788 42 00 Tel: +43 1 4073919
|
Btarapna Chiesi Bulgaria EOOD Tea.: + 359 29201205 |
Luxembourg/Luxemburg Chiesi sa/nv Tél/Tel: + 32 (0)2 788 42 00 |
|
Česká republika Chiesi CZ s.r.o. Tel: +420 261221745 |
Magyarország Chiesi Hungary Kft. Tel.: +36-1-429 1060 |
|
Danmark Chiesi Pharma AB Tlf: +46 8 753 35 20 |
Malta Chiesi Farmaceutici S.p.A. Tel: + 39 0521 2791 |
|
Deutschland Chiesi GmbH Tel: + 49 40 89724-0 |
Nederland Chiesi Pharmaceuticals B.V. Tel: + 31 0 70 413 20 80 |
|
Eesti Chiesi Pharmaceuticals GmbH Tel: +43 1 4073919 |
Norge Chiesi Pharma AB Tlf: +46 8 753 35 20 |
|
EXXáSa Chiesi Hellas AEBE Tnh + 30 210 6179763 |
Osterreich Chiesi Pharmaceuticals GmbH Tel: + 43 1 4073919 |
|
Espaňa Chiesi Espana, S.A Tel: + 34 93 494 8000 |
Polska Chiesi Poland Sp. z.o.o. Tel.: + 48 22 620 1421 |
|
France Chiesi S.A. Tél: + 33 1 47688899 |
Portugal Chiesi Farmaceutici S.p.A. Tel: + 39 0521 2791 |
|
Hrvatska Chiesi Pharmaceuticals GmbH Tel: + 43 1 4073919 |
Románia Chiesi Romania S.R.L. Tel: + 40 212023642 |
|
Ireland Chiesi Ltd Tel: + 44 0161 4885555 |
Slovenija Chiesi Slovenija d.o.o. Tel: + 386-1-43 00 901 |
|
Ísland Chiesi Pharma AB Tlf: +46 8 753 35 20 |
Slovenská republika Chiesi Slovakia s.r.o. Tel: + 421 259300060 |
|
Italia Chiesi Farmaceutici S.p.A. Tel: + 39 0521 2791 |
Suomi/Finland Chiesi Pharma AB Tlf: +46 8 753 35 20 |
|
Kúnpoq Chiesi Farmaceutici S.p.A. Tnh: + 39 0521 2791 |
Sverige Chiesi Pharma AB Tlf: +46 8 753 35 20 |
|
Latvija Chiesi Pharmaceuticals GmbH Tel: + 43 1 4073919 |
United Kingdom Chiesi Ltd Tel: + 44 0161 4885555 |
Tato příbalová informace byla naposledy revidována
Tomuto léčivému přípravku bylo uděleno tzv. podmíněné schválení. Znamená to, že informace o tomto přípravku budou přibývat.
Evropská agentura pro léčivé přípravky nejméně jednou za rok vyhodnotí nové informace o tomto léčivém přípravku a tato příbalová informace bude podle potřeby aktualizována.
Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
<........................................................................................................................>
Následující informace jsou určeny pouze pro zdravotnické pracovníky:
(Úplný souhrn údajů o přípravku bude přiložen k balení léčivého přípravku jako samostatný dokument.)
27