Holmevis 2 Mg/2 Ml Koncentrát Pro Přípravu Infuzního Roztoku
zastaralé informace, vyhledat novějšíPříloha č. 2 k rozhodnutí o změně registrace sp.zn. sukls114584/2012
SOUHRN ÚDAJŮ O PŘÍPRAVKU
1. NÁZEV PŘÍPRAVKU
Holmevis 1 mg/1 ml koncentrát pro přípravu infuzního roztoku Holmevis 2 mg/2 ml koncentrát pro přípravu infuzního roztoku Holmevis 6 mg/6 ml koncentrát pro přípravu infuzního roztoku
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna ampulka s 1 ml koncentrátu pro přípravu infuzního roztoku obsahuje acidum ibandronicum
1 mg (ve formě natrii ibandronas monohydricus 1,125 mg).
Jedna ampulka s 2 ml koncentrátu pro přípravu infuzního roztoku obsahuje acidum ibandronicum
2 mg (ve formě natrii ibandronas monohydricus 2,25 mg).
Jedna injekční lahvička s 6 ml koncentrátu pro přípravu infuzního roztoku obsahuje acidum ibandronicum 6 mg (ve formě natrii ibandronas monohydricus 6,75 mg).
Pomocné látky: Sodík (méně než 1 mmol na dávku).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Koncentrát pro přípravu infuzního roztoku.
Čirý, bezbarvý roztok. 1 doporučení o dávkování a podávání přípravku této skupině pacientů, které je uvedeno v odstavci
Pacienti s onemocněním ledvin (viz bod 4.2).
Léčba hvverkalcémie vyvolané nádorem
Před léčbou kyselinou ibandronovou mají být pacienti adekvátně rehydratováni roztokem chloridu sodného o koncentraci 9 mg/ml (0,9% roztok). Je třeba brát v úvahu stupeň závažnosti hyperkalcémie stejně jako typ nádoru. Pacienti s osteolytickými metastázami kostí potřebují obecně nižší dávky než pacienti s humorálním typem hyperkalcémie. U většiny pacientů s těžkou hyperkalcémií (albuminem korigovaný sérový vápník* >3 mmol/l nebo >12 mg/dl) postačuje jednotlivá dávka 4 mg. U pacientů s mírnou hyperkalcémií (albuminem korigovaný sérový vápník <3 mmol/l nebo <12 mg/dl) je účinná dávka 2 mg. Nejvyšší dávka použitá v klinických studiích byla 6 mg, ale tato dávka se neprojevila vyšším účinkem.
* Poznámka: koncentrace albuminem korigovaného sérového vápníku se vypočte následujícím
|
způsobem: | ||
|
Koncentrace albuminem sérového vápníku (mmol/l) |
korigovaného = |
vápník v séru (mmol/l) - [0,02 x albumin (g/l)] + 0,8 |
|
Nebo | ||
|
Koncentrace albuminem sérového vápníku (mg/dl) |
korigovaného = |
vápník v séru (mg/dl) + 0,8 x [4 -albumin (g/dl)] |
Hodnoty albuminem korigovaného sérového vápníku v mmol/l lze převést na mg/dl vynásobením 4.
Ve většině případů může být zvýšená hladina sérového vápníku snížena na normální hladinu během 7 dnů. Střední doba recidivy (opětovné zvýšení albuminem korigovaného sérového vápníku nad 3 mmol/l) byla 18-19 dnů při dávkách 2 mg a 4 mg. Střední doba recidivy při dávce 6 mg byla 26 dnů.
Omezený počet pacientů (50 pacientů) dostal druhou infuzi z důvodů hyperkalcémie. Léčbu je třeba opakovat v případě vracející se hyperkalcémie nebo při nedostatečné účinnosti.
Pacienti s poruchou jaterních funkcí
Úprava dávkování není nutná (viz bod 5.2).
Pacienti s poruchou renálních funkcí
U pacientů s poruchami funkce ledvin různého stupně nebyly v souvislosti se zvýšenou expozicí vůči ibandronátu pozorovány žádné známky snížení tolerability. V prevenci kostních příhod u pacientů s karcinomem prsu a kostními metastázami je však třeba dodržovat následující doporučení:_
|
Clearance (ml/min) |
kreatininu 1 Dávka / Doba infuze |
Objem infuze 2 |
|
50< Clkr <80 |
6 mg/15 minut |
100 ml |
|
30< Clkr <50 |
4 mg/1 hodina |
500 ml |
|
<30 |
2 mg/1 hodina |
500 ml |
1 Při podávání každé 3 až 4 týdny
Způsob podání Intravenózní podání
Pouze k jednorázovému použití. Použit má být pouze čirý roztok bez jakýchkoli částic.
Holmevis, koncentrát pro přípravu infuzního roztoku, se aplikuje jako intravenózní infuze. K tomuto účelu se přidává obsah injekčních lahviček k 500 ml izotonického roztoku chloridu sodného (nebo 500 ml 5% roztoku glukosy) a infuze se aplikuje po dobu minimálně dvou hodin.
Při neúmyslné intraarteriální aplikaci přípravků, které nejsou pro tento účel výslovně určeny, stejně jako při náhodném paravenózním podání, může dojít k poškození tkáně, a proto je nutno ve zvýšené míře dbát na to, aby byl přípravek Holmevis podáván intravenózně.
4.3 Kontraindikace
• Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku tohoto přípravku
• U pacientů se známou hypersenzitivitou na jiné bisfosfonáty je třeba postupovat se zvýšenou opatrností
• Hypokalcémie
4.4 Zvláštní upozornění a opatření pro použití
Pacienti s poruchami kostního a minerálového metabolismu
Před zahájením léčby přípravkem Holmevis u metastatického postižení kostí má být účinně léčena hypokalcémie a další poruchy kostního a minerálového metabolismu.
U všech pacientů je důležitý dostatečný přísun vápníku a vitaminu D. V případě jejich nedostatečného přísunu ve stravě by pacienti měli užívat doplňky vápníku a/nebo vitaminu D.
Osteonekróza čelisti (ONJ)
U pacientů s onkologickým onemocněním, v jejichž léčebném režimu byly zahrnuty zejména intravenózně podávané bisfosfonáty, byl hlášen výskyt osteonekrózy čelisti, zpravidla spojovaný s extrakcí zubu a/nebo lokální infekcí (včetně osteomyelitidy). Mnozí z těchto pacientů byli také léčeni chemoterapií a kortikosteroidy. Osteonekróza čelisti byla také hlášena u pacientů s osteoporózou, kterým byly bisfosfonáty podávány perorálně.
Před vlastním zahájením léčby bisfosfonáty by měla být u pacientů se souběžnými rizikovými faktory (např. onkologické onemocnění, chemoterapie, radioterapie, podávání kortikosteroidů, špatná ústní hygiena) zvážena nutnost zubní prohlídky včetně odpovídajících preventivních zásahů.
Pokud je to u těchto pacientů možné, neměly by být v průběhu léčby prováděny jakékoliv invazivní stomatologické zákroky. U pacientů, u kterých dojde v průběhu léčby bisfosfonáty k rozvoji osteonekrózy čelisti, by stomatologická operace mohla vést ke zhoršení stavu. Nejsou k dispozici žádné údaje, které by nasvědčovaly tomu, že přerušení léčby bisfosfonáty snižuje riziko výskytu osteonekrózy čelisti u pacientů vyžadujících stomatologické zákroky. Klinické posouzení stavu každého pacienta ošetřujícím lékařem na základě individuálního hodnocení poměru přínosu léčby vůči riziku by mělo být vodítkem ke stanovení léčebného plánu.
Atypické zlomeniny stehenní kosti
V souvislosti s léčbou bisfosfonáty byly hlášeny atypické subtrochanterické a diafyzární zlomeniny femuru, zejména u pacientů dlouhodobě léčených pro osteoporózu. Tyto příčné nebo krátké šikmé zlomeniny se mohou objevit kdekoli v celé délce femuru od oblasti těsně pod malým trochanterem až do části těsně nad suprakondylickým rozšířením. Tyto zlomeniny se objevují po minimálním traumatu nebo bez souvislosti s traumatem a u některých pacientů se mohou projevovat bolestí ve stehně nebo třísle, často spolu s rysy únavové zlomeniny na zobrazovacích vyšetřeních, týdny až měsíce před manifestací kompletní zlomeniny femuru. Zlomeniny jsou často oboustranné, proto je nutné u pacientů léčených bisfosfonáty, kteří utrpěli zlomeninu diafýzy femuru, vyšetřit i kontralaterální femur. Rovněž bylo zaznamenáno špatné hojení těchto zlomenin. U pacientů, u kterých je podezření na atypickou zlomeninu femuru, je třeba při hodnocení jejich stavu zvážit i přerušení léčby bisfosfonáty, a to na základě zhodnocení prospěchu a rizika léčby u jednotlivého pacienta. Pacienty je třeba poučit, aby během léčby bisfosfonáty hlásili jakoukoli bolest v oblasti stehna, boku nebo třísla, a všechny pacienty, u kterých se tyto příznaky objeví, je třeba vyšetřit s ohledem na možnou inkompletní zlomeninu femuru.
Pacienti s poruchou renálních funkcí
Klinické studie neprokázaly, že by při dlouhodobé léčbě kyselinou ibandronovou docházelo ke zhoršení renálních funkcí. Nicméně se doporučuje na základě zhodnocení klinického stavu každého pacienta sledovat při léčbě kyselinou ibandronovou renální funkce, hladinu vápníku, fosfátu a hořčíku v séru.
Pacienti s poruchou jaterních funkcí
Z důvodu nedostačujících klinických údajů není možné doporučit dávkování u pacientů s těžkou jaterní dysfunkcí.
Pacienti se srdeční poruchou
U pacientů ohrožených rozvojem srdečního selhání je třeba zabránit převodnění.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Studie interakcí byly provedeny pouze u dospělých.
Při souběžném podání s melfalanem/prednisolonem u pacientů s mnohočetným myelomem nebyly pozorovány žádné interakce.
Další studie lékových interakcí u žen po menopause prokázaly, že při souběžném podání tamoxifenu nebo hormonální substituční léčby (estrogenu) nevznikají žádné interakce.
Ve vztahu k průniku do tkání se nepředpokládají žádné klinicky významné lékové interakce. Kyselina ibandronová se vylučuje pouze renální sekrecí a neprochází žádnou metabolickou přeměnou. Sekreční dráha se nepřekrývá se žádným ze známých kyselých nebo zásaditých transportních systémů, které se podílejí na vylučování jiných aktivních látek. Kyselina ibandronová dále neinhibuje hlavní lidské hepatální izoenzymy P450 a neindukuje systém hepatálního cytochromu P450 u potkanů. Vazba na plazmatické bílkoviny je po podání terapeutických dávek nízká, a proto se nepředpokládá, že by kyselina ibandronová vytěsňovala jiné aktivní látky.
Zvýšená opatrnost se doporučuje při podávání bisfosfonátů souběžně s aminoglykosidy, neboť obě látky snižují koncentraci vápníku v séru na delší dobu. Je třeba také věnovat pozornost možnému rozvoji souběžné hypomagnesémie.
V klinických studiích byla kyselina ibandronová podávána souběžně s běžně užívanými antineoplastiky, diuretiky, antibiotiky a analgetiky bez klinických projevů interakcí.
4.6 Fertilita, těhotenství a kojení
O podávání kyseliny ibandronové těhotným ženám nejsou k dispozici potřebné údaje. Studie u potkanů prokázaly reprodukční toxicitu (viz bod 5.3). Možné riziko u lidí není známo. Proto by kyselina ibandronová neměla být těhotným ženám podávána.
Kojení
Není známo, zda se kyselina ibandronová vylučuje do lidského mléka. Studie u kojících potkanů prokázaly po nitrožilním podání kyseliny ibandronové přítomnost její nízké koncentrace v mléce. Kojícím ženám by kyselina ibandronová neměla být podávána.
Fertilita
Údaje týkající se účinků kyseliny ibandronové u člověka nejsou k dispozici. V reprodukčních studiích s perorálním podáním u potkanů kyselina ibandronová snižovala fertilitu. Ve studiích s intravenózním podáním u potkanů kyselina ibandronová snižovala fertilitu při vysokých denních dávkách (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Studie sledující možnost ovlivnění schopnosti řídit motorová vozidla a obsluhovat stroje nebyly prováděny.
4.8 Nežádoucí účinky
Nežádoucí účinky jsou uvedeny podle četnosti výskytu, nejčastější jako první, podle následujících konvencí: velmi časté (>1/10), časté (>1/100 až <1/10), méně časté (>1/1000 až <1/100), vzácné (>1/10000 až <1/1000), a velmi vzácné (<1/10000).
Léčba hyperkalcémie vyvolané nádorem
Bezpečnostní profil kyseliny ibandronové je definován na základě kontrolovaných klinických studií, při schválené indikaci a po intravenózním podávání kyseliny ibandronové v doporučených dávkách. Léčba byla nejčastěji spojena se zvýšením tělesné teploty. Příležitostně byly hlášeny příznaky podobné chřipce zahrnující horečku, třesavku, bolesti kostí a/nebo svalů. Ve většině případů nebylo zapotřebí zvláštní léčby a příznaky za pár hodin/dnů odezněly.
Tabulka 1 Nežádoucí účinky po léčbě kyselinou ibandronovou v kontrolovaných klinických studiích u nádorem vyvolané hyperkalcémie____
|
Třída orgánových systémů |
Velmi časté |
Časté |
Méně časté |
Vzácné |
Velmi vzácné |
|
Poruchy imunitního systému |
Přecitlivělost | ||||
|
Poruchy metabolismu a výživy |
Hypokalcémie3 | ||||
|
Respirační, hrudní a mediastinální poruchy |
Bronchospasmus | ||||
|
Poruchy kůže a podkožní tkáně |
Angioneurotický edém | ||||
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Bolesti kostí |
Myalgie | |||
|
Celkové poruchy a reakce v místě aplikace |
Pyrexie |
Onemocnění podobné chřipce3, |
Pozn.: Data pro obě síly 2 mg a 4 mg kyseliny ibandronové byla sloučena.
Prevence kostních _příhod u _pacientů s rakovinou _prsu a kostními metastázami
Bezpečnostní profil nitrožilního podání kyseliny ibandronové u pacientů s karcinomem prsu a kostními metastázami je definován na základě kontrolovaných klinických studií v této indikaci a po intravenózním podávání kyseliny ibandronové v doporučených dávkách.
V tabulce 2 jsou uvedeny nežádoucí účinky z klíčové studie fáze III (152 pacientů léčených kyselinou ibandronovou 6 mg), tj. nežádoucí příhody ve vzdálené, možné nebo pravděpodobné souvislosti s hodnoceným léčivem a po uvedení přípravku na trh.
Tabulka 2 Nežádoucí účinky, které se vyskytovaly u pacientů s kostními metastázami vyvolanými karcinomem prsu, kteří byli léčeni intravenózně podávanou kyselinou ibandronovou v dávce 6 mg.______
|
Třída orgánových systémů |
Velmi časté |
Časté |
Méně časté |
Vzácné |
Velmi vzácné |
|
Infekce a infestace |
Infekce |
Cystitida, vaginitida, ústní kandidóza | |||
|
Novotvary benigní, maligní a blíže neurčené |
Benigní kožní novotvar | ||||
|
Poruchy krve a lymfatického systému |
Anémie, krevní dyskrazie | ||||
|
Endokrinní poruchy |
Poruchy příštítných tělísek | ||||
|
Poruchy metabolismu a výživy |
Hypofosfatémie | ||||
|
Psychiatrické poruchy |
Poruchy spánku, úzkost, afektivní labilita | ||||
|
Poruchy nervového systému |
Bolest hlavy, závratě, dysgeusie (poruchy chuti) |
Cerebrovaskulární onemocnění, léze nervového kořene, amnézie, migréna, neuralgie, hypertonie, hyperestézie, cirkumorální parestézie, parosmie | |||
|
Poruchy oka |
Katarakta |
Zánět okaf** | |||
|
Poruchy ucha a labyrintu |
Hluchota | ||||
|
Srdeční poruchy |
Blokáda Tawarova raménka |
Ischémie myokardu, kardiovaskulární onemocnění, palpitace | |||
|
Respirační, hrudní a mediastinální poruchy |
Faryngitida |
Plicní edém, stridor |
|
Gastrointestinální poruchy |
Průjem, zvracení, dyspepsie, gastrointestinální bolesti, zubní potíže |
Gastroenteritida, gastritida, ulcerace v ústech, dysfagie, cheilitida | |||
|
Poruchy jater a žlučových cest |
Žlučové kameny | ||||
|
Poruchy kůže a podkožní tkáně |
Poruchy kůže, ekchymóza |
Vyrážka, alopecie | |||
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Osteoartróza, myalgie, artralgie, onemocnění kloubů |
Atypické subtrochante rické a diafyzární zlomeniny femuru f (skupinový nežádoucí účinek bisfosfonátů) |
Osteonekróza čelistif** | ||
|
Poruchy ledvin a močových cest |
Močová retence, ledvinná cysta | ||||
|
Poruchy reprodukčního systému a prsu |
Bolesti v pánvi | ||||
|
Celkové poruchy a reakce v místě aplikace |
Onemocnění podobné chřipce, periferní edém, slabost, žízeň |
Hypotermie | |||
|
Vyšetření |
Zvýšení gama-GT, zvýšení kreatininu |
Zvýšení alkalické fosfatázy v krvi, snížení tělesné hmotnosti | |||
|
Poranění, otravy a procedurální komplikace |
Poranění, bolest v místě vpichu |
**Viz další informace níže
f Identifikovány po uvedení přípravku na trh
Osteonekróza čelisti
Osteonekróza čelisti byla hlášena u pacientů léčených bisfosfonáty. Většina těchto hlášení se vztahovala k pacientům s onkologickým onemocněním, ale podobné případy byly také hlášeny u pacientů léčených z důvodu osteoporózy. Osteonekróza čelisti je zpravidla spojována s extrakcí zubu a/nebo lokální infekcí (včetně osteomyelitidy). Diagnóza onkologického onemocnění, chemoterapie, radioterapie, léčba kortikosteroidy a špatná ústní hygiena jsou také považovány za rizikové faktory (viz bod 4.4).
Zánět oka
Při podávání kyseliny ibandronové byly hlášeny oční zánětlivé reakce, jako jsou uveitida, episkleritida a skleritida. V některých případech tyto nežádoucí reakce neustoupily, dokud podávání kyseliny ibandronové nebylo ukončeno.
4.9 Předávkování
Dosud nejsou zkušenosti s akutní otravou kyselinou ibandronovou ve formě koncentrátu pro přípravu infuzního roztoku. Vzhledem k tomu, že v preklinických studiích s vysokými dávkami přípravku byly toxicitou nejvíce postiženy játra a ledviny, je nutné jejich činnost během léčby monitorovat. Klinicky závažnou hypokalcémii lze upravit intravenózním podáním glukonanu vápenatého.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Léčiva ovlivňující stavbu a mineralizaci kostí, bisfosfonáty,
ATC kód: M05BA06
Kyselina ibandronová patří do skupiny bisfosfonátových sloučenin, které mají specifický účinek na kosti. Její selektivní působení na kostní tkáň je založeno na vysoké afinitě bisfosfonátů ke kostním minerálům. Bisfosfonáty inhibují aktivitu osteoklastů, ale přesný mechanizmus není dosud znám.
In vivo kyselina ibandronová zabraňuje pokusně způsobené destrukci kostí, která byla navozena přerušením funkcí gonád, retinoidy, nádory nebo extrakty z nádorů. Potlačení endogenní resorpce kostí bylo dokumentováno studiemi kinetiky 45Ca a uvolněním radioaktivního tetracyklinu předtím zavedeného do kostry.
V dávkách, které byly zřetelně vyšší, než je farmakologicky účinná dávka, neměla kyselina ibandronová žádný účinek na mineralizaci kostí.
Pro resorpci kosti při malignitě je typická nadměrná resorpce, která není vyrovnána odpovídající novotvorbou kosti. Kyselina ibandronová selektivně inhibuje aktivitu osteoklastů, což vede ke zpomalení kostní resorpce a tím ke sníženému výskytu kostních komplikací u maligních onemocnění.
Klinické studie léčby nádorem vyvolané hvverkalcémie
Klinické studie hyperkalcémie u malignit ukázaly, že inhibiční efekt kyseliny ibandronové na tumory indukovanou osteolýzu, a zvláště na nádorem vyvolanou hyperkalcémii, je charakterizován poklesem hladiny sérového vápníku a poklesem vylučování vápníku močí.
V dávkování doporučovaném pro léčbu byly zjištěny následující četnosti odezvy s odpovídajícím intervalem spolehlivosti na základě výsledků klinických studií u pacientů se vstupní koncentrací albuminem korigovaného sérového vápníku >3,0 mmol/l po náležité rehydrataci.
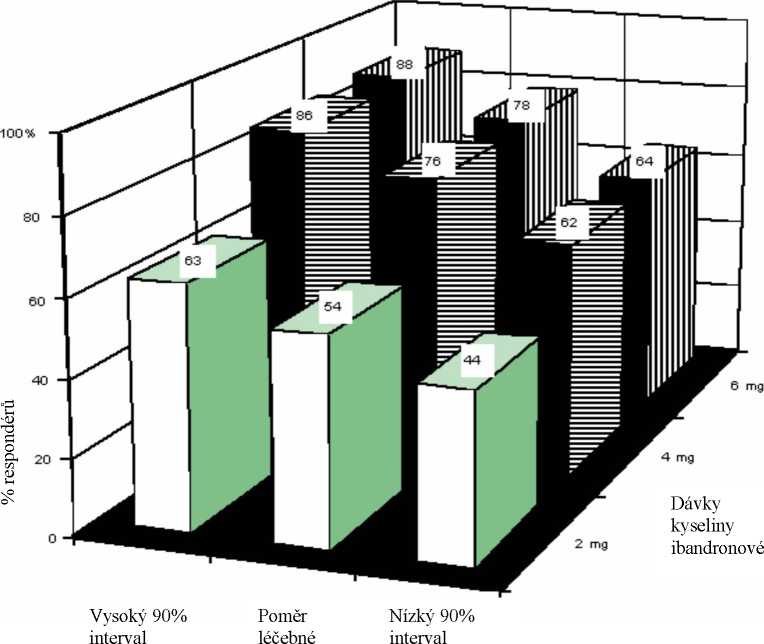
spolehlivosti odoovědi soolehlivosti
U těchto pacientu dostávajících výše uvedené dávky byl průměrný čas pro dosažení normokalcémie 47 dnů. Průměrná doba recidivy (návrat albuminem korigovaného sérového vápníku nad 3,0mmol/l) byla 18 až 26 dnů.
Klinické studie prevence kostních příhod u pacientů s rakovinou prsu a kostními metastázami
Klinické studie u pacientů s karcinomem prsu a kostními metastázami ukázaly, že zde existuje na dávce závislý inhibiční účinek na osteolýzu kosti, který lze hodnotit markery kostní resorpce, a na dávce závislý účinek na kostní příhody.
Prevence kostních komplikací u pacientů s karcinomem prsu a kostními metastázami při intravenózním podání 6 mg kyseliny ibandronové byla hodnocena v jedné randomizované, placebem kontrolované klinické studii fáze III, která trvala 96 týdnů. Pacientky s karcinomem prsu a radiologicky potvrzenými kostními metastázami byly randomizovány do skupin léčených placebem (158 pacientek) nebo 6 mg kyseliny ibandronové (154 pacientek). Výsledky těchto studií jsou shrnuty níže.
Primární výsledný ukazatel účinnosti
Primárním výsledným ukazatelem účinnosti v klinické studii byla doba do objevení se kostní komplikace (skeletal morbidity period rate, SMPR). Jednalo se o sloučený výsledný ukazatel, do kterého patřily následující kostní komplikace (skeletal related events, SREs):
- ozařování kosti z indikace léčby zlomeniny nebo hrozící zlomeniny
- chirurgická léčba zlomenin
- zlomeniny obratlů
- non-vertebrální zlomeniny
Analýza SMPR byla korigovaná na čas a zohledňovala skutečnost, že jedna nebo více příhod, které se staly v odstupu 12 týdnů, mohou navzájem souviset. Vícečetné příhody jsou tedy pro účely analýzy započítány pouze jednou. Údaje z této studie prokázaly významný přínos léčby intravenózně podávanou kyselinou ibandronovou 6 mg oproti placebu z hlediska snížení počtu SRE při hodnocení parametru SMPR (p=0,004). Počet SRE byl také významně nižší při léčbě kyselinou ibandronovou 6 mg a oproti placebu došlo ke 40% snížení rizika SRE (relativní riziko 0,6; p=0,003). Výsledky hodnocení účinnosti jsou shrnuty v tabulce 3.
Tabulka 3 Výsledky hodnocení účinnosti (pacienti s karcinomem prsu a s kostními metastázami)
|
Všechny kostní komplikace (SREs) | |||
|
Placebo |
Kyselina ibandronová |
Hodnota P | |
|
n=158 |
n=154 | ||
|
SMPR (na jednoho pacienta a rok) |
1,48 |
1,19 |
p=0,004 |
|
Počet příhod (na jednu pacientku) |
3,64 |
2,65 |
p=0,025 |
|
Relativní riziko SRE |
- |
0,60 |
p=0,003 |
Sekundární výsledné ukazatele účinnosti
Při léčbě intravenózně podávanou kyselinou ibandronovou v dávce 6 mg bylo ve srovnání s placebem zjištěno statisticky významné zlepšení skóre bolesti kostí. Stupeň zmírnění bolesti byl po celou dobu trvání studie setrvale pod úrovní, která byla při zahájení studie, a při srovnání s placebem byl provázen významným snížením spotřeby analgetik. Zhoršení při hodnocení kvality života bylo ve skupině léčené kyselinou ibandronovou menší než ve skupině léčené placebem. Shrnutí sekundárních výsledných parametrů je uvedeno v tabulce 4.
Tabulka 4 Sekundární výsledné parametry účinnosti (pacienti s karcinomem prsu s kostními metastázami)____
|
Placebo |
Kyselina ibandronová 6 mg |
Hodnota P | |
|
n=158 |
n=154 | ||
|
Bolesti kostí * |
0,21 |
-0,28 |
p<0,001 |
|
Potřeba analgetik * |
0,90 |
0,51 |
p=0,083 |
|
Kvalita života * |
-45,4 |
-10,3 |
p=0,004 |
* Průměrná změna od vstupního vyšetření do posledního hodnocení
U pacientek léčených kyselinou ibandronovou byl také zjištěn významný pokles markerů kostní resorpce v moči (pyridinolin a deoxypyridinolin), který byl ve srovnání s placebem statisticky významný.
Ve studii zahrnující 130 pacientů s metastazujícím karcinomem prsu byla srovnávána bezpečnost kyseliny ibandronové podávané v infuzi trvající 1 hodinu oproti infuzi trvající 15 minut. Z hlediska indikátorů funkce ledvin nebyl pozorován žádný rozdíl. Celkový profil nežádoucích příhod vyskytujících se po 15minutové infuzi kyseliny ibandronové byl ve shodě se známým bezpečnostním profilem po delší době infuze a v souvislosti s infuzí trvající 15 minut nebyly zaznamenány žádné nové skutečnosti týkající se bezpečnosti přípravku.
U pacientů s nádory a s clearance kreatininu <50 ml/min nebyla infuze trvající 15 minut dosud studována.
Pediatrická populace
U dětí a dospívajících mladších než 18 let nebyla bezpečnost a účinnost kyseliny ibandronové stanovena. Žádné údaje nejsou k dispozici.
5.2 Farmakokinetické vlastnosti
Po dvouhodinové infuzi 2, 4 a 6 mg kyseliny ibandronové jsou farmakokinetické parametry přímo úměrné výši podané dávky.
Distribuce
Po prvotním systémovém rozptýlení se kyselina ibandronová rychle váže na kostní tkáň nebo se vylučuje do moči. U lidí je účinný terminální distribuční objem nejméně 90 litrů a množství dávky přípravku, které se dostane do kosti, se odhaduje na 40-50 % množství, které se dostane do krevního oběhu. Vazba na bílkoviny lidské plazmy je v terapeutických koncentracích přibližně 87 % a tak nejsou lékové interakce v důsledku vytěsňování z této vazby pravděpodobné.
Biotransformace
Žádné důkazy nenasvědčují tomu, že by u zvířat nebo lidí podléhala kyselina ibandronová metabolické přeměně.
Vylučování
Rozpětí zjištěných poločasů je široké a závisí na dávce a citlivosti zvolené vyšetřovací metody, ale zjevný terminální poločas se pohybuje v rozpětí 10-60 hodin. Časné plazmatické koncentrace však klesají rychle a dosahují 10 % maximální koncentrace za 3 hodiny po nitrožilním podání a 8 hodin po perorálním podání. Při nitrožilním podání kyseliny ibandronové jednou za 4 týdny po dobu 48 týdnů nebyly zjištěny projevy systémové kumulace.
Celková clearance ibandronové kyseliny je nízká, s průměrnou hodnotou v rozpětí 84-160 ml/min. Renální clearance (přibližně 60 ml/min u zdravých žen po menopause) tvoří 50-60 % celkové clearance a souvisí s clearance kreatininu. Rozdíl mezi zjevnou celkovou a renální clearance odpovídá stupni vychytání látky v kosti.
Farmakokinetika u zvláštních skupin pacientů
Pohlaví
Biologická dostupnost a farmakokinetika kyseliny ibandronové jsou u mužů i žen podobné.
Rasa
Žádné důkazy nenasvědčují tomu, že by chování kyseliny ibandronové v organismu bylo odlišné u různých etnických skupin Asiatů a bělochů. Od pacientů afroamerického původu je k dispozici pouze velmi malé množství údajů.
Pacienti s renální insuficiencí
U pacientů s různým stupněm poškození funkce ledvin souvisí expozice kyselině ibandronové s clearance kreatininu (Clkr). U jedinců s těžkou poruchou funkce ledvin (střední hodnota Clkr=21,2 ml/min) byla střední hodnota AUC0-24h, korigovaná na dávku, o 110 % vyšší v porovnání se zdravými dobrovolníky. V klinické farmakologické studii WP18551 bylo zjištěno, že po jediné intravenózní dávce 6 mg (15minutová infuze) se průměrná AUC0-24 u subjektů s mírným stupněm poškození ledvin zvýšila o 14 % (průměrná odhadovaná Clkr=68,1 ml/min) a u subjektů se středním stupněm poškození ledvin o 86 % (průměrná odhadovaná Clkr=41,2 ml/min) ve srovnání se zdravými dobrovolníky (průměrná odhadovaná Clkr=120 ml/min). Průměrná hodnota Cmax se u pacientů s mírným stupněm poškození ledvin nezvýšila a u pacientů se středním stupněm poškození ledvin byla zvýšena o 12 %. U pacientů s mírnou poruchou funkce ledvin (Clkr>50 a <80 ml/min) není úprava dávkování nutná. U pacientů s rakovinou prsu a kostními metastázami se středně těžkou poruchou funkce ledvin (Clkr>30 a <50 ml/min) nebo těžkou poruchou funkce ledvin (Clkr<30 ml/min), kteří jsou léčeni z důvodu prevence kostních příhod, je úprava dávkování doporučena (viz bod 4.2).
Pacienti s jaterní insuficiencí
U pacientů s poškozením funkce jater nejsou pro kyselinu ibandronovou k dispozici žádné farmakokinetické údaje. Játra se na clearance kyseliny ibandronové zásadním způsobem nepodílejí, neboť tato kyselina se nemetabolizuje, nýbrž se vylučuje ledvinami a vychytává se v kosti. Proto u nemocných s porušenou funkcí jater není úprava dávkování nutná. Kromě toho je vazba kyseliny ibandronové v terapeutických koncentracích na plazmatické bílkoviny přibližně 87 %, takže hypoproteinémie v případě těžkého poškození funkce jater pravděpodobně nepovede k významnému zvýšení koncentrace volné frakce v plazmě.
Starší pacienti
Ve víceproměnné analýze nebyl věk mezi nezávislými faktory, které ovlivňovaly hodnocené farmakokinetické parametry. Vzhledem k tomu, že s narůstajícím věkem dochází ke zhoršování renálních funkcí, je tento faktor třeba vzít v úvahu (viz bod poškození ledvin).
Pediatrická populace
U osob mladších 18 let nejsou pro použití kyseliny ibandronové k dispozici žádné údaje.
5.3 Předklinické údaje vztahující se k bezpečnosti
Účinky v neklinických studiích byly pozorovány pouze po expozicích dostatečně převyšujících maximální expozice u člověka, což svědčí o malém významu pro klinické použití. Podobně jako u ostatních bisfosfonátů bylo prokázáno, že primárním orgánem systémové toxicity jsou ledviny.
Mutagenita/Karcinogenita:
Karcinogenní potenciál nebyl pozorován. Testy genotoxicity nepřinesly žádné důkazy o genetickém účinku kyseliny ibandronové.
Reprodukční toxicita:
U potkanů a králíků, kterým byla podávána kyselina ibandronová intravenózně, nebyly zjištěny žádné důkazy o přímé fetální toxicitě nebo teratogenních účincích. V reprodukčních studiích s perorálním podáním u potkanů se účinky na fertilitu sestávaly ze zvýšených preimplantačních ztrát při dávkách 1 mg/kg/den a vyšších. V reprodukčních studiích s intravenózním podáním u potkanů kyselina ibandronová snižovala počet spermií při dávkách 0,3 a 1 mg/kg/den a snižovala fertilitu u samců při dávkách 1 mg/kg/den a u samic při dávkách 1,2 mg/kg/den. Nežádoucí účinky kyseliny ibandronové pozorované ve studiích reprodukční toxicity u potkanů odpovídaly očekávanému spektru pro tuto lékovou skupinu (bisfosfonáty). Patří mezi ně snížení počtu implantačních míst, narušení přirozené porodní činnosti (dystokie) a zvýšení výskytu viscerálních variací (syndrom ledvinné pánvičky a ureteru) a abnormality zubů u první filiální generace potkanů.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Chlorid sodný Kyselina octová (98%)
Trihydrát octanu sodného Voda na injekci
6.2 Inkompatibility
Aby bylo možno vyloučit inkompatibilitu, může být koncentrát pro přípravu infuzního roztoku Holmevis ředěn pouze izotonickým roztokem chloridu sodného nebo 5% roztokem glukosy.
Informace o pH a osmolaritě rekonstituovaného parenterálního roztoku jsou uvedeny v následující tabulce:
|
pH |
3,5-4,5 |
|
Osmolarita |
0,290 mOsmol/l±5% 0,276-0,305 mOsmol/l |
Holmevis se nesmí míchat s roztoky obsahujícími vápník.
6.3 Doba použitelnosti
30 měsíců
Po naředění: 24 hodin
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje před naředěním žádné zvláštní podmínky uchovávání.
Po naředění: Uchovávejte při 2 °C-8 °C (v chladničce).
Z mikrobiologického hlediska má být roztok spotřebován okamžitě. Pokud není spotřebován okamžitě, jsou doba a podmínky uchovávání plně v odpovědnosti uživatele a neměly by přesáhnout dobu 24 hodin při teplotě 2-8 °C, jestliže naředění neproběhlo za kontrolovaných a validovaných aseptických podmínek.
6.5 Druh obalu a obsah balení
Holmevis 1 mg/1 ml je dodáván v baleních obsahujících 1 ampulku (2ml čirá ampulka ze skla typu I).
Holmevis 2 mg/2 ml je dodáván v baleních obsahujících 1 ampulku (4ml čirá ampulka ze skla typu I).
Holmevis 6 mg/6 ml je dodáván v baleních obsahujících 1, 5 a 10 injekčních lahviček (9ml čirá injekční lahvička ze skla typu I) uzavřených pryžovou zátkou a hliníkovým uzávěrem.
Na trhu nemusí být všechny velikosti balení
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Všechen nepoužitý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Dopad farmaceutických výrobků na životní prostředí je třeba minimalizovat.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
EGIS Pharmaceuticals PLC H-1106 Budapešť, Keresztúri út 30-38.
Maďarsko
8. REGISTRAČNÍ ČÍSLO(A)
87/528/11-C
87/529/11-C
87/530/11-C
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 17.8.2011
10. DATUM REVIZE TEXTU
30.7.2012
Strana 13 (celkem 13)
KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Holmevis je indikován u dospělých pacientů
- k prevenci kostních příhod (patologických zlomenin, kostních komplikací, které vyžadují radiologickou nebo chirurgickou léčbu) u pacientů s rakovinou prsu a kostními metastázami.
- k léčbě hyperkalcémie vyvolané nádorem s výskytem nebo bez výskytu metastáz.
4.2 Dávkování a způsob podání
Léčba přípravkem Holmevis by měla být zahájena pouze lékařem se zkušenostmi s léčbou onkologického onemocnění.
Dávkování
Prevence kostních _příhod u _pacientů s rakovinou _prsu a kostními metastázami
Doporučené dávkování v prevenci kostních příhod u pacientů s karcinomem prsu a kostními metastázami je 6 mg ve formě intravenózní injekce podávané každé 3-4 týdny. Jednotlivá dávka by měla být podána infuzí trvající alespoň 15 minut. Při přípravě infuze má být obsah injekční lahvičky (lahviček) přidán pouze ke 100 ml izotonického roztoku chloridu sodného nebo 100 ml 5% roztoku glukosy.
Kratší doba (tj. 15 min) infuze má být použita pouze u pacientů s normální funkcí ledvin nebo s mírně zhoršenou funkcí ledvin. Nejsou k dispozici žádné údaje charakterizující užití kratší doby infuze u pacientů s clearancí kreatininu pod 50 ml/min. Předepisující lékař by měl věnovat pozornost
0,9% roztok chloridu sodného nebo 5% roztok glukosy
U pacientů s nádorovým onemocněním a Clk <50 ml/min nebyla doba infuze trvající 15 minut dosud studována.
Starší pacienti
Úprava dávkování není nutná.
Pediatrická populace
U dětí a dospívajících mladších než 18 let nebyla bezpečnost a účinnost kyseliny ibandronové stanovena. Žádné údaje nejsou k dispozici.
Viz další informace níže Hypokalcémie
Snížené vylučování kalcia ledvinami může být doprovázeno poklesem hladiny fosfátů v séru, které nevyžaduje terapeutický zásah. Hladina vápníku v séru může klesnout na hypokalcemické hodnoty.
Onemocnění _podobné chřipce
Objevily se příznaky podobné chřipce, zahrnující horečku, třesavku, bolesti kostí a/nebo svalů. Ve většině případů nebylo zapotřebí zvláštní léčby a příznaky za pár hodin/dnů odezněly.