Halaven 0,44 Mg/Ml
SOUHRN ÚDAJŮ O PŘÍPRAVKU
PŘÍLOHA I
HALAVEN 0,44 mg/ml injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden mililitr obsahuje eribulini mesilas odpovídající eribulinum 0,44 mg.
Jedna 2ml injekční lahvička obsahuje eribulini mesilas odpovídající eribulinum 0,88 mg. Jedna 3ml injekční lahvička obsahuje eribulini mesilas odpovídající eribulinum 1,32 mg.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok (injekce). Čirý, bezbarvý vodný roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek HALAVEN je indikován k léčbě dospělých pacientů s lokálně pokročilým nebo metastazujícím karcinomem prsu, jejichž stav se zhoršil po nejméně jednom chemoterapeutickém režimu zaměřeném na pokročilé onemocnění (viz bod 5.1). Předchozí léčba měla zahrnovat antracyklin a taxan buď jako adjuvantní léčbu nebo jako léčbu metastatického onemocnění, s výjimkou případů, kdy u pacientů nebyla léčba těmito přípravky vhodná.
Přípravek HALAVEN je indikován k léčbě dospělých pacientů s neresekovatelným liposarkomem, kteří již podstoupili léčbu obsahující antracyklin (s výjimkou pacientů, u nichž nebyla tato léčba vhodná) zaměřenou na pokročilé nebo metastazující onemocnění (viz bod 5.1).
4.2 Dávkování a způsob podání
Přípravek HALAVEN se má podávat pouze pod dohledem kvalifikovaného lékaře se zkušenostmi s náležitým používáním cytotoxických léčivých přípravků.
Dávkování
Doporučená dávka eribulinu v podobě roztoku k přímému použití je 1,23 mg/m2, který je nutné podávat intravenózně po dobu 2-5 minut v 1. a 8. den každého 21denního cyklu.
Poznámka:
V EU se doporučená dávka vztahuje k bázi léčivé látky (eribulinu). Výpočet individuální dávky, která se má pacientovi podávat, musí vycházet z koncentrace roztoku k přímému použití, který obsahuje 0,44 mg/ml eribulinu a z doporučené dávky 1,23 mg/m2. Doporučená snížení dávky uvedená níže jsou rovněž uvedena jako dávka eribulinu, která má být podána, na základě koncentrace roztoku k přímému použití.
V pivotních studiích, v odpovídajících publikacích a v některých dalších oblastech, např. ve Spojených státech amerických a ve Švýcarsku, vychází doporučená dávka z obsahu ve formě soli (eribulin-mesylát).
Pacienti mohou mít nauzeu či zvracení. Má být zvážena antiemetická profylaxe včetně kortikoidů.
Odložení podání dávky v průběhu léčby
Podání přípravku HALAVEN v 1. den nebo 8. den je nutné odložit, platí-li kterýkoli z následujících bodů:
- Absolutní počet neutrofilů (ANC) < 1 x 109/l
- Krevní destičky < 75 x 109/l
- Nehematologická toxicita stupně 3 nebo 4.
Snížení dávky v průběhu léčby
Doporučení týkající se snížení dávky pro opakovanou léčbu jsou uvedena v následující tabulce. Doporučená snížení dávky__
|
Nežádoucí účinek po předchozím podání přípravku HALAVEN |
Doporučená dávka eribulinu |
|
Hematologický: | |
|
- Absolutní počet neutrofilů (ANC) < 0,5 x 109/l trvající déle než 7 dní |
0,97 mg/m2 |
|
- Absolutní počet neutrofilů (ANC) < 1 x 109/l komplikovaný horečkou nebo infekcí | |
|
- Krevní destičky < 25 x 109/l trombocytopenie | |
|
- Krevní destičky < 50 x 109/l trombocytopenie komplikovaná krvácením nebo požadavkem na transfuzi krve nebo krevních destiček | |
|
Nehematologický: | |
|
Jakýkoli stupeň 3 nebo 4 v předchozím cyklu | |
|
Opětovný výskyt jakýchkoli hematologických či nehematologických nežádoucích účinků dle výše uvedených specifikací | |
|
I při snížení dávky na 0,97 mg/m2 |
0,62 mg/m2 |
|
I při snížení dávky na 0,62 mg/m2 |
Zvažte ukončení léčby |
Po snížení dávky nemá být dávka eribulinu opětovně zvýšena.
Pacienti s poruchou funkce jater Porucha funkce jater v důsledku metastáz:
Doporučená dávka eribulinu u pacientů s mírnou poruchou funkce jater (skóre dle Childa-Pugha A) je 0,97 mg/m2 podávaná intravenózně po dobu 2-5 minut v 1. a 8. den 21denního cyklu. Doporučená dávka eribulinu u pacientů se středně závažnou poruchou funkce jater (skóre dle Childa-Pugha B) je 0,62 mg/m2 podávaná intravenózně po dobu 2-5 minut v 1. a 8. den 21denního cyklu.
Závažná porucha funkce jater (skóre dle Childa-Pugha C) nebyla studována, ale předpokládá se, že je při použití eribulinu u těchto pacientů nutné výraznější snížení dávky.
Porucha funkce jater v důsledku cirhózy:
Tato skupina pacientů nebyla studována. Výše uvedené dávky lze používat v případě mírných a středně závažných poruch, ale doporučuje se důkladné sledování pacientů, neboť může být nutná další úprava dávky.
Někteří pacienti se středně závažnou či závažnou poruchou funkce ledvin (clearance kreatininu <50 ml/min) mohou mít zvýšenou expozici eribulinu a může být nutné snížení dávky. U všech pacientů s poruchou funkce ledvin se doporučuje postupovat s opatrností a pacienty důkladně sledovat (viz bod 5.2).
Starší _ pacienti
V souvislosti s věkem pacienta nejsou doporučeny žádné specifické úpravy dávky (viz bod 4.8).
Pediatrická _ populace
Použití přípravku HALAVEN v indikaci karcinomu prsu u dětí a dospívajících není relevantní.
Bezpečnost a účinnost přípravku HALAVEN u dětí ve věku od narození do 18 let nebyla u sarkomů měkkých tkání dosud stanovena. Nejsou dostupné žádné údaje.
Způsob podání
Přípravek HALAVEN je určen pro intravenózní podání. Dávku je možné naředit až ve 100 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9%). Nesmí se ředit v 5% infuzním roztoku glukózy. Návod k naředění tohoto léčivého přípravku před jeho podáním je uveden v bodě 6.6. Před podáním je nutné zajistit dobrý přístup do periferního žilního systému nebo přístupný centrální žilní katétr. Není známo, že by eribulin mesylát způsoboval puchýře nebo podráždění. V případě extravazace má být léčba symptomatická. Informace týkající se manipulace s cytotoxickými léčivými přípravky viz bod 6.6.
4.3 Kontraindikace
- Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
- Kojení
4.4 Zvláštní upozornění a opatření pro použití
Hematologie
Myelosuprese závisí na dávce a primárně se projevuje formou neutropenie (bod 4.8). U všech pacientů je nutné před každým podáním dávky eribulinu provést vyšetření celého krevního obrazu. Léčba eribulinem má být zahájena pouze u pacientů s hodnotami ANC > 1,5 x 109/l a hodnotami krevních destiček > 100 x 109/l.
Febrilní neutropenie se vyskytla u < 5 % pacientů léčených eribulinem. Pacienti, u nichž se vyskytla febrilní neutropenie, závažná neutropenie nebo trombocytopenie, by měli být léčeni podle doporučení uvedených v bodě 4.2.
Pacienti s hladinami alaninaminotransferázy (ALT) nebo aspartátaminotransferázy(AST) >3 x horní hranice normálu (ULN) zaznamenali vyšší výskyt neutropenie stupně 4 a febrilní neutropenie.
Přestože je množství údajů omezené, také pacienti s bilirubinem >1,5 x ULN zaznamenali vyšší výskyt neutropenie stupně 4 a febrilní neutropenie.
Byly hlášeny fatální případy febrilní neutropenie, neutropenické sepse a septického šoku.
Závažnou neutropenii je možné léčit použitím faktoru stimulujícího kolonie granulocytů (G-CSF) nebo jeho ekvivalentu dle rozhodnutí lékaře, v souladu s příslušnými směrnicemi (viz bod 5.1).
Periferní neuropatie
U pacientů je nutné důkladně sledovat známky periferní motorické a senzorické neuropatie. V případě rozvoje závažné periferní neurotoxicity je nutné odložení nebo snížení dávky (viz bod 4.2).
Pacienti s preexistující neuropatií vyššího než druhého stupně nebyli zařazeni do klinických studií. U pacientů s preexistující neuropatií prvního nebo druhého stupně nebyla pravděpodobnost výskytu nových nebo zhoršení stávajících příznaků větší, než u pacientů, kteří do studie vstoupili bez neuropatie.
Prodloužení QT intervalu
V nekontrolované otevřené studii, která hodnotila EKG u 26 pacientů, bylo v 8. dnu pozorováno prodloužení QT intervalu, které nebylo závislé na koncetraci eribulinu a které nebylo pozorováno v 1. dnu. Doporučuje se monitorovat EKG, pokud je léčba nasazována u pacientů s městnavým selháváním srdce, bradyarytmiemi, abnormitami v koncentracích elektrolytů a u pacientů, kteří současně užívají léky, u nichž může dojít k prodloužení QT intervalu, včetně antiarytmik třídy I a III. Hypokalémie nebo hypomagnezémie musí být korigovány před nasazením přípravku Halaven a musí být periodicky monitorovány v průběhu léčby. Eribulin nemá být ordinován pacientům s vrozeným syndromem prodloužení QT intervalu.
Pomocné látky
Léčivý přípravek obsahuje malé množství ethanolu (alkohol), méně než 100 mg v jedné dávce.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
K eliminaci eribulinu dochází především (až ze 70 %) prostřednictvím žlučové exkrece. Transportní protein zapojený do tohoto procesu není znám. Eribulin není substrátem transportérů proteinu rezistence karcinomu prsu (breast cancer resistance protein, BCRP), organických aniontů (OAT1, OAT3, OATP1B1, OATP1B3), proteinů mnohočetné lékové rezistence (MRP2, MRP4) a exportní pumpy žlučových solí (BSEP).
U inhibitorů a induktorů CYP3A4 se neočekávají žádné mezilékové interakce. Expozice eribulinu (hodnoty AUC a Cmax) nebyla ketokonazolem, inhibitorem CYP3A4 a P glykoproteinu (Pgp), ani rifampicinem, induktorem CYP3A4, ovlivněna.
Účinky eribulinu na farmakokinetiku jiných přípravků
Údaje získané in vitro ukazují, že eribulin je slabým inhibitorem důležitého enzymu CYP3A4, který metabolizuje léky. Ze studií in vivo nejsou k dispozici žádné údaje. Při souběžném používání s látkami, které mají úzké terapeutické okno a vylučují se především prostřednictvím metabolismu enzymem CYP3A4 (např. alfentanil, cyklosporin, ergotamin, fentanyl, pimozid, chinidin, sirolimus, takrolimus), je nutné postupovat s opatrností a doporučuje se sledovat výskyt nežádoucích příhod.
Eribulin neinhibuje CYP enzymy CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6 a 2E1 v relevantních klinických koncentracích.
Eribulin v relevantních klinických koncentracích neinhiboval aktivitu zprostředkovanou transportéry BCRP, OCT1, OCT2, OAT1, OAT3, OATP1B1 a OATP1B3.
4.6 Fertilita, těhotenství a kojení
Údaje o podávání eribulinu těhotným ženám nejsou k dispozici. Eribulin je u potkanů embryotoxický, fetotoxický a teratogenní. Přípravek HALAVEN lze v těhotenství použít pouze tehdy, když je to skutečně nutné a po důkladném zvážení potřeb matky a rizik pro plod.
Ženy ve fertilním věku je nutné informovat, aby v době, kdy používají přípravek HALAVEN, nebo jej používá jejich partner, zamezily těhotenství, a aby v průběhu léčby a ještě 3 měsíce po ukončení terapie používaly účinnou antikoncepci.
Kojení
Není známo, zda se eribulin/metabolity vylučují do lidského nebo zvířecího mateřského mléka. Riziko pro kojené novorozence/děti nelze vyloučit, a proto se přípravek HALAVEN nesmí během kojení podávat (viz bod 4.3).
Fertilita
U potkanů a psů byla pozorována testikulární toxicita (viz bod 5.3). Pacienti mužského pohlaví by se měli před léčbou poradit o možnosti uchování spermatu, a to vzhledem k možné nevratné neplodnosti v důsledku léčby přípravkem HALAVEN.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek HALAVEN může způsobovat nežádoucí účinky, jako například únavu a závratě, které by mohly vést k malému až mírnému vlivu na schopnost řídit nebo obsluhovat stroje. Pacienty je nutné informovat, aby neřídili ani neobsluhovali stroje, pokud cítí únavu nebo závratě.
4.8 Nežádoucí účinky
Shrnutí bezpečnostního profilu
Nejčastěji hlášenými nežádoucími účinky spojenými s přípravkem HALAVEN jsou suprese kostní dřeně, která se projevuje neutropenií, leukopenií, anémií, trombocytopenií a s tím souvisejícími infekcemi. Bylo také hlášeno nové propuknutí nebo zhoršení již přítomné periferní neuropatie. Mezi hlášenými nežádoucími účinky j e gastrointestinální toxicita, která se projevuje anorexií, nauzeou, zvracením, průjmem, zácpou a stomatitidou. Mezi další nežádoucí účinky patří únava, alopecie, zvýšené hladiny jaterních enzymů, sepse a syndrom muskuloskeletální bolesti.
Nežádoucí účinky v tabulce
Pokud není uvedeno jinak, ukazuje tabulka výskyt případů nežádoucích účinků pozorovaných u pacientů s karcinomem prsu a se sarkomem měkké tkáně, kterým byla doporučená dávka podávána ve studiích fáze 2 a fáze 3.
Kategorie frekvencí jsou definovány následovně: velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1 000 až < 1/100), vzácné (> 1/10 000 až < 1/1 000), a velmi vzácné (< 1/10 000).
V každé skupině frekvencí jsou nežádoucí účinky seřazeny podle klesající frekvence výskytu.
V případech, kdy se objevily účinky stupně 3 nebo 4, jsou uvedeny aktuální celkové frekvence a frekvence účinků stupně 3 nebo 4.
|
Třídy orgánových systémů |
Nežádoucí účinky - všechny stupně | |||
|
Velmi časté (Frekvence %) |
Časté (Frekvence %) |
Méně časté (Frekvence %) |
Vzácné nebo frekvence není známa | |
|
Infekce a infestace |
Infekce močového ústrojí (8,5 %), (G 3/4: 0,7 %) Pneumonie (1,6 %), (G 3/4: 1,0 %) Orální kandidóza Herpes labialis Infekce horních cest dýchacích Zánět nosohltanu Rinitida Herpes zoster |
Sepse (0,5 %), (G 3/4: 0,5 %)a Neutropenická sepse (0,2 %) (G 3/4: 0,2 %)a Septický šok (0,2 %) (G 3/4: 0,2 %)a | ||
|
Poruchy krve a lymfatického systému |
Neutropenie (53,6 %), (G 3/4: 46.0 %) Leukopenie (27,9 %), (G 3/4: 17.0 %) Anémie (21,8 %), (G 3/4: 3,0 %) |
Lymfopenie (5,7 %), (G 3/4: 2,1 %) Febrilní neutropenie (4,5 %), (G 3/4: 4,4 %)a T rombocytopenie (4,2 %), (G 3/4: 0,7 %) |
*Diseminovaná intravaskulární koagulaceb | |
|
Poruchy metabolismu a výživy |
Snížená chuť k jídlu (22,5 %), (G 3/4: 0,7 %)d |
Hypokalemie (6,8 %), (G 3/4: 2,0 %) Hypomagnesemie (2,8 %), (G 3/4: 0,3 %) Dehydratace (2,8 %), (G 3/4: 0,5 %)d Hyperglykemie Hypofosfatemie | ||
|
Psychiatrické poruchy | ||||
|
Poruchy nervového systému |
Periferní neuropatiec (35,9 %), (G 3/4: 7,3 %) Bolest hlavy (17,5 %), (G 3/4: 0,7 %) |
Dysgeusie Závratě (9,0 %), (G 3/4: 0,4 %)d Hypoestezie Netečnost Neurotoxicita | ||
|
Poruchy oka |
Zvýšené slzení (5,8 %), (G 3/4: 0,1 %)d Konjuktivitida | |||
|
Poruchy ucha a labyrintu |
Tinitus | |||
|
Srdeční poruchy | ||||
|
Cévní poruchy |
Nával horka Plicní embolie (1,3 %), (G3/4: 1,1 %)a |
Hluboká žilní trombóza | ||
|
Respirační, hrudní a mediastinální poruchy |
Dyspnoe (15,2 %)a (G 3/4: 3,5 %)a Kašel (15,0 %), (G 3/4: 0,5 %)d |
Bolest orofaryngeální oblasti Epistaxe Rinorea |
Intersticiální plicní nemoc (0,2 %), (G3/4: 0,1 %) | |
|
Třídy orgánových systémů |
Nežádoucí účinky - všechny stupně | |||
|
Velmi časté (Frekvence %) |
Časté (Frekvence %) |
Méně časté (Frekvence %) |
Vzácné nebo frekvence není známa | |
|
Gastrointestinální poruchy |
Nauzea (35,7 %), (G 3/4: 1,1 %)d Zácpa (22,3 %), (G 3/4: 0,7 %)d Průjem (18,7 %), (G 3/4: 0,8 %) Zvracení (18,1 %), (G 3/4: 1,0 %) |
Bolesti břicha Stomatitida (11,1 %), (G 3/4: 1,0 %)d Sucho v ústech Dyspepsie (6,5 %), (G 3/4: 0,3 %)d Gastroezofagální reflux Abdominální distenze |
Vředy v dutině ústní Pankreatitida | |
|
Poruchy jater a žlučových cest |
Zvýšená hladina aspartát aminotransferázy (7,7 %), (G 3/4: 1,4 %)d Zvýšená hladina alaninaminotransferázy (7,6 %), (G 3/4: 1,9 %)d Zvýšená hladina gamaglutamyltransferá zy (1,7 %), (G 3/4: 0,9 %)d Hyperbilirubinemie (1,4 %), (G 3/4: 0,4 %) |
Hepatotoxicita (0,8 %), (G 3/4: 0,6 %) | ||
|
Poruchy kůže a podkožní tkáně |
Alopecie |
Vyrážka (4,9 %), (G3/4: 0,1 %) Pruritus (3,9 %), (G 3/4: 0,1 %)d Potíže s nehty Noční pocení Suchá pokožka Erytém Hyperhidróza Palmoplantární erytrodysestezie (1,0 %), (G3/4: 0,1 %)d |
Angioedém |
**Stevens- Johnsonův syndrom / toxická epidermální nekrolýzab |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Artralgie a myalgie (20,4 %), (G 3/4: 1,0 %) Bolest zad (12,8 %), (G 3/4: 1,5 %) Bolesti končetin (10,0 %), (G 3/4: 0,7 %)d |
Bolest kostí (6,7 %), (G 3/4: 1,2 %) Svalové křeče (5,3 %), (G 3/4: 0,1 %)d Muskuloskeletální bolest Muskuloskeletální bolest v oblasti hrudníku Svalová slabost | ||
|
Poruchy ledvin a močových cest |
Hematurie Proteinurie Selhání ledvin | |||
|
Třídy orgánových systémů |
Nežádoucí účinky - všechny stupně | |||
|
Velmi časté (Frekvence %) |
Časté (Frekvence %) |
Méně časté (Frekvence %) |
Vzácné nebo frekvence není známa | |
|
Celkové poruchy a reakce v místě aplikace |
Únava/Astenie (53,2 %), (G 3/4: 7,7 %) Horečka (21,8 %), (G 3/4: 0,7 %) |
Zánět sliznic (6,4 %), (G 3/4: 0,9 %)d Periferní edém Bolest Chřipkovité onemocnění | ||
|
Vyšetření |
Snížení tělesné hmotnosti (11,4 %), (G 3/4: 0,4 %)d | |||
a Zahrnuje příhody stupně 5. b Podle spontánních hlášení
c Zahrnuje preferované termíny periferní neuropatie, periferní motorické neuropatie, polyneuropatie, parestézie, periferní senzorické neuropatie, periferní senzoricko-motorické neuropatie a demyelinizační polyneuropatie d Žádná příhoda stupně 4.
* Vzácné
** Frekvence není známa
Celkově byly bezpečnostní profily obdobné v populacích pacientů s karcinomem prsu a pacientů se sarkomem měkké tkáně.
Popis vybraných nežádoucích účinků
Neutropenie
Pozorovaná neutropenie byla reverzibilní a nebyla kumulativní; průměrná doba do dosažení minima činila 13 dnů a průměrná doba do zotavení ze závažné neutropenie (< 0,5 x109/l) činila 8 dnů.
Počty neutrofilů < 0,5 x109/l, které přetrvávaly déle než 7 dnů, se objevily u 13 % pacientů s karcinomem prsu léčených eribulinem ve studii EMBRACE.
Neutropenie byla hlášena jako nežádoucí příhoda vyvolaná léčbou (Treatment Emergent Adverse Event, TEAE) u 151/404 pacientů (37,4 % pro všechny stupně) v populaci se sarkomem a u 902/1559 pacientů (57,9 % pro všechny stupně) v populaci s karcinomem prsu. Četnosti kombinovaného parametru TEAE a abnormálních laboratorních hodnot neutrofilů v uvedených skupinách byly 307/404 (76,0 %), resp. 1314/1559 (84,3 %). Medián doby trvání léčby byl 12,0 týdne u pacientů se sarkomem a 15,9 týdne u pacientů s karcinomem prsu.
Byly hlášeny fatální případy febrilní neutropenie, neutropenické sepse a septického šoku.
Z 1963 pacientů s karcinomem prsu a sarkomem měkké tkáně, kteří byli v klinických hodnoceních léčeni eribulinem v doporučené dávce, byla zjištěna jedna fatální příhoda neutropenické sepse (0,1 %) a jedna fatální příhoda febrilní neutropenie (0,1 %). Kromě toho byly zjištěny tři fatální příhody sepse (0,2 %) a jedna fatální příhoda septického šoku (0,1 %).
Závažnou neutropenii je možné léčit použitím G-CSF (faktor stimulující kolonie granulocytů) nebo jeho ekvivalentu dle rozhodnutí lékaře v souladu s příslušnými směrnicemi. U 18 %, resp. 13 % pacientů léčených eribulinem byl ve dvou studiích fáze 3 zaměřených na karcinom prsu (studie 305 a studie 301) podán G-CSF. Ve studii fáze 3 zaměřené na sarkom byl G-CSF podán 26 % pacientů léčených eribulinem.
Neutropenie měla za následek ukončení léčby u < 1 % pacientů, kterým byl podáván eribulin.
Diseminovaná intravaskulární koagulace
Byly hlášeny případy diseminované intravaskulární koagulace, typicky v souvislosti s neutropenií a/nebo sepsí.
Periferní neuropatie
U 1559 pacientů s karcinomem prsu byla nejčastějším nežádoucím účinkem, který měl za následek ukončení léčby eribulinem, periferní neuropatie (3,4 %). Medián doby do periferní neuropatie stupně 2 činil 12,6 týdne (po 4 cyklech). U 2 ze 404 pacientů se sarkomem byla léčba eribulinem ukončena v důsledku periferní neuropatie. Medián doby do periferní neuropatie stupně 2 činil 18,4 týdne.
K rozvoji periferní neuropatie stupně 3 nebo 4 došlo u 7,4 % pacientů s karcinomem prsu a u 3,5 % pacientů se sarkomem. V klinických hodnoceních byla u pacientů s již přítomnou neuropatií pravděpodobnost výskytu nových nebo zhoršení stávajících příznaků stejně vysoká jako u pacientů, kteří do studie vstoupili bez neuropatie.
U pacientů s karcinomem prsu s již přítomnou periferní neuropatií stupně 1 nebo 2 byla frekvence výskytu periferní neuropatie stupně 3 vznikající v důsledku léčby 1 4 %.
Hepatotoxicita
Bylo hlášeno, že u některých pacientů s normální/abnormální hladinou jaterních enzymů před léčbou eribulinem došlo se zahájením léčby eribulinem ke zvýšení hladin jaterních enzymů. Zdá se, že k vzestupům hladiny jaterních enzymů u většiny těchto pacientů došlo v časné fázi léčby eribulinem v 1.-2. cyklu, a proto se má za to, že se u většiny pacientů pravděpodobně jedná o projev adaptace jater na léčbu eribulinem, a ne o známku významné jaterní toxicity; hepatotoxicita byla také hlášena.
Zvláštní skupiny pacientů
Starší _ pacienti
Z 1559 pacientů s rakovinou prsu léčených doporučenou dávkou eribulinu bylo 283 pacientů (18,2 %) ve věku > 65 let. Ze 404 pacientů se sarkomem bylo 90 pacientů (22,3 %) léčených eribulinem ve věku > 65 let. Bezpečnostní profil eribulinu u starších pacientů (> 65 let věku) byl podobný bezpečnostnímu profilu u pacientů ve věku< 65 let s výjimkou astenie/únavy, které měly tendenci zvyšovat se s věkem. U starší populace nejsou doporučeny žádné úpravy dávky.
Pacienti s _poruchou _funkce _jater
Pacienti s ALT nebo AST >3 x ULN zaznamenali vyšší výskyt neutropenie stupně 4 a febrilní neutropenie. Přestože je množství údajů omezené, také pacienti s bilirubinem >1,5 x ULN zaznamenali vyšší výskyt neutropenie stupně 4 a febrilní neutropenie (viz také bod 4.2 a 5.2).
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
V jednom případě předávkování bylo pacientovi neúmyslně podáno 7,6 mg eribulinu (přibližně čtyřnásobek plánované dávky) a následně se u pacienta v 3. den objevila hypersenzitivní reakce (stupeň 3) a v 7. den neutropenie (stupeň 3). Oba tyto nežádoucí účinky byly díky podpůrné péči vyřešeny.
Pro případ předávkování eribulinem neexistuje žádné známé antidotum. V případě předávkování je nutné pacienta důkladně sledovat. Léčba předávkování má zahrnovat podpůrné lékařské zákroky k léčbě přítomných klinických projevů.
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Jiná cytostatika, ATC kód: L01XX41
Eribulin mesylát je inhibitor dynamiky mikrotubulů patřící mezi cytostatika do skupiny halichondrinů. Jde o strukturně zjednodušený syntetický analog halichondrinu B, přirozeného produktu izolovaného z mořské houby rodu Halichondria okadai.
Eribulin inhibuje fázi růstu mikrotubulů, aniž by ovlivňoval jejich fázi zkracování a izoluje tubulin do neproduktivních celků. Eribulin své účinky uplatňuje prostřednictvím antimitotického mechanismu na bázi tubulinů, který vede k bloku v G2/M buněčného cyklu, narušení dělicího vřeténka, a nakonec také k apoptotické smrti buňky po delší a nevratné blokádě mitózy.
Klinická účinnost
Karcinom prsu
Účinnost přípravku HALAVEN u karcinomu prsu podporují především dvě randomizované srovnávací studie fáze 3.
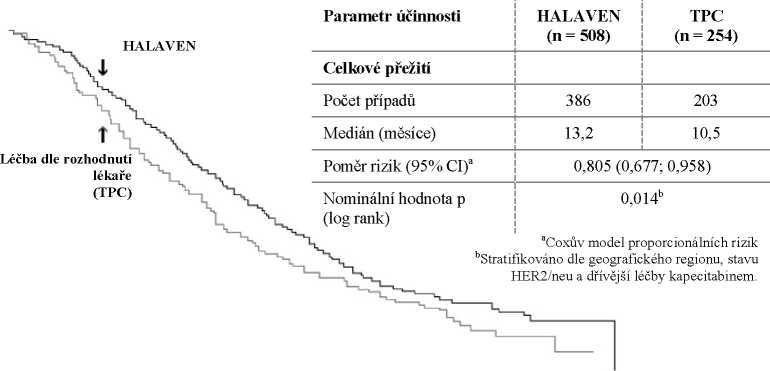
762 pacientů v pivotní studii EMBRACE fáze 3 (studie 305) mělo lokálně rekurentní nebo metastatický karcinom prsu, nebo v minulosti podstoupili minimálně dva a maximálně pět chemoterapeutických režimů, včetně antracyklinového a taxanového (pokud nebyly kontraindikovány). Stav pacientů se musel během 6 měsíců od posledního chemoterapeutického režimu zhoršit. Stav HER2 pacientů byl: 16,1 % pozitivní, 74,2 % negativní a 9,7 % není známo, zatímco 18,9 % bylo trojitě negativní. Pacienti byli randomizováni v poměru 2:1. Dostávali buď přípravek HALAVEN, nebo léčbu dle rozhodnutí lékaře (TPC - treatment of physician's choice), kterou tvořila z 97 % chemoterapie (26 % vinorelbin, 18 % gemcitabin, 18 % kapecitabin, 16 % taxan, 9 % antracyklin, 10 % jiná chemoterapie) a ze 3 % hormonální terapie.
Studie splnila svůj primární cílový ukazatel s výsledkem celkového přežití, který byl statisticky významně lepší u skupiny eribulinu ve srovnání se skupinou TPC v 55 % případů.
Tento výsledek potvrdila aktualizovaná celková analýza přežití provedená v 77 % případů.
Studie 305 - aktualizované celkové přežití (populace ITT)
i,o -
0,9
0,8 -0,7 -0,6 -0,5 -0,4 -0,3 -
0,2 -0,1 -
0,0 -
=£3
H
Z
SĚ
o
2S
.1
>
>P3
0 6 12 S 24 30 36
CAS (měsíce)
POČET PACIENTŮ V OHROŽENÍ ŽIVOTA
|
508 |
406 |
274 |
142 |
54 |
11 |
0 |
|
254 |
178 |
106 |
61 |
26 |
5 |
0 |
HALAVEN
TPC
Dle nezávislé kontroly činil medián doby přežití bez progrese (PFS - progression free survival)
3,7 měsíců u eribulinu ve srovnání s 2,2 měsíci v rameni s TPC (poměr rizik 0,865; 95% CI: 0,714; 1,048; p = 0,137). U pacientů, u nichž bylo možné hodnotit jejich odpověď, činila objektivní odpověď dle kritérií RECIST 12,2 % (95% CI: 9,4 %, 15,5 %) dle nezávislé kontroly v rameni s eribulinem ve srovnání se 4,7 % (95% CI: 2,3 %, 8,4 %) v rameni s TPC.
Pozitivní účinek na celkové přežití byl zaznamenán jak u skupiny pacientů refrakterních k léčbě taxany, tak také u skupiny nerefrakterních pacientů. U aktualizovaného celkového přežití činil poměr rizika eribulinu oproti TPC 0,90 (95% CI: 0,71; 1,14) ve prospěch eribulinu u pacientů refrakterních k léčbě taxany a 0,73 (95% CI: 0,56; 0,96) u pacientů, kteří nejsou refrakterní k léčbě taxany.
Pozitivní účinek na celkové přežití byl zaznamenán jak u skupiny pacientů dosud neléčených kapecitabinem, tak také u skupiny pacientů již léčených kapecitabinem. Analýza aktualizovaného celkového přežití vykazovala benefit v přežití u skupiny eribulinu ve srovnání s TPC, a to jak u pacientů již dříve léčených kapecitabinem s poměrem rizika 0,787 (95% CI: 0,645; 0,961), tak také u pacientů dosud neléčených kapecitabinem s odpovídajícím poměrem rizika 0,865 (95%: CI 0,606; 1,233).
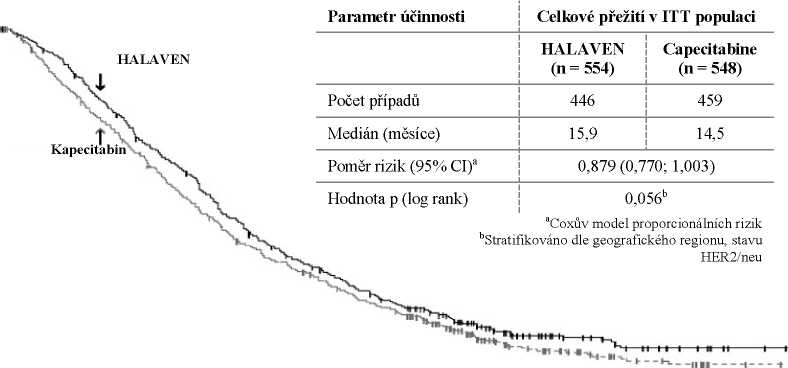
Druhá studie fáze 3 v časnější fázi léčby metastatického karcinomu prsu, studie 301, byla otevřená, randomizovaná studie u pacientů (n = 1102) s lokálně pokročilým nebo metastatickým karcinomem prsu, která zkoumala účinnost monoterapie přípravkem HALAVEN ve srovnání s monoterapií kapecitabinem z hlediska celkového přežití a přežití bez progrese jako společných primárních cílových ukazatelů. Pacienti dříve podstoupili až tři chemoterapeutické režimy, zahrnující jak antracyklin, tak taxan, a maximálně dva kvůli pokročilému onemocnění. Procentuální zastoupení pacientů, kteří dříve podstoupili 0, 1 nebo 2 chemoterapie kvůli metastatickému karcinomu prsu, bylo 20,0 %, 52,0 %, resp. 27,2 %. Stav HER2 pacientů byl: 15,3 % pozitivní, 68,5 % negativní a 16,2 % není známo, zatímco 25,8 % pacientů bylo trojitě negativních.
Studie 301 - Celkové přežití (ITT populace)
1,0
0,9
H
Ph
H
Z
O
ž
oa
o
o
o
Ph
Q
>
0,8
0,7
0,6
0,5
0,4
0,3
0,2
0,1
0,0
0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30 32 34 36 38 40 42 44 46 48 50 52 54 56 58
CAS (měsíce)
POČET PACIENTŮ V OHROŽENÍ ŽIVOTA
HALAVEN 554 530 505 464 423 378 349 320 268 243 214 193 173 151 133 119 99 77 52 38 32 26 22 15 13 9 7 2 2 0
Kapecitabin 548 513 466 426 391 352 308 277 242 214 191 175 155 135 122 108 81 62 42 33 27 23 17 13 12 10 2 2 1 0
Přežití bez progrese hodnocené nezávislou kontrolou bylo u eribulinu a kapecitabinu podobné, s mediány 4,1 měsíců oproti 4,2 měsícům (poměr rizik 1,08 [95% CI: 0,932; 1,250]). Objektivní odpověď hodnocená nezávislou kontrolou byla u eribulinu a kapecitabinu také podobná; 11,0 % (95% CI: 8,5; 13,9) ve skupině s eribulinem a 11,5 % (95% CI: 8,9; 14,5) ve skupině s kapecitabinem.
Níže je uvedeno celkové přežití u pacientů HER2 negativních a HER2 pozitivních ve skupině s eribulinem a v kontrolní skupině ve studii 301 a studii 305:
|
Parametr účinnosti |
Studie 305 Aktualizované celkové přežití v ITT populaci | |||
|
HER2 negativní |
HER2 |
pozitivní | ||
|
HALAVEN (n = 373) |
TPC (n = 192) |
HALAVEN (n = 83) |
TPC (n = 40) | |
|
Počet případů |
285 |
151 |
66 |
37 |
|
Medián v měsících |
13,4 |
10,5 |
11,8 |
8,9 |
|
Poměr rizik (95% CI) |
0,849 (0,695; 1,036) |
0,594 (0,389; 0,907) | ||
|
Hodnota p (log rank) |
0,106 |
0,015 | ||
|
Parametr účinnosti |
Studie 301 Celkové přežití v ITT populaci | |||
|
HER2 negativní |
HER2 |
pozitivní | ||
|
HALAVEN (n = 375) |
Capecitabine (n = 380) |
HALAVEN (n = 86) |
Capecitabine (n = 83) | |
|
Počet případů |
296 |
316 |
73 |
73 |
|
Medián v měsících |
15,9 |
13,5 |
14,3 |
17,1 |
|
Poměr rizik (95% CI) |
0,838 (0,715; 0,983) |
0,965 (0,688; 1,355) | ||
|
Hodnota p (log rank) |
0,030 |
0,837 | ||
Poznámka: Souběžná léčba anti-HER2 nebyla součástí studie 305 a studie 301.
Liposarkom
Účinnost eribulinu u liposarkomu podporuje pivotní studie fáze 3 zaměřená na sarkom (studie 309). Pacienti v této studii (n = 452) měli lokálně rekurentní, neoperovatelný a/nebo metastazující sarkom měkké tkáně jednoho ze dvou podtypů - leiomyosarkom nebo liposarkom. Pacienti v minulosti podstoupili minimálně dva chemoterapeutické režimy, z nichž jeden musel být antracyklinový (pokud nebyl kontraindikován).
Stav pacientů se musel během 6 měsíců od posledního chemoterapeutického režimu zhoršit. Pacienti byli randomizováni v poměru 1:1. Dostávali buď eribulin 1,23 mg/m2 v 1. a 8. den 21denního cyklu, nebo dakarbazin 850 mg/m2, 1000 mg/m2 nebo 1200 mg/m2 (dávka byla stanovena zkoušejícím před randomizací) každých 21 dní.
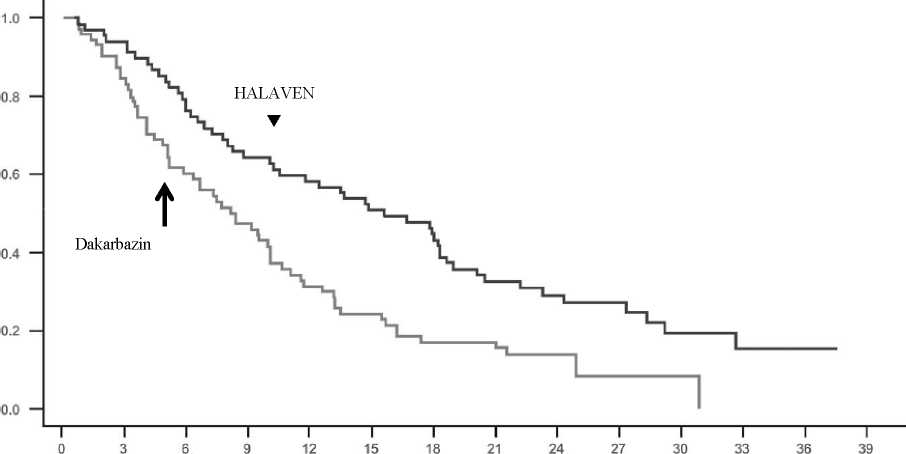
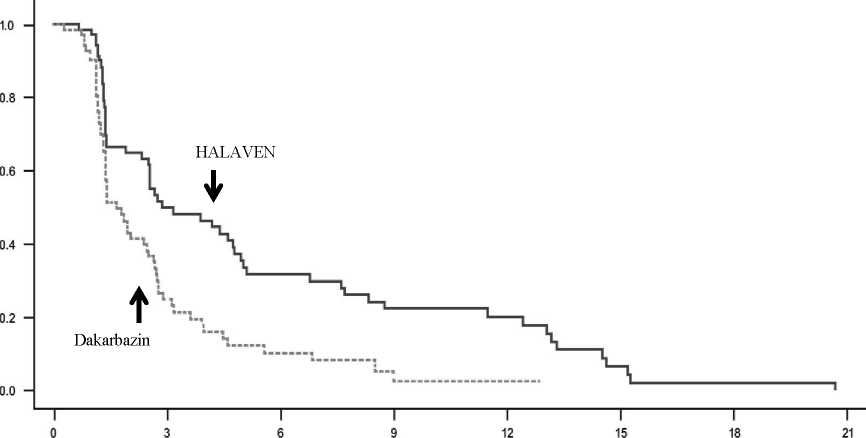
Ve studii 309 bylo pozorováno statisticky významné zlepšení celkového přežití u pacientů randomizovaných do ramene s eribulinem v porovnání s kontrolním ramenem. To představovalo zlepšení mediánu celkového přežití o 2 měsíce (13,5 měsíce u pacientů léčených eribulinem v porovnání s 11,5 měsíce u pacientů léčených dakarbazinem). V celkové populaci nebyl mezi léčebnými rameny zjištěn žádný významný rozdíl v přežití bez progrese nebo celkové odpovědi.
Léčebné účinky eribulinu byly omezeny na pacienty s liposarkomem (45 % nediferencovaným, 37 % myxoidním/z okrouhlých buněk a 18 % pleomorfním ve studii 309) na základě předem plánovaných analýz celkového přežití a přežití bez progrese podle podskupin. Nebyl zjištěn žádný rozdíl v účinnosti mezi eribulinem a dakarbazinem u pacientů s pokročilým nebo metastatickým leiomyosarkomem.
|
Studie 309 Podskupina liposarkomu |
Studie 309 Podskupina leiomyosarkomu |
Studie 309 ITT populace | ||||
|
HALAVEN (n = 71) |
Dakarbazin (n = 72) |
HALAVEN (n = 157) |
Dakarbazin (n = 152) |
HALAVEN (n = 228) |
Dakarbazin (n = 224) | |
|
Celkové přežití | ||||||
|
Počet případů |
52 |
63 |
124 |
118 |
176 |
181 |
|
Medián v měsících |
15,6 |
8,4 |
12,7 |
13,0 |
13,5 |
11,5 |
|
Poměr rizik (95% CI) |
0,511 (0,346; 0,753) |
0,927 (0,714; 1,203) |
0,768 (0,618; 0,954) | |||
|
Nominální hodnota p |
0,0006 |
0,5730 |
0,0169 | |||
|
Přežití bez progrese | ||||||
|
Počet případů |
57 |
59 |
140 |
129 |
197 |
188 |
|
Medián v měsících |
2,9 |
1,7 |
2,2 |
2,6 |
2,6 |
2,6 |
|
Poměr rizik (95% CI) |
0,521 (0,346; 0,784) |
1,072 (0,835; 1,375) |
0,877 (0,710; 1,085) | |||
|
Nominální hodnota p |
0,0015 |
0,5848 |
0,2287 | |||
Studie 309 - Celkové přežití v podskupině s liposarkomem
Čas (měsíce)
POČET PACIENTŮ V OHROŽENÍ ŽIVOTA:
|
HALAVEN |
71 |
63 |
51 |
43 |
39 |
34 |
30 |
20 |
15 |
12 |
7 |
4 |
2 |
0 |
|
Dakarbazin |
72 |
59 |
42 |
33 |
22 |
17 |
12 |
11 |
6 |
3 |
2 |
0 |
0 |
0 |
Čas (měsíce)
POČET PACIENTŮ V OHROŽENÍ ŽIVOTA:
|
HALAVEN |
71 |
28 |
17 |
12 |
9 |
3 |
1 |
0 |
|
Dakarbazin |
72 |
15 |
5 |
2 |
1 |
0 |
0 |
0 |
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s eribulinem u všech podskupin pediatrické populace v indikaci karcinomu prsu (informace o použití u dětí viz bod 4.2).
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s přípravkem HALAVEN u jedné nebo více podskupin pediatrické populace pro léčbu rabdomyosarkomových a nerabdomyosarkomových nádorů měkkých tkání. Informace o použití u dětí viz bod 4.2.
5.2 Farmakokinetické vlastnosti
Distribuce
Farmakokinetika eribulinu je charakterizována rychlou fází distribuce následovanou dlouhou fází eliminace, přičemž průměrný terminální poločas činí přibližně 40 hodin. Má velký distribuční objem (rozmezí průměrů 43 až 114 l/m2).
Eribulin se slabě váže na plazmatické bílkoviny. Vazba na plazmatické bílkoviny se u eribulinu (100 - 1000 ng/ml) pohybovala v rozmezí od 49 % do 65 % v lidské plazmě.
Biotransformace
Nezměněný eribulin byl hlavní formou cirkulující v plazmě po podání 14C-eribulinu pacientům. Koncentrace metabolitů představovaly < 0,6 % mateřské sloučeniny, což potvrzuje, že u člověka nejsou žádné významné metabolity eribulinu.
Eliminace
Clearance eribulinu je nízká (rozmezí průměrů 1,16 až 2,42 l/hod/m2). Při týdenním podávání nebyla zaznamenána žádná významná akumulace eribulinu. Farmakokinetické vlastnosti nejsou závislé na dávce nebo čase v rozmezí dávek eribulinu 0,22 až 3,53 mg/m2.
K eliminaci eribulinu dochází především prostřednictvím žlučové exkrece. Transportní protein zapojený do této exkrece v současné době není znám. Předklinické studie in vitro naznačují, že je eribulin transportován prostřednictvím Pgp. Nicméně bylo prokázáno, že při klinicky relevantních koncentracích není eribulin inhibitorem Pgp in vitro. In vivo navíc souběžné podávání ketokonazolu, inhibitoru Pgp, nijak neovlivňuje expozici eribulinu (AUC a Cmax). Studie in vitro taktéž naznačily, že eribulin není substrátem pro OCT1.
Po podání 14C-eribulinu pacientům se přibližně 82 % dávky eliminovalo ve stolici a 9 % v moči, což naznačuje, že ledvinová clearance nepředstavuje významnou cestu eliminace eribulinu.
Nezměněný eribulin představoval většinu celkové radioaktivity ve stolici a moči.
Porucha funkce jater
Studie hodnotila farmakokinetiku eribulinu u pacientů s mírnou (skóre dle Childa-Pugha A; n=7) a středně závažnou (skóre dle Childa-Pugha B; n=4) poruchou funkce jater v důsledku metastáz na játrech. Ve srovnání s pacienty s normální funkcí jater (n=6) se expozice eribulinu u pacientů s mírnou a středně závažnou poruchou funkce jater zvýšila 1,8násobně, respektive 3násobně. Podávání přípravku HALAVEN v dávce 0,97 mg/m2 pacientům s mírnou poruchou funkce jater a v dávce 0,62 mg/m2 pacientům se středně závažnou poruchou funkce jater mělo za následek poněkud zvýšenou expozici než u dávky 1,23 mg/m2 podané pacientům s normální jaterní funkcí. Přípravek HALAVEN nebyl u pacientů se závažnou poruchou funkce jater (skóre dle Childa-Pugha C) studován. Nebyla provedena žádná studie u pacientů s poruchou funkce jater v důsledku cirhózy. Doporučené dávkování viz bod 4.2.
Porucha funkce ledvin
Zvýšená expozice eribulinu byla pozorována u některých pacientů se středně závažnou či závažnou poruchou funkce ledvin, s vysokou interindividuální variabilitou. Farmakokinetika eribulinu byla hodnocena ve studii fáze 1 u pacientů s normální funkcí ledvin (clearance kreatininu: > 80 ml/min; n=6), se středně závažnou poruchou ledvin (30-50 ml/min, n=7) nebo se závažnou poruchou ledvin (15-<30 ml/min; n=6). Clearance kreatininu byla určena vzorcem dle Cockcrofta a Gaulta. U pacientů se středně závažnou a závažnou poruchou ledvin byla pozorována 1,5krát (90% CI: 0,9-2,5) vyšší normalizovaná dávka AUC(0-inf). Doporučená léčba viz bod 4.2.
5.3 Předklinické údaje vztahující se k bezpečnosti
Eribulin nebyl v testu reverzní mutace u bakterií (Amesův test) provedeném in vitro mutagenní. Eribulin byl pozitivní v testu mutagenity u myšího lymfomu a klastogenní v in vivo mikronukleus testu u potkanů.
Žádné studie karcinogenity nebyly s eribulinem provedeny.
Studie fertility nebyla s eribulinem provedena, ale na základě neklinických zjištění ze studií po opakovaném podání, kde byla zjištěna testikulární toxicita jak u potkanů (hypocelularita seminiformního epitelu s hypospermií/aspermií) tak u psů, lze usuzovat, že mužská plodnost může být léčbou eribulinem ohrožena. Studie embryofetálního vývoje u potkanů potvrdila vývojovou toxicitu a teratogenní potenciál eribulinu. Březím samicím potkanů byly podávány dávky eribulin-mesylátu odpovídajících 0,009; 0,027; 0,088 a 0,133 mg/kg eribulinu v 8., 10. a 12. den březosti. Při dávkách > 0,088 mg/kg byl pozorován s dávkou související zvýšený počet resorpcí a snížená hmotnost plodu a u dávek ve výši 0,133 mg/kg byl zaznamenán zvýšený výskyt malformací (absence dolní čelisti, jazyka, žaludku a sleziny).
FARMACEUTICKÉ ÚDAJE
6.
6.1 Seznam pomocných látek
Bezvodý ethanol Voda na injekci
Kyselina chlorovodíková (na úpravu pH)
Hydroxid sodný (na úpravu pH)
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky s výjimkou těch, které jsou uvedeny v bodě 6.6.
6.3 Doba použitelnosti
Neotevřené injekční lahvičky 4 roky.
Doba použitelnosti otevřeného přípravku
Z mikrobiologického hlediska, pokud způsob otevření nevyloučí riziko mikrobiální kontaminace, přípravek má být použit okamžitě. Pokud není použit okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou v odpovědnosti uživatele.
Pokud není ihned použit, přípravek HALAVEN se nemá uchovávat ve formě nezředěného roztoku v injekční stříkačce za normálních okolností déle než 4 hodiny při teplotě 25 °C a okolním osvětlení, nebo 24 hodin při teplotě 2 až 8 °C.
Naředěné roztoky přípravku HALAVEN (0,018 mg/ml až 0,18 mg/ml eribulinu v injekčním roztoku chloridu sodného 9 mg/ml (0,9 %)) se nemají uchovávat déle než 24 hodin při teplotě 2 až 8 °C, s výjimkou případů, pokud ředění neproběhlo za kontrolovaných a validovaných aseptických podmínek.
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
Podmínky uchovávání tohoto léčivého přípravku po jeho prvním otevření nebo naředění jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
5ml injekční lahvička ze skla typu I, s teflonem potaženou, butylovou pryžovou zátkou a odtrhávacím hliníkovým ochranným krytem, obsahující 2 ml roztoku.
5ml injekční lahvička ze skla typu I, s teflonem potaženou, butylovou pryžovou zátkou a odtrhávacím hliníkovým ochranným krytem, obsahující 3 ml roztoku.
Velikosti balení: krabičky obsahující 1 nebo 6 injekčních lahviček.
Na trhu nemusí být všechny velikosti balení.
HALAVEN je cytotoxický léčivý přípravek proti rakovině a stejně jako u ostatních toxických látek je při manipulaci s ním nutné postupovat s opatrností. Doporučuje se používat rukavice, brýle a ochranné oblečení. Pokud dojde ke kontaktu roztoku s pokožkou, je nutné místo ihned důkladně omýt mýdlem a vodou. Pokud dojde ke kontaktu se sliznicemi, je nutné místo důkladně vypláchnout vodou. Přípravek HALAVEN by měl připravovat a podávat pouze personál náležitě vyškolený v manipulaci s cytotoxickými látkami. Těhotné ženy by s přípravkem HALAVEN neměly manipulovat.
Při použití aseptické techniky je možné přípravek HALAVEN naředit až na 100 ml pomocí injekčního roztoku chloridu sodného 9 mg/ml (0,9%). Po podání se doporučuje propláchnout intravenózní vstup injekčním roztokem chloridu sodného 9 mg/ml (0,9%), aby se zajistilo podání celé dávky. Tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky a nesmí se ředit v 5% infuzním roztoku glukózy.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Eisai Europe Ltd
European Knowledge Centre
Mosquito Way
Hatfield
Hertfordshire
AL10 9SN
Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/11/678/001-004
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 17. března 2011
Datum posledního prodloužení registrace: 19. listopadu 2015
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
PŘÍLOHA II
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného za propouštění šarží
Eisai Manufacturing Ltd.
European Knowledge Centre Mosquito Way Hatfield, Herts AL10 9SN Velká Británie
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
Držitel rozhodnutí o registraci předloží první pravidelně aktualizovanou zprávu o bezpečnosti pro tento léčivý přípravek do 6 měsíců od jeho registrace.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
PŘÍLOHA III
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
HALAVEN 0,44 mg/ml injekční roztok Eribulinum
Jedna 2ml injekční lahvička obsahuje eribulini mesilas odpovídající eribulinum 0,88 mg.
Bezvodý ethanol, voda na injekci, kyselina chlorovodíková, hydroxid sodný Další informace naleznete v příbalové informaci.
Injekční roztok
1 injekční lahvička o obsahu 2 ml 6 injekčních lahviček o obsahu 2 ml
Intravenózní podání
Před použitím si přečtěte příbalovou informaci.
Uchovávejte mimo dohled a dosah dětí.
EXP:
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Eisai Europe Ltd
European Knowledge Centre
Mosquito Way
Hatfield
Hertfordshire
AL10 9SN
Velká Británie
EU/1/11/678/001 1 injekční lahvička EU/1/11/678/002 6 injekčních lahviček
Lot:
Nevyžaduje se - odůvodnění přijato.
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
HALAVEN 0,44 mg/ml injekce
Eribulinum
i.v.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP:
4. ČÍSLO ŠARŽE
Lot:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
Obsahuje eribulinum 0,88 mg ve 2 ml
6. JINÉ
HALAVEN 0,44 mg/ml injekční roztok Eribulinum
Jedna 3ml injekční lahvička obsahuje eribulini mesilas odpovídající eribulinum 1,32 mg.
Bezvodý ethanol, voda na injekci, kyselina chlorovodíková, hydroxid sodný Další informace naleznete v příbalové informaci.
Injekční roztok
1 injekční lahvička o obsahu 3 ml 6 injekčních lahviček o obsahu 3 ml
Intravenózní podání
Před použitím si přečtěte příbalovou informaci.
Uchovávejte mimo dohled a dosah dětí.
EXP:
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Eisai Europe Ltd
European Knowledge Centre
Mosquito Way
Hatfield
Hertfordshire
AL10 9SN
Velká Británie
EU/1/11/678/003 1 injekční lahvička EU/1/11/678/004 6 injekčních lahviček
Lot:
Nevyžaduje se - odůvodnění přijato.
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
HALAVEN 0,44 mg/ml injekce
Eribulinum
i.v.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP:
4. ČÍSLO ŠARŽE
Lot:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
Obsahuje eribulinum 1,32 mg ve 3 ml
6. JINÉ
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro uživatele
HALAVEN 0,44 mg/ml injekční roztok
eribulinum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo zdravotní sestry.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek HALAVEN a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek HALAVEN používat
3. Jak se přípravek HALAVEN používá
4. Možné nežádoucí účinky
5. Jak přípravek HALAVEN uchovávat
6. Obsah balení a další informace
1. Co je přípravek HALAVEN a k čemu se používá
HALAVEN obsahuje léčivou látku eribulin a je to lék proti rakovině, jehož funkcí je zastavovat růst a šíření rakovinných buněk.
Používá se k léčbě lokálně pokročilého nebo metastazujícího karcinomu prsu (tj. karcinom prsu, který se rozšířil mimo původní nádor) u dospělých, kdy byla vyzkoušena nejméně jedna jiná terapie, která však přestala účinkovat.
Používá se také k léčbě pokročilého nebo metastazujícího liposarkomu (tj. typ nádoru vznikající v tukové tkáni) u dospělých, kdy předchozí terapie byla vyzkoušena, ale přestala účinkovat.
2. Čemu musíte věnovat pozornost, než začnete přípravek HALAVEN používat Nepoužívejte přípravek HALAVEN
- jestliže jste alergický(á) na eribulin mesylát nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6)
- jestliže kojíte
Upozornění a opatření
Před použitím přípravku HALAVEN se poraďte se svým lékařem nebo zdravotní sestrou
- jestliže máte problémy s játry
- jestliže máte horečku nebo infekci
- jestliže zaznamenáte necitlivost, brnění, pocit svrbění, citlivost na dotek nebo svalovou slabost
- jestliže máte problémy se srdcem
Máte-li kterýkoli z těchto příznaků, sdělte to svému lékaři, který může rozhodnout o ukončení léčby nebo snížení dávky.
Děti a dospívající
Přípravek HALAVEN se nedoporučuje používat u dětí ve věku do 18 let s dětskými typy sarkomu, neboť dosud není známo, jak tento přípravek v této věkové skupině působí.
Další léčivé přípravky a přípravek HALAVEN
Informujte svého lékaře o všech lécích, které používáte, které jste v nedávné době používal(a) nebo které možná budete používat.
Těhotenství, kojení a plodnost
Přípravek HALAVEN může způsobit vážné vrozené vady a nemá se používat, jestliže jste těhotná, pokud to není po zvážení veškerých rizik pro vás i pro dítě považováno za nezbytně nutné. U mužů může přípravek v budoucnu způsobit trvalé problémy s plodností. Muži by toto téma měli před zahájením léčby prodiskutovat se svým lékařem. Ženy v plodném věku musí během léčby přípravkem HALAVEN a ještě 3 měsíce po ukončení léčby používat účinnou antikoncepci.
Přípravek HALAVEN se nesmí vzhledem k možným rizikům pro dítě během kojení podávat.
Řízení dopravních prostředků a obsluha strojů
Přípravek HALAVEN může způsobovat nežádoucí účinky, jako je únava (velmi časté) a závratě (časté). Neřiďte dopravní prostředek a neobsluhujte žádné stroje, jestliže cítíte únavu nebo závratě.
Přípravek HALAVEN obsahuje alkohol
Tento léčivý přípravek obsahuje malé množství alkoholu, méně než 100 mg v jedné injekční lahvičce.
3. Jak se přípravek HALAVEN používá
Přípravek HALAVEN vám bude podávat lékař nebo zdravotní sestra ve formě injekce do žíly, po dobu 2-5 minut. Dávka, kterou dostanete, je založena na ploše povrchu vašeho těla (vyjádřené ve čtverečních metrech, neboli m2), která se spočítá z vaší hmotnosti a výšky. Obvyklá dávka přípravku HALAVEN je 1,23 mg/m2, ovšem váš lékař ji může upravit na základě výsledků vašich krevních testů nebo jiných faktorů. Aby se zajistilo podání celé dávky přípravku HALAVEN, doporučuje se, aby se po podání přípravku HALAVEN vstříkl do žíly fyziologický (solný) roztok.
Jak často vám bude přípravek HALAVEN podáván?
Přípravek HALAVEN se obvykle podává v 1. a 8. den každého 21denního cyklu. Váš lékař určí, kolik cyklů léčby byste měl(a) podstoupit. V závislosti na výsledcích vašich krevních testů může lékař odložit podání přípravku do doby, než se výsledky vrátí do normálu. Lékař také může rozhodnout o snížení dávky, která je vám podávána.
Máte-li jakékoli další otázky týkající se používání tohoto přípravku, zeptejte se svého lékaře.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí
vyskytnout u každého.
Jestliže se u Vás objeví kterýkoli z následujících závažných příznaků, přestaňte přípravek HALAVEN
používat a okamžitě vyhledejte lékařskou pomoc:
- Horečka s rychlým srdečním tepem, rychlým mělkým dýcháním, studenou, bledou a vlhkou kůží nebo se skvrnami na kůži a/nebo zmatením. Toto mohou být známky stavu, který se označuje jako sepse - závažná a těžká reakce na infekci. Sepse se vyskytuje méně často (může postihnout až 1 ze 100 osob), může ohrozit život a může vést k úmrtí.
- Jakékoli dechové obtíže nebo otok tváře, úst, jazyka nebo krku. Mohlo by se jednat o známky méně často se vyskytující alergické reakce (může postihnout až 1 ze 100 osob).
- Závažné kožní vyrážky s tvorbou puchýřů na kůži, v ústech, očích a na genitáliích. Toto mohou být známky stavu, který se označuje jako Stevens-Johnsonův syndrom / toxická epidermální nekrolýza. Frekvence není známa, ale může jít o život ohrožující stav.
Další nežádoucí účinky:
Velmi časté nežádoucí účinky (mohou postihnout více než 1 z 10 osob) jsou:
- Snížený počet bílých krvinek nebo červených krvinek
- Únava nebo slabost
- Pocit na zvracení, zvracení, zácpa, průjem
- Necitlivost, pocit brnění nebo svrbění
- Horečka
- Ztráta chuti k jídlu, úbytek tělesné hmotnosti
- Dechové obtíže, kašel
- Bolest kloubů, svalů a zad
- Bolest hlavy
- Vypadávání vlasů
Časté nežádoucí účinky (mohou postihnout až 1 z 10 osob) jsou:
- Snížený počet krevních destiček (který může mít za následek vznik podlitin nebo delší dobu do zastavení krvácení)
- Infekce s horečkou, zánět plic, zimnice
- Rychlý srdeční tep, návaly horka
- Závratě, mrákotné stavy
- Zvýšená tvorba slz, konjunktivitida (zčervenání a bolestivost povrchu oka), krvácení z nosu
- Dehydratace, sucho v ústech, opary, moučnivka, trávicí potíže, pálení žáhy, bolest nebo nadmutí břicha
- Otoky měkkých tkání, bolesti (především bolest hrudníku, zad a kostí), svalové křeče nebo slabost
- Infekce úst, dýchacího nebo močového ústrojí, bolest při močení
- Bolesti v krku, bolest nebo výtok z nosu, příznaky podobné chřipce, bolest v krku
- Abnormální výsledky jaterních testů, změny v hladinách krevního cukru, bilirubinu, fosfátů, draslíku nebo hořčíku
- Problémy se spánkem, deprese, změněná chuť
- Vyrážka, svědění, potíže s nehty, suchá nebo zarudlá pokožka
- Nadměrné pocení (včetně nočního pocení)
- Zvonění v uších
- Krevní sraženiny v plicích
- Pásový opar
- Kožní otoky, necitlivost rukou a nohou
Méně časté nežádoucí účinky (mohou postihnout až 1 ze 100 osob) jsou:
- Krevní sraženiny
- Abnormální výsledky jaterních testů (hepatotoxicita)
- Selhání ledvin, krev nebo bílkovina v moči
- Rozsáhlý zánět plic, který může vést k zjizvení plicní tkáně
- Zánět slinivky břišní
- Vředy v ústech
Vzácné nežádoucí účinky (mohou postihnout až 1 z 1 000 osob) jsou:
- Závažná porucha srážení krve, která vede k rozsáhlé tvorbě krevních sraženin a k vnitřnímu krvácení.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové
informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek HALAVEN uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a lahvičce za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Tento přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co přípravek HALAVEN obsahuje
- Léčivou látkou je eribulinum. Jedna 2ml injekční lahvička obsahuje eribulini mesilas odpovídající eribulinum 0,88 mg. Jedna 3ml injekční lahvička obsahuje eribulini mesilas odpovídající eribulinum 1,32 mg.
- Dalšími složkami jsou bezvodý ethanol a voda na injekci, s možnou přítomností kyseliny chlorovodíkové a hydroxidu sodného ve velmi malých množstvích.
Jak přípravek HALAVEN vypadá a co obsahuje toto balení
HALAVEN je čirý, bezbarvý vodný injekční roztok dodávaný ve skleněných injekčních lahvičkách o obsahu 2 ml nebo 3 ml roztoku. Jedna krabička obsahuje buď 1, nebo 6 injekčních lahviček.
Držitel rozhodnutí o registraci a výrobce
Eisai Europe Limited
European Knowledge Centre
Mosquito Way
Hatfield
Hertfordshire
AL10 9SN
Velká Británie
+44 (0) 845 676 1400
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgie/Belgique/Belgien
Eisai SA/NV
Tél/Tel: + 32 (0) 2 502 58 04 EunrapuH PharmaSwiss EOOD Ten: + 359 2 895 21 10
Lietuva
UAB „PharmaSwiss”
Tel: + 370 5 2790 762 Luxembourg/Luxemburg
Eisai SA/NV
Tél/Tel: + 32 (0) 2 502 58 04 (Belgique/Belgien)
Česká republika
Eisai GesmbH organizační složka Tel.: + 420 242 485 839 Danmark Eisai AB
Tlf: + 46 (0) 8 501 01 600 (Sverige)
Deutschland Eisai GmbH
Tel: + 49 (0) 69 66 58 50 Eesti
PharmaSwiss Eesti OU Tel. +372 682 7400
EkXáSa Eisai Ltd.
Tqk + 44 (0) 845 676 1400 (Hvropévo Baoí^sro)
Espaňa
Eisai Farmacéutica, S.A.
Tel: + (34) 91 455 94 55
France
Eisai SAS
Tél: + (33) 1 47 67 00 05
Hrvatska
Eisai Ltd.
Tel: + 44 (0) 208 600 1400 (Velika Britanija)
Ireland Eisai Ltd.
Tel: + 44 (0) 208 600 1400 (United Kingdom)
Ísland
Eisai AB
Sími: + 46 (0)8 501 01 600 (Svífrjóó)
Italia
Eisai S.r.l.
Tel: + 39 02 5181401
Kúnpoq
Eisai Ltd.
TnU +44 (0) 845 676 1400 (Hvropévo Baoí^sro)
Latvija
SIA PharmaSwiss Latvia Tel: + 371 67502185
Magyarország
Valeant Pharma Hungary Ltd.
Tel: +36-1-345-5900 Malta
Associated Drug Company Ltd.
Tel: + 356 22778000
Nederland
Eisai B.V.
Tél/Tel: + 31 (0) 900 575 3340 Norge
Eisai AB
Tlf: + 46 (0) 8 501 01 600 (Sverige)
Osterreich
Eisai GesmbH
Tel: + 43 (0) 1 535 1980-0
Polska
VP Valeant Sp. z o.o. Sp.j.
Tel.: +48 (17) 865 51 00 Portugal
Eisai Farmacéutica, Unipessoal Lda Tel: + 351 214 875 540 Románia
Valeant Pharma S.R.L.
Tel: +40 374 102 600
Slovenija
Pharmaswiss d.o.o.
Tel: + 386 (0) 1 2364 700
Slovenská republika
Eisai GesmbH organizační složka Tel.: + 420 242 485 839 (Česká republika)
Suomi/Finland Eisai AB
Puh/Tel: + 46 (0) 8 501 01 600 (Ruotsi)
Sverige
Eisai AB
Tel: + 46 (0) 8 501 01 600
United Kingdom
Eisai Ltd.
Tel: + 44 (0) 845 676 1400
Tato příbalová informace byla naposledy revidována
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky: http://www.ema.europa.eu.
PŘÍLOHA IV
VĚDECKÉ ZÁVĚRY A ZDŮVODNĚNÍ ZMĚNY V REGISTRACI
S ohledem na hodnotící zprávu výboru PRAC týkající se pravidelně aktualizované zprávy o bezpečnosti (PSUR) eribulinu dospěl výbor CHMP k těmto vědeckým závěrům:
V průběhu kumulativního hodnocení příhod závažných kožních reakcí byly zjištěny tři případy Stevens-Johnsonova syndromu (SJS), u nichž byla příčinná souvislost posouzena jako přinejmenším možná. Dvě z těchto hlášení měly přiměřenou dobu do propuknutí a byly potvrzeny biopsií. Ve všech třech případech byly souběžně podávány medikace, u kterých se v souhrnu údajů o přípravku mezi nežádoucími účinky uvádějí SJS a toxická epidermální nekrolýza (TEN). Vzhledem k okolnostem se však považuje za méně pravděpodobné, že by tyto léky vyvolaly tyto nežádoucí účinky. Na základě těchto tří případů SJS a vzhledem k možnému ohrožení života účinkem SJS/TEN a jeho možným závažným následkům se doporučuje aktualizace bodu 4.8 souhrnu údajů o přípravku.
Vzhledem k údajům uvedeným v revidované PSUR proto zastává výbor PRAC názor, že změny údajů o přípravku léčivých přípravků obsahujících eribulin jsou opodstatněné.
Výbor CHMP souhlasí s vědeckými závěry výboru PRAC.
Zdůvodnění změny v registraci
Na základě vědeckých závěrů týkajících se eribulinu výbor CHMP zastává stanovisko, že poměr přínosů a rizik léčivého přípravku obsahujícího eribulin zůstává nezměněný, a to pod podmínkou, že v informacích o přípravku budou provedeny navrhované změny.
Výbor CHMP doporučuje změnu v registraci.
37