Gazyvaro 1000 Mg
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Gazyvaro 1000 mg koncentrát pro infuzní roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička se 40 ml koncentrátu obsahuje obinutuzumabum 1000 mg, což odpovídá koncentraci 25 mg/ml před naředěním.
Obinutuzumab je humanizovaná monoklonální protilátka anti-CD20 II. typu podtřídy IgG1 získaná humanizací původní myší protilátky B-Ly1 a produkovaná technologií rekombinantní DNA na buněčných liniích ovaria čínského křečka.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Koncentrát pro infuzní roztok.
Čirá, bezbarvá až lehce nahnědlá tekutina.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Chronická lymfocytární leukemie (CLL)
Přípravek Gazyvaro je v kombinaci s chlorambucilem indikován k léčbě dospělých pacientů s dříve neléčenou chronickou lymfocytární leukémií (CLL) a s komorbiditami, v důsledku kterých nemohou podstoupit léčbu na bázi fludarabinu v plné dávce (viz bod 5.1).
Folikulární lymfom (FL)
Přípravek Gazyvaro v kombinaci s bendamustinem následovaný udržovací léčbou přípravkem Gazyvaro je indikován k léčbě pacientů s folikulárním lymfomem (FL), kteří neodpověděli na léčbu nebo u kterých došlo k progresi onemocnění v průběhu léčby nebo do 6 měsíců po léčbě rituximabem nebo režimem obsahujícím rituximab.
4.2 Dávkování a způsob podání
Přípravek Gazyvaro se podává pod pečlivým dohledem zkušeného lékaře a v prostředí, kde je okamžitě dostupné plné vybavení pro resuscitaci.
Dávkování
Profylaxe apremedikace syndromu nádorového rozpadu (TLS)
U pacientů s velkou nádorovou masou a/nebo s vysokým počtem cirkulujících lymfocytů (> 25 x 109/l) a/nebo poruchou funkce ledvin (CrCl < 70 ml/min) je třeba zvážit riziko TLS a má být podána profylaxe.
Profylaxe má spočívat v odpovídající hydrataci a podávání urikostatik (např. alopurinol), nebo v podání jiné vhodné léčby, jako jsou urátoxidázy (např. rasburikáza), počínaje 12-24 hodin před zahájením infuze přípravku Gazyvaro dle běžné praxe (viz bod 4.4). Pokud to bude považováno za vhodné, profylaxe by měla být podávána pacientům před každou následnou infuzí.
Profylaxe a premedikace reakcí souvisejících s infuzí (IRR)
Premedikace ke snížení rizika vzniku reakcí souvisejících s infuzí je uvedena v tabulce 1 a 2 (viz také bod 4.4). Premedikace kortikosteroidy je doporučována u pacientů s FL a povinná u pacientů s CLL v prvním cyklu (viz tabulka 1). Podávání premedikace u následných infuzí a další premedikace je popsáno níže.
V průběhu podávání intravenózní infuze přípravku Gazyvaro se může objevit hypotenze (jako příznak IRR). Proto je třeba po dobu 12 hodin před podáním a v průběhu každé infuze přípravku Gazyvaro a po dobu první hodiny po jejím podání nepodávat antihypertenzní léčbu (viz bod 4.4).
Tabulka 1 Premedikace, která se podává před infuzí přípravku Gazyvaro ke snížení rizika vzniku reakcí souvisejících s infuzí u pacientů s CLL (viz bod 4.4).
|
Den cyklu |
Pacienti, kteří vyžadují premedikaci |
Premedikace |
Podání |
|
Cyklus 1: Den 1 |
Všichni pacienti |
Intravenózní kortikosteroidy1 (povinné) |
Podat (dokončit podávání) alespoň 1 hodinu před podáním infuze přípravku Gazyvaro |
|
Perorální analgetika/antipyretika2 |
Podat alespoň 30 minut před podáním infuze přípravku Gazyvaro | ||
|
Antihistaminika3 | |||
|
Cyklus 1: |
Všichni pacienti |
Intravenózní kortikosteroidy1 (povinné) |
Podat (dokončit podávání) alespoň 1 hodinu před podáním infuze přípravku Gazyvaro |
|
Den 2 |
Perorální analgetika/antipyretika2 |
Podat alespoň 30 minut před podáním infuze přípravku Gazyvaro | |
|
Antihistaminika3 | |||
|
Pacienti bez IRR při předchozí infuzi |
Perorální analgetika/antipyretika2 |
Podat alespoň 30 minut | |
|
Pacienti s IRR (stupeň 1 nebo 2) při předchozí infuzi |
Perorální analgetika/antipyretika2 Antihistaminika3 |
před podáním infuze přípravku Gazyvaro | |
|
Všechny následné infuze |
Pacienti s IRR stupně 3 při předchozí infuzi NEBO Pacienti s počtem lymfocytů >25 x 10 /l před podáním následující léčby |
Intravenózní kortikosteroidy1 |
Podat (dokončit podávání) alespoň 1 hodinu před podáním infuze přípravku Gazyvaro |
|
Perorální analgetika/antipyretika2 Antihistaminika3 |
Podat alespoň 30 minut před podáním infuze přípravku Gazyvaro |
1100 mg prednisonu/prednisolonu nebo 20 mg dexamethasonu nebo 80 mg methylprednisolonu. Hydrokortison se nepodává, protože není při snižování výskytu IRR účinný.
2 např. 1000 mg acetaminofenu/paracetamolu
3 např. 50 mg difenhydraminu
Tabulka 2 Premedikace, která se podává před infuzí přípravku Gazyvaro ke snížení rizika vzniku reakcí souvisejících s infuzí u pacientů s FL (viz bod 4.4).
|
Den léčebného cyklu |
Pacienti, kteří vyžadují premedikaci |
Premedikace |
Podání |
|
Intravenózní kortikosteroidy1 (doporučeno) |
Podat (dokončit podávání) alespoň 1 hodinu před podáním infuze přípravku Gazyvaro | ||
|
Cyklus 1: Den 1 |
Všichni pacienti |
Perorální analgetika/antipyretika2 |
Podat alespoň 30 minut před podáním infuze přípravku Gazyvaro |
|
Antihistaminika3 | |||
|
Pacienti bez IRR při předchozí infuzi |
Perorální analgetika/antipyretika2 |
Podat alespoň 30 minut | |
|
Všechny následné infuze |
Pacienti s IRR (stupeň 1 nebo 2) při předchozí infuzi |
Perorální analgetika/antipyretika2 Antihistaminika3 |
před podáním infuze přípravku Gazyvaro |
|
Pacienti s IRR stupně 3 při předchozí infuzi NEBO Pacienti s počtem lymfocytů >25 x 109/l před podáním následující léčby |
Intravenózní kortikosteroidy1 |
Podat (dokončit podávání) alespoň 1 hodinu před podáním infuze přípravku Gazyvaro | |
|
Perorální analgetika/antipyretika2 Antihistaminika3 |
Podat alespoň 30 minut před podáním infuze přípravku Gazyvaro |
1100 mg prednisonu/prednisolonu nebo 20 mg dexamethasonu nebo 80 mg methylprednisolonu .
Hydrokortison se nepodává, protože není při snižování výskytu IRR účinný.
2 např. 1000 mg acetaminofenu/paracetamolu
3 např. 50 mg difenhydraminu
Dávka
Chronická lymfocytární leukemie (CLL, v kombinaci s chlorambucilem)1
Doporučená dávka přípravku Gazyvaro v kombinaci s chlorambucilem u pacientů s CLL je uvedená v tabulce 3.
Cyklus 1
Doporučená dávka přípravku Gazyvaro v kombinaci s chlorambucilem je 1000 mg podaných v průběhu 1. a 2. dne (den 1 a den 2 nebo při pokračovaní v den 1) a poté 8. a 15. den (den 8 a den 15) prvního 28denního léčebného cyklu. Na infuzi na 1. a 2. den se připraví dva infuzní vaky (100 mg na den 1 a 900 mg na den 2). Pokud je první vak podán bez nutnosti úpravy rychlosti infuze nebo přerušení, je možné podat druhý vak ve stejný den (bez nutnosti odstupu mezi jednotlivými dávkami, bez opakovaného podání premedikace) za předpokladu, že na podání infuze je dostatečná doba a po celou dobu infuze budou k dispozici odpovídající podmínky a lékařský dohled. Pokud v průběhu první 100 mg infuze je rychlost infuze nutné upravit, nebo infuzi přerušit, musí se druhý vak podat následující den.
Cyklus 2 - 6
Doporučená dávka přípravku Gazyvaro v kombinaci s chlorambucilem je 1000 mg podaných 1. den každého cyklu .
Tabulka 3 Dávka přípravku Gazyvaro, která se podává v průběhu šesti cyklů léčby (každý
v délce 28 dnů) u pacientů s CLL
|
Cyklus |
Den léčby |
Dávka přípravku Gazyvaro |
|
Cyklus 1 |
Den 1 |
100 mg |
|
Den 2 (nebo Den 1 pokračování) |
900 mg | |
|
Den 8 |
1000 mg | |
|
Den 15 |
1000 mg | |
|
Cyklus 2-6 |
Den 1 |
1000 mg |
1Další informace o dávce chlorambucilu viz bod 5.1
Trvání léčby
Šest cyklů léčby, každý v délce 28 dnů.
Opoždění nebo vynechání dávky
Pokud dojde k vynechání plánované dávky přípravku Gazyvaro, je třeba ji podat co nejdříve je to možné, nečeká se až do podání další plánované dávky. Je však třeba zachovat plánovaný interval mezi jednotlivými dávkami přípravku Gazyvaro.
Folikulární lymfom (FL)
Doporučená dávka přípravku Gazyvaro v kombinaci s bendamustinem u pacientů s FL je uvedena v tabulce 4.
Dávkování (v kombinaci s bendamustinem2)
Cyklus 1
Doporučená dávka přípravku Gazyvaro v kombinaci s bendamustinem je 1000 mg podaných v 1., 8. a 15. den (den 1, den 8, den 15) prvního 28denního léčebného cyklu.
Cyklus 2 - 6
Doporučená dávka přípravku Gazyvaro v kombinaci s bendamustinem je 1000 mg podaných v 1. den každého 28denního léčebného cyklu.
Udržovací léčba
Pacientům, kteří odpoví na úvodní léčbu (tj. 6 počátečních léčebných cyklů) přípravkem Gazyvaro v kombinaci s bendamustinem nebo kteří mají stabilní onemocnění, má být nadále podáván přípravek Gazyvaro 1000 mg jako udržovací léčba v monoterapii jednou za 2 měsíce po dobu dvou let nebo do progrese onemocnění (podle toho, co nastane dříve).
Tabulka 4 Dávka přípravku Gazyvaro podávaná v průběhu 6 léčebných cyklů, každý v
délce 28 dnů, následně udržovací léčba přípravkem Gazyvaro u pacientů s FL
|
Cyklus |
Den léčby |
Dávka přípravku Gazyvaro |
|
Cyklus 1 |
Den 1 |
1 000 mg |
|
Den 8 |
1 000 mg | |
|
Den 15 |
1 000 mg | |
|
Cykly 2-6 |
Den 1 |
1 000 mg |
|
Udržovací léčba |
Jednou za dva měsíce po dobu dvou let nebo do progrese onemocnění (podle toho, co nastane dříve) |
1,000 mg |
2 Další informace o dávce bendamustinu viz bod 5.1
Délka léčby
Šest cyklů léčby, každý v délce 28 dnů, následně udržovací léčba jednou za 2 měsíce po dobu dvou let nebo do progrese onemocnění (podle toho, co nastane dříve).
Opoždění nebo vynechání dávky
Pokud dojde k vynechání plánované dávky přípravku Gazyvaro, je třeba ji podat co nejdříve, nečeká se až do podání další plánované dávky. Během úvodní léčby je však třeba zachovat plánovaný interval mezi jednotlivými dávkami přípravku Gazyvaro. Během udržovací léčby je třeba zachovat původní dávkovací schéma pro následné dávky.
Úprava dávky v průběhu léčby (všechny indikace)
Snížení dávky přípravku Gazyvaro se nedoporučuje.
Léčba symptomatických nežádoucích účinků (včetně IRR), viz odstavec níže (Léčba IRR nebo bod 4.4).
Zvláštní populace
Starší pacienti
U starších pacientů není nutná žádná úprava dávky (viz bod 5.2).
Porucha funkce ledvin
U pacientů s lehkou až středně těžkou poruchou funkce ledvin (clearance kreatininu [CrCl] 30-89 ml/min) není nutná žádná úprava dávky (viz bod 5.2). Bezpečnost a účinnost přípravku Gazyvaro nebyla stanovena u pacientů s těžkou poruchou funkce ledvin (CrCl < 30 ml/min).
Porucha funkce jater
Bezpečnost a účinnost přípravku Gazyvaro u pacientů s poruchou funkce jater nebyla stanovena.
Nelze proto uvést žádná doporučení pro dávkování.
Pediatrická populace
Bezpečnost a účinnost přípravku Gazyvaro u dětí a dospívajících mladších 18 let nebyla stanovena. Nejsou dostupné žádné údaje.
Způsob podání
Přípravek Gazyvaro je určen k intravenóznímu podání. Podává se po naředění (viz bod 6.6) intravenózní infuzí prostřednictvím speciální linky. Infuze přípravku Gazyvaro se nesmí podávat formou intravenózní injekce nebo bolusu.
Instrukce týkající se ředění přípravku Gazyvaro před podáním jsou uvedené v bodě 6.6.
Instrukce týkající se rychlosti infuze jsou uvedeny v tabulkách 5-6.
Tabulka 5 Standardní rychlost infuze při absenci reakcí souvisejících s infuzí/hypersenzitivity u pacientů s CLL (v případě reakcí souvisejících s infuzí, viz Léčba IRR)
|
Cyklus |
Den léčby |
Rychlost infuze |
|
Cyklus 1 |
Den 1 (100 mg) |
Podává se rychlostí 25 mg/hodinu po dobu 4 hodin. Nezvyšujte rychlost infuze. |
|
Den 2 (nebo Den 1 pokračování) (900 mg) |
Pokud se v průběhu předchozí infuze nevyskytly reakce související s infuzí, podává se rychlostí 50 mg/hodinu. Rychlost infuze lze zvýšit o 50 mg/hodinu každých 30 minut až k maximální rychlosti 400 mg/hodinu. | |
|
Den 8 (1 000 mg) |
Pokud se v průběhu předchozí infuze nevyskytly reakce související s infuzí, kdy konečná rychlost infuze byla 100 mg/hodinu nebo rychlejší, infuzi lze zahájit rychlostí 100 mg/hodinu a zvyšovat o 100 mg/hodinu každých 30 minut až k maximální rychlosti 400 mg/hodinu. | |
|
Den 15 (1 000 mg) | ||
|
Cyklus 2-6 |
Den 1 (1 000 mg) |
Tabulka 6 Standardní rychlosti infuze při absenci reakcí souvisejících s
infuzí/hypersenzitivity u pacientů s FL (v případě reakcí souvisejících s infuzí, viz Léčba IRR)
|
Cyklus |
Den léčby |
Rychlost infuze |
|
Cyklus 1 |
Den 1 (1 000 mg) |
Podává se rychlostí 50 mg/hodinu. Rychlost infuze lze zvýšit o 50 mg/hodinu každých 30 minut až k maximální rychlosti 400 mg/hodinu. |
|
Den 8 (1 000 mg) |
Pokud se v průběhu předchozí infuze nevyskytly reakce související s infuzí, kdy konečná rychlost infuze byla 100 mg/hodinu nebo rychlejší, infuzi lze zahájit rychlostí 100 mg/hodinu a zvyšovat o 100 mg/hodinu každých 30 minut až k maximální rychlosti 400 mg/hodinu. | |
|
Den 15 (1 000 mg) | ||
|
Cykly 2-6 |
Den 1 (1 000 mg) | |
|
Udržovací léčba |
Jednou za dva měsíce po dobu dvou let nebo do progrese onemocnění (podle toho, co nastane dříve) |
Léčba IRR (všechny indikace)
Léčba IRR může vyžadovat dočasné přerušení infuze, snížení rychlosti infuze nebo ukončení léčby
přípravkem Gazyvaro, jak je uvedeno níže (viz rovněž bod 4.4).
• Stupeň 4 (život ohrožující): Infuze musí být zastavena a léčba trvale ukončena.
• Stupeň 3 (závažné): Infuze musí být dočasně zastavena a příznaky léčeny. Po odeznění příznaků může být infuze znovu zahájena s maximálně poloviční rychlostí, než byla rychlost předchozí (rychlost, kterou kapala infuze v době výskytu IRR), a pokud se žádné příznaky IRR znovu neobjeví, může se rychlost infuze postupně zvyšovat v přírůstcích a intervalech odpovídajících dané dávce (viz tabulky 5 a 6). U pacientů s CLL, u kterých se první dávka prvního cyklu rozděluje na dva dny, lze první den po jedné hodině rychlost infuze zvýšit zpět až na 25 mg/hodinu, ale ne více. Pokud se IRR stupně 3 objeví podruhé, infuze musí být zastavena a léčba trvale ukončena.
• Stupeň 1-2 (lehké až středně těžké): Rychlost infuze musí být snížena a příznaky léčeny. Po odeznění příznaků, a pokud se žádné příznaky znovu neobjevují, lze v infuzi pokračovat, rychlost infuze lze zvyšovat v přírůstcích a intervalech odpovídajících dané dávce (viz tabulky 5 a 6). U pacientů s CLL, u kterých se první dávka prvního cyklu rozděluje na dva dny, lze první den po jedné hodině rychlost infuze zvýšit zpět až na 25 mg/hodinu, ale ne více.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Z důvodu snadnější zpětné zjistitelnosti biologických léčivých přípravků mají být obchodní název a
číslo šarže podávaného přípravku zřetelně zaznamenány (nebo vyznačeny) v pacientově dokumentaci.
Reakce související s infuzí (IRR)
Nej častěji pozorované nežádoucí účinky u pacientů léčených přípravkem Gazyvaro jsou IRR, které se objevují převážně v průběhu první 1000mg infuze. IRR mohou mít souvislost se syndromem uvolňování cytokinů, který byl také hlášen u pacientů léčených přípravkem Gazyvaro. U pacientů s CLL, kteří obdrželi kombinovaná opatření k prevenci IRR (adekvátní kortikosteroidy, perorální analgetikum/antihistaminikum, vynechání antihypertenzní léčby ráno první den infuze a podání první dávky prvního cyklu (cyklus 1 den 1) rozděleně ve dvou dnech), jak jsou popsaná v bodě 4.2, bylo pozorováno snížení incidence všech stupňů IRR. Výskyt IRR stupně 3-4 (který byl založený na relativně malém počtu pacientů) byl podobný před i po zavedení zmírňujících opatření. Podle těchto opatření ke zmírnění/snížení výskytu IRR je třeba postupovat (viz bod 4.2). Incidence a závažnost symptomů souvisejících s infuzí výrazně klesá po první 1000mg infuzi, přičemž většina pacientů nemá při podání dalších infuzí přípravku Gazyvaro již žádné IRR (viz bod 4.8).
U většiny pacientů, bez ohledu na indikaci, byly IRR lehké až středně závažné a bylo možné je zvládnout zpomalením nebo dočasným zastavením první infuze. Byly však rovněž hlášeny závažné a život ohrožující IRR, které vyžadovaly symptomatickou léčbu. IRR mohou být klinicky nerozlišitelné od alergických reakcí zprostředkovaných imunoglobulinem E (IgE) (např. anafylaxe). Pacienti s velkou masou tumoru a/nebo vysokým počtem cirkulujících lymfocytů u CLL (> 25 x 109/l) mohou mít vyšší riziko závažných IRR. Pacienti s poruchou funkce ledvin (CrCl < 50 ml/min) a pacienti s CIRS (Cumulative Illness Rating Scale) > 6 a zároveň CrCl < 70 ml/min mají rovněž vyšší riziko IRR, včetně závažných IRR (viz bod 4.8).
U pacientů, u nichž byly zaznamenány IRR, je třeba infuzi podávat podle stupně této reakce. U IRR stupně 4 musí být infuze okamžitě zastavena a léčba trvale ukončena. U pacientů s IRR stupně 3 musí být infuze dočasně zastavena a zavedena odpovídající léčba příznaků. U IRR stupně 1-2 musí být infuze zpomalena a příznaky vhodně léčeny. Po odeznění příznaků je možné infuzi znovu podat (s výjimkou IRR stupně 4) a to maximálně poloviční rychlostí v porovnání s rychlostí původní. Pokud se u pacienta neobjeví znovu stejné IRR stejné závažnosti, je možné rychlost infuze postupně zvyšovat v přírůstcích a intervalech odpovídajících dané dávce. Pokud předchozí rychlost infuze u pacientů s CLL nebyla dobře tolerována, je třeba pro následné cykly postupovat podle instrukcí týkajících se rychlosti infuze pro 1. a 2. den 1. cyklu (cyklus 1, den 1 a den 2) (viz tabulka 5 v bodě 4.2).
Pacientům nesmí být podána další infuze přípravku Gazyvaro, pokud se u nich objeví:
• akutní život ohrožující respirační příznaky
• IRR stupně 4 (tj. život ohrožující) nebo
• druhý výskyt IRR stupně 3 (prolongované/rekurentní) (po obnovení první infuze nebo v průběhu následující infuze).
Pacienti, kteří mají základní srdeční nebo plicní onemocnění, musí být v průběhu podávání infuze i v období po jejím podání pečlivě monitorováni. V průběhu intravenózní infuze přípravku Gazyvaro se může objevit hypotenze. Proto je třeba zvážit vynechání antihypertenzní léčby po dobu 12 hodin před podáním a v průběhu podání infuze a po dobu první hodiny po jejím podání. Pacienti s akutním rizikem hypertenzní krize musí být vyšetření s ohledem na přínos a rizika vynechání jejich antihypertenzní léčby.
Hypersenzitivní reakce včetně anafylaxe
U pacientů léčených přípravkem Gazyvaro byla hlášena anafylaxe. Hypersenzitivita může být obtížně odlišitelná od IRR. Pokud je podezření na hypersenzitivní reakci v průběhu infuze (např. příznaky typicky se vyskytující po předchozí expozici a pouze velmi vzácně při první infuzi), musí být infuze zastavena a léčba trvale ukončena. Pacienti se známou hypersenzitivitou na obinutuzumab zprostředkovanou IgE nesmí být léčeni (viz bod 4.3).
Syndrom nádorového rozpadu (TLS)
Při podávání přípravku Gazyvaro byl hlášen syndrom nádorového rozpadu (TLS). Pacienti, u kterých je pravděpodobné riziko TLS (např. pacienti s velkou nádorovou masou a/nebo s vysokým počtem cirkulujících lymfocytů [ > 25 x 109/l] a/nebo s poruchou funkce ledvin [ CrCl < 70 ml/min]), musí dostat profylaxi. Profylaxe má spočívat v odpovídající hydrataci a podávání urikostatik (např. alopurinol), nebo v podání jiné vhodné léčby, jako jsou urátoxidázy (např. rasburikáza), počínaje 1224 hodin před zahájením infuze přípravku Gazyvaro dle běžné praxe (viz bod 4.2). Všichni pacienti považovaní za rizikové mají být v průběhu prvních dnů léčby pečlivě sledováni se zaměřením obzvláště na funkce ledvin, na hodnoty draslíku a kyseliny močové. Měla by být dodržována veškerá další opatření v souladu s běžnou praxí. Další léčba TLS, v závislosti na indikaci, zahrnuje úpravu elektrolytové rovnováhy, monitorování renálních funkcí a vodní bilance a podání podpůrné léčby.
Neutropenie
Při léčbě přípravkem Gazyvaro byla hlášena závažná a život ohrožující neutropenie, včetně febrilní neutropenie. Pacienti, u kterých se objeví neutropenie, musí být pečlivě monitorováni a podstupovat pravidelné laboratorní testy až do odeznění příznaků. Pokud je léčba nutná, je třeba ji podávat podle místních doporučení a je třeba zvážit podání faktoru stimulujícího kolonie granulocytů (G-CSF). Jakékoli známky současné infekce je třeba odpovídajícím způsobem léčit. V případě závažné nebo život ohrožující neutropenie je třeba zvážit odložení dávky. U pacientů se závažnou neutropenií trvající déle než 1 týden se důrazně doporučuje podání antimikrobiální profylaxe po celou dobu trvání léčby až do dosažení stupně 1 nebo 2. Je třeba také zvážit protivirovou a antimykotickou profylaxi (viz bod 4.2). Byly rovněž zaznamenány případy pozdního nástupu neutropenie (objevující se >28 dní po ukončení léčby) nebo prolongované neutropenie (trvající déle než 28 dní po završení/ukončení léčby). Vyšší riziko výskytu neutropenie je u pacientů s poruchou funkce ledvin (CrCl < 50 ml/min) (viz bod 4.8).
Trombocytopenie
V průběhu léčby přípravkem Gazyvaro byla pozorována závažná a život ohrožující trombocytopenie, včetně akutní trombocytopenie (objevující se během 24 hodin po podání infuze). Vyšší riziko výskytu trombocytopenie je u pacientů s poruchou funkce ledvin (CrCl < 50 ml/min) (viz bod 4.8). V průběhu prvního cyklu léčby přípravkem Gazyvaro byly rovněž zaznamenány případy fatální hemoragie. Zřejmý vztah mezi trombocytopenií a hemoragickými příhodami nebyl stanoven.
Pacienti mají být pro trombocytopenii pečlivě monitorováni, zejména v průběhu prvního cyklu; je třeba provádět pravidelné laboratorní testy až do odeznění příznaků a v případě těžké nebo život ohrožující trombocytopenie je třeba zvážit odložení dávky. Krevní transfuze (např. transfuze krevních destiček) je podle lokální praxe na uvážení ošetřujícího lékaře. Je třeba vzít také v úvahu (zejména u prvního cyklu léčby) použití jakékoli souběžné léčby, která by mohla zhoršit příhody související s trombocytopenií (jako jsou antiagregancia a antikoagulancia).
Zhoršení preexistujícího kardiálního onemocnění
U pacientů se základním srdečním onemocněním byly při léčbě přípravkem Gazyvaro zaznamenány arytmie (jako je fibrilace síní a tachyarytmie), angina pectoris, akutní koronární syndrom, infarkt myokardu a srdeční selhání (viz bod 4.8). Tyto příhody se mohou vyskytnout jako součást IRR a mohou být fatální. Proto je třeba pacienty s kardiální anamnézou pečlivě sledovat. Tito pacienti mají být navíc hydratováni s opatrností, aby se předešlo možné nadměrné zátěži tekutinami (fluid overload).
Infekce
Přípravek Gazyvaro se nemá podávat při známkách akutní infekce a u pacientů s anamnézou rekurentních nebo chronických infekcí je třeba postupovat s opatrností. V průběhu léčby přípravkem Gazyvaro a po jejím ukončení se mohou objevit závažné bakteriální, mykotické a nové nebo reaktivované virové infekce. Byly hlášeny i fatální případy. Vyšší riziko vzniku infekcí, včetně závažných infekcí, je u pacientů s CIRS > 6 a zároveň CrCl < 70 ml/min (viz bod 4.8).
Reaktivace hepatitidy B
U pacientů léčených protilátkami anti-CD20, včetně přípravku Gazyvaro, byla hlášena reaktivace viru hepatitidy B (HBV), která v některých případech vedla k fulminantní hepatitidě, jaternímu selhání a úmrtí (viz bod 4.8). Před zahájením léčby přípravkem Gazyvaro je třeba u všech pacientů provést screening viru hepatitidy B. Ten zahrnuje přinejmenším vyšetření povrchového antigenu viru hepatitidy B (HBsAg) a protilátky proti HBcAg (HBcAb). Tato vyšetření lze doplnit dalšími odpovídajícími markery podle lokálních doporučení. Pacienti s aktivní hepatitidou B nemají být přípravkem Gazyvaro léčeni. Pacienti s pozitivní serologií na hepatitidu B musí před zahájením léčby konzultovat hepatologa a musí být pečlivě monitorováni a léčeni podle lokálních doporučení pro prevenci reaktivace hepatitidy.
Progresivní multifokální leukoencefalopatie (PML)
U pacientů léčených přípravkem Gazyvaro byla hlášena progresivní multifokální leukoencefalopatie (PML) (viz bod 4.8). Diagnózu PML je třeba zvážit u všech pacientů, u kterých se objeví nové neurologické příznaky, nebo se změní příznaky již existujícího neurologického onemocnění. Příznaky PML jsou nespecifické a mohou se lišit v závislosti na postižené oblasti mozku. Běžné jsou motorické příznaky odpovídající zasažení pyramidové dráhy (tractus corticospinalis) (např. svalová slabost, paréza nebo senzorické poruchy), poruchy čití, cerebelární příznaky a poruchy zrakového pole. Mohou se objevit některé subjektivní/objektivní příznaky odpovídající kortikálnímu poškození (např. afázie nebo poruchy vizuálně-prostorové orientace). Vyšetření PML zahrnuje zejména konzultaci neurologa, vyšetření mozku magnetickou rezonancí (MRI) a lumbální punkci (vyšetření cerebrospinálního moku na přítomnost DNA viru JC). Léčba přípravkem Gazyvaro má být v průběhu vyšetřování možného PML přerušena a pokud se diagnóza potvrdí, pak trvale ukončena. Je třeba rovněž zvážit přerušení nebo snížení veškeré souběžně podávané chemoterapie nebo imunosupresivní terapie. Pacienti mají být k vyšetření a léčbě PML odesláni k neurologovi.
Imunizace
Bezpečnost imunizace živými nebo oslabenými virovými vakcínami po léčbě přípravkem Gazyvaro nebyla hodnocena a vakcinace živými virovými vakcínami se proto v průběhu léčby a do upravení stavu B-lymfocytů nedoporučuje.
Expozice obinutuzumabu in utero a vakcinace kojenců živými virovými očkovacími látkami
Vzhledem k možné depleci B-lymfocytů u dětí matek, které byly vystaveny přípravku Gazyvaro v průběhu těhotenství, mají být kojenci monitorováni s ohledem na depleci B-lymfocytů a vakcinace živými virovými očkovacími látkami má být odložena až do úpravy počtu B-lymfocytů. Bezpečnost a načasování očkování má být projednáno s dětským lékařem (viz bod 4.6).
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné formální studie lékových interakcí, i když omezené podstudie lékových interakcí byly provedeny u přípravku Gazyvaro s bendamustinem, CHOP (cyklofosfamid, doxorubicin, vinkristin, prednison), FC (fludarabin, cyklofosfamid) a chlorambucilem. Riziko vzniku interakcí s jinými současně užívanými léčivými přípravky nelze vyloučit.
Farmakokinetické interakce
Obinutuzumab není substrátem, inhibitorem ani induktorem enzymů cytochromu P450 (CYP450), uridin-difosfát-glukuronyltransferázy (UGT) ani transportérů, jako je glykoprotein P. Proto se při podání s léčivými přípravky, o kterých je známo, že jsou metabolizovány těmito enzymatickými systémy, žádné farmakokinetické interakce neočekávají.
Přípravek Gazyvaro neměl při jejich současném podávání žádný účinek na farmakokinetiku bendamustinu, FC, chlorambucilu nebo jednotlivé složky CHOP. Kromě toho neexistují žádné zřetelné účinky bendamustinu, FC, chlorambucilu nebo CHOP na farmakokinetiku přípravku Gazyvaro.
Farmakodynamické interakce
V průběhu léčby a do úpravy počtu B-lymfocytů se nedoporučuje vakcinace živými virovými očkovacími látkami z důvodu imunosupresivního účinku obinutuzumabu (viz bod 4.4).
Kombinace obinutuzumabu s chlorambucilem nebo bendamustinem může zvýraznit neutropenii (viz bod 4.4).
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku.
Ženy ve fertilním věku musí v průběhu léčby přípravkem Gazyvaro a po dobu 18 měsíců po ukončení léčby přípravkem Gazyvaro používat účinnou antikoncepci.
Studie reprodukce u makaků druhu cynomolgus neprokázaly embryofetální toxicitu ani teratogenní účinky, ale vedly ke kompletní depleci B-lymfocytů u mláďat. Počty B-lymfocytů se u mláďat vrátily k normálním hodnotám a imunologické funkce se upravily v průběhu 6 měsíců po porodu. Sérové koncentrace obinutuzumabu u mláďat 28 dnů po porodu byly podobné koncentracím pozorovaným u matek. Koncentrace v mléce byly ve stejný den velmi nízké, což naznačuje, že obinutuzumab prochází placentou (viz bod 5.3). Údaje o podávání obinutuzumabu těhotným ženám nejsou k dispozici. Přípravek Gazyvaro se nemá podávat těhotným ženám, pokud možný prospěch z léčby nepřeváží její potenciální rizika.
Při expozici během těhotenství lze v důsledku farmakologických vlastností přípravku u kojenců očekávat depleci B-lymfocytů. Má být zváženo odložení očkování živými očkovacími látkami u kojenců narozených matkám, které byly vystaveny přípravku Gazyvaro v průběhu těhotenství, dokud se hladina B-lymfocytů u kojenců nedostane do normálního rozmezí (viz bod 4.4).
Kojení
Studie na zvířatech prokázaly vylučování obinutuzumabu do mateřského mléka (viz bod 5.3).
Protože lidský imunoglobulin G (IgG) je vylučován do mateřského mléka a není znám potenciál absorpce a poškození novorozence a kojence, mají být ženy poučeny, aby v průběhu léčby přípravkem Gazyvaro a po dobu 18 měsíců po podání poslední dávky přípravku Gazyvaro přerušily kojení.
Fertilita
Specifické studie hodnotící účinky obinutuzumabu na fertilitu u zvířat nebyly provedeny. Ve studiích toxicity po opakovaném podávání u makaků rodu cynomolgus nebyly pozorovány žádné nežádoucí účinky na samčí ani samičí reprodukční orgány (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek Gazyvaro nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje. V průběhu první infuze přípravku Gazyvaro se velmi často objevují IRR. Pacienti, u kterých se příznaky související s podáním infuze objeví, musí být poučeni, aby neřídili dopravní prostředky ani neobsluhovali stroje, dokud tyto příznaky nevymizí.
4.8 Nežádoucí účinky
Shrnutí bezpečnostního profilu
Nežádoucí účinky popsané v tomto bodě byly zaznamenány v průběhu léčby a následného sledování ve dvou klíčových klinických studiích, BO21004/CLL11, N=781, a GAO48753g, N=396, u dříve neléčených pacientů s CLL a pacientů s indolentním non-Hodgkinským lymfomem (iNHL) (81,1 % pacientů mělo FL), kteří neodpověděli na léčbu nebo u kterých došlo k progresi onemocnění v průběhu léčby nebo až 6 měsíců po ukončení léčby rituximabem nebo režimy obsahujícími rituximab. Tyto studie zkoumaly přípravek Gazyvaro v kombinaci s různými chemoterapeutickými látkami (chlorambucil k léčbě CLL, bendamustin k léčbě iNHL) a jako udržovací monoterapii (pouze u iNHL). Protokol studie GAO4753g definoval pacienty s iNHL, včetně FL, jako sledovanou populaci. Proto analýza nežádoucích účinků uvedených v následujícím textu byla provedena v celé populaci (tj. iNHL), za účelem zajištění co nejkomplexnější informace o bezpečnosti.
Tabulka 7 shrnuje nežádoucí účinky, které se vyskytly s vyšší incidencí (rozdíl > 2 %) u pacientů s CLL léčených kombinací přípravek Gazyvaro plus chlorambucil v porovnání se skupinou léčenou chlorambucilem samotným nebo kombinací rituximab plus chlorambucil (studie BO21004/CLL11) a u pacientů s iNHL léčených přípravkem Gazyvaro plus bendamustin, s následnou udržovací léčbou přípravkem Gazyvaro u některých pacientů, v porovnání se skupinou léčenou bendamustinem samotným (studie GAO4753g).
Četnosti jsou definovány jako velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1000 až < 1/100), vzácné (> 1/10 000 až < 1/1000) a velmi vzácné (< 1/10 000). V každé kategorii četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
Shrnutí nežádoucích účinků do tabulky
Tabulka 7
Souhrn nežádoucích účinků hlášených s vyšší incidencí (rozdíl > 2 %) u pacientů# léčených kombinací přípravek Gazyvaro + chemoterapie
|
Četnost |
Všechny stupně Gazyvaro + chlorambucil nebo Gazyvaro + bendamustin (úvodní léčba) s následnou udržovací léčbou přípravkem Gazyvaro |
Stupně 3-5Ť Gazyvaro + chlorambucil nebo Gazyvaro + bendamustin (úvodní léčba) s následnou udržovací léčbou přípravkem Gazyvaro |
|
Infekce a infestace | ||
|
Velmi časté |
Infekce horních cest dýchacích, sinusitida | |
|
Časté |
Infekce močových cest, nazofaryngitida, herpes simplex, rýma, faryngitida, plicní infekce, chřipka |
Infekce močových cest |
|
Méně časté |
Nazofaryngitida | |
|
Novotvary benigní, maligní a blíže neurčené (zahrnující cysty a polypy) | ||
|
Časté |
Spinocelulární karcinom kůže |
Spinocelulární karcinom kůže |
|
Poruchy krve a lymfatického systému | ||
|
Velmi časté |
Neutropenie, trombocytopenie, anémie |
Neutropenie, trombocytopenie |
|
Časté |
Leukopenie, bolest lymfatické uzliny |
Anémie, leukopenie |
|
Poruchy metabolismu a výživy | ||
|
Časté |
Syndrom nádorového rozpadu, hyperurikémie |
Syndrom nádorového rozpadu |
|
Méně časté |
Hyperurikémie | |
|
Psychiatrické poruchy | ||
|
Časté | ||
|
Poruchy oka | ||
|
Časté |
Oční hyperemie | |
|
Četnost |
Všechny stupně Gazyvaro + chlorambucil nebo Gazyvaro + bendamustin (úvodní léčba) s následnou udržovací léčbou přípravkem Gazyvaro |
Stupně 3-5Ť Gazyvaro + chlorambucil nebo Gazyvaro + bendamustin (úvodní léčba) s následnou udržovací léčbou přípravkem Gazyvaro |
|
Srdeční poruchy | ||
|
Časté |
Fibrilace síní, srdeční selhání | |
|
Méně časté |
Fibrilace síní | |
|
Cévní poruchy | ||
|
Časté |
Hypertenze |
Hypertenze |
|
Respirační, hrudní a mediastinální poruchy | ||
|
Velmi časté | ||
|
Časté |
Otok nosní sliznice rýma | |
|
Gastrointestinální poruchy | ||
|
Velmi časté |
Průjem, zácpa | |
|
Časté |
Dyspepsie, kolitida, hemoroidy | |
|
Poruchy kůže a podkožní tkáně | ||
|
Časté |
Alopecie, pruritus, noční pocení, ekzém | |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně | ||
|
Velmi časté |
Artralgie | |
|
Časté |
Bolest zad, muskuloskeletální bolest na hrudi, bolest končetiny, bolest kostí | |
|
Méně časté |
Artralgie, bolest zad, muskuloskeletální bolest na hrudi | |
|
Poruchy ledvin a močových cest | ||
|
Časté |
Dysurie, močová inkontinence | |
|
Celkové poruchy a reakce v místě aplikace | ||
|
Velmi časté |
Pyrexie, astenie | |
|
Časté | ||
|
Méně časté |
Pyrexie | |
|
Vyšetření | ||
|
Časté |
Pokles počtu bílých krvinek, pokles počtu neutrofilů, nárůst tělesné hmotnosti |
Pokles počtu bílých krvinek, pokles počtu neutrofilů |
|
Poranění, otravy a procedurální komplikace | ||
|
Velmi časté |
Reakce související s infuzí |
Reakce související s infuzí |
* s vyšším výskytem (rozdíl > 2 % mezi jednotlivými rameny léčby). Je hlášena pouze nejvyšší četnost v klinických studiích (na základě studií BO21004/ dříve neléčená CLL a GAO4753g/ rituximab refraktemí iNHL) t Nebyly pozorovány žádné nežádoucí účinky stupně 5 s rozdílem > 2% mezi jednotlivými rameny léčby
Ve studii GAO4753g, dostávali pacienti v bendamustinovém (B) rameni pouze 6 měsíců trvající úvodní léčbu, zatímco pacienti v rameni s Gazyvarem a bendamustinem pokračovali po úvodní léčbě udržovací léčbou přípravkem Gazyvaro.
Během udržovací fáze studie GAO4753g byly nejčastějšími nežádoucími účinky kašel (15 %), záněty horních cest dýchacích (12 %), neutropenie (11 %), sinusitidy (10 %), průjem (8 %), infuzní reakce (8 %), nevolnost (8 %), únava (8 %), bronchitida (7 %), bolesti kloubů (7 %), horečka (6 %), nasopharyngitida (6 %), záněty močových cest (6 %). Nejčastějšími nežádoucími účinky stupně 3-5 byly neutropenie (10 %), anémie, febrilní neutropenie, trombocytopenie, záněty horních cest dýchacích a močové infekce (vše 1 %).
Profil nežádoucích účinků byl v podskupině pacientů s FL obdobný jako v celé populaci pacientů s iNHL.
Popis vybraných nežádoucích účinků
Reakce související s infuzí (IRR)
Nej častěji hlášenými (>5 %) symptomy v souvislosti s IRR byly nauzea, únava, zimnice, hypotenze, pyrexie, zvracení, dyspnoe, návaly horka, hypertenze, bolest hlavy, tachykardie, závratě a průjem.
Byly rovněž hlášeny respirační a kardiální příznaky jako bronchospasmus, podráždění laryngu a hrdla, sípot, otok laryngu a fibrilace síní (viz bod 4.4).
Chronická lymfocytární leukemie
Incidence IRR byla vyšší v rameni s kombinací přípravek Gazyvaro plus chlorambucil ve srovnání s ramenem s kombinací rituximab plus chlorambucil. Incidence IRR byla 65 % během podání první 1000mg infuze přípravku Gazyvaro (20 % pacientů zaznamenalo IRR stupně 3-5, nebyly hlášeny fatální případy). Celkem 7 % pacientů zaznamenalo IRR vedoucí k přerušení léčby přípravkem Gazyvaro. Incidence IRR u následných infuzí byla 3 %, u druhé 1000mg dávky a poté 1 %. Po první 1000mg infuzi v prvním cyklu nebyly již hlášeny žádné stupně 3-5 IRR.
U pacientů, u kterých byla použita kombinovaná opatření pro prevenci IRR (odpovídající kortikosteroidy, perorální analgetika/antihistaminika, vynechání antihyperteziv ráno před první infuzí a rozložení první infuze v prvním cyklu do dvou dnů) popsaná v bodě 4.2, byl pozorován snížený výskyt IRR všech stupňů závažnosti. Výskyt IRR stupně 3-4 (které se objevily u poměrně malého počtu pacientů) byl podobný před i po zavedení opatření pro snížení jejich výskytu.
Indolentní non-Hodgkinský lymfom včetně folikulárního lymfomu
Celková incidence IRR byla v cyklu 1 vyšší u pacientů v rameni s kombinací přípravek Gazyvaro s bendamustinem (G+B) (55 %) ve srovnání s ramenem s bendamustinem samotným (42 %) (příhody IRR stupně 3-5 byly hlášeny v 9 % resp. 2 % a nebyly hlášeny žádné fatální příhody). U pacientů, kterým byl podáván G+B, byla incidence IRR vyšší v den 1 (38 %) a postupně klesala v den 2 (25 %), den 8 (7 %) a 15 (4 %). Během cyklu 2 byla incidence IRR nižší u pacientů, kterým byl podáván G+B (24 %), ve srovnání s pacienty, kterým byl podáván samotný bendamustin (B) (32 %). Incidence IRR u následných infuzí byla srovnatelná v obou ramenech a s každým cyklem se snižovala. IRR byly také pozorovány u 8 % pacientů v průběhu udržovací léčby přípravkem Gazyvaro. Celkem u 3 % pacientů vedl výskyt reakcí související s infuzí k přerušení léčby přípravkem Gazyvaro.
Neutropenie a infekce
Chronická lymfocytární leukemie
Incidence neutropenie byla vyšší v rameni s kombinací přípravek Gazyvaro plus chlorambucil (41 %) ve srovnání s ramenem s kombinací rituximab plus chlorambucil, přičemž neutropenie odezněla spontánně nebo po podání faktorů stimulujících kolonie granulocytů (G-CSF). Incidence infekce byla 38 % v rameni s kombinací přípravek Gazyvaro plus chlorambucil a 37 % v rameni s kombinací rituximab plus chlorambucil (příhody stupně 3-5 byly hlášeny ve 12 % resp. 14 % a fatální příhody byly hlášeny u < 1 % v obou ramenech léčby). Byly rovněž hlášeny případy prolongované neutropenie (2 % v rameni s kombinací přípravek Gazyvaro plus chlorambucil a 4 % v rameni s kombinací rituximab plus chlorambucil) a pozdního nástupu neutropenie (16 % v rameni s kombinací přípravek Gazyvaro plus chlorambucil a 12 % v rameni s kombinací rituximab plus chlorambucil) (viz bod 4.4).
Indolentní non-Hodgkinský lymfom včetně folikulámího lymfomu
Výskyt neutropenie byl vyšší v rameni s kombinací přípravek Gazyvaro plus bendamustin (G+B)
(38 %) ve srovnání s ramenem s bendamustinem samotným (B) (32 %). Incidence infekce byla 65 % v rameni G+B a 56 % v rameni B (příhody stupně 3-5 byly hlášeny v 18 % resp. 17 % a fatální příhody byly hlášeny u 5 pacientů (3 %) v rameni G+B a u 7 pacientů (4 %) v rameni B).
Byly rovněž hlášeny případy prolongované neutropenie (3 % v rameni G+B) a pozdního nástupu neutropenie (7 % v rameni G+B) (viz bod 4.4).
Trombocytopenie
Chronická lymfocytární leukemie
Incidence trombocytopenie byla vyšší v rameni s kombinací přípravek Gazyvaro plus chlorambucil (15 %) ve srovnání s ramenem s kombinací rituximab plus chlorambucil, zejména v průběhu prvního cyklu léčby. Čtyři procenta pacientů léčených kombinací přípravek Gazyvaro plus chlorambucil prodělalo akutní trombocytopenii (objevila se v průběhu 24 hodin po podání infuze přípravku Gazyvaro) (viz bod 4.4). Celková incidence hemoragických příhod byla podobná v rameni léčby s přípravkem Gazyvaro i v rameni léčby s rituximabem. Počet fatálních hemoragických příhod byl podobný v jednotlivých ramenech léčby, všechny příhody u pacientů léčených přípravkem Gazyvaro však byly hlášeny v průběhu Cyklu 1. Zřejmý vztah mezi trombocytopenií a hemoragickými příhodami nebyl stanoven.
Indolentní non-Hodgkinský lymfom včetně folikulárního lymfomu
Incidence trombocytopenie byla nižší v rameni s kombinací přípravek Gazyvaro plus bendamustin (G+B) (15 %) ve srovnání s ramenem se samotným bendamustinem (B) (24 %). Incidence hemoragických příhod (11 % G+B, 10 % B) a hemoragických příhod stupně 3-5 (5 % G+B, 3 % B) byla podobná v obou ramenech léčby bez hlášených fatálních příhod.
Zvláštní populace
Starší pacienti
Chronická lymfocytární leukemie
V klíčové studii bylo 46 % (156 z 336) pacientů s CLL léčených kombinací přípravek Gazyvaro plus chlorambucil ve věku 75 let nebo starších (medián věku 74 let). Tito pacienti zaznamenávali závažnější nežádoucí účinky a nežádoucí účinky vedoucí k úmrtí častěji než pacienti mladší 75 let.
Indolentní non-Hodgkinský lymfom včetně folikulárního lymfomu
V klíčové studii iNHL bylo 44 % (85 ze 194) pacientů s iNHL léčených kombinací přípravek Gazyvaro plus bendamustin ve věku 65 let nebo starších. Nebyly pozorovány žádné klinicky významné rozdíly v bezpečnosti mezi těmito pacienty a pacienty mladšího věku.
Porucha funkce ledvin
Chronická lymfocytární leukemie
Ve studii CLL11 mělo 27 % (90 z 336) pacientů léčených kombinací přípravek Gazyvaro plus chlorambucil středně těžkou poruchu funkce ledvin (CrCl < 50 ml/min). Tito pacienti zaznamenávali závažnější nežádoucí účinky a nežádoucí účinky vedoucí k úmrtí častěji než pacienti s CrCl > 50 ml/min (viz body 4.2, 4.4 a 5.2). Pacienti s CrCL < 30 ml/min byli vyloučeni ze studie (viz bod 5.1).
Indolentní non-Hodgkinský lymfom včetně folikulárního lymfomu
V klíčové studii s iNHL měla malá část 8 % (15 ze 194) pacientů léčených kombinací přípravek Gazyvaro plus bendamustin středně těžkou poruchu funkce ledvin (CrCl < 50 ml/min). Tito pacienti zaznamenávali závažnější nežádoucí účinky a nežádoucí účinky vedoucí k úmrtí častěji než pacienti s CrCl > 50 ml/min (viz body 4.2 a 5.2). Pacienti s CrCL < 40 ml/min byli vyloučeni ze studie (viz bod 5.1).
Doplňující informace týkající se bezpečnosti získané ze zkušeností z klinických studií
Progresivní multifokální leukoencefalopatie (PML)
U pacientů léčených přípravkem Gazyvaro byla hlášena PML (viz bod 4.4).
Reaktivace hepatitidy B
U pacientů léčených přípravkem Gazyvaro byla hlášena reaktivace hepatitidy B (viz bod 4.4).
Gastrointestinální perforace
Byly hlášeny případy gastrointestinální perforace u pacientů, kterým byl podáván přípravek Gazyvaro, zejména u NHL. V klíčové studii GAO4753g se vyskytly gastrointestinální perforace u 1 % pacientů.
Zhoršení preexistujícího kardiálního onemocnění
U pacientů léčených přípravkem Gazyvaro se objevily případy arytmií (jako je fibrilace síní a tachyarytmie), anginy pectoris, akutního koronálního syndromu, infarktu myokardu a srdečního selhání (viz bod 4.4). Tyto příhody mohou být součástí IRR a mohou být fatální.
Laboratorní abnormality
Krátce po první infuzi přípravku Gazyvaro byly pozorovány přechodné elevace jaterních enzymů (aspartátaminotransferázy (AST), alaninaminotransferázy (ALT) a alkalické fosfatázy).
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Z klinických studií u člověka nejsou žádné zkušenosti s předávkováním. V klinických studiích s přípravkem Gazyvaro byly podávány dávky od 50 mg až do (a včetně) 2000 mg v jedné infuzi. Incidence a intenzita nežádoucích účinků hlášených z těchto studií se nezdála být závislá na dávce.
U pacientů, u kterých dojde k předávkování, je nutné okamžitě zastavit podávání infuze nebo snížit její rychlost, a pacienty pečlivě sledovat. Je třeba věnovat pozornost nutnosti kontroly krevního obrazu a zvýšenému riziku infekce u pacientů s deplecí B-lymfocytů.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Cytostatika, monoklonální protilátky, ATC kód: L01XC15 Mechanismus účinku
Obinutuzumab je rekombinantní humanizovaná monoklonální protilátka anti-CD20 II. typu izotypu IgG1 získaná pomocí inženýrství glykoproteinů (IGP). Váže se specificky na extracelulární smyčku transmembránového antigenu CD20 na povrchu nemaligních i maligních pre-B a zralých B-lymfocytů, neváže se však na hematopoetické kmenové buňky, pro-B buňky, normální plazmatické buňky ani další normální tkáně. IGP Fc části obinutuzumabu vede k vyšší afinitě k FcyRIII receptorům na imunitních efektorových buňkách (jako jsou NK buňky, makrofágy a monocyty) ve srovnání s protilátkami získanými bez IGP.
V neklinických studiích obinutuzumab indukuje přímo buněčnou smrt a zprostředkovává cytotoxicitu závislou na protilátce (ADCC) a fagocytózu závislou na protilátce (ADCP) prostřednictvím náboru (recruitment) FcyRIII pozitivních efektorových buněk. In vivo navíc obinutuzumab zprostředkovává nízký stupeň cytotoxicity závislé na komplementu (CDC). Ve srovnání s protilátkami typu I je obinutuzumab, protilátka typu II, charakterizován zvýrazněním indukce přímé buněčné smrti se současným snížením CDC při stejné dávce. Ve srovnání s protilátkami získanými bez IGP obinutuzumab, protilátka získaná IGP, je při stejné dávce charakterizován zvýšenou cytotoxicitou závislou na protilátce (ADCC) a fagocytóza (ADCP). V modelech na zvířatech obinutuzumab zprostředkovává výraznou depleci B-lymfocytů a protinádorový účinek.
V klíčové klinické studii BO21004/CLL11 mělo 91 % (40 ze 44) hodnotitelných pacientů léčených přípravkem Gazyvaro depleci B buněk (definovanou jako počet CD19+ B buněk < 0,07 x 109/l) na konci období léčby a tato deplece přetrvávala po dobu prvních šesti měsíců období následného sledování. Úprava počtu B buněk byla pozorována v průběhu 12-18 měsíců následného sledování u 35 % (14 ze 40) pacientů bez progrese onemocnění a u 13 % (5 ze 40) pacientů s progresivním onemocněním.
Klinická účinnost a bezpečnost Chronická lymfocytární leukemie
Mezinárodní, multicentrická, otevřená, randomizovaná, dvoustupňová studie fáze II se třemi rameny léčby (BO21004/CLL11) hodnotila účinnost a bezpečnost kombinace přípravek Gazyvaro plus chlorambucil (GClb) ve srovnání s kombinací rituximab plus chlorambucil (RClb) a s monoterapií chlorambucilem (Clb) u dosud neléčených pacientů s chronickou lymfocytární leukémií s komorbiditami.
Před zařazením do studie měli pacienti potvrzenou diagnózu CD20+ CLL a splňovali jednu nebo obě následující lékařské podmínky: kumulativní skóre komorbidit (CIRS) vyšší než 6 nebo snížení funkce ledvin s CrCl < 70 ml/min. Pacienti s poruchou funkce jater (funkční jaterní testy stupně 3 dle NCI-CTCAE, National Cancer Institute - Common Terminology Criteria for Adverse Events) (AST, ALT > 5 x ULN po dobu > 2 týdnů, bilirubin > 3x ULN) a s poruchou funkce ledvin (CrCl < 30 ml/min) byli ze studie vyloučeni. Pacienti s poruchou funkce jednoho nebo více orgánů/systémů skóre 4 podle definice CIRS (s výjimkou očí, uší, nosu, hrdla a laryngu) byli ze studie vyloučeni.
Celkem bylo randomizováno 781 pacientů v poměru 2:2:1 k léčbě kombinací přípravek Gazyvaro plus chlorambucil, rituximab plus chlorambucil nebo samotným chlorambucilem. Stupeň 1a porovnával kombinaci přípravek Gazyvaro plus chlorambucil oproti chlorambucilu samotnému u 356 pacientů a stupeň 2 porovnával kombinaci přípravek Gazyvaro plus chlorambucil oproti kombinaci rituximab plus chlorambucil u 663 pacientů. Výsledky účinnosti jsou shrnuty v tabulce 8 a na obrázcích 1-3.
Většina pacientů dostávala přípravek Gazyvaro intravenózně ve formě 1000 mg úvodní dávky podané 1., 8. a 15. den prvního léčebného cyklu (Cyklus 1, Den 1, 8 a 15). Aby se snížil výskyt reakcí souvisejících s infuzí, bylo později zavedeno doplnění doporučení týkající se podání první dávky přípravku Gazyvaro a 140 pacientů dostalo první dávku přípravku Gazyvaro v průběhu 2 dnů (Den 1 (100 mg) a Den 2 (900 mg)) (viz bod 4.2 a 4.4). V následujících cyklech (Cyklus 2 až 6) dostávali pacienti přípravek Gazyvaro v dávce 1000 mg pouze první den (Den 1). Chlorambucil se podával perorálně v dávce 0,5 mg/kg tělesné hmotnosti 1. a 15. den všech léčebných cyklů (Cyklus 1-6, Den 1 a 15).
Demografické údaje a základní charakteristiky byly rovnoměrně rozloženy mezi jednotlivými skupinami léčby. Většina pacientů byla bílé rasy (95 %) a mužského pohlaví (61 %). Medián věku byl
73 let, přičemž 44 % pacientů bylo ve věku 75 let a starších. V úvodu léčby bylo 22 % pacientů ve stadiu A dle Bineta, 42 % ve stadiu B dle Bineta a 36 % ve stadiu C dle Bineta.
Medián skóre komorbidit byl 8 a 76 % pacientů zařazených do studie mělo skóre komorbidit nad 6. Medián odhadované CrCl byl 62 ml/min a 66 % pacientů mělo CrCl < 70 ml/min. Čtyřicet dva procent pacientů zařazených do studie mělo CrCl < 70 ml/min a zároveň skóre komorbidit > 6. Třicet čtyři procent pacientů bylo zařazeno pouze na základě zvýšeného skóre komorbidit a 23 % pacientů bylo zařazeno pouze na základě poruchy funkce ledvin.
Nejčastěji hlášené koexistující zdravotní komplikace (při cut-off 30 % nebo vyšší) podle MedDRA tříd orgánových systémů byly: cévní poruchy (73 %), srdeční poruchy (46 %), gastrointestinální poruchy (38 %), poruchy metabolismu a výživy (40 %), poruchy ledvin a močových cest (38 %) a poruchy svalové a kosterní soustavy a pojivové tkáně (33 %).
Tabulka 8 Shrnutí účinnosti ze studie BO21004/CLL11
|
Stupeň 1a |
Stupeň 2 | |||
|
Chlorambucil |
Gazyvaro + chlorambucil |
Rituximab + chlorambucil |
Gazyvaro + chlorambucil | |
|
n =118 |
n = 238 |
n = 330 |
n = 333 | |
|
Medián doby sledování 22,8 |
Medián doby sledování 18,7 | |||
|
měsíce |
měsíce | |||
|
Primární cílový parametr | ||||
|
PFS hodnocené zkoušejícím (PFS-INV)a | ||||
|
Počet (%) pacientů s příhodou |
96 (81,4 %) |
93 (39,1 %) |
199 (60,3 %) |
104 (31,2 %) |
|
Medián trvání PFS (měsíce) |
11,1 |
26,7 |
15,2 |
26,7 |
|
Poměr rizik (95% CI) |
0,18 [0,13; 0,24] |
0,39 [0,31; 0,49] | ||
|
p-hodnota (log-rank test, stratifikovanýb) |
< 0,0001 |
< 0,0001 | ||
|
Klíčový sekundární cílový parametr | ||||
|
PFS hodnocené IRC (PFS-IRC)a | ||||
|
Počet (%) pacientů s příhodou |
90 (76,3 %) |
89 (37,4 %) |
183 (55,5 %) |
103 (30,9 %) |
|
Medián trvání PFS (měsíce) |
11,2 |
27,2 |
14,9 |
26,7 |
|
Poměr rizik (95% CI) |
0,19 [0,14; 0,27] |
0,42 [0,33; 0,54] | ||
|
p-hodnota (log-rank test, stratifikovaný ) |
<0,0001 |
<0,0001 | ||
|
Výskyt odpovědi na konci léčby | ||||
|
Počet pacientů zařazených do analýzy |
118 |
238 |
329 |
333 |
|
Respondéři (%) |
37 (31,4 %) |
184 (77,3 %) |
214 (65,0 %) |
261 (78,4 %) |
|
Non-respondéři (%) |
81 (68,6 %) |
54 (22,7 %) |
115 (35,0 %) |
72 (21,6 %) |
|
Rozdíl ve výskytu odpovědi (95% CI) |
45,95 [35,6; 56,3] |
13,33 [6,4; 20,3] | ||
|
p-hodnota (Chi-kvadrát test) |
<0,0001 |
0,0001 | ||
|
Počet kompletních respondérůc (%) |
0 (0,0 %) |
53 (22,3 %) |
23 (7,0 %) |
69 (20,7 %) |
|
Stupeň 1a |
Stupeň 2 | |||
|
Chlorambucil |
Gazyvaro + chlorambucil |
Rituximab + chlorambucil |
Gazyvaro + chlorambucil | |
|
n =118 |
n = 238 |
n = 330 |
n = 333 | |
|
Medián doby sledování 22,8 |
Medián doby sledování 18,7 | |||
|
měsíce |
měsíce | |||
|
Molekulárníremise na konci léčbyd | ||||
|
Počet pacientů zařazených do analýzy |
90 |
168 |
244 |
239 |
|
MRD negativníe (%) |
0 (0 %) |
45 (26,8 %) |
6 (2,5 %) |
61 (25,5 %) |
|
MRD pozitivníf (%) |
90 (100 %) |
123 (73,2 %) |
238 (97,5 %) |
178 (74,5 %) |
|
Rozdíl ve výskytu MRD (95% CI) |
26,79 [19,5; 34,1] |
23,06 [17,0; 29,1] | ||
|
Přežití bez příhody | ||||
|
Počet (%) pacientů s příhodou |
103 (87,3 %) |
104 (43,7 %) |
208 (63,0 %) |
118 (35,4 %) |
|
Medián doby do příhody (měsíce) |
10,8 |
26,1 |
14,3 |
26,1 |
|
Poměr rizik (95% CI) |
0,19 [0,14; 0,25] |
0,43 [0,34; 0,54] | ||
|
p-hodnota (log-rank test, stratifikovaný ) |
<0,0001 |
<0,0001 | ||
|
Doba do zahájení nové léčby leukemie | ||||
|
Počet (%) pacientů s příhodou |
65 (55,1 %) |
51 (21,4 %) |
86 (26,1 %) |
55 (16,5 %) |
|
Medián trvání PFS (měsíce) |
14,8 |
- |
30,8 |
- |
|
Poměr rizik (95% CI) |
0,24 [0,16; 0,35] |
0,59 [0,42; 0,82] | ||
|
p-hodnota (log-rank test, stratifikovaný ) |
<0,0001 |
<0,0018 | ||
|
Celkové přežití | ||||
|
Počet (%) pacientů s příhodou |
24 (20,3 %) |
22 (9,2 %) |
41 (12,4 %) |
28 (8,4 %) |
|
Medián doby do příhody (měsíce) |
NR |
NR |
NR** |
NR** |
|
Poměr rizik (95% CI) |
0,41 [0,23; 0,74] |
0,66 [0,41; 1,06] ** | ||
|
p-hodnota (log-rank test, stratifikovanýb) |
0,0022 |
0,0849** | ||
IRC: Výbor pro nezávislý přezkum; PFS: přežití bez progrese; HR: poměr rizik; CI: interval spolehlivosti;
MRD: minimální reziduální onemocnění
a Definován jako doba od randomizace do prvního výskytu progrese, relapsu nebo úmrtí z jakékoli příčiny podle hodnocení zkoušejícím
b stratifikováno podle stadia dle Bineta v úvodu léčby
c Včetně 11 pacientů v rameni GClb s kompletní odpovědí s neúplným zotavením kostní dřeně d Kombinace krve a kostní dřeně
e Negativní MRD definováno jako výsledek nižší než 0,0001
f Zahrnuje MRD pozitivní pacienty a pacienty s progresí nebo úmrtím před ukončením léčby
NR = Nedosaženo
** Údaje zatím nejsou k dispozici
Celkové přežití pro stupeň 1 je shrnuto na obrázku 2. Sledování celkového přežití pro stupeň 2 bude pokračovat, protože ještě nejsou dostupné všechny údaje. Výsledky analýzy podskupiny PFS (tj. pohlaví, věk, stadium dle Bineta, CrCl, skóre CIRS, beta-2-mikroglogulin, stav IGVH, chromozomální abnormality, počet lymfocytů v úvodu) byly konzistentní s výsledky pozorovanými v celkové ITT (intent-to-treat) populaci. Riziko progrese onemocnění nebo úmrtí bylo sníženo v rameni GClb ve srovnání s ramenem RClb a ramenem Clb ve všech podskupinách kromě podskupiny pacientů s delecí 17p. V malé podskupině pacientů s delecí 17p byl pozorován pouze pozitivní trend ve srovnání s Clb (poměr rizik = 0,42; p = 0,0892); prospěch ve srovnání s RClb nebyl pozorován. U jednotlivých
Doba (měsíce)
Počet
Cib: chlorambucil; G-Clb: Gazyvaro-chlorambucil
C b
G-C b
Medián trvaní
mesice)
Poměr rizik
95% interval
0,23-0,74
spolehlivosti
Log-rank p-hodnota
0,0022
0.9-
0.7-
M MU A
0.6-
0.4-
0.3-
0.1 -
G-Clb (n - 238)
— cib (n = 118)
G-Clb
podskupin se redukce rizika progrese onemocnění nebo úmrtí pohybovala v rozmezí od 92 % do 58 % pro GClb oproti Clb a 72 % až 29 % pro GClb oproti RClb.
Obrázek 1 Kaplan-Meierova křivka přežití bez progrese dle hodnocení zkoušejícího ze
stupně 1a
Clb
G-Clb
Medián trvaní (mesice
Poměr rizik
95% interval
(0,13-0,24)
spolehlivosti
Log-rank p-hodnota
< 0,0001
0.9
0.8
0.7
0.6
0.5
0.4
0.2
0.1
G-Clb (n = 238)
— cib (n = ns)
Počet
Doba (měsíce)
v riziku
c b
118
238
220
218
207
188
156
122
v riziku
C b
18
09
03
02
G-C b
238
226
223
2 5
70
44
5
Clb: chlorambucil; G-Clb: Gazyvaro-chlorambucil; NR: nedosazeno
R-C b
G-C b
Medián trvaní
15.2
(mesice)
Poměr rizik
95% interva
(0,31-0,49)
spolehlivosti
Log-rank p-hodnota
< 0.0001
0.9-
0.8-
0.7-
0.6-
0.5-
0.4-
0.3-
0.2-
0.1 -
G-Clb n = 3331
— — — R-Clb n= 3301
* U
Doba(měsice)
Počet
v riziku
R-C b
309
259
3-C b
307
302
278
R-Cib:rituximab-chlorambucil; G-Clb: Gazyvaro-chlorambucil
Kvalita života
V dotaznících QLQC30 a QLQ-CLL-16 prováděných v průběhu léčby nebyl zaznamenán žádný významný rozdíl v žádné z pozorovaných podstupnic. Údaje z následného sledování, zejména
z ramene se samotným chlorambucilem, jsou omezené. Žádné výrazné rozdíly v kvalitě života v průběhu následného sledování však do dnešní doby nebyly zaznamenány.
Hodnocení kvality života v souvislosti se zdravím, zejména s ohledem na únavu v průběhu léčby, neukázalo žádný statisticky významný rozdíl, což naznačuje, že přidání přípravku Gazyvaro k chlorambucilu nezvyšuje výskyt únavy u pacientů.
Folikulární lymfom
V otevřené, multicentrické, randomizované klinické studii fáze III (GAO4753g (GADOLIN)) bylo hodnoceno 396 pacientů s iNHL, kteří neodpověděli na léčbu nebo u kterých došlo k progresi onemocnění během 6 měsíců po poslední dávce rituximabu nebo režimu obsahujícím rituximab (včetně rituximabu v monoterapii jako součást úvodní nebo udržovací léčby). Pacienti byli randomizováni v poměru 1:1 buď k léčbě samotným bendamustinem (B) (n=202) nebo k léčbě přípravkem Gazyvaro v kombinaci s bendamustinem (G+B) v 6 28denních cyklech. Pacienti v rameni G+B, u kterých nedošlo k progresi onemocnění, (tzn. u pacientů s kompletní léčebnou odpovědí (CR, částečnou léčebnou odpovědí (PR) nebo stabilním onemocněním (SD) na konci úvodní léčby) pokračovali v udržovací léčbě přípravek Gazyvaro každé dva měsíce po dobu 2 let nebo do progrese onemocnění (podle toho, co nastalo dříve). Pacienti byli stratifikováni podle regionu, subtypu iNHL(folikulární vs jiné než folikulární), typu refrakterity na rituximab (refrakterní na předchozí monoterapii rituximab vs refrakterní na předchozí kombinaci rituximabu s chemoterapií) a podle počtu předchozích linií léčby (<2 vs >2).
Demografické údaje a vstupní charakteristiky byly vyrovnané (medián věku 63 let, většina pacientů byla bílé rasy (88 %) a mužského pohlaví (58 %). Většina pacientů měla folikulární lymfom (81 %). Medián doby od vstupní diagnózy byl 3 roky a medián počtu předchozí léčby byl 2 (od 1 do 10); 44 % pacientů podstoupilo 1 předchozí léčbu a 34 % 2 předchozí léčby.
Přípravek Gazyvaro byl podáván nitrožilní infuzí v jednotné dávce 1000 mg v den 1, den 8 a en 15 1. cyklu léčby a v den 1 2.-6. cyklu léčby a u pacientů, u kterých nedošlo k progresi onemocnění, každé dva měsíce po dobu 2 let nebo do progrese onemocnění (podle toho, co nastalo dříve). Bendamustin
byl podáván nitrožilně v den 1 a den 2 každé cyklu léčby (cykly 1 až 6) v dávce 90 mg/m2/den, pokud byl podáván v kombinaci s přípravkem Gazyvaro, nebo v dávce 120 mg/m2/den, pokud byl podáván samotný. Všech 6 cyklů léčby podstoupilo 79,4 % pacientů léčených kombinací G+B ve srovnání s 66,7 % v rameni B.
Primární analýza založená na hodnocení Independent Review Committee (IRC) prokázala statisticky významné 45% snížení rizika progrese onemocnění (PD) či úmrtí u pacientů léčených kombinací G+B s následnou udržovací léčbou přípravkem Gazyvaro oproti pacientům léčeným samotným bendamustinem. Snížení rizika progrese onemocnění nebo úmrtí znamenané v populaci pacientů s iNHL je dáno podskupinou pacientů s FL.
Většina pacientů ve studii GAO4753g měla folikulární lymfom (FL) (81,1%). Výsledky účinnosti u populace pacientů s folikulárním lymfomem jsou shrnuty v tabulce 9. 11,6 % pacientů mělo lymfom z marginální zóny (MZL) a 7,1 % mělo lymfom z malých lymfocytů (SLL).
Tabulka 9 Shrnutí výsledků účinnosti u pacientů s FL ve studii GAO4753g (GADOLIN)
|
Bendamustin N=166 |
Gazyvaro + bendamustin s následnou udržovací léčbou přípravkem Gazyvaro N= 155 | |
|
Medián doby sledování: 20 měsíců |
Medián doby sledování: 22 měsíců | |
|
Primární cílový parametr u populace s FL PFS hodnocené IRC (PFS-IRC) Počet (%) pacientů s příhodou Medián trvání (měsíce) PFS (95% CI) HR (95% CI) p-hodnota (log-rank test, stratifikovaný*) |
90 (54,2 %) 54 (34,8 %) 13,8 (11,4; 16,2) NR (22,5;-) 0,48 (0,34; 0,68) <0,0001 | |
|
Sekundární cílový parametr PFS hodnocené zkoušejícím (PFS-INV) Počet (%) pacientů s příhodou Medián trvání (měsíce) PFS (95% CI) HR (95% CI) p-hodnota (log-rank test, stratifikovaný*) |
102 (61,4 %) 62 (40,0 %) 13,7 (11,0; 15,5) 29,2 (17,5;-) 0,48 (0,35; 0,67) <0,0001 | |
|
Nejlepší celková odpověď (BOR) (hodnoceno IRC)§ Počet pacientů zařazených do analýzy Pacienti s léčebnou odpovědí (%) (CR/PR) Rozdíl ve výskytu odpovědi (95% CI) p-hodnota (Cochran-Mantel-Haenszel test) Pacienti s kompletní léčebnou odpovědí (%) Pacienti s částečnou léčebnou odpovědí (%) Pacienti se stabilním onemocněním (%) |
161 153 124 (77,0 %) 122 (79,7 %) 2,72 (-6,74; 12,18) 0,6142 31 (19,3 %) 24 (15,7%) 93 (57,8 %) 98 (64,1%) 18 (11,2 %) 13 (8,5%) | |
|
Trvání odpovědí (DOR) (hodnoceno IRC) | ||
|
Bendamustin N=166 |
Gazyvaro + bendamustin s následnou udržovací léčbou přípravkem Gazyvaro N= 155 | |
|
Medián doby sledování: 20 měsíců |
Medián doby sledování: 22 měsíců | |
|
Počet pacientů zařazených do analýzy Počet (%) pacientů s příhodou Medián trvání (měsíce) DOR (95% CI) HR (95% CI) |
127 122 74 (58,3 %) 36 (29,5 %) 11,9 (8,8; 13,6) NR (25,4;-) 0,36 (0,24; 0,54) | |
|
Celkové přežití (data dosud nejsou zralá) Počet (%) pacientů s příhodou Medián doby do příhody (měsíce) HR (95% CI) p-hodnota (log-rank test, stratifikovaný*) |
36 (21,7 %) 25 (16,1 %) NR NR 0,71 (0,43; 1,19) 0,1976 | |
IRC: Výbor pro nezávislý přezkum; PFS: přežití bez progrese; HR: poměr rizik; CI: interval spolehlivosti,
NR = Nedosaženo
*Pro analýzu byly použity stratifikační faktory typ refrakterity (monoterapie rituximabem vs. kombinace rituximabu s chemoterapií) a počet předchozích linií léčby (< 2 vs >2). Ve studii byl použit také stratifikační faktor folikulární lymfom vs jiné typy lymfomu, ale ten není možno použít v dílčí analýze pacientů s folikulárním lymfomem.
§ Nejlepší odpověď v průběhu 12 měsíců od začátku léčby
Poměr rizik přežití bez progrese hodnocené IRC u populace bez folikulárního lymfomu byl 0,94 [95% interval spolehlivosti: 0,49; 1,90].
Ohledně účinnosti u subpopulací pacientů s MZL a SLL nemohou být učiněny konečné závěry.
Obrázek 4 Kaplan-Meierova křivka přežití bez progrese hodnocené IRC u pacientů s folikulárním lymfomem
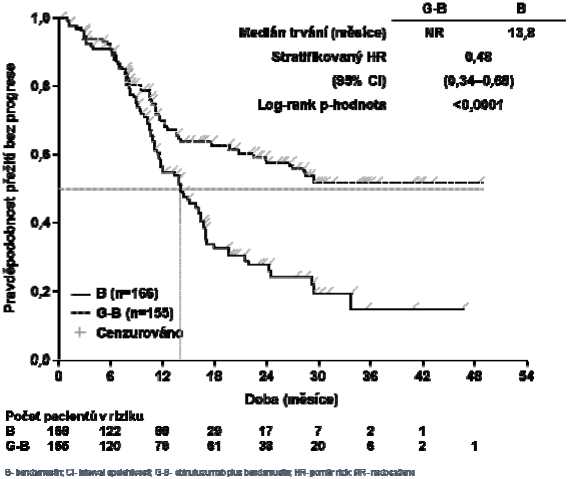
0.6-
0.2-
— G-B (r = 164)
Medián trváni (měsíce)
G-B
--B (n = 171)
Stratifi kovaný HR
0,62
-i- cenzurována
(95% Cl)
(0,39-0.98)
□oba (měsíce)
Počet pacientů v riziku
158
125
107
G-B 164
143
130
b- ^aTdanustjn: LU rrtara spitenrasti: G-B-obruunjTHb rbs HR-porna-rizk KFt- n&3csaieno
Post-hoc analýza byla provedena 8 měsíců po sběru dat primární analýzy. Z počtu pacientů s folikulárním lymfomem s mediánem doby sledování 24,1 měsíce, 48 pacientů (28,1 %) v rameni B a 30 pacientů (18,3 %) v rameni G+B zemřelo. Pozorované zlepšení celkového přežití zaznamenané u G+B v této post-hoc analýze bylo podpořeno stratifikovaným poměrem rizik celkového přežití 0,62 (95% interval spolehlivosti: 0,39; 0,98). Medián celkového přežití nebyl zatím dosažen v žádném z ramen. Výsledky post-hoc analýzy přežití bez progrese jsou v souladu s primární analýzou, statistická významnost se nemění a bezpečnostní profil je v souladu s primární analýzou.
Výsledky analýz podskupin
Výsledky analýz podskupin byly v zásadě shodné s výsledky pozorovanými v populaci pacientů s FL, což podporuje robustnost celkových výsledků.
B g g
(n=1H) (n= 166)
1-ro tni t-nofinl
Cdk. KM KU 96%
|
Vstupní (Mdory |
n |
n |
Příhody |
poměr |
n |
Enrrts |
P«nir |
stranní Cl | |
|
Všichni pacienti |
321 |
166 |
90 |
54,868 |
155 |
54 |
69,219 |
0,49 |
(0,35; 0,68) |
|
Pohlaví | |||||||||
|
Muž] |
iao |
06 |
47 |
66,028 |
86 |
28 |
72,607 |
0,49 |
[0,30; 0,78) |
|
Ženy |
141 |
71 |
43 |
64*683 |
70 |
28 |
66^083 |
0,60 |
(0^t;0,02) |
|
Bulky onemocnění na počátku kóty (hranice: 6 cm] | |||||||||
|
Ano |
107 |
58 |
33 |
53,774 |
49 |
17 |
69,569 |
0,51 |
(0.29; 0,91) |
|
Ne |
212 |
106 |
57 |
55,515 |
106 |
37 |
69,113 |
0,48 |
(0,32; 0,74) |
|
Bumptomy (2t) rwpočíOojláčtay | |||||||||
|
Ano |
47 |
27 |
18 |
54^444 |
20 |
7 |
88,177 |
0,66 |
[0^2; 1,34) |
|
Na |
271 |
137 |
73 |
65,366 |
134 |
47 |
S&37B |
0,48 |
[0,34; 0,70) |
|
ECOG na počátku léčby | |||||||||
|
0-1 |
304 |
157 |
35 |
55,820 |
147 |
50 |
70,512 |
0,47 |
(0,33; 0,67) |
|
2 |
10 |
7 |
4 |
41,667 |
8 |
4 |
50,000 |
1,02 |
<0,25; 4,17) |
|
Počat předchozích tarapl | |||||||||
|
9 |
255 |
13D |
74 |
66,1 DD |
126 |
41 |
88,828 |
0,43 |
[0,29; 0,83) |
|
>2 |
88 |
38 |
18 |
H*DB2 |
30 |
t3 |
16,8® |
0,82 |
[0,38; 1,7?) |
|
Refrakterni na | |||||||||
|
R-mono |
64 |
39 |
20 |
63,962 |
25 |
e |
82,143 |
0,34 |
(0,15; 0,80) |
|
R-c herno v indukční léčbě |
120 |
64 |
34 |
43,448 |
56 |
20 |
72,098 |
0,50 |
(0.28: 0,87) |
|
R-udnovaci léčba pa indukci |
133 |
62 |
35 |
56,561 |
71 |
26 |
60,649 |
0,58 |
(0,35; 0,97) |
|
DoiAls ran nu tury statue | |||||||||
|
Ano |
232 |
133 |
73 |
6t,Z24 |
118 |
42 |
87.441 |
0,61 |
[0,34; 0,74) |
|
Na |
68 |
33 |
17 |
68,168 |
36 |
t2 |
74*828 |
0*43 |
[0,20; 0,91) |
-Ú.ŮS ů.t—53-=f-3-ra—30
Zobnnn iwbuliíikováný poměr řídi. Om X v logaritmické akáte.
B-bandarnuaťrt; BL-východ stav; chemo-diantotarepto; ci-IntsrvM spolehlivosti; ECOG-Eastorn GooparMfra Gncdogy Group;
G-B-oblnutuzumsb plus bendsmusUn; hR-pomftr rizik; KM-Ksplan-Meler R-chsmo- rtfujdmab plus chsmo terapií;
R-udrtovací- udržovací léfibs rHuKtaiabom; R-morws- rtfaixbnab v morut orapll
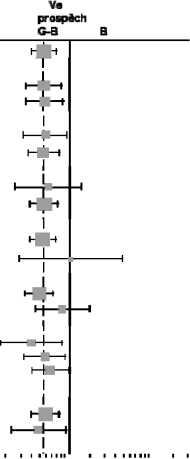
*předem definované analýzy provedené na ITT populaci byly provedeny také na populaci pacientůs FL; analýza dvojitě refrakterních pacientů (tzn. nedostatečná odpověď nebo progrese onemocnění během léčby nebo do 6 měsíců od ukončení léčby režimem obsahujícím alkylační látku) byla exploratorní.
Výsledky hlášené pacienty
Na základě dotazníku FACT-Lym a škály EQ-5D shromažďovaných v průběhu léčby i v následném sledování byla v klíčových studiích kvalita života související se zdravotním stavem zachována bez významných rozdílů mezi léčebnými skupinami.
U pacientů s FL však přidání přípravku Gazyvaro k bendamustinu prodloužilo dobu do zhoršení kvality života související se zdravotním stavem měřeného pomocí FACT-Lym TOI o 2,2 měsíce (medián 5,6 versus 7,8 měsíce pro B a kombinaci G+B HR = 0,83; 95% CI: 0,60; 1,13).
Imunogenita
Výsledky imunologických vyšetření jsou výrazně závislé na několika faktorech včetně senzitivity a specificity testu, metodologie testu, robustnosti testu ke kvantitě přípravku Gazyvaro/protilátka v cirkulaci, zacházení se vzorkem, načasování sběru vzorků, souběžné medikaci a základního onemocnění. Z tohoto důvodu porovnávání incidence protilátek proti přípravku Gazyvaro s incidencí protilátek proti dalším přípravkům může být zavádějící.
V klíčové studii BO21004/CLL11 byli pacienti opakovaně vyšetřováni na přítomnost protilátek proti přípravku Gazyvaro (ATA - anti-therapeutic antibodies). U pacientů léčených přípravkem Gazyvaro mělo prokázanou přítomnost ATA po 12 měsících následného sledování 8 ze 140 pacientů
v randomizační fázi studie a 2 ze 6 ve vstupní fázi studie. U žádného z těchto pacientů se nevyskytla anafylaktická či hypersenzitivní reakce, které by byla přičítána ATA, ani u nich nedošlo k ovlivnění léčebné odpovědi.
V klíčové studii pro iNHL, GAO4753g, měli 2 pacienti ve větvi G+B pozitivní HAHA (Human Anti-Human Antibody) při vstupu do studie a vyskytly se u nich IRR. U žádného pacienta se neobjevily HAHA v průběhu léčby přípravkem Gazyvaro nebo po jejím ukončení.
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Gazyvaro u všech podskupin pediatrické populace ve schválené indikaci chronická lymfocytární leukemie a folikulární lymfom (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
K hodnocení farmakokinetických údajů u 469 pacientů s iNHL, u 342 pacientů s CLL a u 130 pacientů DLBCL ve studiích fáze I, fáze II a fáze III, kteří byli léčeni obinutuzumabem samotným nebo v kombinaci s chemoterapií byl vyvinut populační farmakokinetický model.
Absorpce
Obinutuzumab se podává intravenózně, proto absorpce není relevantní. Pro další způsoby podání nebyly provedeny žádné studie. Z populačního PK modelu byla u pacientů s CLL po infuzi první den
6. cyklu (Cyklus 6 Den 1) odhadovaná hodnota mediánu Cmax 465,7 pg/ml a hodnota AUC(t)
8961 pg^den/ml a u pacientů s iNHL byla odhadovaná hodnota mediánu Cmax 539,3 pg/ml a hodnota AUC(t) 10956 pg^den/ml.
Distribuce
Po intravenózním podání se distribuční objem centrálního kompartmentu (2,98 l u pacientů s CLL a 2,97 u pacientů s iNHL) přibližuje objemu séra, což naznačuje, že distribuce je výrazně omezena na plazmu a intersticiální tekutinu.
Biotransformace
Metabolismus obinutuzumabu nebyl přímo hodnocen. Protilátky jsou většinou odstraňovány katabolicky.
Eliminace
Clearance obinutuzumabu byla přibližně 0,11 l/den u pacientů s CLL a 0,08 l/den u pacientů s iNHL s mediánem eliminace tJ/2 26,4 dne u pacientů s CLL a 36,8 dne u pacientů s iNHL. Vylučování obinutuzumabu utváří dvě paralelní metabolické cesty, které popisují clearance, lineární metabolická cesta clearance a ne-lineární metabolická cesta clearance, které se mění v závislosti na čase. Při zahájení léčby je dominantní ne-lineární cesta clearance měnící se s časem a v důsledku toho je hlavní cestou clearance. Při pokračování v léčbě se dopad této cesty zmenšuje a začíná převažovat lineární cesta clearance. To ukazuje na dispozici léku podle cíle (TMDD, target mediated drug disposition), kde počáteční nadbytek CD20 buněk způsobí rychlé vylučování obinutuzumabu z oběhu. Jakmile je na většinu CD20 buněk navázán obinutuzumab, je minimalizován vliv TMDD na PK.
Farmakokinetický(é)/farmakodynamický(é) vztah(y)
V populační farmakokinetické analýze bylo zjištěno, že pohlaví je spoluproměnná (kovariát), která vysvětluje určitou variabilitu mezi pacienty, s clearance v rovnovážném stavu (Clss) o 22 % vyšší a distribučním objemem (V) o 19 % vyšším u mužů. Výsledky z populační analýzy však ukázaly, že rozdíl v expozici není signifikantní (u pacientů s CLL s odhadovaným mediánem AUC a Cmax 11 282 pg^den/ml resp. 578,9 pg/ml u žen a 8451 pg^den/ml resp. 432,5 pg/ml u mužů v 6. cyklu a u pacientů s iNHL s odhadovaným mediánem AUC a Cmax 13172 pg^den/ml resp. 635,7 pg/ml u žen a 9769 pg^den/ml resp. 481,3 pg/ml u mužů), což naznačuje, že zde není potřeba dávky na základě pohlaví upravovat.
Starší pacienti
Populační farmakokinetická analýza obinutuzumabu ukázala, že věk neovlivňuje farmakokinetiku obinutuzumabu. Mezi pacienty < 65 let (n=375), pacienty ve věku mezi 65-75 lety (n=265) a pacienty > 75 let (n=171) nebyl ve farmakokinetice obinutuzumabu pozorován žádný významný rozdíl.
Pediatrická populace
Studie hodnotící farmakokinetiku obinutuzumabu u pediatrických pacientů nebyly provedeny. Porucha funkce ledvin
Populační farmakokinetická analýza obinutuzumabu ukázala, že clearance kreatininu neovlivňuje farmakokinetiku obinutuzumabu. Farmakokinetika obinutuzumabu u pacientů s lehkou (CrCl 50-89 ml/min, n=464) nebo středně těžkou (CrCl 30-49 ml/min, n=106) poruchou funkce ledvin byla podobná jako u pacientů s normálními renálními funkcemi (CrCl > 90 ml/min, n=383). Farmakokinetické údaje u pacientů s těžkou poruchou funkce ledvin (CrCl 15-29 ml/min) jsou omezené (n=8), proto na jejich základě nelze dát žádná doporučení týkající se dávkování.
Porucha funkce jater
Žádné formální farmakokinetické studie nebyly u pacientů s poruchou funkce jater provedeny.
5.3 Předklinické údaje vztahující se k bezpečnosti
Studie hodnotící kancerogenní potenciál obinutuzumabu nebyly provedeny.
Žádné specifické studie hodnotící účinnost obinutuzumabu na fertilitu u zvířat nebyly provedeny. Ve studiích toxicity po opakovaném podávání u makaků rodu cynomolgus neměl obinutuzumab nežádoucí účinky na samčí ani samičí reprodukční orgány.
Ve studii toxicity zrychleného pre- a postnatálního vývoje (ePPND) u březích makaků rodu cynomolgus nebyly prokázány teratogenní účinky. Týdenní podávání obinutuzumabu od 20. dne po koitu do porodu však vedlo ke kompletní depleci B buněk u mláďat opic při týdenních intravenózních dávkách obinutuzumabu 25 a 50 mg/kg (2- až 5násobek klinické expozice na základě Cmax a AUC). Expozice mláďat 28 dní po porodu naznačuje, že obinutuzumab může procházet placentární bariérou. Koncentrace v séru mláďat 28 dnů po porodu byly v rozmezí koncentrací odpovídajícím koncentraci v séru matky, zatímco koncentrace v mléce ten samý den byly nižší (méně než 0,5 % odpovídajících maternálních sérových hladin), což naznačuje, že expozice mláďat musí být získána in utero. Počty B buněk se vracejí k normálním hodnotám a imunologické funkce se upravují v průběhu 6 měsíců po porodu.
V 26týdenní studii u makaků rodu cynomolgus byly zaznamenány hypersenzitivní reakce, které odpovídají rozeznání cizí humanizované protilátky u makaků rodu cynomolgus (0,7- až 6násobek klinické expozice založené na Cmax a AUC v rovnovážném stavu po týdenním podávání dávek 5, 25 a 50 mg/kg). Nálezy zahrnovaly akutní anafylaktické a anafylaktoidní reakce a zvýšenou prevalenci systémových zánětů a infiltrátů, což odpovídá hypersenzitivní reakci zprostředkované imunitními komplexy, jako jsou arteritida/periarteritida, glomerulonefritida, zánět serózy/adventicie. Tyto reakce vedly k neplánovanému ukončení u 6/36 zvířat léčených obinutuzumabem během léčby i ve fázi zotavování; tyto změny byly částečně reverzibilní.
U člověka nebyla pozorována renální toxicita s kauzálním vztahem k obinutuzumabu.
FARMACEUTICKÉ ÚDAJE
6.
6.1 Seznam pomocných látek
Histidin
Monohydrát histidin-hydrochloridu Dihydrát trehalosy Poloxamer 188 Voda na injekci
6.2 Inkompatibility
Tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky s výjimkou těch, které jsou uvedeny v bodě 6.6.
6.3 Doba použitelnosti
Neotevřená injekční lahvička 3 roky
Po naředění
Po naředění byla chemická a fyzikální stabilita v 0,9% injekčním roztoku chloridu sodného (9 mg/ml) při koncentracích od 0,4 mg/ml do 20 mg/ml prokázána po dobu 24 hodin při teplotě od 2 °C do 8 °C a poté po dobu 48 hodin (včetně doby infuze) při teplotě < 30 °C.
Z mikrobiologického hlediska má být roztok použit okamžitě. Není-li použit okamžitě, doba a podmínky uchovávání přípravku po naředění před použitím jsou v odpovědnosti uživatele a normálně by doba neměla být delší než 24 hodin při teplotě 2 až 8 °C, pokud ředění neproběhlo za kontrolovaných a validovaných aseptických podmínek.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Uchovávejte injekční lahvičku v krabičce, aby byla chráněna před světlem.
Podmínky uchovávání tohoto léčivého přípravku po jeho naředění jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
Čtyřicet ml koncentrátu v 50ml injekční lahvičce (čiré sklo třídy I) se zátkou (butylová pryž).
Balení obsahuje 1 injekční lahvičku.
6.6 Zvláštní opatření pro likvidaci přípravku
Návod k naředění
Přípravek Gazyvaro má připravit zdravotnický pracovník za použití techniky asepse.
Injekční lahvičkou netřeste.
CLL cykly 2-6 a všechny FL cykly
Z injekční lahvičky odeberte 40 ml koncentrátu a nařeďte je v infuzním vaku z polyvinylchloridu (PVC) nebo polyolefinu (non-PVC) obsahujícím 0,9% injekční roztok chloridu sodného (9 mg/ml).
Pouze CLL - cyklus 1
K zajištění odlišení dvou infuzních vaků pro úvodní 1000 mg dávku se doporučuje použít vaky různých velikostí, aby se odlišila dávka 100 mg pro 1. den 1. cyklu (Cyklus 1 Den 1) a dávka 900 mg pro 1. den 1. cyklu (Cyklus 1 Den 1, pokračování) nebo 2. den 1 cyklu (Cyklus 1 Den 2). K přípravě dvou infuzních vaků odeberte z injekční lahvičky 40 ml koncentrátu a 4 ml vstříkněte do 100 ml infuzního vaku z PVC nebo polyolefinu (non-PVC) a zbývajících 36 ml do 250 ml infuzního vaku z PVC nebo polyolefinu (non-PVC) obsahujícího 0,9% injekční roztok chloridu sodného (9 mg/ml). Každý infuzní vak označte viditelně štítkem. Podmínky uchovávání infuzních vaků jsou uvedeny v bodě 6.3.
|
Dávka přípravku Gazyvaro, která se bude podávat |
Požadované množství koncentrátu přípravku Gazyvaro |
Velikost infuzního vaku z PVC nebo polyolefinu (non-PVC) |
|
100 mg |
4 ml |
100 ml |
|
900 mg |
36 ml |
250 ml |
|
1000 mg |
40 ml |
250 ml |
Nepoužívejte jiná rozpouštědla, jako je např. roztok glukózy (5%) (viz bod 6.2).
Vak se jemně obrátí, aby se roztok smísil a zabránilo se nadměrné tvorbě pěny. Naředěným roztokem se nesmí třást a nesmí se zmrazit.
Parenterální léčivé přípravky je nutné před podáním vizuálně zkontrolovat, zda neobsahují částice nebo nejsou zbarvené.
Mezi přípravkem Gazyvaro v koncentracích pohybujících se v rozmezí od 0,4 mg/ml do 20,0 mg/ml po naředění 0,9% injekčním roztokem chloridu sodného (9 mg/ml) a následujícími zdravotnickými prostředky nebyly pozorovány žádné inkompatibility:
- vaky z PVC, polyethylenu (PE), polypropylenu nebo polyolefinu
- infuzní sety z PVC, polyuretanu (PUR) nebo PE
- volitelné in-line filtry s kontaktními povrchy z polyethersulfonu (PES), trojcestné infuzní kohouty z polykarbonátu (PC) a katetry z polyetherurethanu (PEU).
Likvidace
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/14/937/001
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 23. července 2014
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA
PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce biologické léčivé látky
Roche Diagnostics GmbH Nonnenwald 2 823 77 Penzberg Německo
Název a adresa výrobce odpovědného za propouštění šarží
Roche Pharma AG Emil-Barell-Strasse 1 79639 Grenzach-Wyhlen Německo
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE,
• Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předloží první pravidelně aktualizovanou zprávu o bezpečnosti pro tento léčivý přípravek do 8 měsíců od jeho registrace. Držitel rozhodnutí o registraci dále předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Gazyvaro 1000 mg koncentrát pro infuzní roztok Obinutuzumabum
2 OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna injekční lahvička se 40 ml koncentrátu obsahuje obinutuzumabum 1000 mg, což odpovídá koncentraci 25 mg/ml před naředěním.
3. SEZNAM POMOCNÝCH LÁTEK
Histidin
Monohydrát histidin-hydrochloriduDihydrát trehalosy Poloxamer 188 Voda na injekci
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Koncentrát pro infuzní roztok 1000 mg/40 ml 1 injekční lahvička
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci Intravenózní podání po naředění Injekční lahvičkou netřepejte
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do:
Uchovávejte v chladničce Chraňte před mrazem
Uchovávejte injekční lahvičku v krabičce, aby byla chráněna před světlem
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH
PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/14/937/001
13. ČÍSLO ŠARŽE
č.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
Nevyžaduje se - odůvodnění přijato
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
<2D čárový kód s jedinečným identifikátorem.>
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
PC:
SN:
NN:
1 NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Gazyvaro 1000 mg koncentrát pro infuzní roztok
Obinutuzumabum
Intravenózní podání
2. ZPŮSOB PODÁNÍ
Intravenózní podání po naředění
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
1000 mg/40 ml
6. JINÉ
B. PŘÍBALOVÁ INFORMACE
Gazyvaro 1000 mg koncentrát pro infuzní roztok
Obinutuzumabum
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
• Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
• Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo zdravotní sestry.
• Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Gazyvaro a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Gazyvaro používat
3. Jak se přípravek Gazyvaro používá
4. Možné nežádoucí účinky
5. Jak přípravek Gazyvaro uchovávat
6. Obsah balení a další informace
1. Co je přípravek Gazyvaro a k čemu se používá
Co je přípravek Gazyvaro
Přípravek Gazyvaro obsahuje léčivou látku obinutuzumab. Ten patří do skupiny léků zvaných „monoklonální protilátky“. Protilátky účinkují tak, že se naváží na specifické cíle v těle.
K čemu se přípravek Gazyvaro používá
Přípravek Gazyvaro může být používán u dospělých k léčbě dvou různých typů nádorového onemocnění
• Chronická lymfocytární leukemie (také nazývaná „CLL“)
• Přípravek Gazyvaro se používá u pacientů, kteří nepodstoupili žádnou předchozí léčbu CLL a kteří mají další onemocnění, v důsledku kterého nemohou dostávat plnou dávku jiného přípravku zvaného fludarabin k léčbě CLL.
• Přípravek Gazyvaro se používá k léčbě nádorového onemocnění společně s dalším přípravkem zvaným chlorambucil.
• Folikulární lymfom (také nazýván „FL“)
• Přípravek Gazyvaro se používá u pacientů, kteří byli dříve alespoň jednou léčeni přípravkem zvaným rituximab a u kterých se po této léčbě FL znovu objevil nebo zhoršil.
• Na začátku léčby FL se přípravek Gazyvaro používá společně s dalším přípravkem k léčbě nádorového onemocnění zvaným bendamustin.
• Přípravek Gazyvaro může být používán samotný po dobu 2 let jako „udržovací léčba“.
Jak přípravek Gazyvaro účinkuje
• CLL a FL jsou typy nádorového onemocnění, které působí na bílé krvinky zvané „B-lymfocyty“. Tyto postižené „B-lymfocyty“ se množí příliš rychle a přežívají příliš dlouho.
Přípravek Gazyvaro se váže cíleně na povrch postižených B-lymfocytů a způsobuje jejich smrt.
• Podáním přípravku Gazyvaro pacientům s CLL nebo FL společně s dalšími přípravky k léčbě nádorového onemocnění se zpomaluje doba do zhoršení nemoci.
2. Čemu musíte věnovat pozornost, než začnete přípravek Gazyvaro používat Nepoužívejte přípravek Gazyvaro:
• jestliže jste alergický(á) na obinutuzumab nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Pokud si nejste jistý(á), poraďte se se svým lékařem nebo zdravotní sestrou dříve, než Vám bude přípravek Gazyvaro podán.
Upozornění a opatření
Před podáním přípravku Gazyvaro se poraďte se svým lékařem nebo zdravotní sestrou jestliže:
• máte infekci nebo jste měl(a) infekci v minulosti, která trvala dlouho nebo se opakovala
• jste někdy užíval(a) léky nebo Vám byly podány léky, které ovlivňují imunitní systém (jako je např. chemoterapie nebo imunosupresivní léčba)
• užíváte léky k léčbě vysokého krevního tlaku nebo léky na ředění krve - Váš lékař možná bude muset změnit jejich užívání
• jste někdy měl(a) problémy se srdcem
• jste někdy měl(a) neurologické problémy (jako jsou problémy s pamětí, potíže s pohybem nebo čitím Vašeho těla, problémy se zrakem)
• jste někdy měl(a) problémy s dýcháním nebo plicní problémy
• jste někdy měl(a) „hepatitidu B“, typ jaterního onemocnění
• máte podstoupit očkování nebo víte, že je možná budete potřebovat podstoupit v blízké budoucnosti.
Pokud se Vás cokoli z výše uvedeného týká (nebo pokud si nejste jistý(á)), poraďte se se svým lékařem nebo zdravotní sestrou dříve, než Vám bude podán přípravek Gazyvaro.
Věnujte pozornost následujícím nežádoucím účinkům
Přípravek Gazyvaro může způsobit závažné nežádoucí účinky, o kterých musíte ihned informovat svého lékaře nebo zdravotní sestru. Mezi ně patří:
Reakce související s infuzí
• Neprodleně sdělte svému lékaři nebo zdravotní sestře, pokud se u Vás objeví jakákoli reakce související s infuzí uvedená na začátku bodu 4. Reakce související s infuzí se mohou objevit v průběhu infuze nebo až do 24 hodin po podání infuze.
• Pokud se u Vás objeví reakce související s infuzí, je možné, že budete potřebovat další léčbu nebo bude třeba infuzi zpomalit nebo zastavit. Pokud příznaky odezní nebo se zlepší, v infuzi je možné pokračovat. Tyto reakce se při první infuzi objevují s větší pravděpodobností. Pokud se objeví silná reakce na infuzi, může Váš lékař rozhodnout o ukončení léčby přípravkem Gazyvaro.
• Před podáním každé infuze přípravku Gazyvaro Vám budou podány léky, které pomáhají zmírnit možné reakce související s infuzí nebo „ syndrom nádorového rozpadu“. Syndrom nádorového rozpadu je potenciálně život ohrožující komplikací, která je způsobena chemickými změnami v krvi, které vznikají při rozpadu odumírajících nádorových buněk (viz bod 3).
Progresivní multifokální leukoencefalopatie (nazývaná také „PML“)
• PML je velmi vzácná a život ohrožující infekce mozku, která byla hlášena v souvislosti s podáváním přípravku Gazyvaro.
• Pokud zaznamenáte ztrátu paměti, problémy s řečí, obtíže při chůzi nebo problémy se zrakem, sdělte to neprodleně svému lékaři nebo zdravotní sestře.
• Pokud jste kterýkoli z těchto příznaků měl(a) před zahájením léčby přípravkem Gazyvaro, sdělte ihned svému lékaři, pokud zaznamenáte jakékoli změny těchto příznaků. Je možné, že budete potřebovat léčbu.
• Informujte ihned svého lékaře nebo zdravotní sestru, pokud se u Vás objeví jakékoli příznaky infekce po léčbě přípravkem Gazyvaro (viz „Infekce“ v bodě 4).
Děti a dospívající
Nepodávejte přípravek Gazyvaro dětem a osobám mladším 18 let. Důvodem tohoto doporučení je, že o použití v této věkové skupině nejsou k dispozici žádné informace.
Další léčivé přípravky a přípravek Gazyvaro
Informujte svého lékaře nebo zdravotní sestru o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat. To se týká i léků dostupných bez lékařského předpisu nebo rostlinných přípravků.
• Pokud jste těhotná, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, sdělte to svému lékaři nebo zdravotní sestře. Pomohou Vám zvážit prospěch při pokračování v léčbě přípravkem Gazyvaro oproti rizikům pro Vaše dítě.
• Pokud otěhotníte v průběhu léčby přípravkem Gazyvaro, sdělte to svému lékaři nebo zdravotní sestře co nejdříve. To je proto, že léčba přípravkem Gazyvaro může ovlivnit Vaše zdraví nebo zdraví Vašeho dítěte.
Kojení
• V průběhu léčby přípravkem Gazyvaro a po dobu dalších 18 měsíců po ukončení léčby přípravkem Gazyvaro nekojte. Důvodem tohoto doporučení je, že malé množství tohoto léku může procházet do mateřského mléka.
Antikoncepce
• V průběhu léčby přípravkem Gazyvaro používejte účinnou metodu antikoncepce.
• Pokračujte v používání účinné antikoncepce ještě po dobu 18 měsíců po skončení léčby přípravkem Gazyvaro.
Řízení dopravních prostředků a obsluha strojů
Není pravděpodobné, že by přípravek Gazyvaro ovlivnil Vaši schopnost řídit, jezdit na kole nebo obsluhovat stroje a používat nástroje. Pokud se však u Vás reakce na infuzi objeví (viz bod 4), neřiďte dopravní prostředky, nejezděte na kole ani nepoužívejte nástroje nebo neobsluhujte stroje, dokud tato reakce neodezní.
3. Jak se přípravek Gazyvaro používá
Jak bude přípravek Gazyvaro podáván
Přípravek Gazyvaro Vám bude podáván pod dohledem lékaře, který má zkušenosti s touto léčbou.
Podává se do žíly ve formě kapačky (nitrožilní infuze) po dobu několika hodin.
Léčba přípravkem Gazyvaro
Chronická lymfocytární leukemie
• Bude Vám podáno 6 cyklů léčby přípravkem Gazyvaro v kombinaci s dalším lékem k léčbě rakoviny nazývaným chlorambucil. Každý cyklus trvá 28 dní.
• První den prvního cyklu Vám bude podána část Vaší první dávky 100 miligramů (mg) přípravku Gazyvaro velmi pomalu. Váš lékař/zdravotní sestra Vás bude pečlivě sledovat z důvodu reakcí na infuzi.
• Pokud se u Vás neobjeví žádná reakce na infuzi po malé části Vaší první dávky, můžete dostat i zbytek první dávky (900 mg) ve stejný den.
• Pokud se reakce na infuzi objeví po malé části Vaší první dávky, bude Vám zbytek Vaší první dávky podán druhý den.
Typické schéma je následující.
Cyklus 1 - zahrnuje 3 dávky přípravku Gazyvaro po dobu 28 dnů:
• Den 1 - část Vaší první dávky (100 mg)
• Den 2 nebo den 1 (pokračování) - zbytek první dávky (900 mg)
• Den 8 - plná dávka (1000 mg)
• Den 15 - plná dávka (1000 mg)
Cykly 2, 3, 4, 5 a 6 - jedna dávka přípravku Gazyvaro po dobu 28 dnů:
• Den 1 - plná dávka (1000 mg)
Folikulární lymfom
• Bude Vám podáno 6 léčebných cyklů přípravku Gazyvaro v kombinaci s dalším lékem k léčbě rakoviny nazývaným bendamustin - každý cyklus trvá 28 dní.
• Léčba bude následována „udržovací fází“ - v průběhu této doby Vám bude podáván přípravek Gazyvaro jednou za 2 měsíce po dobu až 2 let nebo do doby progrese onemocnění.
• Typické schéma je uvedeno níže.
Úvodní fáze
Cyklus 1 - zahrnuje 3 dávky přípravku Gazyvaro po dobu 28 dnů:
• Den 1 - plná dávka (1000 mg)
• Den 8 - plná dávka (1000 mg)
• Den 15 - plná dávka (1000 mg)
Cykly 2, 3, 4, 5 a 6 - jedna dávka přípravku Gazyvaro po dobu 28 dnů:
• Den 1 - plná dávka (1000 mg)
Udržovací fáze
• Plná dávka (1000 mg) jednou za 2 měsíce po dobu až 2 let nebo do doby progrese onemocnění.
Léky podávané před každou infuzí
Před podáním každé infuze přípravku Gazyvaro Vám lékař podá léky ke zmírnění možné reakce na infuzi nebo syndrom nádorového rozpadu. Mohou to být:
• tekutiny
• léky ke snížení horečky
• léky proti bolesti (analgetika)
• léky ke zmírnění zánětu (kortikosteroidy)
• léky ke zmírnění alergické reakce (antihistaminika)
• léky k zamezení vzniku syndromu nádorového rozpadu (například alopurinol).
Jestliže jste zapomněl(a) na podání dávky přípravku Gazyvaro
Pokud zapomenete na podání dávky, domluvte si další termín co nejdříve. Pokud má být tento léčivý přípravek plně účinný, je důležité postupovat přesně dle dávkovacího schématu.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo zdravotní sestry.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého. U tohoto léčivého přípravku byly hlášeny následující nežádoucí účinky:
Závažné nežádoucí účinky
Reakce na infuzi
Sdělte ihned svému lékaři nebo zdravotní sestře, pokud zaznamenáte jakékoli z následujících příznaků v průběhu infuze nebo do 24 hodin po podání infuze. Tyto příznaky jsou velmi časté (mohou postihnout více než 1 osobu z 10):
Nejčastěji hlášené nežádoucí účinky:
• pocit na zvracení
• únava
• závratě
• bolest hlavy
• průjem
• horečka, návaly horka nebo zimnice
• zvracení
• dušnost
• nízký nebo vysoký krevní tlak
• zrychlený tlukot srdce
Méně často hlášené:
• nepravidelný tlukot srdce
• otok hrdla nebo dýchacích cest
• sípot, obtíže s dýcháním, pocit tíhy na hrudi nebo podráždění hrdla
Pokud zaznamenáte jakýkoli nežádoucí účinek z výše uvedených, sdělte to ihned svému lékaři nebo zdravotní sestře.
Progresivní multifokální leukoencefalopatie (PML)
PML je velmi vzácná a život ohrožující infekce mozku, která byla hlášena při podávání přípravku Gazyvaro.
Sdělte ihned svému lékaři nebo zdravotní sestře, pokud zaznamenáte:
• ztrátu paměti
• problémy s řečí
• potíže s chůzí
• problémy se zrakem
Pokud jste jakékoli z těchto příznaků měl(a) již před léčbou přípravkem Gazyvaro, sdělte ihned svému lékaři, pokud zaznamenáte jakoukoli změnu těchto příznaků. Je možné, že budete potřebovat léčbu.
V průběhu léčby přípravkem Gazyvaro můžete snadněji prodělat infekci. Často je to nachlazení, ale mohou se objevit i závažnější infekce. U pacientů, kteří prodělali hepatitidu B v minulosti, byl rovněž hlášen návrat onemocnění jater zvané „hepatitida B“.
Sdělte ihned svému lékaři nebo zdravotní sestře, pokud se u Vás po podání přípravku Gazyvaro objeví jakékoli známky infekce. Mohou to být:
• horečka
• kašel
• bolest v krku
• pálivá bolest při močení
• pocit slabosti nebo celkové nemoci
V případě, že jste měl(a) opakující se nebo chronické infekce před zahájením léčby přípravkem Gazyvaro, informujte svého lékaře.
Další nežádoucí účinky:
Informujte svého lékaře nebo zdravotní sestru, pokud zaznamenáte jakékoli z následujících nežádoucích účinků:
Velmi časté (mohou postihovat více než 1 osobu z 10)
• horečka
• bolest kloubů
• pocit slabosti
• průjem, zácpa
• změny v krevních testech:
• anémie (nízký počet červených krvinek)
• nízký počet neutrofilů (druh bílých krvinek)
• nízký počet krevních destiček (druh krevních buněk, které pomáhají při srážení krve)
• infekce horních cest dýchacích (infekce nosu, hltanu, hrtanu a vedlejších nosních dutin), kašel.
Časté (mohou postihovat až 1 osobu z 10)
• začervenání oka
• opar rtu
• deprese
• noční pocení
• infekce plic
• chřipka
• zvýšení tělesné hmotnosti
• bolest lymfatických uzlin
• rýma nebo ucpaný nos
• ztráta vlasů, svědění, ekzém
• zánět v nose a/nebo hrdle
• bolest svalů nebo kostí hrudníku
• nádorové onemocnění kůže (spinocelulární karcinom)
• bolest zad, kostí, bolest v rukou a nohou
• nepravidelný srdeční rytmus (fibrilace síní), srdeční příhoda (infarkt)
• infekce močových cest, problémy s močením, inkontinence moči
• problémy s trávením (např. pálení žáhy), zánět střev, hemoroidy
• změny v krevních testech:
• nízký počet všech typů bílých krvinek (kombinováno)
• nízký počet všech druhů lymfocytů (druh bílých krvinek)
• zvýšení hladiny draslíku, fosfátu a kyseliny močové - což může způsobit problémy s ledvinami (součást syndromu nádorového rozpadu)
Méně časté (mohou postihovat až 1 osobu ze 100)
• proděravění žaludku nebo střeva (gastrointestinální perforace, obzvlášť v případech, kdy má
nádorové onemocnění vliv na žaludek a střeva)
Informujte svého lékaře nebo zdravotní sestru, pokud zaznamenáte jakýkoli z nežádoucích účinků uvedených výše.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Gazyvaro uchovávat
Přípravek Gazyvaro budou uchovávat zdravotničtí pracovníci v nemocnici nebo na klinice. Podmínky uchovávání jsou následující:
• Uchovávejte tento přípravek mimo dohled a dosah dětí.
• Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce za „Použitelné do:“. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
• Uchovávejte v chladničce (2 °C - 8 °C). Chraňte před mrazem.
• Uchovávejte injekční lahvičku v krabičce, aby byla chráněna před světlem
Žádné léčivé přípravky se nesmí vyhazovat do odpadních vod nebo domácího odpadu. Váš lékař znehodnotí všechny léky, které již nebude potřebovat. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co přípravek Gazyvaro obsahuje
• Léčivou látkou je obinutuzumabum: 1000 mg/40 ml v jedné injekční lahvičce odpovídá koncentraci 25 mg/ml před naředěním.
• Dalšími složkami jsou histidin, monohydrát histidin-hydrochloridu, dihydrát trehalosy, poloxamer 188, voda na injekci.
Jak přípravek Gazyvaro vypadá a co obsahuje toto balení
Přípravek Gazyvaro je koncentrát pro infuzní roztok a je to bezbarvá až lehce nahnědlá tekutina. Přípravek Gazyvaro je dostupný v balení obsahujícím 1 skleněnou injekční lahvičku.
Držitel rozhodnutí o registraci
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
Výrobce
Roche Pharma AG Emil-Barell-Strasse 1 D-79639 Grenzach-Wyhlen Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci
|
Belgie/Belgique/Belgien N.V. Roche S.A. Tél/Tel: +32 (0) 2 525 82 11 |
Lietuva UAB “Roche Lietuva” Tel: +370 5 2546799 |
|
BtarapHH Pom Etnrapua EOOfl Tea: +359 2 818 44 44 |
Luxembourg/Luxemburg (Voir/siehe Belgique/Belgien) |
|
Česká republika Roche s. r. o. Tel: +420 - 2 20382111 |
Magyarország Roche (Magyarország) Kft. Tel: +36 - 23 446 800 |
|
Danmark Roche a/s Tlf: +45 - 36 39 99 99 |
Malta (See United Kingdom) |
|
Deutschland Roche Pharma AG Tel: +49 (0) 7624 140 |
Nederland Roche Nederland B.V. Tel: +31 (0) 348 438050 |
|
Eesti Roche Eesti OU Tel: + 372 - 6 177 380 |
Norge Roche Norge AS Tlf: +47 - 22 78 90 00 |
|
EXlába Roche (Hellas) A.E. T^k: +30 210 61 66 100 |
Osterreich Roche Austria GmbH Tel: +43 (0) 1 27739 |
|
Espaňa Roche Farma S.A. Tel: +34 - 91 324 81 00 |
Polska Roche Polska Sp.z o.o. Tel: +48 - 22 345 18 88 |
|
France Roche Tél: +33 (0)1 47 61 40 00 |
Portugal Roche Farmaceutica Química, Lda Tel: +351 - 21 425 70 00 |
|
Hrvatska Roche d.o.o. Tel: + 385 1 47 22 333 |
Románia Roche Románia S.R.L. Tel: +40 21 206 47 01 |
|
Ireland Roche Products (Ireland) Ltd. Tel: +353 (0) 1 469 0700 |
Slovenija Roche farmacevtska družba d.o.o. Tel: +386 - 1 360 26 00 |
|
Island Roche a/s c/o Icepharma hf Sími: +354 540 8000 |
Slovenská republika Roche Slovensko, s.r.o. Tel: +421 - 2 52638201 |
|
Italia Roche S.p.A. Tel: +39 - 039 2471 |
Suomi/Finland Roche Oy Puh/Tel: +358 (0) 10 554 500 |
|
Kónpoq r.A.Exapáxn? & Eia AxS. T^: +357 - 22 76 62 76 |
Sverige Roche AB Tel: +46 (0) 8 726 1200 |
|
Latvija Roche Latvija SIA Tel: +371 - 6 7039831 |
United Kingdom Roche Products Ltd. Tel: +44 (0) 1707 366000 |
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Následující informace jsou určeny pouze pro zdravotnické pracovníky:
Dávkování
Přípravek Gazyvaro se podává pod pečlivým dohledem zkušeného lékaře a v prostředí, kde je okamžitě dostupné plné vybavení pro resuscitaci.
Profylaxe a premedikace syndromu nádorového rozpadu (TLS)
U pacientů s velkou nádorovou masou a/nebo s vysokým počtem cirkulujících lymfocytů (> 25 x 109/l) a/nebo poruchou funkce ledvin (CrCl < 70 ml/min) je třeba zvážit riziko TLS a má být podána profylaxe.
Profylaxe má spočívat v odpovídající hydrataci a podávání urikostatik (např. alopurinol), nebo ve vhodné alternativní léčbě, jako jsou urátoxidázy (např. rasburikáza), počínaje 12-24 hodin před zahájením léčby dle běžné praxe. Všichni pacienti považovaní za rizikové mají být v průběhu prvních dnů léčby pečlivě sledováni se zaměřením obzvláště na funkce ledvin, na hodnoty draslíku a kyseliny močové. Měla by být dodržována veškerá další opatření v souladu s běžnou praxí.
Profylaxe a premedikace reakcí souvisejících s infuzí (IRR)
Premedikace ke snížení rizika reakcí souvisejících s infuzí je uvedena v tabulce 1 a 2. Premedikace kortikosteroidy je doporučena u pacientů s folikulárním lymfomem (FL) a povinná u pacientů s CLL v prvním cyklu léčby (viz tabulka 1). Premedikace před následnými infuzemi a další premedikace mají být podány dle popisu níže.
V průběhu podávání intravenózní infuze přípravku Gazyvaro se může objevit hypotenze jako příznak IRR. Proto je třeba po dobu 12 hodin před podáním a v průběhu každé infuze přípravku Gazyvaro a po dobu první hodiny po jejím podání nepodávat antihypertenzní léčbu.
|
Den cyklu |
Pacienti, kteří vyžadují premedikaci |
Premedikace |
Podání |
|
Cyklus 1: Den 1 |
Všichni pacienti |
Intravenózní kortikosteroidy1 (povinné) |
Podat (dokončit podávání) alespoň 1 hodinu před podáním infuze přípravku Gazyvaro |
|
Perorální analgetika/antipyretika2 |
Podat alespoň 30 minut před podáním infuze přípravku Gazyvaro | ||
|
Antihistaminika3 | |||
|
Cyklus 1: |
Všichni pacienti |
Intravenózní kortikosteroidy1 (povinné) |
Podat (dokončit podávání) alespoň 1 hodinu před podáním infuze přípravku Gazyvaro |
|
Den 2 |
Perorální analgetika/antipyretika2 |
Podat alespoň 30 minut před podáním infuze přípravku Gazyvaro | |
|
Antihistaminika3 | |||
|
Pacienti bez IRR při předchozí infuzi |
Perorální analgetika/antipyretika2 |
Podat alespoň 30 minut před podáním infuze přípravku Gazyvaro | |
|
Všechny následné infuze |
Pacienti s IRR (stupeň 1 nebo 2) při předchozí infuzi |
Perorální analgetika/antipyretika2 Antihistaminika3 | |
|
Pacienti s IRR stupně 3 při předchozí infuzi NEBO pacienti s počtem lymfocytů >25 x 109/l před |
Intravenózní kortikosteroidy1 |
Podat (dokončit podávání) alespoň 1 hodinu před podáním infuze přípravku Gazyvaro | |
|
podáním následující léčby |
Perorální analgetika/antipyretika2 Antihistaminika3 |
Podat alespoň 30 minut před podáním infuze přípravku Gazyvaro |
1100 mg prednisonu/prednisolonu nebo 20 mg dexamethasonu nebo 80 mg methylprednisolonu. Hydrokortison se nepodává, protože není při snižování výskytu IRR účinný.
2 např. 1000 mg acetaminofenu/paracetamolu
3 např. 50 mg difenhydraminu
|
Den cyklu |
Pacienti, kteří vyžadují premedikaci |
Premedikace |
Podání |
|
Intravenózní kortikosteroidy1 (doporučeno) |
Podat (dokončit podávání) alespoň 1 hodinu před podáním infuze přípravku Gazyvaro | ||
|
Cyklus 1: Den 1 |
Všichni pacienti |
Perorální analgetika/antipyretika2 |
Podat alespoň 30 minut před podáním infuze přípravku Gazyvaro |
|
Antihistaminika3 | |||
|
Pacienti bez IRR při předchozí infuzi |
Perorální analgetika/antipyretika2 |
Podat alespoň 30 minut | |
|
Všechny následné infuze |
Pacienti s IRR (stupeň 1 nebo 2) při předchozí infuzi |
Perorální analgetika/antipyretika2 Antihistaminika3 |
před podáním infuze přípravku Gazyvaro |
|
Pacienti s IRR stupně 3 při předchozí infuzi NEBO Pacienti s počtem lymfocytů >25 x 109/l před podáním následující léčby |
Intravenózní kortikosteroidy1 |
Podat (dokončit podávání) alespoň 1 hodinu před podáním infuze přípravku Gazyvaro | |
|
Perorální analgetika/antipyretika2 Antihistaminika3 |
Podat alespoň 30 minut před podáním infuze přípravku Gazyvaro |
1100 mg prednisonu/prednisolonu nebo 20 mg dexamethasonu nebo 80 mg methylprednisolonu.
Hydrokortison se nepodává, protože není při snižování výskytu IRR účinný.
2 např. 1000 mg acetaminofenu/paracetamolu
3 např. 50 mg difenhydraminu
Dávka
Chronická lymfocytární leukemie (v kombinaci s chlorambucilem1)
Doporučená dávka přípravku Gazyvaro v kombinaci s chlorambucilem u pacientů s CLL je uvedená v tabulce 3.
Cyklus 1
Doporučená dávka přípravku Gazyvaro v kombinaci s chlorambucilem je 1000 mg podaných v průběhu 1. a 2. dne (den 1 a den 2 nebo při pokračovaní dnu 1) a poté 8. a 15. den (den 8 a den 15) prvního 28denního léčebného cyklu. Na infuzi na 1. a 2. den se připraví dva infuzní vaky (100 mg na den 1 a 900 mg na den 2). Pokud je první vak podán bez nutnosti úpravy rychlosti infuze nebo přerušení, je možné podat druhý vak ve stejný den (bez nutnosti odstupu mezi jednotlivými dávkami, bez opakovaného podání premedikace) za předpokladu, že na podání infuze je dostatečná doba a po celou dobu infuze budou k dispozici odpovídající podmínky a lékařský dohled. Pokud v průběhu první 100 mg infuze je rychlost infuze nutné upravit, nebo infuzi přerušit, musí se druhý vak podat následující den.
Cyklus 2 až 6
Doporučená dávka přípravku Gazyvaro v kombinaci s chlorambucilem je 1000 mg podaných 1. den každého cyklu.
|
Cyklus |
Den léčby |
Dávka přípravku Gazyvaro |
|
Cyklus 1 |
Den 1 |
100 mg |
|
Den 2 (nebo Den 1 pokračování) |
900 mg | |
|
Den 8 |
1000 mg | |
|
Den 15 |
1000 mg | |
|
Cyklus 2-6 |
Den 1 |
1000 mg |
1 Chlorambucil je podáván perorálně v dávce 0,5 mg/kg tělesné hmotnosti v den 1 a den 15 každého léčebného cyklu
Délka léčby
Šest cyklů léčby, každý v délce 28 dnů.
Folikulární lymfom (FL)
Doporučená dávka přípravku Gazyvaro v kombinaci s bendamustinem u pacientů s FL je uvedená v tabulce 4.
Dávkování (v kombinaci s bendamustinem2)
Cyklus 1
Doporučená dávka přípravku Gazyvaro v kombinaci s bendamustinem je 1000 mg podaných v 1., 8. a 15. den (den 1, den 8, den 15) prvního 28denního léčebného cyklu.
Cyklus 2 až 6
Doporučená dávka přípravku Gazyvaro v kombinaci s bendamustinem je 1000 mg podaných v 1. den každého cyklu.
Udržovací léčba
Pacientům, kteří reagují na úvodní léčbu (tj. 6 počátečních léčebných cyklů) přípravkem Gazyvaro v kombinaci s bendamustinem nebo kteří mají stabilní onemocnění, má být nadále podáván přípravek Gazyvaro 1000 mg jako udržovací léčba v monoterapii jednou za 2 měsíce po dobu dvou let nebo do progrese onemocnění (podle toho, co nastane dříve).
|
Cyklus |
Den léčby |
Dávka přípravku Gazyvaro |
|
Cyklus 1 |
Den 1 |
1 000 mg |
|
Den 8 |
1 000 mg | |
|
Den 15 |
1 000 mg | |
|
Cykly 2-6 |
Den 1 |
1 000 mg |
|
Udržovací léčba |
Jednou za dva měsíce po dobu dvou let nebo do progrese onemocnění (podle toho, co nastane dříve) |
1,000 mg |
2 Bendamustin se podává intravenózně v den 1 a 2 každého léčebného cyklu (cykly 1-6) v dávce 90 mg/m2/den Trvání léčby
Šest cyklů léčby, každý v délce 28 dnů, po udržovací léčbě jednou za 2 měsíce po dobu dvou let nebo do progrese onemocnění (podle toho, co nastane dříve).
Způsob podání
Přípravek Gazyvaro je určen k intravenóznímu podání. Podává se intravenózní infuzí prostřednictvím speciální linky po naředění. Infuze přípravku Gazyvaro se nesmí podávat formou intravenózní injekce nebo bolusu.
Instrukce týkající se ředění přípravku Gazyvaro před podáním jsou uvedené níže.
Instrukce týkající se rychlosti infuze jsou uvedeny v tabulkách 5-6.
Tabulka 5 Standardní rychlost infuze při absenci reakcí souvisejících s infuzí/hypersenzitivity u pacientů s CLL (v případě reakcí souvisejících s infuzí, viz Léčba IRR)
|
Cyklus |
Den léčby |
Rychlost infuze |
|
Cyklus 1 |
Den 1 (100 mg) |
Podává se rychlostí 25 mg/hodinu po dobu 4 hodin. Nezvyšujte rychlost infuze. |
|
Den 2 (nebo Den 1 pokračování) (900 mg) |
Pokud se v průběhu předchozí infuze nevyskytly reakce související s infuzí, podává se rychlostí 50 mg/hodinu. Rychlost infuze lze zvýšit o 50 mg/hodinu každých 30 minut až k maximální rychlosti 400 mg/hodinu. | |
|
Den 8 (1 000 mg) |
Pokud se v průběhu předchozí infuze nevyskytly reakce související s infuzí, kdy konečná rychlost infuze byla 100 mg/hodinu nebo rychlejší, infuzi lze zahájit rychlostí 100 mg/hodinu a zvyšovat o 100 mg/hodinu každých 30 minut až k maximální rychlosti 400 mg/hodinu. | |
|
Den 15 (1 000 mg) | ||
|
Cyklus 2-6 |
Den 1 (1 000 mg) |
Folikulární lymfom (FL)
Tabulka 6 Standardní rychlosti infuze při absenci reakcí souvisejících s
infuzí/hypersenzitivity u pacientů s FL (v případě reakcí souvisejících s infuzí, viz Léčba IRR)
|
Cyklus |
Den léčby |
Rychlost infuze |
|
Cyklus 1 |
Den 1 (1 000 mg) |
Podává se rychlostí 50 mg/hodinu. Rychlost infuze lze zvýšit o 50 mg/hodinu každých 30 minut až k maximální rychlosti 400 mg/hodinu. |
|
Den 8 (1 000 mg) | ||
|
Den 15 (1 000 mg) |
Pokud se v průběhu předchozí infuze nevyskytly reakce související s infuzí, kdy konečná rychlost infuze byla 100 mg/hodinu nebo rychlejší, infuzi lze zahájit rychlostí 100 mg/hodinu a zvyšovat o 100 mg/hodinu každých 30 minut až k maximální rychlosti 400 mg/hodinu. | |
|
Cykly 2-6 |
Den 1 (1 000 mg) | |
|
Udržovací léčba |
Jednou za dva měsíce po dobu dvou let nebo do progrese onemocnění (podle toho, co nastane dříve) |
Léčba IRR (všechny indikace)
Léčba IRR může vyžadovat dočasné přerušení infuze, snížení rychlosti infuze nebo ukončení léčby přípravkem Gazyvaro, jak je uvedeno níže.
• Stupeň 4 (život ohrožující): Infuze musí být zastavena a léčba trvale ukončena.
• Stupeň 3 (závažné): Infuze musí být dočasně zastavena a příznaky léčeny. Po odeznění příznaků může být infuze znovu zahájena s maximálně poloviční rychlostí, než byla rychlost předchozí (rychlost, kterou kapala infuze v době výskytu IRR), a pokud se žádné příznaky IRR znovu neobjeví, může se rychlost infuze postupně zvyšovat v přírůstcích a intervalech odpovídajících dané dávce (viz tabulky 5 a 6). U pacientů s CLL, u kterých se první dávka prvního cyklu rozděluje na dva dny, lze první den po jedné hodině rychlost infuze zvýšit zpět až na 25 mg/hodinu, ale ne více. Pokud se IRR stupně 3 objeví podruhé, infuze se musí zastavit a léčba trvale ukončit.
• Stupeň 1-2 (lehké až středně těžké): Rychlost infuze musí být snížena a příznaky léčeny. Po odeznění příznaků, a pokud se žádné příznaky znovu neobjevují, lze v infuzi pokračovat, rychlost infuze lze zvyšovat v přírůstcích a intervalech odpovídajících dané dávce (viz tabulky 5 a 6). U pacientů s CLL, u kterých se první dávka prvního cyklu rozděluje na dva dny, lze první den po jedné hodině rychlost infuze zvýšit zpět až na 25 mg/hodinu, ale ne více.
Návod k naředění
Přípravek Gazyvaro má připravit zdravotnický pracovník za použití techniky asepse.
Injekční lahvičkou netřeste.
CLL cykly 2-6 a všechny FL cykly
Z injekční lahvičky odeberte 40 ml koncentrátu a nařeďte je v infuzním vaku z polyvinylchloridu (PVC) nebo polyolefinu (non-PVC) obsahujícím 0,9% injekční roztok chloridu sodného (9 mg/ml).
Pouze CLL - cyklus 1
K zajištění odlišení dvou infuzních vaků pro úvodní 1000 mg dávku se doporučuje použít vaky různých velikostí, aby se odlišila dávka 100 mg pro 1. den 1. cyklu (Cyklus 1 Den 1) a dávka 900 mg pro 1. den 1. cyklu (Cyklus 1 Den 1, pokračování) nebo 2. den 1. cyklu (Cyklus 1 Den 2). K přípravě dvou infuzních vaků odeberte z injekční lahvičky 40 ml koncentrátu a 4 ml vstříkněte do 100 ml infuzního vaku z PVC nebo polyolefinu (non-PVC) a zbývajících 36 ml do 250 ml infuzního vaku z PVC nebo polyolefinu (non-PVC) obsahujícího 0,9% injekční roztok chloridu sodného (9 mg/ml). Každý infuzní vak označte viditelně štítkem.
|
Dávka přípravku Gazyvaro, která se bude podávat |
Požadované množství koncentrátu přípravku Gazyvaro |
Velikost infuzního vaku z PVC nebo polyolefinu (non-PVC) |
|
100 mg |
4 ml |
100 ml |
|
900 mg |
36 ml |
250 ml |
|
1000 mg |
40 ml |
250 ml |
Mezi přípravkem Gazyvaro v koncentracích pohybujících se v rozmezí od 0,4 mg/ml do 20,0 mg/ml po naředění 0,9% injekčním roztokem chloridu sodného (9 mg/ml) a následujícími zdravotnickými prostředky nebyly pozorovány žádné inkompatibility:
• vaky z PVC, polyethylenu (PE), polypropylenu nebo polyolefinu
• infuzní sety z PVC, polyuretanu (PUR) nebo PE
• volitelné in-line filtry s kontaktními povrchy z polyethersulfonu (PES), trojcestné infuzní kohouty z polykarbonátu (PC) a katetry z polyetherurethanu (PEU).
Nepoužívejte jiná rozpouštědla, jako je např. roztok glukózy (5%).
Vak se jemně obrátí, aby se roztok smísil a zabránilo se nadměrné tvorbě pěny. Naředěným roztokem se nesmí třást a nesmí se zmrazit.
Parenterální léčivé přípravky je nutné před podáním vizuálně zkontrolovat, zda neobsahují částice nebo nejsou zbarvené.
Po naředění byla chemická a fyzikální stabilita v 0,9% injekčním roztoku chloridu sodného (9 mg/ml) při koncentracích od 0,4 mg/ml do 20 mg/ml prokázána po dobu 24 hodin při teplotě od 2 °C do 8 °C a poté po dobu 48 hodin (včetně doby infuze) při teplotě < 30 °C.
Z mikrobiologického hlediska má být roztok použit okamžitě. Není-li použit okamžitě, doba a podmínky uchovávání přípravku po naředění před použitím jsou v odpovědnosti uživatele a normálně by doba neměla být delší než 24 hodin při teplotě 2 až 8 °C, pokud ředění neproběhlo za kontrolovaných a validovaných aseptických podmínek.
Likvidace
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
56