Fuzeon 90 Mg/Ml
SOUHRN ÚDAJŮ O PŘÍPRAVKU
PŘÍLOHA I
Fuzeon 90 mg/ml prášek a rozpouštědlo pro injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička obsahuje 108 mg enfuvirtidu (enfuvirtidum).
Jeden ml rekonstituovaného roztoku obsahuje 90 mg enfuvirtidu.
Pomocná látka se známým účinkem: sodík. Obsahuje méně než 1 mmol sodíku (23 mg) v jedné dávce, tj. je v podstatě bez obsahu sodíku.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční roztok Bílý až téměř bílý lyofilizovaný prášek.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Fuzeon je indikován spolu s dalšími antiretrovirovými léčivými přípravky k léčbě pacientů infikovaných virem HIV-1, u kterých došlo k selhání léčebných režimů obsahujících alespoň po jednom přípravku z následujících skupin antiretrovirových chemoterapeutik: inhibitorů HIV proteázy, nenukleozidových inhibitorů reverzní transkriptázy a nukleozidových inhibitorů reverzní transkriptázy, nebo se u těchto pacientů vyvinula nesnášenlivost k předchozím antiretrovirovým léčebným režimům (viz bod 5.1).
Při rozhodování o novém léčebném režimu u pacientů, u kterých došlo k selhání předchozí antiretrovirové léčby, je nutné pečlivě zvážit všechny údaje o dosavadní léčbě jednotlivého pacienta a charakteru mutací, spojených s různými léčivými přípravky. Je-li to možné, je vhodné provést vyšetření rezistence (viz bod 4.4 a 5.1).
4.2 Dávkování a způsob podání
Fuzeon by měl být předepisován pouze lékařem, který má zkušenosti s léčbou HIV infekce. Dávkování
Dospělí a mladiství > 16 let: Doporučená dávka Fuzeonu je 90 mg, dvakrát denně podávaná formou podkožní injekce do horní části paže, přední plochy stehna nebo břicha.
V případě vynechání dávky přípravku Fuzeon mají být pacienti poučeni, aby si vynechanou dávku podali co nejdříve. Je-li ale zbývající doba do příští dávky kratší než 6 hodin, má být dávka vynechána.
Starší osoby: Dosud nejsou zkušenosti s podáváním Fuzeonu u osob > 65 let.
Děti > 6 let a mladiství: Zkušenosti s podáváním dětem jsou omezené (viz bod 5.2). V klinických studiích bylo používáno dávkování uvedené v Tabulce 1:
Tabulka 1: Dávkování u dělí
|
Hmotnost (kg) |
Dávka na jednu injekci podávanou 2 x denně (mg/dávka) |
Objem injekce (90 mg enfuvirtidu v 1 ml) |
|
11,0 až 15,5 |
27 |
0,3 ml |
|
15,6 až 20,0 |
36 |
0,4 ml |
|
20,1 až 24,5 |
45 |
0,5 ml |
|
24,6 až 29,0 |
54 |
0,6 ml |
|
29,1 až 33,5 |
63 |
0,7 ml |
|
33,6 až 38,0 |
72 |
0,8 ml |
|
38,1 až 42,5 |
81 |
0,9 ml |
|
> 42,6 |
90 |
1,0 ml |
Vzhledem k nedostatku údajů týkajících se bezpečnosti a účinnosti se Fuzeon nedoporučuje podávat dětem do 6 let (viz bod 5.2).
Poškození ledvin: Dávkování není třeba upravovat u pacientů s poškozením ledvin včetně pacientů na dialýze (viz body 4.4 a 5.2).
Poškození jater: Nejsou k dispozici údaje o dávkování u pacientů s poškozením jater (viz body 4.4 a 5.2).
Způsob podání
Fuzeon je podáván pouze formou podkožní injekce. Návod k rekonstituci tohoto léčivého přípravku před jeho podáním je uveden v bodě 6.6.
4.3 Kontraindikace
Hypersensitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Fuzeon musí být podáván pouze jako součást kombinované léčby. Řiďte se prosím také doporučeními uvedenými v souhrnech údajů o přípravcích, které se týkají antiretrovirových léčivých přípravků používaných v kombinaci. Podobně jako u jiných antiretrovirotik, má být enfuvirtid optimálně kombinován s antiretrovirovými přípravky, na které je virus pacienta citlivý (viz bod 5.1).
Pacienty je třeba upozornit, že podávání Fuzeonu nevede k vyléčení HIV infekce. Přestože bylo prokázáno, že účinná virová suprese antiretrovirové terapie výrazně snižuje riziko přenosu infekce pohlavním stykem, tak případné riziko nelze vyloučit. Opatření k prevenci přenosu infekce mají být v souladu s národními požadavky.
Studie se zvířaty ukázaly, že enfuvirtid může zhoršovat některé imunitní funkce (viz bod 5.3).
V klinických hodnoceních byl u pacientů léčených přípravkem Fuzeon pozorován zvýšený výskyt některých bakteriálních infekcí, zejména se jednalo o vyšší výskyt pneumonie; nicméně zvýšené riziko bakteriální pneumonie spojené s užíváním přípravku Fuzeon nebylo potvrzeno následnými epidemiologickými údaji.
V souvislosti s léčbou enfuvirtidem se občas vyskytly případy přecitlivělosti a ve vzácných případech se opakovaly při znovuzahájení léčby. Alergické reakce zahrnovaly vyrážku, horečku, nevolnost a zvracení, zimnici, ztuhlost, pokles krevního tlaku, vzestup jaterních transamináz, a pravděpodobně primární imunokomplexové reakce - poruchy dýchání a glomerulonefritida. U pacientů, u kterých se vyskytly příznaky svědčící pro systémovou alergickou reakci, musí být léčba enfuvirtidem přerušena a pacienti musí ihned vyhledat lékařské ošetření. Při výskytu celkových známek a příznaků svědčících s vysokou pravděpodobností pro alergickou reakci na enfuvirtid, nemá být léčba enfuvirtidem znovu zahajována. Rizikové faktory umožňující předpovědět výskyt a závažnost hypersenzitivity na enfuvirtid nebyly identifikovány.
Jaterní onemocnění: Bezpečnost a účinnost enfuvirtidu nebyla speciálně u pacientů s významnými jaterními chorobami studována. U pacientů s chronickou hepatitidou B a C, kteří užívají antiretrovirovou léčbu, existuje zvýšené riziko výskytu závažných a potenciálně fatálních nežádoucích příhod postihujících játra. Do fáze III klinických studií byl zařazen jen omezený počet pacientů současně infikovaných hepatitidou B/C. U těchto pacientů nevedla léčba Fuzeonem ke zvýšenému výskytu jaterních poruch. V případě souběžné antivirové léčby hepatitidy B nebo C se prosím řiďte doporučeními uvedenými v informacích o přípravku pro příslušné léčivé přípravky.
Podávání Fuzeonu osobám, které nejsou infikovány virem HIV, může navodit tvorbu protilátek proti enfuvirtidu, které zkříženě reagují s HIV gp41. Následkem toho může dojít k falešné pozitivitě ELISA testů na přítomnost protilátek proti HIV.
Nejsou žádné zkušenosti u pacientů s poruchou funkce jater. Omezené zkušenosti jsou u pacientů se středně těžkou až závažnou poruchou renálních funkcí a u pacientů na dialýze. Těmto skupinám pacientů je třeba podávat Fuzeon s opatrností (viz body 4.2 a 5.2).
Syndrom imunitní reaktivace: Při zavedení kombinované antiretrovirové léčby (combination antiretroviral therapy, CART) se u pacientů s infekcí HIV s těžkou imunodeficiencí může vyskytnout zánětlivá reakce na asymptomatické nebo reziduální oportunní patogeny, která může způsobit klinicky závažné stavy nebo zhoršení příznaků onemocnění. Takové reakce byly nej častěji pozorovány během několika prvních týdnů či měsíců od zahájení CART. Jedná se například o cytomegalovirovou retinitidu, generalizované a/nebo fokální mykobakteriální infekce a pneumonii způsobenou Pneumocystis carinii. Jakékoli příznaky zánětu by měly být vyhodnoceny a v případě potřeby by měla být zahájena příslušná léčba.
Při imunitní reaktivaci byl také hlášen výskyt autoimunitních onemocnění (jako je Gravesova choroba), avšak hlášená doba do jejich nástupu byla velmi různá. Tyto stavy se mohou objevit mnoho měsíců po zahájení léčby.
Osteonekróza:
Ačkoli je etiologie považována za multifaktoriální (zahrnující používání kortikosteroidů, konzumaci alkoholu, těžkou imunosupresi a vyšší index tělesné hmotnosti), byly případy osteonekrózy hlášeny především u pacientů s pokročilým onemocněním HIV a/nebo při dlouhodobé expozici kombinované antiretrovirové terapii (CART). Pacienti mají být poučeni, aby vyhledali lékařskou pomoc, pokud zaznamenají bolesti kloubů, ztuhlost kloubů nebo pokud mají pohybové potíže.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Studie interakcí byly provedeny pouze u dospělých.
Není pravděpodobné, že by se mohly objevit klinicky významné farmakokinetické interakce mezi enfuvirtidem a jinými současně podávanými léčivými přípravky, které jsou metabolizovány prostřednictvím enzymů cytochromu CYP450.
Vliv enfuvirtidu na metabolizmus současně užívaných léčivých přípravků: V metabolické studii in vivo na lidech neinhiboval enfuvirtid podávaný v doporučené dávce 90 mg dvakrát denně metabolizmus substrátů CYP3A4 (dapson), CYP2D6 (debrisochin), CYP1A2 (kofein), CYP2C19 (mefenytoin) a CYP2E1 (chlorzoxazon).
Vliv současně užívaných léčivých přípravků na metabolizmus enfuvirtidu: Samostatné farmakokinetické studie neprokázaly, že by současné podávání ritonaviru (účinný inhibitor CYP3A4) nebo saquinaviru v kombinaci s indukčními dávkami ritonaviru či rifampicinu (účinný induktor CYP3A4) mělo klinicky významný vliv na farmakokinetiku enfuvirtidu.
4.6 Fertilita, těhotenství a kojení
Těhotenství: Dosud nebyly provedeny odpovídající kontrolované klinické studie u těhotných žen. Studie na zvířatech neprokázaly škodlivý vliv na vývoj plodu. V těhotenství by měl být enfuvirtid použit jen tehdy, jestliže možný prospěch z léčby převáží případné riziko pro plod.
Kojení: Není známo, zda je enfuvirtid vylučován do mateřského mléka. Matky by měly být poučeny, že nemají kojit, užívají-li enfuvirtid, protože hrozí nebezpečí přenosu HIV a rozvoje možných nežádoucích účinků u kojených dětí.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Nebyly provedeny žádné studie sledující vliv na schopnost řídit a obsluhovat stroje. Nejsou žádné důkazy o tom, že by enfuvirtid mohl jakkoliv ovlivnit schopnost pacienta řídit či ovládat stroje, nicméně je třeba vzít v úvahu možné nežádoucí účinky enfuvirtidu (viz bod 4.8).
4.8 Nežádoucí účinky
a. Shrnutí bezpečnostního profilu
Údaje o bezpečnosti přípravku se vztahují zejména k souhrnným hodnotám získaným v průběhu 48. týdne klinických studiích TORO 1 a TORO 2 (viz bod 5.1). Výsledky jsou vyjádřeny jako počet pacientů, u nichž se projevily nežádoucí účinky (mimo reakcí v místě vpichu), na 100 paciento-roků v průběhu podávání léku.
Nej častěji hlášené příhody byly reakce v místě vpichu injekce, průjem a nauzea. Přidání Fuzeonu k ostatním antiretrovirovým přípravkům obecně nezvýšilo výskyt či závažnost většiny nežádoucích účinků.
b. Shrnutí nežádoucích účinků do tabulky
Tabulka 2 uvádí nežádoucí účinky, které se vyskytovaly častěji u pacientů léčených Fuzeonem v kombinaci s dalšími antiretrovirotiky než u pacientů léčených ostatními antiretrovirotiky bez Fuzeonu. Uvedeny jsou takové nežádoucí účinky, jejichž výskyt se zvýšil v souvislosti s léčbou nejméně u 2 pacientů na 100 paciento-roků. Jako statisticky signifikantní se ukázalo zvýšení výskytu pneumonie a lymfadenopatie. Většina nežádoucích účinků dosahovala mírného nebo středního stupně závažnosti. Nežádoucí účinky jsou uvedené podle MedDRA tříd orgánových systémů a kategorií četností. Kategorie četností jsou definovány za použití následující konvence: velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1000 až < 1/100), vzácné (> 1/10 000 až < 1/1000), velmi vzácné (< 1/10 000) a není známo (z dostupných údajů nelze určit).
Tabulka 2: Nežádoucí účinky vztahující se k léčbě přípravkem Fuzeon ve studii TORO 1 a kombinované studii TORO 2
|
Třídy orgánových systémů Četnost |
Nežádoucí účinek |
|
Infekce and infestace Časté |
Sinusitida, kožní papilom, chřipka, pneumonie, ušní infekce |
|
Poruchy krve a lymfatického systému Časté |
Lymfadenopatie |
|
Poruchy metabolismu a výživy Časté |
Snížení chuti k jídlu, anorexie, hypertriglyceridemie, zvýšení krevních triglyceridů, diabetes mellitus |
|
Psychiatrické poruchy Časté |
Úzkost, noční můry, podrážděnost |
|
Poruchy nervového systému Velmi časté Časté |
Periferní neuropatie Hypestesie, poruchy pozornosti, třes |
|
Poruchy oka Časté |
Konjunktivitida |
|
Poruchy ucha a labyrintu Časté | |
|
Respirační, hrudní a mediastinální poruchy Časté |
Překrvení nosní sliznice |
|
Gastrointestinální poruchy Časté |
Pankreatitida, nemoc gastroesofageálního refluxu |
|
Poruchy kůže a podkožní tkáně Časté |
Suchá kůže, seboroický ekzém, erytém, akné |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně Časté |
Myalgie |
|
Poruchy ledvin a močových cest Časté |
Nefrolitiáza, hematurie |
|
Celkové poruchy a reakce v místě aplikace Velmi časté Časté |
Pokles hmotnosti Příznaky připomínající chřipkovité onemocnění, slabost |
c. Popis vybraných nežádoucích účinků Reakce v místě vpichu injekce
Reakce v místě vpichu injekce (ISRs - Injection site reactions) patřily mezi nej častěji hlášené nežádoucí účinky a vyskytovaly se u 98 % pacientů (Tabulka 3). Převážná většina ISRs se objevila během prvního týdne podání přípravku Fuzeon a byla spojena s mírnou až středně závažnou bolestí
nebo nepříjemnými pocity v místě vpichu injekce, které ovšem nijak neomezovaly běžné denní činnosti. Stupeň závažnosti bolesti a nepříjemných pocitů se v průběhu léčby nezvyšoval. Známky a příznaky obecně trvaly 7 či méně dní. Infekce v místě vpichu injekce (včetně abscesu a celulitidy) se objevila u 1,5 % pacientů.
Tabulka 3: Shrnutí jednotlivých známek/příznaků charakterizující reakce v místě vpichu _injekce ve studii TORO 1 ^ a v kombinované studii TORO 2 (% pacientů)_
|
n=663 | |||
|
Procento ukončení kvůli ISRs |
4% | ||
|
Kategorie příhody |
Fuzeon +optimalizované prostředí3 |
% příhody zahrnující reakce stupně 3 |
% příhody zahrnující reakce stupně 4 |
|
Bolest / nepříjemné pocity |
96,1 % |
11,0 %b |
0 %b |
|
Erytém |
90,8 % |
23,8 %c |
10,5 %c |
|
Indurace |
90,2 % |
43,5 %d |
19,4 %d |
|
Noduly a cysty |
80,4 % |
29,1 %e |
0,2 %e |
|
Pruritus |
65,2 % |
3,9 %f |
NA |
|
Ekchymóza |
51,9 % |
8,7 %g |
4,7 %g |
a Jakýkoli stupeň závažnosti.
b Stupeň 3 = závažná bolest vyžadující analgetika (nebo narkotická analgetika pro < 72 h) a/nebo omezující bežné aktivity; Stupeň 4 = závažná bolest vyžadující hospitalizaci nebo prodloužení hospitalizace, vedoucí k úmrtí, nebo trvalému či významnému postižení/nezpůsobilosti, nebo život ohrožující či lékařsky významné. c Stupeň 3 = střední průměr > 50 mm, ale < 85 mm; Stupeň 4 =střední průměr > 85 mm. d Stupeň 3 = střední průměr > 25 mm, ale < 50 mm; Stupeň 4 = střední průměr > 50 mm. e Stupeň 3 = > 3 cm; Stupeň 4 = v případě vyčerpání.
f Stupeň 3 = refrakterní na topickou léčbu nebo vyžadující perorální či parenterální léčbu; Stupeň 4 = nedefinováno. g Stupeň 3 = > 3 cm, ale < 5 cm; Stupeň 4 = > 5 cm.
Navíc se vyskytl malý počet případů hypersenzitivních reakcí na enfuvirtid, které se někdy opakovaly při pokusech o opětovné zahájení léčby (viz bod 4.4).
Další nežádoucí účinky
Při zahájení kombinované protivirové léčby (CART) se u pacientů infikovaných HIV s těžkou imunodeficiencí může vyskytnout zánětlivá reakce na asymptomatické nebo reziduální oportunní infekce. Byl také hlášen výskyt autoimunitních onemocnění (jako je Gravesova choroba); avšak hlášená doba do jejich nástupu byla velmi různá. Tyto stavy se mohou objevit mnoho měsíců po zahájení léčby (viz bod 4.4).
Byly hlášeny případy osteonekrózy, a to především u pacientů s obecně známými rizikovými faktory, s pokročilým onemocněním HIV nebo při dlouhodobé expozici CART. Jejich frekvence není známa (viz bod 4.4).
Protože enfuvirtid je peptid, může způsobit kožní amyloidózu v místě vpichu injekce.
Laboratorní abnormality
V průběhu studií se u většiny pacientů neobjevily změny stupně toxicity u žádného z laboratorních parametrů, s výjimkou případů uvedených v Tabulce 4. V průběhu 48. týdne studie se častěji vyskytla eozinofilie [hodnoty vyšší nežli horní referenční mez, > 0,7 x 109/l] u skupiny pacientů léčených Fuzeonem (12,4 pacienta s eozinofilií na 100 paciento-roků) ve srovnání s pacienty léčenými pouze ostatními antiretrovirotiky (5,6 pacienta na 100 paciento-roků). Při použití vyššího kritéria pro eozinofilii (> 1,4 x 109/l) je počet pacientů s eozinofilií v obou skupinách srovnatelný (1,8 pacienta s eozinofilií na 100 paciento-roků).
Tabulka 4: Abnormality laboratorních nálezů 3. a 4. stupně souvisejících s léčbou u skupiny
pacientů léčených Fuzeonem v kombinaci s dalšími antiretrovirotiky a u skupiny pacientů léčených dalšími antiretrovirotiky bez podávání Fuzeonu, které se vyskytovaly u více než 2 pacientů na 100 paciento-roků_
|
Stupně laboratorních |
Fuzeon + další antiretrovirotika |
Antiretrovirotika jiná než Fuzeon |
|
parametrů |
na 100 paciento-roků |
na 100 paciento-roků |
|
Počet |
663 |
334 |
|
(Celkový počet léčených |
(557,0) |
(162,1) |
|
pacientů za rok) | ||
|
ALAT | ||
|
St. 3 (> 5-10 x ULN) |
4,8 |
4,3 |
|
St. 4 (> 10 x ULN) |
1,4 |
1,2 |
|
Hemoglobin | ||
|
St. 3 (6,5-7,9 g/dl) |
2,0 |
1,9 |
|
St. 4 (< 6,5 g/dl) |
0,7 |
1,2 |
|
Kreatinin fosfokináza | ||
|
St. 3 (> 5-10 x ULN) |
8,3 |
8,0 |
|
St. 4 (> 10 x ULN) |
3,1 |
8,6 |
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Dosud nebyl hlášen žádný případ předávkování. Nejvyšší jednorázová dávka podaná 12 pacientům v rámci klinické studie bylo 180 mg podkožně. U žádného z těchto pacientů se neobjevily nežádoucí účinky, které by se nevyskytovaly také u osob léčených doporučenými dávkami. Ve studii „Early Access Program” bylo 1 pacientovi podáno 180 mg Fuzeonu v jednorázové dávce. U pacienta se nevyskytly žádné nežádoucí účinky, které byly důsledkem podání léku.
Není k dispozici žádné specifické antidotum, které by bylo možno podat v případě předávkování enfuvirtidem. Při léčbě případného předávkování lze použít jen všeobecná podpůrná opatření.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Jiná antivirotika, ACT kód: J05AX07
Mechanizmus účinku: Enfuvirtid patří do skupiny léků zvaných inhibitory fúze. Inhibuje strukturální přestavbu glykoproteinu gp-41 viru HIV a jeho účinek je založen na specifické vazbě na virový protein, čímž extracelulárně blokuje fúzi mezi membránou viru a membránou cílových buněk a zabrání vstupu virové RNA do těchto buněk.
Antivirová aktivita in vitro: U citlivosti na enfuvirtid testované u 612 rekombinantních kmenů HIV obsahujících env geny ze vzorků HIV RNA odebraných od pacientů na začátku III. fáze klinických studií byl geometrický průměr EC50 = 0,259 pg/ml (geometrický průměr + 2SD = 1,96 pg/ml) při stanovení průniku viru s rekombinantním fenotypem do buňky. Enfúvirtid rovněž inhiboval buněčnou fúzi zprostředkovanou proteinovým obalem HIV-1. Klinické studie sledující účinek enfuvirtidu v kombinaci s ostatními antiretrovirotiky prokázaly jeho zřejmý aditivní až synergický efekt, přičemž antagonizmus mezi podávanými látkami nebyl prokázán. Vztah mezi citlivostí HIV-1 k enfuvirtidu in vitro a inhibicí replikace HIV-1 v lidském organizmu dosud nebyl stanoven.
Rezistence na antiretrovirotika: Neúplná suprese virové replikace může mít za následek vznik lékové rezistence na jednu nebo více složek terapeutického režimu.
Rezistence na enfuvirtid in vitro: Ve studiích in vitro byly vyselektovány izoláty HIV-1 se sníženou citlivostí na enfuvirtid, které se vyznačovaly substitucí aminokyselin na pozicích 36 - 38 vnější domény glykoproteinu gp-41. Tyto substituce korelovaly s různými stupni snížené citlivosti místně cílených HIV mutant na enfuvirtid.
Rezistence na enfuvirtid in vivo: HIV rekombinanty obsahující env geny ze vzorků HIV RNA od 187 pacientů odebraných do 24. týdne III. fáze klinického hodnocení ukázaly více než čtyřikrát nižší citlivost na enfuvirtid ve srovnání se vzorky odebranými před zahájením léčby. U 185 (98,9 %) z nich byla zjištěna substituce aminokyselin v env genech v oblasti 36 - 45 glykoproteinu gp-41. Pozorované substituce se vyskytovaly s klesající četností na těchto pozicích: 38, 43, 36, 40, 42 a 45. Specifické jednotlivé substituce v těchto oblastech glykoproteinu gp-41 měly za následek snížení citlivosti rekombinantních kmenů vůči enfuvirtidu v porovnání s výchozími hodnotami před zahájením léčby. Geometrický průměr změn citlivosti se pohyboval od 15,2 násobně nižší citlivosti u substituce V38M až do 41,6 násobně nižší citlivosti u substituce V38A. Dosud nebylo k dispozici dostatečné množství klinických izolátů s mnohočetnými substitucemi, které by umožnily konzistentně stanovit význam těchto substitucí a jejich vliv na citlivost viru k enfuvirtidu. Vzájemný vztah těchto substitucí a účinnosti enfuvirtidu in vivo nebyl doposud definován. Pokles citlivosti viru koreloval se stupněm rezistence k ostatním antiretrovirotikům před zahájením léčby enfuvirtidem (viz Tabulka 6).
Zkřížená rezistence: Díky zcela novému místu účinku je enfuvirtid in vitro stejně účinný jak u divokých i klinických izolátů HIV, tak i kmenů s rezistencí proti jedné, dvěma nebo dokonce všem třem skupinám ostatních antiretrovirotik (nukleozidové inhibitory reverzní transkriptázy, nenukleozidové inhibitory reverzní transkriptázy a inhibitory proteázy). Naopak, neočekává se, že mutanty se substitucí aminokyselin na pozicích 36 - 45 glykoproteinu gp-41, které jsou rezistentní na enfuvirtid, budou vykazovat zkříženou rezistenci k ostatním skupinám antiretrovirotik.
Klinické farmakodynamické údaje
Studie u pacientů již léčených antiretrovirotiky: Klinický účinek Fuzeonu (v kombinaci s ostatními antiretrovirovými přípravky) na plazmatické hladiny HIV RNA a počet CD4 lymfocytů byl studován ve dvou randomizovaných, multicentrických, kontrolovaných studiích, které trvaly 48 týdnů (TORO 1 a TORO 2). Populace "Intent-to-Treat" zahrnovala 995 pacientů. U této demografické skupiny byl medián výchozí hodnoty HIV-1 RNA 5,2 logi0 kopií/ml a medián výchozí hodnoty počtu CD4+ buněk 88 buněk/mm3 u skupiny léčené Fuzeonem v kombinaci s dalšími antiretrovirotiky resp. 5,1 log10 kopií/ml a 97 buněk/mm3 ve skupině léčené ostatními antiretrovirotiky mimo Fuzeonu. U pacientů před léčbou bylo průměrně aplikováno 12 antiretrovirových přípravků v průběhu průměrně 7 let. Všichni pacienti byli léčeni obvykle 3 - 5 antiretrovirovými přípravky, které byly zvoleny na základě předchozí léčby a podle stanovení genotypové a fenotypové rezistence vůči viru.
Mezi pacienty s počtem virových kopií < 400 kopií/ml v průběhu 48. týdne bylo 30,4 % pacientů léčených Fuzeonem v kombinaci s dalšími antiretrovirotiky, v porovnání s 12 % pacientů léčených pouze ostatními antiretrovirotiky (ARV) bez Fuzeonu. Průměrný nárůst počtu CD4 buněk byl větší u pacientů léčených Fuzeonem v kombinaci s dalšími antiretrovirotiky ve srovnání s pacienty léčenými pouze ostatními antiretrovirotiky (viz Tabulka 5).
Tabulka 5: Výsledné hodnoty randomizované léčby získané ve 48. týdnu (souhrn ze studií
|
TORO 1 a TORO 2, |
[TT) | ||||
|
Výsledky |
Fuzeon + ostatní ARV 90 mg 2x denně (N=661) |
Ostatní ARV (N=334) |
Rozdíly mezi skupinami |
95 % konfidenční interval |
Hodnota p |
|
HIV-1 RNA Logaritmus změny v porovnání s výchozí hodnotou (logio kopií/ml) |
-1,48 |
-0,63 |
LSM -0,85 |
-1,073, -0,628 |
<.0001 |
|
Počet CD4+ buněk Změna v porovnání s výchozí hodnotou (buněk/mm3)# |
+91 |
+45 |
LSM 46,4 |
25,1, 67,8 |
<.0001 |
|
HIV RNA >1 log pod výchozí hodnotou |
247 (37,4 %) |
57 (17,1 %) |
poměr šancí 3,02 |
2,16, 4,20 |
<.0001 |
|
HIV RNA < 400 kopií/ml** |
201 (30,4 %) |
40 (12,0 %) |
poměr šancí 3,45 |
2,36, 5,06 |
<.0001 |
|
HIV RNA < 50 kopií/ml** |
121 (183 %) |
26 (7,8 %) |
poměr šancí 2,77 |
1,76, 4,37 |
<.0001 |
|
Přerušení z důvodu výskytu nežádoucích účinků/přidružených onemocnění/laboratorních t nálezů |
9 % |
11 % | |||
|
Přerušení z důvodu reakce v t místě vpichu |
4 % |
N/A | |||
|
Přerušení z jiných důvodů t$§ |
13 % |
25 % | |||
*
Údaje o počtu kopií viru/ml periferní krve získané ve 48. týdnu studie na základě výsledků souhrnných dat ze studií TORO 1 a TORO 2 u ITT populace jedinců, u kterých neproběhla následná kontrola, přerušili terapii nebo u nich došlo k virologickému selhání - záměně při jejich posledním měření (LOCF).
#
**
t
$
§
Použita poslední hodnota.
M-H test: Přerušení nebo virologická selhání považovaná za selhání.
Procentuální zastoupení určené u populace léčené Fuzeonem + dalšími látkami (N=663) a pouze dalšími látkami (N=334). Jmenovatel u počtu pacientů, kteří nepřešli z jedné skupiny do druhé: N=112.
Dle úsudku řešitele klinické studie.
Zahrnuje přerušení z důvodu vynechání následné kontroly, odmítnutí léčby a z dalších důvodů.
Terapie Fuzeonem a ostatními antiretrovirotiky byla u vyššího průměrného počtu pacientů asociována s dosažením < 400 virových kopií/ml (nebo < 50 kopií/ml) ve všech podskupinách podle výchozích hodnot počtu CD4+, výchozích hodnot HIV-1 RNA, počtu antiretrovirotik (ARV) aplikovaných v předchozí době, nebo počtu aktivních ARV, které byly součástí režimu léčby ostatními antiretrovirotiky. Taktéž u jedinců s výchozími hodnotami CD4+ buněk > 100 buněk/mm3, s výchozími hodnotami HIV-1 RNA < 5,0 log10 kopií/ml, s < 10 předchozími ARV anebo jinými aktivními ARV v režimu léčby ostatními antiretrovirotiky, byly častěji dosahovány hodnoty HIV-1 RNA < 400 virových kopií/ml (nebo < 50 kopií/ml) při téže léčbě (viz Tabulka 6).
Tabulka 6: Procentuální zastoupení pacientů, kteří dosáhli hodnot < 400 virových kopií/ml a
< 50 kopií/ml ve 48. týdnu léčby podle podskupin (souhrnná data ze studií TORO 1 a TORO 2, ITT)_
|
Podskupiny |
HIV-1 RNA < 400 kopií/ml |
HIV-1 RNA < 50 kopií/ml | ||
|
Fuzeon + ostatní ARV 90 mg 2 x denně (N=661) |
ostatní ARV (N=334) |
Fuzeon + ostatní ARV 90 mg 2 x denně (N=661) |
ostatní ARV (N=334) | |
|
BL HIV-1 RNA < 5.0 log101 kopií/ml |
118/269 (43,9 %) |
26/144 (18,1 %) |
77/269 (28,6 %) |
18/144 (12,5 %) |
|
BL HIV-1 RNA > 5.0 logio1 kopií/ml |
83/392 (21,2 %) |
14/190 (7,4 %) |
44/392 (11,2 %) |
8/190 (4,2 %) |
|
Celkový počet předchozích ARV < 101 |
100/215 (46,5 %) |
29/120 (24,2 %) |
64/215 (29,8 %) |
19/120 (15,8 %) |
|
Celkový počet předchozích ARV > 101 |
101/446 (22,6 %) |
11/214 (5,1 %) |
57/446 (12,8 %) |
7/214 (3,3 %) |
|
Žádné podávané aktivní ARV 1,2 |
9/112 (8,0 %) |
0/53 (0 %) |
4/112 (3,5 %) |
0/53 (0 %) |
|
Jedno aktivní podávané ARV 1,2 |
56/194 (28,9 %) |
7/95 (7,4 %) |
34/194 (17,5 %) |
3/95 (3,2 %) |
|
Dvě nebo více než dvě podávaná aktivní ARV 12 |
130/344 (37,8 %) |
32/183 (17,5 %) |
77/334 (22,4 %) |
22/183 (12,0 %) |
'Přerušení a virologická selhání = selhání 2Na základě GSS skóre
5.2 Farmakokinetické vlastnosti
Farmakokinetické vlastnosti enfuvirtidu byly studovány jak u dospělých infikovaných HIV, tak i u dětí.
Absorpce: Absolutní biologická dostupnost po podkožním podání 90 mg enfuvirtidu v oblasti břicha činila 84,3 ± 15,5 %. Průměrná (± směrodatná odchylka) hodnota Cmax byla 4,59 ± 1,5 pg/ml, AUC byla 55,8 ± 12,1 pg *hod/ml. Subkutánní absorpce enfuvirtidu je v rozsahu dávky 45 až 180 mg proporcionální. Absorpce dávky 90 mg podané subkutánně je stejná jak v oblasti břicha tak i na stehnech či pažích. Ve čtyřech různých studiích (N= 9 až 12) činily průměrné hodnoty minimální plazmatické koncentrace v rovnovážném stavu 2,6 až 3,4 pg/ml.
Distribuce: Distribuční objem v rovnovážném stavu po intravenózním podání 90 mg enfuvirtidu byl 5,5 ± 1,1 l. Enfuvirtid je z 92 % vázán na plazmatické bílkoviny v plazmě infikované virem HIV v rozmezí koncentrace od 2 do 10 pg/ml, Enfuvirtid je vázán především na albumin a v menší míře na kyselý glykoprotein a-1. Ve studiích in vitro nebyl enfuvirtid z této vazby vytěsňován jinými léčivými přípravky a stejně tak enfuvirtid nevytěsňoval jiné přípravky z jejich vazebných míst. Hladiny enfuvirtidu zjištěné v mozkomíšním moku pacientů s HIV byly zanedbatelné.
Biotransformace: Enfuvirtid je peptid a lze proto očekávat, že bude katabolizován na jednotlivé aminokyseliny, které budou dále recyklovány v organizmu. Výsledky studií in vitro s lidskými mikrozómy a studií in vivo prokazuji, že enfuvirtid není inhibitorem enzymů CYP450. Ve studiích in vitro s lidskými mikrozómy a hepatocyty měla hydrolýza amidu C-koncové aminokyseliny fenylalaninu za následek tvorbu deamidovaného metabolitu, jehož produkce není závislá na NADPH. Tento metabolit lze po podání enfuvirtidu prokázat v lidské plazmě, jeho množství vyjádřené AUC se pohybuje v rozmezí 2,4 až 15 % AUC samotného enfuvirtidu.
Eliminace: Clearance enfuvirtidu po intravenózním podání 90 mg byla 1,4 ± 0,28 l/h a poločas vylučování byl 3,2 ± 0,42 hod. Po podkožním podání 90 mg enfuvirtidu byl poločas vylučování 3,8 ± 0,6 hod. Studie hmotnostní bilance k určení eliminační cesty/cest enfuvirtidu nebyla u lidí provedena.
Porucha funkce jater: Farmakokinetika enfuvirtidu u pacientů s jaterními poruchami nebyla dosud studována.
Porucha funkce ledvin: Z analýzy dat týkajících se plazmatické koncentrace enfuvirtidu u pacientů účastnících se klinických studií lze usuzovat, že clearance enfuvirtidu není klinicky významně ovlivněna u pacientů s mírnou až středně těžkou poruchou funkce ledvin. Ve výsledcích studie u poruchy funkce ledvin bylo AUC enfuvirtidu zvýšeno v průměru o 43 - 62 % u pacientů se závažnou poruchou funkce ledvin nebo u pacientů v koncovém stádiu renálního onemocnění v porovnání s pacienty s normálními renálními funkcemi. Hemodialýza neměla na clearance enfuvirtidu signifikantní vliv. V průběhu hemodialýzy bylo odstraněno méně než 13 % dávky. U pacientů s poruchou fúnkce ledvin není nutné upravovat dávkování.
Starší pacienti: Farmakokinetika enfuvirtidu nebyla studována u pacientů starších 65 let.
Pohlaví a hmotnost: Analýzy plazmatických koncentrací u pacientů v klinických studiích prokázaly, že clearance enfuvirtidu je u žen o 20 % nižší bez ohledu na váhu a že se vzestupem hmotnosti stoupá bez závislosti na pohlaví (clearance je o 20 % vyšší u osoby s hmotností 100 kg a o 20 % nižší u osoby s hmotností 40 kg než u osoby s hmotností 70 kg). Tyto změny nejsou klinicky významné a není třeba měnit dávku.
Rasa: Analýzy plazmatických koncentrací u pacientů v klinických studiích neprokázaly odlišnosti mezi kavkazskou a afro-americkou rasou v clearance enfuvirtidu. Jiné farmakokinetické studie neprokázaly po přihlédnutí k tělesné hmotnosti odlišnosti mezi asijskou a kavkazskou rasou.
Pediatrická populace: Farmakokinetika enfuvirtidu byla studována u 37 dětských pacientů. Po dávce 2 mg/kg dvakrát denně (maximálně 90 mg dvakrát denně) dosahovaly plazmatické koncentrace podobných hodnot jako u dospělých, kteří dostali dávku 90 mg dvakrát denně. U 25 dětských pacientů ve věku 5 až 16 let, kteří dostávali dávku 2 mg/kg dvakrát denně do horní části paže, přední části stehna nebo břicha, byla v rovnovážném stavu průměrná hodnota AUC 54,3 ± 23,5 pg *hod/ml, Cmax byla 6,14 ± 2,48 pg/ml, Cmin byla 2,93 ± 1,55 pg/ml.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti, toxicity přípravku po opakovaném podávání, genotoxicity a vlivu na vývoj embrya neprokazují zvláštní riziko pro člověka. Dlouhodobé studie kancerogenity u zvířat nebyly prováděny.
Studie na morčatech prokázaly možnost vzniku opožděné kožní přecitlivělosti na enfuvirtid. U zvířat, v krysím modelu rezistence vůči chřipkové infekci, byla pozorována porucha tvorby IFN-y (nebo interferonu-y). Rezistence vůči chřipkové a streptokokové infekci přesto nebyla u těchto krys výrazně snížena. Klinický význam těchto zjištění není znám.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek
Uhličitan sodný Mannitol Hydroxid sodný Kyselina chlorovodíková
Rozpouštědlo Voda na injekci
6.2 Inkompatibility
Tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky s výjimkou těch, které jsou uvedeny v bodě 6.6.
6.3 Doba použitelnosti
Prášek 4 roky
Rozpouštědlo
4 roky
Doba použitelnosti po rozpuštění
Po rozpuštění: Uchovávejte v chladničce (2°C - 8°C)
Chemická a fyzikální stabilita po otevření před použitím byla prokázána na dobu 48 hodin při teplotě
5 °C, za podmínky ochrany před světlem.
Z mikrobiologického hlediska má být přípravek použit okamžitě. Není-li použit okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou v odpovědnosti uživatele a normálně by doba neměla být delší než 24 hodin při teplotě 2 až 8 °C, pokud rekonstituce/ředění neproběhlo za kontrolovaných a validovaných aseptických podmínek.
6.4 Zvláštní opatření pro uchovávání
Prášek
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem. Podmínky uchovávání tohoto léčivého přípravku po jeho rekonstituci jsou uvedeny v bodě 6.3.
Rozpouštědlo
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
6.5 Druh obalu a velikost balení
Prášek
Injekční lahvička: 3 ml injekční lahvička, z bezbarvého skla typu 1 Uzávěr: zátka, pryž (neobsahující latex)
Plomba: hliníkový pertl se snímatelným víčkem
Rozpouštědlo
Lahvička: 2 ml injekční lahvička, z bezbarvého skla typu 1
Uzávěr: pryžová zátka (neobsahující latex)
Plomba: hliníkový pertl se snímatelným víčkem
Velikost balení
60 injekčních lahviček s práškem pro přípravu injekčního roztoku 60 injekčních lahviček s rozpouštědlem 60 injekčních stříkaček o objemu 3 ml 60 injekčních stříkaček o objemu 1 ml 180 tamponů s alkoholem
6.6 Zvláštní opatření pro likvidaci přípravku a jeho rekonstituci
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Před prvním užitím Fuzeonu musí být pacienti o užívání a způsobu podávání poučeni profesionálními zdravotnickými pracovníky.
K rozpouštění Fuzeonu smí být použita výhradně voda na injekci v objemu 1,1 ml. Pacientům je třeba vysvětlit, že po přidání vody na injekce je třeba opatrně poklepávat špičkami prstů na lahvičku, až se prášek začne rozpouštět. V žádném případě nesmějí s lahvičkou třepat, obracet ji nahoru a dolů, neboť by došlo k nadměrné tvorbě pěny. Když se prášek začne rozpouštět, je možné položit lahvičku na bok tak, aby se usnadnilo úplné rozpuštění prášku. Úplné rozpuštění prášku může trvat až 45 minut. Aby se zkrátila doba potřebná k úplnému rozpuštění, může pacient po přidání vody na injekce opatrně otáčet lahvičkou mezi rukama, dokud nedojde k úplnému rozpuštění. Před natažením roztoku do stříkačky musí pacient vizuálně zkontrolovat lahvičku, aby se ujistil, že se celý obsah rozpustil, že je roztok čirý a že neobsahuje bubliny či drobné částice. Obsahuje-li roztok drobné částice, nesmí být použit a lék je třeba zlikvidovat nebo vrátit do lékárny.
Injekční lahvička s rozpouštědlem obsahuje 2 ml vody na injekce, ze které musí být odebráno 1,1 ml pro rozpuštění prášku. Pacienti by měli být poučeni o nutnosti zlikvidovat zbytek vody, která zůstane v injekční lahvičce.
Fuzeon neobsahuje žádné konzervační přísady. Rekonstituovaný roztok má být použit okamžitě. Není-li možné užít připravený roztok okamžitě, je nutné jej uschovat v chladničce a užít během 24 hodin po rozpuštění. Ochlazený roztok je třeba před použitím nechat ohřát na pokojovou teplotu.
1 ml rekonstituovaného roztoku se aplikuje podkožně do horní části paže, břicha či přední části stehna. Injekce je třeba aplikovat do jiného místa než byla aplikována předchozí injekce, kde není přítomna lokální reakce po vpichu. Injekční lahvička je určena k jednomu použití, nepoužité zbytky musí být zlikvidovány.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/03/252/001
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 27. května 2003
Datum posledního prodloužení: 27. května 2008
10. DATUM REVIZE TEXTU
Podrobné informace o tomto přípravku jsou k dispizoci na webových stránkách Evropské lékové agentury pro léčivé přípravky http://www.ema.europa.eu
A. VÝROBCE ODPOVĚDNÝ /VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného za propouštění šarží
Roche Pharma AG, Emil-Barrell-Str. 1, D-79639 Grenzach-Wyhlen, Německo
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz Příloha I: Souhrn údajů o přípravku, bod 4.2)
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci dále předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
Neuplatňuje se.
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
Fuzeon 90 mg/ml prášek a rozpouštědlo pro injekční roztok Enfuvirtidum
2. OBSAH LÉČIVÉ LÁTKY/LÉCIVÝCH LÁTEK
Jedna injekční lahvička obsahuje 108 mg enfuvirtidu.
1 ml připraveného roztoku obsahuje 90 mg enfuvirtidu.
3. SEZNAM POMOCNÝCH LÁTEK
Jedna injekční lahvička s práškem také obsahuje uhličitan sodný, mannitol, hydroxid sodný a kyselinu chlorovodíkovou.
Jedna injekční lahvička s rozpouštědlem obsahuje 2 ml vody na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Prášek a rozpouštědlo pro injekční roztok Balení obsahuje:
60 injekčních lahviček s práškem pro injekční roztok 60 injekčních lahviček s rozpouštědlem 60 injekčních stříkaček o objemu 3 ml 60 injekčních stříkaček o objemu 1 ml 180 tamponů s alkoholem
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Subkutánní podání
Před použitím si přečtěte příbalovou informaci
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do:
9 ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem Po rozpuštění uchovávejte v chladničce
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
Voda na injekce, která zůstane v injekční lahvičce po odebrání 1,1 ml potřebného na rozpuštění prášku, musí být zlikvidována
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/03/252/001
13. ČÍSLO ŠARŽE
č. š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
Nevyžaduje se - odůvodnění přijato
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
2D čárový kód s jedinečným identifikátorem.
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
PC:
SN:
NN:
KRABIČKA PRO INJEKČNÍ LAHVIČKU PŘÍPRAVKU FUZEON
1 NÁZEV LÉČIVÉHO PŘÍPRAVKU
Fuzeon 90 mg/ml prášek pro injekční roztok Enfuvirtidum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna injekční lahvička s přípravkem Fuzeon obsahuje 108 mg enfuvirtidu. 1 ml připraveného roztoku obsahuje 90 mg enfuvirtidu.
3. SEZNAM POMOCNÝCH LÁTEK
Jedna injekční lahvička také obsahuje uhličitan sodný, mannitol, hydroxid sodný a kyselinu chlorovodíkovou.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Prášek pro injekční roztok
60 injekčních lahviček s práškem pro injekční roztok
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Subkutánní podání
Před použitím si přečtěte příbalovou informaci
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem Po rozpuštění uchovávejte v chladničce
10 ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11 NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/03/252/001
13. ČÍSLO ŠARŽE
c. s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léCivého přípravku vázán na lékařský předpis
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
Nevyžaduje se - odůvodnění přijato
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
2D Cárový kód s jedinečným identifikátorem.
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
PC:
SN:
NN:
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Fuzeon 90 mg/ml prášek pro injekční roztok
Enfuvirtidum
Subkutánní podání
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci
3. POUŽITELNOST
Použitelné do:
4. ČÍSLO ŠARŽE č. š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET DÁVEK
108 mg enfuvirtidum
6. JINÉ
KRABIČKA PRO INJEKČNÍ LAHVIČKU S VODOU NA INJEKCE
1 NÁZEV LÉČIVÉHO PŘÍPRAVKU
Rozpouštědlo pro injekční roztok Voda na injekci
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Rozpouštědlo pro parenterální podání
Krabička obsahuje 60 injekčních lahviček s 2 ml vody na injekci
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Tato voda na injekci je určena k rozpuštění přípravku Fuzeon 90 mg/ml prášek pro injekční roztok, získaný roztok je určen k podkožnímu podání Před použitím čtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
Voda na injekce, která zůstane v injekční lahvičce po odebrání 1,1 ml potřebného na rozpuštění prášku, musí být zlikvidována
TT název a adresa držitele rozhodnutí o registraci
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
T2. REGISTRAČNÍ CÍSLO/CÍSLA
EU/1/03/252/001
T3 ČÍSLO ŠARŽE
c. s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
Nevyžaduje se - odůvodnění přijato
T 7. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
2D čárový kód s jedinečným identifikátorem.
T8. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
PC:
SN:
NN:
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA(Y) PODÁNÍ
Rozpouštědlo pro injekční roztok Voda na injekci Subkutánní podání
2. ZPŮSOB PODÁNÍ
Před použitím čtěte příbalovou informaci
3. POUŽITELNOST
Použitelné do:
4. ČÍSLO ŠARŽE č. š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET DÁVEK
2 ml
6. JINÉ
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro pacienta
Fuzeon 90 mg/ml prášek a rozpouštědlo pro injekční roztok
Enfuvirtidum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
• Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
• Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
• Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí
ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
• Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Fuzeon a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Fuzeon používat
3. Jak se přípravek Fuzeon používá
4. Možné nežádoucí účinky
5. Jak přípravek Fuzeon uchovávat
6. Obsah balení a další informace
7. Návod „krok za krokem“ jak přípravek Fuzeon aplikovat
1. Co je přípravek Fuzeon a k čemu se používá
Co je přípravek Fuzeon
Přípravek Fuzeon obsahuje léčivou látku „enfuvirtid“ a patří do skupiny léčivých přípravků zvaných „antiretroviry“.
K čemu se přípravek Fuzeon používá
Přípravek Fuzeon se používá k léčbě viru lidské imunodeficience (HIV) - v kombinaci s dalšími antiretrovirovými léčivými přípravky u osob s infekcí HIV.
• Váš lékař Vám předepsal přípravek Fuzeon, aby se dosáhlo lepšího zvládání infekce HIV.
• Léčba přípravkem Fuzeon nevede k vyléčení HIV infekce.
Jak přípravek Fuzeon působí
HIV napadá buňky ve Vaší krvi zvané CD4 nebo T-lymfocyty. Virus se musí přichytit a vniknout do těchto buněk, aby se mohl množit. Přípravek Fuzeon tomu pomáhá zabránit.
2. Čemu musíte věnovat pozornost, než začnete přípravek Fuzeon používat
Nepoužívejte přípravek Fuzeon, jestliže
• jste alergický(á) na enfuvirtid nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Pokud si nejste jistý(á), poraďte se svým lékařem, lékárníkem nebo zdravotní sestrou před použitím přípravku Fuzeon.
Upozornění a opatření
Před užitím přípravku Fuzeon se poraďte se svým lékařem, lékárníkem nebo zdravotní sestrou, jestliže:
• jste někdy prodělal(a) nějaké plicní onemocnění
• jste někdy prodělal(a) nějaké ledvinové onemocnění
• máte chronickou hepatitidu (žloutenku) typu B nebo C nebo jiné jaterní onemocnění - máte větší pravděpodobnost, že se u Vás vyskytnou závažné jaterní potíže v průběhu používání tohoto léčivého přípravku
Známky z předchozích infekcí
U některých pacientů s HIV infekcí (AIDS) v pokročilém stádiu, kteří dříve prodělali oportunní infekce, se mohou brzy po zahájení antiretrovirové léčby vyskytnout známky a příznaky zánětu z předchozích infekcí. Má se za to, že tyto příznaky jsou důsledkem regenerace imunitního systému organizmu. Toto zlepšení umožní zdolávat infekce, které mohou být bez viditelných příznaků v těle přítomné. Všimnete-li si jakýchkoli příznaků infekce, informujte, prosím, ihned svého lékaře.
Známky autoimunitních onemocnění
Jakmile začnete užívat léčivé přípravky k léčbě HIV infekce, mohou se u Vás kromě oportunních infekcí vyskytnout autoimunitní onemocnění (stavy, které se vyskytují, když imunitní system napadá zdravé tkáně). Autoimunitní onemocnění se mohou objevit mnoho měsíců po zahájení léčby. Pokud zaznamenáte příznaky infekce nebo jiné příznaky jako jsou svalová slabost, slabost začínající v rukách a chodidlech a postupující směrem k tělesnému trupu, bušení srdce, třes nebo hyperaktivita, prosím, informujte ihned svého lékaře a požádejte ho o nezbytnou léčbu.
Pacienti s onemocněním jater
Pacienti s chronickou hepatitidou B nebo C, kteří jsou léčeni antiretrovirovou terapií, mají vyšší riziko závažných jaterních potíží. V případě, že jste prodělal(a) v minulosti jaterní onemocnění, poraďte se se svým lékařem.
Kostní poruchy (osteonekróza)
U některých pacientů se může při užívání kombinované antiretrovirové terapie vyvinout kostní onemocnění zvané osteonekróza. To se vyskytuje při odumírání kostní tkáně z důvodu ztráty zásobování kosti krví (odumírání kostní tkáně způsobené nedostatečným zásobením kosti krví).
• Známky osteonekrózy jsou ztuhlost kloubů, bolesti kloubů (zvláště kyčlí, kolen a ramen) a pohybové potíže. Pokud zpozorujete některé z těchto příznaků, informujte o tom, prosím, svého lékaře.
• Rizikové faktory vedoucí ke vzniku tohoto onemocnění zahrnují: délku kombinované antiretrovirové terapie, zda-li jste užíval(a) kortikosteroidy, množství konzumovaného alkoholu, stav Vašeho imunitního systému a pokud máte nadváhu.
Přenos HIV infekce na jinou osobu
Při užívání tohoto léčivého přípravku můžete stále přenášet infekci HIV, ačkoli toto riziko je nižší díky účinné antiretrovirové terapii. Konzultujte proto se svým lékařem možná opatření, která zabraňují přenosu infekce na ostatní osoby.
Další léčivé přípravky a přípravek Fuzeon
Informujte svého lékaře, lékárníka nebo zdravotní sestru o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat. To zahrnuje i léčivé přípravky běžně dostupné bez lékařského předpisu nebo rostlinné přípravky. Nebyl prokázán jakýkoliv vliv přípravku Fuzeon na ostatní současně užívané antiretrovirové léky či rifampicin (antibiotikum).
Přípravek Fuzeon s jídlem a pitím
Můžete používat Fuzeon s jídlem nebo bez jídla. Nicméně je třeba vždy respektovat doporučení uvedená v příbalových informacích dalších léků, které užíváte.
Těhotenství a kojení
• Pokud jste těhotná, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem dříve, než začnete tento přípravek užívat. Přípravek Fuzeon nesmíte používat s výjimkou případů, kdy Vám byl předepsán a doporučen k používání Vaším lékařem.
• Pokud máte infekci HIV, nesmíte kojit, protože je zde možný přenos HIV infekce na Vaše dítě.
Řízení dopravních prostředků a obsluha strojů
Vliv Fuzeonu na řízení motorového vozidla, používání přístrojů nebo obsluhování strojů nebyl testován. Máte-li při používání přípravku Fuzeon pocit závrati, neřiďte auto, nepracujte s žádnými přístroji ani neobsluhujte stroje.
Přípravek Fuzeon obsahuje sodík
Přípravek Fuzeon obsahuje méně než 1 mmol sodíku (23 mg) v jedné dávce, tj. je v podstatě „bez sodíku“
3. Jak se přípravek Fuzeon používá
Vždy používejte tento přípravek přesně podle pokynů svého lékaře nebo lékárníka. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Jak si připravit a aplikovat injekci přípravku Fuzeon
Přípravek Fuzeon musí být podán injekčně pod kůži - tzv. „subkutánní“ injekcí. V bodě 7 se dozvíte, jak si přípravek Fuzeon připravit a jak si sám(a) aplikovat injekci.
Jak velké množství přípravku použít
• Doporučená dávka přípravku je dvakrát denně 90 mg u dospělých a dospívajících (ve věku 16 let a starší).
• Podává se 1 ml injekce pod kůži.
• Přípravek Fuzeon se má podávat každý den nejlépe ve stejný čas.
• Vyzkoušejte a zvolte si dobu podávání dávek rovnoměrně od sebe, podle toho jak Vám vyhovují - např. první dávku ráno a pak v podvečer.
Další instrukce o tom, jak používat přípravek Fuzeon jsou uvedeny na konci příbalové informace (viz bod 7). Naleznete tam návod, jak přípravek Fuzeon připravit a jak si sám(sama) budete přípravek Fuzeon aplikovat.
Jestliže jste použil(a) více přípravku Fuzeon, než jste měl(a)
Jestliže jste použil(a) vyšší dávku Fuzeonu, než jste měl(a), poraďte se se svým lékařem nebo ihned navštivte nemocnici. Balení léčivého přípravku si vezměte s sebou.
Jestliže jste zapomněl(a) použít přípravek Fuzeon
• Jestliže jste zapomněl(a) použít dávku, aplikujte si ji co nejdříve, jakmile si vzpomenete. Je-li ale zbývající doba do příští dávky kratší než 6 hodin, tuto dávku vynechejte.
• Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku.
Jestliže jste přestal(a) používat přípravek Fuzeon
• Tento přípravek používejte tak dlouho, dokud Vám lékař nedoporučí, abyste léčbu ukončil(a). V případě, že přestanete přípravek používat a dojde k přerušení ve Vaší léčbě, může HIV ve Vaší krvi vyvinout rezistenci na přípravek Fuzeon rychleji. To je méně pravděpodobné, pokud přípravek budete používat pravidelně bez přerušení v průběhu léčby.
• Virus HIV ve Vaší krvi se případně může stát rezistentním na přípravek Fuzeon. Pokud k tomu dojde, může to mít za následek vzestup hladin viru v krvi. V tomto případě se Váš lékař může rozhodnout nepokračovat dále v léčbě přípravkem Fuzeon. O tomto rozhodnutí Vás bude Váš lékař informovat.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Pokud zaznamenáte jakékoli z níže uvedených závažných nežádoucích účinků, přestaňte používat přípravek Fuzeon a ihned navštivte lékaře - je možné, že budete potřebovat akutní lékařské ošetření:
• Alergická reakce (hypersenzitivita) - známky mohou zahrnovat: vyrážku, vysoké teploty nebo třesavku, nevolnost, pocení nebo třes.
Tyto nežádoucí účinky jsou vzácné (postihnou méně než 1 pacienta z 1 000). Tyto známky zcela nepotvrzují, že jste alergický(á) na tento léčivý přípravek.
Informujte svého lékaře, pokud se u Vás vyskytnou nežádoucí účinky v místě aplikace injekce
Nejčastější nežádoucí účinky (postihnou více než 1 pacienta z 10) jsou potíže v místě, kde si aplikujete injekci. Pravděpodobně se u Vás vyskytne některá z následujících lehkých až středně těžkých reakcí:
• zarudnutí
• otok
• pocit svědění
• tvorba modřin
• zatvrdnutí kůže či drobné bulky
• citlivost či bolestivost
Tyto reakce se mohou objevit během prvního týdne léčby Fuzeonem a obvykle trvají pouze 7 nebo méně než 7 dnů. Poté se potíže obvykle dále nezhoršují. Pokud zaznamenáte jakoukoli z těchto reakcí, nepřestávejte používat přípravek Fuzeon a informujte svého lékaře o jakýchkoli potížích, které máte.
Reakce mohou být závažnější, jestliže je injekce aplikována do stejného místa na těle. Také mohou být závažnější, když je injekce aplikována hlouběji než podkožně (např. do svalu). Vzácně může u Vás dojít k rozvoji infekce v místě vpichu jednotlivé injekce. Ke snížení rizika infekce je důležité dodržovat instrukce, které jsou uvedeny v bodě 7.
Přípravek Fuzeon může způsobit pod kůží v místě vpichu amyloidózu (ukládání amyloidu). Cítit to můžete jako bulku pod kůží. V případě výskytu bulky se obraťte, prosím, na svého lékaře.
Další možné nežádoucí účinky
Velmi časté (postihnou více než 1 pacienta z 10)
• průjem
• nevolnost
• úbytek hmotnosti
• bolest a pocit necitlivosti v rukou či nohou
Časté (postihnou méně než 1 pacienta z 10)
• pneumonie
• ušní infekce
• otok žláz (mízních uzlin)
• zanícené oko (konjunktivitida)
• příznaky podobné chřipce
• zanícené vedlejší dutiny nosní
• překrvení nosní sliznice
• anorexie
• pálení žáhy
• zánět slinivky břišní
• snížená chuť k j ídlu
• diabetes (cukrovka)
• noční můry
• pocit závrati
• třesavka (třes)
• úzkost či rozrušení
• neschopnost koncentrace
• snížená citlivost
• akné
• zarudnutí kůže
• ekzém
• suchá kůže
• bradavice
• bolest svalů
• ledvinové kameny
• pocit slabosti
• krev v moči
• změny v krevních testech (zvýšené hodnoty krevních tuků)
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Fuzeon uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené buď na nálepce injekční lahvičky s Fuzeonem nebo na nálepce injekční lahvičky s vodou na injekce za „Použitelné do“. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
Jednou připravený roztok pro injekci musí být ihned aplikován. V případě, že připravený roztok neaplikujete ihned, je třeba jej uchovávat v chladničce (2 °C - 8 °C) a užít jej během 24 hodin.. Neaplikujte si tento přípravek, pokud si všimnete v prášku nebo v roztoku po přidání vody na injekci nějaké částice. Rovněž nepoužívejte vodu na injekci, jestliže jste zpozoroval(a) v injekční lahvičce nějaké částice nebo zákal.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
Obsah balení a další informace
6.
Co přípravek Fuzeon obsahuje
• Léčivou látkou je enfuvirtid. Jedna injekční lahvička obsahuje 108 mg enfuvirtidu. Po připravení roztoku pomocí přiloženého rozpouštědla obsahuje 1 ml roztoku 90 mg enfuvirtidu.
• Dalšími složkami jsou:
Prášek
Uhličitan sodný Mannitol Hydroxid sodný Kyselina chlorovodíková
Rozpouštědlo Voda na injekci
Jak přípravek Fuzeon vypadá a co obsahuje toto balení
Fuzeon prášek a rozpouštědlo pro injekční roztok je dodáván v baleních obsahujících:
60 injekčních lahviček s Fuzeonem
60 injekčních lahviček s vodou na injekci, která slouží jako rozpouštědlo k přípravě roztoku 60 injekčních stříkaček o objemu 3 ml 60 injekčních stříkaček o objemu 1 ml 180 tamponů s alkoholem
Toto balení obsahuje vše, co potřebujete k přípravě a podání injekcí při léčbě Fuzeonem po dobu 30 dnů.
Držitel rozhodnutí o registraci
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
Výrobce odpovědný za propouštění šarží je
Roche Pharma AG Emil-Barell-Str. 1,
D-79639 Grenzach-Wyhlen Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci
|
Belgie/Belgique/Belgien N.V. Roche S.A. Tél/Tel: +32 (0) 2 525 82 11 |
Lietuva UAB “Roche Lietuva” Tel: +370 5 2546799 |
|
Bt^rapnn Pom Etnrapna EOOfl Ten: +359 2 818 44 44 |
Luxembourg/Luxemburg (Voir/siehe Belgique/Belgien) |
|
Česká republika Roche s. r. o. Tel: +420 - 2 20382111 |
Magyarország Roche (Magyarország) Kft. Tel: +36 - 23 446 800 |
|
Danmark Roche a/s Tlf: +45 - 36 39 99 99 |
Malta (See United Kingdom) |
|
Deutschland Roche Pharma AG Tel: +49 (0) 7624 140 |
Nederland Roche Nederland B.V. Tel: +31 (0) 348 438050 |
|
Eesti Roche Eesti OU Tel: + 372 - 6 177 380 |
Norge Roche Norge AS Tlf: +47 - 22 78 90 00 |
|
EkXába Roche (Hellas) A.E. Tr|k: +30 210 61 66 100 |
Osterreich Roche Austria GmbH Tel: +43 (0) 1 27739 |
|
Espaňa Roche Farma S.A. Tel: +34 - 91 324 81 00 |
Polska Roche Polska Sp.z o.o. Tel: +48 - 22 345 18 88 |
|
France Roche Tél: +33 (0) 1 47 61 40 00 |
Portugal Roche Farmaceutica Química, Lda Tel: +351 - 21 425 70 00 |
|
Hrvatska Roche d.o.o. Tel: + 385 1 47 22 333 |
Románia Roche Románia S.R.L. Tel: +40 21 206 47 01 |
|
Ireland Roche Products (Ireland) Ltd. Tel: +353 (0) 1 469 0700 |
Slovenija Roche farmacevtska družba d.o.o. Tel: +386 - 1 360 26 00 |
|
Island Roche a/s c/o Icepharma hf Sími: +354 540 8000 |
Slovenská republika Roche Slovensko, s.r.o. Tel: +421 - 2 52638201 |
|
Italia Roche S.p.A. Tel: +39 - 039 2471 |
Suomi/Finland Roche Oy Puh/Tel: +358 (0) 10 554 500 |
United Kingdom
Latvija
Roche Latvija SIA
Tel: +371 - 6 7039831
Roche Products Ltd.
Tel: +44 (0) 1707 366000
Tato příbalová informace byla naposledy revidována:
Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Na webových stránkách Evropské agentury pro léčivé přípravky je tato příbalová informace k dispozici ve všech úředních jazycích EU/EHP.
7. Návod „krok za krokem“ jak přípravek Fuzeon aplikovat
Tento přípravek používejte vždy přesně tak, jak Vám doporučil Váš lékař nebo lékárník. Pokud si nejste jistý(á), poraďte se se svým lékařem, lékárníkem nebo zdravotní sestrou.
Jak si lék aplikovat, jste-li levák
Na obrázcích v této příbalové informaci je ukázáno, jak si lék aplikují praváci. Jste-li levák(levačka), aplikujte si lék tak, jak Vám to vyhovuje. Pravděpodobně pro Vás bude nejlepší:
• když budete injekční stříkačku držet ve Vaší levé ruce a
• injekční lahvičku budete držet mezi palcem a ukazovákem pravé ruky.
Kdy je vhodné požádat o pomoc
Ze začátku bude asi obtížnější aplikace injekcí do některých míst, např. do horní části paží. Jestliže potřebujete pomoc, požádejte svého partnera, přítele nebo rodinného příslušníka. Případně by bylo dobré požádat někoho, aby s Vámi absolvoval nácvik podání injekce u Vašeho lékaře nebo zdravotní sestry.
Vaše injekční stříkačky
Injekční stříkačky, které jsou přiloženy k Vašemu léku, mají barevný ochranný kryt jehly. Ten je k jehle přiložen a nasazuje se zpětně na použitou jehlu, čímž se sníží riziko poranění dalších osob jehlou. I když je používání takto upravených injekčních stříkaček bezpečnější, je vždy třeba správně zlikvidovat použité injekční stříkačky tak, jak Vám poradil Váš lékař, lékárník nebo zdravotní sestra.
Bezpečnostní doporučení
• Vždy si dobře umyjte ruce. Tím se sníží nebezpečí bakteriální infekce.
• Jakmile máte ruce umyté, nedotýkejte se žádných jiných předmětů kromě těch, které slouží k přípravě a aplikaci léku.
• Při manipulaci s injekční stříkačkou se nikdy nesmíte dotknout jehly.
• Nikdy se nedotýkejte zátky injekční lahvičky poté, co byla dezinfikována alkoholovým tamponem.
• Nikdy nepoužívejte již otevřené balení. Před použitím se ujistěte, že žádná součást balení nebyla otevřená.
• Nikdy nepoužívejte ani nesdílejte použitou jehlu.
• Nikdy nepoužívejte injekční stříkačku s ohnutou nebo poškozenou jehlou.
• Nikdy nepoužívejte k rozpouštění svého léku vodu z vodovodu.
• Nikdy si neaplikujte svůj lék s jinými injekčními léky.
• Přípravek Fuzeon se aplikuje pouze pod kůži („subkutánně“).
• Fuzeon se nesmí aplikovat přímo do žíly („intravenózně“) nebo přímo do svalu („intramuskulárně“).
• Veškerý použitý materiál ukládejte do určeného odpadního kontejneru s uzávěrem. A to včetně injekčních lahviček, které obsahují nespotřebované množství léčivého přípravku nebo vodu na injekci, jelikož jsou určeny pouze k jednorázovému použití. V případě nejasností o bezpečné likvidaci použitého materiálu se poraďte se svým lékařem, lékárníkem nebo zdravotní sestrou.
V následujícím textu jsou krok za krokem probrány základní instrukce pro aplikaci Vašeho léčivého přípravku.
Krok A: Začínáme
1. Připravte si všechny následující pomůcky:
• Jednu injekční lahvičku s přípravkem Fuzeon (skleněná lahvička obsahující bílý prášek).
• Jednu injekční lahvičku s vodou na injekce (skleněná lahvička obsahující čirou bezbarvou tekutinu).
• Jednu injekční stříkačku o objemu 3 ml (větší) s jehlou dlouhou 25 mm.
• Jednu injekční stříkačku o objemu 1 ml (menší) s jehlou dlouhou 13 mm.
• 3 tampony napuštěné alkoholem.
• Víčkem opatřený kontejner určený k bezpečné likvidaci použitého materiálu.
2. Otevřete obaly injekčních stříkaček a sejměte víčka injekčních lahviček.
• Vyhoďte obaly injekčních stříkaček a víčka injekčních lahviček do odpadního kontejneru s uzávěrem.
• Položte injekční stříkačky a injekční lahvičky na čisté místo.
3. Pečlivě si umyjte ruce.
• Po umytí rukou se nedotýkejte žádných jiných předmětů než těch, které potřebujete k přípravě a aplikaci léku ani místa, kam bude injekce aplikována.
4. Očistěte zátky injekčních lahviček.
• Otřete zátku každé injekční lahvičky čerstvým alkoholovým polštářkem. Ponechte zátku oschnout na vzduchu.
• Nedotýkejte se pryžových zátek injekčních lahviček poté, co jste je očistil(a). Jestliže jste se jich dotkl(a), musíte je znovu očistit.
Krok B: Rozpouštění přípravku Fuzeon
Natažení vody na injekci do injekční stříkačky
1. Uchopte velkou injekční stříkačku o objemu 3 ml. Pomocí ukazováčku odstraňte barevný
ochranný kryt jehly směrem od jehly.

2. Ujistěte se, že jehla je bezpečně nasazena na injekční stříkačce:
• přidržte plastový ochranný kryt j ehly
• jemně pootočte jehlou spolu s krytem ve směru hodinových ručiček. Nepoužívejte příliš velké síly, aby nedošlo k poškození jehly.
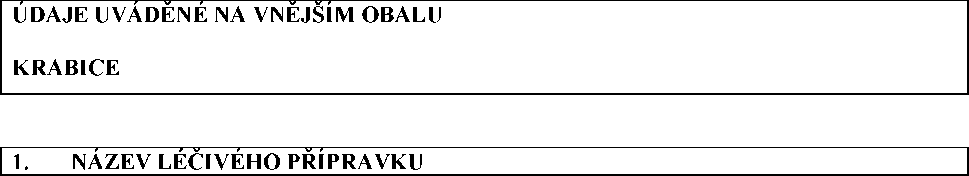
3. Průhledný, plastový, ochranný kryt j ehly odstraníte:
• zatlačte směrem k injekční stříkačce, poté jej stáhněte.
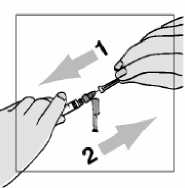
4. Natáhněte 1,1 ml vzduchu.
5. Vpíchněte injekční stříkačku do pryžové zátky injekční lahvičky obsahující vodu na injekci a stlačte píst. Tím vstříknete vzduch do injekční lahvičky.
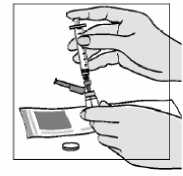
6. Opatrně otočte injekční lahvičku dnem vzhůru. Ujistěte se, že špička jehly je stále pod hladinou roztoku vody na injekci, aby nedošlo ke vniknutí bublin vzduchu do injekční stříkačky.
7. Pomalu táhněte píst zpět tak, aby voda dosáhla značky 1,1 ml. Prosím, uvědomte si, že injekční lahvička obsahuje více roztoku, než potřebujete (2 ml); abyste si správně připravil(a) svou injekci, potřebujete odebrat pouze 1,1 ml.
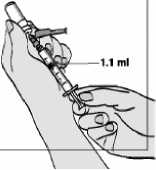
8. Poklepejte opatrně na injekční stříkačku tak, aby bubliny vystoupaly nahoru.
• Jestliže do injekční stříkačky vnikne větší množství vzduchu, stlačte opatrně píst, aby vytlačil všechen vzduch zpět do injekční lahvičky.
• Potom natáhněte vodu znovu.
• Zkontrolujte, zda jste do injekční stříkačky natáhl(a) 1,1 ml vody na injekci.
• Zopakujte tento krok případně znovu, dokud do injekční stříkačky nenatáhnete přesný objem vody na injekci.
9. Vytáhněte jehlu z injekční lahvičky. V žádném případě se jehlou nedotkněte prstů nebo čehokoli jiného.
10. Zlikvidujte injekční lahvičku a vodu na injekci vyhozením do určeného odpadního kontejneru s uzávěrem - tato injekční lahvička je určena pouze pro jednorázové použití.
Vstříknutí vody na injekci do prášku Fuzeon
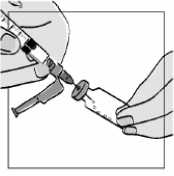
1. Jemně poklepejte na injekční lahvičku s přípravkem Fuzeon, aby došlo k nakypření prášku.
2. Držte injekční stříkačku s vodou na injekci za její hlavní část (tělo) a vpíchněte ji pod mírným úhlem do pryžové zátky injekční lahvičky.
3. Pomalu stiskněte píst injekční stříkačky.
• Vodu nechte pomalu vniknout do injekční lahvičky.
• V žádném případě nesmíte vodu násilím vstříknout do prášku, protože by to mohlo způsobit vznik pěny.
• Dojde-li ke vzniku pěny, může trvat déle, než se prášek úplně rozpustí.
4. Poté, co všechna voda na injekci vnikla do injekční lahvičky s přípravkem Fuzeon, vyjměte injekční stříkačku z injekční lahvičky.
5. Uchopte jednou rukou injekční stříkačku za její hlavní část (tělo) a barevný ochranný kryt opřete o rovnou podložku a mírným tlakem jej zaklopte zpět na jehlu.
• Uslyšíte cvaknutí. Nikdy nezaklápějte ochranný kryt jehly druhou rukou.
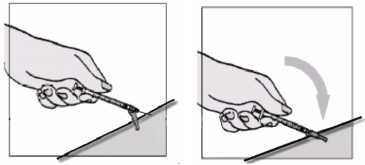
6. Vyhoďte použitou injekční stříkačku do určeného odpadního kontejneru s uzávěrem.
Smíchání vody na injekci s práškem Fuzeon
1. Poklepávejte opatrně špičkami prstů na injekční lahvičku, až se prášek začne rozpouštět.
V žádném případě nesmíte injekční lahvičkou třepat nebo ji obracet nahoru a dolů, protože by došlo k nadměrné tvorbě pěny.
2. Když se prášek začne rozpouštět, je možné položit injekční lahvičku na bok tak, aby se usnadnilo úplné rozpuštění prášku.
• Úplné rozpuštění prásku může trvat až 45 minut.
• Můžete po přidání vody na injekci injekční lahvičkou opatrně otáčet mezi rukama, dokud se prášek úplně nerozpustí.
• Tím zkrátíte dobu potřebnou k úplnému rozpuštění.
3. Po úplném rozpuštění prášku:
• V případě, že se vytvořily bubliny, nechte je, aby se usadily.
• Rychlejšímu usazení bublin může pomoci opatrné poklepávání na stěnu injekční lahvičky.
4. Je třeba zkontrolovat, zda roztok neobsahuje nějaké nerozpuštěné kousky (částice).
• Jestliže zpozorujete v roztoku nějaké nerozpuštěné kousky, nesmíte jej použít.
• Injekční lahvičku vyhoďte do určeného odpadního kontejneru s uzávěrem nebo ji vraťte do lékárny.
Přípravu léku musíte začít znovu s novou injekční lahvičkou obsahující přípravek Fuzeon prášek.
5. V případě, že se náhodně dotknete gumového uzávěru, je třeba jej očistit novým alkoholovým tamponem.
6. Jednou rozpuštěná dávka ve vodě na injekci musí být ihned aplikována. Pokud ji hned neaplikujete, uschovejte ji v chladničce po dobu maximálně 24 hodin.
• Před použitím nechte roztok ohřát na pokojovou teplotu.
7. Připravujete-li si obě denní dávky najednou, ujistěte se, že použijete vždy nové injekční stříkačky, novou injekční lahvičku s vodou na injekce a novou injekční lahvičku s přípravkem Fuzeon pro přípravu každé dávky.
Krok C: Příprava aplikace injekce
Natažení Fuzeonu do injekční stříkačky o objemu 1 ml
1. Otřete znovu povrch zátky injekční lahvičky s přípravkem Fuzeon novým tamponem s alkoholem.
2. Uchopte malou injekční stříkačku o objemu 1 ml. Pomocí ukazováčku odstraňte barevný ochranný kryt jehly směrem od jehly.
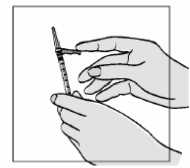
3. Ujistěte se, že jehla je bezpečně nasazena na injekční stříkačce:
• přidržte plastikový ochranný kryt jehly
• utáhněte jehlu spolu s krytem jemným pootočením a zatlačením směrem ke stříkačce ve směru hodinových ručiček. Nepoužívejte příliš velké síly, aby nedošlo k poškození jehly.
4. Průhledný, plastový, ochranný kryt jehly odstraníte:
• zatlačte směrem k injekční stříkačce a poté jej stáhněte.

5. Natáhněte 1 ml vzduchu.
• Dbejte na to, abyste nezatáhl(a) za píst příliš rychle, aby nedošlo k překročení značky 1 ml nebo vytažení pístu z injekční stříkačky.
6. Vpíchněte jehlu injekční stříkačky do pryžové zátky injekční lahvičky s přípravkem Fuzeon a stlačte píst. Tím vstříknete vzduch do injekční lahvičky.
7. Opatrně otočte injekční lahvičku dnem vzhůru.
Zkontrolujte, zda je špička jehly stále pod hladinou roztoku, aby nedošlo ke vniknutí bublin vzduchu do injekční stříkačky.
8. Pomalu táhněte píst zpět tak, aby roztok dosáhl značky 1,0 ml.
• Dbejte na to, abyste nezatáhl(a) za píst příliš rychle, aby nedošlo k překročení značky 1 ml nebo vytažení pístu z injekční stříkačky.
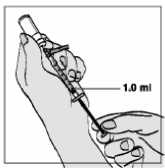
9. Poklepejte opatrně na injekční stříkačku tak, aby bubliny vystoupaly nahoru.
• Jestliže do injekční stříkačky vnikne větší množství vzduchu, stlačte opatrně píst tak, aby vytlačil všechen vzduch zpět do injekční lahvičky.
• Potom roztok natáhněte znovu.
• Zkontrolujte, zda jste do injekční stříkačky natáhl(a) přesně 1,0 ml roztoku (nebo množství, které Vám předepsal Váš lékař).
• Zopakujte tento krok případně znovu, dokud do injekční stříkačky nenatáhnete přesný objem roztoku.
10. Vytáhněte j ehlu z inj ekční lahvičky.
Krok D: Aplikace injekce s přípravkem Fuzeon
Rada: Váš lékař nebo zdravotní sestra Vám může doporučit jinou, pro Vás lepší techniku, jak aplikovat injekce s přípravkem Fuzeon.
Kam se injekce aplikují

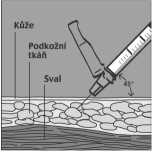
• Přípravek Fuzeon se aplikuje jako 1 ml injekce přímo pod kůži - tzv. „subkutánní“ injekcí.
• Injekci si můžete aplikovat do horní části paže a stehen nebo do míst na břiše (abdomen).
• Vyberte si jiné místo, než kam jste si lék aplikoval(a) naposledy.
• Injekce nesmí být aplikována do místa, kde je dosud patrná reakce po některé předchozí dávce. Zkontrolujte také všechna místa, kde by mohla být lokální reakce v místě vpichu stlačením kůže tak, abyste lépe viděl(a) případné bulky.
• Injekce se nesmí aplikovat do míst, která by mohla být podrážděna páskem nebo v pase oděvem.
• Injekce se nesmí aplikovat do mateřských znamének, jizev, modřin či do pupku.
Očištění místa pro vpich injekce
Očistěte si pečlivě místo určené k aplikaci injekce polštářkem napuštěným alkoholem. Krouživé pohyby provádějte od středu směrem ven. Nechte místo vpichu oschnout.
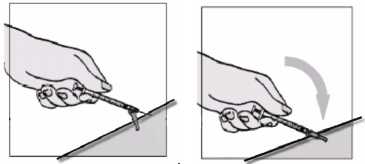
Vpíchnutí jehly a aplikace injekce
1. Zmáčkněte si co nejvíce kůže tak, aby Vám to nebylo nepříjemné.

2. Pod úhlem 45o propíchněte kůži j ehlou.
3. Když je j ehla vpíchnuta:
• uvolněte si kůži
• volnou rukou si přidržte injekční stříkačku za její hlavní část (tělo) tak, aby se injekční stříkačka nemohla posunout.
4. Pomocí palce druhé ruky stlačte píst injekční stříkačky.
• Poté, co byl vytlačen celý objem roztoku, vytáhněte jehlu z kůže.
Po vytáhnutí jehly
1. Uchopte j ednou rukou inj ekční stříkačku za její hlavní část (tělo).
• Potom barevný ochranný kryt opřete o rovnou podložku a mírným tlakem jej zaklopte zpět na jehlu.
• Uslyšíte cvaknutí.
Nikdy nezaklápějte ochranný kryt jehly druhou rukou.
2. Vyhoďte použitou injekční stříkačku do určeného odpadního kontejneru s uzávěrem.
3. Je-li na místě vpichu viditelná krev, zakryjte si místo vpichu náplastí.
Krok E: Likvidace použitého materiálu
• Veškerý použitý materiál vyhazujte přímo do určeného kontejneru s uzávěrem. A to včetně injekčních lahviček, které obsahují nespotřebované množství léčivého přípravku nebo vodu na injekci, jelikož jsou určeny pouze k jednorázovému použití.
• Dbejte na to, aby tento kontejner byl trvale uzavřený a mimo dosah dětí.
• Požádejte svého lékaře, lékárníka nebo zdravotní sestru o radu, jak správně likvidovat použité kontejnery.
• Máte-li nějaké otázky ohledně bezpečné likvidace použitého materiálu, požádejte, prosím, o radu svého lékaře, lékárníka nebo zdravotní sestru.
PŘÍLOHA IV
VĚDECKÉ ZÁVĚRY A ZDŮVODNĚNÍ ZMĚNY PODMÍNEK ROZHODNUTÍ O
REGISTRACI
Vědecké závěry
S ohledem na hodnotící zprávu výboru PRAC týkající se pravidelně aktualizovaných zpráv o bezpečnosti (PSUR) pro enfuvirtid, dospěl výbor CHMP k těmto vědeckým závěrům:
Během průběžného vyhodnocování farmakovigilančních signálů bylo z databáze Eudravigilance vyňato pět přezkoumání termínů vysoké úrovně („HLT“) hlášených případů amyloidózy souvisejících s podáním enfuvirtidu. Hlášení bylo považováno za potenciálně závažné a držitel rozhodnutí o registraci provedl kumulativní přezkoumání termínů vysoké úrovně („HLT“) amyloidózy. Dostupné informace v hlášeních případů lokalizované amyloidózy prokazují důkazy, na základě dvou dobře zdokumentovaných případů, že enfuvirtid může jako peptid způsobit kožní amyloidózu v místě vpichu injekce. S ohledem na výše uvedené bylo vyhodnoceno, že je třeba kožní amyloidózu v místě vpichu injekce zahrnout jako nežádoucí účinek do údajů o přípravku Fuzeon.
Proto s odvoláním na dostupné údaje týkající se přípravku Fuzeon, PRAC považoval tyto změny v údajích o přípravku za nutné.
Výbor CHMP souhlasí s vědeckými závěry vydanými PRAC.
Zdůvodnění doporučující změnu podmínek o rozhodnutí o registraci
Na základě vědeckých závěrů týkajících se enfuvirtidu zastává výbor CHMP stanovisko, že poměr přínosů a rizik léčivých přípravků obsahujících enfuvirtid je příznivý pod podmínkou, že v údajích o přípravku budou provedeny navržené změny.
Výbor CHMP doporučuje změnit podmínky rozhodnutí o registraci.
46