Flixotide Diskus 500
sp.zn. sukls197422/2016
SOUHRN ÚDAJŮ O PŘÍPRAVKU
1. NÁZEV PŘÍPRAVKU
Flixotide Diskus 100 Flixotide Diskus 250 Flixotide Diskus 500 Dávkovaný prášek k inhalaci
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Fluticasoni propionas, 100 mikrogramů v jedné inhalační dávce. Fluticasoni propionas, 250 mikrogramů v jedné inhalační dávce. Fluticasoni propionas, 500 mikrogramů v jedné inhalační dávce.
Pomocná látka se známým účinkem:
Přípravek obsahuje laktosu (ve formě monohydrátu).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Dávkovaný prášek k inhalaci. Bílý prášek.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
4.1.1 Astma
Flutikason-propionát má v plicích výrazný protizánětlivý účinek.
Snižuje výskyt příznaků a exacerbací astmatu u pacientů, kteří byli předtím léčeni samotným bronchodilatačním léčivem nebo v kombinaci s jinými profylaktickými léčivy.
Těžká forma astmatu vyžaduje pravidelné lékařské sledování, protože může dojít k úmrtí.
Pacienti s těžkou formou astmatu mají trvalé příznaky a časté exacerbace, s omezenou fyzickou kapacitou. Hodnoty jejich maximální výdechové rychlosti (peak expiratory flow, PEF) na začátku léčby jsou nižší než 60 % náležité hodnoty, s větší než 30% variabilitou, a po podání bronchodilatačního léčiva se obvykle nevracejí k normálním hodnotám. Tito pacienti potřebují vysokou inhalační dávku (viz pokyny pro dávkování) nebo terapii perorálními kortikosteroidy. Náhlé zhoršení příznaků může vyžadovat aplikaci zvýšených dávek kortikosteroidů, jež mají být podávány pod dohledem lékaře neodkladné péče.
• Dospělí
Profylaktická léčba:
- Mírného astmatu (hodnoty PEF na začátku léčby vyšší než 80 % náležité hodnoty s menší než 20% variabilitou): u pacientů, kteří potřebují častější intermitentní symptomatickou bronchodilatační medikaci;
- Středně těžkého astmatu (hodnoty PEF na začátku léčby v rozmezí 60 až 80 % náležité hodnoty, s 20 až 30% variabilitou): u pacientů, kteří potřebují pravidelnou protiastmatickou medikaci, a u pacientů, jejichž astma je při dosavadní jiné profylaktické farmakoterapii, nebo při aplikaci samotného bronchodilatačního léčiva nestabilní, nebo se zhoršuje;
- Těžkého astmatu (hodnoty PEF na začátku léčby menší než 60 % náležité hodnoty s větší než 30% variabilitou): u pacientů s těžkým chronickým astmatem. Zahájení terapie inhalačním flutikason-propionátem může u mnohých pacientů, kteří byli pro udržení příznaků pod adekvátní kontrolou závislí na systémových kortikosteroidech, významně snížit nebo eliminovat jejich nároky na perorální kortikosteroidy.
• Děti
Každé dítě, které potřebuje profylaktickou protiastmatickou medikaci, včetně pacientů, jejichž příznaky nejsou při dosavadní profylaktické medikaci pod kontrolou.
4.1.2 Chronická obstrukční plicní nemoc (CHOPN)
Flutikason-propionát je indikován k léčbě CHOPN při použití v kombinaci s dlouhodobě působícími bronchodilatátory [např. dlouhodobě působícími beta agonisty (LABA)].
4.2 Dávkování a způsob podání
Pacienti mají být poučeni o profylaktické povaze léčby inhalačním flutikason-propionátem a o tom, že jej mají inhalovat pravidelně, i když jsou asymptomatičtí.
Flutikason-propionát je určen výhradně k perorální inhalaci.
4.2.1 Astma
Nástup terapeutického účinku se dostaví po čtyřech až sedmi dnech podávání, ačkoliv některý přínos může být patrný hned po 24 hodinách u pacientů, kteří předtím neužívali inhalační kortikosteroidy.
Zjistí-li pacienti, že účinek užívaného úlevového krátkodobě působícího bronchodilatačního přípravku slábne, nebo potřebují-li více inhalací než obvykle, musí vyhledat lékaře.
Populace
Dávkování u dospělých a dospívajících starších než 16 let
100 až 1 000 mikrogramů dvakrát denně.
Zahajovací dávka inhalačního flutikason-propionátu má odpovídat závažnosti onemocnění konkrétního pacienta:
Mírná forma bronchiálního astmatu: 100 až 250 mikrogramů dvakrát denně;
Středně těžká forma bronchiálního astmatu: 250 až 500 mikrogramů dvakrát denně;
Těžká forma bronchiálního astmatu: 500 až 1 000 mikrogramů dvakrát denně.
Při pokračování léčby by měla být dávka upravena podle individuální klinické odezvy, nebo snížena na minimální účinnou dávku.
Alternativně lze zahajovací dávku flutikason-propionátu stanovit odhadem ve výši poloviny celkové denní dávky beklometason-dipropionátu, nebo ekvivalentní dávky jiného inhalačního kortikosteroidu, podávaného tlakovým aerosolovým dávkovacím inhalátorem.
• Dávkování u dětí ve věku 4 let a starších
50 až 200 mikrogramů dvakrát denně.
K dobré kontrole astmatu vede u mnohých dětí dávkovací režim 50 až 100 mikrogramů dvakrát denně. U pacientů s nedostatečnou kontrolou astmatu může být vhodné další zvýšení dávkování až na 200 mikrogramů dvakrát denně.
Zahajovací dávka inhalačního flutikason-propionátu má odpovídat závažnosti onemocnění konkrétního dítěte.
Dávka má být upravena podle individuální odezvy tak, aby se dosáhlo adekvátního zvládnutí příznaků, nebo má být snížena na nejnižší dávku, která ještě účinně udrží příznaky pod kontrolou.
• Dávkování u dětí mladších než 4 roky
Tento inhalační prostředek se nedoporučuje používat pro věkovou skupinu dětí mladších než 4 roky.
4.2.2 Chronická obstrukční plicní nemoc (CHOPN)
Populace
• Dávkování u dospělých
500 mikrogramů dvakrát denně, jako přídatná dávka k léčbě dlouhodobě působícími bronchodilatátory (např. LABA).
Pro optimální přínos léčby, což může trvat tři až šest měsíců, musí být přípravek podáván denně,. Nedojde-li však po třech až šesti měsících ke zlepšení, má být pacient znovu podrobně vyšetřen.
K zajištění této dávky jsou vhodné pouze přípravky obsahující 250 nebo 500 mikrogramů léčivé látky.
• Zvláštní skupiny pacientů
Není nutné dávku upravovat u starších pacientů, ani u pacientů s jaterním nebo renálním onemocněním.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Léčba astmatu má být vedená podle stupňového programu a odezvu pacienta je třeba pravidelně posuzovat na základě klinického obrazu a funkčního vyšetření plic.
Zvýšená spotřeba krátkodobě působících inhalačních beta2-agonistů signalizuje zhoršování kontroly astmatu. Za těchto okolností je nutné přehodnotit léčebný plán pacienta.
Náhlé a progresivní zhoršování astmatu potenciálně ohrožuje život a je třeba zvážit zahájení nebo zintenzivnění kortikosteroidní terapie. U pacientů považovaných za rizikové lze zahájit každodenní monitorování maximální výdechové rychlosti (PEF).
Flutikason-propionát není určen k léčbě akutních astmatických záchvatů, ale k pravidelné dlouhodobé léčbě. K úlevě od akutních příznaků astmatu potřebují pacienti rychle a krátkodobě působící inhalační bronchodilatační přípravek.
Nedostatečnou léčebnou odezvu nebo těžké exacerbace astmatu je třeba řešit zvýšením dávky inhalačního flutikason-propionátu a v případě nutnosti aplikací systémového kortikosteroidu a/nebo v přítomnosti infekce podáním antibiotika.
Při podávání každého inhalačního kortikosteroidu se mohou vyskytnout systémové účinky, zejména při vysokých dávkách předepisovaných po dlouhá období. Pravděpodobnost výskytu těchto účinků je mnohem menší než u perorálních kortikosteroidů (viz bod 4.9). K možným systémovým účinkům patří Cushingův syndrom, Cushingova nemoc, suprese adrenální funkce, retardace růstu u dětí a dospívajících, pokles minerální kostní denzity, katarakta a glaukom. Proto je důležité, aby pacienti byli pravidelně sledováni a dávka inhalačního kortikosteroidu byla snížená na nejnižší dávku, která ještě účinně udrží příznaky astmatu pod kontrolou (viz bod 4.8).
U dětí dlouhodobě léčených inhalačním kortikosteroidem se doporučuje pravidelně kontrolovat tělesnou výšku.
Pacienti převádění z terapie perorálními kortikosteroidy na terapii inhalačním flutikason-propionátem musí být vzhledem k možnosti narušené adrenální odezvy léčeni se zvláštní opatrností, a je u nich nutné pravidelně sledovat funkci kůry nadledvin.
Po zahájení léčby inhalačním flutikason-propionátem musí být terapie systémovými kortikosteroidy ukončována postupně a pacient má být vybaven výstražnou průkazkou (kterou musí mít stále při sobě), upozorňující na to, že ve stresových obdobích může potřebovat doplňkovou terapii systémovými kortikosteroidy.
Možnost narušené adrenální odezvy je nutné mít na paměti v každé akutní (včetně chirurgického výkonu) nebo elektivní situaci, která pravděpodobně je nebo bude situací stresovou, zvláště u pacientů užívajících dlouhodobě vysoké dávky. V těchto případech je třeba zvážit náležitou léčbu kortikosteroidy (viz bod 4.9).
Podobně může náhrada systémové kortikosteroidní léčby inhalační terapií demaskovat alergie, např. alergickou rinitidu nebo ekzém, jež byly předtím potlačeny systémově aplikovaným léčivem.
Terapie flutikason-propionátem se nemá náhle přerušit.
Velmi vzácně došlo ke zvýšení krevní hladiny glukózy (viz bod 4.8), proto by se podání pacientům s anamnézou diabetes mellitus mělo zvážit.
Stejně jako při podávání ostatních inhalačních kortikosteroidů je nutné věnovat zvláštní péči pacientům s aktivní i inaktivní plicní tuberkulózou.
V průběhu postmarketingového užití byly pozorovány klinicky signifikantní lékové interakce u pacientů užívajících flutikason-propionát a ritonavir vedoucí ke vzniku systémových účinků léčby kortikosteroidy včetně Cushingova syndromu a adrenální suprese. Současnému užívání flutikason-propionátu a ritonaviru je proto třeba se vyhnout, pokud možný přínos pro pacienta nepřeváží riziko vzniku systémových nežádoucích účinků léčby kortikosteroidy (viz bod 4.5).
Stejně jako při podávání jiných inhalačních přípravků může po inhalaci tohoto přípravku dojít k paradoxnímu bronchospasmu s bezprostředním zhoršením pískotů (hvízdavého dýchání). V takovém případě je nutné okamžitě podat krátkodobě působící inhalační bronchodilatancia s rychlým nástupem účinku. Dále je nutné okamžitě ukončit podávání flutikason-propionátu, znovu zhodnotit stav pacienta a v případě potřeby zavést alternativní terapii.
Pneumonie u pacientů s CHOPN
U pacientů s CHOPN, kterým byly podávány inhalační glukokortikoidy, byl pozorován vyšší výskyt pneumonie, včetně pneumonie vyžadující hospitalizaci. Existují určité důkazy o tom, že zvýšené riziko pneumonie souvisí se zvyšováním dávky steroidu, avšak tuto závislost se nepodařilo definitivně prokázat ve všech studiích.
Neexistují jednoznačné klinické důkazy o rozdílech mezi léčivými přípravky ze skupiny inhalačních glukokortikoidů ohledně výše rizika pneumonie.
Lékaři mají sledovat možný vývoj pneumonie u pacientů s CHOPN, neboť klinické známky těchto infekcí se mohou překrývat se symptomy, které doprovázejí exacerbaci CHOPN.
Rizikovými faktory pro pneumonii u pacientů s CHOPN jsou současné kouření, vyšší věk, nízký index tělesné hmotnosti (BMI) a těžká forma CHOPN.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Za normálních okolností je po inhalačním podání dosaženo nízkých plazmatických koncentrací flutikason-propionátu vzhledem k velmi silnému metabolismu látky během prvního průchodu játry (first pass metabolism) a vysoké systémové clearance zprostředkované cytochromem P450 3A4 ve střevě a játrech. Z toho důvodu jsou klinicky významné interakce s jinými léčivy zprostředkované flutikason-propionátem nepravděpodobné.
Ve studii interakcí u zdravých jedinců se prokázalo, že ritonavir (velmi silný inhibitor cytochromu P450 3A4) může značně zvýšit plazmatickou koncentraci flutikason-propionátu, což vedlo ke značné redukci sérové koncentrace kortizolu. V průběhu postmarketingového užití byly hlášeny klinicky signifikantní lékové interakce u pacientů užívajících intranasální nebo inhalační flutikason-propionát s ritonavirem, což vedlo k systémovým účinkům kortikosteroidů včetně vývoje Cushingova syndromu a adrenální suprese. Současnému podávání flutikason-propionátu a ritonaviru je třeba se vyhnout, pokud možný přínos pro pacienta nepřeváží riziko vzniku systémových nežádoucích účinků léčby kortikosteroidy.
Studie prokázaly, že jiné inhibitory cytochromu P450 3A4 vytvářejí zanedbatelné (erythromycin) a menší (ketokonazol) zvýšení systémové expozice flutikason-propionátu bez pozorovatelné redukce sérové koncentrace kortizolu. Nicméně opatrnosti je zapotřebí u souběžného podávání silných inhibitorů cytochromu P450 3A4 (např. ketokonazolu), kdy je možné zvýšení systémové expozice flutikason-propionátu.
4.6 Fertilita, těhotenství a kojení Fertilita
K dispozici nejsou žádné údaje týkající se účinku na fertilitu u člověka. Studie na zvířatech neprokázaly žádné účinky flutikason-propionátu na samčí nebo samičí fertilitu.
Těhotenství
U těhotných žen jsou omezená data. Podávání flutikason-propionátu v období těhotenství se má zvážit pouze tehdy, pokud očekávaný prospěch pro matku převáží možná rizika pro plod.
Výsledky retrospektivní epidemiologické studie expozice flutikason-propionátem neprokázaly statisticky významně zvýšené riziko výskytu významných vrozených malformací ve srovnání s jinými inhalačními kortikosteroidy podávanými v průběhu prvního trimestru těhotenství (viz bod 5.1 Klinické studie).
V reprodukčních studiích na zvířatech byly pozorovány účinky charakteristické pro glukokortikosteroidy pouze při systémové expozici mnohonásobně překračující doporučenou inhalační terapeutickou dávku.
Kojení
Vylučování flutikason-propionátu do lidského mateřského mléka nebylo zkoumáno. Po podkožním podání dávky, která vyvolala měřitelnou plazmatickou hladinu flutikason-propionátu, laboratorním potkaním samicím v období laktace byl flutikason-propionát prokázán v jejich mléce. Je však pravděpodobné, že plazmatické hladiny flutikason-propionátu u pacientů po jeho inhalační aplikaci v doporučených dávkách jsou velmi nízké.
Podávání flutikason-propionátu v období kojení se má zvážit pouze tehdy, pokud očekávaný prospěch pro matku převáží možná rizika pro plod.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Při užívání tohoto přípravku není známo negativní ovlivnění činnosti vyžadující zvýšenou pozornost, schopnost soustředění a koordinaci pohybů.
4.8 Nežádoucí účinky
V následujícím textu jsou nežádoucí účinky uvedené podle orgánových tříd a frekvence výskytu. Četnost je definována jako: velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1 000 až < 1/100), vzácné (> 1/10 000 až < 1/1 000) a velmi vzácné (< 1/10 000) včetně jednotlivých hlášených případů a není známo (z dostupných údajů nelze určit). Velmi časté, časté a méně časté nežádoucí účinky byly obecně stanoveny z klinických studií. Vzácné a velmi vzácné nežádoucí účinky byly obecně stanoveny ze spontánních hlášeních.
Infekce a infestace
Velmi časté: Kandidóza (soor, moučnivka) orální a faryngeální oblasti.
U některých pacientů se může v orální a faryngeální oblasti vyskytnout kandidóza (soor, moučnivka). Aby se tomuto stavu předešlo, je vhodné si bezprostředně po inhalaci vypláchnout ústa vodou nebo se napít. Symptomatickou kandidózu lze léčit místně antimykotickými přípravky, přičemž se nadále pokračuje v léčbě flutikason-propionátem.
Časté: Pneumonie (u pacientů s CHOPN);
Vzácné: Esofageální kandidóza.
Poruchy imunitního systému
Byly pozorovány tyto reakce přecitlivělosti:
Velmi vzácné: Angioedém (hlavně ve formě faciálního a orofaryngeálního
edému), respirační příznaky (dyspnoe a/nebo bronchospasmus) a anafylaktická reakce.
Endokrinní poruchy
K možným systémovým účinkům patří (viz bod 4.4):
Velmi vzácné: Cushingův syndrom, Cushingova nemoc, suprese adrenální
funkce, retardace růstu, pokles minerální kostní denzity, katarakta, glaukom.
Poruchy metabolismu a výživy
Velmi vzácné: Hyperglykémie.
Psychiatrické poruchy
Velmi vzácné: Úzkost, porucha spánku a změny chování, včetně hyperaktivity
a podrážděnosti (převážně u dětí).
Respirační, hrudní a mediastinální poruchy
Časté: Chrapot.
Není známo: Epistaxe.
U některých pacientů může inhalační flutikason-propionát způsobit chrapot. Pak může být užitečné po každé inhalaci okamžité vykloktání vodou.
Velmi vzácné: Paradoxní bronchospasmus (viz bod 4.4).
Poruchy kůže a podkožní tkáně
Časté: Pohmožděniny.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na adresu:
Státní ústav pro kontrolu léčiv Šrobárova 48 100 41 Praha 10
Webové stránky: www.sukl.cz/nahlasit-nezadouci-ucinek
4.9 Předávkování
Příznaky
Akutní inhalace flutikason-propionátu v dávkách vyšších než schválených může vést k přechodné supresi hypotalamo-hypofyzární osy. Běžně nejsou požadovaná urgentní opatření, protože adrenální funkce se obvykle spontánně zotaví během několika dnů.
Jestliže se ve vyšším než schváleném dávkování pokračuje delší dobu, může dojít k signifikantní supresi adrenokortikální funkce. Při podávání vyšších dávek než schválených dětem (1 000 mikrogramů denně a více) po delší dobu (několik měsíců nebo let) byl velmi vzácně pozorován vznik akutní adrenální krize.
Byly pozorovány charakteristické komplikace včetně hypoglykémie s následnými změnami vědomí a/nebo konvulze. Spouštěcími mechanismy akutních adrenálních krizí jsou: trauma, chirurgický zákrok, infekce nebo rychlý pokles dávek.
Léčba
Pacienti, kterým se podávají vyšší než schválené dávky přípravku, by měli být pravidelně kontrolováni včetně postupného snižování dávek.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Jiná inhalační léčiva onemocnění spojených s obstrukcí dýchacích cest. Antiastmatikum.
ATC kód: R03BA05
Mechanismus účinku
Flutikason-propionát podávaný inhalačně v doporučených dávkách má silný glukokortikoidní protizánětlivý účinek v plicích a jeho výsledkem je omezení příznaků a exacerbací astmatu.
5.2 Farmakokinetické vlastnosti
Absorpce
Absolutní biologická dostupnost flutikason-propionátu byla odhadována pro každou dostupnou inhalační formu ze srovnávacích studií farmakokinetiky inhalačně a intravenózně podaných přípravků. U zdravých dospělých jedinců byla absolutní biologická dostupnost pro flutikason-propionát ve formě diskusu odhadována na 7,8 %, pro flutikason-propionát použitý cestou diskhaleru na 9,0 % a flutikason-propionát v inhaleru na 10,9 %. U pacientů s bronchiálním astmatem nebo CHOPN byl při inhalaci flutikason-propionátu pozorován nižší stupeň systémové expozice. K systémové absorpci dochází hlavně plícemi, zpočátku rychle a později pomaleji. Zbytek inhalační dávky může být spolknut, avšak jeho příspěvek k systémové expozici je minimální, protože perorální biologická dostupnost je z důvodu nízké rozpustnosti ve vodě a presystémové metabolizace menší než 1 %. Systémová expozice roste lineárně se stoupající inhalační dávkou.
Distribuce
Flutikason-propionát má velký distribuční objem v ustáleném stavu (přibližně 300 l). Vazba na plazmatické proteiny je středně vysoká (91 %).
Flutikason-propionát je ze systémové cirkulace odstraňován velmi rychle, hlavně biotransformací na neúčinný metabolit - kyselinu karboxylovou - prostřednictvím izoenzymu cytochromu P450 CYP3A4. Zvláštní opatrnost je nutná při souběžné aplikaci známých inhibitorů izoenzymu CYP3A4, a to z důvodu možného zvýšení systémové expozice flutikason-propionátu.
Eliminace
Dispozice flutikason-propionátu je charakterizována vysokou plazmatickou clearance (1 150 ml/min) a terminálním poločasem přibližně 8 hodin. Renální clearance flutikason-propionátu je zanedbatelná (menší než 0,2 %), renální clearance metabolitu je menší než 5 %.
Klinické studie
CHOPN
Klinické studie prokázaly významné snížení příznaků CHOPN a zlepšení plicních funkcí bez ohledu na věk, pohlaví, základní plicní funkce, úroveň kouření nebo stupeň hypersenzitivity. Tyto poznatky mohou významně přispět ke zlepšení kvality života.
Studie TORCH (Towards a Revolution in COPD Health)
TORCH byla tříletá klinická studie hodnotící účinek léčby salmeterolem/flutikason-propionátem Diskus 50/500 mikrogramů podávaným dvakrát denně, 50 mikrogramy salmeterolu v Diskusu dvakrát denně, 500 mikrogramy flutikason-propionátu v Diskusu dvakrát denně nebo placeba na úmrtnost z jakékoliv příčiny u pacientů s CHOPN. Pacienti se středně těžkou až těžkou formou CHOPN s úvodní (prebronchodilatační) FEVi < 60 % náležité hodnoty byli randomizováni k dvojitě zaslepené medikaci. V průběhu studie byla pacientům povolena obvyklá léčba CHOPN s výjimkou jiných inhalačních kortikosteroidů, bronchodilatancií s dlouhodobým účinkem a dlouhodobého podávání systémových kortikosteroidů. Stav přežití byl zjišťován po 3 letech u všech pacientů bez ohledu na ukončení léčby zahrnuté do studie. Primárním výsledkem byla redukce úmrtnosti z jakékoliv příčiny po 3 letech u salmeterolu/flutikason-propionátu proti placebu.
|
Placebo N = 1 524 |
Salmeterol50 N = 1 521 |
Flutikason-propionát N = 1 534 |
Salmeterol/ flutikason-propionát 50/500 N = 1 533 | |
|
Úmrtnost z jakékoliv příčiny po 3 letech | ||||
|
Počet úmrtí (%) |
231 |
205 |
246 |
193 |
|
(15,2 %) |
(13,5 %) |
(16,0 %) |
(12,6 %) | |
|
Poměrné riziko |
Neuplatňuje |
0,879 |
1,060 |
0,825 |
|
proti placebu (kortikosteroidy) |
se |
(0,73; 1,06) |
(0,89; 1,27) |
(0,68; 1,00) |
|
p hodnota |
0,180 |
0,525 |
0,0521 | |
|
Poměrné riziko |
Neuplatňuje |
0,932 |
0,774 |
Neuplatňuje se |
|
přípravku Seretide 50/500 proti složkám (kortikosteroidy) p hodnota |
se |
(0,77; 1,13) 0,481 |
(0,64; 0,93) 0,007 | |
1. Nesignifikantní p hodnota byla přizpůsobená pro dvě průběžné analýzy srovnání primární účinnosti z long-rank analýzy stratifikované podle kuřáctví.
Salmeterol a flutikason-propionát (FP) snížily riziko úmrtí o 17,5 % kdykoliv během 3 let ve srovnání s placebem [poměrné riziko 0,825 (95% Cl: 0,68; 1,00; p = 0,052); vše přizpůsobené prozatímním analýzám]. Došlo k 12% snížení rizika úmrtí kdykoliv během 3 let z jakýchkoliv příčin u salmeterolu ve srovnání s placebem (p = 0,180) a k 6% zvýšení u flutikason-propionátu ve srovnání s placebem (p = 0,525).
Podpůrná analýza používající Coxův model poměrného rizika udala poměrné riziko 0,811 (95% Cl 0,670; 0,982; p = 0,031) pro salmeterol-FP versus placebo, který představuje 19% snížení rizika úmrtí po dobu 3 let. Model bral v úvahu další faktory (kouření, věk, pohlaví, oblast, bazální FEV1 a index tělesné hmoty). Nebylo prokázáno, že by se účinek léčby měnil dle těchto faktorů.
Podíl pacientů, kteří zemřeli během 3 let z důvodů vztahujících se k CHOPN, byl 6 % u placeba a 6,1 % u salmeterolu, 6,9 % flutikason-propionátu a 4,7 % u salmeterol-FP.
Salmeterol-FP tak snížil výskyt středních až těžkých exacerbací o 25 % (95% Cl: 19 % až 31 %; p < 0,001) ve srovnání s placebem. Salmeterol-FP snižoval počet exacerbací o 12 % ve srovnání se salmeterolem (95% Cl: 5 % až 19 %, p = 0,002) a o 9 % ve srovnání s flutikason-propionátem (95% Cl: 1 % až 16 %, p = 0,024). Salmeterol a flutikason-propionát významně snížily výskyt exacerbací ve srovnání s placebem o 15 % (95% Cl: 7 % až 22 %; p < 0,001) a o 18 % (95% Cl: 11 % až 24 %;
p < 0,001).
Kvalita života vztahující se ke zdravotnímu stavu měřená podle standardizovaného specifického dotazníku SGRQ se zlepšila u všech aktivních léčebných postupů ve srovnání s placebem. Průměrné zlepšení za 3 roky bylo pro salmeterol-FP srovnávané s placebem -3,1 jednotek (95% Cl: -4,1 až -2,1; p < 0,001), ve srovnání se salmeterolem -2,2 jednotek (p < 0,001) a ve srovnání
s flutikason-propionátem bylo -1,2 jednotek (p = 0,017).
Během tříletého léčebného období byly hodnoty FEVi vyšší u pacientů léčených kombinací salmeterol-FP než u pacientů léčených placebem (průměrný rozdíl po dobu 3 let 92 ml, 95% Cl: 75 až 108 ml; p < 0,001). Salmeterol-FP byly rovněž účinnější ve zlepšení hodnot FEVi než salmeterol nebo flutikason-propionát (průměrný rozdíl 50 ml, p < 0,001 pro salmeterol a 44 ml, p < 0,001 pro FP).
Odhadovaná tříletá pravděpodobnost výskytu pneumonie hlášené jako nežádoucí příhoda byla 12,3 % u placeba, 13,3 % u salmeterolu, 18,3 % u flutikason-propionátu, 19,6 % u salmeterolu-FP (poměrné riziko pro salmeterol-FP versus placebo: 1,64; 95% Cl: 1,33 až 2,01 , p < 0 ,001). Nedošlo ke zvýšení úmrtí v souvislostí s pneumonií. Úmrtí během léčby, která byla posuzována jako primární zapříčiněná pneumonií, byla v 7 případech u placeba, 9 u salmeterolu, 13 u flutikason-propionátu a 8 u salmeterolu-FP. Nebyl významný rozdíl v pravděpodobnosti výskytu kostních zlomenin (5,1 % u placeba, 5,1 % u salmeterolu, 5,4 % u flutikason-propionátu a 6,3 % u salmeterolu-FP; poměrné riziko pro salmeterol-FP versus placebo: 1,22; 95% Cl: 0,87 až 1,72, p = 0,248). Výskyt nežádoucích účinků jako onemocnění očí, porucha kostí a porucha osy HPA byl nízký a nebyl pozorován rozdíl mezi léčbou. U léčených skupin užívajících salmeterol nebyl prokázán zvýšený výskyt kardiálních nežádoucích účinků.
Přípravky obsahujícíflutikason-propionát užívané k léčbě bronchiálního astmatu v období těhotenství
Pozorovací retrospektivní epidemiologická studie využívající elektronické zdravotní záznamy z Velké Británie byla provedena za účelem zhodnocení rizika výskytu vrozených malformací (MCM) po expozici samotným inhalačním flutikason-propionátem (FP) a kombinací salmeterol-FP ve srovnání s inhalačními kortikosteroidy neobsahujícími flutikason-propionát v prvním trimestru těhotenství.
Do této studie nebylo pro srovnání zahrnuto placebo.
Ve skupině 5 362 těhotných s bronchiálním astmatem vystavených inhalačním kortikosteroidům v prvním trimestru těhotenství byl u 131 těhotných diagnostikován výskyt vrozených malformací; z 1 612 (30 %) těhotných vystavených FP nebo kombinaci salmeterol-FP byly diagnostikovány vrozené malformace u 42. Přizpůsobené odds ratio pro diagnostikované vrozené malformace po 1 roce bylo 1,1 (95% CI: 0,5 - 2,3) při expozici FP ve srovnání s expozicí non-FP inhalačním kortikosteroidům u žen se středně těžkou formou bronchiálního astmatu a 1,2 (95% CI: 0,7 - 2,0) u žen s prokázanou závažnou formou bronchiálního astmatu. U žen vystavených v prvním trimestru těhotenství samotnému FP nebyl prokázán žádný rozdíl rizika výskytu vrozených malformací ve srovnání s kombinací salmeterol-FP. Absolutní rizika vzniku vrozených malformací napříč všemi formami bronchiálního astmatu byla v rozmezí 2,0 až 2,9 při expozici 100 těhotných žen FP, což je srovnatelné s výsledkem studie u 15 840 těhotných žen, které nebyly vystaveny léčbě používané u bronchiálního astmatu získané z General Practice Research Database (2,8 případů vrozených malformací na 100 těhotných).
5.3 Předklinické údaje vztahující se k bezpečnosti
Při testech toxicity se projevily pouze účinky typické pro silně působící kortikosteroidy, a to jen při dávkách výrazně převyšujících dávky doporučované k terapeutickým účelům. Opakovanými testy toxicity, studiemi reprodukce nebo studiemi teratogenity se žádné jiné účinky neprokázaly.
Flutikason-propionát u hlodavců nevykazuje in vitro ani in vivo mutagenní aktivitu ani onkogenní potenciál. V experimentálních modelech se přípravek osvědčil jako nedráždící a nealergizující látka.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Monohydrát laktosy (s obsahem mléčných bílkovin)
6.2 Inkompatibility Neuplatňuje se.
6.3 Doba použitelnosti
18 měsíců
6.4 Zvláštní opatření pro uchovávání
Přípravek uchovávejte při teplotě do 30 °C.
Uchovávejte na suchém místě.
6.5 Druh obalu a obsah balení
Prášek s flutikason-propionátem a laktosou je uložen v blistrovém stripu skládajícím se z tvarované základní fólie a víčka se stahujícím se fóliovým laminátem.
Blistový strip je vložený v diskovitém plastovém inhaleru s počítadlem dávek. Diskus je zabalen v ochranné hliníkové fólii. Po otevření ochranné hliníkové fólie je nutno ji odstranit.
Velikost balení:
Flixotide Diskus 100: 60 dávek (60x 100 mikrogramů)
Flixotide Diskus 250: 60 dávek (60x 250 mikrogramů)
Flixotide Diskus 500: 60 dávek (60x 500 mikrogramů)
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním Jak používat přípravek Flixotide Diskus:
Informace o Vašem Diskusu:
Diskus je inhalační pomůcka z umělé hmoty zabalená v ochranné hliníkové fólii. Fólie poskytuje ochranu proti vlhkosti a má se otevřít, pokud je pacient připraven k prvnímu podání dávky. Jednou otevřená ochranná hliníková fólie se má pak odstranit.
Popis přístroje:
Obrázek 1. Obrázek 2.
Zavřený Diskus Otevřený Diskus
1
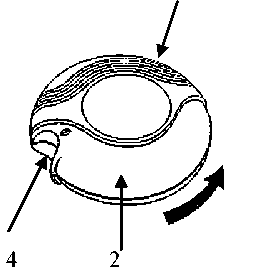
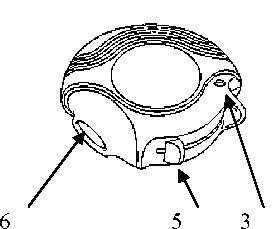
1. základní část
2. posuvná část
3. počítadlo dávek
4. jezdec
5. posuvná páčka
6. náustek
ZAVŘENO:
Po vynětí Diskusu z krabičky a ochranné hliníkové fólie bude v pozici zavřeno.
Nová inhalační pomůcka obsahuje 60 dávek tohoto přípravku. Počítadlo dávek ukazuje, kolik jich ještě zbývá.
Každá dávka je přesně odměřena a hygienicky chráněna. Inhalační pomůcka tedy nevyžaduje žádnou údržbu ani opětovné plnění.
Počítadlo dávek umístěné na horní straně ukazuje, kolik dávek ještě zbývá. Číslice 5 až 0 jsou ČERVENÉ, aby byl pacient upozorněn, že zbývá už jen málo dávek.
Používání tohoto přípravku je snadné. Je třeba se řídit následujícími pěti jednoduchými pokyny:
1. Otevírání inhalační pomůcky
2. Nastavení inhalační pomůcky
3. Inhalování
4. Ukončení inhalace
5. Výplach
Jak Diskus pracuje:
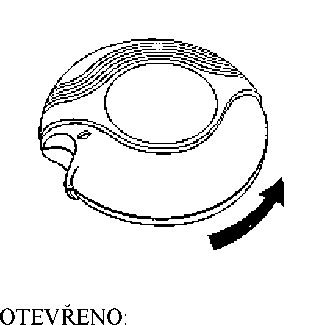
Posunutím páčky na inhalační pomůcce se otevře malý otvor v náustku a současně se uvolní dávka a připraví se k vdechnutí. Po zavření inhalační pomůcky se páčka automaticky vrátí do své původní polohy a pomůcka je připravena na další dávku (až bude potřeba). Vnější kryt chrání inhalační pomůcku, pokud se právě nepoužívá.
1. Otevírání inhalační pomůcky - jak použít Diskus
Inhalační pomůcka se otevře tak, že vnější kryt zařízení se drží v jedné ruce a palec druhé ruky se vloží do jezdce a zatlačí se co nejvíce dozadu.

Inhalační pomůcka se otočí náustkem směrem k pacientovi. Páčka se zatlačí co nejdále směrem od sebe, až cvakne. Pak je Diskus připraven k použití. Po každém zatlačení na páčku je připravena další dávka k vdechnutí. Je to vidět na počítadle dávek. S páčkou se nesmí zbytečně manipulovat, protože se tím uvolní dávka, která se vyplýtvá bez užitku.

Před začátkem vdechování dávky je nutné si pozorně přečíst tuto část textu.
Před přiložením inhalační pomůcky k ústům se musí vydechnout, jak nejvíce to je bez námahy možné. Upozornění - nikdy se nesmí vydechovat do inhalační pomůcky.
Náustek se vloží mezi rty. Začne se ústy, ne nosem, pomalu a zhluboka nadechovat přes inhalační pomůcku.
Inhalační pomůcka se odloží.
Dech se zadrží asi na 10 sekund nebo na tak dlouho, jak je to bez námahy možné.
Pomalu se vydechne.

Inhalační pomůcka se zavře tak, že se palec vloží do jezdce a posune se jím co nejvíce směrem k sobě. Při zavření je slyšet zaklapnutí. Páčka se automaticky vrátí do původní polohy a pomůcka je znovu připravena k aplikaci další dávky.
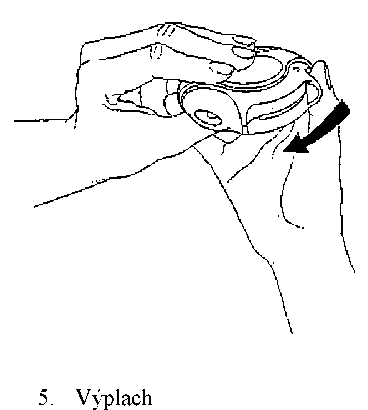
Poté se vypláchnou ústa vodou a voda se vyplivne.
Pokud se užívají dvě dávky, musí se inhalační pomůcka zavřít a pak se opakují všechny uvedené kroky 1 - 4.
Upozornění
- Přípravek Flixotide Diskus se musí uchovávat v suchu.
- Pokud se přípravek Flixotide Diskus nepoužívá, nechává se inhalační pomůcka zavřená.
- Nikdy se do inhalační pomůcky nesmí vydechovat.
- Dávka se uvolní pouhým posunutím páčky tehdy, když je pacient připraven k užití dávky.
- Nepřekračovat stanovené dávky. Uchovávejte mimo dohled a dosah dětí.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Glaxo Group Ltd.,
980 Great West Road,
Brentford, Middlesex, TW8 9GS,
Velká Británie.
8. REGISTRAČNÍ ČÍSLO(A)
Flixotide Diskus 100: 14/074/00-C Flixotide Diskus 250: 14/075/00-C Flixotide Diskus 500: 14/076/00-C
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 2.2.2000
Datum posledního prodloužení registrace: 27.10.2010
10. DATUM REVIZE TEXTU
19.8.2016
16/16