Firmagon 80 Mg
SOUHRN ÚDAJŮ O PŘÍPRAVKU
PŘÍLOHA I
FIRMAGON 80 mg prášek a rozpouštědlo pro injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna lahvička obsahuje degarelixum 80 mg (ve formě degarelixi acetas). Po rozředění obsahuje jeden ml roztoku degarelixum 20 mg.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční roztok
Prášek: bílý až bělavý prášek Rozpouštědlo: čirý, bezbarvý roztok
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
FIRMAGON je antagonista gonadoliberinu (GnRH) indikovaný k léčbě dospělých mužů s pokročilým hormonálně závislým nádorovým onemocněním prostaty.
4.2 Dávkování a způsob podání
Dávkování
|
Úvodní dávka |
Udržovací dávka - podávaná jednou měsíčně |
|
240 mg aplikovaných ve dvou po sobě jdoucích subkutánních injekcích po 120 mg |
80 mg aplikovaných v jedné subkutánní injekci |
První udržovací dávku je třeba podat jeden měsíc po úvodní dávce.
Terapeutické účinky degarelixu je nutno monitorovat podle klinických parametrů a podle sérových hladin prostatického specifického antigenu (PSA). Jak vyplývá z klinických studií, dochází k poklesu hladin testosteronu (T) bezprostředně po podání úvodní dávky; přitom 96% pacientů má sérové hladiny testosteronu odpovídající medikamentózní kastraci (T<0,5 ng/ml) už po třech dnech a 100% pacientů po uplynutí jednoho měsíce. Dlouhodobá léčba formou udržovacích dávek v celkovém trvání až jeden rok ukazuje, že hladiny testosteronu zůstávají setrvale potlačeny u 97% mužů (T<0,5 ng/ml).
V případě nedostatečné klinické odovědi je nutno se ujistit, že hladiny sérového testosteronu zůstávají dostatečně nízké.
Vzhledem k tomu, že degarelix nevede k prudkému vzestupu hladin testosteronu, není na začátku léčby ani nutné podávat navíc antiandrogen jako ochranu proti jeho náhlému zvýšení.
Zvláštní skupiny populace
Starší pacienti, osoby s poškozením jater nebo ledvin:
Starším osobám či pacientům s mírně nebo středně poškozenou funkcí jater nebo ledvin není nutno dávkování upravovat (viz bod 5.2). Pacienti se závažným poškozením jater či ledvin nebyli předmětem zkoumání, a proto je na místě opatrnost (viz bod 4.4).
Pediatrická populace
Neexistuje žádné relevantní použití přípravku FIRMAGON u dětí a dospívajících při léčbě dospělých mužů s pokročilým hormonálně závislým nádorovým onemocněním prostaty.
Způsob podání
FIRMAGON je nutno před podáním rozředit. Pokyny pro rozředění a aplikaci přípravku jsou uvedeny v bodě 6.6.
FIRMAGON se aplikuje POUZE subkutánně, nesmí se podávat intravenózně.
Nedoporučuje se podávat ho intramuskulárně, protože tento způsob aplikace není vyzkoušen.
FIRMAGON se aplikuje formou subkutánní injekce v oblasti břicha. Místa vpichu je třeba periodicky střídat. Pro aplikaci léku je nutno vybírat taková místa na těle pacienta, která nejsou vystavena tlaku, např. nepříliš blízko pasu, opasku a také nikoli v blízkosti žeber.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.
4.4 Zvláštní upozornění a opatření pro použití Účinky na interval QT/QTc
Dlouhodobá léčba formou androgenní deprivace může vyvolat prodloužení intervalu QT. V registrační studii, která srovnávala FIRMAGON s leuprorelinem bylo pravidelně (měsíčně) měřeno elektrokardiogramem ( EKG); obě terapie ukázaly QT/QTc intervaly překračující 450 ms u přibližně 20% pacientů a 500 ms u 1% pacientů léčených degarelixem a 2% pacientů léčených leuprorelinem (viz bod 5.1). FIRMAGON nebyl zkoumán u pacientů, kteří mají v anamnéze prodloužení intervalu QT na více než 450 ms nebo torsades de pointes, kteří mají rizikové faktory pro torsades de pointes, a u pacientů, kteří současně užívají léky, jež by mohly QT prodlužovat. Proto u těchto pacientů se musí důkladně zhodnotit poměr přínos/riziko pro FIRMAGON (viz body 4.5 a 4.8).
Z detailní studie intervalu QT vyplynulo, že degarelix nemá žádný skutečný účinek na interval QT/QTC (viz bod 4.8).
Poruchy funkce jater
Pacienti se známou poruchou funkcí jater nebo s podezřením na ni nebyli zařazováni do dlouhodobých klinických studií s degarelixem. Byly sice pozorovány menší, přechodné vzestupy hodnot ALT a AST, ty však nebyly doprovázeny vzestupem hodnot bilirubinu ani klinickými příznaky. V průběhu léčby se doporučuje sledovat jaterní funkce u pacientů se zjištěnou jaterní poruchou nebo s podezřením na ni. Farmakokinetika degarelixu byla zkoumána po jednodávkovém intravenosním podání u osob s mírným až středně závažným postižením jater (viz bod 5.2).
Poruchy funkce ledvin
Degarelix nebyl testován u pacientů se závažným poškozením ledvin, a proto je třeba dbát zvýšené opatrnosti u této skupiny pacientů.
Hypersenzitivita
Degarelix nebyl zkoumán u pacientů s anamnézou těžkého neléčeného astmatu, anafylaktických reakcí nebo závažné kopřivky či angioedému.
Změny v hustotě kostní hmoty
V lékařské literatuře se uvádějí případy snížené hustoty kostní hmoty u mužů, kteří prodělali orchiektomii nebo se léčili některým z agonistů hormonu GnRH. Dá se předpokládat, že dlouhodobá suprese testosteronu u mužů ovlivňuje hustotu kostní hmoty. Při léčbě degarelixem nebyla hustota kostní hmoty měřena.
Glukózová tolerance
Snížená glukózová tolerance byla pozorována u mužů, kteří podstoupili orchiektomii nebo byli léčeni některým z agonistů GnRH. Vzhledem k tomu, že v takových případech může dojít k rozvoji nebo zhoršení cukrovky, mohou diabetici léčení metodou androgenní deprivace vyžadovat častější kontroly hladin cukru v krvi. Vliv degarelixu na hladinu insulinu a glukosy nebyl zkoumán.
Kardiovaskulární onemocnění
Kardiovaskulární onemocnění, jako jsou cévní mozkové příhody a infarkt myokardu byly hlášeny v lékařské literatuře u pacienta s léčbou androgenní deprivace. Z tohoto důvodu je třeba vzít v úvahu všechny rizikové faktory kardiovaskulárního onemocnění.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Žádné studie interakcí nebyly provedeny.
Vzhledem k tomu, že léčba metodou androgenní deprivace může prodlužovat interval QTc, je třeba důkladně zvažovat podávání degarelixu současně s léčivými přípravky, o nichž je známo, že tento interval prodlužují, nebo s léčivými přípravky schopnými vyvolat torsades de pointes, jako jsou antiarytmika třídy IA (např.chinidin a disopyramid) nebo třídy III (např. amiodaron, sotalol, dofetilid, ibutilid), metadon, moxifloxacin, antipsychotika atd. (viz bod 4.4).
Degarelix není substrátem pro lidský systém CYP450 a neprokázalo se, že by in vitro ve větší míře indukoval nebo tlumil CYP1A2, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1 či CYP3A4/5. Proto není pravděpodobné, že by vzhledem k těmto isoenzymům docházelo v metabolismu ke klinicky významným farmakokinetickým lékovým interakcím.
4.6 Fertilita, těhotenství a kojení
Těhotenství a kojení
Pro podávání přípravku FIRMAGON ženám nejsou žádné odpovídající indikace.
Fertilita
FIRMAGON může inhibovat fertilitu u mužů, pokud je potlačena tvorba testosteronu.
4.7 Účinky na schopnost řídit a obsluhovat stroje
FIRMAGON nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje. Nicméně únava a závratě jsou běžné nežádoucí účinky, které mohou ovlivnit schopnost řídit a obsluhovat stroje.
4.8 Nežádoucí účinky
Shrnutí bezpečnostního profilu
Nežádoucí účinky nejčastěji pozorované při léčbě degarelixem v registrační studii fáze III (N= 409) byly důsledkem očekávaného fyziologického působení suprese testosteronu; patřily mezi ně např. návaly horka a přibývání na váze (u 25%, resp. 7% pacientů léčených po dobu jednoho roku) nebo nežádoucí reakce v místě vpichu. Až několik hodin po podání léku byly hlášeny případy přechodné třesavky (u 3% pacientů), horečky (u 2% pacientů) nebo chřipku připomínajících onemocnění (u 1% pacientů).
Nežádoucí reakce v místě vpichu, převážně v podobě bolesti nebo zarudnutí, se vyskytovaly u 28%, resp. 17% pacientů; méně časté byly případy otoku (6%), indurace (4%) a zduření uzlin (3%). K těmto příhodám docházelo především po podání úvodní dávky, zatímco při udržovací terapii s dávkou 80 mg byla incidence na každých 100 injekcí: 3 případy bolesti a <1 případ erytému, otoku, zduření uzlin a indurace. Hlášené příhody byly vesměs přechodného rázu, malé nebo střední intenzity, a vyžadovaly jen málokdy přerušení léčby (<1%). Velmi vzácně byly hlášeny závažné reakce v místě vpichu, jako je infekce v místě vpichu, absces v místě vpichu nebo nekrózy v místě vpichu, které by mohly vyžadovat chirurgický zákrok /drenáž.
Tabulkový přehled nežádoucích účinků
Nežádoucí účinky jsou podle četnosti klasifikovány takto: velmi časté (> 1/10); časté (> 1/100 až < 1/10); méně časté (> 1/1000 až < 1/100), vzácné (>1/10 000 až <1/1000) a velmi vzácné (<1/10 000).
Uvnitř každé skupiny četnosti jsou nežádoucí účinky uváděny v pořadí klesající závažnosti.
Tab. 1: Četnost nežádoucích účinků léčivého přípravku hlášených u 1259 pacientů léčených po dobu celkem 1781 pacient/roků ( studie fáze II a III) a z postmarketingových hlášení.
|
MedDRA systémová orgánová třída (SOC) |
Velmi časté |
Časté |
Méně časté |
Vzácné |
|
Poruchy krve a lymfatického systému |
Anémie* |
Febrilní neutropenie | ||
|
Poruchy imunitního systému |
Hypersenzitivita |
Anafylaktické reakce | ||
|
Poruchy metabolismu a výživy |
Přibývání na váze* |
Hyperglykemie/diabetes mellitus, zvýšený cholesterol, úbytek na váze, snížená chuť k jídlu, změny hladin vápníku v krvi | ||
|
Psychiatrické poruchy |
Deprese, pokles libida* | |||
|
Poruchy nervového systému |
Závratě, bolesti hlavy |
Mentální porucha, hypestézie | ||
|
Poruchy oka |
Rozmazané vidění | |||
|
Srdeční poruchy |
Srdeční arythmie (včetně fibrilace síní), palpitace, prodloužení QT* (viz body 4.4 a 4.5) |
Infarkt myokardu, srdeční selhání | ||
|
Cévní poruchy |
Návaly horka* |
Hypertenze, vazovagální reakce (včetně hypotenze) | ||
|
Respirační, hrudní a mediastinální poruchy | ||||
|
Gastrointestinální poruchy |
Zácpa, zvracení, bolesti břicha, břišní potíže, sucho v ústech, | |||
|
Poruchy jater a žlučových cest |
Zvýšené jaterní transaminázy |
Zvýšený bilirubin, zvýšená alkalická fosfatáza | ||
|
Poruchy kůže a podkožní tkáně |
Hyperhidróza (včetně nočního pocení)*, vyrážka |
Kopřivka, kožní uzliny, alopecie, svědění erytém | ||
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Muskuloskeletální bolesti, a potíže |
Osteoporóza/osteopenie, bolesti kloubů, svalová ochablost, svalové křeče, otoky/ztuhlost kloubů |
|
Poruchy ledvin a močových cest |
Polakisurie, naléhavé močení, dysurie, nokturie, renální dysfunkce, inkontinence | |||
|
Poruchy reprodukčního systému a prsu |
Gynekomastie*, testikulární atrofie*, erektilní dysfunkce* |
Bolesti varlat, bolesti v prsu, bolesti v pánvi, podráždění genitálu, ejakulační selhání | ||
|
Celkové poruchy a reakce v místě aplikace |
Nežádoucí reakce v místě vpichu injekce |
Třesavka, pyrexie, únava*, chřipku připomínající onemocnění |
Nauzea, periferní edém |
*Známý fyziologický následek snížené hladiny testosteronu
Popis vybraných nežádoucích účinků
Změny laboratorních ukazatelů
Změny laboratorních hodnot zjištěné při jednoleté léčbě v rámci konfirmační studie fáze III (N= 409) se pohybovaly ve stejných mezích u degarelixu i u agonisty GnRH (leuprorelinu) použitého pro srovnání. U 2 -6 % pacientů, kteří vykazovali před léčbou normální hodnoty, byly po léčbě oběma léky zjištěny výrazně abnormální (>3násobek horní meze normy) hodnoty jaterních transamináz (ALT, AST a GMT). Výrazný pokles hematologických hodnot - hematokritu (<0,37) a hemoglobinu (<115g/l) byl nalezen u 40%, resp. 13 - 15% pacientů, s normálními hodnotami naměřenými před léčbou, po léčbě oběma léky. Není známo, do jaké míry byl tento pokles hematologických ukazatelů způsoben přítomností rakoviny prostaty a do jaké míry byl důsledkem terapie formou androgenní deprivace. U pacientů léčených degarelixem a leuprorelinem, kteří vykazovali před léčbou normální hodnoty, byly zjištěny výrazně abnormální hodnoty draslíku (>5,8 mmol/l) u 6% léčených degarelixem a u 3% léčených leuprorelinem, kreatininu (>177 pmol/l ) u 2% léčených degarelixem a u 2% léčených leuprorelinem a močovinového dusíku v krvi (>10,7 mmol/l) u 15% léčených degarelixem a u 14% léčených leuprorelinem.
Změny v EKG měřeních
Změny v EKG měřeních pozorované během jednoroční léčby v rámci konfirmační studie fáze III (N= 409) byly ve stejném rozsahu pro degarelix a agonistu GnRH (leuprorelin) použitého pro srovnání. Tři (<1%) z 409 pacientů léčených degarelixem a čtyři (2%) z 201 pacientů léčených leuprorelinem 7,5 mg mělo QTcF > 500 ms. Od zahájení studie až do jejího ukončení byla střední změna QTcF pro degarelix 12,0 ms a pro leuprorelin 16,7 ms.
Nedostatek skutečného účinku degarelixu na repolarizaci myokardu (QTcF), srdeční frekvenci, AV vedení, depolarizaci myokardu, či na morfologii vlny T nebo V potvrdila důkladná studie QT u zdravých probandů (N - 80) po i.v. infuzi degarelixu v trvání 60 minut, kdy bylo dosaženo 222 ng/ml střední hodnoty Cmax, tedy hodnoty 3 až 4 krát větší než Cmax při léčení karcinomu prostaty.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Klinické zkušenosti s účinky akutního předávkování degarelixem nejsou k dispozici. V případě předávkování je třeba pacienta sledovat a, pokud to bude považováno za nutné, zahájit vhodnou podpůrnou léčbu.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: hormonální léčiva používaná v onkologii, jiní antagonisté hormonů a příbuzné látky, ATC kód: L02BX02
Mechanismus účinku
Degarelix je selektivní antagonista gonadoliberinu (GnRH), který se kompetitivně a reverzibilně váže na hypofyzární receptory GnRH, a tím rychle redukuje uvolňování gonadotropinů, luteinizačního hormonu (LH) a folikulostimulačního hormonu (FSH), čímž snižuje sekreci testosteronu (T) ve varlatech. Je známo, že karcinom prostaty je citlivý na androgeny a reaguje na léčbu, která zdroj androgenů odstraňuje. Na rozdíl od agonistů GnRH nenavozují antagonisté GnRH po zahájení léčby náhlý vzestup LH s následným prudkým vzestupem hladin testosteronu provázeným stimulací nádoru a potenciálním vzplanutím symptomů.
Jedna dávka 240 mg degarelixu následovaná udržovací dávkou 80 mg měsíčně rychle vyvolá pokles hladin LH, FSH a poté i testosteronu. Podobně jako hladiny testosteronu klesají v séru i koncentrace dihydrotestosteronu (DHT).
Degarelix efektivně dosahuje a udržuje snížené koncentrace testosteronu bezpečně pod hladinou medikamentozní kastrace 0,5 ng/ml. Podávání měsíční udržovací dávky 80 mg vedlo u 97% pacientů k setrvale sníženým hladinám testosteronu po dobu nejméně jednoho roku. Během léčby degarelixem nebyly po reinjekci pozorovány žádné náhlé vzestupy hladin testosteronu. Střední hladiny testosteronu po jednoroční léčbě byly 0,087 ng/ml (v interkvartilním rozsahu 0,06-0,15) N=167.
Výsledky registrační studie fáze III:
Účinnost a bezpečnost degarelixu byly hodnoceny v otevřené, multicentrické, randomizované, aktivním komparátorem kontrolované studii s paralelními skupinami. Studie zkoumala účinnost a bezpečnost degarelixu podávaného ve dvou různých režimech při zahajovací dávce 240 mg (40 mg/ml), po níž každý měsíc následovaly subkutánně aplikované dávky 160 mg (40 mg/ml) nebo 80 mg (20 mg/ml) oproti měsíčním, nitrosvalovým dávkám 7,5 mg leuprorelinu u pacientů s rakovinou prostaty vyžadující léčbu metodou androgenní deprivace. Celkem 620 pacientů bylo randomizovaně rozděleno do tří terapeutických skupin; z tohoto počtu 504 (81%) mužů studii dokončilo. Ve skupině léčené degarelixem 240/80 mg přerušilo studii 41 (20%) pacientů ve srovnání s 32 (16%) pacienty ve skupině užívající leuprorelin.
Z těchto 610 léčených pacientů
• 31% mělo lokalizovanou rakovinu prostaty
• 29% mělo místně pokročilou rakovinu prostaty
• 20 % mělo metastatickou rakovinu prostaty
• 7% mělo neznámý stav metastatického postižení
• 13% už dříve prodělalo kurativně cílenou operaci nebo ozařování a mělo stoupající PSA
Základní demografické údaje byly u všech podobné. Střední věk byl 74 let (v rozmezí 47 až 98). Primárním cílem bylo doložit, že degarelix je během 12 měsíců léčby účinný se zřetelem na dosažení a udržení suprese tvorby testosteronu pod hladinou 0,5 ng/ml. Zvolena byla nejnižší účinná udržovací dávka 80 mg degarelixu.
Dosahování sérových hladin testosteronu (T) < 0,5 ng/ml FIRMAGON účinně a rychle potlačuje tvorbu testosteronu, viz Tab.2
Tab. 2: Procento pacientů dosahujících T<0,5 ng/ml po zahájení léčby.
|
Doba |
Degarelix 240/80 mg |
Leuprorelin 7,5 mg |
|
Den 1 |
52% |
0% |
|
Den 3 |
96% |
0% |
|
Den 7 |
99% |
1% |
|
Den 14 |
100% |
18% |
|
Den 28 |
100% |
100% |
Prevence náhlého vzestupu testosteronu
Náhlý vzestup byl definován jako stav, kdy hladina testosteronu přestoupila během prvních dvou týdnů výchozí hodnotu o >15%. U žádného z pacientů léčených degarelixem k náhlému vzestupu testosteronu nedošlo; v den 3 byl zaznamenán průměrný pokles testosteronu o 94%. U většiny pacientů užívajících leuprorelin k náhlému vzestupu hladiny testosteronu došlo; v den 3 byl zaznamenán průměrný vzestup hladiny testosteronu o 65%. Tento rozdíl byl statisticky významný (p<0,001).
Obr.1: Změna hladiny testosteronu v jednotlivých léčebných skupinách (v%) během prvních 28 dnů (střední hodnota s interkvartilními rozsahy).
Změna hladiny testosteronu (v %) ode dne 0 do dne 28
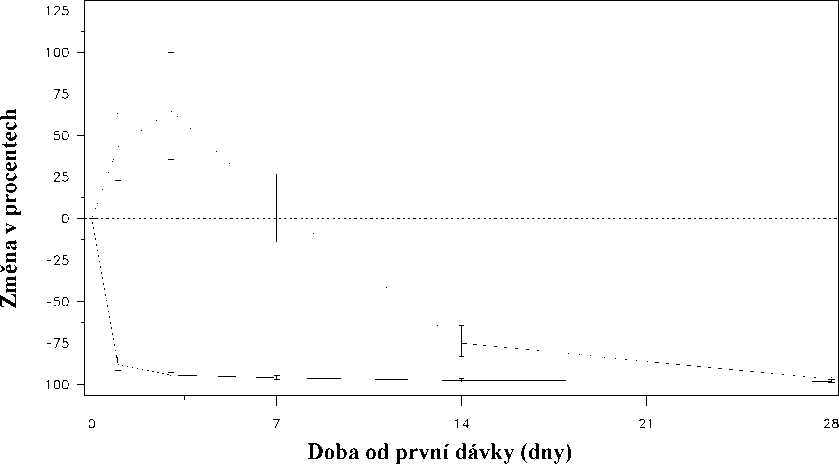
Léčebná skupina_Degarelix 240@40/80@20.......Leuprorelin 7,5 mg
Primární end-point studie bylo potlačení hladiny testosteronu po jednoroční léčbě degarelixem nebo leuprorelinem. Klinický přínos pro degarelix ve srovnání s leuprorelinem plus antiandrogenem v počáteční fázi léčby nebyl zjištěn.
Reverzibilita testosteronu
Ve studii s pacienty, u nichž po lokální léčbě (zejména radikální prostatektomii a ozařování) došlo ke zvýšení antigenu PSA, byl po dobu sedmi měsíců podáván FIRMAGON; poté následovalo dalších sedm měsíců monitorování. Střední čas obnovení hladin testosteronu (>0,5 ng/ml, nad úroveň kastrace) po vysazení léčby byl 112 dnů (počítáno od začátku monitorovacího období, tj. od doby 28 dnů po poslední ínjekci). Střední čas pro dosažení hladiny >1,5 ng/ml (nad dolní hranici normálního rozmezí) byl 168 dnů.
Dlouhodobý účinek
Za úspěšnou léčebnou odpověď bylo v klinickém hodnocení považováno dosažení medikamentózní kastrace ke dni 28 a její udržení až do dne 364, kdy ani jednou hladina testosteronu nepřestoupila hodnotu 0,5 ng/ml.
Tab. 3: Kumulativní pravděpodobnost udržení testosteronu <0,5 ng/ml ode dne 28 do dne 364.
|
Degarelix 240/80 mg N=207 |
Leuprorelin 7,5 mg N=201 | |
|
Počet respondentů |
202 |
194 |
|
Četnost odpovědí (intervaly spolehlivosti)* |
97,2% (93,5; 98,8%) |
96,4% (92,5; 98,2%) |
* Odhad ve skupině podle Kaplana a Meiera
Dosažení redukované hodnoty prostatického specifického antigenu (PSA)
Velikost nádoru se neměřila přímo v rámci programu klinického hodnocení, nicméně nádor nepřímo vykazoval příznivou odezvu na léčbu, jak naznačuje po 12 měsících 95% redukce střední hodnoty PSA u degarelixu.
Výchozí hodnoty PSA (medián) ve studii činil:
• u skupiny léčené degarelixem 240/80 mg - 19,8 ng/ml (interkvartilní rozmezí : P25 9,4 ng/ml, P75 46,4 ng/ml)
• u skupiny léčené leuprorelinem 7,5 mg - 17,4 ng/ml (interkvartilní rozmezí: P25 8,4 ng/ml, P75 56,5 ng/ml)
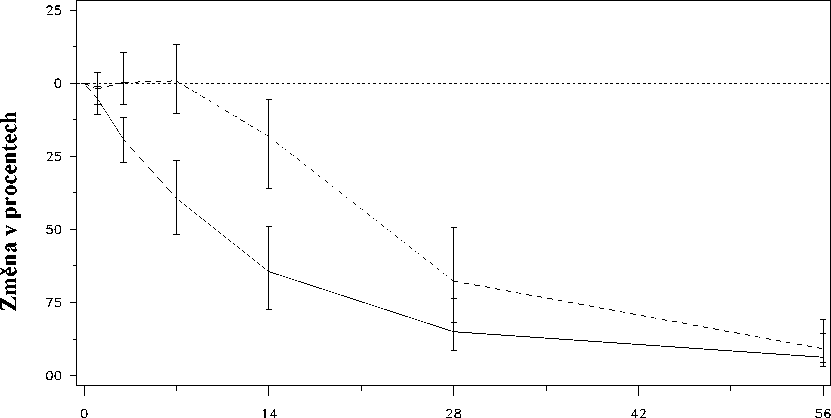
Obr. 2: Změna PSA (v %) v jednotlivých léčebných skupinách v prvních 56 dnech terapie.
Změna PSA ode dne 0 do dne 56 (v%)
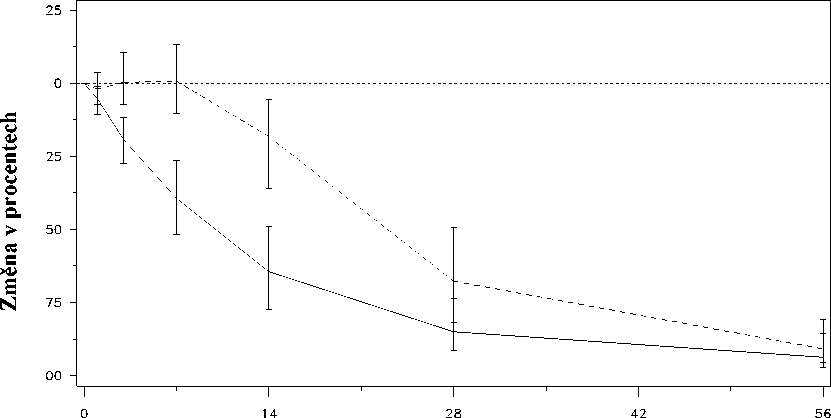
Doba od první dávky (dny)
Léčebná skupina_Degarelix 240@40/80@20.......Leuprorelin 7,5 mg
Tento rozdíl byl při předběžné analýze ke dni 14 a ke dni 28 statisticky významný (p<0,001).
Hladiny prostatického specifického antigenu (PSA) byly po dvou týdnech léčby degarelixem sníženy o 64%, po jednom měsíci o 85%, po třech měsících o 95% a zůstaly potlačené (přibližně o 97%) po celou dobu jednoho roku léčby.
Pokud jde o snížení oproti výchozím hodnotám (v%) nebyly ode dne 56 do dne 364 zjištěny žádné významné rozdíly mezi degarelixem a komparátorem (srovnávací látkou).
Účinek na objem prostaty
Po třech měsících léčby degarelixem (v dávkovacím režimu 240/80 mg) bylo dosaženo 37% redukce objemu prostaty, měřené metodou TRUS (transrektální ultrazvukové snímkování), a to jak u pacientů vyžadujících
9
hormonální léčbu před ozařováním, tak u pacientů vybraných za kandidáty farmakologické kastrace. Zmenšení objemu prostaty bylo podobné hodnotám dosaženým při podávání goserelinu spolu s prevencí androgenního flare-up fenoménu pomocí antiandrogenů.
Vliv na QT/QTc intervaly
V rámci registrační studie porovnávající FIRMAGON s leuprorelinem byly periodicky pořizovány elektrokardiogramy. U obou terapií se ukázalo, že u zhruba 20% pacientů byly QT/QTc intervaly delší než 450 ms. Počítáno od výchozí hodnoty do konce klinického hodnocení, činila střední hodnota změny u přípravku FIRMAGON 12,0 ms, u leuprorelinu 16,7 ms.
Protilátky proti degarelixu
Po jednoroční léčbě přípravkem FIRMAGON byl u 10% pacientů pozorován rozvoj tvorby protilátek proti degarelixu a u 29 % pacientů léčených FIRMAGONEM po dobu až pěti a půl roku.. Nic však nenaznačuje, že tvorbou protilátek je nějak dotčena účinnost či bezpečnost léčby přípravkem FIRMAGON po pěti a půlleté léčbě.
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem FIRMAGON u všech podskupin pediatrické populace (viz bod 4.2 Pediatrická populace).
5.2 Farmakokinetické vlastnosti
Absorpce
Poté, co v pivotní studii CS21 bylo pacientům s rakovinou prostaty podkožně aplikováno 240 mg degarelixu při koncentraci 40 mg/ml, byla zjištěna hodnota AUC0-28 dnů 635 (602-668) den*ng/ml, hodnota Cmax 66,0 (61,0-71,0) ng/ml byla naměřena při tmax 40 (37-42) hodin. Střední minimální hodnoty byly přibližně 11-12 ng/ml po zahajovací dávce a 11-16 ng/ml po udržovací dávce 80 mg při koncentraci 20 mg/ml. Cmax plasmatické hladiny degarelixu se dvoufázově snižuje tak, že střední terminální poločas (tJ/2) při udržovací dávce je 29 dnů.Dlouhý poločas u podkožní aplikace je důsledkem velmi pomalého uvolňování degarelixu z úložiště, které se vytvoří na místě vpichu /místech vpichů. Farmakokinetické chování léku je ovlivňováno jeho koncentrací v injekčním roztoku. V důsledku toho mají Cmax a biologická dostupnost tendenci klesat s rostoucí koncentrací dávky za současného prodlužování poločasu. Z toho důvodu se nemá používat jiných než doporučených koncentrací.
Distribuce
Distribuční objem u zdravých starších mužů je přibližně 1 l/kg. Vazba na plasmatické proteiny je odhadem asi 90%.
Biotransformace
Při průchodu hepatobiliární soustavou podléhá degarelix obvyklé peptidové degradaci a vylučuje se převážně v podobě peptidových fragmentů stolicí. Po subkutánní aplikaci se ve vzorcích plasmatu neobjevily žádné významné metabolity. Jak vyplývá ze studií in vitro, degarelix netvoří substrát pro lidský CYP450 systém.
Eliminace
U zdravých mužů se zhruba 20-30% jednotlivé nitrožilně podané dávky vyloučí močí; z toho plyne, že 70-80% se vyloučí přes hepatobiliární soustavu. Bylo zjištěno, že clearance degarelixu podaného zdravým starším mužům v jednotlivých intravenózních dávkách (0,864-49,4pg/kg) je 35-50 ml/h/kg.
Zvláštní skupiny populace
Pacienti s postižením ledvinných funkcí
Nebyly uskutečněny žádné farmakokinetické studie u pacientů s renálním postižením. Pouze asi 20-30% aplikované dávky degarelixu se v nezměněné podobě vylučuje renální cestou. Z populačně farmakokinetického rozboru údajů z registrační studie fáze III. vyplývá, že pacienti s mírným až středně závažným postižením funkce ledvin mají clearance degarelixu sníženou o přibližně 23%; z toho důvodu se u pacientů s mírným až středně závažným postižením ledvin úprava dávkování nedoporučuje. Vzhledem k tomu, že údajů o pacientech se závažnými poruchami renálních funkcí je málo, je u této skupiny pacientů na místě opatrnost.
Pacienti s postižením jaterních funkcí
Degarelix byl zkoumán v rámci farmakokinetické studie u pacientů s mírným až středně závažným postižením funkcí jater. Ve srovnání se zdravými osobami nebyly u pacientů s postižením jaterních funkcí pozorovány žádné známky zvýšené expozice. U pacientů s mírným nebo středně závažným postižením funkcí jater není nutno dávkování upravovat. Pacienti se závažným postižením jaterních funkcí nebyli předmětem zkoumání, a z toho důvodu je u této skupiny na místě opatrnost.
5.3 Předklinické údaje vztahující se k bezpečnosti
Jak ukázaly reprodukční pokusy na zvířatech, způsobuje degarelix neplodnost u samců. Ta je důsledkem farmakologického působení; tyto účinky jsou reverzibilní.
Studiemi reprodukční toxicity u samic se zjistilo, že degarelix působí tak, jak se při jeho farmakologických vlastnostech dalo očekávat. Vyvolával dávkově závislé prodloužení doby před pářením a zabřeznutím, snížení počtu žlutých tělísek corpora lutea, zvýšení počtu před- a po- implantačních ztrát, potratů, časné smrti zárodků/plodů, předčasných porodů a změn v trvání porodu.
Předklinické studie farmakologické bezpečnosti, toxicity po opakovaném podávání, genotoxicity a kancerogenního potenciálu neodhalily žádná zvláštní rizika pro lidi. Jak in vitro tak i in vivo studie neprokázaly známky prodloužení QT.
Studie akutní, subakutní a chronické toxicity provedené na potkanech a opicích nezjistily po podkožní aplikaci degarelixu žádnou toxicitu pro cílové orgány. V souvislosti s tímto lékem bylo u zvířat pozorováno místní podráždění tam, kde degarelix byl do podkoží aplikován ve velkých dávkách.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek
Mannitol (E421)
Rozpouštědlo Voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto tento léčivý přípravek nesmí být mísen s žádnými dalšími léčivými přípravky.
6.3 Doba použitelnosti
3 roky.
Po naředění
Chemická a fyzikální stabilita přípravku po naředění byla prokázána pro dobu 2 hodin při 25°C. Z mikrobiologického hlediska je přípravek určen k okamžitému použití, pokud způsob rozředění nevylučuje riziko mikrobiální kontaminace. Není-li přípravek použit okamžitě, nese odpovědnost za dobu a podmínky uchování přípravku po naředění sám uživatel.
6.4 Zvláštní opatření pro uchovávání
Tento přípravek nevyžaduje zvláštní podmínky uchovávání.
Podmínky uchovávání rekonstituovaného léčivého přípravku viz bod 6.3.
6.5 Druh obalu a velikost balení
Injekční lahvička ze skla (typ I) s bromobutylovou pryžovou zátkou a hliníkovým flip-off uzávěrem obsahující 80 mg prášku pro přípravu injekčního roztoku
Předplněná injekční stříkačka ze skla (typ I) s elastomerovým pístovým uzávěrem s víčkem a ryskou označující 4 ml obsahující 4,2 ml rozpouštědla Táhlo pístu
Adaptér pro injekční lahvičky Injekční jehla (25G 0,5 x 25 mm)
Velikost balení
FIRMAGON se dodává ve 2 velikostech balení:
Balení s 1 tvarovanou plastovou vložkou obsahuje: 1 injekční lahvičku s práškem, 1 předplněnou stříkačku s rozpuštědlem, 1 táhlo pístu, 1 adaptér pro injekční lahvičku a 1 jehlu.
Balení s 3 tvarovanými plastovýni vložkami obsahuje: 3 injekční lahvičky s práškem, 3 předplněné stříkačky s rozpouštědlem, 3 táhla pístu, 3 adaptéry pro injekční lahvičky a 3 jehly.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Návodem pro rozředění je třeba se pečlivě řídit.
Podávat lék v jiných koncentracích se nedoporučuje, protože tvorbu depotního gelu ovlivňuje právě koncentrace. Rozředěný roztok musí být čirá tekutina bez nerozpuštěného prášku.
POZNÁMKA:
• INJEKČNÍMI LAHVIČKAMI SE NESMÍ TŘEPAT
Balení obsahuje jednu injekční lahvičku prášku a jednu předplněnou stříkačku rozpouštědla, které je nutno připravit pro podkožní injekci.
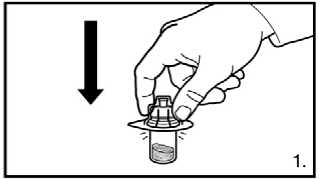
1. Odstraňte uzávěr z balení adaptéru injekční lahvičky. Adaptér na injekční lahvičky s práškem nasaďte stlačením adaptéru, dokud špička neprojde gumovým uzávěrem a adaptér nezapadne na místo.
2. Připravte předplněnou stříkačku nasazením zapuštěného pístu.
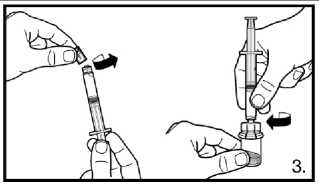
3. Odstraňte víčko z předplněné stříkačky.
Našroubujte injekční lahvičku na adaptér injekční lahvičky s rozpouštědlem. Rozpouštědlo přemístěte do injekční lahvičky s práškem.
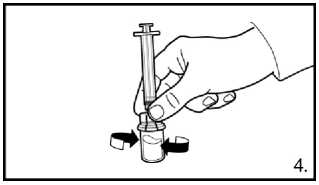
4. S lahvičkou, stále připevněnou na adaptér opatrně kružte, dokud tekutina není čirá a bez nerozpuštěného prášku nebo částic. Pokud prášek přilne ke sklu nad hladinou, lze lahvičku mírně naklonit. Netřepejte, aby se roztok nezpěnil.
Kroužek vzduchových bublin na hladině tekutiny nevadí. Rozředění injekčního roztoku může v některých případech trvat několi minut, ale v některých případech může trvat až 15 minut,
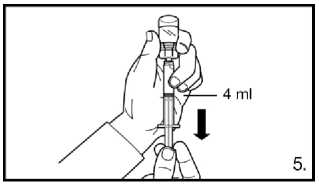
5. Obraťte injekční lahvičku dnem vzhůru a natáhněte k rysce na předplněné stříkačce.
Vždy se ujistěte, že odebíráte přesné množství
bez vzduchových bublin.
6. Oddělte stříkačku od adaptéru lahvičky a na stříkačku nasaďte jehlu pro aplikaci hluboké
podkožní injekce. Opatrně vytlačte vzduchové bublinky._
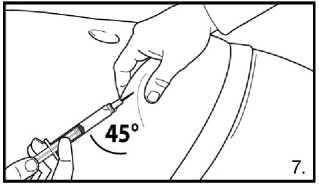
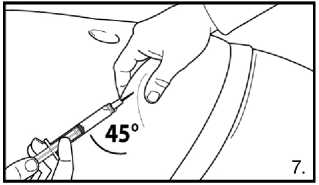
7.Pokračujte hlubokou podkožní injekcí. Uchopte kůži na břiše, povytáhněte podkožní tkáň a zasuňte jehlu hluboko pod úhlem alespoň 45 stupňů. Bezprostředně po rekonstituci pomalu injikujte 4,0 ml přípravku FIRMAGON 80 mg .
8
Injekce se nemá aplikovat do míst, která jsou vystavena tlaku např. kolem pasu, opasku nebo blízko žeber.
Neinjikujte roztok přímo do žíly. Opatrně povytáhněte píst stříkačky a ujistěte se, že se nenasála krev. Objeví-li se ve stříkačce krev, je přípravek dále nepoužitelný. Přerušte proceduru a stříkačku i s jehlou zlikvidujte (rekonstituujte pro pacienta novou dávku).
Žádné zvláštní požadavky pro likvidaci.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Ferring Pharmaceuticals A/S Kay Fiskers Plads 11 DK-2300 Copenhagen S Dánsko
Tel: +45 88 33 88 34
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/08/504/001
EU/1/08/504/003
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace:17/02/2009
Datum posledního prodloužení registrace: 19/09/2013 10. DATUM REVIZE TEXTU
{dd/mm/rrrr}
Podrobné informace o tomto přípravku jsou uveřejněny na webových stránkách Evropské lékové agentury http: //www .ema.europa.eu/.
NÁZEV PŘÍPRAVKU
1.
FIRMAGON 120 mg prášek a rozpouštědlo pro injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna lahvička obsahuje degarelixum 120 mg (ve formě degarelixi acetas). Po rozředění obsahuje jeden ml roztoku degarelixum 40 mg.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční roztok
Prášek: bílý až bělavý prášek Rozpouštědlo: čirý, bezbarvý roztok
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
FIRMAGON je antagonista gonadoliberinu(GnRH) indikovaný k léčbě dospělých mužů s pokročilým hormonálně závislým nádorovým onemocněním prostaty.
4.2 Dávkování a způsob podání
Dávkování
|
Úvodní dávka |
Udržovací dávka - podávaná jednou měsíčně |
|
240 mg aplikovaných ve dvou po sobě jdoucích subkutánních injekcích po 120 mg |
80 mg aplikovaných v jedné subkutánní injekci |
První udržovací dávku je třeba podat jeden měsíc po úvodní dávce.
Terapeutické účinky degarelixu je nutno monitorovat podle klinických parametrů a podle sérových hladin prostatického specifického antigenu (PSA). Jak vyplývá z klinických studií, dochází k poklesu hladin testosteronu (T) bezprostředně po podání úvodní dávky; přitom 96% pacientů má sérové hladiny testosteronu odpovídající medikamentózní kastraci (T<0,5 ng/ml) už po třech dnech a 100% pacientů po uplynutí jednoho měsíce. Dlouhodobá léčba formou udržovacích dávek v celkovém trvání až jeden rok ukazuje, že hladiny testosteronu zůstávají setrvale potlačeny u 97% mužů (T<0,5 ng/ml).
V případě nedostatečné klinické odovědi je nutno se ujistit , že hladiny sérového testosteronu zůstávají dostatečně nízké.
Vzhledem k tomu, že degarelix nevede k prudkému vzestupu hladin testosteronu, není na začátku léčby ani nutné podávat navíc antiandrogen jako ochranu proti jeho náhlému zvýšení.
Zvláštní skupiny populace
Starší pacienti, osoby s poškozením jater nebo ledvin:
Starším osobám či pacientům s mírně nebo středně poškozenou funkcí jater nebo ledvin není nutno dávkování upravovat (viz bod 5.2). Pacienti se závažným poškozením jater či ledvin nebyli předmětem zkoumání, a proto je na místě opatrnost (viz bod 4.4).
Pediatrická populace
Neexistuje žádné relevantní použitíí přípravku FIRMAGON u dětí a dospívajících při léčbě dospělých můžů s pokročilým hormonálně závislým nádorovým onemocněním prostaty.
Způsob podání
FIRMAGON je nutno před podáním rozředit. Pokyny pro rozředění a aplikaci přípravku jsou uvedeny v bodě 6.6.
FIRMAGON se aplikuje POUZE subkutánně, nesmí se podávat intravenózně.
Nedoporučuje se podávat ho intramuskulárně, protože tento způsob aplikace není vyzkoušen.
FIRMAGON se aplikuje formou subkutánní injekce v oblasti břicha. Místa vpichu je třeba periodicky střídat. Pro aplikaci léku je nutno vybírat taková místa na těle pacienta, která nejsou vystavena tlaku, např. nepříliš blízko pasu, opasku a také nikoli v blízkosti žeber.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.
4.4 Zvláštní upozornění a opatření pro použití Účinky na interval QT/QTc
Dlouhodobá léčba formou androgenní deprivace může vyvolat prodloužení intervalu QT. V registrační studii, která srovnávala FIRMAGON s leuprorelinem bylo pravidelně (měsíčně) měřeno elektrokardiogramem (EKG); obě terapie ukázaly QT/QTc intervaly překračující 450 ms u přibližně 20% pacientů a 500 ms u 1% pacientů léčených degarelixem a 2% pacientů léčených leuprorelinem (viz bod 5.1). FIRMAGON nebyl zkoumán u pacientů, kteří mají v anamnéze prodloužení intervalu QT na více než 450 ms nebo torsades de pointes, které mají rizikové faktory pro torsades de pointes, a u pacientů, kteří současně užívají léky, jež by mohly QT prodlužovat. Proto u těchto pacientů se musí důkladně zhodnotit poměr přínos/riziko pro FIRMAGON (viz body 4.5 a 4.8).
Z detailní studie intervalu QT vyplynulo, že degarelix nemá žádný skutečný účinek na interval QT/QTC (viz bod 4.8).
Poruchy funkce jater
Pacienti se známou poruchou funkcí jater nebo s podezřením na ni nebyli zařazováni do dlouhodobých klinických studií s degarelixem. Byly sice pozorovány menší, přechodné vzestupy hodnot ALT a AST, ty však nebyly doprovázeny vzestupem hodnot bilirubinu ani klinickými příznaky. V průběhu léčby se doporučuje sledovat jaterní funkce u pacientů se zjištěnou jaterní poruchou nebo podezřením na ni. Farmakokinetika degarelixu byla zkoumána po jednodávkovém intravenosním podání u osob s mírným až středně závažným postižením jater (viz bod 5.2).
Poruchy funkce ledvin
Degarelix nebyl testován u pacientů se závažným poškozením ledvin a proto je třeba dbát zvýšené opatrnosti u této skupiny pacientů.
Hypersenzitivita
Degarelix nebyl zkoumán u pacientů s anamnézou těžkého neléčeného astmatu, anafylaktických reakcí nebo závažné kopřivky či angioedému.
Změny v hustotě kostní hmoty
V lékařské literatuře se uvádějí případy snížené hustoty kostní hmoty u mužů, kteří prodělali orchiektomii nebo se léčili některým z agonistů hormonu GnRH. Dá se předpokládat, že dlouhodobá suprese testosteronu u mužů ovlivňuje hustotu kostní hmoty. Při léčbě degarelixem nebyla hustota kostní hmoty měřena.
Glukózová tolerance
Snížená glukózová tolerance byla pozorována u mužů, kteří podstoupili orchiektomii nebo byli léčeni některým z agonistů GnRH. Vzhledem k tomu, že v takových případech může dojít k rozvoji nebo zhoršení cukrovky, mohou diabetici léčení metodou androgenní deprivace vyžadovat častější kontroly hladin cukru v krvi. Vliv degarelixu na hladinu insulinu a glukosy nebyl zkoumán.
Kardiovaskulární onemocnění
Kardiovaskulární onemocnění, jako jsou cévní mozkové příhody a infarkt myokardu byly hlášeny v lékařské literatuře u pacienta s léčbou androgenní deprivace. Z tohoto důvodu je třeba vzít v úvahu všechny rizikové faktory kardiovaskulárního onemocnění.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Žádné studie interakcí nebyly provedeny.
Vzhledem k tomu, že léčba metodou androgenní deprivace může prodlužovat interval QTc, je třeba důkladně zvažovat podávání degarelixu současně s léčivými přípravky, o nichž je známo, že tento interval prodlužují, nebo s léčivými přípravky schopnými vyvolat torsades de pointes, jako jsou antiarytmika třídy IA (např.chinidin a disopyramid) nebo třídy III (např. amiodaron, sotalol, dofetilid, ibutilid), metadon,moxifloxacin, antipsychotika atd. (viz bod 4.4).
Degarelix není substrátem pro lidský systém CYP450 a neprokázalo se, že by in vitro ve větší míře indukoval nebo tlumil CYP1A2, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1 či CYP3A4/5. Proto není pravděpodobné, že by vzhledem k těmto isoenzymům docházelo v metabolismu ke klinicky významným farmakokinetickým lékovým interakcím.
4.6 Fertilita, těhotenství a kojení
Těhotenství a kojení
Pro podávání přípravku FIRMAGON ženám nejsou žádné odpovídající indikace.
Fertilita
FIRMAGON může inhibovat fertilitu u mužů, pokud je potlačena tvorba testosteronu.
4.7 Účinky na schopnost řídit a obsluhovat stroje
FIRMAGON nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje. Nicméně únava a závratě jsou běžné nežádoucí účinky, které mohou ovlivnit schopnost řídit a obsluhovat stroje.
4.8 Nežádoucí účinky
Shrnutí bezpečnostního profilu
Nežádoucí účinky nejčastěji pozorované při léčbě degarelixem v registrační studii fáze III (N= 409) byly důsledkem očekávaného fyziologického působení suprese testosteronu; patřily mezi ně např. návaly horka a přibývání na váze (u 25% resp. 7% pacientů léčených po dobu jednoho roku) nebo nežádoucí reakce v místě vpichu. Až několik hodin po podání léku byly hlášeny případy přechodné třesavky (u 3% pacientů), horečky (u 2% pacientů) nebo chřipku připomínajících onemocnění (u 1% pacientů).
Nežádoucí reakce v místě vpichu, převážně v podobě bolesti nebo zarudnutí, se vyskytovaly u 28%, resp. 17% pacientů; méně časté byly případy otoku (6%), indurace (4%) a zduření uzlin (3%). K těmto příhodám docházelo především po podání úvodní dávky, zatímco při udržovací terapii s dávkou 80 mg byla incidence na každých 100 injekcí: 3 případy bolesti a <1 případ erytému, otoku, zduření uzlin a indurace. Hlášené příhody byly vesměs přechodného rázu, malé nebo střední intenzity, a vyžadovaly jen málokdy přerušení léčby (<1%). Velmi vzácně byly hlášeny závažné reakce v místě vpichu, jako je infekce v místě vpichu, absces v místě vpichu nebo nekrózy v místě vpichu, které by mohly vyžadovat chirurgický zákrok /drenáž.
Tabulkový přehled nežádoucích účinků
Nežádoucí účinky jsou podle četnosti klasifikovány takto: velmi časté (> 1/10); časté (> 1/100 až < 1/10); méně časté (> 1/1000 až < 1/100), vzácné (>1/10000 až <1/1 000) a velmi vzácné (<1/10 000).
Uvnitř každé skupiny četnosti jsou nežádoucí účinky uváděny v pořadí klesající závažnosti.
Tab. 1: Četnost nežádoucích účinků léčivého přípravku hlášených u 1259 pacientů léčených po dobu celkem 1781 pacient/roků (studie fáze II a III) a z postmarketingových hlášení
|
MedDRA systémová orgánová třída (SOC) |
Velmi časté |
Časté |
Méně časté |
Vzácné |
|
Poruchy krve a lymfatického systému |
Anémie* |
Febrilní neutropenie | ||
|
Poruchy imunitního systému |
Hypersenzitivita |
Anafylaktické reakce | ||
|
Poruchy metabolismu a výživy |
Přibývání na váze* |
Hyperglykemie/diabetes mellitus, zvýšený cholesterol, úbytek na váze, snížená chuť k jídlu, změny hladin vápníku v krvi | ||
|
Psychiatrické poruchy |
Deprese, pokles libida* | |||
|
Poruchy nervového systému |
Závratě, bolesti hlavy |
Mentální porucha, hypestézie | ||
|
Poruchy oka |
Rozmazané vidění | |||
|
Srdeční poruchy |
Srdeční arythmie (včetně fibrilace síní), palpitace, prodloužení QT* (viz body 4.4 a 4.5) | |||
|
Cévní poruchy |
Návaly horka* |
Hypertenze, vazovagální reakce (včetně hypotenze) |
Infarkt myokardu, srdeční selhání | |
|
Respirační, hrudní a mediastinální poruchy | ||||
|
Gastrointestinální poruchy |
Zácpa, zvracení, bolesti břicha, břišní potíže, sucho v ústech | |||
|
Poruchy jater a žlučových cest |
Zvýšené jaterní transaminázy |
Zvýšený bilirubin, zvýšená alkalická fosfatáza | ||
|
Poruchy kůže a podkožní tkáně |
Hyperhidróza (včetně nočního pocení)*, vyrážka |
Kopřivka, kožní uzliny, alopecie, svědění, erytém | ||
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Muskuloskeletální bolesti a potíže |
Osteoporóza/osteopenie, bolesti kloubů, svalová ochablost, svalové křeče, otoky/ztuhlost kloubů | ||
|
Poruchy ledvin a močových cest |
Polakisurie, naléhavé močení, dysurie, nokturie, renální dysfunkce, inkontinence |
|
Poruchy reprodukčního systému a prsu |
Gynekomastie*, testikulární atrofie*, erektilní dysfunkce* |
Bolesti varlat, bolesti v prsu, ,bolesti v pánvi, podráždění genitálu, ejakulační selhání | ||
|
Celkové poruchy a reakce v místě aplikace |
Nežádoucí reakce v místě vpichu injekce |
Třesavka, pyrexie, únava*, chřipku připomínající onemocnění |
Nevolnost, periferní edém |
*Známý fyziologický následek snížené hladiny testosteronu
Popis vybraných nežádoucích účinků
Změny laboratorních ukazatelů
Změny laboratorních hodnot zjištěné při jednoleté léčbě v rámci konfirmační studie fáze III (N= 409) se pohybovaly ve stejných mezích u degarelixu i u agonisty GnRH(leuprorelinu) použitého pro srovnání. U 2 -6 % pacientů, kteří vykazovali před léčbou normální hodnoty, byly po léčbě oběma léky zjištěny výrazně abnormální (>3násobek horní meze normy) hodnoty jaterních transamináz (ALT, AST a GMT). Výrazný pokles hematologických hodnot - hematokritu (<0,37) a hemoglobinu (<115g/l) byl nalezen u 40%, resp.13 -15% pacientů, s normálními hodnotami naměřenými před léčbou, po léčbě oběma léky. Není známo, do jaké míry byl tento pokles hematologických ukazatelů způsoben přítomností rakoviny prostaty a do jaké míry byl důsledkem terapie formou androgenní deprivace. U pacientů léčených degarelixem a leuprorelinem, kteří vykazovali před léčbou normální hodnoty, byly zjištěny výrazně abnormální hodnoty draslíku (>5,8 mmol/l) u 6% léčených degarelixem a u 3% léčených leuprorelinem, kreatininu (>177 pmol/l ) u 2% léčených degarelixem a u 2% léčených leuprorelinem a močovinového dusíku v krvi (>10,7 mmol/l) u 15% léčených degarelixem a u 14% léčených leuprorelinem.
Změny v EKG měřeních
Změny v EKG měřeních pozorované během jednoroční léčby v rámci konfirmační studie fáze III (N= 409) byly ve stejném rozsahu pro degarelix a agonistu GnRH (leuprorelin) použitého pro srovnání. Tři (<1%) z 409 pacientů léčených degarelixem a čtyři (2%) z 201 pacientů léčených leuprorelinem 7,5 mg mělo QTcF > 500 ms. Od zahájení studie až do jejího ukončení byla střední změna QTcF pro degarelix 12,0 ms a pro leuprorelin 16,7 ms.
Nedostatek skutečného účinku degarelixu na repolarizaci myokardu (QTcF), srdeční frekvenci, AV vedení, depolarizaci myokardu, či na morfologii vlny T nebo V potvrdila důkladná studie QT u zdravých probandů (n - 80) po i.v. infuzi degarelixu v trvání 60 minut, kdy bylo dosaženo 222 ng/ml střední hodnoty Cmax, tedy hodnoty 3 až 4 krát větší než Cmax při léčení karcinomu prostaty.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V
4.9 Předávkování
Klinické zkušenosti s účinky akutního předávkování degarelixem nejsou k dispozici. V případě předávkování je třeba pacienta sledovat a, pokud to bude považováno za nutné, zahájit vhodnou podpůrnou léčbu.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: hormonální léčiva používaná v onkologii, jiní antagonisté hormonů a příbuzné látky, ATC kód: L02BX02
Mechanismus účinku
Degarelix je selektivní antagonista gonadoliberinu (GnRH), který se kompetitivně a reverzibilně váže na hypofyzámí receptory GnRH, a tím rychle redukuje uvolňování gonadotropinů, luteinizačního hormonu (LH) a folikulostimulačního hormonu (FSH), čímž snižuje sekreci testosteronu (T) ve varlatech. Je známo, že karcinom prostaty je citlivý na androgeny a reaguje na léčbu, která zdroj androgenů odstraňuje. Na rozdíl od agonistů GnRH nenavozují antagonisté GnRH po zahájení léčby náhlý vzestup LH s následným prudkým vzestupem hladin testosteronu provázeným stimulací nádoru a potenciálním vzplanutím symptomů.
Jedna dávka 240 mg degarelixu následovaná udržovací dávkou 80 mg měsíčně rychle vyvolá pokles hladin LH, FSH a poté i testosteronu. Podobně jako hladiny testosteronu klesají v séru i koncentrace dihydrotestosteronu (DHT).
Degarelix efektivně dosahuje a udržuje snížené koncentrace testosteronu bezpečně pod hladinou medikamentozní kastrace 0,5ng/ml. Podávání měsíční udržovací dávky 80 mg vedlo u 97% pacientů k setrvale sníženým hladinám testosteronu po dobu nejméně jednoho roku. Během léčby degarelixem nebyly po reinjekci pozorovány žádné náhlé vzestupy hladin testosteronu.
Střední hladiny testosteronu po jednoroční léčbě byly 0,087 ng/ml (v interkvartilním rozsahu 0,06-0,15) N=167.
Výsledky registrační studie fáze III:
Účinnost a bezpečnost degarelixu byly hodnoceny v otevřené, multicentrické, randomizované, aktivním komparátorem kontrolované studii s paralelními skupinami. Studie zkoumala účinnost a bezpečnost degarelixu podávaného ve dvou různých režimech při zahajovací dávce 240 mg (40 mg/ml), po níž každý měsíc následovaly subkutánně aplikované dávky 160 mg (40 mg/ml) nebo 80 mg (20 mg/ml) oproti měsíčním, nitrosvalovým dávkám 7,5 mg leuprorelinu u pacientů s rakovinou prostaty vyžadující léčbu metodou androgenní deprivace. Celkem 620 pacientů bylo randomizovaně rozděleno do tří terapeutických skupin; z tohoto počtu 504 (81%) mužů studii dokončilo. Ve skupině léčené degarelixem 240/80 mg přerušilo studii 41 (20%) pacientů ve srovnání s 32 (16%) pacienty ve skupině užívající leuprorelin.
Z těchto 610 léčených pacientů
• 31% mělo lokalizovanou rakovinu prostaty
• 29% mělo místně pokročilou rakovinu prostaty
• 20 % mělo metastatickou rakovinu prostaty
• 7% mělo neznámý stav metastatického postižení
• 13% už dříve prodělalo kurativně cílenou operaci nebo ozařování a mělo stoupající PSA
Základní demografické údaje byly u všech podobné. Střední věk byl 74 let (v rozmezí 47 až 98). Primárním cílem bylo doložit, že degarelix je během 12 měsíců léčby účinný se zřetelem na dosažení a udržení suprese tvorby testosteronu pod hladinou 0,5 ng/ml. Zvolena byla nejnižší účinná udržovací dávka 80 mg degarelixu.
Dosahování sérových hladin testosteronu (T) < 0,5 ng/ml FIRMAGON účinně a rychle potlačuje tvorbu testosteronu, viz Tab.2
Tab. 2: Procento pacientů dosahujících T<0,5 ng/ml po zahájení léčby.
|
Doba |
Degarelix 240/80 mg |
Leuprorelin 7,5 mg |
|
Den 1 |
52% |
0% |
|
Den 3 |
96% |
0% |
|
Den 7 |
99% |
1% |
|
Den 14 |
100% |
18% |
|
Den 28 |
100% |
100% |
Prevence náhlého vzestupu testosteronu
Náhlý vzestup byl definován jako stav, kdy hladina testosteronu přestoupila během prvních dvou týdnů výchozí hodnotu o >15%. U žádného z pacientů léčených degarelixem k náhlému vzestupu testosteronu nedošlo; v den 3 byl zaznamenán průměrný pokles testosteronu o 94%. U většiny pacientů užívajících leuprorelin k náhlému vzestupu hladiny testosteronu došlo; v den 3 byl zaznamenán průměrný vzestup hladiny testosteronu o 65%. Tento rozdíl byl statisticky významný (p<0,001).
Obr.1: Změna hladiny testosteronu v jednotlivých léčebných skupinách (v%) během prvních 28 dnů (střední hodnota s interkvartilními rozsahy).
Změna hladiny testosteronu (v %) ode dne 0 do dne 28
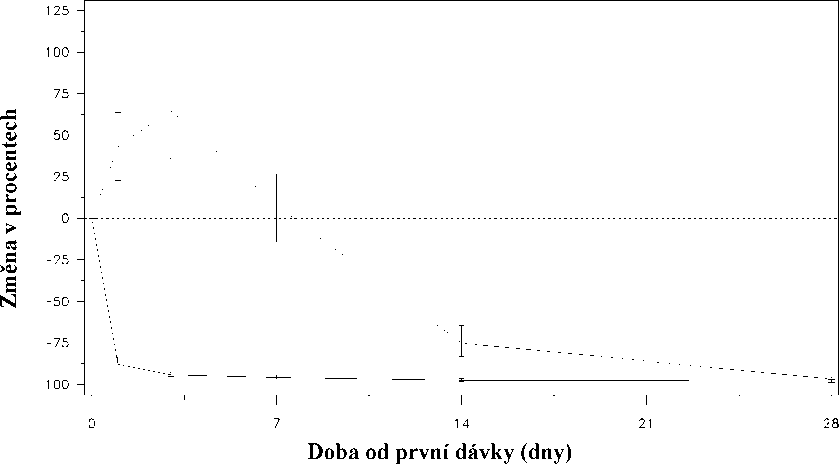
Léčebná skupina_Degarelix 240@40/80@20.......Leuprorelin 7,5 mg
Primární end-point studie bylo potlačení hladiny testosteronu po jednoroční léčbě degarelixem nebo leuprorelinem. Klinický přínos pro degarelix ve srovnání s leuprorelinem plus antiandrogenem v počáteční fázi léčby nebyl zjištěn.
Reverzibilita testosteronu
Ve studii s pacienty, u nichž po lokální léčbě (zejména radikální prostatektomii a ozařování) došlo ke zvýšení antigenu PSA, byl po dobu sedmi měsíců podáván FIRMAGON; poté následovalo dalších sedm měsíců monitorování. Střední čas obnovení hladin testosteronu (>0,5 ng/ml, nad úroveň kastrace) po vysazení léčby byl 112 dnů (počítáno od začátku monitorovacího období, tj. od doby 28 dnů po poslední ínjekci). Střední čas pro dosažení hladiny >1,5 ng/ml (nad dolní hranici normálního rozmezí) byl 168 dnů.
Dlouhodobý účinek
Za úspěšnou léčebnou odpověď bylo v klinickém hodnocení považováno dosažení medikamentózní kastrace ke dni 28 a její udržení až do dne 364, kdy ani jednou hladina testosteronu nepřestoupila hodnotu 0,5 ng/ml.
Tab. 3: Kumulativní pravděpodobnost udržení testosteronu <0,5 ng/ml ode dne 28 do dne 364.
|
Degarelix 240/80 mg N=207 |
Leuprorelin 7,5 mg N=201 | |
|
Počet respondentů |
202 |
194 |
|
Četnost odpovědí |
97,2% |
96,4% |
|
(intervaly spolehlivosti)* |
(93,5; 98,8%) |
(92,5; 98,2%) |
* Odhad ve skupině podle Kaplana a Meiera
Dosažení redukované hodnoty prostatického specifického antigenu (PSA)
Velikost nádoru se neměřila přímo v rámci programu klinického hodnocení, nicméně nádor nepřímo vykazoval příznivou odezvu na léčbu, jak naznačuje po 12 měsících 95% redukce střední hodnoty PSA u degarelixu.
Výchozí hodnoty PSA (medián) ve studii činil:
• u skupiny léčené degarelixem 240/80 mg - 19,8 ng/ml (interkvartilní rozmezí : P25 9,4 ng/ml, P75 46,4 ng/ml)
• u skupiny léčené leuprorelinem 7,5 mg - 17,4 ng/ml (interkvartilní rozmezí: P25 8,4 ng/ml, P75 56,5 ng/ml)
Obr. 2: Změna PSA (v %) v jednotlivých léčebných skupinách v prvních 56 dnech terapie.
Změna PSA ode dne 0 do dne 56 (v%)
Doba od první dávky (dny)
Léčebná skupina_Degarelix 240@40/80@20.......Leuprorelin 7,5 mg
Tento rozdíl byl při předběžné analýze ke dni 14 a ke dni 28 statisticky významný (p<0,001).
Hladiny prostatického specifického antigenu (PSA) byly po dvou týdnech léčby degarelixem sníženy o 64%, po jednom měsíci o 85%, po třech měsících o 95% a zůstaly potlačené (přibližně o 97%) po celou dobu jednoho roku léčby.
Pokud jde o snížení oproti výchozím hodnotám (v%) nebyly ode dne 56 do dne 364 zjištěny žádné významné rozdíly mezi degarelixem a komparátorem (srovnávací látkou).
Účinek na objem prostaty
Po třech měsících léčby degarelixem (v dávkovacím režimu 240/80 mg) bylo dosaženo 37% redukce objemu prostaty, měřené metodou TRUS (transrektální ultrazvukové snímkování), a to jak u pacientů vyžadujících hormonální léčbu před ozařováním, tak u pacientů vybraných za kandidáty farmakologické kastrace. Zmenšení objemu prostaty bylo podobné hodnotám dosaženým při podávání goserelinu spolu s prevencí androgenního flare-up fenoménu pomocí antiandrogenů.
Vliv na QT/QTc intervaly
V rámci registrační studie porovnávající FIRMAGON s leuprorelinem byly periodicky pořizovány elektrokardiogramy. U obou terapií se ukázalo, že u zhruba 20% pacientů byly QT/QTc intervaly delší než 450 ms. Počítáno od výchozí hodnoty do konce klinického hodnocení, činila střední hodnota změny u přípravku FIRMAGON 12,0 ms, u leuprorelinu 16,7 ms.
Protilátky proti degarelixu
Po jednoroční léčbě přípravkem FIRMAGON byl u 10% pacientů pozorován rozvoj tvorby protilátek proti degarelixu a u 29 % pacientů léčených FIRMAGONEM po dobu až pěti a půl roku.. Nic však nenaznačuje, že tvorbou protilátek je nějak dotčena účinnost či bezpečnost léčby přípravkem FIRMAGON po pěti a půlleté léčbě.
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem FIRMAGON u všech podskupin pediatrické populace ( viz bod 4.2 Pediatrická populace).
5.2 Farmakokinetické vlastnosti
Absorpce
Poté, co v pivotní studii CS21 bylo pacientům s rakovinou prostaty podkožně aplikováno 240 mg degarelixu při koncentraci 40 mg/ml, byla zjištěna hodnota AUC0-28 dnů 635 (602-668) den*ng/ml, hodnota Cmax 66,0 (61,0-71,0) ng/ml byla naměřena při tnax 40 (37-42) hodin. Střední minimální hodnoty byly přibližně 11-12 ng/ml po zahajovací dávce a 11-16 ng/ml po udržovací dávce 80 mg při koncentraci 20 mg/ml. Cmax s plazmatické hladiny degarelixu se dvoufázově snižuje tak, že střední terminální poločas (tJ/2) při udržovací dávce je 29 dnů.. Dlouhý poločas u podkožní aplikace je důsledkem velmi pomalého uvolňování degarelixu z úložiště, které se vytvoří na místě vpichu/místech vpichů. Farmakokinetické chování léku je ovlivňováno jeho koncentrací v injekčním roztoku. V důsledku toho mají Cmax a biologická dostupnost tendenci klesat s rostoucí koncentrací dávky za současného prodlužování poločasu. Z toho důvodu se nemá používat jiných než doporučených koncentrací.
Distribuce
Distribuční objem u zdravých starších mužů je přibližně 1 l/kg. Vazba na plasmatické proteiny je odhadem asi 90%.
Biotransformace
Při průchodu hepatobiliární soustavou podléhá degarelix obvyklé peptidové degradaci a vylučuje se převážně v podobě peptidových fragmentů stolicí. Po subkutánní aplikaci se ve vzorcích plasmatu neobjevily žádné významné metabolity. Jak vyplývá ze studií in vitro, degarelix netvoří substrát pro lidský CYP450 systém.
Eliminace
U zdravých mužů se zhruba 20-30% jednotlivé nitrožilně podané dávky vyloučí močí; z toho plyne, že 70-80% se vyloučí přes hepatobiliární soustavu. Bylo zjištěno, že clearance degarelixu podaného zdravým starším mužům v jednotlivých intravenózních dávkách (0,864-49,4pg/kg) je 35-50 ml/h/kg.
Zvláštní skupiny populace:
Pacienti s postižením ledvinných funkcí
Nebyly uskutečněny žádné farmakokinetické studie u pacientů s renálním postižením. Pouze asi 20-30% aplikované dávky degarelixu se v nezměněné podobě vylučuje renální cestou. Z populačně farmakokinetického rozboru údajů z registrační studie fáze III.vyplývá, že pacienti s mírným až středně závažným postižením funkce ledvin mají clearance degarelixu sníženou o přibližně 23%; z toho důvodu se u pacientů s mírným až středně závažným postižením ledvin úprava dávkování nedoporučuje. Vzhledem k tomu, že údajů o pacientech se závažnými poruchami renálních funkcí je málo, je u této skupiny pacientů na místě opatrnost.
Pacienti s postižením jaterních funkcí
Degarelix byl zkoumán v rámci farmakokinetické studie u pacientů s mírným až středně závažným postižením funkcí jater. Ve srovnání se zdravými osobami nebyly u pacientů s postižením jaterních funkcí pozorovány žádné známky zvýšené expozice. U pacientů s mírným nebo středně závažným postižením funkcí jater není nutno dávkování upravovat. Pacienti se závažným postižením jaterních funkcí nebyli předmětem zkoumání, a z toho důvodu je u této skupiny na místě opatrnost.
5.3 Předklinické údaje vztahující se k bezpečnosti
Jak ukázaly reprodukční pokusy na zvířatech, způsobuje degarelix neplodnost u samců. Ta je důsledkem farmakologického působení; tyto účinky jsou reverzibilní.
Studiemi reprodukční toxicity u samic se zjistilo, že degarelix působí tak, jak se při jeho farmakologických vlastnostech dalo očekávat. Vyvolával dávkově závislé prodloužení doby před pářením a zabřeznutím, snížení počtu žlutých tělísek corpora lutea, zvýšení počtu před- a po- implantačních ztrát, potratů, časné smrti zárodků/plodů, předčasných porodů a změn v trvání porodu.
Předklinické studie farmakologické bezpečnosti, toxicity po opakovaném podávání, genotoxicity a kancerogenního potenciálu neodhalily žádná zvláštní rizika pro lidi. Jak in vitro tak i in vivo studie neprokázaly známky prodloužení QT.
Studie akutní, subakutní a chronické toxicity provedené na potkanech a opicích nezjistily po podkožní aplikaci degarelixu žádnou toxicitu pro cílové orgány. V souvislosti s tímto lékem bylo u zvířat pozorováno místní podráždění tam, kde degarelix byl do podkoží aplikován ve velkých dávkách.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek
Manitol (E421)
Rozpouštědlo Voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto tento léčivý přípravek nesmí být mísen s žádnými dalšími léčivými přípravky.
6.3 Doba použitelnosti
3 roky.
Po naředění
Chemická a fyzikální stabilita přípravku po naředění byla prokázána pro dobu 2 hodin při 25°C. Z mikrobiologického hlediska je přípravek určen k okamžitému použití, pokud způsob rozředění nevylučuje riziko mikrobiální kontaminace. Není-li přípravek použit okamžitě, nese odpovědnost za dobu a podmínky uchování přípravku prvním otevření sám uživatel.
6.4 Zvláštní opatření pro uchovávání
Tento přípravek nevyžaduje zvláštní podmínky uchovávání.
Podmínky uchovávání rekonstituovaného léčivého přípravku viz bod 6.3.
6.5 Druh obalu a velikost balení
Injekční lahvička ze skla (typ I) s bromobutylovou pryžovou zátkou a hliníkovým flip-off uzávěrem obsahující 120 mg prášku pro přípravu injekčního roztoku
Předplněná injekční stříkačka ze skla (typ I) s elastomerovým uzávěrem s víčkem a ryskou označující 3 ml obsahující 3 ml rozpouštědla Táhlo pístu
Adaptér pro injekční lahvičky Injekční jehla (25G 0,5 x 25mm)
Velikost balení
Balení s 2 tvarovanými plastovými vložkami obsahuje 2 injekční lahvičky s práškem, 2 předplněné stříkačky s rozpouštedlem, 2 táhla pístu, 2 adaptéry pro injekční lahvičky a 2 jehly.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Návodem pro rozředění je třeba se pečlivě řídit.
Podávat lék v jiných koncentracích se nedoporučuje, protože tvorbu depotního gelu ovlivňuje právě koncentrace. Rozředěný roztok musí být čirá tekutina bez nerozpuštěného prášku.
POZNÁMKA:
• INJEKČNÍMI LAHVIČKAMI SE NESMÍ TŘEPAT
Balení obsahuje dvě injekční lahvičky prášku a dvě předplněné stříkačky s rozpouštědlem, které je nutno připravit pro podkožní injekci.
Z toho důvodu je třeba ještě jednou zopakovat níže uvedené pokyny.
1. Odstraňte uzávěr z balení adaptéru injekční
lahvičky. Adaptéry na injekční lahvičky s práškem nasaďte stlačením adaptéru, dokud špička neprojde gumovým uzávěrem a adapter nezapadne na místo.
2. Připravte předplněnou stříkačku nasazením zapuštěného pístu.
3. Odstraňte víčko z předplněné stříkačky.
Našroubujte injekční lahvičku na adaptér injekční lahvičky s rozpouštědlem. Rozpouštědlo přemístěte do injekční lahvičky s práškem.
4. S lahvičkou, stále připevněnou na adaptér opatrně kružte, dokud tekutina není čirá a bez nerozpuštěného prášku nebo částic. Pokud prášek přilne ke sklu nad hladinou, lze lahvičku mírně naklonit. Netřepejte, aby se roztok nezpěnil.
Kroužek vzduchových bublin na hladině tekutiny nevadí. Rozředění injekčního roztoku může v některých případech trvat několi minut, ale v některých případech může trvat až 15 minut,
5. Obraťte injekční lahvičku dnem vzhůru a natáhněte k rysce na předplněné stříkačce.
Vždy se ujistěte, že odebíráte přesné množství
bez vzduchových bublin.
6. Oddělte stříkačku od adaptéru lahvičky a na stříkačku nasaďte jehlu pro aplikaci hluboké podkožní injekce. Opatrně vytlačte vzduchové bublinky.
7. Pokračujte hlubokou podkožní injekcí. Uchopte kůži na břiše, povytáhněte podkožní tkáň a zasuňte jehlu hluboko pod úhlem alespoň 45 stupňů.
Bezprostředně po rekonstituci injikujte 3,0 ml přípravku FIRMAGON 120 mg.
8. Injekce se nemá aplikovat do míst, která jsou vystavena tlaku např. kolem pasu, opasku nebo blízko žeber.
Neinjikujte roztok přímo do žíly. Opatrně povytáhněte píst stříkačky a zkontrolujte, zda se nenasála krev. Objeví-li se ve stříkačce krev, je přípravek dále nepoužitelný. Přerušte proceduru a stříkačku i s jehlou zlikvidujte (je nutno rekonstituovat pro pacienta novou dávku).
9. Pro přípravu druhé dávky opakujte rekonstituci. Zvolte jiné místo vpichu a injikujte 3,0 ml.
Žádné zvláštní požadavky pro likvidaci.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Ferring Pharmaceuticals A/S Kay Fiskers Plads 11 DK-2300 Copenhagen S Dánsko
Tel: +45 88 33 88 34
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/08/504/002
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace:17/02/2009
Datum posledního prodloužení registrace: 19/09/2013
10. DATUM REVIZE TEXTU
{dd/mm/rrrr}
Podrobné informace o tomto přípravku jsou uveřejněny na webových stránkách Evropské lékové agentury http: //www .ema.europa.eu/.
A. VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného za propouštění šarží
Ferring GmbH Wittland 11 D-24109 Kiel Německo
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis.
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP) Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
• Další opatření k minimalizaci rizik
Držitel rozhodnutí o registraci odsouhlasí s národními autoritami příslušného členského státu podrobnosti vzdělávacího programu a musí jej implementovat tak, že před uvedením přípravku na trh poskytne všem lékařům vzdělávací materiál, který bude obsahovat následující :
• Vzdělávací materiál
• Souhrn údajů o přípravku (SPC), Označení na obalu a Příbalovou informaci
Vzdělávací materiál musí obsahovat následující klíčové informace:
• Dávkování
• Informace o podání
• Informace o tvorbě depotního gelu a možných lokálních reakcích v místě vpichu
• Informace o poznaných a možných rizicích
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
Krabička pro FIRMAGON 80 mg prášek a rozpouštědlo pro injekční roztok
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
FIRMAGON 80 mg prášek a rozpouštědlo pro injekční roztok degarelixum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna injekční lahvička obsahuje degarelixum 80 mg (ve formě degarelixi acetas). Po rekonstituci jeden ml roztoku obsahuje degarelixum 20 mg .
3. SEZNAM POMOCNÝCH LÁTEK
Mannitol (E 421), voda na injekci
4. LÉKOVÁ FORMA A VELIKOST BALENÍ
Prášek a rozpouštědlo pro injekční roztok
Balení s 1 tvarovanou plastovou vložkou obsahuje:
1 injekční lahvička s obsahem 80 mg degarelixu (prášek) 1 předplněná stříkačka s obsahem 4,2 ml rozpouštědla 1 táhlo pístu
1 adaptér pro injekční lahvičky 1 injekční jehla
Balení s 3 tvarovanými plastovými vložkami obsahuje:
3 injekční lahvičky s obsahem 80 mg degarelixu (prášek) 3 předplněné stříkačky s obsahem 4,2 ml rozpouštědla 3 táhla pístu
3 adaptéry pro injekční lahvičky 3 injekční jehly
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Pouze pro subkutánní podání.
6 ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10 ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11 NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Ferring Pharmaceuticals A/S Kay Fiskers Plads 11 2300 Copenhagen S Dánsko
+ 45 88 33 88 34
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/08/504/001 Balení s 1 tvarovanou plastovou vložkou EU/1/08/504/003 Balení s 3 tvarovanými plastovými vložkami
13 ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
Nevyžaduje se - odůvodnění přijato.
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
FIRMAGON 80 mg prášek na injekci
degarelixum
Pouze pro s.c. podání
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci.
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
80 mg
6 JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ Rozpouštědlo pro FIRMAGON
Voda na injekci
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci.
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
4,2 ml
6. JINÉ
Krabička pro FIRMAGON 120 mg prášek a rozpouštědlo pro injekční roztok
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
FIRMAGON 120 mg prášek a rozpouštědlo pro injekční roztok degarelixum
2 OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna injekční lahvička obsahuje degarelixum 120 mg (ve formě degarelixi acetas). Po rekonstituci jeden ml roztoku obsahuje degarelixum 40 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Manitol (E 421), voda na injekci
4. LÉKOVÁ FORMA A VELIKOST BALENÍ
Prášek a roztok pro injekční roztok
Balení s 2 tvarovanými plastovými vložkami obsahuje:
2 injekční lahvičky s obsahem 120 mg degarelixu (prášek) 2 předplněné stříkačky s obsahem 3 ml rozpouštědla 2 táhla pístu
2 adaptéry pro injekční lahvičky 2 injekční jehly
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Pouze pro subkutánní podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8 POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Ferring Pharmaceuticals A/S Kay Fiskers Plads 11 2300 Copenhagen S Dánsko
+ 45 88 33 88 34
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/08/504/002
13 ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
Nevyžaduje se - odůvodnění přijato.
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
FIRMAGON 120 mg prášek na injekci
degarelixum
Pouze pro s.c. podání
|
2. |
ZPŮSOB PODÁNÍ |
|
Před použitím si přečtěte příbalovou informaci. | |
|
3. |
POUŽITELNOST |
|
EXP | |
|
4. |
ČÍSLO ŠARŽE |
|
Lot | |
|
5. |
OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET |
|
120 |
mg |
|
6. |
JINÉ |
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ Rozpouštědlo pro FIRMAGON
Voda na injekci
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci.
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
3 ml
6. JINÉ
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro uživatele
FIRMAGON 80 mg prášek a rozpouštědlo pro injekční roztok
Degarelixum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře..
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte
v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz. bod 4.
Co naleznete v této příbalové informaci:
1. Co je FIRMAGON a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete FIRMAGON používat
3. Jak se FIRMAGON používá
4. Možné nežádoucí účinky
5. Jak FIRMAGON uchovávat
6. Obsah balení a další informace
1. Co je FIRMAGON a k čemu se používá
FIRMAGON obsahuje degarelix.
Degarelix je syntetický hormonální blokátor používaný k léčbě rakoviny prostaty u dospělých mužských pacientů. Zablokováním vazebných míst (receptorů) pro přirozený hormon (gonadoliberin - GnRH) přímo dochází k zablokování jeho účinků a v důsledku toho degarelix bezprostředně snižuje hladinu testosteronu, mužského pohlavního hormonu, který stimuluje rakovinu předstojné žlázy.
2. Čemu musíte věnovat pozornost, než začnete FIRMAGON používat
Nepoužívejte FIRMAGON
- jestliže j ste alergický/á (přecitlivělý/á) na degarelix nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Upozornění a opatření
Informujte svého lékaře před užíváním tohoto přípravku:
- jestliže máte kardiovasculární onemocnění nebo problémy se srdečním rytmem (arytmie) nebo užíváte léky na kontrolu srdečního rytmu. FIRMAGON může zvýšit riziko potíží se srdečním rytmem.
- jestliže máte cukrovku. Může dojít ke zhoršení nebo nástupu cukrovky. Pokud již trpíte cukrovkou, je možné, že si budete muset častěji měřit hladinu cukru v krvi.
- jestliže máte onemocnění jater. Pravědpodobně bude nutné sledovat funkci jater.
- jestliže máte onemocnění ledvin. Použití přípravku FIRMAGON se nezkoumalo u pacientů se závažným onemocněním ledvin.
- jestliže máte osteoporózu nebo jakékoli onemocnění, které ovlivňuje pevnost Vašich kostí._Snížená hladina testosteronu může způsobit snížení vápníku v kostech (řídnutí kostí).
- jestliže trpíte závažnou přecitlivělostí. Použití přípravku FIRMAGON se nezkoumalo u pacientů se závažnými reakcemi přecitlivělosti.
Děti a dospívající
Tento přípravek není určen pro děti a dospívající.
Další léčivé přípravky a FIRMAGON
FIRMAGON může ovlivňovat účinky některých léků určených k léčbě potíží se srdečním rytmem, jako jsou např. chinidin, prokainamid, amiodaron a sotalol, nebo dalších léků, které mohou mít vliv na srdeční rytmus (např. metadon (používaný pro úlevu od bolesti a jako součást léčby drogové závislosti), moxifloxacin (antibiotikum), antipsychotika).
Informujte svého lékaře o všech lécích, které užíváte, nebo jste užíval, nebo které možná budete užívat. Řízení dopravních prostředků a obsluha strojů
Únava a závrať jsou běžné nežádoucí účinky, které mohou snížit Vaši schopnost obsluhovat a řídit stroje. Tyto nežádoucí účinky mohou být způsobeny léčbou nebo samotným onemocněním.
3. Jak se FIRMAGON používá
Tento léčivý přípravek je obvykle aplikován zdravotní sestrou nebo lékařem.
Doporučená zahajovací dávka jsou dvě po sobě jdoucí 120 mg injekce. Poté budete dostávat měsíčně 80 mg injekci. Aplikovaná tekutina se přemění v gel, ze kterého se uvolňuje degarelix po dobu jednoho měsíce.
FIRMAGON lze podávat POUZE ve formě podkožních injekcí (subkutánně), NIKDY ne do krevní cévy (intravenózně). Proto je nutno zajistit, aby náhodou nedošlo ke vstříknutí léku do žíly. Injekci je možno aplikovat do různých míst kůže na břiše.
Jestliže jste zapomněl(a) použít přípravek FIRMAGON
Pokud se domníváte, že Vám nebyla podána Vaše měsíční dávka přípravku FIRMAGON, informujte o tom svého lékaře.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Velmi závažné alergické reakce na tento lék jsou vzácné. Vyhledejte lékařskou pomoc ihned, jestliže se u Vás objeví závažná vyrážka, svědění nebo dušnost a nebo obtížné dýchání. Ty by mohly být příznaky závažné alergické reakce.
Velmi časté (mohou postihnoout více než 1 uživatele z 10)
Návaly horka, bolestivost a zarudnutí v místě vpichu. Nežádoucí účinky v místě vpichu injekce se vyskytují nejčastěji po úvodní dávce a méně často při udržovací dávkách.
Časté (mohou postihnout 1 až 10 uživatelů)
- otoky, zduření mízních uzlin a zatvrdnutí v místě vpichu
- poinjekční třesavka, horečka nebo chřipku připomínající onemocnění
- poruchy spánku, únava, závratě, bolesti hlavy
- přibírání na váze, nevolnost, průjem, zvýšené hladiny některých jaterních enzymů
- nadměrné pocení (včetně nočních potů), vyrážka
- chudokrevnost
- muskuloskeletání bolesti a potíže
- zmenšování varlat, otok prsů, impotence
Méně časté (mohou postihnout 1 až 10 uživatelů z 1 000)
- ztráta zájmu o sex, bolestivá varlata, bolesti v pánevní oblasti, ejakulační selhání, podráždění genitálu, bolest prsu
- deprese, mentální porucha
- zarudnutí kůže, vypadávání vlasů, kožní uzlíky, pocit necitlivosti
- alergické reakce, kopřivka, svědění
- snížená chuť k jídlu, zácpa, zvracení, sucho v ústech, břišní bolesti a potíže, zvýšená hladina cukru v krvi/ cukrovka, zvýšený cholesterol, změny v hladině vápníku v krvi, ubývání na váze
- vysoký krevní tlak, změny srdečního rytmu, změny na EKG (prodloužení QT intervalu), pocit nenormálního tlukotu srdce, dušnost, periferní edém
- svalová slabost, svalové křeče, otoky/ ztuhlost kloubů, osteoporóza/ ubývání kostní tkáně, bolesti v kloubech
- časté močení, naléhavá potřeba močit (nutnost spěchat s močením), ztížené nebo bolestivé močení, noční močení, porucha funkce ledvin, inkontinence
- rozmazané vidění
- potíže při aplikaci injekce včetně snížení krevního tlaku a srdečního tepu (vazovagální reakce)
- nevolnost
Vzácné (mohou postihnout až 1 uživatele z 1 000)
- febrilní neutropenie (velmi nízký počet bílých krvinek v kombinaci s horečkou), infarkt myokardu, srdeční selhání
Velmi vzácné (mohou postihnout až 1 uživatele z 10 000)
- infekce v místě vpichu, absces a nekróza
Nežádoucí účinky v místě vpichu se vyskytují nejčastěji po úvodní dávce, po udržovacích dávkách jsou méně časté.
Hlášení nežádoucích účinků
Pokud se se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku
5. Jak FIRMAGON uchovávat
Uchovávejte tento přípravek mimo dohled a dosah a dohled dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na injekčních lahvičkách, stříkačkách a na krabičce. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Tento přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
Po rekonstituci
Tento přípravek je stabilní po dobu 2 hodin při 25°C.
Vzhledem k riziku mikrobiální kontaminace má být přípravek použit okamžitě.. Není-li přípravek použit okamžitě, nese odpovědnost za dobu a podmínky uchování přípravku po rozředění sám uživatel.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co FIRMAGON obsahuje
- Léčivou látkou je degarelixum. Jedna lahvička obsahuje degarelixum 80 mg ( ve formě acetátu). Po rekonstituci se v jednom ml rekonstituovaného roztoku nachází degarelixum 20 mg.
- Pomocnou látkou prášku j e manitol (E 421).
- Rozpouštědlem j e voda na inj ekce.
Jak přípravek FIRMAGON vypadá a co obsahuje toto balení
FIRMAGON je prášek a rozpouštědlo pro přípravu injekčního roztoku. Prášek je bílý až bělavý. Rozpouštědlem je čirý, bezbarvý roztok.
FIRMAGON se dodává ve 2 velikostech balení.
Balení s 1 tvarovanou plastovou vložkou obsahuje:
1 injekční lahvičku s práškem s obsahem 80 mg degarelixu a 1 předplněnou stříkačku s obsahem 4,2 ml rozpouštědla,1 táhlo pístu, 1 adaptér pro injekční lahvičku a 1 injekční jehlu.
Balení s 3 tvarovanými plastovými vložkami obsahuje:
3 injekční lahvičky s práškem s obsahem 80 mg degarelixu a 3 předplněné stříkačky s obsahem 4,2 ml rozpouštědla,3 táhla pístu, 3 adaptéry pro injekční lahvičky a 3 injekční jehly.
Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci
Ferring Pharmaceuticals A/S Kay Fiskers Plads 11,
DK-2300 Copenhagen S Dánsko
+45 8833 8834
Výrobce:
Ferring GmbH Wittland 11, D-24109 Kiel Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgie/Belgique/Belgien
nv Ferring sa Tel/Tél: +32-53 72 92 00 ferringnvsa@ferring .be
Eunrapnu
AKBaXHM Afl
Ten.: +359 2 807 5022
Česká republika
Ferring Pharmaceuticals CZ s.r.o. Tel: +420 241 041 111 czinfo@ferring.com
Danmark
Ferring L^gemidler A/S Tlf: +45 88 16 88 17
Lietuva
UAB PharmaSwiss
Tel.: +370 5 2790762
lithuania.info@pharmaswiss.com
Luxembourg/Luxemburg
nv Ferring sa,
Belgique/Belgien Tél: +32-53 72 92 00 ferringnvsa@ferring .be
Magyarország
Ferring Magyarország Gyógyszerkereskedelmi Kft.
Tel.: +36 1 236 3800 ferring@ferring .hu
Malta
E.J. Busuttil Ltd.
Tel: +356 21447184
|
Deutschland Ferring Arzneimittel GmbH Tel: + 49-(0)431-5852 0 info-service@ferring. de |
Nederland Ferring BV Tel: +31-235680300 |
|
Eesti PharmaSwiss Eesti OU Tel.: +372 682 7400 estonia.info@Dharmaswiss.com |
Norge Ferring Legemidler AS Tlf: +47 22 02 08 80 mail@oslo.ferring.com |
|
EXXáSa Ferring Effág MEnE Tnf: +30 210 68 43 449 |
Osterreich Ferring Arzneimittel GesmbH Tel: +43 1 60 808 0 office@ferring. at |
|
Espaňa Ferring, S.A.U. Tel: +34 91 799 47 80 es0-Reaistros@ferring.com |
Polska Ferring Pharmaceuticals Poland Sp. z.o.o. Tel: +48 22 246 06 80 ferring@ferring .pl |
|
France Ferring S.A.S. Tél : +33 1 49 08 91 23 |
Portugal Ferring Portuguesa - Produtos Farmaceuticos, Sociedade Unipessoal, Lda. Tel: +351 21 940 5190 geral@ferring.com |
|
Hrvatska PharmaSwiss d.o.o. Tel: +385(1)6311-833 |
Románia Ferring Pharmaceuticals SA Reprezentanta ín Románia Tel: +40 356 113 270 |
|
Ireland Ferring Ireland Ltd. Tel: + 353 (0)1 4637355 enauiries.ireland@ferring.com |
Slovenija SALUS, Veletrgovina, d.o.o. Tel: +386 1 5899 179 regulatorv@salus.si |
|
Ísland Vistor hf. Sími: +354 535 70 00 |
Slovenská republika FERRING Slovakia s.r.o. Tel: +421 2 54 416 010 SK0-Recepcia@ferring.com |
|
Italia Ferring S.p.A. Tel: +39 02 640 00 11 |
Suomi/Finland Ferring Láákkeet Oy Puh/Tel: + 358-207 401440 info@ferring.fi |
|
Kúnpoq A. Potamitis Medicare Ltd Tn^: +357 22583333 |
Sverige Ferring Lákemedel AB Tel: +46 40 691 69 00 info@ferring.se |
|
Latvija PharmaSwiss SIA Latvia Talr.: +371 6 750 2185 |
United Kingdom Ferring Pharmaceuticals Ltd Tel: +44 844 931 0050 |
latvia.info@pharmaswiss.com contact@ferring.co.uk
Tato příbalová informace byla naposledy revidována {mm/rrrr}.
Podrobné informace o tomto přípravku jsou uveřejněny na webových stránkách Evropské lékové agentury : http://www.ema.europa.eu/
Následující informace je určena pouze pro zdravotnické pracovníky:
Návod ke správnému použití POZNÁMKA:
• INJEKČNÍMI LAHVIČKAMI SE NESMÍ TŘEPAT
Balení obsahuje jednu injekční lahvičku prášku a jednu předplněnou stříkačku rozpouštědla, které je nutno připravit pro podkožní injekci .
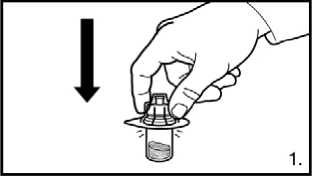
1. Odstraňte uzávěr z balení adaptéru injekční lahvičky. Adaptér na injekční lahvičky s práškem nasaďte stlačením adaptéru, dokud špička neprojde gumovým uzávěrem a adaptér nezapadne na místo.
2. Připravte předplněnou stříkačku nasazením zapuštěného pístu.
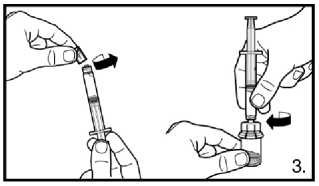
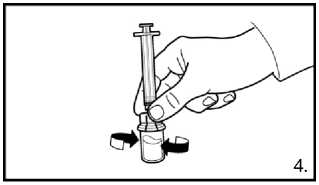
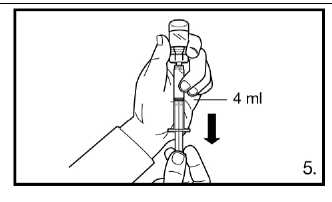
3. Odstraňte víčko z předplněné stříkačky
Našroubujte injekční lahvičku na adaptér injekční lahvičky s rozpouštědlem. Rozpouštědlo přemístěte do injekční lahvičky s práškem.
4. S lahvičkou, stále připevněnou na adaptér opatrně kružte, dokud tekutina není čirá a bez nerozpuštěného prášku nebo částic. Pokud prášek přilne ke sklu nad hladinou, lze lahvičku mírně naklonit.
Netřepejte, aby se roztok nezpěnil.
Kroužek vzduchových bublin na hladině tekutiny nevadí. Rozředění injekčního roztoku může v některých případech trvat několik minut, ale v některých případech může trvat až 15 minut,
5. Obraťte injekční lahvičku dnem vzhůru a natáhněte k rysce na předplněné stříkačce.
Vždy se ujistěte, že odebíráte přesné množství
bez vzduchových bublin.
6. Oddělte stříkačku od adaptéru lahvičky a na stříkačku nasaďte jehlu pro aplikaci hluboké podkožní injekce. Opatrně vytlačte vzduchové bublinky.
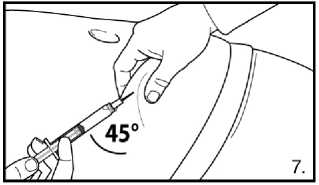
7. Pokračujte hlubokou podkožní injekcí Uchopte kůži na břiše, povytáhněte podkožní tkáň a zasuňte jehlu hluboko pod úhlem alespoň 45 stupňů.
Bezprostředně po rekonstituci injikujte 4,0 ml přípravku FIRMAGON 80 mg .*
9. Injekce se nemá aplikovat do míst, která jsou vystavena tlaku např. kolem pasu, opasku nebo blízko žeber.
Neinjikujte roztok přímo do žíly. Opatrně povytáhněte píst stříkačky a ujistěte se, že se nenasála krev. Objeví-li se ve stříkačce krev, je přípravek dále nepoužitelný. Přerušte proceduru a stříkačku i s jehlou zlikvidujte (rekonstituujte pro pacienta novou dávku).
* Chemická a fyzikální stabilita pro použití byla prokázána pro dobu 2 hodin při 25°C. Z mikrobiologického hlediska je přípravek určen k okamžitému použití, pokud způsob rekonstituce nevylučuje riziko mikrobiální kontaminace. Není-li přípravek použit okamžitě, nese odpovědnost za dobu a podmínky uchování pro použití po naředění sám uživatel.
Příbalová informace: informace pro uživatele
FIRMAGON 120 mg prášek a rozpouštědlo pro injekční roztok
Degarelixum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte
v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci:
1. Co je FIRMAGON a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete FIRMAGON používat
3. Jak se FIRMAGON používá
4. Možné nežádoucí účinky
5. Jak FIRMAGON uchovávat
6. Obsah balení a další informace
1. Co je FIRMAGON a k čemu se používá
FIRMAGON obsahuje degarelix.
Degarelix je syntetický hormonální blokátor používaný k léčbě rakoviny prostaty u dospělých mužských pacientů. Zablokováním vazebných míst (receptorů) pro přirozený hormon (gonadoliberin - GnRH) přímo dochází k zablokování jeho účinků a v důsledku toho degarelix bezprostředně snižuje hladinu testosteronu, mužského pohlavního hormonu, který stimuluje rakovinu předstojné žlázy.
2. Čemu musíte věnovat pozornost, než začnete FIRMAGON používat
Nepoužívejte FIRMAGON
- jestliže jste alergický/á (přecitlivělý/á) na degarelix nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6)..
Upozornění a opatření
Informujte svého lékaře před užíváním tohoto přípravku:
- jestliže máte kariovaskulární onemocnění nebo problémy se srdečním rytmem (arytmie) nebo užíváte léky na kontrolu srdečního rytmu. FIRMAGON může zvýšit riziko potíží se srdečním rytmem.
- jestliže máte cukrovku. Může dojít ke zhoršení nebo nástupu cukrovky. Pokud již trpíte cukrovkou, je možné, že si budete muset častěji měřit hladinu cukru v krvi.
- jestliže máte onemocnění jater. Pravědpodobně bude nutné sledovat funkci jater.
- jestliže máte onemocnění ledvin. Použití přípravku FIRMAGON se nezkoumalo u pacientů se závažným onemocněním ledvin.
- jestliže máte osteoporózu nebo jakékoli onemocnění, které ovlivňuje pevnost Vašich kostí._Snížená hladina testosteronu může způsobit snížení vápníku v kostech (řídnutí kostí).
- jestliže trpíte závažnou přecitlivělostí. Použití přípravku FIRMAGON se nezkoumalo u pacientů se závažnými reakcemi přecitlivělosti.
Děti a dospívající
Tento přípravke není určen pro děti a dospívající.
Další léčivé přípravky a FIRMAGON
FIRMAGON může ovlivňovat účinky některých léků určených k léčbě potíží se srdečním rytmem, jako jsou např. chinidin, prokainamid, amiodaron a sotalol, nebo dalších léků, které mohou mít vliv na srdeční rytmus (např. metadon (používaný pro úlevu od bolesti a jako součást léčby drogové závislosti moxifloxacin (antibiotikum), antipsychotika).
Informujte svého lékaře o všech lécích, které užíváte nebo jste užíval, nebo které možná budete užívat. Řízení dopravních prostředků a obsluha strojů
Únava a závrať jsou běžné nežádoucí účinky, které mohou snížit Vaší schopnost obsluhovat a řídit stroje. Tyto nežádoucí účinky mohou být způsobeny léčbou nebo samotným onemocněním.
3. Jak se FIRMAGON používá
Tento léčivý přípravek je obvykle aplikován zdravotní sestrou nebo lékařem.
Doporučená zahajovací dávka jsou dvě po sobě jdoucí 120 mg injekce. Poté budete dostávat měsíčně 80 mg injekci. Aplikovaná tekutina se přemění v gel, ze kterého se uvolňujet degarelix po dobu jednoho měsíce.
FIRMAGON lze podávat POUZE ve formě podkožních injekcí (subkutánně), NIKDY ne do krevní cévy (intravenózně). Proto je nutno zajistit, aby náhodou nedošlo ke vstříknutí léku do žíly. Injekce je možno aplikovat do různých míst kůže na břiše.
Jestliže jste zapomněl(a) použít přípravek FIRMAGON
Pokud se domníváte, že Vám nebyla podána Vaše měsíční dávka přípravku FIRMAGON, informujte o tom svého lékaře.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Velmi závažné alergické reakce na tento lék jsou vzácné. Vyhledejte lékařskou pomoc ihned, jestliže se u Vás objeví závažná vyrážka, svědění nebo dušnost a nebo obtížné dýchání. Ty by mohly být příznaky závažné alergické reakce.
Velmi časté (mohou postihovat více než 1 uživatele z 10)
Návaly horka, bolestivost a zarudnutí v místě vpichu. Nežádoucí účinky v místě vpichu injekce se vyskytují nejčastěji po úvodní dávce a méně často při udržovací dávkách.
Časté (mohou postihovat 1 až 10 uživatelů)
- otoky, zduření mízních uzlin a zatvrdnutí v místě vpichu
- poinjekční třesavka, horečka nebo chřipku připomínající onemocnění
- poruchy spánku,únava,ochablost, závratě, bolesti hlavy
- přibírání na váze, nevolnost, průjem, zvýšené hladiny některých jaterních enzymů
- nadměrné pocení (včetně nočních potů), vyrážka
- chudokrevnost
- muskuloskeletání bolesti a potíže
- zmenšování varlat, otok prsů, impotence
Méně časté (mohou postihovat 1 až 10 uživatelů z 1 000)
- ztráta zájmu o sex, bolestivá varlata, bolesti v pánevní oblasti, ejakulační selhání, podráždění genitálu, bolest prsu
- deprese, mentální porucha
- zarudnutí kůže, vypadávání vlasů, kožní uzlíky, pocit necitlivosti
- alergické reakce, kopřivka, svědění
- snížená chuť k jídlu, zácpa, zvracení, sucho v ústech, břišní bolesti a potíže, zvýšená hladina cukru v krvi/ cukrovka, zvýšený cholesterol, změny v hladině vápníku v krvi, ubývání na váze
- vysoký krevní tlak, změny srdečního rytmu, změny na EKG (prodloužení QT intervalu), pocit nenormálního tlukotu srdce, dušnost, periferní edém
- svalová slabost, svalové křeče, otoky/ ztuhlost kloubů, osteoporóza/ ubývání kostní tkáně, bolesti v kloubech
- časté močení, naléhavá potřeba močit (nutnost spěchat s močením), ztížené nebo bolestivé močení, noční močení, porucha funkce ledvin, inkontinence
- rozmazané vidění
- potíže při aplikaci injekce včetně snížení krevního tlaku a srdečního tepu (vazovagální reakce)
- nevolnost
Vzácné (mohou postihnout až 1 uživatele z 1 000)
- febrilní neutropenie (velmi nízký počet bílých krvinek v kombinaci s horečkou), infarkt myokardu, srdeční selhání
Velmi vzácné (mohou postihnout až 1 uživatele z 10 000)
- infekce v místě vpichu, absces a nekróza
Nežádoucí účinky v místě vpichu se vyskytují nejčastěji po úvodní dávce, po udržovacích dávkách jsou méně časté.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku
5. Jak FIRMAGON uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na injekčních lahvičkách, stříkačkách a na krabičce. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Tento přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
Po rekonstituci
Tento přípravek je stabilní po dobu 2 hodin při 25°C.
Vzhledem k možnému riziku mikrobiální kontaminace má být přípravek použit okamžitě.Není-li přípravek použit okamžitě, nese odpovědnost za dobu a podmínky uchování přípravku po rozředění sám uživatel.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co FIRMAGON obsahuje
- Léčivou látkou je degarelixum. Jedna lahvička obsahuje degarelixum 120 mg (ve formě acetátu). Po rekonstituci se v jednom ml rekonstituovaného roztoku nachází degarelixum 40 mg.
- Pomocnou látkou prášku j e manitol (E 421).
- Rozpouštědlem j e voda na inj ekce.
Jak přípravek FIRMAGON vypadá a co obsahuje toto balení
FIRMAGON je prášek a rozpouštědlo pro přípravu injekčního roztoku. Prášek je bílý až bělavý. Rozpouštědlem je čirý, bezbarvý roztok.
Balení s 2 tvarovanými plastovými vložkami obsahuje:
2 injekční lahvičky s práškem s obsahem 120 mg degarelixu, 2 předplněné stříkačky s obsahem 3 ml rozpouštědla, 2 táhla pístu, 2 adaptéry pro injekční lahvičky a 2 injekční jehly.
Držitel rozhodnutí o registraci
Ferring Pharmaceuticals A/S Kay Fiskers Plads 11,
DK-2300 Copenhagen S Dánsko
+45 8833 8834
Výrobce:
Ferring GmbH Wittland 11 D-24109 Kiel Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgie/Belgique/Belgien
nv Ferring sa Tel/Tél: +32-53 72 92 00 ferringnvsa@ferring .be
Lietuva
UAB PharmaSwiss
Tel.: +370 5 2790762
lithuania.info@pharmaswiss.com
Luxembourg/Luxemburg
nv Ferring sa, Belgique/Belgien Tél: +32-53 72 92 00 ferringnvsa@ferring.be
Česká republika
Ferring Pharmaceuticals CZ s.r.o. Tel: +420 241 041 111 czinfo@ferring.com
Magyarország
Ferring Magyarország Gyógyszerkereskedelmi Kft.
Tel.: +36 1 236 3800 ferring@ferring.hu
Tlf: +45 88 16 88 17
Deutschland
Ferring Arzneimittel GmbH Tel: + 49-(0)431-5852 0 info-service@ferring.de
Malta
E.J. Busuttil Ltd.
Tel: +356 21447184 admin@ejbusuttil.com
Nederland
Ferring BV
Tel: +31-235680300
|
Eesti PharmaSwiss Eesti OU Tel.: +372 682 7400 estonia.info@pharmaswiss.com |
Norge Ferring Legemidler AS Tlf: +47 22 02 08 80 mail@oslo.ferring.com |
|
EXXáSa Ferring Effág MEnE Tnf: +30 210 68 43 449 |
Osterreich Ferring Arzneimittel GesmbH Tel: +43 1 60 808 0 office@ferring.at |
|
Espaňa Ferring, S.A.U. Tel: +34 91 799 47 80 es0-Reaistros@ferring.com |
Polska Ferring Pharmaceuticals Poland Sp. z.o.o. Tel: +48 22 246 06 80 ferring@ferring.pl |
|
France Ferring S.A.S. Tél : +33 1 49 08 91 23 |
Portugal Ferring Portuguesa - Produtos Farmaceuticos, Sociedade Unipessoal, Lda. Tel: +351 21 940 51-90 geral@ferring.com |
|
Hrvatska PharmaSwiss d.o.o. Tel: +385(1)6311-833 |
Románia Ferring Pharmaceuticals SA Reprezentanta ín Románia Tel: +40 356 113 270 |
|
Ireland Ferring Ireland Ltd. Tel: + 353 (0)1 4637355 enauiries.ireland@ferring.com |
Slovenija SALUS, Veletrgovina, d.o.o. Tel: +386 1 5899 179 regulatory@salus. si |
|
Ísland Vistor hf. Sími: +354 535 70 00 |
Slovenská republika FERRING Slovakia s.r.o. Tel: +421 2 54 416 010 SK0 -Recepcia@ferring. com |
|
Italia Ferring S.p.A. Tel: +39 02 640 00 11 |
Suomi/Finland Ferring Láákkeet Oy Puh/Tel: +358-207 401440 info@ferring.fi |
|
Kúnpoq A. Potamitis Medicare Ltd Tn^: +357 22583333 |
Sverige Ferring Lákemedel AB Tel: +46 40 691 69 00 info@ferring.se |
|
Latvija PharmaSwiss SIA Latvia Talr.: +371 6 750 2185 latvia.info@pharmaswiss.com |
United Kingdom Ferring Pharmaceuticals Ltd Tel: +44 844 931 0050 contact@ferring .co.uk |
Tato příbalová informace byla naposledy revidována {mm/rrrr}.
Podrobné informace o tomto přípravku jsou uveřejněny na webových stránkách Evropské lékové agentury: http://www.ema.europa.eu/.
Následující informace je určena pouze pro zdravotnické pracovníky:
Návod ke správnému použití POZNÁMKA:
• INJEKČNÍMI LAHVIČKAMI SE NESMÍ TŘEPAT
Balení obsahuje dvě injekční lahvičky a dvě předplněné stříkačky s rozpouštědlem, které je nutno připravit pro podkožní injekci. Z toho důvodu je třeba ještě jednou zopakovat níže uvedené pokyny.
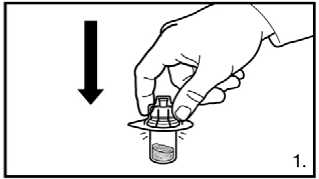
1. Odstraňte uzávěr z balení adapteru injekční lahvičky. Adaptér na injekční lahvičky s práškem nasaďte stlačením adaptéru, dokud špička neprojde gumovým uzávěrem a adapter nezapadne na místo.
2. Připravte předplněnou stříkačku nasazením zapuštěného pístu.
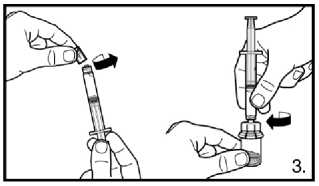
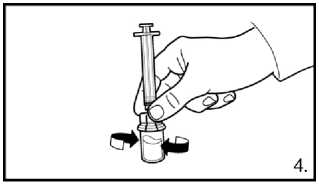
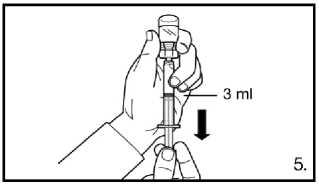
3. Odstraňte víčko z předplněné injekční stříkačky. Našroubujte injekční lahvičku na adaptér injekční lahvičky s rozpouštědlem. Rozpouštědlo přemístěte do injekční lahvičky s práškem.
4. S lahvičkou, stále připevněnou na adaptér opatrně kružte, dokud tekutina není čirá a bez nerozpuštěného prášku nebo částic. Pokud prášek přilne ke sklu nad hladinou, lze lahvičku mírně naklonit. Netřepejte, aby se roztok nezpěnil.
Kroužek vzduchových bublin na hladině tekutiny nevadí. Rozředění injekčního roztoku může v některých případech trvat až 15 minut, obykle však trvá pouze pár minut.
5. Obraťte injekční lahvičku dnem vzhůru a natáhněte k rysce na předplněné stříkačce.
Vždy se ujistěte, že odebíráte přesné množství
bez vzduchovýcg bublin.
6. Oddělte stříkačku od adaptéru lahvičky a na stříkačku nasaďte jehlu pro aplikaci hluboké podkožní injekce. Opatrně vytlačte vzduchové bublinky.
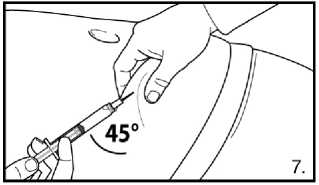
7. Pokračujte hlubokou podkožní injekcí. Uchopte kůži na břiše, povytáhněte podkožní tkáň a zasuňte jehlu pod úhlem alespoň 45 stupňů.
Bezprostředně po rekonstituci injikujte 3,0 ml přípravku FIRMAGON 120 mg. *
8. Injekce se nemá aplikovat do míst, kde je kůže pacienta vystavena tlaku, např. v oblasti pasu nebo opasku nebo blízko žeber.
Neinjikujte roztok přímo do žíly. Opatrně povytáhněte píst stříkačky a zkontrolujte, zda se nenasála krev. Objeví-li se ve stříkačce krev, je přípravek dále nepoužitelný. Přerušte proceduru a stříkačku i s jehlou zlikvidujte (je nutno rekonstituovat pro pacienta novou dávku).
9. Pro přípravu druhé dávky opakujte rekonstituci. Zvolte jiné místo vpichu a injikujte 3,0 ml.
Chemická a fyzikální stabilita pro použití byla prokázána pro dobu 2 hodin při 25°C. Z mikrobiologického hlediska je přípravek určen k okamžitému použití, pokud způsob rekonstituce nevylučuje riziko mikrobiální kontaminace. Není-li přípravek použit okamžitě, nese odpovědnost za dobu a podmínky uchování pro použití po naředění sám uživatel.
PŘÍLOHA IV
VĚDECKÉ ZÁVĚRY A ZDŮVODNĚNÍ ZMĚNY PODMÍNEK ROZHODNUTÍ O REGISTRACI
Vědecké závěry
S ohledem na hodnotící zprávu výboru PRAC týkající se pravidelně aktualizovaných zpráv o bezpečnosti (PSUR) pro Firmagon, dospěl výbor CHMP k těmto vědeckým závěrům:
Pro degarelix bylo hlášeno celkem devět případů nevhodného naplánování podávání léku. Aby se zabránilo nevhodnému naplánování podávání léku, část 4.2 Souhrnu údajů o přípravku a příbalová informace mají být revidovány tak, aby bylo zřejmé, že obě subkutánní injekce, každá po 120 mg, musí být podány po sobě.
Proto s ohledem na dostupné údaje týkající se degarelixu, má PRAC za to, že změny v informacích o přípravku jsou oprávněné.
Výbor CHMP souhlasí s vědeckými závěry, ke kterým dospěl PRAC.
Zdůvodnění doporučující změnu podmínek rozhodnutí o registraci
Na základě vědeckých závěrů týkajících se přípravku Firmagon zastává výbor CHMP stanovisko, že poměr přínosů a rizik léčivého přípravku obsahujícího léčivou látku degarelix je příznivý pod podmínkou, že v údajích o přípravku budou provedeny navržené změny.
Výbor CHMP doporučuje změnit podmínky rozhodnutí o registraci.
56