Firazyr 30 Mg
SOUHRN ÚDAJŮ O PŘÍPRAVKU
PŘÍLOHA I
Firazyr 30 mg injekční roztok v předplněné injekční stříkačce.
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna předplněná injekční stříkačka s objemem 3 ml obsahuje icatibanti acetas odpovídající icatibantum 30 mg.
Jeden ml roztoku obsahuje icatibantum 10 mg .
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok.
Roztok je čirá a bezbarvá tekutina.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Firazyr je určen k symptomatické léčbě akutních atak dědičného angioedému (HAE) u dospělých pacientů (s deficitem inhibitoru esterázy C1).
4.2 Dávkování a způsob podání
Firazyr je určen k použití pod vedením zdravotnického pracovníka.
Dávkování
Doporučená dávka je jedna subkutánní injekce přípravku Firazyr 30 mg.
Ve většině případů stačí k léčbě ataky jediná injekce přípravku Firazyr. Pokud nedojde k úplnému ústupu příznaků nebo se příznaky objeví znovu, po 6 hodinách lze podat druhou injekci přípravku Firazyr. V případě, že ani po podání druhé injekce nedojde k úplnému ústupu příznaků nebo se příznaky objeví znovu, lze po dalších 6 hodinách podat třetí injekci přípravku Firazyr. V průběhu 24 hodin by neměly být podány více než 3 injekce přípravku Firazyr.
V rámci klinických studií nebylo podáváno více než 8 injekcí přípravku Firazyr měsíčně.
Zvláštní populace
Starší osoby
Zkušenosti s podáváním přípravku u pacientů ve věku nad 65 let jsou omezené.
U starších osob byla prokázána zvýšená systémová expozice ikatibantu. Není známo, zda je tato skutečnost významná ve vztahu k bezpečnosti přípravku Firazyr (viz bod 5.2).
Poškození funkce jater
U pacientů s poškozením funkce jater není nutná úprava dávkování.
Poškození funkce ledvin
U pacientů s poškozením funkce ledvin není nutná úprava dávkování.
Pediatrická populace
Bezpečnost a účinnost přípravku Firazyr u dětí ve věku 0-18 let nebyla stanovena.
Údaje nejsou k dispozici.
Způsob podání
Firazyr je určen k subkutánnímu podání, nejlépe do břišní oblasti.
Firazyr může být podáván samotným pacientem nebo ošetřující osobou pouze po proškolení v technice subkutánní injekce provedeném zdravotnickým pracovníkem.
O zahájení podávání přípravku Firazyr samotným pacientem by měl rozhodnout pouze lékař se zkušenostmi v diagnostice a léčbě dědičného angioedému (viz bod 4.4).
Jedna injekční stříkačka přípravku Firazyr je určena pouze pro jedno použití.
Firazyr injekční roztok má být injikován pomalu, a to vzhledem k objemu, který je podáván (3 ml).
4.3 Kontraindikace
Přecitlivělost na léčivou látku nebo na kteroukoli pomocnou látku tohoto přípravku uvedenou v bodě
6.1
4.4 Zvláštní upozornění a opatření pro použití
Laryngeální ataky
Pacienty s laryngeálními atakami je třeba po podání injekce pečlivě sledovat ve vhodném zdravotnickém zařízení, dokud lékař nerozhodne, že pacienta lze bez rizika propustit.
Ischemická choroba srdeční
V případě ischémie může antagonismus bradykininových receptorů II. typu teoreticky způsobit zhoršení srdeční funkce a snížení průtoku krve koronárními cévami. Při podávání přípravku Firazyr pacientům s akutní ischemickou chorobou srdeční nebo nestabilní anginou pectoris je proto zapotřebí opatrnosti (viz bod 5.3).
Mozková příhoda
Ačkoli existují důkazy, které podporují pozitivní vliv blokády B2 receptorů bezprostředně po vzniku mozkové příhody, teoreticky je možné, že by ikatibant mohl oslabit pozitivní neuroprotektivní účinek bradykininu v pozdní fáze. Proto je zapotřebí opatrnosti při podávání ikatibantu pacientům během několika týdnů po vzniku mozkové příhody.
Samostatné podávání pacientem
Pacientům, kteří Firazyr nikdy dříve nedostali, by měla být první dávka podána ve zdravotnickém zařízení nebo pod dohledem lékaře.
Pokud po samostatném podání nedojde k úplnému ústupu příznaků nebo se příznaky objeví znovu, doporučuje se, aby pacient vyhledal lékařskou pomoc a aby byly další dávky podány ve zdravotnickém zařízení (viz bod 4.2).
Pacienti s laryngeální atakou mají vždy vyhledat lékařskou pomoc a být sledováni ve zdravotnickém zařízení, a to i poté, co si aplikovali injekci v domácím prostředí.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Neočekávají se žádné farmakokinetické lékové interakce s postižením CYP450 (viz bod 5.2).
Současné podávání přípravku Firazyr s inhibitory angiotenzin konvertujícího enzymu (ACE) nebylo zkoumáno. ACE inhibitory jsou kontraindikovány u pacientů s dědičným angioedémem vzhledem k možnému zvýšení hladiny bradykininu.
4.6 Fertilita, těhotenství a kojení
Nejsou k dispozici klinické údaje o podávání ikatibantu během těhotenství. Studie na zvířatech prokázaly, že přípravek ovlivňuje uhnízdění zárodku do dělohy a porod (viz bod 5.3), potenciální riziko pro člověka však není známé.
Během těhotenství lze přípravek Firazyr podávat pouze v odůvodněných případech, jestliže potenciální léčebný přínos převáží možná rizika pro plod (např. k léčbě potenciálně život ohrožujících laryngeálních atak).
Kojení
Ikatibant se vylučuje do mléka kojících potkanů v podobných koncentracích, v jakých se nachází v krvi matek. Nebyl zaznamenán žádný vliv na postnatální vývoj potkaních mláďat.
Není známo, zda je ikatibant vylučován do mateřského mléka u lidí, doporučuje se však, aby kojící ženy, které chtějí použít přípravek Firazyr, nekojily po dobu 12 hodin po podání přípravku.
Fertilita
Opakované používání ikatibantu mělo účinky na reprodukční orgány jak u potkanů, tak u psů.
Ikatibant neovlivňoval plodnost samců myší a potkanů (viz bod 5.3). Ve studii u 39 zdravých dospělých mužů a žen, kterým byly podávány 3 dávky po 30 mg každých 6 hodin každé 3 dny do celkového počtu 9 dávek, nebyly zaznamenány žádné klinicky významné změny bazální koncentrace reprodukčních hormonů nebo jejich koncentrace po stimulaci GnRH oproti výchozí koncentraci, a to ani u žen, ani u mužů. Nebyly zjištěny žádné významné účinky ikatibantu na koncentraci progesteronu v luteální fázi a na luteální funkci ani na délku menstruačního cyklu u žen a nebyly zaznamenány žádné významné účinky ikatibantu na počet, motilitu a morfologii spermií u mužů. Není pravděpodobné, že by dávkovací režim použitý v této studii byl udržován v rámci klinické praxe
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek Firazyr má malý vliv na schopnost řídit a obsluhovat stroje. Po použití přípravku Firazyr byla hlášena únava, apatie, vyčerpanost, ospalost a závratě. Tyto příznaky se mohou vyskytnout v důsledku ataky dědičného angioedému. Pacientům je třeba doporučit, aby neřídili a neobsluhovali stroje, jestliže se cítí unaveni nebo mají-li závratě.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
V klinických studiích použitých pro registraci bylo celkem 999 atak dědičného angioedému léčeno 30 mg Firazyru podanými subkutánně zdravotnickým pracovníkem. Firazyr 30 mg s.c. byl podán zdravotnickým pracovníkem 129 zdravým subjektům a 236 pacientům s dědičným angioedémem.
Téměř u všech jedinců, kteří byli v rámci klinických studií léčeni podkožně podávaným ikatibantem, se vyskytly reakce v místě podání injekce (charakterizované podrážděním kůže, otokem, bolestí, svěděním, erytémem, pocitem pálení). Tyto reakce byly zpravidla mírné až středně závažné, přechodné a vymizely bez nutnosti další intervence.
Tabulkový přehled nežádoucích účinků
Četnost nežádoucích reakcí uvedených v tabulce 1 je definována následujícím způsobem:
velmi časté (>1/10); časté (>1/100 až <1/10); méně časté (>1/1 000 až <1/100); vzácné (>1/10 000 až <1/1 000); velmi vzácné (<1/10 000).
Table 1: Nežádoucí reakce hlášené v souvislosti s podáváním ikatibantu
|
Třída orgánových systémů |
Preferovaný termín |
|
(kategorie incidence) | |
|
Poruchy nervového systému (Časté, >1/100 až <1/10) |
Závrať |
|
Gastrointestinální poruchy (Časté, >1/100 až <1/10) | |
|
Poruchy kůže a podkožní tkáně (Časté, >1/100 až <1/10) |
Erytém Pruritus |
|
Celkové poruchy a reakce v místě aplikace (Velmi časté, >1/10) |
Reakce v místě injekce* |
|
(Časté, >1/100 až <1/10) |
Pyrexie |
|
Vyšetření (Časté, >1/100 až <1/10) |
Zvýšení transamináz |
|
* Podlitina v místě injekce, hematom v místě injekce, pálení v místě injekce, erytém v místě injekce, hypestezie místa injekce, podráždění v místě injekce, znecitlivění v místě injekce, edém v místě injekce, bolest v místě injekce, pocit tlaku v místě injekce, pruritus v místě injekce, zduření v místě injekce, kopřivka v místě injekce a teplo v místě injekce. | |
Popis vybraných nežádoucích účinků Imunogenita
Při opakované léčbě v rámci kontrolovaných studií fáze III byla ve vzácných případech pozorována přechodná pozitivita na protilátky proti ikatibantu. U všech pacientů byla zachována účinnost. Jeden pacient léčený Firazyrem měl pozitivní test na protilátky proti ikatibantu před léčbou Firazyrem i po ní. Tento pacient byl sledován po dobu 5 měsíců a další vzorky byly negativní na protilátky proti ikatibantu. U přípravku Firazyr nebyly hlášeny žádné hypersenzitivní ani anafylaktické reakce.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Nejsou k dispozici žádné klinické údaje týkající se předávkování.
Dávka 3,2 mg/kg podaná intravenózně (přibližně 8krát vyšší než terapeutická dávka) způsobila u zdravých jedinců přechodný erytém, svědění nebo hypotenzi. Nebyla zapotřebí žádná terapeutická intervence.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: jiné hematologické látky, léky užívané k léčbě dědičného angioedému, ATC kód: B06AC02.
Mechanismus účinku
Dědičný angioedém (HAE) (autosomálně dominantní onemocnění) je způsoben chybějícím nebo dysfunkčním inhibitorem esterázy C1. Ataky dědičného angioedému jsou provázeny zvýšeným uvolňováním bradykininu, což je klíčový mediátor rozvoje klinických příznaků.
Dědičný angioedém se projevuje jako periodické ataky podkožního anebo submukózního edému, který postihuje horní dýchací cesty, kůži a gastrointestinální trakt. Ataka obvykle trvá 2-5 dní.
Ikatibant je selektivní kompetitivní antagonista bradykininových receptorů typu 2 (B2). Je to syntetický dekapeptid, který je strukturálně podobný bradykininu, obsahuje však 5 neproteinogenních aminokyselin. U dědičného angioedému je zvýšená koncentrace bradykininu klíčovým mediátorem rozvoje klinických příznaků.
Farmakodynamické účinky
U zdravých mladých jedinců zabránil ikatibant podávaný v dávkách 0,8 mg/kg během 4 hodin a 1,5 mg/kg/den nebo 0,15 mg/kg/den po dobu 3 dní rozvoji bradykininem navozené hypotenze, vasodilatace a reflexní tachykardie. Když byla dávka bradykininu zvýšena čtyřikrát, bylo prokázáno, že ikatibant je kompetitivním antagonistou.
Klinické účinky a bezpečnost
Údaje o účinnosti přípravku byly získány v rámci iniciální otevřené studie fáze II a v rámci tří kontrolovaných studií fáze III.
Klinické studie fáze III (FAST-1 a FAST-2) byly randomizované, dvojitě zaslepené, kontrolované studie, které měly stejné uspořádání s výjimkou komparátoru (jedna s perorálně podávanou kyselinou tranexamovou jako komparátorem a jedna kontrolovaná placebem). Celkem 130 pacientů bylo randomizováno do skupin, kterým byl podáván buď ikatibant v dávce 30 mg (63 pacientů), nebo kontrolní látka (kyselina tranexamová u 38 pacientů, nebo placebo u 29 pacientů). Následné epizody dědičného angioedému byly léčeny v rámci otevřené rozšířené studie. Pacientům s příznaky laryngeálního angioedému byl ikatibant podáván v otevřené studii. Primárním cílovým parametrem účinnosti byla doba do začátku odeznívání příznaků určovaná pomocí vizuální analogové škály (VAS). Tabulka 2 uvádí výsledky účinnosti v těchto studiích.
FAST-3 byla randomizovaná, placebem kontrolovaná studie s paralelními skupinami u 98 dospělých pacientů s mediánem věku 36 let. Pacienti byli randomizováni k podávání buď ikatibantu 30 mg nebo placeba subkutánní injekcí. U podskupiny pacientů v této studii se vyskytly akutní ataky dědičného angioedému během užívání androgenů, antifibrinolytik nebo inhibitorů C1. Primárním cílovým parametrem byla doba do začátku odeznívání příznaků hodnocená pomocí kompozitního skóre třípoložkové vizuální analogové škály (VAS-3) sestávající z hodnocení otoku kůže, bolesti kůže a bolesti břicha. Tabulka 3 uvádí výsledky účinnosti ve studii FAST-3.
V těchto studiích byl u pacientů užívajících ikatibant zaznamenán kratší medián doby do začátku odeznívání příznaků (v první studii 2,0 hodiny, ve druhé 2,5 hodiny a ve třetí 2,0 hodiny) ve srovnání s kyselinou tranexamovou (12,0 hodin) a s placebem (4,6 a 19,8 hodiny). Léčebný efekt ikatibantu byl potvrzen sekundárními koncovými body účinnosti.
V integrované analýze těchto kontrolovaných studií fáze III byly doba do začátku odeznívání příznaků a doba do začátku odeznívání primárních příznaků podobné bez ohledu na věkovou skupinu, pohlaví, rasu, tělesnou hmotnost nebo na to, zda pacient užíval androgeny nebo antifibrinolytika nebo ne.
Odpověď na léčbu byla rovněž konzistentní při opakovaných atakách v kontrolovaných studiích fáze III. Celkem bylo u 237 pacientů léčeno 1278 atak akutního dědičného angioedému podáním 1386 dávek 30 mg ikatibantu. V hodnocení prvních 15 atak léčených přípravkem Firazyr (1114 dávek pro 1030 atak) byla střední doba do začátku odeznívání příznaků podobná pro všechny ataky (2,0 až 2,5 hodin). 92,4 % těchto atak dědičného angioedému bylo léčeno jedinou dávkou přípravku Firazyr.
Tabulka 2. Výsledky účinnosti ve studiích FAST-1 a FAST-2
|
Kontrolovaná klinická studie srovnávající přípravek FIRAZYR s kyselinou tranexamovou nebo placebem: výsledky týkající se účinnosti léčby | |||||
|
FAST-2 |
FAST-1 | ||||
|
Ikatibant |
Kyselina tranexamová |
Ikatibant |
Placebo | ||
|
Počet jedinců v ITT populaci* |
36 |
38 |
Počet jedinců v ITT populaci |
27 |
29 |
|
Základní hodnoty na vizuální analogové škále (mm) |
63,7 |
61,5 |
Základní hodnoty na vizuální analogové škále (mm) |
69,3 |
67,7 |
|
Změna ze základních hodnot na 4 hodiny |
-41,6 |
-14,6 |
Změna ze základních hodnot na 4 hodiny |
-44,8 |
-23,5 |
|
Rozdíl mezi způsoby léčby (95% interval spolehlivosti, P-hodnota) |
-27,8 (-39,4, -16,2) p < 0,001 |
Rozdíl mezi způsoby léčby (95% interval spolehlivosti, P-hodnota) |
-23,3 (-37,1, -9,4) p = 0,002 | ||
|
Změna ze základních hodnot na 12 hodin |
-54,0 |
-30,3 |
Změna ze základních hodnot na 12 hodin |
-54,2 |
-42,4 |
|
Rozdíl mezi způsoby léčby (95% interval spolehlivosti, P-hodnota) |
-24,1 (-33,6, -14,6) p < 0,001 |
Rozdíl mezi způsoby léčby (95% interval spolehlivosti, P-hodnota) |
-15,2 (-28,6, -1,7) p = 0,028 | ||
|
Medián doby do začátku odeznívání příznaků (hodiny) |
Medián doby do začátku odeznívání příznaků (hodiny) | ||||
|
Všechny epizody (N = 74) |
2,0 |
12,0 |
Všechny epizody (N = 56) |
2,5 |
4,6 |
|
Míra odezvy (%, CI) po 4 hodinách od zahájení léčby |
Míra odezvy (%, CI) po 4 hodinách od zahájení léčby | ||||
|
Všechny epizody (N = 74) |
80,0 (63.1; 91,6) |
30.6 (16,3; 48,1) |
Všechny epizody (N = 56) |
66,7 (46,0; 83,5) |
46,4 (27,5; 66,1) |
|
Kontrolovaná klinická studie srovnávající přípravek FIRAZYR s kyselinou tranexamovou nebo placebem: výsledky týkající se účinnosti léčby | |||||
|
FAST-2 |
FAST-1 | ||||
|
Ikatibant |
Kyselina tranexamová |
Ikatibant |
Placebo | ||
|
Medián doby do začátku odeznívání |
Medián doby do začátku odeznívání | ||||
|
příznaků: všechny příznaky (hodiny): |
příznaků: všechny příznaky (hodiny): | ||||
|
1,6 |
3,5 |
2,0 |
3,3 | ||
|
Otok kůže |
2,6 |
18,1 |
Otok kůže |
3,1 |
10,2 |
|
Bolestivost kůže |
1,5 |
12,0 |
Bolestivost kůže |
1,6 |
9,0 |
|
Medián doby do téměř úplného odeznění příznaků (hodiny) |
Medián doby do téměř úplného odeznění příznaků (hodiny) | ||||
|
Všechny epizody (N = 74) |
10,0 |
51,0 |
Všechny epizody (N = 56) |
8,5 |
19,4 |
|
Medián doby |
Medián doby | ||||
|
do regrese příznaků, podle pacienta (hodiny) |
do regrese příznaků, podle pacienta (hodiny) | ||||
|
Všechny epizody (N = 74) |
0,8 |
7,9 |
Všechny epizody (N = 56) |
0,8 |
16,9 |
|
Medián doby do celkového zlepšení |
Medián doby do celkového | ||||
|
stavu pacienta, podle lékaře (hodiny) |
zlepšení stavu pacienta, podle lékaře (hodiny) | ||||
|
Všechny epizody (N = 74) |
1,5 |
6,9 |
Všechny epizody (N = 56) |
1,0 |
5,7 |
*populace podle léčebného záměru (intention to treat, ITT)
Tabulka 3. Výsledky účinnosti ve studii FAST-3
|
Výsledky účinnosti: FAST-3; kontrolovaná fáze - ITT populace | ||||
|
Cílový parametr |
Statistika |
Firazyr |
Placebo |
p-hodnota |
|
(n = 43) |
(n = 45) | |||
|
Primární cílový parametr | ||||
|
Doba do začátku odeznívání příznaků - kompozitní V AS (hod.) |
Medián |
2,0 |
19,8 |
<0,001 |
|
Ostatní cílové parametry | ||||
|
Doba do začátku odeznívání primárních příznaků (hod.) |
Medián |
1,5 |
18,5 |
< 0,001 |
|
Změna v kompozitním skóre VAS 2 hodiny po léčbě |
Průměr |
-19,74 |
-7,49 |
< 0,001 |
|
Změna v kompozitním skóre příznaků po 2 hodinách |
Průměr |
-0,53 |
-0,22 |
< 0,001 |
|
Výsledky účinnosti: FAST-3; kontrolovaná fáze - ITT populace | ||||
|
Cílový parametr |
Statistika |
Firazyr |
Placebo |
p-hodnota |
|
CO II |
(n = 45) | |||
|
hodnoceném subjektem | ||||
|
Změna v kompozitním skóre příznaků po 2 hodinách hodnoceném zkoušejícím |
Průměr |
-0,44 |
-0,19 |
< 0,001 |
|
Doba do téměř úplného odeznění příznaků (hod.) |
Medián |
8,0 |
36,0 |
0,012 |
|
Doba do počátečního zlepšení příznaků hodnoceného subjektem (hod.) |
Medián |
0,8 |
3,5 |
< 0,001 |
|
Doba do počátečního viditelného zlepšení příznaků hodnoceného zkoušejícím (hod.) |
Medián |
0,8 |
3,4 |
< 0,001 |
Celkem bylo v těchto kontrolovaných klinických studiích fáze III léčeno 66 pacientů s atakami dědičného angioedému postihujícími hrtan. Výsledky ohledně doby do začátku odeznívání příznaků byly podobné jako u pacientů s nelaryngeálními atakami dědičného angioedému.
5.2 Farmakokinetické vlastnosti
Farmakokinetika ikatibantu byla podrobně popsána studiemi, v nichž byl ikatibant podáván intravenózně a subkutánně zdravým dobrovolníkům a pacientům. Farmakokinetický profil ikatibantu u pacientů s dědičným angioedémem je podobný jako u zdravých dobrovolníků.
Absorpce
Po subkutánním podání je absolutní biologická dostupnost ikatibantu 97 %. Čas potřebný k dosažení maximální sérové koncentrace je přibližně 30 minut.
Distribuce
Distribuční objem ikatibantu v rovnovážném stavu (Vss) je přibližně 20 - 25 l. Na proteiny v plazmě se váže 44 % ikatibantu.
Vylučování
Ikatibant je vylučován převážně ve formě metabolitů, méně než 10% dávky se vylučuje v nezměněné formě močí. Hodnota clearance je přibližně 15 - 20 l/hod. nezávisle na dávce. Plazmatický poločas eliminace je přibližně 1 - 2 hodiny.
Biotransformace
Ikatibant je převážně metabolizován proteolytickými enzymy na inaktivní metabolity, které jsou vylučovány především močí.
In vitro studie potvrdily, že ikatibant není degradován oxidačními metabolickými cestami, není inhibitorem izoenzymů nejvýznamnějšího cytochromu P450 (CYP): CYP 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 a 3A4 a není induktorem CYP 1A2 a 3A4.
Zvláštní populace
Získané údaje naznačují, že dochází k poklesu clearance v souvislosti s věkem, důsledkem čehož je o 50-60 % vyšší expozice u starších osob (ve věku 75-80 let) ve srovnání s pacienty ve věku 40 let.
Z údajů vyplývá, že pohlaví a váha pacienta nemají významný vliv na farmakokinetiku ikatibantu.
Na základě omezených dat se lze domnívat, že expozice ikatibantu není ovlivňována jatemím či renálním poškozením. Vliv rasy na farmakokinetiku ikatibantu nebyl hodnocen. Nejsou k dispozici žádné údaje týkající se farmakokinetiky u dětí.
5.3 Předklinické údaje vztahující se k bezpečnosti
Byly provedeny studie po opakovaném podání dávky trvající až šest měsíců u potkanů a devět měsíců u psů. U potkanů i u psů bylo zaznamenáno na dávce závislé snížení hladin cirkulujících pohlavních hormonů, a opakované podávání ikatibantu reverzibilně zpozdilo pohlavní dozrávání.
V devítiměsíční studii u psů byla maximální denní expozice definovaná plochou pod křivkou (AUC) při dávkách, při kterých nebyly pozorovány žádné nežádoucí účinky (No Observed Adverse Effect Levels, NOAEL), 2,3krát vyšší než AUC u lidí po subkutánním podání dávky 30 mg. Ve studii
u potkanů nebyla hodnota NOAEL měřitelná, nicméně všechna zjištění této studie prokázala buď plně nebo částečně reverzibilní účinky u léčených potkanů. Při všech dávkách testovaných na potkanech byla pozorována hypertrofie nadledvinek. Hypertrofie nadledvinek odezněla po ukončení podávání ikatibantu. Klinický význam nálezů týkajících se nadledvinek není znám.
Ikatibant neměl žádný vliv na plodnost samců myší (maximální dávka 80,8 mg/kg/den) a potkanů (maximální dávka 10 mg/kg/den).
V dvouleté studii, která hodnotila karcinogenní potenciál ikatibantu u potkanů, neměly denní dávky na úrovni přibližně dvojnásobně vyšší expozice, než je expozice dosažená terapeutickou dávkou u lidí, žádný vliv na výskyt či morfologii nádorů. Z výsledků nevyplývá karcinogenní potenciál ikatibantu.
Standardní série in vitro a in vivo testů neprokázala genotoxicitu ikatibantu.
Ikatibant neměl teratogenní vlastnosti při podávání formou subkutánní injekce během časného embryonálního a fetálního vývoje potkanům (v maximální dávce 25 mg/kg/den) a králíkům (v maximální dávce 10 mg/kg/den). Ikatibant je silný antagonista bradykininu, proto ve vysokých dávkách může ovlivňovat uhnízdění zárodku do dělohy a následně děložní stabilitu v časné fázi těhotenství. Tento vliv na dělohu se projevuje také v pozdní fázi těhotenství, kdy se objevují tokolytické účinky ikatibantu, což má u potkanů za následek zpoždění porodu s častějším ohrožením plodu a ve vysokých dávkách (10 mg/kg/den) perinatálním úmrtím.
Ve studii toxicity u mláďat, ve které byly pohlavně nezralým potkanům podávány 3 mg/kg denně po dobu 7 týdnů, byla pozorována atrofie varlat a nadvarlat. Podobné účinky ikatibantu na reprodukční tkáň byly zaznamenány u pohlavně zralých potkanů a psů. Tyto nálezy ve tkáních byly konzistentní s popsanými účinky na gonadotropiny a během následného období bez léčby se jevily jako reverzibilní.
Ikatibant nevyvolal žádné změny převodového systému srdce in vitro (hERG kanál) ani in vivo u zdravých psů, ani u různých psích modelů (programovaná stimulace komor, fyzická námaha a ligatura koronární artérie), kde nebyly pozorovány žádné přidružené hemodynamické změny. Bylo potvrzeno, že ikatibant zhoršuje vyvolanou srdeční ischémii u několika neklinických modelů, ačkoli u akutní ischémie nebyl konzistentně zjišťován škodlivý účinek.
6. FARMACEUTICKÉ ÚDAJE 6.1 Seznam pomocných látek
Chlorid sodný
Koncentrovaná kyselina octová (k úpravě pH) Hydroxid sodný (k úpravě pH)
Voda na injekci
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
2 roky
6.4 Zvláštní opatření pro uchovávání
Uchovávejte při teplotě do 25 °C.
Chraňte před mrazem.
6.5 Druh obalu a velikost balení
3 ml roztoku v 3 ml předplněné injekční stříkačce (sklo typu I) s plunžrovým uzávěrem (bromobutyl potažený fluorokarbonovým polymerem). Součástí balení je subkutánní jehla (25 G; 16 mm).
Velikost balení: jedna předplněná injekční stříkačka s jednou jehlou nebo vícečetné balení obsahující tři předplněné injekční stříkačky se třemi jehlami.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku
Roztok by měl být čirý a bezbarvý, bez jakýchkoli viditelných částic. Pouze pro jedno použití. Všechen nepoužitý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Shire Orphan Therapies GmbH Friedrichstrasse 149 D-10117 Berlín Německo
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/08/461/001
EU/1/08/461/002
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 11. července 2008
Datum posledního prodloužení registrace: 13. března 2013
10. DATUM REVIZE TEXTU
Podrobné informace o tomto přípravku jsou uveřejněny na webových stránkách Evropská agentura pro léčivé přípravky http://www.ema.europa.eu
A VÝROBCE ODPOVĚDNÝ /VÝROBCI ODPOVĚDNÍ ZA
PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ /VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného za propouštění šarží
Shire Pharmaceuticals Ireland Limited 5 Riverwalk
Citywest Business Campus
Dublin 24
Irsko
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis.
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
• Pravidelně aktualizované zprávy o bezpečnosti
•
Držitel rozhodnutí o registraci předloží první pravidelně aktualizovanou zprávu o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Dále je třeba aktualizovaný RMP předložit:
• na žádost Evropské agentury pro léčivé přípravky;
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud jsou data předložení PSUR a aktualizace RMP stejné, je možné je předložit současně.
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
Firazyr 30 mg injekční roztok v předplněné injekční stříkačce Icatibantum
Jedna 3ml předplněná injekční stříkačka obsahuje icatibantum ve formě icatibanti acetas odpovídající 30 mg ikatibantu.
Jeden ml roztoku obsahuje 10 mg ikatibantu.
Obsahuje: koncentrovanou kyselinu octovou, hydroxid sodný, chlorid sodný, vodu na injekce.
Injekční roztok
Jedna předplněná injekční stříkačka Jedna 25G jehla
Subkutánní podání
Před použitím si přečtěte příbalovou informaci Pouze pro jedno použití
Uchovávejte mimo dohled a dosah dětí.
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte při teplotě do 25 °C. Chraňte před mrazem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z TAKOVÝCH LÉČIVÝCH PŘÍPRAVKŮ, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Držitel rozhodnutí o registraci Shire Orphan Therapies GmbH Friedrichstrasse 149 D-10117 Berlín Německo
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/08/461/001
13. ČÍSLO ŠARŽE
c. s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Firazyr 30 mg
Firazyr 30 mg injekční roztok v předplněné injekční stříkačce Icatibantum
Jedna 3 ml předplněná injekční stříkačka obsahuje icatibantum ve formě icatibanti acetas odpovídající 30 mg ikatibantu.
Jeden ml roztoku obsahuje 10 mg ikatibantu.
Obsahuje: koncentrovanou kyselinu octovou, hydroxid sodný, chlorid sodný, vodu na injekci.
Injekční roztok
Vícečetné balení obsahující tři předplněné injekční stříkačky a tři 25G jehly
Subkutánní podání
Před použitím si přečtěte příbalovou informaci Pouze pro jedno použití
Uchovávejte mimo dohled a dohled dětí.
EXP
Neuchovávejte při teplotě nad 25 °C. Chraňte před mrazem.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Držitel rozhodnutí o registraci Shire Orphan Therapies GmbH Friedrichstrasse 149 D-10117 Berlín Německo
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/08/461/002
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Firazyr 30 mg
Firazyr 30 mg injekční roztok v předplněné injekční stříkačce Icatibantum
Jedna 3 ml předplněná injekční stříkačka obsahuje icatibantum ve formě icatibanti acetas odpovídající 30 mg ikatibantu.
Jeden ml roztoku obsahuje 10 mg ikatibantu.
Obsahuje: koncentrovanou kyselinu octovou, hydroxid sodný, chlorid sodný, vodu na injekci.
Injekční roztok
Jedna předplněná injekční stříkačka a jedna 25G jehla.
Součást vícečetného balení, není určeno k jednotlivému prodeji.
Subkutánní podání
Před použitím si přečtěte příbalovou informaci Pouze pro jedno použití
Uchovávejte mimo dohled a dohled dětí.
EXP
Neuchovávejte při teplotě nad 25 °C. Chraňte před mrazem.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Držitel rozhodnutí o registraci Shire Orphan Therapies GmbH Friedrichstrasse 149 D-10117 Berlín Německo
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/08/461/002
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Firazyr 30 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Firazyr 30 mg injekční roztok v předplněné injekční stříkačce.
Icatibantum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Shire Orphan Therapies GmbH
3. POUŽITELNOST
Použitelné do:
4. ČÍSLO ŠARŽE
č. š.:
5. JINÉ
Subkutánní podání
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Firazyr 30 mg
Icatibant
sc
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
30 mg/3 ml
6. JINÉ
Shire Orphan Therapies GmbH
Firazyr 30 mg injekční roztok v předplněné injekční stříkačce
Icatibantum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci :
1. Co je přípravek Firazyr a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Firazyr používat
3. Jak se přípravek Firazyr používá
4. Možné nežádoucí účinky
5. Jak přípravek Firazyr uchovávat
6. Obsah balení a další informace
1. Co je přípravek Firazyr a k čemu se používá
Firazyr obsahuje léčivou látku ikatibant.
Přípravek Firazyr se používá k léčbě příznaků dědičného angioedému u dospělých pacientů.
U dědičného angioedému je v krvi zvýšená hladina látky s názvem bradykinin, což vede k rozvoji příznaků jako je otok, bolest, nucení ke zvracení a průjem.
Přípravek Firazyr blokuje aktivitu bradykininu, proto přeruší rozvoj příznaků ataky dědičného angioedému.
2. Čemu musíte věnovat pozornost, než začnete přípravek Firazyr používat Nepoužívejte přípravek Firazyr
- jestliže j ste alergický/á na ikatibant nebo na kteroukoli další složku přípravku Firazyr (uvedenou v bodě 6).
Upozornění a opatření
Před použitím přípravku Firazyr se poraďte se svým lékařem.
Nežádoucí účinky souvisejících s přípravkem Firazyr jsou podobné příznakům Vašeho onemocnění. Pokud zaznamenáte, že se příznaky ataků zhoršily poté, co Vám byl podán Firazyr, okamžitě informujte svého lékaře.
- pokud trpíte srdeční angínou (snížený průtok krve srdečním svalem);
- pokud jste v nedávné době prodělal mozkovou příhodu.
Dále také:
- Musíte být proškolen(a) v technice podávání subkutánní injekce (pod kůži) předtím, než si začnete Firazyr sám/sama aplikovat.
- Ihned poté, co si podáte injekci přípravku Firazyr během probíhající laryngeální ataky (ucpání horních cest dýchacích), nebo Vám injekci během ataky podá ošetřující osoba, musíte vyhledat lékařskou péči ve zdravotnickém zařízení.
- Pokud příznaky po jedné samostatně podané injekci přípravku Firazyr neustoupí, vyhledejte lékařskou radu kvůli dalším injekcím přípravku Firazyr. Během 24 hodin mohou být podány až 2 další injekce.
Děti a dospívající
Podávání přípravku Firazyr dětem a dospívajícím mladším 18 let se nedoporučuje, protože nebyl u této věkové skupiny studován.
Další léčivé přípravky a přípravek Firazyr
Informujte svého lékaře o všech lécích, které užíváte, které jste v nedávné době užíval(a),nebo které možná budete užívat.
Není známo, že by přípravek Firazyr ovlivňoval působení dalších léčivých přípravků. Pokud užíváte lék známý jako inhibitor angiotenzin konvertujícího enzymu (ACE) (například kaptopril, enalapril, ramipril, chinapril, lisinopril), který se používá ke snižování krevního tlaku nebo z jiného důvodu, měl(a) byste informovat svého lékaře, než začnete užívat přípravek Firazyr.
Těhotenství a kojení
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete těhotenství, poraďte se se svým lékařem dříve, než začnete přípravek Firazyr používat.
Jestliže kojíte, pak byste neměla kojit 12 hodin poté, co vám byl naposledy podán přípravek Firazyr. Řízení dopravních prostředků a obsluha strojů
Pokud se následkem ataky dědičného angioedému nebo po podání přípravku Firazyr cítíte unaven(á) nebo máte závratě, neřiďte motorové vozidlo ani neobsluhujte stroje.
Přípravek Firazyr obsahuje malé množství sodíku
Injekční roztok obsahuje méně než 1 mmol (23 miligramů) sodíku, takže je v podstatě sodíku prostý.
3. Jak se přípravek Firazyr používá
Vždy používejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem. Pokud jste ještě nikdy dříve Firazyr nedostal(a), první dávka přípravku Firazyr Vám bude podána lékařem nebo zdravotní sestrou. Lékař vám sdělí, kdy můžete bezpečně odejít domů.
Je možné, že si po poradě se svým lékařem nebo zdravotní sestrou a proškolení v technice subkutánní injekce (pod kůži) budete moci v případě ataky dědičného angioedému Firazyr injikovat sám/sama nebo Vám injekci Firazyru bude moci podat Vaše ošetřující osoba. Je důležité, aby byl Firazyr injikován subkutánně (pod kůži), jakmile zpozorujete ataku dědičného angioedému. Váš lékař nebo zdravotní sestra Vás a Vaši ošetřující osobu vyškolí, jak bezpečně injikovat Firazyr podle pokynů uvedených v této příbalové informaci.
Kdy a jak často byste měli používat přípravek Firazyr?
- Váš lékař určí přesnou dávku přípravku Firazyr a sdělí vám, jak často má být používán.
Doporučená dávka přípravku Firazyr je jedna injekce (3 ml, 30 mg) podána subkutánně (pod kůži), jakmile zpozorujete ataku dědičného angioedému (například v případě zvýšeného otoku kůže, zejména v obličeji a na krku, nebo v případě zhoršující se bolesti břicha).
Pokud u vás nedojde k ústupu příznaků po uplynutí 6 hodin, vyhledejte lékařskou pomoc ohledně dalších injekcí přípravku Firazyr. Během 24 hodin mohou být podány až 2 další injekce.
V průběhu 24 hodin by neměly být podány více než 3 injekce přípravku Firazyr, a pokud vyžadujete více než 8 injekcí v průběhu jednoho měsíce, poraďte se s lékařem.
Jakým způsobem má být přípravek Firazyr podán?
Přípravek Firazyr je určen k podání formou subkutánní injekce (pod kůži). Injekční stříkačku je možné použít pouze jednou.
Přípravek Firazyr je injikován krátkou jehlou do tukové tkáně pod kůži břicha.
Máte-li jakékoli další otázky týkající se užívání tohoto léku, zeptejte se svého lékaře nebo lékárníka. Následující návod krok za krokem je určen pouze pro samostatné podávání Postup se skládá z následujících hlavních kroků:
1) Důležité všeobecné informace
2) Příprava injekční stříkačky a jehly k injekci
3) Příprava místa pro inj ekci
4) Injikování roztoku
5) Likvidace injekčního setu
Návod k injekci krok za krokem 1) Důležité všeobecné informace
• Než začnete, důkladně si omyjte ruce mýdlem a vodou
• Otevřete blistr odloupnutím fólie
• Vyjměte předplněnou injekční stříkačku z blistrové vaničky
• Odstraňte kryt z konce předplněné injekční stříkačky odšroubováním krytu
• Po odšroubování krytu položte předplněnou injekční stříkačku na rovný povrch
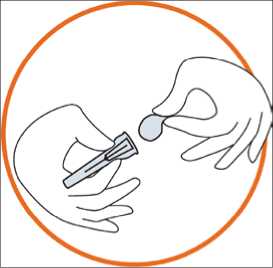
• Vyjměte kryt s jehlou z blistru
• Odstraňte fólii z krytu j ehly (j ehla by měla zůstat v krytu)
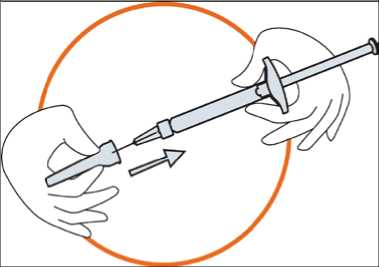
• Uchopte pevně injekční stříkačku. Nasaďte jehlu opatrně na předplněnou injekční stříkačku obsahující bezbarvý roztok
• Našroubujte předplněnou injekční stříkačku na jehlu s jehlou stále ještě v krytu
• Vyjměte jehlu z krytu zatáhnutím za stříkačku. Nevytahujte píst
• Stříkačka j e nyní připravena k inj ekci
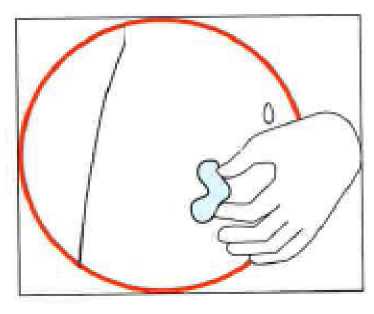
• Zvolte místo pro injekci. Místem vpichu injekce by měl být záhyb kůže na levé či pravé straně břicha přibližně 5-10 cm pod pupkem. Toto místo by mělo být nejméně 5 cm od jakýchkoli jizev. Nevybírejte si místo, které je pohmožděné, oteklé nebo bolestivé
• Očistěte místo injekce alkoholovým tamponem a nechte kůži oschnout
4) Injikování roztoku
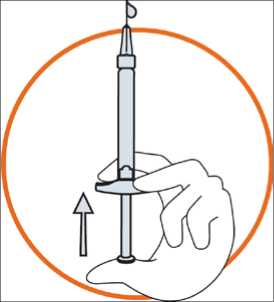
• Držte stříkačku mezi dvěma prsty jedné ruky, s palcem na dolní části pístu
• Zajistěte, aby ve stříkačce nebyly žádné vzduchové bubliny, a to stlačením pístu, dokud se na špičce jehly neobjeví první kapka
4) Injikování roztoku (pokračování)
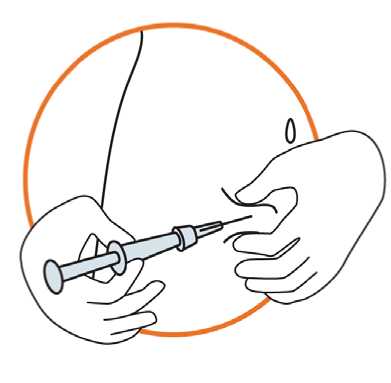
• Držte stříkačku pod úhlem 45-90 stupňů vzhledem ke kůži, s jehlou směřující ke kůži
• Držte stříkačku jednou rukou a druhou rukou jemně uchopte mezi palcem a prsty kožní řasu na dříve vydezinfikovaném místě pro injekci
• Držte kožní řasu, přiložte stříkačku ke kůži a rychlým pohybem vpíchněte jehlu do kožní řasy
• Pomalu a rovnoměrně tlačte na píst stříkačky, dokud není všechen roztok injikován do kůže a ve stříkačce nezbývá žádná tekutina
• Píst stlačujte pomalu, aby injekce trvala přibližně 30 vteřin
• Uvolněte kožní řasu a jemně vytáhněte jehlu
5) Likvidace injekčního setu
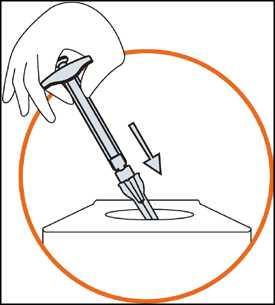
• Injekční stříkačku, jehlu a kryt jehly vyhoďte do nádoby na ostré předměty sloužící k likvidaci odpadu, který by mohl ublížit jiným, pokud s ním není správně zacházeno.
Možné nežádoucí účinky
4.
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého. Téměř u všech pacientů, kterým je podán přípravek Firazyr, se vyskytnou reakce v místě podání injekce (jako např. podráždění kůže, otok, bolest, svědění, zarudnutí kůže a pocit pálení). Tyto účinky jsou obvykle mírné a odezní bez nutnosti další léčby.
Velmi časté nežádoucí účinky (postihují více než 1 pacienta z 10) jsou:
Další reakce v místě podání injekce (pocit tlaku, podlitina, snížená citlivost a/nebo znecitlivění, vyvýšená svědivá kožní vyrážka a pocit tepla).
Časté nežádoucí účinky (postihují až 1 z 10 pacientů) jsou:
Nevolnost Bolest hlavy Závratě Horečka Svědění Kožní vyrážka Zarudnutí kůže
Abnormální hodnoty jaterních testů
Informujte neprodleně svého lékaře, pokud si všimnete, že u Vás došlo ke zhoršení příznaků ataky poté, co Vám byl podán přípravek Firazyr.
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Firazyr uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti, uvedené na štítku za ,EXP‘. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte při teplotě do 25 °C. Chraňte před mrazem.
Nepoužívejte tento přípravek, pokud si všimnete, že je poškozen obal stříkačky nebo jehly, nebo v případě jakýchkoli viditelných známek poškození, například jestliže je roztok zakalený, obsahuje cizorodé částice nebo pokud došlo ke změně jeho zabarvení.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co přípravek Firazyr obsahuje
Léčivou látkou přípravku je ikatibant, přičemž jedna předplněná injekční stříkačka obsahuje 30 miligramů icatibantu (ve formě icatibanti acetas). Další složky přípravku jsou: chlorid sodný, koncentrovaná kyselina octová, hydroxid sodný a voda na injekci.
Jak přípravek Firazyr vypadá a co obsahuje toto balení
Přípravek Firazyr je dodáván jako čirý a bezbarvý injekční roztok v předplněné skleněné injekční stříkačce o objemu 3 ml. Součástí balení je subkutánní jehla.
Firazyr je dostupný v jednodávkovém balení obsahujícím jednu předplněnou injekční stříkačku s jednou jehlou nebo ve vícedávkovém balení obsahujícím tři předplněné injekční stříkačky se třemi jehlami.
Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci
Shire Orphan Therapies GmbH Friedrichstrasse 149 D-10117 Berlín Německo
Výrobce
Shire Pharmaceuticals Ireland Limited 5 Riverwalk
Citywest Business Campus
Dublin 24
Irsko
Tato příbalová informace byla naposledy revidována v Další zdroje informací
Podrobné informace o tomto přípravku jsou uveřejněny na webových stránkách Evropské agentury pro léčivé přípravky: http://www.ema.europa.eu. Na těchto stránkách naleznete též odkazy na další webové stránky, týkající se vzácných onemocnění a jejich léčby.
32