Farydak 10 Mg
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Farydak 10 mg tvrdé tobolky Farydak 15 mg tvrdé tobolky Farydak 20 mg tvrdé tobolky
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Farydak 10 mg tvrdé tobolky
Jedna tvrdá tobolka obsahuje panobinostati lactas odpovídající panobinostatum 10 mg. Farydak 15 mg tvrdé tobolky
Jedna tvrdá tobolka obsahuje panobinostati lactas odpovídající panobinostatum 15 mg. Farydak 20 mg tvrdé tobolky
Jedna tvrdá tobolka obsahuje panobinostati lactas odpovídající panobinostatum 20 mg. Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Tvrdá tobolka (tobolka).
Farydak 10 mg tvrdé tobolky
Světle zelená neprůhledná tvrdá želatinová tobolka (15,6-16,2 mm) obsahující bílý až téměř bílý prášek, s příčným označením “LBH 10 mg” černým inkoustem na víčku a dvěma příčnými pruhy černým inkoustem na těle tobolky.
Farydak 15 mg tvrdé tobolky
Oranžová neprůhledná tvrdá želatinová tobolka (19,1-19,7 mm) obsahující bílý až téměř bílý prášek, s příčným označením “LBH 15 mg” černým inkoustem na víčku a dvěma příčnými pruhy černým inkoustem na těle tobolky.
Farydak 20 mg tvrdé tobolky
Červená neprůhledná tvrdá želatinová tobolka (19,1-19,7 mm) obsahující bílý až téměř bílý prášek, s příčným označením “LBH 20 mg” černým inkoustem na víčku a dvěma příčnými pruhy černým inkoustem na těle tobolky.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Farydak, je v kombinaci s bortezomibem a dexamethazonem určen k léčbě dospělých pacientů s relabujícím a/nebo refrakterním mnohočetným myelomem, kteří dostávali nejméně dva předchozí režimy včetně bortezomibu a imunomodulačního léku.
4.2 Dávkování a způsob podání
Léčba přípravkem Farydak má být zahájena lékařem se zkušeností s použitím protinádorových léčivých přípravků.
Dávkování
Doporučená zahajovací dávka panobinostatu je 20 mg, podávaná perorálně jednou denně ve dnech 1, 3, 5, 8, 10 a 12 během 21denního cyklu. Pacienti by měli být nejdříve léčeni osmi cykly. Doporučuje se, aby pacienti profitující z léčby pokračovali v léčbě dalšími osmi cykly. Celkové trvání léčby je až 16 cyklů (48 týdnů).
Panobinostat se podává v kombinaci s bortezomibem a dexamethazonem podle popisu v Tabulce 1 a 2. Příbalové informace bortezomibu a dexamethazonu mají být konzultovány před zahájením kombinační léčby za účelem posouzení možnosti snížení dávky.
Doporučená dávka bortezomibu je 1,3 mg/m2 podávaných injekcí. Doporučená dávka dexamethazonu je 20 mg užívaných perorálně na plný žaludek.
Tabulka 1 Doporučený rozvrh dávkování panobinostatu v kombinaci s bortezomibem a dexamethazonem (cykly 1-8)
|
Cykly 1-8 (3týdenní cykly) |
Týden 1 Dny_ |
Týden 2 Dny_ |
Týden 3 | ||||||||||||
|
Farydak |
1 |
3 |
5 |
8 |
10 |
12 |
Přestávka | ||||||||
|
Bortezomib |
1 |
4 |
8 |
11 |
Přestávka | ||||||||||
|
Dexamethazon |
1 |
2 |
4 |
5 |
8 |
9 |
11 |
12 |
Přestávka | ||||||
Tabulka 2 Doporučený rozvrh dávkování panobinostatu v kombinaci s bortezomibem a dexamethazonem (cykly 9-16)
|
Cykly 9-16 (3týdenní cykly) |
Týden 1 Dny |
Týden 2 Dny |
Týden 3 | ||||||||||||
|
Farydak |
1 |
3 |
5 |
8 |
10 |
12 |
Přestávka | ||||||||
|
Bortezomib |
1 |
8 |
Přestávka | ||||||||||||
|
Dexamethazon |
1 |
2 |
8 |
9 |
Přestávka | ||||||||||
Doporučení pro sledování Krevní obraz
Před zahájením léčby panobinostatem musí být vyhodnocen krevní obraz. Počet krevních destiček ve výchozím stavu má být >100 x 109/l a celkový počet neutrofilů ve výchozím stavu (ANC) >1,0 x 109/l. Během léčby má být pravidelně sledován krevní obraz (zejména před každou injekcí bortezomibu, tj. ve dnech 1, 4, 8 a 11 v cyklech 1 až 8 a ve dnech 1 a 8 v cyklech 9 až 16), zejména pro trombocytopenii (viz bod 4.4). Před zahájením jakéhokoli cyklu léčby panobinostatem v kombinaci s bortezomibem a dexamethazonem by měl být počet krevních destiček nejméně >100 x 109/l (viz bod 4.4). Další stanovení počtu krevních destiček by mělo být zvažováno během “přestávky” - tj. ve dnech 15 a/nebo 18, zejména u pacientů >65 let a pacientů s nižším počtem krevních destiček než 150 x 109/l ve výchozím stavu.
EKG
Panobinostat může prodloužit QTc interval (viz bod 4.4). Proto by mělo být před zahájením léčby opakovaně periodicky před každým cyklem léčby vyhodnoceno EKG. QTcF má být <480 ms před zahájením léčby panobinostatem (viz bod 4.4 o úpravě dávky níže).
Krevní elektrolyty
Krevní elektrolyty, zejména draslík, hořčík a fosfor, by měly být vyhodnoceny ve výchozím stavu a sledovány periodicky dle klinické potřeby, zejména u pacientů s průjmem. Neobvyklé hodnoty by měly být upraveny dle klinické potřeby (viz bod 4.4).
Testy jaterních funkcí
Před léčbou a pravidelně během léčby mají být sledovány jaterní funkce, jak je klinicky indikováno, především u pacientů s poruchou jaterních funkcí (viz bod 4.4).
Testy funkce štítné žlázy
Mírný hypotyroidismus byl hlášen u pacientů léčených panobinostatem + bortezomibem + dexamethazonem ve studii D2308, z nichž někteří vyžadovali léčbu (viz bod 4.4). Funkce štítné žlázy a hypofýzy by měly být sledovány pomocí hodnocení hodnot hormonů (tj. volný T4 a TSH) dle klinické potřeby.
Úprava dávky
Na základě individuální tolerability může být vyžadována úprava dávky a/nebo rozvrhu léčby. Pokud u pacienta dojde ke vzniku nežádoucích účinků, je nutné klinické rozhodnutí, jak pokračovat v léčbě
Pokud je vyžadováno snížení dávky, má být dávka panobinostatu snížena o 5 mg (tj. z 20 mg na 15 mg nebo z 15 mg na 10 mg). Dávka nemá být snížena pod 10 mg a má být udržován stejný léčebný rozvrh (3týdenní léčebný cyklus).
Trombocytopenie
Před podáním každé dávky bortezomibu má být sledován počet krevních destiček (tj. ve dnech 1, 4, 8 a 11 cyklů 1 - 8, viz Tabulka 1, a ve dnech 1 a 8 cyklů 9-16, viz Tabulka 2). Pokud se u pacientů projeví trombocytopenie, může být panobinostat dočasně vysazen a následná dávka může být snížena (viz Tabulka 3). U pacientů s počtem krevních destiček <50 x 109/l (komplikovaným krvácením) nebo <25 x 109/l, by měla být léčba přípravkem Farydak vysazena a měla by pokračovat sníženou dávkou po návratu počtu krevních destiček na >50 x 109/l. Počty krevních destiček by měly být sledovány nejméně dvakrát týdně až do stavu >50 x 109/l. Podle klinické potřeby může být vyžadovaná transfuze krevních destiček (viz bod 4.4). Má být zvažováno ukončení léčby, pokud se trombocytopenie navzdory úpravám dávky popsaným níže nezlepší a/nebo stav pacienta vyžaduje opakované transfúze krevních destiček. Dále má být zvažována úprava dávky bortezomibu (viz SPC bortezomibu a Tabulka 3).
Tabulka 3 Doporučené úpravy dávky pro trombocytopenii
|
Stupeň trombocytopenie v den léčby |
Úprava zahajovací dávky panobinostat u |
Dávka panobinostatu při zotavení na trombocytopenii stupně 2 (>50 x 109/l) |
Úprava zahajovací dávky bortezomibu |
Dávka bortezomibu při zotavení na trombocytopenii stupně 2 (>50 x 109/l) | |
|
1 vynech aná dávka |
Více než 1 vynecha ná dávka | ||||
|
Stupeň 3 Počet krevních destiček <50 x 109/l s krvácením |
Vynechat dávku |
Pokračovat sníženou dávkou |
Vynechat dávku |
Pokračov at stejnou dávkou |
Pokračova t sníženou dávkou |
|
Stupeň 4 Počet krevních destiček <25 x 109/l |
Vynechat dávku |
Pokračovat sníženou dávkou |
Vynechat dávku |
Pokračov at stejnou dávkou |
Pokračova t sníženou dávkou |
Gastrointestinálni toxicita
Gastrointestinální toxicita je velmi častá u pacientů léčených panobinostatem. Pacienti s průjmem a nauzeou nebo zvracením mohou vyžadovat dočasné přerušení léčby nebo snížení dávek, jak je uvedeno v Tabulce 4.
Tabulka 4 Doporučené úpravy dávky pro gastrointestinální toxicitu
|
Nežádoucí účinek |
Stupeň v den léčby |
Úprava zahajovací dávky panobinostatu |
Dávka panobinostatu při zotavení na stupeň < 1 |
Úprava zahajovací dávky bortezomibu |
Dávka bortezomibu při zotavení na stupeň < 1 |
|
Stupeň 2 navzdory protiprůj movému léčivému přípravku |
Vynechat dávku |
Pokračovat stejnou dávkou |
Vynechat dávku |
Pokračovat sníženou dávkou nebo změna na podání jednou týdně | |
|
Stupeň 3 navzdory protiprůjmovému léčivému přípravku |
Vynechat dávku |
Pokračovat sníženou dávkou |
Vynechat dávku |
Pokračovat sníženou dávkou nebo stejnou dávkou ale podávanou podle jednotýdenního rozpisu | |
|
Stupeň 4 navzdory protiprůj movému léčivému přípravku |
Trvale ukončit |
Trvale ukončit |
Při prvních známkách křečí břicha, řídké stolice nebo nového vzniku průjmu je doporučené, aby byli pacienti léčeni protiprůjmovým léčivým přípravkem (např. loperamid).
Pokud se navzdory podání antiemetik objeví případy nauzey stupně 3 nebo zvracení stupně 3 nebo 4, má být panobinostat dočasně vysazen a pří zotavení na stupeň 1 podávána snížená dávka.
Profalyktická antiemetika má lékař podávat dle svého uvážení v souladu s lokální lékařskou praxí (viz bod 4.4).
Neutropenie
Neutropenie může vyžadovat dočasné nebo trvalé snížení dávky. Pokyny pro přerušení léčby a snížení dávky panobinostatu jsou uvedeny v Tabulce 5.
Tabulka 5 Doporučené úpravy dávky pro neutropenii
|
Stupeň neutropenie v den léčby |
Úprava zahajovací dávky panobinostatu |
Dávka panobinostatu při zotavení na neutropenii stupně 2 (<1,5-1,0 x 109/l) |
Úprava zahajovací dávky bortezomibu |
Dávka bortezomibu při zotavení na neutropenii stupně 2 (<1,5-1,0 x 109/l) |
|
Neutropenie stupně 3 (<1,0-0,5 x 109/l) |
Vysadit dávku |
Pokračovat stejnou dávkou |
Vysadit dávku |
Pokračovat stejnou dávkou |
|
Neutropenie stupně 4 (<0,5 x 109/l) nebo febrilní neutropenie (<1,0 x 109/l a horečka >38,5°C) |
Vysadit dávku |
Pokračovat sníženou dávkou |
Vysadit dávku |
Pokračovat stejnou dávkou |
Při případech neutropenie stupně 3 nebo 4 má lékař zvážit použití růstových faktorů (např. G-CSF) podle lokálních směrnic. Ukončení léčby má být zvažováno, pokud se stav neutropenie nezlepší ani po úpravě dávky a/nebo po zavedení léčby kolonie granulocytů stimulujícím faktorem s přihlédnutím k místní lékařské praxi a k doporučením léčby, a/nebo při případech závažných sekundárních infekcí.
Prodloužení QTc intervalu
Při výskytu případů prodlouženého QT intervalu před zahájením léčby panobinostatem (QTcF >480 ms ve výchozím stavu), má být zahájení léčby odloženo, dokud se průměr QTcF před podáním dávky nevrátí na hodnotu <480 ms. Dále mají být před zahájením léčby přípravkem Farydak upraveny všechny abnormální hodnoty draslíku, hořčíku nebo fosforu v plazmě (viz bod 4.4). V případě prodloužení QT během léčby:
• Dávka má být vynechána, pokud je QTcF >480 ms nebo nad 60 ms od výchozího stavu.
• Pokud je prodloužení QT vyřešeno během 7 dní, má léčba pokračovat předchozí dávkou při prvním výskytu nebo sníženou dávkou, pokud došlo k prodloužení QT opakovaně.
• Pokud není prodloužení QT vyřešeno během 7 dní, má být léčba ukončena.
• Pokud jakákoli QTcF hodnota přesahuje 500 ms, má být léčba přípravkem Farydak trvale ukončena.
Další nežádoucí účinky léku
U pacientů s projevy dalších závažných nežádoucích účinků, jiných než trombocytopenie, gastrointestinální toxicita, neutropenie nebo prodloužení QTc platí následující doporučení:
• Opakovaný výskyt toxicity CTC stupně 2 nebo toxicita CTC stupně 3 a 4 - vynechat dávku do návratu na CTC stupně < 1 a znovu zahájit léčbu sníženou dávkou.
• Opakovaná toxicita CTC stupně 3 nebo 4 - má být zvažováno další snížení dávky, dokud se nežádoucí příhoda nevyřeší na CTC stupeň <_1.
Zvláštní populace
Pacienti s poruchou funkcí ledvin
Plazmatická expozice panobinostatu není pozměněna u pacientů s nádorovým onemocněním a s lehkou až těžkou poruchou funkcí ledvin. Proto není úprava zahajovací dávky nutná. Panobinostat nebyl studován u pacientů v konečné fázi onemocnění ledvin (ESRD) nebo pacientů na dialýze (viz bod 5.2).
Pacienti s poruchou jaterních funkcí
Klinická studie u pacientů s nádorovým onemocněním a s poruchou jaterních funkcí prokázala, že plazmatická expozice panobinostatu vzrostla o 43 % (1,4násobně) u pacientů s lehkou poruchou jaterních funkcí respektive o 105 % (2násobně) u pacientů se středně těžkou poruchou jaterních funkcí. U pacientů s lehkou poruchou jaterních funkcí by se měla léčba zahajovat sníženou dávkou panobinostatu 15 mg během prvního cyklu léčby. Na základě tolerability pacienta může být zvažováno zvýšení dávky z 15 mg na 20 mg. Pacienti se středně těžkou poruchou jaterních funkcí by měli zahájit léčbu panobinostatem během prvního cyklu léčby sníženou dávkou 10 mg. Při pacientově tolerabilitě je možné zvažovat zvýšení dávky z 10 mg na 15 mg. Četnost sledování těchto pacientů má být během léčby panobinostatem zvýšena, zejména během fáze zvýšení dávky. Panobinostat nemá být podáván pacientům s těžkou poruchou funkce jater z důvodu nedostatku zkušeností a bezpečnostních údajů v této populaci. Také by měla být zvážena úprava dávky bortezomibu (viz SmPC bortezomibu a Tabulka 6).
Tabulka 6 Doporučené úpravy zahajovací dávky pro pacienty s poruchou jaterních funkcí
|
Stupeň poruchy jaterních funkcí* |
Hladina bilirubinu |
Hladiny SGOT (AST) |
Úprava zahajovací dávky panobinostatu |
Úprava zahajovací dávky bortezomibu |
|
Lehká |
<1,0 x ULN |
>ULN |
Snižte dávku panobinostatu na 15 mg v prvním léčebném cyklu. Zvažte zvýšení dávky až na 20 mg v následných cyklech na základě tolerability pacienta. |
Žádná |
|
>1,0 x ULN a <1,5 x ULN |
Jakékoli | |||
|
Středně těžká |
>1,5 x ULN a <3,0 x ULN |
Jakékoli |
Snižte dávku panobinostatu na 10 mg v prvním léčebném cyklu. Zvažte zvýšení dávky až na 15 mg v následných cyklech na základě pacientovy tolerability. |
Snižte dávku bortezomibu na 0,7 mg/m2 v prvním léčebném cyklu. Zvažte zvýšení dávky na 1,0 mg/m2 nebo další snížení dávky na 0,5 mg/m2 v následných cyklech na základě pacientovy tolerability. |
SGOT = sérová glutamát-oxalacetát transamináza; AST = aspartátaminotransferáza ULN = horní hranice normálního rozmezí * Na základě NCI-CTEP klasifikace
Starší populace
U pacientů starších 65 let byly vybrané nežádoucí účinky a ukončení léčby z důvodu výskytu nežádoucích účinků četnější. Doporučuje se častěji sledovat pacienty starší 65 let, zejména výskyt trombocytopenie a gastrointestinální toxicity (viz body 4.4 a 4.8).
U pacientů starších 75 let má být podle celkového stavu pacienta a konkomitantních onemocnění zvážena úprava zahajovacích dávek nebo rozvrhu složek kombinovaného režimu. Léčba panobinostatem může být zahájena dávkou 15 mg, a pokud je tolerována v prvním cyklu, může být zvýšena na 20 mg v druhém cyklu. Podávání bortezomibu může být zahájeno dávkou 1,3 mg/m2 jednou týdně ve dnech 1 a 8 a dexamethazon dávkou 20 mg ve dnech 1 a 8.
Pediatrická populace
Nejsou dostupné žádné údaje pro použití panobinostatu u pediatrických pacientů mladších 18 let v indikaci mnohočetný myelom (viz bod 5.2).
Silné inhibitory CYP3A4
U pacientů, kteří užívají konkomitantní léčivé přípravky, které jsou silnými inhibitory CYP3A a/nebo Pgp, jako jsou například ketokonazol, itrakonazol, vorikonazol, ritonavir, sachuinavir, telithromycin, posakonazol a nefazodon, má být dávka panobinostatu snížena na 10 mg (viz bod 4.5). Pokud je vyžadovaná kontinuální léčba silnými inhibitory CYP3A4, může být na základě tolerability pacienta zvažováno zvýšení dávky z 10 mg na 15 mg panobinostatu.
U pacientů s poruchou jaterních funkcí užívajících konkomitantní léčivé přípravky, které jsou silnými inhibitory CYP3A4, má být léčba vyloučena z důvodu nedostatku zkušeností a bezpečnostních dat v této pacientské populaci.
Léčba silnými inhibitory CYP3A nemá být zahajována u pacientů, kteří již užívali sníženou dávku panobinostatu z důvodu výskytu nežádoucích účinků. Pokud je to nevyhnutelné, měli by být pacienti pečlivě sledováni a v případě klinické potřeby může být zvažováno další snížení dávky nebo přerušení léčby (viz bod 4.5).
Způsob podání
Farydak se podává perorálně jednou denně pouze ve stanovených dnech, každý den ve stejnou dobu. Tobolky mají být spolknuty celé s vodou, s jídlem nebo nalačno (viz bod 5.2) a nesmí být otevřeny, drceny nebo rozkousány. Pokud dojde k opomenutí dávky, může být užita až do 12 hodin po specifikované době užívání. Pokud dojde ke zvracení, neměl by pacient užívat doplňkovou dávku, ale měl by užít až další obvyklou předepsanou dávku.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Kojení (viz bod 4.6).
4.4 Zvláštní upozornění a opatření pro použití
Panobinostat se používá při kombinované léčbě, proto musí být před zahájením léčby panobinostatem konzultovány preskripční informace bortezomibu a dexamethazonu.
Pokles počtů krevních elementů
U pacientů léčených panobinostatem byly hlášené hematologické nežádoucí účinky, zahrnující závažné trombocytopenie, neutropenie a anémie (CTC stupně 3 až 4) Proto musí být před zahájením léčby panobinostatem provedeno vyšetření krevního obrazu, s častým sledováním během léčby (zejména před každou injekcí bortezomibu, jak uvádí SPC bortezomibu).
Počet krevních destiček by měl být >100 x 109/l a celkový počet neutrofilů >1,0 x 109/l před zahájením léčby. Počet destiček by měl být >100 x 109/l před zahájením jakéhokoli cyklu léčby (viz bod 4.2).
Ve studii fáze III, se trombocytopenie typicky obnovila na výchozí stav zahájením dalšího 21denního cyklu (viz Obrázek 1). Medián doby do výskytu trombocytopenie stupně 3 a 4 byl jeden měsíc a medián doby do zotavení byl 12 dní.
Obrázek 1
Medián počtu krevních destiček v průběhu času (Studie D2308, Bezpečnostní data, cykly 1-8)
Počet krevních destiček 10 x 109/l
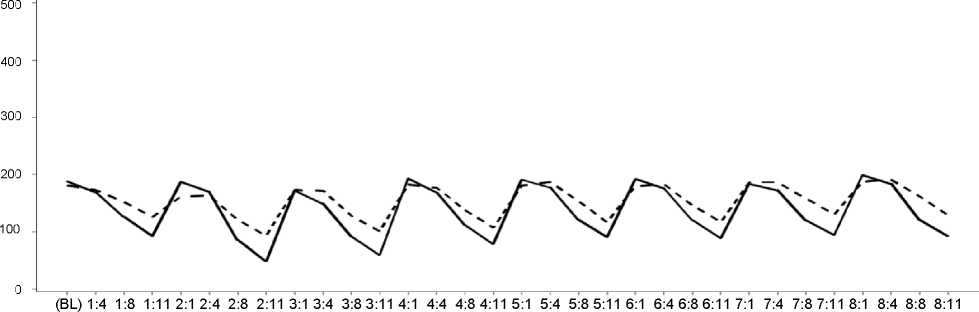
-PAN+BTZ+Dex — — PBO+BTZ+Dex Cyklus:Den
PAN+BTZ+Dex n= 381 370 367 363 351 342 335 320 323 310 298 283 285 265 268 250 253 232 238 225 230 206 206 199 205 183 188 175 185 159 170 155
PBO+BTZ+Dex n= 377 364 368 365 357 349 340 339 335 325 313 302 308 292 292 279 275 255 259 241 251 234 242 203 233 209 217 204 211 192 194 188
PAN=panobino stat BTZ= bortezomib Dex = dexamethazon
U pacientů s trombocytopenií CTC stupně 3 (počet krevních destiček <50 x 109/l s krvácením) může být nezbytné dočasně přerušit léčbu panobinostatem a/nebo snížit následující dávku. Podle klinické potřeby mohou být vyžadovány transfuze krevních destiček (viz body 4.2 a 4.8).
Krvácení
Během léčby panobinostatem bylo u pacientů hlášené krvácení. Krvácení CTC stupně 3 nebo 4 bylo hlášené u 4,2 % pacientů, včetně případů gastrointestinálního a plicního krvácení s fatálními následky. Proto si mají být lékaři a pacienti vědomi zvýšeného rizika trombocytopenie a možností krvácení, zejména u pacientů s poruchou koagulace nebo u pacientů, kteří užívají chronickou antikoagulační terapii.
Infekce
U pacientů užívajících panobinostat byly hlášené lokalizované a systémové infekce, včetně pneumonie, dalších bakteriálních onemocnění, invazivních plísňových infekcí jako je aspergilóza nebo kandidóza a virových infekcí včetně virové hepatitidy B a herpes simplex. Některé z těchto infekcí (např. pneumonie) byly závažné (např. vedly k sepsi, nebo respiračnímu nebo multiorgánovému selhání) a měly velmi závažné následky (viz bod 4.8). Zatímco byla neutropenie stupně 3 a stupně 4 pozorována u 28 %, respektive 7 % pacientů, febrilní neutropenie byla pozorována u 1 % pacientů (viz bod 4.8). Lékaři a pacienti by si měli být vědomi zvýšeného rizika infekce při léčbě panobinostatem.
Léčba přípravkem Farydak nemá být zahájena u pacientů s aktivními infekcemi. Preexistující infekce mají být před zahájením terapie vyléčeny. U pacientů mají být během léčby panobinostatem sledovány příznaky a symptomy infekcí. Pokud je diagnostikována infekce, má být neprodleně zavedena protiinfekční léčba a má být zvažováno vysazení nebo ukončení léčby přípravkem Farydak.
Pokud je diagnostikována invazivní systémová plísňová infekce, má být ukončena léčba panobinostatem a zavedena vhodná protiplísňová léčba.
Gastrointestinální poruchy
Závažná nauzea, průjem, zácpa a zvracení, které někdy vyžadovaly použití antiemetických a protiprůjmových léčivých přípravků, byly hlášené u pacientů léčených přípravkem Farydak (viz bod 4.8). Během léčby má být periodicky sledováno množství tekutin a hladiny elektrolytů v krvi, zejména draslíku, hořčíku a fosfátu a upraveno dle klinické potřeby aby se zamezilo potenciální dehydrataci a poruše rovnováhy elektrolytů (viz bod 4.2).
Lékař může profylakticky použít dle svého uvážení antiemetika (např. prochlorperezin) podle lokální lékařské praxe. Antiemetické přípravky se známým rizikem prodloužení QT intervalu jako je dolasetron, granisetron, ondansetron a tropisetron mají být užívány s opatrností (viz bod 4.5).
Při prvních příznacích břišních křečí, řídké stolice nebo výskytu průjmu je doporučeno, aby byli pacienti léčeni protiprůjmovými léčivými přípravky (např. loperamid) nebo doplňkovou léčbou v souladu místními léčebnými zvyklostmi. Pokud je to vhodné, má být užita intravenózní náhrada tekutin a elektrolytů. Léčivé přípravky s projímavými účinky mají být užívány s opatrností z důvodů potenciálního výskytu prohloubení průjmu. Pacienti by měli být poučeni o možnosti prodiskutovat použití laxativ se svým lékařem.
Změny na elektrokardiografu
Panobinostat může prodloužit srdeční ventrikulární repolarizaci (QT interval) (viz bod 5.3).
V klinické studii fáze III s dávkou 20 mg přípravku Farydak v kombinaci s bortezomibem a dexamethazonem nebyly hlášené žádné epizody prodloužení QTcF >500 ms. Poolovaná klinická data získaná od 500 pacientů léčených pouze panobinostatem ve více indikacích a při různých dávkách prokázala, že výskyt prodloužení QTc CTC stupně 3 (QTcF >500 ms) byl souhrnně přibližně 1 % a 5 % či více při dávce 60 mg nebo vyšší; nebyly pozorovány žádné epizody torsades de pointes.
Další analýzy naznačují, že riziko prodloužení QTc v průběhu času nevzrůstá (viz bod 4.2).
QTcF má být <480 ms před zahájením léčby přípravkem Farydak.
Ve výchozím stavu a periodicky během léčby má být prováděno náležité vyšetření hladin elektrolytů (např. draslík, hořčík a fosfor), zejména u pacientů se závažným gastrointestinálním nežádoucím účinkem (viz bod 4.2).
Přípravek Farydak má být užíván s opatrností u pacientů, kteří již měli nebo kteří jsou ve významném riziku vzniku prodloužení QTc. Zahrnuje pacienty:
• se syndromem prodloužení QT intervalu.
• s nekontrolovaným nebo signifikantním onemocněním srdce, včetně recentního infarktu myokardu, městnavého selhávání srdce, nestabilní anginy nebo klinicky významné bradykardie.
Souběžné podávání léčivých přípravků, u nichž je známý vliv na prodloužení QTc by mělo být užíváno s opatrností (viz bod 4.5).
Při konkomitantním užívání látek, které mohou zvýšit plazmatické koncentrace panobinostatu, jako jsou silné inhibitory CYP3A4, je vyžadovaná úprava dávky (viz body 4.5 a 4.2).
Hepatotoxicita
Během léčby panobinostatem byly u pacientů hlášené jaterní dysfunkce, především mírná přechodná zvýšení aminotransferáz a celkového bilirubinu.
Před léčbou a pravidelně během léčby mají být sledovány jatemí funkce. Pokud výsledky testů jatemích funkcí vykazují abnormality podle klasifikace NCI-CTEP, doporučují se úpravy dávky pro pacienty s mírnou a středně závažnou poruchou jaterních funkcí a pacienti by v jejich užívání měli pokračovat, dokud se hodnoty nevrátí k normálním hladinám nebo hladinám před léčbou. Panobinostat by neměl být podáván pacientům s těžkou poruchou funkce jater, protože o podání v této populaci chybí zkušenostní a bezpečnostní data. Také má být zvažovaná úprava dávky bortezomibu (viz SPC bortezomibu a Tabulka 6).
Starší populace
Doporučuje se sledovat pacienty starší 65 let častěji, především trombocytopenii a gastrointestinální toxicitu (viz bod 4.8 a bod 4.2).
Pro pacienty ve věku >75 let, v závislosti na celkovém stavu pacienta a konkomitantních onemocněních, může být zvažovaná úprava zahajovacích dávek nebo rozvrhu dávkování komponent kombinovaného režimu (viz bod 4.2).
Silné induktory CYP3A4
Silné induktory mohou snížit účinnost panobinostatu, proto má být vyloučeno konkomitantní užívání silných induktorů CYP3A4 zahrnujících například karbamazepin, fenobarbital, fenytoin, rifabutin, rifampicin a třezalkou tečkovanou (Hypericum perforatum) (viz bod 4.5).
Ženy ve fertilním věku
Ženy ve fertilním věku užívající panobinostat v kombinaci s bortezomibem a dexamethazonem musí tři měsíce po ukončení léčby používat vysoce účinnou metodu antikoncepce (viz body 4.5 a 4.6 a SPC bortezomibu a dexamethazonu). Ženy užívající hormonální antikoncepci by měly doplňkově využívat bariérovou metodu antikoncepce.
Hypotyreóza
Známky hypotyreózy vykazovalo 8 z 381 pacientů léčených panobinostatem v kombinaci s bortezomibem a dexamethazonem ve studii D2308, z nichž 2 pacienti se podrobili následné léčbě. Funkce hypofýzy a štítné žlázy by měla být sledována měřením hladiny hormonů (např. volného T4 a TSH), pokud je to klinicky indikováno (viz bod 4.2).
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Metabolismus přípravku Farydak probíhá jak pomocí drah zprostředkovaných CYP, tak i bez účasti CYP. Přibližně 40 % panobinostatu je metabolizováno CYP3A4. Podíl metabolismu pomocí CYP2D6 a 2C19 byl menší. Proto léčivé přípravky, které mohou ovlivnit aktivitu CYP3A4 enzymů, mohou změnit farmakokinetiku panobinostatu. Panobinostat je P-gp substrát.
Látky, které mohou zvýšit plazmatické koncentrace panobinostatu
Souběžné podání jednorázové dávky 20 mg panobinostatu s ketokonazolem, silným inhibitorem CYP3A, zvýšila Cmax a AUC panobinostatu 1,6násobně, respektive 1,8násobně, oproti samotnému podání panobinostatu.
U pacientů, kteří užívají konkomitantní léčivé přípravky, které jsou silnými inhibitory CYP3A a/nebo Pgp, které zahrnují například ketokonazol, itrakonazol, vorikonazol, ritonavir, sachuinavir, telithromycin, posakonazol a nefazodon, má být dávka panobinostatu snížena (viz bod 4.2).
Pacienti by měli být poučeni o tom, aby se vyhýbali konzumaci karamboly, grapefruitu, grapefruitové šťávy, granátového jablka a šťávy z granátových jablek, protože je u nich prokázaná inhibice enzymů cytochromu P450 3A a mohou zvýšit biologickou dostupnost panobinostatu.
Látky, u nichž se očekává snížení koncentrací panobinostatu
Podíl panobinostatu metabolizovaného CYP3A4 je přibližně 40 %. V klinických studiích s mnohočetným myelomem byla expozice panobinostatu snížená přibližně o 20 % souběžným podáním dexamethazonu, což je mírný/středně silný induktor CYP3A4 závislý na dávce. U silných induktorů se očekávají silnější účinky a možnost snížení účinnosti panobinostatu, proto by mělo být vyloučeno konkomitantní užívání silných induktorů CYP3A4 včetně například karbamazepinu, fenobarbitalu, fenytoinu, rifabutinu, rifampicinu a třezalky tečkované (Hypericum perforatum).
Látky, jejich plazmatické koncentrace mohou být panobinostatem zvýšeny
Panobinostat zvýšil Cmax a AUC dextromethorfanu (substrát CYP2D6) 1,8násobně, respektive 1,6násobně a nelze vyloučit, že účinek na citlivější substrát CYP2D6 může být vyšší. Vyhněte se užívání panobinostatu u pacientů, kteří užívají substráty CYP2D6 s úzkým terapeutickým indexem (jde například o pimozid). Při souběžném podávání přípravku Farydak s citlivými substráty CYP2D6 (např. atomoxetin, dextromethorfan, metoprolol, nebivolol, perfenazin, a pimozid) titrujte dávky jednotlivých substrátů CYP2D6 na základě tolerability a častého sledování pacientů z důvodu výskytu nežádoucích účinků.
Látky, jejichž plazmatická expozice může být snížená panobinostatem Přípravky hormonální antikoncepce
V současné době není známo, zda může panobinostat snížit účinnost přípravků hormonální antikoncepce. Pokud je panobinostat podáván spolu s dexamethazonem, který je známým slabým až středně silným induktorem CYP3A4 stejně jako jiných enzymů a transportérů, musí být zvažováno riziko snížené účinnosti antikoncepce. Ženy užívající hormonální antikoncepci by měly doplňkově využívat bariérovou metodu antikoncepce.
Protože nejsou dostupná žádná data, nelze vyloučit, že by panobinostat mohl být slabým induktorem enzymu CYP3A4 v gastrointestinálním traktu. To může potenciálně vést k mírně snížené expozici k citlivým substrátům CYP3A4.
Očekávané farmakodynamické interakce
Prodloužení QT intervalu
Na základě preklinických a klinických dat bylo zjištěno, že panobinostat má potenciál prodloužit QT interval. Souběžné užívání antiarytmických léčivých přípravků (včetně například amiodaronu, disopyramidu, prokainamidu, chinidinu a sotalolu) a dalších látek, o nichž je známo, že prodlužují QT interval (včetně například chlorochinu, halofantrinu, klarithromycinu, metadonu, moxifloxacinu, bepridilu a pimozidu) není doporučeno. Antiemetické léčivé přípravky se známým rizikem prodloužení QT, jako je dolasetron, granisetron, ondansetron a tropisetron mají být užívány s opatrností (viz bod 4.4).
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku/Antikoncepce mužů a žen
Na základě nálezů u zvířat je možné předpovídat s vysokou pravděpodobností, že panobinostat při podání těhotným ženám zvyšuje riziko úmrtí plodu a vývojových abnormalit kostry. Ženám ve fertilním věku má být před zahájením léčby přípravkem Farydak proveden těhotenský test a ženy musí během léčby a tři měsíce po podání poslední dávky přípravku Farydak používat vysoce účinnou metodu antikoncepce. Ženy užívající hormonální antikoncepci by měly doplňkově využívat bariérovou metodu antikoncepce.
Díky svému cytostatickému/cytotoxickému modu působení může panobinostat ovlivnit kvalitu spermií tvořených během léčby. Sexuálně aktivní muži užívající přípravek Farydak a jejich ženské partnerky by měly používat vysoce účinnou metodu antikoncepce během léčby muže a po dobu šesti měsíců od poslední dávky jeho léčby přípravkem Farydak.
Pokud je panobinostat podáván společně s dexamethazonem, který je známým slabým až středně silným induktorem CYP3A4 stejně jako jiných enzymů a transportérů, musí být zváženo riziko snížené účinnosti hormonální antikoncepce. Dále není v současné době známo, zda může panobinostat snížit efektivitu hormonální antikoncepce a proto by měly ženy užívající hormonální antikoncepci používat doplňkovou metodu v podobě bariérové antikoncepce.
Klinické studie zabývající se užíváním přípravku Farydak u těhotných pacientek nebyly provedeny. Studie na zvířatech prokázaly reprodukční a embryofetální toxicitu (viz bod 5.3). Vzhledem k cytostatickému/cytotoxickému modu účinkování panobinostatu je potenciální riziko pro plod vysoké. Přípravek Farydak by měl být užíván během těhotenství pouze, pokud očekávaný prospěch převažuje možná rizika pro plod. Pokud se užívá během těhotenství, nebo pokud pacientka otěhotní během jeho užívání, musí být pacientka informována o potenciálním riziku pro plod.
Kojení
Není známo, zda se panobinostat vylučuje do lidského mateřského mléka. Na základě cytostatického/cytotoxického mechanismu účinku je kojení během léčby přípravkem Farydak kontraindikováno (viz bod 4.3).
Fertilita
Na základě neklinických nálezů lze usuzovat na možné oslabení fertility mužů léčbou přípravkem Farydak (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek Farydak může mít malý vliv na schopnost řídit a obsluhovat stroje. Po podání přípravku Farydak se může objevit závrať (viz bod 4.8).
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Bezpečnostní data panobinostatu byla vyhodnocena na základě zhodnocení 451 pacientů s mnohočetným myelomem léčených panobinostatem v kombinaci s bortezomibem a dexamethazonem a 278 pacientů léčeným panobinostatem v monoterapii.
Níže uvedená bezpečnostní data jsou založená na klinické studii fáze III (Panorama 1) u 381 pacientů s mnohočetným myelomem léčených 20 mg panobinostatu jednou denně třikrát týdně, v režimu 2 týdny léčba a 1 týden vysazení léčby v kombinaci s bortezomibem a dexamethazonem.
Medián trvání expozice ve studii byl 5,0 měsíce. 15,7 % pacientů bylo exponováno studijní léčbě po dobu > 48 týdnů.
Nej častější nehematologické nežádoucí účinky byly průjem, únava, nauzea a zvracení.
Hematologická toxicita závislá na léčbě zahrnuje trombocytopenii, anémii, neutropenii a lymfopenii.
QTcF >480 a <500 ms byl zaznamenán u 1,3 % pacientů, změna od výchozího stavu >60 ms byla pozorována u 0,8 % pacientů. Žádný pacient neměl absolutní QTcF vyšší než 500 ms.
Srdeční příhody (nejčastěji fibrilace síní, tachykardie, palpitace a sinusová tachykardie) byly hlášené u 17,6 % pacientů léčených panobinostatem + bortezomibem + dexamethazonem oproti 9,8 % u pacientů léčených placebem + bortezomibem + dexamethazonem a případy synkopy byly hlášené u 6,0 % oproti 2,4 %.
Ukončení z důvodu výskytu nežádoucích účinků, bez ohledu na kauzalitu, bylo pozorováno u 36,2 % pacientů. Nejčastější nežádoucí účinky (NÚ), které vedly k ukončení léčby, byl průjem (4,5 %), astenie a slabost (2,9 % každý z nich) a pneumonie (1,3 %).
Úmrtí během léčby nezaviněné studijní indikací (mnohočetný myelom) bylo hlášené u 6,8 % pacientů léčených panobinostatem + bortezomibem + dexamethazonem oproti 3,2 % pacientů léčených placebem + bortezomibem + dexamethazonem.
Přehled nežádoucích účinků z klinických studií v tabulce
Nežádoucí účinky ze studie fáze III (Panorama 1) jsou uvedené v Tabulce 7. Nežádoucí účinky jsou seřazeny podle MedDRA systému orgánových tříd. V rámci každého systému orgánových tříd jsou nežádoucí účinky řazené dle četnosti, nejčastější nežádoucí účinky jsou řazeny jako první. V každé skupině četností jsou nežádoucí účinky uvedené v pořadí podle klesající závažnosti. Dále jsou pro každý nežádoucí účinek přiřazeny korespondující kategorie četností klasifikované dle následujících pravidel (CIOMS III): velmi časté (>1/10); časté (>1/100 až <1/10); méně časté (>1/1000 až <1/100); vzácné (>1/10000 až <1/1000); velmi vzácné (<1/10000); a není známo (z dostupných údajů nelze určit).
Tabulka 7 Nežádoucí účinky zjištěné u pacientů s mnohočetným myelomem ve studii fáze III
|
Třídy orgánových systémů |
Četnost |
Nežádoucí účinky |
|
Infekce a infestace |
Velmi časté |
Infekce horních cest dýchacích, pneumonie |
|
Časté |
Septický šok, infekce močových cest, virové infekce, ústní herpes, kolitida způsobená Clostridium difficile, zánět středního ucha, celulitida, sepse, gastroenteritida, infekce dolních cest dýchacích, kandidóza | |
|
Méně časté |
Plísňová pneumonie, hepatitida B, aspergilóza | |
|
Poruchy krve a lymfatického systému a |
Velmi časté |
Pancytopenie, trombocytopenie, anemie, leukopenie, neutropenie, lymfopenie |
|
Endokrinní poruchy |
Časté |
Hypotyroidismus |
|
Poruchy metabolismu a výživy |
Velmi časté |
Snížená chuť k jídlu, hypofosfatémie a, hyponatrémie a, hypokalémie a |
|
Časté |
Hyperglykémie, dehydratace, hypoalbuminémie, zadržování tekutin, hyperurikémie, hypokalcémie, hypomagnezémie | |
|
Psychiatrické poruchy |
Velmi časté |
Insomnie |
|
Poruchy nervového systému |
Velmi časté |
Závrať, bolest hlavy |
|
Časté |
Intrakraniální krvácení, synkopa, třes, dysgeuzie | |
|
Poruchy oka |
Časté |
Krvácení do spojivek |
|
Srdeční poruchy |
Časté |
Bradykardie, fibrilace síní, sinusová tachykardie, tachykardie, palpitace |
|
Méně časté |
Infarkt myokardu | |
|
Cévní poruchy |
Velmi časté | |
|
Časté |
Hypertenze, hematom, ortostatická hypotenze | |
|
Méně časté |
Hemorrhagický šok |
|
Respirační, hrudní a mediastinální poruchy |
Velmi časté | |
|
Common |
Respirační selhání, chrůpky, sípot, epistaxe | |
|
Méně časté |
Plicní krvácení, hemoptýza | |
|
Gastrointestinální poruchy |
Velmi časté | |
|
Časté |
Gastrointestinální krvácení, hematochezie, gastritida, cheilitida, břišní distenze, sucho v ústech, plynatost | |
|
Méně časté |
Kolitida, hematemeza, gastrointestinální bolest | |
|
Poruchy jater a žlučových cest |
Časté |
Neobvyklé výsledky jaterních funkcí, hyperbilirubinémie a |
|
Poruchy kůže a podkožní tkáně |
Časté |
Kožní léze, vyrážka, erytém |
|
Méně časté |
Petechie | |
|
Poruchy svalů a pojivových tkání |
Časté |
Otoky kloubů |
|
Poruchy ledvin a močových cest |
Časté |
Selhání ledvin, hematurie, inkontinence moči |
|
Celkové poruchy a reakce v místě aplikace |
Velmi časté |
Únava, periferní edém, pyrexie, astenie |
|
Časté | ||
|
Vyšetření |
Velmi časté |
Snížení tělesné hmotnosti |
|
Časté |
Zvýšená hladina močoviny v krvi, snížená rychlost glomerulární filtrace, zvýšená hladina alkalické fosfatázy v krvi, prodloužený elektrokardiogram QT, zvýšená hladina kreatininu v krvi a, zvýšená SGPT alanintransaminázy (ALT) a, zvýšená SGOT aspartáttransaminázy (AST) a |
četnost je založená na laboratorních hodnotách
Popis vybraných nežádoucích účinků
Gastrointestinální
Gastrointestinální toxicita, především průjem, nauzea a zvracení, patří mezi nejčastěji hlášené nežádoucí účinky. Nicméně ukončení léčby způsobené těmito účinky bylo hlášené u relativně malé části pacientů, s průjmem u 4,5 % a nauzeou a zvracením u 0,5 % pro každý z nich. Pacienti mají být instruováni tak, aby kontaktovali svého lékaře, pokud se u nich objeví závažná gastrointestinální toxicita a může být vyžadovaná úprava dávky nebo ukončení léčby (viz bod 4.4).
Trombocytopenie
Kvůli vlastnostem mnohočetného myelomu a známé hematotoxicitě panobinostatu a jeho léku v kombinaci bortezomibu byla často hlášená trombocytopenie, která byla často závažná. Trombocytopenie CTC stupně 3 nebo 4 se objevila u 256 pacientů, s mediánem doby výskytu jeden měsíc. Nicméně trombocytopenie je reverzibilní (medián doby do zotavení 12 dní) a může být obvykle zvládnuta úpravou dávky a přerušením léčby s podáním transfúze krevních destiček nebo bez podání krevních destiček (viz bod 4.4). U 33,3 % pacientů v rameni s panobinostatem + bortezomibem + dexamethazonem a u 10,3 % pacientů v rameni s placebem + bortezomibem + dexamethazonem byly podány během léčby transfúze krevních destiček.
Trombocytopenie vzácně vede k ukončení léčby (1,6 % pacientů). U většiny pacientů s trombocytopenií nedošlo ke krvácení. 20,7 % pacientů zažilo krvácení, nejčastěji epistaxi (4,7 %), hematom (2,6 %) a krvácení do spojivek (2,1 %). Krvácení CTC stupně 3 nebo 4 bylo hlášeno u 4,2 % pacientů, včetně nejčastějšího gastrointestinálního krvácení. Pět pacientů (1,3 %) zemřelo na události spojené s krvácením. Mezi pacienty, kteří zemřeli na krvácení, měl jeden pacient trombocytopenii stupně 4, tři pacienti měli trombocytopenii stupně 3 a 1 pacient měl trombocytopenii stupně 1.
Neutropenie
Neutropenie byla často hlášená na základě laboratorních nálezů zjištěných během studie (všechny stupně: 75 %). Většina nově se vyskytujících případů závažné neutropenie byla stupně 3 (28 %), s podstatně nižším počtem případů stupně 4 (6,6 %). Zatímco se u mnoha pacientů vyvinula neutropenie, febrilní neutropenie se vyskytovala pouze u části léčených pacientů (1,0 % u všech CTC stupňů i u stupňů 3 a 4). Pacienti s neutropenií jsou náchylní k infekci, zejména horních cest dýchacích nebo pneumonii. Pouze u 0,3 % pacientů byla ukončena léčba z důvodu výskytu neutropenie.
Únava a astenie
Únava a astenie byla hlášená u 41,2 % respektive 22,0 % pacientů. Slabost CTC stupně 3 byla hlášena u 15,7 % pacientů, stupeň 4 u 1,3 %. Astenie stupně 3 byla pozorována u 9,4 % pacientů, u žádného z pacientů se neprojevila astenie CTC stupně 4. U 2,9 % pacientů byla léčba ukončena z důvodu výskytu únavy a astenie.
Infekce
Pacienti s recidivujícím nebo refrakterním mnohočetným myelomem jsou ohroženi infekcemi. Možné přispívající faktory zahrnují předchozí chemoterapii, transplantaci kmenových buněk, podstatu onemocnění samotného a neutropenii nebo lymfopenii spojené s léčbou přípravkem Farydak. Nejčastěji hlášené nežádoucí účinky zahrnují infekci horních cest dýchacích, pneumonii a nazofaryngitidu. Byly hlášené fatální příhody zahrnující pneumonii nebo sepsi. Ukončení léčby způsobené infekcí bylo hlášeno u 5 % pacientů.
Prodloužení QT intervalu a abnormality EKG
Bylo pozorováno prodloužení QTc, většinou mírné: QTcF interval > 450 ms a < 480 ms byl hlášen u 10,8 % pacientů, s maximem zvýšení z výchozího stavu > 30 ms a < 60 ms u 14,5 % pacientů. QTcF > 500 ms nebyl hlášen u žádného pacienta. Tyto změny nebyly obecně spojené s klinickými příznaky a jejich klinický význam proto není známý.
Abnormality EKG (elektrokardiogramu) byly hlášeny u pacientů léčených panobinostatem+bortezomibem+dexamethazonem, šlo zejména o depresi úseku ST-T (21,7 %) a změny T vlny (39,6 %). Bez ohledu na chronologii událostí byla synkopa hlášená u 9 % pacientů s depresí úseku ST-T, u 7,2 % pacientů se změnou T vlny a u 4,9 % pacientů s žádnou z těchto abnormalit EKG. Podobně ischemické srdeční onemocnění (zahrnující infarkt myokardu a ischemii) bylo hlášené u 4,5 % pacientů s depresí úseku ST-T, u 4,8 % pacientů se změnou T vlny a u 2,7 % pacientů s žádnou z těchto abnormalit EKG.
Zvláštní populace
Starší populace
Výskyt úmrtí nespojených se studijní indikací byl 8,8 % u pacientů ve věku >65 let v porovnání s 5,4 % pacientů ve věku <65 let.
Nežádoucí účinky vedoucí k trvalému ukončení se vyskytovaly u 30 % pacientů ve věku <65 let, u 44 % pacientů ve věku 65-75 let a u 47 % pacientů ve věku >75 let. Případy stupně 3-4 se vyskytovaly častěji u pacientů a zahrnovaly následující: (procenta pro pacienty ve věku <65 let,
65-75 let a >75 let věku): trombocytopenie (60 %, 74 % a 91 %), anemie (16 %, 17 % a 29 %), průjem (21 %, 27 % a 47 %), a únava (18 %, 28 % a 47 %).
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Během klinických studií byly hlášené omezené zkušenosti s předávkováním. Pozorované nežádoucí účinky byly konzistentní s bezpečnostním profilem, s účinky zahrnujícími zejména hematologické a gastrointestinální poruchy jako je trombocytopenie, pancytopenie, průjem, nauzea, zvracení a anorexie. Při případech předávkování by mělo být provedeno srdeční sledování a vyhodnocení elektrolytů a počty krevních destiček a v případě potřeby poskytnuta podpůrná péče. Není známo, zda je panobinostat dialyzovatelný.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Jiné antineoplastické látky, ATC kód: L01XX42 Mechanismus účinku
Přípravek Farydak je inhibitor histonové deacetylázy (HDAC), který inhibuje enzymatickou aktivitu HDAC při nanomolárních koncentracích. Histonové deacetylázy (HDAC) katalyzují odstranění acetylových skupin z lyzinových reziduí histonů a některých nehistonových proteinů. Inhibice aktivity HDAC vede ke zvýšené acetylaci histonových proteinů, epigenetické alteraci, jejímž výsledkem je uvolnění chromatinu, vedoucí k aktivaci transkripce. In vitro způsobil panobinostat akumulaci acetylovaných histonů a jiných proteinů, čímž vyvolal zastavení buněčného cyklu a/nebo apoptózu některých pozměněných buněk. Zvýšené hladiny acetylovaných histonů byly pozorovány na štěpech z myší léčených panobinostatem. Panobinostat vykazuje vyšší toxicitu vůči nádorovým buňkám oproti normálním buňkám.
Farmakodynamické účinky
Léčba tumorových buněk panobinostatem vyústila k na dávce závislému zvýšení acetylace histonů H3 a H4 v modelech in vitro i v preklinických modelech zvířecích štěpů, prokazující tak cílovou inhibici. Navíc byla expozicí panobinostatu spuštěna zvýšená exprese tumor suprimujícího genu p21CDKNIA (cyklin dependentní inhibitor kinázy 1/p21), klíčového mediátoru zástavy a diferenciace fáze G1.
Klinická účinnost a bezpečnost
Klinická účinnost u pacientů s relabujícím a relabujícím a refrakterním mnohočetným myelomem (Studie D2308 - Panorama 1)
Účinnost a bezpečnost panobinostatu v kombinaci s bortezomibem a dexamethazonem byla hodnocena v randomizované, dvojitě zaslepené, placebem kontrolované, multicentrické studii fáze III u pacientů s relabujícím nebo relabujícím a refrakterním mnohočetným myelomem, kteří dostávali 1-3 předchozí linie léčby.
Pacienti užívali panobinostat (20 mg užívaných perorálně jednou denně třikrát za týden, v dávkovacím režimu 2 týdny léčby a 1 týden vysazení léčby) v kombinaci s bortezomibem (1,3 mg/m2 podávaných intravenózní injekcí) a dexamethazonem (20 mg). Léčba byla podávána maximálně v 16 cyklech (viz Tabulka 1 a 2).
Celkem 768 pacientů bylo randomizováno v poměru 1:1 do ramene panobinostat + bortezomib + dexamethazon (n=387) nebo ramene placebo + bortezomib + dexamethazon (n=381) stratifikovaných podle předchozího používání bortezomibu [Ano (n=336 (43,8 %)), Ne (n=432 (56,3 %))] a počtu předchozích linií terapie myelomu [1 předchozí linie (n=352 (45,8 %)), 2 až 3 předchozí linie (n=416 (54,2 %))]. Demografické charakteristiky a charakteristiky onemocnění ve výchozím stavu byly v ramenech studie vyvážené a srovnatelné.
Medián věku byl 63 let, rozmezí 28-84; 42,1 % pacientů bylo starších než 65 let. 53,0 % pacientů tvořili muži. Běloši tvořili 65,0 % studijní populace, Asiaté 30,2 % a černoši 2,9 %. ECOG performance status byl 0-1 u 93 % pacientů. Medián počtu předchozích terapií byl 1,0. Více než polovina (57,2 %) pacientů prodělala předchozí transplantaci kmenových buněk a 62,8 % pacientů recidivovalo po předchozí antineoplastické terapii (např. melfalan 79,6 %, dexamethazon 81,1 %, thalidomid 51,2 %, cyklofosfamid 45,3 %, bortezomib 43,0 %, kombinace bortezomibu a dexamethazonu 37,8 %, lenalidomid 20,4 %). Více než jedna třetina (35,8 %) pacientů byla v recidivě a neodpovídala na předchozí léčbu.
Medián trvání sledování byl 28,75 měsíce v rameni panobinostat + bortezomib + dexamethazon a 29,04 měsíce v rameni placebo + bortezomib + dexamethazon.
Primárním cílovým parametrem bylo přežití bez progrese (progression free survival - PFS) podle modifikovaných kritérií Evropské skupiny pro transplantaci kostní dřeně (mEBMT) a stanovené zkoušejícím. V celkové populaci pacientů se PFS, založené na kompletní sadě analýz (FAS), mezi rameny léčby statisticky významně lišilo (stratifikovaný Log-rank test p<0,0001, s odhadovaným 37% snížením rizika v rameni panobinostat + bortezomib + dexamethazon oproti ramenu placebo + bortezomib + dexamethazon (poměr rizik: 0,63 (95% CI: 0,52; 0,76)). Medián PFS (95% CI) byl 12,0 měsíců (10,3; 12,9) respektive 8,1 měsíců (7,6; 9,2).
Celkové přežití (OS) byl klíčový sekundární cílový parametr. Rozdíl OS nebyl statisticky významný mezi dvěma léčebnými skupinami. Medián OS byl 40,3 měsíců v rameni panobinostat + bortezomib + dexamethazon a 35,8 měsíců v rameni placebo + bortezomib + dexamethazon (poměr rizik: 0,94 (95% CI: 0,78; 1,14)).
Mimo prespecifikovanou podskupinu pacientů s předchozí léčbou bortezomibem a imunomodulačním agens (N=193), užívalo 76 % pacientů nejméně dva předchozí režimy. V této podskupině pacientů (N=147) byl medián trvání léčby 4,5 měsíce v rameni s panobinostatem + bortezomibem + dexamethazonem a 4,8 měsíců v rameni s placebem + bortezomibem + dexamethazonem. Medián PFS (95% CI) byl 12,5 měsíce (7,26; 14,03) v rameni s panobinostatem + bortezomibem + dexamethazonem a 4,7 měsíců (3,71; 6,05) v rameni s placebem + bortezomibem + a dexamethazonem [HR: 0,47 (0,31; 0,72)]. Tito pacienti měli medián počtu přechozích terapií 3. Výsledky účinnosti shrnuje Tabulka 8 a Kaplan-Meierovy křivky pro PFS jsou uvedené na Obrázku 2.
Tabulka 8: Přežití bez progrese u pacientů, kteří dostávali nejméně dva předchozí režimy včetně bortezomibu a imunomodulačního léku
|
F arydak bortezomib a dexamethazon N=73 |
Placebo bortezomib a dexamethazon N=74 | |
|
Přežití bez progrese | ||
|
Medián, měsíce [95% CI] |
12,5 [7,26; 14,03] |
4,7 [3,71, 6,05] |
|
Poměr rizik [95% CI]1 |
0,47 (0,31; 0,72) | |
|
1 Poměr rizik získaný ze stratifikovaného Cox modelu | ||
Obrázek 2: Kaplan-Meierova křivka přežití bez progrese u pacientů s mnohočetným
myelomem, kteří dostávali nejméně dva předchozí režimy včetně bortezomibu a imunomodulačního léku
0
(/)
0
O)
O
100
80
N
0
-Q
>N
0
60
Poměr rizik= 0.47 95 % CI [0.31; 0.72]
Logrank p-hodnota=0.0003 Kaplan Meierovy mediány PAN+BTZ+Dex: 12.48 měsíce PBO+BTZ+Dex: 4.70 měsíce
A
40
_Q
O
~o
20
O
Cl
>0
■0
>
0
Časy cenzorování PAN+BTZ+Dex (n/N=44/73) PBO+BTZ+Dex (n/N=54/74)
0
|
0 |
2 |
4 |
6 |
8 |
10 |
12 |
14 |
16 |
18 |
20 |
22 |
24 |
26 |
28 |
30 | |
|
Počet pacientů v riziku |
Čas (měsíce) | |||||||||||||||
|
Čas (měsíce) |
0 |
2 |
4 |
6 |
8 |
10 |
12 |
14 |
16 |
18 |
20 |
22 |
24 |
26 |
28 |
30 |
|
PAN+BTZ+Dex |
73 |
57 |
42 |
36 |
32 |
25 |
20 |
15 |
10 |
6 |
4 |
3 |
2 |
2 |
1 |
0 |
|
PBO+BTZ+Dex |
74 |
54 |
37 |
23 |
11 |
9 |
5 |
4 |
2 |
2 |
2 |
2 |
2 |
0 |
0 |
0 |
PAN= panobinostat PBO= placebo BTZ= bortezomib Dex = dexamethazon
V podskupině pacientů, kteří dostávali nejméně dva předchozí režimy včetně bortezomibu a imunomodulačního léku (n=147) byl celkový výskyt odpovědi při použití EBMT kritérií 59 % v rameni s panobinostatem + bortezomibem + dexamethazonem a 39 % v rameni s placebem + bortezomibem + dexamethazonem. Výskyt odpovědi je shrnutý v Tabulce 9.
Tabulka 9: Výskyt odpovědi u pacientů s mnohočetným myelomem, kteří dostávali nejméně dva předchozí režimy včetně bortezomibu a imunomodulačního léku
|
F arydak bortezomib a dexamethazon N=73 |
Placebo bortezomib a dexamethazon N=74 | |
|
Celkové přežití |
43 (59 %) |
29 (39 %) |
|
[95% CI] |
(46,8; 70,3) |
(28; 51,2) |
|
Kompletní odpověď |
6 (8 %) |
0 |
|
Téměř kompletní odpověď |
10(14 %) |
6 (8 %) |
|
Částečná odpověď |
27 (37 %) |
23 (31 %) |
|
Klinická účinnost u pacientů s |
mnohočetným myelomem refrakterním k bortezomibu (Studie DUS71 - | |
Panorama 2)
Studie DUS71 byla dvojstupňová, jednoramenná, otevřená multicentrická studie fáze II s perorálním panobinostatem (20 mg) v kombinaci s bortezomibem (1,3 mg/m2) a dexamethazonem (20 mg) u 55 pacientů s relabujícím a refrakterním mnohočetným myelomem, kteří byli refrakterní k bortezomibu a dostávali nejméně dvě předchozí linie léčby. Pacienti byli exponováni IMiDy (lenalidomid nebo thalidomid). Refrakternost vůči bortezomibu byla definována jako progrese onemocnění na léčbě nebo během 60 dní od konce poslední linie léčby obsahující bortezomib.
Primárním cílovým parametrem studie bylo vyhodnocení míry celkového přežití (ORR) po 8 cyklech léčby podle mEBMT kritérií.
Pacienti byli intenzivně předléčeni a dostávali více předchozích režimů (medián: 4; rozmezí: 2-11). Všech 55 pacientů bylo předběžně léčeno bortezomibem a nejméně jedním IMiDem (lenalidomid:
98.2 %, thalidomid: 69,1 %). Většina pacientů podstoupila transplantaci (63,6 %).
Medián trvání expozice k léčbě ve studii byl 4,6 měsíce (rozmezí: 0,1-24,1 měsíce). Pacienti dosáhli ORR (> PR (částečná odpověď) ze 34,5 % a 52,7 % (>MR (minimální odpověď)). Medián doby do odpovědi byl 1,4 měsíce a medián trvání odpovědi byl 6,0 měsíce. Medián celkového přežití byl 17,5 měsíce.
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Farydak u všech podskupin pediatrické populace s mnohočetným myelomem (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Absorpce
Panobinostat je rychle a takřka kompletně absorbován s Tmax dosaženou během 2 hodin po perorálním podání u pacientů s pokročilým nádorovým onemocněním. Absolutní perorální biologická dostupnost panobinostatu byla přibližně 21 %. Po perorálním podání se farmakokinetika panobinostatu zdá být při rozmezí dávek 10-30 mg lineární, ale AUC vzrostla při vysokých dávkách méně než proporcionálně s dávkou.
Celková expozice panobinostatu a mezipacientská variabilita zůstala s jídlem či nalačno nezměněna, zatímco Cmax byla snížena o <45 % a Tmax se prodloužila o 1 až 2,5 hodiny s jídlem (tj. normální snídaně a snídaně s vysokým obsahem tuků). Protože jídlo nezměnilo celkovou biologickou dostupnost (AUC), může být panobinostat podáván u pacientů s nádorovým onemocněním bez ohledu na jídlo.
Distribuce
Panobinostat je mírně (přibližně 90 %) vázán na proteiny v lidské plazmě. Jeho frakce v erytrocytu je 0,60 in vitro, nezávisle na koncentraci. Objem distribuce panobinostatu v klidovém stavu (Vss), založený na odhadech konečných parametrů v analýzách populační farmakokinetiky, je přibližně 1000 litrů.
Biotransformace
Panobinostat je rozsáhle metabolizován a velká část dávky je metabolizována před dosažením systémové cirkulace. Související metabolické dráhy zapojené v biotransformaci panobinostatu jsou redukce, hydrolýza, oxidace a glukuronidace. Oxidativní metabolismus panobinostatu hrál méně významnou roli, touto drahou bylo eliminováno přibližně 40 % dávky. Cytochrom P450 3A4 (CYP3A4) je hlavní oxidační enzym, s potenciálním menším zapojením jsou enzymy CYP2D6 a 2C19.
Panobinostat představoval 6 až 9 % lékové expozice v plazmě. Mateřská látka je dle domněnek zodpovědná za celkovou farmakologickou aktivitu panobinostatu.
Eliminace
Po jednorázovém perorálním podání dávky [14C] panobinostatu pacientům se 29 až 51 % podané radioaktivity vylučovalo močí a 44 až 77 % stolicí. Nezměněný panobinostat tvořil < 2,5 % dávky v moči a < 3,5 % dávky ve stolici. Zbytky jsou metabolity. Clearance panobinostatu v ledvinách (CLr/F) se pohybovala v rozmezí od 2,4 po 5,5 l/h. Panobinostat má poločas terminální eliminace, založený na odhadu konečných parametrů v populaci PK analýzy, přibližně 37 hodin.
Zvláštní populace
Pediatrická populace
Panobinostat nebyl hodnocen u pacientů s mnohočetným myelomem mladších 18 let.
Starší populace
V klinické studii fáze III bylo zahrnuto 162 pacientů ve věku 65 let nebo více z celkového počtu 387 pacientů. Plazmatická expozice panobinostatu u pacientů ve věku 65 let nebo mladších byla podobná jako u pacientů starších než 65 let v poolovaných studiích nebo studiích s monoterapií s dávkami v rozmezí 10 mg až 80 mg.
Pacienti s poruchou jaterních funkcí
Vliv poruchy jaterních funkcí na farmakokinetiku panobinostatu byl vyhodnocen ve studii fáze I u 24 pacientů se solidními tumory a s různými stupni závažnosti poruchy jaterních funkcí. Lehká a středně těžká porucha jaterních funkcí dle NCI-CTEP klasifikace zvýšila plazmatickou expozici panobinostatu o 43 % respektive o 105 %. Pro pacienty s těžkou poruchou jaterních funkcí nejsou k dispozici žádná farmakokinetická data.
Pacienti s poruchou funkce ledvin
Vliv poruchy funkce ledvin na farmakokinetiku panobinostatu byl vyhodnocen ve studii fáze I u 37 pacientů s pokročilými solidními tumory s různými stupni poruchy funkce ledvin. Lehká, středně těžká a těžká porucha funkce ledvin ve výchozím stavu, zjištěná pomocí clearance kreatininu v moči, nezvyšuj e plazmatickou expozici panobinostatu ve skupinách s lehkou, středně těžkou a těžkou poruchou.
5.3 Předklinické údaje vztahující se k bezpečnosti
Studie zabývající se toxicitou opakovaných dávek
Jako primární cílové orgány toxicity po podání panobinostatu potkanům a psům byly identifikovány erytropoetický, myelopoetický a lymfatický systém. Změny tyroidního hormonů zahrnující hormony u psů (snížení trijodotyroninu (T3)) a potkanů (snížení trijodotyroninu (T3), tetrajodotyroninu (T4) (samci) a tyreotropního hormonu (TSH)) byly pozorované při expozicích korespondujících s 0,07-2,2 klinicky sledované AUC u člověka.
Kancerogeneze a mutageneze
Studie kancerogenicity s panobinostatem nebyly provedeny. Panobinostat prokázal mutagenní potenciál v Amesově testu, endoreduplikační účinky v lidských periferních lymfocytech a poškození DNA v in vivo studii COMET v myších lymfomatických buňkách L5178Y, které jsou připisovány farmakologickému způsobu účinku.
Reprodukční toxicita
U samic potkana (dávky >30 mg/kg) bylo pozorováno zvýšení časné resorpce. Atrofie prostaty spojená se zmenšenými sekrečními granuly, degenerací varlat, oligospermií a zvýšeným epididymálním rozpadem byla pozorovaná u psů při expozicích odpovídajících 0,41-0,69 lidské klinické AUC a nebyla zcela reverzibilní po 4týdenní periodě zotavení.
Na základě dat získaných u zvířat, se odhaduje pravděpodobnost, že panobinostat zvyšuje riziko úmrtí zárodku a vývojové abnormality skeletu jako vysoká Při překročení expozic odpovídajících 0,25 lidské klinické AUC byla pozorovaná embryofetální letalita zvýšení výskytu anomálií skeletu (nadbytečné obratle, nadbytečná sternebra a zvýšený počet malých změn skeletu, zpožděná osifikace a změny sterneber).
Účinky panobinostatu na vývoj a postnatální růst a zrání nebyly ve studiích u zvířat vyhodnoceny.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Obsah tobolky
Magnesium-stearát
Mannitol
Mikrokrystalická celulóza Předbobtnalý kukuřičný škrob
Obal tobolky
Farydak 10 mg tvrdé tobolky Želatina
Oxid titaničitý (E171)
Brilantní modř FCF (E133)
Žlutý oxid železitý (E172)
Farydak 15 mg tvrdé tobolky Želatina
Oxid titaničitý (E171)
Žlutý oxid železitý (E172)
Červený oxid železitý (E172)
Farydak 20 mg tvrdé tobolky Želatina
Oxid titaničitý (E171)
Červený oxid železitý (E172)
Inkoust potisku
Černý oxid železitý (E172)
Propylenglykol (E1520)
Šelak
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
3 roky.
6.4 Zvláštní opatření pro uchovávání
Neuchovávejte při teplotě nad 30°C.
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
6.5 Druh obalu a obsah balení
PVC/PCTFE/Al blistr obsahující 6 tobolek.
Balení obsahující 6, 12 nebo 24 tobolek.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
Farydak 10 mg tvrdé tobolky EU/1/15/1023/001-003
Farydak 15 mg tvrdé tobolky EU/1/15/1023/004-006
Farydak 20 mg tvrdé tobolky EU/1/15/1023/007-009
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
28. srpna 2015
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A. VÝROBCE ODPOVĚDNÝ /VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ /VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného/výrobců odpovědných za propouštění šarží
Novartis Pharma GmbH Roonstrasse 25 90429 Norimberk Německo
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předloží první pravidelně aktualizovanou zprávu o bezpečnosti pro tento léčivý přípravek do 6 měsíců od jeho registrace. Držitel rozhodnutí o registraci dále předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
• Další opatření k minimalizaci rizik
Před uvedením přípravku Farydak na trh se musí držitel rozhodnutí o registraci (MAH) v každém členském státě (s kompetentní státní autoritou) dohodnout na obsahu a formě vzdělávacího programu a to včetně sdělovacích médií, způsobu distribuce a s tím souvisejících dalších aspektů programu.
Edukační program slouží ke školení správného použití přípravku a minimalizaci rizik.
Držitel rozhodnutí o registraci musí zajistit, aby v každém členském státě, kde je Farydak uváděn na trh, měl každý pacient/ošetřovatel, který bude ve styku s přípravkem Farydak, přístup k/byl mu poskytnut následující vzdělávací balíček:
• Informační balíček pro pacienta
Informační balíček pro pacienta by měl obsahovat: o Příbalovou informaci pro pacienta
o Kartu pacienta
• Karta pacienta musí obsahovat instrukce týkající se následujících klíčových informací:
o Seznámení se s kartou pacienta: tato část obsahuje obecné informace o kartě pacienta a o jejím účelu.
o Jak používat kartu pacienta: tato část poskytuje celkový přehled o tom, jak používat kartu pacienta.
o Jak užívat léky dle předpisu: tato část poskytuje návod, jak správně vyplnit kartu pacienta.
o Doporučení nosit kartu pacienta neustále u sebe: tato část upozorňuje pacienta na nutnost nosit a předkládat kartu při jakékoli návštěvě lékaře či terapeutické nebo diagnostické proceduře.
o Tabulka popisující léčebný režim pro každý den cyklu s prostorem, kde si pacient může poznamenat, které léky užil.
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Farydak 10 mg tvrdé tobolky panobinostatum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tvrdá tobolka obsahuje panobinostati lactas odpovídající panobinostatum 10 mg.
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Tvrdá tobolka
6 tobolek 12 tobolek 24 tobolek
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/15/1023/001 6 tobolek
EU/1/15/1023/002 12 tobolek
EU/1/15/1023/003 24 tobolek
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Farydak 10 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Farydak 10 mg tobolky panobinostatum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
|
Týden 1 | |
|
Týden 2 | |
|
Týden 3 | |
|
Den |
1 |
|
Den |
2 |
|
Den |
3 |
|
Den |
4 |
|
Den |
5 |
|
Den |
6 |
|
Den |
7 |
|
Den |
8 |
|
Den |
9 |
|
Den |
10 |
|
Den |
11 |
|
Den |
12 |
|
Den |
13 |
|
Den |
14 |
|
Den |
15 |
|
Den |
16 |
|
Den |
17 |
|
Den |
18 |
|
Den |
19 |
|
Den |
20 |
|
Den |
21 |
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Farydak 15 mg tvrdé tobolky panobinostatum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tvrdá tobolka obsahuje panobinostatum lacticum anhydricum odpovídající panobinostatum 15 mg.
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Tvrdá tobolka
6 tobolek 12 tobolek 24 tobolek
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/15/1023/004 6 tobolek
EU/1/15/1023/005 12 tobolek
EU/1/15/1023/006 24 tobolek
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Farydak 15 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Farydak 15 mg tobolky panobinostatum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
|
Týden 1 | |
|
Týden 2 | |
|
Týden 3 | |
|
Den |
1 |
|
Den |
2 |
|
Den |
3 |
|
Den |
4 |
|
Den |
5 |
|
Den |
6 |
|
Den |
7 |
|
Den |
8 |
|
Den |
9 |
|
Den |
10 |
|
Den |
11 |
|
Den |
12 |
|
Den |
13 |
|
Den |
14 |
|
Den |
15 |
|
Den |
16 |
|
Den |
17 |
|
Den |
18 |
|
Den |
19 |
|
Den |
20 |
|
Den |
21 |
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Farydak 20 mg tvrdé tobolky panobinostatum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tvrdá tobolka obsahuje panobinostatum lacticum anhydricum odpovídající panobinostatum 20 mg.
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Tvrdá tobolka
6 tobolek 12 tobolek 24 tobolek
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/15/1023/007 6 tobolek
EU/1/15/1023/008 12 tobolek
EU/1/15/1023/009 24 tobolek
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Farydak 20 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Farydak 20 mg tobolky panobinostatum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
|
Týden 1 | |
|
Týden 2 | |
|
Týden 3 | |
|
Den |
1 |
|
Den |
2 |
|
Den |
3 |
|
Den |
4 |
|
Den |
5 |
|
Den |
6 |
|
Den |
7 |
|
Den |
8 |
|
Den |
9 |
|
Den |
10 |
|
Den |
11 |
|
Den |
12 |
|
Den |
13 |
|
Den |
14 |
|
Den |
15 |
|
Den |
16 |
|
Den |
17 |
|
Den |
18 |
|
Den |
19 |
|
Den |
20 |
|
Den |
21 |
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro pacienta
Farydak 10 mg tvrdé tobolky Farydak 15 mg tvrdé tobolky Farydak 20 mg tvrdé tobolky
panobinostatum
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Farydak a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Farydak užívat
3. Jak se přípravek Farydak užívá
4. Možné nežádoucí účinky
5 Jak přípravek Farydak uchovávat
6. Obsah balení a další informace
1. Co je přípravek Farydak a k čemu se používá Co je přípravek Farydak
Přípravek Farydak je protinádorový lék, který obsahuje léčivou látku panobinostat, která patří do skupiny léčiv nazývaných inhibitory pan-deacetylázy.
K čemu se přípravek Farydak používá
Přípravek Farydak se používá k léčbě dospělých pacientů se vzácným typem rakoviny krve, který se nazývá mnohočetný myelom. Mnohočetný myelom je poškození plazmatických buněk (typ krvinek), které nekontrolovaně rostou v kostní dřeni.
Přípravek Farydak blokuje růst rakovinných plazmatických buněk a snižuje počet rakovinných buněk.
Přípravek Farydak se užívá vždy spolu se dvěma dalšími přípravky: bortezomibem a dexamethazonem.
Jestliže máte jakékoli dotazy týkající se působení přípravku Farydak nebo důvodu, proč Vám byl předepsán, zeptejte se svého lékaře nebo lékárníka.
2. Čemu musíte věnovat pozornost, než začnete přípravek Farydak užívat Neužívejte přípravek Farydak:
- jestliže jste alergický(á) na panobinostat nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
- jestliže kojíte
Upozornění a opatření
Řiďte se pečlivě radami svého lékaře.
Před užitím přípravku Farydak se poraďte se svým lékařem nebo lékárníkem:
• pokud máte, nebo jste měl(a) jaterní potíže.
• pokud máte srdeční potíže nebo potíže se srdečním tepem, jako je nepravidelný srdeční tep nebo stav nazývaný syndrom dlouhého QT intervalu.
• pokud máte bakteriální, virovou nebo plísňovou infekci.
• pokud máte gastrointestinální potíže, jako je průjem, pocit na zvracení nebo zvracení.
• pokud máte problémy se srážlivostí krve (poruchu koagulace).
Během léčby přípravkem Farydak okamžitě informujte svého lékaře nebo lékárníka:
• pokud zaznamenáte jakékoli příznaky gastrointestinálních potíží.
• pokud zaznamenáte j akékoli příznaky j aterních potíží.
• pokud zaznamenáte j akékoli příznaky infekce.
• pokud zaznamenáte jakékoli příznaky srdečních potíží.
Seznam souvisejících příznaků je uveden v bodě 4, Možné nežádoucí účinky.
Váš lékař může upravit, dočasně zastavit nebo zcela ukončit léčbu přípravkem Farydak, pokud se u Vás objeví nežádoucí účinky.
Sledování během Vaší léčby přípravkem Farydak
Během léčby přípravkem Farydak byste se měl(a) podrobit pravidelným testům krve. Jde o tyto testy:
• kontrola správné funkce jater (měřením krevních hladin bilirubinu a transamináz, které jsou látkami tvořenými játry).
• kontrola množství určitého typu buněk v krvi (bílé krvinky, červené krvinky, krevní destičky).
• kontrola množství elektrolytů (jako je draslík, hořčík, fosfát) v těle.
• kontrola funkce štítné žlázy a hypofýzy (vyhodnocením hladiny hormonů štítné žlázy v krvi).
Srdeční tep bude také kontrolován pomocí přístroje, který měří elektrickou aktivitu srdce (nazývá se EKG).
Děti a dospívající
Přípravek Farydak nemá být podáván dětem a dospívajícím do 18 let věku.
Další léčivé přípravky a přípravek Farydak
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat, a to včetně léků, které jsou dostupné bez lékařského předpisu, jako jsou vitaminové nebo rostlinné doplňky, protože se mohou vzájemně ovlivňovat s přípravkem Farydak.
Především informujte svého lékaře nebo lékárníka, pokud užíváte některý z následujících léků:
• léky užívané k léčbě infekcí, včetně plísňových infekcí (jako je ketokonazol, itrakonazol, vorikonazol nebo posakonazol) a některých bakteriálních infekcí (antibiotika jako klarithromycin nebo telithromycin). Léky užívané k léčbě tuberkulózy, jako je rifabutin nebo rifampicin.
• léky užívané k zastavení záchvatů (antiepileptika jako je karbamazepin, ferfenazin, fenobarbital nebo fenytoin).
• léky užívané k léčbě HIV, jako je ritonavir nebo sachuinavir.
• léky užívané k léčbe deprese, jako je nefazodon.
• třezalku tečkovanou, rostlinný lék užívaný k léčbě deprese.
• léky užívané k prevenci tvorby krevních sraženin nazývané antikoagulancia, jako je warfarin nebo heparin.
• léky užívané k léčbě kašle, jako je dextromethorfan.
• léky užívané k léčbě nepravidelného srdečního tepu, jako je amiodaron, disopyramid, prokainamid, chinidin, propafenon nebo sotalol.
• léky, které mohou mít nechtěné účinky na srdce (nazývané dlouhý QT interval), jako je chlorochin, halofantrin, metadon, moxifloxacin, bepridil nebo pimozid.
• léky užívané k léčbě hypertenze, jako je metoprolol nebo nebivolol.
• léky užívané k léčbě závažných duševních potíží, jako je risperidon.
• léky užívané k léčbě rakoviny prsu, jako je tamoxifen.
• léky užívané k léčbě pocitu na zvracení a zvracení jako je dolasetron, granisetron, ondansetron nebo tropisetron; tyto léky mohou mít také nežádoucí vliv na srdce (prodloužení QT intervalu).
• atomoxetin, lék užívaný k léčbě poruch pozornosti.
Tyto léky mají být užívány s opatrností nebo mají být během léčby přípravkem Farydak vysazeny. Pokud užíváte jakýkoliv z nich, může Vám lékař během léčby přípravkem Farydak takový lék zaměnit za jiný lék.
Poraďte se se svým lékařem nebo lékárníkem, pokud si nejste jistý(á), zda je některý z léků, které užíváte, uveden v seznamu výše.
Během léčby přípravkem Farydak byste také měl(a) informovat svého lékaře nebo lékárníka, pokud Vám byl předepsán další lék, který jste dříve neužíval (a).
Přípravek Farydak s jídlem a pitím
Během Vaší léčby přípravkem Farydak byste neměl(a) jíst karambolu, granátové jablko nebo grapefruit nebo pít šťávu z granátového jablka nebo grapefruitu, protože mohou zvýšit množství léku, které se dostane do krve.
Těhotenství a kojení
Z důvodu potenciálního rizika úmrtí nebo malformace plodu by neměl být přípravek Farydak užíván během:
• Těhotenství
Přípravek Farydak nemá být užíván během těhotenství, pokud možný prospěch pro matku nepřeváží možné riziko pro dítě. Pokud jste těhotná, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem. Váš lékař s Vámi prodiskutuje možná rizika užívání přípravku Farydak během těhotenství.
• Kojení
Přípravek Farydak se nesmí užívat během kojení.
Antikoncepce pro ženy a muže
Z důvodu potenciálního rizika úmrtí nebo malformace plodu byste měl(a) při užívání přípravku Farydak využít následující metody antikoncepce:
• Pro ženy užívající přípravek Farydak
Pokud jste sexuálně aktivní žena, měl by Vám být před zahájením léčby proveden těhotenský test a během léčby přípravkem Farydak musíte používat vysoce účinný způsob antikoncepce. Tuto antikoncepci musíte používat ještě po dobu tří měsíců po ukončení léčby přípravkem Farydak. Váš lékař s Vámi prodiskutuje nejlepší metodu. Pokud užíváte hormonální antikoncepci, musíte také jako její doplněk používat bariérovou metodu antikoncepce (jako je kondom nebo diafragma).
• Pro muže užívající přípravek Farydak
Pokud jste sexuálně aktivní muž, měl byste během léčby přípravkem Farydak používat kondomy. Kondomy byste měl používat ještě po dobu šesti měsíců po ukončení léčby přípravkem Farydak. Pokud je Vaše partnerka schopná otěhotnět, měla by během Vaší léčby a tři měsíce po jejím skončení používat vysoce účinný způsob antikoncepce. Pokud Vaše partnerka otěhotní, zatímco užíváte přípravek Farydak nebo během tří měsíců po ukončení léčby přípravkem Farydak, okamžitě informujte svého lékaře.
Řízení dopravních prostředků a obsluha strojů
Přípravek Farydak může mít malý vliv na schopnost řídit dopravní prostředky a obsluhovat stroje. Pokud máte závratě v průběhu užívání tohoto léku, nesmíte řídit dopravní prostředky ani obsluhovat stroje.
3. Jak se přípravek Farydak užívá
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Kolik přípravku užívat
• Přípravek Farydak se užívá 21 dní (2 týdny se užívá a 1 týden se vysadí) - toto období se nazývá léčebný cyklus.
• Neužívejte lék každý den.
• Na základě doporučení Vašeho lékaře má být dávka přípravku Farydak 20 mg, 15 mg nebo 10 mg užívaná jednou denně ve dnech 1, 3, 5, 8, 10 a 12 21denního cyklu.
• Neužívejte přípravek Farydak v Týdnu 3.
• Po Týdnu 3 zahajte nový cyklus znovu podle Tabulky 1 a 2 níže.
Věnujte prosím pozornost Tabulce 1 pro cykly 1 až 8 a Tabulce 2 pro cykly 9-16.
Tabulka 1 Doporučený rozvrh pro užívání přípravku Farydak v kombinaci s bortezomibem a dexamethazonem (cykly 1-8)
|
Cykly 1-8 (3týdenní cykly) |
Týden 1 Dny |
Týden 2 Dny |
Týden 3 | ||||||||||||
|
Farydak |
1 |
3 |
5 |
8 |
10 |
12 |
Přestávka | ||||||||
|
Bortezomib |
1 |
4 |
8 |
11 |
Přestávka | ||||||||||
|
Dexamethazon |
1 |
2 |
4 |
5 |
8 |
9 |
11 |
12 |
Přestávka | ||||||
Tabulka 2 Doporučený rozvrh pro užívání přípravku Farydak vkombinaci s bortezomibem a dexamethazonem (cykly 9-16).
|
Cykly 9-16 (3týdenní cykly) |
Týden 1 Dny |
Týden 2 Dny |
Týden 3 | ||||||||||||
|
Farydak |
1 |
3 |
5 |
8 |
10 |
12 |
Přestávka | ||||||||
|
Bortezomib |
1 |
8 |
Přestávka | ||||||||||||
|
Dexamethazon |
1 |
2 |
8 |
9 |
Přestávka | ||||||||||
Váš lékař Vám sdělí přesný počet tobolek přípravku Farydak, který máte užívat. Neměňte dávku bez konzultace s Vaším lékařem.
Užívejte přípravek Farydak jednou denně pouze ve stanovených dnech ve stejnou dobu.
Užívání tohoto léku
• Polykejte tobolky celé, zapijte je sklenicí vody.
• Tento lék může být užíván s jídlem nebo nalačno.
• Nežvýkejte ani nedrťte tobolky.
Pokud jste po spolknutí tobolek přípravku Farydak zvracel(a), neužívejte žádné další tobolky až do další naplánované dávky.
Jak používat blistr přípravku Farydak
Jeden blistr přípravku Farydak = 3 týdny = 1 cyklus
Příslušné dny cyklu jsou na blistru vyznačeny. Užívejte Farydak ve dnech 1, 3 a 5 a dále 8, 10 a 12.
Vezměte tobolku přípravku Farydak vymáčknutím z blistru ve dnech 1, 3 a 5 v 1. týdnu a dále ve dnech 8, 10 a 12 v období 2. týdne.
Pro lepší sledování Vašeho léčebného plánu si ve dnech kdy Farydak neužíváte, včetně období přestávky ve 3. týdnu, vyznačte příslušné prázdné dutinky na blistru.
Jak dlouho užívat přípravek Farydak
Pokračujte v užívaní přípravku Farydak tak dlouho, jak Vám doporučí Váš lékař. Jde o dlouhodobou léčbu s 16 cykly (48 týdnů). Váš lékař bude sledovat Váš zdravotní stav, aby zjistil, zda má léčba požadovaný účinek. Pokud máte otázky jak dlouho užívat přípravek Farydak, zeptejte se svého lékaře nebo lékárníka.
Jestliže jste užil(a) více přípravku Farydak, než jste měl(a)
Jestliže nedopatřením užijete více tobolek, než máte předepsáno nebo pokud někdo nedopatřením užije Váš lék, okamžitě požádejte o radu svého lékaře nebo vyhledejte zdravotnické zařízení. Přineste sebou balení a tuto příbalovou informaci. Může být nutná lékařská péče.
Jestliže jste zapomněl(a) užít přípravek Farydak
• Pokud uplynulo méně než 12 hodin od chvíle, kdy jste měl(a) lék užít, užijte zmeškanou dávku, jakmile si na to vzpomenete. Poté pokračujte v užívání obvyklým způsobem.
• Pokud uplynulo více než 12 hodin od chvíle, kdy jste měl(a) lék užít, vynechejte zmeškanou dávku. Poté pokračujte v užívání obvyklým způsobem.
Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku.
Nikdy neužívejte zmeškanou dávku přípravku Farydak v jednom z “volných” dní, kdy není podání přípravku Farydak plánováno.
Informujte svého lékaře o všech dávkách, které jste zapomněl(a) užít během kteréhokoli 21denního cyklu léčby.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí
vyskytnout u každého.
Některé nežádoucí účinky mohou být závažné
ZASTAVTE užívání přípravku Farydak a okamžitě vyhledejte lékařskou pomoc, pokud se
u Vás projeví následující:
• potíže s dýcháním nebo polykáním, otoky obličeje, rtů, jazyka nebo krku, silné svědění kůže s červenou vyrážkou nebo vystouplými pupeny (možné příznaky alergické reakce)
• silné bolesti hlavy, pocit slabosti nebo ochrnutí končetin nebo obličeje, potíže s řečí, náhlá ztráta vědomí (možné příznaky potíží nervového systému, jako je nitrolební nebo mozkové krvácení nebo otok)
• zrychlené dýchání, pocit závratě
• náhlá a zdrcující bolest na hrudi, pocit únavy, nepravidelný srdeční tep (možné příznaky srdeční příhody)
• vykašlávání krve, výtok krvavé tekutiny z nosu (příznaky krvácení v plicích)
• zvracení krve, černá nebo krvavá stolice, průchod čerstvé krve konečníkem, obvykle ve stolici nebo se stolicí (příznaky gastrointestinálního krvácení)
• potíže s dýcháním spolu se zmodráním okolí úst, které mohou vést ke ztrátě vědomí (příznaky závažných plicních potíží)
• horečka, bolest na hrudi, zvýšená tepová frekvence, snížený krevní tlak, dušnost nebo zrychlené dýchání (příznaky otravy krve, která se označuje také jako sepse)
• bolest na hrudi nebo nepříjemný pocit na hrudi, změny srdečního tepu (rychlejší nebo pomalejší), pocit bušení srdce, točení hlavy, pocit slabosti, závrať, zmodrání rtů, dušnost, otoky dolních končetin nebo kůže (příznaky srdečních potíží)
Okamžitě informujte svého lékaře nebo lékárníka, pokud si všimnete některého z těchto nežádoucích účinků:
• bolest břicha nebo žaludku, pocit na zvracení, průjem, zvracení, černá nebo krvavá stolice, zácpa, pálení žáhy, otok nebo nadmutí břicha (příznaky potíží v trávicím traktu)
• nový výskyt nebo zhoršení příznaků, jako je kašel s hlenem nebo bez hlenu, horečka, obtížné nebo bolestivé dýchání, sípání, bolest na hrudi při dýchání, dušnost nebo potíže s dýcháním, bolest nebo pocit pálení při močení, přehnaný pocit nucení na močení, krev v moči (příznaky infekcí plic nebo močových cest)
• horečka, bolest v krku nebo vřídky v ústech způsobené infekcí (příznaky nízkého počtu bílých krvinek)
• náhlé krvácení nebo podlitiny na kůži (příznaky nízkého počtu krevních destiček)
• průjem, bolest břicha, horečka (příznaky zánětu tlustého střeva)
• pocit točení hlavy, především ve stoje (příznaky nízkého krevního tlaku)
• pocit žízně, malé množství moči, ztráta tělesné hmotnosti, suchá zarudlá kůže, podrážděnost (příznaky dehydratace (nedostatku tekutin v těle))
• otoky kloubů (příznak nízké hladiny albuminu v krvi, označovaná jako hypoalbuminémie)
• pocit únavy, svědění, zežloutnutí kůže a očního bělma, pocit na zvracení nebo zvracení, ztráta chuti k jídlu, bolest na pravé straně břicha, tmavá nebo hnědá moč, krvácení nebo snadnější tvorba podlitin než je obvyklé (příznaky jaterních potíží)
• výrazně snížené množství moči, otok dolních končetin (příznaky ledvinných potíží)
• svalová slabost, svalové křeče, neobvyklý srdeční tep (příznaky změn hladiny draslíku v krvi)
Další možné nežádoucí účinky
Pokud se některý z níže uvedených nežádoucích účinků projeví v závažné míře, informujte svého lékaře nebo lékárníka.
Velmi časté (mohou postihnout vice než 1 z 10 lidí)
• pocit únavy (únava), bledá pokožka. Může jít o příznaky nízkého počtu červených krvinek.
• snížená chuť k jídlu nebo ztráta tělesné hmotnosti
• potíže s usínáním nebo udržením spánku (nespavost)
• bolest hlavy
• pocit závrati, únavy nebo slabosti
• zvracení, pocit na zvracení, žaludeční nevolnost, trávicí potíže
• otoky dolních nebo horních končetin
• snížená hladina fosfátu nebo sodíku v krvi
Časté (mohou postihnout až 1 z 10 lidí)
• vyrážka malých tekutinou naplněných puchýřků, které se vyskytují na zarudlé kůži, v ústech nebo dásních (příznaky možné závažné virové infekce)
• zánět uší, krvácení z nosu nebo krvácení do bělma očí, podlitiny, kožní zánět způsobený infekcí (vyrážka, zarudnutí kůže, označované jako erytém)
• bolest břicha, průjem, otok nebo nadmutí břicha (příznaky zánětu žaludeční sliznice)
• moučnivka v ústech (kvasinková infekce v ústech)
• pocit žízně, vysoké množství moči, zvýšená chuť k jídlu se ztrátou tělesné hmotnosti (příznaky vysoké hladiny cukru v krvi)
• rychlý přírůstek tělesné hmotnosti, otoky rukou, kotníků, chodidel nebo obličeje (příznaky zadržování tekutin)
• snížená hladina vápníku v krvi, která někdy vede ke křečím
• nekontrolovatelný třes těla
• bušení srdce
• šustivý, chrastivý nebo praskavý zvuk vycházející z plic při dýchání
• popraskané rty
• sucho v ústech nebo změna vnímání chuti
• plynatost
• bolest nebo zánět kloubů
• krev v moči (příznak potíží s ledvinami)
• ztráta schopnosti kontrolovat močení z důvodu ztráty kontroly nebo slabé kontroly nad močovým měchýřem
• třesavka
• nárůst tělesné hmotnosti, pocit únavy, padání vlasů, svalová slabost, pocit chladu (příznaky nedostatečné činnosti štítné žlázy, která je známá jako hypotyroidismus)
• celkový pocit slabosti a únavy
• zvýšená hladina kyseliny močové v krvi
• snížená hladina hořčíku v krvi
• zvýšená hladina odpadní látky kreatininu
• zvýšené krevní hladiny jaterních enzymů alaninaminotransferázy (ALT),
aspartataminotransferázy (AST) nebo alkalické fosfatázy (ALP).
Méně časté (mohou postihnout až 1 ze 100 lidí)
• červené nebo fialové ploché skvrny velikosti špendlíkové hlavičky pod kůží
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Farydak uchovávat
• Uchovávejte tento přípravek mimo dohled a dosah dětí.
• Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a folii blistru.
• Neuchovávejte při teplotě nad 30°C.
• Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
• Neužívejte tento lék, pokud si všimnete jakéhokoli poškození obalu nebo známek manipulace s ním.
• Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co přípravek Farydak obsahuje
- Léčivou látkou přípravku Farydak je panobinostatum.
- Jedna tvrdá tobolka Farydak 10 mg obsahuje panobinostatum 10 mg. Dalšími složkami jsou: magnesium-stearát, mannitol, mikrokrystalická celulóza, předbobtnalý kukuřičný škrob, želatina, oxid titaničitý (E171), brilantní modř FCF (E133), žlutý oxid železitý (E172), černý oxid železitý (E172), propylenglykol (E1520), šelak.
- Jedna tvrdá tobolka Farydak 15 mg obsahuje panobinostatum 15 mg. Dalšími složkami jsou: magnesium-stearát, mannitol, mikrokrystalická celulóza, předbobtnalý kukuřičný škrob, želatina, oxid titaničitý (E171), žlutý oxid železitý (E172), červený oxid železitý (E172), černý oxid železitý (E172), propylenglykol (E1520), šelak.
- Jedna tvrdá tobolka Farydak 20 mg obsahuje panobinostatum 20 mg. Dalšími složkami jsou: magnesium-stearát, mannitol, mikrokrystalická celulóza, přredbobtnalý škrob, želatina, oxid titaničitý (E171), červený oxid železitý (E172), černý oxid železitý (E172), propylenglykol (E1520), šelak.
Jak přípravek Farydak vypadá a co obsahuje toto balení
Farydak 10 mg tvrdé tobolky jsou světle zelené neprůhledné tobolky (15,6-16,2 mm) obsahující bílý až téměř bílý prášek, s příčným označením “LBH 10 mg” černým inkoustem na víčku a dvěma příčnými pruhy značenými černým inkoustem na těle tobolky dodávané dodávané v blistrech. Farydak 15 mg tvrdé tobolky jsou oranžové neprůhledné tobolky (19,1-19,7 mm) obsahující bílý až téměř bílý prášek s příčným označením “LBH 15 mg” černým inkoustem na víčku a dvěma příčnými pruhy značenými černým inkoustem na těle tobolky, dodávané v blistrech.
Farydak 20 tvrdé tobolky jsou červené neprůhledné tobolky (19,1-19,7 mm) obsahující bílý až téměř bílý prášek s příčným označením “LBH 20 mg” černým inkoustem na víčku a dvěma příčnými pruhy značenými černým inkoustem na těle tobolky, dodávané v blistrech.
Dostupné jsou následující velikosti balení: balení s blistry obsahující 6, 12 nebo 24 tobolek.
Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
Výrobce
Novartis Pharma GmbH Roonstrasse 25 90429 Norimberk Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgie/Belgique/Belgien Lietuva
Novartis Pharma N.V. Novartis Pharma Services Inc.
Tél/Tel: +32 2 246 16 11 Tel: +370 5 269 16 50
Bt^rapuu
Novartis Pharma Services Inc. Ten: +359 2 489 98 28
Česká republika
Novartis s.r.o.
Tel: +420 225 775 111
Danmark
Novartis Healthcare A/S Tlf: +45 39 16 84 00
Deutschland
Novartis Pharma GmbH Tel: +49 911 273 0
Eesti
Novartis Pharma Services Inc. Tel: +372 66 30 810
Luxembourg/Luxemburg
Novartis Pharma N.V.
Tél/Tel: +32 2 246 16 11
Magyarország
Novartis Hungária Kft. Pharma Tel.: +36 1 457 65 00
Malta
Novartis Pharma Services Inc. Tel: +356 2122 2872
Nederland
Novartis Pharma B.V.
Tel: +31 26 37 82 555
Norge
Novartis Norge AS Tlf: +47 23 05 20 00
|
Espaňa Novartis Farmacéutica, S.A. Tel: +34 93 306 42 00 |
Polska Novartis Poland Sp. z o.o. Tel.: +48 22 375 4888 |
|
France Novartis Pharma S.A.S. Tél: +33 1 55 47 66 00 |
Portugal Novartis Farma - Produtos Farmaceuticos, S.A Tel: +351 21 000 8600 |
|
Hrvatska Novartis Hrvatska d.o.o. Tel. +385 1 6274 220 |
Románia Novartis Pharma Services Romania SRL Tel: +40 21 31299 01 |
|
Ireland Novartis Ireland Limited Tel: +353 1 260 12 55 |
Slovenija Novartis Pharma Services Inc. Tel: +386 1 300 75 50 |
|
Island Vistor hf. Sími: +354 535 7000 |
Slovenská republika Novartis Slovakia s.r.o. Tel: +421 2 5542 5439 |
|
Italia Novartis Farma S.p.A. Tel: +39 02 96 54 1 |
Suomi/Finland Novartis Finland Oy Puh/Tel: +358 (0)10 6133 200 |
|
Kúnpoq Novartis Pharma Services Inc. Tn^: +357 22 690 690 |
Sverige Novartis Sverige AB Tel: +46 8 732 32 00 |
|
Latvija Novartis Pharma Services Inc. Tel: +371 67 887 070 |
United Kingdom Novartis Pharmaceuticals UK Ltd. Tel: +44 1276 698370 |
EXXáSa
Novartis (Hellas) A.E.B.E. Tnk: +30 210 281 17 12
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
48