Extavia 250 Mikrogramů/Ml
SOUHRN ÚDAJŮ O PŘÍPRAVKU
PŘÍLOHA I
Extavia 250 mikrogramů/ml, prášek a rozpouštědlo pro injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Extavia obsahuje 300 mikrogramů (9,6 milionů IU) rekombinantního interferonu beta-1b v jedné injekční lahvičce*.
Po naředění obsahuje jeden ml 250 mikrogramů (8 milionů IU) rekombinantní interferonum beta-1b.
* vyrobeno genetickým inženýrstvím z kmene Escherichia coli.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční roztok.
Prášek - bílé až špinavě bílé barvy. Rozpouštědlo - čirý/bezbarvý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Extavia je určen pro léčbu:
• Pacientů s jedinou demyelinizační příhodou s aktivním zánětlivým procesem, která byla natolik závažná, že k léčbě bylo nutno podat intravenózně kortikoidy. U těchto pacientů byla vyloučena jiná možná diagnóza a bylo u nich stanoveno vysoké riziko klinicky definitivní roztroušené sklerózy (viz bod 5.1).
• Pacientů s relaps-remitentní formou roztroušené sklerózy se dvěma nebo více relapsy v posledních dvou letech.
• Pacientů se sekundárně progresivní formou roztroušené sklerózy, u aktivního onemocnění projevujícího se relapsy.
4.2 Dávkování a způsob podání
Léčba přípravkem Extavia musí být zahájena pod dohledem lékaře, který má zkušenosti s léčbou tohoto onemocnění.
Dávkování
Dospělí a dospívající ve věku od 12 do 17 let
Doporučená dávka přípravku Extavia je 250 mikrogramů (8 milionů IU) obsažená v 1 ml rekonstituovaného roztoku (viz. bod 6.6) aplikovaná subkutánně obden.
Všeobecně se na začátku léčby doporučuje titrace dávky.
Pacient by měl začít dávkou 62,5 mikrogramů (0,25 ml) subkutánně obden a dávka by se měla pomalu zvyšovat do 250 mikrogramů (1,0 ml) obden (viz tabulka A). Dobu titrace lze upravit, pokud dojde k jakémukoliv významnému nežádoucímu účinku. Pro dosažení adekvátní účinnosti by se mělo dosáhnout dávky 250 mikrogramů (1,0 ml) obden.
Tabulka A: Harmonogram pro titraci dávky*
|
Den léčby |
Dávka |
Objem |
|
1, 3, 5 |
62,5 mikrogramu |
0,25 ml |
|
7, 9, 11 |
125 mikrogramu |
0,5 ml |
|
13, 15, 17 |
187,5 mikrogramu |
0,75 ml |
|
> 19 |
250 mikrogramu |
1,0 ml |
* Dobu titrace lze upravit, jestliže se vyskytne jakýkoliv závažný nežádoucí účinek.
Optimální dávka nebyla ještě zcela určena.
V současné době není známo, jak dlouho by měl být pacient léčen. Existují data z 5ti letého klinicky kontrolovaného sledování u pacientů s relaps - remitentní roztroušenou sklerózou a z 3 letého klinicky kontrolovaného sledování u pacientů se sekundárně progresivní roztroušenou sklerózou. Účinnost léčby byla prokázána po dobu prvních dvou let u pacientů s relaps -remitentní roztroušenou sklerózou. Dostupná data z léčby v dalších třech letech podporují dostatečnou léčebnou účinnost přípravku Extavia po celou dobu sledování.
U pacientů s jedinou klinickou příhodou připomínající roztroušenou sklerózu byla účinnost prokázána po dobu tří let.
Léčba se nedoporučuje u pacientů s relaps-remitentní formou roztroušené sklerózy, kteří prodělali méně než 2 relapsy během posledních dvou let, nebo u pacientů se sekundárně progresivní formou, jejichž onemocnění nebylo aktivní v předchozích dvou letech. O déletrvající léčbě musí v individuálních případech rozhodnout lékař.
Pokud pacient neodpovídá na léčbu, například trvalou progresí v Expanded Disability Status Scale (EDSS) po dobu 6 měsíců, nebo jestliže během jednoho roku i přes léčbu přípravkem Extavia je nezbytné nejméně třikrát aplikovat léčbu adrenokortikotropní hormon (ACTH) nebo kortikosteroidy, léčba přípravkem Extavia by měla být ukončena.
Pediatrická populace
Žádné cílené klinické nebo farmakokinetické studie nebyly u dětí nebo dospívajících provedeny. Avšak omezená publikovaná data naznačují, že bezpečnostní profil u dospívajících ve věku od 12 do 17 let dostávajících obden dávku 8 milionů IU přípravku Extavia subkutánně je podobný jako u dospělých lidí. O podávání přípravku Extavia dětem ve věku do 12 let nejsou dostupné žádné údaje, a proto by přípravek Extavia neměl být u této skupiny pacientů používán.
Způsob podání
Rekonstituovaný roztok má být podáván podkožně obden.
Návod k rekonstituci tohoto léčivého přípravku před jeho podáním je uveden v bodě 6.6.
4.3 Kontraindikace
- Hypersenzitivita na přirozený či rekombinantní interferon beta, na lidský albumin nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
- Zahájení léčby v těhotenství (viz bod 4.6).
- Pacienti trpící závažnou depresivní poruchou a/nebo suicidálním myšlenkami (viz body 4.4 a 4.8).
- Pacienti s dekompenzovaným onemocněním jater (viz body 4.4, 4.5 a 4.8).
4.4 Zvláštní upozornění a opatření pro použití Poruchy imunitního systému
Podávání cytokinů pacientům s již dříve existující monoklonální gamapatií bylo spojeno s rozvojem syndromu systémového kapilárního průsaku s příznaky podobnými šoku a majícímu za následek smrt.
Gastrointestinální poruchy
Při užívání přípravku Extavia byly pozorovány případy pankreatitidy, často spojené s hypertriacylglycerolemií.
Poruchy nervového systému
Přípravek Extavia by měl být podáván s opatrností pacientům, kteří trpěli dříve nebo trpí nyní depresivními poruchami, zvláště těm, u kterých se již projevily suicidální myšlenky (viz bod 4.3). Je známo, že deprese a suicidální myšlenky se vyskytují se zvýšenou frekvencí u pacientů trpících roztroušenou sklerózou a ve spojitosti s použitím interferonů. Pacienty, kteří jsou léčeni přípravkem Extavia je třeba upozornit, že musí okamžitě hlásit svému lékaři jakékoliv známky deprese nebo suicidálních myšlenek. Pacienty trpící depresí je třeba během léčby přípravkem Extavia pečlivě sledovat a náležitě léčit. Případné přerušení léčby přípravkem Extavia by mělo být zváženo (viz body 4.3 a 4.8).
Přípravek Extavia by měl být podáván s opatrností pacientům, kteří mají v anamnéze záchvaty křečí, pacientům léčeným antiepileptiky a zejména pacientům s epilepsií, jejich nemoc není antiepileptiky adekvátně kontrolována (viz body 4.5 a 4.8).
Tento léčivý přípravek obsahuje lidský albumin, a proto představuje možné riziko přenosu virových onemocnění. Riziko přenosu Creutzfeld-Jacobovy nemoci ( CJD) nemůže být vyloučeno.
Laboratorní vyšetření
Doporučuje se pravidelně provádět funkční vyšetření štítné žlázy u pacientů s dysfunkcí štítné žlázy v anamnéze, nebo pokud existuje klinická indikace.
K běžně požadovanému laboratornímu vyšetření u pacientů s roztroušenou sklerózou se doporučuje provést kompletní rozbor krve a stanovení diferenciálního počtu bílých krvinek, stanovení krevních destiček, chemického složení krve, včetně jaterních testů (např. aspartátaminotransferáza/ sérová glutamátoxalacetátransamináza (SGOT), alaninaminotransferáza/ sérová glutamátpyruváttransamináza (SGPT) a
gamaglutamyltransferáza), před zahájením léčby přípravkem Extavia a v pravidelných intervalech po zahájení léčby, následně pak v pravidelných intervalech po vymizení klinických příznaků.
U pacientů s anemii, trombocytopenií nebo leukopenií (samostatnou nebo v jakékoliv kombinaci) je nezbytné intenzivnější sledování celkového počtu krvinek s diferenciálním počtem bílých krvinek a počtu destiček. U pacientů, u nichž dojde ke vzniku neutropenie, by sledování mělo být přísnější kvůli riziku horečky či infekce. Byly hlášeny případy trombocytopenie s výrazným poklesem počtu krevních destiček.
Poruchy jater a žlučových cest
V klinických studiích se u pacientů léčených přípravkem Extavia velmi často vyskytlo asymptomatické zvýšení sérových transamináz, ve většině případů bylo mírné a přechodné. Jako u jiných interferonů beta, byly u pacientů léčených přípravkem Extavia vzácně uváděny případy závažného poškození jater, včetně jaterního selhání. Nejzávažnější případy se vyskytovaly často u pacientů, kteří byli vystaveni jiným léčivým přípravkům nebo látkám se známou spojitostí s hepatotoxicitou nebo v přítomnosti chorobných stavů (např. metastazující maligní onemocnění, závažná infekce a sepse, abusus alkoholu).
U pacientů by měly být sledovány známky jaterního poškození. Zvýšení sérových transamináz je důvodem k pečlivému monitorování a vyšetření. Je-li zvýšení hladin signifikantní nebo je-li spojeno s klinickými příznaky, jako je například žloutenka, mělo by se zvážit přerušení léčby přípravkem Extavia. Nejsou-li klinické projevy poškození jater, lze po úpravě hladin jaterních enzymů zvážit obnovení terapie při řádném průběžném sledování jaterních funkcí.
Trombotická mikroangiopatie (TMA)
V souvislosti s přípravky obsahujícími interferon beta byly hlášeny případy trombotické mikroangiopatie projevující se jako trombotická trombocytopenická purpura (TTP) nebo hemolyticko-uremický syndrom (HUS), včetně fatálních případů. Případy byly hlášeny v různých časových bodech během léčby a mohou se objevit několik týdnů až několik let po zahájení léčby interferonem beta. Mezi časné klinické příznaky patří trombocytopenie, nově nastupující hypertenze, horečka, symptomy týkající se centrální nervové soustavy (např. zmatenost, paréza) a zhoršená funkce ledvin. Mezi laboratorní nálezy naznačující TMA patří snížený počet krevních destiček, zvýšená hladina laktát dehydrogenázy (LDH) v séru v důsledku hemolýzy a schistocyty (fragmentované erytrocyty) v krevním nátěru. Proto, pokud jsou zjištěny klinické příznaky TMA, doporučuje se provést další kontrolu počtu krevních destiček, hladiny LDH v séru, krevních nátěrů a funkce ledvin. Jestliže je diagnostikována TMA, je třeba okamžitě zahájit léčbu (a zvážit výměnu plazmy), přičemž se doporučuje okamžitě přerušit léčbu přípravkem Extavia.
Poruchy ledvin a močových cest
Při podávání interferonu beta je třeba zachovávat opatrnost a pečlivě sledovat pacienty se závažnou poruchou funkce ledvin.
Nefrotický syndrom
Během léčby přípravky obsahujícími interferon beta byly hlášeny případy nefrotického syndromu spojeného s různými základními nefropatiemi, které zahrnují kolabující fokální segmentální glomerulosklerózu (FSGS), nefropatii s minimálními změnami (MCD), membranoproliferativní glomerulonefritidu (MPGN) a membranózní glomerulopatii (MGN). Příhody byly hlášeny v různých časových bodech během léčby a mohou se objevit po několika letech léčby interferonem beta. Doporučuje se pravidelně sledovat časné příznaky nebo symptomy, např. edém, proteinurii a poruchy funkce ledvin, zejména u pacientů s vysokým rizikem onemocnění ledvin. Je vyžadována rychlá léčba nefrotického syndromu a je třeba zvážit ukončení léčby přípravkem Extavia.
Srdeční poruchy
Také při podávání přípravku Extavia pacientům, kteří mají preexistující srdeční onemocnění, je třeba opatrnosti. Pacienti s již existujícím signifikantním srdečním onemocněním, například s kongestivním srdečním selháváním, s onemocněním koronárních artérií nebo s arytmií, by měli být sledováni z hlediska případného zhoršování jejich kardiálních funkcí, zejména při zahájení léčby přípravkem Extavia.
I když Extavia nemá žádnou známou přímo působící kardiální toxicitu, příznaky syndromu připomínajícího chřipku spojované s beta interferony mohou být stresujícím faktorem u pacientů s již dříve existujícím signifikantním kardiálním onemocněním. Po uvedení na trh byly velmi vzácně popsány případy dočasného zhoršení kardiálních funkcí spojené se zahájením léčby přípravkem Extavia u pacientů s již dříve existujícím srdečním onemocněním.
Byly hlášeny případy kardiomyopatie, dojde-li k jejímu výskytu a existuje-li podezření na souvislost s přípravkem Extavia, léčba by měla být ukončena.
Celkové a jinde nezařazené poruchy a lokální reakce po podání
Mohou se objevit závažné hypersenzitivní reakce (závažné akutní reakce, jako například bronchospazmus, anafylaxe a kopřivka). Pokud jsou reakce závažné, léčba přípravkem Extavia by měla být přerušena a měl by být zahájen odpovídající lékařský zásah.
U pacientů léčených přípravkem Extavia (viz bod 4.8) byla hlášena nekróza v místě vpichu injekce. Může být rozsáhlá a může postihnout i svalové fascie společně s tukovou tkání, a proto může způsobit vznik jizvy. Někdy je třeba odstranit mrtvou tkáň z rány, méně často je nutné provést transplantaci kožního štěpu. Zhojení může trvat až 6 měsíců.
Jestliže u pacienta dojde k jakémukoliv porušení kůže, které může být spojeno například s otokem či vytékáním tekutiny z místa vpichu, pacient by měl být poučen, aby o tom informoval svého lékaře předtím, než bude v injekčním podávání přípravku Extavia pokračovat.
Má-li pacient vícečetné léze, léčba přípravkem Extavia by měla být přerušena, dokud nedojde ke zhojení. Pacient s jednou lézí může pokračovat v léčbě přípravkem Extavia v případě, že nekróza není příliš rozsáhlá, protože u některých pacientů došlo ke zhojení nekrózy v místě vpichu i bez přerušení léčby přípravkem Extavia.
Pro minimalizaci rizika nekrózy v místě vpichu injekce je třeba upozornit pacienty na to, aby:
- používali aseptickou injekční techniku,
- obměňovali místa injekčního vpichu po každé dávce.
Výskyt reakcí v místě vpichu může být snížen při použit autoinjektoru. V pivotní studii pacientů s jedinou klinickou příhodou připomínající roztroušenou sklerózu byl u většiny pacientů použit autoinjektor. Reakce v místě vpichu a nekrózy v místě vpichu byly v této studii pozorovány méně často než v jiných pivotních studiích.
Postup při aplikaci samotným pacientem je třeba pravidelně kontrolovat, zvláště pokud se vyskytnou reakce v místě vpichu.
Imunogenita
Jako u všech terapeutických proteinů, i v tomto případě existuje možnost imunogenity. Vzorky séra v kontrolovaných klinických studiích byly odebírány každé 3 měsíce ke sledování vývoje protilátek proti přípravku Extavia.
V různých kontrolovaných klinických studiích došlo u 23 % až 41 % pacientů ke vzniku sérové neutralizační aktivity proti interferonu beta-1b potvrzené nálezem nejméně dvou po sobě následujících pozitivních titrů. U 43 % až 55 % těchto pacientů došlo ke konverzi na stabilní negativní stav protilátek (na základě dvou po sobě jdoucích negativních titrů) během následujícího období sledování příslušných studií.
Rozvoj neutralizační aktivity je spojen se snížením účinnosti jen s ohledem na množství relapsů. Některé analýzy naznačují, že tento efekt může být výraznější u pacientů s vyšším titrem neutralizační aktivity.
Ve studii s pacienty s jedinou klinickou příhodou připomínající roztroušenou sklerózu byla neutralizační aktivita, měřená každých 6 měsíců, pozorována nejméně jednou u 32 % (89) pacientů okamžitě léčených přípravkem Extavia; z nichž se na základě posledního dostupného vyšetření během pětiletého období vrátilo 60 % (53) těchto pacientů do negativního stavu. Během této doby byl rozvoj neutralizační aktivity spojen s významným vzrůstem počtu nových aktivních lézí a objemu T2 lézí při zobrazení magnetickou rezonancí. Nicméně toto nemusí být spojeno se snížením klinické účinnosti (pokud jde o dobu do vzniku klinicky definitivní roztroušené sklerózy (CDMS), doby do potvrzení progrese EDSS a výskytu relapsu).
S rozvojem neutralizační aktivity nebyly spojeny žádné nové nežádoucí účinky.
In vitro byla prokázána zkřížená interakce mezi přípravkem Extavia a přirozeným interferonem beta. Toto testování však neproběhlo in vivo a jeho klinický význam je nejistý.
Údaje získané od pacientů, u kterých došlo k rozvoji neutralizační aktivity a kteří dokončili léčbu přípravkem Extavia, jsou nepříliš četné a neprůkazné.
Rozhodnutí v léčbě pokračovat nebo ji ukončit by mělo být založeno na klinické aktivitě onemocnění spíše než na stavu neutralizační aktivity.
Pomocné látky
Tento přípravek obsahuje méně než 1 mmol (23 mg) sodíku v jedné dávce, tj. v podstatě je „bez sodíku“.
Osoby citlivé na latex
Odnímatelná krytka hrotu předplněné injekční stříkačky Extavia obsahuje derivát přírodního latexu. Ačkoli krytka neobsahuje latex, bezpečné použití předplněné injekční stříkačky Extavia u jedinců citlivých na latex nebylo hodnoceno, a tak potenciální riziko alergické reakce nelze zcela vyloučit.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné studie interakcí.
Účinek podávání 250 mikrogramů (8 milionů IU) přípravku Extavia každý druhý den pacientům s roztroušenou sklerózou na jejich lékový metabolismus je neznámý. Léčba kortikosteroidy nebo ACTH během relapsů po dobu až 28 dnů byla pacienty léčenými přípravkem Extavia dobře tolerována.
Vzhledem k nedostatku klinických zkušeností u pacientů s roztroušenou sklerózou, léčba přípravkem Extavia probíhající současně s podáváním jiných imunomodulancií než kortikosteroidů nebo ACTH se nedoporučuje.
Interferony snižují u lidí i zvířat aktivitu enzymů závislých na jaterním cytochromu P450. Je třeba velké opatrnosti, je-li přípravek Extavia podáván v kombinaci s léčivými přípravky, které mají úzký terapeutický index a jej ichž clearance j e silně závislá na systému j aterního cytochromu P450, např. antiepileptika. Velké opatrnosti je třeba také při současné léčbě léky ovlivňujícími hematopoetický systém.
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku
Ženy ve fertilním věku musí používat vhodnou kontracepční metodu.
O užívání přípravku Extavia v těhotenství existují pouze omezené informace. Dostupné údaje naznačují možné zvýšení rizika spontánních potratů. Zahájení léčby v průběhu těhotenství je kontraindikováno (viz bod 4.3). Pokud pacientka léčená přípravkem Extavia během léčby otěhotní, nebo zamýšlí otěhotnět, musí být informována o možných rizicích a mělo by být zváženo ukončení léčby (viz bod 5.3). U pacientek s velkým počtem relapsů před zahájením léčby by mělo být zváženo riziko závažných relapsů po přerušení léčby přípravkem Extavia z důvodu těhotenství oproti možnému riziku spontánního potratu.
Kojení
Není známo, zda se interferon beta-1b vylučuje do lidského mateřského mléka. Vzhledem k nebezpečí závažných nežádoucích reakcí na Extavia u kojených dětí je třeba rozhodnout, zda ukončit kojení nebo léčbu přípravkem Extavia.
Fertilita
Nebyly provedeny žádné studie vztahující se k fertilitě (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Studie hodnotící účinky na schopnost řídit a používat stroje nebyly provedeny.
Nežádoucí účinky na centrální nervový systém spojené s užíváním přípravku Extavia by mohly u vnímavých pacientů ovlivnit schopnost řídit a používat stroje.
4.8 Nežádoucí účinky
Přehled bezpečnostního profilu
Na začátku léčby je výskyt nežádoucích účinků častý, ale obecně se s další léčbou zmírňují. Nejčastěji pozorovanými nežádoucími účinky je komplex příznaků podobných chřipce (horečka, mrazení, bolest kloubů, malátnost, pocení, bolest hlavy nebo bolest svalů), což je hlavně způsobeno farmakologickými účinky léčivého přípravku a reakcí v místě podání injekce. Reakce v místě vpichu injekce se po podání přípravku Extavia vyskytly často. Zarudnutí, otok, zblednutí, zánět, bolest, hypersenzitivita, nekróza a nespecifické reakce byly signifikantně spojeny s léčbou přípravkem Extavia v dávce 250 mikrogramů (8,0 milionů IU).
Obecně se doporučuje titrace dávky na počátku léčby, aby se zvýšila snášenlivost přípravku Extavia (viz bod 4.2). Symptomy připomínající chřipku mohou být rovněž sníženy podáním nesteroidních protizánětlivých léčivých přípravků. Incidence výskytu reakcí v místě aplikace může být snížena při použití autoinjektoru.
Seznam nežádoucích účinků v tabulce
V následujících tabulkách je k popsání jednotlivých reakcí použit nejvhodnější termín dle MedDRA spolu s jeho synonymy a souvisejícími stavy.
Následující výčet nežádoucích účinků je založen na hlášeních z klinických studií (Tabulka 1, nežádoucí příhody a laboratorní abnormality) a na hlášeních po zavedení přípravku Extavia na trh (Tabulka 2, četnosti - kde jsou známé - založené na poolovaných klinických studiích (velmi časté >1/10, časté >1/100 až <1/10, méně časté >1/1 000 až <1/100, vzácné >1/10 000 až <1/1 000, velmi vzácné <1/10 000)). Zkušenosti s přípravkem Extavia u pacientů s roztroušenou sklerózou (RS) jsou omezené, proto nežádoucí účinky, které se objevují velmi vzácně, nemusely ještě vůbec být zaznamenány.
Tabulka 1 Nežádoucí příhody a laboratorní abnormality s incidencí >10 % a odpovídající procenta při použití placeba; nežádoucí účinky s významnou asociací <10 % na základě hlášení z klinických studií
|
Třídy orgánových systémů Nežádoucí příhoda a laboratorní anomálie |
Jediná příhoda připomínající roztroušenou sklerózu (BENEFIT) |
Sekundárně progresivní roztroušená skleróza (evropská studie) |
Sekundárně progresivní roztroušená skleróza (studie Severní Ameriky) |
Relaps- remitentní roztroušená skleróza |
|
Extavia 250 mikrogra mů (placebo) n=292 (n=176) |
Extavia 250 mikrogra mů (placebo) n=360 (n=358) |
Extavia 250 mikrogra mů (placebo) n=317 (n=308) |
Extavia 250 mikrogra mů (placebo) n=124 (n=123) | |
|
Infekce a infestace | ||||
|
Infekce |
6 % (3 %) |
13 % (11 %) |
11 % (10 %) |
14 % (13 %) |
|
Absces |
0 % (1 %) |
4 % (2 %) |
4 % (5 %) |
1 % (6 %) |
|
Poruchy krve a lymfatického systému | ||||
|
Snížený počet lymfocytů (<1500/mm3) x A ° |
79% (45 %) |
53 % (28 %) |
88 % (68 %) |
82% (67 %) |
|
Snížený celkový počet neutrofilů (<1500/mm3) x A |
11 % (2 %) |
18 % (5 %) |
4 % (10 %) |
18 % (5 %) |
|
Snížený počet leukocytů celkem (<3000/mm3) x A *° |
11 % (2 %) |
13 % (4 %) |
13 % (4 %) |
16 % (4 %) |
|
Lymfadenopatie |
1 % (1 %) |
3 % (1 %) |
11 % (5 %) |
14 % (11 %) |
|
Poruchy metabolismu a výživy | ||||
|
Snížená hladina glukózy v krvi (<55 mg/dl)x |
3 % (5 %) |
27 % (27 %) |
5 % (3 %) |
15 % (13 %) |
|
Psychiatrické poruchy | ||||
|
10 % (11 %) |
24 % (31 %) |
44 % (41 %) |
25 % (24 %) | |
|
Úzkost |
3 % (5 %) |
6 % (5 %) |
10 % (11 %) |
15 % (13 %) |
|
Poruchy nervového systému | ||||
|
27% (17 %) |
47 % (41 %) |
55 % (46 %) |
84 % (77 %) | |
|
Závrať |
3 % (4 %) |
14 % (14 %) |
28 % (26%) |
35 % (28 %) |
|
8 % (4 %) |
12 % (8 %) |
26 % (25 %) |
31 % (33 %) | |
|
Migréna |
2 % (2 %) |
4 % (3 %) |
5 % (4 %) |
12 % (7 %) |
|
Parestezie |
16 % (17 %) |
35% (39 %) |
40 % (43 %) |
19 % (21 %) |
|
Poruchy oka | ||||
|
Konjunktivitis |
1 % (1 %) |
2 % (3 %) |
6 % (6 %) |
12 % (10 %) |
|
Poruchy vidění A |
3 % (1 %) |
11 % (15 %) |
11 % (11 %) |
7 % (4 %) |
|
Poruchy ucha a labyrintu | ||||
|
Bolest uší |
0 % (1 %) |
<1 % (1 %) |
6 % (8 %) |
16 % (15 %) |
|
Srdeční poruchy | ||||
|
Palpitace * |
1 % (1 %) |
2 % (3 %) |
5 % (2 %) |
8 % (2 %) |
|
Cévní poruchy | ||||
|
Vazodilatace |
0 % (0 %) |
6 % (4 %) |
13 % (8 %) |
18 % (17 %) |
|
Hypertenze ° |
2 % (0 %) |
4 % (2 %) |
9 % (8 %) |
7 % (2 %) |
|
Respirační, hrudní a mediastinální poruchy | ||||
|
Infekce horních cest dýchacích |
18 % (19 %) |
3 % (2 %) | ||
|
Sinusitis |
4 % (6 %) |
6 % (6 %) |
16 % (18 %) |
36 % (26 %) |
|
Zhoršení kašle |
2 % (2 %) |
5 % (10 %) |
11 % (15 %) |
31 % (23 %) |
|
Dyspnoe * |
0 % (0 %) |
3 % (2 %) |
8 % (6 %) |
8 % (2 %) |
|
Gastrointestinální poruchy | ||||
|
4 % (2 %) |
7 % (10 %) |
21 % (19 %) |
35 % (29 %) | |
|
Zácpa |
1 % (1 %) |
12 % (12 %) |
22 % (24 %) |
24 % (18 %) |
|
3 % (4 %) |
13 % (13 %) |
32 % (30 %) |
48 % (49 %) | |
|
Zvracení A |
5 % (1 %) |
4 % (6 %) |
10 % (12 %) |
21 % (19 %) |
|
5 % (3 %) |
11 % (6 %) |
18 % (16 %) |
32 % (24 %) | |
|
Poruchy jater a žlučových cest | ||||
|
Zvýšená alaninaminotransferáza (SGPT>5 násobek výchozí hodnoty) x A * o |
18 % (5 %) |
14 % (5 %) |
4 % (2 %) |
19 % (6 %) |
|
Zvýšená aspartátaminotransferáza (SGOT>5 násobek výchozí hodnoty) x A * o |
6 % (1 %) |
4 % (1 %) |
2 % (1 %) |
4 % (0 %) |
|
Poruchy kůže a podkožní tkáně | ||||
|
Kožní poruchy |
1 % (0 %) |
4 % (4 %) |
19 % (17 %) |
6 % (8 %) |
|
Vyrážka A ° |
11 % (3 %) |
20 % (12 %) |
26 % (20 %) |
27 % (32 %) |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně | ||||
|
Hypertonie o |
2 % (1 %) |
41 % (31 %) |
57 % (57 %) |
26 % (24 %) |
|
Myalgie * ° |
8 % (8%) |
23 % (9 %) |
19 % (29 %) |
44 % (28 %) |
|
Myasténie |
2 % (2 %) |
39 % (40 %) |
57 % (60 %) |
13 % (10 %) |
|
10 % (7 %) |
26 % (24 %) |
31 % (32 %) |
36 % (37 %) | |
|
Bolest končetin |
6 % (3 %) |
14 % (12 %) |
0 % (0 %) | |
|
Poruchy ledvin a močových |
cest | |||
|
Retence moče |
1 % (1 %) |
4 % (6 %) |
15 % (13 %) |
- |
|
Pozitivní nález proteinů v moči (>1+)x |
25 % (2 6%) |
14 % (11 %) |
5 % (5 %) |
5 % (3 %) |
|
Časté močení |
1 % (1 %) |
6 % (5%) |
12 % (11 %) |
3 % (5 %) |
|
Inkontinence |
1 % (1 %) |
8 % (15%) |
20 % (19 %) |
2 % (1 %) |
|
Urgentní močení |
1 % (1 %) |
8 % (7%) |
21 % (17 %) |
4 % (2 %) |
|
Poruchy reprodukčního systému a prsu | ||||
|
Dysmenorea |
2 % (0 %) |
<1 % (<1 %) |
6 % (5 %) |
18 % (11 %) |
|
Menstruační poruchy * |
1 % (2 %) |
9 % (13 %) |
10 % (8 %) |
17 % (8 %) |
|
Metroragie |
2 % (0 %) |
12 % (6 %) |
10 % (10 %) |
15 % (8 %) |
|
Impotence |
1 % (0 %) |
7 % (4 %) |
10 % (11 %) |
2 % (1 %) |
|
Celkové poruchy a reakce v místě aplikace | ||||
|
Reakce v místě aplikace (různého druhu) A * ° § |
52 % (11 %) |
78 % (20 %) |
89 % (37 %) |
85 % (37 %) |
|
Nekróza v místě aplikace*° |
1 % (0 %) |
5 % (0 %) |
6 % (0 %) |
5 % (0 %) |
|
Chřipce podobné příznaky& A ^ o |
44 % (18 %) |
61 % (40 %) |
43 % (33 %) |
52 % (48 %) |
|
Horečka A * o |
13 % (5 %) |
40 % (13 %) |
29 % (24 %) |
59 % (41 %) |
|
Bolest |
4 % (4 %) |
31 % (25 %) |
59 % (59 %) |
52 % (48 %) |
|
1 % (0 %) |
5 % (4 %) |
15 % (8 %) |
15 % (15 %) | |
|
Periferní edém |
0 % (0 %) |
7 % (7 %) |
21 % (18 %) |
7 % (8 %) |
|
Asténie * |
22 % (17 %) |
63 % (58 %) |
64 % (58 %) |
49 % (35 %) |
|
Mrazení A* ° |
5 % (1 %) |
23 % (7 %) |
22 % (12 %) |
46 % (19 %) |
|
Pocení |
2 % (1 %) |
6 % (6 %) |
10 % (10 %) |
23 % (11 %) |
|
Slabost * |
0 % (1 %) |
8 % (5 %) |
6 % (2 %) |
15 % (3 %) |
|
Pro popis určitých reakcí a souvisejících stavů se používá nejvhodnější termín dle MedDRA a jeho synonyma. x Laboratorní abnormalita A Signifikantně spojeno s léčbou přípravkem Extavia u pacientů s první příhodou připomínající SM, p<0,05 * Signifikantně spojené s léčbou přípravkem Extavia pro relaps-remitentní RS, p<0,05 ° Signifikantně spojené s léčbou přípravkem Extavia pro sekundárně progresivní RS, p<0,05 § Reakce v místě vpichu (různého druhu) zahrnují všechny nežádoucí příhody, které se vyskytly v místě podání, tj. následující stavy: krvácení v místě aplikace, hypersenzitivita v místě aplikace, zánět v místě aplikace, nahromadění látky v místě aplikace, nekróza v místě aplikace, bolest v místě aplikace, reakce v místě aplikace, edém v místě aplikace a atrofie v místě aplikace & „Chřipce podobné příznaky“ označují chřipkový syndrom a/nebo kombinaci nejméně dvou následujících nežádoucích příhod: horečka, mrazení, myalgie, slabost, pocení. | ||||
Tabulka 2: Nežádoucí účinky (NÚ) zjištěné po zavedení přípravku na trh (četnosti - kde
jsou známé - vypočtené na základě shromážděných dat z klinických studií n=1 093)
|
Třídy orgánových systémů |
Velmi časté (>1/10) |
Časté (>1/100 až <1/10) |
Méně časté (>1/1 000 až <1/100) |
Vzácné (>1/10 000 až <1/1 000) |
Četnost není známo |
|
Poruchy krve a lymfatického systému |
Anemie |
T rombocytopenie |
Trombotická mikroangiopatie včetně trombotické trombocytopenické purpury/hemolyticko -uremického syndromu# | ||
|
Poruchy imunitního systému |
Anafylaktické reakce |
Syndrom zvýšené propustnosti kapilár u již dříve existující monoklonální gamopatie* | |||
|
Endokrinní poruchy |
Hypotyreóza |
Hypertyreóza, porucha štítné žlázy | |||
|
Poruchy metabolismu a výživy |
Zvýšení tělesné hmotnosti, snížení tělesné hmotnosti |
Zvýšení triglyceridů v krvi |
Anorexie* | ||
|
Psychiatrické poruchy |
Stavy zmatenosti |
Suicidální pokusy (viz také bod 4.4), emocionální labilita | |||
|
Poruchy nervového systému |
Křeče | ||||
|
Srdeční poruchy |
Kardiomyopatie* | ||||
|
Respirační, hrudní a mediastinální poruchy |
Bronchospasmus* |
Plicní arteriální hypertenze** | |||
|
Gastrointestinální poruchy |
Pankreatitida | ||||
|
Poruchy jater a žlučových cest |
Zvýšený bilirubin v krvi |
Zvýšená gamaglutamyl- transferáza, hepatitis |
Poškození jater (včetně hepatitidy), selhání jater* | ||
|
Poruchy kůže a podkožní tkáně |
Kopřivka, pruritus, alopecie |
Změny barvy kůže | |||
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Artralgie |
Léky vyvolaný lupus erythematodes | |||
|
Poruchy ledvin a močových cest |
Nefrotický syndrom, glomeruloskleróza (viz bod 4.4)* , # | ||||
|
Poruchy reprodukčního systému a prsu |
Menoragie |
* Nežádoucí účinky získané v postregistračním období.
# Tyto nežádoucí účinky se vztahují k celé skupině přípravků obsahujících interferon beta (viz bod 4.4).
** Znění textu dle druhu přípravků obsahujících interferon viz dále Plicní arteriální hypertenze“
Plicní arteriální hypertenze
U přípravků s interferonem beta byly hlášeny případy plicní arteriální hypertenze (PAH). Příhody byly hlášeny v různých časových bodech až do několika let od zahájení léčby interferonem beta.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Interferon beta-1b byl podáván dospělým pacientům s karcinomem v individuálních dávkách až 5 500 mikrogramů (176 milionů IU) intravenózně třikrát týdně bez závažných nežádoucích účinků ovlivňujících vitální funkce.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Imunostimulancia, interferony, ATC kód: L03AB08
Interferony patří do skupiny cytokinů, což jsou přirozeně se vyskytující proteiny. Interferony mají molekulovou hmotnost od 15 000 do 21 000 daltonů. Jsou známy tři velké skupiny interferonů: alfa, beta a gama. Interferon alfa, interferon beta a interferon gama mají překrývající se a přesto odlišnou biologickou účinnost. Účinky interferonu beta-1b jsou druhově specifické, a proto farmakologické informace, které se nejvíce týkají interferonu beta-1b jsou získány ze studií na kulturách lidských buněk nebo ze studií in vivo na lidech.
Mechanismus účinku
Interferon beta-1b má antivirovou i imunoregulační aktivitu. Mechanismus účinku interferonu beta-1b u roztroušené sklerózy dosud není přesně znám. Jisté však je, že vlastnosti interferonu beta-1b modifikující biologickou odpověď u roztroušené sklerózy jsou zprostředkovány jeho interakcí se specifickými buněčnými receptory nacházejícími se na povrchu lidských buněk. Vazba interferonu beta-1b na tyto receptory indukuje expresi množství genových produktů, které jsou považovány za mediátory biologického účinku interferonu beta-1b. Množství těchto produktů bylo měřeno v séru a v buněčné frakci krve odebrané pacientům léčeným interferonem beta-1b. Interferon beta-1b snižuje vazebnou afinitu a zvyšuje internalizaci a degradaci receptorů interferonu gama. Interferon beta-1b také zesiluje supresorovou aktivitu mononukleárních buněk v periferní krvi.
Klinická účinnost a bezpečnost
Nebyla prováděna žádná zvláštní sledování týkající se vlivu přípravku Extavia na kardiovaskulární systém, respirační systém a funkci endokrinních orgánů.
Recidivující-remitentní roztroušená skleróza (RR-RS)
Byla provedena jedna kontrolovaná klinická studie u pacientů s relaps-remitentní roztroušenou sklerózou, kteří byli schopni chodit sami bez pomoci (počáteční EDSS 0-5,5). U pacientů dostávajících přípravek Extavia došlo ke snížení frekvence (30 %) a závažnosti klinických relapsů a počtu hospitalizací vztahujících se k onemocnění. Navíc se prodloužil interval bez relapsů. Není důkaz o účinku přípravku Extavia na trvání relapsů nebo na příznaky mezi dvěma relapsy a nebyl pozorován významný účinek na progresi onemocnění u relaps-remitentní RS.
Sekundární progresivní roztoušená skleróza (SP-RS)
Byly provedeny dvě klinické studie s přípravkem Extavia u 1657 pacientů trpících sekundárně progresivní formou RS (základní EDSS 3-6,5, tj. pacienti byli schopni chodit). Pacienti s mírným onemocnění a pacienti, kteří nebyli schopni chodit, nebyli studováni. Tyto dvě studie přinesly nekonsistentní výsledky pro primární endpoint potvrzení progrese onemocnění představované dobou opoždění progrese invalidity:
První z těchto dvou studií prokázala statisticky významné oddálení progrese (Hazard Ratio -poměr rizika = 0,69, 95% interval spolehlivosti (0,55, 0,86), p=0,0010, odpovídající 31% redukci rizika přisouzené přípravku Extavia) a oddálení progrese k těžké invaliditě, kdy se pacienti stávají závislí na invalidním vozíku (Hazard Ratio - poměr rizika = 0,61, 95% interval spolehlivosti (0,44, 0,85), p=0,0036, odpovídající 39% redukci rizika přisouzené přípravku Extavia) u pacientů, kteří byli léčeni přípravkem Extavia. Tento účinek pokračoval po celé období pozorování až do doby 33 měsíců. Tento efekt byl pozorován na všech úrovních hodnocených schopností a byl nezávislý na aktivitě relapsů.
Ve druhé studii hodnotící léčbu přípravku Extavia u sekundárně progresivní roztroušené sklerózy nebylo pozorováno žádné časové opoždění progrese postižení. Existují důkazy o tom, že pacienti sledovaní v rámci této studie měli celkově méně aktivní onemocnění než pacienti se sekundárně progresivní RS v první studii.
V retrospektivních meta-analýzách včetně údajů z obou studií byl zjištěn statisticky významný celkový léčebný účinek, který byl (p=0,0076, 8,0 MIU přípravku Extavia versus všichni pacienti léčení placebem).
Retrospektivní analýzy v podskupinách ukázaly, že léčebný efekt na progresi postižení je nejpravděpodobnější u pacientů s aktivním onemocněním před zahájením léčby (Hazard Ratio -poměr rizika = 0,72, 95% interval spolehlivosti (0,59, 0,88), p=0,0011, odpovídající 28% redukce rizika přisouzené účinku přípravku Extavia u pacientů s relapsy nebo s vyjádřenou EDSS progresí, 8,0 MIU přípravku Extavia versus všichni pacienti léšení placebem). Z těchto retrospektivních analýz podskupin je zřejmé, že relapsy stejně jako vyjádřená EDSS progrese (EDSS >1 bod nebo >0,5 bodu pro EDSS >=6 v předchozích dvou letech) mohou pomoci identifikovat pacienty s aktivním onemocněním.
V obou studiích došlo ke snížení (30 %) četnosti klinických relapsů u pacientů se sekundárně progresivní roztroušenou sklerózou léčených přípravkem Extavia. O tom, že by přípravek Extavia měl efekt na trvání relapsů, není důkaz.
Jediná klinická příhoda připomínající roztroušenou sklerózu
Jedno kontrolované klinické hodnocení léčby přípravku Extavia bylo provedeno u pacientů s jedním klinickým projevem připomínajícím roztroušenou sklerózu podpořeným nálezem na magnetické rezonanci (MRI), (nejméně dvě klinicky němé léze v T2 váženém obraze). Do hodnocení byli zahrnuti pacienti s monofokálním či multifokálním nástupem onemocnění ( tj. pacienti s klinickými projevy jedné nebo přinejmenším dvou lézí centrálního nervového systému). Jiné onemocnění, které by mohlo lépe vysvětlit příznaky pacienta, muselo být vyloučeno. Tato studie měla dvě fáze, placebem kontrolovaná fáze následovaná předem naplánovanou fází následného sledování. Placebem kontrolovaná fáze trvala podle toho, které z kritérií nastalo dříve: buď 2 roky nebo do doby, než se u pacienta objevila klinicky jednoznačná roztroušená skleróza (clinically definite multiple sclerosis CDMS). Po placebem kontrolované fázi vstoupili pacienti do předem naplánované fáze sledování s přípravkem Extavia, aby se hodnotily účinky okamžitého zahájení léčby přípravkem Extavia, oproti jejímu opožděnému zahájení. Byli porovnáváni pacienti na začátku randomizovaní do skupiny s přípravkem Extavia („skupina s okamžitou léčbou“) a do skupiny s placebem („skupina s opožděnou léčbou“). Pacienti ani zkoušející neznali počáteční alokaci léčby.
V placebem kontrolované fázi přípravek Extavia zpomalil progresi z první klinické příhody do klinicky definitivní roztroušené sklerózy (CDMS) statisticky signifikantním a klinicky významným způsobem, což odpovídalo snížení rizika o 47 % (poměr rizika = 0,53, 95% interval spolehlivosti (0,39, 0,73), p<0,0001). Během dvou let trvání studie došlo k CDMS u 45 % pacientů ve skupině s placebem v porovnání s 28 % ve skupině s přípravkem Extavia (výpočet odhadu dle Kaplan-Meier). Přípravek Extavia prodloužil dobu do CDMS o 363 dnů, z 255 dnů ve skupině s placebem, a až na 618 dnů ve skupině s přípravkem Extavia (založeno na 25. percentilech). Tento léčebný efekt byl stále patrný po dalším roce sledování a tehdy bylo riziko redukováno o 41 % (poměr rizika = 0,59, 95% interval spolehlivosti (0,42, 0,83), p=0,0011). Během tříletého hodnocení se CDRS objevila u 51 % pacientů ze skupiny s opožděnou léčbou ve srovnání s 37 % pacientů ze skupiny s okamžitou léčbou (výpočet odhadu dle Kaplan-Meier). Bylo pozorováno přetrvávání léčebného účinku, přestože většina pacientů z placebové skupiny byla v třetím roce studie léčena přípravkem Extavia.
Robustnost léčebného účinku se rovněž projevila zpomalením progrese roztroušené sklerózy podle McDonaldových kritérií. Během dvou let bylo riziko 85 % ve skupině s placebem a 69 % ve skupině s přípravkem Extavia (poměr rizika = 0,57, interval spolehlivosti 95% (0,46, 0,71),
p<0,00001).
Po 3 letech ukázala předem plánovaná interim analýza, že k progresi EDSS (potvrzeno zvýšení o více než nebo rovno 1,0 ve srovnání s výchozí hodnotou) došlo u 24 % pacientů ve skupině s opožděnou léčbou ve srovnání s 16 % ve skupině s okamžitou léčbou (poměr rizika = 0,6, 95% interval spolehlivosti (0,39, 0,92), p=0,022). Pro většinu pacientů, kteří dostali „okamžitou“ léčbu, nejsou důkazy o přínosu ve smyslu progrese postižení. Sledování pacientů nadále pokračuje, aby byly získány další údaje. Žádný přínos v kvalitě života, který by bylo možné přisoudit podávání přípravku Extavia, (měřeno FHRS - Funkční Hodnocení RS: Index Výsledku Léčby) nebyl pozorován.
Analýzy podskupin podle výchozích faktorů prokázaly účinnost ve všech hodnocených podskupinách. Signifikantní účinky byly také získány u pacientů s méně rozšířenou a méně aktivní nemocí v době první příhody. Riziko progrese do CDMS během dvou let u pacientů s monofokálním onemocněním bylo 47 % ve skupině s placebem a 24 % ve skupině s přípravkem Extavia, bez zesílení gadoliniem (Gd-) byla rizika 41 % a 20 %, s méně než 9 lézemi v T2 váženém obraze byla rizika 39 % a 18 %. Další analýzy podskupin ukázaly vysoké riziko progrese do CDMS během 2 let u pacientů s monofokálním onemocněním s nejméně 9 lézemi v T2 váženém obraze (55 % pro skupinu s placebem, 26 % pro skupinu s přípravkem Extavia ) nebo při zesílení Gd (63 % oproti 33 %). U pacientů s multifokálním onemocněním nezáviselo riziko CDMS na nálezech MRI při výchozím stavu, což ukazuje na vysoké riziko CDMS pro diseminaci onemocnění, zjištěné klinickými nálezy. Dlouhodobý vliv včasného zahájení léčby přípravkem Extavia avšak i u těchto vysoce rizikových skupin není znám, protože studie byla navržena spíše ke zhodnocení doby do vzniku CDMS, než ke zhodnocení dlouhodobého vývoje onemocnění. Navíc definice vysoce rizikového pacienta není v současné době dostatečně zavedena, přestože spíše konzervativní přístup chápe rizikového pacienta jako pacienta, u kterého je nález alespoň devíti hyperintenzivních lézí v T2 váženém obraze na prvním skenu a alespoň jedné nové léze v T2 váženém obraze nebo jedné nové Gd zesílené léze na následujícím skenu, který nebyl proveden dříve, než za 1 měsíc po prvním skenu. V každém případě by léčba měla být zvažována pouze u pacientů považovaných za vysoce rizikové.
Léčba přípravkem Extavia byla ve studii s pacienty s jednou klinickou příhodou velmi dobře snášena, což indikuje vysoká míra dokončení klinického hodnocení (92,8 % ve skupině s přípravkem Extavia). Pro zvýšení snášenlivosti přípravku Extavia ve studii u pacientů s jednou klinickou příhodou, byla na počátku terapie použita titrace dávky a byly podávány nesteroidní protizánětlivé léčivé přípravky. Navíc většina pacientů ve studii používala autoinjektor.
RR-RS, SP-RS a jedina klinická příhoda připomínající RS
Ve všech studiích roztroušené sklerózy měl přípravek Extavia vliv na snížení aktivity onemocnění (akutní zánět v centrálním nervovém systému a trvalé poškození tkáně) měřeno zobrazením pomocí magnetické rezonance (MRI). Vztah mezi aktivitou RS, měřenou pomocí MRI, a klinickým nálezem není v současnosti ještě plně pochopen.
5.2 Farmakokinetické vlastnosti
Sérové hladiny přípravku Extavia byly sledovány u pacientů a dobrovolníků pomocí biologické zkoušky, která nebyla zcela specifická. Po subkutánní injekci 500 mikrogramů (16 milionů IU) interferonu beta-1b byly naměřeny maximální sérové hladiny asi 40 IU/ml za 1-8 hodin po podání. Z různých studií byly průměrná clearance a poločas dispoziční fáze ze séra odhadnuty maximálně na 30 ml/min/kg a 5 hodin.
Podávání injekcí přípravku Extavia obden nevede ke zvýšení sérové hladiny a farmakokinetika se pravděpodobně během léčby nemění.
Absolutní biologická dostupnost z podkožně podávaného interferonu beta-1b byla přibližně 50 %.
5.3 Předklinické údaje vztahující se k bezpečnosti
Studie akutní toxicity nebyly provedeny. Jelikož hlodavci nereagují na lidský interferon beta, studie opakovaně podávaných dávek byly prováděny na opicích makak rhesus. Byla pozorována přechodná hypertermie, výrazné přechodné zvýšení počtu lymfocytů a výrazný přechodný pokles trombocytů a segmentovaných neutrofilů.
Dlouhodobé studie nebyly prováděny. Reprodukční studie prováděné na opicích makak rhesus prokázaly maternální toxicitu a zvýšenou míru potratů s výslednou prenatální mortalitou. U přežívajících zvířat nebyly pozorovány žádné malformace.
Sledování fertility nebylo prováděno. Nebyl pozorován vliv na estrální cyklus u opic. Zkušenosti s jinými interferony naznačují možnost poškození mužské i ženské fertility.
V jediné studii na genotoxicitu (Ames test) nebyl pozorován žádný mutagenní efekt. Studie kancerogenity nebyly prováděny. Test na transformaci buněk in vitro neprokázal žádné známky tumorigenního potenciálu.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek
Lidský albumin Mannitol (E421)
Rozpouštědlo Chlorid sodný Voda na injekci
6.2 Inkompatibility
Tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky s výjimkou dodávaného rozpouštědla uvedeného v bodě 6.6.
6.3 Doba použitelnosti
2 roky.
Po rekonstituci je doporučeno přípravek bezprostředně použít. Stabilita však byla doložena na
3 hodiny při teplotě 2°C - 8°C.
6.4 Zvláštní opatření pro uchovávání
Neuchovávejte při teplotě nad 25°C.
Chraňte před mrazem.
Podmínky uchovávání rekonstituovaného léčivého přípravku jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
Prášek
Injekční lahvička o objemu 3 ml (čiré sklo typ I) s butylovou pryžovou zátkou (typ I) a hliníkovým pertlem obsahující 300 mikrogramů (9,6 milionů IU) prášku (rekombinantní interferonum beta-1b).
Rozpouštědlo
Předplněná inj. stříkačka opatřená stupnicí (s označením: 0,25 ml, 0,5 ml, 0,75 ml, 1,0 ml) o objemu 2,25 ml (sklo typ I) s 1,2 ml rozpouštědla.
Velikost balení
- Balení obsahující 5 injekčních lahviček s práškem a 5 předplněných inj. stříkaček s rozpouštědlem.
- Balení obsahující 14 injekčních lahviček s práškem a 14 předplněných inj. stříkaček s rozpouštědlem.
- Balení obsahující 15 injekčních lahviček s práškem a 15 předplněných inj. stříkaček s rozpouštědlem.
- Balení obsahující 14 injekčních lahviček s práškem a 15 předplněných inj. stříkaček s rozpouštědlem.
injekčních lahviček s práškem a 42 (3x14) injekčních lahviček s práškem a 45 (3x15) injekčních lahviček s práškem a 45 (3x15)
- 3měsíční vícečetné balení obsahující 42 (3x14) předplněných stříkaček s rozpouštědlem.
- 3měsíční vícečetné balení obsahující 45 (3x15) předplněných stříkaček s rozpouštědlem.
- 3měsíční vícečetné balení obsahující 42 (3x14) předplněných stříkaček s rozpouštědlem.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Krytka hrotu předplněné injekční stříkačky obsahuje derivát přírodního latexu. Proto může krytka hrotu obsahovat přírodní latex a neměla by přijít do styku s osobou, která je citlivá na latex.
Rekonstituce
Pro rekonstituci prášku musí být použita předplněná stříkačka s rozpouštědlem s jehlou nebo lahvičkovým adaptérem ke vstříknutí 1,2 ml rozpouštědla (injekčního roztoku chloridu sodného 5,4 mg/ml (0,54%) do injekční lahvičky s přípravkem Extavia. Prášek se musí zcela rozpustit bez třepání. Po rekonstituci by měl být z injekční lahvičky odebrán 1,0 ml roztoku do inj. stříkačky k podání 250 mikrogramů přípravku Extavia.
Kontrola před použitím
Rekonstituovaný přípravek musí být před použitím opticky zkontrolován. Naředěný přípravek je bezbarvý až mírně nažloutlý, slabě opalescentní až opalescentní.
Pokud naředěný léčivý přípravek obsahuje částečky nebo má změněnou barvu, musí být před použitím zlikvidován.
Likvidace
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/08/454/008-014
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 20. květen 2008
Datum posledního prodloužení registrace: 20. květen 2013
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce biologické léčivé látky
Boehringer Ingelheim RCV GmbH & Co KG Dr.-Boehringer-Gasse 5-11 A-1121 Vídeň Rakousko
Název a adresa výrobce odpovědného za propouštění šarží
Novartis Pharma GmbH Roonstrasse 25 D-90429 Norimberk Německo
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci dále předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky. Cyklus periodicky aktualizované zprávy o bezpečnosti přípravku (PSUR) Extavia je sladěný s referenčním přípravkem Betaferon, pokud nebude stanoveno jinak.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky;
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU KRABIČKA PRO JEDNOTLIVÉ BALENÍ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Extavia 250 mikrogramů/ml, prášek a rozpouštědlo pro injekční roztok interferonum beta-1b
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
1 lahvička obsahuje 300 mikrogramů (9,6 milionů IU) interferonu beta-1b.
1 ml naředěného roztoku obsahuje 250 mikrogramů (8,0 milionů IU) interferonu beta-1b.
3. SEZNAM POMOCNÝCH LÁTEK
Pomocné látky:
Prášek: lidský albumin, mannitol Rozpouštědlo: chlorid sodný, voda na injekci
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Prášek a rozpouštědlo pro injekční roztok
5 injekčních lahviček s práškem a 5 předplněných injekčních stříkaček s 1,2 ml rozpouštědla.
14 injekčních lahviček s práškem a 14 předplněných injekčních stříkaček s 1,2 ml rozpouštědla.
15 injekčních lahviček s práškem a 15 předplněných injekčních stříkaček s 1,2 ml rozpouštědla.
14 injekčních lahviček s práškem a 15 předplněných injekčních stříkaček s 1,2 ml rozpouštědla.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
K subkutánnímu podání po rozpuštění v 1,2 ml rozpouštědla. Pro jednorázové použití.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
Použitelné do:
Po rekonstituci je doporučeno přípravek bezprostředně použít. Stabilita byla doložena na 3 hodiny při teplotě 2°C - 8°C.
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Neuchovávejte při teplotě nad 25°C. Chraňte před mrazem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
|
EU/1/08/454/008 |
15 injekčních lahviček s práškem a 15 předplněných injekčních |
|
stříkaček s rozpouštědlem | |
|
EU/1/08/454/010 |
5 injekčních lahviček s práškem a 5 předplněných injekčních stříkaček |
|
rozpouštědlem | |
|
EU/1/08/454/011 |
14 injekčních lahviček s práškem a 14 předplněných injekčních |
|
stříkaček s rozpouštědlem | |
|
EU/1/08/454/013 |
14 injekčních lahviček s práškem a 15 předplněných injekčních |
stříkaček s rozpouštědlem
13. ČÍSLO ŠARŽE
č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
VNĚJŠÍ KRABIČKA VÍCEČETNÉHO BALENÍ (VČETNĚ „BLUE BOX“)
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Extavia 250 mikrogramů/ml, prášek a rozpouštědlo pro injekční roztok interferonum beta-1b
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
1 lahvička obsahuje 300 mikrogramů (9,6 milionů IU) interferonu beta-1b.
1 ml naředěného roztoku obsahuje 250 mikrogramů (8,0 milionů IU) interferonu beta-1b.
3. SEZNAM POMOCNÝCH LÁTEK
Pomocné látky:
Prášek: lidský albumin, mannitol Rozpouštědlo: chlorid sodný, voda na injekci
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Prášek a rozpouštědlo pro injekční roztok
3měsíční vícečetné balení: 42 (3 balení po 14) injekčních lahviček s práškem a 42 (3 balení po
14) předplněných injekčních stříkaček s 1,2 ml rozpouštědla.
3měsíční vícečetné balení: 45 (3 balení po 15) injekčních lahviček s práškem a 45 (3 balení po
15) předplněných injekčních stříkaček s 1,2 ml rozpouštědla.
3měsíční vícečetné balení: 42 (3 balení po 14) injekčních lahviček s práškem a 45 (3 balení po 15) předplněných injekčních stříkaček s 1,2 ml rozpouštědla.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
K subkutánnímu podání po rozpuštění v 1,2 ml rozpouštědla. Pro jednorázové použití.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
Použitelné do:
Po rekonstituci je doporučeno přípravek bezprostředně použít. Stabilita byla doložena na 3 hodiny při teplotě 2°C - 8°C.
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Neuchovávejte při teplotě nad 25°C. Chraňte před mrazem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/08/454/009 3měsíční vícečetné balení obsahující 45 injekčních lahviček s práškem
a 45 předplněných injekčních stříkaček s rozpouštědlem EU/1/08/454/012 3měsíční vícečetné balení obsahující 42 injekčních lahviček s práškem
a 42 předplněných injekčních stříkaček s rozpouštědlem EU/1/08/454/014 3měsíční vícečetné balení obsahující 42 injekčních lahviček s práškem
a 45 předplněných injekčních stříkaček s rozpouštědlem
13. ČÍSLO ŠARŽE
č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
VNITŘNÍ KRABIČKA VÍCEČETNÉHO BALENÍ (BEZ „BLUE BOX“)
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Extavia 250 mikrogramů/ml, prášek a rozpouštědlo pro injekční roztok interferonum beta-1b
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
1 lahvička obsahuje 300 mikrogramů (9,6 milionů IU) interferonu beta-1b.
1 ml naředěného roztoku obsahuje 250 mikrogramů (8,0 milionů IU) interferonu beta-1b.
3. SEZNAM POMOCNÝCH LÁTEK
Pomocné látky:
Prášek: lidský albumin, mannitol Rozpouštědlo: chlorid sodný, voda na injekci
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Prášek a rozpouštědlo pro injekční roztok
14 injekčních lahviček s práškem a 14 předplněných injekčních stříkaček s 1,2 ml rozpouštědla. Součást 3měsíčního vícečetného balení. Neprodávat samostatně.
15 injekčních lahviček s práškem a 15 předplněných injekčních stříkaček s 1,2 ml rozpouštědla. Součást 3měsíčního vícečetného balení. Neprodávat samostatně.
14 injekčních lahviček s práškem a 15 předplněných injekčních stříkaček s 1,2 ml rozpouštědla. Součást 3měsíčního vícečetného balení. Neprodávat samostatně.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
K subkutánnímu podání po rozpuštění v 1,2 ml rozpouštědla. Pro jednorázové použití.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
Použitelné do:
Po rekonstituci je doporučeno přípravek bezprostředně použít. Stabilita byla doložena na 3 hodiny při teplotě 2°C - 8°C.
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Neuchovávejte při teplotě nad 25°C. Chraňte před mrazem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/08/454/009 3měsíční vícečetné balení obsahující 45 injekčních lahviček s práškem
a 45 předplněných injekčních stříkaček s rozpouštědlem EU/1/08/454/012 3měsíční vícečetné balení obsahující 42 injekčních lahviček s práškem
a 42 předplněných injekčních stříkaček s rozpouštědlem EU/1/08/454/014 3měsíční vícečetné balení obsahující 42 injekčních lahviček s práškem
a 45 předplněných injekčních stříkaček s rozpouštědlem
13. ČÍSLO ŠARŽE
č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA PODÁNÍ
Extavia 250 mikrogramů/ml, prášek pro injekční roztok interferonum beta-1b Subkutánní podání
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
Po rekonstituci je doporučeno přípravek bezprostředně použít. Stabilita byla doložena na 3 hodiny při teplotě 2°C - 8°C.
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
250 mikrogramů (8,0 milionů IU)/ml po rozpuštění.
6. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Rozpouštědlo pro rekonstituci Extavia 1,2 ml roztoku chloridu sodného 5,4 mg/ml
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
Před použitím si přečtěte příbalovou informaci.
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA PODÁNÍ
Rozpouštědlo pro přípravek Extavia K subkutánnímu podání po rekonstituci.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
1,2 ml roztoku chloridu sodného 5,4 mg/ml
6. JINÉ
0,25 / 0,5 / 0,75 / 1,0
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro uživatele
Extavia 250 mikrogramů/ml, prášek a rozpouštědlo pro injekční roztok
interferonum beta-1b
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Extavia a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Extavia užívat
3. Jak se přípravek Extavia užívá
4. Možné nežádoucí účinky
5. Jak přípravek Extavia uchovávat
6. Obsah balení a další informace Dodatek - samostatné podávaní injekcí
1. Co je přípravek Extavia a k čemu se používá Co je Extavia
Extavia je typem přípravku známého jako interferon, používaného k léčbě roztroušené sklerózy. Interferony jsou bílkoviny (proteiny) produkované organismem, které pomáhají bojovat při napadení imunitního systému např. virovými infekcemi.
Jak Extavia působí
Roztroušená skleróza (RS) je dlouhodobý stav, který ovlivňuje centrální nervový systém (CNS), zejména funkci mozku a míchy. Zánět při RS ničí ochranné pouzdro (zvané myelin) kolem nervů CNS a brání nervům ve správné funkci. Toto se nazývá demyelinizace.
Přesná příčina RS je neznámá. Uvažuje se o tom, že v procesu, který poškozuje CNS, hraje důležitou úlohu abnormální odezva imunitního systému organismu.
K poškození CNS může dojít během atak (relapsu) RS. Toto může způsobit dočasné potíže, např. při chůzi. Symptomy mohou zcela nebo částečně vymizet.
Interferon beta-1b upravuje odezvu imunitního systému a pomáhá tak snížit aktivitu onemocnění. Jak Extavia pomáhá bojovat s vaší nemocí
Jediná klinická příhoda indikující vysoké riziko vzniku roztroušené sklerózy: Léčba přípravkem Extavia zpomaluje přechod do definitivní roztroušené sklerózy.
Relaps-remitentní roztroušená skleróza: Pacienti s relaps-remitentní formou RS měli příležitostné ataky nebo relapsy, během nichž se symptomy znatelně zhoršily. Léčba přípravkem Extavia snížila četnost atak a zmírnila je. Snižuje počet dní strávených v nemocnici kvůli onemocnění a prodlužuje čas bez relapsů.
Sekundárně progresivní roztroušená skleróza: V některých případech pacienti s relaps-remitentní RS zjišťují, že jejich symptomy se zhoršují a progredují do další formy RS zvané sekundárně progresivní RS. V těchto případech pacienti pozorují postupné zhoršení, ať s relapsy nebo bez nich. Extavia může snižovat počet a závažnost atak a zpomalovat postup onemocnění.
K čemu se Extavia používá Extavia se používá u pacientů,
► kteří mají poprvé příznaky, jež upozorňují na vysoké riziko rozvoje roztroušené sklerózy. Váš ošetřující lékař před zahájením léčby vyloučí jakékoli další příčiny, které by mohly tyto příznaky vysvětlovat.
► kteří trpí relaps-remitentní formou roztroušené sklerózy nejméně se dvěma relapsy v posledních dvou letech.
► kteří trpí sekundárně progresivní formou roztroušené sklerózy a mají aktivní onemocnění projevující se relapsy.
2. Čemu musíte věnovat pozornost, než začnete přípravek Extavia užívat
Neužívejte přípravek Extavia
- jestliže jste alergický(á) na přirozený nebo rekombinantní interferon beta, lidský albumin nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
- jestliže jste těhotná. Nesmíte zahájit léčbu přípravkem Extavia (viz bod „Těhotenství“ níže).
- jestliže jste otěhotněla nebo plánujete těhotenství. Nesmíte zahájit léčbu přípravkem Extavia a informujte svého lékaře (viz bod „Těhotenství“ níže).
- jestliže trpíte závažným depresivním onemocněním a/nebo máte myšlenky na sebevraždu (viz „Upozornění a opatření“ a bod 4. „Možné nežádoucí účinky“);
- jestliže trpíte závažným onemocněním jater (viz „Upozornění a opatření“, „Další léčivé přípravky a Extavia“ a bod 4. „Možné nežádoucí účinky“).
► Informujte svého lékaře, jestliže se na vás vztahuje něco z výše uvedeného.
Upozornění a opatření
Před použitím přípravku Extavia se poraďte se svým lékařem:
- Zda nemáte monoklonální gamapatii. Jedná se o poruchu imunitního systému, při níž se v krvi nalézají abnormální bílkoviny. Při používání léčiv jako Extavia se mohou vyvinout problémy (syndrom systémového kapilárního průsaku) s malými krevními cévami (kapilárami). Toto může vést k šoku (zhroucení), který může být smrtelný.
- Pokud jste měl(a) nebo máte deprese nebo se dříve u vás objevily sebevražedné myšlenky. Váš lékař Vás bude během léčby pečlivě sledovat. Jestliže jsou deprese a/nebo sebevražedné úmysly vážné, přípravek Extavia Vám nebude předepsán (viz také „Neužívejte Extavia“).
- Jestliže jste prodělal(a) záchvaty křečí nebo pokud užíváte léky na léčbu epilepsie
(antiepileptika, Váš lékař bude léčbu pečlivě sledovat (viz také „Další léčivé přípravky a Extavia“ a bod 4. „Možné nežádoucí účinky“).
- Jestliže máte vážné problémy s ledvinami, Váš lékař může během léčby sledovat funkci ledvin.
- Pokud jste měl(a) alergickou reakci na latex. Krytka hrotu předplněné injekční stříkačky obsahuje derivát přírodního latexu. Proto může krytka hrotu obsahovat přírodní latex.
V době, kdy užíváte Extavia, Váš lékař musí rovněž vědět následující:
- Jestliže zpozorujete příznaky jako svědění po celém těle, otoky obličeje a/nebo jazyka nebo náhlou dusností. Může se jednat o příznaky vážné alergické reakce, která může ohrozit život.
- Pokud budete pociťovat větší smutek nebo beznaděj než před zahájením léčby přípravkem Extavia, nebo objeví-li se u vás sebevražedné myšlenky. Pokud se u vás objevily deprese během léčby přípravkem Extavia, budete možná vyžadovat zvláštní léčbu, Váš lékař vás bude pečlivě sledovat a může také zvážit ukončení léčby. Jestliže trpíte vážnou depresí a/nebo sebevražednými úmysly, nebudete přípravkem Extavia léčen(a) (viz také „Neužívejte Extavia“).
- Jestliže jste si povšimli jakýchkoliv neobvyklých modřin, nadměrného krvácení po poranění nebo se vám zdá, že příliš snadno onemocníte infekcemi. Může se jednat o příznaky poklesu počtu krvinek nebo krevních destiček (buňky pomáhající srážení krve). Budete možná vyžadovat zvláštní sledování svým lékařem.
- Jestliže pociťujete ztrátu chuti k jídlu, únavu, pocit nevolnosti (nauzeu), opakované zvracení a zejména tehdy, kdy zaznamenáte celkové svědění, zežloutnutí kůže nebo očního bělma a snadnou tvorbu modřin. Tyto příznaky mohou poukazovat na problémy s játry. V klinických studiích se u pacientů léčených přípravkem Extavia vyskytly změny hodnot jaterních funkcí. Jako u jiných interferonů beta bylo u pacientů užívajících Extavia vzácně uváděno závažné poškození jater, včetně případů jaterního selhání. Nejzávažnější poškození bylo uváděno u pacientů užívajících jiné léčivé přípravky nebo kteří trpěli nemocemi, které mohou ovlivnit funkci jater (např.nadměrné požívání-abusus alkoholu, závažná infekce).
- Jestliže trpíte příznaky, jako je nepravidelný srdeční tep, otoky, např. kotníků nebo nohou, nebo dušnost. Může to poukazovat na onemocnění srdečního svalu (kardiomyopatie), které bylo hlášeno u pacientů užívajících Extavia.
- Jestliže zaznamenáte bolest břicha vyzařující do zad a/nebo je vám špatně nebo máte horečku. Může to poukazovat na zánět slinivky břišní (pankreatida), který byl hlášený při užívání přípravku Extavia. Toto je často spojeno se zvýšením koncentrace určitých tuků
v krvi (triglyceridů).
► Přestaňte užívat Extavia a okamžitě informujte svého lékaře, jestliže se u vás vyskytne některý z výše uvedených příznaků.
Při užívání Extavia je dále třeba vzít v úvahu následující:
- Budou se muset provést krevní vyšetření, aby byl stanoven počet Vašich krvinek, krevní biochemie a jaterní enzymy. Toto bude provedeno před tím, než začnete přípravek Extavia užívat, pravidelně poté, kdy byla léčba přípravkem Extavia zahájena a poté periodicky během léčby, i když nemáte žádné konkrétní příznaky. Tyto krevní testy se provedou navíc k testům, které se normálně dělají pro kontrolu RS.
- Jestliže trpíte srdeční chorobou, symptomy podobné chřipce, které se často vyskytují na počátku léčby, se mohou ukázat pro vás jako stresující. Extavia se musí užívat
s opatrností a váš lékař musí sledovat, zdali nedochází u vás ke zhoršení srdečního onemocnění, zejména na počátku léčby. Samotný přípravek Extavia srdce přímo neovlivňuje.
- Bude u Vás prováděno funkční vyšetření štítné žlázy, pravidelně nebo kdykoli to bude Váš lékař považovat za nezbytné z jiných důvodů.
- Extavia obsahuje lidský albumin, proto představuje možné riziko přenosu virových onemocnění. Nelze vyloučit riziko přenosu Creutzfeld-Jacobovy nemoci (CJD).
- Během léčby přípravkem Extavia může vaše tělo produkovat látky, které se nazývají neutralizační protilátky, které mohou reagovat s přípravkem Extavia. Není jasné, zdali tyto neutralizační protilátky snižují účinnost léčby. Neutralizační protilátky nejsou vytvářeny u všech pacientů. V současnosti není možné určit předem, kteří pacienti patří do této skupiny.
- Během léčby přípravkem Extavia se mohou objevit poruchy ledvin, které mohou snížit funkci ledvin a to včetně zjizvení (glomerulosklerózy). Váš lékař může provést testy, aby funkci ledvin prověřil.
- Během léčby se v malých cévách mohou vyskytnout krevní sraženiny. Tyto krevní sraženiny by mohly ovlivnit funkci ledvin. K tomu může dojít několik týdnů až několik let po zahájení léčby přípravkem Extavia. Může se stát, že lékař bude chtít zkontrolovat krevní tlak, krevní obraz (počet krevních destiček) a funkci ledvin.
Reakce v místě vpichu injekce
Během léčby přípravkem Extavia je pravděpodobné, že u vás dojde k reakcím v místě vpichu injekce. Příznaky zahrnují zarudnutí, otok, změnu barvy kůže, zánět, bolest a hypersenzitivitu. Rozpad kůže a poškození tkáně (nekróza) kolem místa vpichu jsou uváděny méně často. Výskyt reakcí v místě vpichu obvykle časem klesá.
Porušení kůže a zničení tkáně v místě vpichu může vést k tvorbě jizev. Jestliže je stav závažný, lékař může odstranit cizí hmoty a neživé tkáně (debridement) a (méně často) provést transplantaci kůže. Zhojení může trvat až 6 měsíců.
Aby bylo riziko vzniku reakce v místě injekce co nejmenší, je nutné:
- užít sterilní (aseptickou) techniku při podání injekce,
- střídat místa injekce při podání každé dávky (viz dodatek „Samostatné podávání injekcí“).
Reakce v místě podání se mohou vyskytovat méně často, pokud se používá autoinjektor. Váš lékař nebo zdravotní sestra vám o tom řekne více.
Jestliže se u vás vyskytne jakékoliv porušení kůže spojené s otokem nebo vytékáním tekutiny z místa vpichu:
► Přerušte injekční podávání přípravku Extavia a poraďte se se svým lékařem.
► Jestliže máte pouze jedno bolestivé místo vpichu (lézi) a poškození tkání (nekróza) není příliš rozsáhlé, můžete pokračovat v užívání přípravku Extavia.
► Jestliže máte více bolestivých míst vpichu (vícečetné léze), je třeba přerušit léčbu přípravkem Extavia, dokud nedojde k jejich zhojení.
Váš lékař bude pravidelně kontrolovat způsob, jakým si aplikujete injekci, zvláště tehdy, došlo-li u vás ke vzniku reakcí v místě vpichu.
Děti a dospívající
U dětí nebo dospívajících nebyly provedeny žádné formální klinické studie.
Jsou však k dispozici některá data u dospívajících ve věku od 12 do 17 let, která naznačují, že bezpečnostní profil u této skupiny je podobný jako u dospělých. Přípravek Extavia by neměl být používán u dětí ve věku do 12 let, protože pro tuto věkovou skupinu nejsou dostupné žádné informace.
Další léčivé přípravky a Extavia
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Žádné formální studie interakcí nebyly provedeny za účelem zjištění, zda Extavia působí na jiné léčivé přípravky nebo je nimi ovlivňován.
Léčba přípravkem Extavia probíhající současně s podáváním jiných léčivých přípravků ovlivňujících imunitní systém s výjimkou protizánětlivých přípravků zvaných kortikosteroidy nebo adrenokortikotropního hormonu (ACTH) se nedoporučuje.
Extavia se musí užívat opatrně při:
- podávání léčiv, které potřebují určitý jaterní enzymový systém (známý jako cytochrom P450) k jejich vyloučení z organismu, např. léčivé přípravky užívané pro léčbu epilepsie (např. fenytoin);
- podávání léčiv, které mají účinek na tvorbu krvinek.
Přípravek Extavia s jídlem a pitím
Přípravek Extavia je podáván injekcí pod kůži, neočekává se proto žádný vliv jídla nebo pití na Extavia
Těhotenství
Ženy, které mohou otěhotnět, by měly během léčby přípravkem Extavia užívat vhodnou antikoncepci.
► Pokud jste těhotná, nebo si myslíte, že byste mohla otěhotnět, informujte svého lékaře. Pokud jste těhotná, léčba přípravkem Extavia by neměla být zahájena (viz také „Neužívejte Extavia“).
► Chcete-li otěhotnět, poraďte se před zahájením léčby přípravkem Extavia se svým lékařem.
► Pokud otěhotníte při léčbě přípravkem Extavia, léčbu přerušte a okamžitě informujte svého lékaře. Lékař spolu s vámi rozhodne, zda v léčbě přípravkem Extavia pokračovat.
Poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek užívat.
Kojení
Není známo, zda se interferon beta-1b vylučuje do mateřského mléka. Je však teoreticky možné, že se u kojeného dítěte mohou vyskytnout závažné nežádoucí účinky přípravku Extavia.
► Poraďte se se svým lékařem před zahájením léčby přípravkem Extavia, abyste se mohla rozhodnout, zda přestanete kojit, abyste mohla přípravek Extavia užívat.
Poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek užívat.
Řízení dopravních prostředků a obsluha strojů
Extavia může mít nežádoucí účinky na centrální nervový systém (viz bod 4. Možné nežádoucí účinky). Pokud jste zvlášť citlivý(á), může to ovlivnit vaší schopnost řídit nebo obsluhovat stroje.
Přípravek Extavia obsahuje sodík
Tento přípravek obsahuje méně než 1 mmol (23 mg) sodíku v jedné dávce, tj. v podstatě je „bez sodíku”.
Léčba přípravkem Extavia musí být zahájena pod dohledem lékaře, který má zkušenosti s léčbou roztroušené sklerózy.
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Doporučená dávka je každý druhý den (obden) 1 ml připraveného roztoku přípravku Extavia (viz Dodatek „Samostatné podávání injekcí“ ve druhé části této příbalové informace) podaných podkožní injekcí (subkutánně). Dávka odpovídá 250 mikrogramům (8,0 milionů mezinárodních jednotek IU) interferonu beta-1b.
Obecně platí, že léčba by měla být zahájena nízkou dávkou 0,25 ml (62,5 mikrogramů). Vaše dávky budou poté postupně zvyšovány na celou dávku 1,0 ml (250 mikrogramů).
Dávka by se měla zvyšovat při každé čtvrté injekci čtyřikrát za sebou (0,25 ml, 0,5 ml, 0,75 ml, 1,0 ml). Váš lékař může společně s vámi rozhodnout o změně časových intervalů pro zvýšení dávky v závislosti na nežádoucích účincích, které u vás mohou při zahájení léčby nastat.
Příprava injekce
Před podáním injekce se musí připravit injekční roztok přípravku Extavia z obsahu injekční lahvičky s práškem přípravku Extavia a 1,2 ml roztoku z předplněné injekční stříkačky s rozpouštědlem. Toto bude provedeno buď lékařem, zdravotní sestrou nebo vámi samotnými poté, co budete dostatečně vycvičeni.
Podrobný návod pro samostatné subkutánní podávání injekcí přípravku Extavia je uveden v dodatku na konci této příbalové informace. Tento návod vám také řekne, jak se má injekční roztok přípravku Extavia připravit.
Místo pro aplikaci injekce se musí pravidelně obměňovat. Viz bod 2 „Upozornění a opatření“ a sledujte také instrukce „Střídání míst vpichu“ uvedené v dodatku k této příbalové informaci.
Délka léčby
V současné době není známo, jak dlouho by měla léčba přípravkem Extavia trvat. O délce léčby rozhodne váš lékař společně s vámi.
Jestliže jste užil(a) více přípravku Extavia, než jste měl(a)
Ani podání dávky přípravku Extavia mnohonásobně vyšší, než jsou dávky doporučené pro léčbu roztroušené sklerózy, nevedlo k život ohrožujícímu stavu.
► Promluvte si se svým lékařem, jestliže jste podali více přípravku Extavia nebo častěji. Jestliže jste zapomněl(a) užít přípravek Extavia
Pokud jste zapomněl(a) na podání injekce ve správnou dobu, musíte si ji aplikovat okamžitě, jakmile si chybu uvědomíte. Příští injekce by měla následovat za 48 hodin.
Nezdvojnásobujte následující injekční dávku, abyste nahradil(a) vynechanou jednotlivou dávku.
Jestliže jste přestal(a) užívat přípravek Extavia
Jestliže s léčbou přestanete nebo chcete přestat, promluvte si se svým lékařem. Není známo, že by ukončení podávání přípravku Extavia způsobovalo akutní příznaky z vysazení.
► Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
Možné nežádoucí účinky
4.
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Extavia může způsobit závažné nežádoucí účinky. Pokud se kterýkoli z nežádoucích účinků vyskytne v závažné míře, nebo pokud si všimnete jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci, prosím, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře.
► Okamžitě informujte svého lékaře a přestaňte užívat přípravek Extavia:
- jestliže se objeví příznaky jako svědění po celém těle, otoky obličeje a/nebo jazyka nebo náhlá dušnost.
- jestliže budete pociťovat větší smutek nebo beznaděj než před zahájením léčby přípravkem Extavia, nebo objeví-li se u vás sebevražedné myšlenky.
- jestliže jste si povšimli jakýchkoliv neobvyklých modřin, nadměrného krvácení po poranění nebo se vám zdá, že příliš snadno onemocníte infekcemi.
- jestliže pociťujete ztrátu chuti k jídlu, únavu, máte pocit nevolnosti (nauzea), opakovaně zvracíte, zvláště když zaznamenáte celkové svědění, zežloutnutí kůže nebo očního bělma a snadnou tvorbu modřin.
- jestliže trpíte příznaky, jako je nepravidelný srdeční tep, otoky, např. kotníků nebo nohou nebo dušnost.
- jestliže zaznamenáte bolest břicha vyzařující do zad a/nebo je vám špatně nebo máte horečku.
► Okamžitě informujte svého lékaře:
- jestliže se objeví některý z následujících příznaků: zpěněná moč, únava, otoky, zejména kotníků a očních víček, a přírůstek tělesné hmotnosti, protože může jít o příznaky možných potíží ledvin.
Na začátku léčby jsou nežádoucí účinky časté, ale jejich četnost se obecně s další léčbou snižuje. Nejběžnější nežádoucí účinky jsou:
► Chřipce podobné příznaky, např. horečka, mrazení, bolesti kloubů, malátnost, pocení, bolest hlavy nebo bolest svalů. Tyto příznaky lze snížit podáním paracetamolu nebo nesteroidních protizánětlivých léčiv, např. ibuprofenu.
► Reakce v místě vpichu. Příznaky mohou být zarudnutí, otok, zblednutí, zánět, bolest, hypersenzitivita, poškození tkáně (nekróza). Více informací o tom, co dělat, pokud u vás dojde k reakci v místě vpichu, naleznete v části „Upozornění a opatření“, bod 2. Tyto reakce lze snížit používáním autoinjektoru. Další informace získáte od svého lékaře, lékárníka nebo zdravotní sestry.
Pro snížení rizika nežádoucích účinku na začátku léčby musí lékař začít s nízkou dávkou přípravku Extavia a postupně tuto dávku zvyšovat (viz bod 3 „Jak se přípravek Extavia užívá“).
Následující výčet nežádoucích účinků je založen na hlášeních z klinických studií s přípravkem Extavia (Seznam 1) a na hlášeních po zavedení přípravku na trh (Seznam 2).
Seznam 1: Velmi časté nežádoucí účinky, které se objevily v klinických studiích s přípravkem Extavia (mohou postihnout více než 1 z 10 lidí) a ve vyšších procentech, než bylo jejich pozorování při užívání neúčinné látky (placeba). Seznam také zahrnuje nežádoucí účinky, které se vyskytovaly často (mohou postihnout až 1 z 10 lidí), ale které jsou významně spojené s léčbou.
- infekce, absces
- pokles počtu bílých krvinek v krvi, zduření lymfatických uzlin (lymfadenopatie)
- snížení hladiny cukru v krvi (hypoglykémie)
- deprese, úzkost
- bolest hlavy, závrať, nespavost, migréna, necitlivost nebo pocit brnění (parestézie)
- zánět spojivek (konjunktivitis), poruchy vidění
- bolest uší
- nepravidelný, rychlý tep nebo bušení srdce (palpitace)
- zčervenání a/nebo zarudnutí obličeje způsobené rozšířením krevních cév (vazodilatace), vzestup krevního tlaku (hypertenze)
- rýma, kašel, chrapot způsobený infekcí horních cest dýchacích, záněty dutin (sinusitis), zhoršení kašle, dušnost (dyspnoe)
- průjem, zácpa, nevolnost, zvracení, bolest v krajině břišní
- zvýšení krevních hladin jaterních enzymů (prokáže se krevními testy)
- kožní poruchy, vyrážka
- svalová ztuhlost (hypertonie), bolesti svalů (myalgie), svalová slabost (myastenie), bolest v zádech, bolest v končetinách, např. v prstech a palcích
- zadržování moče (retence moče), bílkovina v moči (prokáže se testy moče), časté močení, neschopnost udržet moč (urinární diskontinence), nucení na močení
- bolestivé periody (dysmenorhea), menstruační poruchy, silné děložní krvácení (metrorhagie) zejména mezi menstruačními periodami, impotence
- reakce v místě vpichu (zahrnuje zarudnutí, otoky, zblednutí, zánět, bolest, alergickou reakci, viz bod 2 „Upozornění a opatření“), rozpad kůže a poškození tkání (nekróza) v místě injekce (viz bod 2 „Upozornění a opatření“)
- chřipce podobné příznaky, horečka, bolest, bolest na hrudi, hromadění tekutiny v rukou, nohou či obličeji (periferní edém), nedostatek/ztráta síly (astenie), mrazení, pocení, celkový pocit nevolnosti.
Dále byly po uvedení přípravku na trh zjištěny následující nežádoucí účinky.
Seznam 2: Nežádoucí účinky hlášené po uvedení přípravku na trh (četnosti - kde je známo - na základě klinických studií):
► Velmi časté (mohou postihnout více než 1 z 10 lidí):
- bolest kloubů (artralgie).
► Časté (mohou postihnout až 1 z 10 lidí):
- může se snížit počet červených krvinek (anémie),
- porucha funkce štítné žlázy (tvoří příliš málo hormonů) (hypothyroidismus),
- zvýšení nebo snížení tělesné hmotnosti,
- zmatenost,
- mimořádné zvýšení srdečního tepu (tachykardie),
- může dojít ke zvýšení hladiny červenožlutého barviva (bilirubinu) produkovaného játry (prokáže se při jaterních testech),
- otoky a obvykle svědivé části kůže nebo mukózní membrány (kopřivka),
- svědění (pruritus),
- ztráta vlasů na hlavě (alopecie),
- menstruční poruchy (menorhagie).
► Méně časté (mohou postihnout až 1 ze 100 lidí):
- může dojít ke snížení počtu krevních destiček (pomáhajících srážení krve) (trombocytopenie),
- možný vzestup určitého typu krevních tuků (triglyceridů) (prokáže se při krevních testech) viz bod 2 “Upozornění a opatření”,
- sebevražedné pokusy,
- změny nálad,
- křeče,
- může dojít ke zvýšení krevní hladiny specifického jaterního enzymu (gama GT), který je tvořen játry (prokáže se při krevních testech),
- zánět jater (hepatitis),
- změna barvy kůže.
► Vzácné (mohou postihnout až 1 z 1 000 lidí):
- závažné alergické (anafylaktické) reakce,
- štítná žláza nefunguje správně (poruchy funkce štítné žlázy), tvoří příliš mnoho hormonů (hypertyroidismus),
- zánět slinivky břišní (pankreatitis), viz bod 2 “Upozornění a opatření”.
- krevní sraženiny v malých cévách mohou ovlivnit funkci ledvin (trombotická trombocytopenická purpura nebo hemolyticko-uremický syndrom). Mezi příznaky může patřit zvýšená tvorba modřin, krvácení, horečka, výrazná slabost, bolest hlavy, závrať či pocit na omdlení. Může se stát, že lékař zjistí, že máte změny v krevním obraze či změny funkce ledvin.
Nežádoucí účinky hlášené v období po uvedení na trh:
- potíže s ledvinami včetně zjizvení (glomeruloskleróza), které mohou omezit funkci Vašich ledvin, (méně časté).
- výrazná ztráta chuti k jídlu vedoucí ke ztrátě hmotnosti (anorexie), (vzácné).
- onemocnění srdečního svalu (kardiomyopatie), (vzácné).
- náhlá dušnost (bronchospazmus), (vzácné).
- porucha funkce jater (jaterní poškození [zahrnující hepatitidu], selhání funkce jater), (vzácné).
- při užívání přípravků, jako je Extavia, se u Vás mohou projevit problémy s malými cévami (kapilárámi) (syndrom zvýšené propustnosti kapilár), (četnost neznámá).
- vyrážka, zarudnutí kůže v obličeji, bolest kloubů, horečka, slabost a nežádoucí účinky způsobené léky (léky vyvolaný lupus erythematodes), (četnost neznámá).
- onemocnění typické silným zúžením krevních cév v plicích což následně vede k vysokému krevnímu tlaku v krevních cévách, kterými proudí krev ze srdce do plic (plicní arteriální hypertenze), (četnost neznámá). Plicní arteriální hypertenze byla pozorována v různých časových odstupech od počátku léčby přípravkem Extavia a to až do několika let od zahájení léčby.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na obalu. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Neuchovávejte při teplotě nad 25°C. Chraňte před mrazem.
Roztok se musí použít bezprostředně po přípravě. Pokud tak nemůžete učinit ihned, roztok lze použít do 3 hodin, pokud se uchovával v chladničce (2°C - 8°C).
Nepoužívejte tento přípravek, pokud si všimnete, že obsahuje částice nebo má změněnou barvu.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co přípravek Extavia obsahuje
- Léčivou látkou je interferon beta-1b. Jedna lahvička obsahuje 300 mikrogramů (9,6milionů IU) interferonu beta-1b. Po přípravě roztoku obsahuje každý mililitr 250 mikrogramů (8,0 milionů IU) interferonu beta-1b.
- Dalšími složkami jsou
- v prášku: mannitol a lidský albumin
- v rozpouštědle: chlorid sodný, voda na injekci.
Krytka hrotu předplněné injekční stříkačky obsahuje derivát přírodního latexu. Proto může krytka hrotu obsahovat přírodní latex.
Jak přípravek Extavia vypadá a co obsahuje toto balení
Extavia je prášek a rozpouštědlo pro injekční roztok.
Prášek má bílou až špinavě bílou barvu.
Prášek Extavia je dodáván ve 3 ml injekčních lahvičkách.
Rozpouštědlo je čitý/bezbarvý roztok.
Rozpouštědlo pro Extavia se dodává v 2,25 ml předplněné injekční stříkačce a obsahuje 1,2 ml roztoku chloridu sodného 5,4 mg/ml (0,54%) injekčního roztoku.
Přípravek Extavia je dostupný v baleních o velikostech:
- 5 injekčních lahviček s interferonem beta-1b a 5 předplněných injekčních stříkaček obsahujících rozpouštědlo.
- 14 injekčních lahviček s interferonem beta-1b a 14 předplněných injekčních stříkaček obsahujících rozpouštědlo.
- 15 injekčních lahviček s interferonem beta-1b a 15 předplněných injekčních stříkaček obsahujících rozpouštědlo.
- 14 injekčních lahviček s interferonem beta-1b a 15 předplněných injekčních stříkaček obsahujících rozpouštědlo.
- 3měsíční vícečetné balení obsahujicí 42 (3x14) injekčních lahviček s interferonem beta-1b a 42 (3x14) předplněných injekčních stříkaček obsahujících rozpouštědlo.
- 3měsíční vícečetné balení obsahujicí 45 (3x15) injekčních lahviček s interferonem beta-1b a 45 (3x15) předplněných injekčních stříkaček obsahujících rozpouštědlo.
- 3měsíční vícečetné balení obsahujicí 42 (3x14) injekčních lahviček s interferonem beta-1b
a 45 (3x15) předplněných injekčních stříkaček obsahujících rozpouštědlo.
Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
Výrobce
Novartis Pharma GmbH Roonstrasse 25 D-90429 Norimberk Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci
|
Belgie/Belgique/Belgien Novartis Pharma N.V. Tél/Tel: +32 2 246 16 11 |
Lietuva Novartis Pharma Services Inc. Tel: +370 5 269 16 50 |
|
Bt^rapnn Novartis Pharma Services Inc. Ten.: +359 2 489 98 28 |
Luxembourg/Luxemburg Novartis Pharma N.V. Tél/Tel: +32 2 246 16 11 |
|
Česká republika Novartis s.r.o. Tel: +420 225 775 111 |
Magyarország Novartis Hungária Kft. Pharma Tel.: +36 1 457 65 00 |
|
Danmark Novartis Healthcare A/S Tlf: +45 39 16 84 00 |
Malta Novartis Pharma Services Inc. Tel: +356 2122 2872 |
|
Deutschland Novartis Pharma GmbH Tel: +49 911 273 0 |
Nederland Novartis Pharma B.V. Tel: +31 26 37 82 111 |
|
Eesti Novartis Pharma Services Inc. Tel: +372 66 30 810 |
Norge Novartis Norge AS Tlf: +47 23 05 20 00 |
|
EkXáda Novartis (Hellas) A.E.B.E. Tq^: +30 210 281 17 12 |
Osterreich Novartis Pharma GmbH Tel: +43 1 86 6570 |
|
Espaňa Novartis Farmacéutica, S.A. Tel: +34 93 306 42 00 |
Polska Novartis Poland Sp. z o.o. Tel.: +48 22 375 4888 |
|
France Novartis Pharma S.A.S. Tél: +33 1 55 47 66 00 |
Portugal Novartis Farma - Produtos Farmaceuticos, S.A Tel: +351 21 000 8600 |
|
Hrvatska Novartis Hrvatska d.o.o. Tel. +385 1 6274 220 |
Románia Novartis Pharma Services Romania SRL Tel: +40 21 31299 01 |
|
Ireland Novartis Ireland Limited Tel: +353 1 260 12 55 |
Slovenija Novartis Pharma Services Inc. Tel: +386 1 300 75 50 |
|
Island Vistor hf. Sími: +354 535 7000 |
Slovenská republika Novartis Slovakia s.r.o. Tel: +421 2 5542 5439 |
|
Italia Novartis Farma S.p.A. Tel: +39 02 96 54 1 |
Suomi/Finland Novartis Finland Oy Puh/Tel: +358 (0)10 6133 200 |
|
Knnpoq Novartis Pharma Services Inc. TpA,: +357 22 690 690 |
Sverige Novartis Sverige AB Tel: +46 8 732 32 00 |
|
Latvija Novartis Pharma Services Inc. Tel: +371 67 887 070 |
United Kingdom Novartis Pharmaceuticals UK Ltd. Tel: +44 1276 698370 |
Tato příbalová informace byla naposledy revidována
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu
Dodatek: SAMOSTATNÉ PODÁVÁNÍ INJEKCÍ
Následující pokyny a obrázky vysvětlí, jak připravit injekci přípravku Extavia a jak postupovat při jejím podání, které si budete provádět sami. Čtěte, prosím, tento návod pozorně a postupujte podle něj krok za krokem. Váš lékař nebo zdravotní sestra vám pomohou naučit se celý postup a techniku samostatného podání injekce. Nepokoušejte se o aplikaci dříve, než si budete zcela jisti, že přesně rozumíte všem požadavkům na přípravu injekčního roztoku a celému postupu při aplikaci injekce.
ČÁST I: NÁVOD KROK ZA KROKEM
Postup se skládá z následujících hlavních kroků:
A) Všeobecné pokyny
B) Příprava k aplikaci (podání) injekce
C) Proces ředění a natažení injekčního roztoku krok za krokem
D) Manuální aplikace injekce (aplikace injekce autoinjektorem ExtaviPro 30G - viz návod k použití poskytovaný s autoinjektorem)
A) Všeobecné pokyny
• Dobře začněte!
Zjistíte, že po několika týdnech se vaše léčba stane přirozenou součástí vašich každodenních činností. Až budete začínat, mohou být následující tipy pro vás užitečné:
- Vytvořte si stálé vhodné místo pro uchovávání, které je mimo dohledu a dosahu dětí tak, abyste Extavia a další pomůcky, které budete potřebovat, vždy snadno našli.
Podmínky uchovávání viz bod 5 příbalové informace „Jak Extavia uchovávat“.
- Snažte se aplikovat injekci vždy ve stejnou denní dobu. Tímto způsobem si snáze zapamatujete a naplánujete čas, kdy nebudete rušeni.
Další podrobnosti o užívání Extavia naleznete v bodě 3 příbalové informace „Jak se Extavia užívá“.
- Každou dávku připravte pouze tehdy, když jste připraveni na aplikaci injekce. Po namíchání Extavia musíte injekci aplikovat okamžitě (jestliže není přípravek použit okamžitě, viz bod 5 příbalové informace „Jak Extavia uchovávat“).
• Důležité tipy, které je nutno mít na paměti
- Buďte důslední - používejte tento přípravek tak, jak je popsáno v bodě 3 příbalové informace „Jak se Extavia užívá“. Dávku vždy dvakrát zkontrolujte.
- Injekční stříkačky a odpadní nádobu na použité stříkačky a jehly uchovávejte mimo dohled a dosah dětí; pokud možno pod zámkem.
- Inj ekční stříkačky nebo j ehly nikdy nepoužívejte opakovaně.
- Vždy použijte zde popsanou sterilní (aseptickou) techniku.
- Použité injekční stříkačky vždy odložte do správné odpadní nádoby na použité stříkačky a jehly.
B) Příprava k aplikaci injekce
• Výběr místa pro injekci
Před přípravou své injekce rozhodněte, do kterého injekčního místa ji budete aplikovat. Přípravek by měl být injekčně podáván do tukové vrstvy mezi kůží a svalem (tj. subkutánně, přibližně 8 mm až 12 mm pod kůži). Nejvhodnějšími místy pro injekce jsou ta, kde je kůže volná a měkká, vždy mimo klouby, nervy a kosti, např. břicho, paže, stehno nebo hýždě.
Krytka hrotu předplněné injekční stříkačky obsahuje derivát přírodního latexu. Proto může krytka hrotu obsahovat přírodní latex. Pokud trpíte alergií na latex, před použitím přípravku Extavia se poraďte se svým lékařem.
Nevolte místa, kde nahmatáte boule, hrboly, pevné uzlíky nebo která jsou bolestivá. Nevolte ani oblasti, kde má kůže změněné zabarvení, prohlubně, strupy nebo je porušená. O každém takovém nebo i jiném neobvyklém nálezu informujte svého lékaře nebo zdravotní sestru.
Místo vpichu byste při každé injekci měli střídat. Pokud se vám některé plochy budou zdát obtížně dosažitelné, požádejte o pomoc s aplikací injekce člena rodiny nebo přítele. Dodržujte posloupnost popsanou v harmonogramu na konci tohoto dodatku (viz Část II „Střídání míst vpichu“) a po 8 injekcích (16 dnech) se vrátíte do místa, kde jste aplikovali injekci poprvé. To poskytne každému místu aplikace příležitost plně se obnovit předtím, než dojde k podání další injekce.
Další informace naleznete v plánu střídání míst vpichu na konci tohoto dodatku, kde se dozvíte, jak volit místa vpichu. Jako příklad je připojena i karta záznamu o léčbě (viz Dodatek Část III). Tento záznam by vám měl poskytnout představu o tom, jak sledovat místa a data svých injekčních podání.
• Léčivý přípravek
Budete potřebovat léčivý přípravek:
• 1 Extavia injekční lahvička (s práškem pro přípravu injekčního roztoku)
• 1 předplněnou injekční stříkačku rozpouštědla pro Extavia
K ředění a aplikaci Vašeho léčivého přípravku budete potřebovat aplikační set ExtaviPro 30G (dodávaný zvlášť), který obsahuje následující součásti, a návod jak je používat:
• Adaptéry na injekční lahvičky určené k ředění Vašeho léčivého přípravku
• Jehly o síle 30 pro injekční podání Vašeho léčivého přípravku
• Tampóny namočené v alkoholu
Budete také potřebovat odpadní nádobu na použité stříkačky a jehly.
Jehly o síle 30 dodávané v aplikační sadě pro podávání tohoto přípravku mohou být použity k manuální injekci NEBO s autoinjektorem ExtaviPro 30G.
Pro dezinfekci kůže použijte vhodné dezinfekční činidlo doporučené Vaším lékárníkem.
C) Ředění a natažení injekčního roztoku krok za krokem
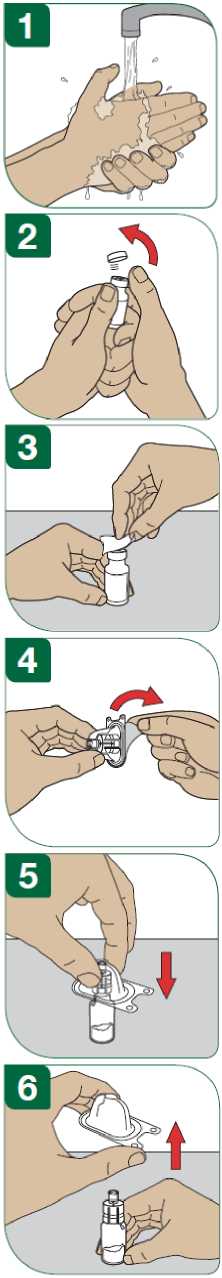
1 - Než začnete, důkladně si omyjte ruce mýdlem a vodou.
2 - Odstraňte krytku z injekční lahvičky s přípravkem Extavia. Je lepší používat palec než nehet, protože nehet by se mohl ulomit. Postavte lahvičku na stůl.
3 - Očistěte horní část injekční lahvičky tampónem s alkoholem, pohybujte tampónem pouze jedním směrem. Tampón ponechte na vršku injekční lahvičky.
4 - Sloupněte a ostraňte kryt z obalu adaptéru na injekční lahvičky.
Nevyjímejte adaptér na injekční lahvičky z balení.
5 - Odstraňte tampon z vrcholu injekční lahvičky.
K uchopení adaptéru na injekční lahvičky využijte jeho obal. Přichyťte adaptér na lahvičku tlakem dolů, dokud se adaptér nepřichytí a nezaklesne okolo ústí injekční lahvičky.
6 - Uchopte obal v rozích, sejměte jej a zlikvidujte, přičemž adaptér musí zůstat na lahvičce.
7 - Vyjměte předplněnou injekční stříkačku s rozpouštědlem z obalu. Odlomte a zlikvidujte ochranný kryt z konce předplněné injekční stříkačky.
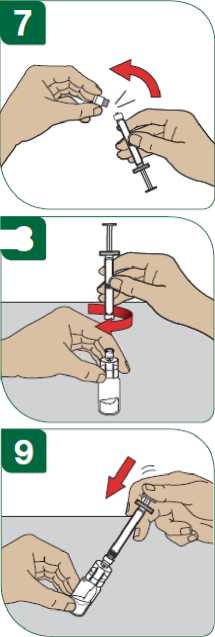
• •
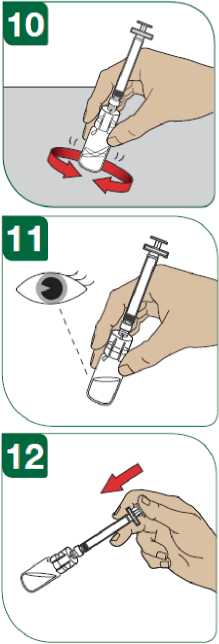
Poznámka: Dejte pozor, abyste se konce stříkačky nedotkli. Netlačte na píst.
8 - Uchopte pevně injekční lahvičku a adaptér, injekční stříkačku našroubujte na adaptér.
Takto se vytvořila soustava injekční stříkačka-injekční lahvička.
9 - Držte soustavu injekční stříkačka-injekční lahvička našikmo. Pomalu stlačujte píst stříkačky tak, aby roztok stékal po její vnitřní stěně.
Přeneste veškeré rozpouštědlo do lahvičky.
Poznámka: Lahvičkou netřeste, protože může dojít k vzniku nadměrné pěny.
10 - Uchopte injekční lahvičku mezi palcem, ukazovákem a prostředníkem. Míchejte lahvičkou opatrným kroužením rukou, dokud se veškerý prášek nerozpustí.
Poznámka: Lahvičkou netřepejte.
11 - Roztok pečlivě zkontrolujte. Měl by být čirý a neměl by obsahovat žádné částečky.
Poznámka: Pokud je roztok zbarvený nebo obsahuje částice, zlikvidujte jej a začněte znovu s novou stříkačkou a lahvičkou z Vašeho balení.
Pokud se vytvoří přebytečná pěna - což se může stát, pokud budete injekční lahvičkou otáčet nebo třepat příliš silně - nechte injekční lahvičku v klidu stát, dokud se pěna neusadí.
12 - Před započetím dalšího kroku se ujistěte, že je píst zcela stlačen, protože mohlo dojít k jeho posunu.
13 - Otočte soupravu injekční stříkačky a lahvičky tak, aby byla lahvička nahoře. Pomalu vytahujte píst, abyste natáhl/a veškerý roztok do injekční stříkačky.
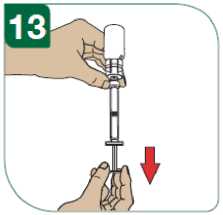
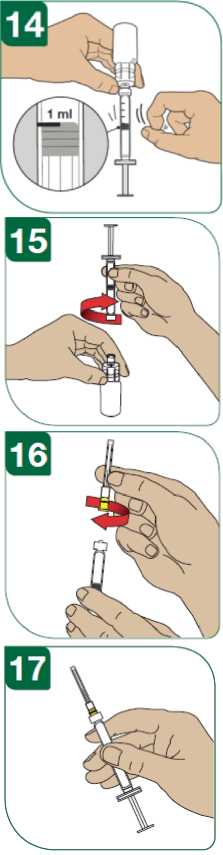
14 - Odstraňte veškeré vzduchové bubliny lehkým poklepáním na injekční stříkačku. Zatlačte píst na značku 1 ml (nebo na objem předepsaný vaším lékařem).
Poznámka: Může být nutné několikrát posunout pístem nahoru a dolů, aby se odstranily vzduchové bubliny a ve stříkačce zůstal pouze 1 ml roztoku.
15 - Odšroubujte injekční stříkačku a ponechejte adaptér na lahvičce.
Vyhoďte injekční lahvičku a nepoužitou část roztoku do nádoby na odpad.
16 - Vyjměte jehlu z obalu a pevně ji našroubujte na konus stříkačky.
17 - Ponechejte nasazený kryt jehly. Nyní jste připraveni k manuálnímu injekčnímu podání nebo k použití autoinjektoru ExtaviPro 30G pro podání přípravku Extavia.
Uchovávání po naredení
Pokud z nějakého důvodu nebudete moci přípravek Extavia okamžitě aplikovat, můžete před použitím naředěný roztok uchovávat v chladničce až 3 hodiny. Roztok nezmrazujte a nečekejte s aplikací déle než 3 hodiny. Jestliže uplynou více než 3 hodiny, léčivý přípravek zlikvidujte a připravte si novou injekci. Před vlastním podáním zahřejte injekci nebo lahvičku v rukou, aby se zabránilo bolesti.
D) Aplikace injekce manuální (aplikace injekce autoinjektorem ExtaviPro 30G -viz návod k použití poskytovaný s autoinjektorem)
1 - Vyberte si místo vpichu (viz část „Výběr místa vpichu“ a diagramy na konci tohoto návodu) a zaznamenejte jej do záznamu o léčbě.
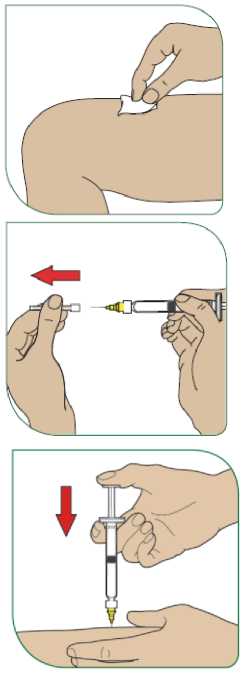
2 - Alkoholovým tampónem si očistěte kůži na místě plánovaného vpichu. Kůži nechejte oschnout na vzduchu. Tampón vyhoďte.
3 - Sejměte kryt z jehly. Kryt sejměte tahem a nekruťte jím.
4 - Na vhodném místě jemně zmáčkněte kůži okolo dezinfikovaného místa (mírně ji vyzdvihněte).
5 - Rychlým, pevným pohybem vbodněte jehlu přímo do kůže pod úhlem 90°. Držte injekční stříkačku jako tužku nebo šipku.
6 - Aplikujte lék (pomalým, stálým tlakem na píst stlačujte píst po celé jeho dráze, dokud nebude injekční stříkačka prázdná).
7 - Použitou injekční stříkačku zlikvidujte vyhozením do odpadní nádoby.
Pro každou injekci si musíte zvolit nové místo vpichu, protože jejich střídání poskytne oblasti čas na zotavení a lze tak předejít infekci. V první části tohoto dodatku je doporučení, která místa zvolit. Je velmi užitečné rozhodnout se, kam si píchnete injekci, dříve než si připravíte stříkačku. Schéma na diagramu vám pomůže pravidelně střídat místa vpichu. Například pokud si píchnete první injekci na pravou stranu břicha, zvolte pak levou stranu břicha pro druhou injekci, při třetí injekci postupte na pravé stehno, a tak pokračujte dále podle diagramu, dokud nevystřídáte všechna vhodná místa na těle. Zaznamenejte si vždy, kdy a kam jste si píchli poslední injekci. Jednou z možností je poznamenat si místo vpichu do přiložené karty pro záznamy o léčbě.
Budete-li se řídit tímto schématem, vrátíte se k první oblasti (např. pravá strana břicha) po 8 injekcích (16 dnech). To se nazývá cyklus obměny. Na našem příkladu harmonogramu je každá plocha rozdělena na 6 míst vpichu (což dohromady dává 48 míst vpichu), vlevo a vpravo v horní, střední a dolní části příslušné oblasti. Pokud se vrátíte do jedné oblasti po jednom cyklu obměny, zvolte si v této oblasti nejvzdálenější místo vpichu. Jestliže bude oblast bolavá, poraďte se se svým lékařem nebo zdravotní sestrou o výběru jiných míst vpichu.
Harmonogram obměny
Doporučujeme si vést jako pomůcku pro řádnou obměnu míst vpichu záznam o dnech a místech podání vašich injekcí. Můžete použít následující harmonogram obměny.
Používejte střídavý cyklus obměn. Každý cyklus bude obsahovat 8 injekcí (16 dní), aplikovaných postupně od oblasti 1 až po oblast 8. Dodržením tohoto sledu poskytnete každému místu aplikace možnost zotavit se před podáním další injekce.
Cyklus obměny 1: Cyklus obměny 2: Cyklus obměny 3: Cyklus obměny 4: Cyklus obměny 5: Cyklus obměny 6:
Levá horní část každé oblasti Pravá dolní část každé oblasti Střední levá část každé oblasti Pravá horní část každé oblasti Levá dolní část každé oblasti Střední pravá část každé oblasti
Pokyny pro vedení záznamů o místech a dnech podání vašich injekcí
- Začněte svojí první injekcí (nebo svojí poslední injekcí, pokud nebudete novým uživatelem Extavia).
- Zvolte místo pro injekci. (Pokud jste si již Extavia podávali, začněte v oblasti, která nebyla použita pro injekci během posledního cyklu obměny (tj. minulých 16 dní).
- Po podání injekce zaškrtněte použité místo vpichu a zapište datum v tabulce vaší karty pro záznamy o léčbě (viz příklad: Vedení záznamů o místech a dnech podání Vašich injekcí).
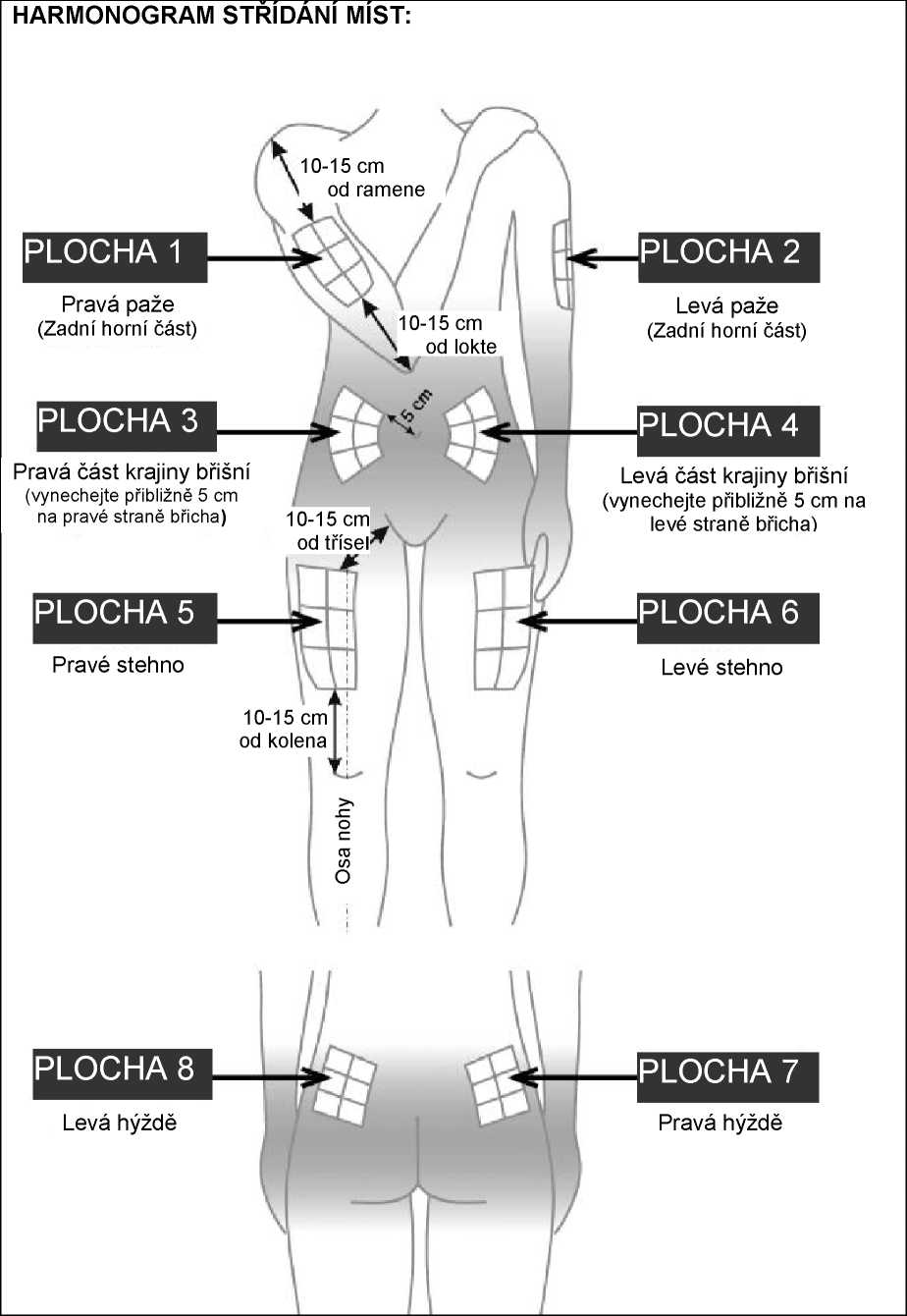
Pravá paže
|
04/A2. | |
Pravá část krajiny břišní
|
Ot>[KL | |
|
Pravé stehno | |
|
KL (42- | |
10-15 cm od kolena
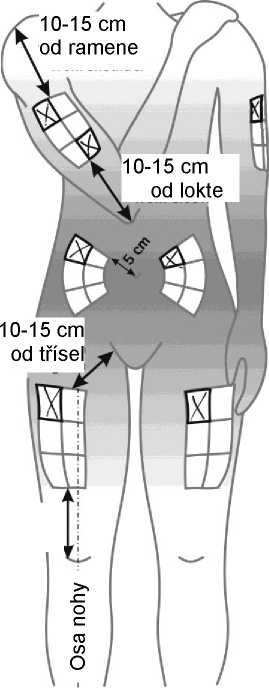
Levá paže
|
os [KL | |
Levá část krajiny břišní
|
Áo[kL | |
|
Levé stehno | |
|
M[A2_ | |
Levá hýždě
|
/8f kL | |
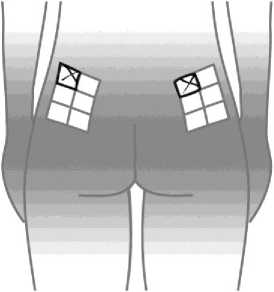
Pravá hýždě
|
A&f KL | |
53