Evra 203 Mikrogramů/24 Hodin + 33,9 Mikrogramů/24 Hodin
SOUHRN ÚDAJŮ O PŘÍPRAVKU
PŘÍLOHA I
EVRA 203 mikrogramů /24 hodin + 33,9 mikrogramů/24 hodin transdermální náplast Norelgestorminum/ethinylestradiolum
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
20 cm2 náplasti obsahuje 6 mg norelgestrominum (NGMN) a 600 mikrogramů ethinylestradiolum (EE).
Za 24 hodin se z jedné náplasti uvolní v průměru 203 mikrogramů NGMN a 33,9 mikrogramů EE. Expozice léčivému přípravku je náležitěji charakterizována jeho farmakokinetickým profilem (viz bod 5.2).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Transdermální náplast.
Tenká, matrixová transdermální náplast sestávající ze tří vrstev.
Zevní strana krycí vrstvy je béžové barvy s vytlačeným názvem „EVRA“.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Antikoncepce pro ženy.
EVRA je určena pro ženy ve fertilním věku. Bezpečnost a účinnost byla stanovena u žen ve věku 18 až 45 let.
Rozhodnutí předepsat přípravek EVRA by mělo být provedeno při zvážení jednotlivých současných rizikových faktorů ženy, zvláště rizikových faktorů pro žilní tromboembolizaci (VTE), a toho, jaké je riziko VTE u přípravku EVRA v porovnání s dalšími přípravky CHC (viz body 4.3 a 4.4).
4.2 Dávkování a způsob podání
Dávkování
K dosažení maximální antikoncepční účinnosti musí ženy používat náplast EVRA přesně dle doporučení lékaře. Pokyny „Jak zahájit podávání náplasti EVRA“ jsou uvedeny níže.
Současně může být aplikována pouze jedna transdermální náplast.
Každou odstraněnou transdermální náplast je nutné okamžitě nahradit novou náplastí týž den v týdnu (den výměny) 8. a 15. den cyklu. Výměna náplasti je možná v libovolnou dobu dne výměny. Čtvrtý týden, začínající 22. dnem cyklu, se neaplikuje žádná transdermální náplast, je to týden bez náplasti.
Nový antikoncepční cyklus začíná den následující po týdnu bez transdermální náplasti; další náplast EVRA musí být nalepena, i když se nedostavilo nebo ještě neskončilo krvácení ze spádu.
Za žádných okolností nesmí být interval bez transdermální náplasti mezi týdny s medikací delší než 7 dní. Pokud je interval bez transdermální náplasti delší než 7 dní, nemusí být žena chráněna před otěhotněním. Po dobu 7 dní musí být proto používána nehormonální antikoncepce. Analogicky stoupá riziko ovulace každým dalším dnem mimo doporučené období bez transdermální náplasti. Jestliže dojde k pohlavnímu styku během prodlouženého období bez transdermální náplasti, je nutné počítat s možností otěhotnění.
Zvláštní populace
Tělesná váha vyšší než 90 kg
Antikoncepční účinek může být snížený u žen s hmotností 90 kg a více.
Poruchy ledvin
Přípravek EVRA nebyl studován u žen s renálním poškozením. Úprava dávkování není nutná, ale jak naznačuje literatura frakce ethinylestradiolu je vyšší, proto by měla EVRA být používána u této populace pod dohledem.
Poruchy jater
EVRA nebyla studována u žen s jaterním poškozením. EVRA je kontraindikována u žen s jaterním poškozením (viz bod 4.3).
Ženy v postmenopauze
EVRA není indikována u žen v postmenopauze a není vhodná jako hormonální substituční léčba.
Pediatrická populace
Bezpečnost a účinnost nebyla hodnocena u adolescentů mladších 18let. Použití přípravku EVRA není relevantní u dětí a adolescentů v premenarché.
Způsob aplikace
EVRA musí být aplikována na čistou, suchou, neochlupenou a neporušenou kůži hýždí, břicha, horní zevní část paže nebo horní část trupu, tj. na místa, kde nedochází ke tření s těsnými oděvy. EVRA nesmí být aplikována na prsa, zarudlou, podrážděnou nebo poraněnou kůži.
K zamezení možného podráždění musí být každá další transdermální náplast nalepena na jiné místo, i když se může jednat o stejnou část těla.
Transdermální náplast musí být pevně přitlačena, dokud se její okraje důkladně nepřilepí.
Na místa stávající nebo budoucí aplikace transdermální náplasti se nesmí aplikovat make-up, krémy, tělová mléka, zásypy nebo jiné lokálně aplikované přípravky, aby nebyla narušena lepivost transdermální náplasti.
Doporučuje se, aby ženy denně kontrolovaly, zda je transdermální náplast stále správně přilepena.
Transdermální náplast EVRA se nesmí stříhat, poškodit nebo jakkoli upravovat, protože to může snížit antikoncepční účinek.
Použité transdermální náplasti musejí být pečlivě likvidovány dle návodu uvedeného v bodu 6.6.
Jak začít používat transdermální náplast EVRA
Pokud nebyla užívána žádná hormonální antikoncepce v předchozím cyklu Antikoncepce s použitím přípravku EVRA se zahajuje první den menstruace. Aplikuje se jedna transdermální náplast a ponechává se po dobu jednoho celého týdne (7 dní). Den aplikace první transdermální náplasti (den 1/den zahájení) určuje následné dny výměny - den výměny). Den výměny transdermální náplasti bude týž den v každém týdnu (8., 15., 22. a 1. den následujícího cyklu). Čtvrtý týden začíná 22. dnem a je bez aplikace transdermální náplasti.
V případě zahájení 1. terapie po prvním dnu menstruačního cyklu musí být po dobu prvních 7 dní současně používána nehormonální antikoncepce.
Přechod z perorální kombinované antikoncepce
Aplikace transdermální náplasti EVRA by měla být zahájena první den krvácení ze spádu. Jestliže se do 5. dne od poslední tablety s účinnou složkou (obsahující hormon) nedostaví krvácení ze spádu, musí být před zahájením aplikace transdermální náplasti EVRA vyloučena gravidita. Pokud je aplikace zahájena po prvním dni krvácení ze spádu, musí být po dobu 7 dní současně používána nehormonální antikoncepce.
Jestliže od užití poslední perorální antikoncepční tablety s účinnou složkou uplyne více než 7 dní, může se u žen dostavit ovulace, a proto by se měly před aplikací transdermální náplasti EVRA poradit s lékařem. Jestliže dojde během tohoto prodlouženého období bez užívání tablet k pohlavnímu styku, je nutné vzít v úvahu možnost těhotenství.
Přechod z čistěprogestogenové antikoncepce
Ženy mohou přejít libovolný den z čistě progestogenových tablet (z implantátu v den jeho odstranění, z injekční formy v den, kdy by měla být aplikována další injekce), ale během prvních 7 dní musí být zajištěna bariérová metoda antikoncepce.
Aplikace po spontánním potratu nebo umělém přerušení těhotenství
Po potratu nebo umělém přerušení těhotenství před 20. gestačním týdnem mohou být transdermální náplasti EVRA aplikovány okamžitě. Pokud je přípravek EVRA aplikován okamžitě, další antikoncepce není nutná. Je nutné si uvědomit, že během 10 dní po potratu nebo přerušení těhotenství může dojít k ovulaci.
Po potratu nebo umělém přerušení těhotenství po 20. gestačním týdnu může být aplikace transdermální náplastí EVRA zahájena buď 21. den po potratu nebo první den první spontánní menstruace, dle časové priority. Výskyt ovulace 21. den po potratu (ve 20. gestačním týdnu) není znám.
Po porodu
Ženy, které se rozhodnou, že nebudou kojit, mohou začít s antikoncepcí transdermálními náplastmi EVRA nejdříve 4 týdny po porodu. Pokud začnou později, měla by jim být na dobu 7 dní doporučena bariérová metoda antikoncepce. Pokud již však došlo k pohlavnímu styku, musí být před zahájením léčby vyloučena gravidita nebo musí ženy počkat do první menstruace.
Informace pro kojící ženy jsou uvedeny v bodu 4.6.
Co dělat, jestliže se transdermální náplast částečně nebo úplně odlepí
Pokud se transdermální náplast EVRA částečně nebo úplně odlepí a zůstane odlepená, je dávkování přípravku nedostatečné.
Pokud zůstává transdermální náplast EVRA částečně odlepená:
- Po dobu kratší než jeden den (méně než 24 hodin): měla by být znovu přilepena na totéž místo nebo neprodleně nahrazena novou transdermální náplastí EVRA. Žádná další antikoncepce není nutná. Další transdermální náplast EVRA by měla být aplikována jako obvykle v den výměny.
- Po dobu delší než jeden den (24 hodin nebo déle) nebo pokud si žena není jistá, jak dlouho je transdermální náplast odlepená: je možné, že žena není chráněna před otěhotněním. Žena by měla ukončit současný antikoncepční cyklus a neprodleně zahájit nový cyklus nalepením nové transdermální náplasti EVRA. Začíná tak nyní nový den 1 a nový den výměny. Po dobu prvních 7 dní nového cyklu musí být používána ještě nehormonální antikoncepce.
Pokud transdermální náplast nelepí, neměla by být znovu aplikována a okamžitě by měla být nalepena nová transdermální náplast. K fixaci náplasti nesmějí být použity náhradní lepidla nebo obvazy.
Opoždění dne výměny transdermální náplasti
Při zahájení kteréhokoli cyklu aplikace transdermální náplasti (týden 1, den 1):
Je možné, že žena není chráněna před otěhotněním. První transdermální náplast nového cyklu musí být nalepena co nejdříve poté, jakmile si žena vzpomene. Tak nastane nový den výměny a nový den 1.
Po dobu prvních 7 dní nového cyklu musí být současně používána i nehormonální antikoncepce. Jestliže dojde během tohoto prodlouženého období bez aplikace transdermální náplasti k pohlavnímu styku, je nutné vzít v úvahu možnost otěhotnění.
Opoždění v průběhu cyklu (2. týden/den 8 nebo 3. týden/den 15):
- O jeden nebo dva dny (do 48 hodin): žena si musí aplikovat okamžitě novou transdermální náplast EVRA. Další transdermální náplast EVRA se pak mění v obvyklý den výměny. Jestliže byla transdermální náplast během předchozích 7 dní před prvním odlepením nalepena správně, není nutná další antikoncepce.
- O více než dva dny (48 hodin a více): žena není chráněna před otěhotněním. Žena by měla okamžitě přerušit současný antikoncepční cyklus a zahájit nový čtyřtýdenní cyklus aplikací nové transdermální náplasti EVRA. Tím je dán nový den 1 a nový den výměny. Během prvních 7 dní nového cyklu musí žena používat souběžně i další nehormonální antikoncepci.
Na konci cyklu (4. Týden/den 22)
- Na konci cyklu (4. týden /den 22): Pokud není transdermální náplast EVRA odstraněna na začátku 4. týdne (den 22), měla by být odstraněna co nejdříve. Další cyklus musí být zahájen v obvyklý den výměny, což je den výměny po 28. dnu. Současné použití další antikoncepce není vyžadováno.
Změna dne výměny
K oddálení menstruace o jeden cyklus si žena musí aplikovat další transdermální náplast na začátku 4. týdne (den 22), a tím vynechá období bez transdermální náplasti. Může se objevit intermenstruační krvácení a špinění. Po 6 po sobě jdoucích týdnech aplikace náplasti by měl nastat interval 7 dní bez transdermální náplasti. Poté se pokračuje v pravidelné aplikaci transdermálních náplastí EVRA.
Pokud si žena přeje změnit den výměny transdermálních náplastí, musí být dokončen současný cyklus a 3. transdermální náplast EVRA odstraněna ve správný den. Během období bez transdermální náplasti může být vybrán nový den výměny tak, že první transdermální náplast EVRA bude nalepena první požadovaný den dalšího cyklu. V žádném případě nesmí být období bez transdermální náplasti delší než 7 po sobě jdoucích dní. Čím kratší je interval bez transdermální náplasti, tím vyšší je riziko, že se nedostaví krvácení ze spádu a během následujícího cyklu se může objevit intermenstruační krvácení a špinění.
Mírné podráždění kůže
Jestliže se v místě nalepení transdermální náplasti objeví nepříjemné podráždění, může být nalepena nová transdermální náplast na jiné vhodné místo do dalšího dne výměny. Současně může být aplikována pouze jedna transdermální náplast.
4.3 Kontraindikace
Kombinovaná hormonální antikoncepce (CHC) by se neměla používat u následujících stavů. Pokud se některá z těchto poruch vyskytne během aplikace náplasti EVRA, musí být aplikace okamžitě ukončena.
• Přítomnost nebo riziko žilní tromboembolizace (VTE)
• žilní tromboembolizace - současná žilní tromboembolizace (léčená pomocí antikoagulancií) nebo anamnéza VTE (např. hluboká žilní trombóza [DVT] nebo plicní embolizace [PE]);
• známá dědičná nebo získaná predispozice pro žilní tromboembolizaci, jako je rezistence na APC (včetně faktoru V Leiden), deficitu antitrombinu III, deficitu proteinu C, deficitu proteinu S;
• velký chirurgický zákrok nebo prodloužená imobilizace (viz bod 4.4);
• vysoké riziko žilní tromboembolizace v důsledku přítomnosti více rizikových faktorů (viz bod 4.4);
• Přítomnost nebo riziko arteriální tromboembolizace (ATE)
• arteriální tromboembolizace - současná arteriální tromboembolizace, anamnéza arteriální tromboembolizace (např. infarkt myokardu) nebo prodromální stav (např. angina pectoris);
• cerebrovaskulární onemocnění - současná cévní mozková příhoda, anamnéza cévní mozkové příhody nebo prodromálního stavu (např. tranzitorní ischemická ataka, TIA);
• známá hereditární nebo získaná predispozice k arteriální tromboembolizaci, jako je hyperhomocysteinemie a antifosfolipidové protilátky (antikardiolipinové protilátky, lupus antikoagulans);
• anamnéza migrény s fekálními neurologickými příznaky;
• vysoké riziko arteriální tromboembolizace v důsledku vícečetných rizikových faktorů (viz bod 4.4) nebo přítomnost jednoho závažného rizikového faktoru, jako je:
- diabetes mellitus s cévními příznaky;
- závažná hypertenze;
- závažná dyslipoproteinemie.
• Hypersenzitivita na léčivé látky nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1 tohoto přípravku.
• Diagnostikovaný nebo suspektní karcinom prsu.
• Karcinom endometria nebo diagnostikované nebo suspektní jiné estrogen-dependentní neoplazie.
• Abnormální funkce jater související s akutním nebo chronickým hepatocelulárním onemocněním.
• Adenom nebo karcinom jater.
• Nediagnostikované abnormální krvácení z genitálu.
4.4 Zvláštní upozornění a opatření pro použití Upozornění
Pokud jsou přítomna jakákoli onemocnění nebo rizikové faktory uvedené níže, měla by být vhodnost přípravku EVRA s ženou prodiskutována.
V případě zhoršení nebo prvního výskytu jakéhokoli z těchto stavů nebo rizikových faktorů by mělo být ženě doporučeno, aby kontaktovala svého lékaře, který stanoví, zda by měla užívání přípravku EVRA ukončit.
Klinicky nebylo prokázáno, že transdermální náplast je v nějakém aspektu bezpečnější než kombinovaná perorální antikoncepce.
EVRA není indikována během těhotenství (viz bod 4.6).
Riziko žilní tromboembolizace (VTE)
Užívání jakékoli kombinované hormonální antikoncepce (CHC) zvyšuje riziko žilní tromboembolizace (VTE) ve srovnání s jejím neužíváním. Přípravky, které obsahují levonorgestrel, norgestimát nebo norethisteron souvisí s nejnižším rizikem VTE. Další přípravky, jako je přípravek EVRA mohou mít až dvakrát vyšší úroveň rizika. Rozhodnutí používat jakýkoli přípravek jiný než ten, který má nejnižší riziko VTE, by mělo být učiněno po diskusi se ženou, aby se zajistilo, že rozumí riziku VTE u přípravku EVRA, jak její současné rizikové faktory toto riziko ovlivňují a že riziko VTE je nejvyšší v prvním roce užívání léku. Existují také některé důkazy, že riziko se zvyšuje, když je CHC opětovně zahájena po pauze v užívání trvající 4 týdny nebo déle.
U žen, které nepoužívají CHC a nejsou těhotné, se asi u 2 z 10 000 vyvine VTE v průběhu jednoho roku. U každé jednotlivé ženy však může být riziko daleko vyšší v závislosti na jejích základních rizikových faktorech (viz níže).
Odhaduje se, že z 10 000 žen, které používají CHC obsahující levonorgestrel se asi u 61 vyvine VTE během jednoho roku. Studie naznačily, že incidence VTE u žen, které používají přípravek EVRA, je až 2násobně vyšší než u uživatelek CHC obsahující levonorgestrel. To odpovídá přibližně 6 až 12 VTE za rok u 10 000 žen, které užívají přípravek EVRA.
V obou případech je tento počet VTE za rok menší než počet očekávaný u žen během těhotenství nebo v období po porodu.
VTE může být fatální v 1-2 % případů.
Počet příhod VTE na 10 000 žen za rok
Number of VTE events
A
12 -10 -
8 -
I
4 -
Uživatelka hormonální antikoncepce nezahrnující CHC (2 příhody)
CHC obsahující levonorgestrel (5-7 příhod)
CHC obsahující norelgestromin (6-12 příhod)
Extrémně vzácně byla hlášena trombóza u uživatelek CHC v dalších krevních cévách, např. jaterních, mezenterických, renálních nebo retinálních žilách a tepnách.
Rizikové faktory VTE
Riziko žilních tromboembolických komplikací u uživatelek CHC se může podstatně zvyšovat u ženy, která má další rizikové faktory, zvláště pokud je přítomno více rizikových faktorů (viz tabulka).
Přípravek EVRA je kontraindikován, pokud má žena více rizikových faktorů, které pro ni představují vysoké riziko žilní trombózy (viz bod 4.3). Pokud má žena více než jeden rizikový faktor, je možné, že zvýšení rizika je vyšší než součet jednotlivých faktorů - v tomto případě by mělo být zváženo její celkové riziko VTE. Pokud je poměr přínosů a rizik považován za negativní, neměla by být CHC předepisována (viz bod 4.3).
Tabulka: Rizikové faktory VTE
|
Rizikový faktor |
Poznámka |
|
Obezita (index tělesné hmotnosti nad 30 kg/m2) |
Při zvýšení BMI se významně zvyšuje riziko. Zvláště důležité je zvážit, zda jsou také přítomny další rizikové faktory. |
1 Střední bod rozmezí 5-7 na 10 000 WY (žen-roků) na základě relativního rizika pro CHC obsahující levonorgestrel oproti jejímu nepoužívání přibližně 2,3 až 3,6
|
Prodloužená imobilizace, velký chirurgický zákrok, jakýkoli chirurgický zákrok na nohách a pánvi, neurochirurgický zákrok nebo větší trauma. Poznámka: dočasná imobilizace, včetně cestování letadlem > 4 hodiny může být také rizikovým faktorem VTE, zvláště u žen s dalšími rizikovými faktory. |
V těchto situacích je doporučeno ukončit používání náplasti (v případě plánovaného chirurgického výkonu minimálně 4 týdny předem) a nezahajovat používání do dvou týdnů po kompletní remobilizaci. Měla by se použít další antikoncepční metoda pro zabránění nechtěnému těhotenství. Antitrombotická léčba by měla být zvážena, pokud přípravek EVRA nebyl předem vysazen. |
|
Pozitivní rodinná anamnéza (žilní tromboembolizace kdykoli u sourozence nebo rodiče, zvláště v relativně nízkém věku) |
Pokud je suspektní hereditární predispozice, měla by být žena před rozhodnutím o používání jakékoli CHC odeslána k odborníkovi na konzultaci. |
|
Další onemocnění související s VTE |
Zhoubné onemocnění, systémový lupus erythematodes, hemolyticko-uremický syndrom, chronické zánětlivé onemocnění střev (Crohnova choroba nebo ulcerózní kolitida) a srpkovitá anemie. |
|
Vyšší věk |
Zvláště nad 35 let. |
Není žádná shoda o možné roli varixů a povrchové tromboflebitidy v nástupu nebo progresi žilní trombózy.
Zvýšené riziko tromboembolizace v těhotenství a zvláště během šestinedělí musí být zváženo (pro informaci o „Těhotenství a kojení “ viz bod 4.6).
Příznaky VTE (hluboká žilní trombóza a plicní embolizace)
V případě příznaků by mělo být ženě doporučeno, aby vyhledala naléhavou lékařskou péči a informovala lékaře, že užívá CHC.
Příznaky hluboké žilní trombózy (DVT) mohou zahrnovat:
- jednostranný otok nohy a/nebo chodidla nebo podél žíly v noze;
- bolest nebo citlivost v noze, která může být pociťována pouze vstoje nebo při chůzi;
- zvýšenou teplotu postižené nohy, zarudnutí nebo změnu barvy kůže nohy.
Příznaky plicní embolizace (PE) mohou zahrnovat:
- náhlý nástup nevysvětlitelné dušnosti nebo rychlého dýchání;
- náhlý kašel, který může souviset s hemoptýzou;
- ostrou bolest na hrudi;
- těžké točení hlavy nebo závrať;
- rychlý nebo nepravidelný srdeční tep.
Některé z těchto příznaků (např. „dušnost“, „kašel“) nejsou specifické a mohou být nesprávně interpretovány jako častější nebo méně závažné příhody (např. infekce dýchacího traktu).
Dalšími známkami cévní okluze mohou být: náhlá bolest, otok a světle modré zbarvení končetin.
Pokud nastane okluze v oku, mohou se příznaky pohybovat od nebolestivého rozmazaného vidění, které může přejít do ztráty zraku. Někdy může nastat ztráta zraku téměř okamžitě.
Riziko arteriální tromboembolizace (ATE)
Epidemiologické studie souvisely s používáním CHC se zvýšeným rizikem arteriální tromboembolizace (infarkt myokardu) nebo cerebrovaskulární příhody (např. tranzitorní ischemická ataka, cévní mozková příhoda). Arteriální tromboembolické příhody mohou být fatální.
Rizikové faktory ATE
Riziko arteriálních tromboembolických komplikací nebo cerebrovaskulámí příhody u uživatelek CHC se zvyšuje u žen s rizikovými faktory (viz tabulka). Přípravek EVRA je kontraindikován, pokud má žena jeden závažný rizikový faktor nebo více rizikových faktorů ATE, které pro ni představují riziko arteriální trombózy (viz bod 4.3). Pokud má žena více než jeden rizikový faktor, je možné, že zvýšení rizika je vyšší než součet jednotlivých faktorů - v tomto případě by mělo být zváženo její celkové riziko. Pokud je poměr přínosů a rizik považován za negativní, neměla by být CHC předepisována (viz bod 4.3).
Tabulka: Rizikové faktory ATE
|
Rizikový faktor |
Poznámka |
|
Vyšší věk |
Zvláště nad 35 let. |
|
Kouření |
Ženě by mělo být doporučeno, aby nekouřila, pokud chce používat CHC. Ženám ve věku nad 35 let, které dále kouří, by mělo být důrazně doporučeno, aby používaly jinou metodu antikoncepce. |
|
Hypertenze | |
|
Obezita (index tělesné hmotnosti nad 30 kg/m2) |
Při zvýšení BMI se významně zvyšuje riziko. Zvláště důležité u žen s dalšími rizikovými faktory. |
|
Pozitivní rodinná anamnéza (arteriální tromboembolizace kdykoli u sourozence nebo rodiče, zvláště v relativně nízkém věku např. do 50 let věku) |
Pokud je suspektní hereditární predispozice, měla by být žena odeslána k odborníkovi na konzultaci před rozhodnutím o používání jakékoli CHC. |
|
Migréna |
Zvýšení frekvence nebo závažnosti migrény během používání CHC (což může být prodromální známka cévní mozkové příhody) může být důvodem okamžitého ukončení léčby. |
|
Další onemocnění související s nežádoucími cévními příhodami |
Diabetes mellitus, hyperhomocysteinemie, chlopenní srdeční vada a fibrilace síní, dyslipoproteinemie a systémový lupus erythematodes. |
Příznaky ATE
V případě příznaků by mělo být ženě doporučeno, aby vyhledala naléhavou lékařskou péči a informovala lékaře, že užívá CHC.
Příznaky cévní mozkové příhody mohou zahrnovat:
- náhlou necitlivost nebo slabost obličeje, paže nebo nohy, zvláště na jedné straně těla;
- náhlé potíže s chůzí, závratě, ztrátu rovnováhy nebo koordinace;
- náhlou zmatenost, problémy s řečí nebo porozuměním;
- náhlé potíže se zrakem na jednom nebo obou očích;
- náhlou, závažnou nebo prodlouženou bolest hlavy neznámé příčiny;
- ztrátu vědomí nebo omdlení s nebo bez záchvatu.
Dočasné příznaky naznačují, že se jedná o tranzitorní ischemickou ataku (TIA).
Příznaky infarktu myokardu mohou zahrnovat:
- bolest, nepohodlí, tlak, těžkost, pocit stlačení nebo plnosti na hrudi, v paži nebo pod hrudní kostí;
- nepohodlí vyzařující do zad, čelisti, hrdla, paže, žaludku;
- pocit plnosti, poruchu trávení nebo dušení;
- pocení, nauzeu, zvracení nebo závratě;
- extrémní slabost, úzkost nebo dušnost;
- rychlý nebo nepravidelný srdeční tep.
Ženy užívající kombinovanou antikoncepci musejí být důrazně upozorněny, že je nutné v případě výskytu příznaků trombózy vyhledat lékaře. V případě suspektní nebo potvrzené trombózy musí být užívání hormonální antikoncepce ukončeno. Současně musí být zahájena jiná adekvátní antikoncepce z důvodu teratogenity antikoagulační léčby (kumariny).
Nádory
V některých epidemiologických studiích bylo u žen dlouhodobě užívajících COC hlášeno zvýšené riziko výskytu cervikálních karcinomů, ale až dosud nepanuje shoda v názoru, do jaké míry jsou tyto nálezy způsobeny kombinací účinku přípravku a způsobu sexuálního života a případně jiných dalších faktorů jako je lidský papiloma virus (HPV).
Meta-analýzou 54 epidemiologických studií bylo zjištěno mírné zvýšení rizika (RR = 1,24) diagnostikovaných karcinomů prsu u žen, které užívají COC. Zvýšené riziko mizí postupně během 10 let po vysazení COC. Karcinom prsu je vzácný u žen mladších 40 let a zvýšení výskytu diagnostikovaného karcinomu prsu u žen, které užívaly a užívají COC, je v porovnání s celkovým rizikem výskytu karcinomu prsu velmi nízké. Karcinom prsu diagnostikovaný u žen, které užívaly COC, je obvykle v méně pokročilém stádiu než u žen, které hormonální antikoncepci nikdy neužívaly. Pozorované zvýšené riziko může být dáno časnější diagnózou karcinomu prsu u žen užívajících COC, biologickým účinkem COC nebo kombinací obojího.
Ve vzácných případech byly pozorovány benigní tumory jater a ještě vzácněji byl hlášen výskyt maligních tumorů jater u žen s hormonální antikoncepcí. Ve výjimečných případech vedly tyto tumory k ohrožení života intra-abdominálním krvácením. Z tohoto důvodu je nutné u žen používajících transdermální náplasti EVRA brát v úvahu nádory jater při diferenciální diagnostice závažných bolestí nadbřišku, zvětšení jater nebo příznaků nitrobřišního krvácení.
Další upozornění
- U žen s hmotností 90 kg nebo více může být snížena antikoncepční účinnost (viz body 4.2 a 5.1).
- U žen s hypertriglycerolemií nebo jejím výskytem v rodinné anamnéze může být při užívání kombinované hormonální antikoncepce zvýšeno riziko výskytu pankreatitidy.
- I když bylo u mnoha žen užívajících hormonální antikoncepci popsáno mírné zvýšení krevního tlaku, je klinicky významné zvýšení jen velmi vzácné. Souvislost mezi klinicky významným zvýšením krevního tlaku a užíváním hormonální antikoncepce nebyla dosud definitivně prokázána. Jestliže během užívání u žen s již existující hypertenzí konstantně zvýšený krevní tlak nebo výrazné zvýšení krevního tlaku neodpovídá očekávaným způsobem na antihypertenzní léčbu, musí být ukončena. Po úpravě krevního tlaku antihypertenzní léčbou může být podávání znovu zahájeno.
- Výskyt nebo zhoršení některých stavů byl popsán během těhotenství i při užívání COC, ale souvislost s COC není prokázána: žloutenka nebo svědění v souvislosti s cholestázou; onemocnění žlučníku včetně cholecystitidy a cholelithiázy; porfyrie, systémový lupus erythematodes, hemolytický uremický syndrom, Sydenhamova chorea, herpes gestationis, ztráta sluchu spojená s otosklerózou.
- Akutní nebo chronické poruchy jaterních funkcí si mohou vynutit přerušení kombinované hormonální antikoncepce do návratu hodnot jaterních funkcí k normálu. Opakovaný pruritus vyvolaný cholestázou, který se objevil v průběhu předchozího těhotenství nebo dřívějšího užívání steroidních hormonů, vyžaduje ukončení užívání kombinované hormonální antikoncepce.
- Přestože kombinovaná hormonální antikoncepce může ovlivňovat periferní inzulínovou rezistenci a glukózovou toleranci, nejsou důkazy o nutnosti změny léčebného režimu
u diabetiků v průběhu užívání kombinované hormonální antikoncepce. Přesto by ženy trpící diabetem měly být pečlivě sledovány především v časné fázi aplikace náplastí EVRA.
- Během podávání COC bylo popsáno zhoršení endogenních depresí, epilepsie, Crohnovy choroby nebo ulcerózní kolitidy.
- Při užívání hormonální antikoncepce se může někdy objevit chloasma, především u žen
s chloasma gravidarum v anamnéze. Ženy náchylné k výskytu chloasma by se při aplikaci náplastí EVRA neměly vystavovat slunečnímu nebo ultrafialovému záření. Chloasma nemusí být často plně reverzibilní.
Lékařské vyšetření/konzultace
Před dalším zahájením léčby přípravkem EVRA by měla být získána kompletní anamnéza (včetně rodinné anamnézy) a musí být vyloučeno těhotenství. Měl by se změřit krevní tlak a mělo by být provedeno tělesné vyšetření při zvážení kontraindikací (viz bod 4.3) a varování (viz bod 4.4). Je důležité, aby byla žena upozorněna na informace o žilní a arteriální trombóze, včetně rizika přípravku EVRA v porovnání s dalšími typy CHC, na příznaky VTE a ATE, známé rizikové faktory a co by měla dělat v případě suspektní trombózy.
Žena by také měla být informována, aby si pečlivě přečetla příbalovou informaci pro uživatele a aby dodržovala uvedené instrukce. Frekvence a povaha vyšetření by měly být založeny na stanovených postupech a upraveny podle individuálních potřeb ženy.
Ženy by měly být informovány, že hormonální antikoncepce nechrání před HIV infekcí (AIDS) a dalšími sexuálně přenosnými chorobami.
Nepravidelné krvácení
Užívání kombinované hormonální antikoncepce může být provázeno nepravidelným krvácením (špiněním, intermenstruálním krvácením), a to především v počátečních měsících používání. Vyšetření lékařem a posouzení celé situace má proto v případě nepravidelného krvácení význam až po přibližně 3 cyklech. Jestliže při správné aplikaci transdermální náplasti EVRA přetrvává nebo se po předchozích pravidelných menstruačních cyklech vyskytne intermenstruální krvácení, je nutné uvažovat i o jiné možné příčině než je aplikace transdermální náplastí EVRA. Musí být posouzena možnost nehormonální příčiny krvácení a provedena příslušná vyšetření vedoucí k vyloučení organického onemocnění nebo těhotenství. K těmto vyšetřením může patřit i kyretáž. U některých žen se během období bez náplasti nemusí dostavit krvácení ze spádu. Pokud je transdermální náplast EVRA aplikována přesně podle návodu uvedeného v bodu 4.2, je nepravděpodobné, že by žena byla těhotná. Pokud však transdermální náplast EVRA není před prvním chybějícím krvácením ze spádu nebo při druhém chybějícím krvácení ze spádu používána přesně dle výše uvedených doporučení, musí být před dalším pokračováním aplikace transdermální náplastí EVRA vyloučena gravidita.
U některých žen se po ukončení hormonální antikoncepce může objevit amenorea nebo oligorea, a to zejména, pokud se tyto obtíže vyskytovaly již dříve.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Pozor: informační texty současně podávaných léčivých přípravků musejí být posuzovány tak, aby odhalily potenciální interakce.
Účinky jiných léčivých přípravků na přípravek EVRA
Mohou se objevit interakce s přípravky, které indukují mikrosomální enzymy, což může vést ke zvýšené clearance pohlavních hormonů a k intermenstruálnímu krvácení a/nebo selhání hormonální antikoncepce. Následující interakce byly popsané v literatuře.
Látky zvyšující clearance CHC (snížená účinnost CHC v důsledku indukce enzymů)
Barbituráty, bosentan, karbamazepin, fenytoin, primidon, rifampicin, modafinil a přípravky proti HIV ritonavir, nevirapin a efavirenz a případně rovněž felbamát, griseofulvin, oxkarbazepin, topiramát a přípravky obsahující třezalku tečkovanou (Hypericum perforatum).
Léčba
Indukci enzymů lze pozorovat po několika dnech léčby. Maximální indukce enzymů se obecně pozoruje za asi 10 dní, nicméně může poté přetrvávat po dobu nejméně 4 týdnů po ukončení léčby léčivým přípravkem.
Krátkodobé užívání
Žena krátkodobě léčená léčivými přípravky, které indukují jaterní enzymy metabolizující léčiva, nebo jednotlivými léčivými látkami, které indukují tyto enzymy, musí vedle přípravku EVRA dočasně používat bariérovou metodu, tj. během doby, kdy se současně podává léčivý přípravek a 28 dní po jeho vysazení.
Pokud se léčivý přípravek podává po konci třítýdenního období, kdy je transdermální náplast aplikována, musí se další transdermální náplast aplikovat bez obvyklého intervalu bez aplikace transdermální náplasti.
Dlouhodobé užívání
U žen dlouhodobě léčených léčivými přípravky indukujícími enzymy se doporučuje jiná spolehlivá nehormonální antikoncepční metoda.
Látky s proměnlivými účinky na clearance CHC
Pokud se podávají spolu s CHC, mohou mnohé kombinace inhibitorů HIV proteázy a nenukleosidových inhibitorů reverzní transkriptázy, včetně kombinací s inhibitory HCV, zvyšovat nebo snižovat plasmatické koncentrace estrogenu nebo gestagenů. V některých případech může být čistý účinek těchto změn klinicky relevantní.
Proto je nutno pročíst informace o současně podávaných léčivých přípravcích proti HIV, aby se zjistily potenciální interakce a všechna související doporučení. V případě jakýchkoli pochybností musí žena léčená inhibitorem proteázy nebo nenukleosidovým inhibitorem reverzní transkriptázy používat dodatečnou bariérovou antikoncepční metodu.
Inhibice metabolismu ethinylestradiolu
Etorikoxib prokazatelně zvyšuje plazmatické hodnoty ethinylestradiolu (50 až 60 %), pokud je užíván souběžně s perorální třífázovu hormonální antikoncepcí. Předpokládá se, že etorikoxib zvyšuje hodnoty ethinylestradiolu tím, že inhibuje aktivitu sulfotransferázy, a tak inhibuje metabolismus ethinylestradiolu.
Účinek přípravku EVRA na jiné léčivé přípravky
Hormonální antikoncepce může ovlivnit metabolismus určitých léčivých látek. Proto jejich plazmatické a tkáňové hladiny mohou být zvýšené (např. cyklosporin) nebo snížené (např. lamotrigin). Může být třeba upravit dávkování souběžně užívaných přípravků.
Lamotrigin: prokázalo se, že kombinovaná hormonální antikoncepce významně snižuje plazmatické koncentrace lamotriginu, pokud je užívaný současně, pravděpodobně z důvodu indukce glukuronidace lamotriginu. Může to omezit kontrolu záchvatů křečí, a proto může být potřebná úprava dávkování lamotriginu.
Laboratorní testy
Užívání hormonální antikoncepce může ovlivnit některé laboratorní testy včetně biochemických jaterních parametrů, endokrinologické testy, noradrenalinové a renální funkce, plazmatické hodnoty proteinů např. globulin vázající steroidní hormony, vazebné sérové proteiny, parametry metabolismu karbohydrátů a parametry koagulace a fibrinolýzy. Změny zůstávají obecně v rozmezí laboratorního rozpětí.
4.6 Fertilita, těhotenství a kojení
EVRA je v těhotenství kontraindikována (viz bod 4.3).
V epidemiologických studiích nebylo zjištěno zvýšené riziko vrozených defektů dětí u žen, které před otěhotněním užívaly kombinovanou perorální hormonální antikoncepci. Ve většině recentních studií nebyl prokázán teratogenní účinek, i když byla kombinovaná perorální hormonální antikoncepce užívána nedopatřením v časném těhotenství.
Omezené údaje u žen, které užívaly přípravek EVRA v těhotenství, neumožňují stanovit závěry o jeho bezpečnosti v průběhu těhotenství.
Studie na zvířatech prokázaly nežádoucí účinky během těhotenství a kojení (viz bod 5.3). Na základě těchto údajů získaných na zvířatech, nelze vyloučit nežádoucí účinky vzhledem k hormonálnímu působení léčivých látek. Nicméně všeobecné zkušenosti s užíváním kombinované perorální antikoncepce v průběhu těhotenství nepřinesly důkaz skutečných nežádoucích účinků u člověka.
Jestliže dojde během aplikace transdermálních náplastí EVRA k otěhotnění, musí být aplikace okamžitě ukončena.
Zvýšené riziko VTE v poporodním období je třeba považovat při opětovném používání přípravku EVRA (viz body 4.2 a 4.4).
Kojení
Kombinovaná hormonální antikoncepce může ovlivnit kojení, především může dojít ke snížení množství a změně složení mléka. Z těchto důvodů se nedoporučuje kojícím matkám do doby definitivního odstavení dítěte aplikovat transdermální náplasti EVRA.
Fertilita
Po ukončení používání přípravku EVRA mohou ženy zaznamenat zpoždění početí.
4.7 Účinky na schopnost řídit a obsluhovat stroje
EVRA nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky Souhrn bezpečnostního profilu
Nejčastěji hlášenými nežádoucími účinky v klinických hodnoceních byly bolest hlavy, nauzea a napětí v prsou, které se vyskytovaly přibližně u 21,0 %, 16,6 %, resp. 15,9 % pacientek.
Nežádoucí účinky, které se mohou objevit v počátcích léčby, ale obvykle vymizí po prvních třech cyklech, zahrnují špinění, citlivost prsů a nauzeu.
Popis vybraných nežádoucích účinků
U žen užívajících CHC bylo pozorováno zvýšené riziko arteriálních a žilních trombotických a tromboembolických příhod, včetně infarktu myokardu, cévní mozkové příhody, tranzitorních ischemických atak, žilní trombózy a plicní embolizace a je podrobněji popsáno v bodě 4.4.
Seznam nežádoucích účinků v tabulce
Bezpečnost byla hodnocena u 3 322 sexuálně aktivních žen, které se účastnily tří klinických studií Fáze III, založených ke zhodnocení antikoncepčních účinků. Tyto ženy dostaly antikoncepci na šest nebo 13 cyklů (přípravek EVRA nebo perorální antikoncepční komparátor), užívaly nejméně jednu dávku studijního přípravku a poskytly bezpečnostní údaje. V Tabulce 1 níže, jsou uvedeny nežádoucí účinky hlášené v klinických studiích a z post-marketingu. Kategorie četnosti dle MedDRA jsou definovány jako: velmi časté (>1/10); časté (> 1/100 až < 1/10); méně časté (> 1/1000 až < 1/100); vzácné (> 1/10000 až < 1/1000); velmi vzácné (< 1/10000); není známo (z dostupných údajů nelze určit).
Tabulka 1: Frekvence nežádoucích účinků
|
Třídy orgánových systémů Frekvence |
Nežádoucí účinek |
|
Infekce a infestace | |
|
Časté |
(Vulvo) vaginální infekce |
|
Vaginální kandidóza | |
|
Vzácné |
Pustulární vyrážka* |
|
Pustulky v místě aplikace | |
a blíže neurcene (zahrnující cysty a
|
Vzácné |
Jatemí neoplasma*f Karcinom prsu*f Karcinom děložního čípku*f Jaterní adenom*f Děložní leiomyom Fibroadenom prsu |
|
Poruchy imunitního systému | |
|
Méně časté |
Hypersenzitivita |
|
Poruchy metabolismu a výživy | |
|
Méně časté |
Hypercholesterolemie Retence tekutin Zvýšení chuti k jídlu |
|
Vzácné |
Hyperglykemie* Insulinová resistence* |
|
Psychiatrické poruchy | |
|
Časté |
Změny nálady a rychlé střídání nálad |
|
Méně časté |
Insomnie Snížení libida |
|
Vzácné |
Zlost* Frustrace* Zvýšení libida |
|
Poruchy nervového systému | |
|
Velmi časté | |
|
Časté |
Migréna Závratě |
|
Vzácné |
Cévní mozková příhoda**f Krvácení do mozku*f Abnormální chuť* |
|
Poruchy oka | |
|
Vzácné |
Nesnášenlivost kontaktních čoček* |
|
Srdeční poruchy | |
|
Vzácné |
Arteriální tromboembolizace (Akutní) infarkt myokardu*f |
|
Cévní poruchy | |
|
Méně časté |
Hypertenze |
|
Vzácné |
Hypertenzní krize* Arteriální trombóza**f Žilní trombóza**f Trombóza*f Žilní tromboembolizace |
|
Vzácné |
Trombóza plicní tepny*f Plicní embolief |
|
Gastrointestinální poruchy | |
|
Velmi časté |
|
Časté Vzácné |
Břišní distenze Kolitida* |
|
Poruchy jater a žlučových cest | |
|
Vzácné |
Cholecystitida Cholelitiázaf Poškození jater* Cholestatická hepatitida*f Cholestáza*f |
|
Poruchy kůže a podkožní tkáně | |
|
Časté |
Akné Vyrážka Pruritus Kožní reakce Podráždění kůže |
|
Méně časté |
Alopecie Alergická dermatitida Ekzém Fotosenzitivní reakce Kontaktní dermatitida Kopřivka Erytém |
|
Vzácné |
Angioedém* Erytém (multiformní, nodózní)* Chloasmaf Exfoliativní vyrážka* Generalizovaný pruritus Vyrážka (erytematózní, svědivá) Seboroická dermatitida* |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně | |
|
Časté |
Svalové spasmy |
|
Poruchy reprodukčního systému a prsu | |
|
Velmi časté |
Citlivost prsů |
|
Časté |
Dysmenorea Vaginální krvácení a poruchy menstruace**f Děložní spasmy Potíže s prsy Vaginální výtok |
|
Méně časté |
Galaktorea Premenstruační syndrom Vulvovaginální suchost |
|
Vzácné |
Dysplazie čípku* Potlačení laktace* Výtok z genitálu |
|
Celkové poruchy a reakce v místě aplikace | |
|
Časté |
Únava Reakce v místě aplikace (erytém, podráždění, pruritus, vyrážka) |
|
Méně časté |
Generalizovaný edém Periferní edém Reakce v místě aplikace** |
|
Vzácné |
Edém obličeje* Edém s ďolíčky* Otok Reakce v místě aplikace* (např. absces, eroze) Lokalizovaný edém* |
|
Vyšetření | |
|
Časté |
Zvýšení tělesné hmotnosti |
|
Méně časté |
Vzestup krevního tlaku Poruchy lipidů** |
|
Vzácné |
Snížení glukózy v krvi*f Abnormální hladina glykemie*f |
* Post-marketingové údaje
** Nežádoucí účinky dle údajů z klinických studií a post-marketingových hlášení f Viz bod 4.4
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Po náhodném požití většího množství perorálních antikoncepčních přípravků nebyly hlášeny závažné nežádoucí účinky. Předávkování může vyvolat nauzeu nebo zvracení. U některých žen se může objevit vaginální krvácení. V případě podezření na předávkování musí být transdermální antikoncepce ukončena a zahájena symptomatická léčba.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: pohlavní hormony a modulátory pohlavního systému, progestogeny a estrogeny, fixní kombinace, norelgestromin a estrogen; ATC kód: G03AA13
Mechanismus účinku
EVRA působí estrogenní a gestagenní aktivitou ethinylestradiolu a norelgestrominu supresi gonadotropinu. Primárním mechanismem účinku je inhibice ovulace, ale k účinnosti přípravku mohou také přispívat změny cervikálního hlenu a endometria.
Klinická účinnost a bezpečnost Pearl Index (viz tabulka):
|
Skupina |
CONT-002 EVRA |
CONT-003 EVRA |
CONT-003 COC* |
CONT-004 EVRA |
CONT-004 COC** |
Všechny EVRA |
|
počty cyklů |
10 743 |
5831 |
4592 |
5095 |
4005 |
21 669 |
|
Celkový Pearl Index (95 % CI) |
0,73 (0,15; 1,31) |
0,89 (0,02; 1,76) |
0,57 (0; 1,35) |
1,28 (0,16; 2,39) |
2,27 (0,59; 3,96) |
0,90 (0,44; 1,35) |
|
Selhání metody Pearl Index (95 % CI) |
0,61 (0,0; 1,14) |
0,67 (0; 1,42) |
0,28 (0; 0,84) |
1,02 (0,02; 2,02) |
1,30 (0,03; 2,57) |
0,72 (0,31; 1,13) |
* DSG 150 + 20 gg EE
** 50 gg LNG + 30 gg po dny 1-6; 75 gg LNG + 40 gg EE po dny 7-11; 125 gg LNG + 30 gg EE po dny 12-21.
Ve III. fázi klinické studie byly u populace žen (n = 3 319) charakterizované věkem, rasou a tělesnou hmotností provedeny explorační analýzy za účelem stanovení, zda v takto charakterizované populaci žen existuje souvislost s graviditou. Analýzou nebyla nalezena souvislost gravidity s věkem a rasou.
S ohledem na tělesnou hmotnost byla však u 5 těhotných žen z 15 těhotných, které používaly transdermální náplasti EVRA, výchozí hmotnost 90 kg nebo více, což tvoří < 3 % sledované populace. U žen s hmotností nižší než 90 kg nebyla zjištěna souvislost mezi tělesnou hmotností a těhotenstvím. Ačkoliv pouze 10 až 20 % variability ve farmakokinetických zjištěních může být vysvětlováno tělesnou hmotností (viz bod 5.2), byla vyšší frekvence gravidit u žen o hmotnosti 90 kg a více statisticky významná, což svědčí o nižší účinnosti transdermálních náplastí EVRA u těchto žen.
Při užití COC s vyšší dávkou ethinylestradiolu (50 gg ethinylestradiolu) bylo riziko nádorů endometria a ovarií sníženo. Možná aplikace těchto výsledků na nízko dávkovanou kombinovanou hormonální antikoncepci nebyla potvrzena.
5.2 Farmakokinetické vlastnosti
Absorpce
Po aplikaci transdermální náplastí EVRA bylo plateau norelgestrominu a ethinylestradiolu v séru dosaženo přibližně za 48 hodin. Koncentrace norelgestrominu v rovnovážném stavu byla po jednotýdenní aplikaci náplasti 0,8 ng/ml a EE 50 pg/ml. Po opakované aplikaci byla koncentrace v séru a AUC pro norelgestromin a EE ve srovnání s 1. týdnem 1. cyklu jen mírně zvýšena.
Absorpce norelgestrominu a ethinylestradiolu byla po aplikaci náplasti studována za různých podmínek při návštěvách fitnes center (sauna, Whirlpool, šlapací kolo a jiná aerobická cvičení) a při koupání ve studené vodě. Na základě výsledků absorpce norelgestrominu nebyl pozorován rozdílný účinek léčby na Css a AUC ve srovnání s normálními podmínkami aplikace. Pro EE byl pozorován mírný rozdíl během cvičení na šlapacím kole a cvičení aerobiku; avšak hodnoty Css byly v rozmezí referenčních hodnot. Koupání ve studené vodě tyto parametry neovlivnilo.
Výsledky prodloužené aplikace jedné antikoncepční náplasti EVRA ze 7 na 10 dní naznačují, že cílové koncentrace Css norelgestrominu a ethinylestradiolu přetrvávají po další 3 dny prodloužené aplikace EVRA (10denní). Tyto výsledky potvrzují předpoklad, že klinická účinnost může přetrvávat, i když není dodržen správný režim výměny náplasti a zpoždění výměny není delší než 2 celé dny.
Distribuce
Norelgestromin a norgestrel (sérový metabolit norelgestrominu) se pevně váží na plazmatické proteiny (> 97 %). Norelgestromin je vázán na albumin a ne na SHBG, zatímco norgestrel je vázán zejména na SHBG, což omezuje jeho biologickou účinnost. Ethinylestradiol se významně váže na sérový albumin.
Biotransformace
Norelgestromin je metabolizován v játrech a mezi jeho metabolity patří norgestrel, který je ve velkém rozsahu vázán na SHBG a různé hydroxylované a konjugované metabolity. Ethinylestradiol je také metabolizován na různé hydroxylované produkty a jejich glukuronidové nebo sulfátové konjugáty.
Eliminace
Po odstranění náplasti byly průměrné eliminační poločasy norelgestrominu přibližně 28 hodin a ethinylestradiolu 17 hodin. Metabolity jsou vylučovány močí nebo stolicí.
Transdermální versus perorální antikoncepce
Farmakokinetické profily transdermálních a perorálních kombinovaných antikoncepčních přípravků jsou rozdílné, a proto je třeba při přímém srovnávání těchto PK parametrů postupovat obezřetně.
Ve studii srovnávající přípravek EVRA s perorálním antikoncepčním přípravkem obsahujícím norgestimát (prekurzor norelgestrominu) 250 pg/ ethinylestradiol 35 pg byly hodnoty Cmax 2x vyšší u NGMN a EE u subjektů s perorální antikoncepcí ve srovnání s přípravkem EVRA, zatímco celková expozice (AUC a Css) byla u subjektů s přípravkem EVRA srovnatelná. Inter-individuální variabilita (% CV) PK parametrů po aplikaci přípravku EVRA byla vyšší ve srovnání s variabilitou zjištěnou u perorální antikoncepce.
Vliv věku, tělesné hmotnosti a tělesného povrchu
Vliv věku, tělesné hmotnosti a tělesného povrchu na farmakokinetiku norelgestrominu a ethinylestradiolu byl hodnocen u 230 zdravých žen v devíti farmakokinetických studiích po jednorázové 7denní aplikaci náplasti EVRA. Pro norelgestromin i pro EE souviselo zvýšení věku, tělesné hmotnosti nebo tělesného povrchu s mírným snížením hodnot Css a AUC. Avšak pouze malá část (10 - 20 %) z celkové variability ve farmakokinetice norelgestrominu a EE může být v souvislosti s aplikací náplastí EVRA spojována s výše uvedenými demografickými parametry.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti, toxicity po opakovaném podání, genotoxicity a hodnocení kancerogenního potenciálu neodhalily žádné zvláštní riziko pro člověka. S ohledem na reprodukční toxicitu norelgestrominu byla u králíků zjištěna fetální toxicita, avšak hranice bezpečnosti byla pro tento jev dostatečně vysoká. Údaje o reprodukční toxicitě kombinace norelgestrominu s ethinylestradiolem nejsou dostupné. Údaje o kombinaci norgestimátu (prekursor norelgestrominu) s ethinylestradiolem prokazují u zvířecích samic snížení fertility a schopnosti implantace u laboratorních potkanů a zvýšení fetální resorpce u laboratorních potkanů a králíků.
Po vyšších dávkách byl pozorován pokles životaschopnosti a fertility samičích mláďat laboratorních potkanů. Relevance těchto zjištění u lidí není známa, i když tyto účinky byly pozorovány v kontextu s dobře známou farmakodynamikou a druhovou specificitou.
Studie provedené za účelem sledování účinku náplastí EVRA na kůži svědčí o tom, že tento systém není schopen vyvolat senzibilizaci, a při aplikaci na kůži králíků bylo zjištěno jen mírné podráždění.
6. FARMACEUTICKÉ ÚDAJE 6.1 Seznam pomocných látek
Zevní krycí vrstva
vnější - pigmentovaný polyethylen nízké hustoty vnitřní - polyesterová fólie
Střední vrstva
adhezivní polyisobutenová/polybutenová vrstva
krospovidon
netkaná polyesterová vrstva lauryl-laktát
Třetí vrstva pegoterát (PET) polydimethylsiloxan
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
2 roky
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem a vlhkostí.
Neukládejte do chladničky nebo nezmrazujte.
6.5 Druh obalu a velikost balení
Vnitřní obal
Sáček je složen ze 4vrstev: polyethylenový film nízké hustoty (nejvnitřnější vrstva), hliníková fólie, polyethylenový film nízké hustoty a zevní vrstva z běleného papíru.
Vnější obal Papírová skládačka.
Velikost balení: 3, 9 nebo 18 transdermálních náplastí EVRA opatřených oddělitelnou fólií, v jednotlivých sáčcích. Sáčky jsou baleny po třech v průhledné perforované plastové fólii a umístěny v papírové skládačce.
Na trhu nemusejí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Náplast musí být nalepena okamžitě po vyndání ze sáčku.
Aby se předešlo narušení lepivosti transdermální náplasti EVRA nesmí se na místa aplikace transdermální náplasti EVRA aplikovat krémy, tělová mléka nebo zásypy.
Po použití transdermální náplast stále obsahuje určité množství léčivé látky, která po kontaktu s vodním prostředím může mít nepříznivé účinky na životní prostředí. Z tohoto důvodu je zapotřebí použitou transdermální náplast pečlivě likvidovat. Odlepte proto z vnější strany sáčku nálepku určenou k likvidaci použité transdermální náplasti. Použitou transdermální náplast vložte do otevřené nálepky k likvidaci transdermální náplasti tak, aby lepicí vrstva překrývala vyznačené místo na sáčku. Nálepku k likvidaci transdermální náplasti s vloženou náplastí potom uzavřete slepením. Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky. Použité transdermální náplasti nesmějí být splachovány do toalety ani jiných tekutých odpadů.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Janssen-Cilag International NV Turnhoutseweg, 30 B-2340 Beerse Belgie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/02/223/001
EU/1/02/223/002
EU/1/02/223/003
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 22. srpna 2002
Datum posledního prodloužení registrace: 22. srpna 2012
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A. VÝROBCE ODPOVĚDNÝ /VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného/výrobců odpovědných za propouštění šarží Janssen Pharmaceutica NV, Turnhoutseweg 30, B-2340 Beerse, Belgie
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis.
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
• Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném ve čl. 107c odst. 7 směrnice 2011/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Neuplatňuje se.
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU PAPÍROVÁ KRABIČKA
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
EVRA 203 mikrogramů /24 hodin + 33,9 mikrogramů/24 hodin transdermální náplast norelgestrominum/ethinylestradiolum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
1 náplast 20 cm2 obsahuje: norelgestrominum 6 mg a ethinylestradiolum 600 mikrogramů.
1 náplast uvolňuje: norelgestrominum 203 mikrogramů za 24 hodin a ethinylestradiolum 33,9 mikrogramů za 24 hodin.
3. SEZNAM POMOCNÝCH LÁTEK
Zevní krycí vrstva: pigmentovaný polyethylen nízké hustoty, polyesterová fólie.
Střední vrstva: adhezivní polyisobutenová/polybutenová vrstva, krospovidon, netkaná polyesterová vrstva, lauryl-laktát.
Třetí vrstva: pegoterát (PET), polydimethylsiloxan.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
3 transdermální náplasti 9 transdermálních náplastí 18 transdermálních náplastí
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Transdermální podání
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem. Chraňte před chladem nebo mrazem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Použité ani nepoužité náplasti nesplachujte do toalety. Přečtěte si příbalovou informaci, kde najdete pokyny k likvidaci náplasti.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Janssen-Cilag International NV Turnhoutseweg 30 B-2340 Beerse, Belgie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/02/223/001: 3 transdermální náplasti EU/1/02/223/002: 9 transdermálních náplastí EU/1/02/223/003: 18 transdermálních náplastí
13. ČÍSLO ŠARŽE
č.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
evra
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
2D čárový kód s jedinečným identifikátorem.
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
PC:
SN:
NN:
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITŘNÍM OBALU NÁLEPKA NA SÁČEK
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
EVRA 203 mikrogramů /24 hodin + 33,9 mikrogramů/24 hodin transdermální náplast norelgestrominum /ethinylestradiolum
2. ZPŮSOB PODÁNÍ
Transdermálm podání
Před použitím si přečtěte příbalovou informaci.
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
č.s.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
Obsahuje 1 transdermální náplast
6. JINÉ
Nálepky pro připomenutí
Použijte tyto nálepky pro označení dne výměny náplasti ve Vašem kalendáři
Další cyklus První náplast
Aktuální cyklus
První náplast Druhá náplast Třetí náplast Odstraňte náplast
(1. týden) (2. týden) (3. týden) Obstarejte si novou
náplast
Nálepka k likvidaci náplasti
NÁLEPKA K LIKVIDACI NÁPLASTI Likvidace použité náplasti:
1. Použitou náplast přiložte lepicí vrstvou na vyznačené místo.
2. Odstraňte ochrannou vrstvu.
3. Nálepku uzavřete a její okraje přitlačte okolo náplasti.
4. Zlikvidujte s komunálním odpadem.
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: Informace pro pacienta
EVRA 203 mikrogramů/24 hodin + 33,9 mikrogramů/24 hodin transdermální náplast
norelgestrominum / ethinylestradiolum
Důležité věci, které je třeba vědět o kombinované hormonální antikoncepci (CHC):
- Jedná se o nejspolehlivější reverzibilní metody antikoncepce, pokud jsou používány správně.
- Mírně zvyšují riziko krevní sraženiny v žilách a tepnách, zvláště v prvním roce nebo při znovuzahájení kombinované hormonální antikoncepce po pauze trvající 4 týdny nebo déle.
- Buďte prosím opatrná a navštivte svého lékaře, pokud si myslíte, že máte příznaky krevní sraženiny (viz bod 2 „Krevní sraženiny“).
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka nebo zdravotní sestry.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je EVRA a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek EVRA používat
3. Jak se EVRA používá
4. Možné nežádoucí účinky
5. Jak přípravek EVRA uchovávat
6. Obsah balení a další informace
1. Co je EVRA a k čemu se používá
EVRA obsahuje dva druhy hormonů, progesteron nazývaný norelgestromin a estrogen nazývaný ethinylestradiol.
Vzhledem k tomu, že obsahuje dva hormony, nazývá se EVRA „kombinovaná hormonální antikoncepce“.
Používá se k zabránění otěhotnění.
2. Čemu musíte věnovat pozornost, než začnete přípravek EVRA používat Obecné poznámky
Předtím, než začnete používat přípravek EVRA, měla byste si přečíst informace o krevních sraženinách v bodě 2. Je zvláště důležité, abyste si přečetla příznaky krevních sraženin - viz bod 2 „Krevní sraženiny“.
Kdy byste neměla používat přípravek EVRA
Neměla byste používat přípravek EVRA, pokud máte jakýkoli ze stavů uvedených níže. Pokud máte níže uvedený stav, musíte informovat svého lékaře. Váš lékař s Vámi prodiskutuje, jaká jiná antikoncepční metoda by pro Vás byla vhodná.
• pokud máte (nebo jste měla) krevní sraženinu v krevní cévě ve svých nohách (hluboká žilní trombóza, DVT), ve Vašich plicích (plicní embolie, PE) nebo v jiných orgánech;
• pokud víte, že máte poruchu postihující Vaši krevní srážlivost - například deficit proteinu C, deficit proteinu S, deficit antitrombinu III, faktor V Leiden nebo protilátky proti fosfolipidu;
• pokud potřebujete operaci nebo dlouhou dobu nechodíte (viz bod „Krevní sraženiny“);
• pokud jste někdy měla srdeční záchvat nebo cévní mozkovou příhodu;
• pokud máte (nebo jste někdy měla) anginu pectoris (stav, který způsobuje těžkou bolest na hrudi a může být první známkou srdečního záchvatu) nebo tranzitorní ischemickou ataku TIA -dočasné příznaky cévní mozkové příhody);
• pokud máte některá z následujících onemocnění, která zvyšují riziko sraženiny v tepnách, jako je:
- těžký diabetes s poškozením krevních cév;
- velmi vysoký krevní tlak;
- velmi vysokou hladinu tuku v krvi (cholesterol nebo triglyceridy);
- onemocnění označované jako hyperhomocysteinemie;
• pokud máte (nebo jste měla) typ migrény označovaný jako „migréna s aurou“;
• jestliže jste alergická na norelgestromin, ethinylestradiol nebo na kteroukoli další složku tohoto přípravku (uvedeny v bodě 6);
• jestliže Vám bylo řečeno, že byste mohla mít rakovinu prsu nebo rakovinu dělohy, čípku nebo pochvy;
• jestliže jste v minulosti měla nádor na játrech, nebo onemocnění jater, kvůli kterému játra nefungují správně;
• jestliže trpíte na nevysvětlené krvácení z pochvy.
Vyskytne-li se u Vás cokoli z výše popsaného, nepoužívejte tento přípravek. Jestliže si nejste jistá, poraďte se se svým lékařem lékárníkem nebo zdravotní sestrou, než začnete tento přípravek používat.
Kdy je třeba zvláštní opatrnost při používání přípravku EVRA_
Kdy byste měla kontaktovat svého lékaře?
Vyhledejte naléhavou lékařskou pomoc
• pokud si všimnete možných známek krevní sraženiny, která může znamenat, že máte krevní sraženinu v noze (tj. hluboká žilní trombóza), krevní sraženinu v plicích (tj. plicní embolizace), srdeční záchvat nebo cévní mozkovou příhodu (viz bod „Krevní sraženina [trombóza]“ níže).
Pro popis příznaků těchto závažných nežádoucích účinků si prosím přečtěte „Jak rozpoznat krevní sraženinu“.
Upozornění a opatření
Dříve než začnete tento přípravek používat, musíte podstoupit lékařské vyšetření.
Informujte svého lékaře, pokud se Vás týká některý z následujících stavů.
Pokud se stav vyvine nebo se zhorší během používání přípravku EVRA, měla byste také informovat
svého lékaře.
• pokud máte Crohnovu chorobu nebo ulcerózní kolitidu (chronické zánětlivé střevní onemocnění);
• pokud máte systémový lupus erythematodes (SLE - onemocnění, které postihuje Váš přirozený obranný systém);
• pokud máte hemolyticko-uremický syndrom (HUS - porucha srážení krve, která vede k selhání ledvin);
• pokud máte srpkovitou anemii (dědičné onemocnění červených krvinek);
• pokud máte zvýšené hladiny tuku v krvi (hypertriglyceridemie) nebo pozitivní rodinnou anamnézu tohoto onemocnění. Hypertriglyceridemie souvisí se zvýšeným rizikem rozvoje pankreatitidy (zánět slinivky břišní);
• pokud potřebujete operaci nebo dlouhou dobu nechodíte (viz bod 2 „Krevní sraženiny“);
• pokud jste právě porodila, máte zvýšené riziko krevních sraženin. Měla byste se zeptat svého lékaře, jak brzy po porodu můžete začít používat přípravek EVRA;
• pokud máte zánět žil pod kůží (povrchová tromboflebitida);
• pokud máte křečové žíly.
KREVNÍ SRAŽENINY
Užívání kombinované hormonální antikoncepce, jako je přípravek EVRA zvyšuje Vaše riziko rozvoje krevní sraženiny v porovnání s jejím neužíváním. Ve vzácných případech může krevní sraženina zablokovat krevní cévy a způsobit vážné problémy.
Krevní sraženiny se mohou vyvinout
• v žilách (označuje se jako „žilní trombóza“, „žilní tromboembolizace“ nebo VTE);
• v tepnách (označuje se jako „arteriální trombóza“, „arteriální tromboembolizace“ nebo ATE).
Zotavení se z krevních sraženin není vždy úplné. Vzácně se mohou vyskytnout závažné, trvalé účinky nebo velmi vzácně mohou být fatální.
Je důležité si pamatovat, že celkové riziko škodlivé krevní sraženiny v důsledku přípravku EVRA je malé.
JAK ROZPOZNAT KREVNÍ SRAŽENINU
Vyhledejte rychlou lékařskou pomoc, pokud si všimnete některé/ho z následujících známek nebo příznaků.
|
Máte některé z těchto příznaků? |
Cím pravděpodobně trpíte? |
|
• otok jedné nohy nebo podél žíly na noze nebo chodidle, zvláště doprovázený: - bolestí nebo citlivostí v noze, která může být pociťována pouze vstoje nebo při chůzi - zvýšenou teplotou postižené nohy - změnou barvy kůže na noze, např. zblednutí, zčervenání nebo zmodrání |
Hluboká žilní trombóza |
|
• náhlý nástup nevysvětlitelné dušnosti nebo rychlého dýchání • náhlý kašel bez zřejmé příčiny, který může způsobit vykašlávání krve • ostrá bolest na hrudi, která se může zvyšovat při hlubokém dýchání • těžké točení hlavy nebo závrať • rychlý nebo nepravidelný srdeční tep • těžká bolest žaludku Pokud si nejste jistá, informujte svého lékaře, protože některé z těchto příznaků, jako je kašel nebo dušnost, mohou být zaměněny za mírnější onemocnění, jako je infekce dýchacího traktu (např. „nachlazení“). |
Plicní embolizace |
|
Příznaky se nejčastěji objevují na jednom oku: • okamžitá ztráta zraku nebo • bezbolestné rozmazané vidění, které může přejít do ztráty zraku |
Trombóza retinální žíly (krevní sraženina v oku) |
|
• bolest na hrudi, nepohodlí, tlak, tíže • pocit stlačení nebo plnosti na hrudi, v paži nebo pod hrudní kostí • plnost, porucha trávení nebo pocit dušení • nepohodlí v horní části těla vyzařující do zad, čelisti, hrdla, paže, žaludku • pocení, nevolnost, zvracení nebo závratě • extrémní slabost, úzkost nebo dušnost • rychlý nebo nepravidelný srdeční tep |
Srdeční záchvat |
|
• náhlá slabost nebo necitlivost obličeje, paže nebo nohy, zvláště na jedné straně těla • náhlá zmatenost, problémy s řečí nebo porozuměním • náhlé potíže se zrakem na jednom nebo obou očích • náhlé potíže s chůzí, závratě, ztráta rovnováhy nebo koordinace • náhlá, závažná nebo prodloužená bolest hlavy neznámé příčiny • ztráta vědomí nebo omdlení s nebo bez záchvatu |
Cévní mozková příhoda |
|
Někdy mohou být příznaky cévní mozkové příhody krátké s téměř okamžitým a plným zotavením, ale měla byste vyhledat okamžitou lékařskou pomoc, protože můžete mít riziko další cévní mozkové příhody. | |
|
• otok a lehké zmodrání končetiny • těžká bolest žaludku (akutní břicho) |
Krevní sraženiny blokující jiné cévy |
KREVNÍ SRAŽENINY V ŽÍLE
Co se může stát, pokud se v žíle vytvoří krevní sraženina?
• Používání kombinované hormonální antikoncepce souviselo se zvýšeným rizikem krevních sraženin v žíle (žilní tromboembolizace). Tyto nežádoucí účinky jsou však vzácné. Nejčastěji se objevují v prvním roce používání kombinované hormonální antikoncepce.
• Pokud se vytvoří krevní sraženina v žíle v noze nebo chodidle, může způsobit hlubokou žilní trombózu (DVT).
• Pokud se krevní sraženina přesune z nohy a usadí se v plicích, může způsobit plicní embolii.
• Velmi vzácně se může krevní sraženina vytvořit v žíle v jiném orgánu, jako je oko (trombóza retinální žíly).
Kdy je riziko rozvoje krevní sraženiny v žíle nejvyšší?
Riziko rozvoje krevní sraženiny v žíle je nejvyšší v prvním roce užívání kombinované hormonální antikoncepce. Riziko může být také vyšší, pokud znovu zahájíte užívání kombinované hormonální antikoncepce (stejný nebo odlišný přípravek) po pauze trvající 4 týdny nebo déle.
Po prvním roce se riziko zmenšuje, ale je vždy mírně vyšší, než kdybyste neužívala kombinovanou hormonální antikoncepci.
Když ukončíte používání přípravku EVRA, vrátí se Vaše riziko krevní sraženiny k normální úrovni během několika týdnů.
Jaké je riziko rozvoje krevní sraženiny?
Riziko závisí na Vašem přirozeném riziku VTE a na typu kombinované hormonální antikoncepce, kterou používáte.
Celkové riziko krevní sraženiny v noze nebo plicích (DVT nebo PE) u přípravku EVRA je malé.
Z 10 000 žen, které nepoužívají žádnou hormonální antikoncepci a nejsou těhotné, se asi u 2 žen vyvine krevní sraženina během jednoho roku.
Z 10 000 žen, které používají kombinovanou hormonální antikoncepci obsahující levonorgestrel, norethisteron nebo norgestimát, se asi u 5-7 žen vyvine krevní sraženina během jednoho roku.
Z 10 000 žen, které používají kombinovanou hormonální antikoncepci obsahující etonorgestrel nebo norelgestromin, jako je přípravek EVRA, se asi u 6-12 žen vyvine krevní sraženina během jednoho roku.
Riziko krevní sraženiny se bude měnit podle Vaší osobní anamnézy (viz níže „Faktory, které zvyšují Vaše riziko krevní sraženiny“).
|
Riziko rozvoje krevní sraženiny za rok | |
|
Ženy, které neužívají/nepoužívají kombinovanou hormonální pilulku/náplast/kroužek a nejsou těhotné |
Asi 2 z 10 000 žen |
|
Ženy, které užívají kombinovanou antikoncepční pilulku obsahující levonorgestrel, norethisteron nebo norgestimát |
Asi 5-7 z 10 000 žen |
|
Ženy, které užívají přípravek EVRA |
Asi 6-12 z 10 000 žen |
Faktory, které zvyšují Vaše riziko krevní sraženiny v žíle
Riziko krevní sraženiny u přípravku EVRA je malé, ale některá onemocnění riziko zvyšují. Vaše riziko je vyšší, pokud:
• máte velkou nadváhu (index tělesné hmotnosti nebo BMI nad 30 kg/m2);
• někdo z Vašich přímých příbuzných měl krevní sraženinu v noze, plicích nebo jiném orgánu v mladém věku (např. do 50 let věku). V tomto případě byste mohla mít dědičnou poruchu srážlivosti krve;
• potřebujete operaci nebo dlouhou dobu nechodíte z důvodu úrazu nebo onemocnění nebo pokud máte nohu v sádře. Může být nutné užívání přípravku EVRA zastavit na několik týdnů před operací, nebo když máte omezenou pohyblivost. Pokud musíte zastavit užívání přípravku EVRA, zeptejte se svého lékaře, kdy jej můžete začít znovu užívat;
• jste ve vyšším věku (zvláště ve věku nad 35 let);
• jste porodila před méně než několika týdny.
Riziko rozvoje krevní sraženiny se zvyšuje s více onemocněními, která máte.
Cestování letadlem (> 4 hodiny) může dočasně zvýšit Vaše riziko krevní sraženiny, zvláště pokud máte některé z dalších uvedených faktorů.
Je důležité, abyste informovala svého lékaře, pokud se Vás některé z těchto stavů týkají, i když si nejste jistá. Váš lékař se může rozhodnout, že je třeba užívání přípravku EVRA zastavit.
Pokud se některý z výše uvedených stavů změní v průběhu užívání přípravku EVRA, například se u přímého příbuzného vyskytne trombóza z neznámého důvodu nebo pokud se zvýší Vaše tělesná hmotnost, sdělte to svému lékaři.
KREVNÍ SRAŽENINY V TEPNĚ
Co se může stát, pokud se v tepně vytvoří krevní sraženina?
Podobně jako krevní sraženina v žíle, může sraženina v tepně způsobit závažné problémy. Může například způsobit srdeční záchvat nebo cévní mozkovou příhodu.
Faktory, které zvyšují Vaše riziko krevní sraženiny v tepně
Je důležité si pamatovat, že riziko srdečního záchvatu nebo cévní mozkové příhody při užívání přípravku EVRA je velmi malé, ale může se zvyšovat:
• se zvyšujícím se věkem (nad 35 let věku);
• pokud kouříte. Při užívání kombinované hormonální antikoncepce, jako je přípravek EVRA je doporučeno, abyste přestala kouřit. Pokud nejste schopna přestat kouřit a je Vám více než
35 let, může Vám lékař poradit, abyste používala jiný typ antikoncepce;
• pokud máte nadváhu;
• pokud máte vysoký krevní tlak;
• pokud měl Váš přímý příbuzný srdeční záchvat nebo cévní mozkovou příhodu v mladém věku (do 50 let věku). V tomto případě máte také vyšší riziko srdečního záchvatu nebo cévní mozkové příhody;
• pokud Vy nebo někdo z Vašich přímých příbuzných má vysokou hladinu tuků v krvi (cholesterol nebo triglyceridy);
• pokud máte migrény, zvláště migrény s aurou;
• pokud máte problém se srdcem (chlopenní vadu, poruchu rytmu označenou jako fibrilace síní);
• pokud máte diabetes.
Pokud máte více než jeden z těchto stavů nebo pokud je některý z nich zvláště závažný, může být riziko rozvoje krevní sraženiny ještě zvýšeno.
Pokud se některý z výše uvedených stavů změní v průběhu užívání přípravku EVRA, například začnete kouřit, u přímého příbuzného se vyskytne trombóza z neznámého důvodu, nebo pokud se zvýší Vaše tělesná hmotnost, sdělte to svému lékaři.
Před používáním přípravku EVRA se poraďte s lékařem, lékárníkem nebo zdravotní sestrou, vyskytuje-li se u Vás cokoli z dále uvedeného nebo se tento stav objeví nebo zhorší během používání přípravku EVRA:
• Myslíte si, že byste mohla být těhotná.
• Máte bolesti hlavy, které se zhoršují nebo se vyskytují častěji než dříve.
• Vážíte 90 kg nebo více.
• Máte vysoký krevní tlak nebo krevní tlak stoupá.
• Máte onemocnění žlučníku včetně žlučových kamenů nebo zánět žlučníku.
• Máte chorobu krve zvanou porfyrie.
• Máte nervovou poruchu zahrnující náhlé pohyby těla nazývanou „Sydenhamova chorea“.
• Měla jste během těhotenství kožní vyrážku s puchýřky (zváno „herpes gestationis“).
• Trpíte ztrátou sluchu.
• Máte cukrovku.
• Máte deprese.
• Máte epilepsii nebo jakýkoli problém, který může působit křeče (záchvaty).
• Máte problémy s játry zahrnující zežloutnutí kůže a očního bělma (žloutenka).
• Máte „těhotenské skvrny“. Jsou to žlutohnědé skvrny zejména na obličeji (zvané „chloasma“). Tyto skvrny nemusí úplně vymizet, ani když přestanete používat přípravek EVRA. Chraňte kůži před slunečním světlem a ultrafialovým zářením. To může zabránit vzniku těchto skvrn nebo jejich zhoršení.
• Máte potíže s ledvinami.
Nejste-li si jistá, zda se Vás cokoli z výše uvedeného týká, poraďte se před použitím přípravku EVRA se svým lékařem nebo lékárníkem.
Pohlavně přenosné nemoci
Tento přípravek Vás nechrání proti infekci HIV (AIDS) nebo jiným pohlavně přenosným nemocem. Ty zahrnují infekce chlamydiemi, genitální opar, genitální bradavice, kapavku, hepatitidu B (žloutenku), syfilis. Abyste se chránila proti těmto nemocem, používejte vždy kondom.
Lékařská laboratorní vyšetření
• Upozorněte lékaře nebo osobu odebírající vzorek při vyšetření krve nebo moči, že používáte přípravek EVRA. Hormonální antikoncepce může ovlivnit výsledky některých testů.
Děti a dospívající
EVRA nebyla sledována u dětí a dospívajících mladších 18let. EVRA se nesmí používat u dětí a dospívajících, pokud ještě neměli první menstruaci.
Další léčivé přípravky a přípravek EVRA
Prosím, informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užívala, nebo které možná budete užívat.
Některé léky a rostlinné přípravky mohou snížit účinnost přípravku EVRA. Pokud by se tak stalo, mohla byste otěhotnět nebo by mohlo dojít k neočekávanému krvácení.
Patří sem léčivé přípravky, které se používají k léčbě:
• některé antiretrovirové léčivé přípravky k léčbě HIV/AIDS a infekce virem hepatitidy C (takzvané inhibitory proteázy a nenukleosidové inhibitory reverzní trankriptázy, jako je ritonavir, nevirapin, efavirenz).
• léčivé přípravky na infekci (jako rifampicin a griseofulvin).
• léčivé přípravky proti křečím (jako jsou barbituráty, topiramát, fenytoin, karbamazepin, primidon, oxkarbazepin a felbamát).
• bosentan (lék na vysoký krevní tlak v plicních cévách).
• třezalku tečkovanou (rostlinný přípravek užívaný na deprese).
Užíváte-li kterýkoli z těchto léčivých přípravků, může být nutné, abyste používala jiný způsob kontroly početí (jako kondom, pesar nebo pěnu). Rušivý účinek některých z těchto přípravků může trvat až 28 dní po skončení jejich užívání. Promluvte si se svým lékařem nebo lékárníkem o použití jiné metody antikoncepce, pokud používáte přípravek EVRA současně s některým z výše uvedených přípravků.
EVRA může snížit účinek některých léků, jako jsou:
• léky obsahující cyklosporin
• lamotrigin na léčbu epilepsie [To může zvýšit riziko křečí (záchvatů)].
Lékař pravděpodobně upraví dávkování tohoto přípravku. Před užíváním jakéhokoli léčivého přípravku se poraďte s lékařem nebo lékárníkem.
Těhotenství a kojení
• Nepoužívejte tento přípravek, pokud jste těhotná nebo si myslíte, že byste mohla být těhotná.
• Přestaňte ihned používat tento přípravek, pokud zjistíte, že jste těhotná.
• Nepoužívejte tento přípravek, pokud kojíte nebo plánujete kojení.
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete těhotenství, poraďte se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek užívat.
Řízení dopravních prostředků a obsluha strojů
Během používání tohoto přípravku můžete řídit dopravní prostředky a obsluhovat stroje.
Rizika užívání kombinované hormonální antikoncepce
Následující informace je založena na znalostech o kombinované antikoncepci v tabletách. Transdermální náplast EVRA obsahuje podobné hormony jako kombinovaná antikoncepce v tabletách, jsou tedy pravděpodobná i obdobná rizika. Užívání jakékoli kombinované hormonální antikoncepce představuje určitá rizika, která mohou vést k invaliditě nebo mít i smrtelné následky.
Nebylo prokázáno, že transdermální náplasti, jako je EVRA, jsou bezpečnější než kombinovaná antikoncepce v tabletách, které se užívají perorálně (ústy).
Kombinovaná hormonální antikoncepce a rakovina Rakovina děložního čípku
Také rakovina děložního čípku se vyskytuje častěji u žen, které užívají kombinovanou hormonální antikoncepci. I tento výskyt však může mít jiné příčiny včetně pohlavně přenosných chorob.
Rakovina prsu
U žen užívajících kombinovanou hormonální antikoncepci byl pozorován zvýšený výskyt nádorů prsu. Je však možné, že kombinovaná hormonální antikoncepce není příčinou tohoto zvýšeného výskytu nádoru prsu. Je možné, že u těchto žen je větší šance na rozpoznání nádoru prsu, protože jsou častěji vyšetřovány. Zvýšené riziko výskytu rakoviny prsu postupně klesá po ukončení užívání kombinované hormonální antikoncepce. Po deseti letech je pravděpodobnost výskytu nádorů prsu stejná jako u žen, které kombinovanou hormonální antikoncepci nikdy neužívaly.
Rakovina jater
U žen užívajících kombinovanou hormonální antikoncepci byly také ve vzácných případech popsány nezhoubné nádory jater. Ještě vzácněji byly zaznamenány zhoubné nádory jater. Ty mohou být příčinou vnitřního krvácení vedoucího k silným bolestem břicha. Pokud by u Vás tyto bolesti nastaly, vyhledejte neprodleně lékaře.
3. Jak se EVRA používá
Vždy používejte tento přípravek přesně podle pokynů svého lékaře nebo lékárníka.
• Pokud tak neučiníte, může dojít ke zvýšení rizika otěhotnění.
• Pokud si nejste jistá, poraďte se s lékařem nebo lékárníkem.
• Nehormonální antikoncepční prostředky (jako kondomy, pěna nebo pesar) slouží jako náhrada při chybě v použití náplastí.
Kolik náplastí použít
• 1., 2. a 3. týden: Nalepte jednu náplast a ponechte ji nalepenou přesně sedm dní.
• 4. týden: Tento týden náplast nenalepujte.
Pokud jste neužívala hormonální antikoncepci během předchozího cyklu
• Můžete začít používat tento přípravek první den následující menstruace.
• Pokud uběhlo několik dní od začátku menstruačního krvácení, poraďte se se svým lékařem o dočasném použití nehormonální antikoncepce.
Převedení z užívání antikoncepčních tablet na používání náplastí EVRA
Pokud měníte způsob antikoncepce z perorálních tablet na tento přípravek:
• Počkejte, až se dostaví menstruační krvácení.
• První náplast nalepte během prvních 24 hodin menstruačního krvácení.
Pokud si náplast nalepíte později než v 1. den vašeho cyklu musíte:
• Po dobu prvních 8 dní cyklu používat ještě doplňkově nehormonální antikoncepci.
Jestliže se menstruační krvácení nedostaví během 5 dní po užití poslední antikoncepční tablety, poraďte se s lékařem dříve, než začnete používat tento přípravek.
Převedení z užívání tablet obsahujících pouze progesteron, implantátu nebo injekční antikoncepce na přípravek EVRA
• Záměna za tento přípravek může být provedena kdykoli po skončení užívání tablet obsahujících progesteron nebo v den odstranění implantátu nebo v době následující injekce.
• První den, kdy jste přestala užívat tablety s progesteronem, kdy Vám byl odstraněn implantát nebo v den, kdy vám byla aplikována injekce, si nalepte náplast.
• Prvních 8 dní cyklu (do dne výměny náplasti) musíte používat ještě i nehormonální antikoncepci.
Po samovolném potratu nebo umělém přerušení těhotenství do 20. týdne těhotenství
• Poraďte se se svým lékařem.
• Přípravek můžete začít používat okamžitě.
Pokud uběhlo několik dní od samovolného potratu nebo po umělém přerušení těhotenství, poraďte se se svým lékařem o dočasném použití nehormonální antikoncepce.
Po samovolném potratu nebo umělém přerušení těhotenství po 20. týdnu těhotenství
• Poraďte se se svým lékařem.
Přípravek můžete začít používat v 21. den po umělém přerušení těhotenství nebo po samovolném potratu, nebo první den následující menstruace, podle toho, co nastane dříve.
Po porodu
• Poraďte se svým lékařem.
• Pokud jste po porodu a nekojíte, neměla byste tento přípravek používat dříve než za 4 týdny po porodu.
• Pokud začnete přípravek používat po více než 4týdnech po porodu, používejte prvních 7 dní spolu s tímto přípravkem dodatečnou nehormonální antikoncepční metodu.
Pokud jste měla po porodu pohlavní styk, počkejte na první menstruaci, nebo navštivte svého lékaře, abyste se před začátkem používání tohoto přípravku ujistila, že nejste těhotná.
Pokud kojíte
• Poraďte se svým lékařem.
• Nepoužívejte tento přípravek, pokud kojíte nebo plánujete kojit (viz také bod 2 Těhotenství a kojení).
Důležitá upozornění při užívání náplastí
• Náplast vyměňujte vždy ve stejný den v týdnu, protože její účinek trvá 7 dní.
• Nezůstávejte nikdy bez náplasti po dobu delší než 7 po sobě jdoucích dní.
• Mějte vždy nalepenu pouze jednu náplast.
• Náplast nestříhejte ani žádným způsobem neupravujte.
• Nikdy nelepte náplast na zarudlou, podrážděnou nebo poraněnou kůži.
• Aby mohla náplast správně působit, musí pevně přilnout na kůži.
• Náplast musí být ke kůži silně přitlačena, aby její okraje pevně držely.
• Na místo nalepení náplasti nebo okolo nalepené náplasti nenanášejte krémy, masti, tělová mléka, zásypy nebo make-up. Toto může být příčinou odlepení náplasti.
• Novou náplast nelepte na stejné místo, odkud jste předchozí náplast odlepila. Zvýšila by se tak možnost podráždění kůže.
• Každý den zkontrolujte, zda se náplast neodlepila.
• Používání náplasti nepřerušujte ani při méně častém pohlavním styku.
Pokud používáte přípravek EVRA poprvé, vyčkejte do Vašeho obvyklého

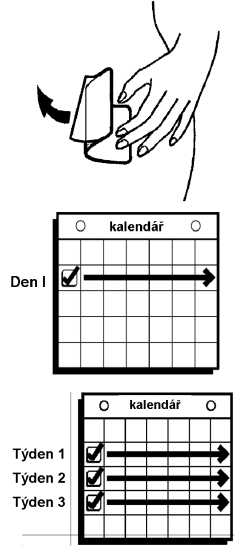
menstruačního krvácení.
• Náplast si nalepte během prvních 24 hodin Vašeho menstruačního krvácení.
• Jestliže náplast nalepíte více než o jeden den později, musíte dalších 8 dní, do dne, kdy budete náplast měnit, používat také nehormonální antikoncepci.
• Den, kdy si nalepíte první náplast, bude 1. den Tento den se stává „Dnem výměny náplasti“ každý další týden.
Vyberte si místo na svém těle, kam náplast nalepíte.
• Náplast nalepujte vždy na čistou suchou neochlupenou pokožku.
• Nalepujte si ji na hýždě, břicho, zevní horní části paže nebo horní část zad, tj. na místa, kde je vyloučeno tření s těsným oděvem.
• Náplast nikdy nelepte na prsa nebo do jejich blízkosti.
Prsty otevřete sáček.
• Sáček otevřete odtržením okraje (nepoužívejte nůžky).
• Pevně uchopte roh náplasti a opatrně vyjměte náplast ze sáčku.
• Na náplasti je průhledná ochranná vrstva.
• Někdy může náplast zůstat přichycena k vnitřní straně sáčku -buďte opatrná, abyste při tom náhodou neodstranila průhlednou vrstvu.
• Potom oddělte polovinu průhledné ochranné vrstvy tak, abyste se nedotkla lepivého povrchu náplasti (jak vidíte na obrázku).
Náplast přiložte na zvolené místo.
• Poté oddělte druhou polovinu ochranné vrstvy.
• Náplast pevně přitlačte dlaní po dobu 10 vteřin.
• Ujistěte se, že okraje dobře přilnuly.
Náplast noste 7 dní (1 týden).
• První „Den výměny náplasti“ je osmý den, odstraňte použitou náplast.
• Okamžitě si nalepte novou náplast.
• 15. den (3. týden) odlepte náplast.
• Okamžitě nalepte novou náplast.
Celkem jsou to tři týdny s náplastmi.
Abyste zabránila podráždění kůže, nelepte novou náplast na stejné místo, odkud jste předchozí náplast odstranila.
Čtvrtý týden (22. až 28. den) nenoste žádnou náplast.
|
| 0 kalendář 0 | ||||||
|
■ | ||||||
|
i Ě Ě |
\ | |||||
|
7 | ||||||
|
\ | ||||||
|
\ | ||||||
|
_ |
_ |
_ |
_ |
_ | ||
Nový čtyřtýdenní cyklus.
• V této době by se mělo dostavit menstruační krvácení.
• Během tohoto týdne jste chráněna proti otěhotnění pouze, pokud si další náplast nalepíte ve správnou dobu.
• Nalepte si novou náplast v pravidelný „Den výměny náplasti“, což je jeden den po 28. dni.
• Udělejte to bez ohledu na to, kdy začalo nebo skončilo menstruační krvácení.
Jestliže jste se rozhodla změnit „Den výměny náplasti“ na jiný den v týdnu, poraďte se o tom se svým lékařem. Budete muset dokončit aktuální cyklus a v příslušný den odstranit třetí náplast. Ve 4. týdnu si můžete zvolit nový „Den výměny náplasti“ a v tento den si aplikovat první náplast. Nesmíte být bez náplasti déle než 7 po sobě jdoucích dní.
Pokud chcete oddálit menstruaci, nalepte si náplast na začátku 4. týdne (22. den) namísto odstranění náplasti ve 4. týdnu. Může se objevit slabé nebo intermenstruační krvácení. Nenalepujte si více jak 6 náplastí (tzn. ne více jak 6 týdnů) za sebou. Po 7denní přestávce bez náplasti si nalepte novou náplast a znovu začnete cyklus, přičemž tento den bude 1. den. Poraďte se se svým lékařem dříve, než se rozhodnete oddálit menstruaci.
Běžné denní činnosti během používání náplastí
• Běžné úkony jako sprchování, koupání, sauna nebo cvičení by neměly ovlivnit účinnost náplasti.
• Náplast je upravena tak, aby při těchto činnostech vydržela přilepená.
• Nicméně se po ukončení těchto činností doporučuje zkontrolovat, zda se náplast neodlepila.
Pokud potřebujete umístit náplast na nové místo na těle v jiný den než v „Den výměny náplasti“
Pokud náplast způsobí podráždění nebo její nošení není pohodlné:
• Náplast můžete odstranit a novou náplast nalepit okamžitě na jiné místo do příštího „Dne výměny náplasti“.
• Současně však smí být nalepena pouze jedna náplast.
Pokud máte potíže si zapamatovat výměnu náplasti
• Požádejte o radu lékaře, lékárníka nebo zdravotní sestru. Ten/ta Vám mohou poradit, jak si snadněji zapamatovat výměnu náplasti. Mohou Vám také poradit, zda musíte užívat jiný způsob ochrany před otěhotněním.
Jestliže jste náplast ztratila, odlepily se okraje nebo náplast odpadla
Po dobu kratší než jeden den (méně než 24 hodin):
• Pokuste se okamžitě nalepit náplast znovu nebo si nalepte novou.
• Další doplňková antikoncepce není nutná.
• Váš „Den výměny náplasti“ zůstane stejný.
• Náplast se nesnažte nalepovat znovu, jestliže:
- již nelepí
- slepila se dohromady nebo se přilepila k jinému povrchu
- přilepilo se na ni něco jiného
- je to již podruhé, co se uvolnila nebo odpadla.
• Nepoužívejte obvazy nebo jiné prostředky k upevnění náplasti na kůži.
• Pokud se Vám nedaří náplast znovu nalepit, použijte okamžitě novou náplast.
Po dobu delší než jeden den (24 hodin a více) nebo pokud si nejste jistá, jak dlouho je náplast odlepena
• Začněte neprodleně nový 4týdenní cyklus tím, že si nalepíte novou náplast.
• Nyní nastává nový „1. den“ a nový „Den výměny náplasti“.
• Po dobu prvního týdne nového cyklu musíte používat ještě další doplňkovou nehormonální antikoncepci.
Nedodržíte-li tyto pokyny, můžete otěhotnět.
Jestliže jste zapomněla náplast vyměnit Na začátku kteréhokoli cyklu [1. týden, (1. den)]:
Pokud jste si náplast zapomněla nalepit, existuje velmi vysoké riziko otěhotnění.
• Po dobu jednoho týdne musíte tedy doplňkově používat ještě nehormonální antikoncepci.
• První náplast nového cyklu nalepte okamžitě, jakmile si vzpomenete.
• Nyní nastává nový „1. den“ i nový „Den výměny náplasti“.
Uprostřed cyklu užívání náplasti (2. nebo 3. týden):
Jestliže se výměna náplasti zpozdila o jeden nebo dva dny (do 48 hodin):
• Nalepte si novou náplast, jakmile si vzpomenete.
• Další náplast nalepte v obvyklý „Den výměny náplasti“.
Další doplňková antikoncepce není nutná.
O více než 2 dny (48 hodin a více):
• Jestliže se výměna náplasti opozdila o více než 2 dny, můžete otěhotnět.
• Jakmile si vzpomenete, začněte nový 4týdenní cyklus nalepením nové náplasti.
• Nyní nastává nový „1. den “ i nový „Den výměny náplasti“.
• Po dobu prvního týdne tohoto nového cyklu musíte používat ještě další doplňkovou antikoncepci.
Na konci cyklu používání náplasti (4. týden):
Pokud jste si zapomněla náplast odstranit:
• Odstraňte ji hned, jakmile si vzpomenete.
• Následující cyklus začněte v obvyklý „Den výměny náplasti“, což je jeden den po 28. dnu.
Další doplňková antikoncepce není nutná.
Pokud při používání přípravku EVRA nekrvácíte nebo krvácíte nepravidelně
Tento přípravek může být příčinou neočekávaného poševního krvácení nebo špinění během týdnů, kdy používáte transdermální náplasti.
• Toto obvykle ustane po několika prvních cyklech.
• Příčinou špinění nebo slabého krvácení může být také nesprávné používání náplastí.
• Pokračujte v používání tohoto přípravku, pokud však krvácení přetrvává déle než první 3 cykly, poraďte se s lékařem nebo lékárníkem.
Pokud se u Vás nedostaví krvácení během 4. týdne, kdy nemáte nalepenou náplast EVRA, použijte novou náplast v obvyklém „Dnu výměny náplasti“.
• Pokud jste používala náplasti EVRA správně a nedostavilo se u Vás krvácení, neznamená to ještě určitě, že jste těhotná.
• Pokud se však u Vás nedostavilo již druhé krvácení v řadě, poraďte se s lékařem nebo lékárníkem, protože by těhotenství mohlo připadat v úvahu.
Jestliže jste použila více přípravku EVRA, než jste měla (více než jednu náplast EVRA najednou)
Odstraňte náplasti a kontaktujte ihned lékaře.
Použití více náplastí může způsobit, že se u Vás vyskytne:
• Nevolnost (nauzea) a zvracení.
• Krvácení z pochvy.
Jestliže jste přestala používat přípravek EVRA
Můžete mít nepravidelné, slabé nebo žádné menstruační krvácení. To se obvykle stává první 3 měsíce a zejména, pokud Vaše menstruace před začátkem používání tohoto přípravku nebyla pravidelná.
Máte-li jakékoli další otázky, týkající se používání tohoto přípravku, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého. Pokud se u Vás vyskytne jakýkoli nežádoucí účinek, zvláště pokud je závažný a trvalý, nebo pokud se vyskytne jakákoli změna Vašeho zdravotního stavu v důsledku užívání přípravku EVRA, informujte prosím svého lékaře.
Zvýšené riziko krevních sraženin ve Vašich žilách [žilní tromboembolizace (VTE) ] nebo krevní sraženiny ve Vašich tepnách (arteriální tromboembolizace, ATE) je přítomné u všech žen, které používají kombinovanou hormonální antikoncepci. Pro podrobnější údaje o různých rizicích v důsledku používání kombinované hormonální antikoncepce si přečtěte bod 2 „Čemu musíte věnovat pozornost, než začnete přípravek EVRA používat“.
Velmi časté nežádoucí účinky (postihují více než 1 z 10 uživatelek)
• Bolest hlavy
• Nauzea
• Napětí v prsou
Časté nežádoucí účinky (mohou postihnout až 1 z 10 žen):
• Kvasinkové infekce pochvy, někdy nazývané kandidóza
• Poruchy nálady jako je deprese, časté střídání nálad, úzkost, plačtivost
• Závratě
• Migréna
• Bolest břicha nebo zvětšení břicha
• Zvracení nebo průjem
• Akné, kožní vyrážka, svědění nebo podráždění kůže
• Svalové křeče
• Problémy s prsy, jako je bolest, zvětšení nebo bulky v prsech
• Změny v menstruačním krvácení, děložní stahy, bolestivá perioda, výtok z pochvy
• Reakce v místě nalepení náplasti, jako zčervenání, podráždění, svědění nebo vyrážka
• Únava nebo pocit celkové zdravotní indispozice
• Zvýšení tělesné hmotnosti
Méně časté nežádoucí účinky (mohou postihnout až 1 ze 100 žen):
• Alergické reakce, kopřivka
• Otoky z důvodu zadržování vody v těle
• Vysoké hladiny tuku v krvi (jako cholesterolu nebo triglyceridů)
• Problémy se spánkem (nespavost)
• Snížení sexuální touhy
• Ekzém, zčervenání kůže
• Abnormální tvorba mléka
• Premenstruační příznaky
• Sucho v pochvě
• Jiné problémy v místě nalepení náplasti
• Otok
• Vysoký krevní tlak nebo zvýšení krevního tlaku
• Zvýšení chuti k jídlu
• Vypadávání vlasů
• Citlivost na sluneční záření
Vzácné nežádoucí účinky (mohou postihnout až 1 z 1 000 žen):
• škodlivé krevní sraženiny v žíle nebo tepně, například:
- v noze nebo chodidle (tj. hluboká žilní trombóza);
- v plicích (tj. plicní embolie);
- srdeční záchvat;
- cévní mozková příhoda;
- příznaky malé cévní mozkové příhody nebo dočasné cévní mozkové příhody známé jako tranzitorní ischemická ataka (TIA);
- krevní sraženiny v játrech, žaludku/střevech, ledvinách nebo oku.
Šance, že se u Vás objeví krevní sraženina, mohou být vyšší, pokud máte jiná onemocnění, která zvyšují riziko (pro více informací o podmínkách, které zvyšují riziko krevních sraženin a příznaků, krevní sraženiny viz bod 2).
• Rakovina prsu, děložního čípku nebo jater
• Potíže v místě, kde byla nalepena náplast, jako je kožní vyrážka s puchýřky nebo vředy
• Nezhoubné (benigní) nádory prsu nebo jater
• Fibromy v děloze (v uteru)
• Vztek nebo pocit frustrace
• Zvýšení sexuální touhy
• Poruchy chuti
• Problémy s nošením kontaktních čoček
• Náhlé výrazné zvýšení krevního tlaku (hypertenzní krize)
• Zánět žlučníku nebo střev
• Neobvyklé buňky na děložním čípku
• Hnědé flíčky nebo skvrny v obličeji
• Žlučové kameny nebo blokáda žlučovodu
• Zežloutnutí kůže a očního bělma
• Abnormální hodnoty krevního cukru nebo inzulinové hodnoty
• Otok obličeje, rtů, hrdla nebo jazyka
• Kožní vyrážka s citlivými rudými uzlinkami na holeních nebo nohách
• Svědící kůže
• Šupinatá, odlupující se a červená kůže
• Potlačení laktace
• Vaginální výtok
• Zadržování tekutin v dolních končetinách
• Zadržování tekutin
• Otok paží, rukou, nohou nebo chodidel.
Pokud máte žaludeční obtíže
• Množství hormonů, které se uvolní z náplasti EVRA, by nemělo být ovlivněno nevolností (zvracením) nebo průjmem
• Pokud máte žaludeční obtíže, není nutné používat další antikoncepci.
Během prvních 3 cyklů můžete špinit nebo lehce krvácet, mít pocit napětí v prsech nebo Vám může být nevolno. Tyto problémy většinou vymizí, ale pokud k tomu nedojde, poraďte se se svým lékařem nebo lékárníkem.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek EVRA uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na obalu za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem a vlhkostí.
Chraňte před chladem nebo mrazem.
Použité náplasti stále obsahují určité množství hormonů a pro ochranu životního prostředí je nutno je likvidovat opatrně. Pro likvidaci použité náplasti:
• Odtrhněte ze sáčku nálepku pro likvidaci.
• Použitou náplast umístěte dovnitř této nálepky tak, aby lepivá vrstva náplasti pokryla vyznačenou oblast.
• Nálepku zavřete, aby použitá náplast zůstala uzavřená uvnitř a poté zlikvidujte mimo dosah dětí.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co EVRA obsahuje
Léčivými látkami jsou norelgestrominum a ethinylestradiolum. Jedna 20 cm2 transdermální náplast obsahuje 6 mg norelgestrominum a 600 mikrogramů ethinylestradiolum. Léčivé látky se uvolňují po dobu 7 dní s tím, že během 24 hodin se uvolní v průměru 203 mikrogramů norelgestrominu a 34 mikrogramů ethinylestradiolu.
Dalšími složkami jsou: zevní krycí vrstva: pigmentovaný polyethylen nízké hustoty; střední vrstva: adhezivní polyisobutenová/polybutenová vrstva, krospovidon, netkaná polyesterová vrstva, lauryl-laktát; třetí vrstva: pegoterát (PET), polydimthylsiloxan ; vnitřní - polyesterová fólie.
Jak EVRA vypadá a co obsahuje toto balení
EVRA je tenká béžová plastická transdermální náplast označená nápisem „EVRA“. Lepivá vrstva přilne k pokožce po odstranění čiré umělohmotné ochranné vrstvy.
EVRA je dostupná v následujících velikostech balení: krabičky obsahující 3, 9 nebo 18 náplastí opatřených oddělitelnou fólií, v jednotlivých sáčcích. Sáčky jsou baleny po třech v průhledné perforované plastové fólii.
Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci a výrobce
Držitel rozhodnutí o registraci: Janssen-Cilag International NV, Tumhoutseweg, 30, B-2340 Beerse, Belgie
Výrobce: Janssen Pharmaceutica NV, Tumhoutseweg 30, B-2340 Beerse, Belgie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
|
Belgie/Belgique/Belgien JANSSEN-CILAG NV Antwerpseweg 15-17 B-2340 Beerse Tel/Tél: + 32 14 64 94 11 |
Lietuva UAB „UAB JOHNSON & JOHNSON Geležinio Vilko g. 18A LT-08104 Vilnius Tel: +370 5 278 68 88 |
|
Btarapna „fl^OHCBH & fl^OHCtH EtarapHa” EOOfl ^.k. Maagocr 4 EH3Hec napK Co$Ha, crpaga 4 Co$Ha 1766 Ten.: +359 2 489 94 00 |
Luxembourg/Luxemburg JANSSEN-CILAG NV Antwerpseweg 15-17 B-2340 Beerse Tél/Tel: + 32 14 64 94 11 |
|
Česká republika JANSSEN-CILAG s.r.o. Karla Engliše 3201/6 CZ-150 00 Praha 5 - Smíchov Tel. +420 227 012 227 |
Magyarország Janssen-Cilag Kft. Nagyenyed u. 8-14 H-Budapest, 1123 Tel: +36 1 884 2858 |
|
Danmark JANSSEN-CILAG A/S Bregnemdvej 133 DK-3460 Birkered Tlf: +45 45 94 82 82 |
Malta A.M.Mangion Ltd Mangion Building, Triq Gdida fi Triq Valletta MT-Hal-Luqa LQA 6000 Tel:+356 2397 6000 |
|
Deutschland JANSSEN-CILAG GmbH Johnson & Johnson Platz 1 D-41470 Neuss Tel: +49 2137 955-955 |
Nederland JANSSEN-CILAG B.V. Dr. Paul Janssenweg 150 NL-5026 RH Tilburg Tel: +31 13 583 73 73 |
|
Eesti „UAB JOHNSON & JOHNSON Eesti filiaal LSStsa 2 EE-11415 Tallinn Tel: +372 617 7410 |
Norge JANSSEN-CILAG AS Postboks 144 NO-1325-Lysaker Tlf: + 47 24 12 65 00 |
|
EXláSa JANSSEN-CILAG Oap^aKeuriKq A.E.B.E. Asro^ópog Erp^vn? 56 GR-151 21 nsÚKn, A0^va T^: +30 210 80 90 000 |
Osterreich JANSSEN-CILAG Pharma GmbH. VorgartenstraBe 206B AT-1020 Wien Tel:+43 1 610 300 |
|
Espaňa JANSSEN-CILAG, S.A. Paseo de las Doce Estrellas, 5-7 E-28042 Madrid Tel: +34 91 722 81 00 |
Polska JANSSEN-CILAG Polska Sp. z o.o. ul. Uzecka 24 PL-02-135 Warszawa Tel.: + 48 22 237 60 00 |
France
JANSSEN-CILAG
1, rue de Camille Desmoulins TSA 91003 F-92787 Issy Les Moulineaux Cedex 9 Tél: 0800 25 50 75 / + 33 1 55 00 40 03
Hrvatska
Johnson & Johnson S.E. d.o.o.
Oreškoviceva 6h 10010 Zagreb Tel: +385 1 6610 700
Ireland
JANSSEN-CILAG Ltd.
50-100 Holmers Farm Way High Wycombe Buckinghamshire HP12 4EG United Kingdom Tel: +44 1 494 567 444
Ísland
JANSSEN-CILAG AB c/o Vistor hf.
Horgatúni 2 IS-210 Garóab^r Iceland
Simi: (+354) 535 7000 Italia
JANSSEN-CILAG SpA Via M.Buonarroti, 23 I-20093 Cologno Monzese MI Tel: +39 02 2510 1
Kúnpoq
BapváPag Xax^nnavay^g AxS Asro^ópog riávvou KpaviSiróxn 226 Aaxoiá
CY-2234 AsuK^oía Tsk: +357 22 207 700
Latvija
„UAB JOHNSON & JOHNSON filiale Latvija Mukusalas iela 101 Riga, LV-1004 Tel: +371 678 93561
Portugal
JANSSEN-CILAG FARMACÉUTICA, LDA Estrada Consiglieri Pedroso, 69 A Queluz de Baixo PT-2734-503 Barcarena Tel: +351 21 43 68 835
Románia
Johnson & Johnson Románia SRL Str. Tipografilor nr.11 - 15 013714 Bucure§ti, ROMÁNIA Tel: +40 21 207 18 00
Slovenija
Johnson & Johnson d.o.o.
Šmartinska cesta 53 SI-1000, Ljubljana Tel. + 386 1 401 18 30
Slovenská republika
Janssen, Johnson & Johnson, s.r.o. CBC III, Karadžičova 12 SK-821 08 Bratislava Tel. +421 232 408 400
Suomi/Finland
JANSSEN-CILAG OY Vaisalantie/Vaisalavágen 2 FI-02130 Espoo/Esbo Puh/Tel: +358 207 531 300
Sverige
JANSSEN-CILAG AB Box 4042 SE-16904 Solna
Tel: +46 8 626 50 00
United Kingdom
JANSSEN-CILAG Ltd.
50-100 Holmers Farm Way High Wycombe
Buckinghamshire HP12 4EG - UK Tel: +44 1 494 567 444
Tato příbalová informace byla naposledy revidována
Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
46