Evarrest
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
▼ Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
EVARREST tkáňové lepidlo
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden cm2 přípravku Evarrest obsahuje
fibrinogenum humanum: 8,1 mg
thrombinum humanum: 40 IU
Pomocná látka/Pomocné látky se známým účinkem: Obsahuje až 3,0 mmol (68,8 mg) sodíku v jedné matrici.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Tkáňové lepidlo
EVARREST je bílý až žlutý biologicky vstřebatelný kombinovaný přípravek vyrobený z pružné kompozitní matrice potažené lidským fibrinogenem a lidským trombinem. Aktivní strana matrice je práškovitá a neaktivní strana má reliéfní vlnovitý vzor.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek EVARREST je indikován u dospělých pro podpůrnou léčbu při chirurgickém zákroku tam, kde nepostačují standardní chirurgické techniky, a to pro zlepšení zástavy krvácení (viz bod 5.1).
4.2 Dávkování a způsob podání
Použití přípravku EVARREST je omezeno pouze na zkušené chirurgy.
Dávkování
Množství přípravku EVARREST, které bude použito, a frekvence použití musejí vždy vycházet z existujících klinických potřeb pacienta.
Množství přípravku EVARREST, které bude použito, záleží na ploše a místu krvácení, které bude ošetřeno. Přípravek EVARREST je nutné aplikovat tak, aby přesahoval cca o 1 až 2 cm okraje cílového krvácejícího místa. Přípravek lze nastříhat na potřebnou velikost a tvar, které odpovídají velikosti krvácející plochy.
Použití přípravku EVARREST u krvácejících míst větších než ta, jež lze překrýt jedním kusem tohoto přípravku, nebylo v klinických studiích hodnoceno. Přípravek EVARREST je nutné aplikovat pouze v jediné vrstvě tak, aby přesahoval cca o 1 až 2 cm na nekrvácející tkáň nebo na přilehlý kus tkáňového lepidla EVARREST.
Přípravkem lze současně ošetřit krvácení z více míst. V těle by nemělo být větší množství přípravku než ekvivalent dvou kusů 10,2 cm x 10,2 cm nebo čtyř kusů 5,1 cm x 10,2 cm, neboť s větším množstvím přípravku existují pouze omezené dlouhodobé zkušenosti. Použití více než čtyř kusů
10,2 cm x 10,2 cm nebo čtyř kusů 5,1 cm x 10,2 cm nebo použití u pacientů, jimž byl již v minulosti přípravek EVARREST aplikován, nebylo hodnoceno.
Pokud není dosaženo hemostázy při jedné aplikaci přípravku EVARREST, lze provést ošetření znovu. Pokyny jsou uvedeny níže.
Pediatrická populace
Bezpečnost a účinnost přípravku EVARREST u dětí od narození do 18 let nebyla dosud stanovena. Nejsou dostupné žádné údaje.
Způsob podání
Pouze pro epilezionální použití. Při aplikaci přípravku EVARREST je nutné přípravek pevně manuálně přitlačit zhruba po dobu 3 minut.
1. Sterilními nůžkami opatrně nastřihejte přípravek EVARREST na potřebnou odpovídající velikost a tvar a přiložte na krvácející ránu tak, aby přípravek překrýval ránu asi o 1 až 2 cm. Dokud je EVARREST na tácku, musí jeho bílá až nažloutlá aktivní strana s práškovým povrchem zůstat ležet směrem dolů.
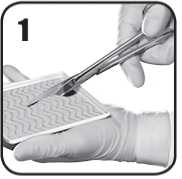
2. Podle potřeby odstraňte nadbytečnou krev či tekutinu z místa aplikace pro zlepšení viditelnosti. Je nutné přesně identifikovat zdroj krvácení. Přípravek EVARREST musí být aplikován přímo na zdroj krvácení a musí ho zcela zakrýt. EVARREST lze použít v aktivně krvácejícím poli.
3. Přiložte aktivní stranu přípravku EVARREST na krvácející místo tak, aby byl v plném kontaktu s tkání. Přípravek se aktivuje kontaktem s tekutinou, přilne ke tkáni a přizpůsobí se jí.

4. Aplikujte kousek přípravku EVARREST vhodné velikosti tak, aby odpovídajícím způsobem zakryl celé krvácející místo, přesahoval jej zhruba o 1 až 2 cm na nekrvácející tkáň a napomohl tak při přilnutí k ráně.
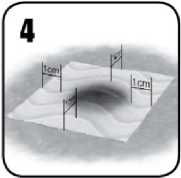
5 a) Podržte suchou či vlhkou chirurgickou gázu nebo laparotomické tampony na přípravku
EVARREST, aby bylo dosaženo plného kontaktu s krvácejícím povrchem.

5b) K dosažení hemostázy okamžitě manuálně zatlačte na celý povrch přípravku EVARREST
(včetně překryvu). Vyviňte při tom dostatečný tlak, který zcela zamezí krvácení. Držte stlačené asi 3 minuty, aby se krvácení zastavilo.

6. Jemně odstraňte chirurgickou gázu nebo laparotomické tampony z místa použití a dávejte při tom pozor, abyste nenarušili či neuvolnili přípravek EVARREST či krevní sraženinu. Zkontrolujte EVARREST a ověřte, zda došlo k hemostáze a zda se přípravek v místě krvácení nezvlnil. Pokud nebudete spokojeni s umístěním přípravku EVARREST, vyjměte jej a použijte nový. EVARREST zůstane na místě a přilne ke tkáni. Přípravek je vstřebatelný.
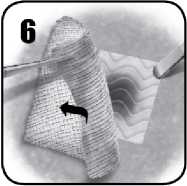
7. Místo aplikace je nutné během chirurgického zákroku sledovat, aby bylo jisté, že došlo k hemostáze.
Opakovaná aplikace
• Pokud se přípravek EVARREST zvlní, vzniknou na něm záhyby nebo se ohne, může být nutná opakovaná aplikace. Pokud nebudete spokojeni s umístěním, odstraňte použitý EVARREST a znovu zopakujte postup aplikace nového kusu tkáňového lepidla EVARREST. uvedený výše.
• Pokud je krvácení způsobeno neúplným zakrytím krvácející rány, lze přiložit další kus přípravku EVARREST. Aplikujte přípravek v jedné vrstvě tak, aby se okraje navzájem překrývaly (asi o 1 až 2 cm) s již přiloženým kusem přípravku EVARREST.
• Jestliže je krvácení způsobeno neúplným přilnutím ke tkáni (v místě, kde krvácení zpod krytí přetrvává), odstraňte EVARREST a použije nový kus přípravku EVARREST.
• Pokud krvácení pokračuje během stanovené doby komprese či poté, vyjměte použitý kus přípravku EVARREST a zkontrolujte místo krvácení. Pokud se zdá, že nejsou nutná další opatření pro primární hemostázu (tj. standardní chirurgické techniky), zopakujte postup aplikace nového kusu přípravku EVARREST, uvedený výše.
4.3 Kontraindikace
• Přípravek EVARREST nesmí být použit pro zástavu těžkého krvácení z rozsáhlých defektů velkých tepen či žil tam, kde poraněná cévní stěna vyžaduje rekonstrukci, při níž musí být zachována průchodnost cévy. Takové použití by mělo za následek trvalou expozici přípravku EVARREST krevnímu proudu nebo tlaku krve během hojení a absorpce přípravku.
• EVARREST nesmí být aplikován intravaskulárně.
• EVARREST nesmí být použit v uzavřených prostorech (např. v otvorech v kostech, okolo nich či v jejich blízkosti nebo v místech omezených kostních prostorů, neboť bobtnání přípravku může způsobit kompresi nervů a cév.
• EVARREST nesmí být použit za přítomnosti aktivní infekce nebo na kontaminovaných plochách v těle, neboť by mohlo dojít k infekci.
• Hypersenzitivita na léčivé látky nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Pouze pro epilezionální použití.
Intravaskulární aplikace
Neaplikujte intravaskulárně. Pokud je přípravek neúmyslně aplikován intravaskulárně, mohou nastat život ohrožující tromboembolické komplikace.
Tepenné krvácení
EVARREST nemá být používán místo sutury či jiných forem mechanické ligace k ošetření rozsáhlého tepenného krvácení.
Použití, _pro něž nejsou k dispozici dostatečné údaje
Nejsou k dispozici dostatečné údaje, které by podpořily použití tohoto přípravku při lepení tkání, neurochirurgických zákrocích nebo při zavedení pomocí flexibilního endoskopu pro zástavu krvácení, v cévní chirurgii nebo v gastrointestinálních anastomózách.
Reakce na přítomnost cizího tělesa
Stejně jako u jiných implantovaných přípravků může v těle dojít k reakci na cizí těleso.
Přípravek EVARREST má být použit pouze v jedné vrstvě tak, aby cca o 1 až 2 cm přesahoval na nekrvácející tkáň, což napomůže jeho přilnutí k ráně. Velikost přípravku EVARREST má být omezena na takovou, která je nezbytná pro hemostázu.
Hypersenzitivní reakce
Stejně jako u jiných bílkovinných přípravků jsou možné hypersenzitivní reakce alergického typu. Známky takových reakcí mohou zahrnovat vyrážku, generalizovanou kopřivku, tíseň na hrudi, sípot, hypotenzi a anafylaxi. Pokud se takové symptomy projeví, je nutné přestat přípravek okamžitě používat. V případě šoku je nutné aplikovat standardní lékařský postup pro léčbu šoku.
Obsahuje sodík.
EVARREST obsahuje až 3,0 mmol (68,8 mg) sodíku v jednom kuse tkáňového lepidla. Tuto skutečnost musejí vzít v úvahu pacienti s dietou s kontrolovaným příjmem sodíku.
Přenosná infekční agens
Standardní opatření, která zabrání vzniku infekcí v důsledku použití léčivých přípravků vyrobených z lidské krve nebo plazmy, zahrnují výběr dárců, screening jednotlivých odebraných dávek krve a směsné plazmy na specifické markery infekce a začlenění účinných kroků pro deaktivaci/odstranění virů do výrobního procesu. Přesto nelze při aplikaci léčivých přípravků vyrobených z lidské krve nebo plazmy zcela vyloučit možnost přenosu infekčních agens. Platí to i pro neznámé či nově se objevující viry a další typy patogenů.
Přijatá opatření jsou považována za účinná pro obalené viry, jako je virus lidské imunodeficience (HIV), virus hepatitidy B (HBV), virus hepatitidy C (HCV) a pro neobalený virus hepatitidy A (HAV). Tato opatření mohou mít omezenou účinnost proti neobaleným virům, jako je parvovirus B19. Infekce parvovirem B19 může být závažná u těhotných žen (fetální infekce) a u jedinců s imunodeficiencí nebo s abnormální erytropoézou (např. hemolytická anémie).
Důrazně se doporučuje, aby vždy, když je přípravek EVARREST aplikován pacientovi, byl zaznamenán název a číslo šarže přípravku, aby bylo možné dosledovat šarži přípravku ke konkrétnímu pacientovi.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné studie interakcí.
Podobně jako u srovnatelných přípravků nebo trombinových roztoků může být přípravek po expozici roztokům obsahujících alkohol, jód či těžké kovy (např. aseptické roztoky) denaturovaný. Takové látky je nutné v maximální možné míře před aplikací přípravku odstranit.
4.6 Fertilita, těhotenství a kojení
Bezpečnost fibrinových tkáňových lepidel/hemostatik při použití během těhotenství u člověka či během kojení nebyla ověřena v kontrolovaných klinických studiích. Experimentální studie na zvířatech jsou nedostatečné k hodnocení bezpečnosti s ohledem na fertilitu, reprodukci, vývoj embrya či plodu, průběh těhotenství, perinatální a postnatální vývoj.
EVARREST by proto měl být těhotným a kojícím ženám aplikován pouze, pokud je to klinicky indikováno.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Není relevantní.
4.8 Nežádoucí účinky
Ve vzácných případech se u pacientů, jimž byla aplikována fibrinová tkáňová lepidla/hemostatika, může projevit hypersensitivita či alergické reakce (mezi něž může patřit angioedém, pálení a štípání v místě aplikace, bronchospasmus, zimnice, zrudnutí, generalizovaná kopřivka, bolest hlavy, vyrážka, hypotenze, letargie, nausea, neklid, tachykardie, tíseň na hrudi, brnění, zvracení a sípot).
V izolovaných případech mohou tyto reakce přejít do těžké anafylaxe. Takové reakce byly zaznamenány zejména, pokud je přípravek aplikován opakovaně nebo je aplikován pacientům, u nichž je známa přecitlivělost na složky přípravku.
Vzácně mohou vzniknout protilátky proti složkám fibrinových tkáňových lepidel/hemostatických přípravků; existuje rovněž riziko anafylaktické reakce (viz bod 4.4).
Pokud je přípravek neúmyslně aplikován intravaskulárně (viz bod 4.4), mohou nastat tromboembolické komplikace.
Informace o bezpečnosti a přenosných agens viz bod 4.4.
Souhrn bezpečnostního profilu
Údaje o bezpečnosti přípravku EVARREST odrážejí typy pooperačních komplikací, které obecně souvisejí s chirurgickým prostředím, v němž byly studie prováděny, a existujícím onemocněním pacientů. V klinických studiích byly nejčastěji hlášenými nežádoucími účinky krvácení a zvýšená hladina fibrinogenu a nejzávažnějšími nežádoucími reakcemi byly aspirace, plicní embolie a krvácení.
Tabulkový přehled nežádoucích účinků
Údaje ze čtyř kontrolovaných klinických studií a jedné nekontrolované klinické studie s přípravkem EVARREST byly sloučeny do integrovaného datového souboru a frekvence výskytu popsaná v dále uvedené tabulce vychází z tohoto integrovaného datového souboru. V integrovaných analýzách bylo přípravkem EVARREST ošetřeno 243 pacientů a 110 pacientům byla aplikována kontrolní léčba.
Pro stanovení pořadí nežádoucích účinků podle četnosti výskytu jsou použity následující kategorie: velmi časté (^ 1/10), časté (^ 1/100 až < 1/10), méně časté (^ 1/1000 až < 1/100), vzácné (^ 1/10 000 a <1 /1000), velmi vzácné (< 1/10 000), není známo (z dostupných údajů nelze určit).
Tabulka 1 Souhrn nežádoucích účinků přípravku EVARREST
|
Třída orgánových systémů MedDRA |
Preferovaný název |
Frekvence |
|
Cévní poruchy |
Hluboká žilní trombóza |
Méně časté |
|
Respirační, hrudní a |
Aspirace |
Méně časté |
|
mediastinální poruchy |
Pleurální efuze |
Méně časté |
|
Méně časté | ||
|
Gastrointestinální poruchy |
Břišní distenze |
Méně časté |
|
Ascites |
Méně časté | |
|
Krvácení |
Méně časté | |
|
• Gastrointestinální krvácení | ||
|
• Nitrobřišní krvácení | ||
|
Lokalizovaný odběr intraabdominální tekutiny |
Méně časté | |
|
Peripankreatická kolekce tekutiny |
Méně časté | |
|
Vyšetření |
Plazmatický fibrinogen zvýšený |
Časté |
|
Poranění, otravy a komplikace výkonů |
Krvácení po provedení výkonu |
Časté |
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Nebyl hlášen žádný případ předávkování.
5 FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antihemoragika, lokální hemostatika, ATC kód: B02BC30
EVARREST obsahuje lidský fibrinogen a lidský trombin jako suchou horní vrstvu na povrchu absorbovatelné kompozitní matrice. Při kontaktu s fyziologickými tekutinami, např. krví, lymfou nebo fyziologickým roztokem, se složky horní vrstvy aktivují a reakce fibrinogenu a trombinu spustí závěrečnou fázi fyziologického srážení krve. Fibrinogen se přeměňuje na monomery fibrinu, jež spontánně polymerizují a vytvářejí fibrinovou sraženinu, která pevně upevní matrici k povrchu rány. Fibrin je poté zesíťován endogenním faktorem XIII za vzniku pevné, mechanicky stabilní fibrinové sítě, která má dobré adhezivní vlastnosti.
Kompozitní matrice se skládá z polyglaktinu 910 a oxidované regenerované celulózy, která je běžně používaným hemostatikem. Matrice poskytuje fyzickou oporu a povrch o velké ploše pro biologické složky, zajišťuje vlastní mechanickou celistvost přípravku a podporuje tvorbu sraženiny. Tvorba sraženiny přípravku EVARREST je integrována s matricí; vytváří mechanickou bariéru proti krvácení a zpevňuje ránu. K přirozenému hojení dochází při rozkladu fibrinu a absorpci přípravku tělem; absorpce podle předpokladů trvá cca 8 týdnů, jak bylo doloženo na zvířecích modelech u hlodavců a prasat.
Klinické studie dokazující hemostázu u mírně až středně krvácejících měkkých tkání byly provedeny celkem u 141 subjektů (111 ošetřeno přípravkem EVARREST a 30 v kontrolní skupině), které podstoupily chirurgický zákrok břicha, retroperitonea, pánve a nekardiální zákrok na hrudníku. Další studie provedená u 91 pacientů, kteří podstoupili chirurgický zákrok břicha, retroperitonea, pánve a nekardiální zákrok na hrudníku (59 ošetřeno přípravkem EVARREST a 32 v kontrolní skupině) doložila hemostázu u těžkého krvácení měkkých tkání. Klinická studie u 104 pacientů, kteří podstoupili chirurgický zákrok jater (59 ošetřeno přípravkem EVARREST a 45 v kontrolní skupině) doložila hemostatickou účinnost u přetrvávajícího parenchymálního krvácení.
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s přípravkem EVARREST u jedné či více podskupin pediatrické populace při ošetření krvácení vzniklého v důsledku chirurgického zákroku (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
EVARREST je určen pouze pro epilezionální použití. Intravaskulární podání je kontraindikováno a může způsobit tromboembolii. V důsledku toho nebyly u lidí provedeny intravaskulární farmakokinetické studie.
Byly provedeny studie u králíků, jejichž cílem bylo hodnocení absorpce a eliminace trombinu aplikovaného na povrch řezné rány v játrech po parciální hepatektomii. Při použití 125I-trombinu bylo prokázáno, že v důsledku rozkladu trombinu došlo k pomalé absorpci biologicky neaktivních peptidů a Cmax v plazmě bylo dosaženo za 6-8 hodin. Při Cmax plazmatická koncentrace představuje pouze 1 až 2 % aplikované dávky.
Fibrinová tkáňová lepidla/hemostatika jsou metabolizována stejným způsobem jako endogenní fibrin, a to fibrinolýzou a fagocytózou.
Po absorbování biologických složek dojde k úplné absorpci složek matrice (polyglaktin 910 a oxidovaná regenerovaná celulóza). Ve studiích na zvířatech byl přípravek EVARREST absorbován do 56 dnů, když byl použit v předpokládané klinické dávce.
5.3 Předklinické údaje vztahující se k bezpečnosti
Hemostatická účinnost přípravku EVARREST byla doložena u řady zvířecích modelů, kdy byla kromě jiných cílových parametrů hodnocena doba do dosažení hemostázy a ztráta krve po ošetření.
Neklinické údaje o složkách matrice neodhalují žádné zvláštní riziko pro člověka na základě studií cytotoxicity, alergizace, intrakutánní reaktivity, akutní systémové toxicity, pyrogenity způsobené materiálem, subchronické toxicity, genotoxicity, implantace a hemokompatibility.
Ve studii na potkanech trvající 90 dnů, jejímž cílem bylo hodnocení subchronické systémové toxicity a imunogenicity přípravku EVARREST po subkutánní implantaci, nebyly zjištěny žádné známky toxických účinků a důkazy o zvýšené imunogenicitě související s fibrinovými tkáňovými lepidly.
6 FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Kompozitní matrice (polyglaktin 910 a oxidovaná regenerovaná celulóza) 20 mg/cm2
Arginin-hydrochlorid
Glycin
Chlorid sodný Citronan sodný Chlorid vápenatý Lidský albumin Mannitol Octan sodný
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
2 roky.
Jakmile je foliový sáček otevřen, přípravek EVARREST může zůstat ve sterilním poli, aby byl k dispozici po celou dobu výkonu.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte při teplotě do 25 °C. Chraňte před mrazem.
6.5 Druh obalu a obsah balení
Matrice 10,2 cm x 10,2 cm na tácku (polyester). Tácek je v sáčku (hliníková folie laminovaná polyesterem) s uzávěrem. Velikost balení: 1 kus matrice 10,2 cm x 10,2 cm.
Matrice 5,1 cm x 10,2 cm na tácku (polyester). Tácek je v sáčku (hliníková folie laminovaná polyesterem) s uzávěrem. Velikost balení: 2 kusy matrice 5,1 cm x 10,2 cm.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Návod k použití přípravku je též popsán v příbalové informaci.
Manipulace s přípravkem EVARREST
• EVARREST se dodává ve sterilních baleních připravený k okamžitému použití a musí se
s ním manipulovat sterilní technikou v aseptickém prostředí. Poškozená balení zlikvidujte.
• Vyjměte foliový sáček z krabičky a opatrně jej otevřete rozloupnutím; zabraňte při tom kontaktu s vnitřní stranou folie nebo bílým sterilním táckem obsahujícím EVARREST.
• Vyjměte bílý sterilní tácek ze sáčku a vložte jej do sterilního pole.
• Držte tácek bezpečně v dlani ruky. Strana s otvory musí směřovat nahoru. Pomocí jazýčků na straně tácku sejměte druhou rukou horní stranu tácku.
• Dolní část tácku obsahuje EVARREST; aktivní strana přípravku je umístěna směrem dolů. Aktivní strana má práškový vzhled. Neaktivní strana má reliéfní vlnovitý vzor.
• Po otevření uchovávejte EVARREST v suchu. Přípravek může zůstat ve sterilním poli, aby byl k dispozici po celou dobu výkonu. EVARREST nepřilne k rukavicím, pinzetě ani chirurgickým nástrojům.
Likvidace
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Omrix Biopharmaceuticals NV Leonardo Da Vincilaan 15 1831 Diegem Belgie
Tel.: +32 2 746 30 00 Telefax: + 32 2 746 30 01
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/13/868/001
EU/1/13/868/002
9. DATUM REGISTRACE/ PRODLOUŽENÍ REGISTRACE
Datum první registrace: 29. září 2013
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/.
A. VÝROBCE/VÝROBCI BIOLOGICKÉ LÉČIVÉ LÁTKY /BIOLOGICKÝCH LÉČIVÝCH LÁTEK A VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE/VÝROBCI BIOLOGICKÉ LÉČIVÉ LÁTKY /BIOLOGICKÝCH LÉČIVÝCH LÁTEK A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa vvrobce/vvrobců biologické léčivé látky/biologických léčivých látek
Fibrinogenum humanum a Thrombinum humanum:
Omrix Biopharmaceuticals Ltd.
Plasma Fractionation Institute (Omrix-PFI), MDA Services Center Sheba Medical Center Ramat Gan 5262000 POB 888
Kiryat Ono 5510801 Izrael
Fibrinogenum humanum:
Omrix Biopharmaceuticals Ltd.
Jerusalem Plant (Omrix-JP)
5 Kiryat Hamada St.,
Ramot Meir Building Har-Hotzvim P.O.B. 45075 Jerusalem 9777605 Izrael
Název a adresa výrobce odpovědného/výrobců odpovědných za propouštění šarží
Omrix Biopharmaceuticals N.V.
Leonardo Da Vincilaan 15
1831 Diegem
Belgie
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz Příloha I:Souhrn údajů o přípravku, bod 4.2).
TTV 1 ř V, V f V V r
• Úřední propouštění sarzí
Úřední propouštění šarží: v souladu s článkem 114 směrnice 2001/83/ES, v platném znění, bude úřední propouštění šarží provedeno státní laboratoří nebo laboratoří určenou k takovému účelu.
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předloží první pravidelně aktualizovanou zprávu o bezpečnosti pro tento léčivý přípravek do šesti měsíců od jeho registrace. Držitel rozhodnutí o registraci dále předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný plán řízení rizik je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU A VNITŘNÍM OBALU Krabička (10,2 cm x 10,2 cm, 5,1 cm x 10,2 cm) a foliový sáček (10,2 cm x 10,2 cm)
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
EVARREST tkáňové lepidlo
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jeden cm2 EVARRESTu obsahuje Fibrinogenum humanum: 8,1 mg
Thrombinum humanum: 40 IU
3. SEZNAM POMOCNÝCH LÁTEK
Pomocné látky:
Kompozitní matrice (polyglaktin 910 a oxidovaná regenerovaná celulóza)
Arginin-hydrochlorid
Glycin
Chlorid sodný Citronan sodný Chlorid vápenatý Lidský albumin Mannitol Octan sodný
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Obsahuje jednu matrici (10,2 cm x 10,2 cm)
Obsahuje dvě matrice (5,1 cm x 10,2 cm)
2 kusy
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Epilezionální použití.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte při teplotě do 25 °C. Chraňte před mrazem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Jakýkoli nepoužitý přípravek nebo odpadní materiál je nutné likvidovat v souladu s místními požadavky.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Omrix Biopharmaceuticals NV Leonardo Da Vincilaan 15 1831 Diegem Belgie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/13/868/001
EU/1/13/868/002
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis s omezením
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Nevyžaduje se - odůvodnění přijato.
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA BLISTRECH NEBO STRIPECH Foliový sáček (5,1 cm x 10,2 cm)_
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
EVARREST tkáňové lepidlo
Jeden cm2 EVARRESTu obsahuje Fibrinogenum humanum: 8,1 mg
Thrombinum humanum: 40 IU
Obsahuje jednu matrici (5,1cm x 10,2 cm)
2 NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Omrix Biopharmaceuticals NV
3. POUŽITELNOST
EXP
4. ČÍSLO SARZE
Lot
5. JINÉ
Epilezionální použití.
Před použitím si přečtěte příbalovou informaci. Uchovávejte při teplotě do 25 °C. Chraňte před mrazem. EU/1/13/868/002
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro uživatele
EVARREST tkáňové lepidlo fibrinogenum humanum, thrombinum humanum
▼ Tento léčivý přípravek podléhá dalšímu sledování. Umožní to rychlé zjištění nových informací o bezpečnosti. Můžete pomoci oznámením nežádoucích účinků, jež se u Vás mohou vyskytnout. Postup pro oznámení nežádoucích účinků viz konec bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než bude tento léčivý prostředek použit pro Vaši léčbu, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je EVARREST a k čemu se používá
2. Čemu musíte věnovat pozornost, než budete léčeni přípravkem EVARREST
3. Jak se EVARREST používá
4. Možné nežádoucí účinky
5. Jak EVARREST uchovávat
6. Obsah balení a další informace
1. Co je EVARREST a k čemu se používá
EVARREST je kombinovaný přípravek vyrobený ze vstřebatelného materiálu (matrice) potaženého lidským fibrinogenem a lidským trombinem.
Fibrinogen je bílkovina extrahovaná z krve, která po působení enzymu trombinu vytvoří fibrinovou sraženinu. Když se suchý práškový povrch přípravku EVARREST navlhčí, trombin působí na fibrinogen, který rychle vytvoří sraženinu. Fibrinová sraženina se pevně ukotví v matrici, což umožní, aby EVARREST pevně přilnul k okolní tkáni.
EVARREST se aplikuje během operačních zákroků u dospělých pacientů, a to pro zástavu krvácení a mokvání během operace. Aplikujte se přímo na tkáň, k níž se pevně přilepí a zastaví krvácení. Po operaci se ponechá na místě a tělo ho vstřebá.
2. Čemu musíte věnovat pozornost, než budete léčeni přípravkem EVARREST:
Váš chirurg Vás nesmí léčit přípravkem EVARREST za následujících okolností:
EVARREST nesmí být použit pro opravu poranění stěny velkých tepen či žil tam, kde je přípravek vystaven nepřetržitému proudu a tlaku krve.
EVARREST nesmí být aplikován uvnitř cév.
Přípravek EVARREST nesmí být použit v uzavřených prostorech (například v otvorech či dutinách v kosti, okolo nich či u nich nebo v jiných omezených oblastech okolo kosti, kde by mohl zvětšit svůj objem a stlačovat nervy či cévy.
EVARREST nesmí být použit za přítomnosti aktivní infekce ani na kontaminovaných plochách v těle, neboť by mohlo dojít ke vzniku infekce.
Přípravkem EVARREST nesmíte být léčen(a), pokud jste alergický(á) na lidský fibrinogen nebo lidský trombin či na kteroukoli z dalších složek tohoto přípravku (jsou uvedeny v bodě 6).
Upozornění a opatření
Použití, _pro něž nejsou k dispozici dostatečné údaje
Použití přípravku EVARREST nebylo hodnoceno u následujících výkonů, a proto nejsou k dispozici žádné informace, jež by doložily, že by bylo účinné:
- slepování tkání,
- chirurgický zákrok v mozku či míše,
- zastavení krvácení v žaludku či střevech zavedením přípravku endoskopem (hadičkou),
- uzavření chirurgických zákroků na střevech.
Reakce na přítomnost cizího tělesa
Stejně jako u jiných implantovaných přípravků může v těle dojít k reakci na cizorodý materiál. Může to mít za následek problémy při hojení. Přípravek EVARREST má být použit pouze v jedné vrstvě tak, aby cca o 1 až 2 cm přesahoval na nekrvácející tkáň, což napomůže jeho přilepení ke krvácející oblasti. Velikost přípravku EVARREST má být omezena na takovou, která je nezbytná pro zástavu krvácení.
Hypersenzitivní reakce (přecitlivělost)
Je možná přecitlivělost (hypersenzitivita) alergického typu. Známky takových reakcí mohou zahrnovat kopřivku, vyrážku, tlak na prsou, sípot, pokles krevního tlaku a anafylaxi (těžkou, rychle nastupující reakci). Pokud takové příznaky nastanou během chirurgického zákroku, je nutné přestat přípravek okamžitě používat.
Přenos infekčních agens
Při výrobě léčivých přípravků z lidské krve či plazmy jsou v praxi provedena určitá opatření, jež mají zabránit přenosu infekce na pacienty. Jedná se mimo jiné o tato pravidla:
- pečlivý výběr dárců krve a plazmy, který zajistí vyloučení osob ohrožujících rizikem přenosu infekce,
- testování každého dárce a směsné plazmy na známky virů/infekce,
- zařazení kroků, které inaktivují nebo eliminují viry, do zpracování krve a plazmy.
I přes tato opatření nelze při podání léčivých přípravků vyrobených z lidské krve nebo plazmy zcela vyloučit možnost přenosu infekce. Platí to i pro jakékoli neznámé či nově se objevující viry či jiné typy infekcí.
Opatření učiněná při výrobě fibrinogenu a trombinu jsou považována za účinná pro obalené viry, jako je virus lidské imunodeficience (HIV), virus hepatitidy B a virus hepatitidy C, a viry neobalené, jako je virus hepatitidy A. Tato opatření mohou mít omezenou účinnost proti neobaleným virům, jako je parvovirus B19. Infekce parvovirem B19 může být závažná u těhotných žen (fetální infekce) a u jedinců se sníženou funkcí imunitního systému nebo s některými typy anémií (např. srpkovitá nemoc nebo hemolytická anémie).
Důrazně se doporučuje, aby vždy, když vám bude aplikován přípravek EVARREST, byl zaznamenán název a číslo šarže přípravku z důvodu evidence použitých šarží.
Děti a dospívající
Použití přípravku EVARREST u dětí a dospívajících do 18 let se nedoporučuje.
Další léčivé přípravky a EVARREST
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Těhotenství, kojení a fertilita
Není k dispozici dostatek informací, aby bylo známo, zda jsou s použitím přípravku EVARREST během těhotenství či kojení spojena nějaká konkrétní rizika, nebo zda by mohl mít vliv na fertilitu. Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem dříve, než začnete tento přípravek užívat.
EVARREST obsahuje sodík
Tento přípravek obsahuje až 3,0 mmol (68,8 mg) sodíku v 1 lepicí matrici EVARREST. Tuto skutečnost by měli vzít v úvahu pacienti s dietou s kontrolovaným příjmem sodíku.
3. Jak se EVARREST používá
Chirurg bude EVARREST aplikovat během operačního zákroku. Aplikuje se pevným přitisknutím na krvácející tkáň po dobu zhruba 3 minut. Přípravek EVARREST se aktivuje kontaktem s krví či jinou tekutinou a pevně přilne ke tkáni. Ponechá se na místě a během cca 8 týdnů ho tělo plně vstřebá.
EVARREST lze nastříhat na potřebnou velikost a tvar, které odpovídají velikosti krvácející plochy. Množství přípravku EVARREST, které bude aplikováno, závisí na ploše a místu krvácení, jež má být během operačního zákroku přípravkem ošetřeno. Přípravek EVARREST má být použit pouze v jedné vrstvě. Podle potřeby lze pro pokrytí celého krvácejícího místa použít až ekvivalent dvou kusů přípravku o rozměru 10,2 cm x 10,2 cm nebo čtyř kusů o rozměru 5,1 cm x 10,2 cm, které budou přesahovat příslušné místo cca o 1 až 2 cm. Pokud krvácení přetrvává, lze přípravek EVARREST odstranit a použít nový.
Celkové množství přípravku EVARREST ponechané v těle po operačním zákroku by nemělo být větší než velikost dvou matric o rozměru 10,2 cm x 10,2 cm nebo čtyř matric o rozměru 5,1 cm x 10,2 cm.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého. Má se za to, že dále uvedené nežádoucí účinky, které se projevily během klinických studií, souvisejí s použitím přípravku EVARREST:
Nejzávažnější nežádoucí účinky Krvácení
- střevní (gastrointestinální krvácení); četnost byla méně častá (může postihovat až 1 ze 100 osob)
- břišní (nitrobřišního krvácení); četnost byla méně častá (může postihovat až 1 ze 100 osob)
- pooperační (krvácení po provedení chirurgického výkonu); četnost byla častá (může postihovat až 1 z 10 osob)
Krevní sraženina (tromboembolie)
- v žilách, zejména nohou (hluboká žilní trombóza)
- v tepnách zásobujících plíce (plicní embolie)
Četnost obou těchto účinků byla méně častá (mohou postihovat až 1 ze 100 osob).
Nezáměrné vniknutí tekutin do dýchacích cest (aspirace); četnost byla méně častá (může postihovat až 1 ze 100 osob).
Pokud se u Vás vyskytnou příznaky, jako je zvracení krve, krev ve stolici, krev v drénu, který máte zavedený do břicha, otok končetin nebo změna barvy kůže na končetinách, bolest na hrudi a dušnost a/nebo jiné příznaky související s chirurgickým zákrokem, který jste podstoupil(a), obraťte se prosím ihned na svého lékaře.
Další nežádoucí účinky
Další nežádoucí účinky, které byly hlášeny během klinických studií s přípravkem EVARREST, zahrnovaly:
Časté (postihují až 1 z 10 osob)
- komplikace v pooperační ráně (včetně krvácení a infekce během operace nebo po ní),
- zvýšená hladina koagulačních bílkovin (fibrinogen),
Méně časté (postihují 1 ze 100 osob až méně než 1 z 10 osob)
- pooperační hromadění tekutiny a vzduchu v břiše (abdomenu) nebo plicích (pleurální výpotek, abdominální distenze, ascites, hromadění tekutiny ve slinivce břišní).
EVARREST obsahuje složky fibrinového tkáňového lepidla. Fibrinová tkáňová lepidla mohou ve vzácných případech (až u 1 z 1 000 osob) způsobit alergickou reakci. Pokud u vás nastane alergická reakce, může se u vás projevit jeden či více z těchto příznaků: podkožní otok (angioedém), vyrážka, kopřivka či kopřivkové pupeny (kopřivka), tlak na prsou, třesavka, zrudnutí, bolest hlavy, nízký krevní tlak, letargie, nevolnost, neklid, zvýšená srdeční frekvence, brnění, zvracení či sípot. Pokud se u vás kterýkoli z těchto příznaků vyskytne po operačním zákroku, poraďte se se svým lékařem či chirurgem.
Existuje i teoretická možnost, že se u Vás mohou vytvořit protilátky proti bílkovinám obsaženým v přípravku EVARREST, což by mohlo případně narušit srážení krve. Četnost tohoto typu příhod není známa (z dostupných údajů ji nelze odhadnout).
Hlášení nežádoucích účinků
Pokud se u vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak EVARREST uchovávat
EVARREST musí být uchováván mimo dohled a dosah dětí.
EVARREST nesmí být použit po uplynutí doby použitelnosti uvedené na foliovém sáčku a krabičce za písmeny EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
EVARREST nesmí být uchováván při teplotě vyšší než 25 °C a nesmí být zmrazen.
EVARREST musí být před použitím vždy uchováván v suchu, aby se zabránilo předčasné aktivaci.
Foliový sáček chrání přípravek EVARREST před vlhkostí a mikrobiologickou kontaminací.
6. Obsah balení a další informace Co EVARREST obsahuje
- Léčivými látkami jsou:
- lidský fibrinogen (8,6 mg/cm2),
- lidský trombin (37,5 IU/ cm2).
- Dalšími složkami jsou:
- kompozitní matrice (polyglaktin 910 a oxidovaná regenerovaná celulóza),
- arginin-hydrochlorid,
- glycin,
- chlorid sodný,
- citronan sodný,
- chlorid vápenatý,
- lidský albumin,
- mannitol,
- octan sodný.
Jak EVARREST vypadá a co obsahuje toto balení
EVARREST má formu matrice o velikosti 10,2 cm x 10,2 cm. Velikost balení 1 kus. V případě matric, které mají velikost 5,1 cm x 10,2 cm, je velikost balení 2 kusy.
Držitel rozhodnutí o registraci a výrobce
Omrix Biopharmaceuticals NV Leonardo Da Vincilaan 15 1831 Diegem Belgie
Telefon: + 32 2 746 30 00 Telefax: + 32 2 746 30 01
Další informace o tomto přípravku získáte u výrobce:
Pharmacovigilance Department Omrix Biopharmaceuticals Ltd
Plasma Fractionation Institute (Omrix-PFI), MDA Services Center Sheba Medical Center Ramat Gan 5262000 POB 888
Kiryat Ono 5510801 Izrael
Telefon: +972-3-5316512 Telefax: +972-3-5316590
Tato příbalová informace byla naposledy revidována MM.RRRR.
Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese: http://www.ema.europa.eu.
Na webových stránkách Evropské agentury pro léčivé přípravky je tato příbalová informace k dispozici ve všech úředních jazycích EU/EHP.
Následující informace jsou určeny pouze pro zdravotnické pracovníky:
Návod k použití
Čtěte tento návod před otevřením balení.
Manipulace s přípravkem EVARREST
EVARREST se dodává ve sterilních baleních připravený k okamžitému použití a musí se s ním manipulovat sterilní technikou v aseptickém prostředí. Poškozená balení zlikvidujte, neboť opakovaná sterilizace není možná.
Vyjměte foliový sáček z krabičky a opatrně jej otevřete rozloupnutím; zabraňte při tom kontaktu s vnitřní stranou folie nebo bílým sterilním táckem obsahujícím EVARREST.
Vyjměte bílý sterilní tácek ze sáčku a vložte jej do sterilního pole.
Držte tácek bezpečně v dlani ruky. Strana s otvory musí směřovat nahoru. Pomocí jazýčků na straně tácku sejměte druhou rukou horní stranu tácku.
Dolní část tácku obsahuje EVARREST; účinná strana přípravku je umístěna směrem dolů. Účinná strana má práškový vzhled. Neúčinná strana má reliéfní vlnovitý vzor.
Po otevření uchovávejte EVARREST v suchu. Přípravek EVARREST může zůstat ve sterilním poli, aby byl k dispozici po celou dobu výkonu. EVARREST nepřilne k rukavicím, pinzetě ani chirurgickým nástrojům.
Uchovávání přípravku EVARREST™
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a na štítku. Uchovávejte mimo dohled a dosah dětí.
Neuchovávejte při teplotě nad 25 °C. Chraňte před mrazem.
Použití přípravku EVARREST™
Pouze pro epilezionální použití. Při aplikaci přípravku EVARREST je nutné přípravek pevně manuálně přitlačit zhruba po dobu 3 minut.
1. Sterilními nůžkami opatrně nastřihejte matrix EVARREST na potřebnou odpovídající velikost a tvar a přiložte na krvácející oblast tak, aby matrice překrývala příslušné místo asi o 1 až 2 cm. Dokud je matrice EVARREST na tácku, musí její bílá až nažloutlá aktivní strana s práškovým povrchem zůstat ležet směrem dolů.
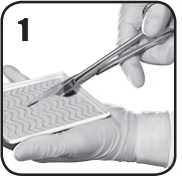
2. Podle potřeby odstraňte nadbytečnou krev či tekutinu z místa aplikace pro zlepšení viditelnosti. Je nutné přesně identifikovat zdroj krvácení. Přípravek EVARREST musí být aplikován přímo na zdroj krvácení a musí ho zcela zakrýt. EVARREST lze použít v aktivně krvácejícím poli.
3. Přiložte aktivní stranu přípravku EVARREST na krvácející plochu tak, aby byla v plném kontaktu s tkání. Přípravek se aktivuje kontaktem s tekutinou, přilne ke tkáni a přizpůsobí se jí.

4. Aplikujte kousek přípravku EVARREST vhodné velikosti tak, aby odpovídajícím způsobem
zakryl celou krvácející oblast, přesahoval ji zhruba o 1 až 2 cm na nekrvácející tkáň a napomohl tak při přilnutí k místu poranění.
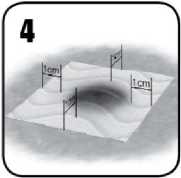
5a) Podržte suchou či vlhkou chirurgickou gázu nebo tampon na přípravku EVARREST, aby bylo dosaženo plného kontaktu s krvácejícím povrchem.

5b) K zajištění hemostáze okamžitě manuálně zatlačte na celý povrch přípravku EVARREST
(včetně překryvu). Vyviňte při tom dostatečný tlak, který zcela zamezí krvácení. Držte stlačené asi 3 minuty, aby se krvácení zastavilo.

6. Jemně odstraňte chirurgickou gázu nebo tampony z místa použití a dávejte při tom pozor, abyste nenarušili či neuvolnili přípravek EVARREST či krevní sraženinu. Zkontrolujte EVARREST a ověřte, zda došlo k hemostáze a zda se přípravek v místě krvácení nezvlnil. Pokud nebudete spokojeni s uložením přípravku EVARREST, vyjměte jej a použijte novou matrici EVARREST. EVARReSt zůstane na místě a přilne ke tkáni. Přípravek je vstřebatelný.
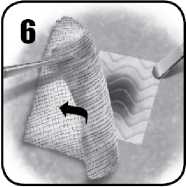
7. Místo aplikace by mělo být během chirurgického zákroku sledováno, aby se ověřilo, zda došlo k hemostáze.
Opakovaná aplikace
• Pokud se přípravek EVARREST zvlní, vzniknou na něm záhyby nebo se ohne, může být nutná opakovaná aplikace. Pokud nebudete spokojeni s umístěním, odstraňte použitý EVARREST a znovu zopakujte postup aplikace nového kusu tkáňového lepidla EVARREST. uvedený výše.
• Pokud je krvácení způsobeno neúplným zakrytím krvácející rány, lze přiložit další kus přípravku EVARREST. Aplikujte přípravek v jedné vrstvě tak, aby se okraje navzájem překrývaly (asi o 1 až 2 cm) s již přiloženým kusem přípravku EVARREST.
• Jestliže je krvácení způsobeno neúplným přilnutím ke tkáni (v místě, kde krvácení zpod krytí přetrvává), odstraňte EVARREST a použije nový kus přípravku EVARREST.
• Pokud krvácení pokračuje během stanovené doby komprese či poté, vyjměte použitý kus přípravku EVARREST a zkontrolujte místo krvácení. Pokud se zdá, že nejsou nutná další opatření pro primární hemostázu (tj. standardní chirurgické techniky), zopakujte postup aplikace nového kusu přípravku EVARREST, uvedený výše.
Likvidace
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními
požadavky.
27