Erivedge 150 Mg
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Erivedge 150 mg tvrdé tobolky
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna tvrdá tobolka obsahuje vismodegibum 150 mg.
Pomocná látka se známým účinkem:
Jedna tvrdá tobolka obsahuje 71,5 mg monohydrátu laktózy.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Tvrdá tobolka (tobolka).
Růžové neprůhledné tělo označené “150 mg” a šedé neprůhledné víčko označené “VISMO” černým inkoustem. Tobolka je velikosti 1 (rozměry 19,0 x 6,6 mm).
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Erivedge je indikován k léčbě dospělých pacientů s:
• symptomatickým metastazujícím bazocelulárním karcinomem
• lokálně pokročilým bazocelulárním karcinomem, který není vhodný k chirurgické léčbě nebo radioterapii (viz bod 5.1).
4.2 Dávkování a způsob podání
Přípravek Erivedge smí být předepsán pouze lékařem specialistou, který má zkušenosti s léčbou schválených indikací, nebo pod jeho dohledem.
Dávkování
Doporučená dávka je jedna 150 mg tobolka jednou denně.
Vynechané dávky
Pokud dojde k vynechání dávky, má být pacient poučen, aby si tuto vynechanou dávku nebral, ale pokračoval v léčbě další pravidelnou dávkou.
Trvání léčby
V klinických hodnoceních léčba přípravkem Erivedge pokračovala až do progrese onemocnění nebo výskytu nepřijatelné toxicity. Přerušení léčby bylo povoleno až do 4 týdnů v závislosti na individuální snášenlivosti.
Prospěch z pokračující léčby má být pravidelně vyhodnocován v rámci určení optimální délky léčby, která je odlišná pro každého pacienta.
Zvláštní skupiny pacientů
Starší lidé
U pacientů ve věku > 65 let není nutná úprava dávky (viz bod 5.2). Z celkového počtu 138 pacientů ve 4 klinických hodnoceních s přípravkem Erivedge u pokročilého bazocelulárního karcinomu bylo přibližně 40 % pacientů ve věku > 65 let a mezi těmito pacienty a pacienty mladšími nebyly pozorovány žádné celkové rozdíly v bezpečnosti a účinnosti.
Porucha funkce ledvin
Nepředpokládá se vliv mírné a středně těžké poruchy funkce ledvin na eliminaci vismodegibu a proto není nutná žádná úprava dávky. U pacientů s těžkou poruchou funkce ledvin jsou dostupné velmi omezené údaje. Pacienti se závažnou poruchou funkce ledvin mají být pečlivě sledováni s ohledem na možný výskyt nežádoucích účinků.
Porucha funkce jater
U pacientů s mírnou, středně těžkou až těžkou poruchou funkce jater definované na základě NCI-ODWG (National Cancer Institute Organ Dysfunction Working Group) - kritéria pro poruchu funkce jater, není nutná žádná úprava dávky:
• mírná: celkový bilirubin (TB) < horní hranice normálu (ULN), aspartátaminotransferáza (AST)>ULN nebo ULN<TB<1,5xULN, jakýkoli AST
• středně těžká: 1,5 x ULN < TB < 3 x ULN, jakýkoli AST
• těžká: 3 x ULN < TB < 10 x ULN, jakýkoli AST (viz bod 5.2)
Pediatrická populace
Bezpečnost a účinnost přípravku Erivedge nebyla u dětí a dospívajících mladších 18 let stanovena.
K dispozici nejsou žádné údaje.
Z důvodu bezpečnosti (viz body 4.4 a 5.3) se tento léčivý přípravek nemá používat u dětí a dospívajících mladších 18 let.
Způsob podání
Přípravek Erivedge je určen k perorálnímu podání. Tobolky se musí polykat celé a zapít vodou, mohou se užívat s jídlem i bez jídla (viz bod 5.2). Tobolky se nesmí otevírat, aby se předešlo nechtěné expozici pacientů a zdravotníků.
4.3 Kontraindikace
• Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1
• Těhotné nebo kojící ženy (viz body 4.4 a 4.6).
• Ženy ve fertilním věku, které nesplňují požadavky Programu prevence početí (Erivedge Pregnancy Prevention Programme) (viz body 4.4 a 4.6).
• Společné podávání s třezalkou tečkovanou (Hypericum perforatum) (viz bod 4.5).
4.4 Zvláštní upozornění a opatření pro použití
Embryo-fetální úmrtí nebo závažné vrozené vady
Přípravek Erivedge může způsobit embryo-fetální úmrtí nebo závažné vrozené vady, pokud se podává těhotným ženám (viz bod 4.6). U inhibitorů signalizační kaskády Hedgehog (viz bod 5.1), jako je vismodegib, byly prokázány embryotoxické a/nebo teratogenní účinky u mnoha zvířecích druhů. Tyto inhibitory mohou způsobovat závažné malformace, včetně kraniofaciálních anomálií, defektů středočárových struktur a defektů končetin (viz bod 5.3). Přípravek Erivedge se nesmí používat v průběhu těhotenství.
Kritéria pro ženu ve fertilním věku
Žena ve fertilním věku je v Programu prevence početí definována následovně:
• pohlavně vyspělá žena, která
• menstruovala kdykoliv v posledních 12 po sobě jdoucích měsících,
• nepodstoupila hysterektomii nebo bilaterální ovarektomii, nebo nemá lékařsky potvrzenou diagnózu syndromu předčasného ovariálního selhání,
• nemá genotyp XY, Turnerův syndrom nebo agenezi uteru,
• má amenoreu po protinádorové léčbě, zahrnující léčbu přípravkem Erivedge.
Poradenství
Pro ženu ve fertilním věku
Přípravek Erivedge je kontraindikován u ženy ve fertilním věku, která nesplňuje kritéria Programu prevence početí.
Žena ve fertilním věku musí být srozuměna s tím, že:
• přípravek Erivedge vystavuje nenarozené dítě teratogennímu riziku,
• nesmí užívat přípravek Erivedge, pokud j e těhotná nebo těhotenství plánuj e,
• musí mít negativní těhotenský test provedený zdravotníkem z období posledních 7 dnů před zahájením léčby přípravkem Erivedge,
• musí mít negativní těhotenský test každý měsíc v průběhu léčby, i když má amenoreu
• nesmí otěhotnět během léčby přípravkem Erivedge a po dobu 24 měsíců po podání poslední dávky,
• musí být schopna používat vhodnou antikoncepci,
• musí během léčby přípravkem Erivedge používat dvě doporučené metody antikoncepce (viz níže bod „Antikoncepce“ a bod 4.6), pokud se nezaváže dodržovat sexuální abstinenci,
• pokud v průběhu léčby nebo v období 24 měsíců po podání poslední dávky dojde k čemukoli z dále uvedeného, musí to sdělit svému lékaři:
• pokud otěhotní nebo pokud se z jakéhokoli důvodu domnívá, že může být těhotná,
• pokud dojde k vynechání pravidelné menstruace,
• pokud přestane užívat antikoncepci a nezaváže se k přerušení sexuálního života
(abstinenci),
• pokud potřebuje změnit antikoncepci v průběhu léčby,
• Během léčby přípravkem Erivedge a po dobu 24 měsíců po podání poslední dávky nesmí kojit.
Pro muže
Vismodegib je přítomen ve spermatu. Z důvodu vyvarování se možné expozice plodu během těhotenství musí být pacient srozuměn s tím, že:
• Pokud má muž nechráněný sex s těhotnou ženou, přípravek Erivedge vystavuje nenarozené dítě teratogennímu riziku,
• Musí vždy používat doporučenou antikoncepci (viz níže bod „Antikoncepce“ a bod 4.6),
• Pokud jeho partnerka otěhotní v době, kdy (on) užívá přípravek Erivedge nebo v průběhu 2 měsíců po podání poslední dávky, sdělí to svému lékaři.
Pro zdravotníky
Zdravotníci musí poučit pacienty, aby rozuměli a potvrdili, že rozumí všem podmínkám Programu prevence početí.
Antikoncepce
Žena ve fertilním věku
Ženy - pacientky musí během léčby přípravkem Erivedge a po dobu 24 měsíců po podání poslední dávky používat dvě doporučené metody antikoncepce, včetně jedné vysoce účinné metody a jedné bariérové metody (viz bod 4.6).
Muži
Pokud mají pacienti během užívání přípravku Erivedge a v období 2 měsíců po podání poslední dávky pohlavní styk s partnerkou, musí vždy používat kondom (se spermicidním přípravkem, je-li to možné), a to i v případě, že mají provedenou vazektomii (viz bod 4.6).
Těhotenské testy
Žena ve fertilním věku musí mít proveden v průběhu 7 dnů před zahájením léčby a pak pravidelně každý měsíc v průběhu léčby lékařem kontrolovaný těhotenský test provedený zdravotníkem. Těhotenský test je nutno provést s citlivostí alespoň 25 mIU/ml, podle místní dostupnosti. U pacientek s amenoreou vzniklou během léčby přípravkem Erivedge se má testování na těhotenství jedenkrát měsíčně opakovat po celou dobu léčby.
Omezení předepisování a výdeje u ženy ve fertilním věku
K prvnímu předepsání a výdeji přípravku Erivedge musí dojít během 7 dnů od provedení negativního těhotenského testu. Přípravek Erivedge má být předepisován na dobu 28 dnů léčby a pokud léčba pokračuje, má být předepsán znovu.
Edukační materiál
Z důvodu podpory prevence expozice embrya a plodu přípravku Erivedge, poskytne držitel rozhodnutí o registraci zdravotníkům a pacientům edukační materiály (Program prevence početí), které upozorní na možná rizika související s léčbou přípravkem Erivedge.
Účinky na postnatální vývoj
U zvířat bylo prokázáno, že vismodegib způsobuje závažné ireverzibilní změny rostoucích zubů (degenerace/nekróza odontoblastů, tvorba cyst naplněných tekutinou v zubní dřeni, osifikace kořenových kanálků a krvácení) a uzavření epifyzální růstové štěrbiny. Tyto nálezy představují možné riziko nízkého vzrůstu a deformit zubů pro kojence a děti (viz bod 5.3).
Dárcovství krve
Pacienti nemají během léčby přípravkem Erivedge a po dobu 24 měsíců po podání poslední dávky darovat krev.
Dárcovství spermatu
Muži - pacienti nemají během léčby přípravkem Erivedge a po dobu 2 měsíců po podání poslední dávky darovat sperma.
Interakce
Je nutné vyhnout se současné léčbě se silným induktorem CYP (např. rifampicin, karbamazepin nebo fenytoin), protože nemůže být vyloučeno riziko snížení koncentrace v plazmě a snížení účinnosti vismodegibu (viz bod 4.5).
Kožní spinocelulární karcinom
Pacienti s pokročilým bazocelulárním karcinomem mají zvýšené riziko rozvoje kožního spinocelulárního karcinomu. Případy tohoto karcinomu byly popsány u pacientů s pokročilým bazocelulárním karcinomem léčených přípravkem Erivedge. Nebylo stanoveno, zda kožní spinocelulární karcinom souvisí s léčbou přípravkem Erivedge. Proto mají být všichni pacienti běžně monitorováni během léčby přípravkem Erivedge a kožní spinocelulární karcinom má být léčen standardní léčbou.
Další opatření
Pacienti mají být instruováni nikdy nepodávat tento léčivý přípravek další osobě. Veškeré nespotřebované tobolky mají být na konci léčby ihned pacientem zlikvidovány v souladu s místními požadavky (vrátit veškeré nespotřebované tobolky zpět do lékárny).
Pomocné látky
Tobolky přípravku Erivedge obsahují monohydrát laktózy. Pacienti se vzácnými dědičnými problémy s intolerancí galaktózy, vrozeným nedostatkem laktázy nebo malabsorpcí glukózy a galaktózy by tento přípravek neměli užívat.
Tento léčivý přípravek obsahuje méně než 1 mmol sodíku (23 mg) v jedné dávce, je tedy v podstatě „bez sodíku“
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Účinky souběžně užívaných léčivých přípravků na vismodegib
Nepředpokládají se klinicky významné farmakokinetické interakce mezi vismodegibem a přípravky zvyšujícími pH. Výsledky z klinické studie prokázaly pokles koncentrací volné látky vismodegibu o 33 % po 7 dnech současné léčby s 20 mg rabeprazolu (inhibitor protonové pumpy) podávaného 2 hodiny před každým podáním vismodegibu. Neočekává se, že by tato interakce byla klinicky významná.
Dále se nepředpokládají klinicky významné farmakokinetické interakce mezi vismodegibem a inhibitory CYP450. Výsledky z klinické studie prokázaly zvýšení koncentrací volné látky vismodegibu o 57 % v den 7 po zahájení současné léčby se 400 mg flukonazolu denně (středně silný inhibitor CYP2C9), ale tato interakce není považována za klinicky významnou. Itrakonazol (silný inhibitor CYP3A4) podávaný v dávce 200 mg denně nemá vliv na vismodegib AUC0-24h po 7 dnech současné léčby u zdravých dobrovolníků.
Nepředpokládají se klinicky významné farmakokinetické interakce mezi vismodegibem a inhibitory P-gp. Výsledky z klinické studie neprokázaly klinicky významné farmakokinetické interakce mezi vismodegibem a itrakonazolem (silný inhibitor P-glykoproteinu) u zdravých dobrovolníků.
Pokud je vismodegib podáván společně s induktory CYP (rifampicin, karbamazepin, fenytoin, třezalka tečkovaná), může být expozice vismodegibu snížena (viz body 4.3 a 4.4).
Vliv vismodegibu na souběžně podávané léčivé přípravky
Antikoncepční steroidy
Výsledky ze studie lékových interakcí provedené u pacientů s nádorovým onemocněním ukázaly, že systémová expozice ethinylestradiolu a norethisteronu není narušena, pokud jsou podávány společně s vismodegibem.
Nicméně studie interakcí trvala pouze 7 dnů a nelze vyloučit, že v průběhu déletrvající léčby je vismodegib induktorem enzymů metabolizujících antikoncepční steroidy. Indukce může vést k snížení systémové expozice antikoncepčních steroidů, a tím ke snížení antikoncepčního účinku.
Vliv na specifické enzymy a transportéry
Studie in vitro naznačují, že vismodegib má potenciál působit jako inhibitor BCRP (breast cancer resistance protein). Údaje týkající se interakcí in vivo nejsou k dispozici. Nelze vyloučit, že vismodegib může vést ke zvýšené expozici léčivých přípravků, které jsou transportovány tímto proteinem, jako jsou rosuvastatin, topotekan a sulfasalazin. Společné podávání je třeba provádět s opatrností a může být nutná úprava dávky.
Neočekávají se klinicky významné farmakokinetické interakce mezi vismodegibem a substráty CYP450. In vitro byl CYP2C8 nejcitlivější izoformmou CYP k inhibici vismodegibu. Nicméně výsledky ze studie lékových interakcí provedené u pacientů s nádorovým onemocněním ukázaly, že systémová expozice rosiglitazonu (substrát CYP2C8) není narušena, pokud je tato látka podávána společně s vismodegibem. Tudíž inhibice CYP enzymů vismodegibem in vivo může být vyloučena.
In vitro inhibuje vismodegib OATP1B1. Nelze vyloučit, že vismodegib může zvyšovat expozici substrátů OATP1B1, např. bosentanu, ezetimibu, glibenklamidu, repaglinidu, valsartanu a statinů. Zvláště je třeba dbát opatrnosti, pokud je vismodegib podáván v kombinaci se statiny.
4.6 Fertilita, těhotenství a kojení
Kritéria pro ženy ve fertilním věku
Vzhledem k riziku embryo-fetálního úmrtí nebo závažných vrozených vad, které způsobuje vismodegib, nesmí být ženy užívající přípravek Erivedge těhotné ani nesmí otěhotnět v průběhu léčby a po dobu 24 měsíců po podání poslední dávky (viz body 4.3 a 4.4).
Přípravek Erivedge je kontraindikován u ženy ve fertilním věku, která nesplňuje kritéria Programu prevence početí.
V případě těhotenství nebo vynechání menstruace
Pokud pacientka otěhotní, dojde k vynechání menstruace nebo pokud má z jakéhokoli důvodu podezření, že může být těhotná, musí ihned informovat svého ošetřujícího lékaře. Přetrvávající vynechání menstruace během léčby přípravkem Erivedge má být považováno za indikátor těhotenství až do lékařského posouzení a potvrzení.
Antikoncepce u mužů a žen
Ženy ve fertilním věku
Žena ve fertilním věku musí být schopna používat účinný způsob antikoncepce. Během léčby přípravkem Erivedge a po dobu 24 měsíců po podání poslední dávky musí používat dvě doporučené metody antikoncepce, včetně jedné vysoce účinné metody a jedné bariérové metody. Žena ve fertilním věku, jejíž menstruace je nepravidelná nebo skončila, se rovněž musí řídit všemi doporučeními o účinné antikoncepci.
Muži
Vismodegib je přítomen ve spermatu. Aby se zabránilo možné expozici plodu během těhotenství, musí pacienti během léčby přípravkem Erivedge a po dobu 2 měsíců po podání poslední dávky při pohlavním styku s partnerkou používat vždy kondom (se spermicidním přípravkem, je-li to možné), a to i v případě, že mají provedenou vazektomii.
Níže jsou uvedeny doporučené formy vysoce účinné antikoncepce:
• Hormonální depotní injekce,
• Tubární sterilizace,
• Vazektomie,
• Nitroděložní tělísko.
Níže jsou uvedeny doporučené formy bariérové antikoncepce:
• Jakýkoli mužský kondom (se spermicidním přípravkem, je-li to možné),
• Pesar (se spermicidním přípravkem, je-li to možné).
Přípravek Erivedge může způsobit embryo-fetální úmrtí nebo závažné vrozené vady, pokud se podává těhotným ženám (viz bod 4.4). U inhibitorů signalizační kaskády Hedgehog (viz bod 5.1), jako je vismodegib, bylo prokázáno, že mají embryotoxické a/nebo teratogenní účinky u mnoha druhů zvířat a mohou způsobovat závažné malformace, včetně kraniofaciálních anomálií, defektů středočárových struktur a defektů končetin (viz bod 5.3). V případě těhotenství u žen léčených přípravkem Erivedge, musí být léčba okamžitě ukončena.
Kojení
Není známo, v jakém rozsahu se vismodegib vylučuje do mateřského mléka. Z důvodu možných závažných vývojových defektů nesmí ženy během léčby přípravkem Erivedge a po dobu 24 měsíců po podání poslední dávky kojit (viz body 4.3 a 5.3).
Fertilita
Léčba přípravkem Erivedge může u žen poškodit fertilitu (viz bod 5.3). Reverzibilita poruchy fertility není známa. V klinických studiích byla navíc u ženy ve fertilním věku pozorována amenorea (viz bod 4.8). S ženou ve fertilním věku je třeba před zahájením léčby přípravkem Erivedge probrat možná opatření k zachování fertility.
Neočekává se porucha fertility u mužů (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek Erivedge nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky Shrnutí bezpečnostního profilu
Nejčastější nežádoucí účinky, které se objevily u > 30 % pacientů, byly svalové spasmy (74,6 %), alopecie (65,9 %), dysgeuzie (58,7 %), pokles tělesné hmotnosti (50,0 %), únava (47,1 %), nauzea (34,8 %) a průjem (33,3 %).
Shrnutí nežádoucích účinků do tabulky
Nežádoucí účinky jsou uvedeny v tabulce 1 níže podle tříd orgánových systémů a absolutní četnosti. Četnosti jsou definovány následovně:
Velmi časté (> 1/10)
Časté (> 1/100 až < 1/10)
Méně časté (> 1/1000 až < 1/100)
Vzácné (> 1/10 000 až <1/1000)
Velmi vzácné (< 1/10 000)
V každé skupině četností jsou nežádoucí účinky řazeny podle klesající závažnosti.
Bezpečnost přípravku Erivedge byla hodnocena v klinických studiích u 138 pacientů léčených pro pokročilý bazocelulární karcinom, který zahrnuje bazocelulární karcinom jak metastazující (mBCC), tak i lokálně pokročilý (laBCC). Ve čtyřech otevřených klinických studiích fáze 1 a 2 dostali pacienti alespoň jednou přípravek Erivedge v monoterapii v dávce > 150 mg. V klinických studiích nevedly dávky >150 mg k vyšším plazmatickým koncentracím a do analýzy byli zařazeni pacienti s dávkou > 150 mg. Pozorovaný bezpečnostní profil byl obecně podobný u pacientů s metastazujícím bazocelulárním karcinomem i lokálně pokročilým bazocelulárním karcinomem, jak je popsáno níže.
Nežádoucí účinky vyskytující se v klinických studiích u pacientů léčených přípravkem Erivedge
|
Třídy orgánových systémů podle databáze MedDRA |
Velmi časté |
Časté |
|
Poruchy metabolismu a výživy |
Snížení chuti k jídlu |
Dehydratace Hyponatremie |
|
Poruchy nervového systému |
Dysgeuzie Ageuzie |
Hypogeuzie |
|
Gastrointestinální poruchy |
Zácpa |
Bolest horní části břicha Bolest břicha |
|
Poruchy kůže a podkožní tkáně |
Alopecie Pruritus |
Madaróza Nadměrné ochlupení |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Svalové spasmy Artralgie Bolest končetin |
Muskuloskeletální bolest hrudníku Myalgie Bolest v boku Muskuloskeletální bolest |
|
Poruchy reprodukčního systému a prsu |
Amenorea* | |
|
Celkové poruchy a reakce v místě aplikace |
Pokles tělesné hmotnosti Únava Bolest |
Astenie |
|
Vyšetření |
Zvýšení hladin jaterních enzymů** | |
|
Všechna hlášení jsou založena na nežádoucích účincích všech stupňů podle NCI-CTCAE (National Cancer Institute - Common Terminology Criteria for Adverse Events), verze 3.0 (s výjimkou případů, kde je uvedeno jinak). *Ze 138 pacientů s pokročilým bazocelulárním karcinomem, bylo 10 žen ve fertilním věku. Mezi těmito ženami byla amenorea pozorována u 3 pacientek (30 %). MedDRA = Medical Dictionary for Regulatory Activities. **Zahrnuje preferované výrazy: abnormální testy jaterních funkcí, zvýšený bilirubin v krvi, zvýšení gama-glutamyl transferázy, zvýšení aspartát aminotransferázy, zvýšení alkalické fosfatázy, zvýšené jaterní enzymy. | ||
Tabulka 1
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Přípravek Erivedge byl podán v dávce 3,6krát vyšší, než je doporučená denní dávka 150 mg.
V průběhu těchto klinických studií nebylo pozorováno zvýšení hladiny vismodegibu v plazmě nebo zvýšení toxicity.
FARMAKOLOGICKÉ VLASTNOSTI
5.
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: cytostatika, jiná cytostatika, ATC kód: L01XX43.
Mechanismus účinku
Vismodegib je perorálně dostupný inhibitor signalizační kaskády Hedgehog o malé molekule. Signalizační kaskáda Hedgehog prostřednictvím transmembránového proteinu SMO (transmembránový protein Smoothened) vede k aktivaci a vstupu transkripčních faktorů onkogenu souvisejícího s gliomem (GLI) do jádra, a tím k indukci cílových genů Hedgehog. Mnoho z těchto genů je zahrnuto do proliferace, přežití a diferenciace. Vismodegib se váže na protein SMO a inhibuje ho, čímž v signalizační kaskádě Hedgehog blokuje přenos signálu do buňky.
Klinická účinnost a bezpečnost
Klíčová studie, ERIVANCE BCC (SHH4476g), byla mezinárodní, jednoramenná, multicentrická studie se 2 kohortami. Metastazující bazocelulární karcinom byl definován jako bazocelulární karcinom, který se rozšířil z kůže dále do jiných částí těla, zahrnujících lymfatické uzliny, plíce, kosti a/nebo vnitřní orgány. Pacienti s lokálně pokročilým bazocelulárním karcinomem měli kožní léze, které nebyly vhodné k chirurgické léčbě (inoperabilní, opakovaně rekurentní, kde kurativní resekce nebyla pravděpodobná nebo kde by chirurgické řešení vedlo k významné deformitě nebo morbiditě) a kde nebyla úspěšná radioterapie, nebo kde tato léčba byla kontraindikovaná nebo nevhodná. Před zařazením do studie byla diagnóza bazocelulárního karcinomu potvrzena histologicky. Pacienti s Gorlinovým syndromem, kteří měli alespoň jednu lézi pokročilého bazocelulárního karcinomu a kteří splňovali vstupní kritéria, byli do studie zařazeni. Pacienti byli léčeni přípravkem Erivedge v perorální denní dávce 150 mg.
Medián věku u populace s hodnotitelnou účinností léčby byl 62 let (46 % pacientů bylo ve věku alespoň 65 let), 61 % pacientů byli muži a 100 % pacientů bylo bílé pleti. V kohortě s metastazujícím bazocelulárním karcinomem absolvovalo 97 % pacientů předchozí léčbu, včetně chirurgické léčby (97 %), radioterapie (58 %) a systémové terapie (30 %). V kohortě s lokálně pokročilým bazocelulárním karcinomem (n = 63) absolvovalo 94 % pacientů předchozí léčbu, včetně chirurgické léčby (89 %), radioterapie (27 %) a systémové/lokální terapie (11 %). Medián trvání léčby byl 12,9 měsíce (rozmezí 0,7 až 47,8 měsíce).
Primárním cílovým parametrem účinnosti byla četnost objektivní odpovědi (ORR) hodnocená nezávislým hodnotitelem, jak je uvedeno v tabulce 2. Objektivní odpověď byla definována jako úplná nebo částečná odpověď zaznamenaná ve dvou po sobě jdoucích vyšetřeních s odstupem alespoň 4 týdnů. V kohortě s metastazujícím bazocelulárním karcinomem byla odpověď tumoru hodnocena podle RECIST (Response Evaluation Critera in Solid Tumours), verze 1.0. V kohortě s lokálně pokročilým bazocelulárním karcinomem byla odpověď tumoru hodnocena na základě vizuálního zhodnocení zevního rozsahu tumoru a ulcerace, pomocí zobrazovacích metod (kde to bylo vhodné) a biopsie tumoru. V kohortě s lokálně pokročilým bazocelulárním karcinomem byli pacienti považováni za pacienty s odpovědí, pokud splnili alespoň jedno z následujících kritérií a pokud nedošlo k progresi onemocnění: (1) > 30% redukce velikosti léze (součet nejdelších průměrů) od výchozích hodnot v cílových lézích dle radiografického vyšetření; (2) > 30% redukce součtu nejdelších průměrů od výchozích hodnot u zevně pozorovatelných rozměrů cílových lézí; (3) kompletní vymizení ulcerace ve všech cílových lézích. Klíčové údaje jsou shrnuty v tabulce 2:
Tabulka 2 Výsledky účinnosti přípravku Erivedge ve studii SHH4776g (IRF 21 měsíců a vyhodnocení sledování investigátorem 39 měsíců poté, kdy byl zapsán poslední pacient): pacienti hodnotitelní s ohledem na účinnost*’1'
|
Nezávislý přezkum (IRF) - vyhodnocení Metastazující Lokálně pokročilý bazocelulární bazocelulární |
Investigátor Metastazující bazocelulární |
- vyhodnocení Lokálně pokročilý bazocelulární | ||
|
karcinom |
karcinom** |
karcinom |
karcinom** | |
|
(n = 33) |
(n = 63) |
(n = 33) |
(n = 63) | |
|
Pacienti s odpovědí 95% interval spolehlivosti pro celkovou odpověď |
11 (33,3 %) (19,2%, 51,8%) |
30 (47,6%) (35,5 %, 60,6%) |
16 (48,5 %) (30,8%, 66,2 %) |
38 (60,3 %) (47,2 %, 71,7 %) |
|
Úplná odpověď Částečná odpověď Stabilní onemocnění |
0 11 (33,3%) 20 |
14 (22,2%) 16 (25,4%) 22 |
0 16 (48,5%) 14 |
20 (31,7%) 18 (28,6%) 15 |
|
Progrese onemocnění* |
1 |
8 |
2 |
6 |
|
Medián trvání |
7,6 |
9,5 |
14,8 |
26,2 |
|
odpovědi (měsíce) (95% interval spolehlivosti) |
(5,5, 9,4) |
(7,4, 21,4) |
(5,6, 17,0) |
(9,0, 37,6) |
|
Medián přežití bez |
9,5 |
9,5 |
9,3 |
12,9 |
|
progrese (měsíce) (95% interval spolehlivosti) |
(7,4,11,1) |
(7,4, 14,8) |
(7,4, 16,6) |
(10,2, 28,0) |
|
Medián celkového |
33,4 |
NE | ||
|
přežití (měsíce) (95% interval spolehlivosti) |
(18,1, NE) |
(NE, NE) | ||
|
Četnost přežití v délce jednoho roku (95% interval spolehlivosti) |
78,7% (64,7, 92,7) |
93,2% (86,8, 99,6) | ||
NE = není odhadnutelné (not estimable)
* Populace pacientů hodnotitelná s ohledem na účinnost je definována jako všichni pacienti zařazení do studie, kteří dostali jakékoli množství přípravku Erivedge a u kterých bylo hodnocení archivního vzorku nebo počáteční tkáňové biopsie nezávislým patologem konzistentní s diagnózou bazocelulárního karcinomu.
f Nehodnotitelné/chybějící údaje zahrnovaly 1 pacienta s metastazujícím bazocelulárním karcinomem a 4 pacienty s lokálně pokročilým bazocelulárním karcinomem.
* Progrese v kohortě s lokálně pokročilým bazocelulárním karcinomem je definována jako splnění jakéhokoli
z následujících kritérií: (1) > 20% zvýšení od nejnižší hodnoty součtu nejdelších průměrů v cílových lézích (dle radiografického vyšetření nebo zevně pozorovatelných rozměrů), (2) Nová ulcerace cílových lézí přetrvávající bez průkazu hojení po dobu alespoň 2 týdnů, (3) Nové léze stanovené radiograficky nebo fyzikálním vyšetřením, (4) Progrese necílových lézí podle RECIST.
** 54 % pacientů s laBCC nemělo histopatologický průkaz BCC ve 24 týdnech.
Podle grafů na obrázcích 1 a 2, které ukazují maximální redukci velikosti cílové léze/cílových lézí u jednotlivých pacientů, vykazovala většina pacientů v obou kohortách zmenšení tumoru podle hodnocení nezávislým hodnotitelem.
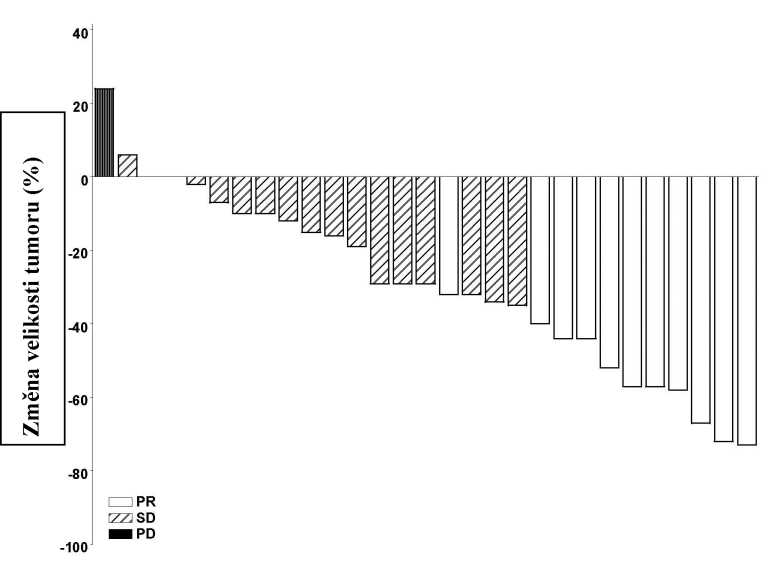
Poznámka: Velikost tumoru odpovídá součtu nejdelších průměrů cílových lézí. PD = progrese onemocnění, SD = stabilní onemocnění, PR = částečná odpověď. Tři pacienti měli nejlepší procentuální odpověď změny velikosti tumoru 0, což odpovídá minimálním pozitivním sloupcům na obrázku. Čtyři pacienti byli z obrázku vyloučeni: 3 pacienti se stabilním onemocněním byli hodnoceni pouze dle necílových lézí a 1 pacient byl nehodnotitelný.
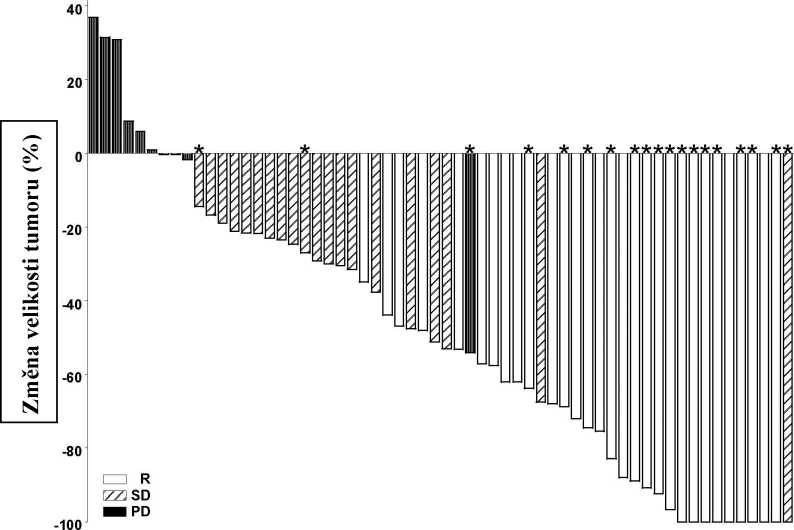
Poznámka: Velikost tumoru odpovídá součtu nejdelších průměrů cílových lézí. PD = progrese onemocnění, SD = stabilní onemocnění, R = odpověď, * = kompletní vymizení ulcerace/ulcerací. Hodnocení odpovědi bylo založeno na složeném cílovém parametru účinnosti definovaném výše. Čtyři pacienti neměli provedeno změření léze a nebyli do grafu zahrnuti.
Doba do maximální redukce tumoru
Mezi pacienty, u kterých došlo k redukci velikosti tumoru, byl medián doby do maximální redukce tumoru 5,6 měsíce u pacientů s laBCC a 5,5 měsíce u pacientů s mBCC, na základě hodnocení nezávislým hodnotitelem. Podle hodnocení zkoušejícím byl medián doby do maximální redukce tumoru 6,7 měsíce u pacientů s laBCC a 5,5 měsíce u pacientů s mBCC.
Elektrofyziologie srdce
Ve studii podrobně hodnotící QTc interval u 60 zdravých subjektů nebyl pozorován žádný vliv terapeutických dávek přípravku Erivedge na QTc interval.
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Erivedge u všech podskupin pediatrické populace s bazocelulárním karcinomem (viz bod
4.2 pro informace o pediatrickém použití).
Tento léčivý přípravek byl registrován postupem tzv. podmíněného schválení. Znamená to, že jsou očekávány další důkazy o jeho přínosech. Evropská agentura pro léčivé přípravky nejméně jednou za rok vyhodnotí nové informace o tomto léčivém přípravku a tento souhrn údajů o přípravku bude podle potřeby aktualizován.
5.2 Farmakokinetické vlastnosti
Absorpce
Přípravek Erivedge je vysoce permeabilní sloučenina s nízkou rozpustností ve vodě (BCS třída 2). Absolutní průměrná biologická dostupnost po jednorázové dávce přípravku Erivedge je 31,8 %
(14,5 %). Absorpce je saturovatelná, což dokazuje nepřítomnost proporcionálního zvýšení po podání jednorázové dávky 270 mg a 540 mg přípravku Erivedge. Za klinicky relevantních podmínek (v
rovnovážném stavu) není farmakokinetika vismodegibu ovlivněna jídlem. Proto se přípravek Erivedge může užívat bez ohledu na jídlo.
Distribuce
Distribuční objem vismodegibu je nízký, pohybující se v rozmezí od 16,4 do 26,6 litru. Při klinicky relevantních koncentracích je vazba vismodegibu na proteiny lidské plazmy in vitro vysoká (97 %). Vismodegib se váže na lidský sérový albumin i alfa-1-kyselý glykoprotein. Při klinicky relevantních koncentracích je in vitro vazba na alfa-1-kyselý glykoprotein saturovatelná. Vazba na plazmatické proteiny ex vivo je u pacientů vyšší než 99 %. Koncentrace vismodegibu výrazně korelují s hladinami alfa-1-kyselého glykoproteinu a vykazují paralelní fluktuace alfa-1-kyselého glykoproteinu a celkové koncentrace vismodegibu v průběhu času a trvale nízkou hladinu nevázaného vismodegibu.
Biotransformace
Vismodegib se pomalu eliminuje kombinací metabolismu a exkrece mateřské látky. Vismodegib převažuje v plazmě, s koncentracemi odpovídajícími více než 98 % celkových cirkulujících koncentrací (včetně přidružených metabolitů). Metabolismus vismodegibu u člověka zahrnuje oxidaci, glukuronizaci a méně časté štěpení pyridinového kruhu. Zdá se, že CYP2C9 přispívá z části na biotransformaci vismodegibu in vivo.
Eliminace
Po perorálním podání radioaktivně značené dávky je vismodegib vstřebáván a pomalu eliminován kombinací metabolismu a exkrece mateřské látky, z níž většina se vyloučí stolicí (82 % podané dávky) a 4,4 % podané dávky se vyloučí močí. Vismodegib a související metabolické produkty jsou eliminovány převážně jaterní cestou.
Po kontinuálním podávání jednou denně se prokázalo, že farmakokinetika vismodegibu je nelineární, v důsledku saturovatelné absorpce a saturovatelné vazby na proteiny. Po jednorázové perorální dávce byl terminální poločas vismodegibu přibližně 12 dnů.
Zdánlivý poločas vismodegibu v rovnovážném stavu je odhadován na 4 dny při kontinuálním denním dávkování. Při kontinuálním denním dávkování dochází k trojnásobnému nárůstu koncentrace vismodegibu v plazmě.
Vismodegib inhibuje in vitro UGT2B7 a nelze vyloučit, že in vivo může k této inhibici docházet ve střevě.
Zvláštní skupiny pacientů
Starší lidé
U starších lidí jsou k dispozici omezené údaje. V klinických hodnoceních s pokročilým bazocelulárním karcinomem bylo přibližně 40 % pacientů v geriatrickém věku (> 65 let). Analýza farmakokinetiky u této populace naznačuje, že věk neměl klinicky významný vliv na rovnovážné koncentrace vismodegibu.
Pohlaví
Na základě populační farmakokinetické analýzy dle kombinovaných údajů od 121 mužů a 104 žen se neprokázalo, že by pohlaví mělo vliv na farmakokinetiku vismodegibu.
Rasa
U pacientů jiné než bílé rasy jsou k dispozici omezené údaje. Protože počet subjektů, kteří byli jiné než bílé rasy, představoval jen < 3 % celé populace (černá rasa 6, bílá rasa 219), nebyla rasa hodnocena jako kovariát při analýze populační farmakokinetiky.
Porucha funkce ledvin
Vylučování perorálně podaného vismodegibu ledvinami je nízké. Proto je nepravděpodobné, že mírná a středně těžká porucha funkce ledvin má klinicky významný účinek na farmakokinetiku vismodegibu. Na základě populační farmakokinetické analýzy u pacientů s mírnou (BSA-indexované CrCl 50 až 80 ml/min, n=58) a středně těžkou (BSA-indexované CrCl 30 až 50 ml/min, n=16) poruchou funkce ledvin neměla mírná a středně těžká porucha funkce ledvin žádný klinicky významný účinek na farmakokinetiku vismodegibu (viz bod 4.2). U pacientů s těžkou poruchou funkce ledvin jsou dostupné velmi omezené údaje.
Porucha funkce jater
Hlavní eliminační cesty vismodegibu zahrnují jaterní metabolismus a biliární/střevní sekreci.
V klinické studii u pacientů s poruchou funkce jater (stupeň poruchy funkce stanoven dle AST subjektu a celkových hladin bilirubinu) se po opakovaných dávkách vismodegibu ukázalo, že u pacientů s mírnou (NCI-ODWG kritéria, n=8), středně těžkou (NCI-ODWG kritéria, n=8) a těžkou (NCI-ODWG kritéria, n=3) poruchou funkce jater byl farmakokinetický profil vismodegibu srovnatelný s jedinci s normální funkcí jater (n=9) (viz bod 4.2).
Pediatrická populace
Pro pediatrickou populaci jsou k dispozici nedostatečné farmakokinetické údaje.
5.3 Předklinické údaje vztahující se k bezpečnosti
Předklinický bezpečnostní profil přípravku Erivedge byl hodnocen u myší, potkanů a psů.
Toxicita po opakovaném podání
Snášenlivost přípravku Erivedge ve studiích toxicity po opakovaném podání na potkanech a psech byla obvykle limitována nespecifickou manifestací toxicity včetně nižšího nárůstu tělesné hmotnosti a spotřeby jídla. Další nálezy při klinicky relevantních expozicích zahrnovaly změny stolice, záškuby kosterního svalstva nebo třes, alopecii, otoky, folikulární hyperkeratózu a zánět polštářků na tlapkách a zvýšení LDL a HDL cholesterolu. Snížení hematokritu nebo počtu trombocytů bylo při klinicky relevantních expozicích pozorováno u některých psů, nebyl však prokázán primární vliv na kostní dřeň u postižených zvířat.
Kancerogenita
Neklinické studie zaměřené na zhodnocení kancerogenity vismodegibu nebyly provedeny. V 26týdenní studii toxicity u potkanů byl však pozorován pilomatrikom (benigní kožní novotvar). V klinických studiích s přípravkem Erivedge nebyl pilomatrikom hlášen a význam tohoto nálezu pro člověka je proto nejasný.
Mutagenita
Ve studiích in vitro (hodnocení vzniku reverzních mutací u bakterií (Amesův test) a test chromozomálních aberací lidských lymfocytů) ani v mikronukleárním testu buněk kostní dřeně u potkanů in vivo nebyla prokázána genotoxicita.
Fertilita
Ve 26týdenní studii fertility u potkanů bylo pozorováno výrazné zvýšení absolutní hmotnosti semenných váčků a snížení absolutní hmotnosti prostaty. Kromě toho byl poměr hmotnosti orgánu k celkové tělesné hmotnosti výrazně zvýšen u nadvarlete, kaudy nadvarlete, varlete a semenných váčků. Ve stejné studii nebyly žádné histopatologické nálezy u samčích reprodukčních orgánů ani žádný vliv vismodegibu na fertilitu vč. motility spermií na konci podávání přípravku při dávce 100 mg/kg/den nebo ve fázi zotavování (koresponduje s 1,3 násobkem ustálené AUC0-24h při doporučené dávce u člověka). Kromě toho ve studiích delších než 26 týdnů, sledujících celkovou toxicitu vismodegibu u pohlavně zralých potkanů a psů, nebyl pozorován žádný účinek na reprodukční orgány. U zvýšeného počtu degenerovaných zárodečných buněk a hypospermie u pohlavně nezralých psů, pozorovaného při dávce > 50 mg/kg/den ve 4týdenní studii sledující celkovou toxicitu, nebyla určena spojitost s podáním vismodegibu.
Ve 26týdenní studii fertility u potkanů byly pozorovány účinky na samičí reprodukční orgány spojené s podáním vismodegibu v dávce 100 mg/kg/den bezprostředně po přerušení léčby, vč. poklesu implantací, zvýšeného procenta preimplantačních ztrát a snížený počet samic s životaschopnými embryi. Tyto nálezy nebyly pozorovány po 16týdenní zotavovací fázi. Nebyly pozorovány žádné korelující histopatologické změny. Expozice samic potkanů dávkou 100 mg/kg koresponduje s 1,2 násobkem ustálené AUC0-24h doporučené dávky u člověka.
Kromě toho ve 26týdenní studii sledující celkovou toxicitu vismodegibu byl pozorován pokles počtu žlutých tělísek při dávce 100 mg/kg/den. Účinek nevymizel na konci 8týdenní zotavovací fáze.
Teratogenita
Studie embryo-fetálního vývoje, ve kterých dostávali březí potkani vismodegib každý den během organogeneze, procházel vismodegib placentou a byl významně toxický s ohledem na početí. U plodů samic, které dostávaly dávky odpovídající 20 % typické expozice v rovnovážném stavu u pacientů, byly pozorovány malformace, včetně kraniofaciálních anomálií, defektů v oblasti perinea a absence a/nebo srůstu prstů a při vyšších dávkách byla pozorována 100% embryoletalita.
Postnatální vývoj
Studie zaměřené na zhodnocení schopnosti vismodegibu ovlivnit postnatální vývoj nebyly provedeny. Ve studiích toxicity u potkanů však byly při klinicky relevantních expozicích pozorovány ireverzibilní defekty rostoucích zubů a předčasný uzávěr femorální epifyzární růstové štěrbiny, což představuje riziko pro postnatální vývoj.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Obsah tobolky Mikrokrystalická celulóza Monohydrát laktózy Natrium-lauryl-sulfát Povidon K29/32
Sodná sůl karboxymetylškrobu (typ A)
Mastek
Magnesium-stearát Obal tobolky
Černý oxid železitý (E172)
Červený oxid železitý (E172)
Oxid titaničitý (E171)
Želatina
Inkoust potisku Šelak
Černý oxid železitý (E172)
6.2 Inkompatibility Neuplatňuje se.
6.3 Doba použitelnosti
3 roky
6.4 Zvláštní opatření pro uchovávání
Uchovávejte při teplotě do 30 °C.
Uchovávejte v dobře uzavřeném vnitřním obalu, aby byl přípravek chráněn před vlhkostí.
6.5 Druh obalu a obsah balení
Lahvička z vysokodenzního polyethylenu s dětským bezpečnostním uzávěrem obsahující 28 tvrdých tobolek. Jedno balení obsahuje jednu lahvičku. Víčko lahvičky je z polypropylenu. Vložka uzávěru lahvičky je z povoskované lepenky potažené hliníkovou fólií.
6.6 Zvláštní opatření pro likvidaci přípravku
Veškerý nespotřebovaný léčivý přípravek musí být na konci léčby pacientem zlikvidován v souladu s místními požadavky (vrátit tobolky do lékárny nebo lékaři).
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/13/848/001
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 12. července 2013
Datum posledního prodloužení registrace: 27. května 2015
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
PŘÍLOHA II
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
E. ZVLÁŠTNÍ POVINNOST USKUTEČNIT POREGISTRAČNÍ OPATŘENÍ PRO PODMÍNĚNOU REGISTRACI PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného za propouštění šarží
Roche Pharma AG Emil-Barell-Strasse 1 D-79639 Grenzach-Wyhlen Německo
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES ajakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
• Další opatření k minimalizaci rizik
Před uvedením léčivého přípravku na trh v členském státě držitel rozhodnutí o registraci odsouhlasí s národní autoritou následující:
• Přeložený text materiálu pro zdravotnické pracovníky
• Metodologii sběru informací o podávání přípravku Erivedge a soulad s požadavky na farmakovigilanci a efektivnost dle Programu prevence početí.
• Formát a obsah materiálu pro zdravotnické pracovníky a pacienty
Držitel rozhodnutí o registraci rozešle při uvedení přípravku na trh dopis určený zdravotnickým pracovníkům, dopis má obsahovat následující:
• Základní text tak, jak byl odsouhlasený raportérem
• Zvláštní národní požadavky tak, jak byly odsouhlaseny národní autoritou, týkající se:
• Distribuce přípravku
• Opatření zajišťujících, že všechny příslušné požadavky byly splněny před tím, než byl přípravek Erivedge předepsán a vydán
Držitel rozhodnutí o registraci bude průběžně zajišťovat, že všem lékařům předepisujícím Erivedge budou poskytovány následující materiály:
Informace o přípravku Edukační materiál pro lékaře Kartu pro lékaře Edukační materiál pro pacienta Kartu pro pacienta
Edukační materiál pro lékaře pro přípravek Erivedge má obsahovat následující základní součásti:
• Stručnou informaci o přípravku Erivedge, jeho schválenou indikaci a dávkování
• Požadavek informovat pacienta o teratogenních rizicích spojených s přípravkem Erivedge a nezbytnost zabránit fetální expozici
• Popis Programu prevence početí a kategorizaci pacientů založenou na pohlaví a fertilním
potenciálu
• Informaci o doporučených formách antikoncepce pro ženy a muže
• Povinnosti lékaře předepisujícího léčivý přípravek Erivedge
• Nezbytnost poskytnout vyčerpávající informace a poradenství pacientům
• Zajistit, že pacienti jsou schopni dodržovat požadavky týkající se bezpečného užití přípravku Erivedge
• Nezbytnost poskytnout pacientům edukační materiály a Kartu pro pacienta
• Bezpečnostní doporučení pro ženy ve fertilním věku
• Nezbytnost odpovídajících antikoncepčních opatření (i v případě, že žena má amenoreu) v průběhu léčby a 24 měsíců po skončení léčby přípravkem Erivedge
• Těhotenský test v průběhu 7 dnů před zahájením léčby a následně v průběhu léčby jednou měsíčně těhotenský test vyhodnocený lékařem
• Nezbytnost ihned ukončit léčbu přípravkem Erivedge při podezření na těhotenství
• Nezbytnost ihned hlásit podezření na těhotenství ošetřujícímu lékaři
• Bezpečnostní doporučení pro muže
• Nezbytnost používat kondom během léčby přípravkem Erivedge a po dobu 2 měsíců po podání poslední dávky (i pokud má muž provedenou vazektomii) v případě, že sexuální partnerka je těhotná nebo žena ve fertilním věku
• Nezbytnost ihned hlásit ošetřujícímu lékaři těhotenství partnerky, pokud k němu dojde v průběhu léčby přípravky Erivedge u muže nebo krátce po ukončení jeho léčby
• Nedarovat sperma v průběhu léčby a 2 měsíce po poslední dávce
• Požadavky v případě těhotenství
• Instrukce ukončit užívání přípravku Erivedge při podezření na těhotenství
• Nezbytnost odeslat pacientku ke specialistovi
• Lokální kontakt pro hlášení jakéhokoli podezření na těhotenství
• Hlášení o těhotenství
• Informovat pacienty, že nemají darovat krev v průběhu léčby přípravkem Erivedge a 24 měsíců po podání poslední dávky
• Kontrolní seznam pro lékaře zajišťující, že pacienti obdrží odpovídající poučení
• Nezbytnost zajistit, aby všichni pacienti vyplnili a podepsali informovaný souhlas pro přípravek Erivedge, formulář má být součástí edukačního materiálu pro lékaře
• Hlášení nežádoucího účinku
• Informace týkající se metodologie sběru informací o podávání přípravku Erivedge a soulad
s požadavky na farmakovigilanci a efektivnost Programu prevence početí, odsouhlasené národní autoritou.
Edukační materiál pro pacienta pro přípravek Erivedge má obsahovat následující základní součásti:
• Informace pro pacienty o teratogenních rizicích spojených s přípravkem Erivedge a nezbytnost zabránit fetální expozici
• Popis Karty pro pacienta
• Nezbytnost odpovídající antikoncepce a určení odpovídající antikoncepce
• Národní nebo jiná odpovídající zvláštní opatření týkající se předepisování a vydávání přípravku Erivedge
• Nedávat přípravek Erivedge žádné další osobě
• Informace o znehodnocení nepoužité medikace
• Nezbytnost uchovávat tobolky přípravku Erivedge mimo dohled a dosah dětí
• Upozornění, že pacient nemá darovat krev v průběhu léčby a 24 měsíců po podání poslední dávky
• Upozornění, že pacientka nemá kojit v průběhu léčby a 24 měsíců po podání poslední dávky
• Upozornění, že pacient má informovat lékaře o jakémkoli nežádoucím účinku
• Informace pro ženy ve fertilním věku
• Popis Programu prevence početí
• Nezbytnost odpovídajících antikoncepčních opatření v průběhu léčby a 24 měsíců po skončení léčby přípravkem Erivedge
• Těhotenský test v průběhu 7 dnů před zahájením léčby a následně v průběhu léčby jednou měsíčně těhotenský test vyhodnocený lékařem
• Nezbytnost ihned ukončit léčbu přípravkem Erivedge při podezření na těhotenství
• Nezbytnost ihned hlásit podezření na těhotenství ošetřujícímu lékaři
• Informace pro muže
• Nezbytnost používat kondom během léčby přípravkem Erivedge a po dobu 2 měsíců po podání poslední dávky (i pokud má muž provedenou vazektomii) v případě, že sexuální partnerka je těhotná nebo žena ve fertilním věku
• Nezbytnost ihned hlásit ošetřujícímu lékaři, pokud dojde k otěhotnění partnerky
• Nedarovat sperma v průběhu léčby a 2 měsíce po poslední dávce
Karta pro lékaře má obsahovat následující základní součásti:
• Informace pro ženy ve fertilním věku
• Nezbytnost provedení těhotenského testu každý měsíc i v případě, že pacientka má amenoreu
• Nezbytnost odpovídajících antikoncepčních opatření v průběhu léčby a 24 měsíců po skončení léčby přípravkem Erivedge
• Nekojit v průběhu léčby a 24 měsíců po skončení léčby přípravkem Erivedge
• Informace pro muže
• Nezbytnost použít kondom v případě pohlavního styku s partnerkou v průběhu léčby a 2 měsíce po skončení léčby přípravkem Erivedge
• Nedarovat sperma v průběhu léčby a 2 měsíce po poslední dávce
• Nezbytnost informovat pacienta o nutnosti ihned hlásit ošetřujícímu lékaři jakékoli podezření na otěhotnění, pokud je pacientem žena nebo partnerka pacienta
• Lékař má vyhodnotit stupeň těhotenství, poučit pacientku o teratogenním riziku a odeslat pacientku ke specialistovi
• Lékař má držiteli rozhodnutí o registraci hlásit potvrzená těhotenství
• Připomenout pacientům vrátit na konci léčby nespotřebované tobolky (likvidace bude záviset na místních požadavcích)
• Připomenout pacientům nedarovat krev v průběhu léčby a 24 měsíců po podání poslední dávky
Karta pro pacienta má obsahovat následující základní součásti:
• Informace pro pacienty o teratogenních rizicích spojených s přípravkem Erivedge a nezbytnost zabránit fetální expozici
• Nedarovat krev v průběhu léčby a 24 měsíců po podání poslední dávky
• Informace pro ženy ve fertilním věku
• Nezbytnost provedení těhotenského testu každý měsíc
• Nezbytnost odpovídajících antikoncepčních opatření
• Nezbytnost ihned kontaktovat lékaře v případě podezření na těhotenství v průběhu léčby nebo 24 měsíců po skončení léčby
• Informace pro muže
• Nezbytnost použít kondom v případě pohlavního styku s partnerkou
• Nedarovat sperma v průběhu léčby a 2 měsíce po podání poslední dávky
• Nezbytnost ihned kontaktovat ošetřujícího lékaře, pokud má partnerka podezření, že otěhotněla v průběhu partnerovy léčby přípravkem Erivedge nebo 2 měsíce po ukončení léčby
• Připomenout pacientům vrátit na konci léčby nespotřebované tobolky (likvidace bude záviset na místních požadavcích)
• Telefonní kontakt v případě nouze
E. ZVLÁŠTNÍ POVINNOST USKUTEČNIT POREGISTRAČNÍ OPATŘENÍ PRO PODMÍNĚNOU REGISTRACI PŘÍPRAVKU
Tato registrace byla schválena postupem tzv. podmíněného schválení, a proto podle článku 14(7) nařízení (ES) č. 726/2004 držitel rozhodnutí o registraci uskuteční v daném termínu následující opatření:
|
Popis |
Termín splnění |
|
Žadatel poskytne další údaje o bezpečnosti a účinnosti u pacientů se symptomatickým metastazujícím BCC z finální analýze MO25616. |
Q1 2016 |
PŘÍLOHA III
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU KRABIČKA
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Erivedge 150 mg tvrdé tobolky vismodegibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tobolka obsahuje vismodegibum 150 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktózu.
Další informace naleznete v příbalové informaci
4. LÉKOVÁ FORMA A OBSAH BALENÍ
28 tobolek
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Tobolku nedrťte, neotvírejte nebo nežvýkejte Před použitím si přečtěte příbalovou informaci Perorální podání
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
Riziko závažných vrozených vad Nepoužívejte v těhotenství nebo při kojení
Musíte dodržovat Program prevence početí pro přípravek Erivedge
8. POUŽITELNOST
Použitelné do:
Uchovávejte při teplotě do 30 °C
Uchovávejte v dobře uzavřeném vnitřním obalu, aby byl přípravek chráněn před vlhkostí
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
Nepoužitelné tobolky na konci léčby vraťte
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/13/848/001
13. ČÍSLO ŠARŽE
č.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
erivedge
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
<2D čárový kód s jedinečným identifikátorem>
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
PC:
SN:
NN:
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITRNÍM OBALU ŠTÍTEK LAHVE
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Erivedge 150 mg tvrdé tobolky vismodegibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tobolka obsahuje vismodegibum 150 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktózu.
Další informace naleznete v příbalové informaci
4. LÉKOVÁ FORMA A OBSAH BALENÍ
28 tobolek
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Tobolku nedrťte, neotvírejte nebo nežvýkejte Před použitím si přečtěte příbalovou informaci Perorální podání
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
Riziko závažných vrozených vad Nepoužívejte v těhotenství nebo při kojení
Musíte dodržovat Program prevence početí pro přípravek Erivedge
8. POUŽITELNOST
Použitelné do:
Uchovávejte při teplotě do 30 °C
Uchovávejte v dobře uzavřeném vnitřním obalu, aby byl přípravek chráněn před vlhkostí
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
Nepoužitelné tobolky na konci léčby vraťte
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/13/848/001
13. ČÍSLO ŠARŽE č.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis
15. NÁVOD K POUŽITÍ 16. INFORMACE V BRAILLOVE PÍSMU
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro pacienta
Erivedge 150 mg tvrdé tobolky
Vismodegibum
Přípravek Erivedge může způsobit závažné vrozené vady. To může vést k úmrtí dítěte před jeho narozením nebo krátce po jeho narození. Nesmíte otěhotnět během léčby tímto přípravkem. Musíte dodržovat opatření týkající se antikoncepce popsaná níže v této příbalové informaci.
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je psáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
• Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
• Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
• Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
• Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Erivedge a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Erivedge užívat
3. Jak se přípravek Erivedge užívá
4. Možné nežádoucí účinky
5. Jak přípravek Erivedge uchovávat
6. Obsah balení a další informace
1. Co je přípravek Erivedge a k čemu se používá Co je přípravek Erivedge
Erivedge je protinádorový lék obsahující léčivou látku vismodegib.
K čemu se přípravek Erivedge používá
Přípravek Erivedge se používá k léčbě dospělých pacientů s určitým typem kožního nádoru nazývaného pokročilý bazocelulární karcinom (bazaliom). Používá se k léčbě, pokud se nádorové onemocnění:
• šíří do dalších částí těla (nazývá se pak “metastazující” bazocelulární karcinom)
• šíří do okolních oblastí (nazývá se pak “lokálně pokročilý” bazocelulární karcinom) a Váš lékař rozhodne, že chirurgické odstranění nebo ozařování nádoru není vhodné.
Jak přípravek Erivedge účinkuje
K rozvoji bazocelulárního karcinomu dochází, pokud dojde k poškození DNA v normálních buňkách a tělo samo není schopno poškození opravit. Toto poškození může změnit funkci určitých bílkovin v těchto buňkách a poškozené buňky se stanou buňkami nádorovými, začnou růst a dělit se. Přípravek Erivedge je protinádorový lék, který účinkuje tak, že kontroluje jednu z klíčových bílkovin, která je obsažena v bazocelulárním karcinomu. To může zpomalit nebo zabránit růstu nádorových buněk, nebo je to může zahubit. Výsledkem je možné zmenšení kožního nádoru.
2. Čemu musíte věnovat pozornost, než začnete přípravek Erivedge užívat
Přečtěte si zvláštní instrukce, které Vám předá Váš lékař, zejména pak informace o vlivu přípravku Erivedge na nenarozené děti.
Pečlivě si přečtěte a řiďte se instrukcemi uvedenými v brožuře pro pacienta a Kartě pro pacienta, které Vám předá Váš lékař.
Neužívejte přípravek Erivedge
• jestliže jste alergický(á) na vismodegib nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
• jestliže jste těhotná, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět během léčby nebo v průběhu 24 měsíců po podání poslední dávky tohoto přípravku. To je proto, že přípravek Erivedge může poškodit nenarozené dítě nebo způsobit jeho úmrtí.
• jestliže kojíte nebo plánujete kojit během léčby nebo v průběhu 24 měsíců po podání poslední dávky tohoto přípravku. To je proto, že není známo, zda přípravek Erivedge může procházet do mateřského mléka a poškodit dítě.
• jestliže můžete otěhotnět, ale nejste schopná nebo ochotná splnit nutná opatření týkající se prevence početí, která jsou uvedena v Programu prevence početí.
• jestliže současně užíváte třezalku tečkovanou (Hypericumperforatum) - bylinný lék užívaný k léčbě deprese (viz „Další léčivé přípravky a přípravek Erivedge).
Více informací o výše uvedené problematice naleznete v bodech „Těhotenství, kojení a plodnost “ a „Antikoncepce - u mužů a žen“
Pokud se Vás cokoli z výše zmíněného týká, neužívejte tento přípravek. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem dříve, než začnete přípravek Erivedge užívat.
Upozornění a opatření
Před užitím přípravku Erivedge se poraďte se svým lékařem nebo lékárníkem, pokud máte otázky týkající se informací uvedených v tomto bodě:
• Kdykoliv během léčby ani v průběhu 24 měsíců po podání poslední dávky tohoto přípravku nesmíte darovat krev.
• Pokud jste muž, nesmíte darovat sperma kdykoliv v průběhu léčby a 2 měsíce po poslední dávce.
• Váš lékař Vám má pravidelně kontrolovat kůži z důvodu výskytu typu karcinomu nazývaného „kožní spinocelulární karcinom“. Není známo, zda kožní spinocelulární karcinom souvisí s léčbou přípravkem Erivedge. Obvykle se tento typ léze objevuje na kůži v místě spáleném sluncem, nerozšiřuje se a může být léčen. Upozorněte svého lékaře v případě, že zaznamenáte jakékoli změny na kůži.
• Nikdy nedávejte lék nikomu jinému. Na konci léčby vraťte nespotřebované tobolky. Domluvte se s lékařem nebo lékárníkem, kde můžete nespotřebované léky vrátit.
Děti a dospívající
Používání přípravku Erivedge u dětí a dospívajících mladších 18 let se nedoporučuje. Je to proto, že není známo, zda je tento přípravek bezpečný nebo účinný u osob této věkové skupiny. Ve studiích na zvířatech hodnotících tento přípravek byly pozorovány problémy s růstem zubů a kostí.
Další léčivé přípravky a přípravek Erivedge
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat. To se týká i léků, které jsou dostupné bez lékařského předpisu, vitamínů a rostlinných přípravků.
Některé léky mohou ovlivnit účinek přípravku Erivedge, nebo mohou zvýšit pravděpodobnost jeho nežádoucích účinků. Přípravek Erivedge může také ovlivnit účinky jiných léků. Sdělte svému lékaři zejména, pokud užíváte některý z následujících léků:
• rifampicin, používaný k léčbě bakteriálních infekcí,
• karbamazepin, fenytoin - používané k léčbě epilepsie,
• ezetimib a statiny, například - atorvastatin, fluvastatin, pravastatin, rosuvastatin, simvastatin -používané ke snížení cholesterolu,
• bosentan, glibenklamid, repaglinid, valsartan,
• topotekan - používaný k léčbě určitých typů rakoviny,
• sulfasalazin - používaný k léčbě určitých zánětlivých poruch, a zvláště pak,
• třezalka tečkovaná (Hypericum perforatum) - bylinný lék užívaný k léčbě deprese, který nesmíte užívat současně s lékem Erivedge.
Těhotenství, kojení a plodnost
Neužívejte přípravek Erivedge, pokud jste těhotná, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět během léčby nebo v průběhu 24 měsíců po podání poslední dávky tohoto přípravku.
Musíte ukončit léčbu a informovat svého lékaře ihned, pokud u Vás dojde k vynechání menstruace, nebo si myslíte, že u Vás došlo k vynechání menstruace, nebo pokud máte neobvyklé menstruační krvácení nebo podezření na těhotenství. Pokud otěhotníte v průběhu léčby přípravkem Erivedge, musíte léčbu okamžitě ukončit a informovat svého lékaře.
Přípravek Erivedge může způsobit závažné vrozené vady. Může rovněž vést k úmrtí nenarozeného dítěte. Od svého lékaře dostanete zvláštní instrukce (Program prevence početí), obsahující informace, které se týkají zejména účinku přípravku Erivedge na nenarozené děti.
Kojení
Během léčby a v průběhu 24 měsíců po podání poslední dávky tohoto přípravku nekojte. Není známo, zda přípravek Erivedge může procházet do mateřského mléka a poškodit dítě.
Plodnost
Přípravek Erivedge může u žen ovlivnit schopnost mít děti. Některé ženy užívající přípravek Erivedge přestaly menstruovat. Pokud k tomu dojde, není známo, zda se pravidelná menstruace znovu vrátí. Pokud si přejete mít v budoucnu děti, promluvte si se svým lékařem.
Antikoncepce - u mužů a žen
Ženy užívající přípravek Erivedge:
Pokud můžete otěhotnět, poraďte se před zahájením léčby se svým lékařem. I v případě, že jste přestala menstruovat, je důležité, abyste se poradila se svým lékařem, zda neexistuje nějaké riziko, že můžete otěhotnět.
Pokud můžete otěhotnět:
• musíte dodržovat příslušná opatření, abyste neotěhotněla v průběhu léčby přípravkem Erivedge
• používejte dvě metody antikoncepce - jednu vysoce účinnou metodu a jednu bariérovou metodu (viz příklady níže)
• musíte pokračovat v používání antikoncepce po dobu 24 měsíců po podání poslední dávky tohoto přípravku - to proto, že přípravek Erivedge může zůstávat v těle až 24 měsíců po podání poslední dávky.
Doporučené metody antikoncepce: o dvou metodách antikoncepce pro Vás nejvhodnějších se poraďte se svým lékařem.
Používejte jednu vysoce účinnou metodu antikoncepce, jako je:
• antikoncepční injekce s prodlouženým uvolňováním
• nitroděložní tělísko
• chirurgická sterilizace.
Musíte rovněž používat jednu bariérovou metodu, jako je:
• kondom (se spermicidním přípravkem, je-li to možné)
• pesar (se spermicidním přípravkem, je-li to možné).
Váš lékař u Vás musí provést těhotenský test:
• nejméně 7 dní před zahájením léčby - aby se ujistil, zda už nejste těhotná
• každý měsíc v průběhu léčby.
Musíte neprodleně informovat svého lékaře během léčby nebo v průběhu 24 měsíců po podání poslední dávky tohoto přípravku, pokud:
• si myslíte, že Vaše antikoncepce z jakéhokoli důvodu selhala,
• jste přestala menstruovat,
• jste přestala používat antikoncepci,
• potřebujete změnit antikoncepci.
Muži užívající přípravek Erivedge:
Přípravek Erivedge může pronikat do spermatu. Vždy, pokud máte pohlavní styk s partnerkou, používejte kondom (se spermicidním přípravkem, je-li to možné), a to i v případě, že jste podstoupil vazektomii (chirurgické podvázání chámovodů). Toto je nutné dodržovat během léčby a po dobu 2 měsíců po podání poslední dávky tohoto přípravku.
Po celou dobu během léčby a po dobu 2 měsíců po podání poslední dávky nesmíte darovat sperma. Řízení dopravních prostředků a obsluha strojů
Není pravděpodobné, že by přípravek Erivedge ovlivňoval Vaši schopnost řídit, obsluhovat jakékoli přístroje nebo stroje. Pokud si nejste jistý(á), poraďte se se svým lékařem.
Přípravek Erivedge obsahuje laktózu a sodík
Přípravek Erivedge obsahuje určitý typ cukru nazývaný laktóza. Pokud Vám lékař řekl, že nesnášíte některé cukry, poraďte se se svým lékařem, než začnete tento léčivý přípravek užívat.
Tento přípravek obsahuje méně než lmmol (23 mg) sodíku v jedné tobolce, tj. v podstatě je „bez sodíku“
3. Jak se přípravek Erivedge užívá
Vždy užívejte přípravek Erivedge přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Užívání tohoto přípravku
Doporučená dávka přípravku je jedna tobolka každý den.
• Tobolku spolkněte celou a zapijte ji vodou.
• Tobolku nedrťte, neotvírejte ani nežvýkejte, abyste zabránil(a) nechtěnému kontaktu s obsahem tobolky.
• Přípravek Erivedge lze užívat s jídlem i bez jídla.
Jestliže jste užil(a) více přípravku Erivedge, než jste měl(a)
Jestliže jste užil(a) více přípravku Erivedge, než jste měl(a), poraďte se se svým lékařem.
Jestliže jste zapomněl(a) užít přípravek Erivedge
Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku, ale pokračujte v léčbě další dávkou podle rozpisu.
Jestliže jste přestal(a) užívat přípravek Erivedge
Nepřestávejte užívat tento přípravek bez předchozí porady se svým lékařem, protože může dojít ke snížení účinku léčby.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i přípravek Erivedge nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Přípravek Erivedge může způsobit závažné vrozené vady. Také může vést k úmrtí dítěte před jeho narozením nebo krátce po jeho narození. Nesmíte otěhotnět během léčby tímto přípravkem (viz bod 2, „Neužívejte přípravek Erivedge“ a „Těhotenství, kojení a plodnost“).
Další nežádoucí účinky jsou uvedeny podle závažnosti a četnosti výskytu:
Velmi časté (mohou postihnout více než 1 osobu z 10):
• ztráta pravidelné menstruace u žen v plodném věku,
• ztráta chuti k jídlu a pokles tělesné hmotnosti,
• pocit únavy,
• svalové křeče,
• průjem,
• vypadávání vlasů (alopecie),
• vyrážka,
• změny vnímání chuti nebo úplná ztráta vnímání chuti,
• zácpa,
• zvracení nebo pocit na zvracení,
• podrážděný žaludek nebo porucha trávení,
• bolest kloubů,
• bolest (obecně) nebo bolest paží, nohou,
• svědění.
Časté (mohou postihnout až 1 osobu z 10):
• bolest hrudníku, zad nebo boku,
• nedostatek energie nebo slabost (astenie),
• ztráta vody z těla (dehydratace),
• bolest svalů, šlach, vazů, kostí,
• bolest žaludku,
• ztráta vnímání chuti,
• nadměrné ochlupení,
• vypadávání řas (madaróza),
• změny v krevních testech, které zahrnují zvýšení hodnot v jaterních testech nebo snížení hodnot sodíku.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Erivedge uchovávat
• Uchovávejte tento přípravek mimo dohled a dosah dětí.
• Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na lahvi a krabičce za „Použitelné do:“ Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
• Uchovávejte při teplotě do 30 °C.
• Uchovávejte v dobře uzavřeném vnitřním obalu, aby byl přípravek chráněn před vlhkostí.
• Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu.
• Na konci Vaší léčby vraťte veškeré nespotřebované tobolky. Tato opatření zamezí zneužití a pomáhají chránit životní prostředí. Domluvte se s lékárníkem nebo s lékařem, kde můžete nespotřebované léky vrátit.
6. Obsah balení a další informace
Co přípravek Erivedge obsahuje
• Léčivou látkou je vismodegibum. Jedna tvrdá tobolka obsahuje vismodegibum 150 mg.
Dalšími složkami jsou:
• Obsah tobolky: mikrokrystalická celulóza, monohydrát laktózy, natrium-lauryl-sulfát, povidon K29/32, sodná sůl karboxymetylškrobu (typ A), mastek a magnesium stearát.
• Obal tobolky: červený oxid železitý (E172), černý oxid železitý (E172), oxid titaničitý, želatina.
• Inkoust potisku: šelak a černý oxid železitý (E172).
Jak přípravek Erivedge vypadá a co obsahuje toto balení
víčko označené “VISMO” černým uzávěrem obsahujících 28 tobolek.
Tobolky mají růžové neprůhledné tělo označené “150 mg” a šedé inkoustem. Jsou dostupné v lahvičkách s dětským bezpečnostním Jedno balení obsahuje jednu lahvičku.
Držitel rozhodnutí o registraci
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
Výrobce
Roche Pharma AG Emil-Barell-Strasse 1 D-79639 Grenzach-Wyhlen Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci
|
Belgie/Belgique/Belgien N.V. Roche S.A. Tél/Tel: +32 (0) 2 525 82 11 |
Lietuva UAB “Roche Lietuva” Tel: +370 5 2546799 |
|
BtnrapHH Pom Etnrapua EOOfl Ten: +359 2 818 44 44 |
Luxembourg/Luxemburg (Voir/siehe Belgique/Belgien) |
|
Česká republika Roche s. r. o. Tel: +420 -2 20382111 |
Magyarország Roche (Magyarország) Kft. Tel: +36 - 23 446 800 |
|
Danmark Roche a/s Tlf: +45 - 36 39 99 99 |
Malta (See United Kingdom) |
|
Deutschland Roche Pharma AG Tel: +49 (0) 7624 140 |
Nederland Roche Nederland B.V. Tel: +31 (0) 348 438050 |
|
Eesti Roche Eesti OU Tel: + 372 -6 177 380 |
Norge Roche Norge AS Tlf: +47 - 22 78 90 00 |
|
EXXáSa Roche (Hellas) A.E. T^: +30 210 61 66 100 |
Osterreich Roche Austria GmbH Tel: +43 (0) 1 27739 |
|
Espaňa Roche Farma S.A. Tel: +34 - 91 324 81 00 |
Polska Roche Polska Sp.z o.o. Tel: +48 -22 345 18 88 |
|
France Roche Tél: +33 (0) 1 47 61 40 00 |
Portugal Roche Farmaceutica Química, Lda Tel: +351 -21 425 70 00 |
|
Hrvatska Roche d.o.o Tel: +385 1 4722 333 |
Románia Roche Románia S.R.L. Tel: +40 21 206 47 01 |
|
Ireland Roche Products (Ireland) Ltd. Tel: +353 (0) 1 469 0700 |
Slovenija Roche farmacevtska družba d.o.o. Tel: +386 - 1 360 26 00 |
|
Ísland Roche a/s c/o Icepharma hf Sími: +354 540 8000 |
Slovenská republika Roche Slovensko, s.r.o. Tel: +421 -2 52638201 |
Suomi/Finland
Italia
Roche S.p.A.
Tel: +39 -039 2471
Roche Oy
Puh/Tel: +358 (0) 10 554 500
Latvija
Roche Latvija SIA
Tel: +371 -6 7 039831
United Kingdom
Roche Products Ltd.
Tel: +44 (0) 1707 366000
Tato příbalová informace byla naposledy revidována {měsíc RRRR}.
Tomuto léčivému přípravku bylo uděleno tzv. podmíněné schválení. Znamená to, že informace o tomto přípravku budou přibývat. Evropská agentura pro léčivé přípravky nejméně jednou za rok vyhodnotí nové informace o tomto léčivém přípravku a tato příbalová informace bude podle potřeby aktualizována.
Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky: http://www.ema.europa.eu.
Na webových stránkách Evropské agentury pro léčivé přípravky je tato příbalová informace k dispozici ve všech úředních jazycích EU/EHP.
Jako součást Programu prevence početí pro přípravek Erivedge, všichni pacienti obdrží:
• Brožuru pro pacienta
• Kartu pro pacienta
V těchto dokumentech naleznete také další informace.
38