Edurant 25 Mg
SOUHRN ÚDAJŮ O PŘÍPRAVKU
PŘÍLOHA I
EDURANT 25 mg potahované tablety
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ"
Jedna tableta obsahuje rilpivirini hydrochloridum ekvivalentní rilpivirinum 25 mg.
Pomocné látky se známým účinkem: Jedna potahovaná tableta obsahuje 56 mg monohydrátu laktosy. Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Potahovaná tableta
Bílá až téměř bílá kulatá bikonvexní potahovaná tableta o průměru 6,4 mm s vyraženým „TMC“ na jedné straně a „25“ na druhé straně.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
EDURANT je indikován v kombinaci s jinými antiretrovirovými léčivými přípravky k léčbě infekce virem lidské imunodeficience typu 1 (HIV-1) u pacientů ve věku 12 let a starších dosud neléčených antiretrovirovými přípravky s virovou náloží < 100 000 HIV-1 RNA kopií/ml.
Při užívání přípravku EDURANT je nutno se řídit testováním genotypové rezistence (viz body 4.4 a
5.1).
4.2 Dávkování a způsob podání
Léčba musí být zahájena lékařem, který má zkušenosti s léčbou HIV infekcí.
Dávkování
Doporučená dávka přípravku EDURANT je jedna 25mg tableta jednou denně. EDURANT se musí užívat spolu s jídlem (viz bod 5.2).
Úprava dávkování
U pacientů užívajících současně rifabutin má být dávka přípravku EDURANT zvýšena na 50 mg (dvě tablety po 25 mg) jednou denně. Pokud je současné podávání rifabutinu ukončeno, dávka přípravku EDURANT má být snížena na 25 mg jednou denně (viz bod 4.5.).
Vynechaná dávka
Jestliže pacient opomine užít dávku přípravku EDURANT a vzpomene si na to do 12 hodin od obvyklého užití, musí přípravek užít co nejdříve spolu s jídlem a dále pokračovat podle normálního dávkovacího schématu. Jestliže pacient zapomene užít dávku přípravku EDURANT a vzpomene si na to po více než 12 hodinách od obvyklého užití, vynechanou dávku neužije, ale pokračuje podle obvyklého dávkovacího režimu.
Zvrací-li pacient během 4 hodin po užití tohoto přípravku, užije se další tableta přípravku EDURANT spolu s jídlem. Zvrací-li pacient po více než 4 hodinách od užití tohoto přípravku, není třeba užít náhradní dávku přípravku EDURANT až do příští pravidelně naplánované dávky.
Zvláštní populace
Starší pacienti
Informace týkající se užití přípravku EDURANT u pacientů ve věku > 65 let jsou omezené. U starších pacientů není nutná úprava dávky přípravku EDURANT (viz bod 5.2). EDURANT musí tato populace užívat s opatrností.
Porucha funkce ledvin
EDURANT byl studován zejména u pacientů s normální funkcí ledvin. U pacientů s lehkou nebo středně těžkou poruchou funkce ledvin není nutná úprava dávky. U pacientů s těžkou poruchou funkce ledvin nebo v terminálním stadiu onemocnění ledvin je nutno rilpivirin užívat s opatrností. U pacientů s těžkou poruchou funkce ledvin nebo v terminálním stadiu onemocnění ledvin lze kombinaci rilpivirinu se silným inhibitorem CYP3A (např. inhibitorem HIV proteázy potencovaným ritonavirem) užít pouze, pokud přínos převýší riziko (viz bod 5.2).
Léčba rilpivirinem vedla k předčasnému malému zvýšení hladin kreatininu v séru; tento nárůst se během času neměnil a nepovažuje se za klinicky významný (viz bod 4.8).
Porucha funkce jater
O užívání přípravku EDURANT u pacientů s lehkou nebo středně těžkou poruchou funkce jater (Child-Pugh třída A nebo B) je omezené množství informací. U pacientů s lehkou nebo středně těžkou poruchou funkce jater není nutná úprava dávky. U pacientů se středně těžkou poruchou funkce jater je nutno užívat EDURANT s opatrností. U pacientů s těžkou poruchou funkce jater (Child-Pugh třída C) nebyl EDURANT studován. Z tohoto důvodu se EDURANT u pacientů s těžkou poruchou funkce jater nedoporučuje (viz bod 5.2).
Pediatrická populace
Bezpečnost a účinnost přípravku EDURANT u dětí ve věku < 12 let nebyla dosud stanovena. Nejsou dostupné žádné údaje.
Způsob podání
EDURANT se užívá perorálně jednou denně spolu s jídlem (viz bod 5.2). Doporučuje se polykat potahované tablety celé s vodou, nekousat je, ani nedrtit.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
EDURANT se nesmí podávat spolu s dále uvedenými léčivými přípravky, protože se může objevit významný pokles koncentrací rilpivirinu v plazmě (vzhledem k indukci enzymů CYP3A nebo zvýšení pH v žaludku), což může vést ke ztrátě terapeutického účinku přípravku EDURANT (viz bod 4.5):
- antiepileptika karbamazepin, oxkarbazepin, fenobarbital, fenytoin;
- antituberkulotika rifampicin, rifapentin;
- inhibitory protonové pumpy jako omerazol, esomeprazol, lansoprazol, pantoprazol, rabeprazol;
- systémově podávaný glukokortikoid dexamethason, s výjimkou léčby jednou dávkou;
- třezalka tečkovaná (Hypericum perforatum).
4.4 Zvláštní upozornění a opatření pro použití
Přestože se prokázalo, že efektivní virová suprese antiretrovirovou léčbou významně snižuje riziko sexuálního přenosu, nelze vyloučit reziduální riziko. Je nutno dodržet opatření k zabránění přenosu v souladu s národními doporučeními.
Virologické selhání a vývoj rezistence
EDURANT nebyl hodnocen u pacientů s předchozím virologickým selháním při jiné antiretrovirové léčbě. Seznam mutací spojených s rezistencí na rilpivirin uvedený v bodu 5.1 je vodítkem pouze pro užití přípravku EDURANT u dosud neléčené populace.
Podle souhrnné analýzy účinnosti z hodnocení fáze III u dospělých po dobu 96 týdnů léčby bylo u pacientů léčených rilpivirinem s počáteční virovou náloží > 100 000 HIV-1 RNA kopií/ml větší riziko virologického selhání (18,2 % u rilpivirinu proti 7,9 % u efavirenzu) ve srovnání s pacienty s počáteční virovou náloží < 100 000 HIV-1 RNA kopií/ml (5,7 % u rilpivirinu proti 3,6 % u efavirenzu). Vyšší riziko virologického selhání bylo pozorováno ve větvi s rilpivirinem v prvních 48 týdnech těchto hodnocení (viz bod 5.1). U pacientů s počáteční virovou náloží > 100 000 HIV-1 RNA kopií/ml, u kterých došlo k virovému selhání, se objevil vyšší podíl rezistence ke třídě nenukleosidových inhibitorů reverzní transkriptázy (NNRTI) vyvolané léčbou. Rezistence spojená s lamivudinem/emtricitabinem se vyvinula více u pacientů, u kterých došlo k virologickému selhání na rilpivirinu, než u pacientů, u kterých došlo k virologickému selhání při léčbě efavirenzem (viz bod
5.1).
Nálezy u dospívajících (ve věku od 12 let do < 18 let) ve studii C213 byly celkově v souladu s těmito daty (viz bod_5.1).
Léčeni rilpivirinem mají být pouze dospívající s pravděpodobně dobrou adherencí k antiretrovirové léčbě, protože suboptimální adherence může vést k rozvoji rezistence a ke ztrátě budoucích možností léčby.
Podobně jako u jiných antiretrovirových léčivých přípravků je nutno se při užívání rilpivirinu řídit testováním rezistence (viz bod 5.1).
Kardiovaskulární systém
V supraterapeutických dávkách (75 a 300 mg jednou denně) byl rilpivirin spojen s prodloužením QTc intervalu na elektrokardiogramu (EKG) (viz body 4.5, 4.8 a 5.2). EDURANT v doporučené dávce 25 mg jednou denně není spojen s klinicky významným účinkem na QTc. EDURANT je nutno užívat s opatrností při současném podávání s léčivými přípravky se známým rizikem torsade de pointes.
Syndrom imunitní reaktivace
U pacientů infikovaných HIV s těžkou imunodeficiencí se může při zahájení CART vyskytnout zánětlivá reakce vyvolaná asymptomatickými nebo reziduálními oportunními patogeny, která může být příčinou závažných klinických stavů nebo zhoršení symptomů. Reakce tohoto typu byly obvykle pozorovány během prvních týdnů nebo měsíců po zahájení CART. Relevantními příklady jsou cytomegalovirová retinitida, generalizované a/nebo fokální mykobakteriální infekce a pneumonie vyvolaná bakterií Pneumocystis jiroveci. Jakékoli příznaky zánětu mají být vyhodnoceny, a pokud je to nutné, zahájena příslušná léčba.
Při imunitní reaktivaci byl také hlášen výskyt autoimunitních onemocnění (jako je Gravesova choroba), avšak hlášená doba do jejich nástupu byla velmi různá. Tyto stavy se mohou objevit mnoho měsíců po zahájení léčby (viz bod 4.8).
Důležité informace o některých složkách přípravku EDURANT
EDURANT obsahuje laktózu. Pacienti se vzácnými dědičnými problémy s intolerancí galaktózy, vrozeným nedostatkem laktázy nebo malabsorpcí glukózy a galaktózy tento léčivý přípravek nemají užívat.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Léčivé přípravky, které ovlivňují expozici rilpivirinu
Rilpivirin je primárně metabolizován cytochromem P450 (CYP)3A. Léčivé přípravky, které indukují nebo inhibují CYP3A, mohou proto ovlivňovat clearance rilpivirinu (viz bod 5.2). Při současném podávání rilpivirinu a léčivých přípravků, které indukují CYP3A, bylo pozorováno snížení koncentrace rilpivirinu v plazmě, což může snížit terapeutický účinek rilpivirinu.
Při současném podávání rilpivirinu a léčivých přípravků, které inhibují CYP3A, bylo pozorováno zvýšení koncentrace rilpivirinu v plazmě.
Současné podávání rilpivirinu a léčivých přípravků, které zvyšují pH v žaludku, může vést ke snížení koncentrací rilpivirinu v plazmě, což může případně snížit terapeutický účinek přípravku EDURANT.
Léčivé přípravky, které jsou ovlivněny užitím rilpivirinu
Nepředpokládá se, že by rilpivirin v dávce 25 mg jednou denně měl klinicky významný vliv na expozici léčivým přípravkům metabolizovaným enzymy CYP.
Rilpivirin inhibuje in vitro P-glykoprotein (IC50 je 9,2 pmol). V klinické studii neovlivňoval rilpivirin významně farmakokinetiku digoxinu. Nelze však úplně vyloučit, že rilpivirin může zvýšit expozici jiným léčivým přípravkům transportovaným P-glykoproteinem, které jsou citlivější k inhibici P-gp ve střevě, např. dabigatran-etexilátu.
Rilpivirin je in vitro inhibitor transportního systému MATE-2K s IC50 < 2,7 nM. Klinický význam tohoto poznatku není v současné době znám.
Známé a teoretické interakce s vybranými antiretrovirovými a neantiretrovirovými léčivými přípravky jsou uvedeny v tabulce 1.
Tabulka interakcí
Studie interakcí byly provedeny jen u dospělých.
Interakce mezi rilpivirinem a současně podávanými přípravky jsou uvedeny v tabulce 1 (zvýšení je označeno jako „j“, snížení „j“, beze změny ,,^“, neaplikovatelné „NA“, interval spolehlivosti jako „CI“).
|
Tabulka 1: INTERAKCE A DOPORUČENÁ DÁVKA S DALŠÍMI LÉČIVÝMI PŘÍPRAVKY | ||
|
Léčivé přípravky podle terapeutických oblastí |
Interakce Průměrná geometrická změna (%) |
Doporučení pro současné podávání |
|
ANTIINFEKTIVA | ||
|
Antiretrovirové látky | ||
|
HIV NRTI/Nft]RTI | ||
|
Didanosin*# 400 mg jednou denně |
AUC didanosinu j 12 % Cmin didanosinu NA Cmax didanosinu ^ AUC rilpivirinu ^ Cmin rilpivirinu ^ Cmax rilpivirinu ^ |
Není nutná úprava dávkování. Didanosin je nutno podávat alespoň dvě hodiny před nebo alespoň čtyři hodiny po podání rilpivirinu. |
|
T enofovir-disoproxyl-fumarát*# 300 mg jednou denně |
AUC tenofoviru j 23 % Cmin tenofoviru j 24 % Cmax tenofoviruj 19 % AUC rilpivirinu ^ Cmin rilpivirinu ^ Cmax rilpivirinu ^ |
Není nutná úprava dávkování. |
|
Další NRTI (abakavir, emtricitabin, lamivudin, stavudin a zidovudin) |
Nebylo studováno. Neočekávají se klinicky relevantní lékové interakce. |
Není nutná úprava dávkování. |
|
HIV NNRTI | ||
|
NNRTI (delavirdin, efavirenz, etravirin, nevirapin) |
Nebylo studováno. |
Současné podávání rilpivirinu s dalšími NNRTI se nedoporučuje. |
|
HIV PI - se současným podáním nízké dávky ritonaviru | ||
|
Darunavir/ritonavir*# 800/100 mg jednou denně |
AUC darunaviru ^ Cmm darunaviru j 11 % Cmax darunaviru ^ AUC rilpivirinu t 130 % Cmm rilpivirinu t 178 % Cmax rilpivirinu t 79 % |
Současné užívání rilpivirinu s PI potencovanými ritonavirem způsobuje zvýšení koncentrace rilpivirinu v plazmě, ale není nutná úprava dávkování. |
|
(inhibice enzymů CYP3A) | ||
|
Lopinavir/ritonavir (měkké želatinové tobolky)*# 400/100 mg dvakrát denně |
AUC lopinaviru ^ Cmin lopinaviru j 11 % Cmax lopinaviru ^ AUC rilpivirinu t 52 % Cmin rilpivirinu t 74 % Cmax rilpivirinu t 29 % | |
|
(inhibice enzymů CYP3A) | ||
|
Další potencované PI (atazanavir/ritonavir, fosamprenavir/ritonavir, sachinavir/ritonavir, tipranavir/ritonavir) |
Nebylo studováno. | |
|
HIVPI - bez současného podání nízké dávky ritonaviru | ||
|
Nepotencované PI (atazanavir, fosamprenavir, indinavir, nelfinavir) |
Nebylo studováno. Očekává se vyšší expozice rilpivirinu. (inhibice enzymů CYP3A) |
Není nutná úprava dávkování. |
|
Antagonisté CCR5 | ||
|
Maravirok |
Nebylo studováno. Neočekávají se klinicky relevantní lékové interakce. |
Není nutná úprava dávkování. |
|
HIV inhibitory integrázy | ||
|
Raltegravir* |
AUC raltegraviru t 9 % Cmin raltegraviru t 27 % Cmax raltegraviru t 10 % AUC rilpivirinu ^ Cmin rilpivirinu ^ Cmax rilpivirinu ^ |
Není nutná úprava dávkování. |
|
Další antiretrovirové látky | ||
|
Ribavirin |
Nebylo studováno. Neočekávají se klinicky relevantní lékové interakce. |
Není nutná úprava dávkování. |
|
Telaprevir* 750 mg každých 8 hodin |
AUC telapreviru j 5 % Cmin telapreviru j 11 % Cmax telapreviru j 3 % AUC rilpivirinu t 78 % Cmin rilpivirinu t 93 % Cmax rilpivirinu t 49 % |
Není nutná úprava dávkování. |
|
DALŠÍ LÁTKY | ||
|
ANTIEPILEPTIKA | ||
|
Karbamazepin Oxkarbazepin Fenobarbital Fenytoin |
Nebylo studováno. Očekávají se významné poklesy koncentrací rilpivirinu v plazmě. (indukce enzymů CYP3A) |
Rilpivirin se nesmí užívat v kombinaci s těmito antiepileptiky, protože současné podání může vést ke ztrátě terapeutického účinku rilpivirinu (viz bod 4.3). |
|
AZOLOVÁ ANTIMYKOTIKA | ||
|
Ketokonazol*# 400 mg jednou denně |
AUC ketokonazolu j 24 % Cmin ketokonazolu j 66 % Cmax ketokonazolu ^ (indukce CYP3A vzhledem k vysoké dávce rilpivirinu v tomto hodnocení) AUC rilpivirinu t 49 % Cmin rilpivirinu t 76 % Cmax rilpivirinu t 30 % (inhibice enzymů CYP3A) |
Při doporučené dávce 25 mg jednou denně není při současném podávání rilpivirinu s ketokonazolem nutná úprava dávkování. |
|
Flukonazol Itrakonazol Posakonazol Vorikonazol |
Nebylo studováno. Současné užívání přípravku EDURANT s azolovými antimykotiky může způsobit zvýšení koncentrací rilipivirinu v plazmě. (inhibice enzymů CYP3A) |
Není nutná úprava dávkování. |
|
TUBERKULOSTATIKA | ||
|
Rifabutin* 300 mg jednou denněŤ 300 mg jednou denně (+25 mg rilpivirinu jednou denně) 300 mg jednou denně (+50 mg rilpivirinu jednou denně) |
AUC rifabutinu ^ Cmin rifabutinu ^ Cmax rifabutinu ^ AUC 25-O-desacetyl-rifabutinu ^ Cmin 25-O-desacetyl-rifabutinu ^ Cmax 25-O-desacetyl-rifabutinu ^ AUC rilpivirinu j 42 % Cmin rilpivirinu j 48 % Cmax rilpivirinu j 31 % AUC rilpivirinu t 16 %* Cmin rilpivirinu ^* Cmax rilpivirinu t 43 %* * ve srovnání s 25 mg rilpivirinu podávaného samostatně jednou denně (indukce enzymů CYP3A) |
Během současného podávání rilpivirinu s rifabutinem má být dávka rilpivirinu zvýšena z 25 mg jednou denně na 50 mg jednou denně. Pokud je současné podávání rifabutinu ukončeno, dávka rilpivirinu má být snížena na 25 mg jednou denně. |
|
Rifampicin*# 600 mg jednou denně |
AUC rifampicinu ^ Cmin rifampicinu NA Cmax rifampicinu ^ AUC 25-desacetyl-rifampicinu j 9 % Cmin 25-desacetyl-rifampicinu NA Cmax 25-desacetyl-rifampicinu ^ AUC rilpivirinu j 80 % Cmin rilpivirinu j 89 % Cmax rilpivirinu j 69 % (indukce enzymů CYP3A) |
Rilpivirin se nesmí užívat v kombinaci s rifampicinem, protože současné podávání pravděpodobně povede ke ztrátě terapeutického účinku rilpivirinu (viz bod 4.3). |
|
Rifapentin |
Nebylo studováno. Očekávají se významné poklesy koncentrací rilpivirinu v plazmě. (indukce enzymů CYP3A) |
Rilpivirin se nesmí užívat v kombinaci s rifapentinem, protože současné podávání pravděpodobně povede ke ztrátě terapeutického účinku rilpivirinu (viz bod 4.3). |
|
MAKROLIDOVÁ ANTIBIOTIKA | ||
|
Klarithromycin Erythromycin |
Nebylo studováno. Očekává se zvýšená expozice rilpivirinu. (inhibice enzymů CYP3A) |
Je-li to možné, je třeba zvážit jinou variantu, jako azithromycin. |
|
GLUKOKORTIKOIDY | ||
|
Dexamethason ( systémově podávaný, s výjimkou užití jednorázové dávky) |
Nebylo studováno. Očekávají se na dávce závislé poklesy koncentrací rilpivirinu v plazmě. (indukce enzymů CYP3A) |
Rilpivirin se nesmí užívat v kombinaci se systémově podávaným dexamethasonem (s výjimkou jednorázové dávky), protože současné podávání může vést ke ztrátě terapeutického účinku rilpivirinu (viz bod 4.3). Zejména pro dlouhodobé použití je nutno uvažovat o jiných variantách. |
|
INHIBITORY PROTONOVÉ PUMPY | ||
|
Omeprazol*# 20 mg jednou denně |
AUC omeprazolu j 14 % Cmin omeprazolu NA Cmax omeprazolu j 14 % AUC rilpivirinu j 40 % Cmin rilpivirinu j 33 % Cmax rilpivirinu j 40 % (snížená absorpce vzhledem ke zvýšení pH v žaludku) |
Rilpivirin se nesmí užívat v kombinaci s inhibitory protonové pumpy, protože současné podávání pravděpodobně povede ke ztrátě terapeutického účinku rilpivirinu (viz bod 4.3). |
|
Lansoprazol Rabeprazol Pantoprazol Esomeprazol |
Nebylo studováno. Očekávají se významné poklesy koncentrací rilpivirinu v plazmě. (snížená absorpce vzhledem ke zvýšení pH v žaludku) | |
|
ANTAGONISTÉ H2-RECEPTORŮ | ||
|
Famotidin*# 40 mg jednorázová dávka užitá 12 hodin před rilpivirinem |
AUC rilpivirinu j 9 % Cmin rilpivirinu NA Cmax rilpivirinu ^ |
Kombinaci rilpivirinu a antagonisty H2-receptorů je nutno užívat se zvláštní opatrností. Je možné užít pouze antagonisty H2-receptorů, které lze dávkovat jednou denně. Je nutné užít přísné dávkovací schema s užitím antagonisty H2-receptorů alespoň 12 hodin před užitím nebo alespoň 4 hodin po užití rilpivirinu. |
|
Famotidin*# 40 mg jednorázová dávka užitá 2 hodiny před rilpivirinem |
AUC rilpivirinu j 76 % Cmin rilpivirinu NA Cmax rilpivirinu j 85 % (snížená absorpce vzhledem ke zvýšení pH v žaludku) | |
|
Famotidin*# 40 mg jednorázová dávka užitá 4 hodiny po rilpivirinu |
AUC rilpivirinu t 13 % Cmin rilpivirinu NA Cmax rilpivirinu t 21 % | |
|
Cimetidin Nizatidin Ranitidin |
Nebylo studováno. (snížená absorpce vzhledem ke zvýšení pH v žaludku) | |
|
ANTACIDA | ||
|
Antacida (např. hydroxid hlinitý nebo hořečnatý, uhličitan vápenatý) |
Nebylo studováno. Očekávají se významné poklesy koncentrací rilpivirinu v plazmě. (snížená absorpce vzhledem ke zvýšení pH v žaludku) |
Kombinaci rilpivirinu a antacida je nutno užívat se zvláštní opatrností. Antacida lze podávat minimálně 2 hodiny před nebo alespoň 4 hodiny po rilpivirinu. |
|
NARKOTICKÁ ANALGETIKA | ||
|
Methadon* 60 - 100 mgjednou denně, individualizovaná dávka |
AUC R(-) methadonu j 16 % Cmin R(-) methadonu j 22 % Cmax R(-) methadonu j 14 % AUC rilpivirinu ^* Cmin rilpivirinu ^* Cmax rilpivirinu ^* * na základě historických kontrol |
Při zahájení současného podávání methadonu s rilpivirinem není nutná úprava dávkování. Doporučuje se však klinické monitorování, protože udržovací léčbu methadonem je u některých pacientů nutno upravovat. |
|
ANTIARYTMIKA | ||
|
Digoxin* |
AUC digoxinu ^ Cmin digoxinu NA Cmax digoxinu ~ |
Není nutná úprava dávkování. |
|
ANTIKOAGULAN CIA | ||
|
Dabigatran-etexilát |
Nebylo studováno. Riziko zvýšení koncentrací dabigatranu v plazmě nelze vyloučit. (inhibice P-gp ve střevě) |
Kombinaci rilpivirinu a dabigatran-etexilátu je nutno užívat s opatrností. |
|
ANTIDIABETIKA | ||
|
Metformin* 850 mg jednorázová dávka |
AUC metforminu ^ Cmin metforminu NA Cmax metforminu ^ |
Není nutná úprava dávkování. |
|
ROSTLINNÉ PŘÍPRAVKY | ||
|
Třezalka tečkovaná (Hypericum perforatum) |
Nebylo studováno. Očekávají se významné poklesy koncentrací rilpivirinu v plazmě. (indukce enzymů CYP3A) |
Rilpivirin se nesmí užívat současně s přípravky s obsahem třezalky tečkované, protože současné podávání může vést ke ztrátě terapeutického účinku rilpivirinu (viz bod 4.3). |
|
ANALGETIKA | ||
|
Paracetamol*# 500 mg jednorázová dávka |
AUC paracetamolu ^ Cmin paracetamolu NA Cmax paracetamolu ^ AUC rilpivirinu ^ Cmin rilpivirinu t 26 % Cmax rilpivirinu ^ |
Není nutná úprava dávkování. |
|
PERORÁLNÍ ANTIKONCEPCE | ||
|
Ethinylestradiol* 0,035 mg jednou denně Norethisteron* 1 mg jednou denně |
AUC ethinylestradiolu ^ Cmin ethinylestradiolu ^ Cmax ethinylestradiolu t 17 % AUC norethisteronu ^ Cmin norethisteronu ^ Cmax norethisteronu ^ AUC rilpivirinu ^* Cmin rilpivirinu ^* Cmax rilpivirinu ^* * na základě historických kontrol |
Není nutná úprava dávkování. |
|
INHIBITORY HMG CO-A REDUKTÁZY | ||
|
Atorvastatin*# 40 mg jednou denně |
AUC atorvastatinu ^ Cmin atorvastatinu j 15 % Cmax atorvastatinu t 35 % AUC rilpivirinu ^ Cmin rilpivirinu ^ Cmax rilpivirinu j 9 % |
Není nutná úprava dávkování. |
|
INHIBITORY FOSFODIESTERÁZY TYPU 5 (PDE-5) | ||
|
Sildenafil*# 50 mg jednorázová dávka |
AUC sildenafilu ^ Cmin sildenafilu NA Cmax sildenafilu ^ AUC rilpivirinu ^ Cmin rilpivirinu ^ Cmax rilpivirinu ^ |
Není nutná úprava dávkování. |
|
Vardenafil Tadalafil |
Nebylo studováno. |
Není nutná úprava dávkování. |
T
#
Interakce mezi rilpivirinem a léčivým přípravkem byly vyhodnoceny v rámci klinického hodnocení. Ostatní zmíněné lékové interakce se předpokládají.
Toto hodnocení interakcí bylo provedeno s vyšší než doporučenou dávkou rilpivirinu, aby se posoudil maximální vliv na současně podávaný léčivý přípravek. Doporučené dávkování je aplikovatelné na doporučenou dávku rilpivirinu 25 mg jednou denně. tato studie interakcí byla provedena s dávkou vyšší než je doporučená dávka rilpivirinu
t
Léčivé přípravky prodlužující QT
O farmakodynamické interakci mezi rilpivirinem a léčivými přípravky, které prodlužují QTc interval na EKG, je dostupné omezené množství informací. V hodnocení se zdravými dobrovolníky se ukázalo, že supraterapeutické dávky rilpivirinu (75 mg jednou denně a 300 mg jednou denně) prodlužují QTc interval na EKG (viz bod 5.2). EDURANT je nutno užívat s opatrností při současném podávání s přípravky s rizikem torsade de pointes.
4.6 Fertilita, těhotenství a kojení
O užívání rilpivirinu u těhotných žen nejsou k dispozici žádné nebo jen omezené údaje (méně než 300 výsledků týkajících se těhotenství).
Studie na zvířatech neukazují přímé či nepřímé škodlivé účinky ve vztahu k reprodukční toxicitě (viz bod 5.3)
Jako preventivní opatření je vhodné se užívání přípravku EDURANT během těhotenství vyhnout. Kojení
Není známo, zda se rilpivirin vylučuje do mateřského mléka. Rilpivirin se vylučuje do mléka potkanů. Z důvodů jak možného přenosu HIV, tak i možných nežádoucích účinků u kojenců, mají být matky poučeny, že nemají kojit, pokud užívají rilpivirin.
Fertilita
U lidí nejsou dostupné žádné údaje o účincích rilpivirinu na fertilitu. Ve studiích na zvířatech nebyly pozorovány klinicky významné účinky na fertilitu (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
EDURANT nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje. Nicméně u některých pacientů užívajících EDURANT byly pozorovány únava, závrať a somnolence, což je nutno vzít v úvahu při posuzování schopnosti pacienta řídit a obsluhovat stroje.
4.8 Nežádoucí účinky
Souhrn profilu bezpečnosti
Během programu klinického vývoje (1 368 pacientů z kontrolovaných studií fáze III TMC278-C209 (ECHO) a TMC278-C215 (THRIVE)) zaznamenalo 55,7 % subjektů nejméně jeden nežádoucí účinek (viz bod 5.1). Nejčastěji hlášenými nežádoucími účinky (NÚ) (> 2 %), které byly nejméně mírné závažnosti, byly deprese (4,1 %), bolesti hlavy (3,5 %), nespavost (3,5 %), vyrážka (2,3 %) a bolesti břicha (2,0 %). Nejčastější závažné NÚ související s léčbou byly hlášeny u 7 (1,0 %) pacientů léčených rilpivirinem. Medián doby expozice u pacientů v rameni s rilpivirinem byl 104,3 týdne a v rameni s efavirenzem 104,1 týdne. Většina nežádoucích účinků se vyskytla během prvních 48 týdnů léčby.
Vybrané klinické laboratorní abnormality (stupně 3 nebo 4), které se objevily u pacientů během léčby přípravkem EDURANT považované za NÚ, byly zvýšení pankreatické amylázy (3,8 %), zvýšení AST (2,3 %), zvýšení ALT (1,6 %), zvýšení LDL cholesterolu (nalačno, 1,5 %), snížení počtu leukocytů (1,2 %), zvýšení lipázy (0,9 %), zvýšení bilirubinu (0,7 %), zvýšení triacylglycerolů (nalačno, 0,6 %), snížení hemoglobinu (0,1 %), snížení počtu trombocytů (0,1 %) a zvýšení celkového cholesterolu (nalačno, 0,1 %).
Přehledný souhrn nežádoucích účinků
Nežádoucí účinky hlášené u dospělých pacientů léčených rilpivirinem jsou uvedeny v Tabulce 2. Nežádoucí účinky jsou uvedeny podle tříd orgánových systémů a četnosti. Četnosti výskytu jsou definovány jako velmi časté (> 1/10), časté (> 1/100 a < 1/10) a méně časté (> 1/1 000 a < 1/100).
V každé skupině četnosti jsou nežádoucí účinky uvedeny s klesající četností.
Tabulka 2: Nežádoucí účinky hlášené u antiretrovirové léčby rilpivirinem dosud neléčených
dospělých pacientů infikovaných HIV-1
(souhrnné údaje z hodnocení po 96 týdnech fáze III studie ECHO a THRIVE) N = 686
|
Třídy orgánových systémů |
Kategorie četnosti |
Nežádoucí účinky (Rilpivirin + BR) |
|
Poruchy krve a lymfatického systému |
časté |
snížení počtu leukocytů snížení hemoglobinu snížení počtu trombocytů |
|
Poruchy imunitního systému |
méně časté |
syndrom imunitní reaktivace |
|
Poruchy metabolismu a výživy |
velmi časté |
zvýšení celkového cholesterolu (nalačno) zvýšení LDL cholesterol (nalačno) |
|
časté |
snížení chuti k jídlu zvýšení triacylglycerolů (nalačno) | |
|
Psychiatrické poruchy |
velmi časté | |
|
časté |
abnormální sny deprese poruchy spánku depresivní nálada | |
|
Poruchy nervového systému |
velmi časté |
bolest hlavy závratě |
|
časté |
somnolence | |
|
Gastrointestinální poruchy |
velmi časté |
zvýšení pankreatické amylázy |
|
časté |
bolest břicha zvracení zvýšení lipázy diskomfort v oblasti břicha sucho v ústech | |
|
Poruchy jater a žlučových cest |
velmi časté |
zvýšení aminotransferáz |
|
časté |
zvýšení bilirubinu | |
|
Poruchy kůže a podkožní tkáně |
časté | |
|
Celkové poruchy a reakce v místě aplikace |
časté |
Laboratorní abnormality
Ve skupině s rilpivirinem byla podle analýzy hodnocení fáze III ECHO a THRIVE po 96 týdnech střední změna oproti výchozí hodnotě u celkového cholesterolu (nalačno) 5 mg/dl, u HDL cholesterolu (nalačno) 4 mg/dl, u LDL cholesterolu (nalačno) 1 mg/dl a u trigliceridů (nalačno) -7 mg/dl.
Popis vybraných nežádoucích účinků
Syndrom imunitní reaktivace
Při zahájení kombinované antiretrovirové léčby (CART) u HIV infikovaných pacientů s těžkou imunodeficiencí se může objevit zánětlivá reakce na asymptomatické nebo reziduální oportunní infekce. Byl také hlášen výskyt autoimunitních onemocnění (jako je Gravesova choroba), avšak hlášená doba do jejich nástupu byla velmi různá. Tyto stavy se mohou objevit mnoho měsíců po zahájení léčby (viz bod 4.4).
Pediatrická populace (ve věku od 12 let do < 18 let)
Posouzení bezpečnosti je založeno na analýze 48týdenní otevřené jednoramenné studie fáze_II, TMC278-C213, ve které 36 dosud neléčených dospívajících pacientů s tělesnou hmotností nejméně 32 kg infikovaných virem HIV-1 dostávalo rilpivirin (25 mg jednou denně) v kombinaci s jinými antiretrovirovými látkami (viz bod_5.1). Medián doby expozice u pacientů byl 63,5 týdne. Žádný pacient neukončil léčbu z důvodu nežádoucích účinků. Nebyly pozorovány žádné nové nežádoucí účinky v porovnání s těmi, které se vyskytly u dospělých.
Většina nežádoucích účinků byla stupně_ 1 nebo 2. Nejčastější nežádoucí účinky (všechny stupně,
> 10 %) byly bolest hlavy (19,4 %), deprese (19,4 %), somnolence (13,9 %) a nauzea (11,1 %). Nebyly hlášeny odchylky laboratorních testů pro AST/ALT stupně 3 - 4 nebo nežádoucí účinky zvýšení aminotransferáz stupně 3 - 4.
Bezpečnost a účinnost rilpivirinu u dětí ve věku < 12 let nebyly dosud stanoveny. K dispozici nejsou žádné údaje.
Další zvláštní populace
Pacienti se souběžnou infekcí virem hepatitidy B a/nebo hepatitidy C
U pacientů nakažených také virem hepatitidy B nebo C léčených rilpivirinem byla incidence zvýšení jatemích enzymů vyšší než u pacientů léčených rilpivirinem, kteří neměli souběžnou nákazu. Stejný nález byl také ve skupině s efavirenzem. Farmakokinetická expozice rilpivirinu u souběžně nakažených pacientů byla srovnatelná s expozicí bez souběžné nákazy.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích učinků uvedeného v Dodatku V.
4.9 Předávkování
Na léčbu předávkování přípravkem EDURANT není specifické antidotum. Zkušenost s předávkováním rilpivirinem u lidí je omezená. Symptomy předávkování mohou zahrnovat bolesti hlavy, nauzeu, závrať a/nebo abnormální sny. Léčba předávkování rilpivirinem spočívá v obecných podpůrných opatřeních, včetně monitorování vitálních funkcí a EKG (interval QT) a sledování klinického stavu pacienta. K odstranění neabsorbované léčivé látky je možné podat aktivní uhlí. Vzhledem k tomu, že se rilpivirin silně váže na bílkoviny plazmy, je nepravděpodobné, že by dialýza vedla k významnému odstranění léčivé látky.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antivirotikum pro systémovou aplikaci, nenukleosidové inhibitory reverzní transkriptázy), ATC kód: J05AG05
Mechanismus účinku
Rilpivirin je diarylpyrimidin NNRTI viru HIV-1. Účinnost rilpivirinu je způsobena nekompetitivní inhibicí HIV-1 reverzní transkriptázy (RT). Rilpivirin neinhibuje lidské celulární DNA polymerázy a,
P a Y.
Antivirová aktivita in vitro
Rilpivirin vykazuje účinnost proti laboratorním kmenům divokého typu viru HIV-1 v akutně infikovaných T-buněčných liniích s mediánem EC50 pro HIV-1/IIIB 0,73 nM (0,27 ng/ml). Ačkoli se u rilpivirinu in vitro prokázala omezená účinnost proti HIV-2 s hodnotou EC50 v rozmezí 2 510 až 10 830 nM (920 až 3 970 ng/ml), léčba infekce HIV-2 rilpivirinem se nedoporučuje, protože neexistují klinické údaje.
U rilpivirinu se prokázala také antivirová účinnost proti širokému spektru HIV-1 primárních izolátů skupiny M (subtypy A, B, C, D, F, G, H) s EC50 v rozmezí 0,07 až 1,01 nM (0,03 až 0,37 ng/ml) a primárních izolátů skupiny O s EC50 v rozmezí 2,88 až 8,45 nM (1,06 až 3,10 ng/ml).
Rezistence
Buněčné kultury
Kmeny rezistentní k rilpivirinu byly vyselektovány v buněčných kulturách počínaje divokým typem HIV-1 různých původů a subtypů až po NNRTI rezistentní HIV-1. Nejčastěji pozorované mutace spojené s rezistencí, které se vyskytly, zahrnovaly L100I, K101E, V108I, E138K, V179F, Y181C, H221Y, F227C a M230I.
Rezistence rilpivirinu byla stanovena jako násobná změna v hodnotě EC50 (FC) nad biologickou limitní hodnotou (rozhraní)(BCO) vzorku.
Dosud neléčení dospělí pacienti
Pro analýzu rezistence byla užita širší definice virologického selhání než pro primární analýzu účinnosti. Podle souhrnné analýzy rezistence ve 48. týdnu z hodnocení fáze III mělo 62 (z celkových 72) virologických selhání ve skupině s rilpivirinem údaje o rezistenci na počátku a v době selhání.
V této analýze byly mutace spojené s rezistencí (RAM) k NNRTI, které se vyvinuly alespoň při
2 virologických selháních rilpivirinu: V90I, K101E, E138K, E138Q, V179I, Y181C, V189I, H221Y a F227C. Ve studiích přítomnost mutací V90I a V189I na počátku neovlivňovala odpověď. Substituce E138K se objevila nejčastěji během léčby rilpivirinem obyčejně v kombinaci se substitucí M184I.
V analýze ve 48. týdnu mělo 31 ze 62 virologických selhání rilpivirinu současně RAM k NNRTI a NRTI; 17 z těchto 31 mělo kombinaci E138K a M184I. Nejčastější mutace byly totožné v analýzách ve 48. týdnu a v 96. týdnu.
Podle souhrnné analýzy rezistence v 96. týdnu byly pozorovány nižší podíly virologického selhání v druhých 48 týdnech než během prvních 48 týdnů léčby. Podle souhrnné analýzy od 48. do 96. týdne se vyskytlo 24 (3,5 %) dalších virologických selhání ve skupině s rilpivirinem a 14 (2,1 %) dalších virologických selhání ve skupině s efavirenzem. Z těchto virologických selhání 9/24, resp. 4/14 bylo u pacientů s počáteční virovou náloží < 100 000 kopií/ml.
Vezmou-li se v úvahu všechny dostupné in vitro a in vivo údaje u dosud neléčených pacientů, jsou mutace spojené s rezistencí přítomné na počátku, které pravděpodobně ovlivňují účinnost rilpivirinu: K101E, K101P, E138A, E138G, E138K, E138R, E138Q, V179L, Y181C, Y181I, Y181V, Y188L, H221Y, F227C, M230I a M230L. Tyto mutace spojené s rezistencí k rilpivirinu by měly ovlivnit pouze použití přípravku EDURANT u dosud neléčených pacientů. Tyto mutace spojené s rezistencí byly odvozeny z in vivo údajů pouze pro dosud neléčené pacienty, a proto je nelze použít pro předpověď účinnosti rilpivirinu u pacientů, u kterých režim obsahující antivirotika již dříve selhal.
Podobně jako u ostatních antivirotik se použití přípravku EDURANT má řídit testováním rezistence.
Zkřížená rezistence
NNRTI mutované viry vázané k místu
V souboru 67 HIV-1 rekombinantních laboratorních kmenů s jednou mutací spojenou s rezistencí na RT pozici spojenou s NNRTI rezistencí, včetně nejčastěji nalezených K103N a Y181C, vykázal rilpivirin účinnost proti 64 (96 %) těmto kmenům. Jediné mutace spojené s rezistencí spojené se ztrátou citlivosti k rilpivirinu byly: K101P, Y181I a Y181V. Substituce K103N sama o sobě nevedla ke snížení citlivosti k rilpivirinu, ale v kombinaci K103N a L100I vedla k 7krát snížené citlivosti
k rilpivirinu.
Rekombinantní klinické izoláty
Rilpivirin si uchoval citlivost (FC < BCO) k 62 % ze 4 786 HIV-1 rekombinantních klinických izolátů rezistentních k efavirenzu a/nebo nevirapinu.
Dosud neléčení dospělí pacienti infikovaní HIV-1
Podle souhrnné analýzy rezistence po 96. týdnu z hodnocení fáze III (ECHO a THRIVE) 42 z 86 subjektů s virologickým selháním k rilpivirinu vykázalo akutní rezistenci k rilpivirinu při léčbě (genotypová analýza). U těchto pacientů byla pozorována dále uvedená fenotypová zkřížená rezistence k dalším NNRTI: etravirin 32/42, efavirenz 30/42 a nevirapin 16/42. U pacientů s počáteční virovou náloží < 100 000 kopií/ml vykázalo 9 z 27 pacientů s virologickým selháním k rilpivirinu akutní rezistenci k rilpivirinu při léčbě (genotypová analýza) s následující četností fenotypové zkřížené rezistence: etravirin 4/9, efavirenz 3/9 a nevirapin 1/9.
Účinky na EKG
Vliv rilpivirinu v doporučené dávce 25 mg jednou denně na interval QTcF byl hodnocen v randomizované placebem a aktivně (moxifloxacin 400 mg jednou denně) kontrolované studii ve zkříženém uspořádání u 60 zdravých dobrovolníků s 13 měřeními v rovnovážném stavu během 24 hodin. EDURANT v doporučené dávce 25 mg jednou denně není spojen s klinicky významným vlivem na QTc.
Při hodnocení supraterapeutických dávek rilpivirinu 75 mg jednou denně a 300 mg jednou denně u zdravých dobrovolníků byly maximální průměrné rozdíly přizpůsobené času (95 % horní interval spolehlivosti) v QTcF oproti placebu, po korekci na výchozí stav, 10,7 (15,3) a 23,3 (28,4) ms. Podání rilpivirinu 75 mg jednou denně a 300 mg jednou denně v rovnovážném stavu vedlo k navýšení střední Cmax přibližně 2,6násobně, resp. 6,7násobně oproti průměrné Cmax pozorované s obvyklou dávkou rilpivirinu 25 mg jednou denně.
Klinická účinnost a bezpečnost
Dosud neléčení dospělí pacienti infikovaní HIV-1
Průkaz účinnosti rilpivirinu je založen na analýze 96týdenních údajů z 2 randomizovaných dvojitě slepých aktivně kontrolovaných studií fáze III TMC278-C209 (ECHO) a TMC278-C215 (THRIVE). Obě studie měly identická uspořádání s výjimkou základní léčby (BR). V analýze účinnosti v týdnu 96 byl hodnocen podíl virologických odpovědí [potvrzená nedetekovatelná virová nálož (< 50 HIV-1 RNA kopií/ml)] u pacientů dostávajících rilpivirin25 mg jednou denně navíc k BR oproti pacientům dostávajícím efavirenz 600 mg jednou denně navíc k BR. V obou studiích byla pozorována podobná účinnost rilpivirinu, což prokazuje noninferioritu vzhledem k efavirenzu.
Byli zahrnuti dosud neléčení pacienti infikovaní virem HIV-1, kteří měli hladinu HIV-1 RNA v plazmě > 5 000 kopií/ml a u nichž se při screeningu ukázala citlivost k N(t)RTI a nepřítomnost specifických mutací spojených s rezistencí k NNRTI. Ve studii ECHO byla BR spojena s N(t)RTI: tenofovir-disoproxyl-fumarát a emtricitabin. Ve studii THRIVE se BR skládala ze 2 investigátorem vybraných N(t)RTI: tenofovir-disoproxyl-fumarát a emtricitabin nebo zidovudin a lamivudin nebo abakavir a lamivudin. Ve studii ECHO byla randomizace provedena podle zjištěné virové nálože. Ve studii THRIVE byla randomizace provedena podle zjištěné virové nálože a podle N(t)RTI BR.
Tato analýza zahrnovala 690 pacientů ve studii ECHO a 678 pacientů ve studii THRIVE, kteří dokončili 96 týdnů léčby nebo ukončili léčbu dříve.
V souhrnné analýze studií ECHO a THRIVE byly demografické a výchozí charakteristiky vyrovnané mezi skupinou s rilpivirinem a skupinou s efavirenzem. Tabulka 3 ukazuje vybrané výchozí charakteristiky pacientů ve skupinách s rilpivirinem a efavirenzem.
|
Tabulka 3: Výchozí charakteristiky onemocnění u dosud neléčených dospělých pacientů nakažených HIV-1 ve studiích ECHO a THRIVE (souhrnná analýza) | ||
|
Souhrnné údaje z hodnocení ECHO a THRIVE | ||
|
Rilpivirin + BR |
Efavirenz + BR | |
|
N = 686 |
N = 682 | |
|
Výchozí charakteristiky onemocnění | ||
|
Medián výchozí HIV-1 RNA v plazmě |
5,0 |
5,0 |
|
(rozmezí), log10 kopií/ml |
(2 - 7) |
(3 - 7) |
|
Medián výchozího počtu CD4+ buněk |
249 |
260 |
|
(rozmezí), x 106 buněk/l |
(1 - 888) |
(1 - 1 137) |
|
Procento subjektů se: | ||
|
současnou infekcí virem hepatitidy B/C |
7,3 % |
9,5 % |
|
léčbou: | ||
|
tenofovir-disoproxyl-fumarát plus |
80,2 % |
80,1 % |
|
emtricitabin | ||
|
zidovudin plus lamivudin |
14,7 % |
15,1 % |
|
abakavir plus lamivudin |
5,1 % |
4,8 % |
BR = základní léčba
Tabulka 4 ukazuje výsledky analýzy účinnosti ve 48. týdnu a v 96. týdnu u pacientů léčených rilpivirinem a pacientů léčených efavirenzem podle souhrnných údajů ze studií ECHO a THRIVE. Podíl odpovědí (potvrzená nedetekovatelná virová nálož < 50 HIV-1 RNA kopií/ml) v 96. týdnu byl srovnatelný ve větvi s rilpivirinem a ve větvi s efavirenzem. Četnost virologického selhání v 96. týdnu byla však vyšší ve větvi s rilpivirinem než ve větvi s efavirenzem; většina virologických selhání se však vyskytla během prvních 48 týdnů léčby. Ukočení léčby kvůli nežádoucím účinkům bylo po 96 týdnech častější ve větvi s efavirenzem než ve větvi s rilpivirinem. Většina těchto ukončení se vyskytla v prvních 48 týdnech léčby.
|
Tabulka 4: Virologické výstupy u dospělých pacientů z hodnocení ECHO a THRIVE (souhrnné údaje z analýz v týdnu 48 (primární) a v týdnu 96; ITT-TLOVR*) | ||||||
|
Výstup podle analýzy ve 48. týdnu |
Výstup podle analýzy v 96. týdnu | |||||
|
Rilpivirin + BR N = 686 |
Efavirenz + BR N = 682 |
Pozorovaný rozdíl (95 % CI) ± |
Rilpivirin + BR N=686 |
Efavirenz + BR N=682 |
Pozorovaný rozdíl (95 % CI) ± | |
|
Odpověď (potvrzených < 50 HIV-1 RNA kopií/ml)§# |
84,3 % (578/686) |
82,3 % (561/682) |
2,0 (-2,0; 6,0) |
77,6 % (532/686) |
77,6 % (529/682) |
0 (-4,4; 4,4) |
|
Bez odpovědi | ||||||
|
Virologické selháníŤ | ||||||
|
Celkem |
9.0 % (62/686) |
4.8 % (33/682) |
ND |
11.5 % (79/686) |
5.9 % (40/682) |
ND |
|
< 100 000 |
3,8 % (14/368) |
3,3 % (11/330) |
ND |
5,7 % (21/368) |
3,6 % (12/329) |
ND |
|
> 100 000 |
15,1 % (48/318) |
6,3 % (22/352) |
ND |
18,2 % (58/318) |
7,9 % (28/353) |
ND |
|
Úmrtí |
0,1 % (1/686) |
0,4 % (3/682) |
ND |
0,1 % (1/686) |
0,9 % (6/682) |
ND |
|
Ukončení léčby kvůli nežádoucím účinkům |
2,0 % (14/686) |
6,7 % (46/682) |
ND |
3,8 % (26/682) |
7,6 % (52/682) |
ND |
|
Ukončení léčby mimo nežádoucí účinky1 |
4,5 % (31/686) |
5,7 % (39/682) |
ND |
7,0 % (48/682) |
8,1 % (55/682) |
ND |
|
Odpověď podle subkategorie | ||||||
|
Podle základních NRTI | ||||||
|
Tenofovir/emtricitabin |
83,5 % (459/550) |
82,4 % (450/546) |
1,0 (-3,4; 5,5) |
76,9 % (423/550) |
77,3 % (422/546) |
-0,4 % (-5,4; 4,6) |
|
Zidovudin/lamivudin |
87,1 % (88/101) |
80,6 % (83/103) |
6,5 (-3,6; 16,7) |
81,2 % (82/101) |
76,7 % (79/103) |
4,5 % (-6,8; 15,7) |
|
Abakavir/lamivudin |
88,6 % (31/35) |
84,8 % (28/33) |
3,7 (-12,7; 20,1) |
77,1 % (27/35) |
84,8 % (28/33) |
-7,7 % (-26,7; 11,3) |
|
Podle počáteční virové nálože (kopie/ml) | ||||||
|
< 100 000 |
90,2 % (332/368) |
83,6 % (276/330) |
6,6 (1,6; 11,5) |
84,0 % (309/368) |
79,9 % (263/329) |
4,0 (-1,7; 9,7) |
|
> 100 000 |
77,4 % (246/318) |
81,0 % (285/352) |
-3,6 (-9,8; 2,5) |
70,1 % (223/318) |
75,4 % (266/353) |
-5,2 (-12,0;1,5) |
|
Podle počátečního počtu CD4 buněk (lďbuněkA) | ||||||
|
< 50 |
58,8 % (20/34) |
80,6 % (29/36) |
-21,7 (-43,0; -0,5) |
55,9 % (19/34) |
69,4 % (25/36) |
-13,6 (-36,4; 9,3) |
|
> 50-< 200 |
80,4 % (156/194) |
81,7 % (143/175) |
-1,3 (-9,3; 6,7) |
71,1 % (138/194) |
74,9 % (131/175) |
-3,7 (-12,8; 5,4) |
|
> 200-< 350 |
86,9 % (272/313) |
82,4 % (253/307) |
4,5 (-1,2; 10,2) |
80,5 % (252/313) |
79,5 % (244/307) |
1,0 (-5,3; 7,3) |
Založeno na normální aproximaci.
|
> 350 |
90,3 % |
82,9 % |
7,4 |
85,4 % |
78,7 % |
6,8 |
|
(130/144) |
(136/164) |
(-0,3; 15,0) |
(123/144) |
(129/164) |
(-1,9; 15,4) |
N
*
±
počet subjektů ve skupině léčby; ND = nebylo stanoveno.
Intent-to-treat doba do ztráty virologické odpovědi.
§
#
Pacienti, kteří dosáhli virologické odpovědi (dvě po sobě jdoucí virové nálože < 50 kopií/ml) a udrželi ji do týdne 48.
Předpokládaný rozdíl podílů odpovědí (95 % CI) v analýze ve 48. týdnu: 1,6 % (-2,2 %; 5,3 %) a v analýze v 96. týdnu: -0,4 %
(-4,6 %; 3,8 %); p-hodnota pro oboje < 0,0001 (non-inferiorita ve 12 % rozmezí) z logistického regresního modelu, včetně stratifikačních faktorů a hodnocení.
Virologické selhání podle souhrnné analýzy účinnosti: zahrnuje pacienty, kteří byli v reboundu (potvrzená virová nálož > 50 kopií/ml po potvrzení statutu odpovídající na léčbu) nebo kteří nikdy nebyli suprimováni (žádná potvrzená virová nálož < 50 kopií/ml, buď pokračující nebo ukončení vzhledem k nedostatku nebo ztrátě účinnosti).
Např. ztráta sledování, non-compliance, zrušení souhlasu.
V 96. týdnu byla průměrná změna od výchozí hodnoty v počtu CD4+ buněk +228x 106 buněk/l u skupiny s rilpivirinem a +219x 106 buněk/l ve skupině s efavirenzem dle souhrnné analýzy ze studií ECHO a THRIVE [předpokládaný rozdíl léčby (95 % CI): 11,3 (-6,8; 29,4)].
Tabulka 5 ukazuje výsledky testů rezistence ze souhrnné analýzy rezistence v 96. týdnu u pacientů s virologickým selháním a párovanými genotypy (počátek a konec léčby) definovanými protokolem klinických studií.
|
Tabulka 5: Hodnocení rezistence podle základního použitého NRTI režimu (souhrnné údaje ze studií ECHO a THRIVE podle analýzy rezistence po 96 týdnech) | ||||
|
tenofovir/ emtricitabin |
zidovudin/ lamivudin |
abakavir/ lamivudin |
Celkem* | |
|
Léčení rilpivirinem | ||||
|
Rezistence# k emtricitabinu/lamivudinu % (n/N) |
6,9 (38/550) |
3,0 (3/101) |
8,6 (3/35) |
6,4 (44/686) |
|
Rezistence k rilpivirinu % (n/N) |
6,5 (36/550) |
3,0 (3/101) |
8,6 (3/35) |
6,1 (42/686) |
|
Léčení efavirenzem | ||||
|
Rezistence k emtricitabinu/lamivudinu % (n/N) |
1,1 (6/546) |
1,9 (2/103) |
3,0 (1/33) |
1,3 (9/682) |
|
Rezistence k efavirenzu % (n/N) |
2,4 (13/546) |
2,9 (3/103) |
3,0 (1/33) |
2,5 (17/682) |
Počet pacientů s virologickým selháním a párovanými genotypy (počátek a selhání léčby) byl 71, 11 a 4 pro rilpivirin a 30, 10 a 2 pro režimy s efavirenzem, tenofovirem/emtricitabinem, zidovudinem/lamivudinem, resp. abakavirem/lamivudinem.
Rezistence byla definována jako objevení se jakékoli mutace spojené s rezistencí při selhání.
U pacientů, u kterých selhala léčba rilpivirinem a u nichž se vyvinula rezistence k rilpivirinu, byla většinou pozorována zkřížená rezistence k ostatním schváleným NNRTI (etravirinu, efavirenzu, nevirapinu).
Studie TMC278-C204 byla randomizovaná aktivně kontrolovaná studie fáze IIb v léčbě dosud neléčených dospělých pacientů nakažených virem HIV-1 sestávající se ze 2 částí: počáteční částečně zaslepené části pro nalezení dávky (zaslepené dávky rilpivirinu) až do 96. týdne následované dlouhodobou otevřenou částí. V otevřené části studie byli pacienti původně randomizovaní do 3 skupin podle dávky rilpivirinu poté, co byla stanovena dávka pro studii fáze III, léčeni rilpivirinem 25 mg 1x denně navíc k BR. Pacienti v kontrolní skupině dostávali efavirenz 600 mg 1x denně navíc k BR v obou částech hodnocení. BR se skládala ze 2 investigátorem vybraných N(t)RTI: zidovudin a lamivudin nebo tenofovir-disoproxyl-fumarát a emtricitabin.
Studie TMC278-C204 zahrnovalo 368 dosud neléčených dospělých pacientů infikovaných HIV-1, kteří měli hladiny HIV-1 RNA v plazmě > 5 000 kopií/ml, dříve dostávali < 2 týdny N(t)RTI nebo inhibitor proteázy, dříve neužívali NNRTI a byli testováni na citlivost k N(t)RTI a na nepřítomnost specifických mutací spojených s rezistencí k NNRTI.
V 96. týdnu byl podíl pacientů s < 50 HIV-1 RNA kopií/ml ve skupině s rilpivirinem (N = 93) 76 % ve srovnání s 71 % ve skupině s efavirenzem (N = 89). Průměrný vzestup od výchozího stavu v počtu CD4+ buněk byl 146x 106 buněk/l u pacientů dostávajících rilpivirin 25 mg a 160x 106 buněk/l u pacientů dostávajících efavirenz.
Z těchto pacientů dostávajících rilpivirin, kteří reagovali v 96. týdnu, zůstala nedetekovatelná virová nálož (< 50 HIV-1 RNA kopií/ml) v týdnu 240 u 74 % ve srovnání s 81 % pacientů dostávajících efavirenz. V analýze ve 240. týdnu nebyly detekovány bezpečnostní problémy.
Pediatrická populace
Farmakokinetika, bezpečnost, snášenlivost a účinnost rilpivirinu 25 mg jednou denně v kombinaci s investigátorem vybranou BR obsahující dva NRTI, byla hodnocena v otevřené jednoramenné studii fáze II TMC278-C213 u dosud neléčených dospívajících pacientů s tělesnou hmotností nejméně 32 kg infikovaných virem HIV-1. Analýza zahrnovala 36 pacientů, kteří dokončili léčbu trvající minimálně 48 týdnů nebo ji přerušili dříve.
U 36 pacientů byl medián věku 14,5 let (rozmezí: 12 až 17 let), 55,6 % bylo žen, 88,9 % černochů a 11,1 % Asiatů. Medián výchozí HIV-1 RNA v plazmě byl 4,8 logJ0 kopií/ml a medián výchozího počtu CD4+ buněk byl 414 x 106 buněk/l (rozmezí: 25 až 983 x 106 buněk/l).
Podíl pacientů s HIV-1 RNA < 50 kopií/ml v týdnu 48 (TLOVR) byl 72,2 % (26/36). Podíl odpovědi byl vyšší u pacientů s počáteční virovou náloží < 100 000 kopií/ml (78,6 %, 22/28) v porovnání s těmi, kteří měli počáteční virovou nálož > 100 000 kopií/ml (50,0 %, 4/8), Podíl virologického selhání byl
22.2 % (8/36). Podíl virologického selhání byl nižší u pacientů s počáteční virovou náloží < 100 000 kopií/ml (17,9 %, 5/28) v porovnání s těmi, kteří měli počáteční virovou nálož
> 100 000 kopií/ml (37,5 %, 3/8). Rezistence rilpivirinových mutací byla pozorována u 62,5 % (5/8) pacientů s virologickým selháním. U 4 z těchto 5 pacientů byla také pozorována rezistence na NRTI. Jeden pacient přerušil kvůli nežádoucímu účinku a 1 pacient přerušil z jiných důvodů než výskyt nežádoucího účinku nebo virologické selhání. V týdnu 48 bylo průměrné zvýšení počtu CD4+ buněk z počáteční hodnoty na 201,2 x 106 buněk/l.
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s rilpivirinem u jedné nebo více podskupin pediatrické populace při léčbě infekce virem lidské imunodeficience (HIV-1) (viz bod 4.2 o použití u pediatrické populace).
5.2 Farmakokinetické vlastnosti
Farmakokinetické vlastnosti rilpivirinu byly hodnoceny u zdravých dospělých dobrovolníků a u dosud neléčených pacientů ve věku 12 let a starších nakažených HIV-1. Expozice rilpivirinu byla obecně nižší u pacientů nakažených HIV-1 než u zdravých dobrovolníků.
Absorpce
Po perorálním podání je maximální koncentrace rilpivirinu v plazmě obvykle dosažena během 4 -5 hodin. Celková biologická dostupnost přípravku EDURANT není známa.
Vliv jídla na absorpci
Po podání přípravku EDURANT nalačno byla expozice rilpivirinu přibližně o 40 % nižší než po podání s jídlem s normálním kalorickým obsahem (533 kcal) nebo s vysoce kalorickým jídlem (928 kcal). Po užití přípravku EDURANT pouze s výživným nápojem bohatým na bílkoviny byly expozice o 50 % nižší než po užití s jídlem. Pro dosažení optimální absorpce je nutno užívat EDURANT s jídlem. Užívání přípravku EDURANT nalačno nebo pouze s výživným nápojem může vést ke snížení koncentrace rilpivirinu v plazmě, což může potenciálně snížit jeho léčivý účinek (viz bod 4.2).
Distribuce v organismu
Rilpivirin se in vitro váže přibližně z 99,7 % na bílkoviny plazmy, primárně na albumin. Distribuce rilpivirinu do jiných kompartmentů než plazma (např. mozkomíšní mok, sekrety genitálu) nebyla u lidí hodnocena.
Biotransformace
In vitro pokusy ukazují, že rilpivirin podléhá primárně oxidačnímu metabolismu zprostředkovanému cytochromy P450 (CYP) 3A systémem.
Eliminace z organismu
Terminální eliminační poločas rilpivirinu je přibližně 45 hodin. Po jednorázovém perorálním podání 14C-rilpivirinu se průměrně 85 % radioaktivity objevilo ve stolici a 6,1 % v moči. Ve stolici tvořil nezměněný rilpivirin průměrně 25 % podané dávky. V moči byly detekovány pouze stopy nezměněného rilpivirinu (< 1 % dávky).
Další informace pro zvláštní populace
Pediatrická populace (ve věku < 18 let)
Farmakokinetika rilpivirinu u dospívajících, dosud neléčených pacientů infikovaných virem HIV-1 dostávajících EDURANT 25 mg jednou denně byla srovnatelná s farmakokinetikou u dospělých, dosud neléčených pacientů infikovaných virem HIV-1 dostávajících EDURANT 25 mg jednou denně. Podobně jako u dospělých, tělesná hmotnost u dospívajících ve studii C213 (33 až 93 kg) neměla vliv na farmakokinetiku rilpivirinu.
Farmakokinetika rilpivirinu u pediatrických pacientů ve věku < 12 let se zkoumá. Doporučení pro dávkování u pediatrických pacientů ve věku < 12 let není možné vytvořit vzhledem k nedostatečným údajům (viz bod 4.2).
Starší osoby
Populační farmakokinetická analýza u pacientů nakažených HIV ukázala, že se farmakokinetika rilpivirinu v hodnoceném věkovém rozmezí (18 až 78 let) neměnila; 3 subjekty byly starší než 65 let.
U starších pacientů není nutná úprava dávkování přípravku EDURANT. U této populace je nutno EDURANT užívat s opatrností (viz bod 4.2).
Pohlaví
Mezi ženami a muži nebyly pozorovány klinicky významné rozdíly ve farmakokinetice rilpivirinu.
Rasa
Populační farmakokinetická analýza u pacientů nakažených HIV ukazuje, že rasa nemá na expozici rilpivirinu klinicky významný vliv.
Porucha funkce jater
Rilpivirin je primárně metabolizován a vylučován játry. Ve studii srovnávající 8 pacientů s lehkou poruchou funkce jater (Child-Pugh skóre A) s 8 kontrolními pacienty a 8 pacientů se středně těžkou poruchou funkce jater (Child-Pugh skóre B) s 8 kontrolními pacienty byla expozice rilpivirinu po opakovaném podání o 47 % vyšší u lehké poruchy funkce jater a o 5 % vyšší u středně těžké poruchy funkce jater. Nelze však vyloučit, že u středně těžké poruchy funkce jater je farmakologicky aktivní expozice nevázanému rilpivirinu významně zvýšena.
U pacientů se středně těžkou poruchou funkce jater není nutná úprava dávkování, ale doporučuje se opatrnost. EDURANT nebyl hodnocen u pacientů s těžkou poruchou funkce jater (Child-Pugh skóre C). Proto se EDURANT nedoporučuje u pacientů s těžkou poruchou funkce jater (viz bod 4.2).
Souběžná infekce virem hepatitidy B a/nebo hepatitidy C
Populační farmakokinetická analýza u pacientů souběžně nakažených virem hepatitidy B a/nebo hepatitidy C neprokázala žádný klinicky významný vliv na farmakokinetiku rilpivirinu.
Porucha funkce ledvin
Farmakokinetika rilpivirinu nebyla studována u pacientů s renální insuficiencí. Vylučování rilpivirinu ledvinami je zanedbatelné. U pacientů s lehkou nebo středně těžkou poruchou funkce ledvin není nutná úprava dávkování. U pacientů s těžkou poruchou funkce ledvin nebo v terminálním stádiu ledvinového onemocnění je nutno EDURANT užívat s opatrností, protože koncentrace v plazmě mohou být zvýšeny kvůli změnám v absorpci léčiva, jeho distribuci a/nebo metabolismu sekundárně k poruše ledvin. U pacientů s těžkou poruchou funkce ledvin nebo v terminálním stádiu ledvinového onemocnění lze kombinaci přípravku EDURANT s účinným inhibitorem CYP3A užívat pouze, pokud přínos převáží riziko. Vzhledem k tomu, že se rilpivirin silně váže na bílkoviny plazmy, není pravděpodobné, že by jej bylo možno odstranit hemodialýzou nebo peritoneální dialýzou (viz bod
4.2).
5.3 Předklinické údaje vztahující se k bezpečnosti
Toxicita po opakovaném podání
U hlodavců byla pozorována hepatotoxicita spojená s indukcí jaterních enzymů. U psů byly zaznamenány účinky podobné cholestáze.
Studie reprodukční toxicity
Studie na zvířatech neprokázaly relevantní embryotoxicitu nebo fetotoxicitu nebo vliv na reprodukční funkce. U potkanů a králíků se neprojevila teratogenita. Expozice v embryo-fetálních No Observed Effects Levels (NOAEL) u potkanů a králíků byly 15krát, resp. 70krát, vyšší než expozice u lidí při doporučeném dávkování 25 mg jednou denně.
Kancerogeneze a mutageneze
Rilpivirin byl hodnocen na kancerogenní potenciál podáváním sondou do žaludku myším a potkanům až po 104 týdny. V nejnižších testovaných dávkách ve studiích kancerogenity byly systémové expozice rilpivirinu (na základě AUC) 21násobné (u myší) a 3násobné (u potkanů) vzhledem k expozicím u lidí při doporučené dávce (25 mg jednou denně). U potkanů nebyla žádná neoplazmata spojená s léčivem. U myší byl rilpivirin pozitivní na hepatocelulární neoplazmata jak u samců, tak i u samic. Pozorované hepatocelulární nálezy u myší mohou být specifické pro hlodavce.
Rilpivirin byl negativní jak bez aktivace metabolického systému, tak s jeho aktivací v in vitro reverzním stanovení mutací podle Amese, tak v in vitro stanovení klastogenního lymfomu u myší. Rilpivirin neindukoval poškození chromozomů v in vivo mikronukleovém testu u myší.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Jádro tablety Monohydrát laktosy Sodná sůl kroskarmelosy Povidon K30 Polysorbát 20
Silicifikovaná mikrokrystalická celulosa Magnesium-stearát
Potahová vrstva tablety Monohydrát laktosy Hypromelosa 2910/6 Oxid titaničitý (E171)
Makrogol 3000 Triacetin
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
3 roky
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v původní lahvičce, aby byl přípravek chráněn před světlem.
6.5 Druh obalu a obsah balení
75ml lahvička z polyethylenu o vysoké hustotě (HDPE) s polypropylenovým (PP) dětským bezpečnostním uzávěrem a garancí neporušenosti obalu. Jedna krabička obsahuje jednu lahvičku se 30 tabletami.
6.6 Zvláštní opatření pro likvidaci přípravku
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Janssen-Cilag International NV Turnhoutseweg 30 B-2340 Beerse Belgie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/11/736/001
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 28. listopadu 2011
Datum posledního prodloužení registrace: DD. měsíc RRRR
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/.
A. VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ /VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného/výrobců odpovědných za propouštění šarží
Janssen-Cilag SpA
Via C. Janssen
Borgo San Michele
04100 Latina
Itálie
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2)."
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
EDURANT 25 mg potahované tablety rilpivirinum
Jedna potahovaná tableta obsahuje rilpivirini hydrochloridum ekvivalentní rilpivirinum 25 mg.
Obsahuje monohydrát laktosy.
Další údaje jsou uvedeny v příbalové informaci.
30 potahovaných tablet
Před použitím si přečtěte příbalovou informaci. Perorální podání
Uchovávejte mimo dohled a dosah dětí.
Použitelné do:
Uchovávejte v původní lahvičce, aby byl přípravek chráněn před světlem.
Janssen-Cilag International NV Tumhoutseweg 30 B-2340 Beerse Belgie
EU/1/11/736/001
c.s.:
Výdej léčivého přípravku vázán na lékařský předpis.
2D čárový kód s jedinečným identifikátorem.
PC: {číslo} [kód přípravku]
SN: {číslo} [sériové číslo]
NN: {číslo} [vnitrostátní úhradové číslo nebo jiné vnitrostátní číslo identifikující léčivý přípravek]
ÚDAJE UVÁDĚNÉ NA VNITŘNÍM OBALU
ŠTÍTEK NA LAHVIČKU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
EDURANT 25 mg potahované tablety rilpivirinum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna potahovaná tableta obsahuje rilpivirini hydrochloridum ekvivalnetní rilpivirinum 25 mg
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje monohydrát laktosy.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
30 potahovaných tablet
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v původní lahvičce, aby byl přípravek chráněn před světlem.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Janssen-Cilag International NV Tumhoutseweg 30 B-2340 Beerse Belgie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/11/736/001
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
B. PŘÍBALOVÁ INFORMACE
Příbalová informace pro uživatele
EDURANT 25 mg potahované tablety
rilpivirinum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je EDURANT a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete EDURANT užívat
3. Jak se EDURANT užívá
4. Možné nežádoucí účinky
5. Jak EDURANT uchovávat
6. Obsah balení a další informace
1. Co je EDURANT a k čemu se používá
EDURANT obsahuje rilpivirin, který se používá k léčbě infekcí způsobených virem lidské imunodeficience (HIV). Patří do skupiny léčivých přípravků proti HIV nazývaných nenukleosidové inhibitory reverzní transkriptázy (NNRTI). EDURANT působí tak, že snižuje množství HIV ve Vašem těle.
EDURANT se užívá v kombinaci s jinými léčivými přípravky proti HIV v léčbě dospělých a dospívajících ve věku 12 let a starších, kteří jsou infikováni HIV a dosud neužívali jiné léky proti HIV.
Váš lékař s Vámi prodiskutuje, která kombinace léčivých přípravků bude pro Vás nejlepší.
2. Čemu musíte věnovat pozornosti, než začnete EDURANT užívat
Neužívejte EDURANT, jestliže jste alergický(á) na rilpivirin nebo na kteroukoli další složku tohoto
přípravku (uvedenou v bodě 6).
Neužívejte EDURANT v kombinaci s kterýmkoli dále uvedeným léčivým přípravkem, protože to
může ovlivnit způsob, jakým EDURANT nebo jiné léčivé přípravky účinkují:
- karbamazepin, oxkarbazepin, fenobarbital, fenytoin (příravky k léčbě epilepsie a prevenci záchvatů);
- rifampicin, rifapentin (přípravky k léčbě bakteriálních infekcí jako tuberkulóza);
- omeprazol, esomeprazol, lansoprazol, pantoprazol, rabeprazol (inhibitory protonové pumpy, což jsou léčivé přípravky k prevenci a léčbě žaludečních vředů, pálení žáhy nebo návratu kyselého obsahu žaludku do jícnu);
- dexamethason (kortikosteroid užívaný u mnoha stavů jako zánět a alergická reakce), je-li užit perorálně nebo injikován, s výjimkou léčby jednorázovou dávkou;
- přípravky obsahující třezalku tečkovanou (Hypericumperforatum) (rostlinný přípravek užívaný proti depresím).
Užíváte-li cokoli z výše uvedeného, poraďte se s lékařem o náhradě.
Upozornění a opatření
Před užitím přípravku EDURANT se poraďte se svým lékařem nebo lékárníkem.
EDURANT HIV infekci nevyléčí. Je to součást léčby, která snižuje množství virů v krvi. I když užíváte tento léčivý přípravek, stále můžete šířit HIV, ačkoli riziko je účinnou antiretrovirovou léčbou sníženo. Poraďte se s lékařem o opatřeních potřebných k zabránění přenosu infekce na další osoby.
EDURANT byl studován u omezeného počtu pacientů ve věku 65 let a starších. Pokud patříte do této věkové skupiny, prosím, poraďte se o užívání přípravku EDURANT se svým lékařem.
Informujte svého lékaře o svém stavu
Přesvědčte se, že jste zkontroloval(a) všechny následující body, a pokud se Vás některý z nich týkal, že jste o nich informoval(a) svého lékaře.
- Řekněte svému lékaři, že máte nebo jste měl(a) problémy s onemocněním jater, včetně zánětu jater typu B a/nebo C a/nebo problémy s ledvinami. Lékař posoudí, jak je onemocnění jater nebo ledvin závažné dříve, než rozhodne, zda budete EDURANT užívat.
- Jestliže zaznamenáte jakékoli příznaky infekce (například horečka, zimnice, pocení), okamžitě navštivte lékaře. U některých pacientů s pokročilou HIV infekcí a výskytem příležitostných infekcí v minulosti se mohou brzy po zahájení anti-HIV léčby objevit známky a příznaky zánětu z předchozích infekcí. Předpokládá se, že tyto projevy jsou způsobeny zlepšením imunitní odpovědi organismu, která umožňuje tělu bojovat s infekcí, která mohla být přítomna bez zjevných příznaků.
- Jakmile začnete užívat léčivé přípravky k léčbě HIV infekce, mohou se u Vás kromě oportunních infekcí vyskytnout autoimunitní onemocnění (stavy, které se vyskytují, když imunitní systém napadá zdravé tkáně). Autoimunitní onemocnění se mohou objevit mnoho měsíců po zahájení léčby. Pokud zaznamenáte příznaky infekce nebo jiné příznaky jako jsou svalová slabost, slabost začínající v rukách a nohách a postupující směrem k trupu, bušení srdce, třes nebo hyperaktivita, prosím, informujte ihned svého lékaře a požádej te o nezbytnou léčbu.
- Informujte svého lékaře, pokud užíváte léky, které mohou vyvolat život ohrožující nepravidelný srdeční tep (torsade de pointe s).
Děti
EDURANT se nepoužívá u dětí ve věku do 12 let, protože tento přípravek nebyl u těchto pacientů dostatečně studován.
Další léčivé přípravky a EDURANT
EDURANT musíte užívat s dalšími přípravky k léčbě HIV. Lékař Vám poradí, které přípravky k léčbě HIV lze s přípravkem EDURANT kombinovat a společně se rozhodnete, která kombinace je pro Vás nejlepší. Pozorně sledujte pokyny lékaře.
Některé léčivé přípravky mohou ovlivnit hladiny přípravku EDURANT v krvi, pokud se s ním užívají zároveň.
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Nedoporučuje se kombinovat EDURANT s jinými nenukleosidovými inhibitory reverzní transkriptázy (NNRTI) jako delavirdin, efavirenz, etravirin a nevirapin.
Účinky přípravku EDURANT nebo jiných léčivých přípravků mohou být ovlivněny, jestliže užíváte EDURANT spolu s některým z následujících léků. Řekněte svému lékaři, že také užíváte:
- rifabutin ( přípravek k léčbě některých bakteriálních infekcí). Jestliže užíváte tento léčivý přípravek současně s přípravkem EDURANT, prosím, čtěte pozorně v bodě 3 jak přípravek EDURANT užívat „Návod ke správnému užívání u dospělých (> 18 let)“.
- klarithromycin, erythromycin (antibiotika);
- cimetidin, famotidin, nizatidin, ranitidin (antagonisté H2-receptorů užívané k léčbě žaludečních nebo dvanáctníkových vředů nebo k úlevě od pálení žáhy v důsledku refluxu). Užíváte-li tyto
léčivé přípravky, přečtěte si pozorně v bodu 3 „Návod ke správnému užívání u dospělých (> 18 let)“, jak je užívat;
- antacida (užívaná k léčbě nemocí spojených s kyselinou v žaludku; např. hydroxid hlinitý/hořečnatý, uhličitan vápenatý). Užíváte-li tyto léčivé přípravky, přečtěte si pozorně v bodu 3 „Návod ke správnému užívání u dospělých (> 18 let)“, jak je užívat;
- methadon (užíváný k léčbě příznaků z vysazení drog a drogové závislosti);
- dabigatran-etexilát (antikoagulans - přípravek zabraňující nadměrnému srážení krve).
Těhotenství a kojení
Jestliže jste těhotná, okamžitě to řekněte svému lékaři. Těhotné ženy nesmějí užívat EDURANT, aniž by jim to lékař speciálně nařídil.
Matky infikované HIV nesmějí kojit, protože je možný přenos HIV infekce na dítě prostřednictvím mateřského mléka.
Poraďte se se svým lékařem nebo lékárníkem dříve, než začnete užívat jakýkoliv lék.
Řízení dopravních prostředků a obsluha strojů
Někteří pacienti mohou během léčby přípravkem EDURANT pocítit únavu, závratě nebo ospalost. Pokud se během užívání přípravku EDURANT cítíte unavený(á), točí se Vám hlava nebo jste ospalý(á), neřiďte nebo neobsluhujte stroje.
EDURANT obsahuje laktosu
Pokud Vám Váš lékař řekl, že nesnášíte některé cukry, poraďte se se svým lékařem, než začnete tento léčivý přípravek užívat.
3. Jak se EDURANT užívá
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře nebo lékárníka. Pokud si nejste
jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Návod ke správnému užívání u dospělých a dospívajících (ve věku od 12 do < 18 let)
Doporučená dávka přípravku EDURANT je jedna tableta jednou denně.
EDURANT je nutno užívat s jídlem. Jídlo je důležité k dosažení správných hladin léčivé látky ve
Vašem těle. Nutriční nápoj (např. bohatý na bílkoviny) sám o sobě jídlo nenahradí.
Čtyři stavy vyžadují zvláštní pozornost:
1. Jestliže užíváte rifabutin (přípravek k léčbě některých bakteriálních infekcí), užívejte 2 tablety přípravku EDURANT jednou denně. Pokud užívání přípravku rifabutin ukončíte, užívejte jednu tabletu přípravku EDURANT jednou denně. Pokud si nejste jistý(á), zeptejte se svého lékaře nebo lékárníka.
2. Užíváte-li antacidum (léčivý přípravek užívaný k léčbě nemocí spojených s kyselinou v žaludku; např. hydroxid hlinitý/hořečnatý, uhličitan vápenatý). Antacidum užijte buď minimálně 2 hodiny před užitím přípravku EDURANT nebo alespoň 4 hodiny po jeho užití (viz bod 2 „Vzájemné působení s dalšími léčivými přípravky“).
3. Užíváte-li antagonistu H2-receptorů [léčivé přípravky užívané k léčbě žaludečních nebo dvanáctníkových vředů nebo k úlevě od pálení žáhy v důsledku refluxu (např. cimetidin, famotidin, nizatidin nebo ranitidin)]. Antagonistu H2-receptorů užijte buď minimálně 12 hodin před užitím přípravku EDURANT nebo alespoň 4 hodiny po jeho užití (viz bod 2 „Vzájemné působení s dalšími léčivými přípravky“). Antagonisty H2-receptorů nelze užívat v režimu dvakrát denně. Domluvte se s lékařem na jiném způsobu užívání.
4. Užíváte-li didanosin (lék k léčbě infekce HIV), není úprava dávky potřebná. Didanosin se musí podávat nalačno nejméně dvě hodiny před nebo nejméně čtyři hodiny po podání přípravku EDURANT (který se musí užívat s jídlem).
Otevření dětského bezpečnostního uzávěru
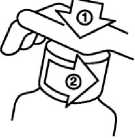
Lahvička se dodává s dětským bezpečnostním uzávěrem. Otevírá se zatlačením na šroubovací uzávěr směrem dolů a současným otáčením uzávěrem proti směru hodinových ručiček.
Jestliže jste užil(a) více přípravku EDURANT, než jste měl(a)
Okamžitě vyhledejte lékaře nebo lékárníka. V případě předávkování můžete mít bolesti hlavy, pocit na zvracení závratě a/nebo mít neobvyklé sny.
Jestliže jste zapomněl(a) užít EDURANT
Pokud si na to vzpomenete do 12 hodin od doby, kdy obvykle EDURANT užíváte, musíte tabletu užít okamžitě. Tableta přípravku EDURANT se musí užít s jídlem. Další dávku užijte v obvyklou dobu. Pokud si na to vzpomenete po 12 hodinách, potom dávku vynechte a další dávku užijte v obvyklém čase. Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku.
Zvracíte-li během 4 hodin po užití přípravku EDURANT, užijte další tabletu spolu s jídlem.
Zvracíte-li po více než 4 hodinách od užití přípravku EDURANT, není nutné užít jinou tabletu až do pravidelně naplánované tablety.
Nejste-li si jistý(á), co dělat v případě vynechání dávky nebo zvracení, kontaktujte lékaře. Nepřestávejte EDURANT užívat
Léčba HIV nevyléčí infekci HIV! Nepřestávejte EDURANT užívat, aniž byste to nejdříve řekl(a) svému lékaři. I když se budete cítit lépe, nepřestávejte užívat EDURANT nebo jiné anti-HIV léčivé přípravky. Pokud to uděláte, mohlo by se zvýšit riziko, že se virus stane rezistentní. Nejdříve to řekněte lékaři.
Máte-li jakékoli další otázky, týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Velmi časté:
- bolesti hlavy
- pocit na zvracení
- potíže s usínáním (nespavost)
- závratě
- změna v některém rutinně prováděném jaterním testu (aminotransferázy);
- zvýšení cholesterolu a/nebo pankreatické amylázy v krvi.
Časté:
- neobvyklé sny
- vyrážka
- bolesti břicha
- deprese
- únava
- zvracení
- ospalost
- snížení chuti k jídlu
- poruchy spánku
- nepříjemné pocity v žaludku
- depresivní nálada
- sucho v ústech
- nízký počet bílých krvinek a/nebo krevních destiček, snížení hladiny hemoglobinu v krvi, zvýšení hladin triacylglycerolů, lipázy a/nebo bilirubinu v krvi;
Méně časté:
- známky nebo příznaky zánětu nebo infekce, jako například horečka, zimnice, pocení (syndrom imunitní reaktivace).
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak EDURANT uchovávat
Uchovávejte mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a lahvičce za „Použitelné do“, resp. „EXP“. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v původní lahvičce, aby byl přípravek chráněn před světlem.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co EDURANT obsahuje
- Léčivou látkou je rilpivirinum ve formě rilpivirini hydrochloridum. Jedna tableta přípravku EDURANT obsahuje rilpivirini hydrochloridum ekvivalentní rilpivirinum 25 mg.
- Dalšími složkami jádra potahované tablety jsou monohydrát laktosy, sodná sůl kroskarmelosy, povidon K30, polysorbát 20, silicifikovaná mikrokrystalická celulosa a magnesium-stearát. Potahová vrstva tablety obsahuje monohydrát laktosy, hypromelosu 2910/6, oxid titaničitý (E171), makrogol 3000 a triacetin.
Jak EDURANT vypadá a co obsahuje toto balení
Bílá až téměř bílá kulatá bikonvexní tableta s vyraženým „TMC“ na jedné straně a „25“ na druhé straně.
Lahvička s dětským bezpečnostním uzávěrem obsahuje 30 potahovaných tablet.
Držitel rozhodnutí o registraci
Janssen-Cilag International NV Turnhoutseweg 30 B-2340 Beerse Belgie
Výrobce
Janssen-Cilag SpA.
Via C. Janssen Borgo San Michele 04100 Latina Itálie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci
|
Belgie/Belgique/Belgien Janssen-Cilag NV Antwerpseweg 15-17 B-2340 Beerse Tel/Tél: +32 14 64 94 11 |
Lietuva UAB "JOHNSON & JOHNSON" Geležinio Vilko g. 18A LT-08104 Vilnius Tel: +370 5 278 68 88 |
|
Btarapna „fl^OHCtH & fl^OHCtH Etarapna” EOOfl ^.k. Maagocr 4 EH3Hec napK Co$hh, crpaga 4 Co$hh 1766 Tea.: +359 2 489 94 00 |
Luxembourg/Luxemburg Janssen-Cilag NV Antwerpseweg 15-17 B-2340 Beerse Belgique/Belgien Tél/Tel: +32 14 64 94 11 |
|
Česká republika Janssen-Cilag s.r.o. Karla Engliše 3201/06 CZ-150 00 Praha 5 - Smíchov Tel: +420 227 012 227 |
Magyarország Janssen-Cilag Kft. Nagyenyed u. 8-14 H-Budapest, 1123 Tel.: +36 1 884 2858 |
|
Danmark Janssen-Cilag A/S Bregneredvej 133 DK-3460 Birkered Tlf: +45 45 94 82 82 |
Malta AM MANGION LTD. Mangion Building, Triq Gdida fi Triq Valletta MT-Hal-Luqa LQA 6000 Tel: +356 2397 6000 |
|
Deutschland Janssen-Cilag GmbH Johnson & Johnson Platz 1 D-41470 Neuss Tel: +49 2137 955-955 |
Nederland Janssen-Cilag B.V. Dr. Paul Janssenweg 150 NL-5026 RH Tilburg Tel: +31 13 583 73 73 |
|
Eesti UAB "JOHNSON & JOHNSON" Eesti filiaal LSStsa 2 EE-11415 Tallinn Tel: +372 617 7410 |
Norge Janssen-Cilag AS Postboks 144 NO-1325-Lysaker Tlf: +47 24 12 65 00 |
|
ELLáSa Janssen-Cilag OappaKsuxiKq A.E.B.E. Asro^ópog Erpqvqg 56 GR-151 21 nsÚKn, AOqva Tq^: +30 210 80 90 000 |
Osterreich Janssen-Cilag Pharma GmbH VorgartenstraBe 206B A-1020 Wien Tel: +43 1 610 300 |
|
Espaňa Janssen-Cilag, S.A. Paseo de las Doce Estrellas, 5-7 E-28042 Madrid Tel: +34 91 722 81 00 |
Polska Janssen-Cilag Polska Sp. z o.o. ul. Uzecka 24 PL-02-135 Warszawa Tel.: +48 22 237 60 00 |
|
France Janssen-Cilag 1, rue Camille Desmoulins, TSA 91003 F-92787 Issy Les Moulineaux, Cedex 9 Tél: 0 800 25 50 75 / +33 1 55 00 40 03 |
Portugal Janssen-Cilag Farmaceutica, Lda. Estrada Consiglieri Pedroso, 69 A Queluz de Baixo PT-2734-503 Barcarena Tel: +351 21 43 68 835 |
|
Hrvatska Johnson & Johnson S.E. d.o.o. Oreškoviéeva 6h 10010 Zagreb Tel: +385 1 6610 700 |
Románia Johnson & Johnson Románia SRL Str. Tipografilor nr. 11-15 Cládirea S-Park, Corp B3-B4, Etaj 3 013714 Bucure§ti, ROMÁNIA Tel: +40 21 207 1800 |
|
Ireland Janssen-Cilag Ltd. 50-100 Holmers Farm Way High Wycombe Buckinghamshire HP12 4EG United Kingdom Tel: +44 1 494 567 444 |
Slovenija Johnson & Johnson d.o.o. Šmartinska cesta 53 SI-1000 Ljubljana Tel: +386 1 401 18 30 |
|
Ísland Janssen-Cilag AB c/o Vistor hf. Horgatúni 2 IS-210 Garóab^r Sími: +354 535 7000 |
Slovenská republika Johnson & Johnson s.r.o. CBC III, Karadžičova 12 SK-821 08 Bratislava Tel: +421 232 408 400 |
|
Italia Janssen-Cilag SpA Via M.Buonarroti, 23 I-20093 Cologno Monzese MI Tel: +39 02 2510 1 |
Suomi/Finland Janssen-Cilag Oy Vaisalantie/Vaisalavágen 2 FI-02130 Espoo/Esbo Puh/Tel: +358 207 531 300 |
|
Kúnpoq BapváPag XarZpnavaypg AtS, Asro^ópog r lávvou KpavrSrróxp 226 Aaxará CY-2234 AsuK^oía TpA,: +357 22 207 700 |
Sverige Janssen-Cilag AB Box 4042 SE-16904 Solna Tel: +46 8 626 50 00 |
|
Latvija UAB "JOHNSON & JOHNSON" filiale Latvija Mukusalas iela 101 Riga, LV-1004 Tel: +371 678 93561 |
United Kingdom Janssen-Cilag Ltd. 50-100 Holmers Farm Way High Wycombe Buckinghamshire HP12 4EG - UK Tel: +44 1 494 567 444 |
Tato příbalová informace byla naposledy revidována {MM/RRRR}.
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/.
36