Ebixa 5 Mg/Dávka
SOUHRN ÚDAJŮ O PŘÍPRAVKU
PŘÍLOHA I
Ebixa 10 mg potahované tablety
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna potahovaná tableta obsahuje memantini hydrochloridum 10 mg, což odpovídá 8,31 mg memantinu.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA Potahovaná tableta.
Světle žlutá až žlutá, oválná potahovaná tableta s půlící rýhou a označením “1 0“ na jedné straně a “M M“ na druhé straně.
Tabletu lze rozdělit na stejné dávky.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba dospělých pacientů se střední až těžkou formou Alzheimerovy choroby.
4.2 Dávkování a způsob podání
Léčbu musí zahájit a dohlížet na ni lékař se zkušeností s diagnostikou a léčbou demence Alzheimerova typu.
Dávkování
Podmínkou zahájení léčby je dostupnost pečovatele, který pravidelně sleduje užívání léčivého přípravku pacientem. Diagnóza musí být stanovena podle soudobých diagnostických postupů. Snášenlivost a dávkování memantinu by měly být pravidelně posuzovány, nejlépe během tří měsíců po zahájení terapie. Klinický přínos memantinu a snášenlivost léčby pacientem by měly být nadále pravidelně vyhodnocovány podle současných doporučení pro léčbu. Udržovací terapie memantinem může pokračovat, dokud je přínosná a pacientem snášená. Ukončení léčby memantinem by mělo být zváženo, pokud není terapeutický účinek již patrný nebo pokud pacient léčbu přestal snášet.
Dospělí:
Titrace dávky
Maximální denní dávka je 20 mg. V zájmu snížení rizika výskytu nežádoucích účinků by se mělo udržovací dávky dosáhnout postupným zvyšováním denní dávky po 5 mg týdně během prvních 3 týdnů léčby takto:
Týden 1 (den 1-7)
Pacient by měl užívat polovinu 10 mg potahované tablety (5 mg) denně po dobu 7 dnů.
Týden 2 (den 8-14)
Pacient by měl užívat jednu 10 mg potahovanou tabletu (10 mg) denně po dobu 7 dnů.
Týden 3 (den 15-21)
Pacient by měl užívat jeden a půl 10 mg potahované tablety (15 mg) denně po dobu 7 dnů.
Od týdne 4 dále
Pacient by měl užívat dvě 10 mg potahované tablety (20 mg) denně.
Udržovací dávka
Doporučená udržovací dávka je 20 mg denně.
Starší osoby
Na základě poznatků z klinických studií je doporučená dávka pro pacienty starší 65 let 20 mg denně (dvě 10 mg potahované tablety jednou denně), jak je uvedeno výše.
Snížená funkce ledvin
U pacientů s mírnou poruchou funkce ledvin (clearance kreatininu 50 - 80 ml/min) není třeba upravovat dávku. U pacientů se středně závažnou poruchou funkce ledvin (clearance kreatininu 30 -49 ml/min) by denní dávka měla být 10 mg. Pokud je tato dávka pacientem minimálně týden dobře snášena, může být dle schématu nastavování dávky zvýšena na 20 mg denně. U pacientů s těžkou poruchou funkce ledvin (clearance kreatininu 5 - 29 ml/min) by denní dávka měla být 10 mg.
Snížená funkce jater
U pacientů s mírně až středně závažnou poruchou funkce jater (Child-Pugh A a Child-Pugh B) není třeba upravovat dávku. Nejsou k dispozici údaje o užívání memantinu u pacientů se závažnou poruchou funkce jater. Podávání přípravku Ebixa není doporučeno u pacientů se závažnou poruchou funkce jater.
Pediatrická populace Nejsou dostupné žádné údaje.
Způsob podání
Přípravek Ebixa by se měl podávat perorálně jednou denně a měl by se užívat ve stejnou dobu každý den. Potahované tablety se mohou užívat s jídlem nebo bez jídla.
4.3 Kontraindikace
Hypersensitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1..
4.4 Zvláštní upozornění a opatření pro použití
Opatrnost je doporučována u pacientů s epilepsií, s předchozí anamnézou křečí nebo u pacientů s predispozičními faktory pro epilepsii.
Neměla by probíhat současná léčba antagonisty N-methyl-D-aspartátu (NMDA), jako jsou amantadin, ketamin nebo dextromethorfan. Tyto léčivé látky působí na stejném receptorovém systému jako memantin, nežádoucí účinky (hlavně související s centrálním nervovým systémem (CNS)) by tudíž mohly být častější nebo výraznější (viz bod 4.5).
Přítomnost některých faktorů, jež mohou zvýšit pH moči (viz bod 5.2 Eliminace), vyžaduje pečlivé sledování pacienta. Tyto faktory zahrnují: zásadní změny stravovacích zvyklostí, např. přechod z masité stravy na vegetariánskou nebo požití velkého množství alkalizujících žaludečních pufrů. Zvýšení pH moči může nastat též při renální tubulární acidóze (RTA) nebo při závažné infekci močových cest způsobené bakterií rodu Proteus.
Z většiny klinických studií byli vyloučeni pacienti s nedávno prodělaným infarktem myokardu, nekompenzovaným městnavým srdečním selháním (NYHA III-IV) nebo neléčenou hypertenzí. Proto jsou u těchto pacientů pouze omezené zkušenosti a případná léčba by měla probíhat za jejich pečlivého sledování.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Vzhledem k farmakologickému působení a mechanismu účinku memantinu mohou nastat tyto interakce:
• Mechanismus účinku naznačuje, že účinky L-dopy, dopaminergních agonistů a anticholinergik se mohou zvýšit při současné léčbě antagonisty NMDA, mezi něž patří memantin. Účinek barbiturátů a neuroleptik se může snížit. Při současném podání memantinu s myorelaxancii dantrolenem nebo baklofenem může dojít k ovlivnění jejich účinku, což může vyžadovat úpravu dávky.
• Současné užití memantinu a amantadinu není vhodné, vzhledem k riziku farmakotoxické psychózy. Obě léčivé látky jsou chemicky podobní antagonisté NMDA. To může platit též pro ketamin a dextromethorfan (viz bod 4.4). Byla publikována jedna kasuistika vztahující se k možnému riziku kombinace memantin a fenytoin.
• Některé další léčivé látky, jako cimetidin, ranitidin, prokainamid, chinidin, chinin a nikotin, které využívají stejný kationtový transportní systém v ledvinách jako amantadin, mohou případně interagovat s memantinem, což vede k možnému riziku zvýšení plazmatických hladin.
• Existuje možnost sníženého vylučování hydrochlorothiazidu v séru, pokud je memantin užíván společně s hydrochlorothiazidem nebo s jakoukoli kombinací, která hydrochlorothiazid obsahuje.
• V postmarketingových studiích bylo zaznamenáno několik ojedinělých případů zvýšení hodnoty mezinárodního normalizovaného poměru (INR) u pacientů užívajících současně warfarin.
Ačkoli nebyla nalezena přímá souvislost, doporučuje se pečlivé sledování protrombinového času nebo INR u pacientů současně léčených perorálními antikoagulancii.
Ve studiích farmakokinetiky (FK) při podávání jednotlivé denní dávky mladým zdravým dobrovolníkům nebyla prokázána interakce léčivá látka - léčivá látka při současném užívání glyburidu/metforminu nebo donepezilu.
V klinických studiích mladých zdravých dobrovolníků nebyl prokázán případný vliv memantinu na farmakokinetiku galantaminu.
Memantin neinhibuje in vitro žádný z těchto systémů: CYP 1A2, 2A6, 2C9, 2D6, 2E1, 3A, monooxygenázu s flavinem, epoxidhydrolázu ani sulfatační pochody.
4.6 Fertilita, těhotenství a kojení
Údaje o podávání memantinu těhotným ženám jsou omezené nebo nejsou k dispozici. Studie na zvířatech naznačují možnost zpomalení nitroděložního růstu při dávkách identických nebo mírně vyšších než těch, které jsou užívány u lidí (viz bod 5.3). Míra případného rizika u lidí není známa. Memantin by se neměl v těhotenství užívat, pokud to není zcela nezbytné.
Kojení
Není známo, zda se memantin vylučuje do mateřského mléka, ovšem pokud se uváží lipofilita léčivé látky, je průnik do mateřského mléka pravděpodobný. Ženy užívající memantin by neměly kojit.
Fertilita
Nebyly zaznamenány žádné nežádoucí účinky memantinu na mužskou a ženskou fertilitu.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Střední až těžká forma Alzheimerovy choroby obvykle narušuje schopnost řízení motorových vozidel a omezuje ovládání strojů. Navíc Ebixa má malý nebo střední vliv na schopnost řídit a obsluhovat stroje, takže by ambulantní pacienti měli být upozorněni, aby věnovali řízení vozidel a ovládání strojů zvýšenou pozornost.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Do klinických studií byli zahrnuti pacienti s mírnou až těžkou demencí; z toho 1784 pacientů bylo léčeno přípravkem Ebixa a 1595 pacientů užívalo placebo. Celkový výskyt nežádoucích účinků se nelišil u pacientů užívajících přípravek Ebixa v porovnání s pacienty užívajícími placebo. Nežádoucí účinky byly mírné až střední závažnosti. Nejčastější nežádoucí účinky, jejichž frekvence výskytu byla vyšší ve skupině léčené přípravkem Ebixa v porovnání se skupinou užívající placebo, byly: závratě (6,3% v porovnání s 5,6%), bolest hlavy (5,2% v porovnání s 3,9%), zácpa (4,6% v porovnání s 2,6%), somnolence (3,4% v porovnání s 2,2%) a hypertenze (4,1% v porovnání s 2,8%).
Tabulkový seznam nežádoucích účinků
Nežádoucí účinky uvedené v tabulce se vyskytly v klinických studiích s přípravkem Ebixa nebo po jeho uvedení na trh.
Nežádoucí účinky jsou seřazeny podle tříd orgánových systémů s použitím následující klasifikace: velmi časté (>1/10), časté (>1/100, <1/10), méně časté (>1/1 000, <1/100), vzácné (>1/10 000, <1/1 000), velmi vzácné (<1/10 000), není známo (z dostupných údajů nelze určit). V každé skupině četnosti výskytu jsou nežádoucí účinky seřazeny dle klesající závažnosti.
|
TŘÍDY ORGÁNOVÝCH SYSTÉMŮ |
ČETNOST |
NEŽÁDOUCÍ ÚČINEK |
|
Infekce a infestace |
Méně časté |
Mykotické infekce |
|
Poruchy imunitního systému |
Časté |
Přecitlivělost na přípravek |
|
Psychiatrické poruchy |
Časté |
Somnolence |
|
Méně časté | ||
|
Méně časté | ||
|
Není známo |
Psychotické reakce2 | |
|
Poruchy nervového systému |
Časté |
Závratě |
|
Časté |
Poruchy rovnováhy | |
|
Méně časté |
Poruchy chůze | |
|
Velmi vzácné | ||
|
Srdeční poruchy |
Méně časté |
Srdeční selhání |
|
Cévní poruchy |
Časté |
Hypertenze |
|
Méně časté |
Žilní trombóza/trombembolismus | |
|
Respirační, hrudní a mediastinální poruchy |
Časté | |
|
Gastrointestinální poruchy |
Časté |
Zácpa |
|
Méně časté | ||
|
Není známo |
Pankreatitida2 | |
|
Poruchy jater a žlučových cest |
Časté |
Zvýšené hodnoty jaterních testů |
|
Není známo |
Hepatitida | |
|
Celkové poruchy a reakce v místě aplikace |
Časté | |
|
Méně časté |
Únava |
'Halucinace byly pozorovány častěji u pacientů s těžkou Alzheimerovou chorobou.
2Ojedinělá hlášení z postmarketingových studií.
Alzheimerova choroba bývá spojována s výskytem deprese, sebevražedných představ a sebevraždy.
V postmarketingových studiích byly tyto účinky hlášeny u pacientů léčených přípravkem Ebixa.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Zkušenosti s předávkováním v klinických studiích nebo po uvedení přípravku na trh jsou pouze omezené.
Známky předávkování:
v případě významného předávkování (200 mg a 105 mg/den, po dobu 3 dnů) se objevily pouze příznaky: únava, slabost a/nebo diarea či předávkování proběhlo bez příznaků. Při požití dávek, které nepřesáhly 140 mg či nebyly známé, se projevilo ovlivnění centrálního nervového systému (zmatenost, otupělost, somnolence, vertigo, agitovanost, agresivita, halucinace a poruchy chůze) a/nebo trávicího traktu (zvracení a diarea).
V případě nejvyššího předávkování pacient přežil požití úhrnné dávky 2000 mg memantinu se známkami ovlivnění centrálního nervového systému (kóma trvající 10 dní, později diplopie a agitovanost). Pacientovi byla poskytnuta symptomatická léčba a plazmaferéza. Pacient přežil bez následků.
V případě jiného významného předávkování pacient požil 400 mg memantinu perorálně a uzdravil se bez následků. U pacienta se objevily příznaky ovlivnění funkce centrálního nervového systému: neklid, psychóza, zrakové halucinace, zvýšená pohotovost ke křečím, somnolence, stupor a bezvědomí.
Léčba:
v případě předávkování je léčba symptomatická. Neexistuje specifické antidotum. Mohou být užity standardní lékařské postupy k odstranění léčivé látky, např. gastrická laváž, podání aktivního uhlí (přerušení případného enterohepatálního oběhu), acidifikace moči a forsírovaná diuréza.
Pokud se projeví známky a příznaky nadměrné stimulace centrálního nervového systému (CNS), měla by být pečlivě zvážena symptomatická léčba.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Psychoanaleptika. Ostatní léky proti demenci, ATC kód: N06DX01
Přibývají důkazy, že narušená činnost glutamátergní neurotransmise, zvláště na NMDA receptorech, přispívá k projevu příznaků a postupné progresi onemocnění v neurodegenerativní demenci.
Memantin je nekompetitivní antagonista receptorů NMDA, závislý na napětí, se středně silnou afinitou. Upravuje účinky patologicky zvýšené excitačně působící hladiny glutamátu, která může vést k dysfunkci neuronů.
Klinické studie
Do stěžejní klinické studie monoterapie memantinem bylo zahrnuto 252 ambulantních pacientů se střední až těžkou formou Alzheimerovy choroby (celkové skóre Mini Mental State Examination -MMSE před léčbou 3 - 14). Studie prokázala příznivý vliv 6měsíční léčby memantinem ve srovnání s placebem (analýza pozorovaných případů dle Clinicians Interview Based Impression of Change (CIBIC-plus): p=0,025; Alzheimeťs Disease Cooperative Study - Activities of Daily Living (ADCS-ADLsev): p=0,003; Severe Impairment Battery (SIB): p=0,002).
Do stěžejní klinické studie léčby pacientů s mírnou až střední formou Alzheimerovy choroby (celkové skóre MMSE před léčbou 10-22) monoterapií memantinem bylo zahrnuto 403 pacientů. Pacienti léčení memantinem vykazovali statisticky významně lepší účinek oproti pacientům užívajícím placebo na primární cílové parametry ve 24. týdnu (Last Observation Carried Forward, LOCF): Alzheimeťs Disease Assessment Scale (ADAS-cog) (p=0,003) a CIBIC-plus (p=0,004). Do jiné monoterapeutické studie mírné až střední formy Alzheimerovy choroby bylo náhodně zařazeno 470 pacientů (celkové skóre MMSE před léčbou 11 - 23). V prospektivně definované primární analýze nebylo ve 24. týdnu dosaženo statistické významnosti v ovlivnění primárních cílových parametrů účinnosti.
Meta-analýza 6 placebem kontrolovaných, 6měsíčních studií fáze III se střední až těžkou formou Alzheimerovy choroby (celkové skóre MMSE před léčbou < 20) prokázala statisticky významný příznivý účinek memantinu ve třech oblastech: kognitivní, celkové a funkční; přičemž v meta-analýze byli zahrnuti pacienti léčeni pouze memantinem nebo současně stabilní dávkou inhibitorů acetylcholinesterázy. Pokud u pacientů docházelo ke zhoršení ve všech třech oblastech, výsledky ukázaly statisticky významný rozdíl účinku; ke zhoršení ve všech třech oblastech docházelo dvakrát častěji u pacientů užívajících placebo ve srovnání s pacienty léčenými memantinem, který působí preventivně proti zhoršení (21% v porovnání s 11%, p<0,0001).
5.2 Farmakokinetické vlastnosti
Absorpce
Memantin má absolutní biologickou dostupnost přibližně 100%. tmax je 3 až 8 hodin. Nic nenasvědčuje ovlivnění absorpce memantinu potravou.
Distribuce
Při denních dávkách 20 mg se plazmatická koncentrace memantinu v ustáleném stavu pohybuje v rozmezí 70-150 ng/ml (0,5-1 pmol) s velkými interindividuálními odchylkami. Při užívání denních dávek v rozmezí 5-30 mg byla vypočítána průměrná hodnota poměru mozkomíšní mok (CSF)/sérum ve výši 0,52. Distribuční objem je zhruba 10 l/kg. Přibližně 45 % memantinu se váže na plazmatické bílkoviny.
Biotransformace
V krevním oběhu člověka se nachází 80 % memantinu v nezměněné formě. Hlavními metabolity v organismu člověka jsou N-3,5-dimethyl-gludantan, směs isomerů 4- a 6-hydroxy-memantinu a 1-nitroso-3,5-dimethyl-adamantan. Žádný z těchto metabolitů nevykazuje aktivitu NMDA antagonisty.
In vitro nebyl zjištěn žádný metabolický pochod katalyzovaný cytochromem P 450.
Ve studii p.o. podání značeného memantinu 14C bylo průměrně 84 % podané dávky detekováno během 20 dnů, více než 99 % se vyloučilo ledvinami.
Eliminace
Eliminace memantinu probíhá podle jednoduché exponenciální křivky s terminálním poločasem t/2 60 až 100 hodin. U dobrovolníků s normální funkcí ledvin činí celková clearance (Cltot)
170 ml/min/1,73 m2 a je částečně dosažena tubulární sekrecí.
V ledvinách dochází též k tubulární reabsorpci, pravděpodobně zprostředkované kationtovými transportními proteiny. Podíl renální eliminace memantinu v prostředí zásadité moči se může snížit o koeficient 7-9 (viz bod 4.4). Zásaditá moč může být následkem zásadní změny stravovacích zvyklostí, např. při přechodu z masité stravy na vegetariánskou nebo při požití velkého množství alkalizujících žaludečních pufrů.
Linearita
Studie u dobrovolníků prokázaly lineární farmakokinetiku v dávkovém rozmezí 10-40 mg. Farmakokinetické/farmakodynamické vztahy
Při dávce 20 mg denně dosahují hladiny memantinu v CSF hodnoty inhibiční konstanty memantinu (ki), která je 0,5 pmol v mozkové kůře čelního laloku člověka.
5.3 Předklinické údaje vztahující se k bezpečnosti
V krátkodobých studiích na potkanech způsobuje memantin podobně jako jiní antagonisté NMDA neuronální vakuolizaci a nekrózu (Olneyovy léze) pouze při dávkách, které vedou k velmi vysokým maximálním sérovým koncentracím. Vakuolizaci a nekróze předcházela ataxie a jiné preklinické známky. Jelikož tyto jevy nebyly pozorovány při dlouhodobých studiích s hlodavci ani s jinými živočišnými druhy, není znám jejich význam pro klinickou praxi.
Oftalmologické nálezy byly rozporně zjištěny ve studiích toxicity po opakovaném podání u hlodavců a psů, nikoli však u opic. Při specifických oftalmoskopických vyšetřeních v rámci klinických studií s memantinem nebyly objeveny žádné oční změny.
U hlodavců byla pozorována fosfolipidóza u plicních makrofágů způsobená hromaděním memantinu v lyzozomech. Tento jev je znám i u jiných léčivých látek s kationtovými amfifilními vlastnostmi. Existuje možnost souvislosti mezi kumulací memantinu a vakuolizací pozorovanou v plicích. Tento jev byl pozorován jen při vysokých dávkách u hlodavců. Klinický význam těchto zjištění není znám.
Standardní testování memantinu neprokázalo jeho genotoxicitu. V dlouhodobých (celoživotních) studiích prováděných na myších a potkanech nebyly nalezeny důkazy pro kancerogenitu. Memantin nebyl teratogenní u potkanů a králíků ani při dávkách toxických pro březí samice a neprokázal žádný nepříznivý vliv na plodnost. U potkanů byl zaznamenán pomalejší růst plodu při dávkách stejných nebo mírně vyšších, než které jsou užívány u lidí.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Jádro tablety:
Mikrokrystalická celulosa Sodná sůl kroskarmelosy Koloidní bezvodý oxid křemičitý Magnesium-stearát
Potah tablety:
Hypromelosa Makrogol 400 Oxid titaničitý Žlutý oxid železitý
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
4 roky.
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
6.5 Druh obalu a velikost balení
Blistr: PVDC/PE/PVC/Al-blistr nebo PP/Al-blistr
Velikosti balení: 14, 28, 30, 42, , 50, 56, , 70, 84, 98, , 100, , 112 potahovaných tablet.
Multipack obsahuje 980 (10 balení každé obsahující 98) nebo 1000 (20 balení každé obsahující 50) potahovaných tablet.
Perforované jednodávkové blistry: PVDC/PE/PVC/Al-blistr nebo PP/Al-blistr Velikosti balení: 49 x 1, 56 x 1, 98 x 1 a 100 x 1 potahovaných tablet.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku
Žádné zvláštní požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
H. Lundbeck A/S Ottiliavej 9 2500 Valby Dánsko
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/02/219/001-003 EU/1/02/219/007-012 EU/1/02/219/014-021
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 15. května 2002 Datum prodloužení registrace: 15. května 2007
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
NÁZEV PŘÍPRAVKU
1.
Ebixa 5 mg/dávka, perorální roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedno stlačení pumpy odměří 0,5 ml roztoku obsahujícího memantini hydrochloridum 5 mg, což odpovídá 4,16 mg memantinu.
Pomocné látky se známým účinkem: Jeden mililitr roztoku obsahuje 100 mg sorbitolu E420 a 0,5 mg draslíku, viz bod 4.4.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Perorální roztok.
Čirý, bezbarvý až slabě nažloutlý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba dospělých pacientů se střední až těžkou formou Alzheimerovy choroby.
4.2 Dávkování a způsob podání
Léčbu musí zahájit a dohlížet na ni lékař se zkušeností s diagnostikou a léčbou demence Alzheimerova typu.
Dávkování
Podmínkou zahájení léčby je dostupnost pečovatele, který pravidelně sleduje užívání léčivého přípravku pacientem. Diagnóza musí být stanovena podle soudobých diagnostických postupů. Snášenlivost a dávkování memantinu by měly být pravidelně posuzovány, nejlépe během tří měsíců po zahájení terapie. Klinický přínos memantinu a snášenlivost léčby pacientem by měly být nadále pravidelně vyhodnocovány podle současných doporučení pro léčbu. Udržovací terapie memantinem může pokračovat, dokud je přínosná a pacientem snášená. Ukončení léčby memantinem by mělo být zváženo, pokud není terapeutický účinek již patrný nebo pokud pacient léčbu přestal snášet.
Dospělí
Titrace dávky
Maximální denní dávka je 20 mg jednou denně. V zájmu snížení rizika výskytu nežádoucích účinků by se mělo udržovací dávky dosáhnout postupným zvyšováním denní dávky po 5 mg týdně během prvních 3 týdnů léčby takto:
Týden 1 (den 1-7)
Pacient by měl užívat 0,5 ml roztoku (5 mg), což odpovídá jednomu stlačení pumpy jednou denně po dobu 7 dnů.
Týden 2 (den 8-14)
Pacient by měl užívat 1 ml roztoku (10 mg), což odpovídá dvěma stlačením pumpy, jednou denně po dobu 7 dnů.
Týden 3 (den 15-21)
Pacient by měl užívat 1,5 ml roztoku (15 mg), což odpovídá třem stlačením pumpy, jednou denně po dobu 7 dnů.
Od týdne 4 dále
Pacient by měl užívat 2 ml roztoku (20 mg), což odpovídá čtyřem stlačením pumpy, jednou denně.
Udržovací dávka
Doporučená udržovací dávka je 20 mg denně.
Starší osoby
Na základě poznatků z klinických studií je doporučená dávka pro pacienty starší 65 let 20 mg denně (2 ml roztoku, což odpovídá čtyřem stlačením pumpy), jak je uvedeno výše.
Snížená funkce ledvin
U pacientů s mírnou poruchou funkce ledvin (clearance kreatininu 50 - 80 ml/min) není třeba upravovat dávku. U pacientů se středně závažnou poruchou funkce ledvin (clearance kreatininu 30 -49 ml/min) by denní dávka měla být 10 mg (1 ml roztoku, což odpovídá dvěma stlačením pumpy). Pokud je tato dávka pacientem minimálně týden dobře snášena, může být dle schématu nastavování dávky zvýšena na 20 mg denně. U pacientů s těžkou poruchou funkce ledvin (clearance kreatininu 5 -29 ml/min) by denní dávka měla být 10 mg (1 ml roztoku, což odpovídá dvěma stlačením pumpy) denně.
Snížená funkce jater
U pacientů s mírně až středně závažnou poruchou funkce jater (Child-Pugh A a Child-Pugh B) není třeba upravovat dávku. Nejsou k dispozici údaje o užívání memantinu u pacientů se závažnou poruchou funkce jater. Podávání přípravku Ebixa není doporučeno u pacientů se závažnou poruchou funkce jater.
Pediatrická populace Nejsou dostupné žádné údaje.
Způsob podání
Přípravek Ebixa by se měl užívat perorálně jednou denně a to ve stejnou dobu každý den. Roztok se může užívat s jídlem nebo bez jídla. Roztok nesmí být aplikován do úst přímo z lahvičky nebo pumpy, ale měl by být dávkován pomocí pumpy na lžičku nebo do sklenice s vodou.
Podrobný návod pro přípravu a zacházení s přípravkem viz bod 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Opatrnost je doporučována u pacientů s epilepsií, s předchozí anamnézou křečí nebo u pacientů s predispozičními faktory pro epilepsii.
Neměla by probíhat současná léčba jinými antagonisty N-methyl-D-aspartátu (NMDA), jako jsou amantadin, ketamin nebo dextromethorfan. Tyto léčivé látky působí na stejném receptorovém systému jako memantin, nežádoucí účinky (hlavně v oblasti centrálního nervového systému (CNS)) by tudíž mohly být častější nebo výraznější (viz bod 4.5).
Přítomnost některých faktorů, jež mohou zvýšit pH moči (viz bod 5.2 Eliminace), vyžaduje pečlivé sledování pacienta. Tyto faktory zahrnují: zásadní změny stravovacích zvyklostí, např. přechod z masité stravy na vegetariánskou nebo požití velkého množství alkalizujících žaludečních pufrů. Zvýšení pH moči může nastat též při renální tubulární acidóze (RTA) nebo při závažné infekci močových cest způsobené bakterií rodu Próteus.
Z většiny klinických studií byli vyloučeni pacienti s nedávno prodělaným infarktem myokardu, nekompenzovaným městnavým srdečním selháním (NYHA III-IV) nebo neléčenou hypertenzí. Proto jsou u těchto pacientů pouze omezené zkušenosti a případná léčba by měla probíhat za jejich pečlivého sledování.
Pomocné látky: perorální roztok obsahuje sorbitol. Pacienti trpící vrozenou intolerancí fruktosy nesmí užívat tento přípravek.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Vzhledem k farmakologickému působení a mechanismu účinku memantinu mohou nastat tyto interakce:
• Mechanismus účinku naznačuje, že účinky L-dopy, dopaminergních agonistů a anticholinergik se mohou zvýšit při současné léčbě antagonisty NMDA, mezi něž patří memantin. Účinek barbiturátů a neuroleptik se může snížit. Při současném podání memantinu s myorelaxancii dantrolenem nebo baklofenem může dojít k ovlivnění jejich účinku, což může vyžadovat úpravu dávkování.
• Současné užití memantinu a amantadinu není vhodné, vzhledem k riziku farmakotoxické psychózy. Obě léčivé látky jsou chemicky podobní antagonisté NMDA. To může platit též pro ketamin a dextromethorfan (viz bod 4.4). Byla publikována jedna kasuistika vztahující se k možnému riziku kombinace memantin a fenytoin.
• Některé další léčivé látky, jako cimetidin, ranitidin, prokainamid, chinidin, chinin a nikotin, které využívají stejný kationtový transportní systém v ledvinách jako amantadin, mohou případně interagovat s memantinem, což vede k možnému riziku zvýšení plazmatických hladin.
• Existuje možnost sníženého vylučování hydrochlorothiazidu v séru, pokud je memantin užíván společně s hydrochlorothiazidem nebo s jakoukoli kombinací, která hydrochlorothiazid obsahuje.
• V postmarketingových studiích bylo zaznamenáno několik ojedinělých případů zvýšení hodnoty mezinárodního normalizovaného poměru (INR) u pacientů užívajících současně warfarin. Ačkoliv nebyla nalezena přímá souvislost, doporučuje se pečlivé sledování protrombinového času nebo INR u pacientů současně léčených perorálními antikoagulancii.
Ve studiích farmakokinetiky (FK) při podávání jednotlivé denní dávky mladým zdravým dobrovolníkům nebyla prokázána interakce účinná látka - účinná látka při současném užívání glyburidu/metforminu nebo donepezilu.
V klinických studiích mladých zdravých dobrovolníků nebyl prokázán případný vliv memantinu na farmakokinetiku galantaminu.
Memantin neinhibuje in vitro žádný z těchto systémů: CYP 1A2, 2A6, 2C9, 2D6, 2E1, 3A, monooxygenázu s flavinem, epoxidhydrolázu ani sulfatační pochody.
4.6 Fertilita, těhotenství a kojení
Údaje o podávání memantinu těhotným ženám jsou omezené nebo nejsou k dispozici. Studie na zvířatech naznačují možnost zpomalení nitroděložního růstu při dávkách identických nebo vyšších než těch, které jsou užívány u lidí (viz bod 5.3). Míra případného rizika u lidí není známa. Memantin by se neměl v těhotenství užívat, pokud to není zcela nezbytné.
Kojení
Není známo, zda se memantin vylučuje do mateřského mléka, ovšem pokud se uváží lipofilita léčivé látky, je průnik do mateřského mléka pravděpodobný. Ženy užívající memantin by neměly kojit.
Fertilita
Nebyly zaznamenány žádné nežádoucí účinky memantinu na mužskou a ženskou fertilitu.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Střední až těžká forma Alzheimerovy choroby obvykle narušuje schopnost řízení motorových vozidel a omezuje ovládání strojů. Navíc Ebixa může mít mírný až střední vliv na schopnost řídit a obsluhovat stroje, takže by ambulantní pacienti měli být upozorněni, aby věnovali řízení vozidel a ovládání strojů zvýšenou pozornost.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Do klinických studií byli zahrnuti pacienti s mírnou až těžkou demencí; z toho 1784 pacientů bylo léčeno přípravkem Ebixa a 1595 pacientů užívalo placebo. Celkový výskyt nežádoucích reakcí se nelišil u pacientů užívajících přípravek Ebixa v porovnání s pacienty užívajícími placebo. Nežádoucí účinky byly mírné až střední závažnosti. Nejčastější nežádoucí účinky, jejichž frekvence výskytu byla vyšší ve skupině léčené přípravkem Ebixa v porovnání se skupinou přijímající placebo, byly: závratě (6,3% v porovnání s 5,6%), bolest hlavy (5,2% v porovnání s 3,9%), zácpa (4,6% v porovnání s 2,6%), somnolence (3,4% v porovnání s 2,2%) a hypertenze (4,1% v porovnání s 2,8%).
Tabulkový seznam nežádoucích účinků
Nežádoucí účinky uvedené v tabulce se vyskytly v klinických studiích s přípravkem Ebixa nebo po jeho uvedení na trh.
Nežádoucí účinky jsou seřazeny podle tříd orgánových systémů s použitím následující klasifikace: velmi časté (>1/10), časté (>1/100, <1/10), méně časté (>1/1 000, <1/100), vzácné (>1/10 000, <1/1 000), velmi vzácné (<1/10 000), není známo (z dostupných údajů nelze určit). V každé skupině četnosti výskytu jsou nežádoucí účinky seřazeny dle klesající závažnosti.
|
TŘÍDY ORGÁNOVÝCH SYSTÉMŮ |
ČETNOST |
NEŽÁDOUCÍ ÚČINEK |
|
Infekce a infestace |
Méně časté |
Mykotické infekce |
|
Poruchy imunitního systému |
Časté |
Přecitlivělost na přípravek |
|
Psychiatrické poruchy |
Časté |
Somnolence |
|
Méně časté | ||
|
Méně časté | ||
|
Není známo |
Psychotické reakce2 | |
|
Poruchy nervového systému |
Časté |
Závratě |
|
Časté |
Poruchy rovnováhy | |
|
Méně časté |
Poruchy chůze | |
|
Velmi vzácné | ||
|
Srdeční poruchy |
Méně časté |
Srdeční selhání |
|
Cévní poruchy |
Časté |
Hypertenze |
|
Méně časté |
Žilní trombóza/trombembolismus | |
|
Respirační, hrudní a mediastinální poruchy |
Časté | |
|
Gastrointestinální poruchy |
Časté |
Zácpa |
|
Méně časté | ||
|
Není známo |
Pankreatitida2 | |
|
Poruchy jater a žlučových cest |
Časté |
Zvýšené hodnoty jaterních testů |
|
Není známo |
Hepatitia | |
|
Celkové poruchy a reakce v místě aplikace |
Časté | |
|
Méně časté |
Únava |
'Halucinace byly pozorovány častěji u pacientů s těžkou Alzheimerovou chorobou.
2Ojedinělá hlášení z postmarketingových studií.
Alzheimerova choroba bývá spojována s výskytem deprese, sebevražedných představ a sebevraždy.
V postmarketingových studiích byly tyto účinky hlášeny u pacientů léčených přípravkem Ebixa.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Zkušenosti s předávkováním v klinických studiích nebo po uvedení přípravku na trh jsou pouze omezené.
Známky předávkování
V případě významného předávkování (200 mg a 105 mg/den, po dobu 3 dnů) se objevily pouze příznaky: únava, slabost a/nebo diarea či předávkování proběhlo bez příznaků. Při požití dávek, které nepřesáhly 140 mg či nebyly známé, se projevilo ovlivnění centrálního nervového systému (zmatenost, otupělost, somnolence, vertigo, agitovanost, agresivita, halucinace a poruchy chůze) a/nebo trávicího traktu (zvracení a diarea).
V případě nejvyššího předávkování pacient přežil požití úhrnné dávky 2000 mg memantinu se známkami ovlivnění centrálního nervového systému (kóma trvající 10 dní, později diplopie a agitovanost). Pacientovi byla poskytnuta symptomatická léčba a plazmaferéza. Pacient přežil bez následků.
V případě jiného významného předávkování pacient požil 400 mg memantinu perorálně a uzdravil se bez následků. U pacienta se objevily příznaky ovlivnění funkce centrálního nervového systému: neklid, psychóza, zrakové halucinace, zvýšená pohotovost ke křečím, somnolence, stupor a bezvědomí.
Opatření při předávkování
V případě předávkování je léčba symptomatická. Neexistuje specifické antidotum. Mohou být užity standardní lékařské postupy k odstranění léčivé látky, např. gastrická laváž, podání aktivního uhlí (přerušení případného enterohepatálního oběhu), acidifikace moči a forsírovaná diuréza.
Pokud se projeví známky a příznaky nadměrné stimulace centrálního nervového systému (CNS), měla by být pečlivě zvážena symptomatická léčba.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Psychoanaleptika. Ostatní léky proti demenci, ATC kód: N06DX01
Přibývají důkazy, že narušená činnost glutamátergní neurotransmise, zvláště na NMDA receptorech, přispívá k projevu příznaků a postupné progresi onemocnění v neurodegenerativní demenci.
Memantin je nekompetitivní antagonista receptorů NMDA, závislý na napětí, se středně silnou afinitou. Upravuje účinky patologicky zvýšené excitačně působící hladiny glutamátu, která může vést k dysfunkci neuronů.
Klinické studie
Do stěžejní klinické studie monoterapie memantinem bylo zahrnuto 252 ambulantních pacientů se střední až těžkou formou Alzheimerovy choroby (celkové skóre Mini Mental State Examination -MMSE před léčbou 3 - 14). Studie prokázala příznivý vliv 6měsíční léčby memantinem ve srovnání s placebem (analýza pozorovaných případů dle Clinicans Interview Based Impression of Change (CIBIC-plus): p=0,025; Alzheimer's Disease Cooperative Study - activities of the daily living (ADCS-ADLsev): p=0,003; Severe Impairment Battery (SIB): p=0,002).
Do stěžejní klinické studie léčby pacientů s mírnou až střední formou Alzheimerovy choroby (celkové skóre MMSE před léčbou 10-22) monoterapií memantinem bylo zahrnuto 403 pacientů. Pacienti léčení memantinem vykazovali statisticky významně lepší účinek oproti pacientům užívajícím placebo na primární cílové parametry ve 24. týdnu (Last Observation Carried Forward, LOCF): Alzheimers disease assessment scale (ADAS-cog) (p=0,003) a CIBIC-plus (p=0,004). Do jiné monoterapeutické studie mírné až střední formy Alzheimerovy choroby bylo náhodně zařazeno 470 pacientů (celkové skóre MMSE před léčbou 11 - 23). V prospektivně definované primární analýze nebylo ve 24. týdnu dosaženo statistické významnosti v ovlivnění primárních cílových parametrů účinnosti.
Meta-analýza 6 placebem kontrolovaných, 6měsíčních studií fáze III se střední až těžkou formou Alzheimerovy choroby (celkové skóre MMSE před léčbou < 20) prokázala statisticky významný příznivý účinek memantinu ve třech oblastech: kognitivní, celkové a funkční; přičemž v meta-analýze byli zahrnuti pacienti léčeni pouze memantinem nebo současně stabilní dávkou inhibitorů acetylcholinesterázy. Pokud u pacientů docházelo ke zhoršení ve všech třech oblastech, výsledky ukázaly statisticky významný rozdíl účinku; ke zhoršení ve všech třech oblastech docházelo dvakrát častěji u pacientů užívajících placebo ve srovnání s pacienty léčenými memantinem, který působí preventivně proti zhoršení (21% v porovnání s 11%, p<0,0001).
5.2 Farmakokinetické vlastnosti
Absorpce
Memantin má absolutní biologickou dostupnost přibližně 100%. tmax je 3 až 8 hodin. Nic nenasvědčuje ovlivnění absorpce memantinu potravou.
Distribuce
Při denních dávkách 20 mg se plazmatická koncentrace memantinu v ustáleném stavu pohybuje v rozmezí 70-150 ng/ml (0,5-1 pmol) s velkými interindividuálními odchylkami. Při užívání denních dávek v rozmezí 5-30 mg byla vypočítána průměrná hodnota poměru mozkomíšní mok (CSF)/sérum ve výši 0,52. Distribuční objem je zhruba 10 l/kg. Přibližně 45 % memantinu se váže na plazmatické bílkoviny.
Biotransformace
V krevním oběhu člověka se nachází 80 % memantinu v nezměněné formě. Hlavními metabolity v organismu člověka jsou N-3,5-dimethyl-gludantan, směs isomerů 4- a 6-hydroxy-memantinu a 1-nitroso-3,5-dimethyl-adamantan. Žádný z těchto metabolitů nevykazuje aktivitu NMDA antagonisty.
In vitro nebyl zjištěn žádný metabolický pochod katalyzovaný cytochromem P 450.
Ve studii p.o. podání značeného memantinu 14C bylo průměrně 84 % podané dávky detekováno během 20 dnů, více než 99 % se vyloučilo ledvinami.
Eliminace
Eliminace memantinu probíhá podle jednoduché exponenciální křivky s terminálním poločasem t/2 60 až 100 hodin. U dobrovolníků s normální funkcí ledvin činí celková clearance (Cltot)
170 ml/min/1,73 m2 a je částečně dosažena tubulární sekrecí.
V ledvinách dochází též k tubulární reabsorpci, pravděpodobně zprostředkované kationtovými transportními proteiny. Podíl renální eliminace memantinu v prostředí zásadité moči se může snížit o koeficient 7-9 (viz bod 4.4). Zásaditá moč může být následkem zásadní změny stravovacích zvyklostí, např. při přechodu z masité stravy na vegetariánskou nebo při požití velkého množství alkalizujících žaludečních pufrů.
Linearita
Studie u dobrovolníků prokázaly lineární farmakokinetiku v dávkovém rozmezí 10-40 mg. Farmakokinetické/farmakodynamické vztahy
Při dávce 20 mg denně dosahují hladiny memantinu v CSF hodnoty inhibiční konstanty memantinu (ki), která je 0,5 pmol v mozkové kůře čelního laloku člověka.
5.3 Předklinické údaje vztahující se k bezpečnosti
V krátkodobých studiích na potkanech způsobuje memantin podobně jako jiní antagonisté NMDA neuronální vakuolizaci a nekrózu (Olneyovy léze) pouze při dávkách, které vedou k velmi vysokým maximálním sérovým koncentracím. Vakuolizaci a nekróze předcházela ataxie a jiné preklinické známky. Jelikož tyto jevy nebyly pozorovány při dlouhodobých studiích s hlodavci ani s jinými živočišnými druhy, není znám jejich význam pro klinickou praxi.
Oftalmologické nálezy byly rozporně zjištěny ve studiích toxicity po opakovaném podání u hlodavců a psů, nikoli však u opic. Při specifických oftalmoskopických vyšetřeních v rámci klinických studií s memantinem nebyly objeveny žádné oční změny.
U hlodavců byla pozorována fosfolipidóza u plicních makrofágů způsobená hromaděním memantinu v lyzozomech. Tento jev je znám i u jiných léčivých látek s kationtovými amfifilními vlastnostmi. Existuje možnost souvislosti mezi kumulací memantinu a vakuolizací pozorovanou v plicích. Tento jev byl pozorován jen při vysokých dávkách u hlodavců. Klinický význam těchto zjištění není znám.
Standardní testování memantinu neprokázalo jeho genotoxicitu. V dlouhodobých (celoživotních) studiích prováděných na myších a potkanech nebyly nalezeny důkazy pro kancerogenitu. Memantin nebyl teratogenní u potkanů a králíků ani při dávkách toxických pro březí samice a neprokázal žádný nepříznivý vliv na plodnost. U potkanů byl zaznamenán pomalejší růst plodu při dávkách stejných nebo mírně vyšších, než které jsou užívány u lidí.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam všech pomocných látek
Sorbitan draselný Sorbitol E420 Čištěná voda
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
4 roky.
Po prvním otevření je třeba obsah lahvičky spotřebovat během 3 měsíců.
6.4 Zvláštní opatření pro uchovávání Uchovávejte při teplotě do 30 °C.
Lahvička s připevněnou pumpou může být uchovávána a přepravována pouze ve svislé poloze.
6.5 Druh obalu a velikost balení
Lahvička z hnědého skla (hydrolytická třída III) obsahující 50 ml, 100 ml nebo 10 x 50 ml roztoku. Na trhu nemusí být všechny velikosti balení
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Žádné zvláštní požadavky.
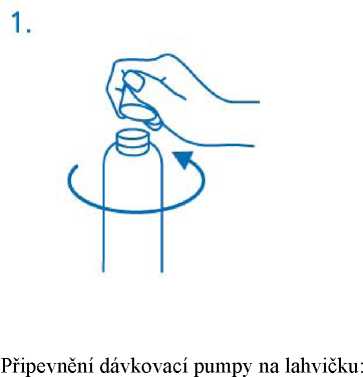
Před prvním použitím musí být dávkovací pumpa našroubována na lahvičku. K odstranění šroubovacího víčka z lahvičky se musí víčkem otočit proti směru hodinových ručiček a úplně odšroubovat (viz obr. 1).
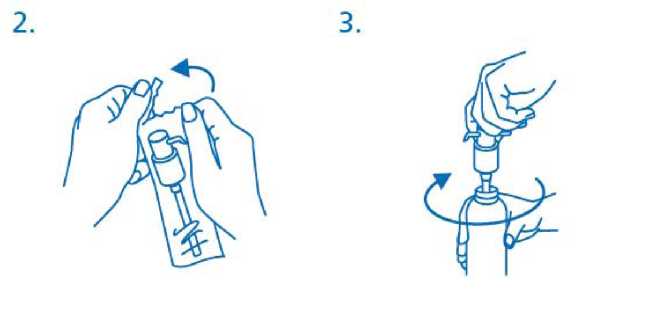
Po vyndání z plastikového sáčku (obr.2) se dávkovači pumpa umístí na vrchol lahvičky a opatrně se vsune plastiková trubička do lahvičky. Potom je třeba podržet dávkovači pumpu na hrdle lahvičky a šroubovat ve směru hodinových ručiček, dokud není pevně připojena (obr.3). Dávkovači pumpa se našroubuje pouze jednou při zahájení používání a neměla by být nikdy odšroubována.
Použití dávkovací pumpy k dávkování:
Hlava dávkovací pumpy má dvě polohy a lze jí snadno otáčet - proti směru hodinových ručiček (neuzamčená poloha) a ve směru hodinových ručiček (uzamčená poloha). Hlava dávkovací pumpy by neměla být stlačována dolů v uzamčené poloze. Roztok lze aplikovat pouze pokud je pumpa v neuzamčené poloze. Aby bylo možno roztok aplikovat, musí se hlava dávkovací pumpy otočit směrem k šipce asi jednu osminu otáčky až nadoraz (obr.4).
Dávkovači pumpa je připravena k použití.
Příprava dávkovači pumpy:
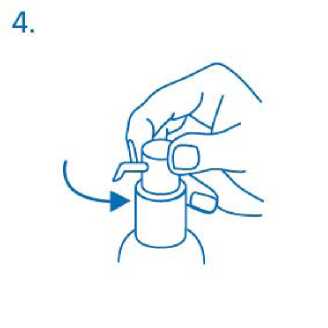
Dávkovači pumpa nedávkuje při prvním použiti přesné množství perorálniho roztoku. Proto musí být pumpa připravena k použiti stlačením hlavy dávkovači pumpy úplně dolů pětkrát za sebou (obr.5).
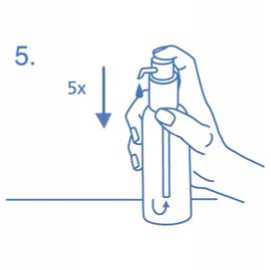
Tento vypumpovaný roztok se nepoužije pro léčbu a má být zlikvidován. Při dalším použiti je hlava dávkovači pumpy stlačena úplně dolů (odpovidá jednomu stlačeni pumpy) a je odměřena správná dávka (1 stlačeni pumpy odpovídá 0,5 ml perorálniho roztoku a obsahuje 5 mg léčivé látky memantini hydrochloridum; obr.6).
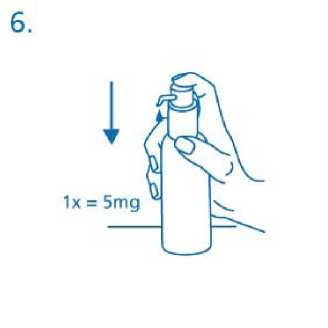
Správné použiti dávkovači pumpy:
Lahvička by měla být umístěna na vodorovnou pločhu, např. na stůl, a použita pouze ve svislé poloze. Skleniče s malým množstvim vody nebo lžička by měla být držena před tryskou a hlava dávkovači pumpy musi být stlačena pevným, ale klidným a stálým stiskem (ne přiliš pomalu) přimo dolů nadoraz (obr.7, obr.8).
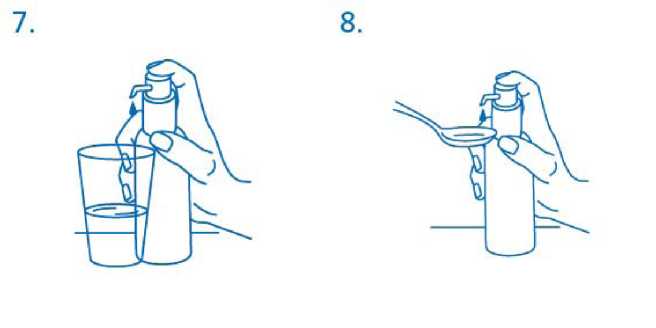
Hlava dávkovači pumpy může být poté uvolněna a je připravena k dalšímu stlačení pumpy.
Dávkovači pumpa může být použita pouze s roztokem memantini hydrochloridum, který je připravený v lahvičce. Nesmi se používat pro jiné látky nebo jiné obaly. Jestliže pumpa nefunguje, jak je popsáno v návodu, pacient by měl informovat svého ošetřujícího lékaře nebo lékárníka. Dávkovači pumpa by měla být po použití nastavena do uzamčené polohy.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
H. Lundbeck A/S Ottiliavej 9 2500 Valby Dánsko
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/02/219/005-006
EU/1/02/219/013
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 15. května 2002 Datum prodloužení registrace: 15. května 2007
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
NÁZEV PŘÍPRAVKU
1.
Ebixa 5 mg potahované tablety Ebixa 10 mg potahované tablety Ebixa 15 mg potahované tablety Ebixa 20 mg potahované tablety
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna potahovaná tableta obsahuje memantini hydrochloridum 5 mg, což odpovídá 4,15 mg memantinu.
Jedna potahovaná tableta obsahuje memantini hydrochloridum 10 mg, což odpovídá 8,31 mg memantinu.
Jedna potahovaná tableta obsahuje memantini hydrochloridum 15 mg, což odpovídá 12,46 mg memantinu.
Jedna potahovaná tableta obsahuje memantini hydrochloridum 20 mg, což odpovídá 16,62 mg memantinu.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Potahovaná tableta
5 mg potahované tablety jsou bílé až téměř bílé oválně podlouhlé potahované tablety s vytištěným „5“ na jedné straně a „MEM“ na straně druhé.
10 mg potahované tablety jsou světle žluté až žluté oválné potahované tablety s půlící rýhou a označením “ 1 0“ na jedné straně a “M M“ na druhé straně. Tabletu je možné rozdělit na stejné dávky.
15 mg potahované tablety jsou oranžové až šedooranžové oválně podlouhlé potahované tablety s vytištěným „15“ na jedné straně a „MEM“ na straně druhé.
20 mg potahované tablety jsou světle červené až šedočervené oválně podlouhlé potahované tablety s vytištěným „20“ na jedné straně a „MEM“ na straně druhé.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba dospělých pacientů se střední až těžkou formou Alzheimerovy choroby.
4.2 Dávkování a způsob podání
Léčbu musí zahájit a dohlížet na ni lékař se zkušeností s diagnostikou a léčbou demence Alzheimerova typu.
Dávkování
Podmínkou zahájení léčby je dostupnost pečovatele, který pravidelně sleduje užívání léčivého přípravku pacientem. Diagnóza musí být stanovena podle soudobých diagnostických postupů. Snášenlivost a dávkování memantinu by měly být pravidelně posuzovány, nejlépe během tří měsíců po zahájení terapie. Klinický přínos memantinu a snášenlivost léčby pacientem by měly být nadále pravidelně vyhodnocovány podle současných doporučení pro léčbu. Udržovací terapie memantinem může pokračovat, dokud je přínosná a pacientem snášená. Ukončení léčby memantinem by mělo být zváženo, pokud není terapeutický účinek již patrný nebo pokud pacient léčbu přestal snášet.
Titrace dávky
Doporučená úvodní dávka je 5 mg denně, která je postupně zvyšována během prvních 4 týdnů léčby a dosahuje doporučenou udržovací dávku následujícím způsobem:
Týden 1 (den 1-7)
Pacient by měl užít jednu 5 mg potahovanou tabletu jednou denně (bílé až téměř bílé oválně podlouhlé) po dobu 7 dnů.
Týden 2 (den 8-14)
Pacient by měl užít jednu 10 mg potahovanou tabletu jednou denně (světle žluté až žluté, oválné) po dobu 7 dnů.
Týden 3 (den 15 - 21)
Pacient by měl užít jednu 15 mg potahovanou tabletu jednou denně (šedooranžové, oválně podlouhlé) po dobu 7 dnů.
Týden 4 (den 22-28)
Pacient by měl užít jednu 20 mg potahovanou tabletu denně (šedočervené, oválně podlouhlé) po dobu 7 dnů
Maximální denní dávka je 20 mg denně.
Udržovací dávka
Doporučená udržovací dávka je 20 mg jednou denně.
Starší osoby
Na základě poznatků z klinických studií je doporučená dávka pro pacienty starší 65 let 20 mg denně (20 mg jednou denně), jak je uvedeno výše.
Snížená funkce ledvin: U pacientů s mírnou poruchou funkce ledvin (clearance kreatininu 50 - 80 ml/min) není třeba upravovat dávku. U pacientů se středně závažnou poruchou funkce ledvin (clearance kreatininu 30 - 49 ml/min) by denní dávka měla být 10 mg. Pokud je tato dávka pacientem minimálně týden dobře snášena, může být dle schématu nastavování dávky zvýšena na 20 mg denně.
U pacientů s těžkou poruchou funkce ledvin (clearance kreatininu 5 - 29 ml/min) by denní dávka měla být 10 mg.
Snížená funkce jater: U pacientů s mírně až středně závažnou poruchou funkce jater (Child-Pugh A a Child-Pugh B) není třeba upravovat dávku. Nejsou k dispozici údaje o užívání memantinu u pacientů se závažnou poruchou funkce jater. Podávání přípravku Ebixa není doporučeno u pacientů se závažnou poruchou funkce jater.
Pediatrická populace Nejsou dostupné žádné údaje.
Způsob podání
Přípravek Ebixa by se měl podávat perorálně jednou denně a měl by se užívat ve stejnou dobu každý den. Potahované tablety se mohou užívat s jídlem bez jídla.
4.3 Kontraindikace
Hypersensitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Opatrnost je doporučována u pacientů s epilepsií, s předchozí anamnézou křečí nebo u pacientů s predispozičními faktory pro epilepsii.
Neměla by probíhat současná léčba antagonisty N-methyl-D-aspartátu (NMDA), jako jsou amantadin, ketamin nebo dextromethorfan. Tyto léčivé látky působí na stejném receptorovém systému jako memantin, nežádoucí účinky (hlavně v oblasti centrálního nervového systému (CNS)) by tudíž mohly být častější nebo výraznější (viz bod 4.5).
Přítomnost některých faktorů, jež mohou zvýšit pH moči (viz bod 5.2 Eliminace), vyžaduje pečlivé sledování pacienta. Tyto faktory zahrnují: zásadní změny stravovacích zvyklostí, např. přechod z masité stravy na vegetariánskou, nebo požití velkého množství alkalizujících žaludečních pufrů. Zvýšení pH moči může nastat též při renální tubulární acidóze (RTA) nebo při závažné infekci močových cest způsobené bakterií rodu Próteus.
Z většiny klinických studií byli vyloučeni pacienti s nedávno prodělaným infarktem myokardu, nekompenzovaným městnavým srdečním selháním (NYHA III-IV) nebo neléčenou hypertenzí. Proto jsou u těchto pacientů pouze omezené zkušenosti, a případná léčba by měla probíhat za jejich pečlivého sledování.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Vzhledem k farmakologickému působení a mechanismu účinku memantinu mohou nastat tyto interakce:
• Mechanismus účinku naznačuje, že účinky L-dopy, dopaminergních agonistů a anticholinergik se mohou zvýšit při současné léčbě antagonisty NMDA, mezi něž patří memantin. Účinek barbiturátů a neuroleptik se může snížit. Při současném podání memantinu s myorelaxancii dantrolenem nebo baklofenem může dojít k ovlivnění jejich účinku, což může vyžadovat úpravu dávky.
• Současné užití memantinu a amantadinu není vhodné, vzhledem k riziku farmakotoxické psychózy. Obě léčivé látky jsou chemicky podobní antagonisté NMDA. To může platit též pro ketamin a dextromethorfan (viz bod 4.4). Byla publikována jedna kasuistika vztahující se k možnému riziku kombinace memantin a fenytoin.
• Některé další léčivé látky, jako cimetidin, ranitidin, prokainamid, chinidin, chinin a nikotin, které využívají stejný kationtový transportní systém v ledvinách jako amantadin, mohou případně interagovat s memantinem, což vede k možnému riziku zvýšení plazmatických hladin.
• Existuje možnost sníženého vylučování hydrochlorothiazidu v séru, pokud je memantin užíván společně s hydrochlorothiazidem nebo s jakoukoli kombinací, která hydrochlorothiazid obsahuje.
• V postmarketingových studiích bylo zaznamenáno několik ojedinělých případů zvýšení hodnoty mezinárodního normalizovaného poměru (INR) u pacientů užívajících současně warfarin. Ačkoliv nebyla nalezena přímá souvislost, doporučuje se pečlivé sledování protrombinového času nebo INR u pacientů současně léčených perorálními antikoagulancii.
Ve studiích farmakokinetiky (FK) při podávání jednotlivé denní dávky mladým zdravým dobrovolníkům nebyla prokázána interakce léčivá látka - léčivá látka při současném užívání glyburidu/metforminu nebo donepezilu.
V klinických studiích mladých zdravých dobrovolníků nebyl prokázán případný vliv memantinu na farmakokinetiku galantaminu.
Memantin neinhibuje in vitro žádný z těchto systémů: CYP 1A2, 2A6, 2C9, 2D6, 2E1, 3A, monooxygenázu s flavinem, epoxidhydrolázu ani sulfatační pochody.
4.6 Fertilita, těhotenství a kojení
Údaje o podávání memantinu těhotným ženám jsou omezené nebo nejsou k dispozici. Studie na zvířatech naznačují možnost zpomalení nitroděložního růstu při dávkách identických nebo mírně vyšších než těch, které jsou užívány u lidí (viz bod 5.3). Míra případného rizika u lidí není známa. Memantin by se neměl v těhotenství užívat, pokud to není zcela nezbytné.
Kojení
Není známo, zda se memantin vylučuje do mateřského mléka, ovšem pokud se uváží lipofilita léčivé látky, je průnik do mateřského mléka pravděpodobný. Ženy užívající memantin by neměly kojit.
Fertilita
Nebyly zaznamenány žádné nežádoucí účinky memantinu na mužskou a ženskou fertilitu.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Střední až těžká forma Alzheimerovy choroby obvykle narušuje schopnost řízení motorových vozidel a omezuje ovládání strojů. Navíc Ebixa má malý nebo střední vliv na schopnost řídit a obsluhovat stroje, takže by ambulantní pacienti měli být upozorněni, aby věnovali řízení vozidel a ovládání strojů zvýšenou pozornost.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Do klinických studií byli zahrnuti pacienti s mírnou až těžkou demencí; z toho 1784 pacientů bylo léčeno přípravkem Ebixa a 1595 pacientů užívalo placebo. Celkový výskyt nežádoucích účinků se nelišil u pacientů užívajících přípravek Ebixa v porovnání s pacienty užívajícími placebo. Nežádoucí účinky byly mírné až střední závažnosti. Nejčastější nežádoucí účinky, jejichž frekvence výskytu byla vyšší ve skupině léčené přípravkem Ebixa v porovnání se skupinou přijímající placebo, byly: závratě (6,3% v porovnání s 5,6%), bolest hlavy (5,2% v porovnání s 3,9%), zácpa (4,6% v porovnání s 2,6%), somnolence (3,4% v porovnání s 2,2%) a hypertenze (4,1% v porovnání s 2,8%).
V každé skupině jsou četnosti výskytu nežádoucích účinků seřazeny dle klesající závažnosti.
Tabulkový seznam nežádoucích účinků
Nežádoucí účinky uvedené v tabulce se vyskytly v klinických studiích s přípravkem Ebixa nebo po jeho uvedení na trh.
Nežádoucí účinky jsou seřazeny podle tříd orgánových systémů s použitím následující klasifikace: velmi časté (>1/10), časté (>1/100, <1/10), méně časté (>1/1 000, <1/100), vzácné (>1/10 000, <1/1 000), velmi vzácné (<1/10 000), není známo (z dostupných údajů nelze určit). V každé skupině četnosti výskytu jsou nežádoucí účinky seřazeny dle klesající závažnosti.
|
TŘÍDY ORGÁNOVÝCH SYSTÉMŮ |
ČETNOST |
NEŽÁDOUCÍ ÚČINEK |
|
Infekce a infestace |
Méně časté |
Mykotické infekce |
|
Poruchy imunitního systému |
Časté |
Přecitlivělost na přípravek |
|
Psychiatrické poruchy |
Časté |
Somnolence |
|
Méně časté | ||
|
Méně časté | ||
|
Není známo |
Psychotické reakce2 | |
|
Poruchy nervového systému |
Časté |
Závratě |
|
Časté |
Poruchy rovnováhy | |
|
Méně časté |
Poruchy chůze | |
|
Velmi vzácné | ||
|
Srdeční poruchy |
Méně časté |
Srdeční selhání |
|
Cévní poruchy |
Časté |
Hypertenze |
|
Méně časté |
Žilní trombóza/trombembolismus | |
|
Respirační, hrudní a mediastinální poruchy |
Časté | |
|
Gastrointestinální poruchy |
Časté |
Zácpa |
|
Méně časté | ||
|
Není známo |
Pankreatitida2 | |
|
Poruchy jater a žlučových cest |
Časté |
Zvýšené hodnoty jaterních testů |
|
Není známo |
Hepatitia | |
|
Celkové poruchy a reakce v místě aplikace |
Časté | |
|
Méně časté |
Únava |
'Halucinace byly pozorovány častěji u pacientů s těžkou Alzheimerovou chorobou.
2Ojedinělá hlášení z postmarketingových studií.
Alzheimerova choroba bývá spojována s výskytem deprese, sebevražedných představ a sebevraždy.
V postmarketingových studiích byly tyto účinky hlášeny u pacientů léčených přípravkem Ebixa.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Zkušenosti s předávkováním v klinických studiích nebo po uvedení přípravku na trh jsou pouze omezené.
Známky předávkování
V případě významného předávkování (200 mg a 105 mg/den, po dobu 3 dnů) se objevily pouze příznaky: únava, slabost a/nebo diarea či předávkování proběhlo bez příznaků. Při požití dávek, které nepřesáhly 140 mg či nebyly známé, se projevilo ovlivnění centrálního nervového systému (zmatenost, otupělost, somnolence, vertigo, agitovanost, agresivita, halucinace a poruchy chůze) a/nebo trávicího traktu (zvracení a diarea).
V případě nejvyššího předávkování pacient přežil požití úhrnné dávky 2000 mg memantinu se známkami ovlivnění centrálního nervového systému (kóma trvající 10 dní, později diplopie a agitovanost). Pacientovi byla poskytnuta symptomatická léčba a plazmaferéza. Pacient přežil bez následků.
V případě jiného významného předávkování pacient požil 400 mg memantinu perorálně a uzdravil se bez následků. U pacienta se objevily příznaky ovlivnění funkce centrálního nervového systému: neklid, psychóza, zrakové halucinace, zvýšená pohotovost ke křečím, somnolence, stupor a bezvědomí.
Léčba
V případě předávkování je léčba symptomatická. Neexistuje specifické antidotum. Mohou být užity standardní lékařské postupy k odstranění léčivé látky, např. gastrická laváž, podání aktivního uhlí (přerušení případného enterohepatálního oběhu), acidifikace moči a forsírovaná diuréza.
Pokud se projeví známky a příznaky nadměrné stimulace centrálního nervového systému (CNS), měla by být pečlivě zvážena symptomatická léčba.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Psychoanaleptika. Ostatní léky proti demenci, ATC kód: N06DX01
Přibývají důkazy, že narušená činnost glutamátergní neurotransmise, zvláště na NMDA receptorech, přispívá k projevu příznaků a postupné progresi onemocnění v neurodegenerativní demenci.
Memantin je nekompetitivní antagonista receptorů NMDA, závislý na napětí, se středně silnou afinitou. Upravuje účinky patologicky zvýšené excitačně působící hladiny glutamátu, která může vést k dysfunkci neuronů.
Klinické studie
Do stěžejní klinické studie monoterapie memantinem bylo zahrnuto 252 ambulantních pacientů se střední až těžkou formou Alzheimerovy choroby (celkové skóre Mini Mental State Examination -MMSE před léčbou 3 - 14). Studie prokázala příznivý vliv 6měsíční léčby memantinem ve srovnání s placebem (analýza pozorovaných případů dle Clinicians Interview Based Impression of Change (CIBIC-plus): p=0,025; Alzheimeťs Disease Cooperative Study - Activities of Daily Living (ADCS-ADLsev): p=0,003; Severe Impairment Battery (SIB): p=0,002).
Do stěžejní klinické studie léčby pacientů s mírnou až střední formou Alzheimerovy choroby (celkové skóre MMSE před léčbou 10-22) monoterapií memantinem bylo zahrnuto 403 pacientů. Pacienti léčení memantinem vykazovali statisticky významně lepší účinek oproti pacientům užívajícím placebo na primární cílové parametry ve 24. týdnu (Last Observation Carried Forward, LOCF): Alzheimeťs Disease Assessment Scale (ADAS-cog) (p=0,003) a CIBIC-plus (p=0,004). Do jiné monoterapeutické studie mírné až střední formy Alzheimerovy choroby bylo náhodně zařazeno 470 pacientů (celkové skóre MMSE před léčbou 11 - 23). V prospektivně definované primární analýze nebylo ve 24. týdnu dosaženo statistické významnosti v ovlivnění primárních cílových parametrů účinnosti.
Metaanalýza 6 placebem kontrolovaných, 6měsíčních studií fáze III se střední až těžkou formou Alzheimerovy choroby (celkové skóre MMSE před léčbou < 20) prokázala statisticky významný příznivý účinek memantinu ve třech oblastech: kognitivní, celkové a funkční; přičemž v meta-analýze byli zahrnuti pacienti léčeni pouze memantinem nebo současně stabilní dávkou inhibitorů acetylcholinesterázy. Pokud u pacientů docházelo ke zhoršení ve všech třech oblastech, výsledky ukázaly statisticky významný rozdíl účinku; ke zhoršení ve všech třech oblastech docházelo dvakrát častěji u pacientů užívajících placebo ve srovnání s pacienty léčenými memantinem, který působí preventivně proti zhoršení (21% v porovnání s 11%, p<0,0001).
5.2 Farmakokinetické vlastnosti
Absorpce
Memantin má absolutní biologickou dostupnost přibližně 100%. tmax je 3 až 8 hodin. Nic nenasvědčuje ovlivnění absorpce memantinu potravou.
Distribuce
Při denních dávkách 20 mg se plazmatická koncentrace memantinu v ustáleném stavu pohybuje v rozmezí 70-150 ng/ml (0,5-1 pmol) s velkými interindividuálními odchylkami. Při užívání denních dávek v rozmezí 5-30 mg byla vypočítána průměrná hodnota poměru mozkomíšní mok (CSF)/sérum ve výši 0,52. Distribuční objem je zhruba 10 l/kg. Přibližně 45 % memantinu se váže na plazmatické bílkoviny.
Biotransformace
V krevním oběhu člověka se nachází 80 % memantinu v nezměněné formě. Hlavními metabolity v organismu člověka jsou N-3,5-dimethyl-gludantan, směs isomerů 4- a 6-hydroxy-memantinu a 1-nitroso-3,5-dimethyl-adamantan. Žádný z těchto metabolitů nevykazuje aktivitu NMDA antagonisty.
In vitro nebyl zjištěn žádný metabolický pochod katalyzovaný cytochromem P 450.
Ve studii p.o. podání značeného memantinu 14C bylo průměrně 84 % podané dávky detekováno během 20 dnů, více než 99 % se vyloučilo ledvinami.
Eliminace
Eliminace memantinu probíhá podle jednoduché exponenciální křivky s terminálním poločasem t/2 60 až 100 hodin. U dobrovolníků s normální funkcí ledvin činí celková clearance (Cltot)
170 ml/min/1,73 m2 a je částečně dosažena tubulární sekrecí.
V ledvinách dochází též k tubulární reabsorpci, pravděpodobně zprostředkované kationtovými transportními proteiny. Podíl renální eliminace memantinu v prostředí zásadité moči se může snížit o koeficient 7-9 (viz bod 4.4). Zásaditá moč může být následkem zásadní změny stravovacích zvyklostí, např. při přechodu z masité stravy na vegetariánskou nebo při požití velkého množství alkalizujících žaludečních pufrů.
Linearita
Studie u dobrovolníků prokázaly lineární farmakokinetiku v dávkovém rozmezí 10-40 mg. Farmakokinetické/farmakodynamické vztahy
Při dávce 20 mg denně dosahují hladiny memantinu v CSF hodnoty inhibiční konstanty memantinu (ki), která je 0,5 pmol v mozkové kůře čelního laloku člověka.
5.3 Předklinické údaje vztahující se k bezpečnosti
V krátkodobých studiích na potkanech způsobuje memantin podobně jako jiní antagonisté NMDA neuronální vakuolizaci a nekrózu (Olneyovy léze) pouze při dávkách, které vedou k velmi vysokým maximálním sérovým koncentracím. Vakuolizaci a nekróze předcházela ataxie a jiné preklinické známky. Jelikož tyto jevy nebyly pozorovány při dlouhodobých studiích s hlodavci ani s jinými živočišnými druhy, není znám jejich význam pro klinickou praxi.
Oftalmologické nálezy byly rozporně zjištěny ve studiích toxicity po opakovaném podání u hlodavců a psů, nikoli však u opic. Při specifických oftalmoskopických vyšetřeních v rámci klinických studií s memantinem nebyly objeveny žádné oční změny.
U hlodavců byla pozorována fosfolipidóza u plicních makrofágů způsobená hromaděním memantinu v lyzozomech. Tento jev je znám i u jiných léčivých látek s kationtovými amfifilními vlastnostmi. Existuje možnost souvislosti mezi kumulací memantinu a vakuolizací pozorovanou v plicích. Tento jev byl pozorován jen při vysokých dávkách u hlodavců. Klinický význam těchto zjištění není znám.
Standardní testování memantinu neprokázalo jeho genotoxicitu. V dlouhodobých (celoživotních) studiích prováděných na myších a potkanech nebyly nalezeny důkazy pro kancerogenitu. Memantin nebyl teratogenní u potkanů a králíků ani při dávkách toxických pro březí samice a neprokázal žádný nepříznivý vliv na plodnost. U potkanů byl zaznamenán pomalejší růst plodu při dávkách stejných nebo mírně vyšších, než které jsou užívány u lidí.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Jádro tablety pro potahované tablety 5/10/15/20 mg:
Mikrokrystalická celulosa Sodná sůl kroskarmelosy Koloidní bezvodý oxid křemičitý Magnesium-stearát
Potah tablety pro potahované tablety 5/10/15/20 mg:
Hypromelosa Makrogol 400 Oxid titaničitý
Další pro potahované tablety 10 mg:
Žlutý oxid železitý
Další pro potahované tablety 15 mg a 20 mg:
Žlutý a červený oxid železitý
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
4 roky.
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
6.5 Druh obalu a velikost balení
Balení obsahuje 28 potahovaných tablet v 4 PVDC/PE/PVC/Al-blistru nebo PP/Al-blistrech se 7 potahovanými tabletami síly 5 mg, 7 potahovanými tabletami síly 10 mg, 7 potahovanými tabletami síly 15 mg a 7 potahovanými tabletami síly 20 mg.
6.6 Zvláštní opatření pro likvidaci přípravku
Žádné zvláštní požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
H. Lundbeck A/S Ottiliavej 9 2500 Valby Dánsko
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/02/219/022
EU/1/02/219/036
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 15. května 2002 Datum prodloužení registrace: 15. května 2007
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/.
NÁZEV PŘÍPRAVKU
1.
Ebixa 20 mg potahované tablety
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna potahovaná tableta obsahuje memantini hydrochloridum 20 mg, což odpovídá 16,62 mg memantinu.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Potahovaná tableta.
Světle červené až šedo-červené, oválné - podlouhlé potahované tablety s vytištěným „20“ na jedné straně a „MEM“ na druhé straně.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba dospělých pacientů se střední až těžkou formou Alzheimerovy choroby.
4.2 Dávkování a způsob podání
Léčbu musí zahájit a dohlížet na ni lékař se zkušeností s diagnostikou a léčbou demence Alzheimerova typu.
Dávkování
Podmínkou zahájení léčby je dostupnost pečovatele, který pravidelně sleduje užívání léčivého přípravku pacientem. Diagnóza musí být stanovena podle soudobých diagnostických postupů. Snášenlivost a dávkování memantinu by měly být pravidelně posuzovány, nejlépe během tří měsíců po zahájení terapie. Klinický přínos memantinu a snášenlivost léčby pacientem by měly být nadále pravidelně vyhodnocovány podle současných doporučení pro léčbu. Udržovací terapie memantinem může pokračovat, dokud je přínosná a pacientem snášená. Ukončení léčby memantinem by mělo být zváženo, pokud není terapeutický účinek již patrný nebo pokud pacient léčbu přestal snášet.
Dospělí
Titrace dávky
Maximální denní dávka je 20 mg denně. V zájmu snížení rizika výskytu nežádoucích účinků by se mělo udržovací dávky dosáhnout postupným zvyšováním denní dávky po 5 mg týdně během prvních 3 týdnů léčby následujícím způsobem. Pro titraci dávky nahoru jsou k dispozici jiné síly tablety.
Týden 1 (den 1-7)
Pacient by měl užívat jednu 5 mg potahovanou tabletu denně po dobu 7 dnů.
Týden 2 (den 8-14)
Pacient by měl užívat jednu 10 mg potahovanou tabletu denně po dobu 7 dnů.
Týden 3 (den 15-21)
Pacient by měl užívat jednu 15 mg potahovanou tabletu denně po dobu 7 dnů.
Od týdne 4 dále
Pacient by měl užívat jednu 20 mg potahovanou tabletu denně.
Udržovací dávka
Doporučená udržovací dávka je 20 mg denně.
Starší osoby
Na základě poznatků z klinických studií je doporučená dávka pro pacienty starší 65 let 20 mg denně, jak je uvedeno výše.
Snížená funkce ledvin
U pacientů s mírnou poruchou funkce ledvin (clearance kreatininu 50 - 80 ml/min) není třeba upravovat dávku. U pacientů se středně závažnou poruchou funkce ledvin (clearance kreatininu 30 -49 ml/min) by denní dávka měla být 10 mg. Pokud je tato dávka pacientem minimálně týden dobře snášena, může být dle schématu nastavování dávky zvýšena na 20 mg denně. U pacientů s těžkou poruchou funkce ledvin (clearance kreatininu 5 - 29 ml/min) by denní dávka měla být 10 mg.
Snížená funkce jater
U pacientů s mírně až středně závažnou poruchou funkce jater (Child-Pugh A a Child-Pugh B) není třeba upravovat dávku. Nejsou k dispozici údaje o užívání memantinu u pacientů se závažnou poruchou funkce jater. Podávání přípravku Ebixa není doporučeno u pacientů se závažnou poruchou funkce jater.
Pediatrická populace Nejsou dostupné žádné údaje.
Způsob podání
Přípravek Ebixa by se měl podávat perorálně jednou denně a měl by se užívat ve stejnou dobu každý den. Potahované tablety se mohou užívat s jídlem nebo bez jídla.
4.3 Kontraindikace
Hypersensitivita na léčivou látku nebo na kteroukoli pomocnou látku tohoto přípravku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Opatrnost je doporučována u pacientů s epilepsií, s předchozí anamnézou křečí nebo u pacientů s predispozičními faktory pro epilepsii.
Neměla by probíhat současná léčba antagonisty N-methyl-D-aspartátu (NMDA), jako jsou amantadin, ketamin nebo dextromethorfan. Tyto léčivé látky působí na stejném receptorovém systému jako memantin, nežádoucí účinky (hlavně v oblasti centrálního nervového systému (CNS)) by tudíž mohly být častější nebo výraznější (viz bod 4.5).
Přítomnost některých faktorů, jež mohou zvýšit pH moči (viz bod 5.2 Eliminace), vyžaduje pečlivé sledování pacienta. Tyto faktory zahrnují: zásadní změny stravovacích zvyklostí, např. přechod z masité stravy na vegetariánskou nebo požití velkého množství alkalizujících žaludečních pufrů. Zvýšení pH moči může nastat též při renální tubulární acidóze (RTA) nebo při závažné infekci močových cest způsobené bakterií rodu Proteus.
Z většiny klinických studií byli vyloučeni pacienti s nedávno prodělaným infarktem myokardu, nekompenzovaným městnavým srdečním selháním (NYHA III-IV) nebo neléčenou hypertenzí. Proto
jsou u těchto pacientů pouze omezené zkušenosti a případná léčba by měla probíhat za jejich pečlivého sledování.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Vzhledem k farmakologickému působení a mechanismu účinku memantinu mohou nastat tyto interakce:
• Mechanismus účinku naznačuje, že účinky L-dopy, dopaminergních agonistů a anticholinergik se mohou zvýšit při současné léčbě antagonisty NMDA, mezi něž patří memantin. Účinek barbiturátů a neuroleptik se může snížit. Při současném podání memantinu s myorelaxancii dantrolenem nebo baklofenem může dojít k ovlivnění jejich účinku, což může vyžadovat úpravu dávkování.
• Současné užití memantinu a amantadinu není vhodné, vzhledem k riziku farmakotoxické psychózy. Obě léčivé látky jsou chemicky podobní antagonisté NMDA. To může platit též pro ketamin a dextromethorfan (viz bod 4.4). Byla publikována jedna kasuistika vztahující se k možnému riziku kombinace memantin a fenytoin.
• Některé další léčivé látky, jako cimetidin, ranitidin, prokainamid, chinidin, chinin a nikotin, které využívají stejný kationtový transportní systém v ledvinách jako amantadin, mohou případně interagovat s memantinem, což vede k možnému riziku zvýšení plazmatických hladin.
• Existuje možnost sníženého vylučování hydrochlorothiazidu v séru, pokud je memantin užíván společně s hydrochlorothiazidem nebo s jakoukoli kombinací, která hydrochlorothiazid obsahuje.
• V postmarketingových studiích bylo zaznamenáno několik ojedinělých případů zvýšení hodnoty mezinárodního normalizovaného poměru (INR) u pacientů užívajících současně warfarin. Ačkoliv nebyla nalezena přímá souvislost, doporučuje se pečlivé sledování protrombinového času nebo INR u pacientů současně léčených perorálními antikoagulancii.
Ve studiích farmakokinetiky (FK) při podávání jednotlivé denní dávky mladým zdravým dobrovolníkům nebyla prokázána interakce účinná látka - účinná látka při současném užívání glyburidu/metforminu nebo donepezilu.
V klinických studiích mladých zdravých dobrovolníků nebyl prokázán případný vliv memantinu na farmakokinetiku galantaminu.
Memantin neinhibuje in vitro žádný z těchto systémů: CYP 1A2, 2A6, 2C9, 2D6, 2E1, 3A, monooxygenázu s flavinem, epoxidhydrolázu ani sulfatační pochody.
4.6 Fertilita, těhotenství a kojení
Údaje o podávání memantinu těhotným ženám jsou omezené nebo nejsou k dispozici. Studie na zvířatech naznačují možnost zpomalení nitroděložního růstu při dávkách identických nebo mírně vyšších než těch, které jsou užívány u lidí (viz bod 5.3). Míra případného rizika u lidí není známa. Memantin by se neměl v těhotenství užívat, pokud to není zcela nezbytné.
Kojení
Není známo, zda se memantin vylučuje do mateřského mléka, ovšem pokud se uváží lipofilita léčivé látky, je průnik do mateřského mléka pravděpodobný. Ženy užívající memantin by neměly kojit.
Fertilita
Nebyly zaznamenány žádné nežádoucí účinky memantinu na mužskou a ženskou fertilitu.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Střední až těžká forma Alzheimerovy choroby obvykle narušuje schopnost řízení motorových vozidel a omezuje ovládání strojů. Navíc Ebixa má malý nebo střední vliv na schopnost řídit a obsluhovat stroje, takže by ambulantní pacienti měli být upozorněni, aby věnovali řízení vozidel a ovládání strojů zvýšenou pozornost.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Do klinických studií byli zahrnuti pacienti s mírnou až těžkou demencí; z toho 1784 pacientů bylo léčeno přípravkem Ebixa a 1595 pacientů užívalo placebo. Celkový výskyt nežádoucích reakcí se nelišil u pacientů užívajících přípravek Ebixa v porovnání s pacienty užívajícími placebo. Nežádoucí účinky byly mírné až střední závažnosti. Nejčastější nežádoucí účinky, jejichž frekvence výskytu byla vyšší ve skupině léčené přípravkem Ebixa v porovnání se skupinou přijímající placebo, byly: závratě (6,3% v porovnání s 5,6%), bolest hlavy (5,2% v porovnání s 3,9%), zácpa (4,6% v porovnání s 2,6%), somnolence (3,4% v porovnání s 2,2%) a hypertenze (4,1% v porovnání s 2,8%).
Tabulkový seznam nežádoucích účinků
Nežádoucí reakce uvedené v tabulce se vyskytly v klinických studiích s přípravkem Ebixa nebo po jeho uvedení na trh.
Nežádoucí účinky jsou seřazeny podle tříd orgánových systémů s použitím následující klasifikace: velmi časté (>1/10), časté (>1/100, <1/10), méně časté (>1/1 000, <1/100), vzácné (>1/10 000, <1/1 000), velmi vzácné (<1/10 000), není známo (z dostupných údajů nelze určit). V každé skupině četnosti jsou nežádoucí účinky seřazeny dle klesající závažnosti.
|
TŘÍDY ORGÁNOVÝCH SYSTÉMŮ |
ČETNOST |
NEŽÁDOUCÍ ÚČINEK |
|
Infekce a infestace |
Méně časté |
Mykotické infekce |
|
Poruchy imunitního systému |
Časté |
Přecitlivělost na přípravek |
|
Psychiatrické poruchy |
Časté |
Somnolence |
|
Méně časté | ||
|
Méně časté | ||
|
Není známo |
Psychotické reakce2 | |
|
Poruchy nervového systému |
Časté |
Závratě |
|
Časté |
Poruchy rovnováhy | |
|
Méně časté |
Poruchy chůze | |
|
Velmi vzácné | ||
|
Srdeční poruchy |
Méně časté |
Srdeční selhání |
|
Cévní poruchy |
Časté |
Hypertenze |
|
Méně časté |
Žilní trombóza/trombembolismus | |
|
Respirační, hrudní a mediastinální poruchy |
Časté | |
|
Gastrointestinální poruchy |
Časté |
Zácpa |
|
Méně časté | ||
|
Není známo |
Pankreatitida2 | |
|
Poruchy jater a žlučových cest |
Časté |
Zvýšené hodnoty jaterních testů |
|
Není známo |
Hepatitia | |
|
Celkové poruchy a reakce v místě aplikace |
Časté | |
|
Méně časté |
Únava |
'Halucinace byly pozorovány častěji u pacientů s těžkou Alzheimerovou chorobou.
2Ojedinělá hlášení z postmarketingových studií.
Alzheimerova choroba bývá spojována s výskytem deprese, sebevražedných představ a sebevraždy.
V postmarketingových studiích byly tyto účinky hlášeny u pacientů léčených přípravkem Ebixa.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Zkušenosti s předávkováním v klinických studiích nebo po uvedení přípravku na trh jsou pouze omezené.
Známky předávkování
V případě významného předávkování (200 mg a 105 mg/den, po dobu 3 dnů) se objevily pouze příznaky: únava, slabost a/nebo diarea či předávkování proběhlo bez příznaků. Při požití dávek, které nepřesáhly 140 mg či nebyly známé, se projevilo ovlivnění centrálního nervového systému (zmatenost, otupělost, somnolence, vertigo, agitovanost, agresivita, halucinace a poruchy chůze) a/nebo trávicího traktu (zvracení a diarea).
V případě nejvyššího předávkování pacient přežil požití úhrnné dávky 2000 mg memantinu se známkami ovlivnění centrálního nervového systému (kóma trvající 10 dní, později diplopie a agitovanost). Pacientovi byla poskytnuta symptomatická léčba a plazmaferéza. Pacient přežil bez následků.
V případě jiného významného předávkování pacient požil 400 mg memantinu perorálně a uzdravil se bez následků. U pacienta se objevily příznaky ovlivnění funkce centrálního nervového systému: neklid, psychóza, zrakové halucinace, zvýšená pohotovost ke křečím, somnolence, stupor a bezvědomí.
Opatření při předávkování
V případě předávkování je léčba symptomatická. Neexistuje specifické antidotum. Mohou být užity standardní lékařské postupy k odstranění léčivé látky, např. gastrická laváž, podání aktivního uhlí (přerušení případného enterohepatálního oběhu), acidifikace moči a forsírovaná diuréza.
Pokud se projeví známky a příznaky nadměrné stimulace centrálního nervového systému (CNS), měla by být pečlivě zvážena symptomatická léčba.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Psychoanaleptika. Ostatní léky proti demenci, ATC kód: N06DX01
Přibývají důkazy, že narušená činnost glutamátergní neurotransmise, zvláště na NMDA receptorech, přispívá k projevu příznaků a postupné progresi onemocnění v neurodegenerativní demenci.
Memantin je nekompetitivní antagonista receptorů NMDA, závislý na napětí, se středně silnou afinitou. Upravuje účinky patologicky zvýšené excitačně působící hladiny glutamátu, která může vést k dysfunkci neuronů.
Klinické studie
Do stěžejní klinické studie monoterapie memantinem bylo zahrnuto 252 ambulantních pacientů se střední až těžkou formou Alzheimerovy choroby (celkové skóre Mini Mental State Examination -MMSE před léčbou 3 - 14). Studie prokázala příznivý vliv 6měsíční léčby memantinem ve srovnání s placebem (analýza pozorovaných případů dle Clinicians Interview Based Impression of Change (CIBIC-plus): p=0,025; Alzheimer's disease cooperative study - activities of daily living (ADCS-ADLsev): p=0,003; Severe Impairment Battery (SIB): p=0,002).
Do stěžejní klinické studie léčby pacientů s mírnou až střední formou Alzheimerovy choroby (celkové skóre MMSE před léčbou 10-22) monoterapií memantinem bylo zahrnuto 403 pacientů. Pacienti léčení memantinem vykazovali statisticky významně lepší účinek oproti pacientům užívajícím placebo na primární cílové parametry ve 24. týdnu (Last Observation Carried Forward, LOCF): Alzheimers Disease Assessment Scale (ADAS-cog) (p=0,003) a CIBIC-plus (p=0,004). Do jiné monoterapeutické studie mírné až střední formy Alzheimerovy choroby bylo náhodně zařazeno 470 pacientů (celkové skóre MMSE před léčbou 11 - 23). V prospektivně definované primární analýze nebylo ve 24. týdnu dosaženo statistické významnosti v ovlivnění primárních cílových parametrů účinnosti.
Meta-analýza 6 placebem kontrolovaných, 6měsíčních studií fáze III se střední až těžkou formou Alzheimerovy choroby (celkové skóre MMSE před léčbou < 20) prokázala statisticky významný příznivý účinek memantinu ve třech oblastech: kognitivní, celkové a funkční; přičemž v meta-analýze byli zahrnuti pacienti léčeni pouze memantinem nebo současně stabilní dávkou inhibitorů acetylcholinesterázy. Pokud u pacientů docházelo ke zhoršení ve všech třech oblastech, výsledky ukázaly statisticky významný rozdíl účinku; ke zhoršení ve všech třech oblastech docházelo dvakrát častěji u pacientů užívajících placebo ve srovnání s pacienty léčenými memantinem, který působí preventivně proti zhoršení (21% v porovnání s 11%, p<0,0001).
5.2 Farmakokinetické vlastnosti
Absorpce
Memantin má absolutní biologickou dostupnost přibližně 100%. tmax je 3 až 8 hodin. Nic nenasvědčuje ovlivnění absorpce memantinu potravou.
Distribuce
Při denních dávkách 20 mg se plazmatická koncentrace memantinu v ustáleném stavu pohybuje v rozmezí 70-150 ng/ml (0,5-1 pmol) s velkými interindividuálními odchylkami. Při užívání denních dávek v rozmezí 5-30 mg byla vypočítána průměrná hodnota poměru mozkomíšní mok (CSF)/sérum ve výši 0,52. Distribuční objem je zhruba 10 l/kg. Přibližně 45 % memantinu se váže na plazmatické bílkoviny.
Biotransformace:
V krevním oběhu člověka se nachází 80 % memantinu v nezměněné formě. Hlavními metabolity v organismu člověka jsou N-3,5-dimethyl-gludantan, směs isomerů 4- a 6-hydroxy-memantinu a 1-nitroso-3,5-dimethyl-adamantan. Žádný z těchto metabolitů nevykazuje aktivitu NMDA antagonisty.
In vitro nebyl zjištěn žádný metabolický pochod katalyzovaný cytochromem P 450.
Ve studii p.o. podání značeného memantinu 14C bylo průměrně 84 % podané dávky detekováno během 20 dnů, více než 99 % se vyloučilo ledvinami.
Eliminace
Eliminace memantinu probíhá podle jednoduché exponenciální křivky s terminálním poločasem t/2 60 až 100 hodin. U dobrovolníků s normální funkcí ledvin činí celková clearance (Cltot)
170 ml/min/1,73 m2 a je částečně dosažena tubulární sekrecí.
V ledvinách dochází též k tubulární reabsorpci, pravděpodobně zprostředkované kationtovými transportními proteiny. Podíl renální eliminace memantinu v prostředí zásadité moči se může snížit o koeficient 7-9 (viz bod 4.4). Zásaditá moč může být následkem zásadní změny stravovacích zvyklostí, např. při přechodu z masité stravy na vegetariánskou nebo při požití velkého množství alkalizujících žaludečních pufrů.
Linearita
Studie u dobrovolníků prokázaly lineární farmakokinetiku v dávkovém rozmezí 10-40 mg. Farmakokinetické/farmakodynamické vztahy
Při dávce 20 mg denně dosahují hladiny memantinu v CSF hodnoty inhibiční konstanty memantinu (ki), která je 0,5 pmol v mozkové kůře čelního laloku člověka.
5.3 Předklinické údaje vztahující se k bezpečnosti
V krátkodobých studiích na potkanech způsobuje memantin podobně jako jiní antagonisté NMDA neuronální vakuolizaci a nekrózu (Olneyovy léze) pouze při dávkách, které vedou k velmi vysokým maximálním sérovým koncentracím. Vakuolizaci a nekróze předcházela ataxie a jiné preklinické známky. Jelikož tyto jevy nebyly pozorovány při dlouhodobých studiích s hlodavci ani s jinými živočišnými druhy, není znám jejich význam pro klinickou praxi.
Oftalmologické nálezy byly rozporně zjištěny ve studiích toxicity po opakovaném podání u hlodavců a psů, nikoli však u opic. Při specifických oftalmoskopických vyšetřeních v rámci klinických studií s memantinem nebyly objeveny žádné oční změny.
U hlodavců byla pozorována fosfolipidóza u plicních makrofágů způsobená hromaděním memantinu v lyzozomech. Tento jev je znám i u jiných léčivých látek s kationtovými amfifilními vlastnostmi. Existuje možnost souvislosti mezi kumulací memantinu a vakuolizací pozorovanou v plicích. Tento jev byl pozorován jen při vysokých dávkách u hlodavců. Klinický význam těchto zjištění není znám.
Standardní testování memantinu neprokázalo jeho genotoxicitu. V dlouhodobých (celoživotních) studiích prováděných na myších a potkanech nebyly nalezeny důkazy pro kancerogenitu. Memantin nebyl teratogenní u potkanů a králíků ani při dávkách toxických pro březí samice a neprokázal žádný nepříznivý vliv na plodnost. U potkanů byl zaznamenán pomalejší růst plodu při dávkách stejných nebo mírně vyšších, než které jsou užívány u lidí.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Jádro tablety:
Mikrokrystalická celulosa Sodná sůl kroskarmelosy Koloidní bezvodý oxid křemičitý Magnesium-stearát
Potah tablety:
Hypromelosa
Makrogol 400
Oxid titaničitý
Žlutý a červený oxid železa
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
4 roky.
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
6.5 Druh obalu a velikost balení
Blistr: PVDC/PE/PVC/Al-blistr nebo PP/Al-blistr Velikosti balení: 14, 28, 42, 56, 70, 84, 98, 112 potahovaných tablet.
Multipack obsahuje 840 (20x42) potahovaných tablet.
Perforovaný jednodávkový blistr: PVDC/PE/PVC/Al-blistr nebo PP/Al-blistr.
Velikosti balení: 49 x 1, 56 x 1, 98 x 1 a 100 x 1 potahovaných tablet.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku
Žádné zvláštní požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
H. Lundbeck A/S Ottiliavej 9 2500 Valby Dánsko
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/02/219/023-035
EU/1/02/219/037-049
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 15. května 2002 Datum prodloužení registrace: 15. května 2007
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného za propouštění šarží
H. Lundbeck A/S Ottiliavej 9 2500 Valby DÁNSKO
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
• PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ, KLADENÉ NA DRŽITELE ROZHODNUTÍ O REGISTRACI
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz Příloha I: Souhrn údajů o přípravku, bod 4.2)
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
• Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které
mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
Ebixa 10 mg potahované tablety Memantini hydrochloridum
Jedna potahovaná tableta obsahuje 10 mg memantini hydrochloridum, což odpovídá 8,31 mg memantinu.
Potahované tablety 14 potahovaných tablet 28 potahovaných tablet 30 potahovaných tablet 42 potahovaných tablet
49 x 1 potahovaná tableta
50 potahovaných tablet 56 potahovaných tablet 56 x 1 potahovaná tableta 70 potahovaných tablet 84 potahovaných tablet 98 potahovaných tablet 98 x 1 potahovaná tableta 100 potahovaných tablet 100 x 1 potahovaná tableta 112 potahovaných tablet
Před použitím si přečtěte příbalovou informaci. Perorální podání.
Uchovávejte mimo dohled a dosah dětí.
Použitelné do: {MM/RRRR}
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z TAKOVÝCH LÉČIVÝCH PŘÍPRAVKŮ, POKUD JE TO VHODNÉ
H. Lundbeck A/S Ottiliavej 9 2500 Valby Dánsko
EU/1/02/219/016 14 potahovaných tablet EU/1/02/219/007 28 potahovaných tablet EU/1/02/219/001 30 potahovaných tablet EU/1/02/219/017 42 potahovaných tablet EU/1/02/219/010 49 x 1 potahovaná tableta EU/1/02/219/002 50 potahovaných tablet EU/1/02/219/008 56 potahovaných tablet EU/1/02/219/014 56 x 1 potahovaná tableta EU/1/02/219/018 70 potahovaných tablet EU/1/02/219/019 84 potahovaných tablet EU/1/02/219/020 98 potahovaných tablet EU/1/02/219/015 98 x 1 potahovaná tableta EU/1/02/219/003 100 potahovaných tablet EU/1/02/219/011 100 x 1 potahovaná tableta EU/1/02/219/009 112 potahovaných tablet
Č.š.: {číslo}
Výdej léčivého přípravku vázán na lékařský předpis.
Ebixa 10 mg tablety
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Ebixa 10 mg potahované tablety Memantini hydrochloridum
2 OBSAH LÉČIVÉ LÁTKY
Jedna potahovaná tableta obsahuje 10 mg memantini hydrochloridum, což odpovídá 8,31 mg memantinu.
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH
Potahované tablety 50 potahovaných tablet 98 potahovaných tablet
Součást většího balení (multipack), nemůže být prodáváno samostatně.
5 ZPŮSOB A CESTA PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do: {MM/RRRR}
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z TAKOVÝCH LÉČIVÝCH PŘÍPRAVKŮ, POKUD JE TO VHODNÉ
H. Lundbeck A/S Ottiliavej 9 2500 Valby Dánsko
12. REGISTRAČNÍ ČÍSLO
EU/1/02/219/021 980 (10 balení každé obsahující 98) potahovaných tablet EU/1/02/219/012 1000 (20 balení každé obsahující 50) potahovaných tablet
13. ČÍSLO ŠARŽE
Č.š.: {číslo}
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16 INFORMACE V BRAILLOVĚ PÍSMU
Ebixa 10 mg tablety
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU
ŠTÍTEK NA FÓLII MULTIPACK (VČETNĚ ,BLUE BOX‘)
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Ebixa 10 mg potahované tablety Memantini hydrochloridum
2 OBSAH LÉČIVÉ LÁTKY
Jedna potahovaná tableta obsahuje 10 mg memantini hydrochloridum, což odpovídá 8,31 mg memantinu.
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH
Potahované tablety
Multipack: 980 (10 balení každé obsahující 98) potahovaných tablet. Multipack: 1000 (20 balení každé obsahující 50) potahovaných tablet.
5 ZPŮSOB A CESTA PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do: {MM/RRRR}
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z TAKOVÝCH LÉČIVÝCH PŘÍPRAVKŮ, POKUD JE TO VHODNÉ
H. Lundbeck A/S Ottiliavej 9 2500 Valby Dánsko
12. REGISTRAČNÍ ČÍSLO
EU/1/02/219/021 980 (10 x 98) potahovaných tablet EU/1/02/219/ 012 1000 (20 x 50) potahovaných tablet
13 ČÍSLO ŠARŽE
Č.š.: {číslo}
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15 NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
Ebixa 10 mg tablety
minimální údaje uvadene na blistrech
BLISTR PRO TABLETY
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Ebixa 10 mg potahované tablety Memantini hydrochloridum
2 NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
H. Lundbeck A/S
3 POUŽITELNOST
Použitelné do: {MM/RRRR} Viz razítko na okraji.
4. ČÍSLO ŠARŽE
Č.š.:{číslo}
Viz razítko na okraji.
5. JINÉ
Ebixa 5 mg/dávka perorální roztok Memantini hydrochloridum
Jedna dávka dávkovací pumpy (jedno stlačení pístu dolů) odměří 0,5 ml roztoku obsahujícího memantini hydrochloridum 5 mg, což odpovídá 4,16 mg memantinu.
Roztok také obsahuje sorbitan draselný a sorbitol E420. Více informací viz příbalová informace.
Perorální roztok. 50 ml 100 ml
Jednou denně.
Před použitím si přečtěte příbalovou informaci. Perorální podání.
Uchovávejte mimo dohled a dosah dětí.
Použitelné do: {MM/RRRR}
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
H. Lundbeck A/S Ottiliavej 9 2500 Valby Dánsko
12. REGISTRAČNÍ ČÍSLO
EU/1/02/219/005 50 ml. EU/1/02/219/006 100 ml.
13. ČÍSLO ŠARŽE
Č.š.: {číslo}
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15 NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
Ebixa 5 mg/dávka roztok
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU A NA VNITŘNÍM OBALU
PAPÍROVÁ KRABIČKA A ŠTÍTEK PRO LAHVIČKU, MEZIOBAL JAKO SOUČÁST VĚTŠÍHO BALENÍ (MULTIPACK) BEZ ,BLUE BOX‘_
Ebixa 5 mg/dávka perorální roztok Memantini hydrochloridum
Jedna dávka dávkovací pumpy (jedno stlačení pístu dolů) odměří 0,5 ml roztoku obsahujícího 5 mg memantini hydrochloridum, což odpovídá 4,16 mg memantinu.
Roztok také obsahuje sorbitan draselný a sorbitol E420. Více informací viz příbalová informace.
Perorální roztok 50 ml
Součást většího balení multipack nemůže být prodáváno samostatně..
Jednou denně.
Před použitím si přečtěte příbalovou informaci. Perorální podání.
Uchovávejte mimo dohled a dosah dětí.
Použitelné do: {MM/RRRR}
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
H. Lundbeck A/S Ottiliavej 9 2500 Valby Dánsko
|
12. |
REGISTRAČNÍ ČÍSLO |
|
EU/1/02/219/013 500 ml (10 lahviček po 50 ml) | |
|
13. |
ČÍSLO ŠARŽE |
|
Č.š.: |
{číslo} |
|
14. |
KLASIFIKACE PRO VÝDEJ |
|
Výdej léčivého přípravku vázán na lékařský předpis. | |
|
15. |
NÁVOD K POUŽITÍ |
|
16. |
INFORMACE V BRAILLOVĚ PÍSMU |
Ebixa 5 mg/dávka roztok
Ebixa 5 mg/dávka perorální roztok Memantini hydrochloridum
Jedna dávka dávkovací pumpy (jedno stlačení pístu dolů) odměří 0,5 ml roztoku obsahujícího 5 mg memantini hydrochloridum, což odpovídá 4,16 mg memantinu.
Roztok také obsahuje sorbitan draselný a sorbitol E420. Více informací viz příbalová informace.
Perorální roztok
Multipack: 500 ml (10 lahviček po 50 ml) perorálního roztoku.
Jednou denně.
Před použitím si přečtěte příbalovou informaci. Perorální podání.
Uchovávejte mimo dohled a dosah dětí.
Použitelné do: {MM/RRRR}
LL NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
H. Lundbeck A/S Ottiliavej 9 2500 Valby Dánsko
|
12. |
REGISTRAČNÍ ČÍSLO |
|
EU/1/02/219/013 500 ml (10 lahviček po 50 ml) | |
|
13. |
ČÍSLO ŠARŽE |
|
Č.š.: |
{číslo} |
|
14. |
KLASIFIKACE PRO VÝDEJ |
|
Výdej léčivého přípravku vázán na lékařský předpis. | |
|
15. |
NÁVOD K POUŽITÍ |
|
16. |
INFORMACE V BRAILLOVĚ PÍSMU |
Ebixa 5 mg/dávka roztok
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Ebixa 5 mg potahované tablety Ebixa 10 mg potahované tablety Ebixa 15 mg potahované tablety Ebixa 20 mg potahované tablety Memantini hydrochloridum
2. OBSAH LÉČIVÉ LÁTKY
Jedna potahovaná tableta obsahuje memantinu.
Jedna potahovaná tableta obsahuje memantinu.
Jedna potahovaná tableta obsahuje memantinu.
Jedna potahovaná tableta obsahuje memantinu.
5 mg memantini hydrochloridum, což odpovídá 4,15 mg 10 mg memantini hydrochloridum, což odpovídá 8,31 mg 15 mg memantini hydrochloridum, což odpovídá 12,46 mg 20 mg memantini hydrochloridum, což odpovídá 16,62 mg
3. SEZNAM POMOCNÝCH LÁTEK
Balení určené k zahájení léčby.
Každé balení s 28 potahovanými tabletami pro 4-týdenní léčebné schéma obsahuje: 7 potahovaných tablet Ebixa 5 mg 7 potahovaných tablet Ebixa 10 mg 7 potahovaných tablet Ebixa 15 mg 7 potahovaných tablet Ebixa 20 mg
5. ZPŮSOB A CESTA PODÁNÍ
Jednou denně.
Před použitím si přečtěte příbalovou informaci.
Perorální podání.
Pro pokračování Vaší léčby se prosím obraťte na svého lékaře.
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
Použitelné do: {MM/RRRR}
9 ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z TAKOVÝCH LÉČIVÝCH PŘÍPRAVKŮ, POKUD JE TO VHODNÉ
11 NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
H. Lundbeck A/S Ottiliavej 9 2500 Valby Dánsko
12. REGISTRAČNÍ ČÍSLO(A)
EU/1/02/219/022 7 x 5 mg + 7 x 10 mg + 7 x 15 mg 7 x 20 mg potahovaných tablet. EU/1/02/219/036 7 x 5 mg + 7 x 10 mg + 7 x 15 mg 7 x 20 mg potahovaných tablet.
13. ČÍSLO ŠARŽE
Č.š.: {číslo}
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15 NÁVOD K POUŽITÍ
Užívejte pouze jednu tabletu denně. Ebixa 5 mg
Memantini hydrochloridum Týden 1, den 1 2 3 4 5 6 7 7 potahovaných tablet Ebixa 5 mg
Ebixa 10 mg
Memantini hydrochloridum Týden 2, den 8 9 10 11 12 13 14 7 potahovaných tablet Ebixa 10 mg
Ebixa 15 mg
Memantini hydrochloridum Týden 3, den 15 16 17 18 19 20 21 7 potahovaných tablet Ebixa 15 mg
Ebixa 20 mg
Memantini hydrochloridum Týden 4, den 22 23 24 25 26 27 28 7 potahovaných tablet Ebixa 20 mg
16. INFORMACE V BRAILLOVE PÍSMU
Ebixa 5 mg, 10 mg, 15 mg, 20 mg tablety
Ebixa 20 mg potahované tablety Memantini hydrochloridum
Jedna potahovaná tableta obsahuje 20 mg memantini hydrochloridum, což odpovídá 16,62 mg memantinu.
Potahované tablety 14 potahovaných tablet 28 potahovaných tablet 42 potahovaných tablet 49 x 1 potahovaná tableta 56 potahovaných tablet 56 x 1 potahovaná tableta 70 potahovaných tablet 84 potahovaných tablet 98 potahovaných tablet 98 x 1 potahovaná tableta 100 x 1 potahovaná tableta 112 potahovaných tablet
Jednou denně.
Před použitím si přečtěte příbalovou informaci. Perorální podání.
Uchovávejte mimo dohled a dosah dětí.
Použitelné do: {MM/RRRR}
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z TAKOVÝCH LÉČIVÝCH PŘÍPRAVKŮ, POKUD JE TO VHODNÉ
H. Lundbeck A/S Ottiliavej 9 2500 Valby Dánsko
EU/1/02/219/023 14 potahovaných tablet EU/1/02/219/024 28 potahovaných tablet EU/1/02/219/025 42 potahovaných tablet EU/1/02/219/026 49 x 1 potahovaných tablet EU/1/02/219/027 56 potahovaných tablet EU/1/02/219/028 56 x 1 potahovaných tablet EU/1/02/219/029 70 potahovaných tablet EU/1/02/219/030 84 potahovaných tablet EU/1/02/219/031 98 potahovaných tablet EU/1/02/219/032 98 x 1 potahovaných tablet EU/1/02/219/033 100 x 1 potahovaných tablet EU/1/02/219/034 112 potahovaných tablet EU/1/02/219/037 14 potahovaných tablet EU/1/02/219/038 28 potahovaných tablet EU/1/02/219/039 42 potahovaných tablet EU/1/02/219/040 49 x 1 potahovaných tablet EU/1/02/219/041 56 potahovaných tablet EU/1/02/219/042 56 x 1 potahovaných tablet EU/1/02/219/043 70 potahovaných tablet EU/1/02/219/044 84 potahovaných tablet EU/1/02/219/045 98 potahovaných tablet EU/1/02/219/046 98 x 1 potahovaných tablet EU/1/02/219/047 100 x 1 potahovaných tablet EU/1/02/219/048 112 potahovaných tablet
Č.š.: {číslo}
Výdej léčivého přípravku vázán na lékařský předpis.
16. INFORMACE V BRAILLOVE PÍSMU
Ebixa 20 mg tablety
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU
PAPÍROVÁ KRABIČKA PRO MEZIOBAL, JAKO SOUČÁST VĚTŠÍHO BALENÍ (MULTIPACK) (BEZ ,BLUE BOX‘)
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Ebixa 20 mg potahované tablety Memantini hydrochloridum
2 OBSAH LÉČIVÉ LÁTKY
Jedna potahovaná tableta obsahuje 20 mg memantini hydrochloridum, což odpovídá 16,62 mg memantinu.
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH
Potahované tablety 42 potahovaných tablet
Součást většího balení (multipack), nemůže být prodáváno samostatně.
5 ZPŮSOB A CESTA PODÁNÍ
Jednou denně.
Před použitím si přečtěte příbalovou informaci. Perorální podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do: {MM/RRRR}
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z TAKOVÝCH LÉČIVÝCH PŘÍPRAVKŮ, POKUD JE TO VHODNÉ
H. Lundbeck A/S Ottiliavej 9 2500 Valby Dánsko
12. REGISTRAČNÍ CISLO(A)
EU/1/02/219/035 840 (20 balení každé obsahující 42) potahovaných tablet EU/1/02/219/049 840 (20 balení každé obsahující 42) potahovaných tablet
13. ČÍSLO ŠARŽE
Č.š.: {číslo}
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16 INFORMACE V BRAILLOVĚ PÍSMU
Ebixa 20 mg tablety
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU
ŠTÍTEK NA FÓLII BALENÍ MULTIPACK (VČETNĚ ,BLUE BOX‘)
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Ebixa 20 mg potahované tablety Memantini hydrochloridum
2 OBSAH LÉČIVÉ LÁTKY/LÁTEK
Jedna potahovaná tableta obsahuje 20 mg memantini hydrochloridum, což odpovídá 16,62 mg memantinu.
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH
Potahované tablety
Multipack : 840 (20 balení každé obsahující 42) potahovaných tablet.
5. ZPŮSOB A CESTA PODÁNÍ
Jednou denně.
Před použitím si přečtěte příbalovou informaci. Perorální podání.
6 ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do: {MM/RRRR}
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z TAKOVÝCH LÉČIVÝCH PŘÍPRAVKŮ, POKUD JE TO VHODNÉ
H. Lundbeck A/S Ottiliavej 9 2500 Valby Dánsko
12. REGISTRAČNÍ CÍSLO(A)
leett
EU/1/02/219/035 840 ( 20 balení každé obsahující 42) potahovaných tabl EU/1/02/219/049 840 (20 balení každé obsahující 42) potahovaných tabl
Č.š.: {číslo}
Výdej léčivého přípravku vázán na lékařský předpis.
15 NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
Ebixa 20 mg tablety
Ebixa 20 mg potahované tablety Memantini hydrochloridum
H. Lundbeck A/S
Použitelné do: {MM/RRRR} Viz razítko na okraji.
Č.š.:{číslo}
Viz razítko na okraji.
Po ^ Út ^ St ^ Čt ^ Pá ^ So ^ Ne
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: Informace pro uživatele
Ebixa 10 mg potahované tablety
Memantini hydrochloridum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí
ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je Ebixa a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Ebixa užívat
3. Jak se přípravek Ebixa užívá
4. Možné nežádoucí účinky
5. Jak přípravek Ebixa uchovávat
6. Obsah balení a další informace
1. Co je Ebixa a k čemu se používá
k léčbě Mozek
Ebixa obsahuje léčivou látku memantin-hydrochlorid a patří do skupiny přípravků užívaných demence.
Ztráta paměti u Alzheimerovy choroby nastává vlivem poruchy přenosu signálů v mozku. obsahuje tzv. N-methyl-D-aspartátové (NMDA) receptory, které zprostředkovávají přenos nervových vzruchů důležitých pro učení a paměť. Ebixa patří do skupiny přípravků tzv. antagonistů NMDA receptorů. Ebixa ovlivňuje tyto NMDA receptory a zlepšuje tak přenos nervových signálů a paměť.
Ebixa se používá k léčbě pacientů se střední až těžkou formou Alzheimerovy choroby.
2. Čemu musíte věnovat pozornost, než začnete přípravek Ebixa užívat Neužívejte přípravek Ebixa
- jestliže jste alergický(á) na memantin nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Upozornění a opatření
Před použitím přípravku Ebixa se poraďte se svým lékařem nebo lékárníkem:
- jestliže se u Vás v minulosti objevily epileptické záchvaty
- jestliže jste nedávno prodělal/a srdeční infarkt, nebo pokud trpíte městnavým selháním srdce nebo neléčenou hypertenzí (vysoký krevní tlak)
V těchto případech by léčba měla být pečlivě sledována a Váš lékař bude pravidelně vyhodnocovat přínos léčby.
Pokud trpíte renálním postižením (potíže s ledvinami), Váš lékař bude činnost ledvin pečlivě sledovat a pokud to bude nutné, upraví dávku memantinu.
Je nutno vyhnout se současnému užívání těchto přípravků: amantadin (k léčbě Parkinsonovy choroby), ketamin (látka používaná jako anestetikum), dextromethorfan (používaný k léčbě kašle) a jiných tzv. antagonistů NMDA receptorů.
Děti a dospívající
U dětí a mladistvých do 18 let se podávání přípravku Ebixa nedoporučuje.
Další léčivé přípravky a Ebixa
Prosím, informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval/a nebo které možná budete užívat.
Přípravek Ebixa může ovlivnit účinky zejména dále uvedených léčivých látek a jejich dávka pak může být Vaším lékařem upravena:
- amantadin, ketamin, dextromethorfan
- dantrolen, baklofen
- cimetidin, ranitidin, prokainamid, chinidin, chinin, nikotin
- hydrochlorothiazid (nebo jakákoli kombinace s hydrochlorothiazidem)
- anticholinergika (látky užívané k léčbě poruch hybnosti nebo křečí zažívacího ústrojí)
- antikonvulziva (látky užívané k předcházení záchvatů křečí a jejich léčbě)
- barbituráty (látky užívané k navození spánku)
- dopaminergní agonisté (L-dopa nebo bromokryptin)
- neuroleptika (látky užívané k léčbě duševních onemocnění)
- perorální antikoagulancia
V případě Vašeho přijetí do nemocnice oznamte lékaři, že užíváte přípravek Ebixa.
Ebixa s jídlem a pitím
Informujte svého lékaře, pokud jste nedávno změnil/a nebo hodláte zásadním způsobem změnit své stravovací návyky (např. přechod z běžné na vegetariánskou stravu) nebo pokud trpíte ledvinovou tubulární acidózou (stav, kdy se vlivem renální dysfunkce (snížená funkce ledvin) dostává do krevního oběhu nadměrné množství kysele reagujících látek), případně závažnou infekcí močových cest (slouží k vylučování moči). Lékař Vám může v takových případech upravit dávku.
Těhotenství a kojení
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek užívat.
Užití memantinu v těhotenství se nedoporučuje.
Kojení
Ženy užívající přípravek Ebixa by neměly kojit.
Řízení dopravních prostředků a obsluha strojů
Váš lékař rozhodne, zda Vám onemocnění umožňuje bezpečné řízení motorových vozidel a ovládání strojů. Ebixa může změnit schopnost reakce natolik, že řízení motorových vozidel a ovládání strojů není vhodné.
3. Jak se přípravek Ebixa užívá
Vždy užívejte přípravek Ebixa přesně podle pokynů svého lékaře. Pokud si nejste jistý/á, poraďte se se svým lékařem nebo lékárníkem.
Doporučená dávka přípravku Ebixa pro dospělé a starší osoby je 20 mg jednou denně. V zájmu snížení rizika výskytu nežádoucích účinků je zapotřebí této dávky dosáhnout pozvolna podle uvedeného postupu:
|
týden 1 |
polovina 10 mg tablety |
|
týden 2 |
jedna 10 mg tableta |
|
týden 3 |
jeden a půl 10 mg tablety |
|
týden 4 a dále |
dvě 10 mg tablety jednou denně |
Obvyklá úvodní dávka je polovina tablety denně (1 x 5 mg) po dobu prvního týdne. Tato dávka se zvyšuje ve druhém týdnu na jednu tabletu jednou denně (1 x 10 mg) a dále ve třetím týdnu na jednu a půl tablety jednou denně. Od čtvrtého týdne je obvyklá dávka 2 tablety jednou denně (1 x 20 mg).
Dávkování u pacientů se sníženou funkcí ledvin
Pokud máte sníženou funkci ledvin, Váš lékař dávku upraví podle Vašeho zdravotního stavu. Bude Vám též pravidelně kontrolovat funkci ledvin.
Podávání
Ebixa se užívá perorálně (ústy) jednou denně. K dosažení příznivého účinku léku je nutno jej užívat pravidelně každý den ve stejnou denní dobu. Tablety polkněte a zapijte vodou. Tablety je možno užít společně s jídlem nebo nalačno.
Délka léčby
Pokračujte v léčbě přípravkem Ebixa tak dlouho, dokud je pro Vás přínosem. Váš lékař bude pravidelně vyhodnocovat léčbu.
Jestliže jste užil/a více přípravku Ebixa, než jste měl/a
- Nadměrná dávka přípravku Ebixa Vám obvykle neublíží. Mohou se u Vás ve zvýšené míře vyskytnout nežádoucí účinky uvedené v bodě 4. “Možné nežádoucí účinky“.
- V případě výrazného předávkování vyhledejte lékaře nebo jej požádejte o radu, protože můžete potřebovat lékařskou péči.
Jestliže jste zapomněl/a užít přípravek Ebixa
- Pokud opomenete užít předepsanou dávku, počkejte a vezměte si následující dávku v obvyklou dobu.
- Nezdvojujte následující dávku, abyste doplnil/a vynechanou dávku.
Máte-li jakékoli další otázky, týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Nežádoucí účinky jsou obvykle mírné až středně těžké.
Časté (postihují 1 až 10 uživatelů ze 100):
• Bolesti hlavy, ospalost, zácpa, zvýšené hodnoty jaterních testů, závratě, poruchy rovnováhy, dušnost, zvýšení krevního tlaku a přecitlivělost na přípravek
Méně časté (postihují 1 až 10 uživatelů z 1 000):
• Únava, mykotické infekce, zmatenost, halucinace, zvracení, poruchy chůze, srdeční selhání a srážení krve v žilách (trombóza/tromboembolismus)
Velmi vzácné (postihují méně než 1 uživatele z 10 000):
• Křeče
Není známo (z dostupných údajů nelze určit)
• Zánět slinivky břišní, zánět jater (hepatitida) a psychotické reakce
Alzheimerova choroba bývá často doprovázena depresí, sebevražednými představami a sebevraždou. Tyto případy se vyskytly též při léčbě přípravkem Ebixa.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Ebixa uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti vyznačené na krabičce a blistru za Použitelné do. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co přípravek Ebixa obsahuje
- Léčivou látkou je memantini hydrochloridum. Jedna tableta obsahuje memantini hydrochloridum 10 mg, což odpovídá 8,31 mg memantinu.
- Další pomocné látky obsažené v jádru tablety jsou: mikrokrystalická celulosa, sodná sůl kroskarmelosy, koloidní bezvodý oxid křemičitý a magnesium-stearát. Potah tablety: hypromelosa, makrogol 400, oxid titaničitý (E 171) a žlutý oxid železitý (E 172).
Jak Ebixa vypadá a co obsahuje toto balení
Ebixa potahované tablety jsou světle žluté až žluté, oválné potahované tablety s půlící rýhou a označením “1 0“ na jedné straně a “M M“ na druhé straně . Tabletu lze rozdělit na stejné dávky.
Ebixa potahované tablety se dodává v blistrech v balení: 14 tablet, 28 tablet, 30 tablet, 42 tablet, 49 x 1 tableta, 50 tablet, 56 tablet, 56 x 1 tableta, 70 tablet, 84 tablet, 98 tablet, 98 x 1 tableta, 100 tablet, 100 x 1 tableta, 112 tablet, 980 (10 x 98) tablet nebo 1000 (20 x 50 tablet). Balení 49 x 1, 56 x 1, 98 x 1 a 100 x 1 potahovaná tableta jsou ve formě blistru jednodávkového.
Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci / výrobce
H. Lundbeck A/S Ottiliavej 9 2500 Valby Dánsko
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci.
EKarapuH Luxembourg/Luxemburg
Lundbeck Export A/S Representative Office Lundbeck S.A.
Tel: +359 2 962 4696 Tél: +32 2 340 2828
Česká republika
Lundbeck Česká republika s.r.o. Tel: +420 225 275 600
Danmark
Lundbeck Pharma A/S Tlf: +45 4371 4270
Deutschland
Lundbeck GmbH Tel: +49 40 23649 0
Eesti
H. Lundbeck A/S, Taani Tel: + 45 36301311
EHúba
Lundbeck Hellas S.A.
T r(k: +30 210 610 5036
Espaňa
Lundbeck Espaňa S.A.
Tel: +34 93 494 9620
Magyarország Lundbeck Hungaria Kft.
Tel: +36 1 4369980
Malta
H. Lundbeck A/S, Denmark Tel: + 45 36301311
Nederland
Lundbeck B.V.
Tel: +31 20 697 1901
Norge
H. Lundbeck AS Tlf: +47 91 300 800
Ósterreich
Lundbeck Austria GmbH Tel: +43 1 331 070
Polska
Lundbeck Poland Sp. z o. o. Tel.: + 48 22 626 93 00
|
France Lundbeck SAS Tél: + 33 1 79 41 29 00 |
Portugal Lundbeck Portugal Lda Tel: +351 21 00 45 900 |
|
Hrvatska Lundbeck Croatia d.o.o. Tel.: + 385 1 3649 210 |
Romania Lundbeck Export A/S Tel: +40 21319 88 26 |
|
Ireland Lundbeck (Ireland) Limited Tel: +353 1 468 9800 |
Slovenija Lundbeck Pharma d.o.o. Tel.: +386 2 229 4500 |
|
Ísland Lundbeck Export A/S, útibú á Íslandi Tel: +354 414 7070 |
Slovenská republika Lundbeck Slovensko s.r.o. Tel: +421 2 5341 42 18 |
|
Italia Lundbeck Italia S.p.A. Tel: +39 02 677 4171 |
Suomi/Finland Oy H. Lundbeck Ab Puh/Tel: +358 2 276 5000 |
|
Kúrcpo^ Lundbeck Hellas A.E Tpk.: +357 22490305 |
Sverige H. Lundbeck AB Tel: +46 4225 4300 |
|
Latvija H. Lundbeck A/S, Danija Tel: + 45 36301311 |
United Kingdom Lundbeck Limited Tel: +44 1908 64 9966 |
Tato příbalová informace byla naposledy revidována
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Příbalová informace: Informace pro uživatele
Ebixa 5 mg/dávka, perorální roztok
Memantini hydrochloridum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí
ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je Ebixa a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Ebixa užívat
3. Jak se přípravek Ebixa užívá
4. Možné nežádoucí účinky
5. Jak přípravek Ebixa uchovávat
6. Obsah balení a další informace
1. Co je Ebixa a k čemu se používá
Ebixa obsahuje léčivou látku memantin-hydrochlorid a patří do skupiny přípravků užívaných k léčbě demence.
Ztráta paměti u Alzheimerovy choroby nastává vlivem poruchy přenosu signálů v mozku. Mozek obsahuje tzv. N-methyl-D-aspartátové (NMDA) receptory, které zprostředkovávají přenos nervových vzruchů důležitých pro učení a paměť. Ebixa patří do skupiny přípravků tzv. antagonistů NMDA receptorů. Ebixa ovlivňuje tyto NMDA receptory a zlepšuje tak přenos nervových signálů a paměť.
Ebixa se používá k léčbě pacientů se střední až těžkou formou Alzheimerovy choroby.
2. Čemu musíte věnovat pozornost, než začnete přípravek Ebixa užívat Neužívejte přípravek Ebixa
- jestliže jste alergický(á) na memantin nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Upozornění a opatření
Před použitím přípravku Ebixa se poraďte se svým lékařem nebo lékárníkem:
- jestliže se u Vás v minulosti objevily epileptické záchvaty
- jestliže jste nedávno prodělal/a srdeční infarkt, nebo pokud trpíte městnavým selháním srdce nebo neléčenou hypertenzí (vysoký krevní tlak)
V těchto případech by léčba měla být pečlivě sledována a Váš lékař bude pravidelně vyhodnocovat přínos léčby.
Pokud trpíte renálním postižením (problém s ledvinami), Váš lékař bude činnost ledvin pečlivě sledovat a pokud to bude nutné, upraví dávku memantinu.
Je nutno vyhnout se současnému užívání těchto přípravků: amantadin, (pro léčbu Parkinsonovy choroby), ketamin (látka používaná jako anestetikum), dextromethorfan (používaný k léčbě kašle) a jiných tzv. antagonistů NMDA receptorů.
Děti a dospívající
U dětí a mladistvých do 18 let se podávání přípravku Ebixa nedoporučuje.
Další léčivé přípravky a Ebixa
Prosím, informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval/a nebo které možná budete užívat.
Přípravek Ebixa může ovlivnit účinky zejména dále uvedených léčivých látek a jejich dávka pak může být Vaším lékařem upravena:
- amantadin, ketamin, dextromethorfan
- dantrolen, baklofen
- cimetidin, ranitidin, prokainamid, chinidin, chinin, nikotin
- hydrochlorothiazid (nebo jakákoli kombinace s hydrochlorothiazidem)
- anticholinergika (látky užívané k léčbě poruch hybnosti nebo křečí zažívacího ústrojí)
- antikonvulziva (látky užívané k předcházení záchvatů křečí a jejich léčbě)
- barbituráty (látky užívané k navození spánku)
- dopaminergní agonisté (L-dopa nebo bromokryptin)
- neuroleptika (látky užívané k léčbě duševních onemocnění)
- perorální antikoagulancia
V případě Vašeho přijetí do nemocnice oznamte lékaři, že užíváte přípravek Ebixa.
Ebixa s jídlem a pitím
Informujte svého lékaře, pokud jste nedávno změnil/a nebo hodláte zásadním způsobem změnit své stravovací návyky (např. přechod z běžné na vegetariánskou stravu) nebo pokud trpíte ledvinovou tubulární acidózou (stav, kdy se vlivem renální dysfunkce (snížená funkce ledvin) dostává do krevního oběhu nadměrné množství kysele reagujících látek), případně závažnou infekcí močových cest (slouží k vylučování moči). Lékař Vám může v takových případech upravit dávku.
Těhotenství a kojení
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek užívat.
Užití memantinu v těhotenství se nedoporučuje.
Kojení
Ženy užívající přípravek Ebixa by neměly kojit.
Řízení dopravních prostředků a obsluha strojů
Váš lékař rozhodne, zda Vám onemocnění umožňuje bezpečné řízení motorových vozidel a ovládání strojů. Ebixa může změnit schopnost reakce natolik, že řízení motorových vozidel a ovládání strojů není vhodné.
Ebixa obsahuje sorbitol
Tento přípravek obsahuje sorbitol. Pokud Vás Váš lékař informoval, že trpíte nesnášenlivostí některého cukru, kontaktujte svého lékaře dříve, než začnete tento přípravek užívat. Váš lékař Vám poradí.
Tento přípravek obsahuje také draslík, ale méně než 1 mmol (39 mg) v jedné dávce, což je zanedbatelné množství.
3. Jak se přípravek Ebixa užívá
Vždy užívejte přípravek Ebixa přesně podle pokynů svého lékaře. Pokud si nejste jistý/á, poraďte se se svým lékařem nebo lékárníkem.
Jedno stlačení pumpy obsahuje 5 mg memantin-hydrochloridu.
Doporučená dávka přípravku Ebixa pro dospělé a starší osoby jsou čtyři dávky, což odpovídá 20 mg jednou denně. V zájmu snížení rizika výskytu nežádoucích účinků je zapotřebí této dávky dosáhnout pozvolna podle uvedeného postupu:
|
týden 1 |
jedno stlačení pumpy |
|
týden 2 |
dvě stlačení pumpy |
|
týden 3 |
tři stlačení pumpy |
|
týden 4 a dále |
čtyři stlačení pumpy |
Obvyklá úvodní dávka se získá jedním stlačením pumpy jednou denně (1 x 5 mg) po dobu prvního týdne. Tato dávka se zvyšuje ve druhém týdnu na dvě stlačení pumpy jednou denně (1 x 10 mg) a dále ve třetím týdnu na tři stlačení pumpy jednou denně (1 x 15 mg). Od čtvrtého týdne je doporučená dávka čtyři stlačení pumpy jednou denně (1 x 20 mg).
Dávkování u pacientů se sníženou funkcí ledvin
Pokud máte sníženou funkci ledvin, Váš lékař dávku upraví podle Vašeho zdravotního stavu. Bude Vám též ve stanovených intervalech kontrolovat funkci ledvin.
Užívání
Ebixa se užívá jednou denně. K dosažení příznivého účinku léku je nutno jej užívat pravidelně každý den ve stejnou denní dobu. Roztok by měl být užíván s malým množstvím vody. Roztok je možno užít společně s jídlem nebo nalačno.
Podrobný návod pro přípravu a zacházení s přípravkem naleznete na konci této příbalové informace. Délka léčby
Pokračujte v léčbě přípravkem Ebixa tak dlouho, dokud je pro Vás přínosem. Váš lékař bude pravidelně vyhodnocovat léčbu.
Jestliže jste užil/a více přípravku Ebixa, než jste měl/a
- Užití nadměrné dávky přípravku Ebixa Vám obvykle neublíží. Mohou se u Vás ve zvýšené míře vyskytnout nežádoucí účinky uvedené v bodě 4 „Možné nežádoucí účinky“.
- V případě výrazného předávkování přípravkem Ebixa vyhledejte svého lékaře nebo jej požádejte o radu, protože můžete potřebovat lékařskou péči.
Jestliže jste zapomněl/a užít přípravek Ebixa
- Pokud opomenete užít předepsanou dávku, počkejte a vezměte si následující dávku v obvyklou dobu.
- Nezdvojujte následující dávku, abyste doplnil/a vynechanou dávku.
Máte-li jakékoli další otázky, týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Nežádoucí účinky jsou obvykle mírné až středně těžké.
Časté (postihují 1 až 10 uživatelů ze 100):
• Bolesti hlavy, ospalost, zácpa, závratě, zvýšené hodnoty jaterních testů, dušnost, poruchy rovnováhy, zvýšení krevního tlaku a přecitlivělost na přípravek
Méně časté (postihují 1 až 10 uživatelů z 1 000):
• Únava, mykotické infekce, zmatenost, halucinace, zvracení, poruchy chůze, srdeční selhání a srážení krve v žilách (trombóza/tromboembolismus)
Velmi vzácné (postihují méně než 1 uživatele z 10 000):
• Křeče
Není známo (z dostupných údajů nelze určit)
• Zánět slinivky břišní, zánět jater (hepatitida) a psychotické reakce
Alzheimerova choroba bývá často doprovázena depresí, sebevražednými představami a sebevraždou. Tyto případy se vyskytly též při léčbě přípravkem Ebixa.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Ebixa uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti vyznačené na krabičce a lahvičce za Použitelné do. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte při teplotě do 30° C.
Po otevření je nutné obsah spotřebovat do 3 měsíců.
Lahvička s připevněnou pumpou musí být uchovávána a přepravována pouze ve svislé poloze.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co obsahuje přípravek Ebixa
- Léčivou látkou je memantini hydrochloridum.
Jedno stlačení pumpy vydá 0,5 ml roztoku obsahujícího 5 mg memantini hydrochloridum, což odpovídá 4,16 mg memantinu.
- Další pomocné látky jsou: kalium-sorbát, sorbitol E420 a čištěná voda.
Jak Ebixa vypadá a co obsahuje toto balení
Ebixa perorální roztok je čirý, bezbarvý až slabě nažloutlý roztok.
Ebixa perorální roztok se dodává v lahvičkách 50 ml, 100 ml nebo 10 x 50 ml.
Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci / výrobce
H. Lundbeck A/S Ottiliavej 9 2500 Valby Dánsko
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci.
Tel: +359 2 962 4696
Česká republika
Lundbeck Česká republika s.r.o. Tel: +420 225 275 600
Danmark
Lundbeck Pharma A/S Tlf: +45 4371 4270
Deutschland
Lundbeck GmbH Tel: +49 40 23649 0
Magyarország Lundbeck Hungaria Kft.
Tel: +36 1 4369980
Malta
H. Lundbeck A/S, Denmark Tel: + 45 36301311
Nederland
Lundbeck B.V.
Tel: +31 20 697 1901
|
Eesti H. Lundbeck A/S, Taani Tel: + 45 36301311 |
Norge H. Lundbeck AS Tlf: +47 91 300 800 |
|
Ekkába Lundbeck Hellas S.A. Tpk: +30 210 610 5036 |
Ósterreich Lundbeck Austria GmbH Tel: +43 1 331 070 |
|
Espaňa Lundbeck Espaňa S.A. Tel: +34 93 494 9620 |
Polska Lundbeck Poland Sp. z o. o Tel.: + 48 22 626 93 00 |
|
France Lundbeck SAS Tél: + 33 1 79 41 29 00 |
Portugal Lundbeck Portugal Lda Tel: +351 21 00 45 900 |
|
Hrvatska Lundbeck Croatia d.o.o. Tel.: + 385 1 3649 210 |
Romania Lundbeck Export A/S Tel: +40 21319 88 26 |
|
Ireland Lundbeck (Ireland) Limited Tel: +353 1 468 9800 |
Slovenija Lundbeck Pharma d.o.o. Tel.: +386 2 229 4500 |
|
Ísland Lundbeck Export A/S, útibú á Íslandi Tel: +354 414 7070 |
Slovenská republika Lundbeck Slovensko s.r.o. Tel: +421 2 5341 42 18 |
|
Italia Lundbeck Italia S.p.A. Tel: +39 02 677 4171 |
Suomi/Finland Oy H. Lundbeck Ab Puh/Tel: +358 2 276 5000 |
|
Kúrcpo^ Lundbeck Hellas A.E Tpk.: +357 22490305 |
Sverige H. Lundbeck AB Tel: +46 4225 4300 |
|
Latvija H. Lundbeck A/S, Danija Tel: + 45 36301311 |
United Kingdom Lundbeck Limited Tel: +44 1908 64 9966 |
Tato příbalová informace byla naposledy revidována
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky: http://www.ema.europa.eu.
Návod pro správné použití pumpy
Užívejte přípravek Ebixa jednou denně a to vždy ve stejnou dobu s malým množstvím vody. Roztok se může užívat s jídlem nebo nalačno.
Roztok nesmí být dávkován do úst přímo z lahvičky nebo pumpy. Dávku odměřte s použitím pumpy na lžičku nebo do sklenice s vodou.
Sejměte šroubovací víčko z lahvičky:
Víčko se musí otočit proti směru hodinových ručiček, úplně odšroubovat a odstranit (viz obr. 1).

Vyjměte dávkovací pumpu z plastikového sáčku (obr.2) a umístěte na vrchol lahvičky. Vsuňte opatrně plastikovou trubičku do lahvičky. Přidržte dávkovací pumpu na hrdle lahvičky a šroubujte ve směru hodinových ručiček, dokud není pevně připojena (obr.3). Dávkovací pumpa je našroubována pouze jednou při zahájení používání a neměla by být nikdy odšroubována.

Jak dávkovací pumpa pracuje:
Hlava dávkovací pumpy má dvě polohy a lze jí snadno otáčet:
- proti směru hodinových ručiček (odblokování)
- ve směru hodinových ručiček (blokování).
Hlava dávkovací pumpy by neměla být stlačována dolů při blokované poloze. Roztok může být dávkován pouze v neblokované poloze. Pokud chcete pumpu odblokovat, otočte hlavu dávkovací pumpy směrem k šipce (asi jednu osminu otáčky, obr.4) tak, že už jí nemůžete dále otočit. Dávkovací pumpa je tak připravena k použití.
Dávkovači pumpa nevydá při prvním použití přesné množství perorálního roztoku. Proto musí být pumpa připravena k použití stlačením hlavy dávkovači pumpy úplně dolů pětkrát za sebou (obr.5).


Tento vypumpovaný roztok se nepoužije pro léčbu a má být zlikvidován. Při dalším použití je hlava dávkovací pumpy stlačena úplně dolů (odpovídá jednomu stlačení pumpy) a je odměřena správná dávka (obr.6).

Správné použití dávkovací pumpy:
Umístěte lahvičku na vodorovnou plochu, např. na stůl, a použijte ji pouze ve svislé poloze. Podržte sklenici s malým množstvím vody nebo lžičku před tryskou. Hlavu dávkovací pumpy stlačte dolů pevným, ale klidným stálým stiskem - ne příliš pomalu (obr.7, obr.8).

Hlava dávkovači pumpy může být poté uvolněna a pumpa je připravena k odměření další dávky stlačením pumpy.
Dávkovací pumpa musí být použita pouze s roztokem přípravku Ebixa, který je připravený v lahvičce. Nesmí se používat pro jiné látky nebo jiné obaly. Jestliže pumpa nefunguje správně, informujte svého lékaře nebo lékárníka. Po užití přípravku Ebixa uveďte dávkovací pumpu do blokované polohy.
Příbalová informace: Informace pro uživatele
Ebixa 5 mg potahované tablety Ebixa 10 mg potahované tablety Ebixa 15 mg potahované tablety Ebixa 20 mg potahované tablety
Memantini hydrochloridum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí
ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je Ebixa a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Ebixa užívat
3. Jak se přípravek Ebixa užívá
4. Možné nežádoucí účinky
5. Jak přípravek Ebixa uchovávat
6. Obsah balení a další informace
1. Co je Ebixa a k čemu se používá
Ebixa obsahuje léčivou látku memantin-hydrochlorid a patří do skupiny přípravků užívaných k léčbě demence.
Ztráta paměti u Alzheimerovy choroby nastává vlivem poruchy přenosu signálů v mozku. Mozek obsahuje tzv. N-methyl-D-aspartátové (NMDA) receptory, které zprostředkovávají přenos nervových vzruchů důležitých pro učení a paměť. Ebixa patří do skupiny přípravků tzv. antagonistů NMDA receptorů. Ebixa ovlivňuje tyto NMDA receptory a zlepšuje tak přenos nervových signálů a paměť.
Ebixa se používá k léčbě pacientů se střední až těžkou formou Alzheimerovy choroby.
2. Čemu musíte věnovat pozornost, než začnete přípravek Ebixa užívat
Neužívejte přípravek Ebixa
- jestliže jste alergický(á) na memantin nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Upozornění a opatření
Před použitím přípravku Ebixa se poraďte se svým lékařem nebo lékárníkem:
- jestliže se u Vás v minulosti objevily epileptické záchvaty
- jestliže jste nedávno prodělal/a srdeční infarkt, nebo pokud trpíte městnavým selháním srdce nebo neléčenou hypertenzí (vysoký krevní tlak)
V těchto případech by léčba měla být pečlivě sledována a Váš lékař bude pravidelně vyhodnocovat přínos léčby.
Pokud trpíte renálním postižením (potíže s ledvinami), Váš lékař bude činnost ledvin pečlivě sledovat a pokud to bude nutné, upraví dávku memantinu.
Je nutno vyhnout se současnému užívání těchto přípravků: amantadin (k léčbě Parkinsonovy choroby), ketamin (látka používaná jako anestetikum), dextromethorfan (používaný k léčbě kašle) a jiných tzv. antagonistů NMDA receptorů.
Děti a dospívající
U dětí a mladistvých do 18 let se podávání přípravku Ebixa nedoporučuje.
Další léčivé přípravky a Ebixa
Prosím, informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval/a nebo které možná budete užívat.
Přípravek Ebixa může ovlivnit účinky zejména dále uvedených léčivých látek a jejich dávka pak může být Vaším lékařem upravena:
- amantadin, ketamin, dextromethorfan
- dantrolen, baklofen
- cimetidin, ranitidin, prokainamid, chinidin, chinin, nikotin
- hydrochlorothiazid (nebo jakákoli kombinace s hydrochlorothiazidem)
- anticholinergika (látky užívané k léčbě poruch hybnosti nebo křečí zažívacího ústrojí)
- antikonvulziva (látky užívané k předcházení záchvatů křečí a jejich léčbě)
- barbituráty (látky užívané k navození spánku)
- dopaminergní agonisté (L-dopa nebo bromokryptin)
- neuroleptika (látky užívané k léčbě duševních onemocnění)
- perorální antikoagulancia
V případě Vašeho přijetí do nemocnice oznamte lékaři, že užíváte přípravek Ebixa.
Ebixa s jídlem a pitím
Informujte svého lékaře, pokud jste nedávno změnil/a nebo hodláte zásadním způsobem změnit své stravovací návyky (např. přechod z běžné na vegetariánskou stravu) nebo pokud trpíte ledvinovou tubulární acidózou (stav, kdy se vlivem renální dysfunkce (snížená funkce ledvin) dostává do krevního oběhu nadměrné množství kysele reagujících látek), případně závažnou infekcí močových cest (slouží k vylučování moči). Lékař Vám může v takových případech upravit dávku.
Těhotenství a kojení
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek užívat.
Užití memantinu v těhotenství se nedoporučuje.
Kojení
Ženy užívající přípravek Ebixa by neměly kojit.
Řízení dopravních prostředků a obsluha strojů
Váš lékař rozhodne, zda Vám onemocnění umožňuje bezpečné řízení motorových vozidel a ovládání strojů. Ebixa může změnit schopnost reakce natolik, že řízení motorových vozidel a ovládání strojů není vhodné.
3. Jak se přípravek Ebixa užívá
Balení přípravku Ebixa pro zahájení léčby se používá pouze na začátku léčby přípravkem Ebixa .
Vždy užívejte přípravek Ebixa přesně podle pokynů svého lékaře. Pokud si nejste jistý/á, poraďte se se svým lékařem nebo lékárníkem.
Doporučená léčebná dávka 20 mg denně je dosažena postupným zvyšováním dávky přípravku Ebixa po dobu prvních 3 týdnů léčby. Léčebné schéma je také určeno pro balení pro zahájení léčby. Užijte jednu tabletu jednou denně.
Týden 1 (den 1-7)
Užijte jednu 5 mg tabletu jednou denně (bílé až téměř bílé, oválně podlouhlé) po dobu 7 dnů.
Týden 2 (den 8-14)
Užijte jednu 10 mg tabletu jednou denně (světle žluté až žluté, oválné ) po dobu 7 dnů.
Týden 3 (den 15 - 21):
Užijte jednu 15 mg tabletu jednou denně (šedooranžové, oválně podlouhlé) po dobu 7 dnů.
Týden 4 (den 22-28):
Užijte jednu 20 mg tabletu denně (šedočervené, oválně podlouhlé) po dobu 7 dnů.
|
týden 1 |
5 mg tableta |
|
týden 2 |
10 mg tableta |
|
týden 3 |
15 mg tableta |
|
týden 4 a dále |
20 mg tablety jednou denně |
Udržovací dávka
Doporučená denní dávka je 20 mg jednou denně.
Pro pokračování v léčbě se prosím obraťte na svého lékaře.
Dávkování u pacientů se sníženou funkcí ledvin
Pokud máte sníženou funkci ledvin, Váš lékař dávku upraví podle Vašeho zdravotního stavu.
V takovém případě by měl Váš lékař provádět pravidelně kontrolu funkce Vašich ledvin.
Užívání
Ebixa se užívá perorálně (ústy) jednou denně. K dosažení příznivého účinku léku je nutno jej užívat pravidelně každý den ve stejnou denní dobu. Tablety polkněte a zapijte vodou. Tablety je možno užít společně s jídlem nebo nalačno.
Délka léčby
Pokračujte v léčbě přípravkem Ebixa tak dlouho, dokud je pro Vás přínosem. Váš lékař bude pravidelně vyhodnocovat léčbu.
Jestliže jste užil/a více přípravku Ebixa, než jste měl/a
- Nadměrná dávka přípravku Ebixa Vám obvykle neublíží. Mohou se u Vás ve zvýšené míře vyskytnout nežádoucí účinky uvedené v bodě 4. “Možné nežádoucí účinky“.
- V případě výrazného předávkování vyhledejte lékaře nebo jej požádejte o radu, protože můžete potřebovat lékařskou péči.
Jestliže jste zapomněl/a užít přípravek Ebixa
- Pokud opomenete užít předepsanou dávku, počkejte a vezměte si následující dávku v obvyklou dobu.
- Nezdvojujte následující dávku, abyste doplnil/a vynechanou dávku.
Máte-li jakékoli další otázky, týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo
lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Nežádoucí účinky jsou obvykle mírné až středně těžké.
Časté (postihují 1 až 10 uživatelů ze 100):
• Bolesti hlavy, ospalost, zácpa, zvýšené hodnoty jaterních testů, závratě, poruchy rovnováhy, dušnost, zvýšení krevního tlaku a přecitlivělost na přípravek
Méně časté (postihují 1 až 10 uživatelů z 1 000):
• Únava, mykotické infekce, zmatenost, halucinace, zvracení, poruchy chůze, srdeční selhání a srážení krve v žilách (trombóza/tromboembolismus).
Velmi vzácné (postihují méně než 1 uživatele z 10 000):
• Křeče
Není známo (z dostupných údajů nelze určit)
• Zánět slinivky břišní, zánět jater (hepatitida) a psychotické reakce
Alzheimerova choroba bývá často doprovázena depresí, sebevražednými představami a sebevraždou. Tyto případy se vyskytly též při léčbě přípravkem Ebixa.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Ebixa uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti vyznačené na krabičce a blistru za Použitelné do. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co obsahuje přípravek Ebixa
- Léčivou látkou je memantini hydrochloridum. Jedna tableta obsahuje 5/10/15/20 mg memantini hydrochloridum (což odpovídá 4,15/8,31/12,46/16,62 mg memantinu).
- Další pomocné látky přípravku Ebixa 5/10/15 a 20 mg potahované tablety jsou mikrokrystalická celulosa, kroskarmelosa sodná, koloidní bezvodý křemík, magnesium stearát, všechny v jádru tablety a hypromelosa, makrogol 400, oxid titaničitý (E 171) a další složkou pro Ebixa 10 mg potahované tablety je žlutý oxid železitý (E 172)a další složkou pro Ebixa 15 mg a Ebixa 20 mg potahované tablety je žlutý a červený oxid železa (E 172), všechny v potahu tablety.
Jak Ebixa vypadá a co obsahuje toto balení
Potahované tablety Ebixa 5 mg jsou k dispozici jako bílé až téměř bílé, oválně podlouhlé tablety s vytištěným „5“ na jedné straně a „MEM“ na druhé straně.
Potahované tablety Ebixa 10 mg jsou k dispozici jako světle žluté až žluté, oválné potahované tablety s půlící rýhou a označením “1 0“ na jedné straně a “M M“ na druhé straně.. Tabletu je možné rozdělit na stejné dávky.
Potahované tablety Ebixa 15 mg jsou k dispozici jako oranžové až šedooranžové, oválně podlouhlé tablety s vytištěným „15“ na jedné straně a „MEM“ na druhé straně.
Potahované tablety Ebixa 20 mg jsou k dispozici jako světle červené až šedočervené, oválně podlouhlé tablety s vytištěným „20“ na jedné straně a „MEM“ na druhé straně.
Jedno balení pro zahájení léčby obsahuje 28 tablet v 4 blistrech s 7 tabletami přípravku Ebixa 5 mg, 7 tablet přípravku Ebixa 10 mg, 7 tablet přípravku Ebixa 15 mg a 7 tablet přípravku Ebixa 20 mg.
Držitel rozhodnutí o registraci / výrobce
H. Lundbeck A/S Ottiliavej 9 2500 Valby Dánsko
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci.
Belgique/Belgie/Belgien
Lundbeck S.A./N.V. Tél/Tel: +32 2 340 2828
Lietuva
H. Lundbeck A/S, Danija Tel: + 45 36301311
Btarapna Luxembourg/Luxemburg
Lundbeck Export A/S Representative Office Lundbeck S.A.
Tel: +359 2 962 4696 Tél: +32 2 340 2828
Magyarország Lundbeck Hungaria Kft.
Tel: +36 1 4369980
Česká republika
Lundbeck Česká republika s.r.o. Tel: +420 225 275 600
|
Danmark Lundbeck Pharma A/S Tlf: +45 4371 4270 |
Malta H. Lundbeck A/S, Denmark Tel: + 45 36301311 |
|
Deutschland Lundbeck GmbH Tel: +49 40 23649 0 |
Nederland Lundbeck B.V. Tel: +31 20 697 1901 |
|
Eesti H. Lundbeck A/S, Taani Tel: + 45 36301311 |
Norge H. Lundbeck AS Tlf: +47 91 300 800 |
|
Ekkába Lundbeck Hellas S.A. Tpk: +30 210 610 5036 |
Ósterreich Lundbeck Austria GmbH Tel: +43 1 331 070 |
|
Espaňa Lundbeck Espaňa S.A. Tel: +34 93 494 9620 |
Polska Lundbeck Poland Sp. z o. o. Tel.: + 48 22 626 93 00 |
|
France Lundbeck SAS Tél: + 33 1 79 41 29 00 |
Portugal Lundbeck Portugal Lda Tel: +351 21 00 45 900 |
|
Hrvatska Lundbeck Croatia d.o.o. Tel.: + 385 1 3649 210 |
Romania Lundbeck Export A/S Tel: +40 21319 88 26 |
|
Ireland Lundbeck (Ireland) Limited Tel: +353 1 468 9800 |
Slovenija Lundbeck Pharma d.o.o. Tel.: +386 2 229 4500 |
|
Ísland Lundbeck Export A/S, útibú á Íslandi Tel: +354 414 7070 |
Slovenská republika Lundbeck Slovensko s.r.o. Tel: +421 2 5341 42 18 |
|
Italia Lundbeck Italia S.p.A. Tel: +39 02 677 4171 |
Suomi/Finland Oy H. Lundbeck Ab Puh/Tel: +358 2 276 5000 |
|
Kúrcpo^ Lundbeck Hellas A.E Tpk.: +357 22490305 |
Sverige H. Lundbeck AB Tel: +46 4225 4300 |
|
Latvija H. Lundbeck A/S, Danija Tel: + 45 36301311 |
United Kingdom Lundbeck Limited Tel: +44 1908 64 9966 |
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Příbalová informace: informace pro uživatele
Ebixa 20 mg potahované tablety
Memantini hydrochloridum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí
ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je Ebixa a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Ebixa užívat
3. Jak se přípravek Ebixa užívá
4. Možné nežádoucí účinky
5. Jak přípravek Ebixa uchovávat
6. Obsah balení a další informace
1. Co je Ebixa a k čemu se používá
k léčbě Mozek
Ebixa obsahuje léčivou látku memantin-hydrochlorid a patří do skupiny přípravků užívaných demence.
Ztráta paměti u Alzheimerovy choroby nastává vlivem poruchy přenosu signálů v mozku. obsahuje tzv. N-methyl-D-aspartátové (NMDA) receptory, které zprostředkovávají přenos nervových vzruchů důležitých pro učení a paměť. Ebixa patří do skupiny přípravků tzv. antagonistů NMDA receptorů. Ebixa ovlivňuje tyto NMDA receptory a zlepšuje tak přenos nervových signálů a paměť.
Ebixa se používá k léčbě pacientů se střední až těžkou formou Alzheimerovy choroby.
2. Čemu musíte věnovat pozornost, než začnete přípravek Ebixa užívat Neužívejte přípravek Ebixa
- jestliže jste alergický(á) na memantin nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Upozornění a opatření
Před použitím přípravku Ebixa se poraďte se svým lékařem nebo lékárníkem:
- jestliže se u Vás v minulosti objevily epileptické záchvaty
- jestliže jste nedávno prodělal/a srdeční infarkt, nebo pokud trpíte městnavým selháním srdce nebo neléčenou hypertenzí (vysoký krevní tlak)
V těchto případech by léčba měla být pečlivě sledována a Váš lékař bude pravidelně vyhodnocovat přínos léčby.
Pokud trpíte renálním postižením (problém s ledvinami), Váš lékař bude činnost ledvin pečlivě sledovat a pokud to bude nutné, upraví dávku memantinu.
Je nutno vyhnout se současnému užívání těchto přípravků: amantadin (k léčbě Parkinsonovy choroby), ketamin (látka používaná jako anestetikum), dextromethorfan (používaný k léčbě kašle) a jiných tzv. antagonistů NMDA receptorů.
Děti a dospívající
U dětí a mladistvých do 18 let se podávání přípravku Ebixa nedoporučuje.
Další léčivé přípravky a Ebixa
Prosím, informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval/a nebo které možná budete užívat.
Přípravek Ebixa může ovlivnit účinky zejména dále uvedených léčivých látek a jejich dávka pak může být Vaším lékařem upravena:
- amantadin, ketamin, dextromethorfan
- dantrolen, baklofen
- cimetidin, ranitidin, prokainamid, chinidin, chinin, nikotin
- hydrochlorothiazid (nebo jakákoli kombinace s hydrochlorothiazidem)
- anticholinergika (látky užívané k léčbě poruch hybnosti nebo křečí zažívacího ústrojí)
- antikonvulziva (látky užívané k předcházení záchvatů křečí a jejich léčbě)
- barbituráty (látky užívané k navození spánku)
- dopaminergní agonisté (L-dopa nebo bromokryptin)
- neuroleptika (látky užívané k léčbě duševních onemocnění)
- perorální antikoagulancia
V případě Vašeho přijetí do nemocnice oznamte lékaři, že užíváte přípravek Ebixa.
Ebixa s jídlem a pitím
Informujte svého lékaře, pokud jste nedávno změnil/a nebo hodláte zásadním způsobem změnit své stravovací návyky (např. přechod z běžné na vegetariánskou stravu) nebo pokud trpíte ledvinovou tubulární acidózou (stav, kdy se vlivem renální dysfunkce (snížená funkce ledvin) dostává do krevního oběhu nadměrné množství kysele reagujících látek), případně závažnou infekcí močových cest (slouží k vylučování moči). Lékař Vám může v takových případech upravit dávku.
Těhotenství a kojení
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek užívat.
Užití memantinu v těhotenství se nedoporučuje.
Kojení
Ženy užívající přípravek Ebixa by neměly kojit.
Řízení dopravních prostředků a obsluha strojů
Váš lékař rozhodne, zda Vám onemocnění umožňuje bezpečné řízení motorových vozidel a ovládání strojů. Ebixa může změnit schopnost reakce natolik, že řízení motorových vozidel a ovládání strojů není vhodné.
3. Jak se přípravek Ebixa užívá
Vždy užívejte přípravek Ebixa přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Doporučená dávka přípravku Ebixa pro dospělé a starší osoby je 20 mg jednou denně.
Za účelem snížení rizika vedlejších účinků je tato dávka dosažena postupně pomocí následujícího denního léčebného schématu. Pro titraci nahoru jsou k dispozici jiné síly tablety.
Na začátku léčby začnete užívat Ebixa 5 mg potahované tablety jednou denně. Tato dávka se bude zvyšovat každý týden o 5 mg do dosažení doporučené (udržovací) dávky. Doporučená udržovací dávka je 20 mg jednou denně, které se dosáhne na začátku 4. týdne.
Dávkování u pacientů se sníženou funkcí ledvin
Pokud máte sníženou funkci ledvin, Váš lékař dávku upraví podle Vašeho zdravotního stavu. Bude Vám též pravidelně kontrolovat funkci ledvin.
Užívání
Ebixa se užívá jednou denně. K dosažení příznivého účinku léku je nutno jej užívat pravidelně každý den ve stejnou denní dobu. Tablety polkněte a zapijte dostatečným množstvím vody. Tablety je možno užít společně s jídlem nebo nalačno.
Délka léčby
Pokračujte v léčbě přípravkem Ebixa tak dlouho, dokud je pro Vás přínosem. Váš lékař bude pravidelně vyhodnocovat léčbu.
Jestliže jste užil(a) více přípravku Ebixa, než jste měl(a)
- Nadměrná dávka přípravku Ebixa Vám obvykle neublíží. Mohou se u Vás ve zvýšené míře vyskytnout nežádoucí účinky uvedené v bodě 4. “Možné nežádoucí účinky“.
- V případě výrazného předávkování vyhledejte lékaře nebo jej požádejte o radu, protože můžete potřebovat lékařskou péči.
Jestliže jste zapomněl(a) užít přípravek Ebixa
- Pokud opomenete užít předepsanou dávku, počkejte a vezměte si následující dávku v obvyklou dobu.
- Nezdvojujte následující dávku, abyste doplnil(a) vynechanou dávku.
Máte-li jakékoli další otázky, týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Nežádoucí účinky jsou obvykle mírné až středně těžké.
Časté (postihují 1 až 10 uživatelů ze 100):
• Bolesti hlavy, ospalost, zácpa, zvýšené hodnoty jaterních testů, závratě, poruchy rovnováhy, dušnost, zvýšení krevního tlaku a přecitlivělost na přípravek
Méně časté (postihují 1 až 10 uživatelů z 1 000):
• Únava, mykotické infekce, zmatenost, halucinace, zvracení, poruchy chůze, srdeční selhání a srážení krve v žilách (trombóza/tromboembolismus).
Velmi vzácné (postihují méně než 1 uživatele z 10 000):
• Křeče
Není známo (z dostupných údajů nelze určit)
• Zánět slinivky břišní, zánět jater (hepatitida) a psychotické reakce
Alzheimerova choroba bývá často doprovázena depresí, sebevražednými představami a sebevraždou. Tyto případy se vyskytly též při léčbě přípravkem Ebixa.
Pokud se kterýkoli z nežádoucích účinků vyskytne v závažné míře, nebo pokud si všimnete jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci, prosím, sdělte to svému lékaři nebo lékárníkovi.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Ebixa uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti vyznačené na krabičce a blistru za Použitelné do. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Tento přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co obsahuje přípravek Ebixa
- Léčivou látkou je memantini hydrochloridum. Jedna tableta obsahuje 20 mg memantini
hydrochloridum, což odpovídá 16,62 mg memantinu.
- Další složky jsou mikrokrystalická celulosa, kroskarmelosa sodná, koloidní bezvodý křemík, magnesium stearát, všechny v jádru tablety a hypromelosa, makrogol 400, oxid titaničitý (E 171), žlutý a červený oxid železa (E 172), všechny v potahu tablety.
Jak Ebixa vypadá a co obsahuje toto balení
Ebixa potahované tablety jsou k dispozici jako světle červené až šedo-červené, oválné - podlouhlé potahované tablety s vytištěným „20“ na jedné straně a „MEM“ na druhé straně.
Ebixa potahované tablety jsou k dispozici v blistrových baleních s 14 tabletami, 28 tabletami,
42 tabletami, 49 x 1 tabletou, 56 tabletami, 70 tabletami, 84 tabletami, 98 tabletami, 98 x 1 tabletou, 100 x 1, 112 tabletami nebo 840 (20 x 42) tabletami. Balení 49 x 1, 56 x 1, 98 x 1 a 100 x 1 potahovaná tableta jsou ve formě blistru jednodávkového.
Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci / výrobce
H. Lundbeck A/S Ottiliavej 9 2500 Valby Dánsko
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci.
Bt^rapuH Luxembourg/Luxemburg
Lundbeck Export A/S Representative Office Lundbeck S.A.
Tel: +359 2 962 4696 Tél: +32 2 340 2828
Česká republika
Lundbeck Česká republika s.r.o. Tel: +420 225 275 600
Danmark
Lundbeck Pharma A/S Tlf: +45 4371 4270
Deutschland
Lundbeck GmbH Tel: +49 40 23649 0
Eesti
H. Lundbeck A/S, Taani Tel: + 45 36301311
Ekkúňa
Lundbeck Hellas S.A.
T qk: +30 210 610 5036
Espaňa
Lundbeck Espaňa S.A.
Tel: +34 93 494 9620
Magyarország Lundbeck Hungaria Kft.
Tel: +36 1 4369980
Malta
H. Lundbeck A/S, Denmark Tel: + 45 36301311
Nederland
Lundbeck B.V.
Tel: +31 20 697 1901
Norge
H. Lundbeck AS Tlf: +47 91 300 800
Ósterreich
Lundbeck Austria GmbH Tel: +43 1 331 070
Polska
Lundbeck Poland Sp. z o. o. Tel.: + 48 22 626 93 00
|
France Lundbeck SAS Tél: + 33 1 79 41 29 00 |
Portugal Lundbeck Portugal Lda Tel: +351 21 00 45 900 |
|
Hrvatska Lundbeck Croatia d.o.o. Tel.: + 385 1 3649 210 |
Romania Lundbeck Export A/S Tel: +40 21319 88 26 |
|
Ireland Lundbeck (Ireland) Limited Tel: +353 1 468 9800 |
Slovenija Lundbeck Pharma d.o.o. Tel.: +386 2 229 4500 |
|
Ísland Lundbeck Export A/S, útibú á Íslandi Tel: +354 414 7070 |
Slovenská republika Lundbeck Slovensko s.r.o. Tel: +421 2 5341 42 18 |
|
Italia Lundbeck Italia S.p.A. Tel: +39 02 677 4171 |
Suomi/Finland Oy H. Lundbeck Ab Puh/Tel: +358 2 276 5000 |
|
Kúrcpo^ Lundbeck Hellas A.E Tpk.: +357 22490305 |
Sverige H. Lundbeck AB Tel: +46 4225 4300 |
|
Latvija H. Lundbeck A/S, Danija Tel: + 45 36301311 |
United Kingdom Lundbeck Limited Tel: +44 1908 64 9966 |
Tato příbalová informace byla naposledy revidována
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
95