Depo-Provera
sp.zn.: sukls193223/2011
SOUHRN ÚDAJŮ O PŘÍPRAVKU
1. NÁZEV PŘÍPRAVKU
DEPO-PROVERA Injekční suspenze
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Léčivá látka: Medroxyprogesteroni acetas 150 mg/ml. Pomocné látky: propylparaben, methylparaben Úplný seznam pomocných látek viz bod 6.1
3. LÉKOVÁ FORMA
Injekční suspenze.
Popis přípravku: bílá suspenze.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
A. Antikoncepce (potlačení ovulace).
B. Endometrióza.
C. Adjuvantní nebo paliativní léčba recidivujícího nebo metastazujícího karcinomu endometria nebo ledvin.
D. Léčba hormon-dependentního recidivujícího karcinomu prsu.
E. Léčba vazomotorických potíží v menopauze.
Protože ztráta kostní hmoty (BMD) se může objevit u žen různých věkových skupin, které dlouhodobě užívají injekce MPA (medroxyprogesteron-acetát) (viz bod 4.4), měl by být zvážen poměr rizika a přínosu a rovněž brán v úvahu úbytek kostní hmoty (BMD) objevující se během těhotenství a/nebo kojení.
Použití u dospívajících (12-18 let)
Vzhledem k tomu, že dlouhodobý účinek ztráty kostní hmoty spojený s podáváním přípravku DEPO-PROVERA během kritického období pro růst kostní hmoty není znám, je použití přípravku DEPO-PROVERA u dospívajících indikováno pouze tehdy, když byly ostatní antikoncepční metody shledány jako nevhodné nebo nepřijatelné (viz bod 4.4).
4.2 Dávkování a způsob podávání
Pediatrická populace
Přípravek DEPO-PROVERA není indikovaný před menarché (viz bod 4.1). Až na obavy týkající se ztráty kostní hmoty se očekává, že bezpečnost a účinnost přípravku DEPO-PROVERA jsou u dospívajících dívek po menarché a u dospělých žen stejné.
Injekční stříkačka přípravku DEPO-PROVERA by měla být těsně před použitím důkladně protřepána, aby bylo zajištěno, že podávaná dávka bude homogenní suspenzí. Léčba by měla být zahájena lékařem nebo zdravotnickým pracovníkem.
A. Antikoncepce (potlačení ovulace): Doporučená dávka je 150 mg přípravku DEPO-PROVERA v hluboké intramuskulární injekci každé tři měsíce.
První injekce: pro zajištění antikoncepce během prvního cyklu použití by měla být podána první dávka během prvních pěti dnů normálního menstruačního cyklu. Je-li injekce podána podle těchto instrukcí, není vyžadována žádná další metoda antikoncepce.
Další dávky: Druhá a další injekce by měly být podávány v intervalu 13 týdnů. Pokud je z jakéhokoli důvodu interval od předchozí injekce delší než 13 týdnů, mělo by být před podáním další injekce vyloučeno těhotenství. Účinnost přípravku závisí na dodržení doporučeného dávkovacího schématu.
Po porodu: Pokud žena nekojí, doporučuje se podat injekci během prvních pěti dnů po porodu (pro větší jistotu že pacientka není gravidní). Pokud je nutné podat injekci v jinou dobu, je nutné vyloučit těhotenství.
Kojí-li pacientka, doporučuje se injekci podat ne dříve než 6 týdnů po porodu, kdy je enzymový systém novorozence již rozvinutější (viz bod 4.6).
Přechod z jiné metody antikoncepce: Při přechodu z jiné metody antikoncepce by měl být přípravek DEPO-PROVERA podáván způsobem, který zajistí průběžnou antikoncepční ochranu na základě mechanismu účinku obou metod (např. pacientky, které přecházejí z perorální antikoncepce, by měly dostat první injekci přípravku DEPO-PROVERA během sedmi dnů poté, co užily poslední antikoncepční tabletku).
B. Endometrióza: Doporučená dávka přípravku DEPO-PROVERA v této indikaci je 50 mg týdně nebo 100 mg každé dva týdny intramuskulárně po dobu nejméně 6 měsíců.
C. Karcinom endometria a ledvin: Jako počáteční léčba se doporučuje intramuskulárně podávat jednou týdně dávky 400 až 1000 mg přípravku DEPO-PROVERA. Pokud během několika týdnů nebo měsíců dojde ke zlepšení a stabilizaci onemocnění, může jako udržovací terapie postačit pouze dávka 400 mg za měsíc.
D. Karcinom prsu: Doporučené dávkovací schéma je 500 mg přípravku DEPO-PROVERA denně intramuskulárně po dobu 28 dnů. Poté by pacientka měla být převedena na udržovací terapii, s dávkami 500 mg 2x týdně tak dlouho, dokud je přítomna odpověď na léčbu.
E. Potíže v menopauze: Doporučená dávka je 150 mg přípravku DEPO-PROVERA intramuskulárně každé tři měsíce.
Hepatální insuficience: Vliv jaterního onemocnění na farmakokinetiku přípravku DEPO-PROVERA není znám. Jelikož je přípravek DEPO-PROVERA téměř výlučně eliminován v játrech, může být nedostatečně metabolizován u pacientek se závažnou jaterní nedostatečností (viz bod 4.3).
Renální insuficience: Vliv onemocnění ledvin na farmakokinetiku přípravku DEPO-PROVERA není znám. Jelikož je přípravek DEPO-PROVERA téměř výlučně eliminován v játrech, není úprava dávkování považována u žen s renální insuficiencí za nutnou.
4.3 Kontraindikace
Přípravek DEPO-PROVERA je kontraindikovaný u pacientek:
• se známou hypersenzitivitou na léčivou látku, na steroidy nebo kteroukoli pomocnou látku uvedenou v bodě 6.1 se závažnou jatemí nedostatečností
• těhotných nebo při podezření na těhotenství
• při vaginálním krvácení z nejasných příčin.
Kontraindikace u pacientek, kterým byl přípravek DEPO-PROVERA předepsán jako antikoncepce nebo při gynekologických obtížích: přípravek DEPO-PROVERA je kontraindikovaný při malignitě v oblasti prsu a pohlavních orgánů, nebo při podezření na ni.
4.4 Zvláštní upozornění a opatření pro použití
Neočekávané krvácení, které se vyskytne během léčby MPA, musí být vyšetřeno.
Riziko vzniku nádoru
Dlouhodobé sledování pacientek, kterým byly podány injekce intramuskulárního medroxyprogesteron-acetátu (DMPA) 150 mg, zjistilo mírné nebo žádné zvýšení celkového rizika nádorů prsu a žádné zvýšení rizika ovariálních, jaterních nebo cervikálních nádorů, dále trvající ochranný účinek snižující riziko karcinomu endometria.
Před použitím přípravku DEPO-PROVERA by měla být pacientka pečlivě vyšetřena. Vyšetření před nasazením léčby by mělo vyloučit přítomnost novotvaru v oblasti genitálu nebo prsů. Toto upozornění se nevztahuje na pacientky, které budou dostávat přípravek DEPO-PROVERA v terapii karcinomu endometria, prsu nebo ledvin.
Výsledky některých epidemiologických studií naznačují, že existuje malý rozdíl v riziku diagnózy karcinomu prsu u žen, které užívají nebo užívaly hormonální antikoncepci, v porovnání s těmi, které ji nikdy neužívaly. Riziko u současných a nedávných uživatelek DMPA je malé, s ohledem na celkové riziko karcinomu prsu, zvláště u mladých žen (viz níže), a po 10 letech od posledního užití není patrné. Délka užívání se nezdá být významná.
Předpokládaný počet dalších případů karcinomu prsu diagnostikovaného po 10 letech po ukončení léčby injekčními progestogeny+
|
Věk při poslední injekci MPA |
Počet případů na 10,000 žen, které nikdy MPA neužívaly |
Předpokládaný počet dalších případů na 10,000 uživatelek MPA |
|
20 |
Méně než 1 |
Výrazně méně než 1 |
|
30 |
44 |
2-3 |
|
40 |
160 |
10 |
+při užívání po dobu 5 let
Tromboembolické poruchy
Ačkoli medroxyprogesteron-acetát nemá zřejmě příčinnou souvislost s indukcí trombotické nebo tromboembolické poruchy, neměl by být u žádné pacientky užívající přípravek DEPO-PROVERA, u které se vyskytne tato příhoda, např. plicní embolie, cévní mozková příhoda, retinální trombóza nebo hluboká žilní trombóza, tento přípravek znovu podáván. Ženy s předchozí anamnézou tromboembolické poruchy nebyly sledovány v klinických hodnoceních a žádné informace, které by dokládaly bezpečnost podání přípravku DEPO-PROVERA této populaci nejsou dostupné.
Anafylaxe a anafylaktická reakce
Pokud se objeví anafylaktická reakce, měla by být ihned zahájena příslušná léčba. Závažné anafylaktické reakce vyžadují okamžitou léčbu.
Vzhledem k tomu, že přípravek obsahuje methylparaben a propylparaben, může vyvolat alergické reakce (i opožděné). V případě výskytu alergické reakce nesmí být přípravek již znovu podán (viz bod 4.3).
Změny tělesné hmotnosti a retence tekutin
DEPO-PROVERA může způsobit, zvláště ve vysokých dávkách, používaných v léčbě u pacientek se zhoubnými nádory, přírůstek tělesné hmotnosti a retenci tekutin. Přípravek musí být proto podáván opatrně pacientkám, jejichž předchozí onemocnění by mohlo být přírůstkem hmotnosti nebo retencí tekutin nepříznivě ovlivněno.
Vysoké dávky přípravku DEPO-PROVERA podávané pacientkám se zhoubným nádorem mohou v některých případech vyvolat cushingoidní příznaky, například měsícovitý obličej, retenci tekutin, poruchy glukózové tolerance a vzestup krevního tlaku.
Uhlohydráty/metabolizmus
U některých pacientek léčených progestiny může dojít ke snížení glukózové tolerance. Během této léčby je třeba pečlivě sledovat pacientky s diabetem.
Psychiatrické poruchy
Pacientky, které byly v minulosti léčeny pro deprese, by měly být během léčby přípravkem DEPO-PROVERA pečlivě sledovány. Některé pacientky si mohou při léčbě přípravkem DEPO-PROVERA stěžovat na depresivní stavy obdobné premenstruální tenzi.
Laboratorní vyšetření
Při odesílání vzorků na patologické vyšetření by měl být patolog informován o tom, že pacientka je léčena přípravkem DEPO-PROVERA. Lékař by měl být informován o tom, že určitá endokrinologická vyšetření a vyšetření jaterních testů a krevních složek mohou být při léčbě progestiny ovlivněna:
a) Snížení hladiny steroidů v plazmě/moči (např. progesteron, estradiol, pregnanediol, testosteron, kortison).
b) Hladiny gonadotropinu v plazmě a moči jsou snížené (např. LH, FSH).
c) Koncentrace pohlavní hormony vázaj ícího globulinu (SHBG) jsou snížené.
Nepravidelný menstruační cyklus:
Většina žen užívajících injekční suspenzi MPA udává poruchy menstruačního cyklu (např. nepravidelné nebo nepředvídatelné krvácení/špinění, vzácně těžké nebo trvalé krvácení). Pacientky by měly být vhodně informovány o možnosti výskytu poruch menstruačního cyklu a možného prodloužení anovulace. S rostoucí dobou užívání injekční suspenze MPA si stále méně pacientek stěžuje na nepravidelné krvácení a více jich udává amenorheu.
Přípravek DEPO-PROVERA má prolongovaný antikoncepční účinek. Střední doba do početí pro ženy, které otěhotní, je 10 měsíců po podání poslední injekce, s rozpětím od 4 do 31 měsíců a nemá souvislost s délkou podávání.
Poruchy očí
Jestliže dojde k částečné nebo úplné ztrátě zraku, k náhlému vzniku exoftalmu, diplopie nebo migrény, je třeba vyčkat s dalším podáváním přípravku až do doby oftalmologického vyšetření. Jestliže vyšetření odhalí edém papily nebo retinální cévní léze, přípravek by neměl být dále podáván.
Předchozí léčba, každoroční anamnéza a fyzikální vyšetření by měly být zaměřeny na prsy a pánevní orgány.
Funkce jater
Pokud se u pacientky užívající přípravek DEPO-PROVERA objeví žloutenka, je vhodné zvážit ukončení podávání léku (viz bod 4.3).
Ochrana před sexuálně přenosnými chorobami
Pacientky by měly být poučeny, že injekce MPA neposkytuje ochranu před HIV infekcí (AIDS) či jinými sexuálně přenosnými chorobami.
Ztráta kostní hmoty (BMD)
Používání injekcí MPA snižuje sérové hladiny estrogenu a je spojeno s významnou ztrátou hustoty kostních minerálů spolu s tím, jak se kostní metabolizmus adaptuje na nižší hladiny estrogenu. Ztráta kostní hmoty je větší při delším užívání, ale zdá se, že se hustota kostní hmota zvyšuje poté, co se podávání přípravku DEPO-PROVERA přeruší a ovariální produkce estrogenů vzroste.
Ztráta kostní hmoty je obzvláště závažná během dospívání a časné dospělosti, kritického období pro růst kostní hmoty. Není známo, zda používání injekcí MPA mladšími ženami snižuje maximální dosaženou hustotu kosti a zvyšuje tak riziko zlomenin ve vyšším věku.
Studie zkoumající účinky i.m. DMPA na hustotu kostní hmoty u dospívajících žen ukázala, že jeho podání bylo spojeno s významným poklesem hustoty kostních minerálů od výchozích hodnot. U malého počtu žen, které byly sledovány po ukončení podávání přípravku DEPO-PROVERA, se střední hustota kostních minerálů vrátila blízko k výchozím hodnotám po 1 - 3 letech po ukončení podávání. U dospívajících může být přípravek DEPO-PROVERA použit, ale jen poté, co byly s pacientkou diskutovány ostatní antikoncepční metody a tyto shledány nedostačující či nevhodné.
U ženy jakéhokoli věku, pokud vyžaduje užívání MPA po dobu delší než 2 roky, by měl být pečlivě přehodnocen poměr přínosu a rizika léčby.
Zvláště u žen se zdravotními rizikovými faktory osteoporózy by měly být před použitím přípravku DEPO-PROVERA zváženy jiné metody antikoncepce.
Významné rizikové faktory osteoporózy jsou:
• Užívání alkoholu a/nebo tabáku
• Chronické užívání léků snižujících kostní hmotu, např. antikonvulziva nebo kortikosteroidy
• Nízký BMI nebo poruchy příjmu potravy, např. anorexie nebo bulimie
• Spontánní zlomenina v anamnéze
• Osteoporóza v rodinné anamnéze
Z výsledků retrospektivní kohortové studie využívající data z General Practice Research Database (GPRD) vyplývá, že ženy používající injekce s MPA (DMPA) jsou vystaveny vyššímu riziku zlomenin v porovnání s uživatelkami kontracepce bez DMPA (riziko výskytu - Incidence Rate Ratio, IRR bylo 1,41, CI 95% 1,35-1,47 pro 5-leté pokračující období). Není známo, zda je to kvůli DMPA, nebo kvůli jiným faktorům souvisejícím se životním stylem, které mají vliv na počet zlomenin. U žen používajících DMPA nebylo riziko zlomenin před používáním DMPA a po jeho používání (relativní riziko 1,08, CI 95% 0,92-1,26) zvýšeno. Důležité je, že tato studie nemůže stanovit, zda má používání DMPA vliv na počet zlomenin v pozdějším věku.
Další informace o změnách denzity kostních minerálů jak u dospělých tak u dospívajících žen, které byly hlášeny v nedávných klinických studiích, jsou uvedeny v bodu 5.1 (Farmakodynamické vlastnosti). Dostatečný přísun vápníku a vitamínu D, ať ve stravě či potravinových doplňcích je důležitý pro zdravé kosti žen v každém věku.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Aminoglutethimid podávaný současně s přípravkem DEPO-PROVERA může výrazně snížit jeho biologickou dostupnost. Ženy užívající přípravek z důvodu antikoncepce by měly být seznámeny s možností snížené účinnosti při současném užívání aminoglutethimidu nebo jakéhokoliv podobného léčiva.
Užíváním přípravku DEPO-PROVERA mohou být ovlivněna následující laboratorní vyšetření:
- hladina gonadotropinů;
- hladina progesteronu v plazmě;
- hladina pregnandiolu v moči;
- hladina testosteronu v plazmě (u mužů);
- hladina estrogenů v plazmě (u žen);
- hladina kortizolu v plazmě;
- glukózový toleranční test;
- metopironový test.
Někteří pacienti užívající MPA mohou mít potlačenou funkci nadledvin. MPA může snížit hladinu ACTH a hydrokortizonu v krvi.
Je vhodné informovat ošetřujícího lékaře/laboratoř, že kromě endokrinních biomarkerů, může použití MPA v onkologii také způsobit částečnou adrenální insuficienci (pokles odpovědi hypofyzoadrenální osy) během metyraponového testu. Tudíž schopnost adrenální kůry reagovat na ACTH by měla být jasně demonstrována předtím, než je podán metyrapon.
4.6 Fertilita, těhotenství a kojení
Přípravek DEPO-PROVERA je kontraindikovaný u těhotných žen.
Některé údaje naznačují souvislost mezi intrauterinní expozicí progestinům v prvním trimestru těhotenství a abnormalitami genitálií u mužských a ženských plodů. Pokud je přípravek DEPO-PROVERA používán v průběhu těhotenství nebo pokud pacientka otěhotní v průběhu léčby tímto přípravkem, měla by být informována o možném riziku pro plod.
Na základě výsledků 1 studie byli novorozenci z neplánovaných těhotenství, která vznikla v průběhu 1
- 2 měsíců po podání injekce medroxyprogesteron-acetátu v dávce 150 mg i.m., vystaveni zvýšenému riziku nízké porodní váhy, což mělo za následek i vyšší riziko novorozenecké úmrtnosti. Celkové riziko je však velmi nízké, neboť otěhotnění při užívání medroxyprogesteron-acetátu v dávce 150 mg
i.m., se nepředpokládá.
U dětí, které byly vystaveny účinkům medroxyprogesteron-acetátu in utero a byly sledovány až do dospívání, nebyly prokázány žádné nežádoucí účinky na zdraví včetně tělesného, intelektuálního, sexuálního nebo sociálního vývoje.
Kojení
Nízká detekovatelná množství léku byla identifikována v mléce matek užívajících medroxyprogesteron-acetát. U kojících matek léčených medroxyprogesteron-acetátem v dávce 150 mg
i.m. nebylo složení mléka a jeho množství nežádoucím způsobem ovlivněno. Novorozenci a děti vystavené působení medroxyprogesteron-acetátu v mateřském mléce byli hodnoceni s ohledem na vývojové a behaviorální účinky během puberty. Nebyly pozorovány žádné nežádoucí účinky. Vzhledem k omezenému počtu údajů o účinku MPA na kojence mladší 6 týdnů se nedoporučuje podání přípravku DEPO-PROVERA dříve než 6 týdnů po porodu, kdy je enzymový systém novorozence rozvinutější.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek DEPO-PROVERA nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky
S použitím gestagenů souvisely příležitostně až vzácně následující nežádoucí účinky, které jsou seřazeny spíše podle své závažnosti než podle četnosti výskytu:
1. Anafylaxe a anafylaktoidní reakce.
2. Tromboembolické poruchy, zvýšení počtu bílých krvinek a destiček.
3. Centrální nervový systém: nervozita, nespavost, somnolence, únava, deprese, závratě a bolesti hlavy, nespavost, zmatenost, křeče, ztráta koncentrace, poruchy zraku.
4. Kůže a sliznice: kopřivka, pruritus, exantém, akné, hirsutismus a alopecie.
5. Urogenitální systém: nepravidelné děložní krvácení, špinění, amenorhea, leukorhea, bolest vpánvi, vaginitida, dlouhodobá anovulace, změna cervikálního sekretu, cervikální eroze.
6. Gastrointestinální systém: nausea, bolest břicha, břišní dyskomfort, nadýmání, porucha jaterních funkcí, žloutenka, zvracení, zácpa, průjem, sucho v ústech.
7. Prsy: zvýšená citlivost, mastodynie a galaktorea.
8. Muskuloskeletální systém: artralgie, astenie, bolest zad, reakce v místě vpichu, křeče nohou, ztráta kostní hmoty.
9. Metabolismus a výživa: snížená glukózová tolerance, adrenergní účinky, kortikoidům podobná reakce, diabetická katarakta, dekompenzace diabetu, glykosurie.
10. Různé: horečka, změny tělesné hmotnosti, retence tekutin, otoky, změněné libido nebo anorgasmie, záchvaty horka, hyperkalcemie, změna chuti k jídlu.
Po uvedení na trh byly u pacientek užívajících přípravek DEPO-PROVERA hlášeny vzácné případy osteoporózy včetně osteoporotických zlomenin.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na adresu:
Státní ústav pro kontrolu léčiv Šrobárova 48 100 41 Praha 10
Webové stránky: www.sukl.cz/nahlasit-nezadouci-ucinek
4.9 Předávkování
Přípravek DEPO-PROVERA je dobře snášen. U žen v reprodukčním věku může jednorázová dávka přípravku DEPO-PROVERA způsobit přechodné změny menstruačního cyklu (tzn. krvácení při vysazení progestinu). Velmi vysoké dávky přípravku DEPO-PROVERA, používané při onkologické léčbě, mohou vyvolat celou řadu symptomů včetně přibývání na váze (spolu s mírnou retencí tekutin), malátnost a v některých případech účinky podobné účinkům kortikoidů. Samozřejmě v těchto případech by docházelo i k anovulaci u žen a snížené endogenní produkci testosteronu u mužů.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: hormony, ATC kód: L02AB02
DEPO-PROVERA je gestagen bez estrogenních účinků a má minimální androgenní účinek. Přípravek DEPO-PROVERA, podaný v příslušných dávkách, potlačuje sekreci hypofyzárních gonadotropinů, což u žen v reprodukčním věku zabraňuje zrání folikulů s následnou anovulací. U mužů přípravek
DEPO-PROVERA, podaný v příslušných dávkách, inhibuje funkci Leydigových buněk, tj. potlačuje produkci endogenního testosteronu.
Dávka 5 nebo 10 mg medroxyprogesteron-acetátu, podávaná denně po dobu 10 dní, má na vyvolání optimální sekreční změny endometria připraveného estrogeny účinek ekvivalentní 20 mg parenterálního progesteronu podávaného denně po dobu l0 dní.
Perorálně podaný medroxyprogesteron-acetát navíc vede k typickým gestagenním změnám cervikálního hlenu (inhibuje arborizaci hlenu) a zvyšuje počet intermediárních buněk v maturačním indexu poševního epitelu.
V případě specifických forem hormonálně-dependentních nádorů může být protinádorový účinek přípravku DEPO-PROVERA ve farmakologických dávkách založen na ovlivnění osy hypothalamus-hypofýza-gonády, dále na účincích na estrogenové receptory a metabolismus steroidů na tkáňové úrovni.
Právě tak jako progesteron je i medroxyprogesteron-acetát termogenní. Při velmi vysokých dávkách (500 mg denně a více), užívaných v léčbě určitých tumorů, se mohou projevit kortikoidní účinky.
Změny hustoty kostní hmoty (BMD) u dospělých žen
V kontrolované klinické studii vykazovaly dospělé ženy užívající MPA injekce (150 mg i.m.) po dobu alespoň 5 let pokles průměrné kostní hustoty v oblasti páteře a kyčle přibližně o 5 - 6% v porovnání s kontrolní skupinou, ve které nedošlo k žádné významné změně. Pokles hustoty kostní hmoty byl výraznější během prvních dvou let užívání, v dalších letech docházelo pouze k menším poklesům. Průměrné změny v oblasti lumbální páteře byly -2,86%, -4,11%, -4,89%, -4,93% a -5,38% po 1, 2, 3, 4 a 5 letech. Průměrný pokles v oblasti celé kyčle a krčku femuru byl podobný.
Po ukončení podávání injekcí MPA (150 mg i.m.) došlo k nárůstu BMD směrem k původním hodnotám. Delší trvání léčby bylo spojeno s pomalejší obnovou BMD.
Změny BMD u dospívajících dívek (12 - 18 let)
Výsledky z otevřené nerandomizované klinické studie zkoumající injekce MPA (150 mg i.m. každých 12 týdnů až po 240 týdnů (4,6 let), s následným sledováním po ukončení podávání u dospívajících dívek (12 - 18 let) také ukázaly, že MPA i.m. byl spojen s významným poklesem BMD oproti výchozímu stavu. Mezi subjekty, kterým byly aplikovány > 4 injekce/60 týdnů byl průměrný pokles v BMD lumbální páteře -2,1% po 240 týdnech (4,6 let); průměrný pokles pro celou kyčel byl -6,4% a pro krček femuru -5,4%. Následné sledování po ukončení podávání ukázalo, že (podle průměrných hodnot) BMD lumbální páteře se vrátila k výchozím hodnotám přibližně za 1 rok po ukončení léčby a BMD kyčle se vrátila k výchozím hodnotám přibližně za 3 roky po ukončení léčby. Je ale nutné vzít v úvahu, že hodně subjektů svoji účast ve studii ukončilo a tyto výsledky jsou tedy založeny na malém počtu subjektů (n=71 v 60. týdnu a n=25 v 240. týdnu po ukončení léčby). Naproti tomu s uživatelkami DMPA neporovnatelná kohorta rozdílných neléčených subjektů s rozdílnými výchozími hodnotami kostních parametrů vykazovala průměrné nárůsty BMD v 240. týdnu o 6,4% u lumbální páteře, 1,7% u celé kyčle a 1,9% u krčku femuru.
5.2 Farmakokinetické vlastnosti
Absorpce
Po intramuskulárním podání se medroxyprogesteron-acetát (MPA) uvolňuje pomalu, čímž se vytváří nízká, ale dlouho přetrvávající hladina přípravku. Po intramuskulární aplikaci dosahují hladiny maxima po 4 až 20 dnech. MPA je v krevním oběhu zjistitelný ještě 7-9 měsíců po intramuskulární injekci.
Distribuce v organismu
Přibližně 90-95% MPA se váže na proteiny. Distribuční objem se uvádí v rozmezí 20 ± 3 litry. MPA prochází hematoencefalickou bariérou a vylučuje se do mateřského mléka.
Biotransformace
MPA je metabolizován v játrech.
Eliminace z organismu
Poločas eliminace po intramuskulární aplikaci je 6 týdnů. MPA je primárně vylučován žlučí do stolice. Přibližně 30% i.m. podané dávky je vyloučeno močí během 4 dnů.
5.3 Předklinické údaje vztahující se k bezpečnosti
Kancerogeneze, mutageneze, poškození fertility
Dlouhodobé intramuskulární podávání medroxyprogesteron-acetátu (DMPA) mělo vliv na tvorbu tumorů mléčných žláz u psů. Není k dispozici důkaz o kancerogenním účinku spojeném s perorálním podáním medroxyprogesteron-acetátu potkanům a myším.
Medroxyprogesteron-acetát nebyl mutagenní v baterii in vitro nebo in vivo testů genetické toxicity. Medroxyprogesteron-acetát ve vysokých dávkách je látkou s antifertilitním účinkem a vysoké dávky mohou poškozovat fertilitu až do ukončení léčby.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Polysorbát 80, methylparaben, propylparaben, makrogol 3000, chlorid sodný, hydroxid sodný a/nebo kyselina chlorovodíková ( na úpravu pH), voda na injekci
6.2 Inkompatibility
Nebyly zjištěny žádné fyzikálně-chemické inkompatibility
6.3 Doba použitelnosti 5 let
6.4 Zvláštní opatření pro uchovávání
Uchovávejte při teplotě do 25°C.
6.5 Druh obalu a obsah balení
a) skleněná lahvička, pryžová zátka, plastový kryt (pertl)
b) jednorázová injekční stříkačka z bezbarvého skla, na jedné straně pryžový kryt, na druhé
straně pryžový píst s plastovým (PP) nástavcem, samostatně sterilně balená jehla na jedno použití s krytem, v zataveném fóliovém blistru a krabičce.
Velikost balení:
Lahvičky o obsahu 150 mg/ml, 500 mg/3,3 ml a 1000 mg/6,7 ml Jednorázová stříkačka o obsahu 150 mg/ml
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním Lahvička
Před použitím je nutno lahvičkou řádně zatřepat, aby podaná dávka vytvořila homogenní suspenzi.
Stříkačka
1. Sejmout kryt stříkačky
2. Nasadit za sterilních podmínek jehlu.
3. Sejmout kryt jehly. Nyní je stříkačka připravená k použití.
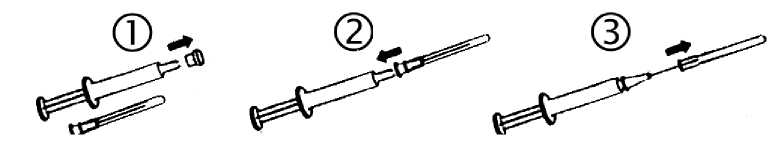
Přípravek je určen k intramuskulárnímu podání.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
PFIZER spol. s r.o., Stroupežnického 17, 150 00 Praha 5, Česká republika
8. REGISTRAČNÍ ČÍSLO
56/111/82-C
9. DATUM PRVNÍ REGISTRACE / PRODLOUŽENÍ REGISTRACE
9.3.1982 / 9.9.2015
10. DATUM REVIZE TEXTU
9.9.2015
10/10