Dacogen 50 Mg
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Dacogen 50 mg prášek pro koncentrát pro infuzní roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička s práškem obsahuje decitabinum 50 mg.
Po rekonstituci 10 ml vody na injekci obsahuje jeden ml koncentrátu decitabinum 5 mg.
Pomocné látky se známým účinkem: Jedna injekční lahvička obsahuje 0,5 mmol draslíku (E340) a 0,29 mmol sodíku (E524).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek pro koncentrát pro infuzní roztok (prášek pro infuzi). Bílý až téměř bílý lyofilizovaný prášek.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Dacogen je indikován k léčbě dospělých pacientů s nově diagnostikovanou de novo nebo sekundární akutní myeloidní leukemií (AML) podle klasifikace Světové zdravotnické organizace (WHO), kteří nemohou podstoupit standardní indukční chemoterapii.
4.2 Dávkování a způsob podání
Podání přípravku Dacogen je nutno zahájit pod dohledem lékaře, který má zkušenosti s používáním chemoterapie.
Dávkování
V léčebném cyklu se Dacogen podává v dávce 20 mg/m2 tělesného povrchu intravenózní infuzí po dobu 1 hodiny každý den po dobu 5 po sobě jdoucích dnů (t.j. celkem 5 dávek za léčebný cyklus). Celková denní dávka nesmí překročit 20 mg/m2 a celková dávka během léčebného cyklu nesmí překročit 100 mg/m2. Dojde-li ke zmeškání dávky, je nutno léčbu obnovit v co nejkratší době. Cyklus se opakuje každé 4 týdny v závislosti na klinické odpovědi pacienta a pozorované toxicitě. Doporučuje se, aby pacienti prodělali alespoň 4 cykly. Dosažení úplné nebo částečné remise však může trvat déle než 4 cykly. Léčba může pokračovat, dokud pacient odpovídá, vykazuje přínos nebo stabilizaci onemocnění, t.j. v nepřítomnosti zřejmé progrese.
Pokud se po 4 cyklech nevrátí pacientovy hematologické hodnoty (např. počet krevních destiček nebo absolutní počet neutrofilů) k hodnotám před léčbou nebo pokud dojde k progresi onemocnění (rostou počty periferních blastů nebo se zhoršují počty blastů v kostní dřeni), je třeba považovat pacienta za neodpovídajícího na léčbu a zvážit jiné terapeutické postupy než Dacogen.
Podávání přípravků k prevenci nauzey a zvracení se rutinně nedoporučuje, ale v případě potřeby je lze podat.
Zvládání myelosuprese a doprovodných komplikací
Myelosuprese a nežádoucí účinky spojené s myelosupresí (trombocytopenie, anemie, neutropenie a febrilní neutropenie) jsou časté jak u léčených tak i neléčených pacientů s AML. Komplikace myelosuprese zahrnují infekce a krvácení. Léčbu může ošetřující lékař odložit, vyskytnou-li se u pacienta komplikace spojené s myelosupresí, např. popsané dále:
• Febrilní neutropenie (teplota > 38,5 °C a absolutní počet neutrofilů < 1 000/^l);
• Aktivní virová, bakteriální nebo plísňová infekce (t.j. vyžadující intravenózní antiinfektiva nebo rozsáhlou podpůrnou péči);
• Krvácení (do gastrointestinálního traktu, do močopohlavního ústrojí, plicní krvácení s počtem krevních destiček < 25 000/^l nebo jakékoli krvácení do centrálního nervového systému).
Léčbu přípravkem Dacogen lze obnovit, jakmile se situace zlepší nebo stabilizuje adekvátní léčbou (antiinfektivy, transfuzí nebo růstovými faktory).
V klinických studiích vyžadovala přibližně třetina pacientů léčených přípravkem Dacogen odložení dávky. Snížení dávky se nedoporučuje.
Pediatrická populace
Bezpečnost a účinnost přípravku Dacogen u dětí ve věku < 18 let nebyly dosud stanoveny. Nejsou dostupné žádné údaje.
Porucha funkce jater
Studie u pacientů s poruchou funkce jater nebyly provedeny. Potřeba úpravy dávky u pacientů s poruchou funkce jater nebyla hodnocena. Vyskytne-li se porucha funkce jater, je nutno pacienty důkladně monitorovat (viz body 4.4 a 5.2).
Porucha funkce ledvin
Studie u pacientů s poruchou funkce ledvin nebyly provedeny. Potřeba úpravy dávky u pacientů s poruchou funkce ledvin nebyla hodnocena (viz body 4.4 a 5.2).
Způsob podání
Dacogen se podává intravenózní infuzí. Centrální venózní katétr není nutný.
Návod k rekonstituci a naředění tohoto léčivého přípravku před jeho podáním je uveden v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na decitabin nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Kojení (viz bod 4.6).
4.4 Zvláštní upozornění a opatření pro použití
Myelosuprese
Myelosuprese a komplikace myelosuprese, včetně infekcí a krvácení, které se vyskytují u pacientů s AML, se léčbou přípravkem Dacogen mohou zhoršit. Proto je u pacientů zvýšené riziko závažných infekcí (v důsledku jakýchkoli patogenů, jako jsou bakteriální, virové a plísňové), s potenciálně smrtelným koncem (viz bod 4.8). Pacienti mají být sledováni pro příznaky a symptomy infekce a být včas léčeni.
V klinických studiích měla většina pacientů počáteční myelosupresi stupně 3/4. U pacientů s počáteční myelosupresí stupně 2 bylo zhoršení myelosuprese pozorováno u většiny pacientů a častěji než
u pacientů s počáteční abnormalitou stupňů 1 nebo 0. Myelosuprese způsobená přípravkem Dacogen je reverzibilní. Pravidelně, dle klinické potřeby a před každým léčebným cyklem, je nutno vyšetřovat krevní obraz a počet krevních destiček. Za přítomnosti myelosuprese nebo jejích komplikací lze léčbu přípravkem Dacogen přerušit a/nebo zavést podpůrná opatření (viz body 4.2 a 4.8).
Porucha funkce jater
Použití příravku Dacogen u pacientů s poruchou funkce jater nebylo hodnoceno. U pacientů s poruchou funkce jater je při podání přípravku Dacogen nutná opatrnost a pacienty je nutno důkladně monitorovat (viz body 4.2 a 5.2).
Porucha funkce ledvin
Použití přípravku Dacogen u pacientů se závažnou poruchou funkce ledvin nebylo hodnoceno.
U pacientů se závažnou poruchou funkce ledvin [clearance kreatininu (CrCl) <30 ml/min] je při podání přípravku Dacogen nutná opatrnost a tato pacienty je nutno důkladně monitorovat (viz bod 4.2).
Srdeční onemocnění
Pacienti s anamnézou závažného městnavého srdečního selhání nebo klinicky nestabilním srdečním onemocněním byli vyloučeni z klinických studií, a proto u těchto pacientů nebyly stanoveny bezpečnost a účinnost přípravku Dacogen.
Pomocné látky
Tento léčivý přípravek obsahuje 0,5 mmol draslíku v injekční lahvičce. Po rekonstituci a naředění roztoku na intravenózní infuzi obsahuje tento léčivý přípravek 1-10 mmol draslíku v dávce v závislosti na infuzním roztoku použitém pro naředění. To je nutno vzít v úvahu u pacientů se sníženou činností ledvin nebo u pacientů na dietě s kontrolovaným obsahem draslíku.
Tento léčivý přípravek obsahuje 0,29 mmol sodíku v injekční lahvičce. Po rekonstituci a naředění roztoku na intravenózní infuzi obsahuje tento léčivý přípravek 0,6 - 6 mmol sodíku v dávce v závislosti na infuzním roztoku použitém pro naředění. To je nutno vzít v úvahu u pacientů na dietě s kontrolovaným obsahem sodíku.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné formální klinické studie lékových interakcí s decitabinem.
Existuje potenciál pro lékové interakce s dalšími látkami, které jsou také aktivovány sekvenční fosforylací (prostřednictvím aktivity intracelulární fosfokinázy) a/nebo metabolizovány enzymy podílejícími se na inaktivaci decitabinu (např. cytidindeamináza). Proto je v případě kombinace přípravku Dacogen s těmito léčivými přípravky nutná opatrnost.
Vliv současně podávaných léčivých přípravků na decitabin
Interakce zprostředkované cytochromem (CYP) 450 se neočekávají, protože metabolismus decitabinu není zprostředkován tímto systémem ale oxidativní deaminací.
Vliv decitabinu na současně podávané léčivé přípravky
Vzhledem ke své nízké vazbě na bílkoviny plazmy (< 1 %) není pravděpodobné, že by decitabin vytěsňoval současně podávané léčivé přípravky z jejich vazby na bílkoviny plazmy. Ukázalo se, že in vitro je decitabin slabým inhibitorem transportu zprostředkovaného P-gp, a proto se neočekává, že by ovlivňoval transport současně podávaných léčivých přípravků, které jsou transportovány prostřednictvím P-gp (viz bod 5.2).
4.6 Fertilita, těhotenství a kojení
Antikoncepce u mužů a žen
Ženy v plodném věku musejí používat účinná antikoncepční opatření a zabránit během léčby přípravkem Dacogen otěhotnění. Odstup, po kterém je možno po ukončení léčby přípravkem Dacogen bezpečně otěhotnět, není znám. Muže je nutno poučit, že při léčbě přípravkem Dacogen a 3 měsíce po jejím ukončení musejí používat účinná antikoncepční opatření a nesmějí počít dítě (viz bod 5.3).
Používání přípravku Dacogen s hormonální antikoncepcí nebylo studováno.
Pro používání přípravku Dacogen těhotnými ženami nejsou k dispozici adekvátní údaje. Studie ukázaly, že decitabin je teratogenní u potkanů a myší (viz bod 5.3). Potenciální riziko pro člověka není známo. Na základě výsledků studií na zvířatech a mechanismu účinku se Dacogen nesmí používat během těhotenství a u žen v plodném věku, které neužívají účinnou antikoncepci. Používá-li se Dacogen během těhotenství nebo pokud pacientka otěhotní během léčby tímto léčivým přípravkem, je nutno ji upozornit na potenciální riziko pro plod.
Kojení
Není známo, zda se decitabin nebo jeho metabolity vylučují do mateřského mléka. Dacogen je během kojení kontraindikován; je-li tedy nutná léčba přípravkem Dacogen, je nutno kojení ukončit (viz bod 4.3).
Fertilita
Nejsou k dispozici žádné údaje o vlivu decitabinu na fertilitu u člověka. V předklinických studiích na zvířatech měnil decitabin samčí plodnost a byl mutagenní. Vzhledem k možnosti neplodnosti v důsledku léčby přípravkem Dacogen by se před zahájením léčby přípravkem Dacogen měli muži poradit o možnosti uchování spermatu a ženy v plodném věku by měly konzultovat možnost uchování zmražených vajíček.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Dacogen má středně významný vliv na schopnost řídit nebo obsluhovat stroje. Pacienty je nutno upozornit, že se u nich během léčby mohou vyskytnout nežádoucí účinky jako anemie. Proto je při řízení nebo obsluze strojů nutná opatrnost.
4.8 Nežádoucí účinky
Souhrn profilu bezpečnosti
Nejčastějšími nežádoucími účinky (> 35 %) během léčby přípravkem Dacogen byly pyrexie, anemie a trombocytopenie.
Nejčastější nežádoucí účinky stupně 3/4 (> 20 %) zahrnovaly pneumonii, trombocytopenii, neutropenii, febrilní neutropenii a anemii.
V klinických studiích se nežádoucí účinky vedoucí k úmrtí během léčby nebo do 30 dnů po podání poslední dávky studovaného přípravku vyskytly u 30 % pacientů léčených přípravkem Dacogen a u 25 % pacientů léčených srovnávacím přípravkem.
Ve skupině s přípravkem Dacogen byl vyšší výskyt ukončení léčby kvůli nežádoucím účinkům u žen ve srovnání s muži (43 % proti 32 %).
Souhrn nežádoucích účinků v tabulce
Nežádoucí účinky hlášené u 293 pacientů s AML léčených přípravkem Dacogen jsou uvedeny v Tabulce 1. Následující tabulka uvádí údaje z klinických studií u AML a z postmarketingových zkušeností. Nežádoucí účinky jsou uvedeny podle tříd orgánových systémů a četnosti. Četnosti výskytu jsou definovány následovně: velmi časté (> 1/10), časté (> 1/100 a < 1/10), méně časté (> 1/1 000 a < 1/100), vzácné (> 1/10 000 až < 1/1 000), velmi vzácné (< 1/10 000), není známo (četnost z dostupných údajů nelze určit).
V každé skupině četnosti jsou nežádoucí účinky uvedeny s klesající četností.
|
Tabul |
ía 1: Nežádoucí účinky pozorované u přípravku Dacogen | |||
|
Třídy orgánových systémů |
Kategorie četnosti (všechny stupně) |
Nežádoucí účinek |
Frekvence | |
|
všechny stupně (%) |
stupně 3 -4a (%) | |||
|
Infekce a infestace |
Velmi časté |
. * |
24 |
20 |
|
infekce močových cest |
15 |
7 | ||
|
všechny ostatní infekce (virové, bakteriální, plísňové)*’ b’ c’ d |
63 |
39 | ||
|
Časté |
septický šok |
6 |
4 | |
|
* sepse |
9 |
8 | ||
|
sinusitida |
3 |
1 | ||
|
Poruchy krve a lymfatického systému |
Velmi časté |
febrilní neutropenie |
34 |
32 |
|
neutropenie |
32 |
30 | ||
|
trombocytopenie , e |
41 |
38 | ||
|
anemie |
38 |
31 | ||
|
leukopenie |
20 |
18 | ||
|
Méně časté |
pancytopenie* |
< 1 |
< 1 | |
|
Poruchy imunitního systému |
Časté |
hypersezitivita včetně anafylaktické reakcef |
1 |
< 1 |
|
Poruchy nervového systému |
Velmi časté |
16 |
1 | |
|
Respirační, hrudní a mediastinální poruchy |
Velmi časté |
epistaxe |
14 |
2 |
|
Gastrointestinální poruchy |
Velmi časté |
31 |
2 | |
|
18 |
1 | |||
|
33 |
< 1 | |||
|
Časté |
stomatitida |
7 |
1 | |
|
Není známo |
enterokolitida, včetně neutropenické kolitidy, zánět slepého střeva* |
není známo |
není známo | |
|
Poruchy kůže a podkožní tkáně |
Méně časté |
akutní febrilní neutrofilní dermatóza (Sweetův syndrom) |
< 1 |
NA |
|
Celkové poruchy a reakce v místě aplikace |
Velmi časté |
Pyrexie |
48 |
9 |
a
b
c
Obecná terminologická kriteria Worst National Cancer Institute pro vyjádření stupně nežádoucích účinků.
Kromě pneumonie, infekcí močových cest, sepse, septického šoku a sinusitidy.
V hodnocení DACO-016 byly nejčastěji hlášené “ostatní infekce”: orální herpes, orální kandidóza, faryngitida, infekce horních cest dýchacích, celulitida, bronchitida, nasofaryngitida.
Včetně infekční enterokolitidy
d
Včetně krvácení spojeného s trombocytopenií, zahrnuje i smrtelné případy.
f
Včetně preferovaných termínů hypersenzitivita, léková hypersenzitivita, anafylaktická reakce, anafylaktický šok, anafylaktoidní reakce, anafylaktoidní šok.
* Zahrnuje účinky s fatálním koncem.
NA=Neaplikovatelné
Popis vybraných nežádoucích účinků
Hematologické nežádoucí účinky
Nejčastěji hlášené hematologické nežádoucí účinky spojené s léčbou přípravkem Dacogen zahrnovaly febrilní neutropenii, trombocytopenii, neutropenii, anemii a leukopenii.
U pacientů léčených přípravkem Dacogen byly v souvislosti se závažnou trombocytopenií hlášeny závažné krvácivé nežádoucí účinky, z nichž některé skončily úmrtím, jako je krvácení do centrálního nervového systému (CNS) (2 %) a krvácení do gastrointestinálního traktu (GIT) (2 %).
Hematologické nežádoucí účinky se zvládají rutinním monitorováním krevního obrazu a včasným zavedením podpůrných opatření. Podpůrná opatření zahrnují profylaktické podání antibiotik a/nebo podpůrného růstového faktoru (např. G-CSF) u neutropenie a transfuze u anemie nebo trombocytopenie podle příslušných doporučení. Situace, kdy je nutno přerušit podávání decitabinu, jsou popsány v bodu 4.2.
Nežádoucí účinky infekce a infestace
U pacientů léčených přípravkem Dacogen byly hlášeny závažné nežádoucí účinky spojené s infekcemi s potenciálně smrtelným koncem, jako septický šok, sepse, pneumonie a jiné infekce (virové, bakteriální a plísňové).
Gastrointestinální poruchy
Během léčby decitabinem byly hlášeny výskyty enterokolitidy, včetně neutropenické kolitidy a zánětu slepého střeva. Enterokolitida může vést k septickým komplikacím a může být spojena se smrtelnými následky.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích učinků uvedeného v Dodatku V.
4.9 Předávkování
S předávkováním u člověka nejsou přímé zkušenosti a neexistuje specifické antidotum. Z prvních údajů z klinických studií publikovaných v literatuře však byla u dávek více než 20krát vyšších, než je současná terapeutická dávka, hlášena zvýšená myelosuprese zahrnující prolongovanou neutropenii a trombocytopenii. Toxicita se pravděpodobně projeví jako prohloubení nežádoucích účinků, primárně myelosuprese. Léčba předávkování je podpůrná.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Cytostatika, antimetabolity, analoga pyrimidinu, ATC kód: L01BC08 Mechanismus účinku
Decitabin (5-aza-2'-deoxycytidin) je analogem deoxynukleosidu cytidinu, který v nízkých dávkách selektivně inhibuje DNA methyltransferázy, což vede k hypomethylaci genových promotorů a dále může vést k reaktivaci genů potlačujících růst tumoru, indukci buněčné diferenciace nebo stárnutí buněk následovanému programovou buněčnou smrtí.
Klinická účinnost a bezpečnost
Používání přípravku Dacogen bylo studováno v otevřené randomizované multicentrické studii fáze 3 (DACO-016) u pacientů s nově diagnostikovanou de novo nebo sekundární AML podle klasifikace WHO. Dacogen (n = 242) byl srovnáván s vybranou léčbou (TC, n = 243), která sestávala podle výběru pacienta, na základě doporučení lékaře, buď pouze z podpůrné léčby (n = 28, 11,5%) nebo 20 mg/m2 cytarabinu subkutánně jednou denně po dobu 10 po sobě jdoucích dnů opakovaně každé 4 týdny (n = 215, 88,5 %). Dacogen byl podáván jako lhodinová intravenózní infuze v dávce 20 mg/m2 jednou denně po dobu 5 po sobě jdoucích dnů opakovaně každé 4 týdny.
Jak ukazují následující výchozí charakteristiky, pacienti, kteří byli považováni za vhodné pro standardní indukční chemoterapii, nebyli v této studii zahrnuti. Medián věku ve vhodné populaci (ITT) byl 73 let (rozmezí 64 až 91 let). Třicet šest procent pacientů mělo na počátku cytogenetiku se špatným rizikem. Zbývající pacienti měli cytogenetiku se středním rizikem. Pacienti s výhodnou cytogenetikou nebyli do této studie zahrnuti. Dvacet pět procent pacientů mělo ECOG skóre > 2. Osmdesát jedno procento pacientů mělo závažná další onemocnění (např. infekci, srdeční onemocnění, onemocnění plic). Rozdělení pacientů léčených přípravkem Dacogen podle rasy bylo 209 (86,4 %) bělochů a 33 (13,6 %) asiatů.
Primárním cílovým parametrem této studie bylo celkové přežití. Sekundárním cílovým parametrem byl výskyt kompletní remise, který byl hodnocen nezávislým odborníkem. Terciárními cílovými parametry byly přežití bez progrese a přežití bez příhod.
Medián doby celkového přežití v této populaci byl 7,7 měsíce u pacientů léčených přípravkem Dacogen ve srovnání s 5,0 měsíci u pacientů v rameni TC (poměr rizika 0,85; 95% CI: 0,69, 1,04; p = 0,1079). Rozdíl nedosáhl statistické významnosti, existoval však trend ke zlepšení přežití s 15% redukcí rizika úmrtí u pacientů léčených v rameni s přípravkem Dacogen (Obrázek 1). Po úpravě kvůli současné léčbě potenciálně modifikující onemocnění (tj. indukční chemoterapii nebo podávání hypomethylující látky) ukázala analýza celkového přežití 20% snížení rizika úmrtí u pacientů v rameni s přípravkem Dacogen [poměr rizika = 0,80; CI: 0,64, 0,99), p = 0,0437)].
Obrázek 1 Celkové přežití (populace ITT)
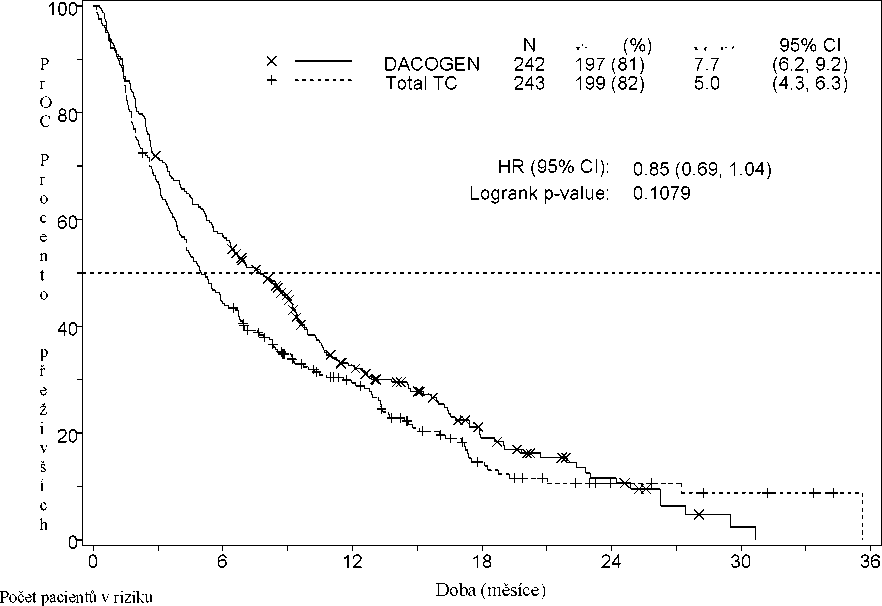
DACOGEN 242 137 65 28 12 1 0
Total TC 243 107 55 19 7 4 0
V analýze údajů o přežití z dalšího 1 roku prokázal účinek přípravku Dacogen na celkové přežití klinické zlepšení ve srovnání s ramenem TC (7,7 měsíce proti 5,0 měsícům, poměr rizika = 0,82; CI: 0,68, 0,99, nominální hodnota p = 0,0373, Obrázek 2).
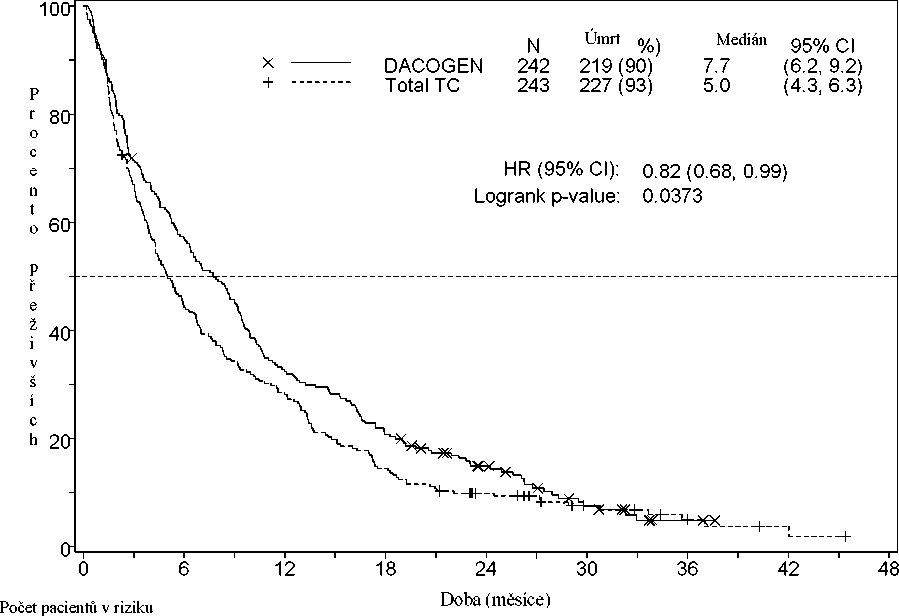
DACOGEN 242 137 78 50 28 11 2 0 0
Total TC 243 107 68 35 20 10 4 2 0
Na základě počáteční analýzy u ITT populace bylo dosaženo statisticky významného rozdílu u výskytu kompletní remise (CR + CRp) ve prospěch pacientů v rameni s přípravkem Dacogen,
17,8 % (43/242), ve srovnání s ramenem TC, 7,8 % (19/243); rozdíl v léčbě 9,9 % (95% CI: 4,07, 15,83), p = 0,0011. Medián doby do nejlepší odpovědi a medián trvání nejlepší odpovědi u pacientů, kteří dosáhli CR nebo CRp, byly 4,3 měsíce, resp. 8,3 měsíce. Doba bez progrese byla signifikantně delší u pacientů v rameni s přípravkem Dacogen, 3,7 měsíce (95% CI: 2,7, 4,6), ve srovnání s pacienty v rameni TC, 2,1 měsíce (95% CI: 1,9, 3,1); poměr rizika 0,75 (95/CI: 0,62, 0,91), p = 0,0031. Tyto výsledky i ostatní výstupy popisuje Tabulka 2.
|
Výstupy |
Dacogen n = 242 |
TC (kombinovaná skupina) n = 243 |
hodnota p |
|
CR + CRp |
43 (17,8 %) |
19 (7,8 %) |
0,0011 |
|
( |
OR = 2,5 1,40; 4,78)b | ||
|
CR |
38 (15,7%) |
18 (7,4%) |
- |
|
EFSa |
3,5 (2,5; 4,1)b |
2,1 (1,9; 2,8)b |
0,0025 |
|
(< |
HR = 0,75 1,62; 0,90)b | ||
|
PFSa |
3,7 (2,7; 4,6)b |
2,1 (1,9; 3,1)b |
0,0031 |
|
(< |
HR = 0,75 1,62; 0,91)b |
CR = kompletní remise; CRp = kompletní remise s nekompletním obnovením krevních destiček, EFS = přežití bez příhod, PFS = přežití bez progrese, OR = odds ratio, HR = poměr rizika - = nehodnotitelné
hlášeno jako medián v měsících b 95% interval spolehlivosti
Celkové přežití a výskyt kompletní remise u předem specifikovaných podskupin spojených s onemocněním [tj. cytogenetické riziko, Eastern Cooperative Oncology Group (ECOG) skóre, věk, typ AML a počáteční hodnota počtu blastů v kostní dřeni] byly konzistentní s výsledky pro celkovou studijní populaci.
U pacientů léčených přípravkem Dacogen (11%, 24/223) došlo ke zhoršení hyperglykemie ve srovnání s pacienty v rameni TC (6 %, 13/212).
Užití přípravku Dacogen jako zahajovací léčby bylo hodnoceno také v otevřené studii fáze 2 s jedním ramenem (DACO-017) u 55 pacientů > 60 let s AML podle klasifikace WHO. Primárním cílovým parametrem byl výskyt kompletní remise (CR), který byl hodnocen nezávislým odborníkem. Sekundárním cílovým parametrem studie bylo celkové přežití. Dacogen byl podáván 1hodinovou intravenózní infuzí v dávce 20 mg/m2 jednou denně po dobu 5 po sobě jdoucích dnů opakovaně každé 4 týdny. V analýze ITT byl u pacientů léčených přípravkem Dacogen pozorovaný podíl CR 23,6 % (95% CI: 13,2, 37) u 13/55 pacientů. Medián doby do CR byl 4,1 měsíce a střední doba trvání CR byla
18.2 měsíce. Medián celkového přežití v populaci ITT byl 7,6 měsíce (95% CI: 5,7, 11,5).
Účinnost a bezpečnost přípravku Dacogen nebyly hodnoceny u pacientů s akutní promyelocytickou leukemií nebo leukemií CNS.
Pediatrická populace
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s přípravkem Dacogen u jedné nebo více podskupin pediatrické populace na léčbu akutní myeloidní leukemie (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Farmakokinetické parametry (PK) decitabinu v populaci byly odvozeny ze 3 klinických studií u 45 pacientů s AML nebo myelodysplastickým syndromem (MDS), ve kterých byl použit 5denní režim. V každé studii byla PK decitabinu hodnocena pátý den prvního léčebného cyklu.
Distribuce v organismu
Farmakokinetika decitabinu po intravenózním podání lhodinovou infuzí byla popsána lineárním dvoukompartmentovým modelem charakterizovaným rychlou eliminací léčiva z centrálního kompartmentu a relativně pomalou distribucí z periferního kompartmentu. Farmakokinetické parametry pro typického pacienta (hmotnost 70 kg/povrch těla 1,73 m2) jsou uvedeny v Tabulce 3 níže.
Tabulka 3: Souhrn analýzy PK v populaci u typického pacienta dostávajícího denně lhodinovou infuzi přípravku Dacogen 20 mg/m2 po dobu 5 dnů každé 4 týdny
|
Parametr3 |
Předpokládaná hodnota |
95% CI |
|
Cmax (ng/ml) |
107 |
88,5 - 129 |
|
AUCcum (ng.h/ml) |
580 |
480 - 695 |
|
t1/2 (min) |
68,2 |
54,2 - 79,6 |
|
Vdss (l) |
116 |
84,1 - 153 |
|
CL (l/hod) |
298 |
249 - 359 |
Celková dávka v cyklu byla 100 mg/m2
Decitabin vykazuje lineární PK a po intravenózní infuzi je rovnovážných koncentrací dosaženo během 0,5 hodiny. Podle modelové simulace byly PK parametry nezávislé na čase (tj. neměnily se během cyklů) a při tomto dávkovacím režimu nebyla pozorována žádná akumulace. Vazba decitabinu na bílkoviny plazmy je zanedbatelná (< 1 %). Vdss decitabinu u pacientů s tumorem ukazuje na distribuci léčiva do periferních tkání. Nebyl pozorován žádný náznak závislosti na věku, clearance kreatininu, celkovém bilirubinu nebo onemocnění.
Biotransformace
Intracelulárně je decitabin aktivován prostřednictvím fosfokinázových aktivit sekvenční fosforylací na odpovídající trifosfát, který je dále inkorporován DNA-polymerázou. In vitro údaje o metabolismu a výsledky ze studie hmotnostní bilance u člověka ukazují, že na biotransformaci decitabinu se nepodílí systém cytochromů P450. Primární cestou metabolismu je pravděpodobně deaminace pomocí cytidin-deaminázy v játrech, ledvinách, sliznici střeva a v krvi. Výsledky ze studie hmotnostní bilance u lidí ukázaly, že nezměněný decitabin v plazmě odpovídá přibližně 2,4 % celkové radioaktivity v plazmě. Předpokládá se, že největší cirkulující metabolity v plazmě nejsou farmakologicky aktivní. Přítomnost těchto metabolitů v moči spolu s velkou celkovou clearance a nízkou urinární sekrecí nezměněného léčiva do moči (~4 % dávky) ukazuje, že decitabin se in vivo patrně biotransformuje. In vitro studie ukazují, že decitabin neinhibuje ani neindukuje enzymy CYP 450 až do více než 20násobku pozorované terapeutické maximální koncentrace v plazmě (Cmax). Z tohoto důvodu se nepředpokládají metabolické lékové interakce zprostředkované CYP a decitabin pravděpodobně neinteraguje s látkami metabolizovanými touto cestou. Dále in vitro údaje ukazují, že decitabin je slabým substrátem P-gp.
Eliminace z organismu
Průměrná plazmatická clearance po intravenózním podání u pacientů s tumorem byla > 200 l/hod se střední interindividuální variabilitou [variační koeficient (CV) je přibližně 50 %]. Zdá se, že vylučování nezměněného léčiva hraje v eliminaci decitabinu pouze malou roli.
Výsledky ze studie hmotnostní bilance s 14C-decitabinem u pacientů s tumorem ukázaly, že 90 % podané dávky decitabinu (4 % nezměněného léčiva) se vylučuje do moči.
Další informace pro zvláštní populace
Účinky poruchy funkce ledvin nebo jater, pohlaví, věku nebo rasy na farmakokinetiku decitabinu nebyly formálně hodnoceny. Informace pro zvláštní populace byly odvozeny z farmakokinetických údajů ze 3 studií citovaných výše a z 1 studie fáze 1 u pacientů s MDS (n = 14, 15 mg/m2 x 3 hodiny 1x za 8 hodin x 3 dny).
Starší osoby
Farmakokinetická analýza v populaci ukázala, že farmakokinetika decitabinu není závislá na věku (hodnocené rozmezí 40 až 87 let; medián 70 let).
Pohlaví
Farmakokinetická analýza v populaci neukázala klinicky významné rozdíly mezi muži a ženami.
Rasa
Většina hodnocených pacientů byli běloši. Farmakokinetická analýza decitabinu v populaci však naznačuje, že rasa nemá na expozici decitabinu pozorovatelný vliv.
Porucha funkce jater
Farmakokinetika decitabinu nebyla u pacientů s poruchou funkce jater formálně hodnocena. Výsledky ze studie hmotnostní bilance u člověka a in vitro experimentů citovaných výše ukazují, že na biotransformaci decitabinu se pravděpodobně nepodílí enzymy CYP. Omezené údaje z farmakokinetiky v populaci navíc neukazují na žádné významné závislosti PK parametrů na koncentraci celkového bilirubinu, přestože hodnoty celkového bilirubinu se dost lišily. Expozice decitabinu tedy u pacientů s poruchou funkce jater není pravděpodobně ovlivněna.
Porucha funkce ledvin
Farmakokinetika decitabinu nebyla u pacientů s poruchou funkce ledvin formálně hodnocena. Farmakokinetická analýza omezených údajů o decitabinu v populaci nenaznačuje žádné závislosti PK parametrů na normalizované clearance kreatininu, indikátoru funkce ledvin. Expozice decitabinu tedy u pacientů s poruchou funkce ledvin není pravděpodobně ovlivněna.
5.3 Předklinické údaje vztahující se k bezpečnosti
S decitabinem nebyly provedeny formální studie kancerogenity. Podklady z literatury naznačují, že decitabin má kancerogenní potenciál. Dostupné údaje z in vitro a in vivo studií dostatečně prokazují, že decitabin má genotoxický potenciál. Údaje z literatury také ukazují, že decitabin má nežádoucí účinky na všechny aspekty reprodukčního cyklu, včetně plodnosti, embryofetálního vývoje a postnatálního vývoje. Studie toxicity ve více cyklech s opakovanou dávkou u potkanů a králíků ukazují, že primární toxicitou byla myelosuprese, včetně účinků na kostní dřeň, která byla po ukončení léčby reverzibilní. Byla pozorována také gastrointestinální toxicita a u samců testikulární atrofie, která se během naplánovaného regeneračního období nezlepšila. Decitabin podaný novorozeným/mladým potkanům vykázal srovnatelný profil celkové toxicity jako u starších potkanů. Při léčbě novorozených/mladých potkanů dávkami vyvolávajícími myeloupresi nebyly neurobehaviorální vývoj a reprodukční kapacita ovlivněny. Informace o použití u dětí viz bod 4.2.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Dihydrogenfosforečnan draselný (E340)
Hydroxid sodný (E524)
Kyselina chlorovodíková (na úpravu pH)
6.2 Inkompatibility
Tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky s výjimkou těch, které jsou uvedeny v bodě 6.6.
6.3 Doba použitelnosti
Neotevřená injekční lahvička
Rekonstituovaný a naředěný roztok
Během 15 minut po rekonstituci je nutno dále zředit koncentrát (v 10 ml sterilní vody na injekci) studeným (2 °C - 8 °C) infuzním roztokem. Takto připravený zředěný roztok pro intravenózní infuzi lze před podáním uchovávat při teplotě 2° C - 8° C maximálně 3 hodiny a poté až 1 hodinu při pokojové teplotě (20° C - 25° C).
Z mikrobiologického hlediska je nutno přípravek použít během výše uvedeného období. Dodržení doporučené doby uchovávání a ujištění se, že k rekonstituci došlo za aseptických podmínek, je v odpovědnosti uživatele.
6.4 Zvláštní opatření pro uchovávání
Neuchovávejte při teplotě nad 25° C.
Podmínky uchovávání tohoto léčivého přípravku po jeho rekonstituci a naředění jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
Čirá bezbarvá 20ml skleněná (Typ I) injekční lahvička s brombutylovou gumovou zátkou a hliníkovým pertlem s plastikovým odtrhovacím krytem obsahující 50 mg decitabinu.
Obsah balení: 1 injekční lahvička.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Doporučení pro bezpečné zacházení
Je nutno se vyvarovat styku kůže s roztokem a používat ochranné rukavice. Je nutno přijmout standardní postupy pro zacházení s cytostatiky.
Postup rekonstituce
Prášek se rekonstituuje asepticky 10 ml vody na injekci. Po rekonstituci obsahuje jeden ml přibližně 5 mg decitabinu a má pH 6,7 až 7,3. Během 15 minut po rekonstituci je nutno roztok dále zředit studeným infuzním roztokem [roztokem chloridu sodného 9 mg/ml (0,9 %) nebo 5% roztokem glukózy na injekci] na výslednou koncentraci 0,1 až 1,0 mg/ml. Doba použitelnosti a podmínky uchovávání po rekonstituci viz bod 6.3.
Pro infuzi přípravku Dacogen se nesmí použít stejný intravenózní vstup/port jako pro další léčivé přípravky.
Likvidace
Tento léčivý přípravek je pouze k jednorázovému užití. Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Janssen-Cilag International NV Turnhoutseweg 30 B-2340 Beerse Belgie
8. REGISTRAČNÍ ČÍSLO(A)
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 20. září 2012
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu.
A. VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného/výrobců odpovědných za propouštění šarží
Janssen Pharmaceutica NV Turnhoutseweg 30 B-2340 Beerse Belgie
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU KRABIČKA
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Dacogen 50 mg prášek pro koncentrát pro infuzní roztok decitabinum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna injekční lahvička obsahuje decitabinum 50 mg.
Po rekonstituci obsahuje 1 ml koncentrátu decitabinum 5 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Pomocné látky: dihydrogenfosforečnan draselný (E340), hydroxid sodný (E524) a kyselina chlorovodíková
Další údaje jsou uvedeny v příbalové informaci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Prášek pro koncentrát pro infuzní roztok 1 injekční lahvička
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Pouze pro jednorázové použití.
Intravenózní podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
Cytotoxické
8. POUŽITELNOST
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Neotevřená injekční lahvička: Neuchovávejte nad 25° C.
Doba použitelnosti rekonstituovaného a naředěného přípravku viz příbalová informace.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Janssen-Cilag International NV Turnhoutseweg 30 B-2340 Beerse Belgie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/12/792/001
13. ČÍSLO ŠARŽE
c.s.
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Nevyžaduje se - odůvodnění přijato
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITŘNÍM OBALU ŠTÍTEK NA INJEKČNÍ LAHVIČKU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Dacogen 50 mg prášek pro infuzi
decitabinum
i.v.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
50 mg
6. JINÉ
Cytotoxické
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro uživatele
Dacogen 50 mg prášek pro koncentrát pro infuzní roztok
Decitabinum
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí
ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je Dacogen a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete Dacogen používat
3. Jak se Dacogen používá
4. Možné nežádoucí účinky
5. Jak Dacogen uchovávat
6. Obsah balení a další informace
1. Co je Dacogen a k čemu se používá Co je Dacogen
Dacogen obsahuje léčivou látku „decitabin“. Jde o léčivý přípravek k léčbě nádorů.
K čemu se Dacogen používá
Dacogen se používá k léčbě nádoru, který se nazývá „akutní myeloidní leukemie“ neboli „AML“. Je to druh nádoru, který postihuje Vaše krvinky. Dacogen dostanete v případě první diagnózy AML. Je určen pouze pro dospělé.
Jak Dacogen účinkuje
Dacogen brání nádorovým buňkám v jejich růstu. Také tyto buňky zabíjí.
Máte-li jakékoli otázky k tomu, jak Dacogen účinkuje nebo proč Vám byl tento léčivý přípravek předepsán, zeptejte se svého lékaře nebo zdravotní sestry.
2. Čemu musíte věnovat pozornost, než začnete Dacogen používat Nepoužívejte Dacogen:
• jestliže jste alergický(á) na decitabin nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
• pokud kojíte.
Nejste-li si jistý(á), zda se Vás cokoli z výše uvedeného týká, poraďte se před používáním přípravku Dacogen s lékařem, lékárníkem nebo zdravotní sestrou.
Upozornění a opatření
Před použitím přípravku Dacogen se poraďte se svým lékařem, lékárníkem nebo zdravotní sestrou pokud máte
• nízký počet krevních destiček, červených krvinek nebo bílých krvinek,
• infekci,
• onemocnění jater,
• závažnou poruchu funkce ledvin,
• onemocnění srdce.
Pokud si nejste jistý/á, že se Vás cokoli z výše uvedeného týká, poraďte se před používáním přípravku Dacogen s lékařem, lékárníkem nebo zdravotní sestrou.
Děti a dospívající
Dacogen není určen k používání u dětí a mladistvých do 18 let.
Další léčivé přípravky a Dacogen
Informujte svého lékaře, lékárníka nebo zdravotní sestru o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat. Zahrnuje to také léčivé přípravky vydávané bez lékařského předpisu a rostlinné přípravky. Je to proto, že Dacogen může ovlivnit způsob, jakým některé jiné léčivé přípravky působí. Také některé jiné léčivé přípravky mohou ovlivnit, jak působí Dacogen.
Vyšetření a kontroly
Před zahájením léčby přípravkem Dacogen a na začátku každého léčebného cyklu podstoupíte vyšetření krve. Toto vyšetření zjišťuje:
• zda máte dostatek krvinek a
• zda Vaše játra a ledviny pracují správně.
Těhotenství a kojení
• Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná nebo plánujete otěhotnět, poraďte se se svým lékařem dříve, než začnete tento přípravek používat.
• Nepoužívejte Dacogen, pokud jste těhotná, protože to může uškodit Vašemu dítěti. Otěhotníte-li během používání přípravku Dacogen, okamžitě to oznamte svému lékaři.
• Nekojte, pokud používáte Dacogen. Je to proto, že není známo, zda se toto léčivo vylučuje do mateřského mléka.
Mužská a ženská plodnost a antikoncepce
• Během používání přípravku Dacogen by muži neměli počít dítě.
• Muži musejí během používání přípravku a po 3 měsíce od ukončení jeho používání používat účinnou antikoncepční metodu.
• Přejete-li si před zahájením léčby uchovat sperma, poraďte se s lékařem.
• Ženy musejí během používání přípravku používat účinnou antikoncepční metodu. Není známo, kdy je pro ženu po ukončení léčby bezpečné otěhotnět.
• Přejete-li si před zahájením léčby zmrazit vajíčka, poraďte se s lékařem.
Řízení dopravních prostředků a obsluha strojů
Při používání přípravku Dacogen se můžete cítit unavený(á) nebo slabý(á). Dojde-li k tomu, neřiďte dopravní prostředky nebo nepoužívejte nástroje a neobsluhujte stroje.
Dacogen obsahuje draslík a sodík
• Tento léčivý přípravek obsahuje 0,5 mmol draslíku v injekční lahvičce. Po rekonstituci a naředění roztoku na intravenózní infuzi obsahuje tento léčivý přípravek 1-10 mmol draslíku v dávce v závislosti na infuzním roztoku použitém pro naředění. To je nutno vzít v úvahu
u pacientů se sníženou činností ledvin nebo u pacientů na dietě s kontrolovaným obsahem draslíku.
• Tento léčivý přípravek obsahuje 0,29 mmol sodíku v injekční lahvičce. Po rekonstituci a naředění roztoku na intravenózní infuzi obsahuje tento léčivý přípravek 0,6 - 6 mmol sodíku v dávce v závislosti na infuzním roztoku použitém pro naředění. To je nutno vzít v úvahu
u pacientů na dietě s kontrolovaným obsahem sodíku.
3. Jak se Dacogen používá
Dacogen Vám bude podán lékařem nebo zdravotní sestrou, kteří jsou školeni v podávání tohoto typu léčivých přípravků.
Kolik se používá
• Dávku přípravku Dacogen stanoví lékař. Závisí na Vaší výšce a tělesné hmotnosti (povrchu těla).
• Dávka je 20 mg/m2 povrchu těla.
• Dacogen budete dostávat po dobu 5 dnů každý den, poté budete 3 týdny bez tohoto léčivého přípravku. To se nazývá „léčebný cyklus“ a opakuje se každé 4 týdny.
• Obvykle proděláte nejméně 4 léčebné cykly.
Na základě Vaší odpovědi na léčbu může lékař odložit dávku a změnit celkový počet cyklů.
Jak se Dacogen podává
Roztok se podává do žíly (jako infuze). Bude to trvat jednu hodinu.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Pokud se u Vás vyskytne cokoli z dále uvedených závažných nežádoucích účinků, okamžitě to oznamte svému lékaři nebo zdravotní sestře:
• Horečka: může být příznakem infekce způsobené nízkou hladinou bílých krvinek (velmi častá).
• Bolest na hrudi nebo dušnost (s horečkou a kašlem nebo bez nich): to může být příznakem infekce plic zvané „pneumonie“ (velmi časté).
• Krvácení: včetně krve ve stolici. To může být známkou krvácení do žaludku nebo střeva (časté).
• Krvácení do hlavy: příznaky mohou zahrnovat obtížnou pohyblivost, problémy s mluvením, rozuměním nebo viděním, náhlou těžkou bolest hlavy, křeče, necitlivost nebo slabost kterékoli části těla (časté).
• Obtíže s dýcháním, otok rtů, svědění nebo vyrážka: Může se jednat o alergickou reakci (reakci přecitlivělosti) (časté).
Pokud se u Vás vyskytne jakýkoli závažný nežádoucí účinek uvedený výše, okamžitě to oznamte svému lékaři nebo zdravotní sestře:
Další nežádoucí účinky přípravku Dacogen zahrnují:
Velmi časté (mohou postihnout více než 1 z 10 osob)
• infekce močových cest;
• jiné infekce na jakékoli části těla způsobené bakteriemi, viry nebo plísněmi;
• krvácení nebo snadná tvorba modřin - může jít o známky poklesu počtu krevních destiček (trombocytopenie);
• pocit únavy nebo bledost - může jít o známky poklesu počtu červených krvinek (anemie);
• bolest hlavy;
• krvácení z nosu;
• průjem;
• zvracení;
• pocit na zvracení;
Časté (mohou postihnout až 1 z 10 osob)
• infekce krve způsobená bakteriemi - může jít o známky poklesu počtu bílých krvinek;
• bolavý nos, rýma, bolest dutin;
• vřídky v ústech nebo na jazyku.
Méně časté (mohou postihnout až 1 ze 100 osob)
• pokles počtu červených krvinek, bílých krvinek a krevních destiček (pancytopenie);
• červené vyvýšené bolestivé skvrny na kůži, horečka, zvýšení počtu bílých krvinek - může jít o známky „akutní febrilní neutrofilové dermatózy“ nebo „Sweetova syndromu“.
Není známo (četnost z dostupných údajů nelze určit)
• infekce střeva (enterokolitida, kolitida a zánět slepého střeva), které jsou provázeny příznaky bolestí břicha, nadmutím břicha nebo průjmem. Enterokolitida může vést k septickým komplikacím a být spojena se smrtelnými následky.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak Dacogen uchovávat
• Za uchovávání je odpovědný lékař, zdravotní sestra nebo lékárník.
• Uchovávejte tento přípravek mimo dohled a dosah dětí.
• Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a lahvičce za Použitelné do/EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
• Neuchovávejte při teplotě nad 25 °C.
• Po rekonstituci je nutno koncentrát dále zředit za použití studeného infuzního roztoku do
15 minut. Takto připravený zředěný roztok pro intravenózní infuzi lze před podáním uchovávat v chladničce při teplotě 2 °C - 8 °C maximálně 3 hodiny a poté až 1 hodinu při pokojové teplotě (20 °C - 25 °C).
• Za správnou likvidaci jakéhokoli nepoužitého přípravku Dacogen jsou odpovědni lékař, zdravotní sestra nebo lékárník.
6. Obsah balení a další informace Co Dacogen obsahuje
• Léčivou látkou je decitabinum. Jedna injekční lahvička s práškem obsahuje decitabinum 50 mg. Po rekonstituci za použití 10 ml vody na injekci obsahuje jeden ml koncentrátu 5 mg decitabinu.
• Dalšími složkami jsou dihydrogenfosforečnan sodný (E340), hydroxid sodný (E254) a kyselina chlorovodíková (na úpravu pH).
Jak Dacogen vypadá a co obsahuje toto balení
Dacogen bílý až téměř bílý prášek pro koncentrát pro infuzní roztok. Dodává se ve 20ml injekční lahvičce obsahující 50 mg decitabinu. Jedno balení obsahuje 1 injekční lahvičku.
Držitel rozhodnutí o registraci
Janssen-Cilag International NV Turnhoutseweg 30 B-2340 Beerse
Belgie
Výrobce
Janssen Pharmaceutica NV Tumhoutseweg 30 B-2340 Beerse Belgie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgie/Belgique/Belgien
Janssen-Cilag NV Tel/Tél: +32 14 64 94 11
Btarapna
„fl^OHCtH & fl^OHCtH Etarapna” EOOfl Ten.: +359 2 489 94 00
Česká republika JANSSEN-CILAG s.r.o.
Tel: +420 227 012 227
Danmark
JANSSEN-CILAG A/S Tlf: +45 45 94 82 82
Deutschland
JANSSEN-CILAG GmbH Tel: +49 2137 955-955
Eesti
UAB "JOHNSON & JOHNSON" Eesti filiaal Tel: +372 617 7410
EXXáSa
JANSSEN-CILAG Oap^aKswix^ A.E.B.E. T^: +30 210 80 90 000
Espaňa
JANSSEN-CILAG, S.A.
Tel: +34 91 722 81 00
France
JANSSEN-CILAG
Tél: 0 800 25 50 75 / +33 1 55 00 40 03 Hrvatska
Johnson & Johnson S.E. d.o.o.
Tel: +385 1 6610 700
Ireland
JANSSEN-CILAG Ltd.
Tel: +44 1 494 567 444
Lietuva
UAB "JOHNSON & JOHNSON"
Tel: +370 5 278 68 88
Luxembourg/Luxemburg
Janssen-Cilag NV Tél/Tel: +32 14 64 94 11
Magyarország JANSSEN-CILAG Kft.
Tel: +36 1 884 2858
Malta
AM MANGION LTD.
Tel: +356 2397 6000
Nederland
JANSSEN-CILAG B.V.
Tel: +31 13 583 73 73
Norge
JANSSEN-CILAG AS Tlf: +47 24 12 65 00
Osterreich
JANSSEN-CILAG Pharma GmbH Tel: +43 1 610 300
Polska
JANSSEN-CILAG Polska Sp. z o.o.
Tel: +48 22 237 60 00
Portugal
JANSSEN-CILAG FARMACÉUTICA, LDA. Tel: +351 21 43 68 835
Románia
Johnson & Johnson Románia SRL Tel: +40 21 207 1800
Slovenija
Johnson & Johnson d.o.o.
Tel: +386 1 401 18 30
Ísland
JANSSEN-CILAG AB c/o Vistor hf.
Sími: +354 535 7000
Italia
JANSSEN-CILAG SpA Tel: +39 02 2510 1
Kúrcpog
BapváPag Xax^nnavay^g AxS,
T^: +357 22 207 700
Latvija
UAB "JOHNSON & JOHNSON" filia Tel: +371 678 93561
Suomi/Finland
JANSSEN-CILAG OY Puh/Tel: +358 207 531 300
Sverige
JANSSEN-CILAG AB Tel: +46 8 626 50 00
United Kingdom
Latvija JANSSEN-CILAG Ltd.
Tel: +44 1 494 567 444
Tato příbalová informace byla naposledy revidována
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu.
Následující informace jsou určeny pouze pro zdravotnické pracovníky:
1. REKONSTITUCE
Je nutno se vyvarovat styku kůže s roztokem a používat ochranné rukavice. Je nutno přijmout standardní postupy pro zacházení s cytostatiky.
Prášek se rekonstituuje asepticky 10 ml vody na injekci. Po rekonstituci obsahuje jeden ml přibližně 5 mg decitabinu a má pH 6,7 až 7,3. Během 15 minut po rekonstituci je nutno roztok dále zředit studeným (2 °C - 8 °C) infuzním roztokem [roztokem chloridu sodného 9 mg/ml (0,9 %) nebo 5% roztokem glukózy na injekci] na výslednou koncentraci 0,1 až 1,0 mg/ml.
Doba použitelnosti a podmínky uchovávání po rekonstituci viz bod 5 příbalové informace.
2. PODÁNÍ
Rekonstituovaný roztok se podává infuzí po dobu 1 hodiny.
3. LIKVIDACE
Injekční lahvička je na jednorázové použití a jakýkoli zbylý roztok je nutno zlikvidovat. Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
28