Cyramza 10 Mg/Ml
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Cyramza 10 mg/ml koncentrát pro infuzní roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden ml koncentrátu pro infuzní roztok obsahuje ramucirumabum 10 mg.
Jedna 10ml injekční lahvička obsahuje ramucirumabum 100 mg.
Jedna 50ml injekční lahvička obsahuje ramucirumabum 500 mg.
Ramucirumab je lidská IgG1 monoklonální protilátka produkovaná v myších (NS0) buňkách technologií rekombinantní DNA.
Pomocná látka se známým účinkem:
Jedna 10ml injekční lahvička obsahuje přibližně 17 mg sodíku.
Jedna 50ml injekční lahvička obsahuje přibližně 85 mg sodíku.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Koncentrát pro infuzní roztok (sterilní koncentrát).
Koncentrát je čirý až mírně opalizující bezbarvý až lehce nažloutlý roztok, pH 6,0.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Cyramza je v kombinaci s paklitaxelem indikován k léčbě dospělých pacientů s pokročilým karcinomem žaludku nebo adenokarcinomem gastroesofageální junkce s progresí choroby po předchozí chemoterapii platinou a fluoropyrimidinem (viz bod 5.1).
Přípravek Cyramza je indikován v monoterapii k léčbě dospělých pacientů s pokročilým karcinomem žaludku nebo adenokarcinomem gastroesofageální junkce s progresí choroby po předchozí chemoterapii platinou nebo fluoropyrimidinem, u kterých není vhodná léčba v kombinaci s paklitaxelem (viz bod 5.1).
Přípravek Cyramza je v kombinaci s FOLFIRI (irinotekan, kyselina folinová a fluoruracil) určen k léčbě dospělých pacientů s metastatickým kolorektálním karcinomem (mCRC) s progresí onemocnění na předchozí léčbě bevacizumabem, oxaliplatinou a fluoropyrimidinem nebo po ní.
Přípravek Cyramza je v kombinaci s docetaxelem indikován k léčbě dospělých pacientů s lokálně pokročilým nebo metastazujícím nemalobuněčným karcinomem plic, u kterých po chemoterapii založené na platině došlo k progresi onemocnění.
4.2 Dávkování a způsob podání
Léčbu ramucirumabem musí zahájit a dohlížet na ni lékař zkušený v oboru onkologie. Dávkování
Karcinom žaludku a adenokarcinom gastroesofageální _junkce (GEJ)
Cyramza v kombinaci s paklitaxelem
Doporučená dávka ramucirumabu je 8 mg/kg 1. a 15. den 28denního cyklu, před infuzí paklitaxelu. Doporučená dávka paklitaxelu je 80 mg/m2, podávaná ve formě intravenózní infuze po dobu přibližně 60 minut 1., 8. a 15. den 28denního cyklu. Před každou infuzí paklitaxelu je třeba u pacienta provést kompletní vyšetření krevního obrazu a biochemický rozbor krve k posouzení funkce jater. Kritéria, která musejí být splněna před každou infuzí paklitaxelu, jsou uvedena v tabulce 1.
Tabulka 1: Kritéria, která musí být splněna před každým podáním paklitaxelu
|
Kritéria | |
|
Neutrofily |
1. den: > 1,5 x 109/l 8. a 15. den: > 1,0 x 109/l |
|
Trombocyty |
1. den: >100 x 109/l 8. a 15. den: >75 x 109/l |
|
Bilirubin |
<1,5násobek horní hranice normálních hodnot (ULN) |
|
Aspartátaminotransferáza (AST) / alaninaminotransferáza (ALT) |
Žádné jaterní metastázy: ALT/AST < 3násobek ULN Jaterní metastázy: ALT/AST < 5násobek ULN |
Cyramza v monoterapii
Doporučená dávka ramucirumabu v monoterapii je 8 mg/kg každé 2 týdny.
Kolorektální karcinom
Doporučená dávka ramucirumabu je 8 mg/kg s podáním jednou za 2 týdny ve formě intravenózní infuze před podáním FOLFIRI. Před chemoterapií je třeba mít k dispozici kompletní krevní obraz pacienta. Kritéria, která je nutné splnit před podáním FOLFIRI, naleznete v tabulce 2.
Tabulka 2: Kritéria, která musí být splněna před podáním FOLFIRI
|
Kritéria | |
|
Neutrofily |
>1,5 x 109/l |
|
Trombocyty |
>100 x 109/l |
|
Gastrointestinální toxicita spojená s chemoterapií |
< stupeň 1 (Obecná terminologická kritéria národního onkologického ústavu pro nežádoucí příhody [NCI CTCAE; National Cancer Institute Common Terminology Criteria for Adverse Events]) |
Nemalobuněčný karcinom plic (NSCLC)
Doporučená dávka ramucirumabu je 10 mg/kg podávaná první den 21denního cyklu před infuzí docetaxelu. Doporučená dávka docetaxelu je 75mg/m2 podávaná jako intravenózní infuze po dobu přibližně 60 minut první den každého 21denního cyklu. U pacientů z Východní Asie má být zváženo použití snížené počáteční dávky docetaxelu 60 mg/m2 první den 21denního cyklu. Pro specifické informace o dávkování docetaxelu viz příslušné informace pro předepisování docetaxelu.
Délka léčby
Doporučuje se, aby léčba pokračovala až do progrese choroby nebo do rozvoje nepřijatelné toxicity.
Premedikace
Před infuzí ramucirumabu se doporučuje premedikace antagonistou histaminových H1 receptorů (např. difenhydraminem). Pokud u pacienta dojde k reakci v souvislosti s podáním infuze 1. nebo 2. stupně závažnosti, musí být premedikace podána při všech dalších infuzích. Pokud u pacienta dojde k reakci v souvislosti s podáním infuze (infusion-related reaction = IRR) 1. nebo 2. stupně závažnosti, podejte dexamethason (nebo jeho ekvivalent) a při následujících infuzích použijte k premedikaci následující léčivé přípravky nebo jejich ekvivalenty: intravenózně podávaný antagonista histaminových H1 receptorů (např. difenhydramin-hydrochlorid), paracetamol a dexamethason.
Požadavky na premedikaci a další informace viz též v příslušných informacích pro předepisování přípravků s paklitaxelem, přípravků se složkami FOLFIRI a docetaxelem.
Úprava dávkování u ramucirumabu
Reakce související s podáním infuze
Pokud u pacienta dojde k IRR 1. nebo 2. stupně závažnosti, je třeba rychlost infuze ramucirumabu snížit o 50 % po celou dobu trvání infuze a u všech následujících infuzí. Pokud dojde k IRR 3. nebo 4. stupně závažnosti, je třeba ramucirumab okamžitě a trvale vysadit (viz bod 4.4).
Hypertenze
Krevní tlak pacientů je třeba monitorovat před každým podáním ramucirumabu a léčit podle klinické indikace. V případě závažné hypertenze je třeba až do doby její kompenzace farmakoterapií léčbu ramucirumabem dočasně přerušit. V případě klinicky významné hypertenze, kterou nelze bezpečně kompenzovat pomocí antihypertenzní léčby, je třeba léčbu ramucirumabem vysadit trvale (viz bod 4.4).
Proteinurie
Během léčby ramucirumabem je třeba u pacientů monitorovat případný rozvoj nebo zhoršení proteinurie. Pokud se na testovacím proužku objeví bílkovina >2, je třeba provést 24hodinový sběr moči. Pokud hladina bílkoviny v moči dosáhne >2 g/24 hodin, je třeba léčbu ramucirumabem dočasně přerušit. Jakmile se hladina bílkoviny v moči vrátí na <2 g/24 hodin, je možné se vrátit k léčbě sníženou dávkou (viz tabulka 3). Pokud znovu dojde ke zvýšení hladiny bílkoviny v moči na >2 g/24 hodin, doporučuje se druhé snížení dávky (viz tabulka 3).
Pokud hladina bílkoviny v moči dosáhne >3 g/24 hodin nebo v případě rozvoje nefrotického syndromu, je třeba léčbu ramucirumabem trvale vysadit.
|
Počáteční dávka ramucirumabu: |
První snížení dávky na: |
Druhé snížení dávky na: |
|
8 mg/kg |
6 mg/kg |
5 mg/kg |
|
10 mg/kg |
8 mg/kg |
6 mg/kg |
Elektivní chirurgický výkon nebo zpomalené hojení ran
Léčbu ramucirumabem je třeba dočasně přerušit nejméně 4 týdny před elektivním chirurgickým zákrokem. Při komplikovaném hojení rány, je třeba léčbu ramucirumabem dočasně přerušit až do úplného zhojení (viz bod 4.4).
Léčbu ramucirumabem je třeba trvale vysadit, v případě:
Závažné arteriální tromboembolické příhody (viz bod 4.4).
Gastrointestinální perforace (viz bod 4.4).
Závažné krvácení: krvácení 3. nebo 4. stupně závažnosti dle NCI CTCAE (viz bod 4.4).
Spontánního vytvoření píštěle (viz bod 4.4).
Úpravy dávky _paklitaxelu
Podle stupně toxicity u daného pacienta lze uplatnit snížení dávky paklitaxelu. U hematologické toxicity 4. stupně nebo nehematologické toxicity 3. stupně závažnosti dle NCI CTCAE související s paklitaxelem se doporučuje snížit dávku paklitaxelu ve všech následujících cyklech o 10 mg/m2. Pokud tato toxicita přetrvává nebo se znovu objeví, doporučuje se druhé snížení o 10 mg/m2.
Úpravy dávky FOLFIRI
U specifických toxicit lze snížit dávky jednotlivých složek FOLFIRI. Úpravy dávek jednotlivých složek FOLFIRI je nutné provádět nezávisle. Bližší informace naleznete v tabulce 4. Tabulka 5 uvádí podrobnosti k odložení dávek nebo snížení dávek složek FOLFIRI v dalším cyklu na základě maximálního stupně specifických nežádoucích příhod.
Tabulka 4: Snížení dávky FOLFIRI
|
Složka FOLFIRIa |
Úroveň dávky | |||
|
Úvodní dávka |
-1 |
-2 |
-3 | |
|
Irinotekan |
180 mg/m2 |
150 mg/m2 |
120 mg/m2 |
100 mg/m2 |
|
Bolus 5-FU |
400 mg/m2 |
200 mg/m2 |
0 mg/m2 |
0 mg/m2 |
|
Infuze 5-FU |
2 000 mg/m2 |
1 600 mg/m2 |
1 200 mg/m2 | |
|
2 400 mg/m2 |
po dobu |
po dobu |
po dobu | |
|
po dobu 46-48 hodin |
46-48 hodin |
46-48 hodin |
46-48 hodin | |
a
5-FU = fluoruracil.
|
AE |
Stupeň NCI CTCAE |
Úprava dávky v den 1 cyklu následujícího po AE | |
|
2 |
Pokud intenzita průjmu klesla na stupeň <1, snižte dávky 5-FU o 1 úroveň. U relabujícího průjmu stupně 2 snižte dávky 5-FU a irinotekanu o 1 úroveň. | ||
|
3 |
Pokud intenzita průjmu klesla na stupeň <1, snižte dávky 5-FU a irinotekanu o 1 úroveň. | ||
|
4 |
Pokud intenzita průjmu klesla na stupeň <1, snižte dávky 5-FU a irinotekanu o 2 úrovně. Pokud průjem stupně 4 neklesne na stupeň <1, vysaďte 5-FU a irinotekan až do poklesu na stupeň <1, maximálně na 28 dní. | ||
|
Neutropenie a trombocytopenie |
Hematoloaická kritéria v tabulce 2 jsou splněna. |
Hematoloaická kritéria v tabulce 2 nejsou splněna. | |
|
2 |
Bez úpravy dávky. |
Snižte dávky 5-FU a irinotekanu o 1 úroveň. | |
|
3 |
Snižte úroveň dávky 5-FU a irinotekanu o 1. |
Odložte 5-FU a irinotekan maximálně o 28* dní až do poklesu na stupeň <1, poté snižte dávky 5-FU a irinotekanu o 1 úroveň. | |
|
4 |
Snižte úroveň dávky 5-FU a irinotekanu o 2. |
Odložte 5-FU a irinotekan maximálně o 28* dní až do poklesu na stupeň <1, poté snižte dávky 5-FU a irinotekanu o 2 úrovně. | |
|
Stomatitida/mukositida |
2 |
Pokud stomatitida/mukositida k 5-FU o 1 úroveň. U relabující stomatitidy stupně |
lesla na stupeň <1, snižte dávky 2 snižte dávky 5-FU o 2 úrovně. |
|
3 |
Pokud stomatitida/mukositida klesla na stupeň <1, snižte dávky 5-FU o 1 úroveň. Pokud mukositida/stomatitida stupně 3 neklesne na stupeň <1, odložte 5-FU maximálně o 28* dní až do poklesu na stupeň <1, poté snižte dávky 5-FU o 2 úrovně. | ||
|
4 |
Vysaďte 5-FU maximálně na 28* dní až do poklesu na stupeň <1, poté snižte dávky 5-FU o 2 úrovně. | ||
|
Febrilní neutropenie |
Hematoloaická kritéria v tabulce 2 jsou splněna a |
Hematoloaická kritéria v tabulce 2 nejsou splněna a | |
|
horečka ustoupila. |
horečka ustoupila. | ||
|
Snižte dávky 5-FU a irinotekanu o 2 úrovně. |
Odložte 5-FU a irinotekan maximálně o 28* dní až do poklesu na stupeň <1, poté snižte dávky 5-FU a irinotekanu o 2 úrovně. Před dalším cyklem zvažte použití růstových faktorů (CSF) | ||
|
*28denní období začíná dnem 1 cyk |
u následujícího po AE. | ||
Úpravy dávkování docetaxelu
Podle stupně toxicity u daného pacienta lze uplatnit snížení dávky docetaxelu. U pacienů, u kterých se během léčby docetaxelem objevily buď febrilní neutopenie, počet neutrofilů <500 buněk/mm3 po dobu
delší než 1 týden, závažné nebo kumulativní kožní reakce nebo nehematologické toxicity 3. nebo 4. stupně, by mělo dojít k přerušení léčby do vymizení toxicity. Doporučuje se snížit dávku docetaxelu ve všech následujících cyklech o 10 mg/m2. Pokud tyto toxicity přetrvávají nebo se znovu objeví, doporučuje se druhé snížení o 15 mg/m2. V tomto případě má být u pacientů z Východní Asie, u kterých byla počáteční dávka docetaxelu 60 mg/m2, podávání docetaxelu přerušeno (viz Dávkování).
Zvláštní _ populace
Starší pacienti
V pivotních studiích se objevily omezené důkazy svědčící pro zvýšené riziko nežádoucích příhod u pacientů ve věku 65 let a více v porovnání s pacienty mladšími 65 let. Není doporučeno snížení dávky (viz body 4.4 a 5.1).
Pacienti s poruchou funkce ledvin
U pacientů s poruchou funkce ledvin neproběhly žádné formální studie s přípravkem Cyramza. Klinické údaje naznačují, že u pacientů s lehkou, středně těžkou nebo těžkou poruchou funkce ledvin není potřeba nijak upravovat dávku (viz body 4.4 a 5.2). Není doporučeno snížení dávky.
Pacienti s poruchou funkce jater
U pacientů s poruchou funkce jater neproběhly žádné formální studie s přípravkem Cyramza. Dle klinických údajů není nutné upravovat dávku u pacientů s mírnou nebo středně těžkou poruchou funkce jater. O podávání ramucirumabu pacientům s těžkou poruchou funkce jater neexistují žádné údaje (viz bod 4.4 a 5.2). Není doporučeno snížení dávky.
Pediatrická populace
Bezpečnost a účinnost přípravku Cyramza u dětí a dospívajících (<18 let) nebyla stanovena.
Nejsou dostupné žádné údaje.
Podávání ramucirumabu pediatrické populaci v indikaci pokročilého karcinomu žaludku nebo gastroesofageální junkce či v léčbě adenokarcinomu kolon a rekta a v indikaci karcinomu plic není relevantní.
Způsob podání
Cyramza se po naředění podává v intravenózní infuzi po dobu přibližně 60 minut. Nepodávejte jako intravenózní bolus nebo injekci. Pro dosažení požadované délky trvání infuze přibližně 60 minut by neměla být překročena maximální rychlost infuze 25 mg/min, jinak je třeba délku trvání infuze prodloužit. U pacienta by během infuze měly být monitorovány známky reakcí souvisejících s podáním infuze (viz bod 4.4) a je třeba zajistit dostupnost vhodného resuscitačního vybavení.
Návod k naředění léčivého přípravku před jeho podáním je uveden v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
U pacientů s NSCLC je ramucirumab kontraindikován v případě kavitace nádoru, nebo v případě, že nádor zahrnuje hlavní cévy (viz bod 4.4).
4.4 Zvláštní upozornění a opatření pro použití
Arteriální tromboembolické příhody
V klinických studiích byly hlášeny závažné, někdy fatální arteriální tromboembolické příhody (arterial thromboembolic event, ATE) včetně infarktu myokardu, srdeční zástavy, cerebrovaskulární příhody a mozkové ischémie. U pacientů, u nichž dojde k závažné ATE, je třeba ramucirumab trvale vysadit (viz bod 4.2).
Gastrointestinální perforace
Ramucirumab je antiangiogenní léčivo a může zvyšovat riziko vzniku gastrointestinálních perforací. U pacientů léčených ramucirumabem byly hlášeny případy gastrointestinální perforace. U pacientů, u nichž dojde ke gastrointestinální perforaci, je třeba ramucirumab trvale vysadit (viz bod 4.2).
Závažné krvácení
Ramucirumab je antiangiogenní léčivo a může zvyšovat riziko vzniku závažného krvácení. U pacientů, u nichž dojde ke krvácení 3. nebo 4. stupně závažnosti, je třeba ramucirumab trvale vysadit (viz bod 4.2). U pacientů se stavy predisponujícími ke krvácení a u pacientů léčených antikoagulancii nebo jinými současně podávanými léčivými přípravky, které zvyšují riziko krvácení, je třeba monitorovat krevní obraz a srážlivost.
U pacientů s karcinomem žaludku léčených ramucirumabem v kombinaci s paklitaxelem a u pacientů s mCRC léčených ramucirumabem v kombinaci s FOLFIRI byly hlášeny případy závažného gastrointestinálního krvácení včetně fatálních případů.
Plicní krvácení u NSCLC
U pacientů s dlaždicobuněčnou histologií je větší riziko, že u nich dojde k závažnému plicnímu krvácení, nicméně u pacientů s dlaždicobuněčnou histologií léčených ramucirumabem ve studii REVEL nebyl pozorován nárůst plicní hemoragie stupně 5. Pacienti s NSCLC s nedávným plicním krvácením (>2,5 ml jasně červené krve), stejně jako pacienti s prokázanou kavitací nádoru před vstupem do studie bez ohledu na histologii, nebo ti, u kterých byla prokázána jakákoliv invaze nádoru do hlavních cév nebo jejich obrůstání nádorem,byli z klinických studií vyloučeni (viz bod 4.3). Pacienti, kteří dostávali jakoukoliv antikoagulační léčbu a/nebo byli dlouhodobě léčeni nesteroidními protizánětlivými léky nebo antiagregačními látkami, byli ze studie REVEL s NSCLC vyloučeni. Bylo povoleno užívání kyseliny acetylsalicylové v dávkách do 325 mg/den (viz bod 5.1).
Reakce související s podáním infuze
V klinických studiích s ramucirumabem byly hlášeny reakce související s podáním infuze. K většině případů došlo v průběhu podání nebo po podání první nebo druhé infuze ramucirumabu. V průběhu podávání infuze je u pacientů třeba monitorovat známky hypersenzitivity. Příznaky zahrnovaly ztuhlost/třes, bolest zad/křeče, bolest na hrudi a/nebo svírání na hrudi, zimnici, návaly, dušnost, sípání, hypoxii a parestezie. V závažných případech příznaky zahrnovaly bronchospasmus, supraventrikulární tachykardii a hypotenzi. U pacientů, u nichž dojde k IRR 3. nebo 4. stupně závažnosti, je třeba ramucirumab okamžitě a trvale vysadit (viz bod 4.2).
Hypertenze
U pacientů, kteří užívali ramucirumab, byla v porovnání s placebem hlášena vyšší incidence závažné hypertenze. Ve většině případů byla hypertenze zvládnuta podáváním standardní antihypertenzní léčby. Pacienti s nekontrolovanou hypertenzí byli ze studií vyloučeni: léčba ramucirumabem by u takových pacientů neměla být zahájena, dokud u nich není preexistující hypertenze kompenzována. U pacientů léčených ramucirumabem je třeba monitorovat krevní tlak. Léčbu ramucirumabem je třeba v případě závažné hypertenze dočasně přerušit, a to až do doby její kompenzace antihypertenzní terapií.
V případě klinicky významné hypertenze, kterou nelze bezpečně kompenzovat pomocí antihypertenzní léčby, je třeba léčbu ramucirumabem trvale vysadit (viz bod 4.2).
Narušené hojení ran
Vliv ramucirumabu nebyl hodnocen u pacientů se závažnými nebo nehojícími se ranami. Ve studii provedené u zvířat ramucirumab nenarušil hojení ran. Nicméně vzhledem k tomu, že je ramucirumab antiangiogenní léčivo a mohl by nežádoucím způsobem ovlivňovat hojení ran, je třeba léčbu ramucirumabem pozastavit nejméně na 4 týdny před plánovanou operací. Rozhodnutí znovu nasadit ramucirumab po chirurgickém zákroku by mělo vycházet z klinického posouzení adekvátního hojení rány.
Pokud u pacienta dojde ke komplikovanému hojení rány během léčby, je třeba ramucirumab vysadit až do úplného zhojení rány (viz bod 4.2).
Porucha funkce jater
Ramucirumab je třeba podávat s opatrností u pacientů se závažnou cirhózou jater (Child-Pugh B nebo C), cirhózou s jaterní encefalopatií, klinicky významným ascitem z důvodu cirhózy nebo s hepatorenálním syndromem. U těchto pacientů by se měl po posouzení ramucirumab podávat pouze v případě, že potenciální přínos léčby převáží potenciální riziko progresivního selhání jater.
Píštěl
Při léčbě přípravkem Cyramza může být u pacientů zvýšeno riziko vzniku píštěle. U pacientů, u nichž se vytvoří píštěl, je třeba ramucirumab vysadit (viz bod 4.2).
Proteinurie
Ve srovnání s placebem byl u pacientů léčených ramucirumabem hlášen zvýšený výskyt proteinurie. Během léčby ramucirumabem je třeba u pacientů monitorovat rozvoj nebo zhoršení proteinurie. Pokud se na testovacím proužku objeví bílkovina >2, je třeba provést 24hodinový sběr moči. Pokud hladina bílkoviny v moči dosáhne >2 g/24 hodin, je třeba léčbu ramucirumabem dočasně přerušit. Jakmile se hladina bílkoviny v moči vrátí na <2 g/24 hodin, je možné se vrátit k léčbě sníženou dávkou. Pokud znovu dojde ke zvýšení hladiny bílkoviny v moči na >2 g/24 hodin, doporučuje se druhé snížení dávky. Pokud hladina bílkoviny v moči dosáhne >3 g/24 hodin nebo v případě rozvoje nefrotického syndromu, je třeba léčbu ramucirumabem trvale vysadit (viz bod 4.2).
Stomatitida
U pacientů léčených ramucirumabem v kombinaci s chemoterapií byla ve srovnání s pacienty užívajícími placebo a chemoterapii hlášena zvýšená incidence stomatitidy. Pokud se stomatitida rozvine, je nutné ihned zahájit symptomatickou léčbu.
Porucha funkce ledvin
U pacientů s těžkou poruchou funkce ledvin (clearance kreatininu 15 až 29 ml/min) léčených ramucirumabem existují pouze omezené údaje o bezpečnosti (viz body 4.2 a 5.2).
Dieta s omezením sodíku
Jedna 10ml injekční lahvička obsahuje přibližně 17 mg sodíku a jedna 50ml injekční lahvička obsahuje přibližně 85 mg sodíku. To je třeba vzít v úvahu u pacientů na dietě s nízkým obsahem sodíku.
Starší pacienti s NSCLC
U pacientů, kteří dostávali ramucirumab s docetaxelem k léčbě pokročilého NSCLC s progresí onemocnění po chemoterapii na bázi platiny, byl pozorován trend k nižší účinnosti s rostoucím věkem (viz bod 5.1). Před započetím léčby starších pacientů musí být tedy důkladně zváženy komorbidity spojené s pokročilým věkem, stav výkonnosti a pravděpodobná tolerance chemoterapie (viz body 4.2 a 5.1).
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Mezi ramucirumabem a paklitaxelem nebyly pozorovány žádné lékové interakce. Farmakokinetika paklitaxelu nebyla narušena současným podáváním ramucirumabu a farmakokinetika ramucirumabu nebyla narušena současným podáváním paklitaxelu. Kombinované podávání s ramucirumabem nemělo vliv na farmakokinetiku irinotekanu a jeho aktivního metabolitu SN-38. Farmakokinetika docetaxelu nebyla ovlivněna současným podáváním ramucirumabu.
4.6 Fertilita, těhotenství a kojení
Ženy v reprodukčním věku/antikoncepce u žen
Ženám v reprodukčním věku je třeba doporučit, aby během užívání přípravku Cyramza neotěhotněly a je třeba je informovat o potenciálním riziku pro těhotenství a plod. Ženy ve fertilním věku by měly během léčby a ještě 3 měsíce po poslední dávce ramucirumabu používat účinnou antikoncepci.
O podávání ramucirumabu u těhotných žen nejsou k dispozici žádné údaje. Data získaná ze studií na zvířatech nejsou z hlediska reprodukční toxicity dostatečná (viz bod 5.3). Vzhledem k tomu, že angiogeneze má zásadní význam pro udržení těhotenství a vývoj plodu, může inhibice angiogeneze po podávání ramucirumabu vést k nežádoucím účinkům na těhotenství včetně plodu. Přípravek Cyrazma by měl být užíván během těhotenství pouze tehdy, pokud potenciální přínos léčby pro matku převýší možné riziko. Pokud pacientka během léčby ramucirumabem otěhotní, má být poučena o potenciálním ohrožení udržení těhotenství a o riziku pro plod. Léčba přípravkem Cyramza se nedoporučuje během těhotenství a u žen v reprodukčním věku, které nepoužívají antikoncepci.
Kojení
Není známo, zda se ramucirumab vylučuje do lidského mateřského mléka. Předpokládá se, že vylučování do mateřského mléka a perorální absorpce jsou malé. Vzhledem k tomu, že nelze vyloučit riziko pro novorozence/kojence, je třeba při léčbě přípravkem Cyramza a alespoň 3 měsíce po poslední dávce kojení přerušit.
Fertilita
Údaje o účinku ramucirumabu na lidskou fertilitu nejsou k dispozici. Na základě studií na zvířatech je pravděpodobné, že fertilita u žen bude během léčby ramucirumabem narušena (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Není známo, že by měl přípravek Cyramza vliv na schopnost řídit nebo obsluhovat stroje. U pacientů s příznaky, které narušují schopnost soustředění a reakce, se doporučuje, aby neřídili a neobsluhovali stroje, dokud tento účinek neodezní.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Nejzávažnější nežádoucí účinky spojené s léčbou ramucirumabem (v monoterapii nebo v kombinaci s cytotoxickou chemoterapií) byly:
gastrointestinální perforace (viz bod 4.4) závažné gastrointestinální krvácení (viz bod 4.4) arteriální tromboembolické příhody (viz bod 4.4)
Nejčastější nežádoucí účinky pozorované u pacientů léčených ramucirumabem jsou: neutropenie, únava/astenie, leukopenie, epistaxe, průjem a stomatitida.
Tabulkový seznam nežádoucích účinků
Nežádoucí účinky, které byly hlášeny u pacientů s pokročilým karcinomem žaludku, mCRC nebo NSCLC, jsou uvedeny níže podle tříd orgánových systémů MedDRA, četnosti a stupně závažnosti.
Pro klasifikaci četnosti výskytu je použita následující frekvence:
Velmi časté (>1/10)
Časté (>1/100 až <1/10)
Méně časté (>1/1 000 až <1/100)
Vzácné (>1/10 000 až <1/1 000)
Velmi vzácné (<1/10 000)
V každé skupině četností jsou nežádoucí účinky uvedeny v pořadí podle klesající závažnosti.
Karcinom žaludku
Ramucirumab v kombinaci s paklitaxelem
Následující tabulka ukazuje četnost výskytu a závažnost nežádoucích účinků na základě výsledků studie fáze 3 RAINBOW u dospělých pacientů s pokročilým karcinomem žaludku randomizovaných k léčbě ramucirumabem v kombinaci s paklitaxelem nebo placebem plus paklitaxelem.
Tabulka 6: Nežádoucí účinky hlášené u >5 % pacientů léčených ramucirumabem ve studii RAINBOW
|
Třídy orgánových systémů |
Frekvence |
Nežádoucí účinek |
Cyramza plus paklitaxel (N=327) |
Placebo plus paklitaxel (N=329) | ||
|
Toxicita všech stupňů (%) |
Toxicita >3. stupně (%) |
Toxicita všech stupňů (%) |
Toxicita >3. stupně (%) | |||
|
Poruchy krve a lymfatického systému |
Velmi časté |
Neutropenie |
54,4 |
40,7 |
31,0 |
18,8 |
|
Velmi časté |
Leukopenie |
33,9 |
17,4 |
21,0 |
6,7 | |
|
Velmi časté |
T rombocytopenie |
13,1 |
1,5 |
6,1 |
1,8 | |
|
Poruchy metabolismu a výživy |
Velmi časté |
Hypoalbuminemie |
11,0 |
1,2 |
4,9 |
0,9 |
|
Cévní poruchy |
Velmi časté |
Hypertenze3 |
25,1 |
14,7 |
5,8 |
2,7 |
|
Respirační, hrudní a mediastinální poruchy |
Velmi časté |
Epistaxe |
30,6 |
0,0 |
7,0 |
0,0 |
|
Gastrointestinální poruchy |
Velmi časté |
Gastrointe stinální krváceníb |
10,1 |
3,7 |
6,1 |
1,5 |
|
Velmi časté |
Stomatitida |
19,6 |
0,6 |
7,3 |
0,6 | |
|
Velmi časté |
32,4 |
3,7 |
23,1 |
1,5 | ||
|
Poruchy ledvin a močových cest |
Velmi časté |
Proteinurie |
16,8 |
1,2 |
6,1 |
0,0 |
|
Celkové poruchy a reakce v místě aplikace |
Velmi časté |
Únava/astenie |
56,9 |
11,9 |
43,8 |
5,5 |
|
Velmi časté |
Periferní edém |
25,1 |
1,5 |
13,7 |
0,6 | |
a Včetně hypertenzní kardiomyopatie.
b Preferované termíny MedDRA zahrnovaly anální krvácení, krvavý průjem, krvácení ze žaludku, gastrointestinální krvácení, hematemezi, hematochezii, hemoroidální krvácení, Mallory-Weissův syndrom, melenu, esofageální krvácení, krvácení z rekta a krvácení z horní části gastrointestinálního traktu.
Klinicky relevantní nežádoucí účinky hlášené u >1 % a <5 % pacientů léčených ramucirumabem plus paklitaxelem ve studii RAINBOW byly gastrointestinální perforace (1,2 % u ramucirumabu plus paklitaxelu versus 0,3 % u placeba plus paklitaxelu) a sepse (3,1 % u ramucirumabu plus paklitaxelu versus 1,8 % u placeba plus paklitaxelu).
Ramucirumab v monoterapii
Následující tabulka ukazuje četnost výskytu a závažnost nežádoucích účinků na základě výsledků studie fáze 3 REGARD u dospělých pacientů s pokročilým karcinomem žaludku randomizovaných k
monoterapii ramucirumabem plus nejlepší podpůrnou péčí (Best Supportive Care, BSC) nebo placebem plus BSC.
Tabulka 7: Nežádoucí účinky hlášené u > 5 % pacientů léčených ramucirumabem ve studii REGARD
|
Třídy orgánových systémů |
F rekvence |
Nežádoucí účineka,b |
Cyramza (N=236) |
Placebo (N=115) | ||
|
Toxicita všech stupňůc (%) |
Toxicita 3.-4. stupně (%) |
Toxicita všech stupňů (%) |
Toxicita 3.-4. stupně (%) | |||
|
Poruchy metabolismu a výživy |
Časté |
Hypokalémied |
5,9 |
2,1 |
5,2 |
0,9 |
|
Časté |
Hyponatrémie |
5,5 |
3,4 |
1,7 |
0,9 | |
|
Poruchy nervového systému |
Časté |
9,3 |
0 |
3,5 |
0 | |
|
Cévní poruchy |
Velmi časté |
Hypertenzee |
16,1 |
7,6 |
7,8 |
2,6 |
|
Gastrointestinální poruchy |
Velmi časté |
Bolest bnchaf |
28,8 |
5,9 |
27,8 |
2,6 |
|
Velmi časté |
14,4 |
0,8 |
8,7 |
1,7 | ||
a
b
Preferovaný MedDRA termín (verze 15.0)
U přípravku Cyramza se neobjevily žádné nežádoucí účinky 5. stupně. Objevil se jeden případ hypokalémie a jeden případ hyponatrémie jako nežádoucí účinky 4. stupně.
c
d
e
f
Jednotlivé stupně toxicity viz kritéria NCI CTCAE (verze 4.0).
Zahrnuté preferované termíny MedDRA jsou: snížení hladiny draslíku v krvi a hypokalémie. Zahrnuté preferované termíny MedDRA jsou: zvýšení krevního tlaku a hypertenze.
Zahrnuté preferované termíny MedDRA jsou: bolest břicha, bolest v podbřišku, bolest v epigastriu a bolest v oblasti jater.
Klinicky relevantní nežádoucí účinky hlášené u >1 % a <5 % pacientů léčených ramucirumabem ve studii REGARD byly: neutropenie, arteriální tromboembolické příhody (viz body 4.2 a 4.4), střevní obstrukce, epistaxe a vyrážka.
Klinicky relevantní reakce (včetně stupně >3) související s antiangiogenní léčbou pozorované u pacientů léčených ramucirumabem napříč klinickými studiemi byly: gastrointestinální perforace, reakce související s podáním infuze a proteinurie (viz body 4.2 a 4.4).
Kolorektální karcinom
Ramucirumab v kombinaci s FOLFIRI
Následující tabulka ukazuje četnost výskytu a závažnost nežádoucích účinků na základě výsledků studie fáze 3 RAISE u dospělých pacientů s mCRC randomizovaných k léčbě ramucirumabem v kombinaci s FOLFIRI nebo placebem plus FOLFIRI.
Tabulka 8: Nežádoucí účinky hlášené u >5 % pacientů léčených ramucirumabem ve studii RAISE
|
Třídy orgánových systémů |
F rekvence |
Nežádoucí účinek |
Cyramza plus FOLFIRI (N=529) |
Placebo plus FOLFIRI (N=528) | ||
|
Toxicita všech stupňů (%) |
Toxicita >3. stupně (%) |
Toxicita všech stupňů (%) |
Toxicita >3. stupně (%) | |||
|
Poruchy krve a lymfatického systému |
Velmi časté |
Neutropenie |
58,8 |
38,4 |
45,6, |
23,3 |
|
Velmi časté |
T rombocytopenie |
28,4 |
3,0 |
13,6 |
0,8 | |
|
Poruchy metabolismu a výživy |
Časté |
Hypalbuminemie |
5,9 |
1,1 |
1,9 |
0,0 |
|
Cévní poruchy |
Velmi časté |
Hypertenze |
26,1 |
11,2 |
8,5 |
2,8 |
|
Respirační, hrudní a mediastinální poruchy |
Velmi časté |
Epistaxe |
33,5 |
0,0 |
15,0 |
0,0 |
|
Gastrointestinální poruchy |
Velmi časté |
Gastrointestinální krvácení |
12,3 |
1,9 |
6,8 |
1,1 |
|
Velmi časté |
Stomatitida |
30,8 |
3,8 |
20,8 |
2,3 | |
|
Poruchy ledvin a močových cest |
Velmi časté |
Proteinurie3 |
17,0 |
3,0 |
4,5 |
0,2 |
|
Poruchy kůže a podkožní tkáně |
Velmi časté |
Syndrom palmární- plantární erythrodysestesie |
12,9 |
1,1 |
5,5 |
0,4 |
|
Celkové poruchy a reakce v místě aplikace |
Velmi časté |
Periferní edém |
20,4 |
0,2 |
9,1 |
0,0 |
Zahrnuje případy nefrotického syndromu
Klinicky relevantní nežádoucí účinky hlášené u >1 % a <5 % pacientů léčených ramucirumabem plus FOLFIRI ve studii RAISE: gastrointestinální perforace (1,7 % u ramucirumabu plus FOLFIRI versus 0,6 % u placeba plus FOLFIRI).
Ve studii RAISE byla u pacientů s mCRC léčených ramucirumabem plus FOLFIRI nejčastějším (>1 %) nežádoucím účinkem, který vedl k vysazení ramucirumabu, proteinurie (1,5 %). Nej častějšími (>1%) nežádoucími účinky vedoucími k vysazení jedné nebo více složek FOLFIRI byly: neutropenie (12,5 %), trombocytopenie (4,2 %), průjem (2,3 %) a stomatitida (2,3 %). Nejčastěji vysazovanou složkou FOLFIRI byl bolus 5-FU.
NSCLC
Ramucirumab v kombinaci s docetaxelem
Tabulka níže uvádí frekvenci a závažnost nežádoucích účinků na základě výsledků studie REVEL, studie fáze 3 u dospělých pacientů s NSCLC randomizovaných k léčbě ramucirumabem v kombinaci s docetaxelem nebo placebem a docetaxelem.
Tabulka 9: Nežádoucí účinky hlášené u > 5 % pacientů léčených ramucirumabem ve studii REVEL
|
Třída orgánového systému |
Frekvence |
Nežádoucí účinek |
Cyramza a docetaxel (N=627) |
Placebo a docetaxel (N=618) | ||
|
Toxicita všech stupňů (%) |
Toxicita 3.-4. stupně (%) |
Toxicita všech stupňů (%) |
Toxicita 3.-4. stupně (%) | |||
|
Poruchy krve a lymfatického systému |
Velmi časté |
Febrilní neutropenie |
15,9 |
15,9 |
10,0 |
10,0 |
|
Velmi časté |
Neutropenie |
55,0 |
48,8 |
46,0 |
39,8 | |
|
Velmi časté |
T rombocytopenie |
13,4 |
2,9 |
5,2 |
0,6 | |
|
Cévní poruchy |
Velmi časté |
Hypertenze |
10,8 |
5,6 |
4,9 |
2,1 |
|
Respirační, hrudní a mediastinální poruchy |
Velmi časté |
Epistaxe |
18,5 |
0,3 |
6,5 |
0,2 |
|
Gastrointe stinální poruchy |
Velmi časté |
Stomatitida |
23,3 |
4,3 |
12,9 |
1,6 |
|
Celkové poruchy a reakce v místě aplikace |
Velmi časté |
Únava/astenie |
54,7 |
14,0 |
50,0 |
10,5 |
|
Velmi časté |
Mukozální zánět |
16,1 |
2,9 |
7,0 |
0,5 | |
|
Velmi časté |
Periferní edém |
16,3 |
0 |
8,6 |
0,3 | |
Klinicky relevantní nežádoucí účinky hlášené u >1% a <5% pacientů léčených ramucirumabem a docetaxelem ve studii REVEL byly hyponatrémie (4,8 % u ramucirumabu s docetaxelem a 2,4 % u placeba s docetaxelem), proteinurie (3,3 % u ramucirumabu s docetaxelem a 0,8 % u placeba s docetaxelem) a gastrointestinální perforace (1 % u ramucirumabu s docetaxelem a 0,3 % u placeba s docetaxelem).
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Neexistují údaje o předávkování u člověka. Přípravek Cyramza byl podáván ve studii 1. fáze až v dávce 10 mg/kg každé dva týdny, aniž by bylo dosaženo maximální tolerované dávky. V případě předávkování je třeba použít podpůrnou léčbu.
FARMAKOLOGICKÉ VLASTNOSTI
5.
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Cytostatika, monoklonální protilátky ATC kód: L01XC21. Mechanismus účinku
Klíčovým mediátorem angiogeneze navozené cévním endoteliálním růstovým faktorem (Vascular Endothelial Growth Factor, VEGF) je VEGF receptor 2. Ramucirumab je lidská protilátka cílená na receptor, která se specificky váže na VEGF receptor 2 a brání navázání VEGF-A, VEGF-C a VEGF-D. To vede k tomu, že ramucirumab inhibuje ligandy stimulovanou aktivaci VEGF receptoru 2 a navazujících signalizačních složek včetně mitogenem aktivovaných proteinkináz p44/p42, a neutralizuje ligandy indukovanou proliferaci a migraci lidských endoteliálních buněk.
Klinická účinnost a bezpečnost
Karcinom žaludku:
RAINBOW
RAINBOW, celosvětová randomizovaná dvojitě zaslepená studie s přípravkem Cyramza plus paklitaxelem versus placebem plus paklitaxelem byla provedena u 665 pacientů s neresekovatelným lokálně recidivujícím nebo metastazujícím karcinomem žaludku (včetně adenokarcinomu GEJ) po chemoterapii obsahující platinu a fluoropyrimidin, s případným přidáním anthracyklinu. Primárním sledovaným parametrem bylo celkové přežití (OS) a sekundární sledované parametry zahrnovaly přežití bez progrese (PFS) a četnost odpovědí na léčbu (ORR). Bylo nutné, aby u pacientů došlo k progresi choroby během první linie léčby nebo během 4 měsíců po podání poslední dávky terapie v první linii a aby měli PS 0-1. Pacienti byli randomizováni v poměru 1:1 k podávání přípravku Cyramza plus paklitaxel (n = 330) nebo placeba plus paklitaxel (n = 335). Randomizace byla stratifikována podle geografické oblasti, doby do progrese od zahájení první linie léčby (<6 měsíců versus >6 měsíců) a měřitelnosti choroby. Přípravek Cyramza v dávce 8 mg/kg nebo placebo byly podávány v intravenózní infuzi každé 2 týdny (1. a 15. den) 28denního cyklu. Paklitaxel v dávce 80 mg/m2 byl podáván v intravenózní infuzi 1., 8. a 15. den každého 28denního cyklu.
Většina (75 %) pacientů randomizovaných ve studii dříve užívala kombinovanou léčbu s platinou a fluoropyrimidinem bez anthracyklinu. Zbývající část (25 %) pacientů užívala kombinovanou léčbu platinou a fluoropyrimidinem s anthracyklinem. U dvou třetin (66,8 %) pacientů došlo k progresi choroby ještě během terapie první linie. Vstupní demografické charakteristiky pacientů i charakteristiky onemocnění byly u obou terapeutických ramen celkově vyvážené: medián věku byl 61 let, 71 % pacientů tvořili muži, 61 % pacientů bylo kavkazské rasy, 35 % Asiatů, ECOG PS byl 0 u 39 % pacientů a 1 u 61 % pacientů, 81 % pacientů mělo měřitelné onemocnění a 79 % pacientů mělo karcinom žaludku, 21 % adenokarcinom GEJ. U většiny (76 %) pacientů došlo k progresi choroby během 6 měsíců od zahájení terapie první linie. U pacientů léčených přípravkem Cyramza plus paklitaxelem byla střední hodnota délky léčby 19 týdnů a u pacientů léčených placebem plus paklitaxelem byla střední hodnota délky léčby 12 týdnů. Medián relativní intenzity dávky přípravku Cyramza byl 98,6 % a u placeba 99,6 %. Medián relativní intenzity dávky paklitaxelu byl 87,7 % v rameni s přípravkem Cyramza plus paklitaxelem a 93,2 % v rameni s placebem plus paklitaxelem. Z důvodu nežádoucích příhod vysadilo léčbu podobné procento pacientů: 12 % pacientů léčených přípravkem Cyramza plus paklitaxelem v porovnání s 11 % pacientů léčených placebem plus paklitaxelem. Systémová protinádorová léčba po vysazení hodnocené léčby byla podávána u 47,9 % pacientů, kteří užívali přípravek Cyramza plus paklitaxel, a u 46,0 % pacientů, kteří dostávali placebo plus paklitaxel.
U pacientů, kteří dostávali přípravek Cyramza plus paklitaxel bylo v porovnání s pacienty, kteří dostávali placebo plus paklitaxel statisticky významně zlepšeno celkové přežití (HR 0,807, 95% CI: 0,678 až 0,962, p = 0,0169). Medián přežití se prodloužil o 2,3 měsíce ve prospěch ramene s přípravkem Cyramza plus paklitaxel: 9,63 měsíce v rameni s přípravkem Cyramza plus paklitaxelem a 7,36 měsíce v rameni s placebem plus paklitaxelem. U pacientů, kteří dostávali přípravek Cyramza plus paklitaxel, v porovnání s pacienty, kteří dostávali placebo plus paklitaxel bylo statisticky významně lepší přežití bez progrese (HR 0,635, 95% CI: 0,536 až 0,752,p < 0,0001). Medián PFS se prodloužil o 1,5 měsíce ve prospěch ramene s přípravkem Cyramza plus paklitaxel: 4,4 měsíce v rameni s přípravkem Cyramza plus paklitaxelem a 2,9 měsíce v rameni s placebem plus paklitaxelem. Podíl pacientů s objektivní odpovědí na léčbu [ORR (kompletní odpověď [CR] + částečná odpověď [PR])] byl statisticky významně lepší u pacientů, kteří dostávali přípravek Cyramza plus paklitaxel, v porovnání s pacienty, kteří dostávali placebo plus paklitaxel (poměr šancí 2,140, 95% CI: 1,499 až 3,160, p = 0,0001). ORR činil v rameni s přípravkem Cyramza plus paklitaxelem 27,9 % a v rameni s placebem plus paklitaxelem 16,1 %. Zlepšení OS a PFS bylo pozorováno i u jednotlivých podskupin předem specifikovaných podle věku, pohlaví, rasy a u většiny dalších předem specifikovaných podskupin. Výsledky týkající se účinnosti jsou uvedeny v tabulce 10.
Tabulka 10: Souhrn údajů o účinnosti - populace s léčebným záměrem (Intent to Treat, ITT)
|
Cyramza plus paklitaxel N=330 |
Placebo plus paklitaxel N=335 | |
|
Celkové přežití, měsíce | ||
|
Medián (95% CI) |
9,6 (8,5-10,8) |
7,4 (6,3-8,4) |
|
Poměr rizik (95% CI) |
0,807 (0,678-0,962) | |
|
Stratifikovaná hodnota p dle log-rank testu |
0,0169 | |
|
Přežití bez progrese, měsíce | ||
|
Medián (95% CI) |
4,4 (4,2-5,3) |
2,9 (2,8-3,0) |
|
Poměr rizik (95% CI) |
0,635 (0,536-0,752) | |
|
Stratifikovaná hodnota p dle log-rank testu |
<0,0001 | |
|
Podíl pacientů s objektivní odpovědí na léčbu (CR + PR) | ||
|
Podíl - procenta (95% CI) |
27,9 (23,3-33,0) |
16,1 (12,6-20,4) |
|
Poměr šancí |
2,14 (1,449-3,16) | |
|
Stratifikovaná hodnota p dle CMH |
0,0001 | |
|
Zkratky: CI = interval spolehlivosti, CR = |
kompletní odpověď, PR = částečná odpověď, CMH = | |
Cochran-Mantel-Haenszel
Cyramza+Paditaxel
Placebo+Pac itaxe
10 11 12 13 14 15 16 17 18 19 20 21 Doba od randomizace (měsíce)
Počet v riziku
Cyramza+Paclitaxel 330 Placebo+Pac litaxel 335
'■i:
* V
Obrázek 2: Kaplan-Meierovy křivky přežití bez progrese u přípravku Cyramza plus paklitaxelu versus placeba plus paklitaxelu ve studii RAINBOW
Cyramza+Paclitaxel Placebo+Paclitaxel
Počet v riziku
Doba od randomizace (měsíce)
Cyramzar-Paclitaxel 330
Placebor-Paclitaxel 335
REGARD
Mezinárodní randomizovaná dvojitě zaslepená studie REGARD s přípravkem Cyramza plus BSC oproti placebu s BSC byla provedena u 355 pacientů s neresekovatelným lokálně recidivujícícm nebo metastazujícím karcinomem žaludku (včetně adenokarcinomu GEJ) po chemoterapii obsahující platinu nebo fluoropyrimidin. Primárním cílovým parametrem bylo OS a sekundární sledované parametry zahrnovaly PFS. Bylo nutné, aby u pacientů došlo k progresi choroby během léčby nebo během 4 měsíců po podání poslední dávky terapie v první linii v případě metastatického onemocnění, nebo během adjuvantní léčby nebo 6 měsíců po poslední dávce adjuvantní léčby, a aby měli PS 0-1.
Aby mohli být pacienti zařazeni do studie, museli mít celkový bilirubin <1,5 mg/dl a AST a ALT <3násobek ULN, nebo <5násobek ULN v případě přítomnosti jaterních metastáz.
Pacienti byli randomizováni v poměru 2:1 k podávání přípravku Cyramza 8 mg/kg v intravenózní infuzi (n = 238) nebo placeba (n = 117) každé 2 týdny. Randomizace byla stratifikována podle úbytku tělesné hmotnosti v průběhu předchozích 3 měsíců (>10 % versus <10 %), geografické oblasti a lokalizace primárního nádoru (žaludek versus GEJ).
Vstupních demografické charakteristiky i charakteristiky onemocnění byly vyvážené. ECOG PS byl 1 u 72 % pacientů. Do studie REGARD nebyl zařazen žádný pacient s jaterní cirhózou Child-Pugh B nebo C. 11 % pacientů léčených přípravkem Cyramza a 6 % pacientů s placebem vysadilo léčbu z důvodu nežádoucích příhod. Celkové přežití bylo statisticky významně lepší u pacientů, kteří dostávali přípravek Cyramza v porovnání s pacienty užívajícími placebo (poměr rizik [HR] 0,776, 95%
CI: 0,603 až 0,998; p = 0,0473), což odpovídá 22% snížení rizika úmrtí a prodloužení střední doby přežití na 5,2 měsíce s přípravkem Cyramza ze 3,8 měsíců u placeba. Přežití bez progrese bylo statisticky významně lepší u pacientů, kteří dostávali přípravek Cyramza v porovnání s pacienty s placebem (poměr rizik [HR] 0,483, 95% CI: 0,376 až 0,620, p < 0,0001), což odpovídá 52% snížení rizika progrese nebo úmrtí a prodloužení střední doby PFS na 2,1 měsíce s přípravkem Cyramza z 1,3 měsíce u placeba. Výsledky týkající se účinnosti jsou uvedeny v tabulce 11.
Tabulka 11: Souhrn údajů o účinnosti - ITT populace
|
Cyramza N=238 |
Placebo N=117 | |
|
Celkové přežití, měsíce | ||
|
Medián (95% CI) |
5,2 (4,4-5,7) |
3,8 (2,8-4,7) |
|
Poměr rizik (95% CI) |
0,776 (0,603-0,998) | |
|
Stratifikovaná hodnota p dle log-rank testu |
0,0473 | |
|
Přežití bez progrese, měsíce | ||
|
Medián (95% CI) |
2,1 (1,5-2,7) |
1,3 (1,3-1,4) |
|
Poměr rizik (95% CI) |
0,483 (0,376-0,62) | |
|
Stratifikovaná hodnota p dle log-rank testu |
<0,0001 | |
|
Podíl pacientů s PFS po 12 týdnech (95% CI) |
40,1 (33,6-46,4) |
15,8 (9,7-23,3) |
Zkratky: CI = interval spolehlivosti
Cyramza
Placebo
Početv riziku
Doba od randomizace (mesice)
Cyramza
Placebo
Na základě omezených dat ze studie REGARD od pacientů s HER2-pozitivním karcinomem žaludku nebo adenokarcinomem GEJ a pacientů s předchozí léčbou trastuzumabem (ve studii RAINBOW) je považováno za nepravděpodobné, že by přípravek Cyramza měl škodlivý účinek, nebo byl zcela bez účinku u pacientů s HER2-pozitivním karcinomem žaludku. Post hoc analýzy nestratifikovaných podskupin pacientů ze studie RAINBOW s předchozí léčbou trastuzumabem (n=39) naznačují u těchto pacientů pozitivní přínos pro přežití (HR 0,679, 95% CI 0,327, 1,419) a demonstrují přínos pro přežití bez progrese (PFS) (HR 0,399, 95% CI 0,194, 0,822).
Kolorektální karcinom
RAISE
RAISE, celosvětová randomizovaná dvojitě zaslepená studie s přípravkem Cyramza plus FOLFIRI versus placebem plus FOLFIRI byla provedena u pacientů s mCRC a progresí onemocnění v průběhu nebo po ukončení terapie první linie s bevacizumabem, oxaliplatinou a fluoropyrimidinem. Pacienti museli mít ECOG PS 0 nebo 1 a k progresi onemocnění muselo dojít do 6 měsíců od poslední dávky terapie první linie. Pacienti museli mít adekvátní jaterní, renální a koagulační funkce. Ze zařazení byli vyloučeni pacienti s anamnézou nekontrolovaného hereditárního nebo získaného krvácivého onemocnění či trombotické poruchy, nedávnou anamnézou těžkého (stupeň >3) krvácení nebo pacienti po arteriální trombotické události (ATE) v období 12 měsíců před randomizací. Pacienti byli také vyloučeni, pokud u nich během terapie bevacizumabem v první linii došlo ke kterékoli z následujících příhod: ATE, hypertenze stupně 4, proteinurie stupně 3, krvácivá událost stupně 3-4 nebo perforace střeva.
Celkem bylo randomizováno 1 072 pacientů (1:1) do skupin užívajících buď přípravek Cyramza (n=536) v dávce 8 mg/kg nebo placebo (n=536), v kombinaci s FOLFIRI. Všechny přípravky byly podávané intravenózně. Režim FOLFIRI sestával z následujících položek: irinotekan 180 mg/m2 podávaný po dobu 90 minut a kyselina folinová 400 mg/m2 podávaná současně po dobu 120 minut; následně bolus fluoruracilu (5-FU) 400 mg/m2 v průběhu 2 až 4 minut a poté kontinuální infuze 5-FU 2 400 mg/m2 podávaná po dobu 46 až 48 hodin. Léčebné cykly v obou ramenech se opakovaly každé 2 týdny. Pacienti, u kterých byla vysazena jedna nebo více složek léčby v důsledku nežádoucích účinků,
mohli pokračovat v léčbě dalšími složkami léčby až do progrese onemocnění nebo nepřijatelné toxicity. Primárním cílovým parametrem bylo OS, sekundární cílové parametry zahrnovaly PFS, četnost odpovědi na léčbu (ORR) a kvality života (QoL) dle dotazníku QLQ-C30 Evropské organizace pro výzkum a léčbu rakoviny (EORTC; European Organisation for Research and Treatment of Cancer). Randomizace byla stratifikována dle geografické oblasti, stavu KRAS v nádoru (mutace nebo nemutovaný „divoký“ typ) a doby do progrese (TTP) od zahájení terapie první linie (<6 měsíců vs. >6 měsíců).
Demografické a výchozí charakteristiky ITT populace byly v léčebných ramenech podobné. Medián věku byl 62 let a 40 % mělo >65 let; 57 % pacientů bylo mužského pohlaví; 76 % bylo kavkazské rasy a 20 % Asiatů; 49 % mělo ECOG PS 0; 49 % pacientů mělo tumory s mutací KRAS a 24 % pacientů mělo TTP <6 měsíců od zahájení terapie první linie. Systémovou protinádorovou léčbu dostalo po vysazení studijní léčby 54 % pacientů léčených přípravkem Cyramza plus FOLFIRI a 56 % pacientů užívajících placebo plus FOLFIRI.
U pacientů léčených přípravkem Cyramza plus FOLFIRI bylo ve srovnání s pacienty užívajícími placebo plus FOLFIRI statisticky významně zlepšeno celkové přežití (HR 0,844; 95% interval spolehlivosti: 0,730 až 0,976; p=0,0219). Došlo k nárůstu mediánu přežití o 1,6 měsíce v prospěch ramene Cyramza plus FOLFIRI; 13,3 měsíců v rameni Cyramza plus FOLFIRI a 11,7 měsíců v rameni placebo plus FOLFIRI. U pacientů užívajících Cyramza plus FOLFIRI ve srovnání s pacienty na kombinaci placebo plus FOLFIRI bylo statisticky významně lepší přežití bez progrese (HR 0,793; 95% interval spolehlivosti: 0,697 až 0,903; p=0,0005). Došlo k nárůstu mediánu PFS o 1,2 měsíce v prospěch ramene Cyramza plus FOLFIRI; 5,7 měsíce v rameni Cyramza plus FOLFIRI a 4,5 měsíce v rameni placebo plus FOLFIRI. Výsledky účinnosti jsou uvedeny v tabulce 12 a na obrázcích 4 a 5.
Byly provedeny předem specifikované analýzy OS a PFS dle stratifikačních faktorů. HR OS bylo 0,82 (95% interval spolehlivosti: 0,67 až 1,0) u pacientů s tumorem s nemutovaným KRAS a 0,89 (95% interval spolehlivosti: 0,73 až 1,09) u pacientů s tumorem s mutací KRAS. HR OS bylo 0,86 (95% interval spolehlivosti: 0,73 až 1,01) u pacientů s TTP >6 měsíců po zahájení terapie první linie a 0,86 (95% interval spolehlivosti: 0,64 až 1,13) u pacientů s TTP <6 měsíců po zahájení terapie první linie. Předem specifikované analýzy podskupin u PFS i OS dle věku (<65 let a >65 let), pohlaví, rasy,
ECOG PS (0 nebo >1), počtu postižených orgánů, pouze jaterních metastáz, místa primárního tumoru (kolon nebo rektum) a hladiny karcinoembryonálního antigenu (<200 pg/l, >200 pg/l) prokázaly všechny efekt léčby ve prospěch kombinace Cyramza plus FOLFIRI ve srovnání s placebem plus FOLFIRI. Ve 32 z 33 předem specifikovaných analýz podskupin pro OS bylo HR <1,0. Jedna podskupina s HR >1 obsahovala pacienty s progresí onemocnění od zahájení léčby bevacizumabem v první linii <3 měsíce (HR 1,02 [95% interval spolehlivosti: 0,068 až 1,55]). Tato podskupina může být považována za skupinu s agresivním onemocněním, které je relativně odolné vůči léčbě v první linii. V obou léčebných ramenech měli pacienti, u kterých se vyskytla neutropenie, delší přežití než ti, u kterých se neutropenie nevyskytla. Nárůst mediánu OS mezi pacienty s neutropenií jakéhokoli stupně byl vyšší u ramene s přípravkem Cyramza plus FOLFIRI (16,1 měsíce) ve srovnání s pacienty v rameni s placebem (12,6 měsíce). Medián OS byl u pacientů bez výskytu neutropenie 10,7 měsíce v obou ramenech.
Tabulka 12: Souhrn údajů o účinnosti - ITT populace
|
Cyramza plus |
Placebo plus | |
|
FOLFIRI |
FOLFIRI | |
|
N=536 |
N=536 | |
|
Celkové přežití, měsíce | ||
|
Medián (95% CI) |
13,3 (12,4, 14,5) |
11,7 (10,8, 12,7) |
|
Poměr rizik (95% CI) |
0,84 (0,73, 0,98) | |
|
Stratifikovaná hodnota p dle log-rank testu |
0,022 | |
|
Přežití bez progrese, měsíce | ||
|
Medián (95% CI) |
5,7 (5,5, 6,2) |
4,5 (4,2, 5,4) |
|
Poměr rizik (95% CI) |
0,79 (0,70, 0,90) | |
|
Stratifikovaná hodnota p dle log-rank testu |
<0,001 | |
Zkratky: CI = interval spolehlivosti
Obrázek 4: Kaplan-Meierovy křivky celkového přežití u přípravku Cyramza plus FOLFIRI versus placebo plus FOLFIRI ve studii RAISE
Cyramza+FOLFIRI
Pacebo+FOLF R
Doba od randomizace (mesice)
Počet v riziku
Cyramza+FOLFIRI
497
421
345
269
95
114
P acebo+FOLF R
536
486
400
329
228
66
108
0.8
0.4
0 0
Cyramza+FOLFIRI
Placebo+FOLFIRI
Doba od randomizace (mesice)
Počet v riziku
Cyramza+FOLFIRI
Placebo+FOLFIRI
ORR bylo podobné v obou léčebných ramenech (13,4 % versus 12,5 %, ramucirumab plus FOLFIRI vs. placebo plus FOLFIRI). Četnost kontroly onemocnění (kompletní odpověď plus parciální odpověď plus stabilizace onemocnění) byla numericky vyšší u pacientů léčených v rameni s ramucirumabem plus FOLFIRI ve srovnání s ramenem placebo plus FOLFIRI (74,1 versus 68,8 %). V dotazníku EORTC QLQ-C30 uváděli pacienti v léčebném rameni ramucirumab plus FOLFIRI přechodný pokles QoL ve srovnání s pacienty v léčebném rameni placebo plus FOLFIRI ve většině škál. Po prvním měsíci léčby bylo uváděno pouze minimum rozdílů mezi rameny.
NSCLC
REVEL
Randomizovaná dvojitě zaslepená studie REVEL s přípravkem Cyramza plus docetaxel versus placebem plus docetaxel byla provedena u 1 253 pacientů s lokálně pokročilým nebo metastatickým dlaždicobuněčným nebo nedlaždicobuněčným NSCLC s progresí onemocnění v průběhu nebo po ukončení jedné léčby na bázi platiny. Primárním cílovým parametrem bylo OS. Pacienti byli randomizováni v poměru 1:1 do skupiny Cyramza plus docetaxel (n=628) nebo placebo plus docetaxel (n=625). Randomizace byla stratifikována dle geografické oblasti, pohlaví, předchozí udržovací léčby a ECOG PS. Cyramza v dávce 10 mg/kg nebo placebo a docetaxel v dávce 75 mg/m2 byly podávány ve formě intravenózní infuze v den 1 21denního cyklu. Pracoviště ve východní Asii podávala nižší dávku docetaxelu, 60 mg/m2 každých 21 dnů. Ze studie byli vyloučeni pacienti s nedávným závažným plicním, gastrointestinálním nebo pooperačním krvácením, známkami krvácení v CNS, úzkým kontaktem nádoru s hlavními dýchacími cestami nebo krevními cévami, kavitacemi uvnitř nádoru a anamnézou závažného krvácení nebo nekontrolovatelnými poruchami srážení krve. Také byli vyloučeni pacienti, kteří dostávali jakoukoliv antikoagulační léčbu a/nebo dlouhodobou léčbu nesteriodními protizánětlivými léky nebo jinými antiagregačními látkami a dále pacienti s neléčenými, klinicky nestabilními metastázami v mozku/CNS (viz bod 4.4). Užíváni kyseliny acetylsalicylové v dávkách do 325 mg/den bylo povoleno (viz bod 4.4). Do studie byl zahrnut omezený počet pacientů nekavkazských ras, především černé rasy (2,6%). Z tohoto důvodu je omezená zkušenost s kombinací
Základní demografické údaje pacientů a charakteristiky onemocnění byly obecně mezi rameny vyváženy: medián věku byl 62 let, 67 % pacientů bylo mužského pohlaví; 82 % bylo kavkazské rasy a 13 % Asiatů; ECOG PS 0 mělo 32 % pacientů, 1 mělo 67 % pacientů; 73 % pacientů mělo nedlaždicobuněčnou histologii a 26 % dlaždicobuněčnou histologii. Nejčastější předchozí léčby zahrnovaly pemetrexed (38 %), gemcitabin (25 %), taxan (24 %) a bevacizumab (14 %); 22 % pacientů absolvovalo předchozí udržovací léčbu. Medián trvání léčby doxetaxelem byl 14,1 týdne v rameni ramucirumab plus docetaxel (s mediánem 4,0 podaných infuzí) a 12,0 týdnů v rameni placebo plus docetaxel (s mediánem 4,0 podaných infuzí).
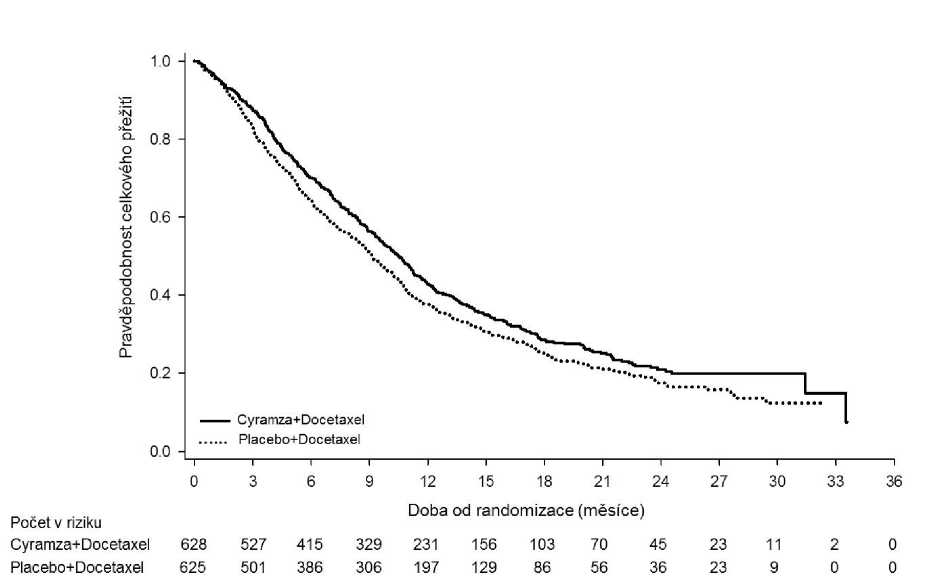
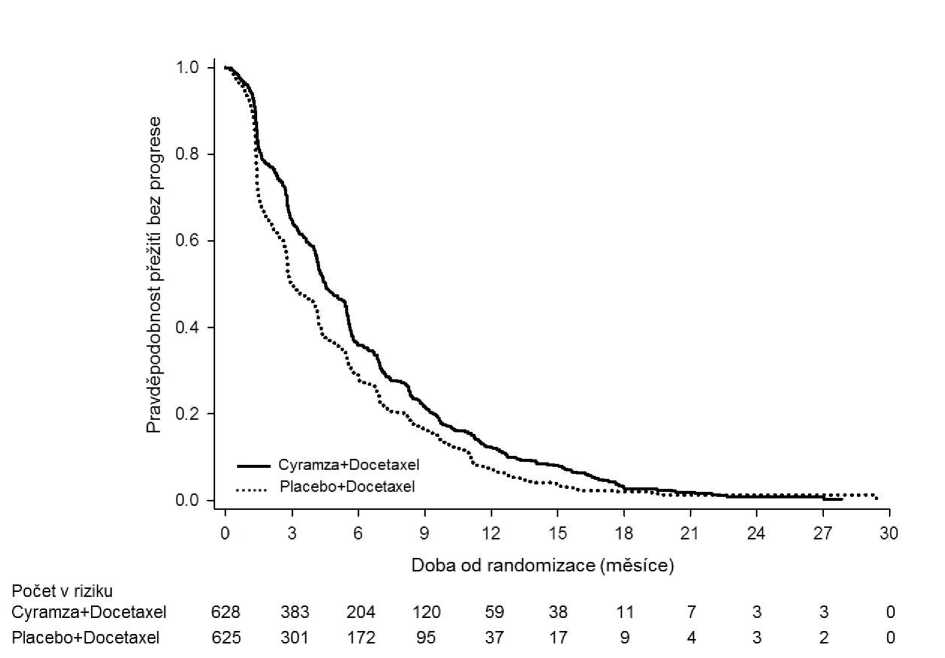
OS bylo statisticky významně zlepšeno u pacientů užívajících kombinaci Cyramza plus docetaxel ve srovnání s pacienty užívajícími placebo plus docetaxel (HR 0,857; 95% CI: 0,751 až 0,979; p=0,024). Došlo k nárůstu mediánu přežití o 1,4 měsíce v prospěch ramene Cyramza plus docetaxel: 10,5 měsíce v rameni Cyramza plus docetaxel a 9,1 měsíce v rameni placebo plus docetaxel. PFS bylo statisticky významně lepší u pacientů léčených kombinací Cyramza plus docetaxel ve srovnání s pacienty užívajícími placebo plus docetaxel (HR 0,762; 95% CI: 0,677 až 0,859; p<0,001). Došlo k nárůstu mediánu PFS o 1,5 měsíce v prospěch ramene Cyramza plus docetaxel: 4,5 měsíce v rameni Cyramza plus docetaxel a 3 měsíce v rameni placebo plus docetaxel. ORR byla významně lepší u pacientů léčených kombinací Cyramza plus docetaxel ve srovnání s pacienty užívajícími placebo plus docetaxel (22,9 % vs 13,6 %, p<0,001). Primární analýza QoL prokázala podobnou dobu do zhoršení všech skóre škály příznaků rakoviny plic (LCSS, Lung Cancer Symptom Scale) mezi rameny léčby. Konzistentní zlepšení (ramucirumab plus docetaxel vs. placebo plus docetaxel) bylo pozorováno v důležitých podskupinách PFS a OS. Výsledky podskupiny OS zahrnovaly následující: nedlaždicobuněčná histologie (HR 0,83; 95% CI: 0,71 až 0,97; medián OS [mOS]: 11,1 vs 9,7 měsíce) a dlaždicobuněčná histologie (HR 0,88; 95% CI: 0,69 až 1,13; mOS: 9,5 vs 8,2 měsíce); pacienti s předchozí udržovací léčbou (HR 0,69; 95% CI: 0,51 až 0,93; mOS: 14,4 vs 10,4 měsíce); doba od zahájení předchozí léčby <9 měsíců (HR 0,75; 95% CI: 0,64 až 0,88; mOS: 9,3 vs 7,0 měsíce); pacienti ve věku <65 let (HR 0,74, 95% CI: 0,62 až 0,87; mOS: 11,3 vs 8,9 měsíce). U pacientů, kteří dostávali ramucirumab s docetaxelem k léčbě pokročilého NSCLC s progresí onemocnění po chemoterapii na bázi platiny byl pozorován trend k nižší účinnost s rostoucím věkem (viz bod 5.1). Nebyly pozorovány žádné rozdíly v účinnosti léčby mezi léčebnými rameny v podskupině pacientů ve věku >65 let (OS HR 1,10, 95% CI: 0,89 až 1,36; medián OS [mOS]: 9,2 vs 9,3 měsíce, viz bod 4.4), pacientů předléčených taxany (HR 0,81; 95% CI: 0,62 až 1,07; mOS 10,8 vs 10,4 měsíce) a těmi, u kterých byla doba od zahájení předchozí léčby >9 měsíců (HR 0,95; 95% CI: 0,75 až 1,2; mOS: 13,7 vs 13,3 měsíce). Výsledky účinnosti jsou uvedeny v tabulce 13.
Tabulka 13: Souhrn údajů o účinnosti - populace s léčebným záměrem (ITT)
|
Cyramza plus docetaxel N=536 |
Placebo plus docetaxel N=536 | |
|
Celkové přežití, měsíce | ||
|
Medián-měsíce (95% CI) |
10,5 (9,5, 11,2) |
9,1 (8,4, 10,0) |
|
Poměr rizik (95% CI) |
0,857 (0,751, 0,979) | |
|
Stratifikovaná hodnota p dle log-rank testu |
0,024 | |
|
Přežití bez progrese, měsíce | ||
|
Medián (95% CI) |
4,5 (4,2, 5,4) |
3,0 (2,8, 3,9) |
|
Poměr rizik (95% CI) |
0,762 (0,677, 0,859) | |
|
Stratifikovaná hodnota p dle log-rank testu |
<0,001 | |
|
Podíl pacientů s objektivní odpovědí na léčbu (CR + PR) | ||
|
Podíl - procenta (95% CI) |
22,9 (19,7, 26,4) |
13,6 (11,0, 16,5) |
|
Stratifikovaná hodnota p dle CMH |
<0,001 | |
Obrázek 6: Kaplan-Meierovy křivky celkového přežití u přípravku Cyramza plus docetaxel versus placebo plus docetaxel ve studii REVEL
Obrázek 7: Kaplan-Meierovy křivky přežití bez progrese u přípravku Cyramza plus docetaxel versus placebo plus docetaxel ve studii REVEL
Imunogenita
U pacientů ve dvou studiích 3. fáze, RAINBOW a REGARD, byla ve více časových okamžicích hodnocena přítomnost protilátek proti léku (ADA). Byly testovány vzorky od 956 pacientů: 527 pacientů léčených ramucirumabem a 429 pacientů s kontrolní léčbou. ADA se vytvořily u jedenácti (2,2 %) pacientů léčených ramucirumabem a u dvou (0,5 %) pacientů s kontrolní léčbou. U žádného pacienta s ADA nedošlo k IRR. Žádný pacient neměl neutralizační protilátky proti ramucirumabu. Pro zhodnocení vlivu ADA na účinnost a bezpečnost ramucirumabu není dostatek údajů.
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Cyramza u všech podskupin pediatrické populace s adenokarcinomem žaludku, adenokarcinomem kolon a rekta a karcinomem plic (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Při režimu dávkování 8 mg/kg každé 2 týdny byl v séru pacientů s pokročilým karcinomem žaludku před podáním čtvrté, resp. sedmé dávky ramucirumabu v monoterapii geometrický medián Cmin u ramucirumabu 49,5 pg/ml (rozmezí 6,3-228 pg/ml), resp. 74,4 pg/ml (rozmezí 13,8-234 pg/ml).
Po dávkovacím režimu 8 mg/kg ramucirumabu každé 2 týdny v kombinaci s FOLFIRI byly v séru u pacientů s mCRC před podáním třetí a páté dávky geometrické průměry ramucirumabu Cmin 46,3 pg/ml (rozsah 7,7-119 pg/ml) a 65,1 pg/ml (rozsah 14,5-205 pg/ml).
Při režimu dávkování ramucirumabu 10 mg/kg každé 3 týdny byl v séru pacientů s NSCLC před podáním třetí, resp. páté dávky ramucirumabu v kombinaci s docetaxelem geometrický medián Cmin u ramucirumabu 28,3 pg/ml (rozmezí 2,5-108 pg/ml), resp. 38,4 pg/ml (rozmezí 3,1-128 pg/ml).
Absorpce
Cyramza se podává v intravenózní infuzi. Nebyly provedeny žádné studie s jinou cestou podání. Distribuce
Podle populačního farmakokinetického přístupu (PopPK) byl průměrný (% variační koeficient [CV%]) distribuční objem ramucirumabu v ustáleném stavu 5,4 l (15 %).
Biotransformace
Metabolismus ramucirumabu nebyl hodnocen. Protilátky se odbourávají převážně katabolicky. Vylučování
Podle PopPK byla průměrná (CV%) clearance ramucirumabu 0,015 l/hod (30 %) a průměrný poločas byl 14 dní (20 %).
Závislost času a dávky
Ve farmakokinetice ramucirumabu nebyla jasná odchylka od proporcionality dávky od 6 mg/kg po 20 mg/kg. U ramucirumabu byl při podávání každé 2 týdny pozorován akumulační koeficient 1,5. Na základě simulací využívajících model PopPK by mělo být ustáleného stavu dosaženo do šesté dávky.
Starší pacienti
Na základě PopPK nebyl rozdíl v expozici ramucirumabu u pacientů ve věku > 65 let v porovnání s pacienty ve věku < 65 let.
Porucha funkce ledvin
Nebyly provedeny žádné formální studie, které by hodnotily vliv poruchy funkce ledvin na farmakokinetiku ramucirumabu. Podle PopPK byla expozice ramucirumabu u pacientů s mírnou poruchou funkce ledvin (clearance kreatininu [CrCl] >60 až <90 ml/min), středním stupněm poruchy funkce ledvin (CrCl >30 až <60 ml/min) nebo těžkou poruchou funkce ledvin (CrCl 15 až 29 ml/min) podobná jako u pacientů s normální funkcí ledvin (CrCl >90 ml/min).
Porucha funkce jater
Nebyly provedeny žádné formální studie, které by hodnotily vliv poruchy jaterních funkcí na farmakokinetiku ramucirumabu. Podle PopPK byla expozice ramucirumabu u pacientů s mírnou poruchou jaterních funkcí (celkový bilirubin 1,0-1,5 násobek horní hranice normálu (ULN) a jakákoli hladina AST nebo celkový bilirubin <1,0 ULN a AST > ULN) nebo středním stupněm poruchy funkce jater (celkový bilirubin > 1,5-3,0 ULN a jakákoli hladina AST) podobná jako u pacientů s normální funkcí jater (celkový bilirubin a AST < ULN). Ramucirumab nebyl hodnocen u pacientů se závažnou poruchou jaterních funkcí (celkový bilirubin > 3,0 násobek ULN a jakákoliv hodnota AST).
Další zvláštní populace
Podle PopPK bylo zjištěno, že stav ramucirumabu v organismu není ovlivňován následujícími nezávislými proměnnými veličinami: věkem, pohlavím, rasou, tělesnou hmotností a hladinou albuminu.
Vztah mezi expozicí a odpovědí:
Účinnost
Analýzy expozice-odpověď ukázaly, že účinnost v pivotních studiích korelovala s expozicí ramucirumabu. Účinnost měřená podle zlepšení OS a PFS souvisela se zvyšující se expozicí ramucirumabu v rozmezí dosaženém při podávání ramucirumabu 8 mg/kg každé 2 týdny a ramucirumabu 10 mg/kg podaného každé 3 týdny.
Bezpečnost
Ve studii RAINBOW byla s vyšší expozicí ramucirumabu zvýšena incidence hypertenze, neutropenie a leukopenie >3. stupně.
Ve studii RAISE se s vyšší s expozicí ramucirumabu zvýšila incidence neutropenie stupně >3.
Ve studii REVEL se s vyšší expozicí ramucirumabu zvýšila incidence febrilní neutropenie a hypertenze >3. stupně.
5.3 Předklinické údaje vztahující se k bezpečnosti
Nebyly provedeny žádné studie se zvířaty, které by hodnotily kancerogenní nebo genotoxický potenciál ramucirumabu.
Cílovými orgány identifikovanými ve studiích toxicity s opakovanými dávkami u makaků jávských byly ledviny (glomerulonefritida), kost (ztluštění a abnormální endochondrální osifikace epifyzeální růstové ploténky) samičí reprodukční orgány (snížená hmotnost ovarií a dělohy). V několika orgánech byl zjištěn minimální stupeň zánětu a/nebo infiltrace mononukleáry.
Studie reprodukční toxicity s ramucirumabem nebyly provedeny, nicméně podle zvířecích modelů existuje souvislost mezi angiogenezí, VEGF a VEGF receptorem 2 a kritickými aspekty reprodukce u samic, embryofetálním vývojem a postnatálním vývojem. Vzhledem k mechanismu účinku ramucirumabu je pravděpodobné, že u zvířat bude ramucirumab inhibovat angiogenezi, což povede k nežádoucím účinkům na fertilitu (ovulaci), vývoj placenty, vývoj plodu a postnatální vývoj.
Jediná dávka ramucirumabu nenarušila hojení rány u opic při použití modelu s incizí v celé tloušťce.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Histidin
Histidin-hydrochlorid Chlorid sodný Glycin (E640)
Polysorbát 80 (E433)
Voda na injekci
6.2 Inkompatibility
Cyramza se nemá podávat ani míchat s roztoky glukózy.
Tento léčivý přípravek se nesmí míchat s jinými léčivými přípravky, kromě přípravků uvedených v bodu 6.6.
6.3 Doba použitelnosti
Neotevřená injekční lavička 3 roky.
Po naředění
Při přípravě podle pokynů neobsahuje roztok přípravku Cyramza žádné antimikrobiální konzervační látky.
Chemická a fyzikální stabilita přípravku Cyramza v injekčním roztoku chloridu sodného 9 mg/ml (0,9 %) při použití byla prokázána na dobu 24 hodin při teplotě 2 °C až 8 °C nebo na dobu 4 hodin při teplotě 25 °C. Z mikrobiologického hlediska má být přípravek použít okamžitě. Není-li použit okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou v odpovědnosti uživatele, normálně by doba neměla být delší než 24 hodin při 2 °C až 8 °C, pokud naředění nebylo provedeno v kontrolovaných a validovaných aseptických podmínkách.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
Podmínky uchovávání tohoto léčivého přípravku po jeho naředění jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
10 ml roztoku v injekční lahvičce (sklo třídy I) s chlorbutylovou pryžovou zátkou, hliníkovou folií a polypropylenovým uzávěrem.
50 ml roztoku v injekční lahvičce (sklo třídy I) s chlorbutylovou pryžovou zátkou, hliníkovou folií a polypropylenovým uzávěrem.
Balení 1 injekční lahvička 10 ml.
Balení 2 injekční lahvičky 10 ml.
Balení 1 injekční lahvička 50 ml.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Injekční lahvičku neprotřepávejte.
Připravte infuzní roztok za použití aseptické metody, aby byla zajištěna sterilita připraveného roztoku.
Každá injekční lahvička je určena pouze pro jedno použití. Před naředěním prohlédněte obsah injekčních lahviček, zda neobsahují částice nebo zabarvení (koncentrát pro infuzní roztok má být čirý až mírně opalizující a bezbarvý až lehce nažloutlý bez viditelných částic). Pokud zjistíte přítomnost částic nebo zabarvení, injekční lahvičku zlikvidujte.
Vypočítejte dávku a objem ramucirumabu potřebné k přípravě infuzního roztoku. Injekční lahvičky obsahují buď 100 mg nebo 500 mg ramucirumabu v roztoku 10 mg/ml. Jako ředicí roztok použijte výhradně injekční roztok chloridu sodného 9 mg/ml (0,9%).
V případě použití předplněných balení k intravenózní infuzi
Na základě vypočítaného objemu ramucirumabu odeberte odpovídající objem injekčního roztoku chloridu sodného 9 mg/ml (0,9 %) z předplněného 250ml balení pro intravenózní aplikaci. Asepticky přeneste vypočítaný objem ramucirumabu do balení pro intravenózní aplikaci. Konečný celkový objem v balení by měl být 250 ml. Obal je třeba jemně převrátit, aby se obsah dostatečně promíchal. Injekční roztok NEZMRAZUJTE ani NEPROTŘEPÁVEJTE. NEŘEĎTE jinými roztoky ani nepodávejte spolu s infuzí s jinými elektrolyty nebo léčivými přípravky.
V případě použití prázdných obalů k intravenózní infuzi
Asepticky přeneste vypočítaný objem ramucirumabu do prázdného obalu pro intravenózní infuzi. Přidejte dostatečné množství injekčního roztoku chloridu sodného 9 mg/ml (0,9 %) do obalu, aby byl konečný objem 250 ml. Obal je třeba jemně převrátit, aby se obsah dostatečně promíchal. Injekční roztok NEZMRAZUJTE ani NEPROTŘEPÁVEJTE. NEŘEĎTE s jinými roztoky ani nepodávejte spolu s infuzí s jinými elektrolyty nebo léčivými přípravky.
Parenterální léčivé přípravky je třeba před podáním vizuálně zkontrolovat, zda neobsahují částice. Pokud zjistíte přítomnost částic, infuzní roztok zlikvidujte.
Zlikvidujte veškerý zbývající objem ramucirumabu, který zůstane v injekční lahvičce, protože přípravek neobsahuje antimikrobiální konzervační látky.
Podejte pomocí infuzní pumpy. K infuzi je nutné použít samostatnou infuzní linku s 0,22 mikronovým filtrem šetrným vůči proteinům a na konci infuze je nutné linku vypláchnout injekčním roztokem chloridu sodného 9 mg/ml (0,9 %).
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Eli Lilly Nederland B.V.
Papendorpseweg 83 3528 BJ Utrecht Nizozemsko
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/14/957/001-003
9. DATUM REGISTRACE/ PRODLOUŽENÍ REGISTRACE
Datum registrace: 19. prosince 2014
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce biologické léčivé látky
ImClone Systems LLC 33 ImClone Drive,
Branchburg New Jersey NJ 08876 USA
Eli Lilly S.A.
Dunderrow Kinsale County Cork Iresko
Název a adresa výrobce odpovědného za propouštění šarží Lilly, S.A.
Avda de la Industria, 30 Alcobendas 28108 Madrid Španělsko
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
• Povinnost uskutečnit poregistrační opatření
Držitel rozhodnutí o registraci uskuteční v daném termínu níže uvedená opatření:
|
Popis |
Termín splnění |
|
Držitel rozhodnutí o registraci předloží výsledky z randomizované studie farmakokinetiky a bezpečnosti dávkových rozmezí ramucirumabu v monoterapii (14T-MC-JVDB). Tato studie fáze 2 bude vyhodnocovat farmakokinetiku a bezpečnost různých dávkovacích schémat ramucirumabu včetně dávek vyšších než schválená dávka 8mg/kg každé 2 týdny ve druhé linii léčby adenokarcinomu žaludku. |
01/04/2017 (PK výsledky) 01/04/2018 (Závěrečná zpráva KH a výsledky bezpečnosti) |
|
Poregistrační studie účinnosti (PAES): za účelem výzkumu potenciální korelace mezi měřenými biomarkery (VEGF-C, VEGF-D, sVEGFR1, sVEGFR2 a sVEGFR3 z plazmy, VEGFR2 IHC, další mutace KRAS, NRAS a BRAF) a celkovou účinností (PFS, OS) předloží držitel rozhodnutí o registraci výsledky testování biomarkerů z populace zkoumané v rámci translačního výzkumu ve studii RAISE. Korelace mezi VEGF-C, VEGF-D, sVEGFR1, sVEGFR2 a sVEGFR3 z plazmy, VEGFR2 IHC bude předložena do Korelace s dalšími mutacemi KRAS, NRAS and BRAF bude předložena do |
30. června 2016 30. září 2016 |
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Cyramza 10 mg/ml koncentrát pro infuzní roztok ramucirumabum
2. OBSAH LÉČIVÉ LÁTKY
1 ml koncentrátu obsahuje ramucirumabum 10 mg
3. SEZNAM POMOCNÝCH LÁTEK
Pomocné látky: histidin, histidin-hydrochlorid, chlorid sodný, glycin, polysorbát 80, voda pro injekci
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Koncentrát pro infuzní roztok
100 mg/10 ml
1 injekční lahvička
2 injekční lahvičky
5. ZPŮSOB A CESTA
Pro intravenózní podání po naředění.
Pouze pro jednorázové podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
Neprotřepávejte.
8. POUŽITELNOST
EXP
Uchovávejte v chladničce.
Chraňte před mrazem.
Uchovávejte lahvičku ve vnějším obalu, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Eli Lilly Nederland B.V. Papendorpseweg 83 3528 BJ Utrecht Nizozemsko
12 REGISTRAČNÍ ČÍSLO
EU/1/14/957/001 - 1 injekční lahvička 10 ml EU/1/14/957/002 - 2 injekční lahvičky 10 ml
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
Nevyžaduje se - odůvodnění přijato
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Cyramza 10 mg/ml koncentrát pro infuzní roztok ramucirumabum
2. OBSAH LÉČIVÉ LÁTKY
1 ml koncentrátu obsahuje ramucirumabum 10 mg
3. SEZNAM POMOCNÝCH LÁTEK
Pomocné látky: histidin, histidin-hydrochlorid, chlorid sodný, glycin, polysorbát 80, voda pro injekci
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Koncentrát pro infuzní roztok
500 mg/50 ml 1 injekční lahvička
5. ZPŮSOB A CESTA
Pro intravenózní podání po naředění.
Pouze pro jednorázové podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
Neprotřepávejte.
8. POUŽITELNOST
EXP
Uchovávejte v chladničce.
Chraňte před mrazem.
Uchovávejte lahvičku ve vnějším obalu, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Eli Lilly Nederland B.V. Papendorpseweg 83 3528 BJ Utrecht Nizozemsko
12 REGISTRAČNÍ ČÍSLO
EU/1/14/957/003
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Nevyžaduje se - odůvodnění přijato
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA PODÁNÍ
Cyramza 10 mg/ml koncentrát pro infuzní roztok
ramucirumabum
i.v. podání po naředění
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci.
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
100 mg/10 ml
6. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA PODÁNÍ
Cyramza 10 mg/ml koncentrát pro infuzní roztok
ramucirumabum
i.v. podání po naředění
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci.
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
500 mg/50ml
6. JINÉ
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro uživatele
Cyramza 10 mg/ml koncentrát pro infuzní roztok
ramucirumabum
"VTento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně tuto příbalovou informaci dříve, než je Vám tento přípravek podán, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou v této příbalové informaci uvedeny. Viz kapitola 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Cyramza a k čemu se používá
2. Čemu musíte věnovat pozornost, než Vám bude přípravek Cyramza podán
3. Jak Vám bude přípravek Cyramza podáván
4. Možné nežádoucí účinky
5. Jak přípravek Cyramza uchovávat
6. Obsah balení a další informace
1. Co je přípravek Cyramza a k čemu se používá
Přípravek Cyramza je protinádorový přípravek, který obsahuje jako účinnou látku ramucirumab, což je monoklonální protilátka. Je to speciální bílkovina, která rozpoznává a váže se na jinou bílkovinu v cévách nazývanou VEGF receptor 2. Tento receptor je nutný pro růst nových cév. Také nádor potřebuje nové cévy, aby mohl růst. Navázáním na VEGF receptor 2 a jeho zablokováním tento lék narušuje krevní zásobení nádorových buněk.
Přípravek Cyramza se podává v kombinaci s paklitaxelem, jiným protinádorovým lékem, k léčbě pokročilé rakoviny žaludku (nebo rakoviny oblasti přechodu jícnu do žaludku) u dospělých, u nichž došlo ke zhoršení choroby po předchozí protinádorové léčbě.
Přípravek Cyramza se používá samostatně k léčbě pokročilé rakoviny žaludku (nebo rakoviny oblasti přechodu jícnu do žaludku) u dospělých, u nichž došlo ke zhoršení choroby po předchozí chemoterapii obsahující platinu nebo fluoropyrimidin a pro které není léčba v kombinaci s paklitaxelem vhodná.
Cyramza se používá k léčbě pokročilé rakoviny tračníku a konečníku (části tlustého střeva) u dospělých. Podává se s jinými přípravky nazývanými „chemoterapie FOLFIRI“, kam patří „fluoruracil“, „kyselina folinová“ a „irinotekan“.
Přípravek Cyramza se podává v kombinaci s docetaxelem, jiným protinádorovým lékem, k léčbě pokročilé rakoviny plic u dospělých, u nichž došlo ke zhoršení choroby po předchozí protinádorové léčbě.
2. Čemu musíte věnovat pozornost, než Vám bude přípravek Cyramza podán Přípravek Cyramza nesmíte dostat
- jestliže j ste alergický(á) na ramucirumab nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
jestliže u Vás bylo rentgenovým vyšetřením zjištěno, že rakovina plic obsahuje dutinu nebo se rakovina plic nachází v blízkosti velkých cév.
Upozornění a opatření
Před podáním přípravku Cyramza informujte svého lékaře nebo zdravotní sestru, pokud:
- máte jakýkoliv stav, který zvyšuje riziko krvácení. Rovněž řekněte lékaři, pokud užíváte léčivé přípravky, které můžou zvyšovat riziko krvácení, nebo které ovlivňují srážení krve. V takovém případě vám bude lékař pravidelně provádět krevní vyšetření a sledovat tak riziko krvácení.
- máte rakovinu plic a měl(a) jste nedávné krvácení z plic (vykašlávání jasně červené krve) nebo pokud pravidelně užíváte nesteroidní protizánětlivé léky, nebo léky ovlivňující srážlivost krve.
- máte vysoký krevní tlak. Cyramza může zvyšovat výskyt vysokého krevního tlaku. Lékař se ujistí, že pokud již máte krevní tlak zvýšený, je před zahájením podávání přípravku Cyramza kompenzován. Lékař bude váš krevní tlak během léčby přípravkem Cyramza sledovat a podle potřeby upraví podávání léčivého přípravku ke snížení krevního tlaku. Je možné, že bude nutné léčbu přípravkem Cyramza dočasně přerušit, dokud nebude váš krevní tlak kompenzován léčivými přípravky, nebo léčbu trvale ukončit, pokud nebude možné krevní tlak odpovídajícím způsobem kompenzovat.
- je u vás plánována operace, pokud jste nedávno prodělal(a) operaci nebo pokud se vám špatně hojí rána po operaci. Cyramza může zvýšit riziko problémů s hojením ran. Neměl(a) byste užívat přípravek Cyramza nejméně 4 týdny před plánovanou operací a lékař následně rozhodne, kdy léčbu znovu zahájit. Pokud se vám během léčby špatně hojí rána, bude léčba přípravkem Cyramza přerušena, dokud se rána zcela nezhojí.
- máte závažné onemocnění jater (‘cirhózu’) a související stavy, např. nadměrné hromadění tekutiny v břiše (‘ascites’). Lékař s vámi probere, zda možný přínos léčby podle jeho úsudku převáží možná rizika vyplývající z léčby.
- máte závažné problémy s ledvinami. K dispozici jsou pouze omezené údaje o užívání přípravku Cyramza u pacientů s těžkou poruchou funkce ledvin.
Ihned lékaři nebo zdravotní sestře sdělte, pokud se během léčby přípravkem Cyramza nebo kdykoli
po léčbě objeví některý z následujících stavů (i když si tím nebudete zcela jistý/á):
- ucpání tepen krevní sraženinou (‘arteriální tromboembolické příhody’):
Cyramza může vyvolat tvorbu krevních sraženin v tepnách. Krevní sraženiny v tepně můžou vyvolat závažné stavy včetně srdečního záchvatu nebo cévní mozkové příhody. Mezi možné příznaky srdečního záchvatu patří bolest na hrudi nebo pocit tíže na hrudi. Mezi možné příznaky cévní mozkové příhody patří náhlá ztráta citlivosti nebo slabost v paži, dolní končetině a na obličeji, pocit zmatenosti, obtíže s mluvením nebo porozuměním ostatním, náhlé problémy s chůzí nebo ztráta rovnováhy nebo koordinace nebo náhlá závrať. Pokud se vám v tepnách vytvoří krevní sraženiny, bude přípravek Cyramza trvale vysazen.
- protržení stěny střeva (‘gastrointestinální perforace’): Cyramza může zvýšit riziko protržení střevní stěny. Mezi příznaky patří silná bolest břicha, nevolnost (zvracení), horečka nebo zimnice; Pokud u vás dojde k protržení střevní stěny, bude přípravek Cyramza trvale vysazen;
- závažné krvácení: Cyramza může zvýšit riziko závažného krvácení. Mezi možné příznaky patří: extrémní únava, slabost, závratě nebo změna barvy stolice. Pokud u vás dojde k závažnému krvácení, bude přípravek Cyramza trvale vysazen.
- reakce související s podáním infuze: Vzhledem k tomu, že se Cyramza podává po kapkách v nitrožilní infuzi, může během léčby dojít k reakcím souvisejícím s podáním infuze (viz bod 3). Během infuze u vás bude lékař nebo zdravotní sestra nežádoucí účinky kontrolovat. Mezi
možné příznaky patří: zvýšené svalové napětí, bolest zad, bolest na hrudi a/nebo svírání na hrudi, zimnice, návaly, obtížné dýchání, sípání a brnění nebo necitlivost v rukou a nohou. V závažných případech můžou příznaky zahrnovat dušnost vyvolanou zúžením dýchacích cest, rychlejší tlukot srdce a pocit na omdlení. Pokud u vás dojde k závažné reakci související s infuzí, bude přípravek Cyramza trvale vysazen;
vytvoření abnormálních trubkovitých spojů nebo průchodů uvnitř těla (‘píštěl’): Přípravek Cyramza může zvyšovat riziko vytvoření abnormálních spojů nebo průchodů uvnitř těla mezi vnitřními orgány a kůží nebo jinými tkáněmi. Pokud se vám vytvoří píštěl, bude přípravek Cyramza trvale vysazen.
abnormální test moči (,proteinurie‘): Přípravek Cyramza může zvyšovat riziko rozvoje nebo zhoršení abnormálních hladin proteinu (bílkoviny) v moči. Léčbu přípravkem Cyramza může být zapotřebí dočasně zastavit, dokud se hladiny bílkovin v moči nesníží a léčba je potom znovu zahájena nižšími dávkami, nebo je léčba zastavena trvale, pokud se hladiny bílkovin dostatečně nesníží.
zánět úst (,stomatitida‘): Podávání přípravku Cyramza v kombinaci s chemoterapií může zvyšovat riziko rozvoje zánětu úst. Příznaky můžou zahrnovat pálení v ústech, vznik vředů, puchýřů nebo otoků. Váš lékař vám může předepsat léčbu, která vám s těmito příznaky pomůže.
horečka nebo infekce: V průběhu léčby se u Vás může vyvinout teplota 38°C nebo vyšší (protože můžete mít méně bílých krvinek než normálně, což je velmi časté). Příznaky mohou zahrnovat pocení nebo další známky infekce, jako je bolest hlavy, bolest končetin nebo snížená chuť k jídlu. Infekce (sepse) může být závažná a může vést k úmrtí.
starší lidé s rakovinou plic: Váš lékař pečlivě zváží léčbu, která je pro Vás nejvhodnější.
Děti a dospívající
Přípravek Cyramza se nemá podávat pacientům do 18 let věku, protože neexistují žádné informace o tom, jak u této věkové skupiny působí.
Další léčivé přípravky a Cyramza
Informujte svého lékaře o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat. Patří sem i léky, které lze získat bez lékařského předpisu a rostlinné přípravky.
Těhotenství, kojení a plodnost
Před zahájením léčby musíte informovat svého lékaře, pokud jste těhotná nebo kojíte, myslíte si, že byste mohla být těhotná nebo plánujete otěhotnět. Během užívání léčivého přípravku a nejméně 3 měsíce po poslední dávce přípravku Cyramza byste neměla otěhotnět. Promluvte si s lékařem o tom, jaká antikoncepce je pro vás nejvhodnější.
Vzhledem k tomu že Cyramza brání vytváření nových krevních cév, může snižovat pravděpodobnost toho, že otěhotníte nebo že těhotenství udržíte. Rovněž může poškodit vaše nenarozené dítě. Tento léčivý přípravek byste neměla užívat během těhotenství. Pokud během léčby přípravkem Cyramza otěhotníte, lékař s vámi probere, zda je pro vás přínos léčby větší než možné riziko pro vaši osobu a vaše nenarozené dítě.
Není známo, zda tento léčivý přípravek přechází do mateřského mléka a zda může ovlivnit kojené dítě. Při léčbě přípravkem Cyramza a alespoň 3 měsíce poté, co dostanete poslední dávku, byste proto své dítě neměla kojit.
Řízení dopravních prostředků a obsluha strojů
Není známo, zda může Cyramza ovlivnit vaši schopnost řídit nebo obsluhovat stroje. Pokud se u vás objeví příznaky, které narušují schopnost soustředění a reakce, neřiďte ani neobsluhujte stroje, dokud tento účinek neodezní.
Cyramza obsahuje sodík
Tento léčivý přípravek obsahuje chlorid sodný.
Jedna 10ml injekční lahvička obsahuje přibližně 17 mg sodíku (méně než 1 mmol). Jedna 50ml injekční lahvička obsahuje přibližně 85 mg sodíku (3,7 mmol).
Nutno vzít v úvahu u pacientů na dietě s nízkým obsahem sodíku.
3. Jak Vám bude přípravek Cyramza podáván
Tuto protinádorovou léčbu vám bude podávat lékař nebo zdravotní sestra.
Dávkování a četnost podávání
Přesné množství přípravku Cyramza potřebné k léčbě vašeho onemocnění vypočítá lékař nebo nemocniční lékárník a závisí na vaší tělesné hmotnosti.
Doporučená dávka přípravku Cyramza k léčbě rakoviny žaludku a léčbě pokročilé rakoviny tračníku a konečníku je 8 mg na kilogram vaší tělesné hmotnosti jednou za 2 týdny.
Doporučená dávka přípravku Cyramza k léčbě rakoviny plic je 10 mg na kilogram vaší tělesné hmotnosti jednou za 3 týdny.
Počet infuzí, které dostanete, bude záviset na tom, jak budete na léčbu reagovat. Lékař to s vámi probere.
Způsob a cesta podání
Cyramza je koncentrát pro infuzní roztok (takzvaný "sterilní koncentrát"). Nemocniční lékárník, zdravotní sestra nebo lékař před použitím obsah injekční lahvičky rozředí v roztoku chloridu sodného 9 mg/ml (0,9%). Tento léčivý přípravek se podává po kapkách v infuzi po dobu přibližně 60 minut.
Premedikace
Před podáním přípravku Cyramza můžete dostat jiný léčivý přípravek ke snížení rizika reakce související s podáním infuze. Pokud u vás během léčby přípravkem Cyramza dojde k reakci související s podáním infuze, dostanete před každou další infuzí premedikaci.
Úpravy dávkování
Během každé infuze u vás bude lékař nebo zdravotní sestra kontrolovat nežádoucí účinky.
Pokud se u vás během léčby objeví reakce související s podáním infuze, bude doba, po kterou budete dostávat infuzi, prodloužena po zbytek dané infuze i u všech dalších infuzí.
Během léčby vám bude pravidelně kontrolováno množství bílkoviny v moči. V závislosti na zjištěné hladině bílkovin může být přípravek Cyramza dočasně vysazen. Jakmile se hladina bílkovin v moči vrátí na určitou úroveň, je možné léčbu znovu zahájit nižší dávkou.
Léčba přípravkem Cyramza bude dočasně vysazena v případě, že:
- se u vás objeví vysoký krevní tlak, dokud nebude kompenzován léčbou na snížení krevního tlaku,
- se u vás objeví problémy s hojením ran, dokud se rána nezhojí
- máte podstoupit plánovanou operaci, a to čtyři týdny před výkonem
Léčba přípravkem Cyramza bude trvale ukončena v případě, že:
- se vám v tepnách vytvoří krevní sraženiny
- u vás dojde k protržení střevní stěny
- u vás dojde k závažnému krvácení
- u vás dojde k závažné reakci související s podáním infuze
- se u vás objeví vysoký krevní tlak, který nelze kompenzovat léčbou
- budete vylučovat více než určité množství bílkovin do moči nebo pokud se u vás rozvine závažné onemocnění ledvin (nefrotický syndrom)
- pokud se vám vytvoří abnormálních spoje nebo průchody uvnitř těla mezi vnitřními orgány a kůží nebo jinými tkáněmi (píštěl)
Při podávání přípravku Cyramza v kombinaci s paklitaxelem nebo docetaxelem
Paklitaxel a docetaxel se rovněž podávají po kapkách do žíly (nitrožilní infuze) po dobu přibližně 60 minut. Pokud budete dostávat ve stejný den přípravek Cyramza v kombinaci buď s paklitaxelem nebo docetaxelem, dostanete přípravek Cyramza jako první.
Množství paklitaxelu nebo docetaxelu, které potřebujete, závisí na povrchu vašeho těla. Lékař nebo nemocniční lékárník spočítá váš tělesný povrch podle vaší výšky a tělesné hmotnosti a určí správnou dávku. Doporučená dávka paklitaxelu je 80 mg na každý čtvereční metr (m2) vašeho tělesného povrchu jednou týdně po dobu 3 týdnů a poté následuje 1 týden bez léčby.
Doporučená dávka docetaxelu je 75 mg na každý čtvereční metr (m2) vašeho tělesného povrchu jednou každé 3 týdny. Jste-li původem z Východní Asie, může Vám být podána snížená počáteční dávka docetaxelu, a to 60 mg na každý m2 vašeho tělesného povrchu jednou za 3 týdny.
Dříve než dostanete infuzi paklitaxelu, bude vám proveden rozbor krve ke kontrole, zda máte dostatek krvinek a zda vám dobře fungují játra.
Další informace jsou uvedeny v příbalové informaci k paklitaxelu nebo docetaxelu.
Při léčbě přípravkem Cyramza v kombinaci s FOLFIRI
Chemoterapie FOLFIRI se podává nitrožilní infuzí po dokončení infuze přípravku Cyramza. Prostudujte si příbalové informace k dalším lékům, ze kterých vaše léčba sestává, a ujistěte se, že jsou pro vás vhodné. Pokud si nejste jistý(á), zeptejte se svého lékaře, lékárníka nebo zdravotní sestry, jestli existují nějaké důvody, kvůli kterým byste tyto léky neměl(a) používat.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Ihned svého lékaře informujte, pokud u vás dojde k některému z následujících závažných nežádoucích účinků, které byly pozorovány během léčby přípravkem Cyramza (viz také Čemu musíte věnovat pozornost, než Vám bude přípravek Cyramza podán):
Časté nežádoucí účinky (mohou postihnout až 1 z 10 osob):
- protržení střevní stěny: jde o protržení stěny žaludku, tenkého nebo tlustého střeva. Mezi příznaky patří velká bolest břicha, nevolnost (zvracení), horečka nebo zimnice;
- závažné krvácení do střev: možné příznaky zahrnují extrémní únavu, slabost, závratě nebo změny barvy stolice;
- krevní sraženiny v tepnách: krevní sraženiny v tepnách můžou vyvolat srdeční záchvat nebo cévní mozkovou příhodu. Mezi možné příznaky srdečního záchvatu patří bolest na hrudi nebo pocit tíže na hrudi. Mezi možné příznaky cévní mozkové příhody patří náhlá ztráta citlivosti nebo slabost v paži, dolní končetině a na obličeji, pocit zmatenosti, obtíže s mluvením nebo porozuměním ostatním, náhlé problémy s chůzí nebo ztráta rovnováhy nebo koordinace nebo náhlá závrať.
Jestliže zaznamenáte některý z následujících dalších nežádoucích účinků, informujte svého lékaře:
Velmi časté nežádoucí účinky (mohou postihnout více než 1 z 10 osob):
- nízké počty bílých krvinek (může zvyšovat riziko infekce)
- pocit únavy nebo slabosti
- krvácení z nosu
- průjem
- zánět sliznice v ústech
- bolest břicha
- nízký počet krevních destiček (krvinky, které pomáhají srážení krve)
- vysoký krevní tlak
- otok rukou, nohou a dolních končetin z důvodu zadržování tekutiny
- bílkovina v moči (abnormální výsledek rozboru moči)
- zánět sliznic, například v zažívacím a dýchacím ústrojí
- horečka provázená nízkým počtem bílých krvinek
- začervenání, otok, necitlivost/brnění nebo bolest a/nebo olupování kůže z rukou a/nebo nohou (nazývané syndrom ruka-noha)
- nízká hladina bílkoviny nazývané albumin v krvi
Časté nežádoucí účinky (mohou postihnout až 1 z 10 osob):
- - bolest hlavy
- nízká hladina draslíku v krvi (hypokalémie), která může vyvolat svalovou slabost, záškuby nebo poruchu srdečního rytmu
- nízká hladina sodíku v krvi (hyponatrémie), která může vést k únavě a zmatenosti nebo svalovým záškubům
- vyrážka
- závažná infekce krve (sepse)
- ucpání střeva, možné příznaky zahrnují zácpu a bolest břicha
Podávání přípravku Cyramza bylo spojeno s reakcemi souvisejícími s podáním infuze.
Cyramza může vyvolat změny laboratorních testů. Z výše uvedených nežádoucích účinků se jedná o: nízký počet bílých krvinek, nízký počet krevních destiček, nízkou hladinu albuminu, draslíku nebo sodíku v krvi, přítomnost bílkoviny v moči.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou v této příbalové informaci uvedeny. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Cyramza uchovávat
Uchovávejte tento léčivý přípravek mimo dohled a dosah dětí.
Nepoužívejte tento léčivý přípravek po uplynutí doby použitelnosti uvedené na krabičce a na štítku na injekční lahvičce za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
Injekční roztok chraňte před mrazem a neprotřepávejte. Nepodávejte roztok, pokud si v něm všimnete jakýchkoliv částic nebo zabarvení.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co Cyramza obsahuje
- Léčivou látkou je ramucirumabum. Jeden ml koncentrátu pro infuzní roztok obsahuje ramucirumabum 10 mg.
- Jedna 10ml injekční lahvička obsahuje 100 mg ramucirumabu.
- Jedna 50ml injekční lahvička obsahuje 500 mg ramucirumabu.
- další složky jsou histidin, histidin-hydrochlorid, chlorid sodný, glycin (E640), polysorbát 80 (E433) a voda na injekce (viz bod 2 “Cyramza obsahuje sodík”).
Jak přípravek Cyramza vypadá a co obsahuje toto balení
Koncentrát pro infuzní roztok (nebo sterilní koncentrát) je čirý až mírně opalizující a bezbarvý až lehce nažloutlý roztok ve skleněné injekční lahvičce s pryžovou zátkou.
Cyramza je k dispozici v baleních:
- 1 injekční lahvička s 10 ml
- 2 injekční lahvičky s 10 ml
- 1 injekční lahvička s 50 ml
Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci
Eli Lilly Nederland B.V.
Papendorpseweg 83 3528 BJ Utrecht Nizozemsko
Výrobce Lilly, S.A.
Avda de la Industria, 30 Alcobendas 28108 Madrid Španělsko
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgique/Belgie/Belgien
Eli Lilly Benelux S.A./N.V.
Tél/Tel: + 32-(0)2 548 84 84
Bt^rapnn
Tn "Enn Hunn HegepnaHg" E.B. - Etnrapna Ten. + 359 2 491 41 40
Česká republika ELI LILLY ČR, s.r.o.
Tel: + 420 234 664 111
Danmark
Eli Lilly Danmark A/S Tlf: +45 45 26 60 00
Deutschland
Lilly Deutschland GmbH Tel. + 49-(0) 6172 273 2222
Eesti
Eli Lilly Holdings Limited Eesti filiaal Tel: +372 6 817 280
EkXáSa
OAPMAIEPB-AIAAY A.E.B.E.
Tr|^: +30 210 629 4600
Espaňa Lilly S.A.
Tel: + 34-91 663 50 00 France
Lilly France SAS
Tél: +33-(0) 1 55 49 34 34
Hrvatska
Eli Lilly Hrvatska d.o.o.
Tel: +385 1 2350 999
Ireland
Eli Lilly and Company (Ireland) Limited Tel: + 353-(0) 1 661 4377
Ísland
Icepharma hf.
Sími + 354 540 8000
Italia
Eli Lilly Italia S.p.A.
Tel: + 39- 055 42571
Lietuva
Eli Lilly Holdings Limited atstovybé Tel. +370 (5) 2649600
Luxembourg/Luxemburg
Eli Lilly Benelux S.A./N.V.
Tél/Tel: + 32-(0)2 548 84 84
Magyarország
Lilly Hungária Kft.
Tel: + 36 1 328 5100
Malta
Charles de Giorgio Ltd.
Tel: + 356 25600 500
Nederland
Eli Lilly Nederland B.V.
Tel: + 31 -(0) 30 60 25 800
Norge
Eli Lilly Norge A.S.
Tlf: + 47 22 88 18 00
Osterreich
Eli Lilly Ges.m.b.H.
Tel: + 43-(0) 1 711 780
Polska
Eli Lilly Polska Sp. z o.o.
Tel: +48 22 440 33 00
Portugal
Lilly Portugal Produtos Farmaceuticos, Lda Tel: + 351-21-4126600
Románia
Eli Lilly Románia S.R.L.
Tel: + 40 21 4023000
Slovenija
Eli Lilly farmacevtska družba, d.o.o.
Tel: +386 (0)1 580 00 10
Slovenská republika
Eli Lilly Slovakia, s.r.o.
Tel: + 421 220 663 111
Suomi/Finland
Oy Eli Lilly Finland Ab Puh/Tel: + 358-(0) 9 85 45 250
Latvija United Kingdom
Eli Lilly Holdings Limited párstavnieciba Latvija Eli Lilly and Company Limited Tel: +371 67364000 Tel: + 44-(0) 1256 315000
Tato příbalová informace byla naposledy revidována .
Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Následující informace jsou určeny pouze pro zdravotnické pracovníky:
Injekční lahvičku neprotřepávejte.
Připravte infuzní roztok za použití aseptické metody, aby byla zajištěna sterilita připraveného roztoku.
Každá injekční lahvička je určena pouze pro jedno použití. Před naředěním prohlédněte obsah injekčních laviček, zda neobsahují částice nebo zabarvení (koncentrát pro infuzní roztok má být čirý až mírně opalizující a bezbarvý až lehce nažloutlý bez viditelných částic). Pokud zjistíte přítomnost částic nebo zabarvení, injekční lahvičku zlikvidujte.
Vypočítejte dávku a objem ramucirumabu potřebné k přípravě infuzního roztoku. Injekční lahvičky obsahují buď 100 mg nebo 500 mg ramucirumabu v roztoku 10 mg/ml. Jako ředicí roztok použijte výhradně injekční roztok chloridu sodného 9 mg/ml (0,9%).
V případě použití předplněných balení k intravenózní infuzi
Na základě vypočítaného objemu ramucirumabu odeberte odpovídající objem injekčního roztoku chloridu sodného 9 mg/ml (0,9 %) z předplněného 250ml balení pro intravenózní aplikaci. Asepticky přeneste vypočítaný objem ramucirumabu do balení pro intravenózní aplikaci. Konečný celkový objem v balení by měl být 250 ml. Obal je třeba jemně převrátit, aby se obsah dostatečně promíchal. Injekční roztok NEZMRAZUJTE ani NEPROTŘEPÁVEJTE. NEŘEĎTE jinými roztoky ani nepodávejte spolu s infuzí s jinými elektrolyty nebo léčivými přípravky.
V případě použití prázdných obalů k intravenózní infuzi
Asepticky přeneste vypočítaný objem ramucirumabu do prázdného obalu pro intravenózní infuzi. Přidejte dostatečné množství injekčního roztoku chloridu sodného 9 mg/ml (0,9 %) do obalu, aby byl konečný objem 250 ml. Obal je třeba jemně převrátit, aby se obsah dostatečně promíchal. Injekční roztok NEZMRAZUJTE ani NEPROTŘEPÁVEJTE. NEŘEĎTE s jinými roztoky ani nepodávejte spolu s infuzí s jinými elektrolyty nebo léčivými přípravky.
Po naředění a přípravě je nutné léčivý přípravek okamžitě použít. Pokud není ihned aplikován, za dobu uchovávání a podmínky před použitím je zodpovědný uživatel, přičemž doba uchovávání by neměla být delší než 24 hodin při teplotě 2 °C až 8 °C.
Parenterální léčivé přípravky je třeba před podáním vizuálně zkontrolovat, zda neobsahují částice. Pokud zjistíte přítomnost částic, infuzní roztok zlikvidujte.
Zlikvidujte veškerý zbývající objem ramucirumabu, který zůstane v injekční lahvičce, protože přípravek neobsahuje antimikrobiální konzervační látky.
Podejte pomocí infuzní pumpy. K infuzi je nutné použít samostatnou infuzní linku s 0,22 mikronovým filtrem šetrným vůči proteinům a na konci infuze je nutné linku vypláchnout injekčním roztokem chloridu sodného 9 mg/ml (0,9 %).
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
51