Cotellic 20 Mg
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Cotellic 20 mg potahované tablety
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna potahovaná tableta obsahuje cobimetinibum 20 mg ve formě cobimetinibi fumaras. Pomocná látka se známým účinkem:
Jedna potahovaná tableta obsahuje 36 mg monohydrátu laktosy.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Potahovaná tableta.
Bílé, kulaté potahované tablety o průměru přibližně 6,6 mm, s vyraženým “COB” na jedné straně.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Cotellic je v kombinaci s vemurafenibem indikován k léčbě dospělých pacientů s neresekovatelným nebo metastazujícím melanomem s pozitivní mutací V600 genu BRAF (viz body 4.4 a 5.1).
4.2 Dávkování a způsob podání
Léčbu přípravkem Cotellic v kombinaci s vemurafenibem má zahajovat a dohlížet na ni pouze kvalifikovaný lékař se zkušenostmi s podáváním protinádorových léčivých přípravků.
Před zahájením léčby musí být u pacientů validovaným testem potvrzena pozitivní mutace V600 genu BRAF (viz body 4.4 a 5.1).
Dávkování
Doporučená dávka přípravku Cotellic je 60 mg (3 tablety po 20 mg) jednou denně.
Přípravek Cotellic se užívá ve 28denním cyklu. Jedna dávka se skládá ze tří 20 mg tablet (60 mg) a má se užívat jednou denně po dobu 21 po sobě jdoucích dní (dny 1 až 21- doba léčby); následováno 7denní přestávkou v léčbě (dny 22 až 28 - přestávka v léčbě). Následující léčebný cyklus přípravku Cotellic má začít po uplynutí 7denní přestávky v léčbě.
Další informace o dávkování vemurafenibu naleznete v příslušném SmPC pro tento přípravek.
V léčbě přípravkem Cotellic se má pokračovat až do doby, kdy pacientovi léčba již nepřináší žádný prospěch nebo do rozvoje nepřijatelné toxicity (viz tabulka 1 níže).
Vynechané dávky
Pokud dojde k vynechání dávky, je možné ji užít nejpozději 12 hodin před další dávkou, aby bylo dodrženo dávkovací schéma jednou denně.
V případě zvracení po podání přípravku Cotellic nemá pacient užít další dávku ve stejný den a léčba má pokračovat následující den, jak je předepsáno.
Obecné úpravy dávkování
Rozhodnutí, zda snížit dávku pro jeden nebo oba léčivé přípravky závisí na posouzení lékaře v závislosti na bezpečnosti nebo snášenlivosti u individuálního pacienta. Úprava dávky přípravku Cotellic je nezávislá na úpravách dávky vemurafenibu.
Pokud jsou dávky z důvodu toxicity vynechány, nemají být tyto dávky nahrazeny. Poté, co byla dávka snížena, nemá být později zvýšena.
Tabulka 1 níže popisuje obecné pokyny týkající se úprav dávkování přípravku Cotellic.
Tabulka 1 Doporučené úpravy dávkování přípravku Cotellic
|
Stupeň (CTC-AE)* |
Doporučená dávka přípravku Cotellic |
|
Stupeň 1 nebo stupeň 2 (tolerovatelný) |
Žádné snížení dávky. Udržení přípravku Cotellic na dávce 60 mg jednou denně (3 tablety) |
|
Stupeň 2 (netolerovatelný) nebo stupeň 3/4 | |
|
1. výskyt |
Přerušení léčby až do dosažení stupně < 1, znovuzahájení léčby dávkou 40 mg jednou denně (2 tablety) |
|
2. výskyt |
Přerušení léčby až do dosažení stupně < 1, znovuzahájení léčby dávkou 20 mg jednou denně (1 tableta) |
|
3. výskyt |
Zvážení trvalého ukončení léčby |
* Intenzita klinických nežádoucích účinků byla stanovena na základě kritérií CTC-AE (Common Terminology Criteria for Adverse Events) verze 4.0.
Doporučení pro úpravu dávkování při dysfunkci levé komory
Je třeba zvážit trvalé přerušení léčby přípravkem Cotellic, pokud jsou kardiální příznaky přisuzovány přípravku Cotellic a nezlepšují se po dočasném přerušení léčby.
Tabulka 2 Doporučené úpravy dávkování přípravku Cotellic u pacientů s ejekční frakcí levé komory (LVEF) při poklesu od počáteční hodnoty
|
Pacient |
LVEF hodnota |
Doporučená úprava dávky přípravku Cotellic |
LVEF hodnota po přestávce v léčbě |
Doporučená denní dávka přípravku Cotellic |
|
Asymptomatic- ký |
> 50% (nebo 40-49% a < 10% absolutní pokles od počáteční hodnoty) |
Pokračování při současné dávce |
N/A |
N/A |
|
< 40% (nebo 40-49% a > 10% absolutní pokles od počáteční hodnoty) |
Přerušení léčby na 2 týdny |
< 10% absolutní pokles od počáteční hodnoty |
1. výskyt: 40 mg 2. výskyt: 20 mg 3. výskyt: trvalé ukončení léčby | |
|
< 40% (nebo > 10% absolutní pokles od počáteční hodnoty) |
Trvalé ukončení léčby | |||
|
Symptomatický |
N/A |
Přerušení léčby na 4 týdny |
Asymptomatické a < 10% absolutní pokles od počáteční hodnoty |
1. výskyt: 40 mg 2. výskyt: 20 mg 3. výskyt: trvalé ukončení léčby |
|
Asymptomatické a < 40% (nebo > 10% absolutní pokles od počáteční hodnoty) |
Trvalé ukončení léčby | |||
|
Symptomatické bez ohledu na LVEF |
Trvalé ukončení léčby |
N/A = Neuplatňuje se
V případě, že je léčba přípravkem Cotellic upravena, může být v léčbě vemurafenibem pokračováno, pokud je tak klinicky stanoveno.
Rady _pro úpravu dávkování _přípravku Cotellic _při užívání s vemurafenibem Abnormality laboratorních jaterních testů
Při abnormalitách laboratorních jaterních testů stupně 1 a 2 se má pokračovat v podávání předepsané dávky přípravku Cotellic a vemurafenibu.
Stupeň 3: V podávání přípravku Cotellic se má dále pokračovat dle předepsané dávky. Dávku vemurafenibu je možné snížit, pokud je to klinicky vhodné. Viz SmPC vemurafenibu.
Stupeň 4:
Léčba přípravkem Cotellic a léčba vemurafenibem má být přerušena. Pokud se abnormality laboratorních jaterních tesů zlepší na stupeň <1 v průběhu 4 týdnů, podávání přípravku Cotellic má být znovu zahájeno v dávce snížené o 20 mg a vemurafenib v klinicky vhodné dávce, jak je uvedeno v příslušném SmPC.
Léčba přípravkem Cotellic a léčba vemurafenibem má být ukončena, pokud se abnormality laboratorních jaterních testů nezlepší na stupeň <1 v průběhu 4 týdnů nebo pokud se stupeň 4 abnormálních laboratorních testů znovu objeví po úvodním zlepšení.
Vzestup hladiny kreatinfosfokinázy (CPK)
Ke zvládnutí asymptomatických vzestupů hladin CPK není třeba úprava dávky přípravku Cotellic ani přerušení jeho podávání.
Fotosenzitivita
Stupeň <2 (tolerovatelný) fotosenzitivity má být upraven podpůrnou léčbou.
Stupeň 2 (netolerovatelný) nebo stupeň >3 fotosenzitivity: Léčba přípravkem Cotellic a vemurafenibem má být přerušena, dokud se nedosáhne stupně <1. Léčba může být znovu zahájena bez úpravy dávky přípravku Cotellic. Dávkování vemurafenibu má být sníženo, pokud je to klinicky vhodné, další informace naleznete v příslušném SmPC.
Při léčbě přípravkem Cotellic nebo při léčbě vemurafenibem se mohou objevit případy vyrážky.
Dávka přípravku Cotellic a/nebo vemurafenibu může být buď dočasně přerušena a/nebo snížena, pokud je to klinicky indikováno.
Dále:
Vyrážka stupně <2 (tolerovatelný) má být léčena podpůrnou léčbou. V podávání přípravku Cotellic se může dále pokračovat bez úpravy dávkování.
Akneiformní vyrážka stupně 2 (netolerovatelný) nebo stupně >3: Mají být dodržována obecná doporučení úpravy dávek uvedená v tabulce 1 přípravku Cotellic. V dávkování vemurafenibu se může pokračovat při úpravě léčby přípravkem Cotellic (dle klinické indikace).
Jiná než akneiformní vyrážka nebo makulopapulózní vyrážka stupně 2 (netolerovatelný) nebo stupně >3: V dávkování přípravku Cotellic je možné pokračovat bez úprav dle klinické indikace. Dávkování vemurafenibu je možné buď dočasně přerušit a/nebo snížit, další informace naleznete v příslušném SmPC.
Prodloužení QT intervalu
Pokud v průběhu léčby QTc přesáhne 500 ms, přečtěte si, prosím, SmPC vemurafenibu (bod 4.2) pro informace o úpravě dávek vemurafenibu. Není vyžadována žádná úprava dávky přípravku Cotellic, pokud je užíván v kombinaci s vemurafenibem.
Zvláštní populace
Starší pacienti
U pacientů ve věku >65 let není nutná žádná zvláštní úprava dávky.
Porucha funkce ledvin
Na základě populační analýzy farmakokinetiky se u pacientů s lehkou až středně těžkou poruchou funkce ledvin nedoporučuje žádná úprava dávky (viz bod 5.2). U pacientů s těžkou poruchou funkce ledvin jsou k dispozici pouze omezené údaje pro přípravek Cotellic, proto jeho účinek nelze vyloučit. Přípravek Cotellic je třeba užívat s opatrností u pacientů s těžkou poruchou ledvin.
Porucha funkce jater
U pacientů s poruchou funkce jater se žádná úprava dávkování nedoporučuje. Pacienti s těžkou poruchou funkce jater mohou mít zvýšené plazmatické koncentrace volného kobimetinibu v porovnání s pacienty s normální funkcí jater (viz bod 5.2). V průběhu léčby přípravkem Cotellic se mohou objevit abnormální laboratorní výsledky vyšetření jater, při podávání pacientům s poruchou funkce jater jakéhokoli stupně je třeba zvýšené opatrnosti (viz bod 4.4).
Pacienti jiné než bílé (kavkazské) rasy
Bezpečnost a účinnost přípravku Cotellic u pacientů jiné než bílé rasy nebyla stanovena.
Pediatrická populace
Bezpečnost a účinnost přípravku Cotellic u dětí a dospívajících mladších 18 let nebyla dosud stanovena. Nejsou dostupné žádné údaje.
Způsob podání
Přípravek Cotellic se užívá perorálně. Tablety se polykají celé a zapíjejí se vodou. Mohou být užívány s jídlem nebo bez jídla.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Před užitím přípravku Cotellic v kombinaci s vemurafenibem musí být u pacientů validovaným testem potvrzena pozitivní mutace V600 genu BRAF.
Přípravek Cotellic v kombinaci s vemurafenibem u pacientů, u kterých došlo k progresi onemocnění po dobu léčby inhibitorem BRAF
K dispozici jsou omezené údaje o užívání kombinace přípravku Cotellic s vemurafenibem u pacientů, u kterých došlo k progresi onemocnění při předchozí léčbě inhibitorem BRAF. Tyto údaje ukazují, že u těchto pacientů bude účinnost kombinace nižší (viz bod 5.1). U pacientů, u kterých došlo k progresi onemocnění po dobu léčby inhibitorem BRAF, se proto před touto léčbou mají zvážit další možnosti léčby. Pořadí, ve kterém se léčby mají podávat po progresi onemocnění po dobu léčby inhibitorem BRAF, nebylo stanoveno.
Přípravek Cotellic v kombinaci s vemurafenibem u pacientů s metastázami v mozku
Bezpečnost a účinnost kombinace přípravku Cotellic a vemurafenibu u pacientů s melanomem s pozitivní mutací V600 genu BRAF, který metastázoval do mozku, nebyla hodnocena. Nitrolebeční aktivita kobimetinibu je v současné době neznámá (viz body 5.1 a 5.2).
Serózní retinopatie
U pacientů léčených MEK-inhibitory, včetně přípravku Cotellic, byla pozorována serózní retinopatie (hromadění tekutiny ve vrstvách sítnice) (viz bod 4.8). Většina případů byla hlášena jako chorioretinopatie nebo odchlípení sítnice.
Střední doba do prvního výskytu případů serózní retinopatie byla 1 měsíc (rozmezí 0-9 měsíců). Většina příhod pozorovaných v klinických studiích odezněla, nebo se zmírnila na asymptomatický stupeň 1 po přerušení podávání dávky nebo jejím snížení.
Pacienti mají být při každé návštěvě vyšetřeni pro zjištění vzniku nových příznaků poruchy zraku nebo jejich zhoršení. Pokud je zjištěn vznik nebo zhoršení příznaků poruchy zraku, doporučuje se oftalmologické vyšetření. Pokud je serózní retinopatie diagnostikována, měla by být léčba přípravkem Cotellic pozastavena, dokud se příznaky nezmírní na stupeň < 1. Serózní retinopatii je možné upravit přerušením léčby, snížením dávky nebo ukončením léčby (viz tabulka 1 v bodě 4.2).
Dysfunkce levé komory
U pacientů užívajících přípravek Cotellic byl hlášen pokles LVEF od výchozí hodnoty (viz bod 4.8). Střední doba do prvního výskytu této příhody byla 4 měsíce (1-13 měsíců).
LVEF má být vyšetřena před zahájením léčby pro stanovení výchozích hodnot, potom po prvním měsíci léčby a alespoň každé 3 měsíce nebo dle klinické indikace až do ukončení léčby. Pokles LVEF od výchozí hodnoty je možné upravit přerušením léčby, snížením dávky nebo ukončením léčby (viz bod 4.2).
Všem pacientům, kteří znovu zahajují léčbu se sníženou dávkou přípravku Cotellic, má být LVEF vyšetřena po přibližně 2 týdnech, 4 týdnech, 10 týdnech a 16 týdnech a poté dle klinické indikace.
Pacienti s výchozí hodnotou LVEF buď pod stanovenou dolní hranicí referenčního rozpětí (LLN), nebo pod 50 %, nebyli zařazeni do studie.
Abnormální laboratorní výsledky vyšetření jater
Abnormální laboratorní výsledky vyšetření jater se mohou objevit, pokud se přípravek Cotellic používá v kombinaci s vemurafenibem a při podávání vemurafenibu jako samotného léčiva (přečtěte si, prosím, příslušné SmPC).
U pacientů léčených přípravkem Cotellic a vemurafenibem byly pozorovány abnormální laboratorní výsledky vyšetření jater, zejména zvýšení alaninaminotransferázy (ALT), aspartátaminotransferázy (AsT) a alkalické fosfatázy (ALP) (viz bod 4.8).
Abnormální jaterní hodnoty mají být monitorovány laboratorními vyšetřeními jater před zahájením kombinované léčby a každý měsíc v průběhu léčby, nebo častěji, pokud je to klinicky indikováno (viz bod 4.2).
Abnormální laboratorní výsledky vyšetření jater stupně 3 lze upravit přerušením léčby vemurafenibem nebo snížením jeho dávky. Abnormální laboratorní výsledky jaterních testů stupně 4 lze upravit přerušením léčby, snížením dávky nebo ukončením léčby přípravkem Cotellic i vemurafenibem (viz bod 4.2).
Byly hlášeny případy průjmu stupně >3 a téžkého průjmu u pacientů léčených přípravkem Cotellic. Průjem lze upravit protiprůjmovými léky a podpůrnou léčbou. U průjmu stupně >3, který přetrvává i přes podpůrnou léčbu, má být léčba přípravkem Cotellic a vemurafenibem přerušena, dokud se průjem nezlepší na stupeň <1. Pokud se průjem stupně >3 opakuje, dávka přípravku Cotellic a vemurafenibu má být snížena (viz bod 4.2).
Intolerance laktosy
Tento léčivý přípravek obsahuje laktosu. Pacienti se vzácnými dědičnými problémy s intolerancí galaktosy, vrozeným nedostatkem laktázy nebo malabsorpcí glukosy a galaktosy se mají poradit se svým lékařem a prodiskutovat, zda z jejich individuálního hlediska přínosy převažují nad riziky.
Lékové interakce: CYP3A inhibitory
Je třeba se vyhnout souběžnému užívání silných CYP3A inhibitorů v průběhu léčby přípravkem Cotellic. Opatrnosti je třeba v případě, že je středně silný CYP3A inhibitor podáván souběžně s přípravkem Cotellic. Pokud se nelze vyhnout souběžnému užívání se silným CYP3A inhibitorem, mají být pacienti z důvodu bezpečnosti a úprav podávaných dávek pečlivě sledováni, pokud je to klinicky indikováno (viz tabulka 1 v bodě 4.2).
Prodloužení QT intervalu
Pokud v průběhu léčby QTc přesáhne 500 ms, přečtěte si, prosím, body 4.2 a 4.4 v SmPC léku obsahujícího vemurafenib.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Vliv jiných léčivých přípravků na kobimetinib
CYP3A inhibitory
Kobimetinib je metabolizován CYP3A a hodnota AUC kobimetinibu se u zdravých osob zvýšila přibližně 7krát v přítomnosti silného CYP3A inhibitoru (itrakonazol). Rozsah interakce by mohl být u pacientů nižší.
Silné CYP3A inhibitory (viz bod 4.4): Vyvarujte se souběžnému podávání se silnými inhibitory CYP3A v průběhu léčby kobimetinibem. Mezi silné CYP3A inhibitory patří, ale není omezeno pouze na uvedené, ritonavir, kobicistat, telaprevir, lopinavir, itrakonazol, vorikonazol, klaritromycin, telithromycin, posakonazol, nefazodon a grepfruitový džus. Pokud se nelze vyhnout souběžnému užívání se silným CYP3A inhibitorem, mají být pacienti z důvodu bezpečnosti pečlivě sledování. U silných CYP3A inhibitorů používaných krátkodobě (7 dní a méně) je třeba zvážit přerušení léčby kobimetinibem v průběhu doby užívání inhibitoru.
Středně silné CYP3A inhibitory (viz bod 4.4): Opatrnosti je třeba v případě, že je kobimetinib podáván souběžně s středně silnými CYP3A inhibitory. Mezi středně silné CYP3A inhibitory patří, ale není omezeno pouze na uvedené, amiodaron, erythromycin, flukonazol, mikonazol, diltiazem, verapamil, delavirdin, amprenavir, fosamprenavir, imatinib. Při souběžném podávání kobimetinibu se středně silným CYP3A inhibitorem mají být pacienti z důvodu bezpečnosti pečlivě sledováni.
Mírné CYP3A inhibitory: Kobimetinib může být souběžně podáván s mírnými inhibitory CYP3A bez úpravy dávkování.
CYP3A induktory
Souběžné podávání kobimetinibu se silnými CYP3A induktory nebylo v klinické studii hodnoceno, je však pravděpodobné snížení expozice kobimetinibu. Proto je třeba se vyhnout souběžnému užívání středně silných a silných CYP3A induktorů (např. karbamazepin, rifampicin, fenytoin a třezalka tečkovaná). Je třeba zvážit použití alternativních léčiv, která nezpůsobují žádnou nebo pouze minimální CYP3A indukci. Vzhledem k tomu, že koncentrace kobimetinibu jsou pravděpodobně významně sníženy při současném podávání se středně silnými a silnými CYP3A induktory, může být účinnost u pacienta oslabena.
Inhibitory P-glykoproteinu
Kobimetinib je substrát P-glykoproteinu (P-gp). Souběžné podávání P-gp inhibitorů, jako jsou cyklosporin a verapamil, může zvýšit plazmatické koncentrace kobimetinibu.
Vliv kobimetinibu na jiné léčivé přípravky
CYP3A a CYP2D6 substráty
Klinická studie lékových interakcí (DDI) u pacientů s karcinomem ukázala, že plazmatické koncentrace midazolamu (citlivý substrát CYP3A) a dextromethorfanu (citlivý substrát CYP2D6) se v přítomnosti kobimetinibu nezměnily.
CYP1A2 substráty
In vitro je kobimetinib možný induktor CYP1A2 a může proto snížit expozici substrátů tohoto enzymu, např. theofilinu. Nebyly provedeny žádné klinické studie lékových interakcí k posouzení klinického významu tohoto zjištění.
BCRP substráty
In vitro je kobimetinib středně silným inhibitorem BCRP (Breast Cancer Resistance Protein - protein zodpovědný za rezistenci při karcinomu prsu). Nebyly provedeny žádné klinické studie lékových interakcí hodnotící toto zjištění, není možné vyloučit klinicky významnou inhibici BCRP ve střevě.
Další protinádorové látky
Vemurafenib
U pacientů s neresekovatelným nebo metastazujícím melanomem neexistují žádné důkazy o jakýchkoli klinicky významných lékových interakcích mezi kobimetinibem a vemurafenibem, a proto nejsou doporučeny žádné úpravy dávek.
Vliv kobimetinibu na transportní systémy látek
In vitro studie ukazují, že kobimetinib není substrátem transportérů hepatálního vychytávání OATP1B1, OATP1B3 a OCT1, tyto transportéry však slabě inhibuje. Klinický význam těchto zjištění nebyl zkoumán.
Pediatrická populace
Studie interakcí byly provedeny pouze u dospělých.
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku.
Ženy ve fertilním věku mají během léčby přípravkem Cotellic a po dobu alespoň 3 měsíců po ukončení léčby používat dvě účinné antikoncepční metody, jako je kondom nebo jiná bariérová metoda (spermicidní, pokud jsou dostupné).
K dispozici nejsou žádné údaje týkající se použití přípravku Cotellic u těhotných žen. Studie na zvířatech ukázaly embryoletalitu a malformace velkých cév a lebky plodu (viz bod 5.3). Přípravek Cotellic se může používat v průběhu těhotenství pouze v nevyhnutelných případech a po pečlivém zvážení potřeby léčby u matky a rizika pro plod.
Kojení
Není známo, zda je kobimetinib vylučován do mateřského mléka. Riziko pro novorozence/kojence nelze vyloučit. Při rozhodování, zda přerušit kojení nebo přerušit léčbu přípravkem Cotellic, je nutné vzít v úvahu prospěch z kojení pro dítě a prospěch z léčby pro matku.
Fertilita
K dispozici nejsou žádné údaje týkající se vlivu kobimetinibu na fertilitu u člověka. Nebyly provedeny žádné studie fertility u zvířat, ale byly pozorovány nežádoucí účinky na reprodukční orgány (viz bod 5.3). Klinický význam tohoto zjištění není znám.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek Cotellic má malý vliv na schopnost řídit nebo obsluhovat stroje. V průběhu klinických studií byly u některých pacientů léčených kobimetinibem hlášeny poruchy zraku (viz body 4.4 a 4.8). Pacienti mají být upozorněni, aby neřídili nebo neobsluhovali stroje, pokud se u nich vyskytnou poruchy zraku nebo jakékoli jiné nežádoucí účinky, které mohou mít vliv na jejich schopnosti.
4.8 Nežádoucí účinky
Shrnutí bezpečnostního profilu
Bezpečnost přípravku Cotellic v kombinaci s vemurafenibem byla hodnocena u 247 pacientů s pokročilým melanomem s mutací V600 genu BRAF ve studii GO28141.Střední doba do výskytu prvních nežádoucích účinků stupně >3 byla 0,6 měsíce ve skupině s přípravkem Cotellic plus vemurafenib v porovnání s 0,8 měsíce ve skupině s placebem plus vemurafenib.
Bezpečnost přípravku Cotellic v kombinaci s vemurafenibem byla také hodnocena u 129 pacientů s pokročilým melanomem s mutací V600 genu BRAF ve studii NO25395. Bezpečnostní profil ve studii NO25395 se shodoval s tím, který byl pozorován ve studii GO28141.
Nejčastějšími nežádoucími účinky (ADR) (>20 %) s vyšší četností výskytu ve studii GO28141 byly průjem, vyrážka, nauzea, pyrexie, fotosenzitivní reakce, zvýšení alaninaminotransferázy, zvýšení aspartátaminotransferázy, zvýšení kreatinfosfokinázy v krvi a zvracení pozorované ve skupině s přípravkem Cotellic plus vemurafenib. Nejčastějšími nežádoucími účinky (>20 %) s vyšší četností výskytu pozorované ve skupině s placebem plus vemurafenib byly artralgie, alopecie a hyperkeratóza. Únava byla pozorována se stejnou frekvencí výskytu v obou skupinách.
Přečtěte si, prosím, SmPC vemurafenibu pro úplný přehled o všech nežádoucích účincích spojených s léčbou vemurafenibem.
Shrnutí nežádoucích účinků do tabulky
Nežádoucí účinky jsou založeny na výsledcích získaných z multicentrické, randomizované, dvojitě zaslepené, placebem kontrolované studie fáze III (GO28141), která hodnotila bezpečnost a účinnost přípravku Cotellic v kombinaci s vemurafenibem v porovnání se samotným vemurafenibem u dříve neléčených pacientů s neresekovatelným lokálně pokročilým (stadium IIIc) nebo metastazujícím melanomem (stadium IV) s pozitivní mutací V600 genu BRAF.
Četnosti nežádoucích účinků jsou založeny na analýze bezpečnosti pacientů léčených kobimetinibem plus vemurafenib se střední dobou sledování 11,2 měsíce (ukončení sběru údajů k datu 19. září 2014).
Nežádoucí účinky, které byly hlášeny u pacientů s melanomem, jsou shrnuty níže podle MedDRA tříd orgánových systémů, četnosti a stupně závažnosti. Ke stanovení četnosti byla použita následující klasifikace četností:
Velmi časté > 1/10 Časté > 1/100 až < 1/10 Méně časté > 1/1000 až < 1/100 Vzácné > 1/10000 až < 1/1000 Velmi vzácné < 1/10000
V tabulce 3 jsou uvedeny nežádoucí účinky, které jsou považovány za související s užíváním přípravku Cotellic. V rámci jednotlivých skupin četností jsou nežádoucí účinky seřazeny podle klesající závažnosti a byly hlášeny v souladu s NCI-CTCAE v 4.0 (všeobecná kritéria toxicity) pro hodnocení toxicity ve studii GO28141.
Tabulka 3 Nežádoucí účinky vyskytující se u pacientů léčených přípravkem Cotellic v kombinaci s vemurafenibem ve studii GO28141A
|
Třída orgánových systémů |
Velmi časté |
Časté |
|
Novotvary benigní, maligní a blíže neurčené (včetně cyst a polypů) |
Bazocelulární karcinom, spinocelulární karcinom kůže**, keratoakantom** | |
|
Poruchy krve a lymfatického systému | ||
|
Poruchy metabolismu a výživy |
Dehydratace, hypofosfatemie, hyponatremie, hyperglykemie | |
|
Poruchy oka |
Serózní retinopatiea, rozmazané vidění |
Poruchy zraku |
|
Cévní poruchy |
Hypertenze, krvácení* | |
|
Respirační, hrudní a mediastinální poruchy |
Pneumonitida | |
|
Gastrointestinální poruchy | ||
|
Poruchy kůže a podkožní tkáně |
Fotosenzitivitab, vyrážka, makulopapulózní vyrážka, akneiformní dermatitis, hyperkeratóza** | |
|
Celkové poruchy a reakce v místě aplikace |
Pyrexie, třesavka | |
|
Vyšetření |
Zvýšení CPK v krvi, zvýšení ALT, zvýšení AST, zvýšení gamma-glutamyltransferazy (GGT), zvýšení ALP v krvi |
Snížení ejekční frakce, zvýšení bilirubinu v krvi |
Ukončení sběru údajů k datu 19. září 2014.
* Přečtěte si, prosím, odstavec Krvácení v bodě „Popis vybraných nežádoucích účinků“
** Přečtěte si, prosím, odstavec Spinocelulární karcinom kůže, keratoakantom a hyperkeratóza v bodě „Popis vybraných nežádoucích účinků“
a Zahrnuje příhody, kterými jsou chorioretinopatie a odchlípení sítnice, poukazující na serózní retinopatii (viz bod 4.4)
b Kombinovaný údaj zahrnující hlášení fotosenzitivních reakcí, spálenin od slunce, solární dermatitidy, aktinické elastózy
Popis vybraných nežádoucích účinků
Krvácení
Příhody krvácení byly hlášeny častěji ve skupině s přípravkem Cotellic plus vemurafenib než ve skupině s placebem plus vemurafenib (všechny typy příhod a stupně závažnosti: 13 % versus 7 %). Ve skupině s přípravkem Cotellic plus vemurafenib byla pozorována vyšší četnost výskytu cerebrálního krvácení (1 % versus 0 %), krvácení z gastrointestinálního traktu (4 % versus 1 %), krvácení v reprodukčním systému (2 % versus <1 %) a hematurie (3 % versus 1 %).
Většina příhod byla stupně 1 nebo 2 a nebyly vážné (12 % pacientů ve skupině s přípravkem Cotellic plus vemurafenib v porovnání s 7 % pacientů ve skupině s placebem plus vemurafenib). Příhody stupně 3-4 se vyskytly u 1 % pacientů v obou skupinách.
Střední doba do výskytu první příhody byla 4,4 měsíce (rozmezí od 0,0 do 12,7 měsíce) ve skupině s přípravkem Cotellic plus vemurafenib.
Fotosenzitivita
Fotosenzitivita byla pozorována s vyšší četností výskytu ve skupině s přípravkem Cotellic plus vemurafenib v porovnání se skupinou s placebem plus vemurafenib (47 % versus 35 %). Většina příhod byla stupně 1 nebo 2, příhody stupně >3 se vyskytly u 4 % pacientů ve skupině s přípravkem Cotellic plus vemurafenib v porovnání s 0 % ve skupině s placebem plus vemurafenib.
Nebyly zjištěny žádné zjevné trendy v době do výskytu příhod stupně >3. Příhody fotosenzitivity stupně >3 ve skupině s přípravkem Cotellic plus vemurafenib byly léčeny primárními topicky podávanými léčivými přípravky, současně s přerušením dávek kobimetinibu i vemurafenibu (viz bod
4.2) .
Při podávání přípravku Cotellic v monoterapii nebyly pozorovány žádné známky fotosenzitivity.
Spinocelulární karcinom kůže, keratoakantom and hyperkeratóza
Spinocelulární karcinom byl hlášen s nižší četností výskytu ve skupině s přípravkem Cotellic plus vemurafenib v porovnání se skupinou s placebem plus vemurafenib (všechny stupně: 3 % versus 13 %). Keratoakantom byl hlášen s nižší četností výskytu ve skupině s přípravkem Cotellic plus vemurafenib v porovnání se skupinou s placebem plus vemurafenib (všechny stupně: 2 % versus 9 %). Hyperkeratóza byla hlášena s nižší četností výskytu ve skupině s přípravkem Cotellic plus vemurafenib v porovnání se skupinou s placebem plus vemurafenib (všechny stupně: 11 % versus 30 %).
Serózní retinopatie
Byly hlášeny případy serózní retinopatie u pacientů léčených přípravkem Cotellic (viz bod 4.4.). U pacientů, kteří zaznamenali vznik nebo zhoršení poruch zraku, se doporučuje oftalmologické vyšetření. Serózní retinopatii je možné upravit přerušením léčby, snížením dávky nebo ukončením léčby (viz tabulka 1 v bodě 4.2).
Dysfunkce levé komory
U pacientů užívajících přípravek Cotellic byl hlášen pokles LVEF od výchozí hodnoty (viz bod 4.4). LVEF má být vyšetřena před zahájením léčby pro stanovení výchozích hodnot, poté po prvním měsíci léčby a alespoň každé 3 měsíce nebo dle klinické indikace až do ukončení léčby. Pokles LVEF od výchozí hodnoty je možné upravit přerušením léčby, snížením dávky nebo ukončením léčby (viz bod
4.2) .
Abnormality laboratorních výsledků Abnormality laboratorních výsledků vyšetření _jater
U pacientů léčených přípravkem Cotellic v kombinaci s vemurafenibem byly pozorovány abnormality laboratorních výsledků vyšetření jater, zejména ALT, AST a ALP (viz bod 4.4). Laboratorní vyšetření jater je třeba provést před zahájením kombinované léčby a každý měsíc v průběhu léčby, nebo častěji, pokud je to klinicky indikováno (viz bod 4.2).
Zvýšení kreatinfosfokinázy v krvi
Ve studii GO28141 (viz bod 4.2) bylo pozorováno asymptomatické zvýšení hladiny CPK v krvi s vyšší četností ve skupině s přípravkem Cotellic plus vemurafenib v porovnání se skupinou s placebem plus vemurafenib. V každé léčebné skupině studie byl pozorován jeden případ rabdomyolýzy se současným zvýšením CPK v krvi.
V tabulce 4 je uvedena četnost výskytu naměřených abnormálních laboratorních výsledků vyšetření jater a zvýšené hladiny kreatinfosfokinázy všech stupňů a stupňů 3-4.
Tabulka 4 Testy funkce jater a další laboratorní testy pozorované ve fázi III studie GO28141
|
Změny v údajích hlášených z laboratorních vyšetření |
kobimetinib plus vemurafenib (n = 247) (%) |
placebo plus vemurafenib (n = 246) (%) | ||
|
Všechny stupně |
Stupně 3-4 |
Všechny stupně |
Stupně 3-4 | |
|
Funkční jaterní testy | ||||
|
Zvýšení ALP |
69 |
7 |
55 |
3 |
|
Zvýšení ALT |
67 |
11 |
54 |
5 |
|
Zvýšení AST |
71 |
7 |
43 |
2 |
|
Zvýšení GGT |
62 |
20 |
59 |
17 |
|
Zvýšení bilirubinu v krvi |
33 |
2 |
43 |
1 |
|
Další laboratorní abnorma |
íty_ | |||
|
Zvýšení CPK v krvi |
70 |
12 |
14 |
<1 |
Zvláštní populace Starší pacienti
Ve studii fáze III s přípravkem Cotellic v kombinaci s vemurafenibem u pacientů s neresekovatelným nebo metastazujícím melanomem (n=247), bylo 183 pacientů (74 %) ve věku <65 let, 44 pacientů (18 %) ve věku 65-74 let, 16 pacientů (6 %) ve věku 75-84 let a 4 pacienti (2 %) byli ve věku >85 let. Podíl pacientů, kteří zaznamenali nežádoucí účinky (AE) byl podobný u pacientů ve věku <65 let jako u pacientů >65 let. U pacientů >65 let byla větší pravděpodobnost výskytu závažných nežádoucích účinků a výskyt nežádoucích účinků vedl u těchto pacientů k ukončení léčby kobimetinibem spíše než u u pacientů <65.
Porucha funkce ledvin
Nebyly provedeny žádné farmakokinetické studie u pacientů s poruchou funkce ledvin. U pacientů s lehkou až středně těžkou poruchou funkce ledvin se na základě výsledků populační analýzy farmakokinetiky nedoporučuje žádná úprava dávky. K dispozici je minimální množství údajů o použití přípravku Cotellic u pacientů s těžkou poruchou funkce ledvin. Přípravek Cotellic je třeba užívat s opatrností u pacientů s těžkou poruchou funkce ledvin.
Porucha funkce jater
U pacientů s poruchou funkce jater se žádná úprava dávkování nedoporučuje (viz bod 5.2).
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Nejsou k dispozici žádné zkušenosti s předávkováním pacientů v klinických studiích. V případě předpokládaného předávkování má být podávání kobimetinibu přerušeno a zahájena podpůrná léčba. Není známo žádné specifické antidotum při předávkování kobimetinibem.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Cytostatika, ATC kód: L01XE38 Mechanismus účinku
Kobimetinib je reverzibilní, selektivní, alosterický, perorální inhibitor, který blokuje dráhu mitogenem aktivované proteinkinázy (MAPK - mitogen-activated protein kinases) tím, že cíleně působí na mitogenem aktivované proteinkinázy regulované extracelulárním signálem (MEK - mitogen-activated extracellular signal regulated kinase) 1 a MEK 2, což vede k inhibici fosforylace proteinkinázy regulovanou extracelulárním signálem (ERK - extracellular signal-related kinase) 1 a ERK 2. Z tohoto důvodu kobimetinib blokuje proliferaci buněk indukovanou dráhou MAPK prostřednictvím inhibice signalizačního uzlu MEK1/2.
V preklinických modelech se ukázalo, že kombinace kobimetinibu s vemurafenibem simultánně cíleně působí na proteinovou mutaci V600 genu BRAF a na proteiny MEK v buňkách melanomu, čímž kombinace těchto dvou léků inhibuje reaktivaci dráhy MAPK prostřednictvím MEK1/2, což vede k silnější inhibici intracelulární signalizace a ke snížení proliferace nádorových buněk.
Klinická účinnost a bezpečnost
K dispozici nejsou žádné údaje o bezpečnosti nebo účinnosti přípravku Cotellic v kombinaci s vemurafenibem u pacientů s metastázami v centrálním nervovém systému ani u pacientů s nekožním maligním melanomem.
Studie GO28141 (coBRIM)
Studie GO28141 je multicentrická, randomizovaná, dvojitě zaslepená, placebem kontrolovaná, studie fáze III k hodnocení bezpečnosti a účinnosti přípravku Cotellic v kombinaci s vemurafenibem ve srovnání s vemurafenibem plus placebo, u dříve neléčených pacientů s neresekovatelným lokálně rozšířeným (stupeň IlIc) nebo metastazujícím melanomem (stupeň IV) s pozitivní mutací V600 genu BRAF.
Do studie GO28141 byli zařazeni pouze pacienti s výkonnostním stavem dle ECOG 0 a 1. Pacienti s výkonnostním stavem dle ECOG 2 nebo vyšším byli ze studie vyloučeni.
Po potvrzení mutace V600 genu BRAF pomocí testu na zjištění přítomnost mutace V600 genu BRAF použitím přístroje cobas® 4800 bylo 495 dříve neléčených pacientů s neresekovatelným lokálně rozšířeným nebo metastazujícím melanomem randomizováno podáním buď:
• Placebo jednou denně ve dnech 1-21 každého 28denního léčebného cyklu a 960 mg vemurafenibu dvakrát denně ve dnech 1-28, nebo
• Přípravek Cotellic 60 mg jednou denně ve dnech 1-21 každého 28denního léčebného cyklu a 960 mg vemurafenibu dvakrát denně ve dnech 1-28
Primárním cílovým parametrem bylo řešitelem hodnocené přežití bez progrese (PFS). Sekundární cílový parametr účinnosti zahrnoval celkové přežití (OS), četnost objektivních odpovědí, řešitelem hodnocené trvání odpovědi (DoR) a nezávisle hodnocené přežití bez progrese (IRF).
Klíčové vstupní charakteristiky zahrnovaly: 58 % pacientů byli muži, medián věku byl 55 let (rozsah od 23 do 88 let), 60 % mělo metastazující melanom stupně M1c a podíl pacientů se zvýšenou LDH byl 46,3 % v rameni kobimetinib plus vemurafenib a 43,0 % v rameni placebo plus vemurafenib.
Ve studii GO28141 bylo 89 pacientů (18,1 %) ve věku 65-74 let, 38 pacientů (7,7 %) ve věku 75-84 let a 5 pacientů (1,0 %) ve věku 85 let a starší.
Výsledky účinnosti jsou shrnuty v tabulce 5.
Tabulka 5 Výsledky účinnosti ^ ze studie GO28141 (coBRIM)
|
Cotellic + vemurafenib N=247 |
Placebo + vemurafenib N=248 | |
|
Primární cílový parametr2^ | ||
|
Přežití bez progrese (PFS) | ||
|
Medián (měsíce) |
12,3 |
7,2 |
|
(95 % interval spolehlivosti) |
(9,5; 13,4) |
(5,6; 7,5) |
|
Poměr rizik (95% interval |
0,58 (0,46; 0,72) | |
|
spolehlivosti)b | ||
|
Klíčové sekundární cílové parametry^ | ||
|
Celkové přežití (OS)g | ||
|
Medián (měsíce) |
22,3 |
17,4 |
|
(95% interval spolehlivosti) |
(20,3; NE) |
(15,0; 19,8) |
|
Poměr rizik (95% interval |
0,70 (95% interval spolehlivosti: 0,55; 0,90) | |
|
spolehlivosti)b |
(p-hodnota |
= 0,0050e) |
|
Četnost objektivních odpovědí (ORR) |
172 (69,6 %) |
124 (50,0 %) |
|
(95% interval spolehlivosti) pro četnost objektivních odpovědí0 |
(63,5%; 75,3%) |
(43,6%; 56,4%) |
|
Rozdíl v četnosti objektivních | ||
|
odpovědí % |
19,6 (11,0; 28,3) | |
|
(95% interval spolehlivosti)d | ||
|
Nejlepší celková odpověď | ||
|
Úplná odpověď |
39 (15,8 %) |
26 (10,5 %) |
|
Částečná odpověď |
133 (53,8 %) |
98 (39,5 %) |
|
Stabilizace nemoci |
44 (17,8 %) |
92 (37,1 %) |
|
Trvání odpovědi (DoR) | ||
|
Medián trvání odpovědi (měsíce) |
13 |
9,2 |
|
(95% interval spolehlivosti) |
(11,1; 16,6) |
(7,5; 12,8) |
NE = nehodnotitelné
a Hodnocené a potvrzené vyšetřujícím lékařem (INV) za použití RECIST v1.1 b Stratifikovaná analýza podle geografické oblasti a klasifikace metastáz (stupeň onemocnění) c Za použití Clopper-Pearsonovy metody d Za použití Hauck-Andersonovy metody
e P-hodnota OS (0,0050) překročila předem stanovenou hranici (p-hodnota <0,0499) f Ukončení sběru dat této aktualizované analýzy PFS a sekundárních cílových parametrů ORR, BOR a DoR k datu 16. ledna 2015. Medián doby sledování byl 14,2 měsíce.
g Ukončení sběru dat konečné analýzy OS k datu 28. sprna 2015 a medíán doby sledování byl 18,5 měsíce.
Primární analýza studie GO28141 byla provedena k 9. květnu 2014. Výrazné zlepšení primárního cílového parametru, řešitelem hodnocené přežití bez progrese, bylo pozorováno u pacientů přiřazených do skupiny s přípravkem Cotellic plus vemurafenib v porovnání se skupinou s placebem plus vemurafenib (poměr rizik 0,51 (0,39; 0,68); p-hodnota < 0,0001).
Střední odhad řešitelem hodnoceného přežití bez progrese byl 9,9 měsíce ve skupině s přípravkem Cotellic plus vemurafenib versus 6,2 měsíce ve skupině s placebem plus vemurafenib. Střední odhad nezávislého hodnocení přežití bez progrese byl 11,3 měsíce ve skupině s přípravkem Cotellic plus vemurafenib versus 6,0 měsíců ve skupině s placebem plus vemurafenib (poměr rizik 0,60 (0,45; 0,79); p-hodnota = 0,0003). Četnost objektivní odpovědi (ORR) ve skupině s přípravkem Cotellic plus vemurafenib byla 67,6 % versus 44,8 % ve skupině s placebem plus vemurafenib. Rozdíl v četnosti objektivní odpovědi byl 22,9 % (p-hodnota < 0,0001).
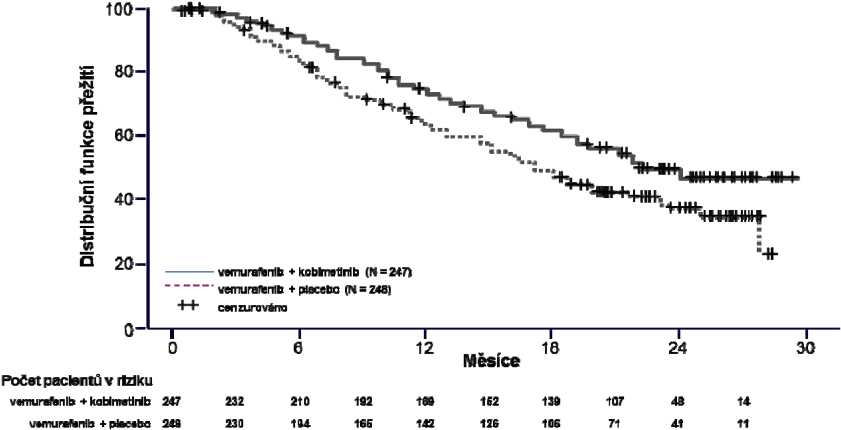
Konečná analýza celkového přežití ve studii GO28141 byla provedena k 28. srpnu 2015. Výrazné zlepšení celkového přežití bylo pozorováno u pacientů přiřazených do skupiny s přípravkem Cotellic plus vemurafenib, v porovnání se skupinou s placebem plus vemurafenib (obrázek 1). Odhady 1letého (75 %) a 2letého (48 %) celkového přežití ve skupině s přípravkem Cotellic plus vemurafenib byly vyšší (64 %) než ve skupině s placebem plus vemurafenib (38 %).
Obrázek 1 Kaplan-Meierovy křivky doby konečného celkového přežití - populace všech randomizovaných pacientů (Intent to Treat Population) (ukončení sběru údajů: 28. srpna 2015)
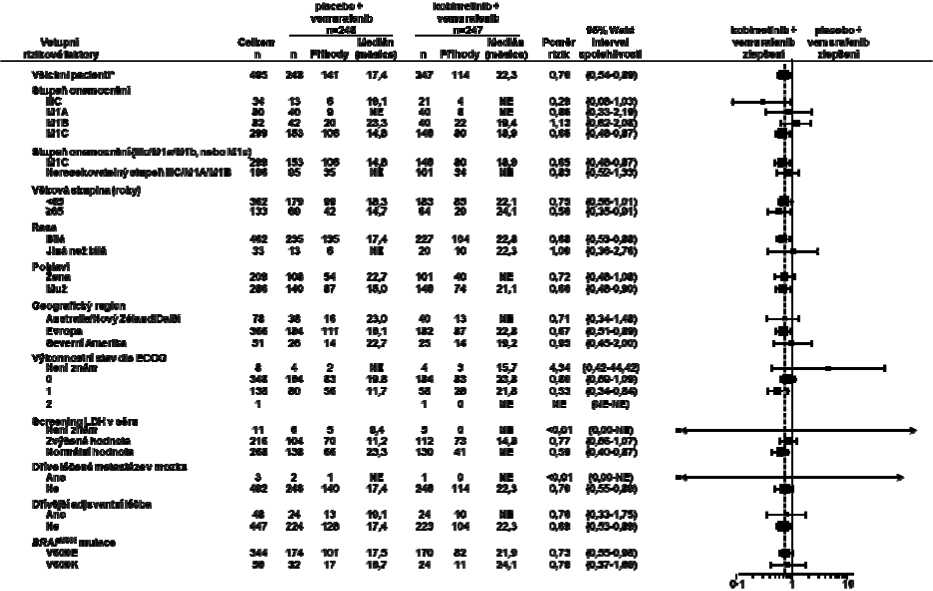
Obrázek 2: Stromový graf (forest plot) pro srovnání poměrů rizik konečného celkového přežití na základě analýz podskupiny - populace všech randomizovaných pacientů (Intent To Treat Population) (ukončení sběru údajů: 28. srpna 2015)
Celkový zdravotní stav/kvalita života související se zdravím udávané pacienty byly hodnoceny pomocí dotazníku kvality života EORTC (European Organisation for Research and Treatment of Cancer ) - 30 hlavních položek (QLQ-C30). Hodnocení všech funkčních oblastí a většina příznaků (nechutenství, zácpa, nauzea a zvracení, dyspnoe, bolest, únava) ukázala, že průměrná odchylka od výchozí hodnoty byla mezi oběma léčebnými skupinami podobná a nebyla prokázána klinicky významná změna (všechna hodnocení byla < 10bodů v porovnání se vstupními hodnotami).
Studie NO25395 (BRIM7)
Účinnost přípravku Cotellic byla hodnocena ve studii fáze Ib, NO25395, která byla zaměřena tak, aby hodnotila bezpečnost, snášenlivost, farmakokinetiku a účinnost přípravku Cotellic při jeho přidání k vemurafenibu pro léčbu pacientů s neresekovatelným nebo metastazujícím melanomem s pozitivitou mutace V600 genu BRAF (zjištěnou pomocí testu na přítomnost mutace V600 genu BRAF provedeného na přístroji Cobas® 4800).
V této studii bylo léčeno 129 pacientů přípravkem Cotellic a vemurafenibem: 63 pacientů předtím nepodstoupilo léčbu inhibitorem BRAF (BRAFi) a u 66 pacientů došlo k progresi nemoci v průběhu předcházející léčby vemurafenibem. Dvacet pacientů z uvedených 63 pacientů nepředléčených BRAFi podstoupilo předcházející systémovou léčbu pokročilého melanomu, ve většině případů (80 %) šlo o imunoterapii.
Výsledky týkající se populace pacientů nepředléčených BRAFi ze studie NO25395 se celkově shodovaly s výsledky ze studie GO28141. U pacientů nepředléčených BRAFi (n=63) byl výskyt objektivní odpovědi na léčbu 87 %, včetně kompletní remise dosažené u 16 % pacientů. Střední doba trvání odpovědi na léčbu byla 14,3 měsíce. U pacientů nepředléčených BRAFi byla střední doba přežití bez progrese (PFS) 13,8 měsíce, přičemž střední doba sledování po léčbě byla 20,6 měsíce.
U pacientů, u kterých došlo k progresi onemocnění po čas léčby vemurafenibem (n=66), byl výskyt objektivní odpovědi na léčbu 15 %. Střední doba trvání odpovědi na léčbu byla 6,8 měsíce. U pacientů, u kterých došlo k progresi nemoci v průběhu léčby vemurafenibem, byla střední doba přežití bez progrese (PFS) 2,8 měsíce a střední doba sledování byla 8,1 měsíce.
U pacientů, kteří nepodstoupili předchozí léčbu inhibitorem BRAF, byla střední doba celkového přežití 28,5 měsíce (95% interval spolehlivosti: 23,3-34,6). U pacientů, u kterých došlo k progresi onemocnění po dobu léčby inhibitorem BRAF, byla střední doba celkového přežití 8,4 měsíce (95% interval spolehlivosti: 6,7-11,1).
Pediatrická populace
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s přípravkem Cotellic u jedné nebo více podskupin pediatrické populace s maligními solidními nádory (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Absorpce
Po perorálním podání dávky 60 mg pacientům s nádorovým onemocněním vykazoval kobimetinib střední rychlost absorpce s mediánem Tmax 2,4 hodiny. Průměrná hodnota Cmax v rovnovážném stavu byla 273 ng/ml a průměrná hodnota AUC0.24 v rovnovážném stavu byla 4340 ng.h/ml. Průměrný poměr akumulace v rovnovážném stavu byl přibližně 2,4násobný.
Kobimetinib má lineární farmakokinetiku v rozmezí dávek ~3,5 mg až 100 mg.
U zdravých osob byla absolutní biologická dostupnost kobimetinibu 45,9 % (90% CI: 39,7 %; 53,1 %). Studie hmotnostní rovnováhy byla provedena u zdravých osob a ukázala, že kobimetinib se intenzivně metabolizuje a vylučuje stolicí. Absorbovaná frakce byla ~88 %, což naznačuje vysokou absorpci a metabolismus při prvním průchodu játry.
Farmakokinetika kobimetinibu se u zdravých osob nemění, pokud se podává spolu s jídlem (jídlo s vysokým obsahem tuku) ve srovnání s podáním na lačno. Vzhledem k tomu, že jídlo nemění farmakokinetiku kobimetinibu, může být kobimetinib podáván s jídlem nebo bez jídla.
Distribuce
In vitro je vazba kobimetinibu na lidské plazmatické bílkoviny 94,8 %. Nebyly pozorovány žádné preferenční vazby na lidské erytrocyty (poměr léčiva v krvi a plazmě je 0,93).
U zdravých osob, kterým byla intravenózně podána dávka 2mg, byl distribuční objem 1050 l. Na základě populační farmakokinetické analýzy byl u pacientů s nádorovým onemocněním zdánlivý distribuční objem 806 l.
V podmínkách in vitro je kobimetinib substrátem P-gp. Přechod hematoencefalickou bariérou není znám.
Biotransformace
Oxidace prostřednictvím CYP3A a glukuronidace prostřednictvím UGT2B7 se jeví jako hlavní metabolické dráhy kobimetinibu. Kobimetinib je převládající složkou v plazmě. V plazmě se nezjistily žádné oxidační metabolity ve větším množství než 10 % z celkové cirkulující izotopem označené látky nebo metabolitů specifických pro člověka. Nezměněný léčivý přípravek představoval 6,6 % z podané dávky ve stolici a 1,6 % z podané dávky v moči, což svědčí o tom, že kobimetinib je primárně metabolizován s minimální renální eliminací. In vitro údaje ukazují, že kobimetinib není inhibitorem OAT1, OAT3 nebo OCT2.
Eliminace
Kobimetinib a jeho metabolity byly charakterizované ve studii hmotnostní rovnováhy provedené u zdravých osob. V průměru 94 % dávky bylo vyloučeno během 17 dní. Kobimetinib se intenzivně metabolizoval a vylučoval stolicí; žádný jednotlivý metabolit nepřevládal.
Po intravenózním podání 2mg dávky kobimetinibu byla průměrná plazmatická clearance (CL)
10,7 l/h. Průměrná zdánlivá CL po perorálním podání 60 mg u pacientů s karcinomem byla 13,8 l/h. Průměrný poločas rozpadu po perorálním podání kobimetinibu byl 43,6 hodin (rozmezí: 23,1 až 69,6 hodiny). Proto eliminace může trvat až 2 týdny po ukončení léčby k dosažení úplného odstranění kobimetinibu ze systémové cirkulace.
Zvláštní populace
Na základě populační analýzy farmakokinetiky se zjistilo, že pohlaví, rasa, etnický původ, výchozí hodnota výkonnostního stavu dle ECOG, lehká až středně těžká porucha funkce ledvin nemají žádný vliv na farmakokinetiku kobimetinibu. Věk a tělesná hmotnost na začátku studie byli identifikované jako statisticky významné kovarianty ovlivňující clearance a distribuční objem kobimetinibu, v uvedeném pořadí. Analýzy citlivosti však ukazují na to, že ani jedna z těchto kovariant nemá významný vliv na expozici v rovnovážném stavu.
Pohlaví
Na základě populační farmakokinetické analýzy zahrnující 210 žen a 277 mužů bylo zjištěno, že pohlaví nemá žádný vliv na expozici kobimetinibu.
Starší lidé
Na základě populační farmakokinetické analýzy zahrnující 133 pacientů ve věku > 65 let bylo zjištěno, že věk nemá žádný vliv na expozici kobimetinibu.
Na základě preklinických údajů a studie hmotnostní rovnováhy je kobimetinib hlavně metabolizován s minimální renální eliminací. Nebyla provedena žádná formální studie farmakokinetiky u pacientů s poruchou funkce ledvin.
Populační analýza farmakokinetiky využívající údaje získané od 151 pacientů s lehkou poruchou funkce ledvin (clearance kreatininu (CRCL) 60 až méně než 90 ml/min), od 48 pacientů se středně těžkou poruchou funkce ledvin (CRCL 30 až méně než 60 ml/min) a od 286 pacientů s normální funkcí ledvin (CRCL vyšší než nebo rovna 90 ml/min) ukázala, že CRCL nemá žádný významný vliv na expozici kobimetinibu. Na základě populační farmakokinetické analýzy bylo zjištěno, že lehká až středně těžká porucha funkce ledvin nemá vliv na expozici kobimetinibu. K dispozici je minimální množství údajů o použití přípravku Cotellic u pacientů s těžkou poruchou funkce ledvin.
Porucha funkce jater
Farmakokinetika kobimetinibu byla hodnocena u 6 pacientů s lehkou poruchou funkce jater (Child Pugh A), 6 pacientů se středně těžkou poruchou funkce jater (Child Pugh B), 6 pacientů s těžkou poruchou funkce jater (Child Pugh C) a 10 zdravých dobrovolníků. Celkové systémové expozice kobimetinibu po jedné dávce byly podobné u pacientů s lehkou nebo středně těžkou poruchou funkce jater, v porovnání se zdravými dobrovolníky, zatímco pacienti s těžkou poruchou funkce jater měli nižší celkovou expozici kobimetinibu (AUC0-<X) geometrický průměr 0,69 v porovnání se zdravými dobrovolníky), což není považováno za klinicky významné. Expozice volnému kobimetinibu byly podobné u pacientů s lehkou a středně těžkou poruchou funkce jater v porovnání s pacienty s normální funkcí jater, zatímco expozice u pacientů s těžkou poruchou funkce jater byla přibližně 2krát vyšší (viz bod 4.2).
Pediatrická populace
Žádné studie hodnotící farmakokinetiku kobimetinibu u pediatrických pacientů nebyly provedeny.
5.3 Předklinické údaje vztahující se k bezpečnosti
Studie kancerogenity s kobimetinibem nebyly provedeny. Standardní studie genotoxicity s kobimetinibem byly negativní.
Nebyly provedeny žádné studie s kobimetinibem na zvířatech specificky zaměřené na fertilitu. Ve studiích toxicity byly pozorovány degenerativní změny v reprodukčních tkáních včetně zvýšené apoptózy/nekrózy žlutého tělíska a epitelových buněk semenných váčků, nadvarlat a pochvy u potkanů a epitelových buněk nadvarlat u psů. Klinický význam těchto jevů není znám.
Při podání kobimetinibu březím samicím potkanů došlo k embryoletalitě a malformaci velkých cév a lebky plodu při systémových expozicích podobných expozici dosahované u člověka při podávání doporučené dávky.
Kardiovaskulární bezpečnost kobimetinibu v kombinaci s vemurafenibem nebyla hodnocena in vivo.
In vitro kobimetinib způsobil středně silnou inhibici hERG iontového kanálu (IC50 0,5 pM
[266 ng/ml]), která je přibližně 18krát vyšší než maximální plazmatické koncentrace (Cmax) dosažené při dávce 60 mg, která má být registrována (Cmax nevázaného kobimetinibu 14 ng/ml [0,03 pM]).
Ve studiích toxicity na potkanech a psech byly zjištěny zpravidla reverzibilní degenerativní změny v kostní dřeni, gastrointestinálním traktu, kůži, thymu, nadledvinách, játrech, slezině, lymfatických uzlinách, ledvinách, srdci, ovariích a pochvě při expozicích v plazmě nižších než jsou klinicky účinné hladiny. Toxické účinky omezující velikost dávku zahrnují kožní ulcerace, povrchové exsudáty a akantózu u potkanů a chronický aktivní zánět a degeneraci jícnu související s různými stupni gastroenteropatie u psů.
Ve studii toxicity po opakovaném podávání mláďatům potkanů byla systémová expozice kobimetinibu v postnatálním dni 10 2krát až 11krát vyšší než v postnatálním dni 38, kdy expozice byla podobná expozici u dospělých potkanů. U mláďat potkanů vedlo podávání kobimetinibu k podobným změnám, které byly pozorovány v klíčových studiích toxicity u dospělých potkanů, včetně reverzibilních degenerativních změn v thymu a játrech, snížení hmotnosti sleziny a štítné žlázy/příštítných tělísek, zvýšeného fosforu, bilirubinu a množství erytrocytů a sníženého množství triglyceridů. Úmrtnost se u mláďat zvířat vyskytovala při dávce (3 mg/kg), která u dospělých zvířat k úmrtnosti nevedla.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Jádro tablety Monohydrát laktosy Mikrokrystalická celulosa (E460)
Sodná sůl kroskarmelosy (E468)
Magnesium-stearát (E470b)
Potahová vrstva Polyvinylalkohol Oxid titaničitý (E171)
Makrogol 3350 Mastek (E 553b)
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
4 roky.
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
6.5 Druh obalu a obsah balení
Průhledné PVC/PVDC blistry obsahující 21 tablet. Jedno balení obsahuje 63 tablet.
6.6 Zvláštní opatření pro likvidaci přípravku
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/15/1048/001
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
20. listopadu 2015
10. DATUM REVIZE TEXTU
1. června 2016
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/.
A. VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného za propouštění šarží
Roche Pharma AG Emil-Barell-Strasse 1 D-79639 Grenzach-Wyhlen Německo
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU KRABIČKA
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Cotellic 20 mg potahované tablety Cobimetinibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna potahovaná tableta obsahuje cobimetinibum 20 mg ve formě cobimetinibi fumaras.
3. SEZNAM POMOCNÝCH LÁTEK
Tablety obsahují také laktosu. Další viz příbalová informace.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
63 potahovaných tablet
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci Peroralní podání
Lék má být užíván po dobu 21 po sobě jdoucích dnů s následující 7denní přestávkou
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Použitelné do:
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/15/1048/001
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
cotellic
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
2D čárový kód s jedinečným identifikátorem.
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
PC:
SN:
NN:
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA BLISTRECH NEBO STRIPECH BLISTR
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Cotellic 20 mg potahované tablety Cobimetinibum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Roche Registration Ltd
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro pacienta
Cotellic 20 mg potahované tablety cobimetinibum
▼ Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je psáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
• Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
• Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
• Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
• Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Cotellic a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Cotellic užívat
3. Jak se přípravek Cotellic užívá
4. Možné nežádoucí účinky
5. Jak přípravek Cotellic uchovávat
6. Obsah balení a další informace
1. Co je přípravek Cotellic a k čemu se používá
Co je přípravek Cotellic
Přípravek Cotellic je protinádorový léčivý přípravek, který obsahuje léčivou látku kobimetinib.
K čemu se přípravek Cotellic používá
Přípravek Cotellic se používá k léčbě dospělých pacientů s určitým typem nádoru kůže nazývaným melanom, který se rozšířil do dalších částí těla nebo který nelze odstranit chirurgicky.
• Používá se v kombinaci s dalším protinádorovým léčivým přípravkem nazývaným vemurafenib.
• Lze ho použít pouze u pacientů, jejichž zhoubný nádor má změnu (mutaci) v bílkovině zvané “BRAF”. Váš lékař provede test ke zjištění této mutace před zahájením léčby. Tato změna mohla vést k rozvoji melanomu.
Jak přípravek Cotellic působí
Přípravek Cotellic cíleně působí na bílkovinu nazývanou “MEK”, která je důležitá při regulaci růstu nádorových buněk. Při použití přípravku Cotellic v kombinaci s vemurafenibem (který cíleně působí na změnu bílkoviny “BRAF”) se zpomaluje nebo zastavuje další růst zhoubného nádoru.
2. Čemu musíte věnovat pozornost, než začnete přípravek Cotellic užívat
Neužívejte přípravek Cotellic:
• jestliže jste alergický(á) na kobimetinib nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Pokud si nejste jistý(á), poraďte se se svým lékařem, lékárníkem nebo zdravotní sestrou před užitím přípravku Cotellic.
Upozornění a opatření
Před užitím přípravku Cotellic se poraďte se svým lékařem, lékárníkem nebo zdravotní sestrou, jestliže máte:
• problémy s očima
• problémy se srdcem
• problémy s játry
Pokud se Vás cokoli z výše uvedeného týká (nebo si nejste jistý(á)), poraďte se se svým lékařem, lékárníkem nebo zdravotní sestrou před užitím přípravku Cotellic.
• Problémy s očima
Přípravek Cotellic může způsobit problémy s očima (viz také „Problémy s očima (se zrakem) “ v bodě 4). Sdělte ihned svému lékaři, pokud se u Vás vyskytnou následující příznaky: rozmazané vidění, zkreslené vidění, částečná ztráta zraku nebo jiné změny zraku v průběhu léčby. Váš lékař Vám provede oční vyšetření, pokud se u Vás v průběhu užívání přípravku Cotellic vyskytnou nové nebo zhoršující se problémy se zrakem.
• Problémy se srdcem
Přípravek Cotellic může snížit množství krve čerpané srdcem (viz také „Problémy se srdcem “ v bodě 4). Před začátkem a v průběhu léčby přípravkem Cotellic Vám Váš lékař provede vyšetření a zkontroluje, jak dobře je srdce schopno čerpat krev. Sdělte ihned svému lékaři, pokud máte pocit bušení srdce, zrychleného nebo nepravidelného tlukotu srdce nebo pokud zaznamenáte závrať, točení hlavy, dušnost, únavu nebo otoky nohou.
• Problémy s játry
Přípravek Cotellic může zvýšit hodnoty některých jaterních enzymů v krvi v průběhu léčby. Váš lékař provede krevní testy ke zjištění hladin těchto enzymů a bude sledovat správnou funkci jater.
• Průjem
Informujte ihned svého lékaře, pokud se u Vás vyskytne průjem. Těžký průjem může způsobit ztrátu tělních tekutin (dehydrataci). Postupujte podle instrukcí lékaře, který Vám poradí, co dělat pro prevenci nebo léčbu průjmu.
Děti a dospívající
Přípravek Cotellic se nedoporučuje podávat dětem a dospívajícím. Účinky přípravku Cotellic u osob mladších 18 let nejsou známy.
Další léčivé přípravky a přípravek Cotellic
Informujte svého lékaře, lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat. Důvodem je, že přípravek Cotellic může mít vliv na účinek některých dalších léčivých přípravků. Zároveň některé další léčivé přípravky mohou mít vliv na účinek přípravku Cotellic.
Informujte před zahájením léčby přípravkem Cotellic svého lékaře, pokud užíváte:
|
Léčivý přípravek nebo léčivá látka |
Účel léčivého přípravku |
|
itrakonazol, klaritromycin, erythromycin, telithromycin, vorikonazol, rifampicin, posakonazol, flukonazol, mikonazol |
k léčbě určitých plísňových a bakteriálních infekcí |
|
ritonavir, kobicistat, lopinavir, delavirdin, amprenavir, fosamprenavir |
k léčbě HIV |
|
telaprevir |
k léčbě žloutenky C |
|
nefadozon |
k léčbě deprese |
|
amiodaron |
k léčbě nepravidelného srdečního tepu |
|
diltiazem, verapamil |
k léčbě vysokého tlaku |
|
imatinib |
k léčbě nádorového onemocnění |
|
karbamazepin, fenytoin |
k léčbě epileptických záchvatů |
|
Třezalka tečkovaná |
bylinný přípravek používaný k lečbě deprese. Je dostupný bez lékařského předpisu. |
Přípravek Cotellic s jídlem a pitím
Vyhněte se užívání přípravku Cotellic s grepfruitovým džusem. Je to z důvodu, že by mohlo dojít ke zvýšení množství přípravku Cotellic v krvi.
Těhotenství a kojení
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek užívat.
• Přípravek Cotellic se nedoporučuje užívat během těhotenství - ačkoliv účinky přípravku Cotellic nebyly u těhotných žen studovány, přípravek může způsobit trvalé poškození nebo vrozené vady u nenarozeného dítěte.
• Pokud otěhotníte během léčby přípravkem Cotellic nebo do 3 měsíců od užití Vaší poslední dávky, informujte ihned svého lékaře.
• Není známo, zda je přípravek Cotellic vylučován do mateřského mléka. Pokud kojíte, Váš lékař Vás bude informovat o prospěchu a rizicích užívání přípravku Cotellic.
Antikoncepce
Ženy, které mohou otěhotnět, mají během léčby přípravkem Cotellic a po dobu alespoň 3 měsíců po ukončení léčby používat dvě účinné antikoncepční metody, jako je kondom nebo jiná bariérová metoda (spermicidní, pokud jsou dostupné). Zeptejte se svého lékaře, která antikoncepce je pro Vás nejvhodnější.
Řízení dopravních prostředků a obsluha strojů
Přípravek Cotellic může ovlivnit schopnost řídit nebo obsluhovat stroje. Vyhněte se řízení nebo obsluze strojů, pokud máte problémy se zrakem nebo další problémy, které mohou ovlivnit Vaše schopnosti, např. pokud máte závrať nebo jste unaven(a). Pokud si nejste jistý(á), poraďte se se svým lékařem.
Přípravek Cotellic obsahuje laktosu
Tablety obsahují laktosu (určitý typ cukru). Pokud Vám lékař řekl, že nesnášíte některé cukry, poraďte se se svým lékařem, než začnete tento přípravek užívat.
3. Jak se přípravek Cotellic užívá
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře nebo lékárníka. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Kolik tablet je třeba užívat
Doporučená dávka jsou 3 tablety (celkem 60 mg) jedenkrát denně.
• Užívejte tablety každý den po dobu 21 dnů (nazýváno “období léčby”).
• Po 21 dnech neužívejte žádné tablety přípravku Cotellic po dobu 7 dnů. Během této 7denní přestávky v léčbě přípravkem Cotellic je třeba, abyste pokračoval(a) v užívání vemurafenibu podle pokynů svého lékaře.
• Další 21denní období léčby s přípravkem Cotellic začněte po uvedené 7denní přestávce.
• Pokud se u Vás objeví nežádoucí účinky, Váš lékař může rozhodnout o snížení dávky, dočasném přerušení nebo trvalém ukončení léčby. Vždy užívejte přípravek Cotellic přesně podle pokynů svého lékaře nebo lékárníka.
Užívání tablet
• Tablety spolkněte celé a zapijte sklenicí vody.
• Přípravek Cotellic se může užívat s jídlem nebo bez jídla
Pokud je Vám špatně
Pokud je Vám špatně (zvracíte) po užití přípravku Cotellic, neužívejte navíc další dávku přípravku Cotellic v ten samý den. Pokračujte s užíváním přípravku Cotellic jako obvykle, následující den.
Jestliže jste užil(a) více přípravku Cotellic, než jste měl(a)
Jestliže jste užil(a) více přípravku Cotellic, než jste měl(a), sdělte to okamžitě svému lékaři. S sebou si vezměte balení tohoto léčivého přípravku a tuto příbalovou informaci.
Jestliže jste zapomněl(a) užít přípravek Cotellic
• Jestliže do další dávky zbývá více než 12 hodin, užijte vynechanou dávku, jakmile si vzpomenete.
• Jestliže do další dávky zbývá méně než 12 hodin, zapomenutou dávku vynechejte. Poté užijte následující dávku v obvyklý čas.
• Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku.
Jestliže jste přestal(a) užívat přípravek Cotellic
Je důležité pokračovat v léčbě přípravkem Cotellic tak dlouho, jak Vám jej předepsal Váš lékař. Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého. Pokud se u Vás objeví nežádoucí účinky, Váš lékář může rozhodnout o snížení dávky, dočasném přerušení nebo trvalém ukončení léčby.
Prosím, přečtěte si také příbalovou informaci pro léčivý přípravek obsahující vemurafenib, který se užívá v kombinaci s přípravkem Cotellic.
Závažné nežádoucí účinky
Informujte ihned svého lékaře, pokud zaznamenáte jakýkoli z nežádoucích účinků uvedený níže nebo pokud se tyto příznaky zhorší v průběhu léčby.
Problémy s očima (se zrakem) (velmi časté: mohou postihnout více než 1 osobu z 10) Přípravek Cotellic může způsobit problémy s očima. Některé z těchto problémů se zrakem mohou být důsledkem onemocnění zvaného “serózní retinopatie” (hromadění tekutiny pod sítnicí v oku). Mezi příznaky serózní retinopatie patří:
• rozmazané vidění
• zkreslené vidění
• částečná ztráta zraku
• jakékoli jiné změny zraku.
Problémy se srdcem (časté: mohou postihnout až 1 z 10 osob)
Přípravek Cotellic může snížit množství krve čerpané srdcem. Mezi příznaky patří:
• pocit závratě
• pocit točení hlavy
• pocit dušnosti
• pocit únavy
• pocit bušení srdce, zrychleného nebo nepravidelného tlukotu srdce
• otoky nohou.
Průjem (velmi časté: mohou postihnout více než 1 osobu z 10)
Informujte ihned svého lékaře, pokud se u Vás průjem vyskytne a postupujte podle instrukcí lékaře, který Vám poradí, co dělat pro prevenci nebo léčbu průjmu.
Další nežádoucí účinky
Informujte svého lékaře, lékárníka nebo zdravotní sestru, jestliže zaznamenáte kterýkoli z následujících nežádoucích účinků:
Velmi časté (mohou postihnout více než 1 osobu z 10)
• zvýšená citlivost kůže na sluneční záření
• kožní vyrážka
• nevolnost (pocit na zvracení)
• horečka
• třesavka
• zvýšené hladiny jaterních enzymů (prokázané krevními testy)
• abnormální výsledky krevních testů vztahující se ke kreatinfosfokináze, což je enzym vyskytující se zejména v srdci, mozku a kosterním svalstvu
• zvracení
• kožní vyrážka s plochými skvrnami na kůži se změněnou barvou nebo zvýšené hrbolky na kůži připomínající akné
• vysoký krevní tlak
• anémie (nízké hodnoty červených krvinek)
• krvácení
• abnormální zesílení vrsty kůže.
Časté (mohou postihnout až 1 z 10 osob)
• n ěkteré typy kožních nádorů, jako jsou bazocelulární karcinom, spinocelulární karcinom kůže a keratoakantom
• dehydratace, kdy v těle není dostatečné množství tekutin
• snížená hladina fosfátu nebo sodíku v krvi (prokázaná krevními testy)
• zvýšená hladina cukru v krvi (prokázaná krevními testy)
• zvýšená hladina jaterního barviva (nazývaného „bilirubin“) v krvi. Mezi příznaky patří zežloutnutí kůže nebo očí
• zápal plic, který může způsobit dýchací potíže a může být život ohrožující (nazývaný „pneumonitida“).
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Cotellic uchovávat
• Uchovávejte tento přípravek mimo dohled a dosah dětí.
• Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na blistru za „EXP“ nebo na krabičce za „Použitelné do:“. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
• Tento přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
• Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co přípravek Cotellic obsahuje
• Léčivou látkou je cobimetinibum. Jedna potahovaná tableta obsahuje cobimetinibum 20 mg ve formě cobimetinibi fumaras.
• Dalšími složkami jsou:
• monohydrát laktosy, mikrokrystalická celulosa, sodná sůl kroskarmelosy a magnesium-stearát v jádru tablety; a
• polyvinylalkohol, oxid titaničitý, makrogol a mastek v potahové vrstvě.
Jak přípravek Cotellic vypadá a co obsahuje toto balení
Přípravek Cotellic jsou bílé, kulaté potahované tablety s vyraženým “COB” na jedné straně. Dostupná velikost balení: 63 tablet (3 blistry po 21 tabletách).
Držitel rozhodnutí o registraci
Roche Registration Limited 6 Falcon Way Shire Park
Welwyn Garden City AL7 1TW Velká Británie
Výrobce
Roche Pharma AG Emil-Barell-Strasse 1 D-79639
Grenzach-Wyhlen
Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci.
Belgie/Belgique/Belgien
N.V. Roche S.A.
Tél/Tel: +32 (0) 2 525 82 11
Bt^rapuu
Pom Etarapna EOOfl Tem +359 2 818 44 44
Magyarország
Česká republika
Roche s. r. o.
Tel: +420 - 2 20382111
Danmark
Roche a/s
Tlf: +45 - 36 39 99 99
Deutschland
Roche Pharma AG Tel: +49 (0) 7624 140
Eesti
Roche Eesti OU Tel: + 372 - 6 177 380
EXlába
Roche (Hellas) A.E.
TnU +30 210 61 66 100
Espaňa
Roche Farma S.A.
Tel: +34 - 91 324 81 00
France
Roche
Tél: +33 (0) 1 47 61 40 00
Hrvatska Roche d.o.o.
Tel: +385 1 4722 333
Ireland
Roche Products (Ireland) Ltd. Tel: +353 (0) 1 469 0700
Ísland
Roche a/s c/o Icepharma hf Sími: +354 540 8000 Italia
Roche S.p.A.
Tel: +39 - 039 2471
Kúrcpog
r.A.Exapáxn? & Eia AxS. Tn^: +357 - 22 76 62 76
Latvija
Roche Latvija SIA Tel: +371 - 6 7039831
Roche (Magyarország) Kft.
Tel: +36 - 23 446 800
Malta
(ara United Kingdom )
Nederland
Roche Nederland B.V.
Tel: +31 (0) 348 438050
Norge
Roche Norge AS Tlf: +47 - 22 78 90 00
Osterreich
Roche Austria GmbH Tel: +43 (0) 1 27739
Polska
Roche Polska Sp.z o.o.
Tel: +48 - 22 345 18 88
Portugal
Roche Farmaceutica Química, Lda Tel: +351 - 21 425 70 00
Románia
Roche Románia S.R.L.
Tel: +40 21 206 47 01
Slovenija
Roche farmacevtska družba d.o.o. Tel: +386 - 1 360 26 00
Slovenská republika
Roche Slovensko, s.r.o.
Tel: +421 - 2 52638201
Suomi/Finland
Roche Oy
Puh/Tel: +358 (0) 10 554 500
Sverige
Roche AB
Tel: +46 (0) 8 726 1200
United Kingdom
Roche Products Ltd.
Tel: +44 (0) 1707 366000
Tato příbalová informace byla naposledy revidována {měsíc RRRR}.
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
38