Cosentyx 150 Mg
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Cosentyx 150 mg prášek pro injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna lahvička prášku obsahuje secukinumabum* 150 mg. Po rekonstituci 1 ml roztoku obsahuje secukinumabum* 150 mg .
*Secukinumab je rekombinantní plně humánní monoklonální protilátka selektivní pro interleukin-17A. Secukinumab patří do IgG1/K-třídy produkované v buňkách ovarií čínského křečka (CHO).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek pro injekční roztok Prášek je bílý pevný lyofilizát.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Ložisková psoriáza
Přípravek Cosentyx je indikován k léčbě středně těžké až těžké ložiskové psoriázy dospělých, kteří jsou kandidáty pro systémovou léčbu.
Psoriatická artritida
Přípravek Cosentyx, samotný nebo v kombinaci s metotrexátem (MTX), je indikován k léčbě aktivní psoriatické artritidy u dospělých pacientů, u nichž se nedostavila adekvátní odpověď na předchozí léčbu chorobu modifikujícími antirevmatiky (DMARD) (viz bod 5.1).
Ankvlozující spondylitida
Přípravek Cosentyx je indikován k léčbě aktivní ankylozující spondylitidy u dospělých, kteří nedostatečně reagovali na konvenční léčbu.
4.2 Dávkování a způsob podání
Přípravek Cosentyx je určen k použití pod vedením a dohledem lékaře obeznámeného s diagnostikou a léčbou stavů, u nichž je přípravek Cosentyx indikován.
Dávkování
Ložisková _ psoriáza
Doporučená dávka je 300 mg secukinumabu ve formě subkutánní injekce s iniciálním podáním v týdnech 0, 1, 2 a 3, následovaná měsíční udržovací dávkou od týdne 4. Každá dávka 300 mg je podána ve dvou dílčích subkutánních injekcích po 150 mg.
Psoriatická artritida
U pacientů se současně přítomnou středně těžkou až těžkou ložiskovou psoriázou nebo u pacientů nedostatečně odpovídajících na anti-TNFa (IR), je doporučená dávka 300 mg ve formě subkutánní injekce s iniciálním podáním v týdnech 0, 1, 2 a 3, následovaná měsíční udržovací dávkou od týdne 4. Každá dávka 300 mg je podána ve dvou dílčích subkutánních injekcích po 150 mg.
U ostatních pacientů je doporučená dávka 150 mg ve formě subkutánní injekce s iniciálním podáním v týdnech 0, 1, 2 a 3, následovaná měsíční udržovací dávkou od týdne 4.
Ankylozující spondylitida
Doporučená dávka je 150 mg ve formě subkutánní injekce s iniciálním podáním v týdnech 0, 1, 2 a 3, následovaná měsíční udržovací dávkou od týdne 4.
Dostupná data naznačují, že klinická odpověď ve všech shora uvedených indikacích se obvykle dostaví během 16 týdnů. U pacientů, u nichž se do 16. týdne nedostaví žádná terapeutická odpověď, je nutné zvážit ukončení léčby. U některých pacientů s počáteční částečnou odpovědí může dojít k následnému zlepšení při pokračování léčby nad 16 týdnů.
Zvláštní populace
Starší_pacienti (ve věku 65 let a více)
Není nutná úprava dávky (viz bod 5.2).
Zhoršená _ funkce ledvin / Zhoršená _ funkce _ jater
Přípravek Cosentyx nebyl u těchto pacientských populací studován. Nelze učinit žádná doporučení ohledně dávkování.
Pediatrická populace
Bezpečnost a účinnost přípravku Cosentyx u dětí ve věku do 18 let nebyla dosud stanovena. Nejsou dostupné žádné údaje.
Způsob podání
Přípravek Cosentyx je určen k podání ve formě subkutánní injekce. Pokud je to možné, oblasti pokožky s projevy psoriázy by neměly být použity k podání injekce. Prášek pro injekční roztok musí být před použitím rekonstituován. Návod k rekonstituci tohoto léčivého přípravku před jeho podáním je uveden v bodě 6.6 a Návodu k použití v příbalové informaci.
4.3 Kontraindikace
Těžké reakce z přecitlivělosti na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Klinicky významné aktivní infekce (např. aktivní tuberkulóza; viz bod 4.4).
4.4 Zvláštní upozornění a opatření pro použití
Infekce
Přípravek Cosentyx má potenciál zvyšovat riziko infekcí. V klinických studiích byly u pacientů léčených přípravkem Cosentyx infekce pozorovány (viz bod 4.8). Většina z nich byly mírně nebo středně závažné infekce horních cest dýchacích jako nasopharyngitis a nevyžadovaly přerušení léčby.
V souladu s mechanizmem účinku přípravku Cosentyx byly u secukinumabu v porovnání s placebem v klinických studiích s psoriázou mnohem častěji hlášeny nezávažné mukokutánní kandidové infekce (3,55 na 100 pacientoroků u secukinumabu 300 mg versus 1,00 na 100 pacientoroků u placeba) (viz bod 4.8).
Opatrnosti je zapotřebí, pokud se uvažuje o použití přípravku Cosentyx u pacientů s chronickou infekcí nebo opakovanou infekcí v anamnéze.
Pacienty je nutné poučit, aby vyhledali lékařskou pomoc, pokud se objeví známky nebo příznaky naznačující přítomnost infekce. Pokud se u pacienta rozvine závažná infekce, je nutné pacienta pečlivě sledovat a nepodávat přípravek Cosentyx, dokud infekce neodezní.
V klinických studiích nebyla hlášena zvýšená citlivost vůči tuberkulóze. Přípravek Cosentyx však nesmí být podáván pacientům s aktivní tuberkulózou. U pacientů s latentní tuberkulózou je nutné zvážit před zahájením léčby přípravkem Cosentyx antituberkulózní léčbu.
Opatrnosti je zapotřebí při předepisování přípravku Cosentyx pacientům s Crohnovou chorobou, protože v klinických hodnoceních byly pozorovány exacerbace Crohnovy choroby, v některých případech závažné, v obou skupinách s přípravkem Cosentyx a skupině s placebem. Pacienty s Crohnovou chorobou léčené přípravkem Cosentyx je nutné pečlivě sledovat.
Reakce z přecitlivělosti
V klinických studiích byly u pacientů léčených přípravkem Cosentyx vzácně pozorovány případy anafylaktických reakcí. Pokud se objeví anafylaktická nebo jiné závažné alergické reakce, musí se podávání přípravku Cosentyx okamžitě přerušit a zahájit vhodnou léčbu.
Očkování
Živé vakcíny nesmí být podávány současně s přípravkem Cosentyx.
Pacienti léčení přípravkem Cosentyx mohou současně absolvovat současné očkování inaktivovanými nebo neživými vakcínami. Ve studii po podání meningokokové a inaktivované chřipkové vakcíny, byla podobná část zdravých dobrovolníků léčených secukinumabem150 mg a těch léčených placebem schopna dosáhnout adekvátní imunitní odpovědi nejméně 4násobného zvýšení titru protilátek při meningokokové a chřipkové vakcíně. Tyto údaje naznačují, že přípravek Cosentyx nepotlačuje látkovou imunitní odpověď na meningokokovou nebo chřipkovou vakcínu.
Současná imunosupresivní léčba
Ve studiích s lupénkou nebyly bezpečnost a účinnost přípravku Cosentyx v kombinaci s imunosupresivy, včetně biologické léčby, nebo fototerapií vyhodnocovány (viz též bod 4.5).
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Živé vakcíny nesmí být podávány současně s přípravkem Cosentyx (viz též bod 4.4).
U člověka nebyly provedeny žádné studie interakcí. Přímý důkaz role IL-17A v expresi CYP450 enzymů není k dispozici. Tvorba některých CYP450 enzymů je u chronických zánětů potlačována zvýšenými hladinami cytokinů. Proto může protizánětlivá léčba, například IL-17A inhibitorem secukinumab, znamenat normalizaci hladin CYP450 s doprovodnou nižší expozicí souběžnou medikací metabolizovanou prostřednictvím CYP450. Proto nelze vyloučit klinicky relevantní účinek na substráty CYP450 s úzkým terapeutickým indexem a individuálně nastavenou dávkou (např. warfarin). U pacientů léčených tímto typem léčivých přípravků je při zahájení léčby secukinumabem nutné zvážit terapeutické monitorování.
Při současném podávání přípravku Cosentyx s metotrexátem (MTX) a/nebo s kortikosteroidy nebyly v artritických studiích (včetně pacientů s psoriatickou artritidou a ankylozující spondylitidou) pozorovány žádné interakce.
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku
Ženy ve fertilním věku musí během léčby a po dobu nejméně 20 týdnů od ukončení léčby používat účinnou metodu kontracepce.
Odpovídající údaje o podávání secukinumabu těhotným ženám nejsou k dispozici. Studie reprodukční toxicity na zvířatech nenaznačují přímé nebo nepřímé škodlivé účinky na těhotenství, embryofetální vývoj, porod nebo pstnatální vývoj (viz bod 5.3). Podávání přípravku Cosentyx v těhotenství se z preventivních důvodů nedoporučuje.
Kojení
Není známo, zda se secukinumab vylučuje do lidského mateřského mléka. Imunoglobuliny se do lidského mateřského mléka vylučují a není známo, zda se secukinumab po požití systémově absorbuje. Vzhledem k možným nežádoucím účinkům secukinumabu na kojené dítě je nutno na základě posouzení prospěšnosti kojení pro dítě a prospěšnosti léčby pro matku rozhodnout, zda během léčby a po dobu až 20 týdnů od ukončení léčby přerušit kojení nebo přerušit léčbu přípravkem Cosentyx..
Fertilita
Vliv secukinumabu na fertilitu u člověka nebyl hodnocen. Studie na zvířatech nenaznačují přímé nebo nepřímé škodlivé účinky na fertilitu (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek Cosentyx nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Přípravkem Cosentyx bylo léčeno celkem 6804 pacientů v zaslepených a otevřených klinických studiích s různými indikacemi (ložisková psoriáza, psoriatická artritida, ankylozující spondylitida a jiné autoimunitní stavy). Z tohoto počtu bylo 3671 pacientů exponováno přípravku Cosentyx po dobu nejméně jednoho roku, což reprezentuje expozici 6450 pacientoroků.
Nežádoucí účinky u ložiskové psoriázy
Čtyři placebem kontrolované studie fáze III ložiskové psoriázy byly sloučeny k vyhodnocení bezpečnosti přípravku Cosentyx v porovnání s placebem až do 12 týdnů po zahájení léčby. Celkem bylo hodnoceno 2076 pacientů (692 pacientů s dávkou 150 mg, 690 pacientů s dávkou 300 mg a 694 pacientů na placebu).
Nejčastěji hlášenými nežádoucími účinky (ADRs) byly infekce horních cest dýchacích (nejčastěji nasofaryngitida, rinitida). Většina reakcí byla mírná nebo středně závažná.
Nežádoucí účinky u psoriatické artritidy
Přípravek Cosentyx byl studován ve dvou placebem kontrolovaných studiích s psoriatickou artritidou s 1003 pacienty (703 pacientů léčených přípravkem Cosentyx a 300 pacientů na placebu) s celkovou dobou expozice 1061 pacientoroků (medián trvání expozice secukinumabem léčených pacientů:
456 dní u PsA studie 1 a 245 dní u PsA studie 2). Bezpečnostní profil pozorovaný u pacientů s psoriatickou artritidou léčených přípravkem Cosentyx je v souladu s bezpečnostním profilem u psoriázy.
Nežádoucí účinky u ankylozující spondvlitidv
Přípravek Cosentyx byl studován ve dvou placebem kontrolovaných studiích s ankylozující spondylitidou s 590 pacienty (394 pacientů léčených přípravkem Cosentyx a 196 pacientů na placebu) s celkovou dobou expozice 755 pacientoroků (medián trvání expozice secukinumabem léčených pacientů: 469 dní u AS studie 1 a 460 dní u AS studie 2). Bezpečnostní profil pozorovaný u pacientů s ankylozující spondylitidou léčených přípravkem Cosentyx je v souladu s bezpečnostním profilem u psoriázy.
Tabulkový seznam nežádoucích účinků
ADRs z klinických studií s psoriázou, psoriatickou artritidou a ankylozující spondylitidou (Tabulka 1) jsou řazeny podle systému orgánových tříd MedDRA. V každé třídě orgánových systémů jsou ADRs řazeny podle frekvence, s nejčastějšími reakcemi nejdřív. V rámci každé skupiny četnosti jsou nežádoucí účinky řazeny podle klesající závažnosti. Navíc jsou odpovídající frekvenční kategorie pro všechny nežádoucí účinky založeny na následující konvenci: velmi časté (>1/10); časté (>1/100 až <1/10); méně časté (>1/1,000 až <1/100); vzácné (>1/10,000 až <1/1,000); velmi vzácné (<1/10,000).
Tabulka 1 Seznam nežádoucích účinků z klinických studií1)
|
Systém orgánových tříd |
Frekvence |
Nežádoucí účinek |
|
Infekce a infestace |
Velmi časté |
Infekce horních cest dýchacích |
|
Časté |
Orální herpes | |
|
Méně časté |
Orální kandidóza | |
|
Tinea pedis | ||
|
Otitis externa | ||
|
Poruchy krve a lymfatického systému |
Méně časté |
Neutropenie |
|
Poruchy imunitního systému |
Vzácné |
Anafylaktické reakce |
|
Poruchy oka |
Méně časté |
Konjunktivitida |
|
Respirační, hrudní a mediastinální poruchy |
Časté |
Rhinorrhoea |
|
Gastrointestinální poruchy |
Časté |
Diarhea |
|
Poruchy kůže a podkožní tkáně |
Méně časté |
Urtikaria |
|
Placebem kontrolované klinické studie (fáze III) u pacientů s ložiskovou psoriázou, s PsA a AS exponovaných dávkám 300 mg, 150 mg nebo placebu s dobou léčby až 12 týdnů (psoriáza) nebo 16 týdnů (PsA a AS). | ||
Popis vybraných nežádoucích účinků Infekce
V placebem kontrolovaném období klinických studií s ložiskovou psoriázou (celkem 1382 pacientů léčených přípravkem Cosentyx a 694 pacientů na placebu po dobu až 12 týdnů), byly infekce hlášeny u 28,7% pacientů léčených přípravkem Cosentyx v porovnání se 18,9% pacientů na placebu. Většina infekcí se skládala z nezávažných až mírných infekcí horních cest dýchacích, jako je nasofaryngitida, které nevyžadovaly přerušení léčby. Objevil se nárůst mukózních nebo kožních kandidóz, konzistentních s mechanizmem účinku, jednalo se však o případy mírné nebo střední závažnosti, nezávažné, reagující na standardní léčbu a nevyžadující přerušení léčby. Závažné infekce se objevily u 0,14% pacientů léčených přípravkem Cosentyx a 0,3% u pacientů na placebu (viz bod 4.4).
Během celé léčebné periody (celkem 3430 léčených přípravkem Cosentyx po dobu až 52 týdnů u většiny pacientů) byly infekce hlášeny u 47,5% pacientů léčených přípravkem Cosentyx (0,9 na pacientorok dalšího sledování). Závažné infekce byly hlášeny u 1,2% pacientů léčených přípravkem Cosentyx (0,015 na pacientorok dalšího sledování).
Počet infekcí pozorovaných ve studiích psoriatické artritidy a ankylozující spondylitidy byl podobný počtu infekcí pozorovaných u psoriatických studií.
Neutropenie
V klinických studiích fáze 3 u psoriázy byla neutropenie mnohem častěji pozorována u secukinumabu než u placeba, nicméně většina případů byla mírná, přechodná a reversibilní. Neutropenie <1,0-0,5x109/l (CTCAE stupeň 3) byla hlášena u 18 z 3430 (0,5%) pacientů léčených secukinumabem, bez závislosti na dávce a bez časové souvislosti s infekcí u 15 z 18 případů. Případy závažnější neutropenie nebyly. Nezávažné infekce s obvyklou odpovědí na standardní léčbu a nevyžadující přerušení léčby přípravkem Cosentyx byly hlášeny ve zbývajících 3 případech.
Frekvence výskytu neutropenie u psoriatické artritidy a ankylozující spondylitidy je podobná jako u psoriázy.
Byly hlášeny vzácné případy neutropenie <0,5x109/l (CTCAE stupeň 4).
Reakce z přecitlivělosti
V klinických studiích byla ve vztahu k přípravku Cosentyx pozorována kopřivka a vzácné případy anafylaktické reakce (viz též bod 4.4).
Imunogenicita
V klinických studiích u psoriázy, psoriatické artritidy a ankylozující spondylitidy si méně než 1% pacientů léčených přípravkem Cosentyx vyvinulo protilátky proti secukinumabu po dobu až 52 týdnů léčby. Zhruba polovina protilátek objevivších se v souvislosti s léčbou byla neutralizujících, nicméně to nebylo spojeno se ztrátou účinnosti nebo farmakokinetickými abnormalitami.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
V klinických studiích nebyly hlášeny případy předávkování.
Dávky až do 30 mg/kg (přibližně 2000 až 3000 mg) byly v klinických studiích podány intravenózně bez známek na dávce závislé toxicity. V případě předávkování se doporučuje monitorovat pacienta kvůli známkám nebo příznakům nežádoucích účinků a je potřeba neprodleně zahájit vhodnou symptomatickou léčbu.
FARMAKOLOGICKÉ VLASTNOSTI
5.
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Imunosupresiva, inhibitory interleukinu, ATC kód: L04AC10 Mechanismus účinku
Secukinumab je plně humánní IgG1/K monoklonální protilátka, která se selektivně váže na prozánětlivý cytokin interleukin-17A (IL-17A) a neutralizuje ho. Secukinumab působí zacílením na IL-17A a inhibicí jeho interakce s receptorem pro IL-17, který je exprimován různými typy buněk, včetně keratinocytů. Jako výsledek secukinumab inhibuje uvolňování prozánětlivých cytokinů, chemokinů a mediátorů tkáňového poškození a snižuje IL-17A-zprostředkovaný příspěvek k autoimunitním a zánětlivým chorobám. V pokožce jsou dosaženy klinicky relevantní hladiny secukinumabu a jsou redukovány lokální zánětlivé markery. Jako přímý důsledek léčby secukinumabem dochází k redukci erytému, ztvrdnutí a odlupování pokožky přítomných v ložiskových psoriatických lézích.
IL-17A je přirozeně se vyskytující cytokin, který se účastní normální zánětlivé a imunitní odpovědi. IL-17A hraje klíčovou roli v patogenezi ložiskové psoriázy, psoriatické artritidy a ankylozující spondylitidy a jeho množství je zvýšeno v kožních lézích v porovnání s pokožkou bez kožních lézí u pacientů s ložiskovou psoriázou a v synoviální tekutině pacientů s psoriatickou artritidou. Četnost výskytu IL-17-produkujících buněk byla též významně vyšší v subchondrální kostní dřeni intervertebrálních kloubů u pacientů s ankylozující spondylitidou.
Farmakodynamické účinky
Sérové hladiny celkového IL-17A (volný a IL-17As navázaným secukinumabem) u pacientů léčených secukinumabem zpočátku rostou. To je následováno pomalým poklesem kvůli snížené clearance IL-17A s navázaným secukinumabem, což naznačuje, že secukinumab selektivně vychytává volný IL-17A, který hraje klíčovou roli v patogenezi ložiskové psoriázy.
Ve studii se secukinumabem došlo po jednom až dvou týdnech léčby k signifikantnímu snížení infiltrujících epidermálních neutrofilů a různých s neutrofily spojovaných markerů, které jsou u pacientů s ložiskovou psoriázou v kožních lézích zvýšeny.
Secukinumab snižoval (během 1 až 2 týdnů léčby) hladiny C-reaktivního proteinu, který je markerem zánětu.
Klinická účinnost a bezpečnost
Ložisková _ psoriáza
Bezpečnost a účinnost přípravku Cosentyx byly hodnoceny ve čtyřech randomizovaných, dvojitě zaslepených, placebem kontrolovaných studiích fáze III u pacientů se středně závažnou až závažnou ložiskovou psoriázou, kteří byli kandidáty pro fototerapii nebo systémovou terapii [ERASURE, FIXTURE, FEATURE, JUNCTURE]. Účinnost a bezpečnost přípravku Cosentyx 150 mg a 300 mg byla hodnocena buď proti placebu nebo proti etanerceptu. Navíc jedna studie hodnotila chronický léčebný režim v porovnání s ‘přeléčením podle potřeby’ [SCULPTURE].
Z 2403 pacientů, kteří byli zařazeni do placebem kontrolovaných studií bylo 79% biologicky naivních, 45% bylo nebiologických selhání a 8% byla biologická selhání (6% anti-TNF selhání a 2% anti-p40 selhání). Přibližně 15 až 25% pacientů ve studiích fáze III mělo při zahájení léčby psoriatickou artritidu (PsA).
Psoriatická studie 1 (ERASURE) hodnotila 738 pacientů. Pacienti randomizovaní na přípravek Cosentyx dostávali dávku 150 mg nebo 300 mg v týdnech 0, 1, 2, a 3, následované stejnou dávkou každý měsíc počínaje týdnem 4. Psoriatická studie 2 (FIXTURE) hodnotila 1,306 pacientů. Pacienti randomizovaní na přípravek Cosentyx dostávali dávku 150 mg nebo 300 mg v týdnech 0, 1, 2, a 3, následované stejnou dávkou každý měsíc počínaje týdnem 4. Pacienti randomizovaní na etanercept dostávali dávku 50 mg dvakrát týdně po dobu 12 týdnů, následované dávkou 50 mg každý týden. V obou studiích 1 a 2 byli pacienti randomizovaní na placebo a neodpovídající na léčbu v týdnu 12 převedeni na přípravek Cosentyx (buď 150 mg nebo 300 mg) v týdnech 12, 13, 14, a 15, následované stejnou dávkou každý měsíc počínaje týdnem 16. Všichni pacienti byli sledováni po dobu až 52 týdnů od prvního podání medikace ve studii.
Psoriatická studie 3 (FEATURE) hodnotila 177 pacientů používajících předplněné injekční stříkačky v porovnání s placebem po 12 týdnech léčby pro zhodnocení bezpečnosti, tolerability a použitelnosti samoaplikace přípravku Cosentyx pomocí předplněné injekční stříkačky. Psoriatická studie 4 (JUNCTURE) hodnotila 182 pacientů používajících předplněné pero v porovnání s placebem po 12 týdnech léčby pro zhodnocení bezpečnosti, tolerability a použitelnosti samoaplikace přípravku Cosentyx pomocí předplněného pera. V obou studiích 3 a 4 pacienti randomizovaní na přípravek Cosentyx dostávali dávku 150 mg nebo 300 mg v týdnech 0, 1, 2, a 3, následované stejnou dávkou každý měsíc počínaje týdnem 4. Pacienti byli též randomizováni na placebo v týdnech 0, 1, 2, a 3, následované stejnou dávkou každý měsíc počínaje týdnem 4.
Psoriatická studie 5 (SCULPTURE) hodnotila 966 pacientů. Všichni pacienti dostávali přípravek Cosentyx v dávce 150 mg nebo 300 mg v týdnech 0, 1, 2, 3, 4, 8 a 12 a poté byli randomizováni buď k udržovací léčbě stejnou dávkou každý měsíc počínaje týdnem 12, nebo na režim s “přeléčením v čas potřeby“ stejnou dávkou. U pacientů randomizovaných k “přeléčení v čas potřeby“ nebylo dosaženo odpovídající udržení odpovědi a proto se doporučuje fixní měsíční udržovací režim.
Koprimamí cíl placebem a aktivním komparátorem kontrolovaných studií činil podíl pacientů s dosaženou odpovědí PASI 75 a IGA mód 2011 odpovědi “čistý” nebo “téměř čistý” v porovnání s placebem v týdnu 12 (viz Tabulku 2 a 3). Dávka 300 mg poskytovala zlepšenou clearance pokožky zejména pro “čistou” nebo “téměř čistou” pokožku v rozmezí cílů účinnosti PASI 90, PASI 100, a IGA mód 2011 0 nebo 1 odpověď u všech studií s největším účinkem pozorovaným v týdnu 16, proto se doporučuje tato dávka.
Tabulka 2 Souhrnná PASI 50/75/90/100 & IGA* mód 2011 “čistá” nebo “téměř čistá” klinická odpověď v psoriatických studiích 1, 3 a 4 (ERASURE, FEATURE and JUNCTURE)
Týden 12_Týden 16_Týden 52
|
Placebo |
150 mg |
300 mg |
150 mg |
300 mg |
150 mg |
300 mg | |
|
Studie 1 | |||||||
|
Počet pacientů |
246 |
244 |
245 |
244 |
245 |
244 |
245 |
|
Odpověď PASI 50 n (%) |
22 |
203 |
222 |
212 |
224 |
187 |
207 |
|
(8,9%) |
(83,5%) |
(90,6%) |
(87,2%) |
(91,4%) |
(77%) |
(84,5%) | |
|
Odpověď PASI 75 n (%) |
11 |
174 |
200 |
188 |
211 |
146 |
182 |
|
(4,5%) |
(71,6%)* * |
(81,6%)* * |
(77,4%) |
(86,1%) |
(60,1%) |
(74,3%) | |
|
Odpověď PASI 90 n (%) |
3 |
95 |
145 |
130 |
171 |
88 |
147 |
|
(1,2%) |
(39,1%)* * |
(59,2%)* * |
(53,5%) |
(69,8%) |
(36,2%) |
(60,0%) | |
|
Odpověď PASI 100 |
2 |
31 |
70 |
51 |
102 |
49 |
96 |
|
Odpověď n (%) |
(0,8%) |
(12,8%) |
(28,6%) |
(21,0%) |
(41,6%) |
(20,2%) |
(39,2%) |
|
Odpověď IGA mód 2011 |
6 |
125 |
160 |
142 |
180 |
101 |
148 |
|
“čistý” nebo “téměř |
(2,40%) |
(51,2%)* |
(65,3%)* |
(58,2%) |
(73,5%) |
(41,4%) |
(60,4%) |
|
čistý” n (%) Studie 3 |
* |
* | |||||
|
Počet pacientů |
59 |
59 |
58 |
- |
- |
- |
- |
|
Odpověď PASI 50 n (%) |
3 |
51 |
51 |
- |
- |
- |
- |
|
(5,1%) |
(86,4%) |
(87,9%) | |||||
|
Odpověď PASI 75 n (%) |
0 |
41 |
44 |
- |
- |
- |
- |
|
(0,0%) |
(69,5%)* * |
(75,9%)* * | |||||
|
Odpověď PASI 90 n (%) |
0 |
27 |
35 |
- |
- |
- |
- |
|
(0,0%) |
(45,8%) |
(60,3%) | |||||
|
Odpověď PASI 100 n |
0 |
5 |
25 |
- |
- |
- |
- |
|
(%) |
(0,0%) |
(8,5%) |
(43,1%) | ||||
|
Odpověď IGA mód 2011 |
0 |
31 |
40 |
- |
- |
- |
- |
|
“čistý” nebo “téměř |
(0,0%) |
(52,5%)* |
(69,0%)* | ||||
|
čistý” n (%) Studie 4 |
* |
* | |||||
|
Počet pacientů |
61 |
60 |
60 |
- |
- |
- |
- |
|
Odpověď PASI 50 n (%) |
5 |
48 |
58 |
- |
- |
- |
- |
|
(8,2%) |
(80,0%) |
(96,7%) | |||||
|
Odpověď PASI 75 n (%) |
2 |
43 |
52 |
- |
- |
- |
- |
|
(3,3%) |
(71,7%)* * |
(86,7%)* * | |||||
|
Odpověď PASI 90 n (%) |
0 |
24 |
33 |
- |
- |
- |
- |
|
(0,0%) |
(40,0%) |
(55,0%) | |||||
|
Odpověď PASI 100 n |
0 |
10 |
16 |
- |
- |
- |
- |
|
(%) |
(0,0%) |
(16,7%) |
(26,7%) | ||||
|
Odpověď IGA mód 2011 |
0 |
32 |
44 |
- |
- |
- |
- |
|
“čistý” nebo “téměř |
(0,0%) |
(53,3%)* |
(73,3%)* | ||||
|
čistý” n (%) |
* |
* | |||||
* IGA mód 2011 je 5 bodová škála zahrnující “0 = čistý ”, “1 = téměř čistý”, “2 = mírný”, “3 = středně závažný” nebo “4 = závažný”, indikující lékařovo celkové hodnocení závažnosti psoriázy zaměřené na ztvrdnutí, erytém a odlupování. Léčebný úspěch “ čistý ” nebo “ téměř čistý ”znamenaly nepřítomnost známek psoriázy nebo normální až růžové zbarvené léze, netloustnutí ložisek a žádné nebo minimální místní odlupování.
** p hodnoty versus placebo a nastavené na multiplicitu: p<0,0001._
Tabulka 3 Souhrn klinické odpovědi v psoriatické studii 2 (FIXTURE)
Týden 12_Týden 16_Týden 52
|
Placeb |
150 |
300 |
Etanerc |
150 |
300 |
Etanerce |
150 |
300 |
Etanerce | |
|
o |
mg |
mg |
ept |
mg |
mg |
pt |
mg |
mg |
pt | |
|
Počet |
324 |
327 |
323 |
323 |
327 |
323 |
323 |
327 |
323 |
323 |
|
pacientů Odpověď |
49 |
266 |
296 |
226 |
290 |
302 |
257 |
249 |
274 |
234 |
|
PASI 50 n |
(15,1% |
(81,3% |
(91,6% |
(70,0%) |
(88,7% |
(93,5% |
(79,6%) |
(76,1% |
(84,8% |
(72,4%) |
|
(%) |
) |
) |
) |
) |
) |
) |
) | |||
|
Odpověď |
16 |
219 |
249 |
142 |
247 |
280 |
189 |
215 |
254 |
179 |
|
PASI 75 n |
(4,9%) |
(67,0% |
(77,1% |
(44,0%) |
(75,5% |
(86,7% |
(58,5%) |
(65,7% |
(78,6% |
(55,4%) |
|
(%) |
)2 |
) |
) |
) |
) | |||||
|
Odpověď |
5 |
137 |
175 |
67 |
176 |
234 |
101 |
147 |
210 |
108 |
|
PASI 90 n |
(1,5%) |
(41,9% |
(54,2% |
(20,7%) |
(53,8% |
(72,4% |
(31,3%) |
(45,0% |
(65,0% |
(33,4%) |
|
(%) |
) |
) |
) |
) |
) |
) | ||||
|
Odpověď |
0 (0%) |
47 |
78 |
14 |
84 |
119 |
24 (7,4%) |
65 |
117 |
32 (9,9%) |
|
PASI 100 |
(14,4% |
(24,1% |
(4,3%) |
(25,7% |
(36,8% |
(19,9% |
(36,2% | |||
|
n (%) |
) |
) |
) |
) |
) |
) | ||||
|
Odpověď |
9 |
167 |
202 |
88 |
200 |
244 |
127 |
168 |
219 |
120 |
|
IgA mód |
(2,8%) |
(51,1% |
(62,5% |
(27,2%) |
(61,2% |
(75,5% |
(39,3%) |
(51,4% |
(67,8% |
(37,2%) |
|
2011 |
)2 |
)2 |
) |
) |
) |
) |
“čistý”
nebo
“téměř
čistý”
Týden 4_Týden 16
|
Secukinumab |
Ustekinumab* |
Secukinumab |
Ustekinumab | |
|
Počet pacientů |
300 mg 334 |
335 |
300 mg 334 |
335 |
|
Odpověď PASI 75 |
167 (50,0%)2 |
69 (20,6%) |
311 (93,1%) |
277 (82,7%) |
|
n (%) Odpověď PASI 90 |
70 (21,0%) |
18 (5,4%) |
264 (79,0%)2 |
193 (57,6%) |
|
n (%) Odpověď PASI 100 |
14 (4,2%) |
3 (0,9%) |
148 (44,3%) |
95 (28,4%) |
|
n (%) Odpověď IGA mód |
126 (37,7%) |
41 (12,2%) |
277 (82,9%) |
226 (67,5%) |
2011 „čistý“ nebo
„téměř čistý“ n (%)_
* Pacienti léčení secukinumabem dostávali dávku 300 mg v týdnech 0, 1, 2 a 3 následovanou stejnou dávkou v týdnech 4, 8 a 12. Pacienti léčení ustekinumabem dostávali dávku 45 mg nebo 90 mg v týdnech 0 a 4 (dávka podle hmotnosti a schváleného dávkování)
** p hodnoty versus ustekinumab: p<0,0001
Přípravek Cosentyx byl účinný u pacientů bez předchozí systémové léčby, biologicky naivních, biologicky/anti-TNF-exponovaných a pacientů s biologickým/anti-TNF selháním. Zlepšení PASI 75 u pacientů se souběžnou psoriatickou artritidou při zahájení léčby byly podobné těm u celé populace s ložiskovou psoriázou.
Přípravek Cosentyx byl spojován s rychlým nástupem účinku s 50% redukcí průměrného PASI do týdne 3 při dávce 300 mg.
Obrázek 1 časový průběh procentní změny od výchozích hodnot průměrného PASI skóre ve Studii 1 (ERASURE)
PASI % změna od základních hodnot
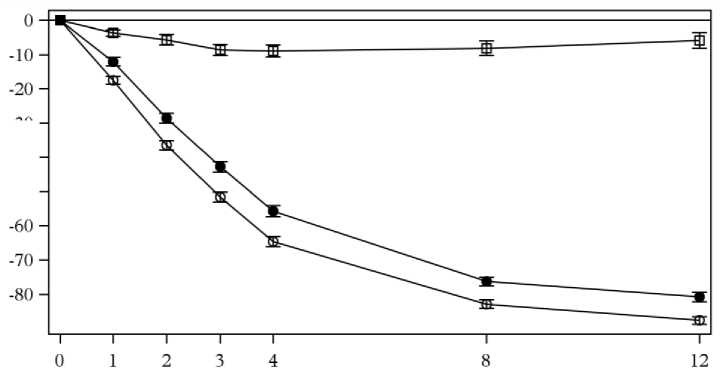
Týdny léčby
n = po čet vyhodnotitelných pacientů
• Cosentyx 150 mg (n=243) o Cosentyx 300 mg (n=245) □ Placebo (n=245)
Specifické lokalizace/formy ložiskové psoriázy
Ve dvou dalších placebem kontrolovaných studiích bylo pozorováno zlepšení jak u nehtové psoriázy (studie TRANSFIGURE, 198 pacientů), tak u palmoplantární ložiskové psoriázy (studie GESTURE, 205 pacientů). Ve studii TRANSFIGURE byla prokázána superiorita secukinumabu oproti placebu v 16. týdnu (46,1% pro 300 mg, 38,4% pro 150 mg a 11,7% pro placebo) ve výrazném zlepšení oproti počátečnímu stavu podle indexu závažnosti psoriázy nehtů (NAPSI %) u pacientů se středně těžkou až těžkou ložiskovou psoriázou s postižením nehtů. Ve studii GESTURE byla prokázána superiorita secukinumabu oproti placebu v 16. týdnu (33,3% pro 300 mg, 22,1% pro 150 mg a 1,5% pro placebo) ve výrazném zlepšení oproti počátečnímu stavu podle odpovědi ppIGA 0 nebo 1 („čistý“ nebo „téměř čistý“) u pacientů se středně těžkou až těžkou palmoplantární ložiskovou psoriázou.
Kvalita života/výsledky hlášené pacienty
Statisticky zlepšení hodnot v týdnu 12 (Studie 1-4) oproti hodnotám před léčbou v porovnání s placebem bylo demonstrováno pomocí DLQI (Dermatology Life Quality Index). Průměrný pokles (zlepšení) DLQI z hodnot před léčbou kolísalo od -10,4 do -11,6 u secukinumabu 300 mg, od -7,7 do -10,1 u secukinumabu 150 mg, v porovnání s -1,1 až -1,9 u placeba v týdnu 12. Toto zlepšení přetrvávalo 52 týdnů (Studie 1 a 2).
Čtyřicet procent účastníků ve studiích 1 a 2 vyplnilo deník Psoriasis Symptom Diary©. U účastníků, kteří vyplnili tento deník, bylo v každé z těchto studií v porovnání s placebem prokázáno statisticky významné zlepšení z hodnot před léčbou v týdnu 12 u pacienty hlášených známek a příznaků svědění, bolesti a odlupování.
Psoriatická artritida
Bezpečnost a účinnost přípravku Cosentyx byla hodnocena u 1003 pacientů ve dvou randomizovaných, dvojitě zaslepených, placebem kontrolovaných studiích fáze III u pacientů s aktivní psoriatickou artritidou (>3 oteklé a >3 bolestivé klouby) přes léčbu nesteroidními protizánětlivými léky (NSAID), kortikosteroidy nebo chorobu modifikujícími antirevmatiky (DMARD). Do těchto studií byli zařazeni pacienti všech subtypů PsA, včetně polyartikulární artritidy bez průkazu revmatoidních nodulů, spondylitidy s periferní artritidou, asymetrickou periferní artritidou, postižení distálních interfalangeálních kloubů a arthritis mutilans. Pacienti v těchto studiích měli stanovenu diagnózu PsA s mediánem 3,9 až 5,3 roku. Většina pacientů měla též aktivní psoriatické kožní léze nebo dokumentovanou psoriázu v anamnéze. Přes 62% a 47% PsA pacientů mělo před zahájením enthesitidu, respektive daktylitidu. V obou studiích bylo dosaženo primárního cíle - odpovědi ACR20 (American College of Rheumatology) v týdnu 24.
Ve studii psoriatické artritidy 1 (PsA studie 1) a studii psoriatické artritidy 2 (PsA studie 2) 29% pacientů, respektive 35% pacientů, bylo již léčeno anti-TNFa přípravkem a ukončilo užívání anti-TNFa přípravku buď pro nedostatek účinku nebo nesnášenlivost (anti-TNFa-IR pacienti).
PsA studie 1 (FUTURE 1) hodnotila 606 pacientů, z nichž 60,7% užívalo současně MTX. Pacienti randomizovaní na přípravek Cosentyx dostávali dávku 10 mg/kg intravenózně v týdnech 0, 2 a 4, následovanou buď 75 mg nebo 150 mg subkutánně každý měsíc počínaje týdnem 8. Pacienti randomizovaní na placebo byli převedeni na přípravek Cosentyx (buď 75 mg nebo 150 mg subkutánně) s následnou stejnou dávkou každý měsíc, non-respondéři v 16. týdnu (časná záchrana) a ostatní ve 24. týdnu.
PsA studie 2 (FUTURE 2) hodnotila 397 pacientů, z nichž 46,6% užívalo současně MTX. Pacienti randomizovaní na přípravek Cosentyx dostávali dávku 75 mg, 150 mg nebo 300 mg subkutánně v týdnech 0, 1, 2 a 3, s následnou stejnou dávkou každý měsíc, počínaje týdnem 4. Pacienti randomizovaní na placebo, kteří v týdnu 16 neodpovídali na léčbu byli v 16. týdnu (časná záchrana) převedeni na přípravek Cosentyx (buď 150 mg nebo 300 mg subkutánně) s následnou stejnou dávkou každý měsíc. Pacienti randomizovaní na placebo, kteří v 16. týdnu měli odpověď na léčbu, byli v 24. týdnu převedeni na přípravek Cosentyx (buď 150 mg nebo 300 mg subkutánně) s následnou stejnou dávkou každý měsíc.
Známky a příznaky
Léčba přípravkem Cosentyx v porovnání s placebem znamenala významné zlepšení v hodnocení aktivity choroby v týdnu 24 (viz Tabulka 5).
Tabulka 5 Klinická odpověď u PsA studie 2 v týdnu 24
|
Týden 24 | ||||
|
Placebo |
75 mg |
150 mg |
300 mg | |
|
Počet randomizovaných pacientů |
98 |
99 |
100 |
100 |
|
ACR20 odpověď n (%) |
15 (15,3%) |
29 (29,3%*) |
51 (51,0%***) |
54 (54,0%***) |
|
ACR50 odpověď n (%) |
7 (7,1%) |
18 (18,2%) |
35 (35,0%) |
35 (35,0%**) |
|
ACR70 odpověď n (%) |
1 (1,0%) |
6 (6,1%) |
21 (21,0%**) |
20 (20,0%**) |
|
DAS28-CRP |
-0,96 |
-1,12 |
-1,58** |
-1,61** |
|
Počet pacientů s >3% BSA psoriatickým postižením pokožky před zahájením |
43 (43,9%) |
50 (50,5%) |
58 (58,0%) |
41 (41,0%) |
|
PASI 75 odpověď n (%) |
7 (16,3%) |
14 (28,0%) |
28 (48,3%**) |
26 (63,4%***) |
|
PASI 90 odpověď n (%) |
4 (9,3%) |
6 (12,0%) |
19 (32,8%**) |
20 (48,8%***) |
|
Vymizení daktylitidy n (%) f |
4 (14,8%) |
10 (30,3%) |
16 (50,0%**) |
26 (56,5%**) |
|
Vymizení entesitidy n (%) $ |
14 (21,5%) |
22 (32,4%) |
27 (42,2%*) |
27 (48,2%**) |
* p<0,05, ** p<0,01, *** p<0,001; oproti placebu
Všechny p-hodnoty j sou nastaveny na mnohočetné testování založené na předem definované hierarchii, s výjimkou ACR70, daktylitidy a entesitidy, což byly výzkumné cíle.
Pacienti s chybějícími údaji o léčebné odpovědi byly považováni za non-respondéry.
ACR: American College of Rheumatology; PASI: Psoriasis Area and Severity Index; DAS: Disease Activity Score (skóre aktivity nemoci); BSA: Body Surface Area (povrch těla) fU pacientů s daktylitidou při zahájení (n=27, 33, 32, 46)
t U pacientů s entesitidou při zahájení (n=65, 68, 64, 56)_
Nástup účinku přípravku Cosentyx byl pozorován již ve 2. týdnu. Statisticky významného rozdílu v ACR 20 oproti placebu bylo dosaženo ve 3. týdnu. V 16. týdnu vykazovali pacienti léčení přípravkem Cosentyx významné zlepšení známek a příznaků s významně vyšší odpovědí v ACR 20 (33,3%, 60,0% a 57,0% u 75 mg, respektive 150 mg a 300 mg) v porovnání s placebem (18,4%).
Procento pacientů s dosaženou ACR 20 odpovědí je na obrázku 2.
Obrázek 2 ACR20 odpověď v PsA studii 2 v závislosti na čase do týdne 24
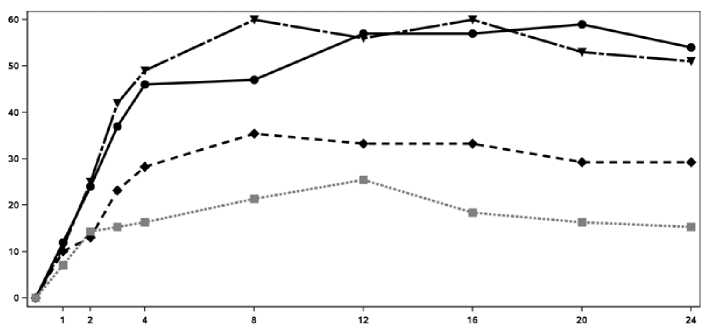
Čas (týdny)
■ Cosentyx 150 mg Cosentyx 300 mg ■ ■ ■ » Placebo
Procento respondérů
Podobné odpovědi pro primární a klíčový sekundární cíl byly pozorovány u PsA pacientů bez ohledu na to, zda byli současně léčeni MTX či nikoliv. V týdnu 24 měli pacienti léčení přípravkem Cosentyx a současně MTX vyšší ACR 20 odpověď (44,7%, 47,7% a 54,4% u 75 mg, respektive 150 mg a 300 mg, v porovnání s placebem 20,0%) a ACR 50 odpověď (27,7%, 31,8% a 38,6% u 75 mg, respektive 150 mg a 300 mg v porovnání s placebem 8,0%). Pacienti léčení přípravkem Cosentyx bez současně podávaného MTX měli vyšší ACR 20 odpověď (15,4%, 53,6% a 53,6% u 75 mg, respektive 150 mg a 300 mg, v porovnání s placebem 10,4%) a ACR 50 odpověď (9,6%, 37,5% a 32,1% u 75 mg, respektive 150 mg a 300 mg, v porovnání s placebem 6,3%).
Jak anti-TNFa-naivní, tak anti-TNFa-IR pacienti léčení přípravkem Cosentyx měli významně vyšší ACR 20 odpověď v porovnání s placebem v týdnu 24, s mírně vyšší odpovědí u anti-TNFa-naivní skupiny (anti-TNFa-naivní: 37%, 64% a 58% u 75 mg, respektive 150 mg and 300 mg, v porovnání s placebem 15,9%; anti-TNFa-IR: 15%, 30% a 46% u 75 mg, respektive 150 mg a 300 mg, v porovnání s placebem 14,3%). V anti-TNFa-IR podskupině pacientů vykázala významně vyšší ACR 20 odpověď v porovnání s placebem (p<0,05) pouze dávka 300 mg ademonstrovala klinicky smysluplný prospěch oproti 150 mg také ve více sekundárních cílech. Zlepšení PASI 75 odpovědi bylo pozorováno v obou podskupinách a dávka 300 mg prokázala statisticky signifikantní přínos u anti-TNFa-IR pacientů.
Počet PsA pacientů s axiálním postižením byl příliš malý na to, aby bylo možné stanovit vypovídající hodnocení.
Zlepšení se objevilo u všech komponent ACR skóre, včetně hodnocení bolesti pacientem. Podíl pacientů s dosaženou modifikovanou PsA Response Criteria (PsARC) odpovědí byl větší ve skupině pacientů léčených přípravkem Cosentyx (38,4%, 62,0% a 63,0% u 75 mg, respektive 150 mg a 300 mg) v porovnání s placebem (29,6%) v týdnu 24.
V PsA studii 1 a PsA studii 2 přetrvávala účinnost do týdne 52. V PsA studii 2 bylo v 52. týdnu stále léčeno 178 pacientů (89%) z 200 pacientů iniciálně randomizovaných na Cosentyx 150 mg a 300 mg. Ze 100 pacientů randomizovaných na Cosentyx 150 mg, 64, respektive 39 a 20, vykázalo ACR 20/50/70 odpověď. Ze 100 pacientů randomizovaných na Cosentyx 300 mg, 64, respektive 44 a 24 vykázalo ACR 20/50/70 odpověď.
Radiografická odpověď
Inhibice progrese strukturálního poškození u PsA nebyla dosud prokázána za použití podkožního zaváděcího dávkovacího režimu schváleného pro klinické použití.
V PsA studii 1 byla inhibice progrese strukturálního poškození hodnocena radiograficky a vyjádřena jako změna modifikovaného celkového Sharp Score (mTSS) a jeho komponent, Erosion Score (ES) a Joint Space Narrowing skóre (JSN) v týdnech 24 a 52, v porovnání s výchozími hodnotami. Data z týdne 24 jsou uvedena v Tabulce 6.
Tabulka 6 Změna modifikovaného Total Sharp skóre u psoriatické artritidy
|
Placebo N=179 |
Cosentyx 75 mg1 N=181 |
Cosentyx 150 mg1 N=185 | |
|
Celkové skóre | |||
|
Počáteční hodnoty |
28,4 |
20,4 |
22,3 |
|
(SD) |
(63,5) |
(39,4) |
(48,0) |
|
Průměrná změna v týdnu 24 |
0,57 |
0,02* |
0,13* |
*p<0,05 založená na nominální, ale neupravené p-hodnotě 110 mg/kg v týdnech 0, 2 a 4 následovaná 75 mg nebo150 mg
Inhibice strukturálního poškození přetrvávala při léčbě přípravkem Cosentyx do týdne 52.
Procento pacientů bez progrese choroby (definované jako změna od výchozích hodnot mTSS <0,5) od randomizace do týdne 24 bylo 92,3% ve větvi se secukinumabem s dávkovacím schématem 10 mg/kg intravenózního bolusu - 75 mg subkutánně (udržovací dávka), 82,3% ve větvi se secukinumabem s dávkovacím schematem 10 mg/kg intravenózního bolusu - 150 mg subkutánně (udržovací dávka) a 75,7% ve větvi s placebem. Procento pacientů bez progrese choroby od týdne 24 do týdne 52 u secukinumabu 10 mg/kg ve formě intravenózního bolusu - následovaného buď 75 mg nebo 150 mg subkutánně (udržovací dávka) a u pacientů na placebu, kteří byli v týdnu 16 nebo 24 převedeni na 75 mg nebo 150 mg subkutánně (udržovací dávka) každé 4 týdny, bylo 85,8%, respektive 85,7% a 86,8%.
Funkční stav a kvalita života
V PsA studii 2, pacienti léčení přípravkem Cosentyx 150 mg (p=0,0555) a 300 mg (p=0,0040) vykázali zlepšení funkčního stavu v porovnání s pacienty na placebu hodnocené podle Health Assessment Questionnaire-Disability Index (HAQ-DI) v týdnu 24. Zlepšení HAQ-DI skóre bylo pozorováno bez ohledu na předchozí expozici anti-TNFa. Podobná odpověď byla pozorována v PsA studii 1.
Pacienti léčení přípravkem Cosentyx hlásili významné zlepšení se zdravím související kvality života měřené pomocí Short Form-36 Health Survey Physical Component Summary (SF-36 PCS) skóre (p<0,001). Prokázalo se též statisticky významé zlepšení, což bylo demonstrováno ve výzkumných cílech hodnocených Functional Assessment of Chronic Illness Therapy - Fatigue (FACIT-F) skóre u dávky 150 mg a 300 mg v porovnání s placebem (7,97, respektive 5,97 versus 1,63). Podobné odpovědi byly pozorovány v PsA studii 1 a účinnost přetrvávala do týdne 52.
Ankylozující spondylitida
Bezpečnost a účinnost přípravku Cosentyx byla hodnocena u 590 pacientů ve dvou randomizovaných, dvojitě zaslepených, placebem kontrolovaných studiích fáze III u pacientů s aktivní ankylozující spondylitidou (AS) s Bath Ankylosing Spondylitis Disease Activity indexem (BASDAI) >4 přes léčbu nesteroidními protizánětlivými léky (NSAID), kortikosteroidy nebo chorobu modifikujícími antirevmatiky (DMARD). Pacienti v těchto studiích měli stanovenu diagnózu AS s mediánem 2,7 až 5,8 roku. V obou studiích bylo primárním cílem nejméně 20% zlepšení Assessment of Spondyloarthritis International Society kritéria (ASAS 20) v týdnu 16.
Ve studii ankylozující spondylitidy 1 (AS studie 1) a studii ankylozující spondylitidy 2 (AS studie 2) 27,0%, respektive 38,8% pacientů, bylo dříve léčeno anti-TNFa přípravkem a léčba anti-TNFa přípravkem byla ukončena buď pro nedostatek účinku nebo nesnášenlivost (anti-TNFa-IR pacienti).
AS Studie 1 (MEASURE 1) hodnotila 371 pacientů, z nichž užívalo současně 14,8%, respektive 33,4%, MTX nebo sulfasalazin. Pacienti randomizovaní na přípravek Cosentyx dostávali dávku 10 mg/kg intravenózně v týdnech 0, 2 a 4, následovanou buď 75 mg nebo 150 mg subkutánně každý měsíc počínaje týdnem 8. Pacienti randomizovaní na placebo byli převedeni na přípravek Cosentyx (buď 75 mg nebo 150 mg subkutánně) s následnou stejnou dávkou každý měsíc, non respondéři v 16. týdnu (časná záchrana) a všichni ostatní ve 24. týdnu.
AS studie 2 (MEASURE 2) hodnotila 219 pacientů, z nichž užívalo současně 11,9%, respektive 14,2%, MTX nebo sulfasalazin. Pacienti randomizovaní na přípravek Cosentyx dostávali dávku 75 mg nebo 150 mg subkutánně v týdnech 0, 1, 2 a 3, s následnou stejnou dávkou každý měsíc, počínaje týdnem 4. V týdnu 16 byli pacienti při zahájení randomizovaní na placebo re-randomizováni na přípravek Cosentyx (buď 75 mg nebo 150 mg subkutánně) každý měsíc.
Známky a příznaky
V AS studii 2 znamenala léčba přípravkem Cosentyx 150 mg v porovnání s placebem větší zlepšení v hodnocení aktivity choroby v týdnu 16 (viz Tabulka 7).
Tabulka 7 Klinická odpověď v AS studii 2 v týdnu 16
|
Výsledek (p-hodnota versus placebo) |
Placebo (n = 74) |
75 mg (n = 73) |
150 mg (n = 72) |
|
ASAS 20 odpověď, % |
28,4 |
41,1 |
61,1*** |
|
ASAS 40 20 odpověď, % |
10,8 |
26,0 |
36,1*** |
|
hsCRP, (post-BSL/BSL ratio) |
1,13 |
0,61 |
0,55*** |
|
ASAS 5/6, % |
8,1 |
34,2 |
43 1*** |
|
ASAS částečná remise, % |
4,1 |
15,1 |
13,9 |
|
BASDAI 50, % |
10,8 |
24,7* |
30,6** |
|
ASDAS-CRP velké zlepšení |
4,1 |
15,1* |
25,0*** |
|
* p<0,05, ** p<0,01, *** p<0,001; versus placebo Všechny p-hodnoty j sou nastaveny na mnohočetné testování založené na předem definované hierarchii, s výjimkou BASDAI 50 a ASDAS-CRP Pacienti s chybějícími údaji o léčebné odpovědi byli považováni za non-respondéry. | |||
|
ASAS: Assessment of SpondyloArthritis International Society Criteria; BASDAI: Bath Ankylosing Spondylitis Disease Activity Index; hsCRP: high-sensitivity C-reactive protein; ASDAS: Ankylosing Spondylitis Disease Activity Score; BSL: baseline | |||
V AS studii 2 se nástup účinku přípravku Cosentyx 150 mg projevil již v týdnu 1 (superiorita Cosentyxu vůči placebu u odpovědi ASAS 20) a v týdnu 2 (superiorita Cosentyxu vůči placebu u odpovědi ASAS 40).
ASAS 20 se zlepšila v týdnu 16 jak u anti-TNFa-naivních pacientů (68,2% versus 31,1%; p<0,05), tak u anti-TNFa-IR pacientů (50,0% versus 24,1%; p<0,05) u přípravku Cosentyx 150 mg v porovnání s placebem.
V obou AS studiích, u pacientů léčených přípravkem Cosentyx (150 mg v AS Studii 2 a oběma režimy AS studie 1) došlo v 16. týdnu k významnému zlepšení známek a příznaků se srovnatelným rozsahem odpovědi. Účinnost přetrvávala do 52. týdne jak u anti-TNFa-naivních, tak u anti-TNFa-IR pacientů.
V AS studii 2 bylo v 52. týdnu stále léčeno 61 pacientů (84,7%) ze 72 pacientů iniciálně randomizovaných na Cosentyx 150 mg. Ze 72 pacientů randomizovaných na Cosentyx 150 mg, 45, respektive 35, vykázalo ASAS 20/40 odpověď.
Mobilita páteře
Pacienti léčení přípravkem Cosentyx v dávce 150 mg vykázali zlepšenou mobilitu páteře hodnocenou jako změnu BASMI v týdnu 16 v porovnání s hodnotami před léčbou v AS studii 1 (-0,40 versus -0,12 pro placebo; p=0,0114) a v AS studii 2 (-0,51 versus -0,22 pro placebo; p=0,0533). Tato zlepšení přetrvávala do týdne 52.
Funkční stav a se zdravím související kvalita života
V AS studii 1 a studii 2, vykázali pacienti léčení přípravkem Cosentyx 150 mg zlepšení zdravotního stavu a kvality života, což bylo měřeno AS Quality of Life Dotazníkem (ASQoL) (p=0,001) a SF-36 Physical Component Summary (SF-36PCS) (p<0,001). Pacienti léčení přípravkem Cosentyx 150 mg také vykázali statisticky významné zlepšení ve fyzické funkci ve výzkumných cílech, což bylo zhodnoceno Bath Ankylosing Spondylitis Functional Indexem (BASFI) v porovnání s placebem (-2,15 versus -0,68), a v únavě, což bylo zhodnoceno Functional Assessment of Chronic Illness Therapy-Fatigue (FACIT-Fatigue) škálou v porovnání s placebem (8,10 versus 3,30). Tato zlepšení přetrvávala do týdne 52.
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Cosentyx u ložiskové psoriázy u pediatrických pacientů ve věku od narození do 6 let a u chronické idiopatické artritidy u dětských pacientů do 2 let. (informace o použití u dětí viz bod 4.2).
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s přípravkem Cosentyx u ložiskové psoriázy u pediatrických pacientů ve věku od 6 let do 18 let a u chronické idiopatické artritidy u dětských pacientů od 2 do 18 let (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Ložisková psoriáza
Absorpce
Po jednorázové podkožní dávce 300 mg ve formě roztoku u zdravých dobrovolníků dosáhl secukinumab nejvyšší sérové koncentrace 43,2±10,4 pg/ml mezi 2 a 14 dny po podání dávky.
Na základě výsledků populační farmakokinetické analýzy secukinumab po jednorázové subkutánní dávce buď 150 mg nebo 300 mg u pacientů s ložiskovou psoriázou dosáhl nejvyšší sérové koncentrace 13,7±4,8 pg/ml a 27,3±9,5 pg/ml mezi 5 a 6 dny od podání dávky.
Na základě výsledků populační farmakokinetické analýzy byl po počátečním týdenním dávkování během prvního měsíce čas potřebný k dosažení maximální koncentrace mezi 31 a 34 dny.
Podle simulovaných údajů byla nejvyšší koncentrace v rovnovážném stavu (Cmax,ss) po subkutánním podání dávky 150 mg nebo 300 mg 27,6 pg/ml and 55,2 pg/ml. Populační farmakokinetická analýza naznačuje, že rovnovážného stavu je dosaženo po 20 týdnech při měsíčním dávkovacím režimu.
V porovnání s expozicí po jednorázovém podání ukázala populační farmakokinetická analýza, že pacienti vykazovali 2násobné zvýšení nejvyšší sérové koncentrace a plochy pod křivkou (AUC) po opakovaném měsíčním podávání během udržovacího režimu.
Populační farmakokinetická analýza ukázala že secukinumab byl u pacientů s ložiskovou psoriázou absorbován s průměrnou absolutní biologickou dostupností 73%. Napříč studiemi byla vypočítaná absolutní biologická dostupnost v rozmezí 60 až 77%.
Distribuce
Průměrný distribuční objem během terminální fáze (Vz) po jednorázovém intravenózním podání kolísal u pacientů s ložiskovou psoriázou v rozmezí od 7,10 do 8,60 litrů, což naznačuje, že secukinumab se omezeně distribuuje do periferních kompartmentů.
Biotransformace
Největší část IgG eliminace probíhá prostřednictvím intracelulárního katabolismu, následovaného fluidní fází nebo receptory zprostředkované endocytózy.
Eliminace
Průměrná systémová clearance (CL) po jednorázovém intravenózním podání kolísala u pacientů s ložiskovou psoriázou od 0,13 do 0,36 l/den. V populační farmakokinetické analýze byla průměrná systémová clearance (CL) u pacientů s ložiskovou psoriázou 0,19 l/den. CL nebyla ovlivněna pohlavím. Clearance byla závislá na dávce a čase.
Průměrný eliminační poločas zjištěný v populační farmakokinetické analýze byl u pacientů s ložiskovou psoriázou 27 dní, a kolísal od 18 do 46 dní napříč psoriatickými studiemi s intravenózním podáním.
Linearita/nelinearita
Farmakokinetika secukinumabu u pacientů s ložiskovou psoriázou po jednotlivé a opakovaných dávkách byla stanovena v četných studiích s intravenózně podanými dávkami v rozmezí od 1x 0,3 mg/kg do 3x 10 mg/kg a subkutánními dávkami v rozmezí od 1x 25 mg až opakovanými dávkami 300 mg. Expozice ve všech dávkovacích režimech odpovídala dávce.
Psoriatická artritida
Farmakokinetické vlastnosti secukinumabu pozorované u pacientů s psoriatickou artritidou byly podobné těm projevujícím se u pacientů s ložiskovou psoriázou. Biologická dostupnost secukinumabu u PsA pacientů činila 85% na základě populačního farmakokinetického modelu.
Ankylozující spondylitida
Farmakokinetické vlastnosti secukinumabu pozorované u pacientů s ankylozující spondylitidou byly podobné těm projevujícím se u pacientů s ložiskovou psoriázou.
Zvláštní populace
Starší pacienti
Z 3430 pacientů s ložiskovou psoriázou exponovaných přípravku Cosentyx v klinických studiích bylo 230 ve věku 65 let nebo starších a 32 pacientů bylo ve věku 75 nebo starších.
Z 974 pacientů s psoriatickou artritidou exponovaných přípravku Cosentyx v klinických studiích, celkem 85 pacientů bylo ve věku 65 let nebo starších a 4 pacienti byli ve věku 75 let nebo starší.
Z 571 pacientů s ankylozující spondylitidou exponovaných přípravku Cosentyx v klinických studiích, celkem 24 pacientů bylo ve věku 65 let nebo starších a 3 pacienti byli ve věku 75 let nebo starší.
Na základě populační farmakokinetické analýzy s omezeným počtem starších pacientů (n=71 pro věk >65 let a n=7 pro věk >75 let) byla clearance u starších pacientů a pacientů mladších 65 let věku podobná.
Pacienti s _poruchou _funkce ledvin nebo _jater
U pacientů s poruchou funkce ledvin nebo jater nejsou k dispozici farmakokinetická data. Předpokládá se, že renální eliminace nedotčeného přípravku Cosentyx, IgG monoklonální protilátky bude nízká a nevýznamná. IgG jsou většinou katabolizovány a neočekává se, že by porucha funkce jater ovlivnila clearance přípravku Cosentyx.
Vliv tělesné hmotnosti na farmakokinetiku
S rostoucí tělesnou hmotností rostou clearance secukinumabu a jeho distribuční objem.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě tkáňových testů zkřížené reaktivity, farmakologických studií bezpečnosti, toxicity po opakovaném podávání a reprodukční toxicity provedené u secukinumabu nebo myších protilátek proti myšímu IL-17A neodhalily žádné zvláštní riziko pro člověka.
Protože secukinumab váže opičí (cynomolgus) a lidský IL-17A, byla jeho bezpečnost studována u opic rodu cynomolgus. Po subkutánním podání secukinumabu opicím rodu cynomolgus po dobu až 13 týdnů a intravenózním podání až 26 týdnů (včetně vyhodnocení farmakokinetiky, farmakodynamiky, imunogenicity a imunotoxicity (tj. na T-buňkách závislá protilátková odpověď a aktivita NK buněk) nebyly pozorovány nežádoucí účinky. Průměrná sérová koncentrace pozorovaná u opic po 13 týdenních subkutánních dávkách 150 mg/kg byla značně vyšší než odhadovaná průměrná sérová koncentrace u psoriatických pacientů při nejvyšší klinické dávce. Protilátky proti secukinumabu byly detekovány pouze u jednoho z exponovaných zvířat. Po aplikaci secukinumabu do normální lidské tkáně nebyla pozorována nespecifická tkáňová zkřížená reaktivita.
Studie na zvířatech k posouzení karcinogenního potenciálu secukinumabu nebyly provedeny.
V embryofetální vývojové studii u opic rodu cynomolgus nevykazoval secukinumab toxicitu pro matku, embryotoxicitu nebo teratogenitu při podání během organogeneze a v pozdní gestaci.
Ve fertilitních a časných embryonálních a pre- a postnatálních studiích u myší nebyly pozorovány nežádoucí účinky myších protilátek proti myšímu IL-17A. Vyšší dávky v těchto studiích převyšovaly maximální účinnou dávku ve smyslu suprese IL-17A a účinku (viz bod 4.6).
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Sacharóza
Histidin
Monohydrát histidin-hydrochloridu Polysorbát 80
6.2 Inkompatibility
Tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky s výjimkou těch, které jsou uvedeny v bodě 6.6
3 roky
Po rekonstituci
Chemická a fyzikální in-use stabilita byla doložena na 24 hodin při 2°C až 8°C.
Z mikrobiologického hlediska musí být přípravek použit neprodleně, pokud způsob rekonstituce nevyloučí riziko mikrobiální kontaminace.
Pokud není použit okamžitě, doba a uchovávání před použitím jsou na zodpovědnosti uživatele.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2°C až 8°C).
Podmínky uchovávání tohoto léčivého přípravku po jeho rekonstituci jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
Přípravek Cosentyx je dodáván v injekční lahvičce z bezbarvého skla s gumovou zátkou šedé barvy a hliníkovou pertlí s bílou odlamovací částí.
Injekční lahvička obsahuje secukinumabum 150 mg.
Přípravek Cosentyx je dostupný v balení obsahujícím jednu lahvičku.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Jednorázová injekční lahvička obsahující 150 mg secukinumabu pro rekonstituci vodou na injekci. Připravený roztok musí být čirý a bezbarvý až mírně nažloutlý. Nepoužívejte, pokud se lyofilizovaný prášek zcela nerozpustí nebo pokud roztok obsahuje zřetelně viditelné částice, je zakalený nebo zřetelně hnědý. Podrobný návod k použití najdete v příbalové informaci.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/14/980/001
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
15.ledna 2015
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Cosentyx 150 mg injekční roztok v předplněné injekční stříkačce
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna předplněná injekční stříkačka obsahuje secukinumabum* 150 mg v 1 ml.
*Secukinumab je rekombinantní plně humánní monoklonální protilátka selektivní pro interleukin-17A. Secukinumab patří do IgG1/K-třídy produkované v buňkách ovarií čínského křečka (CHO).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok v předplněné injekční stříkačce (injekce) Roztok je čirý a bezbarvý až mírně nažloutlý.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Ložisková psoriáza
Přípravek Cosentyx je indikován k léčbě středně těžké až těžké ložiskové psoriázy dospělých, kteří jsou kandidáty pro systémovou léčbu.
Psoriatická artritida
Přípravek Cosentyx, samotný nebo v kombinaci s metotrexátem (MTX), je indikován k léčbě aktivní psoriatické artritidy u dospělých pacientů, u nichž se nedostavila adekvátní odpověď na předchozí léčbu chorobu modifikujícími antirevmatiky (DMARD) (viz bod 5.1).
Ankylozující spondylitida
Přípravek Cosentyx je indikován k léčbě aktivní ankylozující spondylitidy u dospělých, kteří nedostatečně reagovali na konvenční léčbu.
Přípravek Cosentyx je určen k použití pod vedením a dohledem lékaře obeznámeného s diagnostikou a léčboustavů, u nichž je přípravek Cosentyx indikován.
Dávkování
Ložisková _ psoriáza
Doporučená dávka je 300 mg secukinumabu ve formě subkutánní injekce s iniciálním podáním v týdnech 0, 1, 2 a 3, následovaná měsíční udržovací dávkou od týdne 4. Každá dávka 300 mg je podána ve dvou dílčích subkutánních injekcích po 150 mg.
Psoriatická artritida
U pacientů se současně přítomnou středně těžkou až těžkou ložiskovou psoriázou nebo u pacientů nedostatečně odpovídajících na anti-TNFa (IR), je doporučená dávka 300 mg ve formě subkutánní injekce s iniciálním podáním v týdnech 0, 1, 2 a 3, následovaná měsíční udržovací dávkou od týdne 4. Každá dávka 300 mg je podána ve dvou dílčích subkutánních injekcích po 150 mg.
U ostatních pacientů je doporučená dávka 150 mg ve formě subkutánní injekce s iniciálním podáním v týdnech 0, 1, 2 a 3, následovaná měsíční udržovací dávkou od týdne 4.
Ankylozující spondylitida
Doporučená dávka je 150 mg ve formě subkutánní injekce s iniciálním podáním v týdnech 0, 1, 2 a 3, následovaná měsíční udržovací dávkou od týdne 4.
Dostupná data naznačují, že klinická odpověď ve všech shora uvedených indikacích se obvykle dostaví během 16 týdnů. U pacientů, u nichž se do 16. týdne nedostaví žádná terapeutická odpověď, je nutné zvážit ukončení léčby. U některých pacientů s počáteční částečnou odpovědí může dojít k následnému zlepšení při pokračování léčby nad 16 týdnů.
Zvláštní populace
Starší_pacienti (ve věku 65 let a více)
Není nutná úprava dávky (viz bod 5.2).
Zhoršená _ funkce ledvin / Zhoršená _ funkce _ jater
Přípravek Cosentyx nebyl u těchto pacientských populací studován. Nelze učinit žádná doporučení ohledně dávkování.
Pediatrická populace
Bezpečnost a účinnost přípravku Cosentyx u dětí ve věku do 18 let nebyla dosud stanovena. Nejsou dostupné žádné údaje.
Způsob podání
Přípravek Cosentyx je určen k podání ve formě subkutánní injekce. Pokud je to možné, oblasti pokožky s projevy psoriázy by neměly být použity k podání injekce.
Po vhodném nácviku subkutánní injekční techniky, si může pacient sám aplikovat přípravek Cosentyx, pokud to lékař uzná za vhodné. Lékař však musí zajistit přiměřené sledování pacientů. Pacienti musí být instruováni injikovat celé množství přípravku Cosentyx podle instrukcí v příbalové informaci. Kompletní informace o podávání jsou uvedeny v příbalové informaci.
4.3 Kontraindikace
Těžké reakce z přecitlivělosti na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Klinicky významné aktivní infekce (např. aktivní tuberkulóza; viz bod 4.4).
4.4 Zvláštní upozornění a opatření pro použití
Infekce
Přípravek Cosentyx má potenciál zvyšovat riziko infekcí. V klinických studiích byly u pacientů léčených přípravkem Cosentyx infekce pozorovány (viz bod 4.8). Většina z nich byly mírně nebo středně závažné infekce horních cest dýchacích jako nasopharyngitis a nevyžadovaly přerušení léčby.
V souladu s mechanizmem účinku přípravku Cosentyx byly u secukinumabu v porovnání s placebem v klinických studiích s psoriázou mnohem častěji hlášeny nezávažné mukokutánní kandidové infekce (3,55 na 100 pacientoroků u secukinumabu 300 mg versus 1,00 na 100 pacientoroků u placeba) (viz bod 4.8).
Opatrnosti je zapotřebí, pokud se uvažuje o použití přípravku Cosentyx u pacientů s chronickou infekcí nebo opakovanou infekcí v anamnéze.
Pacienty je nutné poučit, aby vyhledali lékařskou pomoc, pokud se objeví známky nebo příznaky naznačující přítomnost infekce. Pokud se u pacienta rozvine závažná infekce, je nutné pacienta pečlivě sledovat a nepodávat přípravek Cosentyx, dokud infekce neodezní.
V klinických studiích nebyla hlášena zvýšená citlivost vůči tuberkulóze. Přípravek Cosentyx však nesmí být podáván pacientům s aktivní tuberkulózou. U pacientů s latentní tuberkulózou je nutné zvážit před zahájením léčby přípravkem Cosentyx antituberkulózní léčbu.
Opatrnosti je zapotřebí při předepisování přípravku Cosentyx pacientům s Crohnovou chorobou, protože v klinických hodnoceních byly pozorovány exacerbace Crohnovy choroby, v některých případech závažné, v obou skupinách s přípravkem Cosentyx a skupině s placebem. Pacienty s Crohnovou chorobou léčené přípravkem Cosentyx je nutné pečlivě sledovat.
Reakce z přecitlivělosti
V klinických studiích byly u pacientů léčených přípravkem Cosentyx vzácně pozorovány případy anafylaktických reakcí. Pokud se objeví anafylaktická nebo jiné závažné alergické reakce, musí se podávání přípravku Cosentyx okamžitě přerušit a zahájit vhodnou léčbu.
Osoby citlivé na latex
Snímatelný kryt jehly předplněné injekční stříkačky přípravku Cosentyx obsahuje derivát přirozeně se vyskytujícího gumového latexu. Ve snímatelném krytu jehly nebyl dosud detekován přirozeně se vyskytující gumový latex. Nicméně použití předplněné injekční stříkačky přípravku Cosentyx u osob citlivých na latex nebylo studováno a proto nelze možné riziko reakcí z přecitlivělosti zcela vyloučit.
Očkování
Živé vakcíny nesmí být podávány současně s přípravkem Cosentyx.
Pacienti léčení přípravkem Cosentyx mohou současně absolvovat současné očkování inaktivovanými nebo neživými vakcínami. Ve studii po podání meningokokové a inaktivované chřipkové vakcíny, byla podobná část zdravých dobrovolníků léčených secukinumabem150 mg a těch léčených placebem schopna dosáhnout adekvátní imunitní odpovědi nejméně 4násobného zvýšení titru protilátek při meningokokové a chřipkové vakcíně. Tyto údaje naznačují, že přípravek Cosentyx nepotlačuje látkovou imunitní odpověď na meningokokovou nebo chřipkovou vakcínu.
Současná imunosupresivní léčba
Ve studiích s lupénkou nebyly bezpečnost a účinnost přípravku Cosentyx v kombinaci s imunosupresivy, včetně biologické léčby, nebo fototerapií vyhodnocovány (viz též bod 4.5).
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Živé vakcíny nesmí být podávány současně s přípravkem Cosentyx (viz též bod 4.4).
U člověka nebyly provedeny žádné studie interakcí. Přímý důkaz role IL-17A v expresi CYP450 enzymů není k dispozici. Tvorba některých CYP450 enzymů je u chronických zánětů potlačována zvýšenými hladinami cytokinů. Proto může protizánětlivá léčba, například IL-17A inhibitorem secukinumab, znamenat normalizaci hladin CYP450 s doprovodnou nižší expozicí souběžnou medikací metabolizovanou prostřednictvím CYP450. Proto nelze vyloučit klinicky relevantní účinek na substráty CYP450 s úzkým terapeutickým indexem a individuálně nastavenou dávkou (např. warfarin). U pacientů léčených tímto typem léčivých přípravků je při zahájení léčby secukinumabem nutné zvážit terapeutické monitorování.
Při současném podávání přípravku Cosentyx s metotrexátem (MTX) a/nebo s kortikosteroidy nebyly v artritických studiích (včetně pacientů s psoriatickou artritidou a ankylozující spondylitidou) pozorovány žádné interakce.
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku
Ženy ve fertilním věku musí během léčby a po dobu nejméně 20 týdnů od ukončení léčby používat účinnou metodu kontracepce.
Odpovídající údaje o podávání secukinumabu těhotným ženám nejsou k dispozici. Studie reprodukční toxicity na zvířatech nenaznačují přímé nebo nepřímé škodlivé účinky na těhotenství, embryofetální vývoj, porod nebo pstnatální vývoj (viz bod 5.3). Podávání přípravku Cosentyx v těhotenství se z preventivních důvodů nedoporučuje.
Kojení
Není známo, zda se secukinumab vylučuje do lidského mateřského mléka. Imunoglobuliny se do lidského mateřského mléka vylučují a není známo, zda se secukinumab po požití systémově absorbuje. Vzhledem k možným nežádoucím účinkům secukinumabu na kojené dítě je nutno na základě posouzení prospěšnosti kojení pro dítě a prospěšnosti léčby pro matku rozhodnout, zda během léčby a po dobu až 20 týdnů od ukončení léčby přerušit kojení nebo přerušit léčbu přípravkem Cosentyx..
Fertilita
Vliv secukinumabu na fertilitu u člověka nebyl hodnocen. Studie na zvířatech nenaznačují přímé nebo nepřímé škodlivé účinky na fertilitu (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek Cosentyx nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky Souhrn bezpečnostního profilu
Přípravkem Cosentyx bylo léčeno celkem 6804 pacientů v zaslepených a otevřených klinických studiích s různými indikacemi (ložisková psoriáza, psoriatická artritida, ankylozující spondylitida a jiné autoimunitní stavy). Z tohoto počtu bylo 3671 pacientů exponováno přípravku Cosentyx po dobu nejméně jednoho roku, což reprezentuje expozici 6450 pacientoroků.
Nežádoucí účinky u ložiskové psoriázy
Čtyři placebem kontrolované studie fáze III ložiskové psoriázy byly sloučeny k vyhodnocení bezpečnosti přípravku Cosentyx v porovnání s placebem až do 12 týdnů po zahájení léčby. Celkem bylo hodnoceno 2076 pacientů (692 pacientů s dávkou 150 mg, 690 pacientů s dávkou 300 mg a 694 pacientů na placebu).
Nejčastěji hlášenými nežádoucími účinky (ADRs) byly infekce horních cest dýchacích (nejčastěji nasofaryngitida, rinitida). Většina reakcí byla mírná nebo středně závažná.
Nežádoucí účinky u psoriatické artritidy
Přípravek Cosentyx byl studován ve dvou placebem kontrolovaných studiích s psoriatickou artritidou s 1003 pacienty (703 pacientů léčených přípravkem Cosentyx a 300 pacientů na placebu) s celkovou dobou expozice 1061 pacientoroků (medián trvání expozice secukinumabem léčených pacientů:
456 dní u PsA studie 1 a 245 dní u PsA studie 2). Bezpečnostní profil pozorovaný u pacientů s psoriatickou artritidou léčených přípravkem Cosentyx je v souladu s bezpečnostním profilem u psoriázy.
Nežádoucí účinky u ankylozující spondvlitidv
Přípravek Cosentyx byl studován ve dvou placebem kontrolovaných studiích s ankylozující spondylitidou s 590 pacienty (394 pacientů léčených přípravkem Cosentyx a 196 pacientů na placebu) s celkovou dobou expozice 755 pacientoroků (medián trvání expozice secukinumabem léčených pacientů: 469 dní u AS studie 1 a 460 dní u AS studie 2). Bezpečnostní profil pozorovaný u pacientů s ankylozující spondylitidou léčených přípravkem Cosentyx je v souladu s bezpečnostním profilem u psoriázy.
Tabulkový seznam nežádoucích účinků
ADRs z klinických studií s psoriázou, psoriatickou artritidou a ankylozující spondylitidou (Tabulka 1) jsou řazeny podle systému orgánových tříd MedDRA. V každé třídě orgánových systémů jsou ADRs řazeny podle frekvence, s nejčastějšími reakcemi nejdřív. V rámci každé skupiny četnosti jsou nežádoucí účinky řazeny podle klesající závažnosti. Navíc jsou odpovídající frekvenční kategorie pro všechny nežádoucí účinky založeny na následující konvenci: velmi časté (>1/10); časté (>1/100 až <1/10); méně časté (>1/1,000 až <1/100); vzácné (>1/10,000 až <1/1,000); velmi vzácné (<1/10,000).
Tabulka 1 Seznam nežádoucích účinků z klinických studií1)
|
Systém orgánových tříd |
Frekvence |
Nežádoucí účinek |
|
Infekce a infestace |
Velmi časté |
Infekce horních cest dýchacích |
|
Časté |
Orální herpes | |
|
Méně časté |
Orální kandidóza | |
|
Tinea pedis | ||
|
Otitis externa | ||
|
Poruchy krve a lymfatického systému |
Méně časté |
Neutropenie |
|
Poruchy imunitního systému |
Vzácné |
Anafylaktické reakce |
|
Poruchy oka |
Méně časté |
Konjunktivitida |
|
Respirační, hrudní a mediastinální poruchy |
Časté |
Rhinorrhoea |
|
Gastrointestinální poruchy |
Časté |
Diarhea |
|
Poruchy kůže a podkožní tkáně |
Méně časté |
Urtikaria |
|
1) Placebem kontrolované klinické studie (fáze III) u pacientů s ložiskovou psoriázou, s PsA a AS exponovaných dávkám 300 mg, 150 mg nebo placebu s dobou léčby až 12 týdnů (psoriáza) nebo 16 týdnů (PsA a AS). | ||
Popis vybraných nežádoucích účinků
Infekce
V placebem kontrolovaném období klinických studií s ložiskovou psoriázou (celkem 1382 pacientů léčených přípravkem Cosentyx a 694 pacientů na placebu po dobu až 12 týdnů), byly infekce hlášeny u 28,7% pacientů léčených přípravkem Cosentyx v porovnání se 18,9% pacientů na placebu. Většina infekcí se skládala z nezávažných až mírných infekcí horních cest dýchacích, jako je nasofaryngitida, které nevyžadovaly přerušení léčby. Objevil se nárůst mukózních nebo kožních kandidóz, konzistentních s mechanizmem účinku, jednalo se však o případy mírné nebo střední závažnosti, nezávažné, reagující na standardní léčbu a nevyžadující přerušení léčby. Závažné infekce se objevily u 0,14% pacientů léčených přípravkem Cosentyx a 0,3% u pacientů na placebu (viz bod 4.4).
Během celé léčebné periody (celkem 3430 léčených přípravkem Cosentyx po dobu až 52 týdnů u většiny pacientů) byly infekce hlášeny u 47,5% pacientů léčených přípravkem Cosentyx (0,9 na pacientorok dalšího sledování). Závažné infekce byly hlášeny u 1,2% pacientů léčených přípravkem Cosentyx (0,015 na pacientorok dalšího sledování).
Počet infekcí pozorovaných ve studiích psoriatické artritidy a ankylozující spondylitidy byl podobný počtu infekcí pozorovaných u psoriatických studií.
Neutropenie
V klinických studiích fáze 3 u psoriázy byla neutropenie mnohem častěji pozorována u secukinumabu než u placeba, nicméně většina případů byla mírná, přechodná a reversibilní. Neutropenie <1,0-0,5x109/l (CTCAE stupeň 3) byla hlášena u 18 z 3430 (0,5%) pacientů léčených secukinumabem, bez závislosti na dávce a bez časové souvislosti s infekcí u 15 z 18 případů. Případy závažnější neutropenie nebyly. Nezávažné infekce s obvyklou odpovědí na standardní léčbu a nevyžadující přerušení léčby přípravkem Cosentyx byly hlášeny ve zbývajících 3 případech.
Frekvence výskytu neutropenie u psoriatické artritidy a ankylozující spondylitidy je podobná jako u psoriázy.
Byly hlášeny vzácné případy neutropenie <0,5x109/l (CTCAE stupeň 4).
Reakce z přecitlivělosti
V klinických studiích byla ve vztahu k přípravku Cosentyx pozorována kopřivka a vzácné případy anafylaktické reakce (viz též bod 4.4).
Imunogenicita
V klinických studiích u psoriázy, psoriatické artritidy a ankylozující spondylitidy si méně než 1% pacientů léčených přípravkem Cosentyx vyvinulo protilátky proti secukinumabu po dobu až 52 týdnů léčby. Zhruba polovina protilátek objevivších se v souvislosti s léčbou byla neutralizujících, nicméně to nebylo spojeno se ztrátou účinnosti nebo farmakokinetickými abnormalitami.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
V klinických studiích nebyly hlášeny případy předávkování.
Dávky až do 30 mg/kg (přibližně 2000 až 3000 mg) byly v klinických studiích podány intravenózně bez známek na dávce závislé toxicity. V případě předávkování se doporučuje monitorovat pacienta kvůli známkám nebo příznakům nežádoucích účinků a je potřeba neprodleně zahájit vhodnou symptomatickou léčbu.
FARMAKOLOGICKÉ VLASTNOSTI
5.
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Imunosupresiva, inhibitory interleukinu, ATC kód: L04AC10 Mechanismus účinku
Secukinumab je plně humánní IgG1/K monoklonální protilátka, která se selektivně váže na prozánětlivý cytokin interleukin-17A (IL-17A) a neutralizuje ho. Secukinumab působí zacílením na IL-17A a inhibicí jeho interakce s receptorem pro IL-17, který je exprimován různými typy buněk, včetně keratinocytů. Jako výsledek secukinumab inhibuje uvolňování prozánětlivých cytokinů, chemokinů a mediátorů tkáňového poškození a snižuje IL-17A-zprostředkovaný příspěvek k autoimunitním a zánětlivým chorobám. V pokožce jsou dosaženy klinicky relevantní hladiny secukinumabu a jsou redukovány lokální zánětlivé markery. Jako přímý důsledek léčby secukinumabem dochází k redukci erytému, ztvrdnutí a odlupování pokožky přítomných v ložiskových psoriatických lézích.
IL-17A je přirozeně se vyskytující cytokin, který se účastní normální zánětlivé a imunitní odpovědi. IL-17A hraje klíčovou roli v patogenezi ložiskové psoriázy, psoriatické artritidy a ankylozující spondylitidy a jeho množství je zvýšeno v kožních lézích v porovnání s pokožkou bez kožních lézí u pacientů s ložiskovou psoriázou a v synoviální tekutině pacientů s psoriatickou artritidou. Četnost výskytu IL-17-produkujících buněk byla též významně vyšší v subchondrální kostní dřeni intervertebrálních kloubů u pacientů s ankylozující spondylitidou.
Farmakodynamické účinky
Sérové hladiny celkového IL-17A (volný a IL-17As navázaným secukinumabem) u pacientů léčených secukinumabem zpočátku rostou. To je následováno pomalým poklesem kvůli snížené clearance IL-17A s navázaným secukinumabem, což naznačuje, že secukinumab selektivně vychytává volný IL-17A, který hraje klíčovou roli v patogenezi ložiskové psoriázy.
Ve studii se secukinumabem došlo po jednom až dvou týdnech léčby k signifikantnímu snížení infiltrujících epidermálních neutrofilů a různých s neutrofily spojovaných markerů, které jsou u pacientů s ložiskovou psoriázou v kožních lézích zvýšeny.
Secukinumab snižoval (během 1 až 2 týdnů léčby) hladiny C-reaktivního proteinu, který je markerem zánětu.
Klinická účinnost a bezpečnost
Ložisková _ psoriáza
Bezpečnost a účinnost přípravku Cosentyx byly hodnoceny ve čtyřech randomizovaných, dvojitě zaslepených, placebem kontrolovaných studiích fáze III u pacientů se středně závažnou až závažnou ložiskovou psoriázou, kteří byli kandidáty pro fototerapii nebo systémovou terapii [ERASURE, FIXTURE, FEATURE, JUNCTURE]. Účinnost a bezpečnost přípravku Cosentyx 150 mg a 300 mg byla hodnocena buď proti placebu nebo proti etanerceptu. Navíc jedna studie hodnotila chronický léčebný režim v porovnání s ‘přeléčením podle potřeby’ [SCULPTURE].
Z 2403 pacientů, kteří byli zařazeni do placebem kontrolovaných studií bylo 79% biologicky naivních, 45% bylo nebiologických selhání a 8% byla biologická selhání (6% anti-TNF selhání a 2% anti-p40 selhání). Přibližně 15 až 25% pacientů ve studiích fáze III mělo při zahájení léčby psoriatickou artritidu (PsA).
Psoriatická studie 1 (ERASURE) hodnotila 738 pacientů. Pacienti randomizovaní na přípravek Cosentyx dostávali dávku 150 mg nebo 300 mg v týdnech 0, 1, 2, a 3, následované stejnou dávkou každý měsíc počínaje týdnem 4. Psoriatická studie 2 (FIXTURE) hodnotila 1,306 pacientů. Pacienti randomizovaní na přípravek Cosentyx dostávali dávku 150 mg nebo 300 mg v týdnech 0, 1, 2, a 3, následované stejnou dávkou každý měsíc počínaje týdnem 4. Pacienti randomizovaní na etanercept dostávali dávku 50 mg dvakrát týdně po dobu 12 týdnů, následované dávkou 50 mg každý týden. V obou studiích 1 a 2 byli pacienti randomizovaní na placebo a neodpovídající na léčbu v týdnu 12 převedeni na přípravek Cosentyx (buď 150 mg nebo 300 mg) v týdnech 12, 13, 14, a 15, následované stejnou dávkou každý měsíc počínaje týdnem 16. Všichni pacienti byli sledováni po dobu až 52 týdnů od prvního podání medikace ve studii.
Psoriatická studie 3 (FEATURE) hodnotila 177 pacientů používajících předplněné injekční stříkačky v porovnání s placebem po 12 týdnech léčby pro zhodnocení bezpečnosti, tolerability a použitelnosti samoaplikace přípravku Cosentyx pomocí předplněné injekční stříkačky. Psoriatická studie 4 (JUNCTURE) hodnotila 182 pacientů používajících předplněné pero v porovnání s placebem po 12 týdnech léčby pro zhodnocení bezpečnosti, tolerability a použitelnosti samoaplikace přípravku Cosentyx pomocí předplněného pera. V obou studiích 3 a 4 pacienti randomizovaní na přípravek Cosentyx dostávali dávku 150 mg nebo 300 mg v týdnech 0, 1, 2, a 3, následované stejnou dávkou každý měsíc počínaje týdnem 4. Pacienti byli též randomizováni na placebo v týdnech 0, 1, 2, a 3, následované stejnou dávkou každý měsíc počínaje týdnem 4.
Psoriatická studie 5 (SCULPTURE) hodnotila 966 pacientů. Všichni pacienti dostávali přípravek Cosentyx v dávce 150 mg nebo 300 mg v týdnech 0, 1, 2, 3, 4, 8 a 12 a poté byli randomizováni buď k udržovací léčbě stejnou dávkou každý měsíc počínaje týdnem 12, nebo na režim s “přeléčením v čas potřeby“ stejnou dávkou. U pacientů randomizovaných k “přeléčení v čas potřeby“ nebylo dosaženo odpovídající udržení odpovědi a proto se doporučuje fixní měsíční udržovací režim.
Koprimamí cíl placebem a aktivním komparátorem kontrolovaných studií činil podíl pacientů s dosaženou odpovědí PASI 75 a IGA mód 2011 odpovědi “čistý” nebo “téměř čistý” v porovnání s placebem v týdnu 12 (viz Tabulku 2 a 3). Dávka 300 mg poskytovala zlepšenou clearance pokožky zejména pro “čistou” nebo “téměř čistou” pokožku v rozmezí cílů účinnosti PASI 90, PASI 100, a IGA mód 2011 0 nebo 1 odpověď u všech studií s největším účinkem pozorovaným v týdnu 16, proto se doporučuje tato dávka.
Tabulka 2 Souhrnná PASI 50/75/90/100 & IGA* mód 2011 “čistá” nebo “téměř čistá” klinická odpověď v psoriatických studiích 1, 3 a 4 (ERASURE, FEATURE and JUNCTURE)
Týden 12_Týden 16_Týden 52
|
Placebo |
150 mg |
300 mg |
150 mg |
300 mg |
150 mg |
300 mg | |
|
Studie 1 | |||||||
|
Počet pacientů |
246 |
244 |
245 |
244 |
245 |
244 |
245 |
|
Odpověď PASI 50 n (%) |
22 |
203 |
222 |
212 |
224 |
187 |
207 |
|
(8,9%) |
(83,5%) |
(90,6%) |
(87,2%) |
(91,4%) |
(77%) |
(84,5%) | |
|
Odpověď PASI 75 n (%) |
11 |
174 |
200 |
188 |
211 |
146 |
182 |
|
(4,5%) |
(71,6%)* * |
(81,6%)* * |
(77,4%) |
(86,1%) |
(60,1%) |
(74,3%) | |
|
Odpověď PASI 90 n (%) |
3 |
95 |
145 |
130 |
171 |
88 |
147 |
|
(1,2%) |
(39,1%)* * |
(59,2%)* * |
(53,5%) |
(69,8%) |
(36,2%) |
(60,0%) | |
|
Odpověď PASI 100 |
2 |
31 |
70 |
51 |
102 |
49 |
96 |
|
Odpověď n (%) |
(0,8%) |
(12,8%) |
(28,6%) |
(21,0%) |
(41,6%) |
(20,2%) |
(39,2%) |
|
Odpověď IGA mód 2011 |
6 |
125 |
160 |
142 |
180 |
101 |
148 |
|
“čistý” nebo “téměř |
(2,40%) |
(51,2%)* |
(65,3%)* |
(58,2%) |
(73,5%) |
(41,4%) |
(60,4%) |
|
čistý” n (%) Studie 3 |
* |
* | |||||
|
Počet pacientů |
59 |
59 |
58 |
- |
- |
- |
- |
|
Odpověď PASI 50 n (%) |
3 |
51 |
51 |
- |
- |
- |
- |
|
(5,1%) |
(86,4%) |
(87,9%) | |||||
|
Odpověď PASI 75 n (%) |
0 |
41 |
44 |
- |
- |
- |
- |
|
(0,0%) |
(69,5%)* * |
(75,9%)* * | |||||
|
Odpověď PASI 90 n (%) |
0 |
27 |
35 |
- |
- |
- |
- |
|
(0,0%) |
(45,8%) |
(60,3%) | |||||
|
Odpověď PASI 100 n |
0 |
5 |
25 |
- |
- |
- |
- |
|
(%) |
(0,0%) |
(8,5%) |
(43,1%) | ||||
|
Odpověď IGA mód 2011 |
0 |
31 |
40 |
- |
- |
- |
- |
|
“čistý” nebo “téměř |
(0,0%) |
(52,5%)* |
(69,0%)* | ||||
|
čistý” n (%) Studie 4 |
* |
* | |||||
|
Počet pacientů |
61 |
60 |
60 |
- |
- |
- |
- |
|
Odpověď PASI 50 n (%) |
5 |
48 |
58 |
- |
- |
- |
- |
|
(8,2%) |
(80,0%) |
(96,7%) | |||||
|
Odpověď PASI 75 n (%) |
2 |
43 |
52 |
- |
- |
- |
- |
|
(3,3%) |
(71,7%)* * |
(86,7%)* * | |||||
|
Odpověď PASI 90 n (%) |
0 |
24 |
33 |
- |
- |
- |
- |
|
(0,0%) |
(40,0%) |
(55,0%) | |||||
|
Odpověď PASI 100 n |
0 |
10 |
16 |
- |
- |
- |
- |
|
(%) |
(0,0%) |
(16,7%) |
(26,7%) | ||||
|
Odpověď IGA mód 2011 |
0 |
32 |
44 |
- |
- |
- |
- |
|
“čistý” nebo “téměř |
(0,0%) |
(53,3%)* |
(73,3%)* | ||||
|
čistý” n (%) |
* |
* | |||||
* IGA mód 2011 je 5 bodová škála zahrnující “0 = čistý”, “1 = téměř čistý”, “2 = mírný”, “3 = středně závažný” nebo “4 = závažný”, indikující lékařovo celkové hodnocení závažnosti psoriázy zaměřené na ztvrdnutí, erytém a odlupování. Léčebný úspěch “ čistý ” nebo “ téměř čistý ” znamenaly nepřítomnost známek psoriázy nebo normální až růžové zbarvené léze, netloustnutí ložisek a žádné nebo minimální místní odlupování.
** p hodnoty versus placebo a nastavené na multiplicitu: p<0,0001._
Tabulka 3 Souhrn klinické odpovědi v psoriatické studii 2 (FIXTURE)
Týden 12_Týden 16_Týden 52
|
Placeb |
150 |
300 |
Etanerc |
150 |
300 |
Etanerce |
150 |
300 |
Etanerce | |
|
o |
mg |
mg |
ept |
mg |
mg |
pt |
mg |
mg |
pt | |
|
Počet |
324 |
327 |
323 |
323 |
327 |
323 |
323 |
327 |
323 |
323 |
|
pacientů Odpověď |
49 |
266 |
296 |
226 |
290 |
302 |
257 |
249 |
274 |
234 |
|
PASI 50 n |
(15,1% |
(81,3% |
(91,6% |
(70,0%) |
(88,7% |
(93,5% |
(79,6%) |
(76,1% |
(84,8% |
(72,4%) |
|
(%) |
) |
) |
) |
) |
) |
) |
) | |||
|
Odpověď |
16 |
219 |
249 |
142 |
247 |
280 |
189 |
215 |
254 |
179 |
|
PASI 75 n |
(4,9%) |
(67,0% |
(77,1% |
(44,0%) |
(75,5% |
(86,7% |
(58,5%) |
(65,7% |
(78,6% |
(55,4%) |
|
(%) |
)4 |
) |
) |
) |
) | |||||
|
Odpověď |
5 |
137 |
175 |
67 |
176 |
234 |
101 |
147 |
210 |
108 |
|
PASI 90 n |
(1,5%) |
(41,9% |
(54,2% |
(20,7%) |
(53,8% |
(72,4% |
(31,3%) |
(45,0% |
(65,0% |
(33,4%) |
|
(%) |
) |
) |
) |
) |
) |
) | ||||
|
Odpověď |
0 (0%) |
47 |
78 |
14 |
84 |
119 |
24 (7,4%) |
65 |
117 |
32 (9,9%) |
|
PASI 100 |
(14,4% |
(24,1% |
(4,3%) |
(25,7% |
(36,8% |
(19,9% |
(36,2% | |||
|
n (%) |
) |
) |
) |
) |
) |
) | ||||
|
Odpověď |
9 |
167 |
202 |
88 |
200 |
244 |
127 |
168 |
219 |
120 |
|
IgA mód |
(2,8%) |
(51,1% |
(62,5% |
(27,2%) |
(61,2% |
(75,5% |
(39,3%) |
(51,4% |
(67,8% |
(37,2%) |
|
2011 |
)4 |
)4 |
) |
) |
) |
) |
“čistý”
nebo
“téměř
čistý”
Týden 4_Týden 16
|
Secukinumab |
Ustekinumab* |
Secukinumab |
Ustekinumab | |
|
Počet pacientů |
300 mg 334 |
335 |
300 mg 334 |
335 |
|
Odpověď PASI 75 |
167 (50,0%)4 |
69 (20,6%) |
311 (93,1%) |
277 (82,7%) |
|
n (%) Odpověď PASI 90 |
70 (21,0%) |
18 (5,4%) |
264 (79,0%)4 |
193 (57,6%) |
|
n (%) Odpověď PASI 100 |
14 (4,2%) |
3 (0,9%) |
148 (44,3%) |
95 (28,4%) |
|
n (%) Odpověď IGA mód |
126 (37,7%) |
41 (12,2%) |
277 (82,9%) |
226 (67,5%) |
2011 „čistý“ nebo
„téměř čistý“ n (%)_
* Pacienti léčení secukinumabem dostávali dávku 300 mg v týdnech 0, 1, 2 a 3 následovanou stejnou dávkou v týdnech 4, 8 a 12. Pacienti léčení ustekinumabem dostávali dávku 45 mg nebo 90 mg v týdnech 0 a 4 (dávka podle hmotnosti a schváleného dávkování)
** p hodnoty versus ustekinumab: p<0,0001
Přípravek Cosentyx byl účinný u pacientů bez předchozí systémové léčby, biologicky naivních, biologicky/anti-TNF-exponovaných a pacientů s biologickým/anti-TNF selháním. Zlepšení PASI 75 u pacientů se souběžnou psoriatickou artritidou při zahájení léčby byly podobné těm u celé populace s ložiskovou psoriázou.
Přípravek Cosentyx byl spojován s rychlým nástupem účinku s 50% redukcí průměrného PASI do týdne 3 při dávce 300 mg.
Obrázek 1 časový průběh procentní změny od výchozích hodnot průměrného PASI skóre ve Studii 1 (ERASURE)
PASI % změna od základních hodnot
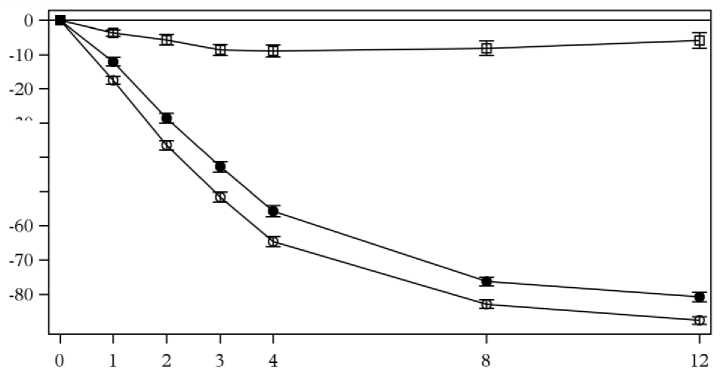
Týdny léčby
n = po čet vyhodnotitelných pacientů
• Cosentyx 150 mg (n=243) o Cosentyx 300 mg (n=245) □ Placebo (n=245)
Specifické lokalizace/formy ložiskové psoriázy
Ve dvou dalších placebem kontrolovaných studiích bylo pozorováno zlepšení jak u nehtové psoriázy (studie TRANSFIGURE, 198 pacientů), tak u palmoplantární ložiskové psoriázy (studie GESTURE, 205 pacientů). Ve studii TRANSFIGURE byla prokázána superiorita secukinumabu oproti placebu v 16. týdnu (46,1% pro 300 mg, 38,4% pro 150 mg a 11,7% pro placebo) ve výrazném zlepšení oproti počátečnímu stavu podle indexu závažnosti psoriázy nehtů (NAPSI %) u pacientů se středně těžkou až těžkou ložiskovou psoriázou s postižením nehtů. Ve studii GESTURE byla prokázána superiorita secukinumabu oproti placebu v 16. týdnu (33,3% pro 300 mg, 22,1% pro 150 mg a 1,5% pro placebo) ve výrazném zlepšení oproti počátečnímu stavu podle odpovědi ppIGA 0 nebo 1 („čistý“ nebo „téměř čistý“) u pacientů se středně těžkou až těžkou palmoplantární ložiskovou psoriázou.
Kvalita života/výsledky hlášené pacienty
Statisticky zlepšení hodnot v týdnu 12 (Studie 1-4) oproti hodnotám před léčbou v porovnání s placebem bylo demonstrováno pomocí DLQI (Dermatology Life Quality Index). Průměrný pokles (zlepšení) DLQI z hodnot před léčbou kolísalo od -10,4 do -11,6 u secukinumabu 300 mg, od -7,7 do -10,1 u secukinumabu 150 mg, v porovnání s -1,1 až -1,9 u placeba v týdnu 12. Toto zlepšení přetrvávalo 52 týdnů (Studie 1 a 2).
Čtyřicet procent účastníků ve studiích 1 a 2 vyplnilo deník Psoriasis Symptom Diary©. U účastníků, kteří vyplnili tento deník, bylo v každé z těchto studií v porovnání s placebem prokázáno statisticky významné zlepšení z hodnot před léčbou v týdnu 12 u pacienty hlášených známek a příznaků svědění, bolesti a odlupování.
Psoriatická artritida
Bezpečnost a účinnost přípravku Cosentyx byla hodnocena u 1003 pacientů ve dvou randomizovaných, dvojitě zaslepených, placebem kontrolovaných studiích fáze III u pacientů s aktivní psoriatickou artritidou (>3 oteklé a >3 bolestivé klouby) přes léčbu nesteroidními protizánětlivými léky (NSAID), kortikosteroidy nebo chorobu modifikujícími antirevmatiky (DMARD). Do těchto studií byli zařazeni pacienti všech subtypů PsA, včetně polyartikulární artritidy bez průkazu revmatoidních nodulů, spondylitidy s periferní artritidou, asymetrickou periferní artritidou, postižení distálních interfalangeálních kloubů a arthritis mutilans. Pacienti v těchto studiích měli stanovenu diagnózu PsA s mediánem 3,9 až 5,3 roku. Většina pacientů měla též aktivní psoriatické kožní léze nebo dokumentovanou psoriázu v anamnéze. Přes 62% a 47% PsA pacientů mělo před zahájením enthesitidu, respektive daktylitidu. V obou studiích bylo dosaženo primárního cíle - odpovědi ACR20 (American College of Rheumatology) v týdnu 24.
Ve studii psoriatické artritidy 1 (PsA studie 1) a studii psoriatické artritidy 2 (PsA studie 2) 29% pacientů, respektive 35% pacientů, bylo již léčeno anti-TNFa přípravkem a ukončilo užívání anti-TNFa přípravku buď pro nedostatek účinku nebo nesnášenlivost (anti-TNFa-IR pacienti).
PsA studie 1 (FUTURE 1) hodnotila 606 pacientů, z nichž 60,7% užívalo současně MTX. Pacienti randomizovaní na přípravek Cosentyx dostávali dávku 10 mg/kg intravenózně v týdnech 0, 2 a 4, následovanou buď 75 mg nebo 150 mg subkutánně každý měsíc počínaje týdnem 8. Pacienti randomizovaní na placebo byli převedeni na přípravek Cosentyx (buď 75 mg nebo 150 mg subkutánně) s následnou stejnou dávkou každý měsíc, non-respondéři v 16. týdnu (časná záchrana) a ostatní ve 24. týdnu.
PsA studie 2 (FUTURE 2) hodnotila 397 pacientů, z nichž 46,6% užívalo současně MTX. Pacienti randomizovaní na přípravek Cosentyx dostávali dávku 75 mg, 150 mg nebo 300 mg subkutánně v týdnech 0, 1, 2 a 3, s následnou stejnou dávkou každý měsíc, počínaje týdnem 4. Pacienti randomizovaní na placebo, kteří v týdnu 16 neodpovídali na léčbu byli v 16. týdnu (časná záchrana) převedeni na přípravek Cosentyx (buď 150 mg nebo 300 mg subkutánně) s následnou stejnou dávkou každý měsíc. Pacienti randomizovaní na placebo, kteří v 16. týdnu měli odpověď na léčbu, byli v 24. týdnu převedeni na přípravek Cosentyx (buď 150 mg nebo 300 mg subkutánně) s následnou stejnou dávkou každý měsíc.
Známky a příznaky
Léčba přípravkem Cosentyx v porovnání s placebem znamenala významné zlepšení v hodnocení aktivity choroby v týdnu 24 (viz Tabulka 5).
Tabulka 5 Klinická odpověď u PsA studie 2 v týdnu 24
|
Týden 24 | ||||
|
Placebo |
75 mg |
150 mg |
300 mg | |
|
Počet randomizovaných pacientů |
98 |
99 |
100 |
100 |
|
ACR20 odpověď n (%) |
15 (15,3%) |
29 (29,3%*) |
51 (51,0%***) |
54 (54,0%***) |
|
ACR50 odpověď n (%) |
7 (7,1%) |
18 (18,2%) |
35 (35,0%) |
35 (35,0%**) |
|
ACR70 odpověď n (%) |
1 (1,0%) |
6 (6,1%) |
21 (21,0%**) |
20 (20,0%**) |
|
DAS28-CRP |
-0,96 |
-1,12 |
-1,58** |
-1,61** |
|
Počet pacientů s >3% BSA psoriatickým postižením pokožky před zahájením |
43 (43,9%) |
50 (50,5%) |
58 (58,0%) |
41 (41,0%) |
|
PASI 75 odpověď n (%) |
7 (16,3%) |
14 (28,0%) |
28 (48,3%**) |
26 (63,4%***) |
|
PASI 90 odpověď n (%) |
4 (9,3%) |
6 (12,0%) |
19 (32,8%**) |
20 (48,8%***) |
|
Vymizení daktylitidy n (%) f |
4 (14,8%) |
10 (30,3%) |
16 (50,0%**) |
26 (56,5%**) |
|
Vymizení entesitidy n (%) $ |
14 (21,5%) |
22 (32,4%) |
27 (42,2%*) |
27 (48,2%**) |
* p<0,05, ** p<0,01, *** p<0,001; oproti placebu
Všechny p-hodnoty jsou nastaveny na mnohočetné testování založené na předem definované hierarchii, s výjimkou ACR70, daktylitidy a entesitidy, což byly výzkumné cíle.
Pacienti s chybějícími údaji o léčebné odpovědi byly považováni za non-respondéry.
ACR: American College of Rheumatology; PASI: Psoriasis Area and Severity Index; DAS: Disease Activity Score (skóre aktivity nemoci); BSA: Body Surface Area (povrch těla) fU pacientů s daktylitidou při zahájení (n=27, 33, 32, 46)
t U pacientů s entesitidou při zahájení (n=65, 68, 64, 56)_
Nástup účinku přípravku Cosentyx byl pozorován již ve 2. týdnu. Statisticky významného rozdílu v ACR 20 oproti placebu bylo dosaženo ve 3. týdnu. V 16. týdnu vykazovali pacienti léčení přípravkem Cosentyx významné zlepšení známek a příznaků s významně vyšší odpovědí v ACR 20 (33,3%, 60,0% a 57,0% u 75 mg, respektive 150 mg a 300 mg) v porovnání s placebem (18,4%).
Procento pacientů s dosaženou ACR 20 odpovědí je na obrázku 2.
Obrázek 2 ACR20 odpověď v PsA studii 2 v závislosti na čase do týdne 24

Čas (týdny)
■ Cosentyx 150 mg Cosentyx 300 mg ■ ■ ■ » Placebo
Procento respondérů
Podobné odpovědi pro primární a klíčový sekundární cíl byly pozorovány u PsA pacientů bez ohledu na to, zda byli současně léčeni MTX či nikoliv. V týdnu 24 měli pacienti léčení přípravkem Cosentyx a současně MTX vyšší ACR 20 odpověď (44,7%, 47,7% a 54,4% u 75 mg, respektive 150 mg a 300 mg, v porovnání s placebem 20,0%) a ACR 50 odpověď (27,7%, 31,8% a 38,6% u 75 mg, respektive 150 mg a 300 mg v porovnání s placebem 8,0%). Pacienti léčení přípravkem Cosentyx bez současně podávaného MTX měli vyšší ACR 20 odpověď (15,4%, 53,6% a 53,6% u 75 mg, respektive 150 mg a 300 mg, v porovnání s placebem 10,4%) a ACR 50 odpověď (9,6%, 37,5% a 32,1% u 75 mg, respektive 150 mg a 300 mg, v porovnání s placebem 6,3%).
Jak anti-TNFa-naivní, tak anti-TNFa-IR pacienti léčení přípravkem Cosentyx měli významně vyšší ACR 20 odpověď v porovnání s placebem v týdnu 24, s mírně vyšší odpovědí u anti-TNFa-naivní skupiny (anti-TNFa-naivní: 37%, 64% a 58% u 75 mg, respektive 150 mg and 300 mg, v porovnání s placebem 15,9%; anti-TNFa-IR: 15%, 30% a 46% u 75 mg, respektive 150 mg a 300 mg, v porovnání s placebem 14,3%). V anti-TNFa-IR podskupině pacientů vykázala významně vyšší ACR 20 odpověď v porovnání s placebem (p<0,05) pouze dávka 300 mg ademonstrovala klinicky smysluplný prospěch oproti 150 mg také ve více sekundárních cílech. Zlepšení PASI 75 odpovědi bylo pozorováno v obou podskupinách a dávka 300 mg prokázala statisticky signifikantní přínos u anti-TNFa-IR pacientů.
Počet PsA pacientů s axiálním postižením byl příliš malý na to, aby bylo možné stanovit vypovídající hodnocení.
Zlepšení se objevilo u všech komponent ACR skóre, včetně hodnocení bolesti pacientem. Podíl pacientů s dosaženou modifikovanou PsA Response Criteria (PsARC) odpovědí byl větší ve skupině pacientů léčených přípravkem Cosentyx (38,4%, 62,0% a 63,0% u 75 mg, respektive 150 mg a 300 mg) v porovnání s placebem (29,6%) v týdnu 24.
V PsA studii 1 a PsA studii 2 přetrvávala účinnost do týdne 52. V PsA studii 2 bylo v 52. týdnu stále léčeno 178 pacientů (89%) z 200 pacientů iniciálně randomizovaných na Cosentyx 150 mg a 300 mg. Ze 100 pacientů randomizovaných na Cosentyx 150 mg, 64, respektive 39 a 20, vykázalo ACR 20/50/70 odpověď. Ze 100 pacientů randomizovaných na Cosentyx 300 mg, 64, respektive 44 a 24 vykázalo ACR 20/50/70 odpověď.
Radiografická odpověď
Inhibice progrese strukturálního poškození u PsA nebyla dosud prokázána za použití podkožního zaváděcího dávkovacího režimu schváleného pro klinické použití.
V PsA studii 1 byla inhibice progrese strukturálního poškození hodnocena radiograficky a vyjádřena jako změna modifikovaného celkového Sharp Score (mTSS) a jeho komponent, Erosion Score (ES) a Joint Space Narrowing skóre (JSN) v týdnech 24 a 52, v porovnání s výchozími hodnotami. Data z týdne 24 jsou uvedena v Tabulce 6.
Tabulka 6 Změna modifikovaného Total Sharp skóre u psoriatické artritidy
|
Placebo N=179 |
Cosentyx 75 mg1 N=181 |
Cosentyx 150 mg1 N=185 | |
|
Celkové skóre | |||
|
Počáteční hodnoty |
28,4 |
20,4 |
22,3 |
|
(SD) |
(63,5) |
(39,4) |
(48,0) |
|
Průměrná změna v týdnu 24 |
0,57 |
0,02* |
0,13* |
*p<0,05 založená na nominální, ale neupravené p-hodnotě 110 mg/kg v týdnech 0, 2 a 4 následovaná 75 mg nebo150 mg
Inhibice strukturálního poškození přetrvávala při léčbě přípravkem Cosentyx do týdne 52.
Procento pacientů bez progrese choroby (definované jako změna od výchozích hodnot mTSS <0,5) od randomizace do týdne 24 bylo 92,3% ve větvi se secukinumabem s dávkovacím schématem 10 mg/kg intravenózního bolusu - 75 mg subkutánně (udržovací dávka), 82,3% ve větvi se secukinumabem s dávkovacím schematem 10 mg/kg intravenózního bolusu - 150 mg subkutánně (udržovací dávka) a 75,7% ve větvi s placebem. Procento pacientů bez progrese choroby od týdne 24 do týdne 52 u secukinumabu 10 mg/kg ve formě intravenózního bolusu - následovaného buď 75 mg nebo 150 mg subkutánně (udržovací dávka) a u pacientů na placebu, kteří byli v týdnu 16 nebo 24 převedeni na 75 mg nebo 150 mg subkutánně (udržovací dávka) každé 4 týdny, bylo 85,8%, respektive 85,7% a 86,8%.
Funkční stav a kvalita života
V PsA studii 2, pacienti léčení přípravkem Cosentyx 150 mg (p=0,0555) a 300 mg (p=0,0040) vykázali zlepšení funkčního stavu v porovnání s pacienty na placebu hodnocené podle Health Assessment Questionnaire-Disability Index (HAQ-DI) v týdnu 24. Zlepšení HAQ-DI skóre bylo pozorováno bez ohledu na předchozí expozici anti-TNFa. Podobná odpověď byla pozorována v PsA studii 1.
Pacienti léčení přípravkem Cosentyx hlásili významné zlepšení se zdravím související kvality života měřené pomocí Short Form-36 Health Survey Physical Component Summary (SF-36 PCS) skóre (p<0,001). Prokázalo se též statisticky významé zlepšení, což bylo demonstrováno ve výzkumných cílech hodnocených Functional Assessment of Chronic Illness Therapy - Fatigue (FACIT-F) skóre u dávky 150 mg a 300 mg v porovnání s placebem (7,97, respektive 5,97 versus 1,63). Podobné odpovědi byly pozorovány v PsA studii 1 a účinnost přetrvávala do týdne 52.
Ankylozující spondylitida
Bezpečnost a účinnost přípravku Cosentyx byla hodnocena u 590 pacientů ve dvou randomizovaných, dvojitě zaslepených, placebem kontrolovaných studiích fáze III u pacientů s aktivní ankylozující spondylitidou (AS) s Bath Ankylosing Spondylitis Disease Activity indexem (BASDAI) >4 přes léčbu nesteroidními protizánětlivými léky (NSAID), kortikosteroidy nebo chorobu modifikujícími antirevmatiky (DMARD). Pacienti v těchto studiích měli stanovenu diagnózu AS s mediánem 2,7 až
5,8 roku. V obou studiích bylo primárním cílem nejméně 20% zlepšení Assessment of Spondyloarthritis International Society kritéria (ASAS 20) v týdnu 16.
Ve studii ankylozující spondylitidy 1 (AS studie 1) a studii ankylozující spondylitidy 2 (AS studie 2) 27,0%, respektive 38,8% pacientů, bylo dříve léčeno anti-TNFa přípravkem a léčba anti-TNFa přípravkem byla ukončena buď pro nedostatek účinku nebo nesnášenlivost (anti-TNFa-IR pacienti).
AS Studie 1 (MEASURE 1) hodnotila 371 pacientů, z nichž užívalo současně 14,8%, respektive 33,4%, MTX nebo sulfasalazin. Pacienti randomizovaní na přípravek Cosentyx dostávali dávku 10 mg/kg intravenózně v týdnech 0, 2 a 4, následovanou buď 75 mg nebo 150 mg subkutánně každý měsíc počínaje týdnem 8. Pacienti randomizovaní na placebo byli převedeni na přípravek Cosentyx (buď 75 mg nebo 150 mg subkutánně) s následnou stejnou dávkou každý měsíc, non respondéři v 16. týdnu (časná záchrana) a všichni ostatní ve 24. týdnu.
AS studie 2 (MEASURE 2) hodnotila 219 pacientů, z nichž užívalo současně 11,9%, respektive 14,2%, MTX nebo sulfasalazin. Pacienti randomizovaní na přípravek Cosentyx dostávali dávku 75 mg nebo 150 mg subkutánně v týdnech 0, 1, 2 a 3, s následnou stejnou dávkou každý měsíc, počínaje týdnem 4. V týdnu 16 byli pacienti při zahájení randomizovaní na placebo re-randomizováni na přípravek Cosentyx (buď 75 mg nebo 150 mg subkutánně) každý měsíc.
Známky a příznaky
V AS studii 2 znamenala léčba přípravkem Cosentyx 150 mg v porovnání s placebem větší zlepšení v hodnocení aktivity choroby v týdnu 16 (viz Tabulka 7).
Tabulka 7 Klinická odpověď v AS studii 2 v týdnu 16
|
Výsledek (p-hodnota versus placebo) |
Placebo (n = 74) |
75 mg (n = 73) |
150 mg (n = 72) |
|
ASAS 20 odpověď, % |
28,4 |
41,1 |
61,1*** |
|
ASAS 40 20 odpověď, % |
10,8 |
26,0 |
36,1*** |
|
hsCRP, (post-BSL/BSL ratio) |
1,13 |
0,61 |
0,55*** |
|
ASAS 5/6, % |
8,1 |
34,2 |
43 1*** |
|
ASAS částečná remise, % |
4,1 |
15,1 |
13,9 |
|
BASDAI 50, % |
10,8 |
24,7* |
30,6** |
|
ASDAS-CRP velké zlepšení |
4,1 |
15,1* |
25,0*** |
|
* p<0,05, ** p<0,01, *** p<0,001; versus placebo Všechny p-hodnoty j sou nastaveny na mnohočetné testování založené na předem definované hierarchii, s výjimkou BASDAI 50 a ASDAS-CRP Pacienti s chybějícími údaji o léčebné odpovědi byli považováni za non-respondéry. | |||
|
ASAS: Assessment of SpondyloArthritis International Society Criteria; BASDAI: Bath Ankylosing Spondylitis Disease Activity Index; hsCRP: high-sensitivity C-reactive protein; ASDAS: Ankylosing Spondylitis Disease Activity Score; BSL: baseline | |||
V AS studii 2 se nástup účinku přípravku Cosentyx 150 mg projevil již v týdnu 1 (superiorita Cosentyxu vůči placebu u odpovědi ASAS 20) a v týdnu 2 (superiorita Cosentyxu vůči placebu u odpovědi ASAS 40).
ASAS 20 se zlepšila v týdnu 16 jak u anti-TNFa-naivních pacientů (68,2% versus 31,1%; p<0,05), tak u anti-TNFa-IR pacientů (50,0% versus 24,1%; p<0,05) u přípravku Cosentyx 150 mg v porovnání s placebem.
V obou AS studiích, u pacientů léčených přípravkem Cosentyx (150 mg v AS Studii 2 a oběma režimy AS studie 1) došlo v 16. týdnu k významnému zlepšení známek a příznaků se srovnatelným rozsahem odpovědi. Účinnost přetrvávala do 52. týdne jak u anti-TNFa-naivních, tak u anti-TNFa-IR pacientů.
V AS studii 2 bylo v 52. týdnu stále léčeno 61 pacientů (84,7%) ze 72 pacientů iniciálně randomizovaných na Cosentyx 150 mg. Ze 72 pacientů randomizovaných na Cosentyx 150 mg, 45, respektive 35, vykázalo ASAS 20/40 odpověď.
Mobilita páteře
Pacienti léčení přípravkem Cosentyx v dávce 150 mg vykázali zlepšenou mobilitu páteře hodnocenou jako změnu BASMI v týdnu 16 v porovnání s hodnotami před léčbou v AS studii 1 (-0,40 versus -0,12 pro placebo; p=0,0114) a v AS studii 2 (-0,51 versus -0,22 pro placebo; p=0,0533). Tato zlepšení přetrvávala do týdne 52.
Funkční stav a se zdravím související kvalita života
V AS studii 1 a studii 2, vykázali pacienti léčení přípravkem Cosentyx 150 mg zlepšení zdravotního stavu a kvality života, což bylo měřeno AS Quality of Life Dotazníkem (ASQoL) (p=0,001) a SF-36 Physical Component Summary (SF-36PCS) (p<0,001). Pacienti léčení přípravkem Cosentyx 150 mg také vykázali statisticky významné zlepšení ve fyzické funkci ve výzkumných cílech, což bylo zhodnoceno Bath Ankylosing Spondylitis Functional Indexem (BASFI) v porovnání s placebem (-2,15 versus -0,68), a v únavě, což bylo zhodnoceno Functional Assessment of Chronic Illness Therapy-Fatigue (FACIT-Fatigue) škálou v porovnání s placebem (8,10 versus 3,30). Tato zlepšení přetrvávala do týdne 52.
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Cosentyx u ložiskové psoriázy u pediatrických pacientů ve věku od narození do 6 let a u chronické idiopatické artritidy u dětských pacientů do 2 let (informace o použití u dětí viz bod 4.2).
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s přípravkem Cosentyx u ložiskové psoriázy u pediatrických pacientů ve věku od 6 let do 18 let a u chronické idiopatické artritidy u dětských pacientů do 2 let do 18 let (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Ložisková psoriáza
Absorpce
Po jednorázové podkožní dávce 300 mg ve formě roztoku u zdravých dobrovolníků dosáhl secukinumab nejvyšší sérové koncentrace 43,2±10,4 pg/ml mezi 2 a 14 dny po podání dávky.
Na základě výsledků populační farmakokinetické analýzy secukinumab po jednorázové subkutánní dávce buď 150 mg nebo 300 mg u pacientů s ložiskovou psoriázou dosáhl nejvyšší sérové koncentrace 13,7±4,8 pg/ml a 27,3±9,5 pg/ml mezi 5 a 6 dny od podání dávky.
Na základě výsledků populační farmakokinetické analýzy byl po počátečním týdenním dávkování během prvního měsíce čas potřebný k dosažení maximální koncentrace mezi 31 a 34 dny.
Podle simulovaných údajů byla nejvyšší koncentrace v rovnovážném stavu (Cmax,ss) po subkutánním podání dávky 150 mg nebo 300 mg 27,6 pg/ml and 55,2 pg/ml. Populační farmakokinetická analýza naznačuje, že rovnovážného stavu je dosaženo po 20 týdnech při měsíčním dávkovacím režimu.
V porovnání s expozicí po jednorázovém podání ukázala populační farmakokinetická analýza, že pacienti vykazovali 2násobné zvýšení nejvyšší sérové koncentrace a plochy pod křivkou (AUC) po opakovaném měsíčním podávání během udržovacího režimu.
Populační farmakokinetická analýza ukázala že secukinumab byl u pacientů s ložiskovou psoriázou absorbován s průměrnou absolutní biologickou dostupností 73%. Napříč studiemi byla vypočítaná absolutní biologická dostupnost v rozmezí 60 až 77%.
Distribuce
Průměrný distribuční objem během terminální fáze (Vz) po jednorázovém intravenózním podání kolísal u pacientů s ložiskovou psoriázou v rozmezí od 7,10 do 8,60 litrů, což naznačuje, že secukinumab se omezeně distribuuje do periferních kompartmentů.
Biotransformace
Největší část IgG eliminace probíhá prostřednictvím intracelulárního katabolismu, následovaného fluidní fází nebo receptory zprostředkované endocytózy.
Eliminace
Průměrná systémová clearance (CL) po jednorázovém intravenózním podání kolísala u pacientů s ložiskovou psoriázou od 0,13 do 0,36 l/den. V populační farmakokinetické analýze byla průměrná systémová clearance (CL) u pacientů s ložiskovou psoriázou 0,19 l/den. CL nebyla ovlivněna pohlavím. Clearance byla závislá na dávce a čase.
Průměrný eliminační poločas zjištěný v populační farmakokinetické analýze byl u pacientů s ložiskovou psoriázou 27 dní, a kolísal od 18 do 46 dní napříč psoriatickými studiemi s intravenózním podáním.
Linearita/nelinearita
Farmakokinetika secukinumabu u pacientů s ložiskovou psoriázou po jednotlivé a opakovaných dávkách byla stanovena v četných studiích s intravenózně podanými dávkami v rozmezí od 1x 0,3 mg/kg do 3x 10 mg/kg a subkutánními dávkami v rozmezí od 1x 25 mg až opakovanými dávkami 300 mg. Expozice ve všech dávkovacích režimech odpovídala dávce.
Psoriatická artritida
Farmakokinetické vlastnosti secukinumabu pozorované u pacientů s psoriatickou artritidou byly podobné těm projevujícím se u pacientů s ložiskovou psoriázou. Biologická dostupnost secukinumabu u PsA pacientů činila 85% na základě populačního farmakokinetického modelu.
Ankylozující spondylitida
Farmakokinetické vlastnosti secukinumabu pozorované u pacientů s ankylozující spondylitidou byly podobné těm projevujícím se u pacientů s ložiskovou psoriázou.
Zvláštní populace
Starší _ pacienti
Z 3430 pacientů s ložiskovou psoriázou exponovaných přípravku Cosentyx v klinických studiích bylo 230 ve věku 65 let nebo starších a 32 pacientů bylo ve věku 75 nebo starších.
Z 974 pacientů s psoriatickou artritidou exponovaných přípravku Cosentyx v klinických studiích, celkem 85 pacientů bylo ve věku 65 let nebo starších a 4 pacienti byli ve věku 75 let nebo starší.
Z 571 pacientů s ankylozující spondylitidou exponovaných přípravku Cosentyx v klinických studiích, celkem 24 pacientů bylo ve věku 65 let nebo starších a 3 pacienti byli ve věku 75 let nebo starší.
Na základě populační farmakokinetické analýzy s omezeným počtem starších pacientů (n=71 pro věk >65 let a n=7 pro věk >75 let) byla clearance u starších pacientů a pacientů mladších 65 let věku podobná.
Pacienti s _poruchou _funkce ledvin nebo _jater
U pacientů s poruchou funkce ledvin nebo jater nejsou k dispozici farmakokinetická data. Předpokládá se, že renální eliminace nedotčeného přípravku Cosentyx, IgG monoklonální protilátky bude nízká a nevýznamná. IgG jsou většinou katabolizovány a neočekává se, že by porucha funkce jater ovlivnila clearance přípravku Cosentyx.
Vliv tělesné hmotnosti na _farmakokinetiku
S rostoucí tělesnou hmotností rostou clearance secukinumabu a jeho distribuční objem.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě tkáňových testů zkřížené reaktivity, farmakologických studií bezpečnosti, toxicity po opakovaném podávání a reprodukční toxicity provedené u secukinumabu nebo myších protilátek proti myšímu IL-17A neodhalily žádné zvláštní riziko pro člověka.
Protože secukinumab váže opičí (cynomolgus) a lidský IL-17A, byla jeho bezpečnost studována u opic rodu cynomolgus. Po subkutánním podání secukinumabu opicím rodu cynomolgus po dobu až 13 týdnů a intravenózním podání až 26 týdnů (včetně vyhodnocení farmakokinetiky, farmakodynamiky, imunogenicity a imunotoxicity (tj. na T-buňkách závislá protilátková odpověď a aktivita NK buněk) nebyly pozorovány nežádoucí účinky. Průměrná sérová koncentrace pozorovaná u opic po 13 týdenních subkutánních dávkách 150 mg/kg byla značně vyšší než odhadovaná průměrná sérová koncentrace u psoriatických pacientů při nejvyšší klinické dávce. Protilátky proti secukinumabu byly detekovány pouze u jednoho z exponovaných zvířat. Po aplikaci secukinumabu do normální lidské tkáně nebyla pozorována nespecifická tkáňová zkřížená reaktivita.
Studie na zvířatech k posouzení karcinogenního potenciálu secukinumabu nebyly provedeny.
V embryofetální vývojové studii u opic rodu cynomolgus nevykazoval secukinumab toxicitu pro matku, embryotoxicitu nebo teratogenitu při podání během organogeneze a v pozdní gestaci.
Ve fertilitních a časných embryonálních a pre- a postnatálních studiích u myší nebyly pozorovány nežádoucí účinky myších protilátek proti myšímu IL-17A. Vyšší dávky v těchto studiích převyšovaly maximální účinnou dávku ve smyslu suprese IL-17A a účinku (viz bod 4.6).
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Dihydrát trehalosy Histidin
Monohydrát histidin-hydrochloridu
Methionin
Polysorbát 80
Voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti
18 měsíců
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2°C to 8°C). Chraňte před mrazem.
Uchovávejte injekční stříkačky v krabičce, aby byly chráněny před světlem.
6.5 Druh obalu a obsah balení
Přípravek Cosentyx je dodáván v předplněné skleněné injekční stříkačce o objemu 1 ml s Fluortecem potaženým pístem, připojenou 27G x íV' jehlou a pevným krytem jehly ze styrenbutadienové gumy, vše v pasivním bezpečnostním obalu z polykarbonátu.
Přípravek Cosentyx je dostupný v jednotkovém balení obsahujícím 1 nebo 2 předplněné injekční stříkačky a vícečetném balení obsahujícím 6 (3x2) předplněných injekčních stříkaček.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Přípravek Cosentyx 150 mg injekční roztok je dodáván v předplněné injekční stříkačce pro individuální použití. S injekční stříkačkou netřepejte a chraňte ji před mrazem. Injekční stříkačku je nutné vyjmout z chladničky 20 minut před aplikací injekce, aby dosáhla pokojové teploty.
Před použitím předplněné injekční stříkačky se doporučuje provést vizuální kontrolu. Roztok musí být čirý. Jeho barva může být od bezbarvé po mírně nažloutlou. Můžete pozorovat malé vzduchové bublinky, což je normální. Nepoužívejte, pokud roztok obsahuje zřetelně viditelné částice, je zakalený nebo zřetelně hnědý. Podrobný návod k použití najdete v příbalové informaci.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/14/980/002
EU/1/14/980/003
EU/1/14/980/006
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
15. ledna 2015
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Cosentyx 150 mg injekční roztok v předplněném peru
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedno předplněné pero obsahuje secukinumabum* 150 mg v 1 ml.
*Secukinumab je rekombinantní plně humánní monoklonální protilátka selektivní pro interleukin-17A. Secukinumab patří do IgG1/K-třídy produkované v buňkách ovarií čínského křečka (CHO).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok v předplněném peru (pero SensoReady)
Roztok je čirý a bezbarvý až mírně nažloutlý.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Ložisková psoriáza
Přípravek Cosentyx je indikován k léčbě středně těžké až těžké ložiskové psoriázy dospělých, kteří jsou kandidáty pro systémovou léčbu.
Psoriatická artritida
Přípravek Cosentyx, samotný nebo v kombinaci s metotrexátem (MTX), je indikován k léčbě aktivní psoriatické artritidy u dospělých pacientů, u nichž se nedostavila adekvátní odpověď na předchozí léčbu chorobu modifikujícími antirevmatiky (DMARD) (viz bod 5.1).
Ankylozující spondylitida
Přípravek Cosentyx je indikován k léčbě aktivní ankylozující spondylitidy u dospělých, kteří nedostatečně reagovali na konvenční léčbu.
Přípravek Cosentyx je určen k použití pod vedením a dohledem lékaře obeznámeného s diagnostikou a léčbou a stavů, u nichž je přípravek Cosentyx indikován.
Dávkování
Ložisková _ psoriáza
Doporučená dávka je 300 mg secukinumabu ve formě subkutánní injekce s iniciálním podáním v týdnech 0, 1, 2 a 3, následovaná měsíční udržovací dávkou od týdne 4. Každá dávka 300 mg je podána ve dvou dílčích subkutánních injekcích po 150 mg.
Psoriatická artritida
U pacientů se současně přítomnou středně těžkou až těžkou ložiskovou psoriázou nebo u pacientů nedostatečně odpovídajících na anti-TNFa (IR), je doporučená dávka 300 mg ve formě subkutánní injekce s iniciálním podáním v týdnech 0, 1, 2 a 3, následovaná měsíční udržovací dávkou od týdne 4. Každá dávka 300 mg je podána ve dvou dílčích subkutánních injekcích po 150 mg.
U ostatních pacientů je doporučená dávka 150 mg ve formě subkutánní injekce s iniciálním podáním v týdnech 0, 1, 2 a 3, následovaná měsíční udržovací dávkou od týdne 4.
Ankylozující spondylitida
Doporučená dávka je 150 mg ve formě subkutánní injekce s iniciálním podáním v týdnech 0, 1, 2 a 3, následovaná měsíční udržovací dávkou od týdne 4.
Dostupná data naznačují, že klinická odpověď ve všech shora uvedených indikacích se obvykle dostaví během 16 týdnů. U pacientů, u nichž se do 16. týdne nedostaví žádná terapeutická odpověď, je nutné zvážit ukončení léčby. U některých pacientů s počáteční částečnou odpovědí může dojít k následnému zlepšení při pokračování léčby nad 16 týdnů.
Zvláštní populace
Starší_pacienti (ve věku 65 let a více)
Není nutná úprava dávky (viz bod 5.2).
Zhoršená _ funkce ledvin / Zhoršená _ funkce _ jater
Přípravek Cosentyx nebyl u těchto pacientských populací studován. Nelze učinit žádná doporučení ohledně dávkování.
Pediatrická populace
Bezpečnost a účinnost přípravku Cosentyx u dětí ve věku do 18 let nebyla dosud stanovena. Nejsou dostupné žádné údaje.
Způsob podání
Přípravek Cosentyx je určen k podání ve formě subkutánní injekce. Pokud je to možné, oblasti pokožky s projevy psoriázy by neměly být použity k podání injekce.
Po vhodném nácviku subkutánní injekční techniky, si může pacient sám aplikovat přípravek Cosentyx, pokud to lékař uzná za vhodné. Lékař však musí zajistit přiměřené sledování pacientů. Pacienti musí být instruováni injikovat celé množství přípravku Cosentyx podle instrukcí v příbalové informaci. Kompletní informace o podávání jsou uvedeny v příbalové informaci.
4.3 Kontraindikace
Těžké reakce z přecitlivělosti na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Klinicky významné aktivní infekce (např. aktivní tuberkulóza; viz bod 4.4).
4.4 Zvláštní upozornění a opatření pro použití
Infekce
Přípravek Cosentyx má potenciál zvyšovat riziko infekcí. V klinických studiích byly u pacientů léčených přípravkem Cosentyx infekce pozorovány (viz bod 4.8). Většina z nich byly mírně nebo středně závažné infekce horních cest dýchacích jako nasopharyngitis a nevyžadovaly přerušení léčby.
V souladu s mechanizmem účinku přípravku Cosentyx byly u secukinumabu v porovnání s placebem v klinických studiích s psoriázou mnohem častěji hlášeny nezávažné mukokutánní kandidové infekce (3,55 na 100 pacientoroků u secukinumabu 300 mg versus 1,00 na 100 pacientoroků u placeba) (viz bod 4.8).
Opatrnosti je zapotřebí, pokud se uvažuje o použití přípravku Cosentyx u pacientů s chronickou infekcí nebo opakovanou infekcí v anamnéze.
Pacienty je nutné poučit, aby vyhledali lékařskou pomoc, pokud se objeví známky nebo příznaky naznačující přítomnost infekce. Pokud se u pacienta rozvine závažná infekce, je nutné pacienta pečlivě sledovat a nepodávat přípravek Cosentyx, dokud infekce neodezní.
V klinických studiích nebyla hlášena zvýšená citlivost vůči tuberkulóze. Přípravek Cosentyx však nesmí být podáván pacientům s aktivní tuberkulózou. U pacientů s latentní tuberkulózou je nutné zvážit před zahájením léčby přípravkem Cosentyx antituberkulózní léčbu.
Opatrnosti je zapotřebí při předepisování přípravku Cosentyx pacientům s Crohnovou chorobou, protože v klinických hodnoceních byly pozorovány exacerbace Crohnovy choroby, v některých případech závažné, v obou skupinách s přípravkem Cosentyx a skupině s placebem. Pacienty s Crohnovou chorobou léčené přípravkem Cosentyx je nutné pečlivě sledovat.
Reakce z přecitlivělosti
V klinických studiích byly u pacientů léčených přípravkem Cosentyx vzácně pozorovány případy anafylaktických reakcí. Pokud se objeví anafylaktická nebo jiné závažné alergické reakce, musí se podávání přípravku Cosentyx okamžitě přerušit a zahájit vhodnou léčbu.
Osoby citlivé na latex
Snímatelný kryt jehly předplněného pera Cosentyx obsahuje derivát přirozeně se vyskytujícího gumového latexu. Ve snímatelném krytu jehly nebyl dosud detekován přirozeně se vyskytující gumový latex. Nicméně použití předplněného pera přípravku Cosentyx u osob citlivých na latex nebylo studováno a proto nelze možné riziko reakcí z přecitlivělosti zcela vyloučit.
Živé vakcíny nesmí být podávány současně s přípravkem Cosentyx.
Pacienti léčení přípravkem Cosentyx mohou současně absolvovat současné očkování inaktivovanými nebo neživými vakcínami. Ve studii po podání meningokokové a inaktivované chřipkové vakcíny, byla podobná část zdravých dobrovolníků léčených secukinumabem150 mg a těch léčených placebem schopna dosáhnout adekvátní imunitní odpovědi nejméně 4násobného zvýšení titru protilátek při meningokokové a chřipkové vakcíně. Tyto údaje naznačují, že přípravek Cosentyx nepotlačuje látkovou imunitní odpověď na meningokokovou nebo chřipkovou vakcínu.
Současná imunosupresivní léčba
Ve studiích s lupénkou nebyly bezpečnost a účinnost přípravku Cosentyx v kombinaci s imunosupresivy, včetně biologické léčby, nebo fototerapií vyhodnocovány (viz též bod 4.5).
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Živé vakcíny nesmí být podávány současně s přípravkem Cosentyx (viz též bod 4.4).
U člověka nebyly provedeny žádné studie interakcí. Přímý důkaz role IL-17A v expresi CYP450 enzymů není k dispozici. Tvorba některých CYP450 enzymů je u chronických zánětů potlačována zvýšenými hladinami cytokinů. Proto může protizánětlivá léčba, například IL-17A inhibitorem secukinumab, znamenat normalizaci hladin CYP450 s doprovodnou nižší expozicí souběžnou medikací metabolizovanou prostřednictvím CYP450. Proto nelze vyloučit klinicky relevantní účinek na substráty CYP450 s úzkým terapeutickým indexem a individuálně nastavenou dávkou (např. warfarin). U pacientů léčených tímto typem léčivých přípravků je při zahájení léčby secukinumabem nutné zvážit terapeutické monitorování.
Při současném podávání přípravku Cosentyx s metotrexátem (MTX) a/nebo s kortikosteroidy nebyly v artritických studiích (včetně pacientů s psoriatickou artritidou a ankylozující spondylitidou) pozorovány žádné interakce.
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku
Ženy ve fertilním věku musí během léčby a po dobu nejméně 20 týdnů od ukončení léčby používat účinnou metodu kontracepce.
Odpovídající údaje o podávání secukinumabu těhotným ženám nejsou k dispozici. Studie reprodukční toxicity na zvířatech nenaznačují přímé nebo nepřímé škodlivé účinky na těhotenství, embryofetální vývoj, porod nebo pstnatální vývoj (viz bod 5.3). Podávání přípravku Cosentyx v těhotenství se z preventivních důvodů nedoporučuje.
Kojení
Není známo, zda se secukinumab vylučuje do lidského mateřského mléka. Imunoglobuliny se do lidského mateřského mléka vylučují a není známo, zda se secukinumab po požití systémově absorbuje. Vzhledem k možným nežádoucím účinkům secukinumabu na kojené dítě je nutno na základě posouzení prospěšnosti kojení pro dítě a prospěšnosti léčby pro matku rozhodnout, zda během léčby a po dobu až 20 týdnů od ukončení léčby přerušit kojení nebo přerušit léčbu přípravkem Cosentyx..
Fertilita
Vliv secukinumabu na fertilitu u člověka nebyl hodnocen. Studie na zvířatech nenaznačují přímé nebo nepřímé škodlivé účinky na fertilitu (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek Cosentyx nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky Souhrn bezpečnostního profilu
Přípravkem Cosentyx bylo léčeno celkem 6804 pacientů v zaslepených a otevřených klinických studiích s různými indikacemi (ložisková psoriáza, psoriatická artritida, ankylozující spondylitida a jiné autoimunitní stavy). Z tohoto počtu bylo 3671 pacientů exponováno přípravku Cosentyx po dobu nejméně jednoho roku, což reprezentuje expozici 6450 pacientoroků.
Nežádoucí účinky u ložiskové psoriázy
Čtyři placebem kontrolované studie fáze III ložiskové psoriázy byly sloučeny k vyhodnocení bezpečnosti přípravku Cosentyx v porovnání s placebem až do 12 týdnů po zahájení léčby. Celkem bylo hodnoceno 2076 pacientů (692 pacientů s dávkou 150 mg, 690 pacientů s dávkou 300 mg a 694 pacientů na placebu).
Nejčastěji hlášenými nežádoucími účinky (ADRs) byly infekce horních cest dýchacích (nejčastěji nasofaryngitida, rinitida). Většina reakcí byla mírná nebo středně závažná.
Nežádoucí účinky u psoriatické artritidy
Přípravek Cosentyx byl studován ve dvou placebem kontrolovaných studiích s psoriatickou artritidou s 1003 pacienty (703 pacientů léčených přípravkem Cosentyx a 300 pacientů na placebu) s celkovou dobou expozice 1061 pacientoroků (medián trvání expozice secukinumabem léčených pacientů:
456 dní u PsA studie 1 a 245 dní u PsA studie 2). Bezpečnostní profil pozorovaný u pacientů s psoriatickou artritidou léčených přípravkem Cosentyx je v souladu s bezpečnostním profilem u psoriázy.
Nežádoucí účinky u ankylozující spondvlitidv
Přípravek Cosentyx byl studován ve dvou placebem kontrolovaných studiích s ankylozující spondylitidou s 590 pacienty (394 pacientů léčených přípravkem Cosentyx a 196 pacientů na placebu) s celkovou dobou expozice 755 pacientoroků (medián trvání expozice secukinumabem léčených pacientů: 469 dní u AS studie 1 a 460 dní u AS studie 2). Bezpečnostní profil pozorovaný u pacientů s ankylozující spondylitidou léčených přípravkem Cosentyx je v souladu s bezpečnostním profilem u psoriázy.
Tabulkový seznam nežádoucích účinků
ADRs z klinických studií s psoriázou, psoriatickou artritidou a ankylozující spondylitidou (Tabulka 1) jsou řazeny podle systému orgánových tříd MedDRA. V každé třídě orgánových systémů jsou ADRs řazeny podle frekvence, s nejčastějšími reakcemi nejdřív. V rámci každé skupiny četnosti jsou nežádoucí účinky řazeny podle klesající závažnosti. Navíc jsou odpovídající frekvenční kategorie pro všechny nežádoucí účinky založeny na následující konvenci: velmi časté (>1/10); časté (>1/100 až <1/10); méně časté (>1/1,000 až <1/100); vzácné (>1/10,000 až <1/1,000); velmi vzácné (<1/10,000).
Tabulka 1 Seznam nežádoucích účinků z klinických studií1)
|
Systém orgánových tříd |
Frekvence |
Nežádoucí účinek |
|
Infekce a infestace |
Velmi časté |
Infekce horních cest dýchacích |
|
Časté |
Orální herpes | |
|
Méně časté |
Orální kandidóza | |
|
Tinea pedis | ||
|
Otitis externa | ||
|
Poruchy krve a lymfatického systému |
Méně časté |
Neutropenie |
|
Poruchy imunitního systému |
Vzácné |
Anafylaktické reakce |
|
Poruchy oka |
Méně časté |
Konjunktivitida |
|
Respirační, hrudní a mediastinální poruchy |
Časté |
Rhinorrhoea |
|
Gastrointestinální poruchy |
Časté |
Diarhea |
|
Poruchy kůže a podkožní tkáně |
Méně časté |
Urtikaria |
|
1) Placebem kontrolované klinické studie (fáze III) u pacientů s ložiskovou psoriázou, s PsA a AS exponovaných dávkám 300 mg, 150 mg nebo placebu s dobou léčby až 12 týdnů (psoriáza) nebo 16 týdnů (PsA a AS). | ||
Popis vybraných nežádoucích účinků Infekce
V placebem kontrolovaném období klinických studií s ložiskovou psoriázou (celkem 1382 pacientů léčených přípravkem Cosentyx a 694 pacientů na placebu po dobu až 12 týdnů), byly infekce hlášeny u 28,7% pacientů léčených přípravkem Cosentyx v porovnání se 18,9% pacientů na placebu. Většina infekcí se skládala z nezávažných až mírných infekcí horních cest dýchacích, jako je nasofaryngitida, které nevyžadovaly přerušení léčby. Objevil se nárůst mukózních nebo kožních kandidóz, konzistentních s mechanizmem účinku, jednalo se však o případy mírné nebo střední závažnosti, nezávažné, reagující na standardní léčbu a nevyžadující přerušení léčby. Závažné infekce se objevily u 0,14% pacientů léčených přípravkem Cosentyx a 0,3% u pacientů na placebu (viz bod 4.4).
Během celé léčebné periody (celkem 3430 léčených přípravkem Cosentyx po dobu až 52 týdnů u většiny pacientů) byly infekce hlášeny u 47,5% pacientů léčených přípravkem Cosentyx (0,9 na pacientorok dalšího sledování). Závažné infekce byly hlášeny u 1,2% pacientů léčených přípravkem Cosentyx (0,015 na pacientorok dalšího sledování).
Počet infekcí pozorovaných ve studiích psoriatické artritidy a ankylozující spondylitidy byl podobný počtu infekcí pozorovaných u psoriatických studií.
Neutropenie
V klinických studiích fáze 3 u psoriázy byla neutropenie mnohem častěji pozorována u secukinumabu než u placeba, nicméně většina případů byla mírná, přechodná a reversibilní. Neutropenie <1,0-0,5x109/l (CTCAE stupeň 3) byla hlášena u 18 z 3430 (0,5%) pacientů léčených secukinumabem, bez závislosti na dávce a bez časové souvislosti s infekcí u 15 z 18 případů. Případy závažnější neutropenie nebyly. Nezávažné infekce s obvyklou odpovědí na standardní léčbu a nevyžadující přerušení léčby přípravkem Cosentyx byly hlášeny ve zbývajících 3 případech.
Frekvence výskytu neutropenie u psoriatické artritidy a ankylozující spondylitidy je podobná jako u psoriázy.
Byly hlášeny vzácné případy neutropenie <0,5x109/l (CTCAE stupeň 4).
Reakce z přecitlivělosti
V klinických studiích byla ve vztahu k přípravku Cosentyx pozorována kopřivka a vzácné případy anafylaktické reakce (viz též bod 4.4).
Imunogenicita
V klinických studiích u psoriázy, psoriatické artritidy a ankylozující spondylitidy si méně než 1% pacientů léčených přípravkem Cosentyx vyvinulo protilátky proti secukinumabu po dobu až 52 týdnů léčby. Zhruba polovina protilátek objevivších se v souvislosti s léčbou byla neutralizujících, nicméně to nebylo spojeno se ztrátou účinnosti nebo farmakokinetickými abnormalitami.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
V klinických studiích nebyly hlášeny případy předávkování.
Dávky až do 30 mg/kg (přibližně 2000 až 3000 mg) byly v klinických studiích podány intravenózně bez známek na dávce závislé toxicity. V případě předávkování se doporučuje monitorovat pacienta kvůli známkám nebo příznakům nežádoucích účinků a je potřeba neprodleně zahájit vhodnou symptomatickou léčbu.
FARMAKOLOGICKÉ VLASTNOSTI
5.
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Imunosupresiva, inhibitory interleukinu, ATC kód: L04AC10 Mechanismus účinku
Secukinumab je plně humánní IgG1/K monoklonální protilátka, která se selektivně váže na prozánětlivý cytokin interleukin-17A (IL-17A) a neutralizuje ho. Secukinumab působí zacílením na IL-17A a inhibicí jeho interakce s receptorem pro IL-17, který je exprimován různými typy buněk, včetně keratinocytů. Jako výsledek secukinumab inhibuje uvolňování prozánětlivých cytokinů, chemokinů a mediátorů tkáňového poškození a snižuje IL-17A-zprostředkovaný příspěvek k autoimunitním a zánětlivým chorobám. V pokožce jsou dosaženy klinicky relevantní hladiny secukinumabu a jsou redukovány lokální zánětlivé markery. Jako přímý důsledek léčby secukinumabem dochází k redukci erytému, ztvrdnutí a odlupování pokožky přítomných v ložiskových psoriatických lézích.
IL-17A je přirozeně se vyskytující cytokin, který se účastní normální zánětlivé a imunitní odpovědi. IL-17A hraje klíčovou roli v patogenezi ložiskové psoriázy, psoriatické artritidy a ankylozující spondylitidy a jeho množství je zvýšeno v kožních lézích v porovnání s pokožkou bez kožních lézí u pacientů s ložiskovou psoriázou a v synoviální tekutině pacientů s psoriatickou artritidou. Četnost výskytu IL-17-produkujících buněk byla též významně vyšší v subchondrální kostní dřeni intervertebrálních kloubů u pacientů s ankylozující spondylitidou.
Farmakodynamické účinky
Sérové hladiny celkového IL-17A (volný a IL-17As navázaným secukinumabem) u pacientů léčených secukinumabem zpočátku rostou. To je následováno pomalým poklesem kvůli snížené clearance IL-17A s navázaným secukinumabem, což naznačuje, že secukinumab selektivně vychytává volný IL-17A, který hraje klíčovou roli v patogenezi ložiskové psoriázy.
Ve studii se secukinumabem došlo po jednom až dvou týdnech léčby k signifikantnímu snížení infiltrujících epidermálních neutrofilů a různých s neutrofily spojovaných markerů, které jsou u pacientů s ložiskovou psoriázou v kožních lézích zvýšeny.
Secukinumab snižoval (během 1 až 2 týdnů léčby) hladiny C-reaktivního proteinu, který je markerem zánětu.
Klinická účinnost a bezpečnost
Ložisková _ psoriáza
Bezpečnost a účinnost přípravku Cosentyx byly hodnoceny ve čtyřech randomizovaných, dvojitě zaslepených, placebem kontrolovaných studiích fáze III u pacientů se středně závažnou až závažnou ložiskovou psoriázou, kteří byli kandidáty pro fototerapii nebo systémovou terapii [ERASURE, FIXTURE, FEATURE, JUNCTURE]. Účinnost a bezpečnost přípravku Cosentyx 150 mg a 300 mg byla hodnocena buď proti placebu nebo proti etanerceptu. Navíc jedna studie hodnotila chronický léčebný režim v porovnání s ‘přeléčením podle potřeby’ [SCULPTURE].
Z 2403 pacientů, kteří byli zařazeni do placebem kontrolovaných studií bylo 79% biologicky naivních, 45% bylo nebiologických selhání a 8% byla biologická selhání (6% anti-TNF selhání a 2% anti-p40 selhání). Přibližně 15 až 25% pacientů ve studiích fáze III mělo při zahájení léčby psoriatickou artritidu (PsA).
Psoriatická studie 1 (ERASURE) hodnotila 738 pacientů. Pacienti randomizovaní na přípravek Cosentyx dostávali dávku 150 mg nebo 300 mg v týdnech 0, 1, 2, a 3, následované stejnou dávkou každý měsíc počínaje týdnem 4. Psoriatická studie 2 (FIXTURE) hodnotila 1,306 pacientů. Pacienti randomizovaní na přípravek Cosentyx dostávali dávku 150 mg nebo 300 mg v týdnech 0, 1, 2, a 3, následované stejnou dávkou každý měsíc počínaje týdnem 4. Pacienti randomizovaní na etanercept dostávali dávku 50 mg dvakrát týdně po dobu 12 týdnů, následované dávkou 50 mg každý týden. V obou studiích 1 a 2 byli pacienti randomizovaní na placebo a neodpovídající na léčbu v týdnu 12 převedeni na přípravek Cosentyx (buď 150 mg nebo 300 mg) v týdnech 12, 13, 14, a 15, následované stejnou dávkou každý měsíc počínaje týdnem 16. Všichni pacienti byli sledováni po dobu až 52 týdnů od prvního podání medikace ve studii.
Psoriatická studie 3 (FEATURE) hodnotila 177 pacientů používajících předplněné injekční stříkačky v porovnání s placebem po 12 týdnech léčby pro zhodnocení bezpečnosti, tolerability a použitelnosti samoaplikace přípravku Cosentyx pomocí předplněné injekční stříkačky. Psoriatická studie 4 (JUNCTURE) hodnotila 182 pacientů používajících předplněné pero v porovnání s placebem po 12 týdnech léčby pro zhodnocení bezpečnosti, tolerability a použitelnosti samoaplikace přípravku Cosentyx pomocí předplněného pera. V obou studiích 3 a 4 pacienti randomizovaní na přípravek Cosentyx dostávali dávku 150 mg nebo 300 mg v týdnech 0, 1, 2, a 3, následované stejnou dávkou každý měsíc počínaje týdnem 4. Pacienti byli též randomizováni na placebo v týdnech 0, 1, 2, a 3, následované stejnou dávkou každý měsíc počínaje týdnem 4.
Psoriatická studie 5 (SCULPTURE) hodnotila 966 pacientů. Všichni pacienti dostávali přípravek Cosentyx v dávce 150 mg nebo 300 mg v týdnech 0, 1, 2, 3, 4, 8 a 12 a poté byli randomizováni buď k udržovací léčbě stejnou dávkou každý měsíc počínaje týdnem 12, nebo na režim s “přeléčením v čas potřeby“ stejnou dávkou. U pacientů randomizovaných k “přeléčení v čas potřeby“ nebylo dosaženo odpovídající udržení odpovědi a proto se doporučuje fixní měsíční udržovací režim.
Koprimarní cíl placebem a aktivním komparátorem kontrolovaných studií činil podíl pacientů s dosaženou odpovědí PASI 75 a IGA mód 2011 odpovědi “čistý” nebo “téměř čistý” v porovnání s placebem v týdnu 12 (viz Tabulku 2 a 3). Dávka 300 mg poskytovala zlepšenou clearance pokožky zejména pro “čistou” nebo “téměř čistou” pokožku v rozmezí cílů účinnosti PASI 90, PASI 100, a IGA mód 2011 0 nebo 1 odpověď u všech studií s největším účinkem pozorovaným v týdnu 16, proto se doporučuje tato dávka.
Týden 12_Týden 16_Týden 52
|
Placebo |
150 mg |
300 mg |
150 mg |
300 mg |
150 mg |
300 mg | |
|
Studie 1 | |||||||
|
Počet pacientů |
246 |
244 |
245 |
244 |
245 |
244 |
245 |
|
Odpověď PASI 50 n (%) |
22 |
203 |
222 |
212 |
224 |
187 |
207 |
|
(8,9%) |
(83,5%) |
(90,6%) |
(87,2%) |
(91,4%) |
(77%) |
(84,5%) | |
|
Odpověď PASI 75 n (%) |
11 |
174 |
200 |
188 |
211 |
146 |
182 |
|
(4,5%) |
(71,6%)* * |
(81,6%)* * |
(77,4%) |
(86,1%) |
(60,1%) |
(74,3%) | |
|
Odpověď PASI 90 n (%) |
3 |
95 |
145 |
130 |
171 |
88 |
147 |
|
(1,2%) |
(39,1%)* * |
(59,2%)* * |
(53,5%) |
(69,8%) |
(36,2%) |
(60,0%) | |
|
Odpověď PASI 100 |
2 |
31 |
70 |
51 |
102 |
49 |
96 |
|
Odpověď n (%) |
(0,8%) |
(12,8%) |
(28,6%) |
(21,0%) |
(41,6%) |
(20,2%) |
(39,2%) |
|
Odpověď IGA mód 2011 |
6 |
125 |
160 |
142 |
180 |
101 |
148 |
|
“čistý” nebo “téměř |
(2,40%) |
(51,2%)* |
(65,3%)* |
(58,2%) |
(73,5%) |
(41,4%) |
(60,4%) |
|
čistý” n (%) Studie 3 |
* |
* | |||||
|
Počet pacientů |
59 |
59 |
58 |
- |
- |
- |
- |
|
Odpověď PASI 50 n (%) |
3 |
51 |
51 |
- |
- |
- |
- |
|
(5,1%) |
(86,4%) |
(87,9%) | |||||
|
Odpověď PASI 75 n (%) |
0 |
41 |
44 |
- |
- |
- |
- |
|
(0,0%) |
(69,5%)* * |
(75,9%)* * | |||||
|
Odpověď PASI 90 n (%) |
0 |
27 |
35 |
- |
- |
- |
- |
|
(0,0%) |
(45,8%) |
(60,3%) | |||||
|
Odpověď PASI 100 n |
0 |
5 |
25 |
- |
- |
- |
- |
|
(%) |
(0,0%) |
(8,5%) |
(43,1%) | ||||
|
Odpověď IGA mód 2011 |
0 |
31 |
40 |
- |
- |
- |
- |
|
“čistý” nebo “téměř |
(0,0%) |
(52,5%)* |
(69,0%)* | ||||
|
čistý” n (%) Studie 4 |
* |
* | |||||
|
Počet pacientů |
61 |
60 |
60 |
- |
- |
- |
- |
|
Odpověď PASI 50 n (%) |
5 |
48 |
58 |
- |
- |
- |
- |
|
(8,2%) |
(80,0%) |
(96,7%) | |||||
|
Odpověď PASI 75 n (%) |
2 |
43 |
52 |
- |
- |
- |
- |
|
(3,3%) |
(71,7%)* * |
(86,7%)* * | |||||
|
Odpověď PASI 90 n (%) |
0 |
24 |
33 |
- |
- |
- |
- |
|
(0,0%) |
(40,0%) |
(55,0%) | |||||
|
Odpověď PASI 100 n |
0 |
10 |
16 |
- |
- |
- |
- |
|
(%) |
(0,0%) |
(16,7%) |
(26,7%) | ||||
|
Odpověď IGA mód 2011 |
0 |
32 |
44 |
- |
- |
- |
- |
|
“čistý” nebo “téměř |
(0,0%) |
(53,3%)* |
(73,3%)* | ||||
|
čistý” n (%) |
* |
* | |||||
* IGA mód 2011 je 5 bodová škála zahrnující “0 = čistý”, “1 = téměř čistý”, “2 = mírný”, “3 = středně závažný” nebo “4 = závažný”, indikující lékařovo celkové hodnocení závažnosti psoriázy zaměřené na ztvrdnutí, erytém a odlupování. Léčebný úspěch “ čistý ” nebo “ téměř čistý ” znamenaly nepřítomnost známek psoriázy nebo normální až růžové zbarvené léze, netloustnutí ložisek a žádné nebo minimální místní odlupování.
** p hodnoty versus placebo a nastavené na multiplicitu: p<0,0001._
“čistý”
Týden 12_Týden 16_Týden 52
|
Placeb |
150 |
300 |
Etanerc |
150 |
300 |
Etanerce |
150 |
300 |
Etanerce | |
|
o |
mg |
mg |
ept |
mg |
mg |
pt |
mg |
mg |
pt | |
|
Počet |
324 |
327 |
323 |
323 |
327 |
323 |
323 |
327 |
323 |
323 |
|
pacientů Odpověď |
49 |
266 |
296 |
226 |
290 |
302 |
257 |
249 |
274 |
234 |
|
PASI 50 n |
(15,1% |
(81,3% |
(91,6% |
(70,0%) |
(88,7% |
(93,5% |
(79,6%) |
(76,1% |
(84,8% |
(72,4%) |
|
(%) |
) |
) |
) |
) |
) |
) |
) | |||
|
Odpověď |
16 |
219 |
249 |
142 |
247 |
280 |
189 |
215 |
254 |
179 |
|
PASI 75 n |
(4,9%) |
(67,0% |
(77,1% |
(44,0%) |
(75,5% |
(86,7% |
(58,5%) |
(65,7% |
(78,6% |
(55,4%) |
|
(%) |
)6 |
) |
) |
) |
) | |||||
|
Odpověď |
5 |
137 |
175 |
67 |
176 |
234 |
101 |
147 |
210 |
108 |
|
PASI 90 n |
(1,5%) |
(41,9% |
(54,2% |
(20,7%) |
(53,8% |
(72,4% |
(31,3%) |
(45,0% |
(65,0% |
(33,4%) |
|
(%) |
) |
) |
) |
) |
) |
) | ||||
|
Odpověď |
0 (0%) |
47 |
78 |
14 |
84 |
119 |
24 (7,4%) |
65 |
117 |
32 (9,9%) |
|
PASI 100 |
(14,4% |
(24,1% |
(4,3%) |
(25,7% |
(36,8% |
(19,9% |
(36,2% | |||
|
n (%) |
) |
) |
) |
) |
) |
) | ||||
|
Odpověď |
9 |
167 |
202 |
88 |
200 |
244 |
127 |
168 |
219 |
120 |
|
IgA mód |
(2,8%) |
(51,1% |
(62,5% |
(27,2%) |
(61,2% |
(75,5% |
(39,3%) |
(51,4% |
(67,8% |
(37,2%) |
|
2011 |
)6 |
)6 |
) |
) |
) |
) |
nebo
“téměř
čistý”
Týden 4_Týden 16
|
Secukinumab |
Ustekinumab* |
Secukinumab |
Ustekinumab | |
|
Počet pacientů |
300 mg 334 |
335 |
300 mg 334 |
335 |
|
Odpověď PASI 75 |
167 (50,0%)6 |
69 (20,6%) |
311 (93,1%) |
277 (82,7%) |
|
n (%) Odpověď PASI 90 |
70 (21,0%) |
18 (5,4%) |
264 (79,0%)6 |
193 (57,6%) |
|
n (%) Odpověď PASI 100 |
14 (4,2%) |
3 (0,9%) |
148 (44,3%) |
95 (28,4%) |
|
n (%) Odpověď IGA mód |
126 (37,7%) |
41 (12,2%) |
277 (82,9%) |
226 (67,5%) |
2011 „čistý“ nebo
„téměř čistý“ n (%)_
* Pacienti léčení secukinumabem dostávali dávku 300 mg v týdnech 0, 1, 2 a 3 následovanou stejnou dávkou v týdnech 4, 8 a 12. Pacienti léčení ustekinumabem dostávali dávku 45 mg nebo 90 mg v týdnech 0 a 4 (dávka podle hmotnosti a schváleného dávkování)
** p hodnoty versus ustekinumab: p<0,0001
Přípravek Cosentyx byl účinný u pacientů bez předchozí systémové léčby, biologicky naivních, biologicky/anti-TNF-exponovaných a pacientů s biologickým/anti-TNF selháním. Zlepšení PASI 75 u pacientů se souběžnou psoriatickou artritidou při zahájení léčby byly podobné těm u celé populace s ložiskovou psoriázou.
Přípravek Cosentyx byl spojován s rychlým nástupem účinku s 50% redukcí průměrného PASI do týdne 3 při dávce 300 mg.
Obrázek 1 časový průběh procentní změny od výchozích hodnot průměrného PASI skóre ve Studii 1 (ERASURE)
PASI % změna od základních hodnot

Týdny léčby
n = po čet vyhodnotitelných pacientů
• Cosentyx 150 mg (n=243) o Cosentyx 300 mg (n=245) □ Placebo (n=245)
Specifické lokalizace/formy ložiskové psoriázy
Ve dvou dalších placebem kontrolovaných studiích bylo pozorováno zlepšení jak u nehtové psoriázy (studie TRANSFIGURE, 198 pacientů), tak u palmoplantární ložiskové psoriázy (studie GESTURE, 205 pacientů). Ve studii TRANSFIGURE byla prokázána superiorita secukinumabu oproti placebu v 16. týdnu (46,1% pro 300 mg, 38,4% pro 150 mg a 11,7% pro placebo) ve výrazném zlepšení oproti počátečnímu stavu podle indexu závažnosti psoriázy nehtů (NAPSI %) u pacientů se středně těžkou až těžkou ložiskovou psoriázou s postižením nehtů. Ve studii GESTURE byla prokázána superiorita secukinumabu oproti placebu v 16. týdnu (33,3% pro 300 mg, 22,1% pro 150 mg a 1,5% pro placebo) ve výrazném zlepšení oproti počátečnímu stavu podle odpovědi ppIGA 0 nebo 1 („čistý“ nebo „téměř čistý“) u pacientů se středně těžkou až těžkou palmoplantární ložiskovou psoriázou.
Kvalita života/výsledky hlášené pacienty
Statisticky zlepšení hodnot v týdnu 12 (Studie 1-4) oproti hodnotám před léčbou v porovnání s placebem bylo demonstrováno pomocí DLQI (Dermatology Life Quality Index). Průměrný pokles (zlepšení) DLQI z hodnot před léčbou kolísalo od -10,4 do -11,6 u secukinumabu 300 mg, od -7,7 do -10,1 u secukinumabu 150 mg, v porovnání s -1,1 až -1,9 u placeba v týdnu 12. Toto zlepšení přetrvávalo 52 týdnů (Studie 1 a 2).
Čtyřicet procent účastníků ve studiích 1 a 2 vyplnilo deník Psoriasis Symptom Diary©. U účastníků, kteří vyplnili tento deník, bylo v každé z těchto studií v porovnání s placebem prokázáno statisticky významné zlepšení z hodnot před léčbou v týdnu 12 u pacienty hlášených známek a příznaků svědění, bolesti a odlupování.
Psoriatická artritida
Bezpečnost a účinnost přípravku Cosentyx byla hodnocena u 1003 pacientů ve dvou randomizovaných, dvojitě zaslepených, placebem kontrolovaných studiích fáze III u pacientů s aktivní psoriatickou artritidou (>3 oteklé a >3 bolestivé klouby) přes léčbu nesteroidními protizánětlivými léky (NSAID), kortikosteroidy nebo chorobu modifikujícími antirevmatiky (DMARD). Do těchto studií byli zařazeni pacienti všech subtypů PsA, včetně polyartikulární artritidy bez průkazu revmatoidních nodulů, spondylitidy s periferní artritidou, asymetrickou periferní artritidou, postižení distálních interfalangeálních kloubů a arthritis mutilans. Pacienti v těchto studiích měli stanovenu diagnózu PsA s mediánem 3,9 až 5,3 roku. Většina pacientů měla též aktivní psoriatické kožní léze nebo dokumentovanou psoriázu v anamnéze. Přes 62% a 47% PsA pacientů mělo před zahájením enthesitidu, respektive daktylitidu. V obou studiích bylo dosaženo primárního cíle - odpovědi ACR20 (American College of Rheumatology) v týdnu 24.
Ve studii psoriatické artritidy 1 (PsA studie 1) a studii psoriatické artritidy 2 (PsA studie 2) 29% pacientů, respektive 35% pacientů, bylo již léčeno anti-TNFa přípravkem a ukončilo užívání anti-TNFa přípravku buď pro nedostatek účinku nebo nesnášenlivost (anti-TNFa-IR pacienti).
PsA studie 1 (FUTURE 1) hodnotila 606 pacientů, z nichž 60,7% užívalo současně MTX. Pacienti randomizovaní na přípravek Cosentyx dostávali dávku 10 mg/kg intravenózně v týdnech 0, 2 a 4, následovanou buď 75 mg nebo 150 mg subkutánně každý měsíc počínaje týdnem 8. Pacienti randomizovaní na placebo byli převedeni na přípravek Cosentyx (buď 75 mg nebo 150 mg subkutánně) s následnou stejnou dávkou každý měsíc, non-respondéři v 16. týdnu (časná záchrana) a ostatní ve 24. týdnu.
PsA studie 2 (FUTURE 2) hodnotila 397 pacientů, z nichž 46,6% užívalo současně MTX. Pacienti randomizovaní na přípravek Cosentyx dostávali dávku 75 mg, 150 mg nebo 300 mg subkutánně v týdnech 0, 1, 2 a 3, s následnou stejnou dávkou každý měsíc, počínaje týdnem 4. Pacienti randomizovaní na placebo, kteří v týdnu 16 neodpovídali na léčbu byli v 16. týdnu (časná záchrana) převedeni na přípravek Cosentyx (buď 150 mg nebo 300 mg subkutánně) s následnou stejnou dávkou každý měsíc. Pacienti randomizovaní na placebo, kteří v 16. týdnu měli odpověď na léčbu, byli v 24. týdnu převedeni na přípravek Cosentyx (buď 150 mg nebo 300 mg subkutánně) s následnou stejnou dávkou každý měsíc.
Známky a příznaky
Léčba přípravkem Cosentyx v porovnání s placebem znamenala významné zlepšení v hodnocení aktivity choroby v týdnu 24 (viz Tabulka 5).
Tabulka 5 Klinická odpověď u PsA studie 2 v týdnu 24
|
Týden 24 | ||||
|
Placebo |
75 mg |
150 mg |
300 mg | |
|
Počet randomizovaných pacientů |
98 |
99 |
100 |
100 |
|
ACR20 odpověď n (%) |
15 (15,3%) |
29 (29,3%*) |
51 (51,0%***) |
54 (54,0%***) |
|
ACR50 odpověď n (%) |
7 (7,1%) |
18 (18,2%) |
35 (35,0%) |
35 (35,0%**) |
|
ACR70 odpověď n (%) |
1 (1,0%) |
6 (6,1%) |
21 (21,0%**) |
20 (20,0%**) |
|
DAS28-CRP |
-0,96 |
-1,12 |
-1,58** |
-1,61** |
|
Počet pacientů s >3% BSA psoriatickým postižením pokožky před zahájením |
43 (43,9%) |
50 (50,5%) |
58 (58,0%) |
41 (41,0%) |
|
PASI 75 odpověď n (%) |
7 (16,3%) |
14 (28,0%) |
28 (48,3%**) |
26 (63,4%***) |
|
PASI 90 odpověď n (%) |
4 (9,3%) |
6 (12,0%) |
19 (32,8%**) |
20 (48,8%***) |
|
Vymizení daktylitidy n (%) f |
4 (14,8%) |
10 (30,3%) |
16 (50,0%**) |
26 (56,5%**) |
|
Vymizení entesitidy n (%) $ |
14 (21,5%) |
22 (32,4%) |
27 (42,2%*) |
27 (48,2%**) |
* p<0,05, ** p<0,01, *** p<0,001; oproti placebu
Všechny p-hodnoty jsou nastaveny na mnohočetné testování založené na předem definované hierarchii, s výjimkou ACR70, daktylitidy a entesitidy, což byly výzkumné cíle.
Pacienti s chybějícími údaji o léčebné odpovědi byly považováni za non-respondéry.
ACR: American College of Rheumatology; PASI: Psoriasis Area and Severity Index; DAS: Disease Activity Score (skóre aktivity nemoci); BSA: Body Surface Area (povrch těla) fU pacientů s daktylitidou při zahájení (n=27, 33, 32, 46)
t U pacientů s entesitidou při zahájení (n=65, 68, 64, 56)_
Nástup účinku přípravku Cosentyx byl pozorován již ve 2. týdnu. Statisticky významného rozdílu v ACR 20 oproti placebu bylo dosaženo ve 3. týdnu. V 16. týdnu vykazovali pacienti léčení přípravkem Cosentyx významné zlepšení známek a příznaků s významně vyšší odpovědí v ACR 20 (33,3%, 60,0% a 57,0% u 75 mg, respektive 150 mg a 300 mg) v porovnání s placebem (18,4%).
Procento pacientů s dosaženou ACR 20 odpovědí je na obrázku 2.
Obrázek 2 ACR20 odpověď v PsA studii 2 v závislosti na čase do týdne 24

Čas (týdny)
■ Cosentyx 150 mg Cosentyx 300 mg ■ ■ ■ » Placebo
Procento respondérů
Podobné odpovědi pro primární a klíčový sekundární cíl byly pozorovány u PsA pacientů bez ohledu na to, zda byli současně léčeni MTX či nikoliv. V týdnu 24 měli pacienti léčení přípravkem Cosentyx a současně MTX vyšší ACR 20 odpověď (44,7%, 47,7% a 54,4% u 75 mg, respektive 150 mg a 300 mg, v porovnání s placebem 20,0%) a ACR 50 odpověď (27,7%, 31,8% a 38,6% u 75 mg, respektive 150 mg a 300 mg v porovnání s placebem 8,0%). Pacienti léčení přípravkem Cosentyx bez současně podávaného MTX měli vyšší ACR 20 odpověď (15,4%, 53,6% a 53,6% u 75 mg, respektive 150 mg a 300 mg, v porovnání s placebem 10,4%) a ACR 50 odpověď (9,6%, 37,5% a 32,1% u 75 mg, respektive 150 mg a 300 mg, v porovnání s placebem 6,3%).
Jak anti-TNFa-naivní, tak anti-TNFa-IR pacienti léčení přípravkem Cosentyx měli významně vyšší ACR 20 odpověď v porovnání s placebem v týdnu 24, s mírně vyšší odpovědí u anti-TNFa-naivní skupiny (anti-TNFa-naivní: 37%, 64% a 58% u 75 mg, respektive 150 mg and 300 mg, v porovnání s placebem 15,9%; anti-TNFa-IR: 15%, 30% a 46% u 75 mg, respektive 150 mg a 300 mg, v porovnání s placebem 14,3%). V anti-TNFa-IR podskupině pacientů vykázala významně vyšší ACR 20 odpověď v porovnání s placebem (p<0,05) pouze dávka 300 mg ademonstrovala klinicky smysluplný prospěch oproti 150 mg také ve více sekundárních cílech. Zlepšení PASI 75 odpovědi bylo pozorováno v obou podskupinách a dávka 300 mg prokázala statisticky signifikantní přínos u anti-TNFa-IR pacientů.
Počet PsA pacientů s axiálním postižením byl příliš malý na to, aby bylo možné stanovit vypovídající hodnocení.
Zlepšení se objevilo u všech komponent ACR skóre, včetně hodnocení bolesti pacientem. Podíl pacientů s dosaženou modifikovanou PsA Response Criteria (PsARC) odpovědí byl větší ve skupině pacientů léčených přípravkem Cosentyx (38,4%, 62,0% a 63,0% u 75 mg, respektive 150 mg a 300 mg) v porovnání s placebem (29,6%) v týdnu 24.
V PsA studii 1 a PsA studii 2 přetrvávala účinnost do týdne 52. V PsA studii 2 bylo v 52. týdnu stále léčeno 178 pacientů (89%) z 200 pacientů iniciálně randomizovaných na Cosentyx 150 mg a 300 mg. Ze 100 pacientů randomizovaných na Cosentyx 150 mg, 64, respektive 39 a 20, vykázalo ACR 20/50/70 odpověď. Ze 100 pacientů randomizovaných na Cosentyx 300 mg, 64, respektive 44 a 24 vykázalo ACR 20/50/70 odpověď.
Radiografická odpověď
Inhibice progrese strukturálního poškození u PsA nebyla dosud prokázána za použití podkožního zaváděcího dávkovacího režimu schváleného pro klinické použití.
V PsA studii 1 byla inhibice progrese strukturálního poškození hodnocena radiograficky a vyjádřena jako změna modifikovaného celkového Sharp Score (mTSS) a jeho komponent, Erosion Score (ES) a Joint Space Narrowing skóre (JSN) v týdnech 24 a 52, v porovnání s výchozími hodnotami. Data z týdne 24 jsou uvedena v Tabulce 6.
Tabulka 6 Změna modifikovaného Total Sharp skóre u psoriatické artritidy
|
Placebo N=179 |
Cosentyx 75 mg1 N=181 |
Cosentyx 150 mg1 N=185 | |
|
Celkové skóre | |||
|
Počáteční hodnoty |
28,4 |
20,4 |
22,3 |
|
(SD) |
(63,5) |
(39,4) |
(48,0) |
|
Průměrná změna v týdnu 24 |
0,57 |
0,02* |
0,13* |
*p<0,05 založená na nominální, ale neupravené p-hodnotě 110 mg/kg v týdnech 0, 2 a 4 následovaná 75 mg nebo150 mg
Inhibice strukturálního poškození přetrvávala při léčbě přípravkem Cosentyx do týdne 52.
Procento pacientů bez progrese choroby (definované jako změna od výchozích hodnot mTSS <0,5) od randomizace do týdne 24 bylo 92,3% ve větvi se secukinumabem s dávkovacím schématem 10 mg/kg intravenózního bolusu - 75 mg subkutánně (udržovací dávka), 82,3% ve větvi se secukinumabem s dávkovacím schematem 10 mg/kg intravenózního bolusu - 150 mg subkutánně (udržovací dávka) a 75,7% ve větvi s placebem. Procento pacientů bez progrese choroby od týdne 24 do týdne 52 u secukinumabu 10 mg/kg ve formě intravenózního bolusu - následovaného buď 75 mg nebo 150 mg subkutánně (udržovací dávka) a u pacientů na placebu, kteří byli v týdnu 16 nebo 24 převedeni na 75 mg nebo 150 mg subkutánně (udržovací dávka) každé 4 týdny, bylo 85,8%, respektive 85,7% a 86,8%.
Funkční stav a kvalita života
V PsA studii 2, pacienti léčení přípravkem Cosentyx 150 mg (p=0,0555) a 300 mg (p=0,0040) vykázali zlepšení funkčního stavu v porovnání s pacienty na placebu hodnocené podle Health Assessment Questionnaire-Disability Index (HAQ-DI) v týdnu 24. Zlepšení HAQ-DI skóre bylo pozorováno bez ohledu na předchozí expozici anti-TNFa. Podobná odpověď byla pozorována v PsA studii 1.
Pacienti léčení přípravkem Cosentyx hlásili významné zlepšení se zdravím související kvality života měřené pomocí Short Form-36 Health Survey Physical Component Summary (SF-36 PCS) skóre (p<0,001). Prokázalo se též statisticky významé zlepšení, což bylo demonstrováno ve výzkumných cílech hodnocených Functional Assessment of Chronic Illness Therapy - Fatigue (FACIT-F) skóre u dávky 150 mg a 300 mg v porovnání s placebem (7,97, respektive 5,97 versus 1,63). Podobné odpovědi byly pozorovány v PsA studii 1 a účinnost přetrvávala do týdne 52.
Ankvlozující spondylitida
Bezpečnost a účinnost přípravku Cosentyx byla hodnocena u 590 pacientů ve dvou randomizovaných, dvojitě zaslepených, placebem kontrolovaných studiích fáze III u pacientů s aktivní ankylozující spondylitidou (AS) s Bath Ankylosing Spondylitis Disease Activity indexem (BASDAI) >4 přes léčbu nesteroidními protizánětlivými léky (NSAID), kortikosteroidy nebo chorobu modifikujícími antirevmatiky (DMARD). Pacienti v těchto studiích měli stanovenu diagnózu AS s mediánem 2,7 až
5,8 roku. V obou studiích bylo primárním cílem nejméně 20% zlepšení Assessment of Spondyloarthritis International Society kritéria (ASAS 20) v týdnu 16.
Ve studii ankylozující spondylitidy 1 (AS studie 1) a studii ankylozující spondylitidy 2 (AS studie 2) 27,0%, respektive 38,8% pacientů, bylo dříve léčeno anti-TNFa přípravkem a léčba anti-TNFa přípravkem byla ukončena buď pro nedostatek účinku nebo nesnášenlivost (anti-TNFa-IR pacienti).
AS Studie 1 (MEASURE 1) hodnotila 371 pacientů, z nichž užívalo současně 14,8%, respektive 33,4%, MTX nebo sulfasalazin. Pacienti randomizovaní na přípravek Cosentyx dostávali dávku 10 mg/kg intravenózně v týdnech 0, 2 a 4, následovanou buď 75 mg nebo 150 mg subkutánně každý měsíc počínaje týdnem 8. Pacienti randomizovaní na placebo byli převedeni na přípravek Cosentyx (buď 75 mg nebo 150 mg subkutánně) s následnou stejnou dávkou každý měsíc, non respondéři v 16. týdnu (časná záchrana) a všichni ostatní ve 24. týdnu.
AS studie 2 (MEASURE 2) hodnotila 219 pacientů, z nichž užívalo současně 11,9%, respektive 14,2%, MTX nebo sulfasalazin. Pacienti randomizovaní na přípravek Cosentyx dostávali dávku 75 mg nebo 150 mg subkutánně v týdnech 0, 1, 2 a 3, s následnou stejnou dávkou každý měsíc, počínaje týdnem 4. V týdnu 16 byli pacienti při zahájení randomizovaní na placebo re-randomizováni na přípravek Cosentyx (buď 75 mg nebo 150 mg subkutánně) každý měsíc.
Známky a příznaky
V AS studii 2 znamenala léčba přípravkem Cosentyx 150 mg v porovnání s placebem větší zlepšení v hodnocení aktivity choroby v týdnu 16 (viz Tabulka 7).
Tabulka 7 Klinická odpověď v AS studii 2 v týdnu 16
|
Výsledek (p-hodnota versus placebo) |
Placebo (n = 74) |
75 mg (n = 73) |
150 mg (n = 72) |
|
ASAS 20 odpověď, % |
28,4 |
41,1 |
61,1*** |
|
ASAS 40 20 odpověď, % |
10,8 |
26,0 |
36,1*** |
|
hsCRP, (post-BSL/BSL ratio) |
1,13 |
0,61 |
0,55*** |
|
ASAS 5/6, % |
8,1 |
34,2 |
43 1*** |
|
ASAS částečná remise, % |
4,1 |
15,1 |
13,9 |
|
BASDAI 50, % |
10,8 |
24,7* |
30,6** |
|
ASDAS-CRP velké zlepšení |
4,1 |
15,1* |
25,0*** |
|
* p<0,05, ** p<0,01, *** p<0,001; versus placebo Všechny p-hodnoty j sou nastaveny na mnohočetné testování založené na předem definované hierarchii, s výjimkou BASDAI 50 a ASDAS-CRP Pacienti s chybějícími údaji o léčebné odpovědi byli považováni za non-respondéry. | |||
|
ASAS: Assessment of SpondyloArthritis International Society Criteria; BASDAI: Bath Ankylosing Spondylitis Disease Activity Index; hsCRP: high-sensitivity C-reactive protein; ASDAS: Ankylosing Spondylitis Disease Activity Score; BSL: baseline | |||
V AS studii 2 se nástup účinku přípravku Cosentyx 150 mg projevil již v týdnu 1 (superiorita Cosentyxu vůči placebu u odpovědi ASAS 20) a v týdnu 2 (superiorita Cosentyxu vůči placebu u odpovědi ASAS 40).
ASAS 20 se zlepšila v týdnu 16 jak u anti-TNFa-naivních pacientů (68,2% versus 31,1%; p<0,05), tak u anti-TNFa-IR pacientů (50,0% versus 24,1%; p<0,05) u přípravku Cosentyx 150 mg v porovnání s placebem.
V obou AS studiích, u pacientů léčených přípravkem Cosentyx (150 mg v AS Studii 2 a oběma režimy AS studie 1) došlo v 16. týdnu k významnému zlepšení známek a příznaků se srovnatelným rozsahem odpovědi. Účinnost přetrvávala do 52. týdne jak u anti-TNFa-naivních, tak u anti-TNFa-IR pacientů.
V AS studii 2 bylo v 52. týdnu stále léčeno 61 pacientů (84,7%) ze 72 pacientů iniciálně randomizovaných na Cosentyx 150 mg. Ze 72 pacientů randomizovaných na Cosentyx 150 mg, 45, respektive 35, vykázalo ASAS 20/40 odpověď.
Mobilita páteře
Pacienti léčení přípravkem Cosentyx v dávce 150 mg vykázali zlepšenou mobilitu páteře hodnocenou jako změnu BASMI v týdnu 16 v porovnání s hodnotami před léčbou v AS studii 1 (-0,40 versus -0,12 pro placebo; p=0,0114) a v AS studii 2 (-0,51 versus -0,22 pro placebo; p=0,0533). Tato zlepšení přetrvávala do týdne 52.
Funkční stav a se zdravím související kvalita života
V AS studii 1 a studii 2, vykázali pacienti léčení přípravkem Cosentyx 150 mg zlepšení zdravotního stavu a kvality života, což bylo měřeno AS Quality of Life Dotazníkem (ASQoL) (p=0,001) a SF-36 Physical Component Summary (SF-36PCS) (p<0,001). Pacienti léčení přípravkem Cosentyx 150 mg také vykázali statisticky významné zlepšení ve fyzické funkci ve výzkumných cílech, což bylo zhodnoceno Bath Ankylosing Spondylitis Functional Indexem (BASFI) v porovnání s placebem (-2,15 versus -0,68), a v únavě, což bylo zhodnoceno Functional Assessment of Chronic Illness Therapy-Fatigue (FACIT-Fatigue) škálou v porovnání s placebem (8,10 versus 3,30). Tato zlepšení přetrvávala do týdne 52.
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Cosentyx u ložiskové psoriázy u pediatrických pacientů ve věku od narození do 6 let a u chronické idiopatické artritidy u dětských pacientů do 2 let (informace o použití u dětí viz bod 4.2).
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s přípravkem Cosentyx u ložiskové psoriázy u pediatrických pacientů ve věku od 6 let do 18 let a u chronické idiopatické artritidy u dětských pacientů od 2 do 18 let (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Ložisková psoriáza Absorpce
Po jednorázové podkožní dávce 300 mg ve formě roztoku u zdravých dobrovolníků dosáhl secukinumab nejvyšší sérové koncentrace 43,2±10,4 pg/ml mezi 2 a 14 dny po podání dávky.
Na základě výsledků populační farmakokinetické analýzy secukinumab po jednorázové subkutánní dávce buď 150 mg nebo 300 mg u pacientů s ložiskovou psoriázou dosáhl nejvyšší sérové koncentrace 13,7±4,8 pg/ml a 27,3±9,5 pg/ml mezi 5 a 6 dny od podání dávky.
Na základě výsledků populační farmakokinetické analýzy byl po počátečním týdenním dávkování během prvního měsíce čas potřebný k dosažení maximální koncentrace mezi 31 a 34 dny.
Podle simulovaných údajů byla nejvyšší koncentrace v rovnovážném stavu (Cmaxss) po subkutánním podání dávky 150 mg nebo 300 mg 27,6 pg/ml and 55,2 pg/ml. Populační farmakokinetická analýza naznačuje, že rovnovážného stavu je dosaženo po 20 týdnech při měsíčním dávkovacím režimu.
V porovnání s expozicí po jednorázovém podání ukázala populační farmakokinetická analýza, že pacienti vykazovali 2násobné zvýšení nejvyšší sérové koncentrace a plochy pod křivkou (AUC) po opakovaném měsíčním podávání během udržovacího režimu.
Populační farmakokinetická analýza ukázala že secukinumab byl u pacientů s ložiskovou psoriázou absorbován s průměrnou absolutní biologickou dostupností 73%. Napříč studiemi byla vypočítaná absolutní biologická dostupnost v rozmezí 60 až 77%.
Distribuce
Průměrný distribuční objem během terminální fáze (Vz) po jednorázovém intravenózním podání kolísal u pacientů s ložiskovou psoriázou v rozmezí od 7,10 do 8,60 litrů, což naznačuje, že secukinumab se omezeně distribuuje do periferních kompartmentů.
Biotransformace
Největší část IgG eliminace probíhá prostřednictvím intracelulárního katabolismu, následovaného fluidní fází nebo receptory zprostředkované endocytózy.
Eliminace
Průměrná systémová clearance (CL) po jednorázovém intravenózním podání kolísala u pacientů s ložiskovou psoriázou od 0,13 do 0,36 l/den. V populační farmakokinetické analýze byla průměrná systémová clearance (CL) u pacientů s ložiskovou psoriázou 0,19 l/den. CL nebyla ovlivněna pohlavím. Clearance byla závislá na dávce a čase.
Průměrný eliminační poločas zjištěný v populační farmakokinetické analýze byl u pacientů s ložiskovou psoriázou 27 dní, a kolísal od 18 do 46 dní napříč psoriatickými studiemi s intravenózním podáním.
Linearita/nelinearita
Farmakokinetika secukinumabu u pacientů s ložiskovou psoriázou po jednotlivé a opakovaných dávkách byla stanovena v četných studiích s intravenózně podanými dávkami v rozmezí od 1x 0,3 mg/kg do 3x 10 mg/kg a subkutánními dávkami v rozmezí od 1x 25 mg až opakovanými dávkami 300 mg. Expozice ve všech dávkovacích režimech odpovídala dávce.
Psoriatická artritida
Farmakokinetické vlastnosti secukinumabu pozorované u pacientů s psoriatickou artritidou byly podobné těm projevujícím se u pacientů s ložiskovou psoriázou. Biologická dostupnost secukinumabu u PsA pacientů činila 85% na základě populačního farmakokinetického modelu.
Ankylozující spondylitida
Farmakokinetické vlastnosti secukinumabu pozorované u pacientů s ankylozující spondylitidou byly podobné těm projevujícím se u pacientů s ložiskovou psoriázou.
Zvláštní populace
Starší pacienti
Z 3430 pacientů s ložiskovou psoriázou exponovaných přípravku Cosentyx v klinických studiích bylo 230 ve věku 65 let nebo starších a 32 pacientů bylo ve věku 75 nebo starších.
Z 974 pacientů s psoriatickou artritidou exponovaných přípravku Cosentyx v klinických studiích, celkem 85 pacientů bylo ve věku 65 let nebo starších a 4 pacienti byli ve věku 75 let nebo starší.
Z 571 pacientů s ankylozující spondylitidou exponovaných přípravku Cosentyx v klinických studiích, celkem 24 pacientů bylo ve věku 65 let nebo starších a 3 pacienti byli ve věku 75 let nebo starší.
Na základě populační farmakokinetické analýzy s omezeným počtem starších pacientů (n=71 pro věk >65 let a n=7 pro věk >75 let) byla clearance u starších pacientů a pacientů mladších 65 let věku podobná.
Pacienti s _poruchou _funkce ledvin nebo _jater
U pacientů s poruchou funkce ledvin nebo jater nejsou k dispozici farmakokinetická data. Předpokládá se, že renální eliminace nedotčeného přípravku Cosentyx, IgG monoklonální protilátky bude nízká a nevýznamná. IgG jsou většinou katabolizovány a neočekává se, že by porucha funkce jater ovlivnila clearance přípravku Cosentyx.
Vliv tělesné hmotnosti na farmakokinetiku
S rostoucí tělesnou hmotností rostou clearance secukinumabu a jeho distribuční objem.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě tkáňových testů zkřížené reaktivity, farmakologických studií bezpečnosti, toxicity po opakovaném podávání a reprodukční toxicity provedené u secukinumabu nebo myších protilátek proti myšímu IL-17A neodhalily žádné zvláštní riziko pro člověka.
Protože secukinumab váže opičí (cynomolgus) a lidský IL-17A, byla jeho bezpečnost studována u opic rodu cynomolgus. Po subkutánním podání secukinumabu opicím rodu cynomolgus po dobu až 13 týdnů a intravenózním podání až 26 týdnů (včetně vyhodnocení farmakokinetiky, farmakodynamiky, imunogenicity a imunotoxicity (tj. na T-buňkách závislá protilátková odpověď a aktivita NK buněk) nebyly pozorovány nežádoucí účinky. Průměrná sérová koncentrace pozorovaná u opic po 13 týdenních subkutánních dávkách 150 mg/kg byla značně vyšší než odhadovaná průměrná sérová koncentrace u psoriatických pacientů při nejvyšší klinické dávce. Protilátky proti secukinumabu byly detekovány pouze u jednoho z exponovaných zvířat. Po aplikaci secukinumabu do normální lidské tkáně nebyla pozorována nespecifická tkáňová zkřížená reaktivita.
Studie na zvířatech k posouzení karcinogenního potenciálu secukinumabu nebyly provedeny.
V embryofetální vývojové studii u opic rodu cynomolgus nevykazoval secukinumab toxicitu pro matku, embryotoxicitu nebo teratogenitu při podání během organogeneze a v pozdní gestaci.
Ve fertilitních a časných embryonálních a pre- a postnatálních studiích u myší nebyly pozorovány nežádoucí účinky myších protilátek proti myšímu IL-17A. Vyšší dávky v těchto studiích převyšovaly maximální účinnou dávku ve smyslu suprese IL-17A a účinku (viz bod 4.6).
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Dihydrát trehalosy Histidin
Monohydrát histidin-hydrochloridu
Methionin
Polysorbát 80
Voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti
18 měsíců
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce ( 2°C to 8°C). Chraňte před mrazem.
Uchovávejte pera v krabičce, aby byly chráněny před světlem.
6.5 Druh obalu a obsah balení
Přípravek Cosentyx je dodáván v jednorázové předplněné injekční stříkačce vložené do pera trojúhelníkového tvaru s průhledným okénkem a štítkem (pero SensoReady). Předplněná injekční stříkačka uvnitř pera je skleněná injekční stříkačka o objemu 1 ml s Fluortecem potaženým pístem, připojenou 27G x íV' jehlou a pevným krytem jehly ze styrenbutadienové gumy.
Přípravek Cosentyx je dostupný v jednotkovém balení obsahujícím 1 nebo 2 předplněná pera a vícečetném balení obsahujícím 6 (3x2) předplněných per.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Přípravek Cosentyx 150 mg injekční roztok je dodáván v předplněném peru pro individuální použití. S perem netřepejte a chraňte ho před mrazem. Pero je nutné vyjmout z chladničky 20 minut před aplikacíinjekce, aby dosáhlo pokojové teploty.
Před použitím předplněného pera se doporučuje provést vizuální kontrolu. Roztok musí být čirý. Jeho barva může být od bezbarvé po mírně nažloutlou. Můžete pozorovat malé vzduchové bublinky, což je normální. Nepoužívejte, pokud roztok obsahuje zřetelně viditelné částice, je zakalený nebo zřetelně hnědý. Podrobný návod k použití najdete v příbalové informaci.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/14/980/004
EU/1/14/980/005
EU/1/14/980/007
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
15.ledna 2015
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce biologické léčivé látky
Novartis Pharma S.A.S.
Centre de Biotechnologie 8, rue de l’Industrie F-68330 Huningue Francie
Název a adresa výrobce odpovědného za propouštění šarží
Novartis Pharma GmbH Roonstrasse 25 D-90429 Norimberk Německo
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
• Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předloží první pravidelně aktualizovanou zprávu o bezpečnosti pro tento léčivý přípravek do 6 měsíců od jeho registrace. Držitel rozhodnutí o registraci dále předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení PSUR a aktualizovaného RMP, je možné je předložit současně.
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU KRABIČKA - lahvička
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Cosentyx 150 mg prášek pro injekční roztok Secukinumabum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK_
Jedna lahvička obsahuje secukinumabum150 mg. Po rekonstituci 1 ml roztoku obsahuje secukinumabum 150 mg.
3. SEZNAM POMOCNÝCH LÁTEK_
Též obsahuje: Sacharosa, Histidin, monohydrát histidin-hydrochloridu, polysorbát 80.
4. LÉKOVÁ FORMA A OBSAH BALENÍ_
Prášek pro injekční roztok 1 lahvička
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Subkutánní podání
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/14/980/001
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Cosentyx 150 mg
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITŘNÍM OBALU ŠTÍTEK LAHVIČKY
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Cosentyx 150 mg prášek pro injekční roztok
Secukinumabum
s.c.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
KRABIČKA JEDNOTKOVÉHO BALENÍ - předplněná injekční stříkačka
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Cosentyx 150 mg injekční roztok v předplněné injekční stříkačce Secukinumabum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna předplněná injekční stříkačka obsahuje secukinumabum 150 mg v 1 ml roztoku.
3. SEZNAM POMOCNÝCH LÁTEK
Též obsahuje: Dihydrát trehalosy, histidin, monohydrát histidin-hydrochloridu, methionin, polysorbát 80, voda na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Injekční roztok
1 předplněná injekční stříkačka
2 předplněné injekční stříkačky
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Subkutánní podání
Před použitím si přečtěte příbalovou informaci. K jednorázovému použití.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Uchovávejte v chladničce. Chraňte před mrazem.
Uchovávejte předplněnou injekční stříkačku v krabičce, aby byla chráněna před světlem. Uchovávejte předplněnou injekční stříkačku v krabičce, aby byla chráněna před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/14/980/002 Balení obsahující 1 předplněnou injekční stříkačku
EU/1/14/980/003 Balení obsahující 2 předplněné injekční stříkačky
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Cosentyx 150 mg
VNĚJŠÍ KRABIČKA VÍCEČETNÉHO BALENÍ (VČETNĚ BLUE BOX) - předplněná injekční stříkačka
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Cosentyx 150 mg injekční roztok v předplněné injekční stříkačce Secukinumabum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna předplněná injekční stříkačka obsahuje secukinumabum 150 mg v 1 ml roztoku.
3. SEZNAM POMOCNÝCH LÁTEK
Též obsahuje: Dihydrát trehalosy, L-histidin, monohydrát histidin-hydrochloridu, L-methionin, polysorbát 80, voda na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Injekční roztok
Vícečetné balení: 6 (3x2) předplněných injekčních stříkaček
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Subkutánní podání
Před použitím si přečtěte příbalovou informaci. K jednorázovému použití.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Uchovávejte v chladničce. Chraňte před mrazem.
Uchovávejte předplněnou injekční stříkačku v krabičce, aby byla chráněna před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/14/980/006 Vícečetné balení obsahující 6 (3x2) předplněných injekčních
stříkaček
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Cosentyx 150 mg
VNITŘNÍ KRABIČKA VÍCEČETNÉHO BALENÍ (BEZ BLUE BOXU) - předplněná injekční stříkačka
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Cosentyx 150 mg injekční roztok v předplněné injekční stříkačce Secukinumabum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna předplněná injekční stříkačka obsahuje secukinumabum 150 mg v 1 ml roztoku.
3. SEZNAM POMOCNÝCH LÁTEK
Též obsahuje: Dihydrát trehalosy, histidin, monohydrát histidin-hydrochloridu,methionin, polysorbát 80, voda na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Injekční roztok
2 předplněné injekční stříkačky. Součást vícečetného balení. Nesmí být prodáváno samostatně.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Subkutánní podání
Před použitím si přečtěte příbalovou informaci. K jednorázovému použití.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Uchovávejte v chladničce. Chraňte před mrazem.
Uchovávejte předplněnou injekční stříkačku v krabičce, aby byla chráněna před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/14/980/006 Vícečetné balení obsahující 6 (3x2) předplněných injekčních
stříkaček
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Cosentyx 150 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Cosentyx 150 mg injekční roztok v předplněné injekční stříkačce Secukinumabum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Cosentyx 150 mg injekce
Secukinumabum
s.c.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
KRABIČKA JEDNOTKOVÉHO BALENÍ - předplněné pero
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Cosentyx 150 mg injekční roztok v předplněném peru Secukinumabum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedno předplněné pero obsahuje secukinumabum 150 mg v 1 ml roztoku.
3. SEZNAM POMOCNÝCH LÁTEK
Též obsahuje: Dihydrát trehalosy, histidin, monohydrát histidin-hydrochloridu, methionin, polysorbát 80, voda na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Injekční roztok
1 předplněné pero SensoReady
2 předplněná pera SensoReady
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Subkutánní podání
Před použitím si přečtěte příbalovou informaci. K jednorázovému použití.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Uchovávejte v chladničce. Chraňte před mrazem.
Uchovávejte předplněné pero SensoReady v krabičce, aby bylo chráněno před světlem. Uchovávejte předplněné pero SensoReady v krabičce, aby bylo chráněno před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/14/980/004 Balení obsahující 1 předplněné pero SensoReady
EU/1/14/980/005 Balení obsahující 2 předplněná pera SensoReady
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Cosentyx 150 mg
VNĚJŠÍ KRABIČKA VÍCEČETNÉHO BALENÍ (VČETNĚ BLUE BOX) - předplněné pero
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Cosentyx 150 mg injekční roztok v předplněném peru Secukinumabum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedno předplněné pero obsahuje secukinumabum 150 mg v 1 ml roztoku.
3. SEZNAM POMOCNÝCH LÁTEK
Též obsahuje: Dihydrát trehalosy, histidin, monohydrát histidin-hydrochloridu, methionin, polysorbát 80, voda na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Injekční roztok
Vícečetné balení: 6 (3x2) předplněných per SensoReady
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Subkutánní podání
Před použitím si přečtěte příbalovou informaci. K jednorázovému použití.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Uchovávejte v chladničce. Chraňte před mrazem.
Uchovávejte předplněné pero SensoReady v krabičce, aby bylo chráněno před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/14/980/007
Vícečetné balení obsahující 6 (3x2) předplněných per SensoReady
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Cosentyx 150 mg
VNITŘNÍ KRABIČKA VÍCEČETNÉHO BALENÍ (BEZ BLUE BOX) - předplněné pero
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Cosentyx 150 mg injekční roztok v předplněném peru Secukinumabum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedno předplněné pero obsahuje secukinumabum 150 mg v 1 ml roztoku.
3. SEZNAM POMOCNÝCH LÁTEK
Též obsahuje: Dihydrát trehalosy, histidin, monohydrát histidin-hydrochloridu, methionin, polysorbát 80, voda na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Injekční roztok
2 předplněná pera SensoReady. Součást vícečetného balení. Nesmí být prodáváno samostatně.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Subkutánní podání
Před použitím si přečtěte příbalovou informaci. K jednorázovému použití.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
Uchovávejte v chladničce. Chraňte před mrazem.
Uchovávejte předplněné pero SensoReady v krabičce, aby bylo chráněno před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/14/980/007
Vícečetné balení obsahující 6 (3x2) předplněných per SensoReady
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Cosentyx 150 mg
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITŘNÍM OBALU ŠTÍTEK PERA
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Cosentyx 150 mg injekční roztok v předplněném peru
Secukinumabum
s.c.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
Pero SensoReady
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro pacienta Cosentyx 150 mg prášek pro injekční roztok
Secukinumabum
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Cosentyx a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Cosentyx používat
3. Jak se přípravek Cosentyx používá
4. Možné nežádoucí účinky
5. Jak přípravek Cosentyx uchovávat
6. Obsah balení a další informace
1. Co je přípravek Cosentyx a k čemu se používá
Přípravek Cosentyx obsahuje léčivou látku secukinumab. Secukinumab je monoklonální protilátka. Monoklonální protilátky jsou bílkoviny, které rozpoznávají a specificky se vážou na některé bílkoviny v těle.
Přípravek Cosentyx patří do skupiny léků zvaných inhibitory interleukinu (IL). Tyto léky neutralizují účinky bílkoviny nazývané IL-17A, která je přítomna ve zvýšeném množství u chorob jako je psoriáza, psoriatická artritida a ankylozující spondylitida.
Přípravek Cosentyx je určen k léčbě následujích zánětlivých chorob:
• Ložisková psoriáza
• Psoriatická artritida
• Ankylozující spondylitida
Ložisková psoriáza
Přípravek Cosentyx se používá k léčbě kožních projevů nazývaných “ložisková psoriáza”, která vyvolává zánět ovlivňující pokožku. Přípravek Cosentyx redukuje zánět a další příznaky choroby. Přípravek Cosentyx se používá u dospělých se středně závažnou až závažnou ložiskovou psoriázou.
Používání přípravku Cosentyx u ložskové psoriázy Vám pomůže tím, že vede ke zlepšení postižení pokožky a redukci příznaků jako odlupování, svědění a bolest.
Psoriatická artritida
Přípravek Cosentyx se používá k léčbě stavu nazývaného“psoriatická artritida”. Tento stav představuje zánětlivou chorobu kloubů, často doprovázenou psoriázou. Pokud trpíte aktivní psoriatickou artirtidou, budete nejdříve léčen(a) jinými léčivými přípravky. Pokud dostatečně neodpovídáte na tyto přípravky, bude Vám předepsán přípravek Cosentyx pro omezení známek a příznaků aktivní psoriatické artiritdy,
zlepšení funkčního stavu a zpomalení poškození chrupavky a kosti kloubů postižených touto chorobou.
Přípravek Cosentyx užívají dospělí s aktivní psoriatickou artritidou a smí se užívat samostatně nebo v kombinaci s jiným přípravkem nazývaným metotrexát.
Užívání přípravku Cosentyx u psoriatické artritidy je pro Vás prospěšné omezením známek a příznaků choroby, zpomalením poškození chrupavky a kosti kloubů a zlepšením schopnosti vykonávat běžné denní aktivity.
Ankylozující spondylitida
Přípravek Cosentyx se používá k léčbě stavu nazývaného“ankylozující spondylitida”. Tento stav představuje zánětlivou chorobu postihující primárně páteř, kdy dochází k zánětu páteřních kloubů. Pokud trpíte ankylozující spondylitidou, budete nejdříve léčen(a) jinými léčivými přípravky. Pokud dostatečně neodpovídáte na tyto přípravky, bude Vám předepsán přípravek Cosentyx pro omezení známek a příznaků této choroby, omezení zánětu a zlepšení fyzické funkce.
Přípravek Cosentyx užívají dospělí s aktivní ankylozující spondylitidou.
Užívání přípravku Cosentyx u ankylozující spondylitidy je pro Vás prospěšné omezením známek a příznaků choroby a a zlepšením fyzické funkce.
2. Čemu musíte věnovat pozornost, než začnete přípravek Cosentyx používat Nepoužívejte přípravek Cosentyx:
• jestliže jste alergický(á) na secukinumab nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Pokud se domníváte, že můžete být alergický(á), poraďte se před použitím přípravku Cosentyx s lékařem.
• Jestliže trpíte aktivní infekcí, kterou Váš lékař považuje za důležitou.
Upozornění a opatření
Poraďte se s lékařem nebo lékárníkem předtím, než začnete přípravek Cosentyx používat:
• pokud trpíte v současnosti infekcí.
• pokud trpíte dlouhodobou nebo opakovaně se vyskytující infekci.
• pokud máte tuberkulózu.
• pokud máte Crohnovu chorobu.
• Pokud jste byl(a) v nedávné době očkován(a) nebo se máte podrobit očkování během léčby přípravkem Cosentyx.
• pokud se podrobujete jiné léčbě psoriázy, jako jsou imunosupresiva nebo fototerapie ultrafialovým světlem (UV).
Sledování infekcí a alergických reakcí
Přípravek Cosentyx může případně vyvolat závažné nežádoucí účinky, včetně infekcí a alergických reakcí. Pokud používáte přípravek Cosentyx, musíte sledovat příznaky těchto stavů.
Přestaňte používat přípravek Cosentyx a informujte neprodleně svého lékaře nebo vyhledejte lékařskou pomoc, pokud zaznamenáte jakékoliv známky naznačující možné závažné infekci nebo alergické reakci. Tyto známky jsou uvedeny v oddíle “Závažné nežádoucí účinky” v bodě 4.
Děti a dospívající
Přípravek Cosentyx se nedoporučuje u dětí a dospívajících (do 18 let věku), protože v této věkové skupině nebyl studován.
Další léčivé přípravky a přípravek Cosentyx
Informujte svého lékaře nebo lékárníka:
• o všech lécích, které používáte, které jste v nedávné době používal(a) nebo které možná budete používat.
• že jste byl(a) v nedávné době očkován(a) nebo se máte podrobit očkování. Při používání přípravku Cosentyx Vám nesmí být podán určitý typ vakcín (živé vakcíny).
Těhotenství, kojení a plodnost
• Je doporučeno vyvarovat se užívání přípravku Cosentyx během těhotenství. Účinky tohoto přípravku na těhotné ženy nejsou známy. Pokud jste žena ve fertilním věku, doporučuje se zabránit otěhotnění a během léčby přípravkem Cosentyx a po dobu nejméně 20 týdnů od poslední dávky přípravku Cosentyx musíte užívat vhodnou antikoncepci. Poraďte se s lékařem, pokud jste těhotná, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět.
• Poraďte se s lékařem, pokud kojíte nebo plánujete kojení. Společně s lékařem rozhodnete, zda budete kojit nebo užívat přípravek Cosentyx. Nemůžete dělat obojí. Po užití přípravku Cosentyx nesmíte kojit po dobu nejméně 20 týdnů od poslední dávky.
Řízení dopravních prostředků a obsluha strojů
Přípravek Cosentyx nemá žádný nebo má zanedbatelný vliv na schopnost řídit a obsluhovat stroje.
3. Jak se přípravek Cosentyx používá
Přípravek Cosentyx podává injekcí pod kůži (což je známo jako podkožní injekce) zdravotnický pracovník.
Poraďte se s lékařem o době podání injekce a o následných návštěvách.
Podrobný návod, jak rekonstituovat a podat přípravek Cosentyx, naleznete v “Návodu k použití přípravku Cosentyx prášek pro injekční roztok” na konci této příbalové informace.
Jaké množství přípravku Cosentyx se podává a jak dlouho
Váš lékař rozhodne, jaku dávku přípravku Cosentyx potřebujete a jak dlouho.
Ložisková psoriáza
• Doporučená dávka přípravku je 300 mg ve formě podkožní injekce.
• Každá dávka 300 mg je podána ve dvou injekcích 150 mg.
Po podání první dávky budete dostávat další týdenní injekce v týdnu 1, 2 a 3. Od týdne 4 budete dostávat injekci měsíčně. Pokaždé dostanete dávku 300 mg rozdělenou do dvou injekcí po 150 mg.
Psoriatická artritida
U pacientů s psoriatickou artritidou, kteří současně trpí středně těžkou až těžkou ložiskovou psoriázou nebo u pacientů, kteří neodpovídají dostatečně na léčbu přípravky „tumour necrosis factor“ (TNF) blokátory:
• Doporučená dávka je 300 mg ve formě podkožní injekce.
• Každá dávka 300 mg je podána ve dvou injekcích 150 mg.
Po první dávce budou následovat další týdenní injekce v týdnech 1, 2 a 3. Od týdne 4 dostanete jednu injekci měsíčně. Vždy dostanete dávku 300 mg podanou ve dvou injekcích po 150 mg.
U ostatních pacientů s psoriatickou artritidou:
• Doporučená dávka je 150 mg ve formě podkožní injekce.
Po první dávce budou následovat další týdenní injekce v týdnech 1, 2 a 3. Od týdne 4 dostanete jednu injekci měsíčně.
Ankylozující spondylitida
• Doporučená dávka je 150 mg ve formě podkožní injekce.
Po první dávce budou následovat další týdenní injekce v týdnech 1, 2 a 3. Od týdne 4 dostanete jednu injekci měsíčně.
Přípravek Cosentyx je určen k dlouhodobému podávání. Lékař bude pravidelně monitorovat Váš stav, aby zjistil, zda má léčba požadovaný účinek.
Jestliže jste použil(a) více přípravku Cosentyx než jste měl(a)
Pokud jste použil(a) více přípravku Cosentyx než jste měl(a) nebo pokud byla dávka podána dříve než lékař předepsal, poraďte se s lékařem.
Jestliže jste zapomněl(a) použít přípravek Cosentyx
Pokud jste zapomněl(a) injekci přípravku Cosentyx, poraďte se s lékařem.
Jestliže jste přestal(a) používat přípravek Cosentyx
Přestat používat přípravek Cosentyx není nebezpečné. Nicméně, pokud přestanete, příznaky psoriázy, psoriatické artritidy nebo ankylozující spondylitidy se mohou vrátit.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
T-W r V r Vil i i V* 1
Závažné nežádoucí účinky
Přestaňte používat přípravek Cosentyx a informujte neprodleně svého lékaře nebo vyhledejte lékařskou pomoc, pokud zaznamenáte jakékoliv následující nežádoucí účinky.
Možné závažné infekce - známky mohou zahrnovat:
• horečka, příznaky podobné chřipce, noční pocení
• pocit únavy nebo dýchavičnosti, přetrvávající kašel
• teplá, rudá a bolestivá pokožka, nebo bolestivá vyrážka s puchýři
• pocit pálení při močení.
Závažná alergická reakce - známky mohou zahrnovat:
• obtížné dýchání nebo polykání
• nízký krevní tlak, který může působit závratě nebo omámení
• otok tváře, rtů, jazyka nebo hrdla
• silné svědění pokožky s červenou vyrážkou nebo vyvýšenými hrbolky .
Váš lékař rozhodne, zda a kdy můžete pokračovat v léčbě.
Jiné nežádoucí účinky
Většina následujících nežádoucích účinků je mírná až středně závažná. Pokud se kterýkoli z těchto nežádoucích účinků stane závažným, sdělte to lékaři, lékárníkovi nebo zdravotní sestře.
Některé nežádoucí účinky jsou velmi časté (mohou postihnout více než 1 z 10 osob):
• infekce horních cest dýchacích s příznaky jako bolest v krku a ucpaný nos (nasofaryingitida, rinitida)
Některé nežádoucí účinky jsou časté (mohou postihnout až 1 z 10 osob):
• opar (na rtech)
• průjem
• výtok z nosu (rýma)
Některé nežádoucí účinky jsou méně časté (mohou postihnout až 1 ze 100 osob):
• ústní povlak (ústní kandidóza)
• známky nízké hladiny bílých krvinek, jako jsou horečka, bolest v krku nebo vředy v ústech kvůli infekci (neutropenie)
• atletická noha (tinea pedis)
• infekce vnějšího ucha (otitis media)
• výtok z očí se svěděním, zarudnutím a otokem (konjunktivitida).
• svědivá vyrážka (kopřivka)
Některé nežádoucí účinky jsou vzácné (mohou postihnout až 1 z 1 000 osob)
• závažná alergická reakce s šokem (anafylaktická reakce)
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Cosentyx uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce nebo lahvičce za „EXP“.
Před rekonstitucí: Lahvičky přípravku uchovávejte v chladničce při teplotě od 2°C do 8°C.
Po rekonstituci: Roztok musí být použit okamžitě nebo může být uchováván při teplotě od 2°C do 8°C po dobu až 24 hodin. Chraňte před mrazem. Roztok musí být podán během jedné hodiny od vyjmutí z prostředí s teplotou od 2°C do 8°C.
Tento přípravek je určen k jednorázovému použití. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte.
6. Obsah balení a další informace
Co přípravek Cosentyx obsahuje
- Léčivou látkou je secukinumabum. Jedna lahvička prášku pro injekční roztok obsahuje 150 mg secukinumabum. Po rekonstituci 1 ml roztoku obsahuje 150 mg secukinumabum.
- Dalšími složkami jsou sacharosa, histidin, monohydrát histidin-hydrochloridu a polysorbát 80.
Jak přípravek Cosentyx vypadá a co obsahuje toto balení
Přípravek Cosentyx prášek pro injekční roztok je pevný bílý prášek ve skleněné lahvičce. Nepoužívejte, pokud není lyofilizovaný prášek zcela rozpuštěn nebo pokud tekutina obsahuje dobře viditelné částice, je zakalená nebo zřetelně hnědá. Přípravek Cosentyx je dodáván v balení obsahujícím jednu lahvičku.
Držitel rozhodnutí o registraci
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
Výrobce
Novartis Pharma GmbH Roonstrasse 25 D-90429 Norimberk Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgie/Belgique/Belgien Lietuva
Novartis Pharma N.V. Novartis Pharma Services Inc.
Tél/Tel: +32 2 246 16 11 Tel: +370 5 269 16 50
Bt^rapuu
Novartis Pharma Services Inc. Tem +359 2 489 98 28
Česká republika
Novartis s.r.o.
Tel: +420 225 775 111
Danmark
Novartis Healthcare A/S Tlf: +45 39 16 84 00
Deutschland
Novartis Pharma GmbH Tel: +49 911 273 0
Eesti
Novartis Pharma Services Inc. Tel: +372 66 30 810
EkXúba
Novartis (Hellas) A.E.B.E. Tqk +30 210 281 17 12
Luxembourg/Luxemburg
Novartis Pharma N.V.
Tél/Tel: +32 2 246 16 11
Magyarország
Novartis Hungária Kft. Pharma Tel.: +36 1 457 65 00
Malta
Novartis Pharma Services Inc. Tel: +356 2122 2872
Nederland
Novartis Pharma B.V.
Tel: +31 26 37 82 111
Norge
Novartis Norge AS Tlf: +47 23 05 20 00
Osterreich
Novartis Pharma GmbH Tel: +43 1 86 6570
|
Espaňa Novartis Farmacéutica, S.A. Tel: +34 93 306 42 00 |
Polska Novartis Poland Sp. z o.o. Tel.: +48 22 375 4888 |
|
France Novartis Pharma S.A.S. Tél: +33 1 55 47 66 00 |
Portugal Novartis Farma - Produtos Farmaceuticos, S.A Tel: +351 21 000 8600 |
|
Hrvatska Novartis Hrvatska d.o.o. Tel. +385 1 6274 220 |
Románia Novartis Pharma Services Romania SRL Tel: +40 21 31299 01 |
|
Ireland Novartis Ireland Limited Tel: +353 1 260 12 55 |
Slovenija Novartis Pharma Services Inc. Tel: +386 1 300 75 50 |
|
Island Vistor hf. Sími: +354 535 7000 |
Slovenská republika Novartis Slovakia s.r.o. Tel: +421 2 5542 5439 |
|
Italia Novartis Farma S.p.A. Tel: +39 02 96 54 1 |
Suomi/Finland Novartis Finland Oy Puh/Tel: +358 (0)10 6133 200 |
|
Knnpoq Novartis Pharma Services Inc. TpA.: +357 22 690 690 |
Sverige Novartis Sverige AB Tel: +46 8 732 32 00 |
|
Latvija Novartis Pharma Services Inc. Tel: +371 67 887 070 |
United Kingdom Novartis Pharmaceuticals UK Ltd. Tel: +44 1276 698370 |
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Návod k použití přípravku Cosentyx prášek pro injekční roztok Následující informace je určena pouze pro zdravotnické pracovníky.
Injekční lahvičku uchovávejte v chladničce při teplotě 2°C až 8°C.
Lahvička k jednorázovému použití obsahuje 150 mg secukinumabu pro rekonstituci vodou na injekci. Nepoužívejte lékovku po době použitelnosti uvedené na krabičce nebo lékovce. Pokud uplynula doba použitelnosti, vraťte celé balení do lékárny.
Příprava roztoku pro subkutánní injekci musí proběhnout bez přerušení a musí být zajištěno použití aseptické techniky. Doba přípravy od propíchnutí zátky do konce rekonstituce trvá v průměru 20 minut a neměla by přesáhnout 90 minut.
Při přípravě přípravku Cosentyx 150 mg roztok pro injekci dodržujte prosím následující instrukce:
Instrukce pro rekonstituci přípravku Cosentyx 150 mg prášek pro injekční roztok:
1. Umístěte lékovku s práškem do prostředí s pokojovou teplotou a zajistěte, že voda na injekci je rovněž v prostředí s pokojovou teplotou.
2. Natáhněte do kalibrované 1 ml jednorázové injekční stříkačky o něco více než 1,0 ml sterilní vody na injekci a upravte objem na 1,0 ml.
3. Sejměte z lahvičky kryt z plastické hmoty.
4. Zasuňte jehlu do lékovky obsahující prášek středem gumové zátky a rekonstituujte prášek pomalou injekcí sterilní vody na injekci do lékovky. Proud sterilní vody na injekci musí
směřovat na prášek.

5. Nakloňte lékovku pod úhlem přibližně 45° a jemně jí rotujte mezi konečky prstů po dobu přibližně 1 minuty. Lékovkou netřeste ani ji neotáčejte.

6. Ponechte lékovku stát při pokojové teplotě nejméně 10 minut, aby došlo k rozpuštění. Roztok přitom může pěnit.
7. Nakloňte lékovku pod úhlem přibližně 45° a jemně jí rotujte mezi konečky prstů po dobu přibližně 1 minuty. Lékovkou netřeste ani ji neotáčejte.

8. Ponechte lékovku v klidu stát při pokojové teplotě nejméně 5 minut. Vzniklý roztok musí být čirý. Jeho barva může být od bezbarvé do mírně nažloutlé. Nepoužívejte, pokud se lyofilizovaný prášek zcela nerozpustí nebo pokud roztok obsahuje zřetelně viditelné částice, je zakalený nebo zřetelně hnědý.
9. Připravte požadované množství lékovek (2 lékovky pro dávku 300 mg).
Po přípravě může být roztok pro subkutánní injekci použit okamžitě nebo může být uchováván při 2°C až 8°C pod dobu až 24 hodin. Chraňte před mrazem. Po uchovávání při teplotě 2°C až 8°C musí být roztok ponechán při pokojové teplotě přibližně 20 minut před podáním. Roztok musí být podán během jedné hodiny od vyjmutí z prostředí s teplotou od 2°C do 8°C.
Instrukce pro podání roztoku přípravku Cosentyx
1. Nakloňte lékovku pod úhlem přibližně 45° a při natahování roztoku do injekční stříkačky umístěte hrot jehly na úplné dno lékovky s roztokem. Lékovku NEOBRACEJTE.

2. Opatrně natáhněte o něco více než 1,0 ml roztoku pro subkutánní injekci z lékovky do 1 ml kalibrované jednorázové injekční stříkačky s použitím vhodné jehly (t. j. 21G x 2"). Tuto jehlu použijte pouze pro natažení přípravku Cosentyx do jednorázové injekční stříkačky. Připravte požadovaní množství injekčních stříkaček (2 injekční stříkačky pro dávku 300 mg).
3. S jehlou směřující vzhůru, jemně poklepejte na stříkačku, aby se vzduchové bubliny dostaly nahoru.

5. Vytlačte vzduchové bubliny a nastavte píst na značku 1,0 ml.
6. Očistěte místo vpichu alkoholovým tampónem.
7. Injikujte podkožně roztok přípravku Cosentyx do přední strany stehen, podbřišku (ne však do oblasti 5 centimetrů okolo pupku) nebo zboku do horní části paže. Při každém podání injekce zvolte odlišné místo vpichu. Neinjikujte do oblastí, kde je pokožka jemná, s podlitinami, zarudlá, odlupuje se neboje tvrdá. Vyvarujte se oblastí s jizvami nebo vytahanou pokožkou.

4.
Vyměňte připojenou jehlu za jehlu 27G x Vi".

8. Zbylý roztok v lékovce nesmí být již použit a musí být znehodnocen v souladu s místními
požadavky. Lékovka je určena pouze k jednorázovému použití. Použité jehly odložte do nádoby na ostré předměty (uzavíratelná schránka, odolná proti propíchnutí). Kvůli bezpečnosti Vás i ostatních nesmí být jehly a injekční stříkačky nikdy použity opakovaně.
Příbalová informace: informace pro pacienta Cosentyx 150 mg injekční roztok v předplněné injekční stříkačce
Secukinumabum
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Cosentyx a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Cosentyx používat
3. Jak se přípravek Cosentyx používá
4. Možné nežádoucí účinky
5. Jak přípravek Cosentyx uchovávat
6. Obsah balení a další informace
1. Co je přípravek Cosentyx a k čemu se používá
Přípravek Cosentyx obsahuje léčivou látku secukinumab. Secukinumab je monoklonální protilátka. Monoklonální protilátky jsou bílkoviny, které rozpoznávají a specificky se vážou na některé bílkoviny v těle.
Přípravek Cosentyx patří do skupiny léků zvaných inhibitory interleukinu (IL). Tyto léky neutralizují účinky bílkoviny nazývané IL-17A, která je přítomna ve zvýšeném množství u chorob jako je psoriáza, psoriatická artritida a ankylozující spondylitida.
Přípravek Cosentyx je určen k léčbě následujích zánětlivých chorob:
• Ložisková psoriáza
• Psoriatická artritida
• Ankylozující spondylitida
Ložisková psoriáza
Přípravek Cosentyx se používá k léčbě kožních projevů nazývaných “ložisková psoriáza”, která vyvolává zánět ovlivňující pokožku. Přípravek Cosentyx redukuje zánět a další příznaky choroby. Přípravek Cosentyx se používá u dospělých se středně závažnou až závažnou ložiskovou psoriázou.
Používání přípravku Cosentyx u ložskové psoriázy Vám pomůže tím, že vede ke zlepšení postižení pokožky a redukci příznaků jako odlupování, svědění a bolest.
Psoriatická artritida
Přípravek Cosentyx se používá k léčbě stavu nazývaného “psoriatická artritida”. Tento stav představuje zánětlivou chorobu kloubů, často doprovázenou psoriázou. Pokud trpíte aktivní psoriatickou artirtidou, budete nejdříve léčen(a) jinými léčivými přípravky. Pokud dostatečně neodpovídáte na tyto přípravky, bude Vám předepsán přípravek Cosentyx pro omezení známek a příznaků aktivní psoriatické artiritdy,
zlepšení funkčního stavu a zpomalení poškození chrupavky a kosti kloubů postižených touto chorobou.
Přípravek Cosentyx užívají dospělí s aktivní psoriatickou artritidou a smí se užívat samostatně nebo v kombinaci s jiným přípravkem nazývaným metotrexát.
Užívání přípravku Cosentyx u psoriatické artritidy je pro Vás prospěšné omezením známek a příznaků choroby, zpomalením poškození chrupavky a kosti kloubů a zlepšením schopnosti vykonávat běžné denní aktivity.
Ankylozující spondylitida
Přípravek Cosentyx se používá k léčbě stavu nazývaného“ankylozující spondylitida”. Tento stav představuje zánětlivou chorobu postihující primárně páteř, kdy dochází k zánětu páteřních kloubů. Pokud trpíte ankylozující spondylitidou, budete nejdříve léčen(a) jinými léčivými přípravky. Pokud dostatečně neodpovídáte na tyto přípravky, bude Vám předepsán přípravek Cosentyx pro omezení známek a příznaků této choroby, omezení zánětu a zlepšení fyzické funkce.
Přípravek Cosentyx užívají dospělí s aktivní ankylozující spondylitidou.
Užívání přípravku Cosentyx u ankylozující spondylitidy je pro Vás prospěšné omezením známek a příznaků choroby a a zlepšením fyzické funkce.
2. Čemu musíte věnovat pozornost, než začnete přípravek Cosentyx používat Nepoužívejte přípravek Cosentyx:
• jestliže jste alergický(á) na secukinumab nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Pokud se domníváte, že můžete být alergický(á), poraďte se před použitím přípravku Cosentyx s lékařem.
• Jestliže trpíte aktivní infekcí, kterou Váš lékař považuje za důležitou.
Upozornění a opatření
Poraďte se s lékařem nebo lékárníkem předtím, než začnete přípravek Cosentyx používat:
• pokud trpíte v současnosti infekcí
• pokud trpíte dlouhodobou nebo opakovaně se vyskytující infekcí.
• pokud máte tuberkulózu.
• Pokud jste někdy měl(a) alergickou reakci na latex.
• pokud máte Crohnovu chorobu.
• Pokud jste byl(a) v nedávné době očkován(a) nebo se máte podrobit očkování během léčby přípravkem Cosentyx.
• pokud se podrobujete jiné léčbě psoriázy, jako jsou imunosupresiva nebo fototerapie ultrafialovým světlem (UV).
Sledování infekcí a alergických reakcí
Přípravek Cosentyx může případně vyvolat závažné nežádoucí účinky, včetně infekcí a alergických reakcí. Pokud používáte přípravek Cosentyx, musíte sledovat příznaky těchto stavů.
Přestaňte používat přípravek Cosentyx a informujte neprodleně svého lékaře nebo vyhledejte lékařskou pomoc, pokud zaznamenáte jakékoliv známky naznačující možné závažné infekci nebo alergické reakci. Tyto známky jsou uvedeny v oddíle “Závažné nežádoucí účinky” v bodě 4.
Děti a dospívající
Přípravek Cosentyx se nedoporučuje u dětí a dospívajících (do 18 let věku), protože v této věkové skupině nebyl studován.
Další léčivé přípravky a přípravek Cosentyx
Informujte svého lékaře nebo lékárníka:
• o všech lécích, které používáte, které jste v nedávné době používal(a) nebo které možná budete používat.
• že jste byl(a) v nedávné době očkován(a) nebo se máte podrobit očkování. Při používání přípravku Cosentyx Vám nesmí být podán určitý typ vakcín (živé vakcíny).
Těhotenství, kojení a plodnost
• Je doporučeno vyvarovat se užívání přípravku Cosentyx během těhotenství. Účinky tohoto přípravku na těhotné ženy nejsou známy. Pokud jste žena ve fertilním věku, doporučuje se zabránit otěhotnění a během léčby přípravkem Cosentyx a po dobu nejméně 20 týdnů od poslední dávky přípravku Cosentyx musíte užívat vhodnou antikoncepci. Poraďte se s lékařem, pokud jste těhotná, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět.
• Poraďte se s lékařem, pokud kojíte nebo plánujete kojení. Společně s lékařem rozhodnete, zda budete kojit nebo užívat přípravek Cosentyx. Nemůžete dělat obojí. Po užití přípravku Cosentyx nesmíte kojit po dobu nejméně 20 týdnů od poslední dávky.
Řízení dopravních prostředků a obsluha strojů
Přípravek Cosentyx nemá žádný nebo má zanedbatelný vliv na schopnost řídit a obsluhovat stroje.
3. Jak se přípravek Cosentyx používá
Vždy používejte tento přípravek přesně podle pokynů Vašeho lékaře. Pokud si nejste jistý(á), poraďte se svým lékařem, lékárníkem nebo zdravotní sestrou.
Přípravek Cosentyx se podává injekcí pod kůži (což je známo jako podkožní injekce). Společně s Vaším lékařem se musíte rozhodnout, zda si budete přípravek Cosentyx inikovat sám (sama).
Je důležité, nesnažit se o injekci sám (sama), dokud nebudete proškolen(a) Vaším lékařem, zdravotní sestrou nebo lékárníkem. Po vhodném proškolení Vám může přípravek Cosentyx injekce podat i Váš opatrovník.
Podrobný návod, jak injikovat přípravek Cosentyx, naleznete v “Návodu k použití přípravku Cosentyx předplněná injekční stříkačka” na konci této příbalové informace.
Jaké množství přípravku Cosentyx se podává a jak dlouho
Váš lékař rozhodne, jaku dávku přípravku Cosentyx potřebujete a jak dlouho.
Ložisková psoriáza
• Doporučená dávka přípravku je 300 mg ve formě podkožní injekce.
• Každá dávka 300 mg je podána ve dvou injekcích 150 mg.
Po podání první dávky budete dostávat další týdenní injekce v týdnu 1, 2 a 3. Od týdne 4 budete dostávat injekci měsíčně. Pokaždé dostanete dávku 300 mg rozdělenou do dvou injekcí po 150 mg.
Psoriatická artritida
U pacientů s psoriatickou artritidou, kteří současně trpí středně těžkou až těžkou ložiskovou psoriázou nebo u pacientů, kteří neodpovídají dostatečně na léčbu přípravky „tumour necrosis factor“ (TNF) blokátory:
• Doporučená dávka je 300 mg ve formě podkožní injekce.
• Každá dávka 300 mg je podána ve dvou injekcích 150 mg.
Po první dávce budou následovat další týdenní injekce v týdnech 1, 2 a 3. Od týdne 4 dostanete jednu injekci měsíčně. Vždy dostanete dávku 300 mg podanou ve dvou injekcích po 150 mg.
U ostatních pacientů s psoriatickou artritidou:
• Doporučená dávka je 150 mg ve formě podkožní injekce.
Po první dávce budou následovat další týdenní injekce v týdnech 1, 2 a 3. Od týdne 4 dostanete jednu injekci měsíčně.
Ankylozující spondylitida
• Doporučená dávka je 150 mg ve formě podkožní injekce.
Po první dávce budou následovat další týdenní injekce v týdnech 1, 2 a 3. Od týdne 4 dostanete jednu injekci měsíčně.
Přípravek Cosentyx je určen k dlouhodobému podávání. Lékař bude pravidelně monitorovat Váš stav, aby zjistil, zda má léčba požadovaný účinek.
Jestliže jste použil(a) více přípravku Cosentyx než jste měl(a)
Pokud jste použil(a) více přípravku Cosentyx než jste měl(a) nebo pokud byla dávka podána dříve než lékař předepsal, poraďte se s lékařem.
Jestliže jste zapomněl(a) použít přípravek Cosentyx
Pokud jste zapomněl injikovat dávku přípravku Cosentyx, injikujte další dávku co nejdříve poté, co si vzpomenete. Následně se poraďte s lékařem, kdy máte injikovat následující dávku.
Jestliže jste přestal(a) používat přípravek Cosentyx
Přestat používat přípravek Cosentyx není nebezpečné. Nicméně, pokud přestanete, příznaky psoriázy, psoriatické artritidy nebo ankylozující spondylitidy se mohou vrátit.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
T-W r V r Vil i i V* 1
Závažné nežádoucí účinky
Přestaňte používat přípravek Cosentyx a informujte neprodleně svého lékaře nebo vyhledejte lékařskou pomoc, pokud zaznamenáte jakékoliv následující nežádoucí účinky.
Možné závažné infekce - známky mohou zahrnovat:
• horečka, příznaky podobné chřipce, noční pocení
• pocit únavy nebo dýchavičnosti, přetrvávající kašel
• teplá, rudá a bolestivá pokožka, nebo bolestivá vyrážka s puchýři
• pocit pálení při močení.
Závažná alergická reakce - známky mohou zahrnovat:
• obtížné dýchání nebo polykání
• nízký krevní tlak, který může působit závratě nebo omámení
• otok tváře, rtů, jazyka nebo hrdla
• silné svědění pokožky s červenou vyrážkou nebo vyvýšenými hrbolky .
Váš lékař rozhodne, zda a kdy můžete pokračovat v léčbě.
Jiné nežádoucí účinky
Většina následujících nežádoucích účinků je mírná až středně závažná. Pokud se kterýkoli z těchto nežádoucích účinků stane závažným, sdělte to lékaři, lékárníkovi nebo zdravotní sestře.
Některé nežádoucí účinky jsou velmi časté (mohou postihnout více než 1 z 10 osob):
• infekce horních cest dýchacích s příznaky jako bolest v krku a ucpaný nos (nasofaryingitida, rinitida)
Některé nežádoucí účinky jsou časté (mohou postihnout až 1 z 10 osob):
• opar (na rtech)
• průjem
• výtok z nosu (rýma)
Některé nežádoucí účinky jsou méně časté (mohou postihnout až 1 ze 100 osob):
• ústní povlak (ústní kandidóza)
• známky nízké hladiny bílých krvinek, jako jsou horečka, bolest v krku nebo vředy v ústech kvůli infekci (neutropenie)
• atletická noha (tinea pedis)
• infekce vnějšího ucha (otitis media)
• výtok z očí se svěděním, zarudnutím a otokem (konjunktivitida).
• svědivá vyrážka (kopřivka)
Některé nežádoucí účinky jsou vzácné (mohou postihnout až 1 z 1 000 osob)
• závažná alergická reakce s šokem (anafylaktická reakce)
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Cosentyx uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek:
• po uplynutí doby použitelnosti uvedené na krabičce nebo na štítku injekční stříkačky po „EXP“.
• pokud roztok obsahuje zřetelně viditelné částice, je zakalený nebo zřetelně hnědý.
Uchovávejte injekční stříkačku uzavřenou v krabičce, aby byla chráněna před světlem. Uchovávejte v chladničce od 2°C do 8°C. Chraňte před mrazem. Netřepejte.
Tento přípravek je určen k jednorázovému použití. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte.
6. Obsah balení a další informace Co přípravek Cosentyx obsahuje
- Léčivou látkou je secukinumabum. Jedna předplněná injekční stříkačka obsahuje 150 mg secukinumabum.
- Dalšími složkami jsou dihydrát trehalosy, histidin, monohydrát histidin-hydrochloridu, methionin, polysorbát 80 a voda na injekci.
Jak přípravek Cosentyx vypadá a co obsahuje toto balení
Přípravek Cosentyx injekční roztok je čirá tekutina. Její barva může být od bezbarvé do mírně nažloutlé. Nepoužívejte, pokud tekutina obsahuje dobře viditelné částice, je zakalená nebo zřetelně hnědá. Přípravek Cosentyx je dostupný v jednotkovém balení obsahujícím 1 nebo 2 předplněné injekční stříkačky a ve vícečetném balení obsahujícím 6 (3x2) předplněných injekčních stříkaček. Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
Výrobce
Novartis Pharma GmbH Roonstrasse 25 D-90429 Norimberk Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgie/Belgique/Belgien
Novartis Pharma N.V.
Tél/Tel: +32 2 246 16 11
Eunrapnu
Novartis Pharma Services Inc. Ten: +359 2 489 98 28
Česká republika
Novartis s.r.o.
Tel: +420 225 775 111
Danmark
Novartis Healthcare A/S Tlf: +45 39 16 84 00
Deutschland
Novartis Pharma GmbH Tel: +49 911 273 0
Eesti
Novartis Pharma Services Inc. Tel: +372 66 30 810
EkXúba
Novartis (Hellas) A.E.B.E. Tqk: +30 210 281 17 12
Espaňa
Novartis Farmacéutica, S.A. Tel: +34 93 306 42 00
Lietuva
Novartis Pharma Services Inc. Tel: +370 5 269 16 50
Luxembourg/Luxemburg
Novartis Pharma N.V.
Tél/Tel: +32 2 246 16 11
Magyarország
Novartis Hungária Kft. Pharma Tel.: +36 1 457 65 00
Malta
Novartis Pharma Services Inc. Tel: +356 2122 2872
Nederland
Novartis Pharma B.V.
Tel: +31 26 37 82 111
Norge
Novartis Norge AS Tlf: +47 23 05 20 00
Osterreich
Novartis Pharma GmbH Tel: +43 1 86 6570
Polska
Novartis Poland Sp. z o.o.
Tel.: +48 22 375 4888
|
France Novartis Pharma S.A.S. Tél: +33 1 55 47 66 00 |
Portugal Novartis Farma - Produtos Farmaceuticos, S.A Tel: +351 21 000 8600 |
|
Hrvatska Novartis Hrvatska d.o.o. Tel. +385 1 6274 220 |
Románia Novartis Pharma Services Romania SRL Tel: +40 21 31299 01 |
|
Ireland Novartis Ireland Limited Tel: +353 1 260 12 55 |
Slovenija Novartis Pharma Services Inc. Tel: +386 1 300 75 50 |
|
Island Vistor hf. Sími: +354 535 7000 |
Slovenská republika Novartis Slovakia s.r.o. Tel: +421 2 5542 5439 |
|
Italia Novartis Farma S.p.A. Tel: +39 02 96 54 1 |
Suomi/Finland Novartis Finland Oy Puh/Tel: +358 (0)10 6133 200 |
|
Kórcpog Novartis Pharma Services Inc. Tn^: +357 22 690 690 |
Sverige Novartis Sverige AB Tel: +46 8 732 32 00 |
|
Latvija Novartis Pharma Services Inc. Tel: +371 67 887 070 |
United Kingdom Novartis Pharmaceuticals UK Ltd. Tel: +44 1276 698370 |
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Instrukce pro použití předplněné injekční stříkačky přípravku Cosentyx
Přečtěte si VŠECHNY následující instrukce před podáním injekce. Je důležité, nesnažit se o injekci sám (sama), dokud nebudete proškolen(a) Vaším lékařem, zdravotní sestrou nebo lékárníkem. Krabička obsahuje předplněné injekční stříkačky přípravku Cosentyx individuálně zapečetěné v plastovém blistru.
Vaše předplněná injekční stříkačka Cosentyx
Ochranné pouzdro injekční stříkačky
Držák
Píst
Průhledové okénko se štítkem a dobou použitelnosti
Křidélka ochranného pouzdra
Hlavice pístu

Kryt jehly
Po podání injekce se aktivuje ochranné pouzdro a skryje injekční jehlu. To pomáhá chránit zdravotnické pracovníky, pacienty samostatně si injikující lékařem předepsané přípravky a osoby asistující při samopodání pacientů před náhodným zraněním jehlou.

Co dále potřebujete pro injekci:
• Alkoholový polštářek.
• Smotek vaty nebo gázy.
• Nádobu na ostré předměty.
Důležité bezpečnostní informace
Upozornění: Uchovávejte injekční stříkačky mimo dohled a dosah dětí.
1. Kryt jehly může obsahovat suchou gumu (latex), nesmí s ním zacházet osoby citlivé na tuto látku.
2. Neotevírejte zapečetěný vnější obal, dokud nejste připraveni tento přípravek použít.
3. Nepoužívejte tento přípravek, pokud j e poškozena pečeť na vněj ším obalu nebo na blistru, protože použití nemusí být pro Vás bezpečné.
4. Nikdy nenechávejte volně ležet injekční stříkačku tam, kde by ji někdo mohl poškodit.
5. Netřeste injekční stříkačkou.
6. Nedotýkejte se křidélek ochranného pouzdra injekční stříkačky před použitím. Při doteku by se mohla ochrana aktivovat předčasně.
7. Nesnímejte kryt jehly dříve než těsně před injekcí.
8. Injekční stříkačka nesmí být opakovaně použita. Odložte použitou injekční stříkačku bezprostředně po použití do schránky na ostré předměty.
Uchovávání předplněné injekční stříkačky přípravku Cosentyx
1. Uchovávejte tento přípravek v zapečetěné krabičce, aby byl přípravek chráněn před světlem. Uchovávejte v chladničce při teplotě mezi 2°C až 8°C. CHRAŇTE PŘED MRAZEM.
2. Pamatujte na vyjmutí injekční stříkačky z chladničky, aby dosáhla pokojové teploty před její přípravou k injekci (15-30 minut).
3. Nepoužívejte injekční stříkačku po uplynutí doby použitelnosti uvedené na krabičce nebo štítku injekční stříkačky za „EXP“. Pokud uplynula doba použitelnosti, vraťte celé balení do lékárny.
Místa injekce


Místo injekce je místo na těle, na němž hodláte použít injekční stříkačku.
• Doporučené místo je přední část Vašich stehen. Můžete též použít podbřišek, nikoliv však oblast 5 centimetrů okolo pupku.
• Při samopodávání injekce zvolte vždy jiné místo injekce.
• Neinjikujte do oblastí, kde je pokožka jemná,
s podlitinami, zarudlá, odlupuje se nebo je tvrdá. Vyvarujte se oblastí s jizvami nebo vytahanou pokožkou. Pokud podává injekci opatrovník, lze též použít vnější stranu paží.
Příprava použití předplněné injekční stříkačky přípravku Cosentyx
Poznámka: Pro dávku 300 mg připravte 2 předplněné injekční stříkačky a injikujte obsah obou.
1. Vyjměte krabičku s injekční stříkačkou z chladničky a ponechte ji neotevřenou 15-30 minut, aby se ohřála na pokojovou teplotu.
2. Když jste připraven(a) použít injekční stříkačku, umyjte si důkladně ruce mýdlem a vodou.
3. Očistěte místo injekce alkoholovým tampónem.
4. Vyjměte injekční stříkačku z krabičky a z blistru.
5. Prohlédněte injekční stříkačku. Tekutina musí být čirá. Její barva může být od bezbarvé do mrně nažloutlé. Můžete vidět malé bublinky vzduchu, což je normální. NEPOUŽÍVEJTE, pokud tekutina obsahuje zřetelně viditelné částice, je zakalená nebo zřetelně hnědá. NEPOUŽÍVEJTE, pokud je pečeť injekční stříkačky porušena. Ve všech těchto případech vraťte celé balení do lékárny.
Jak použít předplněnou injekční stříkačku přípravku Cosentyx

Z injekční stříkačky sejměte kryt jehly. Kryt jehly odhoďte do odpadu. Na hrotu jehly můžete vidět kapku tekutiny. To je normální.
Jemně stiskněte pokožku v místě injekce a vsuňte jehlu podle obrázku. Zatlačte úplně jehlu, aby bylo zajištěno podání celé dávky.


Instrukce k odstranění

Držte injekční stříkačku podle obrázku. Pomalu tiskněte píst na doraz, takže je hlava pístu zcela mezi křidélky ochranného pouzdra.
Držte píst plně stisknutý v dolní poloze, současně držte injekční stříkačku na místě po dobu 5 vteřin.
Držte píst plně stisknutý, zatímco opatrně zvedáte jehlu z místa injekce.
Pomalu uvolněte píst a umožněte,aby ochranné pouzdro zakrylo odkrytou jehlu.
Na místě injekce se může objevit malé množství krve. Můžete na místo injekce přitisknout smotek vaty nebo gázu a podržte 10 vteřin. Místa injekce se nedotýkejte. Pokud je to zapotřebí, můžete místo injekce překrýt malou adhezivní náplastí.
Odložte použitou injekční stříkačku do nádoby na ostrý odpad (uzavíratelná schránka, odolná proti propíchnutí). Kvůli bezpečnosti Vás i ostatních nesmí být jehly a injekční stříkačky nikdy použity opakovaně.
Příbalová informace: informace pro pacienta Cosentyx 150 mg injekční roztok v předplněném peru
Secukinumabum
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Cosentyx a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Cosentyx používat
3. Jak se přípravek Cosentyx používá
4. Možné nežádoucí účinky
5. Jak přípravek Cosentyx uchovávat
6. Obsah balení a další informace
1. Co je přípravek Cosentyx a k čemu se používá
Přípravek Cosentyx obsahuje léčivou látku secukinumab. Secukinumab je monoklonální protilátka. Monoklonální protilátky jsou bílkoviny, které rozpoznávají a specificky se vážou na některé bílkoviny v těle.
Přípravek Cosentyx patří do skupiny léků zvaných inhibitory interleukinu (IL). Tyto léky neutralizují účinky bílkoviny nazývané IL-17A, která je přítomna ve zvýšeném množství u chorob jako je psoriáza, psoriatická artritida a ankylozující spondylitida.
Přípravek Cosentyx je určen k léčbě následujích zánětlivých chorob:
• Ložisková psoriáza
• Psoriatická artritida
• Ankylozující spondylitida
Ložisková psoriáza
Přípravek Cosentyx se používá k léčbě kožních projevů nazývaných “ložisková psoriáza”, která vyvolává zánět ovlivňující pokožku. Přípravek Cosentyx redukuje zánět a další příznaky choroby. Přípravek Cosentyx se používá u dospělých se středně závažnou až závažnou ložiskovou psoriázou.
Používání přípravku Cosentyx u ložskové psoriázy Vám pomůže tím, že vede ke zlepšení postižení pokožky a redukci příznaků jako odlupování, svědění a bolest.
Psoriatická artritida
Přípravek Cosentyx se používá k léčbě stavu nazývaného“psoriatická artritida”. Tento stav představuje zánětlivou chorobu kloubů, často doprovázenou psoriázou. Pokud trpíte aktivní psoriatickou artirtidou, budete nejdříve léčen(a) jinými léčivými přípravky. Pokud dostatečně neodpovídáte na tyto přípravky, bude Vám předepsán přípravek Cosentyx pro omezení známek a příznaků aktivní psoriatické artiritdy,
zlepšení funkčního stavu a zpomalení poškození chrupavky a kosti kloubů postižených touto chorobou.
Přípravek Cosentyx užívají dospělí s aktivní psoriatickou artritidou a smí se užívat samostatně nebo v kombinaci s jiným přípravkem nazývaným metotrexát.
Užívání přípravku Cosentyx u psoriatické artritidy je pro Vás prospěšné omezením známek a příznaků choroby, zpomalením poškození chrupavky a kosti kloubů a zlepšením schopnosti vykonávat běžné denní aktivity.
Ankylozující spondylitida
Přípravek Cosentyx se používá k léčbě stavu nazývaného“ankylozující spondylitida”. Tento stav představuje zánětlivou chorobu postihující primárně páteř, kdy dochází k zánětu páteřních kloubů. Pokud trpíte ankylozující spondylitidou, budete nejdříve léčen(a) jinými léčivými přípravky. Pokud dostatečně neodpovídáte na tyto přípravky, bude Vám předepsán přípravek Cosentyx pro omezení známek a příznaků této choroby, omezení zánětu a zlepšení fyzické funkce.
Přípravek Cosentyx užívají dospělí s aktivní ankylozující spondylitidou.
Užívání přípravku Cosentyx u ankylozující spondylitidy je pro Vás prospěšné omezením známek a příznaků choroby a a zlepšením fyzické funkce.
2. Čemu musíte věnovat pozornost, než začnete přípravek Cosentyx používat Nepoužívejte přípravek Cosentyx:
• jestliže jste alergický(á) na secukinumab nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Pokud se domníváte, že můžete být alergický(á), poraďte se před použitím přípravku Cosentyx s lékařem.
• Jestliže trpíte aktivní infekcí, kterou Váš lékař považuje za důležitou.
Upozornění a opatření
Poraďte se s lékařem nebo lékárníkem předtím, než začnete přípravek Cosentyx používat:
• pokud trpíte v současnosti infekcí
• pokud trpíte dlouhodobou nebo opakovaně se vyskytující infekcí.
• pokud máte tuberkulózu.
• Pokud jste někdy měl(a) alergickou reakci na latex.
• pokud máte Crohnovu chorobu.
• Pokud jste byl(a) v nedávné době očkován(a) nebo se máte podrobit očkování během léčby přípravkem Cosentyx.
• pokud se podrobujete jiné léčbě psoriázy, jako jsou imunosupresiva nebo fototerapie ultrafialovým světlem (UV).
Sledování infekcí a alergických reakcí
Přípravek Cosentyx může případně vyvolat závažné nežádoucí účinky, včetně infekcí a alergických reakcí. Pokud používáte přípravek Cosentyx, musíte sledovat příznaky těchto stavů.
Přestaňte používat přípravek Cosentyx a informujte neprodleně svého lékaře nebo vyhledejte lékařskou pomoc, pokud zaznamenáte jakékoliv známky naznačující možné závažné infekci nebo alergické reakci. Tyto známky jsou uvedeny v oddíle “Závažné nežádoucí účinky” v bodě 4.
Děti a dospívající
Přípravek Cosentyx se nedoporučuje u dětí a dospívajících (do 18 let věku), protože v této věkové skupině nebyl studován.
Další léčivé přípravky a přípravek Cosentyx
Informujte svého lékaře nebo lékárníka:
• o všech lécích, které používáte, které jste v nedávné době používal(a) nebo které možná budete používat.
• že jste byl(a) v nedávné době očkován(a) nebo se máte podrobit očkování. Při používání přípravku Cosentyx Vám nesmí být podán určitý typ vakcín (živé vakcíny).
Těhotenství, kojení a plodnost
• Je doporučeno vyvarovat se užívání přípravku Cosentyx během těhotenství. Účinky tohoto přípravku na těhotné ženy nejsou známy. Pokud jste žena ve fertilním věku, doporučuje se zabránit otěhotnění a během léčby přípravkem Cosentyx a po dobu nejméně 20 týdnů od poslední dávky přípravku Cosentyx musíte užívat vhodnou antikoncepci. Poraďte se s lékařem, pokud jste těhotná, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět.
• Poraďte se s lékařem, pokud kojíte nebo plánujete kojení. Společně s lékařem rozhodnete, zda budete kojit nebo užívat přípravek Cosentyx. Nemůžete dělat obojí. Po užití přípravku Cosentyx nesmíte kojit po dobu nejméně 20 týdnů od poslední dávky.
Řízení dopravních prostředků a obsluha strojů
Přípravek Cosentyx nemá žádný nebo má zanedbatelný vliv na schopnost řídit a obsluhovat stroje.
3. Jak se přípravek Cosentyx používá
Vždy používejte tento přípravek přesně podle pokynů Vašeho lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem, lékárníkem nebo zdravotní sestrou.
Přípravek Cosentyx se podává injekcí pod kůži (což je známo jako podkožní injekce). Společně s Vaším lékařem se musíte rozhodnout, zda si budete přípravek Cosentyx injikovat sám (sama).
Je důležité, nesnažit se o injekci sám (sama), dokud nebudete proškolen(a) Vaším lékařem, zdravotní sestrou nebo lékárníkem. Po vhodném proškolení Vám může přípravek Cosentyx injekce podat i Váš opatrovník.
Podrobný návod, jak injikovat přípravek Cosentyx, naleznete v “Návodu k použití Cosentyx SensoReady pen” na konci této příbalové informace.
Jaké množství přípravku Cosentyx se podává a jak dlouho
Váš lékař rozhodne, jakou dávku přípravku Cosentyx potřebujete a jak dlouho.
Ložisková psoriáza
• Doporučená dávka přípravku je 300 mg ve formě podkožní injekce.
• Každá dávka 300 mg je podána ve dvou injekcích 150 mg.
Po podání první dávky budete dostávat další týdenní injekce v týdnu 1, 2 a 3. Od týdne 4 budete dostávat injekci měsíčně. Pokaždé dostanete dávku 300 mg rozdělenou do dvou injekcí po 150 mg.
Psoriatická artritida
U pacientů s psoriatickou artritidou, kteří současně trpí středně těžkou až těžkou ložiskovou psoriázou nebo u pacientů, kteří neodpovídají dostatečně na léčbu přípravky „tumour necrosis factor“ (TNF) blokátory:
• Doporučená dávka je 300 mg ve formě podkožní injekce.
• Každá dávka 300 mg je podána ve dvou injekcích 150 mg.
Po první dávce budou následovat další týdenní injekce v týdnech 1, 2 a 3. Od týdne 4 dostanete jednu injekci měsíčně. Vždy dostanete dávku 300 mg podanou ve dvou injekcích po 150 mg.
U ostatních pacientů s psoriatickou artritidou:
• Doporučená dávka je 150 mg ve formě podkožní injekce.
Po první dávce budou následovat další týdenní injekce v týdnech 1, 2 a 3. Od týdne 4 dostanete jednu injekci měsíčně.
Ankylozující spondylitida
• Doporučená dávka je 150 mg ve formě podkožní injekce.
Po první dávce budou následovat další týdenní injekce v týdnech 1, 2 a 3. Od týdne 4 dostanete jednu injekci měsíčně.
Přípravek Cosentyx je určen k dlouhodobému podávání. Lékař bude pravidelně monitorovat Váš stav, aby zjistil, zda má léčba požadovaný účinek.
Jestliže jste použil(a) více přípravku Cosentyx než jste měl(a)
Pokud jste použil(a) více přípravku Cosentyx než jste měl(a) nebo pokud byla dávka podána dříve než lékař předepsal, poraďte se s lékařem.
Jestliže jste zapomněl(a) použít přípravek Cosentyx
Pokud jste zapomněl injikovat dávku přípravku Cosentyx, injikujte další dávku co nejdříve poté, co si vzpomenete. Následně se poraďte s lékařem, kdy máte injikovat následující dávku.
Jestliže jste přestal(a) používat přípravek Cosentyx
Přestat používat přípravek Cosentyx není nebezpečné. Nicméně, pokud přestanete, příznaky psoriázy, psoriatické artritidy nebo ankylozující spondylitidy se mohou vrátit.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
T-W r V r Vil i i V* 1
Závažné nežádoucí účinky
Přestaňte používat přípravek Cosentyx a informujte neprodleně svého lékaře nebo vyhledejte lékařskou pomoc, pokud zaznamenáte jakékoliv následující nežádoucí účinky.
Možné závažné infekce - známky mohou zahrnovat:
• horečka, příznaky podobné chřipce, noční pocení
• pocit únavy nebo dýchavičnosti, přetrvávající kašel
• teplá, rudá a bolestivá pokožka, nebo bolestivá vyrážka s puchýři
• pocit pálení při močení.
Závažná alergická reakce - známky mohou zahrnovat:
• obtížné dýchání nebo polykání
• nízký krevní tlak, který může působit závratě nebo omámení
• otok tváře, rtů, jazyka nebo hrdla
• silné svědění pokožky s červenou vyrážkou nebo vyvýšenými hrbolky .
Váš lékař rozhodne, zda a kdy můžete pokračovat v léčbě.
Jiné nežádoucí účinky
Většina následujících nežádoucích účinků je mírná až středně závažná. Pokud se kterýkoli z těchto nežádoucích účinků stane závažným, sdělte to lékaři, lékárníkovi nebo zdravotní sestře.
Některé nežádoucí účinky jsou velmi časté (mohou postihnout více než 1 z 10 osob):
• infekce horních cest dýchacích s příznaky jako bolest v krku a ucpaný nos (nasofaryingitida, rinitida)
Některé nežádoucí účinky jsou časté (mohou postihnout až 1 z 10 osob):
• opar (na rtech)
• průjem
• výtok z nosu (rýma)
Některé nežádoucí účinky jsou méně časté (mohou postihnout až 1 ze 100 osob):
• ústní povlak (ústní kandidóza)
• známky nízké hladiny bílých krvinek, jako jsou horečka, bolest v krku nebo vředy v ústech kvůli infekci (neutropenie)
• atletická noha (tinea pedis)
• infekce vnějšího ucha (otitis media)
• výtok z očí se svěděním, zarudnutím a otokem (konjunktivitida).
• svědivá vyrážka (kopřivka)
Některé nežádoucí účinky jsou vzácné (mohou postihnout až 1 z 1 000 osob)
• závažná alergická reakce s šokem (anafylaktická reakce)
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Cosentyx uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek:
• po uplynutí doby použitelnosti uvedené na krabičce nebo na štítku pera po „EXP“.
• pokud roztok obsahuje zřetelně viditelné částice, je zakalený nebo zřetelně hnědý.
Uchovávejte pero uzavřené v krabičce, aby bylo chráněno před světlem. Uchovávejte v chladničce od 2°C do 8°C. Chraňte před mrazem. Netřepejte.
Tento přípravek je určen k jednorázovému použití. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte.
6. Obsah balení a další informace
Co přípravek Cosentyx obsahuje
- Léčivou látkou je secukinumabum. Jedno předplněné pero obsahuje 150 mg secukinumabum.
- Dalšími složkami jsou dihydrát trehalosy, histidin, monohydrát histidin-hydrochloridu, metionin, polysorbát 80 a voda na injekci.
Jak přípravek Cosentyx vypadá a co obsahuje toto balení
Přípravek Cosentyx injekční roztok je čirá tekutina. Její barva může být od bezbarvé do mírně nažloutlé. Nepoužívejte, pokud tekutina obsahuje dobře viditelné částice, je zakalená nebo zřetelně hnědá. Přípravek Cosentyx je dostupný v jednotkovém balení obsahujícím 1 nebo 2 předplněná pera a ve vícečetném balení obsahujícím 6 (3x2) předplněných per. Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
Výrobce
Novartis Pharma GmbH Roonstrasse 25 D-90429 Norimberk Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgie/Belgique/Belgien Lietuva
Novartis Pharma N.V. Novartis Pharma Services Inc.
Tél/Tel: +32 2 246 16 11
Tel: +370 5 269 16 50
Bunrapun
Novartis Pharma Services Inc. Ten: +359 2 489 98 28
Luxembourg/Luxemburg
Novartis Pharma N.V. Tél/Tel: +32 2 246 16 11
Magyarország
Novartis Hungária Kft. Pharma
Česká republika
Novartis s.r.o.
Tel: +420 225 775 111
Tel.: +36 1 457 65 00
Danmark
Novartis Healthcare A/S Tlf: +45 39 16 84 00
Malta
Novartis Pharma Services Inc. Tel: +356 2122 2872
Deutschland
Novartis Pharma GmbH Tel: +49 911 273 0
Nederland
Novartis Pharma B.V. Tel: +31 26 37 82 111
Norge
Novartis Norge AS Tlf: +47 23 05 20 00
Eesti
Novartis Pharma Services Inc. Tel: +372 66 30 810
EkXúba
Novartis (Hellas) A.E.B.E. Tqk +30 210 281 17 12
Osterreich
Novartis Pharma GmbH Tel: +43 1 86 6570
|
Espaňa Novartis Farmacéutica, S.A. Tel: +34 93 306 42 00 |
Polska Novartis Poland Sp. z o.o. Tel.: +48 22 375 4888 |
|
France Novartis Pharma S.A.S. Tél: +33 1 55 47 66 00 |
Portugal Novartis Farma - Produtos Farmaceuticos, S.A Tel: +351 21 000 8600 |
|
Hrvatska Novartis Hrvatska d.o.o. Tel. +385 1 6274 220 |
Románia Novartis Pharma Services Romania SRL Tel: +40 21 31299 01 |
|
Ireland Novartis Ireland Limited Tel: +353 1 260 12 55 |
Slovenija Novartis Pharma Services Inc. Tel: +386 1 300 75 50 |
|
Ísland Vistor hf. Sími: +354 535 7000 |
Slovenská republika Novartis Slovakia s.r.o. Tel: +421 2 5542 5439 |
|
Italia Novartis Farma S.p.A. Tel: +39 02 96 54 1 |
Suomi/Finland Novartis Finland Oy Puh/Tel: +358 (0)10 6133 200 |
|
Kónpoq Novartis Pharma Services Inc. Tn^: +357 22 690 690 |
Sverige Novartis Sverige AB Tel: +46 8 732 32 00 |
|
Latvija Novartis Pharma Services Inc. Tel: +371 67 887 070 |
United Kingdom Novartis Pharmaceuticals UK Ltd. Tel: +44 1276 698370 |
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Instrukce pro použití předplněného pera Cosentyx SensoReady

Pero Cosentyx SensoReady 150 mg
Injekční roztok v předplněném peru Secukinumabum
Návod k použití pro pacienty

Přečtěte si VŠECHNY následující instrukce před podáním injekce.
Tyto instrukce Vám pomohou správně injikovat s použitím pera Cosentyx SensoReady.
Je důležité, nesnažit se o injekci sám (sama), dokud nebudete proškolen(a) Vaším lékařem, zdravotní sestrou nebo lékárníkem.
Vaše pero Cosentyx SensoReady:
Jehla Ochrana jehly. Kryt
I

V--'
Inspekční okénko Kryt vnitřní jehly
Pero Cosentyx SensoReady znázorněné se sejmutým krytem. Nesnímejte kryt, dokud nejste připraven(a) k injekci.
Uchovávejte pero v krabičce v chladničce při teplotě 2°C až 8°C a mimo dosah dětí.
• Chraňte pero před mrazem.
• Netřeste perem.
• Nepoužívejte pero, pokud Vám vypadlo se sejmutým krytem.
Pro komfortnější injekci, vyjměte pero z chladničky 15-30 minut před injekcí, aby dosáhlo pokojové teploty.
Co potřebujete pro injekci:
Obsaženo v balení:
Neobsaženo v balení:
Nové a nepoužité pero Cosentyx SensoReady (pro dávku 300 mg jsou zapotřebí (2 pera).

• Alkoholový tampón.
• Smotek vaty nebo gázy.
• Nádoba na ostré předměty.





1. Důležitá bezpečnostní kontrola před injekcí:
Tekutina musí být čirá. Její barva může být od bezbarvé do mírně
nažloutlé.
Nepoužívejte, pokud tekutina obsahuje zřetelně viditelné částice, je
zakalená nebo zřetelně hnědá. Můžete vidět malé bublinky vzduch, což je
normální.
Nepoužívejte pero po uplynutí doby použitelnosti.
Nepoužívejte, pokud byla porušena pečeť.
Poraďte se s lékárníkem, pokud pero nevyhoví některé z těchto kontrol.
2a. Zvolte místo injekce:
• Doporučené místo je přední část Vašich stehen. Můžete též použít podbřišek, nikoliv však oblast 5 centimetrů okolo pupku.
• Při samopodávání injekce zvolte vždy jiné místo injekce.
• Neinjikujte do oblastí, kde je pokožka jemná, s podlitinami, zarudlá, odlupuje se nebo je tvrdá. Vyvarujte se oblastí s jizvami nebo vytahanou pokožkou.
2b. Pouze opatrovníci a zdravotničtí pracovníci:
• Pokud Vám podává injekci opatrovník nebo zdravotnický pracovník, lze též použít vnější stranu paží.
3. Očištění místa injekce:
• Umyjte si ruce mýdlem a horkou vodou.
• Krouživým pohybem očistěte místo injekce alkoholovým tampónem. Před injekcí nechte místo vyschnout.
• Před injekcí se nedotýkejte očištěného místa.
Injekce:


4. Sejmutí krytu:
• Kryt jehly sejměte teprve tehdy, pokud jste připraven(a) použít pero.
• Odšroubujte kryt ve směru šipky.
• Po sejmutí odhoďte kryt jehly do odpadu. Nesnažte se kryt znovu nasadit.
• Pero použijte během 5 minut od sejmutí krytu jehly.
5. Držte pero:
• Držte pero pod úhlem 90 stupňů nad očištěným místem injekce.
PŘEČTETE SI PŘED PODÁNÍM INJEKCE.
/\
Během injekce uslyšíte 2 hlasitá cvaknutí.
První cvaknutí znamená, že injekce začala. O několik vteřin později druhé cvaknutí znamená, že injekce je téměř podána.
Musíte držet pero pevně proti pokožce, dokud neuvidíte, že zelený indikátor vyplnil okénko a přestal se pohybovat.

6. Zahájení injekce:
• Přitiskněte pero pevně proti pokožce, abyste zahájil(a) injekci.
• První cvaknutí znamená, že injekce začala.
• Držte stále pero pevně proti pokožce.
• Zelený indikátor ukazuje postup injekce.
7. Ukončení injekce:
• Uslyšíte druhé cvaknutí. To znamená, že injekce je téměř podána.
• Zkontrolujte, zda zelený indikátor vyplnil okénko a přestal se pohybovat.
• Pero lze nyní vyjmout.

8. Zkontrolujte, zda zelený indikátor vyplnil okénko:
• To znamená, že přípravek byl podán. Pokud zelený indikátor není vidět, obraťte se na lékaře.
• Na místě injekce se může objevit malé množství krve. Můžete na místo injekce přitisknout smotek vaty nebo gázu a podržte 10 vteřin. Místa injekce se nedotýkejte. Pokud je to zapotřebí, můžete místo injekce překrýt malou adhezivní náplastí.
9. Odstranění pera Cosentyx SensoReady:
• Odložte použité pero do nádoby na ostrý odpad (tj. proti propíchnutí odolná uzavíratelná schránka nebo podobná)
• Pero se nikdy nesnažte opakovaně použít.
121
n (%)_
p hodnoty versus etanercept: p=0,0250
V další psoriatické studii (CLEAR) bylo hodnoceno 676 pacientů. V této studii byla s ohledem na primární a sekundární cílové parametry prokázána superiorita secukinumabu v dávce 300 mg oproti ustekinumabu v odpovědích PASI 90 v 16. týdnu a v rychlosti nástupu odpovědi PASI 75 ve 4. týdnu. Po celou dobu, od začátku do 16. týdne byla pozorována vyšší účinnost secukinumabu v porovnání s ustekinumabem pro cílové parametry PASI 75/90/100 a odpověď IGA mod 2011 0 nebo 1 („čistý“ nebo „téměř čistý“).
Tabulka 4 Souhrn klinické odpovědi ve studii CLEAR
n (%)_
p hodnoty versus etanercept: p=0,0250
V další psoriatické studii (CLEAR) bylo hodnoceno 676 pacientů. V této studii byla s ohledem na primární a sekundární cílové parametry prokázána superiorita secukinumabu v dávce 300 mg oproti ustekinumabu v odpovědích PASI 90 v 16. týdnu a v rychlosti nástupu odpovědi PASI 75 ve 4. týdnu. Po celou dobu, od začátku do 16. týdne byla pozorována vyšší účinnost secukinumabu v porovnání s ustekinumabem pro cílové parametry PASI 75/90/100 a odpověď IGA mod 2011 0 nebo 1 („čistý“ nebo „téměř čistý“).
Tabulka 4 Souhrn klinické odpovědi ve studii CLEAR
n (%)_
p hodnoty versus etanercept: p=0,0250
V další psoriatické studii (CLEAR) bylo hodnoceno 676 pacientů. V této studii byla s ohledem na primární a sekundární cílové parametry prokázána superiorita secukinumabu v dávce 300 mg oproti ustekinumabu v odpovědích PASI 90 v 16. týdnu a v rychlosti nástupu odpovědi PASI 75 ve 4. týdnu. Po celou dobu, od začátku do 16. týdne byla pozorována vyšší účinnost secukinumabu v porovnání s ustekinumabem pro cílové parametry PASI 75/90/100 a odpověď IGA mod 2011 0 nebo 1 („čistý“ nebo „téměř čistý“).
Tabulka 4 Souhrn klinické odpovědi ve studii CLEAR