Constella 290 Mikrogramů
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Constella 290 mikrogramů tvrdé tobolky.
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna tobolka obsahuje linaclotidum 290 mikrogramů.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Tvrdá tobolka.
Neprůhledná tobolka bílé až bělavo-oranžové barvy (18 mm x 6,35 mm), označená údajem „290“ šedou barvou.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Constella je indikován k symptomatické léčbě středně těžké až těžké formy syndromu dráždivého tračníku spojeného se zácpou (IBS-C, irritable bowel syndrome with constipation) u dospělých pacientů.
4.2 Dávkování a způsob podání
Dávkování
Doporučená dávka je jedna tobolka (290 mikrogramů) jednou denně.
Lékař musí pravidelně hodnotit nutnost pokračování léčby. Účinnost linaklotidu byla stanovena ve dvojitě zaslepených placebem kontrolovaných studiích trvajících až šest měsíců. Pokud se u pacienta po 4 týdnech léčby neprojeví zlepšení příznaků, je nutno jej znovu vyšetřit a znovu posoudit přínos a rizika pokračující léčby.
Zvláštní _ populace
Pacienti s poruchou funkce jater nebo ledvin
U pacientů s poruchou funkce jater nebo ledvin není zapotřebí žádná úprava dávky (viz bod 5.2).
Starší _ pacienti
Ačkoli u starších pacientů není zapotřebí žádná úprava dávky, léčbu je třeba pečlivě monitorovat a pravidelně přehodnocovat (viz bod 4.4).
Pediatrická _ populace
Bezpečnost a účinnost linaklotidu u dětí ve věku 0 až 18 let nebyla dosud stanovena. Nejsou dostupné žádné údaje.
Přípravek Constella se nemá používat u dětí a dospívajících (viz bod 4.4 a 5.1).
Způsob podání
Perorální podání. Tobolka se má užívat nejméně 30 minut před jídlem (viz bod 4.5).
4.3 Kontraindikace
Hypersenzitivita na linaklotid nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1 tohoto přípravku.
Pacienti s potvrzenou nebo suspektní mechanickou obstrukcí zažívacího traktu.
4.4 Zvláštní upozornění a opatření pro použití
Přípravek Constella se smí použít pouze po vyloučení organického onemocnění a po stanovení diagnózy středně těžké až těžké formy IBS-C (viz bod 5.1).
Pacienti musí vzít na vědomí, že se během léčby může vyskytnout průjem a/ nebo krvácení z dolní části trávicího traktu. Musí být poučeni, aby v případě výskytu závažného nebo déletrvajícího průjmu či krvácení z dolní části trávicího traktu informovali svého lékaře (viz bod 4.8).
Vyskytne-li se závažný nebo déletrvající (např. déle než 1 týden) průjem, pacient musí vyhledat lékařskou pomoc a je nutno zvážit dočasné vysazení linaklotidu, dokud průjem neustoupí. Zvláštní opatrnost je nutná u pacientů náchylných k poruchám vodní či elektrolytové rovnováhy (např. u starších pacientů, pacientů s kardiovaskulárním onemocněním, s diabetem nebo s hypertenzí); je třeba zvážit kontroly elektrolytů.
Linaklotid nebyl hodnocen u pacientů s chronickým zánětlivým onemocněním střevního traktu, jako je Crohnova choroba a ulcerózní kolitida; proto se použití přípravku Constella u těchto pacientů nedoporučuje.
Starší pacienti
Ohledně použití u starších pacientů jsou k dispozici jen omezené údaje (viz bod 5.1). Vzhledem k vyššímu riziku průjmu, sledovanému v klinických hodnoceních (viz bod 4.8), je nutno těmto pacientům věnovat zvýšenou pozornost a pečlivě a pravidelně vyhodnocovat poměr přínosů a rizik léčby.
Pediatrická populace
Přípravek Constella se nemá používat u dětí a dospívajících, protože u této populace nebyl hodnocen. Je známo, že v raném věku dochází k nadměrné expresi receptoru GC-C, a proto mohou být děti ve věku do 2 let obzvláště citlivé na účinky linaklotidu.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné studie interakcí s jinými léčivými přípravky. Linaklotid je vzácně detekovatelný v plazmě po podání doporučených klinických dávek a studie in vitro prokázaly, že linaklotid není ani substrát, ani inhibitor či induktor enzymatického systému cytochromu P450 a neinteraguje s řadou obvyklých efluxních transportérů a přenašečových proteinů (viz bod 5.2).
Klinické hodnocení interakce léčivého přípravku s jídlem u zdravých subjektů ukázalo, že linaklotid v terapeutických dávkách není detekovatelný v plazmě po jídle ani nalačno. Při podávání přípravku Constella po jídle došlo k tvorbě častější a řidší stolice a také k více nežádoucím příhodám v zažívacím traktu v porovnání s podáváním nalačno (viz bod 5.1). Tobolka se má užívat 30 minut před jídlem (viz bod 4.2).
Při současné léčbě inhibitory protonové pumpy, laxativy nebo NSAID se může zvýšit riziko vzniku průjmu.
Při závažném nebo déletrvajícím průjmu může být ovlivněna absorpce jiných perorálních léčivých přípravků. Může být snížena účinnost perorální antikoncepce; doporučuje se používat doplňkové metody antikoncepce k prevenci možného selhání perorální antikoncepce (viz informace k předepisování perorální antikoncepce). Je nutno postupovat opatrně při předepisování léčivých přípravků s úzkým terapeutickým indexem, absorbovaných ve střevě, například levothyroxinu, protože jejich účinnost může být snížená.
4.6 Fertilita, těhotenství a kojení
O použití linaklotidu u těhotných žen jsou k dispozici omezené údaje. Studie reprodukční toxicity na zvířatech nenaznačují přímé nebo nepřímé škodlivé účinky (viz bod 5.3). Jako preventivní opatření se doporučuje vyvarovat se použití přípravku během těhotenství.
Kojení
Vzhledem k tomu, že systémová expozice linaklotidu je minimální, není vylučování do mateřského mléka pravděpodobné, ačkoli dosud nebylo hodnoceno. I když se při použití terapeutických dávek nepředpokládá žádný vliv na kojené novorozence či kojence, vzhledem k nedostupnosti údajů týkajících se použití u lidí se přípravek nedoporučuje používat během kojení.
Fertilita
Studie na zvířatech ukazují, že přípravek nemá žádný vliv na mužskou ani ženskou fertilitu.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Constella nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky Souhrn bezpečnostního profilu
Linaklotid byl podáván perorálně 1 166 pacientům s IBS-C v kontrolovaných klinických studiích. 892 z těchto pacientů dostávalo linaklotid v doporučené dávce 290 mikrogramů denně. Celková expozice v klinickém vývojovém plánu překročila 1 500 pacientoroků. Nejčastěji hlášeným nežádoucím účinkem spojeným s léčbou přípravkem Constella byl průjem, většinou mírné až střední intenzity, který se vyskytoval u méně než 20 % pacientů. Ve vzácných a závažnějších případech může -sekundárně - vést k výskytu dehydratace, hypokalémie, snížení hladiny bikarbonátu v krvi, závratí a ortostatické hypotenze.
Další časté nežádoucí účinky (> 1 %) byly bolest břicha, distenze břicha a plynatost.
Souhrn nežádoucích účinků v tabulce
V kontrolovaných klinických studiích byly při doporučeném dávkování 290 mikrogramů denně hlášeny následující nežádoucí účinky, a to s následující frekvencí: velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1000 až < 1/100), vzácné (> 1/10000 až < 1/1000), velmi vzácné (< 1/10000) a není známo (z dostupných údajů nelze určit).
|
Třída orgánových systémů podle MedDRa |
Velmi časté |
Časté |
Méně časté |
Vzácné |
Není známo |
|
Infekce a infestace |
Virová gastroenteritida | ||||
|
Gastrointestinální poruchy |
Bolest břicha Plynatost Distenze břicha |
Fekální inkontinence Nutkání na stolici Krvácení z dolní části trávicího traktu včetně hemoroidálního krvácení a rektálního krvácení Nauzea Zvracení | |||
|
Poruchy kůže a podkožní tkáně | |||||
|
Poruchy metabolismu a výživy |
Hypokalémie Dehydratace Snížená chuť k jídlu | ||||
|
Poruchy nervového systému |
Závrať | ||||
|
Cévní poruchy |
Ortostatická | ||||
|
Vyšetření |
Snížení hladiny bikarbonátu v krvi |
Popis vybraných nežádoucích účinků
Průjem, který je nejčastějším nežádoucím účinkem přípravku, odpovídá farmakologickému účinku léčivé látky. V klinických studiích měla 2 % léčených pacientů silný průjem a 5 % pacientů ukončilo léčbu kvůli průjmu.
Většina hlášených případů průjmu byla mírné (43 %) až střední (47 %) intenzity; 2 % léčených pacientů měla silný průjem. Přibližně polovina epizod průjmu začala v prvním týdnu léčby.
Přibližně u jedné třetiny pacientů průjem ustoupil během sedmi dnů, u 80 pacientů (50 %) však průjem trval déle než 28 dnů (což představovalo 9,9 % všech pacientů léčených linaklotidem).
V klinických studiích ukončilo léčbu kvůli průjmu pět procent pacientů. U pacientů, u kterých bylo nutno kvůli průjmu přerušit léčbu, průjem ustoupil za několik dnů po přerušení léčby.
U starších pacientů (> 65 let), hypertoniků a diabetiků byla vyšší četnost hlášení případů průjmu v porovnání s celkovou populací IBS-C zařazenou do klinických hodnocení.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Při předávkování se mohou objevit příznaky vyplývající ze zesílení známých farmakodynamických účinků léčivého přípravku, zejména průjem. Ve studii se zdravými dobrovolníky, kterým byla podána jedna dávka 2 897 mikrogramů (až 10násobek doporučené terapeutické dávky), odpovídal bezpečnostní profil subjektů bezpečnostnímu profilu u celkové populace, přičemž průjem byl nejčastěji hlášeným nežádoucím účinkem.
Při případném předávkování musí být pacient léčen symptomaticky a podle potřeby s použitím podpůrných opatření.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Jiné léky proti zácpě, ATC kód: A06AX04 Mechanismus účinku
Linaklotid je agonista receptoru guanylátcyklázy (GC-C) s viscerálním analgetickým účinkem a sekretorickým účinkem.
Linaklotid je syntetický peptid tvořený 14 aminokyselinami, který je strukturálně příbuzný se skupinou endogenních guanylin peptidů. Linaklotid i jeho aktivní metabolit se vážou na receptor GC-C na luminálním povrchu střevního epitelu. Je prokázáno, že linaklotid svým působením na GC-C snižuje viscerální bolest a zvyšuje pasáž zažívacím traktem na zvířecích modelech a zvyšuje pasáž tračníkem u člověka. V důsledku aktivace GC-C se zvyšují koncentrace cyklického guanosinmonofosfátu (cGMP), a to extracelulárně i intracelulárně. Extracelulární cGMP snižuje aktivitu nervových vláken vedoucích bolest, což má za následek snížení viscerální bolesti na zvířecích modelech. Intracelulární cGMP způsobuje sekreci chloridů a bikarbonátů do střevního lumen, a to aktivací transmembránového regulátoru vodivosti (CFTR), což vede ke zvýšení množství tekutin ve střevě a ke zrychlení střevní pasáže.
Farmakodynamické účinky
Ve studii zkřížených interakcí s jídlem byl přípravek Constella podáván 18 zdravým subjektům v dávce 290 mikrogramů po dobu 7 dnů, a to jak nalačno, tak po jídle. Při podání přípravku Constella ihned po vysokotučné snídani docházelo k tvorbě častější a řidší stolice a také k více nežádoucím příhodám v zažívacím traktu v porovnání s podáním nalačno.
Klinická účinnost a bezpečnost
Účinnost linaklotidu byla stanovena ve dvou randomizovaných, dvojitě zaslepených, placebem kontrolovaných klinických studiích fáze 3 u pacientů s IBS-C. V jedné klinické studii (studii 1) bylo 802 pacientů léčeno přípravkem Constella v dávce 290 mikrogramů nebo placebem jednou denně po dobu 26 týdnů. Ve druhé studii (studii 2) bylo 800 pacientů léčeno po dobu 12 týdnů a poté byla provedena další randomizace k následné léčbě trvající 4 týdny. Během 2týdenní vstupní fáze před léčbou měli pacienti průměrné skóre bolesti břicha 5,6 (na stupnici 0-10) s 2,2 % dnů bez bolesti břicha, se středním skóre nadýmání 6,6 (na stupnici 0-10) a s průměrným počtem 1,8 spontánních stolic (spontaneous bowel movements, SBM) za týden.
Charakteristika populace pacientů zařazených do klinických hodnocení fáze 3 byla následující: střední věk 43,9 let [rozmezí 18-87 let, přičemž 5,3 % bylo starších 65 let]; 90,1 % žen. Všichni pacienti splňovali kritéria Rome II pro IBS-C a požadavek hlášení skóre bolesti břicha > 3 na číselné hodnotící stupnici 0-10 bodů (kritéria odpovídají populaci se středně těžkou až těžkou formou IBS), < 3 úplné spontánní stolice a < 5 SBM za týden během 2týdenní vstupní fáze.
Ko-primární cílové parametry v obou klinických studiích zahrnovaly počet pacientů, u kterých došlo ke značné nebo úplné úlevě po dobu nejméně 50 % trvání léčby (tzv. responder stupně úlevy IBS) a dále pak počet pacientů, u kterých došlo ke zlepšení o 30 % nebo více po dobu nejméně 50 % trvání léčby (tzv. responder bolesti břicha/břišního diskomfortu). Oba tyto ko-primámí cílové parametry byly vyhodnoceny po 12 týdnech léčby.
Ve studii 1 se podle dat shromážděných za 12 týdnů projevila u 39 % pacientů léčených linaklotidem (v porovnání s 17 % pacientů léčených placebem) odpověď v oblasti stupeň úlevy IBS (p < 0,0001) a u 54 % pacientů léčených linaklotidem (v porovnání s 39 % pacientů léčených placebem) se projevila odpověď v oblasti bolest bncha/bnšní diskomfort (p < 0,0001). Ve studii 2 se u 37 % pacientů léčených linaklotidem (v porovnání s 19 % pacientů léčených placebem) projevila odpověď v oblasti stupeň úlevy IBS (p < 0,0001) a u 55 % pacientů léčených linaklotidem (v porovnání s 42 % pacientů léčených placebem) se projevila odpověď v oblasti bolest bncha/bnšní diskomfort (p < 0,0002).
Ve studii 1 se podle dat shromážděných za 26 týdnů projevila u 37 % pacientů léčených linaklotidem (v porovnání s 17 % pacientů léčených placebem) odpověď v oblasti stupeň úlevy IBS (p < 0,0001) a u 54 % pacientů léčených linaklotidem (v porovnání s 36 % pacientů léčených placebem) se projevila odpověď v oblasti bolest bncha/bnšní diskomfort (p = 0,0001).
V obou studiích bylo toto zlepšení pozorováno v průběhu 1. týdne a zůstalo zachováno po celou dobu léčby (obrázky 1 a 2). Ukázalo se, že u linaklotidu nedochází k tzv. rebound efektu (zhoršení příznaků po vysazení léku) po ukončení 3měsíční kontinuální léčby.
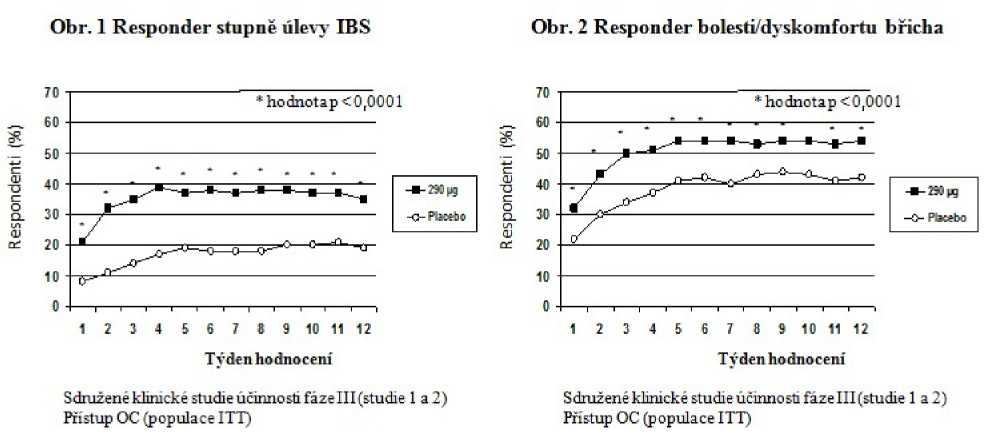
Ostatní známky a příznaky IBS-C včetně nadýmání, frekvence úplné spontánní stolice (complete spontaneous bowel movement, CSBM), úsilí, konzistence stolice se zlepšily u pacientů léčených linaklotidem v porovnání s pacienty léčenými placebem (p < 0,0001), jak ukazuje následující tabulka. Tohoto účinku bylo dosaženo v 1. týdnu a zůstal zachován po celou dobu léčby.
Vliv přípravku Constella na příznaky IBS-C během prvních 12 týdnů léčby ve sdružených klinických studiích účinnosti fáze 3 (studie 1 a 2) __
|
Hlavní sekundární parametry účinnosti |
Placebo (N = 797) |
Linaklotid (N = 805) | |||||
|
Výchozí hodnota, průměr |
12 týdnů, průměr |
Změna oproti výchozí hodnotě, průměr |
Výchozí hodnota, průměr |
12 týdnů, průměr |
Změna oproti výchozí hodnotě, průměr |
Průměrný rozdíl LS | |
|
Nadýmání (11bodová NRS) |
6,5 |
5,4 |
-1,0 |
6,7 |
4,6 |
-1,9 |
-0,9* |
|
CSBM/týden |
0,2 |
1,0 |
0,7 |
0,2 |
2,5 |
2,2 |
1,6* |
|
Konzistence stolice (skóre BSFS) |
2,3 |
3,0 |
0,6 |
2,3 |
4,4 |
2,0 |
1,4* |
|
Úsilí (5bodová ordinální stupnice) |
3,5 |
2,8 |
- 0,6 |
3,6 |
2,2 |
-1,3 |
-0,6* |
*p < 0,0001; linaklotid vs placebo. LS: nejmenší čtverec CSBM: úplná spontánní stolice
Léčba linaklotidem také způsobila významné zlepšení výsledků ve validovaných dotaznících hodnotících kvalitu života (IBS-QoL; p < 0,0001) a EuroQoL (p = 0,001), specifických pro dané onemocnění. Klinicky významná odpověď v celkovém skóre IBS-QoL (rozdíl > 14 bodů) byla dosažena u 54 % pacientů léčených linaklotidem v porovnání s 39 % pacienty léčenými placebem.
Pediatrická _ populace
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky klinických studií s přípravkem Constella u jedné nebo více podskupin pediatrické populace s funkční zácpou. Informace o použití u dětí viz bod 4.2.
5.2 Farmakokinetické vlastnosti
Absorpce
Linaklotid je obecně minimálně detekovatelný v plazmě po podání terapeutických perorálních dávek, a proto nelze vypočítat standardní farmakokinetické parametry.
Po jednotlivých dávkách až 966 mikrogramů a opakovaných dávkách až 290 mikrogramů linaklotidu nebyly zjistitelné plazmatické koncentrace mateřské složky ani aktivního metabolitu (des-tyrosin). Po podání 2 897 mikrogramů 8. den, po 7denním podávání 290 mikrogramů/den, byl linaklotid detekovatelný pouze u 2 z 18 subjektů v koncentracích dosahujících pouze těsně nad dolní limit stanovitelnosti 0,2 ng/ml (rozsah koncentrací byl od 0,212 do 0,735 ng/ml). Ve dvou pivotních studiích fáze 3, ve kterých bylo pacientům podáváno 290 mikrogramů linaklotidu jednou denně, byl linaklotid detekován pouze u 2 ze 162 pacientů přibližně 2 hodiny po počáteční dávce linaklotidu (koncentrace byly 0,241 ng/ml až 0,239 ng/ml); po 4 týdnech léčby nebyl detekován u žádného ze 162 léčených pacientů. Aktivní metabolit nebyl detekován u žádného ze 162 pacientů v žádném časovém okamžiku.
Distribuce v organismu
Vzhledem k tomu, že linaklotid je po podání terapeutických dávek vzácně detekovatelný v plazmě, nebyly provedeny standardní studie distribuce. Předpokládá se, že systémová distribuce linaklotidu je nulová nebo zanedbatelná.
Biotransformace
Linaklotid je metabolizován lokálně v gastrointestinálním traktu na svůj primární aktivní metabolit, des-tyrosin. V gastrointestinálním traktu probíhá redukce a enzymatická proteolýza linaklotidu i aktivního metabolitu des-tyrosinu na menší peptidy a přirozeně se vyskytující aminokyseliny.
In vitro byla zkoumána potenciální inhibiční aktivita linaklotidu a jeho aktivního primárního metabolitu MM-419447 na lidské efluxní transportéry BCRP, MRP2, MRP3 a MRP4 a na lidské přenašečové proteiny OATP1B1, OATP1B3, OATP2B1, PEPT1 a OCTN1. Výsledky této studie ukázaly, že ani peptid není inhibitorem běžných efluxních transportérů a přenašečových proteinů studovaných v klinicky relevantních koncentracích.
In vitro byl zkoumán vliv linaklotidu a jeho metabolitů na inhibici běžných střevních enzymů (CYP2C9 a CYP3A4) a jaterních enzymů (CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 a 3A4) nebo na indukci jaterních enzymů (CYP1A2, 2B6 a 3A4/5). Výsledky těchto studií ukázaly, že linaklotid a jeho metabolit des-tyrosin nejsou inhibitory ani induktory enzymatického systému cytochromu P450.
Eliminace z organismu
Po jedné perorální dávce 2 897 mikrogramů linaklotidu 8. den, po 7denním podávání
290 mikrogramů/den, 18 zdravým dobrovolníkům bylo přibližně 3-5 % dávky vyloučeno stolicí;
prakticky vše byl aktivní metabolit des-tyrosin.
Věk a pohlaví
Klinické studie ke zjištění vlivu věku a pohlaví na klinické farmakokinetické vlastnosti linaklotidu nebyly provedeny vzhledem k jeho vzácné detekovatelnosti v plazmě. Není pravděpodobné, že by pohlaví mělo jakýkoli vliv na dávkování. Informace související s věkem naleznete v bodech 4.2, 4.4 a 4.8.
Porucha funkce ledvin
Přípravek Constella nebyl hodnocen u pacientů s poruchou funkce ledvin. Linaklotid je vzácně detekovatelný v plazmě, proto se nepředpokládá, že by porucha funkce ledvin ovlivňovala clearance mateřské složky nebo jejích metabolitů.
Porucha funkce jater
Přípravek Constella nebyl hodnocen u pacientů s poruchou funkce jater. Linaklotid je vzácně detekovatelný v plazmě a není metabolizován jaterními enzymy cytochromu P450, a proto se nepředpokládá, že by porucha funkce jater ovlivňovala clearance mateřského léku nebo jeho metabolitu.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti, toxicity po opakovaném podávání, genotoxicity, hodnocení kancerogenního potenciálu a reprodukční a vývojové toxicity neodhalily žádné zvláštní riziko pro člověka.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Obsah tobolky Mikrokrystalická celulóza Hypromelóza
Dihydrát chloridu vápenatého Leucin
Obal tobolky Oxid titaničitý (E171)
Želatina
Červený oxid železitý (E172) Žlutý oxid železitý (E172)
Inkoust
Šelak
Propylenglykol
Koncentrovaný roztok amoniaku Hydroxid draselný Oxid titaničitý (E171)
Černý oxid železitý (E172)
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
Neotevřená lahvička: 3 roky.
Po otevření lahvičky se tobolky musí použít do 18 týdnů.
6.4 Zvláštní opatření pro uchovávání
Neuchovávejte při teplotě nad 30 °C. Uchovávejte v dobře uzavřené lahvičce, aby byl přípravek chráněn před vlhkostí.
Lahvička obsahuje jednu nebo více uzavřených nádobek s obsahem silikagelu, který udržuje tobolky v suchu. Tyto nádobky ponechejte v lahvičce.
6.5 Druh obalu a obsah balení
Bílá polyethylenová lahvička (HDPE) s bezpečnostním šroubovacím uzávěrem a s dětskou pojistkou, s jednou nebo více nádobkami obsahujícími vysoušedlo (silikagel).
Velikosti balení: 10, 28, 60, 90 tobolek a multipack obsahující 112 (4x28) tobolek. Na trhu nemusí být k dispozici všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Allergan Pharmaceuticals International Limited
Clonshaugh Industrial Estate
Coolock
Dublin 17
Irsko
8. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/12/801/001 EU/1/12/801/002 EU/1/12/801/003 EU/1/12/801/004 EU/1/12/801/005
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 26. listopad 2012
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu.
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
Název a adresa výrobce
Industrias Farmacéuticas Almirall, S.A.
Ctra. Nacional II, Km. 593
08740 Sant Andreu de la Barca
Barcelona
Španělsko
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis.
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
Constella 290 mikrogramů tvrdé tobolky Linaclotidum
Jedna tobolka obsahuje linaclotidum 290 mikrogramů
Tvrdá tobolka. 10 tobolek 28 tobolek 60 tobolek 90 tobolek
Před použitím si přečtěte příbalovou informaci. Perorální podání
Uchovávejte mimo dohled a dosah dětí.
EXP
Po otevření lahvičky se tobolky musí použít do 18 týdnů.
Allergan Pharmaceuticals International Limited
Clonshaugh Industrial Estate
Coolock
Dublin 17
Irsko
EU/1/12/801/001 10 tobolek EU/1/12/801/002 28 tobolek EU/1/12/801/003 60 tobolek EU/1/12/801/004 90 tobolek
c. s.:
Výdej léčivého přípravku vázán na lékařský předpis.
constella 290 mikrogramů
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU
VNĚJŠÍ KRABIČKA OBSAHUJÍCÍ 4 x LAHVIČKU S 28 TOBOLKAMI (MULTIPACK) S „BLUE BOXEM“
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Constella 290 mikrogramů tvrdé tobolky Linaclotidum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tobolka obsahuje linaclotidum 290 mikrogramů
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Tvrdá tobolka.
112 (4x28) tobolek (multipack).
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
Po otevření lahvičky se tobolky musí použít do 18 týdnů.
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Allergan Pharmaceuticals International Limited
Clonshaugh Industrial Estate
Coolock
Dublin 17
Irsko
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/12/801/005 112 (4x28) kapslí (multipack) 13. ČÍSLO ŠARŽE
č. š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
constella 290 mikrogramů
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU
VNITŘNÍ KRABIČKA OBSAHUJÍCÍ JEDNU LAHVIČKU S 28 TOBOLKAMI (MULTIPACK)
BEZ „BLUE BOXU“_
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Constella 290 mikrogramů tvrdé tobolky Linaclotidum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tobolka obsahuje linaclotidum 290 mikrogramů
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Tvrdá tobolka.
28 tobolek. Součást multipacku, samostatně neprodejné.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
Po otevření lahvičky se tobolky musí použít do 18 týdnů.
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Allergan Pharmaceuticals International Limited
Clonshaugh Industrial Estate
Coolock
Dublin 17
Irsko
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/12/801/005 112 (4x28) kapslí (multipack) 13. ČÍSLO ŠARŽE
č. š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
constella 290 mikrogramů
Constella 290 mikrogramů tvrdé tobolky Linaclotidum
Jedna tobolka obsahuje linaclotidum 290 mikrogramů
Tvrdá tobolka. 10 tobolek 28 tobolek 60 tobolek 90 tobolek
Před použitím si přečtěte příbalovou informaci. Perorální podání
Uchovávejte mimo dohled a dosah dětí.
EXP
Allergan Pharmaceuticals International Limited
Clonshaugh Industrial Estate
Coolock
Dublin 17
Irsko
EU/1/12/801/001 10 tobolek EU/1/12/801/002 28 tobolek EU/1/12/801/003 60 tobolek EU/1/12/801/004 90 tobolek EU/1/12/801/005 112 (4x28) tobolek (multipack)
c. s.:
Výdej léCivého přípravku vázán na lékařský předpis.
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro pacienta
Constella 290 mikrogramů tvrdé tobolky
Linaclotidum
'VTento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně tuto příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechejte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Constella a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Constella užívat
3. Jak se přípravek Constella užívá
4. Možné nežádoucí účinky
5. Jak přípravek Constella uchovávat
6. Obsah balení a další informace
1. Co je Constella a k čemu se používá K čemu se přípravek Constella používá
Přípravek Constella obsahuje léčivou látku linaklotid. Používá se u dospělých pacientů k léčbě příznaků středně těžké až těžké formy syndromu dráždivého tračníku (často nazývaného jen „IBS“) se zácpou.
IBS je častá porucha střev. Hlavní příznaky IBS se zácpou jsou následující:
• bolest v oblasti žaludku nebo břicha,
• pocit nadmutí,
• málo častá, tvrdá, málo objemná nebo „bobkovitá“ stolice.
Tyto příznaky se mohou u jednotlivých osob lišit.
Jak Constella funguje
Constella působí lokálně ve střevě a pomáhá snižovat bolest a nadýmání a obnovovat normální funkci střev. Přípravek se nevstřebává do těla, ale váže se na receptor zvaný guanylátcykláza C na povrchu střeva. Přípravek po navázání na tento receptor blokuje pocit bolesti a umožňuje pronikání tekutiny z těla do střeva, čímž se stolice naředí a zvýší se pohyb střev.
2. Čemu musíte věnovat pozornost, než začnete přípravek Constella užívat Neužívejte přípravek Constella
- jestliže jste alergický(á) na linaklotid nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6),
- jestliže je Vám nebo Vašemu lékaři známo, že máte překážku pasáže v žaludku nebo ve střevě. Upozornění a opatření
Váš lékař Vám předepsal tento lék poté, co vyloučil jiná onemocnění (zvláště onemocnění střev) a usoudil, že trpíte IBS doprovázeným zácpou. Protože tato jiná onemocnění mohou mít stejné příznaky jako IBS, je důležité, abyste svému lékaři neprodleně nahlásil(a) jakékoli změny nebo nepravidelný výskyt příznaků.
Pokud se u Vás vyskytne závažný nebo déletrvající průjem (častá vodnatá stolice po dobu 7 dnů nebo déle), přestaňte přípravek Constella užívat a poraďte se se svým lékařem (viz bod 4). Pijte dostatečné množství tekutin, abyste nahradil(a) ztrátu vody a elektrolytů, jako je draslík, vzniklou průjmem.
Pokud se u Vás vyskytne krvácení z konečníku, uvědomte svého lékaře.
Se zvláštní opatrností postupujte, je-li Vám více než 65 let, protože existuje vyšší riziko, že dostanete průjem.
Se zvláštní opatrností postupujte také v případě, pokud se u Vás vyskytne závažný nebo déletrvající průjem a pokud trpíte dalším onemocněním (jako je například vysoký krevní tlak nebo diabetes) nebo jste v minulosti prodělal(a) srdeční onemocnění nebo onemocnění cév (například pokud jste prodělal(a) srdeční příhodu (infarkt)).
Pokud se u Vás vyskytne zánětlivé onemocnění střev (například Crohnova choroba nebo ulcerózní kolitida), poraďte se se svým lékařem, protože přípravek Constella není u těchto pacientů doporučován.
Děti a dospívající
Tento lék nepodávejte dětem ani dospívajícím do 18 let věku, protože bezpečnost a účinnost přípravku Constella u této věkové skupiny ještě nebyla stanovena.
Další léčivé přípravky a přípravek Constella
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
■ Máte-li silný nebo dlouhotrvající průjem, může být snížená účinnost některých léků, jako například:
- Perorální antikoncepce. Máte-li velmi silný průjem, může se stát, že nemusí správně fungovat antikoncepční pilulky, a proto se doporučuje použít další metodu antikoncepce. Přečtěte si pokyny v příbalové informaci pro pacienty k antikoncepčním pilulkám, které užíváte.
- Léky vyžadující pečlivé a přesné dávkování, jako je například levothyroxin (hormon používaný k léčbě snížené funkce štítné žlázy).
■ Některé léky, pokud se užívají spolu s přípravkem Constella, mohou zvyšovat riziko vzniku průjmu; patří k nim například:
- Léky používané k léčbě žaludečních vředů nebo nadměrného vylučování žaludeční kyseliny, zvané inhibitory protonové pumpy.
- Léky k léčbě bolesti a zánětů, zvané NSAID (nesteroidní protizánětlivé léky).
- Laxativa (projímadla).
Constella s jídlem
Je-li přípravek Constella podáván s jídlem, způsobuje častější stolice a průjem (řidší stolice), než když je podáván nalačno (viz bod 3).
Těhotenství a kojení
O účincích přípravku Constella u těhotných a kojících žen jsou k dispozici pouze omezené údaje.
Neužívejte tento léčivý přípravek, pokud jste těhotná, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, pokud Vám to nedoporučí Váš lékař.
Pokud kojíte, neužívejte přípravek Constella, pokud Vám to nedoporučí lékař.
Řízení dopravních prostředků a obsluha strojů
Constella nebude mít žádný vliv na schopnost řídit nebo obsluhovat stroje.
3. Jak se přípravek Constella užívá
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Doporučená dávka přípravku j e jedna tobolka j ednou denně. T obolka se má užívat nej méně 30 minut před jídlem.
Jestliže jste užil(a) více přípravku Constella než jste měl(a)
Nejpravděpodobnějším účinkem při předávkování přípravkem Constella je průjem. Pokud jste užil(a) příliš velké množství tohoto léčivého přípravku, poraďte se se svým lékařem nebo lékárníkem.
Jestliže jste zapomněl(a) užít přípravek Constella
Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku. Pouze užijte následující dávku v plánovaném čase a pokračujte jako obvykle.
Jestliže jste přestal(a) užívat přípravek Constella
Před ukončením léčby se doporučuje poradit se s lékařem. Léčbu přípravkem Constella však lze bezpečně kdykoli ukončit.
Máte-li jakékoli další otázky, týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Velmi časté nežádoucí účinky (mohou se vyskytnout u více než 1 osoby z 10)
• Průjem
Průjem je obvykle krátkodobý, pokud se však u Vás vyskytne závažný nebo déletrvající průjem (častá vodnatá stolice po dobu 7 dnů nebo déle) a pociťujete točení hlavy, závratě nebo slabost, přestaňte přípravek Constella užívat a poraďte se se svým lékařem.
Časté nežádoucí účinky (mohou se vyskytnout až u 1 osoby z 10)
• Bolest v oblasti žaludku nebo břicha
• Pocit nadmutí
• Plynatost
• Střevní chřipka (virová gastroenteritida)
• Pocity závrati
Méně časté nežádoucí účinky (mohou se vyskytnout až u 1 osoby ze 100)
• Mimovolný odchod stolice (inkontinence stolice)
• Naléhavé nutkání na stolici
• Pocit točení hlavy při náhlém postavení
• Dehydratace (nedostatek vody v organismu)
• Nízká hladina draslíku v krvi
• Snížená chuť k jídlu
• Krvácení z konečníku
• Krvácení ze střev nebo konečníku včetně krvácení z hemoroidů
• Nevolnost
• Zvracení
Vzácné nežádoucí účinky (mohou se vyskytnout až u 1 osoby z 1 000)
• Snížený obsah hydrogenuhličitanu (bikarbonátu) v krvi Nežádoucí účinky s neznámou četností
• Vyrážka
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Constella uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na vnějším obalu a na lahvičce za „EXP“. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Po otevření lahvičky se tobolky musí použít do 18 týdnů.
Neuchovávejte při teplotě nad 30 °C. Uchovávejte v dobře uzavřené lahvičce, aby byl přípravek chráněn před vlhkostí.
Varování: Lahvička obsahuje jednu nebo více uzavřených nádobek obsahujících silikagel, který udržuje tobolky v suchu. Tyto nádobky ponechejte v lahvičce. Nepolykejte je._
Nepoužívejte tento léčivý přípravek, pokud zpozorujete jakékoli známky poškození lahvičky nebo jakoukoli změnu vzhledu tobolek.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co přípravek Constella obsahuje
- Léčivou látkou je linaclotidum. Jedna tobolka obsahuje 290 mikrogramů linaklotidu.
- Dalšími složkami jsou:
Obsah tobolky: mikrokrystalická celulóza, hypromelóza, dihydrát chloridu vápenatého a leucin.
Obal tobolky: červený oxid železitý (E172), oxid titaničitý (E171), žlutý oxid železitý (E172) a želatina.
Inkoust: šelak, propylenglykol, koncentrovaný roztok amoniaku, hydroxid draselný, oxid titaničitý (E171) a černý oxid železitý (E172).
Jak Constella vypadá a co obsahuje toto balení
Constella tobolky jsou tvrdé neprůhledné tobolky bílé až bělavo-oranžové barvy, označené údajem „290“ šedým inkoustem.
Jsou zabaleny v bílé polyethylenové lahvičce (HDPE) s bezpečnostním šroubovacím uzávěrem a s dětskou pojistkou, s jednou nebo více nádobkami obsahujícími vysoušedlo (silikagel).
Constella je k dispozici v baleních obsahujících 10, 28, 60 nebo 90 tobolek a multipacku 112 tobolek obsahujícím 4 krabičky po 28 tobolkách. Na trhu nemusí být k dispozici všechny velikosti balení.
Držitel rozhodnutí o registraci
Allergan Pharmaceuticals International Limited
Clonshaugh Industrial Estate
Coolock
Dublin 17
Irsko
Výrobce
Industrias Farmacéuticas Almirall, S.A.
Ctra. Nacional II, Km. 593,
E-08740 Sant Andreu de la Barca, Barcelona Španělsko
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgie/Belgique/Belgien/Luxembourg/Luxemburg/ Nederland
Allergan n.v
Tél/Tel: +32 (0)2 351 2424
Ireland/Malta/ United Kingdom
Allergan Ltd
Tel: + 44 (0) 1628 494026 Bt^rapnn
AaepraH Etarapua EOOfl
Ten.: +359 (0) 800 20 280
Island
Actavis ehf.
Sími: +354 550 3300
Česká republika
Allergan CZ s.r.o.
Tel: +420 800 188 818
Italia
Allergan S.p.A Tel: + 39 06 509 561
Danmark/ Norge/ Suomi/Finland/Sverige
Allergan Norden AB Tlf/Puh/Tel: + 46 (0)8 594 100 00
Magyarország
Allergan Hungary Kft.
Tel.: +36 80 100 101
Deutschland Pharm-Allergan GmbH Tel: + 49 69 92038-10
Eesti
Allergan Baltics UAB Tel: + 372 56955144
Latvija
Allergan Baltics UAB Tel: +371 27331152
Lietuva
Allergan Baltics UAB Tel: +370 62660247
Osterreich
Pharm-Allergan GmbH Tel: +43 1 99460 6355
Polska
Allergan Sp. z o.o.
Tel: +48 22 256 37 00
EXXáSa/Knnpoq
Allergan Hellas Pharmaceuticals S.A.
Tni: +30 210 74 73 300
Portugal
Profarin Lda.
Tel: + 351 21 425 3242
Espaňa
Allergan S.A.
Tel: + 34 91 807 6130
Románia
Allergan S.R.L.
Tel: +40 21 301 53 02
France
Allergan France SAS Tél: +33 (0)1 49 07 83 00
Slovenija
Ewopharma d.o.o.
Tel: + 386 (0) 590 848 40
Hrvatska
Ewopharma d.o.o.
Tel: +385 1 6646 563
Slovenská republika
Allergan SK s.r.o.
Tel: +421 800 221 223
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu.
31