Cometriq 20 Mg
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
COMETRIQ 20 mg tvrdé tobolky
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna tvrdá tobolka obsahuje cabozantinibi malas, který je ekvivalentní s cabozantinibum 20 mg. Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Tvrdé tobolky.
Tvrdé tobolky jsou šedé s černým vytištěným nápisem „XL184 20mg“ na těle tobolky. Tobolka obsahuje bílý až téměř bílý prášek.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
COMETRIQ je indikován k léčbě dospělých pacientů s progresivním, inoperabilním lokálně pokročilým nebo metastatickým medulárním karcinomem štítné žlázy.
U pacientů, u kterých není znám stav mutace RET (Rearranged during Transfection) nebo je negativní, se před individuálním rozhodnutím o léčbě musí zohlednit možnost nižšího přínosu (viz důležité informace v bodech 4.4 a 5.1).
4.2 Dávkování a způsob podání
Léčbu přípravkem COMETRIQ by měl zahájit lékař, který má zkušenosti s podáváním protinádorových léčivých přípravků.
Dávkování
Doporučená dávka přípravku COMETRIQ je 140 mg jednou denně užitá jako jedna 80mg oranžová tobolka a tři 20mg šedé tobolky. Léčba má trvat, dokud pacient nepřestane vykazovat klinický přínos z léčby nebo se nevyskytne nepřijatelná toxicita.
Je potřebné očekávat, že většina pacientů léčených přípravkem COMETRIQ bude z důvodu toxicity vyžadovat jednu nebo více úprav dávky (snížení a/nebo přerušení). Proto mají být pacienti během prvních osmi týdnů léčby pečlivě sledováni (viz bod 4.4).
Léčba suspektních nežádoucích účinků si může vyžádat dočasné přerušení léčby přípravkem COMETRIQ a/nebo snížení jeho dávky. Pokud je snížení dávky nevyhnutelné, doporučuje se ji nejdřív snížit na 100 mg denně užitých ve formě jedné 80mg oranžové tobolky a jedné 20mg šedé tobolky, a potom na 60 mg denně, užitých ve formě tří 20mg šedých tobolek.
Přerušení dávkování se doporučuje při léčbě toxicity 3. nebo vyššího stupně podle CTCAE nebo nezvladatelné toxicity 2. stupně.
Snížit dávku se doporučuje při takových příhodách, které by se v případě přetrvávání mohly stát závažnými nebo neúnosnými.
Kvůli možnému výskytu většiny příhod na začátku léčby je důležité, aby lékař během prvních osmi týdnů léčby pozorně sledoval stav pacienta s cílem stanovit, zda je potřebné dávku upravit. Příhody, které se obvykle projeví na začátku léčby, zahrnují hypokalcémii, hypokalémii, trombocytopenii, hypertenzi, syndrom palmoplantární erytrodysestézie (PPES) a gastrointestinální (GI) příhody (bolest břicha a ústní dutiny, zánět sliznic, zácpa, průjem, zvracení).
Výskyt některých závažných nežádoucích účinků (jako například gastrointestinální fistuly) může záviset na kumulativní dávce a mohou se vyskytnout v pozděj ší fázi léčby.
Pokud pacient vynechá dávku a do další zbývá méně než 12 hodin, vynechaná dávka se nemá užít.
Konkomitantní léčivé _přípravky
Konkomitantní léčivé přípravky, které jsou silnými inhibitory CYP3A4, mají být užívány s opatrností, a je potřeba se vyhnout chronickému používání konkomitantních léčivých přípravků, které jsou silnými induktory CYP3A4 (viz body 4.4 a 4.5).
Je potřebné zvážit výběr alternativních konkomitantních léčivých přípravků, které nemají žádný nebo jen minimální potenciál indukovat či inhibovat CYP3A4.
Starší pacienti
Při použití cabozantinibu u starších pacientů (> 65 let) se nedoporučuje žádná specifická úprava dávky. Avšak u pacientů ve věku 75 let a starších byla pozorována tendence vyššího výskytu závažných nežádoucích příhod (SAE).
Rasa
Zkušenosti s cabozantinibem u jiných než bělošských pacientů jsou malé.
Pacienti s poruchou funkce ledvin
U pacientů s mírnou až středně závažnou poruchou funkce ledvin se má cabozantinib používat s opatrností.
Cabozantinib se nedoporučuje používat u pacientů se závažnou poruchou funkce ledvin, protože u této populace nebyly dosud stanoveny bezpečnost a účinnost.
Pacienti s poruchou funkce jater
U pacientů s mírnou až středně závažnou poruchou funkce jater je doporučená dávka cabozantinibu 60 mg jednou denně. Je nutné sledování výskytu nežádoucích příhod a podle potřeby úprava dávky nebo přerušení užívání léku (viz bod 4.2). Cabozantinib se nedoporučuje u pacientů se závažnou poruchou funkce jater, protože u této populace nebyly dosud stanoveny bezpečnost a účinnost.
Pacienti se srdečními _poruchami
O pacientech se srdečními poruchami jsou jen omezené údaje. K dávkování není možné dát žádná specifická doporučení.
Pediatrická populace
Bezpečnost a účinnost cabozantinibu u dětí ve věku < 18 let nebyla dosud stanovena. Nejsou dostupné žádné údaje.
Způsob podání
Tobolky se mají spolknout celé a neotevřené. Pacienti mají být poučeni, aby nic nejedli alespoň 2 hodiny před a hodinu po užití přípravku COMETRIQ.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
3
4.4 Zvláštní upozornění a opatření pro použití
V pivotním klinickém hodnocení se u pacientů léčených cabozantinibem vyskytlo snížení dávky u 79 % a přerušení podávání dávky u 72 % pacientů. U 41 % pacientů bylo potřebné dávku snížit dvakrát. Střední čas do prvního snížení dávky byl 43 dní a do prvního přerušení dávky 33 dní. Proto mají být pacienti během prvních osmi týdnů léčby pečlivě sledováni (viz bod 4.2).
Perforace, fistuly a intraabdominální abscesy
Při používání cabozantinibu byly pozorovány závažné, v některých případech fatální perforace gastrointestinálního traktu a fistuly a intrabdominální abscesy. Před začátkem léčby cabozantinibem je potřeba pečlivě zhodnotit a následně pozorně sledovat zdravotní stav pacientů, kteří v nedávné době podstoupili radioterapii, mají zánětlivé onemocnění střev (např. Crohnovu chorobu, ulcerózní kolitidu, peritonitidu nebo divertikulitidu), mají infiltrace tumoru do trachey, bronchů nebo jícnu, kteří mají komplikace z předcházejícího chirurgického zákroku v GIT (zejména pokud jsou spojeny s prodlouženým nebo neúplným hojením) nebo mají komplikace způsobené předcházející radiační terapií v oblasti hrudníku (včetně mediastina). Je potřebné sledovat, zda se u nich nevyskytnou příznaky perforací a fistul. Pokud se po začátku léčby vyskytne mukozitida, musí se dle potřeby vyloučit jiné než gastrointestinální fistuly. Cabozantinib se musí vysadit u pacientů s GI perforací nebo fistulí v gastrointestinálním traktu nebo mimo něj.
Tromboembolické příhody
Při používání cabozantinibu byly pozorovány příhody venózního tromboembolizmu a příhody arteriálního tromboembolizmu. Cabozantinib se má používat s opatrností u pacientů s rizikem těchto příhod nebo u pacientů, kteří mají takové příhody v anamnéze. Podávání cabozantinibu se má přerušit u pacientů, u kterých došlo k akutnímu infarktu myokardu nebo k jiné klinicky signifikantní arteriální tromboembolické komplikaci.
Hemoragie
Při používaní cabozantinibu byla pozorována hemoragie. Před začátkem léčby cabozantinibem je potřeba pečlivě posoudit zdravotní stav pacientů s prokázaným postižením trachey a bronchů tumorem nebo s hemoptýzou v anamnéze. Cabozantinib se nesmí podávat pacientům se závažnou hemoragií nebo nedávnou hemoptýzou.
Komplikace s hojením ran
Při používání cabozantinibu byly pozorovány komplikace s hojením ran. Pokud je to možné, má se léčba cabozantinibem přerušit alespoň na 28 dní před plánovanou operací. Rozhodnutí o pokračování v léčbě cabozantinibem se má učinit na základě klinického posouzení adekvátnosti hojení rány. Cabozantinib se má přestat podávat pacientům s komplikacemi hojení rány, které si vyžadují lékařský zásah.
Hypertenze
Při používání cabozantinibu byla pozorována hypertenze. Všechny pacienty je potřeba sledovat, zda se u nich nevyskytne hypertenze, a podle potřeby ji léčit standardní antihypertenzní terapií. Pokud hypertenze přetrvává i přes použití antihypertenziv, má se snížit dávka cabozantinibu. Pokud je hypertenze závažná a přetrvává i přes nasazení antihypertenzní léčby a snížení dávky cabozantinibu, musí se cabozantinib vysadit. V případě hypertenzní krize se má cabozantinib vysadit.
Osteonekróza
Při použití cabozantinibu byly pozorovány případy osteonekrózy čelisti (ONJ). Před začátkem léčby cabozantinibem a pravidelně během léčby se má vyšetřovat ústní dutina. Pacienti mají být poučeni o postupech při vykonávání ústní hygieny. Pokud je to možné, při invazivních dentálních procedurách se má cabozantinib vysadit alespoň na 28 dní před plánovanou operací. U pacientů, kteří užívají léčiva spojovaná s ONJ jako například bisfosfonáty, je potřebná opatrnost. U pacientů s ONJ se má užívání cabozantinibu přerušit.
Syndrom palmoplantámí ervtrodvsestézie
Při používání cabozantinibu byl pozorován syndrom palmoplantámí erytrodysestézie (PPES).
V případě závažného PPES by se mělo zvážit přerušení léčby cabozantinibem. Léčba s nižší dávkou cabozantinibu by měla být znovu zahájena až po zlepšení PPES na stupeň 1.
Proteinurie
Při používání cabozantinibu byla pozorována proteinurie. Během léčby cabozantinibem se mají pravidelně sledovat proteiny v moči. Pokud se u pacienta vyvine nefrotický syndrom, cabozantinib se musí přestat podávat.
Syndrom reverzibilní posteriorní leukoencefalopatie
Při používání cabozantinibu byl pozorován syndrom reverzibilní posteriorní leukoencefalopatie (RPLS) známý i jako syndrom posteriorní reverzibilní encefalopatie (PRES). Pacientům s RPLS se má cabozantinib přestat podávat.
Prodloužení QT intervalu
Cabozantinib se má používat s opatrností u pacientů s prodloužením QT intervalu v anamnéze, u pacientů užívajících antiarytmika nebo u pacientů s relevantním preexistujícím srdečním onemocněním, bradykardií nebo výkyvy hladin elektrolytů. Během používání cabozantinibu se má zvážit pravidelné sledování EKG a elektrolytů (sérového vápníku, draslíku a hořčíku). Konkomitantní léčbu silnými inhibitory CYP3A4, které mohou zvýšit plazmatické koncentrace cabozantinibu, je nutné používat opatrně.
Induktory a inhibitory CYP3A4
Cabozantinib je substrátem CYP3A4. Souběžné podávání cabozantinibu se silným inhibitorem CYP3A4 ketokonazolem vedlo ke zvýšení plazmatické expozice cabozantinibu. Vyžaduje se opatrnost při podávání cabozantinibu spolu se silnými inhibitory CYP3A4. Souběžné podávání cabozantinibu se silným induktorem CYP3A4 rifampicinem vedlo ke snížení plazmatické expozice cabozantinibu.
Proto je nutné se vyhnout dlouhodobému podávání léčiv, které jsou silnými induktory CYP3A4 (viz body 4.2 a 4.5).
Substráty P-glykoproteinu
Cabozantinib byl inhibitorem (IC50 = 7,0 gM), ale ne substrátem, P-glykoproteinových (P-gp) transportních aktivit v dvousměrném testovacím systému, který používal MDCK-MDR1 buňky. Cabozantinib proto může mít potenciál zvýšit plazmatické koncentrace souběžně podaných substrátů P-gp. Při užívání cabozantinibu musejí být pacienti upozorněni na užívání substrátů P-gp (např. fexofenadin, aliskiren, ambrisentan, dabigatran etexilát, digoxin, kolchicin, maravirok, posakonazol, ranolazin, saxagliptin, sitagliptin, talinolol, tolvaptan).
Inhibitory MRP2
Podávání inhibitorů MRP2 může způsobit zvýšení plazmatických koncentrací cabozantinibu. Proto se má k souběžnému podávání inhibitorů MRP2 (napr. cyklosporin, efavirenz, emtricitabin) přistupovat s opatrností.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Účinek jiných léčivých přípravků na cabozantinib
Inhibitory a induktory CYP3A4
Podávání silného inhibitoru CYP3A4 ketokonazolu (400 mg denně po dobu 27 dní) zdravým dobrovolníkům snížilo hodnotu clearance cabozantinibu (o 29 %) a zvýšilo plazmatickou expozici cabozantinibu po jednorázové dávce (AUC) o 38 %. Proto se má k souběžnému podávání silných inhibitorů CYP3A4 (např. ritonavir, itrakonazol, erythromycin, klarithromycin, grapefruitový džus) s cabozantinibem přistupovat opatrně.
Podávání silného induktoru CYP3A4 rifampicinu (600 mg denně po dobu 31 dní) zdravým dobrovolníkům zvýšilo hodnotu clearance cabozantinibu (4,3krát) a snížilo plazmatickou expozici
5
cabozantinibu (AUC) o 77 %. Je potřeba se vyhnout chronickému souběžnému podávání induktorů CYP3A4 (např. fenytoinu, karbamazepinu, rifampicinu, fenobarbitalu nebo rostlinným přípravkům obsahujícím třezalku tečkovanou [Hypericum perforatum]) s cabozantinibem.
Látky měnící žaludeční pH
Souběžné podání inhibitoru protonové pumpy (PPI) esomeprazolu (40 mg denně po dobu 6 dní) spolu s jednou dávkou cabozantinibu 100 mg zdravým dobrovolníkům nevedlo ke klinicky signifikantním účinkům na plazmatickou expozici cabozantinibu (AUC). Při podávání látek měnících žaludeční pH (tj. PPI, antagonistů H2 receptorů a antacid) souběžně s cabozantinibem není indikována úprava dávky.
Inhibitory MRP2
Data in vitro prokázala, že cabozantinib je substrátem MRP2. Podávání inhibitorů MRP2 proto může způsobit zvýšení plazmatických koncentrací cabozantinibu.
Sekvestranty žlučových kyselin
Sekvestranty žlučových kyselin, jako například cholestyramin a cholestagel, mohou interagovat s cabozantinibem a mohou ovlivnit absorpci (nebo reabsorpci), což může vést ke snížené expozici (viz bod 5.2). Klinický význam těchto potenciálních interakcí není znám.
Účinek cabozantinibu na jiné léčivé přípravky
Účinek cabozantinibu na farmakokinetiku antikoncepčních steroidů se nezjišťoval. Protože není možné garantovat nezměněný antikoncepční účinek, doporučuje se používat další antikoncepční metodu, jako například bariérovou.
Substráty P-glykoproteinu
Cabozantinib byl inhibitorem (IC50 = 7,0 pM), ale ne substrátem, P-gp transportních aktivit v dvousměrném testovacím systému, který používal buňky MDCK-MDR1. Proto může mít cabozantinib potenciál zvýšit plazmatické koncentrace souběžně podaných substrátů P-gp. Při užívaní cabozantinibu musí být pacienti upozorněni na užívání substrátů P-gp (např. fexofenadin, aliskiren, ambrisentan, dabigatran etexilát, digoxin, kolchicin, maravirok, posakonazol, ranolazin, saxagliptin, sitagliptin, talinolol, tolvaptan).
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku/Antikoncepce u mužů a žen
Ženy ve fertilním věku musejí být poučeny, aby předcházely těhotenství, dokud užívají cabozantinib. Partnerky pacientů užívajících cabozantinib se musejí také vyhnout těhotenství. Účinné metody antikoncepce musejí používat pacienti i pacientky a jejich partnerky/partneři během léčby a alespoň po dobu 4 měsíců po ukončení léčby. Protože není možné považovat perorální antikoncepci za „účinnou metodu antikoncepce“, je nutné ji používat současně s další metodou, jako například bariérovou (viz bod 4.5).
Studie s těhotnými ženami užívajícími cabozantinib nebyly provedeny. Studie na zvířatech ukázaly embryofetální a teratogenní účinky (viz bod 5.3). Potenciální riziko pro člověka není známé. Cabozantinib se nesmí používat během těhotenství kromě případů, kdy klinický stav ženy vyžaduje léčbu cabozantinibem.
Kojení
Není známo, zda se cabozantinib a/nebo jeho metabolity vylučují do mateřského mléka. Kvůli možnému poškození dítěte musí matka kojení přerušit během léčby cabozantinibem a alespoň po dobu 4 měsíců po ukončení léčby.
Fertilita
Nejsou údaje o fertilitě u člověka. Při neklinických bezpečnostních hodnoceních se zjistilo, že fertilita mužů i žen může být zhoršená léčbou cabozantinibem (viz bod 5.3). Muži i ženy musejí být poučeni, aby vyhledali odborníka a před léčbou zvážili zachovaní fertility.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Cabozantinib má malý vliv na schopnost řídit nebo obsluhovat stroje. S cabozantinibem se však pojí nežádoucí účinky jako únava a slabost. Proto se doporučuje opatrnost při řízení a obsluze strojů.
4.8 Nežádoucí účinky
Shrnutí bezpečnostního profilu
Nejčastějšími závažnými nežádoucími účinky spojenými s cabozantinibem jsou pneumonie, zánět sliznic, hypokalcémie, dysfagie, dehydratace, plicní embolie a hypertenze. Nejčastější nežádoucí účinky všech stupňů (postihující alespoň 20 % pacientů) zahrnují průjem, PPES, úbytek tělesné hmotnosti, snížení chuti k jídlu, nauzeu, únavu, dysgeuzii, změny barvy vlasů, hypertenzi, stomatitidu, zácpu, zvracení, zánět sliznic, astenii a dysfonii.
Nej častějšími laboratorními abnormalitami byla zvýšená aspartátaminotransferáza (AST), zvýšená alaninaminotransferáza (ALT), zvýšená alkalická fosfatáza (ALP), lymfopenie, hypokalcémie, neutropenie, trombocytopenie, hypofosfatémie, hyperbilirubinémie, hypomagnezémie a hypokalémie.
Shrnutí nežádoucích účinků v tabulce
Nežádoucí účinky jsou uvedeny v Tabulce 1 podle MedDRA tříd orgánových systémů a frekvence výskytu. Frekvence jsou založeny na všech stupních a jsou definovány následovně: velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1 000 až < 1/100). V každé skupině četností jsou nežádoucí účinky řazeny v pořadí podle klesající závažnosti.
Tabulka 1: Nežádoucí účinky hlášené při užívání cabozantinibu
|
Třída orgánových systémů podle MedDRA |
Velmi časté (>1/10) |
Časté (>1/100 až <1/10) |
Méně časté (>1/1 000 až <1/100) |
|
Infekce a infestace |
abscesy (včetně viscerálních, kožních, zubních), pneumonie, folikulitida, mykotické infekce (včetně kožní, orální a genitální) |
aspergilom | |
|
Endokrinní poruchy |
hypotyreóza | ||
|
Poruchy metabolismu a výživy |
snížená chuť k jídlu, hypokalcémie, hypofosfatémie, hyperbilirubinémie, hypokalémie, hypomagnezémie |
dehydratace, hypalbuminémie | |
|
Psychiatrické poruchy |
abnormální sny, delirium |
|
Třída orgánových systémů podle MedDRA |
Velmi časté (>1/10) |
Časté (>1/100 až <1/10) |
Méně časté (>1/1 000 až <1/100) |
|
Poruchy nervového systému |
dysgeuzie, bolest hlavy, závratě |
periferní neuropatie, parestezie, ageuzie, třes |
ataxie, poruchy pozornosti, hepatální encefalopatie, ztráta vědomí, poruchy řeči, tranzitorní ischemická ataka, syndrom posteriorní reverzibilní encefalopatie |
|
Poruchy oka |
rozmazané vidění |
katarakta, konjunktivitida | |
|
Poruchy ucha a labyrintu |
bolest ucha, tinitus |
hypakuzie | |
|
Srdeční poruchy |
atriální fibrilace |
angina pectoris, supraventrikulární | |
|
Cévní poruchy |
hypertenze |
hypotenze, žilní trombóza, bledost, chladné končetiny |
arteriální trombóza |
|
Respirační, hrudní a mediastinální poruchy |
dysfonie, orofaryngeální bolest |
píštěl mimo gastrointestinálního traktu (včetně tracheální, pneumomediastinální, tracheo-ezofageální), plicní embolie, krvácení do dýchacích cest (včetně pulmonárních, bronchiálních, tracheálních), plicní aspirace |
atelektáze, faryngeální edém, pneumonitida |
|
Gastrointestinální poruchy |
průjem, nauzea, stomatitida, zácpa, zvracení, bolest břicha, dyspepsie, dysfagie, glosodynie |
gastrointestinální perforace, gastrointestinální krvácení, pankreatitida, hemeroidy, anální fisura, zánět konečníku, cheilitida |
gastrointestinální píštěl, ezofagitida |
|
Poruchy jater a žlučových cest |
cholelitiáza | ||
|
Poruchy kůže a podkožní tkáně |
palmoplantární erytrodysestézie, změny barvy vlasů, vyrážka, suchá pokožka, alopecie, erytém |
hyperkeratóza, akné, puchýř, abnormální růst vlasů, exfoliace kůže, snížení pigmentace kůže |
kožný vřed, telangiektázie |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
artralgie, svalové křeče |
muskuloskeletální bolest hrudníku, osteonekróza čelisti |
rabdomyolýza |
|
Třída orgánových systémů podle MedDRA |
Velmi časté (>1/10) |
Časté (>1/100 až <1/10) |
Méně časté (>1/1 000 až <1/100) |
|
Poruchy ledvin a močových cest |
proteinurie, dysurie, hematurie |
akutní selhání funkce ledvin | |
|
Poruchy reprodukčního systému a prsu |
amenorea, vaginální krvácení | ||
|
Celkové poruchy a reakce v místě aplikace |
únava, zánět sliznic, astenie |
zhoršené hojení ran, zimnice, otok tváře |
cysta, bolest v tváři, lokalizovaný otok |
|
Vyšetření |
snížená tělesná hmotnost, zvýšená hladina ALT, AST a ALP v séru, zvýšená hladina LDH v krvi, zvýšená hladina TSH v krvi, lymfopenie, neutropenie, trombocytopenie |
zvýšená hladina kreatininfosfokinázy v krvi |
zkrácení aktivovaného parciálního tromboplastinového času, zvýšený počet eozinofilů, zvýšený počet krevných destiček |
Popis vybraných nežádoucích reakcí
Zvýšená hodnota tyreotropního hormonu (TSH) po první dávce byla pozorována u 57 % pacientů na cabozantinibu versus 19 % pacientů na placebu (bez ohledu na výchozí hodnoty). Devadesát dva procent pacientů v ramenu s cabozantinibem předtím podstoupilo tyreoidektomii a 89 % užívalo hormony štítné žlázy před první dávkou.
Dvacátý devátý den (nikoli první den) po zahájení léčby cabozantinibem (dávkou 140 mg jednou denně) bylo v kontrolované klinické studii s pacienty s nádorovým onemocněním pozorováno zvýšení o 10-15 ms v porovnání s výchozím stavem v korigovaném QT intervalu podle Fridericia (QTcF). Tento účinek nebyl spojen se změnou morfologie tvaru srdeční křivky nebo s novým rytmem. Žádný subjekt léčený cabozantinibem neměl QTcF > 500 ms.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Neexistuje specifická léčba předávkování cabozantinibem a nebyly stanoveny možné příznaky předávkování.
V případě podezření na předávkování se musí cabozantinib vysadit a začít s podpůrnou léčbou. Metabolické klinické laboratorní parametry je třeba monitorovat alespoň v týdenních intervalech, nebo na základě klinických požadavků, aby bylo možné hodnotit jejich případný vývoj. Nežádoucí účinky spojené s předávkováním se mají léčit symptomaticky.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: cytostatika, inhibitory proteinkinázy, ATC kód: L01XE26
Mechanismus účinku
Cabozantinib je malá molekula, která inhibuje víc receptorových tyrozinkináz (RTKs) zapojených do růstu tumoru a angiogeneze, patologického remodelování kostí a metastatické progrese nádorového onemocnění. U cabozantinibu se hodnotila jeho inhibiční aktivita proti různým kinázám a byl identifikovaný jako inhibitor MET (receptorový protein růstového faktoru hepatocytů) a VEGF (růstový faktor vaskulárního endotelu) receptorů. Cabozantinib navíc inhibuje jiné tyrozinkinázy včetně RET, receptoru GAS6 (AXL), receptoru pro faktor kmenových buněk (KIT) a FLT3 (Fms-like tyrosine kinase-3).
Farmakodynamické účinky
Cabozantinib vykazuje na dávce závislou inhibici růstu tumoru, regresi tumoru a/nebo inhibuje metastázy v širokém spektru předklinických modelů tumorů.
Účinnost cabozantinibu byla pozorována u pacientů s medulárním karcinomem štítné žlázy bez mutací (divokého typu) nebo s mutací RET.
Klinické údaje o medulárním karcinomu štítné žlázy
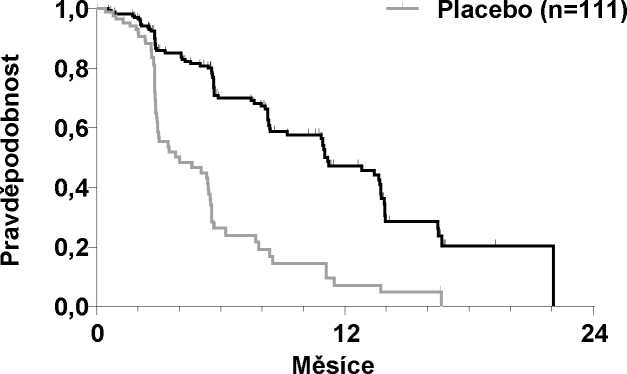
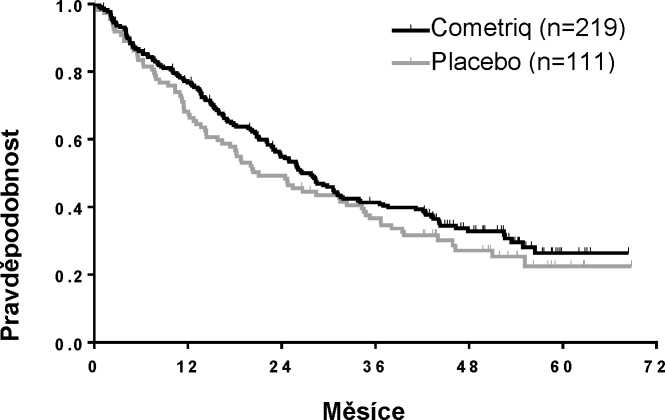
Multicentrická randomizovaná dvojitě zaslepená studie porovnávající cabozantinib (N = 219) s placebem (N = 111) byla provedena s pacienty s inoperabilním lokálně pokročilým nebo metastatickým MTC (medulárním karcinomem štítné žlázy) a zdokumentovanou radiograficky prokázanou progresí choroby během 14 měsíců před zařazením do studie. Primárním cílem bylo porovnat přežívání bez progrese (PFS) u pacientů užívajících cabozantinib v porovnání s pacienty užívajícími placebo. Sekundárními cíli bylo porovnání výskytu celkové odpovědi (ORR) a celkového přežití (OS). Centralizované, zaslepené, nezávislé hodnocení údajů ze zobrazovacích postupů bylo použito při hodnocení PFS a ORR. Pacienti byli léčeni, dokud nedošlo k progresi choroby nebo k nepřijatelné toxicitě.
Výsledky PFS analýzy, které vycházejí z hodnocení RECIST, vykázaly statisticky signifikantní rozdíl v době PFS s cabozantinibem v porovnání s placebem: medián této doby byl 11,2 měsíce u pacientů v rameni s cabozantinibem v porovnání se 4,0 měsíci u pacientů v rameni s placebem (stratifikovaný poměr rizik [HR] = 0,28; 95% IS: 0,19, 0,40; p < 0,0001; Obrázek 1). Výsledky PFS byly konzistentní napříč všemi hodnocenými podskupinami podle výchozího stavu a demografických kritérií včetně podskupiny s předcházející terapií inhibitory tyrozinkinázy (která mohla zahrnovat léčiva působící na dráhy související s anti-angiogenezí), podskupin podle stavu RET mutací (včetně subjektů se zdokumentovanou absencí RET mutací), podskupin podle předcházející protinádorové léčby nebo radioterapie nebo existence kostních metastáz.
ORR byl 27,9 % a 0 % u pacientů v rameni s cabozantinibem a rameni s placebem (p < 0,0001; Tabulka 2). Medián trvání objektivních odpovědí byl 14,6 měsíce (95% IS: 11,1; 17,5) u pacientů v rameni s cabozantinibem.
Cometriq (n=219)
|
Počet pacientů s rizikem | ||||||||
|
Měsíc |
0 |
3 |
6 |
9 |
12 |
15 |
18 |
21 |
|
Cometriq |
219 |
121 |
78 |
55 |
31 |
12 |
2 |
1 |
|
Placebo |
111 |
35 |
11 |
6 |
3 |
2 |
0 |
0 |
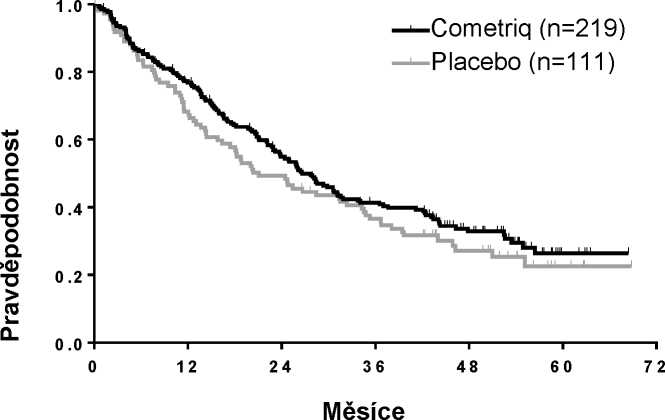
Závěrečná analýza OS vykonaná potom, co nastalo 218 příhod (úmrtí), ukázala tendenci k zvýšení v mediánu přežití o 5,5 měsíce v rameni s cabozantinibem: medián (měsíce) 26,6 cabozantinib versus
21,1 placebo (HR = 0,85 [95% IS: 0,64, 1,12], p = 0,2409).
Obrázek 2: Kaplan-Mayerova křivka celkového přežití
Tabulka 2: Shrnutí klíčových zjištění účinnosti
|
Cabozantinib |
Placebo | |
|
Medián přežití bez progrese |
11,2 měsíce |
4,0 měsíce |
|
HR: 0,28 (0,19, 0,40) p < 0,0001 | ||
|
Medián celkového přežití |
26,6 měsíce |
21,1 měsíce |
|
HR: 0,85 (0,64, 1,12) p = 0,2409 | ||
|
Výskyt celkové odpovědia (95% IS) |
27,9 % (21,9 %, 34,5 %) |
0 % |
|
p < 0,0001 | ||
|
Trvání odpovědi; Medián (95% IS) |
14,6 měsíce (11,1, 17,5) |
N/A |
|
Výskyt kontroly onemocnění b (95% IS) |
55,3 % (48,3 %, 62,2 %) |
13,5 % (7,6 %, 21,6 %) |
|
Kalcitoninová odpověďa |
47 % (49/104)c |
3 % (1/40) c |
|
CEA odpověďa |
33 % (47/143) c |
2 % (1/55)c |
a Odpověď = CR + PR
b Výskyt kontroly onemocnění = SD + ORR
c Zahrnuje pacienty, u kterých byla odpověď hodnotitelná
Stav mutací RET
Z 215 subjektů s dostatečnými údaji na určení stavu mutace bylo 78,6 % (n = 169) klasifikováno jako pozitivní na mutaci RET (z kterých bylo 126 pozitivních na mutaci M918T) a 21,4 % (n = 46) bylo klasifikováno jako negativní na mutaci RET. U dalších 115 subjektů nebylo možno určit stav mutací RET nebo byl tento stav nejasný. Všechny tři podskupiny vykazovaly zvýšení PFS v rameni s cabozantinibem v porovnání s ramenem s placebem (HR byl v podskupině s pozitivní RET mutací 0,23, v podskupině s negativní RET mutací 0,53 a v podskupině s neznámým stavem RET mutací 0,30). Výskyt objektivní odpovědi měřený v těchto podskupinách byl obvykle konzistentní s PFS výsledky, přičemž výskyt reakce nádoru byl 32 % v podskupině s pozitivní mutací RET, 22 % v podskupině s negativní RET mutací a 25 % v podskupině s neznámým stavem této mutace.
Další genetické analýzy ukázaly, že malý podíl pacientů nese somatické mutace tumoru v HRAS, KRAS nebo NRAS. Tito pacienti (n = 16) vykazovali signifikantní prodloužení PFS (HR 0,15) a výskyt objektivní odpovědi 31 %. Pacienti s negativními RET mutacemi bez důkazu mutace RAS (n = 33) ukázali při užívaní cabozantinibu snížený přínos v PFS (HR 0,87) a nižší výskyt odpovědi, 18 %, v porovnání s podskupinami s jinými mutacemi.
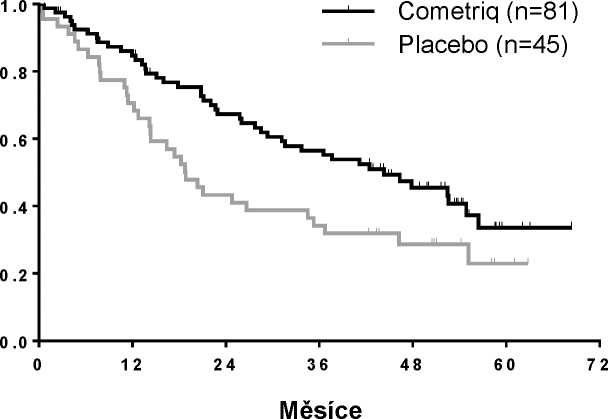
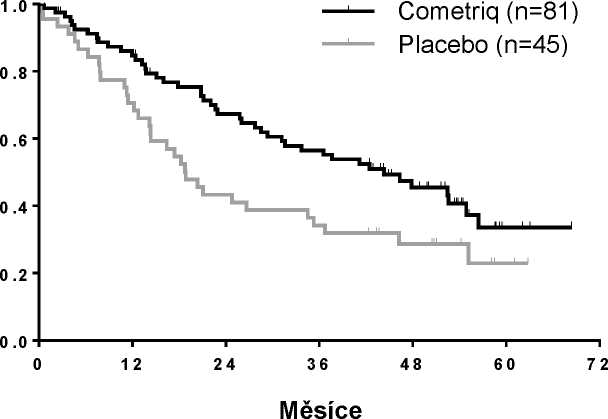
Signifikantní zlepšení v OS bylo pozorováno v podskupině pacientů s pozitivním stavem mutace RET M918T (n=81/219 rameno s cabozantinibem): 44,3 měsíců v rameni s cabozantinibem versus
18,9 měsíců v rameni s placebem (HR = 0,60, p = 0,0255). Nebylo zjištěno žádné zlepšení v OS v podskupinách s negativním a neznámým stavem mutace RET M918T.
Pediatrická populace
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s cabozantinibem u jedné nebo více podskupin pediatrické populace pro léčbu maligních solidních tumorů (informace o použití u dětí viz bod 4.2).
Tento léčivý přípravek byl registrován postupem tzv. podmíněného schválení. Znamená to, že j sou očekávány další důkazy o jeho přínosech.
Evropská agentura pro léčivé přípravky nejméně jednou za rok vyhodnotí nové informace o tomto léčivém přípravku a tento souhrn údajů o přípravku bude podle potřeby aktualizován.
5.2 Farmakokinetické vlastnosti
Absorpce
Po perorálním podání cabozantinibu se jeho maximální koncentrace v plazmě dosáhne 2 až 5 hodin po podání dávky. Profily plazmatické koncentrace v čase ukázaly druhou maximální absorpci přibližně 24 hodin po podání, což naznačuje možnost, že cabozantinib podstupuje enterohepatální recirkulaci.
Podávání opakované denní dávky cabozantinibu 140 mg po dobu 19 dní vedlo k přibližně 4- až 5násobné průměrné akumulaci cabozantinibu (založené na hodnotách AUC) v porovnání s podáním jednorázové dávky; rovnovážný stav byl dosažen přibližně 15. den.
Jídlo bohaté na tuky mírně zvýšilo hodnoty Cmax a AUC (41 % a 57 %) v porovnání se stavem na lačno u zdravých dobrovolníků, kterým byl cabozantinib podán v jednorázové perorální dávce 140 mg. Informace o přesném účinku jídla požitého hodinu po podání cabozantinibu nejsou k dispozici.
Distribuce
Cabozantinib je in vitro v lidské plazmě vysoce vázaný na proteiny (> 99,7 %). Na základě farmakokinetického (FK) populačního modelu byl stanoven distribuční objem (V/F) přibližně 349 l (SE: ± 2,73 %). U pacientů s mírnou až středně závažnou poruchou funkce ledvin nebo jater nebyla změněna vazba na proteiny.
Biotransformace
Cabozantinib byl metabolizován in vivo. V plazmě byly přítomny čtyři metabolity s expozicí (AUC) vyšší než 10 % AUC mateřské látky: XL184-N-oxid, rozkladný produkt XL184-amid, hydroxosíran XL184 a rozkladný produkt 6-desmethyl-amid síran. Oba nekonjugované metabolity (XL184-N-oxid a rozkladný produkt XL184-amid), které mají < 1 % schopnosti inhibice cílové kinázy v porovnání s mateřským cabozantinibem, představují < 10 % celkové plazmatické expozice spojené s léčivem.
Cabozantinib je in vitro substrátem CYP3A4 metabolismu jako neutralizační protilátka CYP3A4 inhibující tvorbu metabolitu XL184 N-oxidu o > 80 % při inkubaci katalyzované NADPH v lidských jaterních mikrozomech; naproti tomu neutralizační protilátky CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C19, CYP2D6 a CYP2E1 nemají žádný účinek na tvorbu metabolitů cabozantinibu. Neutralizační protilátky CYP2C9 vykazovaly minimální účinek na tvorbu metabolitů cabozantinibu (tj. < 20% snížení).
Eliminace
Terminální plazmatický eliminační poločas cabozantinibu ve studiích s jednorázovým podáním zdravým dobrovolníkům byl přibližně 120 hodin. Průměrná hodnota clearance (CL/F) v rovnovážném stavu u pacientů s nádorovým onemocněním byla v populační FK analýze odhadnuta na 4,4 l/hod. Během 48denního období sběru po jednorázové dávce 14C-cabozantinibu zdravým dobrovolníkům bylo zachyceno přibližně 81 % celkové podané radioaktivity, přičemž 54 % v stolici a 27 % v moči.
Farmakokinetika u zvláštních populací pacientů
Porucha funkce ledvin
Výsledky studie s pacienty s poruchou funkce ledvin ukazují, že poměry geometrického průměru cabozantinibu v plazmě metodou nejmenších čtverců, Cmax a AUC0-mf byly o 19 % a 30 % vyšší u pacientů s mírnou poruchou funkce ledvin (90% IS pro Cmax od 91,60 % do 155,51 %; AUC0-mf 98,79 % do 171,26 %) a o 2 % a 6-7 % vyšší pacientů se středně závažnou poruchou funkce ledvin (90% IS pro Cmax 78,64 % do 133,52 %; AUC0-mf 79,61 % až 140,11 %) než u pacientů s normální funkcí ledvin. Pacienti se závažnou poruchou funkce ledvin nebyli předmětem zkoumání.
Porucha _ funkce _ jater
Výsledky studie s pacienty s poruchou funkce jater ukazují, že expozice_(AUC0-mf) se zvyšuje o 81 % u pacientů s mírnou poruchou funkce jater a o 63 % u pacientů se středně závažnou poruchou funkce jater (90% IS pro AUC0-mf: 121,44 % až 270,34 % pro mírné a 107,37 % až 246,67 % pro středně závažné poruchy). Pacienti se závažnou poruchou funkce jater nebyli předmětem zkoumání.
Rasa
Nejsou k dispozici žádné údaje na určení rozdílů ve FK podmíněné rasou.
5.3 Předklinické údaje vztahující se k bezpečnosti
Nežádoucí účinky, které nebyly pozorovány v klinických studiích, avšak vyskytly se ve studiích na zvířatech při systémové expozici podobné expozici při klinickém podávání, a které mohou být důležité pro klinické použití:
Ve studiích toxicity po opakované dávce u potkanů a psů v délce trvání až 6 měsíců byly cílovými orgány toxicity gastrointestinální trakt, kostní dřeň, lymfoidní tkáně, ledviny, tkáně nadledvin a reprodukčního systému. Hodnota, po které nebyly pozorovány žádné nepříznivé účinky (NOAEL), byla při těchto zjištěních nižší než hladiny klinické expozice u člověka při zamýšlené terapeutické dávce.
Ve standardní sadě genotoxických testů nevykazoval cabozantinib žádný mutagenní nebo klastogenní potenciál. Studie kancerogenity nebyly provedeny.
Studie fertility u potkanů ukázaly sníženou samčí i samičí plodnost. Navíc po expozicích nižších než hladiny klinické expozice u člověka při určené terapeutické dávce byla u samců psů pozorována hypospermatogeneze.
Studie embryofetálního vývoje byly provedeny u potkanů a králíků. U potkanů cabozantinib způsoboval postimplantační ztráty, fetální edém, rozštěp patra/rtů, dermální aplazii a deformovaný nebo rudimentární chvost. U králíků cabozantinib způsoboval změny fetálních měkkých tkání (zmenšená velikost sleziny, malé nebo chybějící střední laloky plic) a zvýšenou fetální incidenci celkových malformací. NOAEL při embryo-fetální toxicitě a teratogenních zjištěních byly nižší než hladiny klinické expozice u člověka při zamýšlené terapeutické dávce.
Mláďata potkanů (porovnatelné s pediatrickou populací >2 roky), kterým byl podán cabozantinib, vykazovala zvýšené hodnoty leukocytů, sníženou hematopoézu, pubescentní/nevyzrálý samičí reprodukční systém (bez opožděného vaginálního otevírání), abnormality zubů, snížený obsah minerálů v kostech a sníženou kostní denzitu, pigmentaci jater a hyperplazii žlučovodu. Nálezy na děloze/vaječnících a snížení hematopoézy se zdály být přechodné, zatímco účinky na kostní parametry a pigmentaci jater byly trvalé. Hodnocení mláďat potkanů (porovnatelné s pediatrickou populací <2 roky) nebylo provedeno.
6. FARMACEUTICKÉ INFORMACE
6.1 Seznam pomocných látek
Obsah tobolky mikrokrystalická celulóza sodná sůl kroskarmelózy sodná sůl karboxymethylškrobu koloidní bezvodý oxid křemičitý kyselina stearová
Obal tobolky želatina
černý oxid železitý (E172) oxid titaničitý (E171)
Inkoust na potisk šelak
černý oxid železitý (E172) propylenglykol
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
3 roky.
6.4 Zvláštní opatření pro uchovávání
Neuchovávejte při teplotě nad 25°C.
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
6.5 Druh obalu a obsah balení
PVC/PE/PCTFE-Al blistry s fólií na zadní straně, zatavené do sekundárního kartového obalu uzavřeného teplem.
Blistrové karty obsahují:
21 x 20 mg tobolek (při dávce 60 mg/den je to zásoba na 7 dní)
Balení na 28 dní obsahuje:
84 tobolek (4 blistrové karty 21 x 20 mg) (při dávce 60 mg/den je to zásoba na 28 dní)
6.6 Zvláštní opatření pro likvidaci přípravku
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
TMC Pharma Services Ltd.
Lodge Farm Barn Elvetham Park Estate Fleet Road Hartley Wintney Hampshire RG27 8AS Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/13/890/001 21 x 20 mg tobolek (při dávce 60 mg/den je to zásoba na 7 dní)
EU/1/13/890/004 84 tobolek (4 blistrové karty 21 x 20 mg) (při dávce 60 mg/den je to zásoba na
28 dní)
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 21. března 2014
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
COMETRIQ 20 mg tvrdé tobolky COMETRIQ 80 mg tvrdé tobolky
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna tvrdá tobolka obsahuje cabozantinibi malas, který je ekvivalentní s cabozantinibum 20 mg nebo 80 mg.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Tvrdé tobolky.
Tvrdé tobolky jsou šedé s černým vytištěným nápisem „XL184 20mg“ na těle tobolky. Tobolka obsahuje bílý až téměř bílý prášek.
Tvrdé tobolky jsou oranžové s černým vytištěným nápisem „XL184 80mg“ na těle tobolky. Tobolka obsahuje bílý až téměř bílý prášek.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
COMETRIQ je indikován k léčbě dospělých pacientů s progresivním, inoperabilním lokálně pokročilým nebo metastatickým medulárním karcinomem štítné žlázy.
U pacientů, u kterých není znám stav mutace RET (Rearranged during Transfection) nebo je negativní, se před individuálním rozhodnutím o léčbě musí zohlednit možnost nižšího přínosu (viz důležité informace v bodech 4.4 a 5.1).
4.2 Dávkování a způsob podání
Léčbu přípravkem COMETRIQ by měl zahájit lékař, který má zkušenosti s podáváním protinádorových léčivých přípravků.
Dávkování
Doporučená dávka přípravku COMETRIQ je 140 mg jednou denně užitá jako jedna 80mg oranžová tobolka a tři 20mg šedé tobolky. Léčba má trvat, dokud pacient nepřestane vykazovat klinický přínos z léčby nebo se nevyskytne nepřijatelná toxicita.
Je potřebné očekávat, že většina pacientů léčených přípravkem COMETRIQ bude z důvodu toxicity vyžadovat jednu nebo více úprav dávky (snížení a/nebo přerušení). Proto mají být pacienti během prvních osmi týdnů léčby pečlivě sledováni (viz bod 4.4).
Léčba suspektních nežádoucích účinků si může vyžádat dočasné přerušení léčby přípravkem COMETRIQ a/nebo snížení jeho dávky. Pokud je snížení dávky nevyhnutelné, doporučuje se ji nejdřív snížit na 100 mg denně užitých ve formě jedné 80mg oranžové tobolky a jedné 20mg šedé tobolky, a potom na 60 mg denně, užitých ve formě tří 20mg šedých tobolek.
Přerušení dávkování se doporučuje při léčbě toxicity 3. nebo vyššího stupně podle CTCAE nebo nezvladatelné toxicity 2. stupně.
Snížit dávku se doporučuje při takových příhodách, které by se v případě přetrvávání mohly stát závažnými nebo neúnosnými.
Kvůli možnému výskytu většiny příhod na začátku léčby je důležité, aby lékař během prvních osmi týdnů léčby pozorně sledoval stav pacienta s cílem stanovit, zda je potřebné dávku upravit. Příhody, které se obvykle projeví na začátku léčby, zahrnují hypokalcémii, hypokalémii, trombocytopenii, hypertenzi, syndrom palmoplantární erytrodysestézie (PPES) a gastrointestinální (GI) příhody (bolest břicha a ústní dutiny, zánět sliznic, zácpa, průjem, zvracení).
Výskyt některých závažných nežádoucích účinků (jako například gastrointestinální fistuly) může záviset na kumulativní dávce a mohou se vyskytnout v pozdější fázi léčby.
Pokud pacient vynechá dávku a do další zbývá méně než 12 hodin, vynechaná dávka se nemá užít.
Konkomitantní léčivé přípravky
Konkomitantní léčivé přípravky, které jsou silnými inhibitory CYP3A4, mají být užívány s opatrností, a je potřeba se vyhnout chronickému používání konkomitantních léčivých přípravků, které jsou silnými induktory CYP3A4 (viz body 4.4 a 4.5).
Je potřebné zvážit výběr alternativních konkomitantních léčivých přípravků, které nemají žádný nebo jen minimální potenciál indukovat či inhibovat CYP3A4.
Starší _ pacienti
Při použití cabozantinibu u starších pacientů (> 65 let) se nedoporučuje žádná specifická úprava dávky. Avšak u pacientů ve věku 75 let a starších byla pozorována tendence vyššího výskytu závažných nežádoucích příhod (SAE).
Rasa
Zkušenosti s cabozantinibem u jiných než bělošských pacientů jsou malé.
Pacienti s poruchou funkce ledvin
U pacientů s mírnou až středně závažnou poruchou funkce ledvin se má cabozantinib používat s opatrností.
Cabozantinib se nedoporučuje používat u pacientů se závažnou poruchou funkce ledvin, protože u této populace nebyly dosud stanoveny bezpečnost a účinnost.
Pacienti s _poruchou _funkce _jater
U pacientů s mírnou až středně závažnou poruchou funkce jater je doporučená dávka cabozantinibu 60 mg jednou denně. Je nutné sledování výskytu nežádoucích příhod a podle potřeby úprava dávky nebo přerušení užívání léku (viz bod 4.2). Cabozantinib se nedoporučuje u pacientů se závažnou poruchou funkce jater, protože u této populace nebyly dosud stanoveny bezpečnost a účinnost.
Pacienti se srdečními _poruchami
O pacientech se srdečními poruchami jsou jen omezené údaje. K dávkování není možné dát žádná specifická doporučení.
Pediatrická _ populace
Bezpečnost a účinnost cabozantinibu u dětí ve věku < 18 let nebyla dosud stanovena. Nejsou dostupné žádné údaje.
Způsob podání
Tobolky se mají spolknout celé a neotevřené. Pacienti mají být poučeni, aby nic nejedli alespoň 2 hodiny před a hodinu po užití přípravku COMETRIQ.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
V pivotním klinickém hodnocení se u pacientů léčených cabozantinibem vyskytlo snížení dávky u 79 % a přerušení podávání dávky u 72 % pacientů. U 41 % pacientů bylo potřebné dávku snížit dvakrát. Střední čas do prvního snížení dávky byl 43 dní a do prvního přerušení dávky 33 dní. Proto mají být pacienti během prvních osmi týdnů léčby pečlivě sledováni (viz bod 4.2).
Perforace, fistuly a intraabdominální abscesy
Při používání cabozantinibu byly pozorovány závažné, v některých případech fatální perforace gastrointestinálního traktu a fistuly a intrabdominální abscesy. Před začátkem léčby cabozantinibem je potřeba pečlivě zhodnotit a následně pozorně sledovat zdravotní stav pacientů, kteří v nedávné době podstoupili radioterapii, mají zánětlivé onemocnění střev (např. Crohnovu chorobu, ulcerózní kolitidu, peritonitidu nebo divertikulitidu), mají infiltrace tumoru do trachey, bronchů nebo jícnu, kteří mají komplikace z předcházejícího chirurgického zákroku v GIT (zejména pokud jsou spojeny s prodlouženým nebo neúplným hojením) nebo mají komplikace způsobené předcházející radiační terapií v oblasti hrudníku (včetně mediastina). Je potřebné sledovat, zda se u nich nevyskytnou příznaky perforací a fistul. Pokud se po začátku léčby vyskytne mukozitida, musí se dle potřeby vyloučit jiné než gastrointestinální fistuly. Cabozantinib se musí vysadit u pacientů s GI perforací nebo fistulí v gastrointestinálním traktu nebo mimo něj.
Tromboembolické příhody
Při používání cabozantinibu byly pozorovány příhody venózního tromboembolizmu a příhody arteriálního tromboembolizmu. Cabozantinib se má používat s opatrností u pacientů s rizikem těchto příhod nebo u pacientů, kteří mají takové příhody v anamnéze. Podávání cabozantinibu se má přerušit u pacientů, u kterých došlo k akutnímu infarktu myokardu nebo k jiné klinicky signifikantní arteriální tromboembolické komplikaci.
Hemoragie
Při používaní cabozantinibu byla pozorována hemoragie. Před začátkem léčby cabozantinibem je potřeba pečlivě posoudit zdravotní stav pacientů s prokázaným postižením trachey a bronchů tumorem nebo s hemoptýzou v anamnéze. Cabozantinib se nesmí podávat pacientům se závažnou hemoragií nebo nedávnou hemoptýzou.
Komplikace s hojením ran
Při používání cabozantinibu byly pozorovány komplikace s hojením ran. Pokud je to možné, má se léčba cabozantinibem přerušit alespoň na 28 dní před plánovanou operací. Rozhodnutí o pokračování v léčbě cabozantinibem se má učinit na základě klinického posouzení adekvátnosti hojení rány. Cabozantinib se má přestat podávat pacientům s komplikacemi hojení rány, které si vyžadují lékařský zásah.
Hypertenze
Při používání cabozantinibu byla pozorována hypertenze. Všechny pacienty je potřeba sledovat, zda se u nich nevyskytne hypertenze, a podle potřeby ji léčit standardní antihypertenzní terapií. Pokud hypertenze přetrvává i přes použití antihypertenziv, má se snížit dávka cabozantinibu. Pokud je hypertenze závažná a přetrvává i přes nasazení antihypertenzní léčby a snížení dávky cabozantinibu, musí se cabozantinib vysadit. V případě hypertenzní krize se má cabozantinib vysadit.
Osteonekróza
Při použití cabozantinibu byly pozorovány případy osteonekrózy čelisti (ONJ). Před začátkem léčby cabozantinibem a pravidelně během léčby se má vyšetřovat ústní dutina. Pacienti mají být poučeni o postupech při vykonávání ústní hygieny. Pokud je to možné, při invazivních dentálních procedurách se má cabozantinib vysadit alespoň na 28 dní před plánovanou operací. U pacientů, kteří užívají léčiva spojovaná s ONJ jako například bisfosfonáty, je potřebná opatrnost. U pacientů s ONJ se má užívání cabozantinibu přerušit.
Syndrom palmoplantární ervtrodvsestézie
Při používání cabozantinibu byl pozorován syndrom palmoplantární erytrodysestézie (PPES).
V případě závažného PPES by se mělo zvážit přerušení léčby cabozantinibem. Léčba s nižší dávkou cabozantinibu by měla být znovu zahájena až po zlepšení PPES na stupeň 1.
Proteinurie
Při používání cabozantinibu byla pozorována proteinurie. Během léčby cabozantinibem se mají pravidelně sledovat proteiny v moči. Pokud se u pacienta vyvine nefrotický syndrom, cabozantinib se musí přestat podávat.
Syndrom reverzibilní posteriorní leukoencefalopatie
Při používání cabozantinibu byl pozorován syndrom reverzibilní posteriorní leukoencefalopatie (RPLS) známý i jako syndrom posteriorní reverzibilní encefalopatie (PRES). Pacientům s RPLS se má cabozantinib přestat podávat.
Prodloužení QT intervalu
Cabozantinib se má používat s opatrností u pacientů s prodloužením QT intervalu v anamnéze, u pacientů užívajících antiarytmika nebo u pacientů s relevantním preexistujícím srdečním onemocněním, bradykardií nebo výkyvy hladin elektrolytů. Během používání cabozantinibu se má zvážit pravidelné sledování EKG a elektrolytů (sérového vápníku, draslíku a hořčíku). Konkomitantní léčbu silnými inhibitory CYP3A4, které mohou zvýšit plazmatické koncentrace cabozantinibu, je nutné používat opatrně.
Induktorv a. inhibitory CYP3A4
Cabozantinib je substrátem CYP3A4. Souběžné podávání cabozantinibu se silným inhibitorem CYP3A4 ketokonazolem vedlo ke zvýšení plazmatické expozice cabozantinibu. Vyžaduje se opatrnost při podávání cabozantinibu spolu se silnými inhibitory CYP3A4. Souběžné podávání cabozantinibu se silným induktorem CYP3A4 rifampicinem vedlo ke snížení plazmatické expozice cabozantinibu.
Proto je nutné se vyhnout dlouhodobému podávání léčiv, které jsou silnými induktory CYP3A4 (viz body 4.2 a 4.5).
Substráty P-glykoproteinu
Cabozantinib byl inhibitorem (IC50 = 7,0 gM), ale ne substrátem, P-glykoproteinových (P-gp) transportních aktivit v dvousměrném testovacím systému, který používal MDCK-MDR1 buňky. Cabozantinib proto může mít potenciál zvýšit plazmatické koncentrace souběžně podaných substrátů P-gp. Při užívání cabozantinibu musejí být pacienti upozorněni na užívání substrátů P-gp (např. fexofenadin, aliskiren, ambrisentan, dabigatran etexilát, digoxin, kolchicin, maravirok, posakonazol, ranolazin, saxagliptin, sitagliptin, talinolol, tolvaptan).
Inhibitory MRP2
Podávání inhibitorů MRP2 může způsobit zvýšení plazmatických koncentrací cabozantinibu. Proto se má k souběžnému podávání inhibitorů MRP2 (napr. cyklosporin, efavirenz, emtricitabin) přistupovat s opatrností.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Účinek jiných léčivých přípravků na cabozantinib
Inhibitory a induktory CYP3A4
Podávání silného inhibitoru CYP3A4 ketokonazolu (400 mg denně po dobu 27 dní) zdravým dobrovolníkům snížilo hodnotu clearance cabozantinibu (o 29 %) a zvýšilo plazmatickou expozici cabozantinibu po jednorázové dávce (AUC) o 38 %. Proto se má k souběžnému podávání silných inhibitorů CYP3A4 (např. ritonavir, itrakonazol, erythromycin, klarithromycin, grapefruitový džus) s cabozantinibem přistupovat opatrně.
Podávání silného induktoru CYP3A4 rifampicinu (600 mg denně po dobu 31 dní) zdravým dobrovolníkům zvýšilo hodnotu clearance cabozantinibu (4,3krát) a snížilo plazmatickou expozici cabozantinibu (AUC) o 77 %. Je potřeba se vyhnout chronickému souběžnému podávání induktorů CYP3A4 (např. fenytoinu, karbamazepinu, rifampicinu, fenobarbitalu nebo rostlinným přípravkům obsahujícím třezalku tečkovanou [Hypericum perforatum]) s cabozantinibem.
Látky měnící žaludeční pH
Souběžné podání inhibitoru protonové pumpy (PPI) esomeprazolu (40 mg denně po dobu 6 dní) spolu s jednou dávkou cabozantinibu 100 mg zdravým dobrovolníkům nevedlo ke klinicky signifikantním účinkům na plazmatickou expozici cabozantinibu (AUC). Při podávání látek měnících žaludeční pH (tj. PPI, antagonistů H2 receptorů a antacid) souběžně s cabozantinibem není indikována úprava dávky.
Inhibitory MRP2
Data in vitro prokázala, že cabozantinib je substrátem MRP2. Podávání inhibitorů MRP2 proto může způsobit zvýšení plazmatických koncentrací cabozantinibu.
Sekvestranty žlučových kyselin
Sekvestranty žlučových kyselin, jako například cholestyramin a cholestagel, mohou interagovat s cabozantinibem a mohou ovlivnit absorpci (nebo reabsorpci), což může vést ke snížené expozici (viz bod 5.2). Klinický význam těchto potenciálních interakcí není znám.
Účinek cabozantinibu na jiné léčivé přípravky
Účinek cabozantinibu na farmakokinetiku antikoncepčních steroidů se nezjišťoval. Protože není možné garantovat nezměněný antikoncepční účinek, doporučuje se používat další antikoncepční metodu, jako například bariérovou.
Substráty P-glykoproteinu
Cabozantinib byl inhibitorem (IC50 = 7,0 pM), ale ne substrátem, P-gp transportních aktivit v dvousměrném testovacím systému, který používal buňky MDCK-MDR1. Proto může mít cabozantinib potenciál zvýšit plazmatické koncentrace souběžně podaných substrátů P-gp. Při užívaní cabozantinibu musí být pacienti upozorněni na užívání substrátů P-gp (např. fexofenadin, aliskiren, ambrisentan, dabigatran etexilát, digoxin, kolchicin, maravirok, posakonazol, ranolazin, saxagliptin, sitagliptin, talinolol, tolvaptan).
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku/Antikoncepce u mužů a žen
Ženy ve fertilním věku musejí být poučeny, aby předcházely těhotenství, dokud užívají cabozantinib. Partnerky pacientů užívajících cabozantinib se musejí také vyhnout těhotenství. Účinné metody antikoncepce musejí používat pacienti i pacientky a jejich partnerky/partneři během léčby a alespoň po dobu 4 měsíců po ukončení léčby. Protože není možné považovat perorální antikoncepci za „účinnou metodu antikoncepce“, je nutné ji používat současně s další metodou, jako například bariérovou (viz bod 4.5).
Studie s těhotnými ženami užívajícími cabozantinib nebyly provedeny. Studie na zvířatech ukázaly embryofetální a teratogenní účinky (viz bod 5.3). Potenciální riziko pro člověka není známé. Cabozantinib se nesmí používat během těhotenství kromě případů, kdy klinický stav ženy vyžaduje léčbu cabozantinibem.
Kojení
Není známo, zda se cabozantinib a/nebo jeho metabolity vylučují do mateřského mléka. Kvůli možnému poškození dítěte musí matka kojení přerušit během léčby cabozantinibem a alespoň po dobu 4 měsíců po ukončení léčby.
Fertilita
Nejsou údaje o fertilitě u člověka. Při neklinických bezpečnostních hodnoceních se zjistilo, že fertilita mužů i žen může být zhoršená léčbou cabozantinibem (viz bod 5.3). Muži i ženy musejí být poučeni, aby vyhledali odborníka a před léčbou zvážili zachovaní fertility.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Cabozantinib má malý vliv na schopnost řídit nebo obsluhovat stroje. S cabozantinibem se však pojí nežádoucí účinky jako únava a slabost. Proto se doporučuje opatrnost při řízení a obsluze strojů.
4.8 Nežádoucí účinky
Shrnutí bezpečnostního profilu
Nejčastějšími závažnými nežádoucími účinky spojenými s cabozantinibem jsou pneumonie, zánět sliznic, hypokalcémie, dysfagie, dehydratace, plicní embolie a hypertenze. Nejčastější nežádoucí účinky všech stupňů (postihující alespoň 20 % pacientů) zahrnují průjem, PPES, úbytek tělesné hmotnosti, snížení chuti k jídlu, nauzeu, únavu, dysgeuzii, změny barvy vlasů, hypertenzi, stomatitidu, zácpu, zvracení, zánět sliznic, astenii a dysfonii.
Nej častějšími laboratorními abnormalitami byla zvýšená aspartátaminotransferáza (AST), zvýšená alaninaminotransferáza (ALT), zvýšená alkalická fosfatáza (ALP), lymfopenie, hypokalcémie, neutropenie, trombocytopenie, hypofosfatémie, hyperbilirubinémie, hypomagnezémie a hypokalémie.
Shrnutí nežádoucích účinků v tabulce
Nežádoucí účinky jsou uvedeny v Tabulce 1 podle MedDRA tříd orgánových systémů a frekvence výskytu. Frekvence jsou založeny na všech stupních a jsou definovány následovně: velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1 000 až < 1/100). V každé skupině četností jsou nežádoucí účinky řazeny v pořadí podle klesající závažnosti.
Tabulka 1: Nežádoucí účinky hlášené při užívání cabozantinibu
|
Třída orgánových systémů podle MedDRA |
Velmi časté (>1/10) |
Časté (>1/100 až <1/10) |
Méně časté (>1/1 000 až <1/100) |
|
Infekce a infestace |
abscesy (včetně viscerálních, kožních, zubních), pneumonie, folikulitida, mykotické infekce (včetně kožní, orální a genitální) |
aspergilom | |
|
Endokrinní poruchy |
hypotyreóza |
|
Třída orgánových systémů podle MedDRA |
Velmi časté (>1/10) |
Časté (>1/100 až <1/10) |
Méně časté (>1/1 000 až <1/100) |
|
Poruchy metabolismu a výživy |
snížená chuť k jídlu, hypokalcémie, hypofosfatémie, hyperbilirubinémie, hypokalémie, hypomagnezémie |
dehydratace, hypalbuminémie | |
|
Psychiatrické poruchy |
abnormální sny, delirium | ||
|
Poruchy nervového systému |
dysgeuzie, bolest hlavy, závratě |
periferní neuropatie, parestezie, ageuzie, třes |
ataxie, poruchy pozornosti, hepatální encefalopatie, ztráta vědomí, poruchy řeči, tranzitorní ischemická ataka, syndrom posteriorní reverzibilní encefalopatie |
|
Poruchy oka |
rozmazané vidění |
katarakta, konjunktivitida | |
|
Poruchy ucha a labyrintu |
bolest ucha, tinitus |
hypakuzie | |
|
Srdeční poruchy |
atriální fibrilace |
angina pectoris, supraventrikulární | |
|
Cévní poruchy |
hypertenze |
hypotenze, žilní trombóza, bledost, chladné končetiny |
arteriální trombóza |
|
Respirační, hrudní a mediastinální poruchy |
dysfonie, orofaryngeální bolest |
píštěl mimo gastrointestinálního traktu (včetně tracheální, pneumomediastinální, tracheo-ezofageální), plicní embolie, krvácení do dýchacích cest (včetně pulmonárních, bronchiálních, tracheálních), plicní aspirace |
atelektáze, faryngeální edém, pneumonitida |
|
Gastrointestinální poruchy |
průjem, nauzea, stomatitida, zácpa, zvracení, bolest břicha, dyspepsie, dysfagie, glosodynie |
gastrointestinální perforace, gastrointestinální krvácení, pankreatitida, hemeroidy, anální fisura, zánět konečníku, cheilitida |
gastrointestinální píštěl, ezofagitida |
|
Poruchy jater a žlučových cest |
cholelitiáza |
|
Třída orgánových systémů podle MedDRA |
Velmi časté (>1/10) |
Časté (>1/100 až <1/10) |
Méně časté (>1/1 000 až <1/100) |
|
Poruchy kůže a podkožní tkáně |
palmoplantární erytrodysestézie, změny barvy vlasů, vyrážka, suchá pokožka, alopecie, erytém |
hyperkeratóza, akné, puchýř, abnormální růst vlasů, exfoliace kůže, snížení pigmentace kůže |
kožný vřed, telangiektázie |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
artralgie, svalové křeče |
muskuloskeletální bolest hrudníku, osteonekróza čelisti |
rabdomyolýza |
|
Poruchy ledvin a močových cest |
proteinurie, dysurie, hematurie |
akutní selhání funkce ledvin | |
|
Poruchy reprodukčního systému a prsu |
amenorea, vaginální krvácení | ||
|
Celkové poruchy a reakce v místě aplikace |
únava, zánět sliznic, astenie |
zhoršené hojení ran, zimnice, otok tváře |
cysta, bolest v tváři, lokalizovaný otok |
|
Vyšetření |
snížená tělesná hmotnost, zvýšená hladina ALT, AST a ALP v séru, zvýšená hladina LDH v krvi, zvýšená hladina TSH v krvi, lymfopenie, neutropenie, trombocytopenie |
zvýšená hladina kreatininfosfokinázy v krvi |
zkrácení aktivovaného parciálního tromboplastinového času, zvýšený počet eozinofilů, zvýšený počet krevných destiček |
Popis vybraných nežádoucích reakcí
Zvýšená hodnota tyreotropního hormonu (TSH) po první dávce byla pozorována u 57 % pacientů na cabozantinibu versus 19 % pacientů na placebu (bez ohledu na výchozí hodnoty). Devadesát dva procent pacientů v ramenu s cabozantinibem předtím podstoupilo tyreoidektomii a 89 % užívalo hormony štítné žlázy před první dávkou.
Dvacátý devátý den (nikoli první den) po zahájení léčby cabozantinibem (dávkou 140 mg jednou denně) bylo v kontrolované klinické studii s pacienty s nádorovým onemocněním pozorováno zvýšení o 10-15 ms v porovnání s výchozím stavem v korigovaném QT intervalu podle Fridericia (QTcF).Tento účinek nebyl spojen se změnou morfologie tvaru srdeční křivky nebo s novým rytmem. Žádný subjekt léčený cabozantinibem neměl QTcF > 500 ms.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Neexistuje specifická léčba předávkování cabozantinibem a nebyly stanoveny možné příznaky předávkování.
V případě podezření na předávkování se musí cabozantinib vysadit a začít s podpůrnou léčbou. Metabolické klinické laboratorní parametry je třeba monitorovat alespoň v týdenních intervalech, nebo na základě klinických požadavků, aby bylo možné hodnotit jejich případný vývoj. Nežádoucí účinky spojené s předávkováním se mají léčit symptomaticky.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: cytostatika, inhibitory proteinkinázy, ATC kód: L01XE26 Mechanismus účinku
Cabozantinib je malá molekula, která inhibuje víc receptorových tyrozinkináz (RTKs) zapojených do růstu tumoru a angiogeneze, patologického remodelování kostí a metastatické progrese nádorového onemocnění. U cabozantinibu se hodnotila jeho inhibiční aktivita proti různým kinázám a byl identifikovaný jako inhibitor MET (receptorový protein růstového faktoru hepatocytů) a VEGF (růstový faktor vaskulárního endotelu) receptorů. Cabozantinib navíc inhibuje jiné tyrozinkinázy včetně RET, receptoru GAS6 (AXL), receptoru pro faktor kmenových buněk (KIT) a FLT3 (Fms-like tyrosine kinase-3).
Farmakodynamické účinky
Cabozantinib vykazuje na dávce závislou inhibici růstu tumoru, regresi tumoru a/nebo inhibuje metastázy v širokém spektru předklinických modelů tumorů.
Účinnost cabozantinibu byla pozorována u pacientů s medulárním karcinomem štítné žlázy bez mutací (divokého typu) nebo s mutací RET.
Klinické údaje o medulárním karcinomu štítné žlázy
Multicentrická randomizovaná dvojitě zaslepená studie porovnávající cabozantinib (N = 219) s placebem (N = 111) byla provedena s pacienty s inoperabilním lokálně pokročilým nebo metastatickým MTC (medulárním karcinomem štítné žlázy) a zdokumentovanou radiograficky prokázanou progresí choroby během 14 měsíců před zařazením do studie. Primárním cílem bylo porovnat přežívání bez progrese (PFS) u pacientů užívajících cabozantinib v porovnání s pacienty užívajícími placebo. Sekundárními cíli bylo porovnání výskytu celkové odpovědi (ORR) a celkového přežití (OS). Centralizované, zaslepené, nezávislé hodnocení údajů ze zobrazovacích postupů bylo použito při hodnocení PFS a ORR. Pacienti byli léčeni, dokud nedošlo k progresi choroby nebo k nepřijatelné toxicitě.
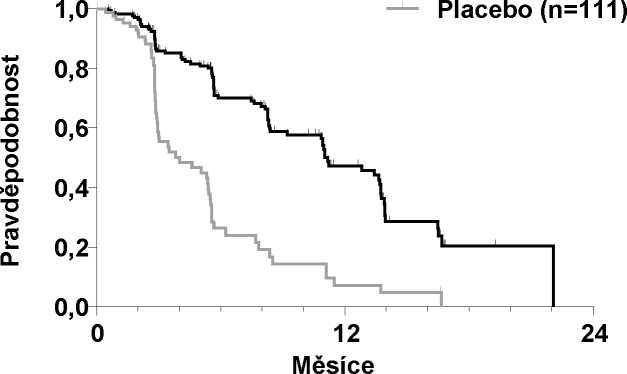
Výsledky PFS analýzy, které vycházejí z hodnocení RECIST, vykázaly statisticky signifikantní rozdíl v době PFS s cabozantinibem v porovnání s placebem: medián této doby byl 11,2 měsíce u pacientů v rameni s cabozantinibem v porovnání se 4,0 měsíci u pacientů v rameni s placebem (stratifikovaný poměr rizik [HR] = 0,28; 95% IS: 0,19, 0,40; p < 0,0001; Obrázek 1). Výsledky PFS byly konzistentní napříč všemi hodnocenými podskupinami podle výchozího stavu a demografických kritérií včetně podskupiny s předcházející terapií inhibitory tyrozinkinázy (která mohla zahrnovat léčiva působící na dráhy související s anti-angiogenezí), podskupin podle stavu RET mutací (včetně subjektů se zdokumentovanou absencí RET mutací), podskupin podle předcházející protinádorové léčby nebo radioterapie nebo existence kostních metastáz.
ORR byl 27,9 % a 0 % u pacientů v rameni s cabozantinibem a rameni s placebem (p < 0,0001; Tabulka 2). Medián trvání objektivních odpovědí byl 14,6 měsíce (95% IS: 11,1; 17,5) u pacientů v rameni s cabozantinibem.
Cometriq (n=219)
|
Počet pacientů s rizikem | ||||||||
|
Měsíc |
0 |
3 |
6 |
9 |
12 |
15 |
18 |
21 |
|
Cometriq |
219 |
121 |
78 |
55 |
31 |
12 |
2 |
1 |
|
Placebo |
111 |
35 |
11 |
6 |
3 |
2 |
0 |
0 |
Závěrečná analýza OS vykonaná potom, co nastalo 218 příhod (úmrtí), ukázala tendenci k zvýšení v mediánu přežití o 5,5 měsíce v rameni s cabozantinibem: medián (měsíce) 26,6 cabozantinib versus
21,1 placebo (HR = 0,85 [95% IS: 0,64, 1,12], p = 0,2409).
Obrázek 2: Kaplan-Mayerova křivka celkového přežití
Tabulka 2: Shrnutí klíčových zjištění účinnosti
|
Cabozantinib |
Placebo | |
|
Medián přežití bez progrese |
11,2 měsíce |
4,0 měsíce |
|
HR: 0,28 (0,19, 0,40) p < 0,0001 | ||
|
Medián celkového přežití |
26,6 měsíce |
21,1 měsíce |
|
HR: 0,85 (0,64, 1,12) p = 0,2409 | ||
|
Výskyt celkové odpovědia (95% IS) |
27,9 % (21,9 %, 34,5 %) |
0 % |
|
p < 0,0001 | ||
|
Trvání odpovědi; Medián (95% IS) |
14,6 měsíce (11,1, 17,5) |
N/A |
|
Výskyt kontroly onemocnění b (95% IS) |
55,3 % (48,3 %, 62,2 %) |
13,5 % (7,6 %, 21,6 %) |
|
Kalcitoninová odpověďa |
47 % (49/104)c |
3 % (1/40) c |
|
CEA odpověďa |
33 % (47/143) c |
2 % (1/55)c |
a Odpověď = CR + PR
b Výskyt kontroly onemocnění = SD+ ORR
c Zahrnuje pacienty, u kterých byla odpověď hodnotitelná
Stav mutací RET
Z 215 subjektů s dostatečnými údaji na určení stavu mutace bylo 78,6 % (n = 169) klasifikováno jako pozitivní na mutaci RET (z kterých bylo 126 pozitivních na mutaci M918T) a 21,4 % (n = 46) bylo klasifikováno jako negativní na mutaci RET. U dalších 115 subjektů nebylo možno určit stav mutací RET nebo byl tento stav nejasný. Všechny tři podskupiny vykazovaly zvýšení PFS v rameni s cabozantinibem v porovnání s ramenem s placebem (HR byl v podskupině s pozitivní RET mutací 0,23, v podskupině s negativní RET mutací 0,53 a v podskupině s neznámým stavem RET mutací 0,30). Výskyt objektivní odpovědi měřený v těchto podskupinách byl obvykle konzistentní s PFS výsledky, přičemž výskyt reakce nádoru byl 32 % v podskupině s pozitivní mutací RET, 22 % v podskupině s negativní RET mutací a 25 % v podskupině s neznámým stavem této mutace.
Další genetické analýzy ukázaly, že malý podíl pacientů nese somatické mutace tumoru v HRAS, KRAS nebo NRAS. Tito pacienti (n = 16) vykazovali signifikantní prodloužení PFS (HR 0,15) a výskyt objektivní odpovědi 31 %. Pacienti s negativními RET mutacemi bez důkazu mutace RAS (n = 33) ukázali při užívaní cabozantinibu snížený přínos v PFS (HR 0,87) a nižší výskyt odpovědi, 18 %, v porovnání s podskupinami s jinými mutacemi.
Signifikantní zlepšení v OS bylo pozorováno v podskupině pacientů s pozitivním stavem mutace RET M918T (n=81/219 rameno s cabozantinibem): 44,3 měsíců v rameni s cabozantinibem versus
18,9 měsíců v rameni s placebem (HR = 0,60, p = 0,0255). Nebylo zjištěno žádné zlepšení v OS v podskupinách s negativním a neznámým stavem mutace RET M918T.
Pediatrická populace
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s cabozantinibem u jedné nebo více podskupin pediatrické populace pro léčbu maligních solidních tumorů (informace o použití u dětí viz bod 4.2).
Tento léčivý přípravek byl registrován postupem tzv. podmíněného schválení. Znamená to, že j sou očekávány další důkazy o jeho přínosech.
Evropská agentura pro léčivé přípravky nejméně jednou za rok vyhodnotí nové informace o tomto léčivém přípravku a tento souhrn údajů o přípravku bude podle potřeby aktualizován.
5.2 Farmakokinetické vlastnosti
Absorpce
Po perorálním podání cabozantinibu se jeho maximální koncentrace v plazmě dosáhne 2 až 5 hodin po podání dávky. Profily plazmatické koncentrace v čase ukázaly druhou maximální absorpci přibližně 24 hodin po podání, což naznačuje možnost, že cabozantinib podstupuje enterohepatální recirkulaci.
Podávání opakované denní dávky cabozantinibu 140 mg po dobu 19 dní vedlo k přibližně 4- až 5násobné průměrné akumulaci cabozantinibu (založené na hodnotách AUC) v porovnání s podáním jednorázové dávky; rovnovážný stav byl dosažen přibližně 15. den.
Jídlo bohaté na tuky mírně zvýšilo hodnoty Cmax a AUC (41 % a 57 %) v porovnání se stavem na lačno u zdravých dobrovolníků, kterým byl cabozantinib podán v jednorázové perorální dávce 140 mg. Informace o přesném účinku jídla požitého hodinu po podání cabozantinibu nejsou k dispozici.
Distribuce
Cabozantinib je in vitro v lidské plazmě vysoce vázaný na proteiny (> 99,7 %). Na základě farmakokinetického (FK) populačního modelu byl stanoven distribuční objem (V/F) přibližně 349 l (SE: ± 2,73 %). U pacientů s mírnou až středně závažnou poruchou funkce ledvin nebo jater nebyla změněna vazba na proteiny.
Biotransformace
Cabozantinib byl metabolizován in vivo. V plazmě byly přítomny čtyři metabolity s expozicí (AUC) vyšší než 10 % AUC mateřské látky: XL184-N-oxid, rozkladný produkt XL184-amid, hydroxosíran XL184 a rozkladný produkt 6-desmethyl-amid síran. Oba nekonjugované metabolity (XL184-N-oxid a rozkladný produkt XL184-amid), které mají < 1 % schopnosti inhibice cílové kinázy v porovnání s mateřským cabozantinibem, představují < 10 % celkové plazmatické expozice spojené s léčivem.
Cabozantinib je in vitro substrátem CYP3A4 metabolismu jako neutralizační protilátka CYP3A4 inhibující tvorbu metabolitu XL184 N-oxidu o > 80 % při inkubaci katalyzované NADPH v lidských jaterních mikrozomech; naproti tomu neutralizační protilátky CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C19, CYP2D6 a CYP2E1 nemají žádný účinek na tvorbu metabolitů cabozantinibu. Neutralizační protilátky CYP2C9 vykazovaly minimální účinek na tvorbu metabolitů cabozantinibu (tj. < 20% snížení).
Eliminace
Terminální plazmatický eliminační poločas cabozantinibu ve studiích s jednorázovým podáním zdravým dobrovolníkům byl přibližně 120 hodin. Průměrná hodnota clearance (CL/F) v rovnovážném stavu u pacientů s nádorovým onemocněním byla v populační FK analýze odhadnuta na 4,4 l/hod. Během 48denního období sběru po jednorázové dávce 14C-cabozantinibu zdravým dobrovolníkům bylo zachyceno přibližně 81 % celkové podané radioaktivity, přičemž 54 % v stolici a 27 % v moči.
Farmakokinetika u zvláštních populací pacientů
Porucha funkce ledvin
Výsledky studie s pacienty s poruchou funkce ledvin ukazují, že poměry geometrického průměru cabozantinibu v plazmě metodou nejmenších čtverců, Cmax a AUC0-mf byly o 19 % a 30 % vyšší u pacientů s mírnou poruchou funkce ledvin (90% IS pro Cmax od 91,60 % do 155,51 %; AUCo-mf 98,79 % do 171,26 %) a o 2 % a 6-7 % vyšší pacientů se středně závažnou poruchou funkce ledvin (90% IS pro Cmax 78,64 % do 133,52 %; AUC0-mf 79,61 % až 140,11 %) než u pacientů s normální funkcí ledvin. Pacienti se závažnou poruchou funkce ledvin nebyli předmětem zkoumání.
Porucha _ funkce _ jater
Výsledky studie s pacienty s poruchou funkce jater ukazují, že expozice_(AUC0-mf) se zvyšuje o 81 % u pacientů s mírnou poruchou funkce jater a o 63 % u pacientů se středně závažnou poruchou funkce jater (90% IS pro AUC0-mf: 121,44 % až 270,34 % pro mírné a 107,37 % až 246,67 % pro středně závažné poruchy). Pacienti se závažnou poruchou funkce jater nebyli předmětem zkoumání.
Rasa
Nejsou k dispozici žádné údaje na určení rozdílů ve FK podmíněné rasou.
5.3 Předklinické údaje vztahující se k bezpečnosti
Nežádoucí účinky, které nebyly pozorovány v klinických studiích, avšak vyskytly se ve studiích na zvířatech při systémové expozici podobné expozici při klinickém podávání, a které mohou být důležité pro klinické použití:
Ve studiích toxicity po opakované dávce u potkanů a psů v délce trvání až 6 měsíců byly cílovými orgány toxicity gastrointestinální trakt, kostní dřeň, lymfoidní tkáně, ledviny, tkáně nadledvin a reprodukčního systému. Hodnota, po které nebyly pozorovány žádné nepříznivé účinky (NOAEL), byla při těchto zjištěních nižší než hladiny klinické expozice u člověka při zamýšlené terapeutické dávce.
Ve standardní sadě genotoxických testů nevykazoval cabozantinib žádný mutagenní nebo klastogenní potenciál. Studie kancerogenity nebyly provedeny.
Studie fertility u potkanů ukázaly sníženou samčí i samičí plodnost. Navíc po expozicích nižších než hladiny klinické expozice u člověka při určené terapeutické dávce byla u samců psů pozorována hypospermatogeneze.
Studie embryofetálního vývoje byly provedeny u potkanů a králíků. U potkanů cabozantinib způsoboval postimplantační ztráty, fetální edém, rozštěp patra/rtů, dermální aplazii a deformovaný nebo rudimentární chvost. U králíků cabozantinib způsoboval změny fetálních měkkých tkání (zmenšená velikost sleziny, malé nebo chybějící střední laloky plic) a zvýšenou fetální incidenci celkových malformací. NOAEL při embryo-fetální toxicitě a teratogenních zjištěních byly nižší než hladiny klinické expozice u člověka při zamýšlené terapeutické dávce.
Mláďata potkanů (porovnatelné s pediatrickou populací >2 roky), kterým byl podán cabozantinib, vykazovala zvýšené hodnoty leukocytů, sníženou hematopoézu, pubescentní/nevyzrálý samičí reprodukční systém (bez opožděného vaginálního otevírání), abnormality zubů, snížený obsah minerálů v kostech a sníženou kostní denzitu, pigmentaci jater a hyperplazii žlučovodu. Nálezy na děloze/vaječnících a snížení hematopoézy se zdály být přechodné, zatímco účinky na kostní parametry a pigmentaci jater byly trvalé. Hodnocení mláďat potkanů (porovnatelné s pediatrickou populací <2 roky) nebylo provedeno.
6. FARMACEUTICKÉ INFORMACE
6.1 Seznam pomocných látek
Obsah tobolky mikrokrystalická celulóza sodná sůl kroskarmelózy sodná sůl karboxymethylškrobu koloidní bezvodý oxid křemičitý kyselina stearová
Obal tobolky želatina
černý oxid železitý (E172) (jen 20mg tobolky) červený oxid železitý (E172) (jen 80mg tobolky) oxid titaničitý (E171)
Inkoust na potisk šelak
černý oxid železitý (E172) propylenglykol
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
3 roky.
6.4 Zvláštní opatření pro uchovávání
Neuchovávejte při teplotě nad 25°C.
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
6.5 Druh obalu a obsah balení
PVC/PE/PCTFE-Al blistry s fólií na zadní straně, zatavené do sekundárního kartového obalu uzavřeného teplem.
Blistrové karty obsahují buď:
7 x 20 mg a 7 x 80 mg tobolek (při dávce 100 mg/den je to zásoba na 7 dní)
21 x 20 mg a 7 x 80 mg tobolek (při dávce 140 mg/den je to zásoba na 7 dní)
Balení na 28 dní obsahuje:
56 tobolek (4 blistrové karty: 7 x 20 mg a 7 x 80 mg) (při dávce 100 mg/den je to zásoba na 28 dní) 112 tobolek (4 blistrové karty: 21 x 20 mg a 7 x 80 mg) (při dávce 140 mg/den je to zásoba na 28 dní)
6.6 Zvláštní opatření pro likvidaci přípravku
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
TMC Pharma Services Ltd.
Lodge Farm Barn Elvetham Park Estate Fleet Road Hartley Wintney Hampshire RG27 8AS Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/13/890/002
EU/1/13/890/003
EU/1/13/890/005
EU/1/13/890/006
7 x 20 mg a 7 x 80 mg tobolek (při dávce 100 mg/den je to zásoba na 7 dní)
21 x 20 mg a 7 x 80 mg tobolek (při dávce 140 mg/den je to zásoba na 7 dní) 56 tobolek (4 blistrové karty: 7 x 20 mg a 7 x 80 mg) (při dávce 100 mg/den je to zásoba na 28 dní)
112 tobolek (4 blistrové karty: 21 x 20 mg a 7 x 80 mg) (při dávce 140 mg/den je to zásoba na 28 dní)
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 21. března 2014
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
PŘÍLOHA II
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
E. ZVLÁŠTNÍ POVINNOST USKUTEČNIT POREGISTRAČNÍ OPATŘENÍ PRO PODMÍNĚNOU REGISTRACI PŘÍPRAVKU
VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
A.
Název a adresa výrobce odpovědného za propouštění šarží Catalent UK Packaging Limited Lancaster Way, Wingates Industrial Park,
Westhoughton, Bolton,
Lancashire, BL5 3XX,
Velká Británie
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
• Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
• Další opatření k minimalizaci rizik
Neuplatňuje se.
ZVLÁŠTNÍ POVINNOST USKUTEČNIT POREGISTRAČNÍ OPATŘENÍ PRO PODMÍNĚNOU REGISTRACI PŘÍPRAVKU
E.
Tato registrace byla schválena postupem tzv. podmíněného schválení, a proto podle článku 14(7) nařízení (ES) č. 726/2004 držitel rozhodnutí o registraci uskuteční v daném termínu následující opatření:
|
Popis |
Termín splnění |
|
Studie na porovnání dávek (XL-184-401) (140 mg versus 60 mg) u 112 pacientů s dědičným nebo vzácně se vyskytujícím medulárním karcinomem štítné žlázy. Podmínky na účast ve studii budou splňovat pacienti se vzácně se vyskytující i dědičnou formou MTC. U pacientů zapojených do studie na porovnání dávek by se z nejnovějšího metastatického ložiska měly odebírat čerstvé vzorky nádoru na genetickou analýzu nádoru. Vzorky budou podrobeny pečlivému hodnocení na přítomnost mutací RET a RAS. Vzorky z tkáně nádoru budou na úvod podrobeny histologickému hodnocení, manuálnímu obohacení nádoru a izolaci DNA. Kvalita získaných vzorůk DNA bude hodnocena amplifikačním PCR testem a Sangerovým sekvenováním na RET M918T. Pokud některý z originálních vzorků nevyhoví během kvalitativního PCR nebo Sangerova sekvenovacího testu, bude požadováno dodání dalšího vzorku. Vykoná se další generace sekvenování RET exonů 10, 11 a 13-16, čímž se pokryje velká většina známých RET mutací. Navíc budou vzorky hodnoceny na přítomnost mutací v „hot spots“ RAS genu (HRAS, KRAS a NRAS geny). Hodnocení FK bude požadováno u všech účastníků (obou skupin vytvořených podle dávky). Výsledky budou použity na vyhodnocení expozice cabozantinibu při dávkách na úrovni 60 a 140 mg a na další popis populačních FK modelů a vztahu expozice-odezva u cabozantinibu a možných metabolitů v této populaci. |
31. březen 2019 |
PŘÍLOHA III
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU
BLISTROVÁ KARTA, 60mg dávka_
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU_
COMETRIQ 20 mg tvrdé tobolky Cabozantinibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK_
Jedna tvrdá tobolka obsahuje cabozantinibi malas ekvivalentní cabozantinibum 20 mg.
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Tvrdé tobolky
20 mg
Dávka 60 mg
Balení pro denní dávku 60 mg
21 x 20mg tobolka (při dávce 60 mg/den je to zásoba na 7 dní) Každá 60 mg denní dávka obsahuje tři šedé 20 mg tobolky.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání.
Před použitím si přečtěte příbalovou informaci.
Příbalová informace pro pacienta uvnitř sáčku.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
Pokyny při výdeji
Každý den užijte všechny tobolky v jedné řadě bez jídla (pacienti nesmí jíst alespoň 2 hodiny před a hodinu po užití tobolek). Vyznačte datum první dávky.
1. Vtlačte záložku
3. Vytlačte tobolku přes folii
EXP
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí. Neuchovávejte při teplotě nad 25°C.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
TMC Pharma Services Ltd.
Lodge Farm Barn
Elvetham Park Estate
Fleet Road
Hartley Wintney
Hampshire
RG27 8AS
Velká Británie
Tel: +44 1252 842255
EU/1/13/890/001
Č. šarže
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
COMETRIQ 20 mg Dávka 60 mg/den
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU
VNĚJŠÍ KRABICE S BALENÍM NA 28 DNÍ, 60 mg dávka (S BLUE BOXEM)
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU_
COMETRIQ 20 mg tvrdé tobolky Cabozantinibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK_
Jedna tvrdá tobolka obsahuje cabozantinibi malas ekvivalentní cabozantinibum 20 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Dávka 60 mg
Balení na 28 dní: 84 tobolek (4 blistrové karty: 21 x 20 mg tobolky) při denní dávce 60 mg je to zásoba na 28 dní).
Každá 60 mg denní dávka obsahuje tři šedé 20 mg tobolky.
Perorální podání.
Před použitím si přečtěte příbalovou informaci.
Uchovávejte mimo dohled a dosah dětí.
Dispenzační pokyny najdete na jednotlivých blistrových kartách.
EXP
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí. Neuchovávejte při teplotě nad 25°C.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Nepoužitelné léčivo vraťte do lékárny.
TMC Pharma Services Ltd.
Lodge Farm Barn, Elvetham Park Estate
Fleet Road
Hartley Wintney
Hampshire
RG27 8AS
Velká Británie
Tel: +44 1252 842255
EU/1/13/890/004 84 tobolek (4 blistrové karty 21 x 20 mg) (při dávce 60 mg/den je to zásoba na
28 dní)
Č. šarže
Výdej léčivého přípravku vázán na lékařský předpis.
COMETRIQ 20 mg Dávka 60 mg/den
ÚDAJE UVÁDĚNÉ NA VNITŘNÍM OBALU
BLISTROVÁ KARTA VÍCENÁSOBNÉHO BALENÍ, 60 mg dávka (BEZ BLUE BOXU)
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU_
COMETRIQ 20 mg tvrdé tobolky Cabozantinibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK_
Jedna tvrdá tobolka obsahuje cabozantinibi malas ekvivalentní cabozantinibum 20 mg.
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Tvrdé tobolky
20 mg
Dávka 60 mg
21 x 20 mg tobolka (při dávce 60 mg/den je to zásoba na 7 dní). Součásti balení na 28 dní není možné prodávat samostatně.
Balení pro denní dávku 60 mg
Každá 60 mg denní dávka obsahuje tři šedé 20 mg tobolky.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání.
Před použitím si přečtěte příbalovou informaci. Příbalová informace pro pacienta uvnitř sáčku.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
Pokyny při výdeji
Každý den užijte všechny tobolky v jedné řadě bez jídla (pacienti nesmí jíst alespoň 2 hodiny před a hodinu po užití tobolek). Vyznačte datum první dávky.
1. Vtlačte záložku
3. Vytlačte tobolku přes folii
EXP
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí. Neuchovávejte při teplotě nad 25°C.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
TMC Pharma Services Ltd.
Lodge Farm Barn
Elvetham Park Estate
Fleet Road
Hartley Wintney
Hampshire
RG27 8AS
Velká Británie
Tel: +44 1252 842255
EU/1/13/890/004 84 tobolek (4 blistrové karty 21 x 20 mg) (při dávce 60 mg/den je to zásoba na
28 dní)
Č. šarže
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
COMETRIQ 20 mg tvrdé tobolky COMETRIQ 80 mg tvrdé tobolky Cabozantinibum
Jedna tvrdá tobolka obsahuje cabozantinibi malas ekvivalentní cabozantinibum 20 mg nebo 80 mg.
Tvrdé tobolky 20 mg a 80 mg Dávka 100 mg
Balení pro denní dávku 100 mg
7 x 20 mg tobolka a 7 x 80 mg tobolka (při dávce 100 mg/den je to zásoba na 7 dní)
Každá 100 mg denní dávka obsahuje kombinaci jedné šedé 20 mg tobolky a jedné oranžové 80 mg tobolky.
Perorální podání.
Před použitím si přečtěte příbalovou informaci. Příbalová informace pro pacienta uvnitř sáčku.
Uchovávejte mimo dohled a dosah dětí.
Pokyny při výdeji
Každý den užijte všechny tobolky v jedné řadě bez jídla (pacienti nesmí jíst alespoň 2 hodiny před a hodinu po užití tobolek). Vyznačte datum první dávky.
1. Vtlačte záložku
3. Vytlačte tobolku přes folii
EXP
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí. Neuchovávejte při teplotě nad 25°C.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
TMC Pharma Services Ltd.
Lodge Farm Barn
Elvetham Park Estate
Fleet Road
Hartley Wintney
Hampshire
RG27 8AS
Velká Británie
Tel: +44 1252 842255
EU/1/13/890/002
Č. šarže
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
COMETRIQ 20 mg COMETRIQ 80 mg Dávka 100 mg/den
COMETRIQ 20 mg tvrdé tobolky COMETRIQ 80 mg tvrdé tobolky Cabozantinibum
Jedna tvrdá tobolka obsahuje cabozantinibi malas ekvivalentní cabozantinibum 20 mg nebo 80 mg.
4. LÉKOVÁ FORMA A OBSAH BALENÍ_
Dávka 100 mg
Balení na 28 dní obsahuje: 56 tobolek (4 blistrové karty: 7 x 20 mg tobolky a 7 x 80 mg tobolky) při denní dávce 100 mg je to zásoba na 28 dní.
Každá 100 mg denní dávka obsahuje kombinaci jedné šedé 20 mg tobolky a jedné oranžové 80 mg tobolky.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Dispenzační pokyny najdete na jednotlivých blistrových kartách.
EXP
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí. Neuchovávejte při teplotě nad 25°C.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Nepoužitelné léčivo vraťte do lékárny.
TMC Pharma Services Ltd.
Lodge Farm Barn, Elvetham Park Estate
Fleet Road
Hartley Wintney
Hampshire
RG27 8AS
Velká Británie
Tel: +44 1252 842255
EU/1/13/890/005 56 tobolek (4 blistrové karty: 7 x 20 mg a 7 x 80 mg) (při dávce 100 mg/den je
to zásoba na 28 dní)
Č. šarže
Výdej léčivého přípravku vázán na lékařský předpis.
COMETRIQ 20 mg COMETRIQ 80 mg Dávka 100 mg/den
COMETRIQ 20 mg tvrdé tobolky COMETRIQ 80 mg tvrdé tobolky Cabozantinibum
Jedna tvrdá tobolka obsahuje cabozantinibi malas ekvivalentní cabozantinibum 20 mg nebo 80 mg.
Tvrdé tobolky 20 mg a 80 mg Dávka 100 mg
7 x 20 mg tobolka a 7 x 80 mg tobolka (při dávce 100 mg/den je to zásoba na 7 dní). Součásti balení na 28 dní není možné prodávat samostatně.
Balení pro denní dávku 100 mg
Každá 100 mg denní dávka obsahuje kombinaci jedné šedé 20 mg tobolky a jedné oranžové 80 mg tobolky.
Perorální podání.
Před použitím si přečtěte příbalovou informaci.
Příbalová informace pro pacienta uvnitř sáčku.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Pokyny při výdeji
Každý den užijte všechny tobolky v jedné řadě bez jídla (pacienti nesmí jíst alespoň 2 hodiny před a hodinu po užití tobolek). Vyznačte datum první dávky.
1. Vtlačte záložku
3. Vytlačte tobolku přes folii
EXP
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí. Neuchovávejte při teplotě nad 25°C.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
TMC Pharma Services Ltd.
Lodge Farm Barn
Elvetham Park Estate
Fleet Road
Hartley Wintney
Hampshire
RG27 8AS
Velká Británie
Tel: +44 1252 842255
EU/1/13/890/005 56 tobolek (4 blistrové karty: 7 x 20 mg a 7 x 80 mg) (při dávce 100 mg/den je
to zásoba na 28 dní)
Č. šarže
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
COMETRIQ 20 mg tvrdé tobolky COMETRIQ 80 mg tvrdé tobolky Cabozantinibum
Jedna tvrdá tobolka obsahuje cabozantinibi malas ekvivalentní cabozantinibum 20 mg nebo 80 mg.
Tvrdé tobolky
20 mg a 80 mg Dávka 140 mg
Balení pro denní dávku 140 mg
21 x 20 mg tobolka a 7 x 80 mg tobolka (při dávce 140 mg/den je to zásoba na 7 dní)
Každá 140mg denní dávka obsahuje kombinaci třech šedých 20 mg tobolek a jedné oranžové 80 mg tobolky.
Perorální podání.
Před použitím si přečtěte příbalovou informaci. Příbalová informace pro pacienta uvnitř sáčku.
Uchovávejte mimo dohled a dosah dětí.
Pokyny při výdeji
Každý den užijte všechny tobolky v jedné řadě bez jídla (pacienti nesmí jíst alespoň 2 hodiny před a hodinu po užití tobolek). Vyznačte datum první dávky.
1. Vtlačte záložku
3. Vytlačte tobolku přes folii
EXP
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí. Neuchovávejte při teplotě nad 25°C.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
TMC Pharma Services Ltd.
Lodge Farm Barn, Elvetham Park Estate
Fleet Road
Hartley Wintney
Hampshire
RG27 8AS
Velká Británie
Tel: +44 1252 842255
EU/1/13/890/003
Č. šarže
Výdej léčivého přípravku vázán na lékařský předpis.
COMETRIQ 20 mg COMETRIQ 80 mg Dávka 140 mg/den
COMETRIQ 20 mg tvrdé tobolky COMETRIQ 80 mg tvrdé tobolky Cabozantinibum
Jedna tvrdá tobolka obsahuje cabozantinibi malas ekvivalentní cabozantinibum 20 mg nebo 80 mg.
4. LÉKOVÁ FORMA A OBSAH BALENÍ_
Dávka 140 mg
Balení na 28 dní: 112 tobolek (4 blistrové karty: 21 x 20 mg tobolky a 7 x 80 mg tobolky) při denní dávce 140 mg je to zásoba na 28 dní.
Každá 140 mg denní dávka obsahuje kombinaci třech šedých 20 mg tobolek a jedné oranžové 80 mg tobolky.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Dispenzační pokyny najdete na jednotlivých blistrových kartách.
EXP
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí. Neuchovávejte při teplotě nad 25°C.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Nepoužitelné léčivo vraťte do lékárny.
TMC Pharma Services Ltd.
Lodge Farm Barn, Elvetham Park Estate
Fleet Road
Hartley Wintney
Hampshire
RG27 8AS
Velká Británie
Tel: +44 1252 842255
EU/1/13/890/006 112 tobolek (4 blistrové karty: 21 x 20 mg a 7 x 80 mg) (při dávce 140 mg/den
je to zásoba na 28 dní)
Č. šarže
Výdej léčivého přípravku vázán na lékařský předpis.
COMETRIQ 20 mg COMETRIQ 80 mg
Dávka 140 mg/den
COMETRIQ 20 mg tvrdé tobolky COMETRIQ 80 mg tvrdé tobolky Cabozantinibum
Jedna tvrdá tobolka obsahuje cabozantinibi malas ekvivalentní cabozantinibum 20 mg nebo 80 mg.
Tvrdé tobolky
20 mg a 80 mg Dávka 140 mg
21 x 20 mg tobolka a 7 x 80 mg tobolka (při dávce 140 mg/den je to zásoba na 7 dní). Součásti balení na 28 dní není možné prodávat samostatně.
Balení pro denní dávku 140 mg
Každá 140 mg denní dávka obsahuje kombinaci třech šedých 20 mg tobolek a jedné oranžové 80 mg tobolky.
Perorální podání.
Před použitím si přečtěte příbalovou informaci.
Příbalová informace pro pacienta uvnitř sáčku.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Pokyny při výdeji
Každý den užijte všechny tobolky v jedné řadě bez jídla (pacienti nesmí jíst alespoň 2 hodiny před a hodinu po užití tobolek). Vyznačte datum první dávky.
1. Vtlačte záložku
3. Vytlačte tobolku přes folii
EXP
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí. Neuchovávejte při teplotě nad 25°C.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
TMC Pharma Services Ltd.
Lodge Farm Barn, Elvetham Park Estate
Fleet Road
Hartley Wintney
Hampshire
RG27 8AS
Velká Británie
Tel: +44 1252 842255
EU/1/13/890/006 112 tobolek (4 blistrové karty: 21 x 20 mg a 7 x 80 mg) (při dávce 140 mg/den
je to zásoba na 28 dní)
Č. šarže
Výdej léčivého přípravku vázán na lékařský předpis.
16. INFORMACE V BRAILLOVĚ PÍSMU
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro pacienta
COMETRIQ 20 mg tvrdé tobolky COMETRIQ 80 mg tvrdé tobolky
Cabozantinibi malas
'VTento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek COMETRIQ a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek COMETRIQ užívat
3. Jak se přípravek COMETRIQ užívá
4. Možné nežádoucí účinky
5. Jak přípravek COMETRIQ uchovávat
6. Obsah balení a další informace
1. Co je přípravek COMETRIQ a k čemu se používá
COMETRIQ je lék, který se používá k léčbě medulárního karcinomu štítné žlázy, což je vzácný typ rakoviny štítné žlázy, který není možné chirurgicky odstranit nebo který se rozšířil do jiných částí těla.
COMETRIQ může zpomalit nebo zastavit růst medulárního karcinomu štítné žlázy. Může pomoci zmenšit nádory spojené s tímto typem karcinomu.
2. Čemu musíte věnovat pozornost, než začnete přípravek COMETRIQ užívat Neužívejte přípravek COMETRIQ
- jestliže jste alergický(á) na cabozantinib nebo na kteroukoli další složku tohoto přípravku
(uvedenou v bodě 6).
Upozornění a opatření
Před užitím přípravku COMETRIQ se poraďte se svým lékařem nebo lékárníkem,
- jestliže máte vysoký krevní tlak,
- jestliže máte průjem,
- jestliže jste v nedávné době vykašlával(a) krev nebo jste měl(a) závažnější krvácení,
- jestliže jste během posledního měsíce podstoupil(a) chirurgický zákrok (nebo jestliže máte chirurgické zákroky naplánované), včetně zubních zákroků,
- jestliže jste během posledních 3 měsíců podstoupil(a) radioterapii,
- jestliže trpíte zánětlivým onemocněním střev (například Crohnovou chorobou nebo ulcerózní kolitidou nebo divertikulitidou),
- jestliže vás informovali, že se u vás rakovina rozšířila do dýchacích cest nebo jícnu,
- jestliže jste v nedávné době prodělal (a) cévní mozkovou příhodu, srdeční infarkt nebo jste měl(a) problém s krevní sraženinou v noze,
- jestliže užíváte léky ke kontrole srdečního rytmu, máte pomalý tep srdce, máte problémy se srdcem nebo s hladinou vápníku, draslíku nebo hořčíku v krvi
- jestliže máte závažné onemocnění jater nebo ledvin.
Informuje svého lékaře, jestliže se Vás něco z výše uvedeného týká. Možná u Vás bude potřebné tyto stavy léčit, nebo lékař rozhodne změnit dávku přípravku COMETRIQ, případně léčbu úplně ukončit. Viz také bod 4 „Možné nežádoucí účinky“.
O tom, že užíváte COMETRIQ, byste měl(a) informovat i Vašeho zubaře. Je pro Vás důležité, abyste během léčby přípravkem COMETRIQ dodržoval(a) správnou ústní hygienu.
Děti a dospívající
COMETRIQ není doporučen pro děti nebo dospívající. Účinky léku COMETRIQ u osob mladších než 18 let nejsou známé.
Další léčivé přípravky a přípravek COMETRIQ
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat, včetně léků dostupných bez lékařského předpisu.
Je to proto, že COMETRIQ může ovlivnit způsob, jak některé léky fungují. Také jiné léky mohou ovlivnit způsob, jak funguje COMETRIQ. Může to znamenat, že lékař bude potřebovat změnit dávku(y), kterou užíváte.
- Léky k léčbě plísňových onemocnění, jako například itrakonazol, ketokonazol a posakonazol
- Léky k léčbě bakteriálních infekcí (antibiotika) jako například erythromycin, klarithromycin a rifampicin
- Léky na alergii jako například fexofenadin a ranolazin
- Steroidy, které se používají na utlumení zánětu nebo k léčbě množství různých onemocnění imunitního systému
- Léky k léčbě epilepsie nebo záchvatů jako například fenytoin, karbamazepin a fenobarbital
- Rostlinné přípravky s obsahem třezalky tečkované (Hypericum perforatum), někdy používané k léčbě depresí nebo stavů spojených s depresí, jako například úzkosti
- Léky používané na ředění krve jako například warfarin
- Léky k léčbě vysokého krevního tlaku nebo jiných srdečních onemocnění jako například aliskiren, ambrisentan, dabigatran-etexilát, digoxin, talinolol a tolvaptan
- Léky na diabetes jako například saxagliptin a sitagliptin
- Léky používané k léčbě dny jako například kolchicin
- Léky používané k léčbě HIV nebo AIDS, jako například ritonavir, maravirok a emtricitabin
- Léky používané k léčbě virových infekcí jako například efavirenz
- Léky používané k prevenci odmítnutí transplantátu (cyklosporin) a léčebné režimy s cyklosporinem používané při revmatoidní artritidě a psoriáze
Perorální antikoncepce
Jestliže užíváte perorální antikoncepci a přitom užijete COMETRIQ, perorální antikoncepce může být neúčinná. Během užívání přípravku COMETRIQ a alespoň 4 měsíce po skončení léčby byste měl(a) používat i bariérovou antikoncepci (např. kondom nebo pesar).
COMETRIQ nesmíte užívat s jídlem. Nesmíte nic jíst alespoň 2 hodiny před užitím přípravku COMETRIQ a 1 hodinu po užití tohoto přípravku. Během užívání tohoto přípravku se vyhněte konzumaci výrobků s obsahem grepfruitu, protože grepfruit může zvyšovat hladinu přípravku COMETRIQ v krvi.
Těhotenství, kojení a plodnost
Během léčby přípravkem COMETRIQ se vyhněte otěhotnění. Pokud Vy nebo Vaše partnerka můžete otěhotnět, používejte vhodnou antikoncepci během léčby a po dobu alespoň 4 měsíců po skončení léčby. Obraťte se na Vašeho lékaře, aby vám poradil, které metody antikoncepce jsou vhodné při užívání přípravku COMETRIQ. Viz bod 2.
Informujte svého lékaře, jestliže otěhotníte nebo Vaše partnerka otěhotní, nebo jestliže plánujete těhotenství během léčby přípravkem COMETRIQ.
Obraťte se na Vašeho lékaře PŘED TÍM, než budete užívat COMETRIQ, jestliže Vy nebo Vaše partnerka plánujete mít dítě po skončení léčby. Je možné, že vaše plodnost bude léčbou přípravkem COMETRIQ ovlivněná.
Ženy užívající COMETRIQ nesmí během léčby a alespoň 4 měsíce po jejím skončení kojit, protože cabozantinib a/nebo jeho metabolity se mohou vylučovat do mateřského mléka a mohou být škodlivé pro dítě.
Řízení dopravních prostředků a obsluha strojů
Při řízení dopravních prostředků a obsluze strojů buďte opatrní. Mějte na paměti, že léčba přípravkem COMETRIQ může způsobovat, že se budete cítit unavený(á) nebo slabý(á).
3. Jak se přípravek COMETRIQ užívá
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře nebo lékárníka. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Měl(a) byste pokračovat v užívání tohoto přípravku, až dokud váš lékař nerozhodne Vaši léčbu zastavit. Lékař se může rozhodnout změnit Vám dávku nebo zastavit léčbu dříve, než se původně plánovalo, jestliže se u Vás vyskytnou závažné nežádoucí účinky. Lékař určí, jestli je potřebná úprava dávky, zejména během prvních osmi týdnů léčby přípravkem COMETRIQ.
COMETRIQ se má užívat jedenkrát denně. Podle předepsané dávky bude třeba užívat následující počet tobolek:
• 140 mg (1 oranžová 80 mg tobolka a 3 šedé 20 mg tobolky)
• 100 mg (1 oranžová 80 mg tobolka a 1 šedá 20 mg tobolka)
• 60 mg (3 šedé 20 mg tobolky)
Lékař Vám určí správnou dávku.
Tobolky se dodávají v blistrových kartách a jsou uspořádané podle předepsané dávky. Každá blistrová karta má počet tobolek postačující na sedm dní (jeden týden). Tobolky jsou dostupné také v balení na 28 dní, které obsahuje počet tobolek postačující na 28 dní. Tobolky jsou zde uspořádané ve čtyřech blistrových kartách, přičemž každá karta obsahuje tobolky na sedm dní.
Každý den užijte všechny tobolky z jedné řady. Další informace o blistrových kartách, včetně informace o tom, kolik tobolek budete užívat a kolik tobolek je v každé blistrové kartě celkem, jsou
uvedeny níže v bodě 6. Na lepší zapamatování Vaší dávky si do prostoru vedle tobolek napište datum, kdy jste užil(a) první dávku. Když chcete vybrat tobolku pro Vaši dávku:
1. Vtlačte záložku
2. Odstraňte zadní papírový kryt
3. Vytlačte tobolku přes folii
COMETRIQ se nesmí užívat s jídlem. Nesmíte nic jíst alespoň 2 hodiny před užitím přípravku COMETRIQ a 1 hodinu po užití tohoto přípravku. Tobolky spolkněte po jedné a každou zapijte vodou. Neotvírejte je.
Jestliže jste užil(a) více přípravku COMETRIQ, než jste měl(a)
Jestliže jste užil(a) více přípravku COMETRIQ, než vám bylo doporučeno, informujte o tom svého lékaře nebo jeďte přímo do nemocnice a vezměte s sebou tobolky i tuto příbalovou informaci.
Jestliže jste zapomněl(a) užít přípravek COMETRIQ
- Jestliže zůstává ještě 12 nebo více hodin do užití další dávky, pak vynechanou dávku užijte ihned, jakmile si vzpomenete. Další dávku užijte v obvyklou dobu.
- Jestliže vám do užití další dávky zůstává méně než 12 hodin, pak vynechanou dávku neužívejte. Další dávku užijte v obvyklou dobu.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého. Pokud se u Vás vyskytnou nežádoucí účinky, lékař Vám může sdělit, abyste užíval(a) nižší dávku přípravku COMETRIQ. Na zvládnutí nežádoucích účinků Vám lékař možná předepíše jiné léky.
Pokud se u Vás vyskytnou následující nežádoucí účinky, ihned to sdělte svému lékaři - možná budete potřebovat okamžitou lékařskou péči:
• Příznaky zahrnující bolest břicha, pocit na zvracení, zvracení, zácpu nebo horečku. Mohou to být příznaky gastrointestinální perforace - otvoru, který se vytvoří v žaludku nebo ve střevě a který může ohrožovat život.
• Otok, bolest v rukou a v nohou nebo dušnost.
• Rána, která se nehojí.
• Zvracení nebo vykašlávání krve, která může být světlečervená nebo může vypadat jako kávová sedlina.
• Bolest v ústech, zubech a/nebo v čelisti, otok nebo bolest v ústech, necitlivost nebo pocit těžké čelisti, nebo uvolnění zubu. Mohlo by se jednat o příznaky poškození kosti v čelisti (osteonekróza).
• Záchvaty, bolesti hlavy, zmatenost nebo těžkosti se soustředěním. Mohou to být příznaky onemocnění nazývaného syndrom reverzibilní posteriorní leukoencefalopatie (RPLS). RPLS je málo častý (postihuje méně než 1 osobu ze 100).
Velmi časté nežádoucí účinky (mohou postihovat více než 1 osobu z 10)
• Podrážděný žaludek včetně průjmu, pocitu na zvracení, zvracení, zácpy, poruch trávení a bolesti břicha
• Puchýř, bolest rukou nebo spodní části nohou, vyrážka nebo zčervenání kůže, suchá kůže
• Snížená chuť k jídlu, ztráta tělesné hmotnosti, změněné vnímání chuti
• Únava, slabost, bolest hlavy, závratě
• Změny zbarvení vlasů (zesvětlení), vypadávání vlasů
• Hypertenze (zvýšení krevního tlaku)
• Zčervenání, otok nebo bolest v ústech nebo v krku, těžkosti při mluvení, chrapot
• Změny v krevních testech používaných na sledování celkového zdravotního stavu a funkce jater, nízké hladiny elektrolytů (jako hořčík, vápník nebo draslík)
• Bolest kloubů, svalové křeče
• Otok lymfatických uzlin
Časté nežádoucí účinky (mohou postihovat až 1 osobu z 10)
• Celková bolest, bolest hrudníku nebo svalů, bolest ucha, zvonění v uších
• Slabost, snížená citlivost nebo mravenčení v končetinách
• Třes, zimnice
• Dehydratace
• Zánět břicha nebo slinivky břišní
• Zánět rtů nebo ústních koutků
• Zánět kořínků vlasů, akné, puchýře (na jiných částech těla než jsou ruce a chodidla)
• Otok tváře a jiných částí těla
• Ztráta chuti
• Hypotenze (snížení krevního tlaku)
• Síňová fibrilace (rychlý a nesprávný srdeční tep)
• Zesvětlení kůže, šupinatá kůže, nezvykle bledá kůže
• Nezvyklý růst vlasů
• Hemoroidy
• Pneumonie
• Bolest v ústech, zubů a/nebo čelisti, otok nebo bolest v ústech, necitlivost nebo pocit těžké čelisti nebo uvolnění zubu
• Snížení činnosti štítné žlázy; příznaky mohou zahrnovat: únavu, nárůst tělesné hmotnosti, zácpu, pocit studené a suché kůže
• Natržení, otvor nebo krvácení ze žaludku nebo střev, zánět nebo natržení konečníku, krevní sraženiny v plicích, krvácení do plic nebo průdušnice (dýchacích cest)
• Neobvyklé spojení tkání v průdušnici (dýchacích cestách), jícnu nebo plicích
• Absces (nahromadění hnisu s otokem a zánětem) v oblasti břicha nebo pánve nebo v zubech/dásních
• Krevní sraženiny v žilách
• Plísňové infekce, které mohou být na kůži, v ústech nebo na genitáliích
• Rány, které se těžce hojí
• Bílkovina nebo krev v moči, žlučníkové kameny, bolestivé močení
• Rozmazané vidění
• Zvýšená hladina bilirubinu v krvi (což může vést k žloutence/zežloutnutí kůže nebo očí)
• Snížení hladin bílkovin v krvi
Méně časté nežádoucí účinky (mohou postihovat až 1 osobu ze 100)
• Zánět jícnu, příznaky mohou zahrnovat pálení žáhy, bolest na hrudníku, pocit na zvracení, změněnou chuť, plynatost, říhání a poruchu trávení
• Natržení nebo neobvyklé spojení tkání v trávicím systému, příznaky mohou zahrnovat těžké nebo přetrvávající bolesti žaludku
• Infekce a zánět v plicích, splasknutí plic
• Kožní vředy, cysty, červené tečky na tváři nebo na stehnech
• Bolest tváře
• Změny ve výsledcích testů na měření krevní srážlivosti nebo krevních buněk
• Ztráta koordinace svalů, poškození kosterních svalů
• Ztráta pozornosti, ztráta vědomí, změny v řeči, delirium, nezvyklé sny
• Malá cévní mozková příhoda, srdeční infarkt, rychlý srdeční tep
• Poškození jater, selhání ledvin
• Poškození sluchu
• Zánět v oku, katarakta
• Zástava menstruace, krvácení z pochvy
• Onemocnění nazývané syndrom posteriorní reverzibilní encefalopatie (PRES) nebo syndrom reverzibilní posteriorní leukoencefalopatie (RPLS), k jehož příznakům patří křeče, bolesti hlavy, zmatenost nebo pocit těžkostí se soustředěním
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek COMETRIQ uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na blistrové kartě za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Neuchovávejte při teplotě nad 25°C. Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co COMETRIQ obsahuje
Léčivou látkou je cabozantinibi malas.
COMETRIQ 20 mg tobolky obsahují cabozantinibi malas, který je ekvivalentní cabozantinibum 20 mg.
COMETRIQ 80 mg tobolky obsahují cabozantinibi malas, který je ekvivalentní cabozantinibum 80 mg.
Dalšími složkami jsou:
- Tobolka obsahuje: mikrokrystalickou celulózu, sodnou sůl kroskarmelózy, sodnou sůl karboxymethylškrobu, koloidní bezvodý oxid křemičitý a kyselinu stearovou
- Obal tobolky: želatina a oxid titaničitý (E171)
- 20 mg tobolky obsahují také černý oxid železitý (E172)
67
- 80 mg tobolky obsahují také červený oxid železitý (E172)
- Barva na potisk: šelak, černý oxid železitý (E172) a propylenglykol
Jako COMETRIQ vypadá a co obsahuje toto balení
COMETRIQ 20 mg tobolky jsou šedé a na jedné straně mají vytištěný nápis „XL184 20mg“ COMETRIQ 80 mg tobolky jsou oranžové a na jedné straně mají vytištěný nápis „XL184 80mg“
COMETRIQ tobolky j sou balené v blistrových kartách uspořádaných podle předepsané dávky. Každá blistrová karta obsahuje množství léku postačující na 7 dní. Každý řádek blistrové karty obsahuje denní dávku.
Blistrová karta pro denní dávku 60 mg obsahuje dvacet jedna 20 mg tobolek pro celkem 7 denních dávek. Každá denní dávka je umístěna v jednom řádku a obsahuje tři 20 mg tobolky:
60 mg
tři šedé 20 mg
Blistrová karta pro denní dávku 100 mg obsahuje sedm 80 mg tobolek a sedm 20 mg tobolek pro celkem 7 denních dávek. Každá denní dávka je umístěna v jednom řádku a obsahuje jednu 80 mg tobolku a jednu 20 mg tobolku:
|
w |
+ |
100 mg
jedna oranžová 80 mg + jedna šedá 20 mg
Blistrová karta pro denní dávku 140 mg obsahuje sedm 80 mg tobolek a dvacet jedna 20 mg tobolek pro celkem 7 denních dávek. Každá denní dávka je umístěna v jednom řádku a obsahuje jednu 80 mg tobolku a tři 20 mg tobolky:
s~\
140 mg
jedna oranžová 80 mg + tři šedé 20 mg
COMETRIQ tobolky jsou dostupné také v balení na 28 dní:
84 tobolek (4 blistrové karty 21 x 20 mg) (dávka 60 mg/den)
56 tobolek (4 blistrové karty: 7 x 20 mg a 7 x 80 mg) (dávka 100 mg/den) 112 tobolek (4 blistrové karty: 21 x 20 mg a 7 x 80 mg) (dávka 140 mg/den)
Jedno balení na 28 dní obsahuje takové množství léku, které postačuje na 28 dní.
Držitel rozhodnutí o registraci
TMC Pharma Services Ltd. Lodge Farm Barn Elvetham Park Estate Fleet Road
Hartley Wintney Hampshire RG27 8AS Velká Británie
Výrobce
Catalent UK Packaging Limited
Lancaster Way
Wingates Industrial Park
Westhoughton
Bolton
Lancashire
BL5 3XX
Velká Británie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci.
Belgie/Belgique/Belgien
Swedish Orphan Biovitrum Ltd
Tél/Tel: + 44 1638 722380 e-mail: mail.uk@sobi.com
Btarapna
Hgnm Op^aH EnoBmpyM KaoH Etarapna OOfl Tea.: +420 257 222 034 e-mail: mail.bg@sobi.com
Česká republika
Swedish Orphan Biovitrum s.r.o.
Tel: +420 257 222 034 e-mail: mail.cz@sobi.com
Danmark
Swedish Orphan Biovitrum A/S Tlf: + 45 32 96 68 69 e-mail: mail.dk@sobi.com
Deutschland
Swedish Orphan Biovitrum GmbH Tel: +49 6103 20269-0 e-mail: mail.de@sobi.com
Eesti
Oy Swedish Orphan Biovitrum AB c/o CentralPharma Communications OU Tel. +372 6 015 540
e-mail: centralpharma@centralpharma.ee
Lietuva
Oy Swedish Orphan Biovitrum AB c/o UAB CentralPharma Communications Tel: +370 5 2430444 e-mail: centralpharma@centralpharma.lt
Luxembourg/Luxemburg
Swedish Orphan Biovitrum Ltd
Tél/Tel: + 44 1638 722380 e-mail: mail.uk@sobi.com
Magyarország
Swedish Orphan Biovitrum s.r.o. Magyarországi Fióktelepe
Tel: +420 257 222 034 e-mail: mail.hu@sobi.com
Malta
Swedish Orphan Biovitrum S.r.l.
Tel: +39 0521 19 111 e-mail: mail.it@sobi.com
Nederland
Swedish Orphan Biovitrum Ltd Tel: + 44 1638 722380 e-mail: mail.uk@sobi.com
Norge
Swedish Orphan Biovitrum AS
Tlf: +47 66 82 34 00 e-mail: mail.no@sobi.com
Eiiáda
Swedish Orphan Biovitrum S.r.l.
Tni: +39 0521 19 111 e-mail: mail.it@sobi.com
Espaňa
Swedish Orphan Biovitrum S.L
Tel: + 34 913 91 35 80 e-mail: mail.es@sobi.com
France
Swedish Orphan Biovitrum SARL Tél: +33 1 85 78 03 40 e-mail: mail.fr@sobi.com
Hrvatska
SWEDISH ORPHAN BIOVITRUM, Glavna Podružnica Zagreb Tel: +420 257 222 034 e-mail: mail.hr@sobi.com
Ireland
Swedish Orphan Biovitrum Ltd
Tel: + 44 1638 722380 e-mail: mail.uk@sobi.com
Island
Swedish Orphan Biovitrum A/S Tlf: + 45 32 96 68 69 e-mail: mail.dk@sobi.com
Italia
Swedish Orphan Biovitrum S.r.l.
Tel: +39 0521 19 111 e-mail: mail.it@sobi.com
Kórcpog
Swedish Orphan Biovitrum S.r.l.
Tni: +39 0521 19 111 e-mail: mail.it@sobi.com
Latvija
Oy Swedish Orphan Biovitrum AB c/o CentralPharma Communications SIA Tel. +371 67 450 497 e-mail: centralpharma@centralpharma.lv
Osterreich
Swedish Orphan Biovitrum GmbH Tel: +49 6103 20269-0 e-mail: mail.de@sobi.com
Polska
Swedish Orphan Biovitrum Sp. z o.o. Oddzial w Polsce
Tel: +420 257 222 034 e-mail: mail.pl@sobi.com
Portugal
Swedish Orphan Biovitrum S.L Tel: + 34 913 91 35 80 e-mail: mail.es@sobi.com
Románia
Swedish Orphan Biovitrum s.r.o. Praga - Sucursala Bucuresti
Tel: +420 257 222 034 e-mail: mail.ro@sobi.com
Slovenija
Swedish Orphan Biovitrum s.r.o. - Podružnica v Sloveniji
Tel: +420 257 222 034 e-mail : mail.si@sobi.com
Slovenská republika
Swedish Orphan Biovitrum o.z.
Tel: +420 257 222 034 e-mail: mail.sk@sobi.com
Suomi/Finland
Oy Swedish Orphan Biovitrum AB Puh/Tel: +358 201 558 840 e-mail: mail.fi@sobi.com
Sverige
Swedish Orphan Biovitrum AB (publ)
Tel: +46 8 697 20 00 e-mail: mail.se@sobi.com
United Kingdom
Swedish Orphan Biovitrum Ltd
Tel: + 44 1638 722380 e-mail: mail.uk@sobi.com
Tato příbalová informace byla naposledy revidována
Tomuto léčivému přípravku bylo uděleno tzv. podmíněné schválení. Znamená to, že informace o tomto přípravku budou přibývat.
Evropská agentura pro léčivé přípravky nejméně jednou za rok vyhodnotí nové informace o tomto léčivém přípravku a tato příbalová informace bude podle potřeby aktualizována.
Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
71