Caprelsa 100 Mg
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Caprelsa 100 mg potahované tablety
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna potahovaná tableta obsahuje vandetanibum 100 mg. Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Potahovaná tableta
Kulaté bikonvexní bílé potahované tablety s vyraženým „Z100“ na jedné straně.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Caprelsa je indikována k léčbě agresivního a symptomatického medulárního karcinomu štítné žlázy (MTC) u pacientů s neresekovatelným lokálně pokročilým nebo metastatickým onemocněním.
U pacientů, u kterých není znám stav mutace RET (rearranged during transfection) nebo mutace není přítomna, je třeba počítat s možností menšího prospěchu z léčby předtím, než se učiní individuální léčebné rozhodnutí (viz důležité informace v bodech 4.4 a 5.1).
4.2 Dávkování a způsob podání
Léčbu má zahájit a vést lékař, který má zkušenosti s léčbou MTC, s používáním protinádorových léčiv a s vyhodnocováním elektrokardiogramu (EKG).
Na lékařském předpisu může být předepsáno pouze 1 balení přípravku. Pro vydání dalšího balení je třeba vystavit nový předpis.
Dávkování
Doporučená dávka je 300 mg jednou denně, užívá se s jídlem, nebo bez jídla v přibližně stejnou denní dobu.
Pokud dojde k vynechání dávky, je třeba ji užít, jakmile si pacient vzpomene. Pokud do další dávky zbývá méně než 12 hodin, pacient si vynechanou dávku nevezme. Pacient by neměl užívat dávku dvojnásobnou (dvě dávky ve stejnou dobu), aby nahradil zapomenutou dávku.
Pacienti léčení přípravkem Caprelsa musí obdržet „Pohotovostní kartu pro pacienta“ a musí být informováni o rizicích přípravku Caprelsa (viz též příbalová informace pro pacienta).
Doba léčby
Vandetanib lze podávat do té doby, dokud má pacient s MTC z léčby prospěch.
Úprava dávky
Před zahájením léčby je nutné pečlivě zhodnotit QTc interval. V případě toxicity stupně 3 nebo vyšší podle obecných terminologických kritérií pro nežádoucí účinky (CTCAE) nebo prodloužení intervalu QTc na EKG je třeba dávkování vandetanibu alespoň dočasně přerušit a znovu zahájit sníženou dávkou po odeznění toxických projevů nebo zlepšení toxicity na stupeň 1 podle CTCAE (viz též bod 4.4). Dávku 300 mg lze snížit na 200 mg (dvě 100 mg tablety) a dále na 100 mg, pokud je třeba. Pacienta je třeba vhodným způsobem monitorovat. Nežádoucí účinky včetně prodloužení intervalu QTc nemusí vzhledem k 19dennímu poločasu odeznít rychle (viz bod 4.4).
Zvláštní populace Pediatrická populace
Bezpečnost a účinnost nebyla u dětí stanovena. Z tohoto důvodu není vandetanib indikován u pediatrické populace.
Starší pacienti
U starších pacientů není třeba upravovat zahajovací dávku. U pacientů s MTC starších než 75 let jsou pouze omezené klinické zkušenosti týkající se vandetanibu.
Poškození ledvin
Farmakokinetická studie u dobrovolníků s mírným, středním a závažným poškozením ledvin prokázala, že expozice vandetanibu po jednorázové dávce je zvýšena až 1,5krát; 1,6krát a 2krát u pacientů s výchozím mírným, středně závažným (clearance kreatininu > 30 až < 50 ml/min) a závažným poškozením (clearance kreatininu nižší než 30 ml/min) (viz bod 5.2). Podle klinických údajů není třeba měnit zahajovací dávku u pacientů s mírným poškozením ledvin. U pacientů se středně závažným poškozením ledvin existují pouze omezené údaje s dávkou 300 mg: dávku bylo potřeba snížit na 200 mg u 5 ze 6 pacientů. U pacientů se středně závažným poškozením ledvin lze zahajovací dávku snížit na 200 mg; bezpečnost a účinnost dávky 200 mg však nebyla stanovena (viz bod 4.4). U pacientů se závažným poškozením funkce ledvin se nedoporučuje vandetanib podávat, neboť existují pouze omezené údaje u pacientů se závažným poškozením ledvin a v této skupině nebyla stanovena bezpečnost a účinnost.
Poškození jater
Nedoporučuje se podávat vandetanib pacientům s poškozením jater (sérový bilirubin vyšší než 1,5násobek horní hranice normy), neboť existují pouze omezené údaje u pacientů s poškozením jater a bezpečnost a účinnost nebyla v této skupině stanovena (viz bod 4.4).
Farmakokinetické údaje od dobrovolníků naznačují, že u pacientů s mírným, středně závažným či závažným poškozením jater není třeba upravovat zahajovací dávku (viz bod 5.2).
Způsob podání
U pacientů, kteří mají potíže s polykáním, lze tablety s vandetanibem rozptýlit v polovině sklenky nesycené pitné vody. Neměla by se používat jiná tekutina. Tableta se vhodí do vody a bez drcení se míchá, až se rozpadne (asi 10 minut) a vzniklá disperze se ihned vypije. Ulpělé částice se znovu dispergují v polovině sklenky vody a vypijí. Tuto disperzi je možné podat též nazogastrickou sondou nebo přes gastrostomickou trubici.
4.3 Kontraindikace
• Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
• Vrozený syndrom dlouhého QTc.
• Pacienti s intervalem QTc delším než 480 ms.
• Souběžné podávání vandetanibu a následujících léčivých přípravků známých tím, že prodlužují interval QTc a/nebo vyvolávají torsades de pointes: arsen, cisaprid, intravenózní erythromycin (i.v.), toremifen, mizolastin, moxifloxacin, antiarytmika třídy IA a III (viz bod 4.5).
• Kojení (viz bod 4.6).
4.4 Zvláštní upozornění a opatření pro použití
Vzhledem k doprovodným rizikům je důležité, aby byla léčba vandetanibem omezena na pacienty, kteří tuto léčbu skutečně potřebují, tj. pacienty se symptomatickou agresivní formou nemoci. Samotná symptomatická nemoc nebo progresivní nemoc nejsou dostatečným důvodem pro potřebu zahájit léčbu vandetanibem. Výskyt změn biomarkerů, např. kalcitoninu (CTN) a/nebo karcinoembryogenního antigenu (CEA), stejně tak jako výskyt změn objemu nádoru v průběhu pečlivého sledování mohou pomoci identifikovat nejen pacienty, kteří potřebují léčbu, ale též vhodný okamžik k zahájení léčby vandetanibem.
Prodloužení intervalu QTc a Torsades de pointes
Vandetanib v dávce 300 mg je spojen s podstatným a na koncentraci závislým prodloužením intervalu QTc (v průměru 28 ms, medián 35 ms). K prvnímu prodloužení intervalu QTc dochází nej častěji v průběhu prvních 3 měsíců léčby, ale může se objevit i později. Prodloužení intervalu QTc je zvláště problematické s ohledem na poločas vandetanibu (19 dnů) (viz bod 4.8). Prodloužení intervalu QTc na EKG na 500 ms při dávkování 300 mg denně u pacientů s MTC ve studii fáze III bylo pozorováno u 11 % pacientů. Prodloužení intervalu QTc na EKG se zdá být závislé na dávce. Méně často byly hlášeny torsades de pointes a komorové tachykardie u pacientů, kterým byl podáván vandetanib v dávce 300 mg denně. Riziko Torsades může být zvýšené u pacientů s elektrolytovou dysbalancí (viz bod 4.8).
Léčba vandetanibem se nesmí zahajovat u pacientů s hodnotou intervalu QTc na EKG větší než 480 ms. Vandetanib by se neměl podávat ani pacientům s anamnézou torsades de pointes, pokud nebyly korigovány všechny rizikové faktory, které přispívaly k výskytu torsades de pointes. Vandetanib nebyl studován u pacientů s komorovými arytmiemi a recentním infarktem myokardu.
Před léčbou, 1, 3, 6 a 12 týdnů po zahájení léčby a každé 3 měsíce nejméně po dobu jednoho roku je třeba provádět vyšetření EKG a stanovení sérové koncentrace draslíku, vápníku a hořčíku a hormonu stimulujícího štítnou žlázu (TSH). Toto časové schéma je třeba uplatnit i po snížení dávky v důsledku prodloužení intervalu QTc a po přerušení dávkování na dobu delší než 2 týdny.
V průběhu léčby i po léčbě je třeba provádět monitorování EKG a krevní testy, pokud je to klinicky indikováno. V častém monitorování intervalu QTc na EKG je třeba pokračovat.
Sérové koncentrace draslíku, hořčíku a vápníku je třeba udržovat v rozmezí normálních koncentrací, aby se snížilo riziko prodloužení intervalu QTc na EKG. Další monitorování intervalu QTc, elektrolytů a funkce ledvin se požaduje zvláště v případě průjmu, zhoršení průjmu/dehydrataci, elektrolytové dysbalanci a/nebo zhoršené funkci ledvin. Pokud dojde ke značnému prodloužení intervalu QTc, ale interval je do 500 ms, měl by být konzultován kardiolog.
Souběžné podávání vandetanibu s látkami, které jsou známy tím, že prodlužují interval QTc na EKG, je kontraindikováno nebo se nedoporučuje (viz body 4.3 a 4.5).
Souběžné podávání vandetanibu a ondansetronu se nedoporučuje (viz bod 4.5).
Pacienti, u kterých se vyvine jednotlivá hodnota intervalu QTc na EKG > 500 ms, musí přerušit užívání vandetanibu. V podávání snížené dávky lze pokračovat až po potvrzení návratu hodnoty intervalu QTc na EKG na hodnotu před léčbou a po korekci možné elektrolytové dysbalance.
Syndrom reverzibilní zadní encefalopatie, PRES (Reversible posterior leukoencephalopathy syndrome - RPLS)
PRES je syndrom subkortikálního vazogenního edému diagnostikovaný při MRI mozku, byl méně často pozorován při léčbě vandetanibem v kombinaci s chemoterapií. PRES byl též pozorován u pacientů léčených vandetanibem v monoterapii. Na tento syndrom je třeba myslet u všech pacientů, kteří mají epileptické záchvaty, bolest hlavy, poruchy vidění, jsou zmatení nebo jinak mentálně alterovaní. U každého pacienta, který má epileptické záchvaty, je zmatený nebo mentálně alterovaný, je třeba provést MRI mozku.
RET (Rearranged during transfection“) status
Pacienti bez přítomnosti mutace RET mohou mít snížený prospěch z léčby vandetanibem a poměr prospěchu/rizika se u této skupiny pacientů může lišit od skupiny s mutací RET.
U pacientů, u kterých může být status mutace RET negativní, je třeba vzít v úvahu možnost menšího prospěchu z léčby ještě před individuálním léčebným rozhodnutím a použití vandetanibu je nutné pečlivě zvážit s ohledem na léčebná rizika. Proto se doporučuje testování RET mutace. Pokud je to možné, je při stanovení statusu mutace RET žádoucí odebrat vzorky tkáně v době zahajování léčby a nikoliv v době stanovení diagnózy (viz body 4.1 a 5.1).
Kožní reakce
U pacientů, kterým byl podáván vandetanib, byla pozorována vyrážka a jiné kožní reakce včetně fotosenzitivní reakce a syndromu palmo-plantární erytrodysestézie. Mírné až středně závažné kožní reakce lze léčit symptomaticky, nebo snížením dávky či přerušením léčby. U závažnějších kožních reakcí (např. Stevens-Johnsonův syndrom) se doporučuje, aby pacient neodkladně vyhledal lékařskou pomoc.
Vzhledem k možnému riziku výskytu fototoxických reakcí spojených s léčbou vandetanibem je třeba opatrnosti při vystavení se slunečnímu záření a doporučuje se použít ochranný oděv a/nebo chránit se před přímým sluncem.
Průjem je příznakem souvisejícím s nemocí samotnou a dobře známým nežádoucím účinkem vandetanibu. K léčbě průjmu se doporučuje podávat obvyklé protiprůjmové prostředky. Častěji je nutné monitorovat QTc a hladiny sérových elektrolytů. Pokud se rozvine závažný průjem (CTCAE stupeň 3 - 4), je třeba léčbu vandetanibem přerušit, dokud průjem neustane. Po zlepšení stavu lze léčbu znovu zahájit se sníženou dávkou (viz body 4.2 a 4.8).
Krvácení
U pacientů s metastázami do mozku, kterým je podáván vandetanib, je třeba opatrnosti, neboť bylo hlášeno nitrolební krvácení.
Srdeční selhání
U pacientů léčených vandetanibem bylo pozorováno srdeční selhání. U pacientů se srdečním selháním může být nezbytné dočasné nebo i trvalé přerušení léčby. Přerušení léčby vandetanibem nemusí vést k reverzibilitě srdečního selhání. Některé případy byly smrtelné.
Hypertenze
U pacientů léčených vandetanibem byla pozorována hypertenze včetně hypertenzní krize. Pacienti by měli být sledováni pro možný výskyt hypertenze a podle potřeby kontrolováni. Pokud nelze vysoký krevní tlak kontrolovat režimovými opatřeními, nelze znovu zahájit léčbu vandetanibem, dokud není krevní tlak pod farmakologickou kontrolou. Snížení dávky může být nezbytné (viz bod 4.8).
Pacienti s poškozením ledvin
U pacientů se středně závažným a závažným poškozením ledvin se nedoporučuje podávat vandetanib, neboť existují pouze omezené údaje a v této skupině nebyla bezpečnost a účinnost stanovena (viz body 4.2, 5.1 a 5.2).
Pacienti s jaterním poškození
U pacientů s poškozením jater (sérový bilirubin vyšší než 1,5násobek horní hranice normy) se nedoporučuje podávat vandetanib, neboť existují pouze omezené údaje u pacientů s poškozením jater a bezpečnost a účinnost nebyla stanovena. Farmakokinetické údaje od dobrovolníků ukazují, že není potřebné upravovat zahajovací dávku u pacientů s mírným, středně závažným nebo závažným poškozením jater (viz body 4.2 a 5.2).
Vzestup koncentrací alaninaminotransferázy
U pacientů léčených vandetanibem často dochází k vzestupu koncentrací alaninaminotransferázy.
S pokračováním léčby dojde ve většině případů k obnovení původního stavu, v ostatních případech pak po přerušení léčby na dobu 1 - 2 týdnů. Doporučuje se pravidelně sledovat koncentraci alaninaminotransferázy.
Intersticiální plicní nemoc
U pacientů léčených vandetanibem byla pozorována intersticiální plicní nemoc (ILD) a některé její případy byly smrtelné. Pokud dojde u pacienta k rozvoji dušnosti, kašle a horečky, je třeba léčbu vandetanibem přerušit a ihned provést vyšetření. Pokud je ILD potvrzena, je třeba léčbu vandetanibem trvale vysadit a pacienta adekvátně léčit.
Induktory CYP3A4
Je třeba vyloučit souběžné podávání vandetanibu a silných induktorů CYP3A4 (např. rifampicin, třezalka tečkovaná, karbamazepin, fenobarbital) (viz bod 4.5).
Koncentrace CTN nižší než 500 pg/ml
Prospěch z léčby vandetanibem u pacientů s koncentrací CTN nižší než 500 pg/ml nebyl stanoven, a proto se doporučuje opatrnost u pacientů s hladinou CTN < 500 pg/ml vzhledem k rizikům spojeným s léčbou vandetanibem.
Pohotovostní karta pro pacienta
Všichni, kteří předepisují přípravek Caprelsa, musí být seznámeni s informacemi určenými pro lékaře a doporučeními k léčbě. Lékaři musí diskutovat rizika léčby přípravkem Caprelsa s pacientem a při každém předpisu přípravku vybavit pacienta „Pohotovostní kartou pro pacienta“.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Farmakokinetické interakce
Vliv vandetanibu na jiné léčivé přípravky
Expozice midazolamu (substrát pro CYP3A4) u zdravých dobrovolníků nebyla ovlivněna současným podáním jednorázové dávky 800 mg vandetanibu.
Vandetanib je inhibitorem transportéru 2 organických kationtů (OCT-2). Při souběžném podávání vandetanibu a metforminu zdravým dobrovolníkům s divokým typem OCT2 se AUC(0-t) a Cmax metforminu (substrát pro OCT2) zvýšila o 74 %, resp. 50 %, a Clr metforminu se snížila o 52 %.
U pacientů, kterým je souběžně podáván vandetanib a metformin, se doporučuje vhodné klinické a/nebo laboratorní monitorování a u těchto pacientů může být potřebná nižší dávka metforminu.
Při souběžném podávání digoxinu (substrát pro P-gp) zdravým dobrovolníkům se hodnoty AUC(0-t) a Cmax zvýšily o 23 %, resp. 29 %, v důsledku inhibice P-gp vandetanibem. Bradykardizující účinek digoxinu může dále zvyšovat riziko prodloužení intervalu QTc a Torsades de Pointes vandetanibem.
U pacientů souběžně léčených digoxinem a vandetanibem se doporučuje vhodné klinické monitorování (např. EKG) a/nebo laboratorní monitorování a u těchto pacientů může být potřebná nižší dávka digoxinu. (K monitorování vandetanibu viz bod 4.2 Dávkování a způsob podání a bod 4.4 Zvláštní upozornění a opatření pro podávání).
V případě kombinace s vandetanibem se doporučuje klinické monitorování u dalších substrátů pro P-gp, jako je např. dabigatran.
Vliv jiných léčivých přípravků na vandetanib
U zdravých dobrovolníků nebyla pozorována klinicky významná interakce mezi vandetanibem (jednorázová dávka 300 mg) a účinným inhibitorem CYP3A4 itrakonazolem (opakované dávky 200 mg jednou denně). U zdravých dobrovolníků mužů byla expozice vandetanibu snížena o 40 %, pokud byl podáván souběžně s účinným induktorem CYP3A4 rifampicinem. Podávání vandetanibu s účinnými induktory CYP3A4 je třeba vyloučit.
Při souběžném podávání omeprazolu zdravým dobrovolníkům se Cmax vandetanibu snížila o 15 % a hodnota AUC(0-t) vandetanibu se nezměnila. Při současném podávání s ranitidinem se hodnoty Cmax a AUC(0-t) vandetanibu nezměnily. Pokud je vandetanib podáván souběžně s omeprazolem nebo ranitidinem, není třeba upravovat dávkování vandetanibu.
Farmakodynamické interakce
Biliární exkrece nezměněného vandetanibu je jednou z vylučovacích cest pro vandetanib. Vandetanib není substrátem pro „multidrug resistance protein 2“ (MRP2), p-glykoprotein (P-gp) nebo „breast cancer resistance protein“ (BCRP).
Léčivé přípravky známé tím, že prodlužují QTc interval
Bylo prokázáno, že vandetanib prodlužuje QTc interval na EKG, méně často byly hlášeny torsades de pointes. Souběžné podávání vandetanibu s léčivými přípravky, které prodlužují QTc interval a/nebo indukují torsades de pointes, je buďto kontraindikováno nebo se nedoporučuje a to v závislosti na dostupnosti jiné alternativní léčby.
• Kombinace kontraindikované (viz bod 4.3): cisaprid, intravenózní erythromycin (i.v.), toremifen, mizolastin, moxifloxacin, arsen, antiarytmika třídy IA a III.
• Kombinace, které se nedoporučují: methadon, haloperidol, amisulprid, chlorpromazin, sulpirid, zuklopenthixol, halofantrin, pentamidin a lumefantrin.
Pokud neexistuje vhodná alternativní léčba, lze nedoporučované kombinace podávat souběžně s vandetanibem při dodatečném sledování QTc intervalu na EKG, hodnocení elektrolytů a další kontrole při nástupu nebo zhoršení průjmu.
Výsledky farmakodynamických a farmakokinetických interakčních studií ukazují, že souběžné podávání s ondansetronem zdravým pacientům má pouze malý vliv na farmakokinetiku vandetanibu, ale má malý aditivní vliv na prodloužení intervalu QTc o přibližně 10 ms. Z tohoto důvodu se nedoporučuje souběžné podávání ondansetronu a vandetanibu. Pokud je ondansetron podáván spolu s vandetanibem, je třeba pečlivě monitorovat hladiny elektrolytů v séru a EKG a důsledně korigovat jakékoliv abnormality.
Antagonisté vitaminu K
Vzhledem ke zvýšenému riziku trombotických příhod u pacientů s nádorovým onemocněním je použití antikoagulancií časté. Vzhledem k vysoké intraindividuální variabilitě odpovědi na podání antikoagulancia a možnosti interakce mezi antagonisty vitaminu K a chemoterapeutiky se doporučuje provádět častější hodnocení INR (International Normalised Ratio), pokud bylo rozhodnuto o léčbě antagonisty vitaminu K.
4.6 Fertilita, těhotenství a kojení
Ženy v reprodukčním věku
Ženy v reprodukčním věku musí používat účinnou antikoncepci v průběhu léčby a alespoň 4 měsíce po podání poslední dávky vandetanibu.
Existují pouze omezené údaje o použití vandetanibu v průběhu těhotenství. Vandetanib vykazuje významný vliv na všechna stádia samičí reprodukce u laboratorních potkanů (viz bod 5.3), což je v souladu s ohledem na farmakologický účinek vandetanibu.
Pokud je vandetanib podáván v průběhu těhotenství nebo pokud pacientka otěhotní v průběhu léčby vandetanibem, je třeba pacientku seznámit s možným nebezpečím vzniku vývojových vad plodu nebo rizikem potratu. V léčbě by se mělo pokračovat pouze tehdy, pokud potenciální prospěch pro matku převáží nad rizikem pro plod.
Kojení
Neexistují údaje o použití vandetanibu u kojících žen. Vandetanib a/nebo jeho metabolity se vylučují do mateřského mléka u laboratorních potkanů a byly nalezeny v plazmě mláďat, pokud byl vandetanib podáván v průběhu kojení (viz bod 5.3).
V průběhu léčby vandetanibem je kojení kontraindikováno.
Fertilita
Vandetanib nemá žádný vliv na samčí plodnost, ale zhoršuje samičí plodnost u laboratorních potkanů (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Nebyly provedeny žádné studie, které by stanovily vliv vandetanibu na schopnost řídit a ovládat stroje. Byly hlášeny případy únavy a neostrého vidění a tito pacienti, kteří mají tyto příznaky, by měli být opatrní, pokud řídí či ovládají stroje.
4.8 Nežádoucí účinky
Celkové shrnutí nežádoucích účinků
Nejčastěji hlášenými nežádoucími účinky byly průjem, vyrážka, nauzea, hypertenze a bolest hlavy. Nežádoucí účinky v průběhu klinických hodnocení
Následující nežádoucí účinky byly identifikovány v průběhu klinických studií u pacientů, kterým byl podáván vandetanib k léčbě MTC. Frekvence nežádoucích účinků jsou uvedeny v Tabulce 1, nežádoucí účinky podle Rady pro mezinárodní organizace lékařských věd (CIOMS III), jsou seřazeny podle tříd orgánových systémů (SOC) MedDRA a na podkladě preferenčních termínů a dále podle frekvence. Frekvence nežádoucích účinků jsou definovány takto: velmi časté (> 1/10); časté (> 1/100 až < 1/10); méně časté (> 1/1000 až < 1/100); vzácné (> 1/10 000 až < 1/1000); velmi vzácné (< 1/10 000); není známo (z dostupných údajů nelze určit). Tento bod zahrnuje pouze data z dokončených klinických studií se známou expozicí pacientů.
|
Tabulka 1 Nežádoucí účinky přípravku a třídy orgánových systémů | |||
|
Třída orgánových systémů |
Velmi časté |
Časté |
Méně časté |
|
Infekce a infestace |
Zánět nosohltanu, zánět průdušek, infekce horních dýchacích cest, infekce močových cest |
Zánět plic, sepse, chřipka, zánět močového měchýře, zánět vedlejšívch nosních dutin, zánět hrtanu, folikulitida, furunkl, plísňové infekce, pyelonefritida |
Zánět slepého střeva, stafylokokové infekce, diverkulitida, celulitida, absces břišní stěny |
|
Endokrinní poruchy |
Hypothyreóza | ||
|
Poruchy metabolismu a výživy |
Snížená chuť k jídlu, hypokalcémie |
Hypokalémie, hyperkalcémie, hyperglykémie, dehydratace, hyponatrémie |
Podvýživa |
|
Psychiatrické poruchy |
Úzkost | ||
|
Poruchy nervového systému |
Bolest hlavy, parestezie, dysestezie, závrať |
Třes, letargie, ztráta vědomí, poruchy rovnováhy, porucha chuti |
Epileptické záchvaty, klonus, otok mozku |
|
Poruchy oka |
Neostré vidění, strukturální změny rohovky (včetně depozit v rohovce a |
Poruchy vidění, vidění zářivého kruhu okolo předmětů/osob („halo vision“), fotopsie, |
Šedý zákal oční čočky, poruchy akomodace |
|
zákalu rohovky) |
zelený zákal oční čočky, zánět spojivky, sucho v očích, keratopatie | ||
|
Srdeční poruchy |
Prodloužení intervalu QTc na EKG (*) (**) |
Srdeční selhání, akutní srdeční selhání, poruchy srdečního rytmu, poruchy vedení srdce, komorové arytmie a srdeční zástava | |
|
Cévní poruchy |
Hypertenze |
Hypertenzní krize, ischemické cerebrovaskulární stavy | |
|
Respirační, hrudní a mediastinální poruchy |
Epistaxe, hemoptýza, intersticiální zánět plic |
Respirační selhání, aspirační pneumonie | |
|
Gastrointestinální poruchy |
Bolest břicha, průjem, nevolnost, zvracení, dyspepsie |
Zánět tlustého střeva, sucho v ústech, stomatitida, porucha polykání, zácpa, gastritida, gastrointestinální krvácení |
Pankreatitida, peritonitida, ileus, perforace střeva, inkontinence stolice |
|
Poruchy jater a žlučových cest |
Cholelithiáza | ||
|
Poruchy kůže a podkožní tkáně |
Fotosenzitivní reakce, vyrážka a jiné kožní reakce (včetně akné, suché kůže, dermatitidy a svědění), poruchy nehtů |
Syndrom palmo-plantární erytrodysestézie, alopecie |
Bulózní dermatitida |
|
Poruchy ledvin a močových cest |
Proteinurie, nefrolithiáza |
Obtíže při močení, hematurie, selhání ledvin, časté močení, nucení na močení |
Zbarvená moč, anurie |
|
Celkové poruchy a reakce v místě aplikace |
Astenie, únava, bolest, otoky |
Zpomalení hojení | |
|
Vyšetření |
Prodloužení intervalu QTc na EKG |
Zvýšení sérových koncentrací ALT a AST, pokles tělesné hmotnosti, zvýšení kreatitinu v krvi |
Zvýšené koncentrace hemoglobinu, zvýšené koncentrace sérové amylázy |
* 13,4 % pacientů léčených vandetanibem mělo QTc (podle Bazetta) > 500 ms ve srovnání s 1 % pacientů, kterým bylo podáváno placebo. U více než 91 % pacientů bylo prodloužení QTcF o > 20 ms, u 35 % o >60 ms a u 1,7 % o >100 ms. U 8 % pacientů došlo ke snížení dávky v důsledku prodloužení QTc.
** včetně 2 úmrtí u pacientů s QTc > 550 ms (jedno v důsledku sepse a jedno v důsledku srdečního selhání).
U pacientů léčených vandetanibem v monoterapii byly pozovány příhody jako torsades de pointes, Stevens-Johnsonův syndrom, erythema multiforme, intersticiální plicní nemoc (někdy fatální) a PRES (RPLS). Předpokládá se, že jde o méně časté nežádoucí účinky u pacientů, kterým je podáván vandetanib k léčbě MTC.
U pacientů, kterým byl podáván vandetanib k léčbě MTC, se často vyskytovaly oční příhody, jako je neostré vidění. Plánovaná vyšetření štěrbinovou lampou odhalila u léčených pacientů zákal rohovky (keratopatie); ovšem pravidelná vyšetření štěrbinovou lampou u pacientů léčených vandetanibem se nevyžadují.
Střední hodnoty koncentrací hemoglobinu při různých délkách expozice u pacientů léčených vandetanibem byly zvýšeny o 0,5 - 1,5 g/dl ve srovnání s výchozí hodnotou.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
V případě předávkování vandetanibem neexistuje specifická léčba a nebyly určeny ani možné příznaky předávkování. Ve studii u zdravých dobrovolníků a pacientů po opakovaných dávkách 300 mg a vyšších byla pozorována vyšší frekvence a závažnost některých nežádoucích účinků, jako je vyrážka, průjem a hypertenze. Je třeba uvažovat i o možnosti prodloužení intervalu QTc a torsades de pointes.
Nežádoucí účinky spojené s předávkováním je třeba léčit symptomaticky; zvláště správně musí být léčen závažný průjem. V případě předávkování je třeba přerušit podávání vandetanibu a je třeba přijmout opatření k vyloučení výskytu nežádoucích účinků, tj. monitorování EKG v průběhu 24 hodin ke stanovení prolongace QTc intervalu. Nežádoucí účinky spojené s předávkováním mohou být prolongované vzhledem k dlouhému poločasu vandetanibu (viz bod 5.2).
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antineoplastika, inhibitory protein kinázy, ATC kód: L01XE12 Mechanismus účinku a farmakodynamické účinky
Vandetanib je silným inhibitorem receptoru-2 vaskulárního endoteliálního růstového faktoru (VEGFR-2, též známého jako receptor s vazným místem pro kinázu [KDR]), receptoru epidermálního růstového faktoru (EGFR) a RET tyrosin kináz. Vandetanib je též sub-mikromolárním inhibitorem tyrosinkinázy pro vaskulární endoteliální receptor-3.
Vandetanib inhibujeVEGF stimulovanou migraci endoteliálních buněk, proliferaci, přežívání a tvorbu nových buněk krevních cév v in vitro modelech angiogeneze. Vandetanib dále inhibuje epidermálním růstovým faktorem (EGF) stimulovanou receptorovou tyrozin kinázu v nádorových a endoteliálních buňkách. Vandetanib inhibuje na EGFR závislou proliferaci a přežívání buněk in vitro. Vandetanib inhibuje jak divoký typ, tak většinu mutovaných aktivovaných forem RET a významně inhibuje proliferaci buněčných linií MTC in vitro.
Podávání vandetanibu v podmínkách in vivo snižuje angiogenezi indukovanou buňkami nádoru, permeabilitu nádorových cév, hustotu nádorových mikrokapilár a inhibuje růst nádoru u celé řady lidských modelů štěpu u athymických myší. Vandetanib též inhibuje růst nádorového štěpu MTC in
vivo.
Přesný mechanismus účinku vandetanibu u lokálně pokročilého nebo metastatického MTC není znám. Klinická účinnost a bezpečnost
Klinická data u MTC
Randomizovaná, dvojitě zaslepená studie kontrolovaná placebem (studie 58) byla provedena za účelem průkazu bezpečnosti a účinnosti vandetanibu 300 mg ve srovnání s placebem. Studie zahrnovala 331 pacientů s neresekovatelným lokálně pokročilým nebo metastatickým MTC. Zařazeni byli pouze pacienti s CTN > 500 pg/ml (konvenční jednotky) nebo > 146,3 pmol/l (standardizované mezinárodní jednotky). Z pacientů zařazených do studie mělo 10 pacientů ve větvi s vandetanibem a 4 pacienti ve větvi s placebem (4 % všech pacientů) performance status podle Světové zdravotnické organizace (WHO PS) > 2 a 28 pacientů (12,1 %) ve větvi s vandetanibem a 10 (10,1 %) ve větvi s placebem mělo srdeční poruchu. Srdeční porucha byla definována jako pacienti s anamnézou kardiovaskulární abnormality.
Primárním cílovým parametrem této studie bylo prokázat zlepšení v parametru přežití bez progrese (PFS) ve větvi s vandetanibem ve srovnání s placebem. Sekundárním cílovým parametrem bylo hodnocení celkového výskytu objektivní odpovědi (ORR), výskytu kontroly nemoci (DCR) definované jako, částečná odpověď (PR) nebo kompletní odpověď (CR) nebo stabilní nemoc (SD) po dobu nejméně 24 týdnů, trvání odpovědi (DOR), čas do zhoršení bolesti na podkladě stručné příručky bolesti (BPI) ve škále nejhorší bolesti a celkové přežití (OS). Primární cílový parametr PFS, ORR a DCR byly vyhodnocovány centrálním nezávislým zaslepeným posouzením obrazových dat. Jako sekundární cílový parametr byla hodnocena i biochemická odpověď na vandetanib ve srovnání s placebem měřením CTN a CEA.
Pacienti byli léčeni vandetanibem nebo placebem až do doby, kdy došlo k objektivní progresi nemoci. Když došlo, podle posouzení řešitele, k objektivní progresi nemoci, pacient byl vyřazen ze zaslepené studijní léčby a byla mu nabídnuta možnost podávání vandetanibu v otevřené fázi studie. V důsledku nežádoucích účinků přerušilo léčbu 28 z 231 pacientů (12,1 %) ve větvi s vandetanibem a 3 z 99 (3,0 %) v placebové větvi. Čtrnáct z 28 pacientů (50 %), kteří ukončili léčbu vandetanibem pro nežádoucí účinek, přerušilo léčbu bez snížení dávky. Pět pacientů ze šesti (83 %) se středně závažným selháním ledvin léčených vandetanibem mělo sníženu dávku na 200 mg v důsledku nežádoucího účinku; u jednoho pacienta bylo nutné snížit dávku na 100 mg.
Výsledky primární analýzy PFS prokázaly statisticky významné zlepšení PFS u pacientů randomizovaných do větve s vandetanibem ve srovnání s placebem (poměr rizik (HR) = 0,46; 95% interval spolehlivosti (CI) = 0,31-0,69; p = 0,0001).
Nebyl dosažen medián PFS u pacientů randomizovaných do větve s vandetanibem; ovšem na podkladě statistického modelování dat pozorovaných až do 43. percentilu je predikovaná hodnota mediánu PFS 30,5 měsíce a při 95% intervalu spolehlivosti 25,5 až 36,5 měsíce. Medián PFS u pacientů randomizovaných do placebové větve byl 19,3 měsíce. U pacientů randomizovaných do větve s vandetanibem byl po 12 měsících podíl pacientů žijících a bez progrese 192 (83 %) a v placebové větvi 63 (63 %). Ve větvi s vandetanibem celkem progredovalo 73 (32 %) pacientů; 64 (28 %) progresí podle kritérií hodnocení odpovědi u solidních nádorů (RECIST) a 9 (4 %) zemřelo bez známek progrese. Zbývajících 158 pacientů (68 %) bylo v analýze PFS cenzurováno. Ve větvi s placebem celkově progredovalo 51 (51 %) pacientů; 46 (46 %) progresí RECIST a 5 (5 %) zemřelo bez známek progrese. Zbývajících 49 pacientů (49 %) bylo v analýze PFS cenzurováno.
|
měsíce |
0 |
6 |
12 |
18 |
24 |
30 |
36 |
|
n-vandetanib |
231 |
196 |
169 |
140 |
40 |
1 |
0 |
|
n-placebo |
100 |
71 |
57 |
45 |
13 |
0 |
0 |
Obrázek 1. Kaplan-Meierova křivka PFS
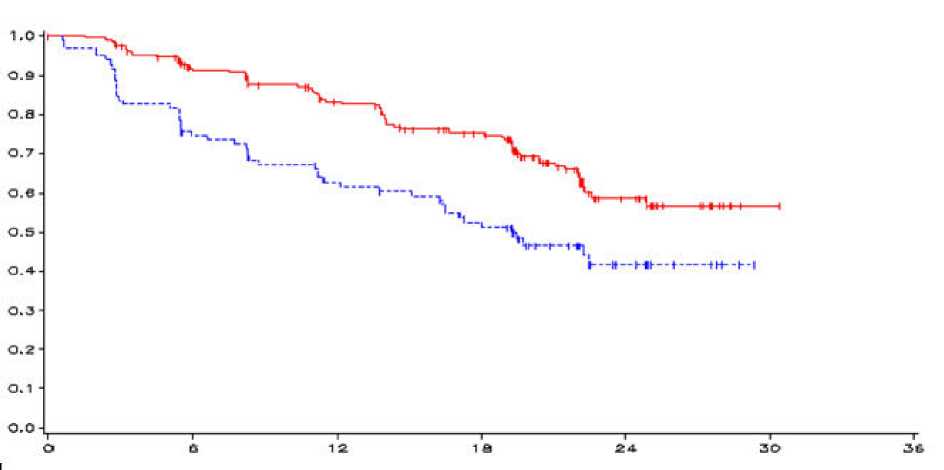
-vandetanib 300 mg, --------- placebo, osa-y = PFS, osa-x = čas v měsících, n-vandetanib = počet
rizikových pacientů vandetanib, n-placebo = počet rizikových pacientů placebo
HR = 0,46, 95% CI (0,36-0,69), p = 0,0001
|
PFS |
N |
Medián PFS |
HRa |
95% CI |
hodnota p |
|
Vandetanib 300 mg |
73/231 (32 %) |
Nebylo dosaženo (předpověď 30,5 měsíce) |
0,46 |
0,31; 0,69 |
0,0001 |
|
Placebo |
51/100 (51 %) |
19,3 měsíce |
V době primární analýzy PFS zemřelo 48 (15 %) pacientů a mezi léčebnými skupinami nebyl významný rozdíl v celkovém přežití (HR = 0,89; 99,98% CI = 0,28-2,85; p = 0,712). V době této analýzy zemřelo 32 pacientů ve větvi s vandetanibem (14 %) a 16 pacientů (16 %) ve větvi s placebem.
Většina (95 % pacientů) ^nělo ^netastazující one^nocnění. Čtrnáct pacientů léčených vandetanibe^n a tři pacienti, kterým bylo podáváno placebo, mělo pouze neresekovatelné lokálně pokročilé onemocnění. Existují pouze omezené údaje o použití vandetanibu u pacientů s neresekovatelným lokálně pokročilým onemocněním bez metastáz.
Statisticky významná výhoda byla pozorována ve větvi s vandetanibem v sekundárních cílových parametrech výskyt odpovědi, výskyt kontroly nemoci a biochemická odpověď.
Tabulka 2: Souhrn klíčových údajů účinnosti ve studii 58
|
ORRa |
N |
Výskyt odpovědi |
ORb |
95% CI |
Hodnota p |
|
Vandetanib 300 mg |
104/231 |
45 % |
5,48 |
2,99; 10,79 |
< 0,0001 |
|
Placebo |
13/100 |
13 % | |||
|
DCRa |
N |
Výskyt odpovědi |
ORb |
95% CI |
Hodnota p |
|
Vandetanib 300 mg |
200/231 |
87 % |
2,64 |
1,48; 4,69 |
0,001 |
|
Placebo |
71/100 |
71 % | |||
|
CTN ODPOVĚĎ |
N |
Výskyt odpovědi |
ORb |
95% CI |
Hodnota p |
|
Vandetanib 300 mg |
160/231 |
69 % |
72,9 |
26,2; 303,2 |
< 0,0001 |
|
Placebo |
3/100 |
3 % | |||
|
ODPOVĚĎ CEA |
N |
Výskyt odpovědi |
ORb |
95% CI |
Hodnota p |
|
Vandetanib 300 mg |
119/231 |
52 % |
52,0 |
16,0; 320,3 |
< 0,0001 |
|
Placebo |
2/100 |
2 % |
a Celkový výskyt odpovědi = kompletní + nekompletní odpověď. Výskyt kontroly nemoci = výskyt odpovědi + stabilní nemoc po 24 týdnech. Intent-to-treat (ITT) analýza zahrnuje pacienty, kterým byl podáván vandetanib v otevřené fázi studie předtím, než byla progrese stanovena centrálním posouzením. b OR = poměr šancí. Hodnota > 1 upřednostňuje vandetanib. Analýza byla provedena za použití logistického regresního modelu s léčbou jako jediným faktorem.
N = počet příhod/počet randomizovaných pacientů.
Statisticky významná výhoda byla pozorována u vandetanibu v sekundárním cílovém parametru času do zhoršení bolesti (odvozen od složeného cílového parametru za použití nejhoršího skóre bolesti v BPI a použití opioidů k analgézii hlášených pacientem) (vandetanib 49 %, placebo 57 %, HR 0,61; 97,5% CI (0,43 - 0,87, p < 0,006: 8 vs 3 měsíce). Neexistují statisticky významné rozdíly pozorované v exploratorním cílovém parametru průjem (hlášen jako frekvence stolic).
Stav mutace RET ve studii 58
Ve studii 58 byla mutace RET určována za použití metody polymerázové řetězové reakce (PCR) založené na metodě amplifikace refraktorního mutačního systému (ARMS) mutace M918T a přímého sekvencování DNA pro mutace v exonech 10, 11, 13, 14, 15 a 16 (místo mutace M918T) u všech sporadických pacientů, kde byla dostupná DNA (297/298).
Status RET nemohl být testován u velkého podílu pacientů (především pro nedostupnost výsledků přímého sekvencování DNA) a výskyt odpovědi byl poněkud nižší u pacientů s neznámým statutem mutace RET ve srovnání s přítomnou mutací RET: 51,8 % vs. 35,9 %. V zaslepeném srovnání vandetanibu vs. placebo byli pouze 2 pacienti se známou nepřítomností mutace RET na všech 6 exonech, kterým byl podáván vandetanib a kteří nevykazovali odpověď.
Byla provedena post-hoc analýza pivotní studie 58 na nepřítomnost mutace RET v podskupinách na základě nepřítomnosti mutace M918T. Mutace RET byla u pacienta považována za přítomnou, pokud byla v nádoru přítomna mutace M918T určená metodou ARMS, nebo mutace RET na kterémkoliv exonu zjištěná sekvencováním. Bylo identifikováno 79 pacientů bez přítomnosti mutace M918T a mutace RET identifikované na kterémkoliv ze 6 testovaných exonů, ale u 71 z těchto pacientů bylo sekvencování 6 exonů neúplné. Mutace M918T je nejčastější mutací pozorovanou u pacientů se sporadickou formou MTC; ovšem nelze vyloučit, že někteří pacienti bez přítomnosti mutace M918T nemají přítomnu mutaci na jiných exonech.
Výsledky podle statusu RET (přítomna, neznámý stav a nepřítomná mutace RET M918T) jsou uvedeny v Tabulce 3
Tabulka 3: Souhrn výsledků účinnosti ve skupině pacientů podle statusu mu tace RET
|
Pacienti s dokumentovanou mutací RET (n=187) |
Pacienti bez mutace M918T a jiných mutací buďto netestovaných nebo nepřítomných (n=79)* | |
|
Objektivní výskyt odpovědi (větev s vandetanibem) |
52 % |
35 % |
|
Cílový parametr |
0,45 (0,26; 0,78) |
0,57 (0,29; 1,13) |
|
účinnosti PFS HR | ||
|
(95%) interval | ||
|
spolehlivosti |
* Stav mutace RET byl u většiny pacientů určen v době diagnózy a mohl se od té doby změnit.
Pediatrická populace
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s vandetanibem u jedné nebo více podskupin pediatrické populace dědičným medulárním karcinomem štítné žlázy (informace o použití u dětí viz bod 4.2).
Tento léčivý přípravek byl registrován postupem tzv. podmíněného schválení. Znamená to, že jsou očekávány další důkazy o jeho přínosech. Evropská agentura pro léčivé přípravky (EMA) nejméně jednou za rok vyhodnotí nové informace o tomto léčivém přípravku a tento souhrn údajů o přípravku bude podle potřeby aktualizován.
5.2 Farmakokinetické vlastnosti
Absorpce
Po perorálním podání vandetanibu je absorpce pomalá a maximálních plazmatických koncentrací je typicky dosaženo s mediánem 6 hodin, rozmezí 4 - 10 hodin, po podání. Vandetanib se po opakovaném podání kumuluje přibližně 8násobně a rovnovážného stavu je dosaženo od přibližně 2 měsíců.
Distribuce
Vandetanib se váže na lidský sérový albumin a alfa-1 kyselý glykoprotein s vazebností in vitro přibližně 90 %. V ex vivo vzorcích od pacientů s kolorektálním karcinomem po podávání 300 mg jednou denně v rovnovážném stavu, byla průměrná vazba na bílkoviny 93,7 % (rozmezí 92,2 až 95,7 %). Farmakokinetika vandetanibu při dávce 300 mg u pacientů s MTC je charakterizována distribučním objemem přibližně 7450 l.
Biotransformace
Po perorálním podání 14C-vandetanibu byl v plazmě, moči a stolici detekován nezměněný vandetanib a metabolity vandetanibu N-oxid a N-desmetyl-vandetanib. Konjugát glukuronid byl pozorován jako minoritní metabolit pouze v exkretech. N-desmetyl-vandetanib se primárně vytváří CYP3A4 a vandetanib-N-oxid monooxygenázami (FMO1 a FMO3), které obsahují flavin. N-desmetyl-vandetanib a vandetanib-N-oxid cirkulují v koncentracích přibližně 11 % a 1,4 % koncentrací vandetanibu.
Eliminace
Farmakokinetika vandetanibu při dávce 300 mg u pacientů s MTC je charakterizována clearance přibližně 13,2 l/h a plazmatickým poločasem přibližně 19 dnů. V průběhu 21denního sběru po jednorázovém podání 14C-vandetanibu se vyloučí přiližně 69 %, z toho 44 % do stolice a 25 % do moči. Vylučování dávky bylo pomalé a je třeba počítat s dalším vylučováním po 21 dnech s ohledem na plazmatický poločas.
Zvláštní populace Poškození ledvin
Farmakokinetická studie po jednorázovém podání dávky dobrovolníkům ukázala, že expozice vandetanibu je u pacientů s mírným, středně závažným a závažným poškozením ledvin zvýšená (až 1,5krát; 1,6krát a 2krát) ve srovnání se subjekty s normální funkcí ledvin (viz body 4.2, 4.4 a 4.5).
Poškození jater
Farmakokinetická studie po jednorázovém podání dávky dobrovolníkům ukázala, že poškození jater neovlivňuje expozici vandetanibu. Existují pouze omezené údaje u pacientů s poškozením jater (sérový bilirubin vyšší než 1,5násobek horní hranice normy) (viz body 4.2 a 4.4).
Vliv _ potravy
Potrava nemá vliv na expozici vandetanibu.
5.3 Předklinické údaje vztahující se k bezpečnosti
Vandetanib nemá mutagenní ani klastogenní potenciál.
V toxikologických studiích s opakovanými dávkami trvajícími až 9 měsíců bylo zaznamenáno zvracení, ztráta tělesné hmotnosti a průjem u psů a porucha vývoje a růstu kostí u mladých psů a laboratorních potkanů s otevřenými růstovými ploténkami. U laboratorních potkanů byl zaznamenán vliv na zuby, ledviny a kůži. Tyto nálezy, které byly učiněny při klinicky relevantních plazmatických koncentracích, byly většinou reverzibilní v průběhu 4 týdnů po přerušení dávkování a byly důsledkem inhibice receptoru vaskulárního endoteliálního růstového faktoru (VEGFR) nebo EGFR.
Účinky pozorované v jiných studiích zahrnovaly inhibici hERG (ether-a-go-go gene) a prodloužení QTc intervalu u psů. U laboratorních potkanů a psů bylo pozorováno zvýšení systolického a diastolického krevního tlaku. U myší vandetanib zpomaloval, ale nevylučoval, hojení ran. Ve studii na cytotoxicitu byl prokázán fototoxický potenciál vandetanibu v podmínkách in vitro. Na zvířecím modelu hojení ran bylo zjištěno, že myši, kterým byl podáván vandetanib, mají sníženou pevnost kůže ve srovnání s kontrolami. Předpokládá se, že vandetanib zpomaluje, ale nevylučuje, hojení ran. Nebyl stanoven vhodný interval mezi přerušením léčby vandetanibem a elektivním chirurgickým zákrokem s ohledem na vyloučení rizika zhoršeného hojení ran. V klinických studiích podstoupil malý počet pacientů užívajících vandetanib chirurgický zákrok a nebyly hlášeny žádné komplikace při hojení ran.
Reprodukční toxikologie
Vandetanib nemá vliv na plodnost u samců laboratorních potkanů. Ve studii fertility u samic laboratorních potkanů byl zaznamenán trend ke zvýšené nepravidelnosti estrálního cyklu, mírné snížení výskytu březosti a zvýšení počtu implantačních ztrát. Ve studii s opakovaným podáním dávky u laboratorních potkanů byl zaznamenán snížený počet žlutých tělísek ve vaječnících potkanů, kterým byl podáván vandetanib po dobu 1 měsíce.
U laboratorních potkanů byla pozorována embryofetální toxicita jako ztráta plodu, opožděný vývoj plodu, abnormality srdečních cév a předčasná osifikace lebečních kostí. Ve studii na prenatální a postnatální vývoj u laboratorních potkanů vandetanib v dávkách vyvolávajících toxické příznaky u matky v průběhu gestace a/nebo laktace zvyšoval počet potratů a zpomaloval postnatální růst mláďat. Vandetanib se vylučoval do mateřského mléka u laboratorních potkanů a byl nalezen v plazmě mláďat při dávkování v průběhu laktace u laboratorních potkanů.
Kancerogenita
Vandetanib neměl kancerogenní potenciál ve studii na transgenních myších.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Jádro tablety
Dihydrát hydrogenfosforečnanu vápenatého
Mikrokrystalická celulosa
Krospovidon typ A
Povidon K 29-32
Magnesium-stearát
Potahová vrstva Hypromelosa Makrogol 300 Oxid titaničitý (E171)
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
4 roky
6.4 Zvláštní opatření pro uchovávání
Uchovávejte při teplotě do 30 °C.
6.5 Druh obalu a velikost balení
PVC/PVDC/Al blistry, uzavřené hliníkovou fólií, každý obsahuje 30 potahovaných tablet.
6.6 Zvláštní opatření pro likvidaci přípravku
Žádné zvláštní požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Genzyme Europe B.V., Gooimeer 10, 1411 DD Naarden, Nizozemsko
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/11/749/001
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 17. února 2012
Datum posledního prodloužení registrace: 15. ledna 2015
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou uveřejněny na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Caprelsa 300 mg potahované tablety
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna potahovaná tableta obsahuje vandetanibum 300 mg. Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Potahovaná tableta.
Oválné bikonvexní bílé potahované tablety s vyraženým „Z300“ na jedné straně.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Caprelsa je indikována k léčbě agresivního a symptomatického medulárního karcinomu štítné žlázy (MTC) u pacientů s neresekovatelným lokálně pokročilým nebo metastatickým onemocněním.
U pacientů, u kterých není znám stav mutace RET (rearranged during transfection) nebo je negativní, je třeba počítat s možností menšího prospěchu z léčby dříve, než se učiní individuální léčebné rozhodnutí (viz důležité informace v bodech 4.4 a 5.1).
4.2 Dávkování a způsob podání
Léčbu má zahájit a vést lékař, který má zkušenosti s léčbou MTC, s používáním protinádorových léčiv a s vyhodnocováním elektrokardiogramu (EKG).
Na lékařském předpisu může být předepsáno pouze 1 balení přípravku. Pro vydání dalšího balení je třeba vystavit nový předpis.
Dávkování
Doporučená dávka je jedna 300 mg tableta jednou denně, užívá se s jídlem, nebo bez jídla v přibližně stejnou denní dobu.
Pokud dojde k vynechání dávky, je třeba ji užít, jakmile si pacient vzpomene. Pokud do další dávky zbývá méně než 12 hodin, pacient si vynechanou dávku nevezme. Pacient by neměl užívat dávku dvojnásobnou (dvě dávky ve stejnou dobu), aby nahradil zapomenutou dávku.
Pacienti léčení přípravkem Caprelsa musí obdržet „Pohotovostní kartu pro pacienta“ a musí být informováni o rizicích přípravku Caprelsa (viz též příbalová informace pro pacienta).
Doba léčby
Vandetanib lze podávat do té doby, dokud má pacient s MTC z léčby prospěch.
Úprava dávky
Před zahájením léčby je nutné pečlivě zhodnotit QTc interval. V případě toxicity stupně 3 nebo vyšší podle obecných terminologických kritérií pro nežádoucí účinky (CTCAE) nebo prodloužení intervalu QTc na EKG je třeba dávkování vandetanibu alespoň dočasně přerušit a znovu zahájit sníženou dávkou po odeznění toxických projevů nebo zlepšení toxicity na stupeň 1 podle CTCAE (viz též bod 4.4). Dávku 300 mg lze snížit na 200 mg (dvě 100 mg tablety) a dále na 100 mg, pokud je třeba. Pacienta je třeba vhodným způsobem monitorovat. Nežádoucí účinky včetně prodloužení intervalu QTc nemusí vzhledem k 19dennímu poločasu odeznít rychle (viz bod 4.4).
Zvláštní populace Pediatrická populace
Bezpečnost a účinnost nebyla u dětí stanovena. Z tohoto důvodu není vandetanib indikován u pediatrické populace.
Starší pacienti
U starších pacientů není třeba upravovat zahajovací dávku. U pacientů s MTC starších než 75 let jsou pouze omezené klinické zkušenosti týkající se vandetanibu.
Poškození ledvin
Farmakokinetická studie u dobrovolníků s mírným, středním a závažným poškozením ledvin prokázala, že expozice vandetanibu po jednorázové dávce je zvýšena až 1,5krát, 1,6krát a 2krát u pacientů s výchozím mírným, středně závažným (clearance kreatininu > 30 až < 50 ml/min) a závažným poškozením (clearance kreatininu nižší než 30 ml/min) (viz bod 5.2). Podle klinických údajů není třeba měnit zahajovací dávku u pacientů s mírným poškozením ledvin. U pacientů se středně závažným poškozením ledvin existují pouze omezené údaje s dávkou 300 mg: dávku bylo potřeba snížit na 200 mg u 5 ze 6 pacientů. U pacientů se středně závažným poškozením ledvin lze zahajovací dávku snížit na 200 mg; bezpečnost a účinnost dávky 200 mg však nebyla stanovena (viz bod 4.4). U pacientů se závažným poškozením funkce ledvin se nedoporučuje vandetanib podávat, neboť existují pouze omezené údaje u pacientů se závažným poškozením ledvin a v této skupině nebyla stanovena bezpečnost a účinnost.
Poškození jater
Nedoporučuje se podávat vandetanib pacientům s poškozením jater (sérový bilirubin vyšší než 1,5násobek horní hranice normy), neboť existují pouze omezené údaje u pacientů s poškozením jater a bezpečnost a účinnost nebyla v této skupině stanovena (viz bod 4.4).
Farmakokinetické údaje od dobrovolníků naznačují, že u pacientů s mírným, středně závažným či závažným poškozením jater není třeba upravovat zahajovací dávku (viz bod 5.2).
Způsob podání
U pacientů, kteří mají potíže s polykáním, lze tablety s vandetanibem rozplýtil v polovině sklenky nesycené pitné vody. Neměla by se používat jiná tekutina. Tableta se vhodí do vody a bez drcení se míchá, až se rozpadne (asi 10 minut) a vzniklá disperze se ihned vypije. Ulpělé částice se znovu dispergují v polovině sklenky vody a vypijí. Tuto disperzi je možné podat též nazogastrickou sondou nebo přes gastrostomickou trubici.
4.3 Kontraindikace
• Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
• Vrozený syndrom dlouhého QTc.
• Pacienti s intervalem QTc delším než 480 ms.
• Souběžné podávání vandetanibu a následujících léčivých přípravků známých tím, že prodlužují interval QTc a/nebo vyvolávají torsades de pointes: arsen, cisaprid, intravenózní erythromycin (i.v.), toremifen, mizolastin, moxifloxacin, antiarytmika třídy IA a III (viz bod 4.5).
• Kojení (viz bod 4.6).
4.4 Zvláštní upozornění a opatření pro použití
Vzhledem k doprovodným rizikům je důležité, aby byla léčba vandetanibem omezena na pacienty, kteří tuto léčbu skutečně potřebují, tj. pacienty se symptomatickou agresivní formou nemoci. Samotná symptomatická nemoc nebo progresivní nemoc nejsou dostatečným důvodem pro potřebu zahájit léčbu vandetanibem. Výskyt změn biomarkerů, např. kalcitoninu (CTN) a/nebo karcinoembryogenního antigenu (CEA), stejně tak jako výskyt změn objemu nádoru v průběhu pečlivého sledování mohou pomoci identifikovat nejen pacienty, kteří potřebují léčbu, ale též vhodný okamžik k zahájení léčby vandetanibem.
Prodloužení intervalu QTc a Torsades de pointes
Vandetanib v dávce 300 mg je spojen s podstatným a na koncentraci závislým prodloužením intervalu QTc (v průměru 28 ms, medián 35 ms). K prvnímu prodloužení intervalu QTc dochází nej častěji v průběhu prvních 3 měsíců léčby, ale může se objevit i později. Prodloužení intervalu QTc je zvláště problematické s ohledem na poločas vandetanibu (19 dnů) (viz bod 4.8). Prodloužení intervalu QTc na EKG na 500 ms při dávkování 300 mg denně u pacientů s MTC ve studii fáze III bylo pozorováno u 11 % pacientů. Prodloužení intervalu QTc na EKG se zdá být závislé na dávce.
Méně často byly hlášeny torsades de pointes a komorové tachykardie u pacientů, kterým byl podáván vandetanib v dávce 300 mg denně. Riziko Torsades může být zvýšené u pacientů s elektrolytovou dysbalancí (viz bod 4.8).
Léčba vandetanibem se nesmí zahajovat u pacientů s hodnotou intervalu QTc na EKG větší než 480 ms. Vandetanib by se neměl podávat ani pacientům s anamnézou torsades de pointes, pokud nebyly korigovány všechny rizikové faktory, které přispívaly k výskytu torsades de pointes. Vandetanib nebyl studován u pacientů s komorovými arytmiemi a recentním infarktem myokardu.
Před léčbou, 1, 3, 6 a 12 týdnů po zahájení léčby a každé 3 měsíce nejméně po dobu jednoho roku je třeba provádět vyšetření EKG a stanovení sérové koncentrace draslíku, vápníku a hořčíku a hormonu stimulujícího štítnou žlázu (TSH). Toto časové schéma je třeba uplatnit i po snížení dávky v důsledku prodloužení intervalu QTc a po přerušení dávkování na dobu delší než 2 týdny.
V průběhu léčby i po léčbě je třeba provádět monitorování EKG a krevní testy, pokud je to klinicky indikováno. V častém monitorování intervalu QTc na EKG je třeba pokračovat.
Sérové koncentrace draslíku, hořčíku a vápníku je třeba udržovat v rozmezí normálních koncentrací, aby se snížilo riziko prodloužení intervalu QTc na EKG. Další monitorování intervalu QTc, elektrolytů a funkce ledvin se požaduje zvláště v případě průjmu, zhoršení průjmu/dehydrataci, elektrolytové dysbalanci a/nebo zhoršené funkci ledvin. Pokud dojde ke značnému prodloužení intervalu QTc, ale interval je do 500 ms, měl by být konzultován kardiolog.
Souběžné podávání vandetanibu s látkami, které jsou známy tím, že prodlužují interval QTc na EKG, je kontraindikováno nebo se nedoporučuje (viz body 4.3 a 4.5).
Souběžné podávání vandetanibu a ondansetronu se nedoporučuje (viz bod 4.5).
Pacienti, u kterých se vyvine jednotlivá hodnota intervalu QTc na EKG > 500 ms, musí přerušit užívání vandetanibu. V podávání snížené dávky lze pokračovat až po potvrzení návratu hodnoty intervalu QTc na EKG na hodnotu před léčbou a po korekci možné elektrolytové dysbalance.
Syndrom reverzibilní zadní encefalopatie, PRES (Reversible posterior leukoencephalopathy syndrome - RPLS)
PRES je syndrom subkortikálního vazogenního edému diagnostikovaný při MRI mozku, byl méně často pozorován při léčbě vandetanibem v kombinaci s chemoterapií. PRES byl též pozorován u pacientů léčených vandetanibem v monoterapii. Na tento syndrom je třeba myslet u všech pacientů,
kteří mají epileptické záchvaty, bolest hlavy, poruchy vidění, jsou zmatení nebo jinak mentálně alterovaní. U každého pacienta, který má epileptické záchvaty, je zmatený nebo mentálně alterovaný, je třeba provést MRI mozku.
RET (Rearranged during transfection) status
Pacienti bez přítomnosti mutace RET mohou mít snížený prospěch z léčby vandetanibem a poměr prospěchu/rizika se u této skupiny pacientů může lišit od skupiny s mutací RET.
U pacientů, u kterých může být status mutace RET negativní, je třeba vzít v úvahu možnost menšího prospěchu z léčby ještě před individuálním léčebným rozhodnutím a použití vandetanibu je nutné pečlivě zvážit s ohledem na léčebná rizika. Proto se doporučuje testování RET mutace. Pokud je to možné, je při stanovení statusu mutace RET žádoucí odebrat vzorky tkáně v době zahajování léčby a nikoliv v době stanovení diagnózy (viz body 4.1 a 5.1).
Kožní reakce
U pacientů, kterým byl podáván vandetanib, byla pozorována vyrážka a jiné kožní reakce včetně fotosenzitivní reakce a syndromu palmo-plantární erytrodysestézie. Mírné až středně závažné kožní reakce lze léčit symptomaticky a/nebo úpravou dávky. U závažnějších kožních reakcí (např. Stevens-Johnsonův syndrom) se doporučuje, aby pacient neodkladně vyhledal lékařskou pomoc.
Vzhledem k možnému riziku výskytu fototoxických reakcí spojených s léčbou vandetanibem je třeba opatrnosti při vystavení se slunečnímu záření a doporučuje se použít ochranný oděv a/nebo chránit se před přímým sluncem.
Průjem je příznakem souvisejícím s nemocí samotnou a dobře známým nežádoucím účinkem vandetanibu. K léčbě průjmu se doporučuje podávat obvyklé protiprůjmové prostředky. Častěji je nutné monitorovat QTc a hladiny sérových elektrolytů. Pokud se rozvine závažný průjem (CTCAE stupeň 3-4), je třeba léčbu vandetanibem přerušit, dokud průjem neustane. Po zlepšení stavu lze léčbu znovu zahájit se sníženou dávkou (viz body 4.2 a 4.8).
Krvácení
U pacientů s metastázami do mozku, kterým je podáván vandetanib, je třeba opatrnosti, neboť bylo hlášeno nitrolební krvácení.
Srdeční selhání
U pacientů léčených vandetanibem bylo pozorováno srdeční selhání. U pacientů se srdečním selháním může být nezbytné dočasné nebo i trvalé přerušení léčby. Přerušení léčby vandetanibem nemusí vést k reverzibilitě srdečního selhání. Některé případy byly smrtelné.
Hypertenze
U pacientů léčených vandetanibem byla pozorována hypertenze včetně hypertenzní krize. Pacienti by měli být sledováni pro možný výskyt hypertenze a podle potřeby kontrolováni. Pokud nelze vysoký krevní tlak kontrolovat režimovými opatřeními, nelze znovu zahájit léčbu vandetanibem, dokud není krevní tlak pod farmakologickou kontrolou. Snížení dávky může být nezbytné (viz bod 4.8).
Pacienti s poškozením ledvin
U pacientů se středně závažným a závažným poškozením ledvin se nedoporučuje podávat vandetanib, neboť existují pouze omezené údaje a v této skupině nebyla bezpečnost a účinnost stanovena (viz body 4.2, 5.1 a 5.2).
Pacienti s jaterním poškození
U pacientů s poškozením jater (sérový bilirubin vyšší než 1,5násobek horní hranice normy) se nedoporučuje podávat vandetanib, neboť existují pouze omezené údaje u pacientů s poškozením jater a bezpečnost a účinnost nebyla stanovena. Farmakokinetické údaje získané od dobrovolníků ukazují, že není potřebné upravovat zahajovací dávku u pacientů s mírným, středně závažným nebo závažným poškozením jater (viz body 4.2 a 5.2).
Vzestup koncentrací alaninaminotransferázv
U pacientů léčených vandetanibem často dochází k vzestupu koncentrací alaninaminotransferázy.
S pokračováním léčby dojde ve většině případů k obnovení původního stavu, v ostatních případech pak po přerušení léčby na dobu 1-2 týdnů. Doporučuje se pravidelně sledovat koncentraci alaninaminotransferázy.
Intersticiální plicní nemoc
U pacientů léčených vandetanibem byla pozorována intersticiální plicní nemoc (ILD) a některé její případy byly smrtelné. Pokud dojde u pacienta k rozvoji dušnosti, kašle a horečky, je třeba léčbu vandetanibem přerušit a ihned provést vyšetření. Pokud je ILD potvrzena, je třeba léčbu vandetanibem trvale vysadit a pacienta adekvátně léčit.
Induktory CYP3A4
Je třeba vyloučit souběžné podávání vandetanibu a silných induktorů CYP3A4 (např. rifampicin, třezalka tečkovaná, karbamazepin, fenobarbital) (viz bod 4.5).
Koncentrace CTN nižší než 500 pg/ml
Prospěch z léčby vandetanibem u pacientů s koncentrací CTN nižší než 500 pg/ml nebyl stanoven, a proto se doporučuje opatrnost u pacientů s hladinou CTN < 500 pg/ml vzhledem k rizikům spojeným s léčbou vandetanibem.
Pohotovostní karta pro pacienta
Všichni, kteří předepisují přípravek Caprelsa, musí být seznámeni s informacemi určenými pro lékaře a doporučeními k léčbě. Lékaři musí diskutovat rizika léčby přípravkem Caprelsa s pacientem a vybavit pacienta „Pohotovostní kartou pro pacienta“ při každém předpisu přípravku.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Farmakokinetické interakce
Vliv vandetanibu na jiné léčivé přípravky
Expozice midazolamu (substrát pro CYP3A4) u zdravých dobrovolníků nebyla ovlivněna současným podáním jednorázové dávky 800 mg vandetanibu.
Vandetanib je inhibitorem transportéru 2 organických kationtů (OCT-2). Při souběžném podávání vandetanibu a metforminu zdravým dobrovolníkům s divokým typem OCT2 se AUC(0-t) a Cmax metforminu (substrát pro OCT2) zvýšila o 74 %, resp. 50 %, a Clr metforminu se snížila o 52 %.
U pacientů, kterým je souběžně podáván vandetanib a metformin, se doporučuje vhodné klinické a/nebo laboratorní monitorování a u těchto pacientů může být potřebná nižší dávka metforminu.
Při souběžném podávání digoxinu (substrát pro P-gp) zdravým dobrovolníkům se hodnoty AUC(0-t) a Cmax zvýšily o 23 %, resp. 29 %, v důsledku inhibice P-gp vandetanibem. Bradykardizující účinek digoxinu může dále zvyšovat riziko prodloužení intervalu QTc a Torsades de Pointes vandetanibem.
U pacientů souběžně léčených digoxinem a vandetanibem se doporučuje vhodné klinické monitorování (např. EKG) a/nebo laboratorní monitorování a u těchto pacientů může být potřebná nižší dávka digoxinu. (K monitorování vandetanibu viz bod 4.2 Dávkování a způsob podání a bod 4.4 Zvláštní upozornění a opatření pro podávání).
V případě kombinace s vandetanibem se doporučuje klinické monitorování u dalších substrátů pro P-gp, jako je např. dabigatran.
Vliv jiných léčivých přípravků na vandetanib
U zdravých dobrovolníků nebyla pozorována klinicky významná interakce mezi vandetanibem (jednorázová dávka 300 mg) a účinným inhibitorem CYP3A4 itrakonazolem (opakované dávky 200 mg jednou denně). U zdravých dobrovolníků mužů byla expozice vandetanibu snížena o 40 %, pokud byl podáván souběžně s účinným induktorem CYP3A4 rifampicinem. Podávání vandetanibu s účinnými induktory CYP3A4 je třeba vyloučit.
Při souběžném podávání omeprazolu zdravým dobrovolníkům se Cmax vandetanibu snížila o 15 % a hodnota AUC(0-t) vandetanibu se nezměnila. Při současném podávání s ranitidinem se hodnoty Cmax a AUC(0-t) vandetanibu nezměnily. Pokud je vandetanib podáván souběžně s omeprazolem nebo ranitidinem, není třeba upravovat dávkování vandetanibu.
Farmakodynamické interakce
Biliární exkrece nezměněného vandetanibu je jednou z vylučovacích cest pro vandetanib. Vandetanib není substrátem pro „multidrug resistance protein 2“ (MRP2), p-glykoprotein (P-gp) nebo „breast cancer resistance protein“ (BCRP).
Léčivé přípravky známé tím, že prodlužují QTc interval
Bylo prokázáno, že vandetanib prodlužuje QTc interval na EKG, méně často byly hlášeny torsades de pointes. Souběžné podávání vandetanibu s léčivými přípravky, které prodlužují QTc interval a/nebo indukují torsades de pointes, je buďto kontraindikováno nebo se nedoporučuje a to v závislosti na dostupnosti jiné alternativní léčby.
• Kombinace kontraindikované (viz bod 4.3): cisaprid, intravenózní erythromycin (i.v.), toremifen, mizolastin, moxifloxacin, arsen, antiarytmika třídy IA a III.
• Kombinace, které se nedoporučují: methadon, haloperidol, amisulprid, chlorpromazin, sulpirid, zuklopenthixol, halofantrin, pentamidin a lumefantrin.
Pokud neexistuje vhodná alternativní léčba, lze nedoporučované kombinace podávat souběžně s vandetanibem při dodatečném sledování QTc intervalu na EKG, hodnocení elektrolytů a další kontrole při nástupu nebo zhoršení průjmu.
Výsledky farmakodynamických a farmakokinetických interakčních studií ukazují, že souběžné podávání s ondansetronem zdravým pacientům má pouze malý vliv na farmakokinetiku vandetanibu, ale má malý aditivní vliv na prodloužení intervalu QTc o přibližně 10 ms. Z tohoto důvodu se nedoporučuje souběžné podávání ondansetronu a vandetanibu. Pokud je ondansetron podáván spolu s vandetanibem, je třeba pečlivě monitorovat hladiny elektrolytů v séru a EKG a důsledně korigovat jakékoliv abnormality.
Antagonisté vitaminu K
Vzhledem ke zvýšenému riziku trombotických příhod u pacientů s nádorovým onemocněním je použití antikoagulancií časté. Vzhledem k vysoké intraindividuální variabilitě odpovědi na podání antikoagulancia a možnosti interakce mezi antagonisty vitaminu K a chemoterapeutiky se, doporučuje provádět častější hodnocení INR (International Normalised Ratio), pokud bylo rozhodnuto o léčbě antagonisty vitaminu K.
4.6 Fertilita, těhotenství a kojení
Ženy v reprodukčním věku
Ženy v reprodukčním věku musí používat účinnou antikoncepci v průběhu léčby a alespoň 4 měsíce po podání poslední dávky vandetanibu.
Existují pouze omezené údaje o použití vandetanibu v průběhu těhotenství. Vandetanib vykazuje významný vliv na všechna stádia samičí reprodukce u laboratorních potkanů (viz bod 5.3), což je v souladu s ohledem na farmakologický účinek vandetanibu.
Pokud je vandetanib podáván v průběhu těhotenství nebo pokud pacientka otěhotní v průběhu léčby vandetanibem, je třeba pacientku seznámit s možným nebezpečím vzniku vývojových vad plodu nebo rizikem potratu. V léčbě by se mělo pokračovat pouze tehdy, pokud potenciální prospěch pro matku převáží nad rizikem pro plod.
Kojení
Neexistují údaje o použití vandetanibu u kojících žen. Vandetanib a/nebo jeho metabolity se vylučují do mateřského mléka u laboratorních potkanů a byly nalezeny v plazmě mláďat, pokud byl vandetanib podáván v průběhu kojení (viz bod 5.3).
V průběhu léčby vandetanibem je kojení kontraindikováno.
Fertilita
Vandetanib nemá žádný vliv na samčí plodnost, ale zhoršuje samičí plodnost u laboratorních potkanů (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Nebyly provedeny žádné studie, které by stanovily vliv vandetanibu na schopnost řídit a ovládat stroje. Byly hlášeny případy únavy a neostrého vidění a tito pacienti, kteří mají tyto příznaky, by měli být opatrní, pokud řídí či ovládají stroje.
4.8 Nežádoucí účinky
Celkové shrnutí nežádoucích účinků
Nejčastěji hlášenými nežádoucími účinky byly průjem, vyrážka, nauzea, hypertenze a bolest hlavy. Nežádoucí účinky v průběhu klinických hodnocení
Následující nežádoucí účinky byly identifikovány v průběhu klinických studií u pacientů, kterým byl podáván vandetanib k léčbě MTC. Frekvence nežádoucích účinků jsou uvedeny v Tabulce 1, nežádoucí účinky podle Rady pro mezinárodní organizace lékařských věd (CIOMS III), jsou seřazeny podle tříd orgánových systémů (SOC) MedDRA a na podkladě preferenčních termínů a dále podle frekvence. Frekvence nežádoucích účinků jsou definovány takto: velmi časté (> 1/10); časté (> 1/100 až < 1/10); méně časté (> 1/1000 až < 1/100); vzácné (> 1/10 000 až < 1/1000); velmi vzácné (< 1/10 000); není známo (z dostupných údajů nelze určit). Tento bod zahrnuje pouze data z dokončených klinických studií se známou expozicí pacientů.
Tabulka 1 Nežádoucí účinky přípravku a třídy prgánových systémů
|
Třída orgánových systémů |
Velmi časté |
Časté |
Méně časté |
|
Infekce a infestace |
Zánět nosohltanu, zánět průdušek, infekce horních dýchacích cest, infekce močových cest |
Zánět plic, sepse, chřipka, zánět močového měchýře, zánět vedlejšívch nosních dutin, zánět hrtanu, folikulitida, furunkl, plísňové infekce, pyelonefritida |
Zánět slepého střeva, stafylokokové infekce, diverkulitida, celulitida, absces břišní stěny |
|
Endokrinní poruchy |
Hypothyreóza | ||
|
Poruchy metabolismu a výživy |
Snížená chuť k jídlu, hypokalcémie |
Hypokalémie, hyperkalcémie, hyperglykémie, dehydratace, hyponatrémie |
Podvýživa |
|
Psychiatrické poruchy |
Úzkost | ||
|
Poruchy nervového systému |
Bolest hlavy, parestezie, dysestezie, závrať |
Třes, letargie, ztráta vědomí, poruchy rovnováhy, porucha chuti |
Epileptické záchvaty, klonus, otok mozku |
|
Poruchy oka |
Neostré vidění, strukturální změny rohovky (včetně depozit v rohovce a |
Poruchy vidění, vidění zářivého kruhu okolo předmětů/osob („halo vision“), fotopsie, |
Šedý zákal oční čočky, poruchy akomodace |
|
zákalu rohovky) |
zelený zákal oční čočky, zánět spojivky, sucho v očích, keratopatie | ||
|
Srdeční poruchy |
Prodloužení intervalu QTc na EKG (*) (**) |
Srdeční selhání, akutní srdeční selhání, poruchy srdečního rytmu, poruchy vedení srdce, komorové arytmie a srdeční zástava | |
|
Cévní poruchy |
Hypertenze |
Hypertenzní krize, ischemické cerebrovaskulární stavy | |
|
Respirační, hrudní a mediastinální poruchy |
Epistaxe, hemoptýza, intersticiální zánět plic |
Respirační selhání, aspirační pneumonie | |
|
Gastrointestinální poruchy |
Bolest břicha, průjem, nevolnost, zvracení, dyspepsie |
Zánět tlustého střeva, sucho v ústech, stomatitida, porucha polykání, zácpa, gastritida, gastrointestinální krvácení |
Pankreatitida, peritonitida, ileus, perforace střeva, inkontinence stolice |
|
Poruchy jater a žlučových cest |
Cholelithiáza | ||
|
Poruchy kůže a podkožní tkáně |
Fotosenzitivní reakce, vyrážka a jiné kožní reakce (včetně akné, suché kůže, dermatitidy a svědění), poruchy nehtů |
Syndrom palmo-plantární erytrodysestézie, alopecie |
Bulózní dermatitida |
|
Poruchy ledvin a močových cest |
Proteinurie, nefrolithiáza |
Obtíže při močení, hematurie, selhání ledvin, časté močení, nucení na močení |
Zbarvená moč, anurie |
|
Celkové poruchy a reakce v místě aplikace |
Astenie, únava, bolest, otoky |
Zpomalení hojení | |
|
Vyšetření |
Prodloužení intervalu QTc na EKG |
Zvýšení sérových koncentrací ALT a AST, pokles tělesné hmotnosti, zvýšení kreatitinu v krvi |
Zvýšené koncentrace hemoglobinu, zvýšené koncentrace sérové amylázy |
* 13,4 % pacientů léčených vandetanibem mělo QTc (podle Bazetta) > 500 ms ve srovnání s 1 % pacientů, kterým bylo podáváno placebo. U více než 91 % pacientů bylo prodloužení QTcF o > 20 ms, u 35 % o >60 ms a u 1,7 % o >100 ms. U 8 % pacientů došlo ke snížení dávky v důsledku prodloužení QTc.
** včetně 2 úmrtí u pacientů s QTc > 550 ms (jedno v důsledku sepse a jedno v důsledku srdečního selhání).
U pacientů léčených vandetanibem v monoterapii byly pozovány příhody jako torsades de pointes, Stevens-Johnsonův syndrom, erythema multiforme, intersticiální plicní nemoc (někdy fatální) a PRES (RPLS). Předpokládá se, že jde o méně časté nežádoucí účinky u pacientů, kterým je podáván vandetanib k léčbě MTC.
U pacientů, kterým byl podáván vandetanib k léčbě MTC, se často vyskytovaly oční příhody, jako je neostré vidění. Plánovaná vyšetření štěrbinovou lampou odhalila u léčených pacientů zákal rohovky (keratopatie); ovšem pravidelná vyšetření štěrbinovou lampou u pacientů léčených vandetanibem se nevyžadují.
Střední hodnoty koncentrací hemoglobinu při různých délkách expozice u pacientů léčených vandetanibem byly zvýšeny o 0,5-1,5 g/dl ve srovnání s výchozí hodnotou.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
V případě předávkování vandetanibem neexistuje specifická léčba a nebyly určeny ani možné příznaky předávkování. Ve studii u zdravých dobrovolníků a pacientů po opakovaných dávkách 300 mg a vyšších byla pozorována vyšší frekvence a závažnost některých nežádoucích účinků, jako je vyrážka, průjem a hypertenze. Je třeba uvažovat i o možnosti prodloužení intervalu QTc a torsades de pointes.
Nežádoucí účinky spojené s předávkováním je třeba léčit symptomaticky; zvláště správně musí být léčen závažný průjem. V případě předávkování je třeba přerušit podávání vandetanibu a je třeba přijmout opatření k vyloučení výskytu nežádoucích účinků, tj. monitorování EKG v průběhu 24 hodin ke stanovení prolongace QTc intervalu. Nežádoucí účinky spojené s předávkováním mohou být prolongované vzhledem k dlouhému poločasu vandetanibu (viz bod 5.2).
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antineoplastika, inhibitory protein kinázy, ATC kód: L01XE12 Mechanismus účinku a farmakodynamické účinky
Vandetanib je silným inhibitorem receptoru-2 vaskulárního endoteliálního růstového faktoru (VEGFR-2, též známého jako receptor s vazným místem pro kinázu [KDR]), receptoru epidermálního růstového faktoru (EGFR) a RET tyrosin kináz. Vandetanib je též sub-mikromolárním inhibitorem tyrosinkinázy pro vaskulární endoteliální receptor-3.
Vandetanib inhibujeVEGF stimulovanou migraci endoteliálních buněk, proliferaci, přežívání a tvorbu nových buněk krevních cév v in vitro modelech angiogeneze. Vandetanib dále inhibuje epidermálním růstovým faktorem (EGF) stimulovanou receptorovou tyrozin kinázu v nádorových a endoteliálních buňkách. Vandetanib inhibuje na EGFR závislou proliferaci a přežívání buněk in vitro. Vandetanib inhibuje jak divoký typ, tak většinu mutovaných aktivovaných forem RET a významně inhibuje proliferaci buněčných linií MTC in vitro.
Podávání vandetanibu v podmínkách in vivo snižuje angiogenezi indukovanou buňkami nádoru, permeabilitu nádorových cév, hustotu nádorových mikrokapilár a inhibuje růst nádoru u celé řady lidských modelů štěpu u athymických myší. Vandetanib též inhibuje růst nádorového štěpu MTC in
vivo.
Přesný mechanismus účinku vandetanibu u lokálně pokročilého nebo metastatického MTC není znám. Klinická účinnost a bezpečnost
Klinická data u MTC
Randomizovaná, dvojitě zaslepená studie kontrolovaná placebem (studie 58) byla provedena za účelem průkazu bezpečnosti a účinnosti vandetanibu 300 mg ve srovnání s placebem. Studie zahrnovala 331 pacientů s neresekovatelným lokálně pokročilým nebo metastatickým MTC. Zařazeni byli pouze pacienti s CTN > 500 pg/ml (konvenční jednotky) nebo > 146,3 pmol/l (standardizované mezinárodní jednotky). Z pacientů zařazených do studie mělo 10 pacientů ve větvi s vandetanibem a 4 pacienti ve větvi s placebem (4 % všech pacientů) performance status podle Světové zdravotnické organizace (WHO PS) > 2 a 28 pacientů (12,1 %) ve větvi s vandetanibem a 10 (10,1 %) ve větvi s placebem mělo srdeční poruchu. Srdeční porucha byla definována jako pacienti s anamnézou kardiovaskulární abnormality.
Primárním cílovým parametrem této studie bylo prokázat zlepšení v parametru přežití bez progrese (PFS) ve větvi s vandetanibem ve srovnání s placebem. Sekundárním cílovým parametrem bylo hodnocení celkového výskytu objektivní odpovědi (ORR), výskytu kontroly nemoci (DCR) definované jako, částečná odpověď (PR) nebo kompletní odpověď (CR) nebo stabilní nemoc (SD) po dobu nejméně 24 týdnů, trvání odpovědi (DOR), čas do zhoršení bolesti na podkladě stručné příručky bolesti (BPI) ve škále nejhorší bolesti a celkové přežití (OS). Primární cílový parametr PFS, ORR a DCR byly vyhodnocovány centrálním nezávislým zaslepeným posouzením obrazových dat. Jako sekundární cílový parametr byla hodnocena i biochemická odpověď na vandetanib ve srovnání s placebem měřením CTN a CEA.
Pacienti byli léčeni vandetanibem nebo placebem až do doby, kdy došlo k objektivní progresi nemoci. Když došlo, podle posouzení řešitele, k objektivní progresi nemoci, pacient byl vyřazen ze zaslepené studijní léčby a byla mu nabídnuta možnost podávání vandetanibu v otevřené fázi studie. V důsledku nežádoucích účinků přerušilo léčbu 28 z 231 pacientů (12,1 %) ve větvi s vandetanibem a 3 z 99 (3,0 %) v placebové větvi. Čtrnáct z 28 pacientů (50 %), kteří ukončili léčbu vandetanibem pro nežádoucí účinek, přerušilo léčbu bez snížení dávky. Pět pacientů ze šesti (83 %) se středně závažným selháním ledvin léčených vandetanibem mělo sníženu dávku na 200 mg v důsledku nežádoucího účinku; u jednoho pacienta bylo nutné snížit dávku na 100 mg.
Výsledky primární analýzy PFS prokázaly statisticky významné zlepšení PFS u pacientů randomizovaných do větve s vandetanibem ve srovnání s placebem (poměr rizik (HR) = 0,46; 95% interval spolehlivosti (CI) = 0,31-0,69; p = 0,0001).
Nebyl dosažen medián PFS u pacientů randomizovaných do větve s vandetanibem; ovšem na podkladě statistického modelování dat pozorovaných až do 43. percentilu je predikovaná hodnota mediánu PFS 30,5 měsíce a při 95% intervalu spolehlivosti 25,5 až 36,5 měsíce. Medián PFS u pacientů randomizovaných do placebové větve byl 19,3 měsíce. U pacientů randomizovaných do větve s vandetanibem byl po 12 měsících podíl pacientů žijících a bez progrese 192 (83 %) a v placebové větvi 63 (63 %). Ve větvi s vandetanibem celkem progredovalo 73 (32 %) pacientů; 64 (28 %) progresí podle kritérií hodnocení odpovědi u solidních nádorů (RECIST) a 9 (4 %) zemřelo bez známek progrese. Zbývajících 158 pacientů (68 %) bylo v analýze PFS cenzurováno. Ve větvi s placebem celkově progredovalo 51 (51 %) pacientů; 46 (46 %) progresí RECIST a 5 (5 %) zemřelo bez známek progrese. Zbývajících 49 pacientů (49 %) bylo v analýze PFS cenzurováno.
|
měsíce |
0 |
6 |
12 |
18 |
24 |
30 |
36 |
|
n-vandetanib |
231 |
196 |
169 |
140 |
40 |
1 |
0 |
|
n-placebo |
100 |
71 |
57 |
45 |
13 |
0 |
0 |
Obrázek 1. Kaplan-Meierova křivka PFS
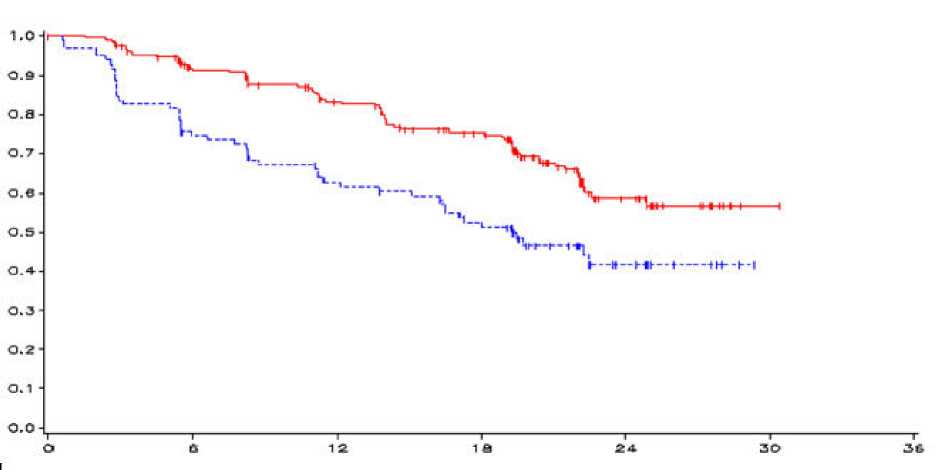
-vandetanib 300 mg, --------- placebo, osa-y = PFS, osa-x = čas v měsících, n-vandetanib = počet
rizikových pacientů vandetanib, n-placebo = počet rizikových pacientů placebo
HR = 0,46, 95% CI (0,36-0,69), p = 0,0001
|
PFS |
N |
Medián PFS |
HRa |
95% CI |
hodnota p |
|
Vandetanib 300 mg |
73/231 (32 %) |
Nebylo dosaženo (předpověď 30,5 měsíce) |
0,46 |
0,31; 0,69 |
0,0001 |
|
Placebo |
51/100 (51 %) |
19,3 měsíce |
V době primární analýzy PFS zemřelo 48 (15 %) pacientů a mezi léčebnými skupinami nebyl významný rozdíl v celkovém přežití (HR = 0,89; 99,98% CI = 0,28-2,85; p = 0,712). V době této analýzy zemřelo 32 pacientů ve větvi s vandetanibem (14 %) a 16 pacientů (16 %) ve větvi s placebem.
Většina (95 % pacientů) ^nělo ^netastazující one^nocnění. Čtrnáct pacientů léčených vandetanibe^n a tři pacienti, kterým bylo podáváno placebo, mělo pouze neresekovatelné lokálně pokročilé onemocnění. Existují pouze omezené údaje o použití vandetanibu u pacientů s neresekovatelným lokálně pokročilým onemocněním bez metastáz.
Statisticky významná výhoda byla pozorována ve větvi s vandetanibem v sekundárních cílových parametrech výskyt odpovědi, výskyt kontroly nemoci a biochemická odpověď.
Tabulka 2: Souhrn klíčových údajů účinnosti ve studii 58
|
ORRa |
N |
Výskyt odpovědi |
ORb |
95% CI |
Hodnota p |
|
Vandetanib 300 mg |
104/231 |
45 % |
5,48 |
2,99; 10,79 |
< 0,0001 |
|
Placebo |
13/100 |
13 % | |||
|
DCRa |
N |
Výskyt odpovědi |
ORb |
95% CI |
Hodnota p |
|
Vandetanib 300 mg |
200/231 |
87 % |
2,64 |
1,48; 4,69 |
0,001 |
|
Placebo |
71/100 |
71 % | |||
|
CTN ODPOVĚĎ |
N |
Výskyt odpovědi |
ORb |
95% CI |
Hodnota p |
|
Vandetanib 300 mg |
160/231 |
69 % |
72,9 |
26,2; 303,2 |
< 0,0001 |
|
Placebo |
3/100 |
3 % | |||
|
ODPOVĚĎ CEA |
N |
Výskyt odpovědi |
ORb |
95% CI |
Hodnota p |
|
Vandetanib 300 mg |
119/231 |
52 % |
52,0 |
16,0; 320,3 |
< 0,0001 |
|
Placebo |
2/100 |
2 % |
a Celkový výskyt odpovědi = kompletní + nekompletní odpověď. Výskyt kontroly nemoci = výskyt odpovědi + stabilní nemoc po 24 týdnech. Intent-to-treat (ITT) analýza zahrnuje pacienty, kterým byl podáván vandetanib v otevřené fázi studie předtím, než byla progrese stanovena centrálním posouzením.
b OR = poměr šancí. Hodnota > 1 upřednostňuje vandetanib. Analýza byla provedena za použití logistického regresního modelu s léčbou jako jediným faktorem.
N = počet příhod/počet randomizovaných pacientů.
Statisticky významná výhoda byla pozorována u vandetanibu v sekundárním cílovém parametru času do zhoršení bolesti (odvozen od složeného cílového parametru za použití nejhoršího skóre bolesti v BPI a použití opioidů k analgézii hlášených pacientem) (vandetanib 49 %, placebo 57 %, HR 0,61, 97,5% CI (0,43-0,87, p < 0,006: 8 vs 3 měsíce). Neexistují statisticky významné rozdíly pozorované v exploratorním cílovém parametru průjem (hlášen jako frekvence stolic).
Stav mutace RET ve studii 58
Ve studii 58 byla mutace RET určována za použití metody polymerázové řetězové reakce (PCR) založené na metodě amplifikace refraktorního mutačního systému (ARMS) mutace M918T a přímého sekvencování DNA pro mutace v exonech 10, 11, 13, 14, 15 a 16 (místo mutace M918T) u všech sporadických pacientů, kde byla dostupná DNA (297/298).
Status RET nemohl být testován u velkého podílu pacientů (především pro nedostupnost výsledků přímého sekvencování DNA) a výskyt odpovědi byl poněkud nižší u pacientů s neznámým statusem mutace RET ve srovnání s přítomnou mutací RET: 51,8 % vs. 35,9 %. V zaslepeném srovnání vandetanibu vs. placebo byli pouze 2 pacienti se známou nepřítomností mutace RET na všech 6 exonech, kterým byl podáván vandetanib a kteří nevykazovali odpověď.
Byla provedena post-hoc analýza pivotní studie 58 na nepřítomnost mutace RET v podskupinách na základě nepřítomnosti mutace M918T. Mutace RET byla u pacienta považována za přítomnou, pokud byla v nádoru přítomna mutace M918T určená metodou ARMS, nebo mutace kteréhokoliv exonu zjištěná sekvencováním. Bylo identifikováno 79 pacientů bez přítomnosti mutace M918T a mutace RET identifikované na kterémkoliv ze 6 testovaných exonů, ale u 71 z těchto pacientů bylo sekvencování 6 exonů neúplné. Mutace M918T je nejčastější mutací pozorovanou u pacientů se sporadickou formou MTC; ovšem nelze vyloučit, že někteří pacienti bez přítomnosti mutace M918T nemají přítomnu mutaci na jiných exonech.
Výsledky podle statusu RET (přítomna, neznámý stav a nepřítomná mutace RET M918T) jsou uvedeny v Tabulce 3
Tabulka 3: Souhrn výsledků účinnosti ve skupině pacientů podle statusu mu tace RET
|
Pacienti s dokumentovanou |
Pacienti bez mutace | |
|
mutací RET |
M918T a jiných | |
|
(n=187) |
mutací buďto netestovaných nebo nepřítomných (n=79)* |
|
Objektivní výskyt odpovědi (větev s vandetanibem) |
52 % |
35 % |
|
Cílový parametr účinnosti PFS HR (95%) interval spolehlivosti |
0,45 (0,26; 0,78) |
0,57 (0,29; 1,13) |
* Stav mutace RET byl u většiny pacientů určen v době diagnózy a mohl se od té doby změnit. Pediatrická populace
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s vandetanibem u jedné nebo více podskupin pediatrické populace dědičným medulárním karcinomem štítné žlázy (informace o použití u dětí viz bod 4.2).
Tento léčivý přípravek byl registrován postupem tzv. podmíněného schválení. Znamená to, že jsou očekávány další důkazy o jeho přínosech. Evropská agentura pro léčivé přípravky (EMA) nejméně jednou za rok vyhodnotí nové informace o tomto léčivém přípravku a tento souhrn údajů o přípravku bude podle potřeby aktualizován.
5.2 Farmakokinetické vlastnosti
Absorpce
Po perorálním podání vandetanibu je absorpce pomalá a maximálních plazmatických koncentrací je typicky dosaženo s mediánem 6 hodin, rozmezí 4-10 hodin, po podání. Vandetanib se po opakovaném podání kumuluje přibližně 8násobně a rovnovážného stavu je dosaženo od přibližně 2 měsíců.
Distribuce
Vandetanib se váže na lidský sérový albumin a alfa-1 kyselý glykoprotein s vazebností in vitro přibližně 90 %. V ex vivo vzorcích od pacientů s kolorektálním karcinomem po podávání 300 mg jednou denně v rovnovážném stavu, byla průměrná vazba na bílkoviny 93,7 % (rozmezí 92,2 až 95,7 %). Farmakokinetika vandetanibu při dávce 300 mg u pacientů s MTC je charakterizována distribučním objemem přibližně 7450 l.
Biotransformace
Po perorálním podání 14C-vandetanibu byl v plazmě, moči a stolici detekován nezměněný vandetanib a metabolity vandetanibu N-oxid a N-desmetyl-vandetanib. Konjugát glukuronid byl pozorován jako minoritní metabolit pouze v exkretech. N-desmetyl-vandetanib se primárně vytváří CYP3A4 a vandetanib-N-oxid monooxygenázami (FMO1 a FMO3), které obsahují flavin. N-desmetyl-vandetanib a vandetanib-N-oxid cirkulují v koncentracích přibližně 11 % a 1,4 % koncentrací vandetanibu.
Eliminace
Farmakokinetika vandetanibu při dávce 300 mg u pacientů s MTC je charakterizována clearance přibližně 13,2 l/h a plazmatickým poločasem přibližně 19 dnů. V průběhu 21denního sběru po jednorázovém podání 14C-vandetanibu se vyloučí přiližně 69 %, z toho 44 % do stolice a 25 % do moči. Vylučování dávky bylo pomalé a je třeba počítat s dalším vylučováním po 21 dnech s ohledem na plazmatický poločas.
Zvláštní _ populace Poškození ledvin
Farmakokinetická studie po jednorázovém podání dávky dobrovolníkům ukázala, že expozice vandetanibu je u pacientů s mírným, středně závažným a závažným poškozením ledvin zvýšená (až 1,5krát; 1,6krát a 2krát) ve srovnání se subjekty s normální funkcí ledvin (viz body 4.2, 4.4 a 4.5).
Poškození jater
Farmakokinetická studie po jednorázovém podání dávky dobrovolníkům ukázala, že poškození jater neovlivňuje expozici vandetanibu. Existují pouze omezené údaje u pacientů s poškozením jater (sérový bilirubin vyšší než 1,5násobek horní hranice normy) (viz body 4.2 a 4.4).
Vliv _ potravy
Potrava nemá vliv na expozici vandetanibu.
5.3 Předklinické údaje vztahující se k bezpečnosti
Vandetanib nemá mutagenní ani klastogenní potenciál.
V toxikologických studiích s opakovanými dávkami trvajícími až 9 měsíců bylo zaznamenáno zvracení, ztráta tělesné hmotnosti a průjem u psů a porucha vývoje a růstu kostí u mladých psů a laboratorních potkanů s otevřenými růstovými ploténkami. U laboratorních potkanů byl zaznamenán vliv na zuby, ledviny a kůži. Tyto nálezy, které byly učiněny při klinicky relevantních plazmatických koncentracích, byly většinou reverzibilní v průběhu 4 týdnů po přerušení dávkování a byly důsledkem inhibice receptoru vaskulárního endoteliálního růstového faktoru (VEGFR) nebo EGFR.
Účinky pozorované v jiných studiích zahrnovaly inhibici genu hERG (human ether-a-go-go gene) a prodloužení QTc intervalu u psů. U laboratorních potkanů a psů bylo pozorováno zvýšení systolického a diastolického krevního tlaku. U myší vandetanib zpomaloval, ale nevylučoval, hojení ran. Ve studii na cytotoxicitu byl prokázán fototoxický potenciál vandetanibu v podmínkách in vitro. Na zvířecím modelu hojení ran bylo zjištěno, že myši, kterým byl podáván vandetanib, mají sníženou pevnost kůže ve srovnání s kontrolami. Předpokládá se, že vandetanib zpomaluje, ale nevylučuje, hojení ran. Nebyl stanoven vhodný interval mezi přerušením léčby vandetanibem a elektivním chirurgickým zákrokem s ohledem na vyloučení rizika zhoršeného hojení ran. V klinických studiích podstoupil malý počet pacientů užívajících vandetanib chirurgický zákrok a nebyly hlášeny žádné komplikace při hojení ran.
Reprodukční toxikologie
Vandetanib nemá vliv na plodnost u samců laboratorních potkanů. Ve studii fertility u samic laboratorních potkanů byl zaznamenán trend ke zvýšené nepravidelnosti estrálního cyklu, mírné snížení výskytu březosti a zvýšení počtu implantačních ztrát. Ve studii s opakovaným podáním dávky u laboratorních potkanů byl zaznamenán snížený počet žlutých tělísek ve vaječnících potkanů, kterým byl podáván vandetanib po dobu 1 měsíce.
U laboratorních potkanů byla pozorována embryofetální toxicita jako ztráta plodu, opožděný vývoj plodu, abnormality srdečních cév a předčasná osifikace lebečních kostí. Ve studii na prenatální a postnatální vývoj u laboratorních potkanů vandetanib v dávkách vyvolávajících toxické příznaky u matky v průběhu gestace a/nebo laktace zvyšoval počet potratů a zpomaloval postnatální růst mláďat. Vandetanib se vylučoval do mateřského mléka u laboratorních potkanů a byl nalezen v plazmě mláďat při dávkování v průběhu laktace u laboratorních potkanů.
Kancerogenita
Vandetanib neměl kancerogenní potenciál ve studii na transgenních myších.
6. FARMACEUTICKÉ ÚDAJE 6.1 Seznam pomocných látek
Jádro tablety
Dihydrát hydrogenfosforečnanu vápenatého
Mikrokrystalická celulosa
Krospovidon typ A
Povidon K 29-32
Magnesium-stearát
Potahová vrstva Hypromelosa Makrogol 300 Oxid titaničitý (E171)
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
4 roky
6.4 Zvláštní opatření pro uchovávání
Uchovávejte při teplotě do 30 °C.
6.5 Druh obalu a velikost balení
PVC/PVDC/Al blistry, uzavřené hliníkovou fólií, každý obsahuje 30 potahovaných tablet.
6.6 Zvláštní opatření pro likvidaci přípravku
Žádné zvláštní požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Genzyme Europe B.V., Gooimeer 10, 1411 DD Naarden, Nizozemsko
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/11/749/002
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 17. února 2012
Datum posledního prodloužení registrace: 15. ledna 2015
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou uveřejněny na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu
PŘÍLOHA II
A. VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
E. ZVLÁŠTNÍ POVINNOST USKUTEČNIT POREGISTRAČNÍ OPATŘENÍ PRO PODMÍNĚNOU REGISTRACI PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného za propouštění šarží
AstraZeneca UK Ltd.
Silk Road Business Park Macclesfield, Cheshire SK10 2NA Velká Británie
V příbalové informaci k léčivému přípravku musí být uveden název a adresa výrobce odpovědného za propouštění dané šarže.
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 schválené registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
Aktualizovaný RMP se předkládá každý rok až do prodloužení registrace.
• Další opatření k minimalizaci rizik
Držitel rozhodnutí o registraci musí odsouhlasit formát a obsah edukačních materiálů s národní lékovou agenturou v každé členské zemi.
Držitel rozhodnutí o registraci (MAH) musí zajistit, že všichni lékaři, u kterých lze předpokládat používání a/nebo předepisování přípravku Caprelsa, obdrží Edukační balíček při a po uvedení přípravku na trh.
Edukační balíček musí obsahovat následující:
• Souhrn údajů o přípravku a příbalovou informaci
• Edukační materiál pro lékaře
• Pohotovostní kartu pacienta (text odsouhlasený CHMP)
Edukační materiál pro lékaře musí obsahovat následující klíčové informace:
• Vandetanib prodlužuje interval QTc a může vyvolat torsades de pointes a náhlé úmrtí
• Léčba vandetanibem se nesmí zahajovat u pacientů:
o jejichž interval QTc na EKG je delší než 480 ms o kteří mají vrozený syndrom dlouhého QTc
o kteří mají v anamnéze torsades de pointes, pokud nebyly všechny rizikové faktory
přispívající k torsades de pointes korigovány.
• Nezbytnost monitorovat EKG, sérové koncentrace draslíku, vápníku a hořčíku a hormonu stimulujícího štítnou žlázu (TSH) a časy a situace, kdy by měly být prováděny
• Pacienti, u kterých se vyvine jednotlivá korigovaná hodnota intervalu QTc na EKG alespoň 500 ms, musí přestat užívat vandetanib. Podávání vandetanibu lze znovu zahájit s nižší dávkou teprve po potvrzení návratu intervalu QTc na EKG na hodnoty před léčbou a pokud byla provedena korekce možné elektrolytové dysbalance.
• Pokud se hodnota intervalu QTc zásadně prodlouží, ale zůstane pod hodnotou 500 ms, je třeba žádat o radu kardiologa.
• Detaily léčivých přípravků, u kterých je souběžné podávání s vandetanibem buďto kontraindikováno nebo se nedoporučuje.
• Informaci, že vandetanib může vyvolat syndrom reverzibilní zadní encefalopatie (PRES) známý též jako syndrom reverzibilní zadní leukoencefalopatie (PRLS).
• Na PRES je třeba myslet u každého pacienta, který si stěžuje na epileptické záchvaty, bolest hlavy, poruchy vidění, zmatenost nebo alteraci mentálních funkcí. U každého pacienta, který si stěžuje na epileptické záchvaty, zmatenost nebo alteraci mentálních funkcí, musí být provedeno MRI vyšetření mozku.
• Nutnost informovat pacienty o riziku prodloužení intervalu QTc a PRES a informovat je o příznacích a projevech, kterých by si měli všímat, a jak se zachovat.
• Význam a použití Pohotovostní karty pacienta.
E. ZVLÁŠTNÍ POVINNOST USKUTEČNIT POREGISTRAČNÍ OPATŘENÍ PRO
PODMÍNĚNOU REGISTRACI PŘÍPRAVKU
Tato registrace byla schválena postupem tzv. podmíněného schválení, a proto podle článku 14(7)
nařízení (ES) č. 726/2004 držitel rozhodnutí o registraci uskuteční v daném termínu následující
opatření:
|
Popis |
Termín splnění |
|
Otevřená klinická studie srovnávající pacienty se sporadickou formou medulárního karcinomu štítné žlázy bez přítomnosti mutace RET a pacienty s přítomnou mutací RET léčené vandetanibem na podkladě protokolu schváleného CHMP. Studie bude zahrnovat přibližně 60 % pacientů, kterým bude podáván vandetanib v rámci EU. |
3Q 2020 |
|
Vstupní kritéria: splnění kritérií indikací uvedených v souhrnu údajů o přípravku. Dále budou zahrnuti a sledováni i pacienti bez přítomnosti mutace RET, kterým by nebyl podáván vandetanib v důsledku stavu mutace RET nebo kontraindikace. | |
|
Vylučovací kritéria: omezené na kontraindikace uvedené v bodě 4.3 souhrnu údajů o přípravku. | |
|
Údaje shromažďované ve studii: |
• Anamnéza a fyzikální vyšetření
• Status mutace RET
• Pacienti bez požadavku na provedení tkáňové biopsie k určení statusu RET pro zařazení do studie
Status mutace RET:
Před zařazením do studie nebude požadováno, aby byla u pacienta provedena nová tkáňová biopsie k určení statusu mutace RET. Řešitelé by však měli být velmi podpořeni v tom, aby získávali recentní vzorky tkáně k určení stavu mutace RET tak často, jak je to možné, dokonce i u pacientů testovaných v časnějších stádiích nemoci. Určení stavu mutace RET by mělo přednostně proběhnout právě před zahájením léčby. Budou shromažďovány údaje o typu tkáně pro vyšetření, datum tkáňové biopsie, druh vyšetření a definice použitá pro přítomnost, resp. nepřítomnost mutace RET. Pacientům, kterým by nebyl podáván vandetanib v důsledku nepřítomnosti mutace RET nebo kontraindikace, bude umožněno zařazení do studie a sledování.
Status mutace RET se vyšetřuje podle předem definované mutační analýzy, kdy je v protokolu předem specifikován druh testu a exony, které je třeba analyzovat.
• Hodnocení bezpečnosti proběhne při každé návštěvě včetně informace o prodloužení intervalu QT
• Objektivní odpověď nádoru, doba trvání odpovědi/progrese
• Hodnocení proběhne podle normální lékařské praxe řešitele v rámci výzkumného centra, pacienti budou vyšetřováni konzistentním způsobem na účinnost v předem definovaných časových intervalech a bez ohledu na stav mutace RET
• Metody používané k hodnocení (např. CT, MRI)
Stav nemoci při každé návštěvě: objektivní odpověď, stabilizovaná nemoc nebo progredující nemoc
• Konečná analýza bude provedena, pokud bude do studie zahrnuto nejméně 40 pacientů s přítomnou mutací RET a 40 pacientů bez průkazu přítomnosti mutace RET a vandetanib jim bude podáván po dobu 14 měsíců. Očekává se celková doba trvání studie v délce 38 měsíců.
Analýzy:
• Studie bude probíhat 2 roky a v předem určených časových intrvalech budou údaje shromážděny a analyzovány (např. 12 měsíců a 24 měsíců)
• Výskyt objektivní odpovědi, stav progrese a DCR v celkové populaci, u pacientů s přítomností mutace RET a bez přítomnosti mutace RET
• Analýza bezpečnosti v celkové populaci, u pacientů s přítomností mutace
_RET a bez přítomnosti mutace RET_
PŘÍLOHA III
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Caprelsa 100 mg potahované tablety vandetanibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tableta obsahuje vandetanibum 100 mg.
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A VELIKOST BALENÍ_
30 potahovaných tablet
5. ZPŮSOB A CESTA/CESTY PODÁNÍ_
Před použitím si přečtěte příbalovou informaci.
Perorální podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ_
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte při teplotě do 30 °C.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z TAKOVÝCH LÉČIVÝCH PŘÍPRAVKŮ, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Genzyme Europe B.V., Gooimeer 10, 1411 DD Naarden, Nizozemsko
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
13. ČÍSLO ŠARŽE
č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis. 15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
caprelsa 100 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU_
Caprelsa 100 mg tablety vandetanibum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Genzyme Europe B.V.
3. POUŽITELNOST_
EXP
4. ČÍSLO ŠARŽE_
Lot
5. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU_
Caprelsa 300 mg potahované tablety vandetanibum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tableta obsahuje vandetanibum 300 mg.
3. SEZNAM POMOCNÝCH LÁTEK
4. LÉKOVÁ FORMA A VELIKOST BALENÍ_
30 potahovaných tablet
5. ZPŮSOB A CESTA/CESTY PODÁNÍ_
Před použitím si přečtěte příbalovou informaci.
Perorální podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ_
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ_
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte při teplotě do 30 °C.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z TAKOVÝCH LÉČIVÝCH PŘÍPRAVKŮ, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Genzyme Europe B.V., Gooimeer 10, 1411 DD Naarden, Nizozemsko 12 REGISTRAČNÍ ČÍSLO/ČÍSLA
13. ČÍSLO ŠARŽE
č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis. 15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
caprelsa 300 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU_
Caprelsa 300 mg tablety vandetanibum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Genzyme Europe B.V.
3. POUŽITELNOST_
EXP
4. ČÍSLO ŠARŽE_
Lot
5. JINÉ
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro pacienta Caprelsa 100 mg potahované tablety
vandetanibum
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Kromě této příbalové informace obdržíte „Pohotovostní kartu pro pacienta“, která obsahuje důležité bezpečnostní informace, které byste měl(a) znát předtím, než začnete užívat přípravek Caprelsa a v průběhu léčby přípravkem Caprelsa.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat,
protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci a „Pohotovostní kartu pro pacienta“ pro případ, že si ji budete potřebovat přečíst znovu.
- Ponechte „Pohotovostní kartu pro pacienta“ u sebe po celou dobu léčby, je to důležité.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Caprelsa a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Caprelsa užívat
3. Jak se přípravek Caprelsa užívá
4. Možné nežádoucí účinky
5. Jak přípravek Caprelsa uchovávat
6. Obsah balení a další informace
1. Co je přípravek Caprelsa a k čemu se používá
Přípravek Caprelsa je určen k léčbě medulárního karcinomu štítné žlázy, který nelze odstranit chirurgicky nebo se rozšířil do jiných částí těla.
Přípravek Caprelsa účinkuje tak, že zpomaluje růst nových krevních cév v nádorech (karcinomech). Tím se přeruší dodávka živin a kyslíku do nádoru. Přípravek Caprelsa účinkuje také přímo na nádorové buňky tím, že je zabíjí nebo zpomaluje jejich růst.
2. Čemu musíme věnovat pozornost, než začnete přípravek Caprelsa užívat
Neužívejte přípravek Caprelsa:
• jestliže jste alergický(á) na vandetanib nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
• jestliže máte vrozené problémy se srdcem označované jako „vrozený syndrom dlouhého QTc“. To lze vidět na elektrokardiogramu (EKG).
• jestliže kojíte.
• jestliže užíváte následující léčivé přípravky: arsen, cisaprid (k léčbě pálení žáhy), nitrožilní erythromycin a moxifloxacin (k léčbě infekcí), toremifen (k léčbě nádoru prsu), mizolastin (k léčbě alergií), antiarytmika třídy IA a III (ke kontrole srdečního rytmu).
Neužívejte přípravek Caprelsa, pokud se Vás cokoli z výše zmíněného týká. Pokud si nejste jistý(á), zeptejte se svého lékaře.
Upozornění a opatření
Před užitím přípravku Caprelsa se poraďte se svým lékařem nebo lékárníkem, jestliže jste citlivý(á) na slunce.
Někteří lidé, kteří užívají přípravek Caprelsa, jsou citliví na slunce. Mohou vzniknout popáleniny po slunění. Když užíváte přípravek Caprelsa, chraňte se při pobytu venku před přímým sluncem a noste vhodné oblečení, abyste zabránil(a) vystavení slunci.
Monitorování Vaší krve a srdce:
Váš lékař nebo zdravotní sestra by měli provádět kontrolní vyšetření hladin draslíku, vápníku, hořčíku a hormonu stimulujícího štítnou žlázu (TSH) v krvi a kontrolu elektrické aktivity Vašeho srdce vyšetřením elektrokardiogramu (EKG). Tato vyšetření byste měl(a) podstoupit:
• před zahájením léčby přípravkem Caprelsa;
• pravidelně v průběhu léčby přípravkem Caprelsa;
• 1, 3 a 6 týdnů po zahájení léčby přípravkem Caprelsa;
• 12 týdnů po zahájení léčby přípravkem Caprelsa;
• dále každé 3 měsíce;
• jestliže lékař nebo lékárník změní dávku přípravku Caprelsa;
• jestliže začnete užívat léky, které ovlivňují činnost srdce;
• podle pokynů Vašeho lékaře nebo lékárníka.
Další léčivé přípravky a přípravek Caprelsa
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a), nebo které možná budete užívat, a to i o lécích, které jsou dostupné bez lékařského předpisu a lécích s obsahem rostlinných léčiv. Důvodem je okolnost, že přípravek Caprelsa může ovlivnit účinek jiných léků a jiné léky mohou mít vliv na účinek přípravku Caprelsa.
Informujte svého lékaře nebo lékárníka, pokud užíváte následující léky:
• itrakonazol, ketokonazol, ritonavir, klarithromycin, rifampicin a moxifloxacin (léky k léčbě infekcí);
• karbamazepin a fenobarbital (k předcházení záchvatům křečí);
• ondansetron (k léčbě pocitu na zvracení a zvracení);
• cisaprid (k léčbě pálení žáhy), pimozid (k léčbě nekontrolovaných opakovaných pohybů těla a přehnaných slovních projevů) a halofantrin a lumefantrin (k léčbě malárie);
• methadon (k odvykací léčbě), haloperidol, chlorpromazin, sulpirid, amisulprid a zuklopenthixol (k léčbě psychických onemocnění);
• pentamidin (k léčbě infekcí);
• antagonisty vitaminu K a dabigatran často označované jako léky „k ředění krve“;
• cyklosporin a takrolimus (k předcházení odhojování štěpu po transplantaci), digoxin (k léčbě nepravidelného srdečního rytmu) a metformin (ke kontrole hladiny cukru v krvi);
• inhibitory protonové pumpy (k léčbě pálení žáhy).
Tyto informace naleznete také na „Pohotovostní kartě pacienta“, kterou jste obdržel(a) od lékaře. Tuto „Pohotovostní kartu“ si ponechte u sebe a ukažte ji i Vašemu partnerovi nebo opatrovníkovi.
Těhotenství a kojení
Pokud jste těhotná nebo se snažíte otěhotnět, poraďte se se svým lékařem dříve, než začnete užívat přípravek Caprelsa. Důvodem je okolnost, že přípravek Caprelsa může mít škodlivý vliv na ještě nenarozené dítě. Lékař s Vámi bude diskutovat o prospěchu a rizicích užívání přípravku Caprelsa v tomto období.
• Pokud můžete otěhotnět, musíte v průběhu léčby přípravkem Caprelsa a ještě alespoň 4 měsíce po podání poslední dávky přípravku Caprelsa užívat účinnou antikoncepci.
V průběhu léčby přípravkem Caprelsa nesmíte kojit s ohledem na bezpečnost Vašeho dítěte.
Řízení dopravních prostředků a obsluha strojů
Buďte opatrní, než budete řídit nebo obsluhovat stroje. Mějte na paměti, že přípravek Caprelsa může vyvolat pocit únavy, slabost nebo neostré vidění.
3. Jak se přípravek Caprelsa užívá
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
• Doporučená dávka přípravku je 300 mg každý den.
• Užívejte přípravek Caprelsa ve stejnou denní dobu.
• Přípravek Caprelsa můžete užívat s jídlem nebo mimo jídlo.
Pokud máte problémy s polykáním tablety
Pokud máte problémy s polykáním tablety, můžete ji rozmíchat ve vodě takto:
• Sklenku naplňte do poloviny vodou bez oxidu uhličitého (nesycená voda). Použijte pouze vodu, nepoužívejte jiné tekutiny.
• Vložte tabletu do vody.
• Míchejte, dokud se tableta nerozpadne. To může trvat asi 10 minut.
• Potom ihned vypijte.
Naplňte ještě jednou sklenku do poloviny vodou a obsah vypijte, abyste užil(a) veškerou léčivou látku. Pokud se objeví nežádoucí účinky
Pokud se objeví nežádoucí účinky, vždy to řekněte svému lékaři. Lékař Vám může předepsat nižší dávku přípravku Caprelsa (např. dvě 100 mg tablety nebo jednu 100 mg tabletu). Lékař Vám může předepsat i jiné léky ke kontrole nežádoucích účinků. Nežádoucí účinky přípravku Caprelsa jsou uvedeny v bodě 4.
Jestliže jste užil(a) více přípravku Caprelsa, než jste měl(a)
Pokud jste užil(a) více tablet přípravku Caprelsa, než Vám předepsal lékař, obraťte se na lékaře nebo jděte přímo do nemocnice.
Jestliže jste zapomněl(a) užít přípravek Caprelsa
Pokud jste zapomněl(a) užít dávku, záleží na tom, kolik času zbývá do další pravidelné dávky.
• Pokud zbývá 12 hodin a více: užijte vynechanou dávku ihned, jakmile si vzpomenete. Další dávku užijte v pravidelnou dobu.
• Pokud zbývá méně než 12 hodin do další dávky: vynechte zapomenutou dávku a pokračujte další dávkou v pravidelnou dobu.
Nezdvojnásobujte následující dávku (dvě dávky ve stejnou dobu), abyste nahradil(a) vynechanou tabletu.
Máte-li jakékoli další otázky, týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého. Pokud se objeví nežádoucí účinky, Váš lékař Vám může předepsat přípravek Caprelsa v nižší dávce. Váš lékař Vám může také předepsat jiné léky ke kontrole nežádoucích účinků.
Lékaře kontaktujte ihned, pokud se objeví některý z následujících nežádoucích účinků - je možné, že budete potřebovat neodkladnou lékařskou pomoc:
• Slabost, závrať nebo změny srdečního rytmu. Může jít o příznaky změněné elektrické aktivity Vašeho srdce. Jsou pozorovány u 8 % pacientů s medulárním karcinomem štítné žlázy, kteří užívají přípravek Caprelsa. Lékař může doporučit užívání přípravku Caprelsa v nižší dávce nebo ukončit užívání přípravku Caprelsa. Léčba přípravkem Caprelsa byla méně často spojena se změnami srdečního rytmu, které ohrožovaly život.
• Závažné kožní reakce postihující velké plochy Vašeho těla. Příznaky mohou zahrnovat zčervenání, bolest, vředy, puchýře a odlupování kůže. Postiženy mohou být i rty, oči a pohlavní orgány. Podle typu kožní reakce může být výskyt častý (postihuje méně než 1 z 10 pacientů) nebo méně častý (postihuje méně než 1 ze 100 pacientů).
• Závažný průjem.
• Závažná dušnost, nebo náhlé zhoršení dušnosti, někdy doprovázené kašlem nebo vysokou teplotou (horečka). Důvodem může být zánět plic, který se nazývá „intersticiální plicní nemoc“. Tento zánět je méně častý (postihuje méně než 1 ze 100 pacientů), ale může ohrožovat život.
• Záchvaty křečí, bolest hlavy, zmatenost nebo potíže s koncentrací. Toto mohou být příznaky stavu, který se označuje jako RPLS (syndrom reverzibilní zadní leukoencefalopatie). Obvykle vymizí po přerušení léčby přípravkem Caprelsa. RPLS je méně častý (postihuje méně než 1 ze 100 pacientů).
Pokud si všimnete kteréhokoliv nežádoucího účinku, který je uveden výše, kontaktujte ihned svého
lékaře.
Další nežádoucí účinky zahrnují:
Velmi časté (postihují více než 1 z 10 pacientů)
• Průjem. Lékař Vám může předepsat jiný lék k léčbě průjmu. Pokud se průjem zhoršuje, informujte ihned svého lékaře.
• Bolest břicha.
• Kožní vyrážka nebo akné.
• Deprese.
• Únava.
• Pocit na zvracení.
• Podrážděný žaludek (porucha trávení).
• Poruchy nehtů.
• Zvracení.
• Ztráta chuti k jídlu (anorexie).
• Slabost.
• Vysoký krevní tlak. Lékař Vám může předepsat další lék k léčbě vysokého krevního tlaku.
• Bolest hlavy.
• Únava, vyčerpání.
• Potíže s usínáním (nespavost).
• Zánět nosních dutin.
• Zánět hlavních dýchacích cest vedoucích do plic.
• Infekce horních dýchacích cest.
• Infekce močových cest.
• Necitlivost nebo píchání v kůži.
• Abnormální citlivost pokožky.
• Závrať.
• Bolest.
• Otoky v důsledku nahromaděných tekutin.
• Kameny nebo ukládání vápníku v močových cestách (ledvinné kaménky).
• Neostré vidění včetně mírných změn oka, které mohou vést k neostrému vidění (zákal rohovky).
• Citlivost kůže na oslunění. Po dobu užívání přípravku Caprelsa chraňte svoji pokožku při pobytu venku opalovacím krémem a vhodným oblečením, abyste vyloučil(a) vystavení slunečnímu záření.
Časté (postihují méně než 1 pacienta z 10)
• Dehydratace (odvodnění organismu).
• Velmi vysoký krevní tlak.
• Ztráta tělesné hmotnosti.
• Cévní mozková příhoda nebo jiné příhody, kdy se mozku nedostává krve.
• Druh vyrážky, která postihuje ruce a chodidla (syndrom ruka noha).
• Zánět dutiny ústní.
• Sucho v ústech.
• Zánět plic.
• Toxiny v krvi jako komplikace probíhající infekce.
• Chřipka.
• Zánět močového měchýře.
• Zánět vedlejších nosních dutin.
• Zánět hlasivek (hrtanu).
• Zánět folikulů (cibulek), zvláště vlasových folikulů.
• Vřídek (furunkl).
• Plísňové infekce.
• Infekce ledvin.
• Ztráta tělesných tekutin (dehydratace).
• Úzkost.
• Třes.
• Ospalost.
• Slabost.
• Pocit nerovnováhy.
• Zvýšený nitrooční tlak (zelený zákal).
• Vykašlávání krve.
• Zánět plicní tkáně.
• Obtíže při polykání.
• Zácpa.
• Zánět sliznice žaludku (gastritida).
• Krvácení do zažívacího traktu.
• Žlučové kameny (cholelithiáza).
• Bolestivé močení.
• Selhání ledvin.
• Časté močení.
• Náhlá potřeba močení.
• Horečka.
• Krvácení z nosu (epistaxe).
• Sucho v očích.
• Podráždění očí (zánět spojivky).
• Zhoršené vidění.
• Vidění zářivého kruhu okolo předmětů/osob.
• Vidění záblesků světla (fotopsie).
• Poruchy oční rohovky (keratopatie).
• Určitý druh průjmu (kolitida).
• Ztráta vlasů na hlavě a chlupů na těle (plešatost).
• Změny vnímání chuti potravin.
Méně časté (postihují méně než 1 pacienta ze 100)
• Srdeční selhání.
• Zánět slepého střeva.
• Bakteriální infekce.
• Zánět divertiklů (malá vychlípenina, která se může vytvořit v trávicím traktu).
• Kožní bakteriální infekce.
• Absces (dutina) v břišní stěně.
• Zhoršený stav výživy.
• Mimovolní svalové stahy/záškuby.
• Rychlé se střídající se stahy a uvolnění svalů (klonus).
• Otok mozku.
• Zákal oční čočky.
• Poruchy srdečního rytmu.
• Zástava srdce.
• Neschopnost plic pracovat normálně.
• Zánět plic po vdechnutí cizího tělesa do plic.
• Neprůchodnost střeva.
• Proděravění (perforace) střeva.
• Ztráta kontroly nad vylučováním stolice.
• Neobvyklá barva moče.
• Nedostatečná tvorba moče.
• Zpomalené hojení.
• Zánět slinivky břišní (pankreatitida).
• Zpuchýřovatění kůže (bulózní dermatitida).
Následující nežádoucí účinky lze zjistit při kontrolních vyšetřeních prováděných lékařem:
• Bílkovina nebo krev ve Vaší moči (při kontrole moči).
• Změny srdečního rytmu (prokázané na EKG). Lékař může ukončit léčbu přípravkem Caprelsa nebo předepsat nižší dávku přípravku Caprelsa.
• Změny funkce Vašich jater nebo slinivky břišní (při kontrole krve). Obvykle se nijak neprojevují, ale lékař může chtít, aby se pravidelně kontrolovaly.
• Snížení hladiny vápníku ve Vaší krvi. Lékař může předepsat či změnit léčbu hormony štítné žlázy.
• Snížená hladina draslíku ve Vaší krvi.
• Zvýšená hladina vápníku ve Vaší krvi.
• Zvýšená hladina glukózy ve Vaší krvi.
• Snížená hladina sodíku ve Vaší krvi.
• Snížení funkce štítné žlázy.
• Zvýšená hodnota červených krvinek ve Vaší krvi.
Pokud se kterýkoli z nežádoucích účinků vyskytne v závažné míře, nebo pokud si všimnete jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci, sdělte to ihned svému lékaři nebo lékárníkovi.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, nebo lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Caprelsa uchovávat
Uchovávejte mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti, uvedené na blistru a krabičce za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte při teplotě do 30 °C.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co přípravek Caprelsa obsahuje
- Léčivou látkou je vandetanibum. Jedna tableta obsahuje 100 mg vandetanibu.
- Pomocnými látkami jsou dihydrát hydrogenfosforečnanu vápenatého, mikrokrystalická celulosa, krospovidon typ A, povidon K29-32, magnesium-stearát, hypromelosa, makrogol, oxid titaničitý (E171).
Jak přípravek Caprelsa vypadá a co obsahuje toto balení
Přípravek Caprelsa 100 mg jsou bílé kulaté tablety s vyraženým „Z100“ na jedné straně.
Přípravek Caprelsa se dodává v blistrech po 30 tabletách.
Držitel rozhodnutí o registraci
Genzyme Europe B.V., Gooimeer 10, 1411 DD Naarden, Nizozemsko Výrobce
AstraZeneca UK Limited, Macclesfield, Cheshire, SK10 2NA, Velká Británie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
|
Belgie/Belgique/Belgien/ |
Magyarország |
|
Luxembourg/Luxemburg |
SANOFI-AVENTIS Zrt. |
|
Sanofi Belgium |
Tel: +36 1 505 0050 |
|
Tél/Tel: + 32 2 710 54 00 | |
|
Bt^rapnn |
Malta |
|
sanofi-aventis Bulgaria EOOD |
Sanofi Malta Ltd |
|
Ten: +359 2 9705300 |
Tel: +356 21493022 |
|
Česká republika |
Nederland |
|
sanofi-aventis, s.r.o. |
Genzyme Europe B.V. |
|
Tel: +420 233 086 111 |
Tel: +31 35 699 1200 |
|
Danmark |
Norge |
|
sanofi-aventis Denmark A/S |
sanofi-aventis Norge AS |
|
Tlf: +45 45 16 70 00 |
Tlf: + 47 67 10 71 00 |
|
Deutschland |
Osterreich |
|
Genzyme GmbH |
sanofi-aventis GmbH |
|
Tel: +49 (0)6102 3674 0 |
Tel: + 43 1 80 185 - 0 |
|
Eesti |
Polska |
|
sanofi-aventis Estonia OU |
sanofi-aventis Sp. z o.o. |
|
Tel. +372 6 273 488 |
Tel.: +48 22 280 00 00 |
|
EXláda/Kónpoq |
Portugal |
|
sanofi-aventis AEBE (EAláSa) |
Sanofi - Produtos Farmaceuticos, Lda. Tel: +351 |
|
T^k: +30 210 900 1600 |
21 422 0100 |
|
Espaňa |
Románia |
|
Genzyme, S.L.U. Tel: +34 93 485 94 00 sanofi-aventis, S.A. Tel: +34 93 485 94 00 |
Sanofi Romania SRL Tel: +40 (0) 21 317 31 36 |
|
Hrvatska |
Slovenská republika |
|
sanofi-aventis Croatia d.o.o. |
sanofi-aventis Pharma Slovakia s.r.o. |
|
Tel: +385 1 600 34 00 |
Tel.: +421 2 33 100 100 |
|
Ísland |
Suomi/Finland |
|
Vistor hf. |
Sanofi Oy |
|
Sími: +354 535 7000 |
Puh/Tel: + 358 201 200 300 |
|
Italia |
Sverige |
|
Genzyme Srl |
Sanofi AB |
|
Tel: +39 059 349 811 |
Tel: +46 (0)8 634 50 00 |
|
Latvija |
United Kingdom/Ireland |
|
sanofi-aventis Latvia SIA |
Genzyme Therapeutics Ltd. (United Kingdom) |
|
Tel: +371 67 33 24 51 |
Tel: +44 (0) 1865 405200 |
|
Lietuva UAB „SANOFI-AVENTIS LIETUVA“ Tel. +370 5 275 5224 |
Tato příbalová informace byla naposledy revidována
Tomuto léčivému přípravku bylo uděleno tzv. podmíněné schválení. Znamená to, že informace o tomto přípravku budou přibývat.
Evropská agentura pro léčivé přípravky nejméně jednou za rok vyhodnotí nové informace o tomto léčivém přípravku a tato příbalová informace bude podle potřeby aktualizována.
Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou uveřejněny na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/
Příbalová informace: informace pro pacienta Caprelsa 300 mg potahované tablety
vandetanibum
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Kromě této příbalové informace obdržíte „Pohotovostní kartu pro pacienta“, která obsahuje důležité bezpečnostní informace, které byste měl(a) znát předtím, než začnete užívat přípravek Caprelsa a v průběhu léčby přípravkem Caprelsa.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat,
protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci a „Pohotovostní kartu pro pacienta“ pro případ, že si ji budete potřebovat přečíst znovu.
- Ponechte „Pohotovostní kartu pro pacienta“ u sebe po celou dobu léčby, je to důležité.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Caprelsa a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Caprelsa užívat
3. Jak se přípravek Caprelsa užívá
4. Možné nežádoucí účinky
5. Jak přípravek Caprelsa uchovávat
6. Obsah balení a další informace
1. Co je přípravek Caprelsa a k čemu se používá
Přípravek Caprelsa je určen k léčbě medulárního karcinomu štítné žlázy, který nelze odstranit chirurgicky nebo se rozšířil do jiných částí těla.
Přípravek Caprelsa účinkuje tak, že zpomaluje růst nových krevních cév v nádorech (karcinomech). Tím se přeruší dodávka živin a kyslíku do nádoru. Přípravek Caprelsa účinkuje také přímo na nádorové buňky tím, že je zabíjí nebo zpomaluje jejich růst.
2. Čemu musíme věnovat pozornost, než začnete přípravek Caprelsa užívat
Neužívejte přípravek Caprelsa:
• jestliže jste alergický(á) na vandetanib nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
• jestliže máte vrozené problémy se srdcem označované jako „vrozený syndrom dlouhého QTc“. To lze vidět na elektrokardiogramu (EKG).
• jestliže kojíte.
• jestliže užíváte následující léčivé přípravky: arsen, cisaprid (k léčbě pálení žáhy), nitrožilní erythromycin a moxifloxacin (k léčbě infekcí), toremifen (k léčbě nádoru prsu), mizolastin (k léčbě alergií), antiarytmika třídy IA a III (ke kontrole srdečního rytmu).
Neužívejte přípravek Caprelsa, pokud se Vás cokoliv z výše zmíněného týká. Pokud si nejste jistý/á, zeptejte se svého lékaře.
Upozornění a opatření
Před užitím přípravku Caprelsa se poraďte se svým lékařem nebo lékárníkem, jestliže jste citlivý(á) na slunce.
Někteří lidé, kteří užívají přípravek Caprelsa, jsou citliví na slunce. Mohou vzniknout popáleniny po slunění. Když užíváte přípravek Caprelsa, chraňte se při pobytu venku před přímým sluncem a noste vhodné oblečení, abyste zabránil(a) vystavení slunci.
Monitorování Vaší krve a srdce:
Váš lékař nebo zdravotní sestra by měli provádět kontrolní vyšetření hladin draslíku, vápníku, hořčíku a hormonu stimulujícího štítnou žlázu (TSH) v krvi a kontrolu elektrické aktivity Vašeho srdce vyšetřením elektrokardiogramu (EKG). Tato vyšetření byste měl(a) podstoupit:
• před zahájením léčby přípravkem Caprelsa;
• pravidelně v průběhu léčby přípravkem Caprelsa;
• 1, 3 a 6 týdnů po zahájení léčby přípravkem Caprelsa;
• 12 týdnů po zahájení léčby přípravkem Caprelsa;
• dále každé 3 měsíce;
• jestliže lékař nebo lékárník změní dávku přípravku Caprelsa;
• jestliže začnete užívat léky, které ovlivňují činnost srdce;
• podle pokynů Vašeho lékaře nebo lékárníka.
Další léčivé přípravky a přípravek Caprelsa
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte nebo jste v nedávné době užíval(a), nebo které možná budete užívat, a to i o lécích, které jsou dostupné bez lékařského předpisu a lécích s obsahem rostlinných léčiv. Důvodem je okolnost, že přípravek Caprelsa může ovlivnit účinek jiných léků a jiné léky mohou mít vliv na účinek přípravku Caprelsa.
Informujte svého lékaře nebo lékárníka, pokud užíváte následující léky:
• itrakonazol, ketokonazol, ritonavir, klarithromycin, rifampicin a moxifloxacin (léky k léčbě infekcí);
• karbamazepin a fenobarbital (k předcházení záchvatům);
• ondansetron (k léčbě pocitu na zvracení a zvracení);
• cisaprid (k léčbě pálení žáhy), pimozid (k léčbě nekontrolovaných opakovaných pohybů těla a přehnaných slovních projevů) a halofantrin a lumefantrin (k léčbě malárie);
• methadon (k odvykací léčbě), haloperidol, chlorpromazin, sulpirid, amisulprid a zuklopenthixol (k léčbě psychických onemocnění);
• pentamidin (k léčbě infekcí);
• antagonisty vitaminu K a dabigatran často označované jako léky „k ředění krve“;
• cyklosporin a takrolimus (k předcházení odhojování štěpu po transplantaci), digoxin (k léčbě nepravidelného srdečního rytmu) a metformin (ke kontrole hladiny cukru v krvi);
• inhibitory protonové pumpy (k léčbě pálení žáhy).
Tyto informace naleznete také na „Pohotovostní kartě pacienta“, kterou jste obdržel(a) od lékaře. Tuto „Pohotovostní kartu“ si ponechte u sebe a ukažte ji i Vašemu partnerovi nebo opatrovníkovi.
Těhotenství a kojení
Pokud jste těhotná nebo se snažíte otěhotnět, poraďte se se svým lékařem dříve, než začnete užívat přípravek Caprelsa. Důvodem je okolnost, že přípravek Caprelsa může mít škodlivý vliv na ještě nenarozené dítě. Lékař s Vámi bude diskutovat o prospěchu a rizicích užívání přípravku Caprelsa v tomto období.
• Pokud můžete otěhotnět, musíte v průběhu léčby přípravkem Caprelsa a ještě alespoň 4 měsíce po podání poslední dávky přípravku Caprelsa užívat účinnou antikoncepci.
V průběhu léčby přípravkem Caprelsa nesmíte kojit s ohledem na bezpečnost Vašeho dítěte.
Řízení dopravních prostředků a obsluha strojů
Buďte opatrní, než budete řídit nebo obsluhovat stroje. Mějte na paměti, že přípravek Caprelsa může vyvolat pocit únavy, slabost nebo neostré vidění.
3. Jak se přípravek Caprelsa užívá
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
• Doporučená dávka přípravku je jedna 300 mg tableta každý den.
• Užívejte přípravek Caprelsa ve stejnou denní dobu.
• Přípravek Caprelsa můžete užívat s jídlem nebo mimo jídlo.
Pokud máte problémy s polykáním tablety
Pokud máte problémy s polykáním tablety, můžete ji rozmíchat ve vodě takto:
• Sklenku naplňte do poloviny vodou bez oxidu uhličitého (nesycená voda). Použijte pouze vodu, nepoužívejte jiné tekutiny.
• Vložte tabletu do vody.
• Míchejte, dokud se tableta nerozpadne. To může trvat asi 10 minut.
• Potom ihned vypijte.
Naplňte ještě jednou sklenku do poloviny vodou a obsah vypijte, abyste užil(a) veškerou léčivou látku. Pokud se objeví nežádoucí účinky
Pokud se objeví nežádoucí účinky, vždy to řekněte svému lékaři. Lékař Vám může předepsat nižší dávku přípravku Caprelsa (např. dvě 100 mg tablety nebo jednu 100 mg tabletu). Lékař Vám může předepsat i jiné léky ke kontrole nežádoucích účinků. Nežádoucí účinky přípravku Caprelsa jsou uvedeny v bodě 4.
Jestliže jste užil(a) více přípravku Caprelsa, než jste měl(a)
Pokud jste užil(a) více tablet přípravku Caprelsa než Vám předepsal lékař, obraťte se na lékaře nebo jděte přímo do nemocnice.
Jestliže jste zapomněl(a) užít přípravek Caprelsa
Pokud jste zapomněl(a) užít dávku, záleží na tom, kolik času zbývá do další pravidelné dávky.
• Pokud zbývá 12 hodin a více: užijte vynechanou dávku ihned, jakmile si vzpomenete. Další dávku užijte v pravidelnou dobu.
• Pokud zbývá méně než 12 hodin do další dávky: vynechte zapomenutou dávku a pokračujte další dávkou v pravidelnou dobu.
Nezdvojnásobujte následující dávku (dvě dávky ve stejnou dobu), abyste nahradil(a) vynechanou tabletu.
Máte-li jakékoli další otázky, týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého. Pokud se objeví nežádoucí účinky, Váš lékař Vám může předepsat přípravek Caprelsa v nižšídávce. Váš lékař Vám může také předepsat jiné léky ke kontrole nežádoucích účinků.
Lékaře kontaktujte ihned, pokud se objeví některý z následujících nežádoucích účinků - je možné, že budete potřebovat neodkladnou lékařskou pomoc:
• Slabost, závrať nebo změny srdečního rytmu. Může jít o příznaky změněné elektrické aktivity Vašeho srdce. Jsou pozorovány u 8 % pacientů s medulárním karcinomem štítné žlázy, kteří užívají přípravek Caprelsa. Lékař může doporučit užívání přípravku Caprelsa v nižší dávce nebo ukončit užívání přípravku Caprelsa. Léčba přípravkem Caprelsa byla méně často spojena se změnami srdečního rytmu, které ohrožovaly život.
• Závažné kožní reakce postihující velké plochy Vašeho těla. Příznaky mohou zahrnovat zčervenání, bolest, vředy, puchýře a odlupování kůže. Postiženy mohou být i rty, oči a pohlavní orgány. Podle typu kožní reakce může být výskyt častý (postihuje méně než 1 z 10 pacientů) nebo méně častý (postihuje méně než 1 ze 100 pacientů).
• Závažný průjem.
• Závažná dušnost, nebo náhlé zhoršení dušnosti, někdy doprovázené kašlem nebo vysokou teplotou (horečka). Důvodem může být zánět plic, který se nazývá „intersticiální plicní nemoc“. Tento zánět je méně častý (postihuje méně než 1 ze 100 pacientů), ale může ohrožovat život.
• Záchvaty křečí, bolest hlavy, zmatenost nebo potíže s koncentrací. Toto mohou být příznaky stavu, který se označuje jako RPLS (syndrom reverzibilní zadní leukoencefalopatie). Obvykle vymizí po přerušení léčby přípravkem Caprelsa. RPLS je méně častý (postihuje méně než 1 ze 100 pacientů).
Pokud si všimnete kteréhokoliv nežádoucího účinku, který je uveden výše, kontaktujte ihned svého
lékaře.
Další nežádoucí účinky zahrnují:
Velmi časté (postihují více než 1 z 10 pacientů)
• Průjem. Lékař Vám může předepsat jiný lék k léčbě průjmu. Pokud se průjem zhoršuje, informujte ihned svého lékaře.
• Bolest břicha.
• Kožní vyrážka nebo akné.
• Deprese.
• Únava.
• Pocit na zvracení.
• Podrážděný žaludek (porucha trávení).
• Poruchy nehtů.
• Zvracení.
• Ztráta chuti k jídlu (anorexie).
• Slabost.
• Vysoký krevní tlak. Lékař Vám může předepsat další lék k léčbě vysokého krevního tlaku.
• Bolest hlavy.
• Únava, vyčerpání.
• Potíže s usínáním (nespavost).
• Zánět nosních dutin.
• Zánět hlavních dýchacích cest vedoucích do plic.
• Infekce horních dýchacích cest.
• Infekce močových cest.
• Necitlivost nebo píchání v kůži.
• Abnormální citlivost pokožky.
• Závrať.
• Bolest.
• Otoky v důsledku nahromaděných tekutin.
• Kameny nebo ukládání vápníku v močových cestách (ledvinné kaménky).
• Neostré vidění včetně mírných změn oka, které mohou vést k neostrému vidění (zákal rohovky).
• Citlivost kůže na oslunění. Po dobu užívání přípravku Caprelsa chraňte svoji pokožku při pobytu venku opalovacím krémem a vhodným oblečením, abyste vyloučil(a) vystavení slunečnímu záření.
Časté (postihují méně než 1 pacienta z 10)
• Dehydratace (odvodnění organismu).
• Velmi vysoký krevní tlak.
• Ztráta tělesné hmotnosti.
• Cévní mozková příhoda nebo jiné příhody, kdy se mozku nedostává krve.
• Druh vyrážky, která postihuje ruce a chodidla (syndrom ruka noha).
• Zánět dutiny ústní.
• Sucho v ústech.
• Zánět plic.
• Toxiny v krvi jako komplikace probíhající infekce.
• Chřipka.
• Zánět močového měchýře.
• Zánět vedlejších nosních dutin.
• Zánět hlasivek (hrtanu).
• Zánět folikulů (cibulek), zvláště vlasových folikulů.
• Vřídek (furunkl).
• Plísňové infekce.
• Infekce ledvin.
• Ztráta tělesných tekutin (dehydratace).
• Úzkost.
• Třes.
• Ospalost.
• Slabost.
• Pocit nerovnováhy.
• Zvýšený nitrooční tlak (zelený zákal).
• Vykašlávání krve.
• Zánět plicní tkáně.
• Obtíže při polykání.
• Zácpa.
• Zánět sliznice žaludku (gastritida).
• Krvácení do zažívacího traktu.
• Žlučové kameny (cholelithiáza).
• Bolestivé močení.
• Selhání ledvin.
• Časté močení.
• Náhlá potřeba močení.
• Horečka.
• Krvácení z nosu (epistaxe).
• Sucho v očích.
• Podráždění očí (zánět spojivky).
• Zhoršené vidění.
• Vidění zářivého kruhu okolo předmětů/osob.
• Vidění záblesků světla (fotopsie).
• Poruchy oční rohovky (keratopatie).
• Určitý druh průjmu (kolitida).
• Ztráta vlasů na hlavě a chlupů na těle (plešatost).
• Změny vnímání chuti potravin.
Méně časté (postihují méně než 1 pacienta ze 100)
• Srdeční selhání.
• Zánět slepého střeva.
• Bakteriální infekce.
• Zánět divertiklů (malá vychlípenina, která se může vytvořit v trávicím traktu).
• Kožní bakteriální infekce.
• Absces (dutina) v břišní stěně.
• Zhoršený stav výživy.
• Mimovolní svalové stahy/záškuby.
• Rychlé se střídající se stahy a uvolnění svalů (klonus).
• Otok mozku.
• Zákal oční čočky.
• Poruchy srdečního rytmu.
• Zástava srdce.
• Neschopnost plic pracovat normálně.
• Zánět plic po vdechnutí cizího tělesa do plic.
• Neprůchodnost střeva.
• Proděravění (perforace) střeva.
• Ztráta kontroly nad vylučováním stolice.
• Neobvyklá barva moče.
• Nedostatečná tvorba moče.
• Zpomalené hojení.
• Zánět slinivky břišní (pankreatitida).
• Zpuchýřovatění kůže (bulózní dermatitida).
Následující nežádoucí účinky lze zjistit při kontrolních vyšetřeních prováděných lékařem:
• Bílkovina nebo krev ve Vaší moči (při kontrole moči).
• Změny srdečního rytmu (prokázané na EKG). Lékař může ukončit léčbu přípravkem Caprelsa nebo předepsat nižší dávku přípravku Caprelsa.
• Změny funkce Vašich jater nebo slinivky břišní (při kontrole krve). Obvykle se nijak neprojevují, ale lékař může chtít, aby se pravidelně kontrolovaly.
• Snížení hladiny vápníku ve Vaší krvi. Lékař může předepsat či změnit léčbu hormony štítné žlázy.
• Snížená hladina draslíku ve Vaší krvi.
• Zvýšená hladina vápníku ve Vaší krvi.
• Zvýšená hladina glukózy ve Vaší krvi.
• Snížená hladina sodíku ve Vaší krvi.
• Snížení funkce štítné žlázy.
• Zvýšená hodnota červených krvinek ve Vaší krvi.
Pokud se kterýkoli z nežádoucích účinků vyskytne v závažné míře, nebo pokud si všimnete jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci, sdělte to ihned svému lékaři nebo lékárníkovi.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, nebo lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Caprelsa uchovávat
Uchovávejte mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti, uvedené na blistru a krabičce za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte při teplotě do 30 °C.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak máte naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co přípravek Caprelsa obsahuje
- Léčivou látkou je vandetanibum. Jedna tableta obsahuje 300 mg vandetanibu.
- Pomocnými látkami jsou dihydrát hydrogenfosforečnanu vápenatého, mikrokrystalická celulosa, krospovidon typ A, povidon K29-32, magnesium-stearát, hypromelosa, makrogol, oxid titaničitý (E171).
Jak přípravek Caprelsa vypadá a co obsahuje toto balení
Caprelsa 300 mg jsou bílé oválné tablety s vyraženým „Z300“ na jedné straně.
Přípravek Caprelsa se dodává v blistrech po 30 tabletách.
Držitel rozhodnutí o registraci
Genzyme Europe B.V., Gooimeer 10, NL-1411 DD, Naarden, The Netherlands Výrobce
AstraZeneca UK Limited, Macclesfield, Cheshire, SK10 2NA, Velká Británie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
|
Belgie/Belgique/Belgien/ |
Magyarország |
|
Luxembourg/Luxemburg |
SANOFI-AVENTIS Zrt. |
|
Sanofi Belgium |
Tel: +36 1 505 0050 |
|
Tél/Tel: + 32 2 710 54 00 | |
|
Bt^rapnn |
Malta |
|
sanofi-aventis Bulgaria EOOD |
Sanofi Malta Ltd |
|
Ten: +359 2 9705300 |
Tel: +356 21493022 |
|
Česká republika |
Nederland |
|
sanofi-aventis, s.r.o. |
Genzyme Europe B.V. |
|
Tel: +420 233 086 111 |
Tel: +31 35 699 1200 |
|
Danmark |
Norge |
|
sanofi-aventis Denmark A/S |
sanofi-aventis Norge AS |
|
Tlf: +45 45 16 70 00 |
Tlf: + 47 67 10 71 00 |
|
Deutschland |
Osterreich |
|
Genzyme GmbH |
sanofi-aventis GmbH |
|
Tel: +49 (0)6102 3674 0 |
Tel: + 43 1 80 185 - 0 |
|
Eesti |
Polska |
|
sanofi-aventis Estonia OU |
sanofi-aventis Sp. z o.o. |
|
Tel. +372 6 273 488 |
Tel.: +48 22 280 00 00 |
|
EXláda/Kónpoq |
Portugal |
|
sanofi-aventis AEBE (EAláSa) |
Sanofi - Produtos Farmaceuticos, Lda. Tel: +351 |
|
T^k: +30 210 900 1600 |
21 422 0100 |
|
Espaňa |
Románia |
|
Genzyme, S.L.U. Tel: +34 93 485 94 00 sanofi-aventis, S.A. Tel: +34 93 485 94 00 |
Sanofi Romania SRL Tel: +40 (0) 21 317 31 36 |
|
France |
Slovenija |
|
Genzyme S.A.S. |
sanofi-aventis d.o.o. |
|
Information médicale: tél:+33(0) 800 100 499 |
Tel: +386 1 560 4800 |
|
Hrvatska |
Slovenská republika |
|
sanofi-aventis Croatia d.o.o. |
sanofi-aventis Pharma Slovakia s.r.o. |
|
Tel: +385 1 600 34 00 |
Tel.: +421 2 33 100 100 |
|
Ísland |
Suomi/Finland |
|
Vistor hf. |
Sanofi Oy |
|
Sími: +354 535 7000 |
Puh/Tel: + 358 201 200 300 |
|
Italia |
Sverige |
|
Genzyme Srl |
Sanofi AB |
|
Tel: +39 059 349 811 |
Tel: +46 (0)8 634 50 00 |
|
Latvija |
United Kingdom/Ireland |
|
sanofi-aventis Latvia SIA |
Genzyme Therapeutics Ltd. (United Kingdom) |
|
Tel: +371 67 33 24 51 |
Tel: +44 (0) 1865 405200 |
|
Lietuva UAB „SANOFI-AVENTIS LIETUVA“ Tel. +370 5 275 5224 |
Tato příbalová informace byla naposledy revidována
Tomuto léčivému přípravku bylo uděleno tzv. podmíněné schválení. Znamená to, že informace o tomto přípravku budou přibývat.
Evropská agentura pro léčivé přípravky nejméně jednou za rok vyhodnotí nové informace o tomto léčivém přípravku a tato příbalová informace bude podle potřeby aktualizována.
Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou uveřejněny na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu
60