Bydureon 2 Mg
SOUHRN ÚDAJŮ O PŘÍPRAVKU
PŘÍLOHA I
Bydureon 2 mg prášek a rozpouštědlo pro injekční suspenzi s prodlouženým účinkem
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička obsahuje exenatidum 2 mg.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční suspenzi s prodlouženým účinkem. Prášek: bílý až téměř bílý prášek.
Rozpouštědlo: čirý, bezbarvý až lehce nažloutlý či nahnědlý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Bydureon je indikován k léčbě diabetes mellitus 2. typu v kombinaci s:
• metforminem
• deriváty sulfonylmočoviny
• thiazolidindiony
• metforminem a sulfonylmočovinou
• metforminem a thiazolidindionem
u dospělých pacientů, u kterých není dosaženo dostatečné kontroly glykemie při podávání maximálních tolerovaných dávek těchto perorálních přípravků.
4.2 Dávkování a způsob podání
Dávkování
Doporučená dávka je 2 mg exenatidu podaná jednou týdně.
U pacientů převáděných z exenatidu s okamžitým účinkem (Byetta) na exenatid s prodlouženým účinkem (Bydureon) může dojít k přechodnému zvýšení hladiny glukózy v krvi, která se zpravidla upraví během prvních dvou týdnů po zahájení léčby.
Jestliže je exenatid s prodlouženým účinkem přidán k léčbě metforminem a/nebo thiazolidindionem, je možné pokračovat v dosavadní dávce metforminu a/nebo thiazolidindionu. Jestliže je exenatid s prodlouženým účinkem přidán k sulfonylmočovině, měla by být zvážena redukce dávky sulfonylmočoviny, aby se snížilo riziko hypoglykemie (viz bod 4.4).
Exenatid s prodlouženým účinkem se podává jednou týdně ve stejný den. Tento den podání může být v případě potřeby změněn, pokud je další dávka podána nejméně o jeden den (24 hodin) později. Exenatid s prodlouženým účinkem může být podán kdykoli v průběhu dne nezávisle na jídle.
V případě opomenutí podání dávky by tato měla být podána co nejdříve je to možné. Při další injekci se pacient může vrátit k původnímu dni podání injekce. Vždy však lze podat pouze jednu injekci v průběhu 24 hodin.
Užívání exenatidu s prodlouženým účinkem nevyžaduje další každodenní měření glykemie prováděné pacientem. Toto měření může být ovšem nezbytné pro úpravu dávky sulfonylmočoviny.
Pokud je zahájena jiná léčba snižující hladinu glukózy v krvi po ukončení podávání exenatidu s prodlouženým účinkem, má se vzít prodloužený účinek přípravku v úvahu (viz bod 5.2).
Zvláštní populace
Starší pacienti
Úprava dávky v závislosti na věku není nutná. Nicméně vzhledem ke snižování renálních funkcí s věkem by měla být renálním funkcím pacienta věnována pozornost (viz Pacienti s renální nedostatečností). Klinické zkušenosti u pacientů nad 75 let jsou velmi omezené (viz bod 5.2).
Renální insuficience
U pacientů s mírnou renální nedostatečností (clearance kreatininu 50 až 80 ml/min) není zapotřebí úprava dávky. U pacientů se středně závažnou renální nedostatečností (clearance kreatininu 30 až 50 ml/min) jsou klinické zkušenosti velmi omezené (viz bod 5.2). Podávání exenatidu s prodlouženým účinkem se těmto pacientům nedoporučuje.
Exenatid s prodlouženým účinkem se nedoporučuje podávat pacientům v konečném stádiu renálního selhávání nebo se závažnou renální nedostatečností (clearance kreatininu <30 ml/min) (viz bod 4.4).
Hepatální insuficience
U pacientů s hepatální nedostatečností není zapotřebí úprava dávky (viz bod 5.2).
Pediatrická populace
Bezpečnost a účinnost podávání exenatidu s prodlouženým účinkem nebyla stanovena u dětí a dospívajících mladších 18 let. Dostupné údaje jsou popsány v bodě 5.2, avšak nelze učinit žádná doporučení pro dávkování.
Způsob podání
Exenatid s prodlouženým účinkem je určen k aplikaci pacientem. Každá souprava by měla být použita pouze jednou osobou a je určena k jednorázovému použití.
Před zahájením léčby exenatidem s prodlouženým účinkem se důrazně doporučuje, aby pacienti a jejich opatrovníci byli vyškoleni odborným zdravotnickým pracovníkem v aplikaci přípravku.
„Pokyny pro uživatele“, které jsou součástí balení přípravku, se musí pečlivě dodržovat.
Jednotlivá dávka musí být aplikována subkutánně do oblasti břicha, stehna nebo zadní části paže ihned po přípravě suspenze prášku v rozpouštědle.
Pokyny pro přípravu suspenze léčivého přípravku před jeho podáním naleznete v bodě 6.6 a v „Pokynech pro uživatele“.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Exenatid s prodlouženým účinkem by neměl být použit u pacientů s diabetes mellitus 1. typu nebo k léčbě diabetické ketoacidózy.
Exenatid s prodlouženým účinkem nesmí být podán intravenózně nebo intramuskulárně.
Renální insuficience
U dialyzovaných pacientů v konečném stádiu renálního selhávání jednotlivé dávky exenatidu s okamžitým účinkem zvýšily frekvenci výskytu a závažnost gastrointestinálních nežádoucích účinků, exenatid s prodlouženým účinkem se proto nedoporučuje podávat pacientům v konečném stádiu renálního selhávání nebo se závažnou renální nedostatečností (clearance kreatininu <30 ml/min).
U pacientů se středně závažnou renální nedostatečností jsou klinické zkušenosti velmi omezené a použití exenatidu s prodlouženým účinkem se proto nedoporučuje.
Méně často byly spontánně hlášeny případy změny renálních funkcí, včetně zvýšení sérového kreatininu, poruchy funkce ledvin, zhoršení chronického renálního selhání a akutní renální selhání, někdy vyžadující hemodialýzu. K některým z těchto příhod došlo u pacientů se stavy, které mohou ovlivňovat hydrataci, včetně nauzey, zvracení a/nebo průjmu, a/nebo u pacientů užívajících přípravky, u kterých je znám jejich vliv na renální funkce/celkovou hydrataci. Současně podávané přípravky zahrnovaly inhibitory angiotenzin konvertujícího enzymu, antagonisty angiotenzinu-II, nesteroidní protizánětlivé přípravky a diuretika. Reverzibilita porušených renálních funkcí byla pozorována při podpůrné léčbě a při vysazení potenciálně působících agens včetně exenatidu.
Závažná gastrointestinální onemocnění
Exenatid s prodlouženým účinkem nebyl hodnocen u pacientů se závažným gastrointestinálním onemocněním včetně gastroparézy. Použití přípravku je často spojeno s nežádoucími účinky v oblasti gastrointestinálního traktu, včetně nauzey, zvracení a průjmu. Z tohoto důvodu se exenatid s prodlouženým účinkem nedoporučuje u pacientů se závažným gastrointestinálním onemocněním.
Akutní pankreatitida
Použití agonistů GLP-1 je spojeno s rizikem vývoje akutní pankreatitidy. U léčivého exenatidu s prodlouženým účinkem byly spontánně hlášeny případy akutní pankreatitidy. Při použití podpůrné léčby byl pozorován ústup pankreatitidy, ale velmi vzácně byly hlášeny případy nekrotizující nebo hemoragické pankreatitidy a/nebo úmrtí. Pacienti by měli být informováni o charakteristických symptomech akutní pankreatitidy: přetrvávající, silná bolest břicha. V případě podezření na pankreatitidu by měla být léčba exenatidem s prodlouženým účinkem ukončena, pokud je akutní pankreatitida potvrzena, léčba exenatidem s prodlouženým účinkem nesmí být znovu zahájena. Opatrnosti je třeba u pacientů s anamnézou pankreatitidy.
Souběžně podávané léčivé přípravky
Souběžné podávání exenatidu s prodlouženým účinkem s inzulínem, deriváty D-fenylalaninu (meglitinidy), inhibitory alfa-glukosidázy, inhibitory dipeptidyl peptidázy-4 nebo jinými agonisty receptoru pro GLP-1 nebylo studováno. Souběžné podávání exenatidu s prodlouženým účinkem a exenatidu s okamžitým účinkem nebylo studováno, a proto se nedoporučuje.
Interakce s warfarinem
Spontánně byly hlášeny případy zvýšení INR (International Normalized Ratio), někdy spojené s krvácením, když byl warfarin podáván souběžně s exenatidem (viz bod 4.5).
Hypoglykemie
Výskyt hypoglykemie byl v klinických studiích vyšší při použití exenatidu s prodlouženým účinkem v kombinaci se sulfonylmočovinou. V klinických studiích měli pacienti s mírnou renální insufuciencí užívající kombinaci se sulfonylmočovinu zvýšený výskyt hypoglykemie v porovnání s pacienty s normální renální funkcí. Pro snížení rizika hypoglykemie spojeného s užíváním sulfonylmočoviny by měla být zvážena redukce dávky sulfonylmočoviny.
Rychlé snížení tělesné hmotnosti
U pacientů užívajících exenatid bylo pozorováno rychlé snížení hmotnosti o více než 1,5 kg za týden. Takto vysoký hmotnostní úbytek může mít škodlivé důsledky. Pacienti s rychlým hmotnostním úbytkem mají být pečlivě sledováni na známky a příznaky cholelithiázy.
Ukončení léčby
Po ukončení léčby může účinek přípravku Bydureon přetrvávat v závislosti na snižování plazmatických hladin exenatidu až po dobu 10 týdnů. Při výběru jiného léčivého přípravku a nastavení dávky by mělo být toto vzato do úvahy také vzhledem k tomu, že mohou přetrvávat nežádoucí účinky a částečně i účinnost do doby úplného snížení hladin exenatidu.
Pomocné látky
Obsah sodíku: Tento léčivý přípravek obsahuje méně než 1 mmol sodíku (23 mg) v jedné dávce, tj. v podstatě je bez sodíku.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Sulfonylmočovina
Dávkování sulfonylmočoviny může vyžadovat úpravu vzhledem ke zvýšenému riziku hypoglykemie spojené s terapií sulfonylmočovinou (viz body 4.2 a 4.4).
Vyprazdňování žaludku
Výsledky studie používající paracetamol jako modelové léčivo k hodnocení vyprazdňování žaludku naznačují, že účinek exenatidu s prodlouženým účinkem na zpomalení vyprazdňování žaludku je malý a neočekává se, že způsobí klinicky významné snížení rychlosti a rozsahu absorpce současně perorálně podaných léčivých přípravků. Z tohoto důvodu nejsou vyžadovány úpravy dávek léčivých přípravků senzitivních na zpomalení vyprazdňování žaludku.
Po 14týdenní terapii exenatidem s prodlouženým účinkem nebyly po podání tablet s obsahem 1000 mg paracetamolu ať už nalačno, nebo po jídle, pozorovány významné změny AUC paracetamolu v porovnání s kontrolním obdobím. Cmax paracetamolu se snížila o 16 % (nalačno) a o 5 % (po jídle) a tmax se zvýšila oproti kontrolnímu období z přibližně 1 hodiny na 1,4 hodiny (nalačno) a na 1,3 hodiny (po jídle).
Byly provedeny následující studie interakcí za použití 10 ^g exenatidu s okamžitým účinkem, avšak nikoli s exenatidem s prodlouženým účinkem:
Warfarin
Při podání warfarinu 35 minut po aplikaci exenatidu okamžitým účinkem bylo pozorováno zpoždění tmax přibližně o 2 hodiny. Nebyl pozorován žádný klinicky relevantní vliv na Cmax nebo AUC. Spontánně byly hlášeny případy zvýšení INR během současného užívání warfarinu a exenatidu s prodlouženým uvolňováním. Během zahájení podávání exenatidu s prodlouženým uvolňováním se má u pacientů užívajících warfarin a/nebo kumarinové deriváty hodnota INR pečlivě monitorovat (viz body 4.4 a 4.8).
Inhibitory HMG-CoA reduktázy
Jestliže byl exenatid s okamžitým účinkem podán společně s jednorázovou dávkou lovastatinu (40 mg), AUC a Cmax lovastatinu byly sníženy o 40 %, resp. 28 % a tmax byl zpožděn asi o 4 hodiny ve srovnání s podáním samotného lovastatinu. V 30týdenních placebem kontrolovaných klinických studiích nebylo současné podávání exenatidu s okamžitým účinkem a inhibitorů HMG CoA reduktázy spojeno se stabilními změnami v lipidovém profilu (viz bod 5.1). Ačkoliv není dopředu vyžadována žádná úprava dávky, lipidové profily se mají pravidelně monitorovat.
Digoxin a lisinopril
Ve studiích interakcí účinku exenatidu s okamžitým účinkem na digoxin a lisinopril nebyl pozorován žádný klinicky významný účinek na Cmax nebo AUC, nicméně bylo pozorováno zpoždění tmax přibližně o 2 hodiny.
Ethinylestradiol a levonorgestrel
Podání kombinovaných perorálních kontraceptiv (30 ^g ethinylestradiolu a 150 ^g levonorgestrelu) hodinu před aplikací exenatidu s okamžitým účinkem neovlivnilo AUC, Cmax nebo Cmin ethinylestradiolu ani levonorgestrelu. Podání perorálních kontraceptiv 35 minut po exenatidu neovlivnilo AUC, ale mělo za následek snížení Cmax ethinylestradiolu o 45 % a Cmax levonorgestrelu o 27-41 % a prodloužilo tmax o 2- 4 hodiny v důsledku zpomaleného vyprazdňování žaludku. Snížení maximální koncentrace Cmax není klinicky významné a úprava dávky perorálních kontraceptiv není nutná.
Pediatrická populace
Interakční studie s exenatidem byly provedeny pouze u dospělých.
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku
Vzhledem k dlouhému poločasu eliminace exenatidu s prodlouženým účinkem by měly ženy ve fertilním věku užívat v průběhu léčby exenatidem s prodlouženým účinkem antikoncepci. Léčba exenatidem s prodlouženým účinkem by měla být ukončena nejméně 3 měsíce před plánovaným těhotenstvím.
Adekvátní údaje o podávání exenatidu s prodlouženým účinkem těhotným ženám nejsou k dispozici. Studie na zvířatech prokázaly reprodukční toxicitu (viz bod 5.3). Případné riziko pro člověka není známé. V průběhu těhotenství nesmí být exenatid s prodlouženým účinkem podáván a doporučuje se podávat inzulín.
Kojení
Není známo, zda se exenatid vylučuje do lidského mateřského mléka. Exenatid s prodlouženým účinkem se nemá podávat kojícím ženám.
Fertilita
Studie fertility nebyly u lidí provedeny.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Jestliže je exenatid s prodlouženým účinkem podáván v kombinaci se sulfonylmočovinou, pacienti by měli být poučeni o preventivních opatřeních k zabránění hypoglykemie v průběhu řízení nebo obsluhy strojů.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Nejčastější nežádoucí účinky byly převážně spojeny s gastrointestinálním traktem (nauzea, která byla nejčastějším nežádoucím účinkem a byla spojena se zahajováním léčby a postupně se její výskyt snižoval, a průjem). Mimo to se vyskytly také reakce v místě vpichu (pruritus, uzlíky, erytém), hypoglykemie (se sulfonylmočovinou) a bolest hlavy. Většina nežádoucích účinků spojená s podáváním exenatidu s prodlouženým účinkem byla mírné až střední intenzity.
Od doby uvedení exenatidu s okamžitým účinkem na trh byla hlášena akutní pankreatitida s frekvencí není známo a akutní renální selhání s frekvencí méně často (viz bod 4.4).
Tabulkový souhrn nežádoucích účinků
Frekvence nežádoucích účinků exenatidu s prodlouženým účinkem identifikované v klinických studiích a ze spontánních hlášení (které nebyly pozorovány v klinickém hodnocení, frekvence není známo) jsou shrnuty níže v Tabulce 1.
Zdrojem údajů pro klinická hodnocení s exenatidem je 18 placebem kontrolovaných klinických studií, 21 studií s aktivním komparátorem a 2 otevřené klinické studie. Základní léčba zahrnovala dietní opatření a pohybové aktivity, metformin, sulfonylmočovinu, thiazolidindion, nebo kombinaci perorálních látek snižujících hladinu glukózy v krvi.
Účinky jsou uvedeny níže za použití terminologie MedDRA a řazeny podle tříd orgánových systémů a četnosti výskytu. Frekvence četností jsou definovány následovně: velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1000 až < 1/100), vzácné (> 1/10000 až < 1/1000), velmi vzácné (< 1/10000) a není známo (z dostupných údajů nelze určit).
Tabulka 1: Nežádoucí účinky exenatidu s prodlouženým účinkem identifikované v klinických
|
studiích a ze spontánníc |
i hlášení | |||||
|
Třídy orgánových systémů/nežádoucí účinek |
Frekvence výskytu | |||||
|
Velmi časté |
Časté |
Méně časté |
Vzácné |
Velmi vzácné |
Není známo | |
|
Poruchy imunitního systému | ||||||
|
Anafylaktická reakce |
X1 | |||||
|
Poruchy metabolismu a výživy | ||||||
|
Hypoglykemie (se sulfonylmočovinou) |
X1 | |||||
|
Snížená chuť k jídlu |
X1 | |||||
|
Dehydratace |
X1 | |||||
|
Poruchy nervového systému | ||||||
|
X1 | ||||||
|
Závrať |
X1 | |||||
|
Porucha chuti |
X1 | |||||
|
Somnolence |
X1 | |||||
|
Gastrointestinální poruchy | ||||||
|
Střevní obstrukce |
X1 | |||||
|
Akutní pankreatitida (viz bod 4.4) |
X2 | |||||
|
X1 | ||||||
|
X1 | ||||||
|
X1 | ||||||
|
X1 | ||||||
|
X1 | ||||||
|
Gastroesofageální refluxní nemoc |
X1 | |||||
|
Abdominální distenze |
X1 | |||||
|
Říhání |
X1 | |||||
|
Zácpa |
X1 | |||||
|
Plynatost |
X1 | |||||
|
Poruchy kůže a podkožní tkáně | ||||||
|
Makulární a papulární vyrážka |
X2 | |||||
|
Svědění a/nebo kopřivka |
X1 | |||||
|
Angioneurotický edém |
X2 | |||||
|
Abscesy a celulitida v místě vpichu |
X2 | |||||
|
Nadměrné pocení |
X1 | |||||
|
Alopecie |
X1 | |||||
|
Poruchy ledvin a močových cest | ||||||
|
Zhoršená funkce ledvin včetně akutního selhání ledvin, zhoršení chronického selhání ledvin, poškození ledvin, zvýšený sérový kreatinin (viz bod 4.4) |
X1 | |||||
1 Frekvence odvozená z databáze dokončených dlouhodobých klinických studií účinnosti a bezpečnosti s exenatidem s prodlouženým účinkem, celkově n = 2868 (n = 1002 pacientů na sulfonylmočovině).
|
Celkové poruchy a rea |
íce v místě aplikace | |||||
|
Svědění v místě aplikace |
X1 | |||||
|
Únava |
X1 | |||||
|
Erytém v místě aplikace |
X1 | |||||
|
Vyrážka v místě aplikace |
X1 | |||||
|
Astenie |
X1 | |||||
|
Pocit nervozity |
X1 | |||||
|
Vyšetření | ||||||
|
Zvýšení hodnot INR (viz bod 4.4) |
X2 | |||||
2 Frekvence odvozená ze spontánních hlášení u exenatidu s prodlouženým účinkem (neznámá četnost). Popis vybraných nežádoucích účinků
Hypoglykemie
Incidence hypoglykemie byla vyšší, pokud byl exenatid s prodlouženým účinkem podáván v kombinaci se sulfonylmočovinou (24.0 % oproti 5,4 %) (viz bod 4.4). Ke snížení rizika hypoglykemie spojeného s podáváním sulfonylmočoviny může být zváženo snížení její dávky (viz body 4.2 a 4.4).
Podávání exenatidu s prodlouženým účinkem bylo spojeno se signifikantně nižším výskytem epizod hypoglykemie oproti bazálnímu inzulinu u pacientů užívajících také metformin (3 % oproti 19 %) a u pacientů užívajících metformin společně se sulfonylmočovinou (20 % oproti 42 %).
Většina případů hypoglykemie (99,9 %, n = 649) byla ve všech 11 studiích s exenatidem s prodlouženým účinkem mírné intenzity a byly vyřešeny perorálním podáním sacharidů. U jednoho pacienta byla hlášena závažná hypoglykemie s nízkou hodnotou glukózy v krvi (2,2 mmol/l) a vyžadovala pomoc s perorálním podáním sacharidů, kdy došlo k vyřešení příhody.
Nejčastěji hlášeným nežádoucím účinkem byla nauzea. U pacientů léčených exenatidem s prodlouženým účinkem byl obecně hlášen ve 20 % případů výskyt nejméně jedné epizody nauzey ve srovnání s 34 % případů u pacientů s exenatidem s okamžitým účinkem. Ve většině případů byla nauzea mírná až středně těžká. U většiny pacientů s nauzeou na počátku léčby se frekvence s dalším pokračováním léčby snižovala.
V 30týdenní kontrolované klinické studii došlo k ukončení léčby z důvodu nežádoucích účinků u 6 % pacientů léčených exenatidem s prodlouženým účinkem, u 5 % pacientů léčených exenatidem
s okamžitým účinkem.
Nejčastějšími nežádoucími účinky vedoucími k vyřazení ze studie byla u obou skupin pacientů nauzea a zvracení. Mezi pacienty léčenými exenatidem s prodlouženým účinkem došlo k vyřazení z důvodu nauzey nebo zvracení u méně než 1 % pacientů, u exenatidu s okamžitým účinkem u 1 % pacientů.
Reakce v místě vpichu
V průběhu 6měsíční kontrolní fáze klinických studií byly lokální reakce v místě podání u pacientů léčených exenatidem s prodlouženým účinkem pozorovány častěji ve srovnání s pacienty léčenými komparátorem (16 % oproti rozmezí 2-7 %). Obecně byly tyto lokální reakce v místě podání mírné a obvykle nevedly k ukončení účasti ve studii. U pacientů mohou být příznaky ošetřením mírněny, zatímco se pokračuje v léčbě. Následující injekce by měly být potom podány každý týden do jiného místa. V poregistračním období byly hlášeny případy abscesů a celulitidy v místě vpichu.
Velmi často byly v klinických studiích pozorovány v místě injekce malé podkožní uzlíky, což je následkem známých vlastností mikročástic z PLGA. Většina jednotlivých uzlíků byla asymptomatická, neměla vliv na účast ve studii a vymizela v průběhu 4-8 týdnů.
Imunogenicita
V souladu s potenciálními imunogenními vlastnostmi léčivých přípravků na bázi proteinů a peptidů se u pacientů léčených exenatidem s prodlouženým účinkem mohou vytvořit protilátky proti exenatidu.
U většiny pacientů, u kterých k tvorbě protilátek došlo, se titr protilátek časem snižoval.
Přítomnost protilátek (vysoký nebo nízký titr) nepredikuje úroveň glykemické kontroly u jednotlivých pacientů.
V klinických studiích s exenatidem s prodlouženým účinkem mělo na konci studie nízký titr protilátek přibližně 45 % pacientů. Průměrné procento pacientů s pozitivním titrem protilátek bylo stálé ve všech klinických hodnoceních. Úroveň kontroly glykemie (HbA1c) byla u této skupiny celkově srovnatelná jako u skupiny bez nalezených protilátek. Vyšší titr protilátek mělo ve studiích fáze 3 průměrně 12 % pacientů. Úměrně tomu chyběla na konci kontrolovaného období glykemická odpověď na léčbu exenatidem s prodlouženým účinkem; glykemická odpověď se neprojevila u 2,6 % pacientů s vyšším titrem protilátek, zatímco u pacientů s negativním nálezem protilátek se odpověď neprojevila u 1,6 %.
U pacientů, u kterých se vytvořily protilátky na exenatid, byla pozorována větší tendence k lokálním reakcím po podání přípravku (např.: zčervenání kůže a svědění), frekvence a typy ostatních nežádoucích příhod byly srovnatelné s pacienty bez protilátek na exenatid.
V 30týdenní a dvou 26týdenních klinických studiích byla u pacientů léčených exenatidem
s prodlouženým účinkem frekvence výskytu potenciálně imunogenních reakcí v místě podání (nejčastěji pruritus s nebo bez erytému) 9 %. Tyty reakce byly méně často pozorovány u pacientů bez protilátek (4 %) ve srovnání s pacienty s přítomností protilátek (13 %), s větším výskytem u pacientů s vyšším titrem protilátek.
Vyšetření vzorků s pozitivním nálezem protilátek neodhalilo signifikantní zkříženou reaktivitu s podobnými endogenními peptidy (glukagon nebo GLP-1).
Rychlé snížení tělesné hmotnosti
V 30týdenní studii došlo k rychlému snížení tělesné hmotnosti v minimálně jednom časovém období u přibližně 3 % pacientů (n=4/148) léčených exenatidem s prodlouženým účinkem (zaznamenané snížení tělesné hmotnosti v období mezi dvěma následujícími studijními návštěvami větší než
1,5 kg/týden).
Zvýšení srdeční frekvence
Při celkové analýze klinických studií s exenatidem s prodlouženým účinkem bylo pozorováno průměrné zvýšení srdeční frekvence (HR) o 2,6 stahu za minutu (bpm) ve srovnání s výchozí hodnotou (74 bpm). U patnácti procent pacientů léčených exenatidem s prodlouženým účinkem bylo průměrné zvýšení srdeční frekvence HR > 10 bpm; přibližně 5 % až 10 % subjektů v jiných léčebných skupinách mělo průměrné zvýšení HR > 10 bpm.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Projevy předávkování exenatidem (založené na klinických studiích exenatidu s okamžitým účinkem) zahrnovaly závážnou nauzeu, silné zvracení a rychlé snížení koncentrace glukózy v krvi. V případě předávkování by měla být s ohledem na pacientovy klinické projevy a příznaky zahájena odpovídající podpůrná léčba.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Léčiva k terapii diabetu, jiná antidiabetika, kromě inzulinů, ATC kód: A10BX04.
Mechanismus účinku
Exenatid je agonista receptoru peptidu podobného glukagonu 1 (GLP-1), který vykazuje některé antihyperglykemické účinky jako peptid podobný glukagonu 1 (glucagon-like peptid-1, GLP-1). Sekvence aminokyselin exenatidu se částečně překrývá se sekvencí lidského GLP-1. U exenatidu se prokázala vazba a aktivace známého lidského GLP-1 receptoru in vitro, mechanismus účinku je zprostředkován cyklickou AMP a/nebo dalšími nitrobuněčnými signálními cestami.
Exenatid zvyšuje, v závislosti na hladině glukózy, sekreci inzulínu v beta-buňkách pankreatu. Se snižováním koncentrace glukózy klesá i produkce inzulínu. Při podávaní exenatidu v kombinaci se samotným metforminem a/nebo thiazolidindionem nebylo pozorováno žádné zvýšení výskytu hypoglykemie v porovnání s placebem v kombinaci s metforminem a/nebo thiazolidindionem, což může být způsobeno tímto glukózo-dependentním inzulinotropním mechanismem účinku (viz bod 4.4).
Exenatid potlačuje sekreci glukagonu, jejíž nepřiměřené zvýšení je známé u diabetu 2. typu. Nižší hladiny glukagonu vedou ke snížení tvorby glukózy v játrech. Exenatid však nemá vliv na normální reakci glukagonu a dalších hormonů v odpovědi na hypoglykémii.
Exenatid zpomaluje vyprazdňování žaludečního obsahu a snižuje tedy rychlost absorpce glukózy z potravy do krevního oběhu.
Bylo prokázáno, že podání exenatidu snižuje příjem potravy prostřednictvím snížené chuti k jídlu a zvýšením pocitu sytosti.
Farmakodynamické účinky
Exenatid zlepšuje kontrolu glykemie prostřednictvím trvalého působení, kdy snižuje postprandiální hladiny glukózy i hladiny glukózy nalačno u pacientů s diabetem 2. typu. Na rozdíl od přirozeného GLP-1, má exenatidem s prodlouženým účinkem u člověka farmakokinetický a farmakodynamický profil vhodný pro podání jednou týdně.
Farmakodynamická studie exenatidu prokázala u pacientů s diabetem 2. typu (n = 13) obnovení první fáze sekrece inzulínu a zlepšení sekrece inzulínu ve druhé fázi jako odpověď na intravenózní bolus glukózy.
Klinická účinnost a bezpečnost
Výsledky dlouhodobých klinických hodnocení exenatidu s prodlouženým účinkem jsou uvedeny níže, tato hodnocení zahrnovala 1628 osob (804 léčených exenatidem s prodlouženým účinkem), 54 % mužů a 46 % žen, 281 osob (141 léčených exenatidem s prodlouženým účinkem) bylo ve věku > 65 let.
Kontrola glykemie
Ve dvou studiích byl exenatid s prodlouženým účinkem 2 mg podávaný jednou týdně srovnáván s 5 ^g exenatidu s okamžitým účinkem následovaného 10 ^g exenatidu s okamžitým účinkem. Délka trvání jedné studie byla 24 týdnů (n = 252) a druhé 30 týdnů (n = 295), následovaných nezaslepeným
pokračováním v délce dalších 22 týdnů, kdy všichni pacienti užívali exenatid s prodlouženým účinkem 2 mg podávaný jednou týdně (n = 243). V obou hodnoceních bylo snížení HbA1c zjevné v obou léčebných skupinách již při prvním měření HbA1c po zahájení léčby (týden 4 nebo 6).
Podávání exenatidu s prodlouženým účinkem vedlo ke statisticky významnému snížení HbA1c ve srovnání s pacienty užívajícími exenatid s okamžitým účinkem (Tabulka 2).
Klinicky významný účinek exenatidu s prodlouženým účinkem a exenatidu s okamžitým účinkem na HbA1c byl v obou studiích pozorován bez ohledu na druh základní antidiabetické terapie.
V těchto dvou klinických studiích dosáhlo snížení HbA1c na hodnoty < 7 % nebo < 7 % klinicky a statisticky významně více subjektů na exenatidu s prodlouženým účinkem ve srovnání s exenatidem s okamžitým účinkem (p < 0,05; resp. p = < 0,0001).
Obě skupiny pacientů užívajících exenatid s prodlouženým účinkem a exenatid s okamžitým účinkem dosáhly snížení tělesné hmotnosti oproti počátečním hodnotám, ačkoli rozdíl mezi oběma rameny nebyl významný.
Další snížení HbA1c a pokračující úbytek hmotnosti byl pozorován minimálně po dobu 52 týdnů u pacientů, kteří dokončili kontrolovanou 30týdenní studii i další nekontrolované pokračování. Hodnotitelní pacienti, kteří byli převedeni z exenatidu s okamžitým účinkem na exenatid s prodlouženým účinkem (n = 121) dosáhli stejného zlepšení HbA1c - 2,0 % na konci 22. týdne pokračování ve srovnání s počátečními hodnotami jako pacienti léčení exenatidem s prodlouženým účinkem po dobu 52 týdnů.
Tabulka 2: Výsledky dvou klinických hodnocení exenatidu s prodlouženým účinkem oproti exenatidu s okamžitým účinkem v kombinaci s dietou a cvičením samotným, metforminem a/nebo sulfonylmočovinou a metforminem a/nebo thiazolidindionem (intent-to-treat pacienti).
|
24týdenní studie |
Exenatid s prodlouženým účinkem 2 mg |
Exenatid s okamžitým účinkem 10 ^g dvakrát denně |
|
N |
129 |
123 |
|
Průměr HbA1c (%) | ||
|
Výchozí hodnota |
8,5 |
8,4 |
|
Změna oproti výchozí hodnotě (±SE) |
-1,6 (±0,1)** |
-0,9 (±0,1) |
|
Průměrný rozdíl mezi léčbami ve změně oproti výchozí hodnotě (95% CI) |
-0,67 (-0,94 |
; -0,39)** |
|
Podíl pacientů (%) dosahujících HbA1c < 7 % |
58 |
30 |
|
Změna v plazmatické hladině glukózy na lačno (mmol/l) (±SE) |
-1,4 (±0,2) |
-0,3 (±0,2) |
|
Průměrná tělesná hmotnost (kg) | ||
|
Výchozí hodnota |
97 |
94 |
|
Změna oproti výchozí hodnotě (±SE) |
-2,3 (±0,4) |
-1,4 (±0,4) |
|
Průměrný rozdíl mezi léčbami ve změně oproti výchozí hodnotě (95% CI) |
-0,95 (-1,91; 0,01) | |
|
30týdenní studie | ||
|
N |
148 |
147 |
|
Průměr HbA1c (%) | ||
|
Výchozí hodnota |
8,3 |
8,3 |
|
Změna oproti výchozí hodnotě (±SE) |
-1,9 (±0,1)* |
-1,5 (±0,1) |
|
Průměrný rozdíl ve změně oproti výchozí hodnotě mezi léčbami (95% CI) |
-0,33 (-0,54; -0,12)* | |
|
Podíl pacientů (%) dosahujících HbA1c < 7 % |
73 |
57 |
|
Změny hladiny glukózy v plazmě nalačno (mmol/l) ( ± SE) |
-2,3 (±0,2) |
-1,4( ±0,2) |
S
|
Průměrná tělesná hmotnost (kg) | ||
|
Výchozí hodnota |
102 |
102 |
|
Změna oproti výchozí hodnotě (±SE) |
-3,7 (±0,5) |
-3,6 (±0,5) |
|
Průměrný rozdíl mezi léčbami ve změně oproti výchozí hodnotě (95% CI) |
-0,08 (-1,29; 1,12) | |
E=standardní chyba, CI=interval spolehlivosti, * p< 0,05, **p< 0,0001
Ve 26týdenní studii byl exenatid s prodlouženým účinkem srovnáván s inzulínem glargin podávaným jednou denně. Exenatid s prodlouženým účinkem prokázal významnější změnu HbA1c ve srovnání s inzulinem glargin. Ve srovnání s léčbou inzulinem glargin exenatid s prodlouženým účinkem významně snižoval průměrnou tělesnou hmotnost a jeho podávání bylo spojeno s menším počtem hypoglykemických příhod (Tabulka 3).
Tabulka 3: Výsledky 26týdenního klinického hodnocení exenatid s prodlouženým účinkem ve srovnání s inzulinem glargin v kombinaci s metforminem samotným nebo metforminem a sulfonylmočovinou (intent to treat pacienti).___
|
Exenatid s prodlouženým účinkem 2 mg |
Inzulin glargin1 | |
|
N |
233 |
223 |
|
Průměr HbA1c (%) | ||
|
Výchozí hodnota |
8,3 |
8,3 |
|
Změna oproti výchozí hodnotě (±SE) |
-1,5 (±0,1)* |
-1,3 (±0,1)* |
|
Průměrný rozdíl mezi léčbami ve změně oproti výchozí hodnotě (95% CI) |
-0,16 (-0,29; -0,03)* | |
|
Podíl pacientů (%) dosahujících HbA1c < 7 % |
62 |
54 |
|
Změny hladiny glukózy v séru nalačno (mmol/l) (±SE) |
-2,1 (±0,2) |
-2,8 (±0,2) |
|
Průměrná tělesná hmotnost (kg) | ||
|
Výchozí hodnota |
91 |
91 |
|
Změna oproti výchozí hodnotě (±SE) |
-2,6 (±0,2) |
+1,4 (±0,2) |
|
Průměrný rozdíl mezi léčbami ve změně oproti výchozí hodnotě (95% CI) |
-4,05 (-4,57; -3,52)* | |
|
E=standardní chyba, CI=interval spolehlivosti, * p<0,05 | ||
1 Inzulin glargin byl dávkován tak, aby se cílová koncentrace glukózy pohybovala v rozmezí od 4,0 do 5,5 mmol/l (72 až 100 mg/dl). Průměrná dávka inzulinu glargin byla na začátku léčby 10,1 IU/den a zvýšila se na 31,1 IU/den u pacientů léčených inzulinem glargin.
Výsledky 156týdenní byly v souladu s výsledky již dříve publikovanými ve 26týdenní zprávě. Léčba exenatidem s prodlouženým účinkem ve srovnání s léčbou inzulinem glarginem trvale významně zlepšovala glykemickou kontrolu a kontrolu tělesné hmotnosti. Bezpečnostní údaje po 156 týdnech byly v souladu s výsledky hlášenými po 26 týdnech.
Ve 26týdenní studii byl exenatid s prodlouženým účinkem srovnáván s maximálními dávkami sitagliptinu a pioglitazonu u pacientů užívajících také metformin. Ve všech léčebných skupinách došlo k významnému snížení HbAJc ve srovnání s počáteční hodnotou. U exenatidu s prodlouženým účinkem byla prokázána superiorita oproti sitagliptinu i pioglitazonu s ohledem na na změnu HbAJc ve srovnání s počáteční hodnotou.
Exenatid s prodlouženým účinkem prokázal významně vyšší snížení tělesné hmotnosti ve srovnání se sitagliptinem. U pacientů užívajících pioglitazon došlo ke zvýšení tělesné hmotnosti (Tabulka 4).
Tabulka 4: Výsledky 26týdenního klinického hodnocení exenatidu s prodlouženým účinkem ve
|
srovnání se sitagliptinem a pioglitazonem v kombinaci s met |
orminem (intent to treat pacienti). | ||
|
Exenatid s prodlouženým účinkem 2 mg |
Sitagliptin 100 mg |
Pioglitazon 45 mg | |
|
N |
160 |
166 |
165 |
|
Průměr HbA1c (%) | |||
|
Výchozí hodnota |
8,6 |
8,5 |
8,5 |
|
Změna oproti výchozí hodnotě (±SE) |
-1,6 (±0,1)* |
-0,9 (±0,1)* |
-1,2 (±0,1)* |
|
Průměrný rozdíl mezi léčbami ve změně oproti výchozí hodnotě (95% CI) sitagliptin |
-0,63 (-0,89; -0,37)** | ||
|
Průměrný rozdíl mezi léčbami ve změně oproti výchozí hodnotě (95% CI) pioglitazon |
-0,32 (-0,57; -0,06)* | ||
|
Podíl pacientů (%) dosahujících HbA1c < 7 % |
62 |
36 |
49 |
|
Změny hladiny glukózy v séru nalačno (mmol/l) (±Se) |
-1,8 (±0,2) |
-0,9 (±0,2) |
-1,5 (±0,2) |
|
Průměrná tělesná hmotnost (kg) | |||
|
Výchozí hodnota |
89 |
87 |
88 |
|
Změna oproti výchozí hodnotě (±SE) |
-2,3 (±0,3) |
-0,8 (±0,3) |
+2,8 (±0,3) |
|
Průměrný rozdíl mezi léčbami ve změně oproti výchozí hodnotě (95% CI) sitagliptin |
-1,54 (-2,35; -0,72)* | ||
|
Průměrný rozdíl mezi léčbami ve změně oproti výchozí hodnotě (95% CI) pioglitazon |
-5,10 (-5,91; -4,28)** | ||
SE=standardní chyba, CI=interval spolehlivosti, * p < 0,05, **p < 0,0001
Tělesná hmotnost
Snížení tělesné hmotnosti oproti počátečním hodnotám bylo pozorováno ve všech klinických hodnoceních exenatidu s prodlouženým účinkem. Toto snížení bylo pozorováno u pacientů užívajících exenatid s prodlouženým účinkem bez ohledu na to, zda se u nich vyskytovala nevolnost nebo ne, ačkoli snížení bylo větší ve skupině pacientů s nauzeou (průměrné snížení o -2,9 kg až -5,2 kg v přítomnosti nauzey oproti -2,2 kg až -2,9 kg bez nauzey).
Podíl pacientů, u kterých došlo ke snížení tělesné hmotnosti i HbA1c, se pohyboval od 70 do 79 % (podíl pacientů u kterých došlo ke snížení HbA1c se pohyboval od 88 do 96 %).
Plazmatické/sérové hladiny glukózy
Léčba exenatidem s prodlouženým účinkem vedla k signifikantnímu snížení plazmatických/sérových hladin glukózy nalačno, tato snížení byla pozorována již po 4 týdnech. Byla pozorována také další snížení postprandiálních koncentrací. Zlepšení hladin glukózy nalačno přetrvávalo po dobu 52 týdnů.
Funkce beta-buněk
Klinická hodnocení exenatidu s prodlouženým účinkem ukázala při použití homeostatického modelu (HOMA-B) zlepšení funkce beta-buněk. Tyto účinky na funkci beta-buněk byly trvalé po dobu 52 týdnů.
Krevní tlak
V klinických hodnoceních exenatidu s prodlouženým účinkem bylo pozorováno snížení systolického krevního tlaku (2,9 mmHg až 4,7 mmHg). Ve 30týdenním srovnávacím klinickém hodnocení s exenatidem s okamžitým účinkem, exenatid s prodlouženým účinkem i exenatid s okamžitým účinkem signifikantně snižovaly systolický krevní tlak oproti výchozím hodnotám (4,7±1,1mmHg, resp. 3,4±1,1_mmHg), rozdíl mezi oběma rameny nebyl významný. Zlepšení krevního tlaku bylo trvalé po dobu 52 týdnů.
Hladiny lipidů nalačno
Exenatid s prodlouženým účinkem nevykazuje nežádoucí účinky na lipidové parametry.
Pediatrická populace
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s exenatidem s prodlouženým účinkem u jedné nebo více podskupin pediatrické populace s diabetes mellitus 2.typu (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Absorpce exenatidu je ovlivněna vlastnostmi lékové formy exenatidu s prodlouženým účinkem. Po absorpci do krevního oběhu je exenatid distribuován a eliminován v souladu s jeho známými farmakokinetickými vlastnostmi (tak jak je popsáno v tomto bodě).
Absorpce
Po podávání exenatidu s prodlouženým účinkem 2 mg jednou týdně přesáhly průměrné koncentrace exenatidu minimální účinné koncentrace (~ 50 pg/ml) v průběhu 2 týdnů s postupným zvyšováním průměrné plazmatické koncentrace exenatidu v průběhu 6 až 7 týdnů. Následně bylo dosaženo koncentrace exenatidu přibližně 300 pg/ml indikující dosažení rovnovážného stavu. Rovnovážné koncentrace exenatidu jsou udržovány v průběhu týdenního intervalu mezi dávkami s minimálními výkyvy nahoru či dolů od této průměrné terapeutické koncentrace.
Distribuce
Průměrný zdánlivý distribuční objem exenatidu po subkutánním podání jednorázové dávky exenatidu je 28 litrů.
Biotrasnformace a eliminace
Neklinické studie ukázaly, že exenatid je eliminován převážně glomerulární filtrací s následnou proteolytickou degradací. V klinických studiích je průměrná zdánlivá clearance exenatidu 9 l/h. Tyto farmakokinetické parametry exenatidu jsou nezávislé na dávce. Střední plazmatické koncentrace exenatidu klesnou pod detekovatelnou hranici přibližně za 10 týdnů po ukončení léčby exenatidem s prodlouženým účinkem.
Zvláštní populace
Renální insuficience
Farmakokinetické populační analýzy pacientů s poškozením ledvin, kterým byl podáván exenatid s prodlouženým účinkem 2 mg, naznačují zvýšení systémové expozice o 74 % a o 23 % (odhad střední hodnoty v každé skupině) u pacientů se středním (N = 10) a mírným (N = 56) poškozením ledvin, ve srovnání s pacienty s normálními renálními funkcemi (N = 84).
Hepatální insuficience
U pacientů s hepatální nedostatečností nebyly prováděny žádné farmakokinetické studie. Exenatid je primárně eliminován ledvinami, proto se neočekává, že by jaterní dysfunkce ovlivňovala koncentrace exenatidu v krvi.
Pohlaví, rasa a tělesná hmotnost
Pohlaví, rasa a tělesná hmotnost nemají na farmakokinetiku exenatidu klinicky významný vliv.
Starší pacienti
Data o starších pacientech jsou omezená, nenaznačují však žádné výrazné změny v expozici exenatidu s rostoucím věkem až do věku 75 let.
Ve farmakokinetické studii exenatidu s okamžitým účinkem u pacientů s diabetem 2.typu vedlo podávání exenatidu (10 ^g) k průměrnému zvýšení celkové plochy pod křivkou exenatidu o 36 %
u 15 starších subjektů ve věku 75—85 let ve srovnání s 15 subjekty ve věku 45—65 let pravděpodobně z důvodu snížené renální funkce u starší věkové skupiny (viz bod 4.2).
Pediatrická populace
Ve farmakokinetické studii jednorázového podání exenatidu s okamžitým účinkem provedené u 13 pacientů ve věku 12 až 16 let s diabetem 2. typu mělo podání exenatidu (5 ^g) za následek mírné snížení průměrné celkové plochy pod křivkou (nižší o 16 %) a maximální plazmatické koncentrace Cmax (nižší o 25 %) ve srovnání s hodnotami pozorovanými u dospělých. U pediatrické populace nebyla provedena žádná klinická studie s exenatidem s prodlouženým účinkem.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti, toxicity po opakovaném podání a genotoxicity provedené s exenatidem s okamžitým účinkem, nebo exenatidem s prodlouženým účinkem, neodhalily žádné zvláštní riziko pro člověka.
Ve 104týdenní studii kancerogenity exenatidu s prodlouženým účinkem byl u laboratorních potkanů pozorován statisticky významný zvýšený výskyt thyreoidního tumoru z C-buněk (adenom a/nebo karcinom) při všech dávkách (1,4 až 26násobek klinické expozice exenatidu s prodlouženým účinkem u člověka). Relevance těchto nálezů pro člověka není v současné době známá.
Studie na zvířatech nenaznačují přímé škodlivé účinky na plodnost nebo těhotenství, vysoké dávky exenatidu způsobovaly poruchy vývoje kostry a zpomalení fetálního a neonatálního růstu.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek
polyglaktin (1:1) sacharóza
Rozpouštědlo sodná sůl karmelosy chlorid sodný polysorbát 20
monohydrát dihydrogenfosforečnanu sodného heptahydrát hydrogenfosforečnanu sodného voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto tento léčivý přípravek nesmí být mísen s žádnými dalšími léčivými přípravky.
6.3 Doba použitelnosti
3 roky
Po rekonstituci suspenze
Suspenze musí být aplikována okamžitě po smísení prášku a rozpouštědla.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C). Chraňte před mrazem.
Před použitím může být souprava uchovávána při teplotě do 30 °C až po dobu 4 týdnů.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Podmínky uchovávání přípravku po smísení viz bod 6.3.
6.5 Druh obalu a obsah balení
Prášek je balen v 3 ml skleněné (typ I) injekční lahvičce s chlorobutylovým pryžovým uzávěrem a hliníkovým pertlem s plastovým víčkem.
Rozpouštědlo je baleno v 1,5 ml skleněné (typ I) předplněné injekční stříkačce s pryžovým bromobutylovým uzávěrem a pryžovým pístem.
Každá souprava pro jednorázovou dávku obsahuje jednu injekční lahvičku se 2 mg exenatidu, jednu předplněnou injekční stříkačku s 0,65 ml rozpouštědla, jednu spojku lahvičky a dvě injekční jehly (jedna rezervní).
Balení obsahující 4 soupravy pro jednorázové podání nebo multipack obsahující 12 (3 balení po 4 soupravách) pro jednorázové podání.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a zacházení s ním
Pacienti by měli být poučeni, aby znehodnotili po každé injekci injekční stříkačku i se stále nasazenou jehlou. Pacient nepotřebuje uchovávat žádnou část použité soupravy pro jednorázové podání.
Před použitím se rozpouštědlo vizuálně zkontroluje. Rozpouštědlo může být použito pouze v případě, že je čiré a neobsahuje žádné částice. Po vytvoření suspenze může být suspenze použita pouze tehdy, je-li skoro bílá až bílá a zakalená.
Exenatid s prodlouženým účinkem se aplikuje ihned po přípravě suspenze prášku v rozpouštědle.
Nesmí být použit exenatid s prodlouženým účinkem, který byl zmrazen.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
AstraZeneca AB SE-151 85 Sodertálje Švédsko
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/11/696/001-002
9. DATUM PRVNÍ REGISRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 17. června 2011 Datum posledního prodloužení registrace:
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu
NÁZEV PŘÍPRAVKU
1.
Bydureon 2 mg prášek a rozpouštědlo pro injekční suspenzi s prodlouženým účinkem v předplněném peru
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedno předplněné pero obsahuje exenatidum 2 mg. Dávka po rekonstituci suspenze podaná z pera je 2 mg v 0,65 ml.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční suspenzi s prodlouženým účinkem. Prášek: bílý až téměř bílý prášek.
Rozpouštědlo: čirý, bezbarvý až lehce nažloutlý či nahnědlý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Bydureon je indikován k léčbě diabetes mellitus 2. typu v kombinaci s:
• metforminem
• deriváty sulfonylmočoviny
• thiazolidindiony
• metforminem a sulfonylmočovinou
• metforminem a thiazolidindionem
u dospělých pacientů, u kterých není dosaženo dostatečné kontroly glykemie při podávání maximálních tolerovaných dávek těchto perorálních přípravků.
4.2 Dávkování a způsob podání
Dávkování
Doporučená dávka je 2 mg exenatidu podaná jednou týdně.
U pacientů převáděných z exenatidu s okamžitým účinkem (Byetta) na exenatid s prodlouženým účinkem může dojít k přechodnému zvýšení hladiny glukózy v krvi, která se zpravidla upraví během prvních dvou týdnů po zahájení léčby.
Jestliže je exenatid s prodlouženým účinkem přidán k léčbě metforminem a/nebo thiazolidindionem, je možné pokračovat v dosavadní dávce metforminu a/nebo thiazolidindionu. Jestliže je exenatid s prodlouženým účinkem přidán k sulfonylmočovině, měla by být zvážena redukce dávky sulfonylmočoviny, aby se snížilo riziko hypoglykemie (viz bod 4.4).
Exenatid s prodlouženým účinkem se podává jednou týdně ve stejný den. Tento den podání může být v případě potřeby změněn, pokud je další dávka podána nejméně o jeden den (24 hodin) později. Exenatid s prodlouženým účinkem může být podán kdykoli v průběhu dne nezávisle na jídle.
V případě opomenutí podání dávky by tato měla být podána co nejdříve je to možné. Při další injekci se pacient může vrátit k původnímu dni pro podání injekce. Ovšem vždy si může podat pouze jednu injekci v průběhu 24 hodin.
Užívání exenatidu s prodlouženým účinkem nevyžaduje další každodenní měření glykemie prováděné pacientem. Toto měření může být ovšem nezbytné pro úpravu dávky sulfonylmočoviny.
Pokud je zahájena jiná léčba snižující hladinu glukózy v krvi po ukončení podávání exenatidu s prodlouženým účinkem, mělo by být prodloužené uvolňování exenatidu s prodlouženým účinkem vzato do úvahy (viz bod 5.2).
Zvláštní populace
Starší pacienti
Úprava dávky v závislosti na věku není nutná. Nicméně vzhledem ke snižování renálních funkcí s věkem by měla být renálním funkcím pacienta věnována pozornost (viz Pacienti s renální nedostatečností). Klinické zkušenosti u pacientů nad 75 let jsou velmi omezené (viz bod 5.2).
Renální insuficience
U pacientů s mírnou renální nedostatečností (clearance kreatininu 50 až 80 ml/min) není zapotřebí úprava dávky. U pacientů se středně závažnou renální nedostatečností (clearance kreatininu 30 až 50 ml/min) jsou klinické zkušenosti velmi omezené (viz bod 5.2). Podávání exenatidu s prodlouženým účinkem se těmto pacientům nedoporučuje.
Exenatid s prodlouženým účinkem se nedoporučuje podávat pacientům v konečném stádiu renálního selhávání nebo se závažnou renální nedostatečností (clearance kreatininu <30 ml/min) (viz bod 4.4).
Hepatální insuficience
U pacientů s hepatální nedostatečností není zapotřebí úprava dávky (viz bod 5.2).
Pediatrická populace
Bezpečnost a účinnost podávání exenatidu s prodlouženým účinkem nebyla stanovena u dětí a dospívajících mladších 18 let. Dostupné údaje jsou popsány v bodě 5.2, avšak nelze učinit žádná doporučení pro dávkování.
Způsob podání
Exenatid s prodlouženým účinkem je určen k aplikaci pacientem. Každé pero by mělo být použito pouze jednou osobou a je určeno k jednorázovému použití.
Před zahájením léčby exenatidem s prodlouženým účinkem se důrazně doporučuje, aby pacienti a jejich opatrovníci byli vyškoleni odborným zdravotnickým pracovníkem v aplikaci přípravku.
„Pokyny pro uživatele“, které jsou součástí balení přípravku, se musí pečlivě dodržovat.
Jednotlivá dávka musí být aplikována subkutánně do oblasti břicha, stehna nebo zadní části paže ihned po přípravě suspenze prášku v rozpouštědle.
Pokyny pro přípravu suspenze léčivého přípravku před jeho podáním naleznete v bodě 6.6 a v „Pokynech pro uživatele“.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Exenatid s prodlouženým účinkem se nemápoužívat u pacientů s diabetes mellitus 1. typu nebo k léčbě diabetické ketoacidózy.
Exenatid s prodlouženým účinkem nesmí být podán intravenózně nebo intramuskulámě.
Renální nedostatečnost
U dialyzovaných pacientů v konečném stádiu renálního selhávání jednotlivé dávky exenatidu s okamžitým účinkem zvýšily frekvenci výskytu a závažnost gastrointestinálních nežádoucích účinků, exenatid s prodlouženým účinkem se proto nedoporučuje podávat pacientům v konečném stádiu renálního selhávání nebo se závažnou renální nedostatečností (clearance kreatininu <30 ml/min).
U pacientů se středně závažnou renální nedostatečností jsou klinické zkušenosti velmi omezené a použití exenatidu s prodlouženým účinkem se proto nedoporučuje.
Méně často byly spontánně hlášeny případy změny renálních funkcí, včetně zvýšení sérového kreatininu, poruchy funkce ledvin, zhoršení chronického renálního selhání a akutní renální selhání, někdy vyžadující hemodialýzu. K některým z těchto příhod došlo u pacientů se stavy, které mohou ovlivňovat hydrataci, včetně nauzey, zvracení a/nebo průjmu, a/nebo u pacientů užívajících přípravky, u kterých je znám jejich vliv na renální funkce/celkovou hydrataci. Současně podávané přípravky zahrnovaly inhibitory angiotenzin konvertujícího enzymu, antagonisty angiotenzinu-II, nesteroidní protizánětlivé přípravky a diuretika. Reverzibilita porušených renálních funkcí byla pozorována při podpůrné léčbě a při vysazení potenciálně působících agens včetně exenatidu.
Závažná gastrointestinální onemocnění
Exenatid s prodlouženým účinkem nebyl hodnocen u pacientů se závažným gastrointestinálním onemocněním včetně gastroparézy. Použití přípravku je často spojeno s nežádoucími účinky v oblasti gastrointestinálního traktu, včetně nauzey, zvracení a průjmu. Z tohoto důvodu se exenatid s prodlouženým účinkem nedoporučuje u pacientů se závažným gastrointestinálním onemocněním.
Akutní pankreatitida
Použití agonistů GLP-1 je spojeno s rizikem vývoje akutní pankreatitidy. U exenatidu s prodlouženým účinkem byly spontánně hlášeny případy akutní pankreatitidy. Při použití podpůrné léčby byl pozorován ústup pankreatitidy, ale velmi vzácně byly hlášeny případy nekrotizující nebo hemoragické pankreatitidy a/nebo úmrtí. Pacienti by měli být informováni o charakteristických symptomech akutní pankreatitidy: přetrvávající, silná bolest břicha. V případě podezření na pankreatitidu by měla být léčba exenatidem s prodlouženým účinkem ukončena, pokud je akutní pankreatitida potvrzena, léčba exenatidem s prodlouženým účinkem nesmí být znovu zahájena. Opatrnosti je třeba u pacientů s anamnézou pankreatitidy.
Souběžně podávané léčivé přípravky
Souběžné podávání exenatidu s prodlouženým účinkem s inzulínem, deriváty D-fenylalaninu (meglitinidy), inhibitory alfa-glukosidázy, inhibitory dipeptidyl peptidázy-4 nebo jinými agonisty receptoru pro GLP-1 nebylo studováno. Současné podávání exenatidu s prodlouženým účinkem a exenatidu s okamžitým účinkem (Byetta) nebylo studováno, a proto se nedoporučuje.
Interakce s warfarinem
Spontánně byly hlášeny případy zvýšení INR (International Normalized Ratio), někdy spojené s krvácením, během současného užívání warfarinu a exenatidu s prodlouženým účinkem (viz bod 4.5).
Hypoglykemie
Výskyt hypoglykemie byl v klinických studiích vyšší při použití exenatidu s prodlouženým účinkem v kombinaci se sulfonylmočovinou. V klinických studiích měli pacienti s mírnou renální insufuciencí užívající kombinaci se sulfonylmočovinu zvýšený výskyt hypoglykemie v porovnání s pacienty s normální renální funkcí. Pro snížení rizika hypoglykemie spojeného s užíváním sulfonylmočoviny by měla být zvážena redukce dávky sulfonylmočoviny.
Rychlé snížení tělesné hmotnosti
U pacientů užívajících exenatid bylo pozorováno rychlé snížení hmotnosti o více než 1,5 kg za týden. Takto vysoký hmotnostní úbytek může mít škodlivé důsledky. Pacienti s rychlým hmotnostním úbytkem se mají sledovat na známky a projevy cholelithiázy.
Ukončení léčby
Po ukončení léčby může účinek exenatidu s prodlouženým účinkem přetrvávat v závislosti na snižování plazmatických hladin exenatidu až po dobu 10 týdnů. Při výběru jiného léčivého přípravku a nastavení dávky by mělo být toto vzato do úvahy také vzhledem k tomu, že mohou přetrvávat nežádoucí účinky a částečně i účinnost do doby úplného snížení hladin exenatidu.
Pomocné látky
Obsah sodíku: Tento léčivý přípravek obsahuje v jedné dávce méně než 1 mmol sodíku (23 mg), tj. v podstatě je bez sodíku.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Sulfonylmočoviny
Dávkování sulfonylmočoviny může vyžadovat úpravu vzhledem ke zvýšenému riziku hypoglykemie spojené s terapií sulfonylmočovinou (viz body 4.2 a 4.4).
Vyprazdňování žaludku
Výsledky studie používající paracetamol jako modelové léčivo k hodnocení vyprazdňování žaludku naznačují, že účinek exenatidu s prodlouženým účinkem na zpomalení vyprazdňování žaludku je malý a neočekává se, že způsobí klinicky významné snížení rychlosti a rozsahu absorpce současně perorálně podaných léčivých přípravků. Z tohoto důvodu nejsou vyžadovány úpravy dávek léčivých přípravků senzitivních na zpomalení vyprazdňování žaludku.
Po 14týdenní terapii exenatidem s prodlouženým účinkem nebyly po podání tablet s obsahem 1000 mg paracetamolu ať už nalačno, nebo po jídle, pozorovány významné změny AUC paracetamolu v porovnání s kontrolním obdobím. Cmax paracetamolu se snížila o 16 % (nalačno) a o 5 % (po jídle) a tmax se zvýšila oproti kontrolnímu období z přibližně 1 hodiny na 1,4 hodiny (nalačno) a na 1,3 hodiny (po jídle).
Byly provedeny následující studie interakcí za použití 10 ^g exenatidu s okamžitým účinkem, nikoli exenatidu s prodlouženým účinkem:
Warfarin
Při podání warfarinu 35 minut po aplikaci exenatidu s okamžitým účinkem bylo pozorováno zpoždění tmax přibližně o 2 hodiny. Nebyl pozorován žádný klinicky relevantní vliv na Cmax nebo AUC. Spontánně byly hlášeny případy zvýšení INR během současného užívání warfarinu a exenatidu s prodlouženým účinkem. Během zahájení podávání exenatidu s prodlouženým účinkem se má u pacientů užívajících warfarin a/nebo kumarinové deriváty hodnota INR pečlivě monitorovat (viz bod 4.8).
Inhibitory HMG CoA reduktázy
Jestliže byl exenatid s okamžitým účinkem podán společně s jednorázovou dávkou lovastatinu (40 mg), AUC a Cmax lovastatinu byly sníženy o 40 %, resp. 28 % a tmax byl zpožděn asi o 4 hodiny ve srovnání s podáním samotného lovastatinu. V 30týdenních placebem kontrolovaných klinických studiích nebylo současné podávání exenatidu s okamžitým účinkem a inhibitorů HMG CoA reduktázy spojeno se stabilními změnami v lipidovém profilu (viz bod 5.1). Ačkoliv není dopředu vyžadována žádná úprava dávky, lipidové profily se mají pravidelně monitorovat.
Digoxin a lisinopril
Ve studiích interakcí účinku exenatidu podávaného dvakrát denně na digoxin a lisinopril nebyl pozorován žádný klinicky významný účinek na Cmax nebo AUC, nicméně bylo pozorováno zpoždění tmax přibližně o 2 hodiny.
Ethinylestradiol a levonorgestrel
Podání kombinovaných perorálních kontraceptiv (30 ^g ethinylestradiolu a 150 ^g levonorgestrelu) hodinu před aplikací exenatidu s okamžitým účinkem neovlivnilo AUC, Cmax nebo Cmin ethinylestradiolu ani levonorgestrelu. Podání perorálních kontraceptiv 35 minut po exenatidu neovlivnilo AUC, ale mělo za následek snížení Cmax ethinylestradiolu o 45 % a Cmax levonorgestrelu o 27—41 % a prodloužilo tmax o 2 až 4 hodiny v důsledku zpomaleného vyprazdňování žaludku. Snížení maximální koncentrace Cmax není klinicky významné a úprava dávky perorálních kontraceptiv není nutná.
Pediatrická populace
Interakční studie s exenatidem byly provedeny pouze u dospělých.
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku
Vzhledem k dlouhému poločasu eliminace exenatidu s prodlouženým účinkem by měly ženy ve fertilním věku užívat v průběhu léčby exenatidem s prodlouženým účinkem antikoncepci. Léčba exenatidem s prodlouženým účinkem má být ukončena nejméně 3 měsíce před plánovaným těhotenstvím.
Adekvátní údaje o podávání exenatidu s prodlouženým účinkem těhotným ženám nejsou k dispozici. Studie na zvířatech prokázaly reprodukční toxicitu (viz bod 5.3). Případné riziko pro člověka není známé. V průběhu těhotenství nesmí být exenatid s prodlouženým účinkem podáván a doporučuje se podávat inzulín.
Kojení
Není známo, zda se exenatid vylučuje do lidského mateřského mléka. Exenatid s prodlouženým účinkem by kojícím ženám neměl být podáván.
Fertilita
Studie fertility nebyly u lidí provedeny.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Exenatid s prodlouženým účinkem má malý vliv na schopnost řídit a obsluhovat stroje. Jestliže je exenatid s prodlouženým účinkem podáván v kombinaci se sulfonylmočovinou, pacienti by měli být poučeni o preventivních opatřeních k zabránění hypoglykemie v průběhu řízení nebo obsluhy strojů.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Nejčastější nežádoucí účinky byly převážně spojeny s gastrointestinálním traktem (nauzea, která byla nejčastějším nežádoucím účinkem a byla spojena se zahajováním léčby a postupně se její výskyt snižoval, a průjem). Mimo to se vyskytly také reakce v místě vpichu (pruritus, uzlíky, erytém), hypoglykemie (se sulfonylmočovinou) a bolest hlavy. Většina nežádoucích účinků spojená s podáváním exenatidu s prodlouženým účinkem byla mírné až střední intenzity.
Od doby uvedení exenatidu s okamžitým účinkem na trh byla hlášena akutní pankreatitida s frekvencí není známo a akutní renální selhání s frekvencí méně často (viz bod 4.4).
Tabulkový souhrn nežádoucích účinků
Frekvence nežádoucích účinků exenatidu s prodlouženým účinkem identifikovaných v klinických studiích a ze spontánních hlášení (které nebyly pozorovány v klinickém hodnocení, frekvence není známo) jsou shrnuty níže v Tabulce 1.
Zdrojem údajů pro klinická hodnocení s exenatidem je 18 placebem kontrolovaných klinických studií, 21 studií s aktivním komparátorem a 2 otevřené klinické studie. Základní léčba zahrnovala dietní opatření a pohybová opatření, metformin, sulfonylmočovinu, thiazolidindion, nebo kombinaci perorálních látek snižujících hladinu glukózy v krvi.
Účinky jsou uvedeny níže za použití terminologie MedDRA a řazeny podle tříd orgánových systémů a četnosti výskytu. Frekvence četností jsou definovány následovně: velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1000 až < 1/100), vzácné (> 1/10000 až < 1/1000), velmi vzácné (< 1/10000) a není známo (z dostupných údajů nelze určit).
V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
Tabulka 1: Nežádoucí účinky exenatidu s prodlouženým účinkem identifikované v klinických
|
studiích a ze spontánníc |
i hlášení | |||||
|
Třídy orgánových systémů/nežádoucí účinek |
Frekvence výskytu | |||||
|
Velmi časté |
Časté |
Méně časté |
Vzácné |
Velmi vzácné |
Není známo | |
|
Poruchy imunitního systému | ||||||
|
Anafylaktická reakce |
X1 | |||||
|
Poruchy metabolismu a výživy | ||||||
|
Hypoglykemie (se sulfonylmočovinou) |
X1 | |||||
|
Snížená chuť k jídlu |
X1 | |||||
|
Dehydratace |
X1 | |||||
|
Poruchy nervového systému | ||||||
|
X1 | ||||||
|
Závrať |
X1 | |||||
|
Porucha chuti |
X1 | |||||
|
Gastrointestinální poruchy | ||||||
|
Střevní obstrukce |
X1 | |||||
|
Akutní pankreatitida (viz bod 4.4) |
X2 | |||||
|
X1 | ||||||
|
X1 | ||||||
|
X1 | ||||||
|
X1 | ||||||
|
X1 | ||||||
|
Gastroesofageální refluxní nemoc |
X1 | |||||
|
Abdominální distenze |
X1 | |||||
|
Říhání |
X1 | |||||
|
Zácpa |
X1 | |||||
|
Plynatost |
X1 | |||||
|
Poruchy kůže a podkožní tkáně | ||||||
|
Makulární a papulární vyrážka |
X2 | |||||
|
Svědění a/nebo kopřivka |
X1 | |||||
|
Angioneurotický edém |
X2 | |||||
|
Absces a celulitida v místě vpichu |
X2 | |||||
|
Nadměrné pocení |
X1 | |||||
|
Alopecie |
X1 | |||||
|
Poruchy ledvin a močových cest | ||||||
|
Zhoršená funkce ledvin včetně akutního selhání ledvin, |
X1 | |||||
1 Frekvence odvozená z databáze dokončených dlouhodobých klinických studií účinnosti a bezpečnosti s exenatidem s prodlouženým účinkem, celkové n = 2868 (n = 1002 pacientů na sulfonylmočovině).
|
zhoršení chronického selhání ledvin, poškození ledvin, zvýšený sérový kreatinin (viz bod 4.4) | ||||||
|
Celkové poruchy a rea |
íce v místě aplikace | |||||
|
Svědění v místě aplikace |
X1 | |||||
|
Únava |
X1 | |||||
|
Erytém v místě aplikace |
X1 | |||||
|
Vyrážka v místě aplikace |
X1 | |||||
|
Astenie |
X1 | |||||
|
Pocit nervozity |
X1 | |||||
|
Vyšetření | ||||||
|
Zvýšení hodnot INR (viz bod 4.4) |
X2 | |||||
2 Frekvence odvozená od dat ze spontánních hlášení pro exenatid s prodlouženým účinkem (neznámá četnost).
Popis vybraných nežádoucích účinků
Hypoglykemie
Incidence hypoglykemie byla vyšší, pokud byl exenatid s prodlouženým účinkem podáván v kombinaci se sulfonylmočovinou (24,0 % oproti 5,4 %) (viz bod 4.4). Ke snížení rizika hypoglykemie spojeného s podáváním sulfonylmočoviny může být zváženo snížení její dávky (viz body 4.2 a 4.4).
Podávání exenatidu s prodlouženým účinkem bylo spojeno se signifikantně nižším výskytem epizod hypoglykemie oproti bazálnímu inzulinu u pacientů užívajících také metformin (3 % oproti 19 %) a u pacientů užívajících metformin společně se sulfonylmočovinou (20 % oproti 42 %).
Většina případů hypoglykemie (99,9 %, n = 649) byla ve všech 11 studiích s exenatidem s prodlouženým účinkem mírné intenzity a byly vyřešeny perorálním podáním sacharidů. U jednoho pacienta byla hlášena závažná hypoglykemie s nízkou hodnotou glukózy v krvi (2,2 mmol/l) a vyžadovala pomoc s perorálním podáním sacharidů, kdy došlo k vyřešení příhody.
Nejčastěji hlášeným nežádoucím účinkem byla nauzea. U pacientů léčených exenatidem s prodlouženým účinkem byl obecně hlášen ve 20 % případů výskyt nejméně jedné epizody nauzey ve srovnání s 34 % případů u pacientů s exenatidem s okamžitým účinkem. Ve většině případů byla nauzea mírná až středně těžká. U většiny pacientů s nauzeou na počátku léčby se frekvence s dalším pokračováním léčby snižovala.
V 30týdenní kontrolované klinické studii došlo k ukončení léčby z důvodu nežádoucích účinků u 6 % pacientů léčených exenatidu s prodlouženým účinkeem, u 5 % pacientů léčených exenatidem s okamžitým účinkem.
Nejčastějšími nežádoucími účinky vedoucími k vyřazení ze studie byla u obou skupin pacientů nauzea a zvracení. Mezi pacienty léčenými exenatidem s prodlouženým účinkem došlo k vyřazení z důvodu nauzey nebo zvracení u méně než 1 % pacientů, u exenatidu s okamžitým účinkem u 1 % pacientů.
Reakce v místě vpichu
V průběhu 6měsíční kontrolní fáze klinických studií byly lokální reakce v místě podání u pacientů léčených exenatidem s prodlouženým účinkem pozorovány častěji ve srovnání s pacienty léčenými komparátorem (16 % oproti rozmezí 2-7 %). Obecně byly tyto lokální reakce v místě podání mírné a obvykle nevedly k ukončení účasti ve studii. U pacientů mohou být příznaky ošetřením mírněny, zatímco se pokračuje v léčbě. Následující injekce by měly být potom podány každý týden do jiného místa. V poregistračním období byly hlášeny případy abscesů a celulitidy v místě vpichu.
Velmi často byly v klinických studiích pozorovány v místě injekce malé podkožní uzlíky, což je následkem známých vlastností mikročástic z PLGA. Většina jednotlivých uzlíků byla asymptomatická, neměla vliv na účast ve studii a vymizela v průběhu 4-8 týdnů.
Imunogenicita
V souladu s potenciálními imunogenními vlastnostmi léčivých přípravků na bázi proteinů a peptidů se u pacientů léčených exenatidem s prodlouženým účinkem mohou vytvořit protilátky proti exenatidu.
U většiny pacientů, u kterých k tvorbě protilátek došlo, se titr protilátek časem snižoval.
Přítomnost protilátek (vysoký nebo nízký titr) nepredikuje úroveň glykemické kontroly u jednotlivých pacientů.
V klinických studiích s exenatidem s prodlouženým účinkem mělo na konci studie nízký titr protilátek přibližně 45 % pacientů. Průměrné procento pacientů s pozitivním titrem protilátek bylo stálé ve všech klinických hodnoceních. Úroveň kontroly glykemie (HbA1c) byla u této skupiny celkově srovnatelná jako u skupiny bez nalezených protilátek. Vyšší titr protilátek mělo ve studiích fáze 3 průměrně 12 % pacientů. Úměrně tomu chyběla na konci kontrolovaného období glykemická odpověď na léčbu exenatidem s prodlouženým účinkem; glykemická odpověď se neprojevila u 2,6 % pacientů s vyšším titrem protilátek, zatímco u pacientů s negativním nálezem protilátek se odpověď neprojevila u 1,6 %.
U pacientů, u kterých se vytvořily protilátky na exenatid, byla pozorována větší tendence k lokálním reakcím po podání přípravku (např.: zčervenání kůže a svědění), frekvence a typy ostatních nežádoucích příhod byly srovnatelné s pacienty bez protilátek na exenatid.
V 30týdenní a dvou 26týdenních klinických studiích byla u pacientů léčených přípravkem Bydureon frekvence výskytu potenciálně imunogenních reakcí v místě podání (nejčastěji pruritus s nebo bez erytému) 9 %. Tyty reakce byly méně často pozorovány u pacientů bez protilátek (4 %) ve srovnání s pacienty s přítomností protilátek (13 %), s větším výskytem u pacientů s vyšším titrem protilátek.
Vyšetření vzorků s pozitivním nálezem protilátek neodhalilo signifikantní zkříženou reaktivitu s podobnými endogenními peptidy (glukagon nebo GLP-1).
Rychlé snížení tělesné hmotnosti
V 30týdenní studii došlo k rychlému snížení tělesné hmotnosti v minimálně jednom časovém období u přibližně 3 % pacientů (n=4/148) léčených exenatidem s prodlouženým účinkem (zaznamenané snížení tělesné hmotnosti v období mezi dvěma následujícími studijními návštěvami větší než
1,5 kg/týden).
Zvýšení srdeční frekvence
Při celkové analýze klinických studií s exenatidem s prodlouženým účinkem bylo pozorováno průměrné zvýšení srdeční frekvence (HR) o 2,6 stahu za minutu (bpm) ve srovnání s výchozí hodnotou (74 bpm). U patnácti procent pacientů léčených exenatidem s prodlouženým účinkem bylo průměrné zvýšení srdeční frekvence HR > 10 bpm; přibližně 5 % až 10 % subjektů v jiných léčebných skupinách mělo průměrné zvýšení HR > 10 bpm.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky,
aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Projevy předávkování exenatidem (založené na klinických studiích exenatidu s okamžitým účinkem) zahrnovaly závážnou nauzeu, silné zvracení a rychlé snížení koncentrace glukózy v krvi. V případě předávkování by měla být s ohledem na pacientovy klinické projevy a příznaky zahájena odpovídající podpůrná léčba.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Léčiva k terapii diabetu, jiná antidiabetika, kromě inzulinů, ATC kód: A10BX04.
Mechanismus účinku
Exenatid je agonista receptoru peptidu podobného glukagonu 1 (GLP-1), který vykazuje některé antihyperglykemické účinky jako peptid podobný glukagonu 1 (glucagon-like peptid-1, GLP-1). Sekvence aminokyselin exenatidu se částečně překrývá se sekvencí lidského GLP-1. U exenatidu se prokázala vazba a aktivace známého lidského GLP-1 receptoru in vitro, mechanismus účinku je zprostředkován cyklickou AMP a/nebo dalšími nitrobuněčnými signálními cestami.
Exenatid zvyšuje, v závislosti na hladině glukózy, sekreci inzulínu v beta-buňkách pankreatu. Se snižováním koncentrace glukózy klesá i produkce inzulínu. Při podávaní exenatidu v kombinaci se samotným metforminem a/nebo thiazolidindionem nebylo pozorováno žádné zvýšení výskytu hypoglykemie v porovnání s placebem v kombinaci s metforminem a/nebo thiazolidindionem, což může být způsobeno tímto glukózo-dependentním inzulinotropním mechanismem účinku (viz bod 4.4).
Exenatid potlačuje sekreci glukagonu, jejíž nepřiměřené zvýšení je známé u diabetu 2. typu. Nižší hladiny glukagonu vedou ke snížení tvorby glukózy v játrech. Exenatid však nemá vliv na normální reakci glukagonu a dalších hormonů v odpovědi na hypoglykémii.
Exenatid zpomaluje vyprazdňování žaludečního obsahu a snižuje tedy rychlost absorpce glukózy z potravy do krevního oběhu.
Bylo prokázáno, že podání exenatidu snižuje příjem potravy prostřednictvím snížené chuti k jídlu a zvýšením pocitu sytosti.
Farmakodynamické účinky
Exenatid zlepšuje kontrolu glykemie prostřednictvím trvalého působení, kdy snižuje postprandiální hladiny glukózy i hladiny glukózy nalačno u pacientů s diabetem 2. typu. Na rozdíl od přirozeného GLP-1, má exenatid s prodlouženým účinkem u člověka farmakokinetický a farmakodynamický profil vhodný pro podání jednou týdně.
Farmakodynamická studie exenatidu prokázala u pacientů s diabetem 2. typu (n = 13) obnovení první fáze sekrece inzulínu a zlepšení sekrece inzulínu ve druhé fázi jako odpověď na intravenózní bolus glukózy.
Klinická účinnost a bezpečnost
Výsledky dlouhodobých klinických hodnocení exenatidu s prodlouženým účinkem jsou uvedeny níže, tato hodnocení zahrnovala 1628 osob (804 léčených exenatidem s prodlouženým účinkem), 54 % mužů a 46 % žen, 281 osob (141 léčených exenatidem s prodlouženým účinkem) bylo ve věku > 65 let.
Kontrola glykemie
Ve dvou studiích byl exenatid s prodlouženým účinkem 2 mg podávaný jednou týdně srovnáván s 5 ^g exenatidu s okamžitým účinkem dvakrát denně následovaného 10 ^g exenatidu s okamžitým účinkemdvakrát denně. Délka trvání jedné studie byla 24 týdnů (n = 252) a druhé 30 týdnů (n = 295), následovaných nezaslepeným pokračováním v délce dalších 22 týdnů, kdy všichni pacienti užívali exenatid s prodlouženým účinkem 2 mg podávaný jednou týdně (n = 243). V obou hodnoceních bylo snížení HbA1c zjevné v obou léčebných skupinách již při prvním měření HbA1c po zahájení léčby (týden 4 nebo 6).
Podávání exenatidu s prodlouženým účinkem vedlo ke statisticky významnému snížení HbAJc ve srovnání s pacienty užívajícími exenatid s okamžitým účinkem (Tabulka 2).
Klinicky významný účinek exenatidu s prodlouženým účinkem a exenatidu s okamžitým účinkem na HbA1c byl v obou studiích pozorován bez ohledu na druh základní antidiabetické terapie.
V těchto dvou klinických studiích dosáhlo snížení HbA1c na hodnoty < 7 % nebo < 7 % klinicky a statisticky významně více subjektů na exenatidu s prodlouženým účinkem ve srovnání s exenatidem s okamžitým účinkem (p < 0,05; resp. p = < 0,0001).
Obě skupiny pacientů užívajících exenatid s prodlouženým účinkem a exenatid s okamžitým účinkem dosáhly snížení tělesné hmotnosti oproti počátečním hodnotám, ačkoli rozdíl mezi oběma rameny nebyl významný.
Další snížení HbA1c a pokračující úbytek hmotnosti byl pozorován minimálně po dobu 52 týdnů u pacientů, kteří dokončili kontrolovanou 30týdenní studii i další nekontrolované pokračování. Hodnotitelní pacienti, kteří byli převedeni z exenatidu s okamžitým účinkem na exenatid s prodlouženým účinkem (n = 121) dosáhli stejného zlepšení HbA1c - 2,0 % na konci 22. týdne pokračování ve srovnání s počátečními hodnotami jako pacienti léčení exenatidem s prodlouženým účinkem po dobu 52 týdnů.
Tabulka 2: Výsledky dvou klinických hodnocení exenatidu s prodlouženým účinkem oproti exenatidu s okamžitým účinkem v kombinaci s dietou a cvičením samotným, metforminem a/nebo sulfonylmočovinou a metforminem a/nebo thiazolidindionem (intent-to-treat pacienti).
|
24týdenní studie |
Exenatid s prodlouženým účinkem 2 mg |
Exenatid s okamžitým účinkem 10 ^g dvakrát denně |
|
N |
129 |
123 |
|
Průměr HbA1c (%) | ||
|
Výchozí hodnota |
8,5 |
8,4 |
|
Změna oproti výchozí hodnotě (±SE) |
-1,6 (±0,1)** |
-0,9 (±0,1) |
|
Průměrný rozdíl mezi léčbami ve změně oproti výchozí hodnotě (95% CI) |
-0,67 (-0,94 |
; -0,39)** |
|
Podíl pacientů (%) dosahujících HbA1c < 7 % |
58 |
30 |
|
Změna v plazmatické hladině glukózy na lačno (mmol/l) (±SE) |
-1,4 (±0,2) |
-0,3 (±0,2) |
|
Průměrná tělesná hmotnost (kg) | ||
|
Výchozí hodnota |
97 |
94 |
|
Změna oproti výchozí hodnotě (±SE) |
-2,3 (±0,4) |
-1,4 (±0,4) |
|
Průměrný rozdíl mezi léčbami ve změně oproti výchozí hodnotě (95% CI) |
-0,95 (-1,91; 0,01) | |
|
30týdenní studie | ||
|
N |
148 |
147 |
|
Průměr HbA1c (%) | ||
|
Výchozí hodnota |
8,3 |
8,3 |
S
|
Změna oproti výchozí hodnotě (±SE) |
-1,9 (±0,1)* |
-1,5 (±0,1) |
|
Průměrný rozdíl ve změně oproti výchozí hodnotě mezi léčbami (95% CI) |
-0,33 (-0,54; -0,12)* | |
|
Podíl pacientů (%) dosahujících HbA1c < 7 % |
73 |
57 |
|
Změny hladiny glukózy v plazmě nalačno (mmol/l) ( ± SE) |
-2,3 (±0,2) |
-1,4( ±0,2) |
|
Průměrná tělesná hmotnost (kg) | ||
|
Výchozí hodnota |
102 |
102 |
|
Změna oproti výchozí hodnotě (±SE) |
-3,7 (±0,5) |
-3,6 (±0,5) |
|
Průměrný rozdíl mezi léčbami ve změně oproti výchozí hodnotě (95% CI) |
-0,08 (-1,29; 1,12) | |
3=standardní chyba, CI=interval spolehlivosti, * p< 0,05, **p< 0,0001
Ve 26týdenní studii byl exenatid s prodlouženým účinkem srovnáván s inzulínem glargin podávaným jednou denně. Exenatid s prodlouženým účinkem prokázal významnější změnu HbAJc ve srovnání s inzulinem glargin. Ve srovnání s léčbou inzulinem glargin exenatid s prodlouženým účinkem významně snižoval průměrnou tělesnou hmotnost a jeho podávání bylo spojeno s menším počtem hypoglykemických příhod (Tabulka 3).
Tabulka 3: Výsledky 26týdenního klinického hodnocení exenatidu s prodlouženým účinkem ve srovnání s inzulinem glargin v kombinaci s metforminem samotným nebo metforminem a
|
Exenatid s prodlouženým účinkem 2 mg |
Inzulin glargin1 | |
|
N |
233 |
223 |
|
Průměr HbA1c (%) | ||
|
Výchozí hodnota |
8,3 |
8,3 |
|
Změna oproti výchozí hodnotě (±SE) |
-1,5 (±0,1)* |
-1,3 (±0,1)* |
|
Průměrný rozdíl mezi léčbami ve změně oproti výchozí hodnotě (95% CI) |
-0,16 (-0,29; -0,03)* | |
|
Podíl pacientů (%) dosahujících HbA1c < 7 % |
62 |
54 |
|
Změny hladiny glukózy v séru nalačno (mmol/l) (±SE) |
-2,1 (±0,2) |
-2,8 (±0,2) |
|
Průměrná tělesná hmotnost (kg) | ||
|
Výchozí hodnota |
91 |
91 |
|
Změna oproti výchozí hodnotě (±SE) |
-2,6 (±0,2) |
+1,4 (±0,2) |
|
Průměrný rozdíl mezi léčbami ve změně oproti výchozí hodnotě (95% CI) |
-4,05 (-4,57; -3,52)* | |
SE=standardní chyba, CI=interval spolehlivosti, * p<0,05
Inzulin glargin byl dávkován tak, aby se cílová koncentrace glukózy pohybovala v rozmezí od 4,0 do 5,5 mmol/l (72 až 100 mg/dl). Průměrná dávka inzulinu glargin byla na začátku léčby 10,1 IU/den a zvýšila se na 31,1 IU/den u pacientů léčených inzulinem glargin.
Výsledky 156týdenní byly v souladu s výsledky již dříve publikovanými ve 26týdenní zprávě. Léčba exenatidem s prodlouženým účinkem ve srovnání s léčbou inzulinem glarginem trvale významně zlepšovala glykemickou kontrolu a kontrolu tělesné hmotnosti. Bezpečnostní údaje po 156 týdnech byly v souladu s výsledky hlášenými po 26 týdnech.
Ve 26týdenní studii byl exenatid s prodlouženým účinkem srovnáván s maximálními dávkami sitagliptinu a pioglitazonu u pacientů užívajících také metformin. Ve všech léčebných skupinách došlo k významnému snížení HbA1c ve srovnání s počáteční hodnotou. U exenatidu s prodlouženým účinkem byla prokázána superiorita oproti sitagliptinu i pioglitazonu s ohledem na na změnu HbAJc ve srovnání s počáteční hodnotou.
Exenatid s prodlouženým účinkem prokázal významně vyšší snížení tělesné hmotnosti ve srovnání se sitagliptinem. U pacientů užívajících pioglitazon došlo ke zvýšení tělesné hmotnosti (Tabulka 4).
Tabulka 4: Výsledky 26týdenního klinického hodnocení exenatidu s prodlouženým účinkem ve
|
srovnání se sitagliptinem a pioglitazonem v kombinaci s met |
orminem (intent to treat pacienti). | ||
|
Exenatid s prodlouženým účinkem 2 mg |
Sitagliptin 100 mg |
Pioglitazon 45 mg | |
|
N |
160 |
166 |
165 |
|
Průměr HbA1c (%) | |||
|
Výchozí hodnota |
8,6 |
8,5 |
8,5 |
|
Změna oproti výchozí hodnotě (±SE) |
-1,6 (±0,1)* |
-0,9 (±0,1)* |
-1,2 (±0,1)* |
|
Průměrný rozdíl mezi léčbami ve změně oproti výchozí hodnotě (95% CI) sitagliptin |
-0,63 (-0,89; -0,37)** | ||
|
Průměrný rozdíl mezi léčbami ve změně oproti výchozí hodnotě (95% CI) pioglitazon |
-0,32 (-0,57; -0,06)* | ||
|
Podíl pacientů (%) dosahujících HbA1c < 7 % |
62 |
36 |
49 |
|
Změny hladiny glukózy v séru nalačno (mmol/l) (±Se) |
-1,8 (±0,2) |
-0,9 (±0,2) |
-1,5 (±0,2) |
|
Průměrná tělesná hmotnost (kg) | |||
|
Výchozí hodnota |
89 |
87 |
88 |
|
Změna oproti výchozí hodnotě (±SE) |
-2,3 (±0,3) |
-0,8 (±0,3) |
+2,8 (±0,3) |
|
Průměrný rozdíl mezi léčbami ve změně oproti výchozí hodnotě (95% CI) sitagliptin |
-1,54 (-2,35; -0,72)* | ||
|
Průměrný rozdíl mezi léčbami ve změně oproti výchozí hodnotě (95% CI) pioglitazon |
-5,10 (-5,91; -4,28)** | ||
SE=standardní chyba, CI=interval spolehlivosti, * p < 0,05, **p < 0,0001
Tělesná hmotnost
Snížení tělesné hmotnosti oproti počátečním hodnotám bylo pozorováno ve všech klinických hodnoceních exenatidu s prodlouženým účinkem. Toto snížení bylo pozorováno u pacientů užívajících exenatid s prodlouženým účinkem bez ohledu na to, zda se u nich vyskytovala nevolnost nebo ne, ačkoli snížení bylo větší ve skupině pacientů s nauzeou (průměrné snížení o -2,9 kg až -5,2 kg v přítomnosti nauzey oproti -2,2 kg až -2,9 kg bez nauzey).
Podíl pacientů, u kterých došlo ke snížení tělesné hmotnosti i HbA1c, se pohyboval od 70 do 79 % (podíl pacientů u kterých došlo ke snížení HbA1c se pohyboval od 88 do 96 %).
Plazmatické/sérové hladiny glukózy
Léčba exenatidem s prodlouženým účinkem vedla k signifikantnímu snížení plazmatických/sérových hladin glukózy nalačno, tato snížení byla pozorována již po 4 týdnech. Byla pozorována také další snížení postprandiálních koncentrací. Zlepšení hladin glukózy nalačno přetrvávalo po dobu 52 týdnů.
Funkce beta-buněk
Klinická hodnocení exenatidu s prodlouženým účinkem ukázala při použití homeostatického modelu (HOMA-B) zlepšení funkce beta-buněk. Tyto účinky na funkci beta-buněk byly trvalé po dobu 52 týdnů.
Krevní tlak
V klinických hodnoceních exenatidu s prodlouženým účinkem bylo pozorováno snížení systolického krevního tlaku (2,9 mmHg až 4,7 mmHg). Ve 30týdenním srovnávacím klinickém hodnocení
s exenatidem s okamžitým účinkem exenatid s prodlouženým účinkem i exenatid s okamžitým účinkem signifikantně snižoval systolický krevní tlak oproti výchozím hodnotám (4,7±1,1_mmHg, resp. 3,4±1,1mmHg), rozdíl mezi oběma rameny nebyl významný. Zlepšení krevního tlaku bylo trvalé po dobu 52 týdnů.
Hladiny lipidů nalačno
Exenatid s prodlouženým účinkem nevykazuje nežádoucí účinky na lipidové parametry.
Pediatrická populace
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s exenatidem s prodlouženým účinkem u jedné nebo více podskupin pediatrické populace s diabetes mellitus 2.typu (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Absorpce exenatidu je ovlivněna vlastnostmi lékové formy exenatidu s prodlouženým účinkem. Po absorpci do krevního oběhu je exenatid distribuován a eliminován v souladu s jeho známými farmakokinetickými vlastnostmi (tak jak je popsáno v tomto bodu).
Absorpce
Po podávání exenatidu s prodlouženým účinkem 2 mg přesáhly průměrné koncentrace exenatidu minimální účinné koncentrace (~ 50 pg/ml) v průběhu 2 týdnů s postupným zvyšováním průměrné plazmatické koncentrace exenatidu v průběhu 6 až 7 týdnů. Následně bylo dosaženo koncentrace exenatidu přibližně 300 pg/ml indikující dosažení rovnovážného stavu. Rovnovážné koncentrace exenatidu jsou udržovány v průběhu týdenního intervalu mezi dávkami s minimálními výkyvy nahoru či dolů od této průměrné terapeutické koncentrace.
Distribuce
Průměrný zdánlivý distribuční objem exenatidu po subkutánním podání jednorázové dávky exenatidu je 28 litrů.
Biotrasnformace a eliminace
Neklinické studie ukázaly, že exenatid je eliminován převážně glomerulární filtrací s následnou proteolytickou degradací. V klinických studiích je průměrná zdánlivá clearance exenatidu 9 l/h. Tyto farmakokinetické parametry exenatidu jsou nezávislé na dávce. Střední plazmatické koncentrace exenatidu klesnou pod detekovatelnou hranici přibližně za 10 týdnů po ukončení léčby exenatidem s prodlouženým účinkem.
Zvláštní populace
Renální insuficience
Farmakokinetické populační analýzy pacientů s poškozením ledvin, kterým byl podáván exenatid s prodlouženým účinkem 2 mg, naznačují zvýšení systémové expozice o 74 % a 23 % (odhad střední hodnoty v každé skupině) u pacientů se středním (N = 10) a mírným (N=56) poškozením ledvin, ve srovnání s pacienty s normálními renálními funkcemi (N = 84).
Hepatální insuficience
U pacientů s hepatální nedostatečností nebyly prováděny žádné farmakokinetické studie. Exenatid je primárně eliminován ledvinami, proto se neočekává, že by jaterní dysfunkce ovlivňovala koncentrace exenatidu v krvi.
Pohlaví, rasa a tělesná hmotnost
Pohlaví, rasa a tělesná hmotnost nemají na farmakokinetiku exenatidu klinicky významný vliv.
Starší pacienti
Data o starších pacientech jsou omezená, nenaznačují však žádné výrazné změny v expozici exenatidu s rostoucím věkem až do věku 75 let.
Ve farmakokinetické studii exenatidu s okamžitým účinkem u pacientů s diabetem 2.typu vedlo podávání exenatidu (10 ^g) k průměrnému zvýšení celkové plochy pod křivkou exenatidu o 36 % u 15 starších subjektů ve věku 75-85 let ve srovnání s 15 subjekty ve věku 45-65 let pravděpodobně z důvodu snížené renální funkce u starší věkové skupiny (viz bod 4.2).
Pediatrická populace
Ve farmakokinetické studii jednorázového podání exenatidu s okamžitým účinkem provedené u 13 pacientů ve věku 12 až 16 let s diabetem 2. typu mělo podání exenatidu (5 ^g) za následek mírné snížení průměrné celkové plochy pod křivkou (nižší o 16 %) a maximální plazmatické koncentrace Cmax (nižší o 25 %) ve srovnání s hodnotami pozorovanými u dospělých. U pediatrické populace nebyla provedena žádná farmakokinetická studie s exenatidem s prodlouženým účinkem.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti, toxicity po opakovaném podání a genotoxicity provedené s exenatidem s okamžitým účinkem, nebo exenatidem s prodlouženým účinkem, neodhalily žádné zvláštní riziko pro člověka.
Ve 104týdenní studii kancerogenity exenatidu s prodlouženým účinkem byl u laboratorních potkanů pozorován statisticky významný zvýšený výskyt thyreoidního tumoru z C-buněk (adenom a/nebo karcinom) při všech dávkách (1,4 až 26násobek klinické expozice exenatidu s prodlouženým účinkem u člověka). Relevance těchto nálezů pro člověka není v současné době známá.
Studie na zvířatech nenaznačují přímé škodlivé účinky na plodnost nebo těhotenství, vysoké dávky exenatidu způsobovaly poruchy vývoje kostry a zpomalení fetálního a neonatálního růstu.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek
polyglaktin (1:1) sacharóza
Rozpouštědlo sodná sůl karmelosy chlorid sodný polysorbát 20
monohydrát dihydrogenfosforečnanu sodného heptahydrát hydrogenfosforečnanu sodného voda na injekci hydroxid sodný (k úpravě pH)
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto tento léčivý přípravek nesmí být mísen s žádnými dalšími léčivými přípravky.
6.3 Doba použitelnosti
3 roky
Po rekonstituci suspenze
Suspenze musí být aplikována okamžitě po smísení prášku a rozpouštědla.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C-8 °C).
Chraňte před mrazem.
Před použitím mohou být pera uchovávána při teplotě do 30 °C až po dobu 4 týdnů. Na konci této doby se musí pera použít nebo zlikvidovat.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Podmínky uchovávání přípravku po smísení viz bod 6.3.
6.5 Druh obalu a obsah balení
Jedno pero s dvojitou komorou obsahuje prášek s exenatidem a rozpouštědlo ve skleněném zásobníku Typu I na jedné straně uzavřeném pryžovým chlorobutylovým uzávěrem a hliníkovým pertlem a na druhé straně chlorobutylovým pryžovým pístem. Obě komory jsou odděleny druhým chlorobutylovým pryžovým pístem. Spolu s perem je dodávána jedna injekční jehla. V krabičce je též jedna náhradní jehla. Používejte pouze injekční jehly dodávané s perem.
Balení obsahující 4 předplněná pera nebo multipack obsahující 12 (3 balení po 4) předplněných per pro jednorázové podání.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a zacházení s ním
Předplněné pero je určeno pro jednorázové použití.
Prášek, který je obsažen v jedné komoře předplněného pera, musí být smísen s rozpouštědlem z druhé komory předplněného pera. Před použitím se rozpouštědlo vizuálně zkontroluje. Rozpouštědlo může být použito pouze v případě, že je čiré a neobsahuje žádné částice. Po rekonstituci suspenze lze směs použít pouze tehdy, je-li směs skoro bílá až bílá a zakalená. Další informace o suspenzi a podání viz Příbalová informace a Pokyny pro uživatele.
Exenatid s prodlouženým účinkem se aplikuje subkutánně ihned po smísení prášku a rozpouštědla. Použijte pouze zakázkové injekční jehly dodané spolu s perem.
Exenatid s prodlouženým účinkem se nesmí použít, pokud byl zmrazen.
Pacient by měl být poučen o bezpečné likvidaci pera po každé aplikaci, s jehlou stále nasazenou.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
AstraZeneca AB SE-151 85 Sodertálje Švédsko
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/11/696/003-004
9. DATUM PRVNÍ REGISRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 17. června 2011 Datum posledního prodloužení registrace:
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu
A. VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresy výrobců odpovědných za propouštění šarží AstraZeneca UK Limited Silk Road Business Park,
Macclesfield, Cheshire, SK10 2NA Velká Británie
Swords Laboratories T/A Lawrence Laboratories
Unit 12 Distribution Centre, Shannon Industrial Estate, Shannon, Co. Clare Irsko
V příbalové informaci k léčivému přípravku musí být uveden název a adresa výrobce odpovědného za propouštění dané šarže.
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis.
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU KRABIČKA - 4 soupravy pro jednorázové podání
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Bydureon 2 mg prášek a rozpouštědlo pro injekční suspenzi s prodlouženým účinkem exenatidum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna injekční lahvička obsahuje exenatidum 2 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Pomocné látky:
Prášek
polyglaktin (1:1) sacharóza
Rozpouštědlo
sodná sůl karmelosy chlorid sodný polysorbát 20
monohydrát dihydrogenfosforečnanu sodného heptahydrát hydrogenfosforečnanu sodného voda na injekci
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Prášek a rozpouštědlo pro injekční suspenzi s prodlouženým účinkem. Jedno balení obsahuje 4 soupravy pro jednorázové podání.
1 souprava pro jednorázové podání obsahuje:
1 inječkní lahvička obsahující 2 mg exenatidu 1 předplněná injekční stříkačka obsahující 0,65 ml rozpouštědla
1 spojka lahvičky
2 injekční jehly
5. ZPŮSOB A CESTA /CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci.
Dodržujte pokyny pro uživatele pro přípravu a podání dávky.
Subkutánní podání
Přípravek Bydureon musí být aplikován ihned po přípravě suspenze prášku v rozpouštědle. Jednou týdně
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce.
Chraňte před mrazem.
Před použitím může být souprava uchovávána při teplotě do 30 °C až po dobu 4 týdnů. Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
AstraZeneca AB SE-151 85 Sodertalje Švédsko
12 REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/11/696/001 13 ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis. 15 NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
bydureon
ÚDAJE UVÁDĚNÉ NA INTERMEDIÁRNÍM OBALU
VNITŘNÍ KRABIČKA - VÍCENÁSOBNÉ BALENÍ - 3 x (4 soupravy pro jednorázové podání) -bez blue boxu
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Bydureon 2 mg prášek a rozpouštědlo pro injekční suspenzi s prodlouženým účinkem exenatidum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna injekční lahvička obsahuje exenatidum 2 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Pomocné látky:
Prášek
polyglaktin (1:1) sacharóza
Rozpouštědlo
sodná sůl karmelosy chlorid sodný polysorbát 20
monohydrát dihydrogenfosforečnanu sodného heptahydrát hydrogenfosforečnanu sodného voda na injekci
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Prášek a rozpouštědlo pro injekční suspenzi s prodlouženým účinkem.
Jedno balení obsahuje 3 x (4 soupravy pro jednorázové podání). Neprodávejte zvlášť. 1 souprava pro jednorázové podání obsahuje:
1 inječkní lahvička obsahující 2 mg exenatidu 1 předplněná injekční stříkačka obsahující 0,65 ml rozpouštědla
1 spojka lahvičky
2 injekční jehly
5. ZPŮSOB A CESTA /CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci.
Dodržujte pokyny pro uživatele pro přípravu a podání dávky.
Subkutánní podání
Přípravek Bydureon musí být aplikován ihned po přípravě suspenze prášku v rozpouštědle. Jednou týdně
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce.
Chraňte před mrazem.
Před použitím může být souprava uchovávána při teplotě do 30° C až po dobu 4 týdnů. Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
AstraZeneca AB SE-151 85 Sodertalje Švédsko
12 REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/11/696/002 13 ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis. 15 NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
bydureon
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU
KRABIČKA - VÍCENÁSOBNÉ BALENÍ - 3 x (4 soupravy pro jednorázové podání) - včetně blue boxu
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Bydureon 2 mg prášek a rozpouštědlo pro injekční suspenzi s prodlouženým účinkem exenatidum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna injekční lahvička obsahuje exenatidum 2 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Pomocné látky:
Prášek
polyglaktin (1:1) sacharóza
Rozpouštědlo
sodná sůl karmelosy chlorid sodný polysorbát 20
monohydrát dihydrogenfosforečnanu sodného heptahydrát hydrogenfosforečnanu sodného voda na injekci
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Prášek a rozpouštědlo pro injekční suspenzi s prodlouženým účinkem.
Jedno balení obsahuje 3x4 soupravy pro jednorázové podání. Neprodávejte zvlášť.
5. ZPŮSOB A CESTA /CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci.
Dodržujte pokyny pro uživatele pro přípravu a podání dávky.
Subkutánní podání
Přípravek Bydureon musí být aplikován ihned po přípravě suspenze prášku v rozpouštědle. Jednou týdně
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce.
Chraňte před mrazem.
Před použitím může být souprava uchovávána při teplotě do 30 °C až po dobu 4 týdnů. Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
AstraZeneca AB SE-151 85 Sodertalje Švédsko
12 REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/11/696/002 13 ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis. 15 NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
bydureon
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Bydureon 2 mg prášek pro injekční suspenzi
exenatidum
s.c.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4 ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
2 mg
6 JINÉ
AstraZeneca AB
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Rozpouštědlo pro přípravek Bydureon 2 ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4 ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
0,65 ml 6 JINÉ
AstraZeneca AB
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU
KRABIČKA (balení po 4 jednodávkových předplněných perech)
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Bydureon 2 mg prášek a rozpouštědlo pro injekční suspenzi s prodlouženým účinkem v předplněném peru
exenatidum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedno předplněné pero obsahuje exenatidum 2 mg. Po vytvoření suspenze je podaná dávka 2 mg/0,65 ml.
3. SEZNAM POMOCNÝCH LÁTEK
Pomocné látky:
Prášek
polyglaktin (1:1) sacharóza
Rozpouštědlo
sodná sůl karmelosy chlorid sodný polysorbát 20
monohydrát dihydrogenfosforečnanu sodného heptahydrát hydrogenfosforečnanu sodného voda na injekci hydroxid sodný
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Prášek a rozpouštědlo pro injekční suspenzi s prodlouženým účinkem. 4 jednodávková předplněná pera 1 náhradní injekční jehla
5. ZPŮSOB A CESTA /CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci.
Dodržujte pokyny pro uživatele pro přípravu a podání dávky.
Subkutánní podání
Pouze pro jednorázové podání
Přípravek Bydureon musí být aplikován ihned po smísení. Jednou týdně
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce.
Chraňte před mrazem.
Před použitím mohou být předplněná pera uchovávána při teplotě do 30 °C až po dobu 4 týdnů. Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
AstraZeneca AB SE-151 85 Sodertalje Švédsko
12 REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/11/696/003 13 ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis. 15 NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
bydureon
VNITŘNÍ KRABIČKA - VÍCENÁSOBNÉ BALENÍ - 3 x (4 jednodávková předplněná pera) -bez blue boxu
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Bydureon 2 mg prášek a rozpouštědlo pro injekční suspenzi s prodlouženým účinkem v předplněném peru
exenatidum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedno předplněné pero obsahuje exenatidum 2 mg. Po vytvoření suspenze je podaná dávka 2 mg/0,65 ml.
3. SEZNAM POMOCNÝCH LÁTEK
Pomocné látky:
Prášek
polyglaktin (1:1) sacharóza
Rozpouštědlo
sodná sůl karmelosy chlorid sodný polysorbát 20
monohydrát dihydrogenfosforečnanu sodného heptahydrát hydrogenfosforečnanu sodného voda na injekci hydroxid sodný
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Prášek a rozpouštědlo pro injekční suspenzi s prodlouženým účinkem.
4 jednodávková předplněná pera. Součást vícenásobného balení, nelze prodat zvlášť. 1 náhradní injekční jehla
5. ZPŮSOB A CESTA /CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci.
Dodržujte pokyny pro uživatele pro přípravu a podání dávky.
Subkutánní podání
Pouze pro jednorázové podání
Přípravek Bydureon musí být aplikován ihned po smísení. Jednou týdně
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce.
Chraňte před mrazem.
Před použitím mohou být předplněná pera uchovávána při teplotě do 30° C až po dobu 4 týdnů. Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
AstraZeneca AB SE-151 85 Sodertalje Švédsko
12 REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/11/696/004 13 ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis. 15 NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
bydureon
VNĚJŠÍ KRABIČKA - VÍCENÁSOBNÉ BALENÍ - 3 x (4 jednodávková předplněná pera) -včetně blue boxu
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Bydureon 2 mg prášek a rozpouštědlo pro injekční suspenzi s prodlouženým účinkem v předplněném peru
exenatidum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedno předplněné pero obsahuje exenatidum 2 mg. Po vytvoření suspenze je podaná dávka 2 mg/0,65 ml.
3. SEZNAM POMOCNÝCH LÁTEK
Pomocné látky:
Prášek
polyglaktin (1:1) sacharóza
Rozpouštědlo
sodná sůl karmelosy chlorid sodný polysorbát 20
monohydrát dihydrogenfosforečnanu sodného heptahydrát hydrogenfosforečnanu sodného voda na injekci hydroxid sodný
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Prášek a rozpouštědlo pro injekční suspenzi s prodlouženým účinkem Vícenásobné balení: 12 (3 balení po 4) jednodávkových předplněných per
5. ZPŮSOB A CESTA /CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci.
Dodržujte pokyny pro uživatele pro přípravu a podání dávky.
Subkutánní podání
Pouze pro jednorázové podání
Přípravek Bydureon musí být aplikován ihned po smísení. Jednou týdně
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce.
Chraňte před mrazem.
Předplněné pera mohou být uchovávána při teplotě do 30 °C až po dobu 4 týdnů. Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
AstraZeneca AB SE-151 85 Sodertalje Švédsko
12 REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/11/696/004 13 ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis. 15 NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
bydureon
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITRNÍM OBALU ŠTÍTEK NA TĚLE PERA
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Bydureon 2 mg prášek a rozpouštědlo pro injekční suspenzi s prodlouženým účinkem v předplněném peru
exenatidum
s.c.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4 ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
2 mg
6 JINÉ
AstraZeneca AB
B. PŘÍBALOVÁ INFORMACE
Příbalová informace:- informace pro uživatele
Bydureon 2 mg prášek a rozpouštědlo pro injekční suspenzi s prodlouženým účinkem
exenatidum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité informace.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoliv další otázky, zeptejte se svého lékaře, lékárníka nebo diabetologické sestry.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo diabetologické sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Bydureon a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Bydureon používat
3. Jak se přípravek Bydureon používá
4. Možné nežádoucí účinky
5. Jak přípravek Bydureon uchovávat
6. Obsah balení a další informace
1. Co je přípravek Bydureon a k čemu se používá
Přípravek Bydureon obsahuje léčivou látku exenatid. Bydureon je injekční léčivý přípravek používaný ke zlepšení kontroly hladiny cukru v krvi u dospělých pacientů s cukrovkou 2. typu (diabetes mellitus nezávislý na inzulinu).
Tento léčivý přípravek se používá v kombinaci s následujícími přípravky používanými k léčbě cukrovky: metformin, deriváty sulfonylmočoviny a thiazolidindiony. Lékař Vám nyní předepsal tento léčivý přípravek jako doplňující lék k zlepšení kontroly hladiny cukru v krvi. Pokračujte v dodržování svého dosavadního dietetického programu a programu cvičení.
Trpíte cukrovkou, protože vaše tělo nevytváří dostatek inzulinu ke kontrole hladiny cukru v krvi nebo Vaše tělo není schopno využívat vlastní inzulín. Léčivá látka v tomto léčivém přípravku pomáhá tělu zvýšit tvorbu inzulinu v případě, kdy je hladina cukru v krvi vysoká.
2. Čemu musíte věnovat pozornost, než začnete přípravekBydureon používat Nepoužívejte přípravek Bydureon:
- jestliže jste alergický(á) na exenatid nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Upozornění a opatření
Před použitím přípravku Bydureon se poraďte se svým lékařem, lékárníkem nebo diabetologickou sestrou:
- jestliže užíváte tento léčivý přípravek v kombinaci se sulfonylmočovinou, může dojít
k přílišnému snížení hladiny cukru v krvi (hypoglykemie). Pravidelně si kontrolujte hladinu
cukru v krvi. Zeptejte se svého lékaře, nebo lékárníka, nebo diabetologické sestry v případě, kdy si nejste jisti, zda Vaše další léky obsahují sulfonylmočovinu.
- pokud trpíte cukrovkou 1. typu nebo diabetickou ketoacidózou, tento léčivý přípravek se nemá používat.
- jak podat tento léčivý přípravek. Měl by být podán pod kůži a ne do žíly nebo do svalu.
- jestliže máte vážné problémy s vyprazdňováním žaludku (včetně gastroparézy) nebo se zažíváním potravy, podávání tohoto léčivého přípravku se nedoporučuje. Léčivá látka v tomto přípravku zpomaluje vyprazdňování žaludku, takže potrava prochází žaludkem pomaleji.
- pokud jste někdy prodělal(a) zánět slinivky břišní (pankreatitida) (viz bod 4).
- pokud u Vás dojde k příliš rychlému úbytku tělesné hmotnosti (více než 1,5 kg za týden), oznamte to svému lékaři, protože takto rychlý úbytek může způsobit problémy se žlučovými kameny.
- jestliže máte závažné onemocnění ledvin nebo chodíte na dialýzu (umělá ledvina), nedoporučuje se podávat tento přípravek. S použitím tohoto léčivého přípravku u pacientů s onemocněním
s ledvin jsou jen malé zkušenosti.
Děti a dospívající
Nepodávejte tento léčivý přípravek dětem a dospívajícím do 18 let, proto se u této věkové skupiny nejsou zkušenosti s tímto přípravkem.
Další léčivé přípravky a přípravek Bydureon
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat, zvláště:
- léčivé přípravky používané k léčbě diabetu 2. typu, jako je inzulin a další léčivé přípravky, které účinkují jako Bydureon (např. liraglutid a Byetta [exenatid s okamžitým účinkem], neboť jejich souběžné podávání s přípravkem Bydureon se nedoporučuje.
- léčivé přípravky používané k ředění krve (antikoagulancia), např. warfarin, neboť bude třeba, aby se u Vás provádělo dodatečné sledování změn INR (měření ředění krve) na počátku léčby tímto léčivým přípravkem.
Těhotenství a kojení
Není známo, zda tento léčivý přípravek poškozuje Vaše nenarozené dítě, a proto nesmíte tento léčivý přípravek používat v průběhu těhotenství a nejméně 3 měsíce před plánovaným otěhotněním.Není známo, zda exenatid přechází do mateřského mléka. V období kojení nesmíte tento léčivý přípravek používat.
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek používat.
Pokud můžete v průběhu léčby tímto léčivým přípravkem otěhotnět, měla byste používat antikoncepci.
Řízení dopravních prostředků a obsluha strojů
Jestliže užíváte tento léčivý přípravek v kombinaci se sulfonylmočovinou, může dojít k přílišnému snížení hladiny cukru v krvi (hypoglykemie). Hypoglykemie může snížit Vaši schopnost se soustředit. Berte, prosím, tento možný problém v úvahu ve všech situacích, které mohou být pro Vás nebo pro ostatní riskantní (např. řízení auta nebo obsluha strojů).
Důležité informace o některých složkách přípravku Bydureon
Tento přípravek obsahuje méně než 1 mmol (23 mg) sodíku v jedné dávce, tj. v podstatě je „bez sodíku“.
3. Jak se přípravek Bydureon používá
Vždy používejte tento léčivý přípravek přesně podle pokynů svého lékaře nebo diabetologické sestry. Pokud si nejste jistý(á), poraďte se se svým lékařem, nebo lékárníkem nebo diabetologickou sestrou.
Tento léčivý přípravek byste si měl(a) podat injekcí jednou týdně, kdykoli v průběhu dne nezávisle na jídle.
Tento léčivý přípravek byste si měl(a) podat injekcí pod kůži (subkutánní injekce) do oblasti břicha, horní části nohy (stehna), nebo do zadní horní části paže.
Každý týden můžete použít stejnou oblast těla. Ujistěte se, že v této oblasti pro injekci vyberete odlišné místo.
Pravidelně si kontrolujte hladinu cukru v krvi, obzvláště je to pro Vás důležité, pokud užíváte také sulfonylmočovinu.
Při aplikaci přípravku Bydureon dodržujte „Pokyny pro uživatele“ přiložené k balení.
Lékař nebo diabetologická sestra Vás musí naučit, jak správně podávat tento léčivý přípravek před tím, než ho použijete poprvé.
Nejprve zkontrolujte, zda je roztok v injekční stříkačce čirý a neobsahuje žádné částice. Po smíchání může být suspenze použita pouze tehdy, je-li směs skoro bílá až bílá a zakalená. Pokud na stěnách nebo dně lahvičky vidíte shluky částic nebo suchý prášek, přípravek NENÍ dobře promíchán.
Důkladně jej protřepejte, dokud není dobře promíchán.
Přípravek musíte aplikovat ihned po přípravě suspenze prášku v rozpouštědle.
Ke každé injekci použijte novou injekční jehlu, kterou po podání bezpečně zlikvidujte tak, jak Vás poučil lékař nebo diabetologická sestra.
Jestliže jste použil(a) více přípravku Bydureon, než jste měl(a)
Jestliže použijete příliš mnoho tohoto léčivého přípravku, můžete potřebovat lékařskou pomoc. Podání příliš velkého množství tohoto léčivého přípravku může způsobit pocit na zvracení, zvracení, závratě nebo příznaky související s nízkou hladinou cukru v krvi (viz bod 4).
Jestliže jste zapomněl(a) použít přípravek Bydureon
Můžete si vybrat den v týdnu, kdy si naplánujete pravidelně aplikovat svou injekci přípravku Bydureon. Pokud zapomenete na injekci, aplikujte injekci co nejdříve, jakmile je to možné provést. Další injekci si můžete podat v původně vybraném dni v týdnu. V průběhu 24 hodin lze podat pouze jednu injekci. Můžete také vybraný den aplikace injekce změnit. Ve stejný den neužívejte dvě injekce.
Jestliže si nejste jistý(á), jestli jste si podal(a) celou dávku přípravku Bydureon
Pokud si nejste jistý(á), jestli jste si podal(a) celou dávku, další dávku tohoto léčivého přípravku si neaplikujte, pokračujte až další týden podle plánu.
Jestliže jste přestal(a) používat přípravek Bydureon
Jestliže se domníváte, že byste měl(a) přestat používat tento léčivý přípravek, poraďte se svým lékařem. Jestliže přestanete používat tento léčivý přípravek, může to ovlivnit hladinu Vašeho cukru v krvi.
Máte-li jakékoli další otázky týkající se používání tohoto přípravku, zeptejte se svého lékaře, nebo lékárníka nebo diabetologické sestry.
4. MOŽNÉ NEŽÁDOUCÍ ÚČINKY
Podobně jako všechny léky, může vyvolávat i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Vzácně byly hlášeny závažné alergické reakce (anafylaxe) (mohou postihnout až 1 z 1000 lidí).
Kontaktujte neprodleně lékaře v případě výskytu následujících příznaků:
• otékání oblasti obličeje, jazyka nebo hrdla (angioedém)
• přecitlivělost (vyrážka, svědění a rychlé otékání krku, obličeje, úst nebo hrdla)
• obtížné polykání
• vyrážka a potíže s dýcháním
Případy zánětu slinivky (pankreatitida) byly hlášeny u pacientů užívajících tento léčivý přípravek (frekvence není známo). Pankreatitida může být závažný, potenciálně život ohrožující stav.
• Informujte svého lékaře, pokud jste prodělal(a) pankreatitidu, trpíte žlučovými kameny, alkoholismem nebo vysokou hladinou tuků v krvi. Tyto stavy mohou zvýšit riziko, že u Vás dojde k onemocnění pankreatitidou, nebo se pankreatitida znovu projeví, a to nezávisle na tom, zda používáte tento léčivý přípravek, nebo ne.
• PŘESTAŇTE používat tento léčivý přípravek a kontaktujte ihned svého lékaře v případě, že se u Vás objeví silná a přetrvávající bolest v oblasti břicha, ať už se zvracením, nebo bez něj, protože se může jednat o zánět slinivky břišní (pankreatitida).
Velmi časté nežádoucí účinky přípravku Bydureon (mohou postihnout více než 1 z 10 lidí):
• pocit na zvracení (pocit na zvracení je nejčastější při zahájení podávání tohoto léčivého přípravku, u většiny pacientů se časem snižuje)
• průjem
• hypoglykemie (snížená hladina cukru v krvi)
Jestliže se tento léčivý přípravek podává společně s léky obsahujícími sulfonylmočovinu, velmi často může dojít k případům přílišného snížení hladiny cukru v krvi (hypoglykemie, obvykle mírná až střední). V případě, že užíváte tento léčivý přípravek, může být zapotřebí snížit dávku léku se sulfonylmočovinou. Známky a příznaky nízké hladiny cukru v krvi mohou zahrnovat bolest hlavy, ospalost, slabost, závratě, zmatek, podrážděnost, hlad, zrychlení srdečního tepu, pocení a pocit nervozity. Váš lékař by Vám měl poradit, jak nízkou hladinu cukru v krvi léčit.
Časté nežádoucí účinky přípravku Bydureon (mohou se vyskytnout až u 1 z 10 lidí):
• závratě
• bolest hlavy
• ztráta energie a síly
• únava (vyčerpání)
• zvracení
• zácpa
• bolest v žaludeční oblasti
• nadýmání
• trávicí potíže
• plynatost
• pálení žáhy
• snížení chuti k jídlu
Tento léčivý přípravek může snižovat Vaši chuť k jídlu, množství přijaté potravy a tělesnou hmotnost. Pokud u Vás dojde k příliš rychlému úbytku tělesné hmotnosti (více než 1,5 kg za týden), oznamte to svému lékaři, protože takto rychlý úbytek může způsobit problémy jako jsou žlučové kameny.
• reakce v místě injekce
Pokud u Vás dojde k reakci v místě injekce (červenání, kopřivka nebo svědění), můžete požádat svého lékaře o pomoc při mírnění těchto potíží a příznaků. Po podání injekce můžete pod kůží vidět nebo cítit malé bulky, měly by zmizet po 4 až 8 týdnech. Není nutné přerušovat léčbu.
Méně časté nežádoucí účinky nebo frekvence není známa přípravku Bydureon (mohou se vyskytnout až u 1 ze 100 lidí):
• snížená funkce ledvin
• dehydratace (ztráta tělesných tekutin), někdy doprovázená sníženou funkcí ledvin
• neprůchodnost střeva
• říhání
• nezvyklá chuť v ústech
• zvýšené pocení
• vypadávání vlasů
• ospalost
Vzácné nežádoucí účinky přípravku Bydureon (mohou se vyskytnout až u 1 z 1000 lidí):
• pocit nervozity
Byly hlášeny některé další nežádoucí účinky (frekvence není známa, z dostupných údajů ji nelze určit).
• změny INR (měření míry srážlivosti krve) byly hlášeny při souběžném podávání warfarinu
• reakce v místě vpichu. Po podání injekce s exenatidem byly hlášeny kožní reakce v místě vpichu. Jedná se o: dutiny obsahující hnis (absces) a oteklé červené plochy kůže na pohmat horké a měkké (celulitida)
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, nebo lékárníkovi nebo diabetologické sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Bydureon uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na štítku a krabičce za zkratkou EXP. Datum použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v chladničce (2 °C až 8 °C). Chraňte před mrazem.
Před použitím může být souprava uchovávána při teplotě do 30 °C až po dobu 4 týdnů.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Přípravek Bydureon, který byl vystaven mrazu, musí být zlikvidován.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co přípravek Bydureon obsahuje
- Léčivou látkou je exenatidum. Jedna injekční lahvička obsahuje exenatidum 2 mg.
- Pomocnými látkami jsou:
- Prášek: polyglaktin (1:1), sacharóza.
- Rozpouštědlo: sodná sůl karmelosy, chlorid sodný, polysorbát 20, monohydrát dihydrogenfosforečnanu sodného, heptahydrát hydrogenfosforečnanu sodného, voda na injekci.
Jak přípravek Bydureon vypadá a co obsahuje toto balení
Prášek a rozpouštědlo pro přípravu injekční suspenze s prodlouženým účinkem.
Prášek je bílý až téměř bílý, rozpouštědlo je čirý bezbarvý až lehce nažloutlý či nahnědlý roztok.
Jedna souprava pro jednorázové podání obsahuje jednu injekční lahvičku obsahující 2 mg exenatidu, jednu předplněnou injekční stříkačku obsahující 0,65 ml rozpouštědla, jednu spojku lahvičky a dvě injekční jehly. Jedna injekční jehla je záložní.
Balení obsahuje 4 soupravy pro jednorázové podání nebo vícenásobné balení obsahující 3x4 soupravy pro jednorázové podání. Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci:
AstraZeneca AB SE-151 85 Sodertalje Švédsko
Výrobce:
AstraZeneca UK Limited Silk Road Business Park,
Macclesfield, Cheshire, SK10 2NA Velká Británie
Swords Laboratories T/A Lawrence Laboratories
Unit 12 Distribution Centre, Shannon Industrial Estate, Shannon, Co. Clare Irsko
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgie/Belgique/Belgien Lietuva
AstraZeneca S.A./N.V. UAB AstraZeneca Lietuva
Tel: +32 2 370 48 11
Tel: +370 5 2660550
Česká republika
AstraZeneca Czech Republic s.r.o.
Tel: +420 222 807 111
Malta
Associated Drug Co. Ltd
Tel: +356 2277 8000
Deutschland
AstraZeneca GmbH Tel: +49 41 03 7080
Eesti
AstraZeneca
Nederland
AstraZeneca BV Tel: +31 79 363 2222
Norge
AstraZeneca AS
Tel: +372 6549 600
Tlf: +47 21 00 64 00
|
EXláSa AstraZeneca A.E. Tn^: +30 2 10 6871500 |
Osterreich AstraZeneca Osterreich GmbH Tel: +43 1 711 31 0 |
|
Espaňa AstraZeneca Farmacéutica Spain, S.A. Tel: +34 91 301 91 00 |
Polska AstraZeneca Pharma Poland Sp. z o.o. Tel.: +48 22 245 73 00 |
|
France AstraZeneca Tél: +33 1 41 29 40 00 |
Portugal AstraZeneca Produtos Farmaceuticos, Lda. Tel: +351 21 434 61 00 |
|
Hrvatska AstraZeneca d.o.o. Tel: +385 1 4628 000 |
Románia AstraZeneca Pharma SRL Tel: +40 21 317 60 41 |
|
Ireland AstraZeneca Pharmaceuticals (Ireland) Ltd Tel: +353 1609 7100 |
Slovenija AstraZeneca UK Limited Tel: +386 1 51 35 600 |
|
Ísland Vistor hf. Sími: +354 535 7000 |
Slovenská republika AstraZeneca AB, o.z. Tel: +421 2 5737 7777 |
|
Italia AstraZeneca S.p.A. Tel: +39 02 9801 1 |
Suomi/Finland AstraZeneca Oy Puh/Tel: +358 10 23 010 |
|
Kúnpoq A^skt^p Oap^aKeuxiKq ArS Tn^: +357 22490305 |
Sverige AstraZeneca AB Tel: +46 8 553 26 000 |
|
Latvija SIA AstraZeneca Latvija Tel: +371 67377100 |
United Kingdom AstraZeneca UK Ltd Tel: +44 1582 836 836 |
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou uveřejněny na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu
Příbalová informace: informace pro uživatele
Bydureon 2 mg prášek a rozpouštědlo pro injekční suspenzi s prodlouženým účinkem
v předplněném peru
exenatidum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité informace.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoliv další otázky, zeptejte se svého lékaře, lékárníka nebo diabetologické sestry.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo diabetologické sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Bydureon a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Bydureon používat
3. Jak se přípravek Bydureon používá
4. Možné nežádoucí účinky
5. Jak přípravek Bydureon uchovávat
6. Obsah balení a další informace
1. Co je přípravek Bydureon a k čemu se používá
Přípravek Bydureon obsahuje léčivou látku exenatid. Bydureon je injekční léčivý přípravek používaný ke zlepšení kontroly hladiny cukru v krvi u dospělých pacientů s cukrovkou 2. typu (diabetes mellitus nezávislý na inzulinu).
Tento léčivý přípravek se používá v kombinaci s následujícími přípravky používanými k léčbě cukrovky: metformin, deriváty sulfonylmočoviny a thiazolidindiony. Lékař Vám nyní předepsal tento léčivý přípravek jako doplňující lék k zlepšení kontroly hladiny cukru v krvi. Pokračujte v dodržování svého dosavadního dietetického programu a programu cvičení.
Trpíte cukrovkou, protože Vaše tělo nevytváří dostatek inzulinu ke kontrole hladiny cukru v krvi nebo Vaše tělo není schopno využívat vlastní inzulín. Léčivá látka v tomto léčivém přípravku pomáhá tělu zvýšit tvorbu inzulinu v případě, kdy je hladina cukru v krvi vysoká.
2. Čemu musíte věnovat pozornost, než začnete přípravek Bydureon používat Nepoužívejte přípravek Bydureon:
- jestliže jste alergický(á) na exenatid nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Upozornění a opatření
Před použitím přípravku Bydureon se poraďte se svým lékařem, nebo lékárníkem nebo diabetologickou sestrou:
- používáte tento léčivý přípravek v kombinaci se sulfonylmočovinou, neboť může dojít
k přílišnému snížení hladiny cukru v krvi (hypoglykemie). Pravidelně si kontrolujte hladinu
cukru v krvi. Zeptejte se svého lékaře, nebo lékárníka, nebo diabetologické sestry v případě, kdy si nejste jisti, zda Vaše další léky obsahují sulfonylmočovinu.
- pokud trpíte cukrovkou 1. typu nebo diabetickou ketoacidózou, tento léčivý přípravek se nemá používat.
- jak podávat tento léčivý přípravek. Měl by být podán pod kůži a ne do žíly nebo do svalu.
- jestliže máte vážné problémy s vyprazdňováním žaludku (včetně gastroparézy) nebo s trávením potravy, podávání tohoto léčivého přípravku se nedoporučuje. Léčivá látka v tomto léčivém přípravku zpomaluje vyprazdňování žaludku, takže potrava prochází žaludkem pomaleji.
- pokud jste někdy prodělal(a) zánět slinivky břišní (pankreatitida) (viz bod 4).
- pokud u Vás dojde k příliš rychlému úbytku tělesné hmotnosti (více než 1,5 kg za týden), oznamte to svému lékaři, protože takto rychlý úbytek může způsobit problémy se žlučovými kameny.
- jestliže máte závažné onemocnění ledvin nebo chodíte na dialýzu (umělá ledvina), nedoporučuje se podávat přípravek Bydureon. S použitím tohoto léčivého přípravku u pacientů
s onemocněním ledvin jsou jen malé zkušenosti.
Děti a dospívající
Nepodávejte tento léčivý přípravek dětem a dospívajícím do 18 let, neboť nejsou zkušenosti s tímto léčivým přípravkem u této věkové kategorie.
Další léčivé přípravky a přípravek Bydureon
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat, zvláště:
- léčivé přípravky používané k léčbě diabetu 2. typu, jako je inzulin a další léčivé přípravky, které účinkují jako Bydureon (např. liraglutid a Byetta [exenatid s okamžitým účinkem], neboť jejich souběžné podávání s přípravkem Bydureon se nedoporučuje.
- léčivé přípravky používané k ředění krve (antikoagulancia), např. warfarin, neboť bude třeba, aby se u Vás provádělo dodatečné sledování změn INR (měření ředění krve) na počátku léčby tímto léčivým přípravkem.
Těhotenství a kojení
Není známo, zda tento léčivý přípravek poškozuje Vaše nenarozené dítě, a proto nesmíte tento léčivý přípravek používat v průběhu těhotenství a nejméně 3 měsíce před plánovaným otěhotněním.
Není známo, zda exenatid přechází do mateřského mléka. V období kojení nesmíte tento léčivý přípravek používat.
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento léčivý přípravek používat.
Pokud můžete v průběhu léčby tímto léčivým přípravkem otěhotnět, měla byste používat antikoncepci.
Řízení dopravních prostředků a obsluha strojů
Jestliže užíváte tento léčivý přípravek v kombinaci se sulfonylmočovinou, může dojít k přílišnému snížení hladiny cukru v krvi (hypoglykemie). Hypoglykemie může snížit Vaši schopnost se soustředit. Berte, prosím, tento možný problém v úvahu ve všech situacích, které mohou být pro Vás nebo pro ostatní riskantní (např. řízení auta nebo obsluha strojů).
Důležité informace o některých složkách přípravku Bydureon
Tento léčivý přípravek obsahuje méně než 1 mmol (23 mg) sodíku v jedné dávce, tj. v podstatě je „bez sodíku“.
3. Jak se přípravek Bydureon používá
Vždy používejte tento léčivý přípravek přesně podle pokynů svého lékaře nebo diabetologické sestry. Pokud si nejste jistý(á), poraďte se se svým lékařem, nebo lékárníkem nebo diabetologickou sestrou.
Tento léčivý přípravek byste si měl(a) podat injekcí jednou týdně, kdykoli v průběhu dne nezávisle na jídle.
Tento léčivý přípravek byste si měl(a) podat injekcí pod kůži (subkutánní injekce) do oblasti břicha, horní části nohy (stehna), nebo do zadní horní části paže.
Každý týden můžete použít stejnou oblast těla. Ujistěte se, že v této oblasti pro injekci vyberete odlišné místo.
Pravidelně si kontrolujte hladinu cukru v krvi, obzvláště je to pro Vás důležité, pokud užíváte také sulfonylmočovinu.
Při aplikaci přípravku Bydureon dodržujte „Pokyny pro uživatele“ přiložené k balení.
Lékař nebo diabetologická sestra Vás musí naučit, jak správně podávat injekci tohoto léčivého přípravku před tím, než jej použijete poprvé.
Nejprve zkontrolujte, zda je roztok v peru čirý a neobsahuje žádné částice. Po smíchání roztoku a prášku může být suspenze použita pouze tehdy, je-li směs skoro bílá až bílá a zakalená. Pokud na stěnách pera vidíte shluky částic nebo suchý prášek, přípravek NENÍ dobře promíchán. Důkladně znovu protřepejte, dokud není dobře promíchán.
Tento léčivý přípravek musíte aplikovat ihned po přípravě suspenze prášku v rozpouštědle.
Ke každé injekci použijte nové pero. Pero musíte po použití bezpečně zlikvidovat, s jehlou stále nasazenou, tak, jak Vám poradil lékař nebo diabetologická sestra.
Jestliže jste použil(a) více přípravku Bydureon, než jste měl(a)
Jestliže použijete příliš mnoho tohoto léčivého přípravku, můžete potřebovat lékařskou pomoc. Podání příliš velkého množství přípravku může způsobit pocit na zvracení, zvracení, závratě nebo příznaky související s nízkou hladinou cukru v krvi (viz bod 4).
Jestliže jste zapomněl(a) použít přípravek Bydureon
Můžete si vybrat den v týdnu, kdy si naplánujete pravidelně aplikovat svou injekci tohoto léčivého přípravku. Pokud zapomenete na injekci v tento den, aplikujte injekci co nejdříve, jakmile je možné to provést. Další injekci si můžete podat v původně vybraném dni v týdnu. V průběhu 24 hodin lze podat pouze jednu injekci. Můžete také vybraný den aplikace injekce změnit. Ve stejný den neužívejte dvě injekce.
Jestliže si nejste jistý(á), jestli jste si podal(a) celou dávku přípravku Bydureon
Pokud si nejste jistý(á), jestli jste si podal(a) celou dávku, další dávku tohoto léčivého přípravkusi neaplikujte, pokračujte pouze další týden podle plánu.
Jestliže jste přestal(a) používat přípravek Bydureon
Jestliže se domníváte, že byste měl(a) přestat používat tento léčivý přípravek, poraďte se svým lékařem. Jestliže přestanete používat tento léčivý přípravek, může to ovlivnit hladinu Vašeho cukru v krvi.
Máte-li jakékoli další otázky týkající se používání tohoto přípravku, zeptejte se svého lékaře, nebo lékárníka nebo diabetologické sestry.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může vyvolávat i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Vzácně byly hlášeny závažné alergické reakce (anafylaxe) (mohou postihnout až 1 z 1000 lidí).
Kontaktujte neprodleně lékaře v případě výskytu následujících příznaků:
• otekání obličeje, jazyka nebo hrdlo (angioedém)
• přecitlivělost (vyrážka, svědění a rychlé otékání oblasti krku, obličeje, úst nebo hrdla)
• obtížné polykání
• vyrážka a potíže s dýcháním
Byly hlášeny případy zánětu slinivky (pankreatitida) u pacientů užívajících tento léčivý přípravek (frekvence není známa). Pankreatitida může být závažný, potenciálně život ohrožující stav.
• Informujte svého lékaře, pokud jste prodělal(a) pankreatitidu, trpíte žlučovými kameny, alkoholismem nebo vysokou hladinou tuků v krvi. Tyto stavy mohou zvýšit riziko, že u Vás dojde k onemocnění pankreatitidou, nebo se pankreatitida znovu projeví, a to nezávisle na tom, zda používáte tento léčivý přípravek, nebo ne.
• PŘESTAŇTE používat tento léčivý přípravek a kontaktujte ihned svého lékaře v případě, že se u Vás objeví silná a přetrvávající bolest v oblasti břicha, ať už se zvracením, nebo bez něj, protože se může jednat o zánět slinivky břišní (pankreatitida).
Velmi časté nežádoucí účinky přípravku Bydureon (mohou postihnout více než 1 z 10 lidí):
• pocit na zvracení (pocit na zvracení je nejčastější při zahájení podávání přípravku Bydureon, u většiny pacientů časem ustupuje)
• průjem
• hypoglykemie (snížená hladina cukru v krvi)
Jestliže je tento léčivý přípravek podáván společně s léky obsahujícími sulfonylmočovinu, velmi často může dojít k případům přílišného snížení hladiny cukru v krvi (hypoglykemie, obvykle mírná až střední). V případě, že užíváte tento léčivý přípravek, může být zapotřebí snížit dávku léku se sulfonylmočovinou. Známky a příznaky nízké hladiny cukru v krvi mohou zahrnovat bolest hlavy, ospalost, slabost, závratě, zmatek, podrážděnost, hlad, zrychlení srdečního tepu, pocení a pocit nervozity. Váš lékař by Vám měl poradit, jak nízkou hladinu cukru v krvi léčit.
Časté nežádoucí účinky přípravku Bydureon (mohou se vyskytnout až u 1 z 10 lidí):
• závratě
• bolest hlavy
• zvracení
• ztráta energie a síly
• únava (vyčerpání)
• zácpa
• bolest v žaludeční oblasti
• nadýmání
• trávicí potíže
• plynatost
• pálení žáhy
• snížení chuti k jídlu
Tento léčivý přípravek může snižovat Vaši chuť k jídlu, množství přijaté potravy a tělesnou hmotnost.Pokud u Vás dojde k příliš rychlému úbytku tělesné hmotnosti (více než 1,5 kg za týden), oznamte to svému lékaři, protože takto rychlý úbytek může způsobit problémy jako jsou žlučové kameny.
• reakce v místě injekce
Pokud u Vás dojde k reakci v místě injekce (červenání, kopřivka nebo svědění), můžete požádat svého lékaře o pomoc při mírnění těchto potíží a příznaků. Po podání injekce můžete pod kůží vidět nebo cítit malé bulky, měly by zmizet po 4 až 8 týdnech. Není nutné přerušovat léčbu.
Méně časté nežádoucí účinky nebo frekvence není známa přípravku Bydureon (mohou se vyskytnout až u 1 ze 100 lidí):
• snížená funkce ledvin
• dehydratace (ztráta tělesných tekutin), někdy doprovázená sníženou funkcí ledvin
• neprůchodnost střeva
• říhání
• nezvyklá chuť v ústech
• zvýšené pocení
• vypadávání vlasů
• ospalost
Vzácné nežádoucí účinky přípravku Bydureon (mohou se vyskytnout až u 1 z 1000 lidí):
• pocit nervozity
Byly hlášeny některé další nežádoucí účinky (frekvence není známa, z dostupných údajů ji nelze určit).
• změny INR (měření míry srážlivosti krve) byly hlášeny při souběžném podávání warfarinu reakce v místě vpichu. Po podání injekce s exenatidem byly hlášeny kožní reakce v místě vpichu. Jedná se o: dutiny obsahující hnis (absces) a oteklé červené plochy kůže na pohmat horké a měkké (celulitida)
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, nebo lékárníkovi nebo diabetologické sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Bydureon uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na štítku a krabičce za zkratkou EXP. Datum použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v chladničce (2 °C až 8 °C). Chraňte před mrazem.
Před použitím může být pero uchováváno při teplotě do 30 °C až po dobu 4 týdnů.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Přípravek Bydureon pero, který byl vystaven mrazu, musí být zlikvidován.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co přípravek Bydureon obsahuje
- Léčivou látkou je exenatidum. Jedno předplněné pero obsahuje 2 mg exenatidu. Po smísení je podaná dávka 2 mg/0,65 ml.
- Pomocnými látkami jsou:
- Prášek: polyglaktin (1:1), sacharóza.
- Rozpouštědlo: sodná sůl karmelosy, chlorid sodný, polysorbát 20, monohydrát dihydrogenfosforečnanu sodného, heptahydrát hydrogenfosforečnanu sodného, voda na injekci a hydroxid sodný (k úpravě pH).
Jak přípravek Bydureon vypadá a co obsahuje toto balení
Tento léčivý přípravek je dodáván jako prášek a rozpouštědlo (tekutina) pro přípravu injekční suspenze v předplněném peru.
Prášek (2 mg) v jedné komoře je bílý až téměř bílý a rozpouštědlo (0,65 ml) ve druhé komoře je čirý bezbarvý až lehce nažloutlý či nahnědlý roztok. Jedno jednodávkové předplněné pero je dodáváno s vhodnou jehlou. Každá krabička obsahuje jednu náhradní jehlu.
Tento léčivý přípravek se dodává v balení po 4 jednodávkových předplněných perech a jako vícenásobné balení po 12 (3 balení po 4) jednodávkových předplněných perech. Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci
AstraZeneca AB SE-151 85 Sodertálje Švédsko
Výrobce
AstraZeneca UK Limited Silk Road Business Park,
Macclesfield, Cheshire, SK10 2NA Velká Británie
Swords Laboratories T/A Lawrence Laboratories
Unit 12 Distribution Centre, Shannon Industrial Estate, Shannon, Co. Clare Irsko
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Česká republika
AstraZeneca Czech Republic s.r.o.
Tel: +420 222 807 111
Danmark
AstraZeneca A/S Tlf: +45 43 66 64 62
Malta
Associated Drug Co. Ltd Tel: +356 2277 8000
|
Deutschland AstraZeneca GmbH Tel: +49 41 03 7080 |
Nederland AstraZeneca BV Tel: +31 79 363 2222 |
|
Eesti AstraZeneca Tel: +372 6549 600 |
Norge AstraZeneca AS Tlf: +47 21 00 64 00 |
|
EXláSa AstraZeneca A.E. Tn^: +30 2 10 6871500 |
Osterreich AstraZeneca Osterreich GmbH Tel: +43 1 711 31 0 |
|
Espaňa AstraZeneca Farmacéutica Spain, S.A. Tel: +34 91 301 91 00 |
Polska AstraZeneca Pharma Poland Sp. z o.o. Tel.: +48 22 245 73 00 |
|
France AstraZeneca Tél: +33 1 41 29 40 00 |
Portugal AstraZeneca Produtos Farmaceuticos, Lda. Tel: +351 21 434 61 00 |
|
Hrvatska AstraZeneca d.o.o. Tel: +385 1 4628 000 |
Románia AstraZeneca Pharma SRL Tel: +40 21 317 60 41 |
|
Ireland AstraZeneca Pharmaceuticals (Ireland) Ltd Tel: +353 1609 7100 |
Slovenija AstraZeneca UK Limited Tel: +386 1 51 35 600 |
|
Ísland Vistor hf. Sími: +354 535 7000 |
Slovenská republika AstraZeneca AB, o.z. Tel: +421 2 5737 7777 |
|
Italia AstraZeneca S.p.A. Tel: +39 02 9801 1 |
Suomi/Finland AstraZeneca Oy Puh/Tel: +358 10 23 010 |
|
Kúnpoq A^skt^p Oap^aKeuxiKq ArS Tn^: +357 22490305 |
Sverige AstraZeneca AB Tel: +46 8 553 26 000 |
|
Latvija SIA AstraZeneca Latvija Tel: +371 67377100 |
United Kingdom AstraZeneca UK Ltd Tel: +44 1582 836 836 |
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou uveřejněny na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu
POKYNY PRO UŽIVATELE
Váš průvodce krok za krokem
Bydureon 2 mg prášek a rozpouštědlo pro injekční suspenzi s prodlouženým účinkem Pokud máte otázky týkající se přípravku Bydureon
• Podívejte se do části Často kladené otázky a odpovědi Užitečné tipy
• Věnujte pokynům dostatek času.
• Dodržujte pokyny krok za krokem.
• Budete potřebovat dostatek času pro dokončení všech kroků bez přestávky.
• Méně času budete potřebovat, jakmile se sám (sama) důkladně naučíte aplikovat injekci.
DŮLEŽITÉ:
Čtěte a pečlivě dodržujte každý krok v těchto pokynech pokaždé, když si budete aplikovat přípravek Bydureon. Žádné kroky nevynechávejte. Přečtěte si také Příbalovou informaci obsaženou v balení přípravku.
Průvodce jednotlivými součástmi
• Souprava pro jednorázové podání
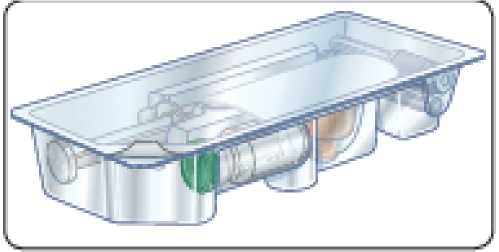
Zde nadzvedněte pro podrobnější seznámení s jednotlivými součástmi.
Ponechejte tuto záložku otevřenou, abyste se později mohl(a) v průběhu všech kroků kdykoli vrátit.
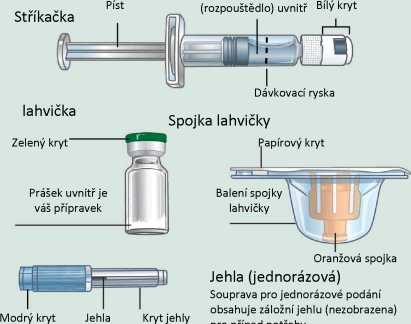
pro případ potřeby.
Průvodce jednotlivými součástmi Souprava pro jednorázové podání
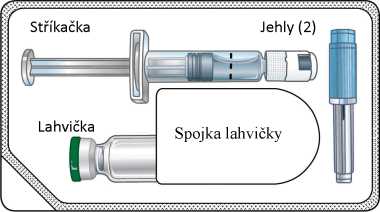
Co balení obsahuje
Pro podání přesné dávky čtěte každou část tak, abyste mohli postupně provádět každý krok. Tento návod je rozdělen do následujících částí:
1 Začínáme
2 Spojení součástí
3 Promíchání přípravku a naplnění stříkačky
4 Aplikace přípravku Často kladené otázky a odpovědi.
1. Začínáme
1a Vyndejte soupravu pro jednorázové podání dávky z ledničky.
Připravte se na bezpečné zlikvidování použité jehly a stříkačky. Připravte si vše, co budete pro bezpečnou likvidaci použitých jehel a stříkaček potřebovat.
1b Umyjte si ruce.
1c

Pro otevření odtrhněte vrchní obal.
Vytáhněte stříkačku. Tekutina ve stříkačce by měla být čirá a bez částic. Je v pořádku, pokud obsahuje vzduchové bubliny.
Vyndejte jehlu, spojku lahvičky, lahvičku a stříkačku na čistý, rovný povrch.
1d
j
, HW í y /
Vytáhněte jehlu a odšroubujte modrý kryt.
Jehlu v pouzdře odložte. Jehla je nyní připravena. Budete ji potřebovat později. Pro případ potřeby je v balení záložní jehla.
Vezměte lahvičku.
Poklepejte s ní několikrát o tvrdý povrch pro uvolnění prášku.
Palcem odstraňte zelený kryt.
Lahvičku odložte.
2. Spojení součástí
Vezměte spojku lahvičky a odtrhněte papírovou fólii. Nedotýkejte se oranžové spojky uvnitř.
Z
Uchopte balení spojky lahvičky. Do druhé ruky uchopte lahvičku.
Zatlačte pevně vršek lahvičky do oranžové spojky.
Nyní vytáhněte lahvičku s připojenou oranžovou spojkou z balení.

Takto by nyní měla injekční lahvička vypadat.
Odložte ji nyní na pozdější použití.
Vezměte injekční stříkačku.
Druhou rukou pevně uchopte 2 šedé čtverečky na bílém krytu.
2g Křup!
\//
Buďte opatrní, abyste nezatlačili píst dovnitř. Kryt odlomte, jako byste lámali větvičku.

Takto vypadá odlomený kryt.
Kryt nebudete potřebovat, můžete jej vyhodit.
Takto nyní vypadá injekční stříkačka.
Nyní uchopte injekční lahvičku s připojenou oranžovou spojkou.
Našroubujte oranžovou spojku na stříkačku, dokud se nepřipojí. Při otáčení držte oranžovou spojku. Neutahujte přespříliš. Buďte opatrní, abyste nezatlačili píst dovnitř.
2k
Takto nyní vypadají součásti po spojení.
3. Promíchání přípravku a naplnění stříkačky DŮLEŽITÉ:
Během těchto kroků smícháte přípravek a naplníte jej do stříkačky. Jakmile smícháte přípravek, musíte jej ihned aplikovat. Nesmíte uchovávat smíchaný přípravek pro pozdější aplikaci injekce.

Palcem zatlačte píst, dokud se nezastaví a držte jej palcem na místě.
Můžete cítit, jako by píst pružil poněkud zpět.

Držte dál píst stisknutý palcem dole a důkladně třepejte. Protřepávejte, dokud nejsou tekutina a prášek dobře promíchány.
Neobávejte se, že se lahvička uvolní. Oranžová spojka udrží lahvičku připojenou ke stříkačce. Protřepávejte opravdu důkladně.
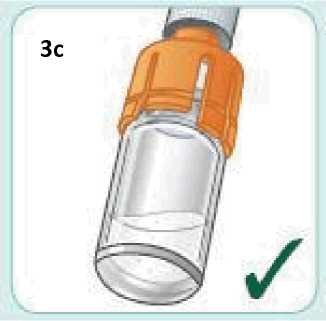
Pokud je přípravek dobře promíchán, měl by vypadat zakalený.
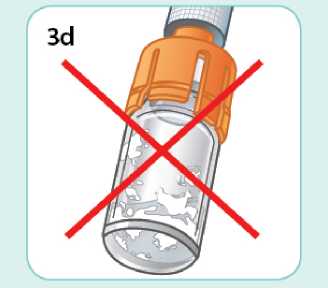
Pokud na stěnách nebo dně lahvičky vidíte shluky částic nebo suchý prášek, přípravek NENÍ dobře promíchán.
Důkladně jej znovu protřepejte, dokud není dobře promíchán.
V průběhu protřepávání držte píst palcem stále stisknutý.
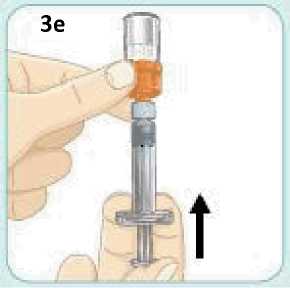
Nyní uchopte lahvičku tak, aby byla stříkačka otočena vzhůru. Stiskněte dál píst palcem, dokud se nezastaví, a držte jej na místě.
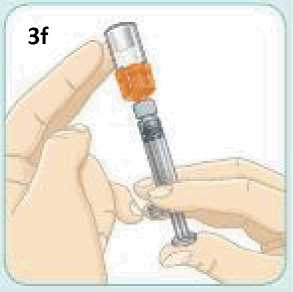
Jemně poklepejte lahvičku druhou rukou. Tiskněte dál píst palcem a držte jej na místě.
Poklepání napomůže přípravku stéct dolů po stěnách lahvičky. Je v pořádku, pokud obsahuje vzduchové bubliny.
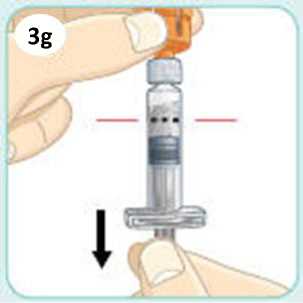
Vytáhněte píst dolů až za černě označenou dávkovači rysku.
Tímto nasajete přípravek z lahvičky do stříkačky. Můžete vidět vzduchové bubliny. To je normální. Na stěnách lahvičky může ulpět malé množství tekutiny. To je také normální.
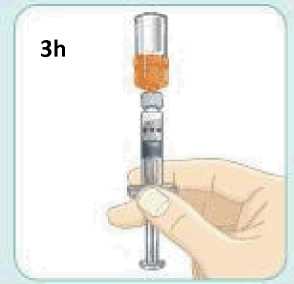
Držte jednou rukou píst na místě tak, aby se nemohl pohybovat.
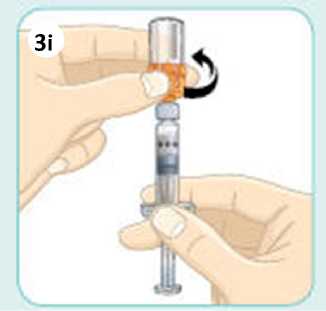
Druhou rukou odšroubujte oranžovou spojku.
Po odstranění spojky dejte pozor, abyste nestiskli píst.

Injekční stříkačka by nyní měla vypadat takto.
4. Aplikace přípravku DŮLEŽITÉ:
Přečtěte si pečlivě následující kroky a důkladně si prohlédněte obrázky. Pomůže Vám to aplikovat si přesnou dávku přípravku.
4a
Našroubujte jehlu na stříkačku do dotažení. Zatím neodstraňujte kryt jehly. Dejte pozor, abyste nestiskli píst.
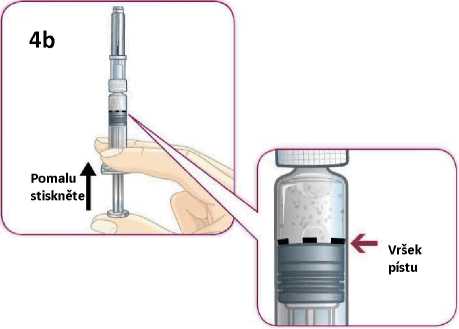
Pomalu tiskněte píst tak, aby se vršek pístu dostal do úrovně černě označené dávkovači rysky.
Nyní uvolněte píst.
Je důležité v tomto okamžiku ukončit stlačování pístu, jinak budete plýtvat přípravkem a nepodáte si přesnou dávku.
Vršek pístu musí zůstat na úrovni s černě značenou dávkovači linkou pro následující kroky.
Toto Vám pomůže zajistit, že si podáte správnou dávku přípravku.
DŮLEŽITÉ:
Je normální, pokud ve směsi vidíte několik vzduchových bublin.
Vzduchové bubliny Vám neublíží ani neovlivní dávku.
|
f | ||
|
4d | ||
|
Zadní strana horní | ||
|
kí! |
j jf ; | |
|
■ Břicho /J <í, JUT | | | ||
|
___- |
^ 1’^frl li ^Stehno U Jí i y | |
|
! H v i ij :ě ni / |
I | |
|
M Zepředu |
CJ lL Zezadu | |
|
Oblasti těla pro aplikaci injekce. |
J | |
|
V | ||
Jednotlivé dávky přípravku můžete aplikovat do oblasti břicha, stehna nebo zadní strany horní části paže.
Každý týden můžete použít stejnou oblast těla. Ujistěte se však, že v této oblasti pro injekci vyberete odlišné místo.
|
4e | |
|
■ U rj | |
Držte stříkačku poblíž černě značené dávkovací rysky.
Stáhněte kryt jehly. Neotáčejte.
Dejte pozor, abyste nestiskli píst.
Při odstraňování krytu můžete vidět 1 nebo 2 kapky tekutiny. To je normální.
Ujistěte se, že používáte injekční techniku doporučenou Vaším lékařem nebo diabetologickou sestrou. Připomenutí: Přípravek Bydureon musíte aplikovat ihned po smíchání.
Vbodněte jehlu do kůže (subkutánně). Abyste si aplikovali celou dávku, zatlačte na píst palcem, dokud se nezastaví.
Vytáhněte jehlu.
V příbalové informaci se dozvíte (bod 3) co dělat, pokud si nejste jistá(ý), jestli jste si podal(a) celou dávku.
4h. Stříkačku s nasazenou jehlou zlikvidujte podle doporučení lékaře nebo diabetologické sestry. NESNAŽTE se jehlu uvolnit nebo znovu použít.
Nemusíte uchovávat žádnou součást. Každá souprava pro jednorázové podání obsahuje všechny součásti nutné pro podání týdenní dávky přípravku Bydureon.
Léčivé přípravky se nesmí vyhazovat do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak máte likvidovat přípravky, které již nepotřebujete. Tato opatření pomáhají chránit životní prostředí.
Když nastane čas pro další týdenní dávku, začněte opět krokem 1.
Často kladené otázky a odpovědi
Pokud se vaše otázka týká: Viz číslo otázky:
Jak brzy aplikovat injekci po smíchání 1
Vzduchové bubliny ve stříkačce 3
Píst není v úrovni černě označené dávkovací rysky 6
Není možné stlačit píst při aplikaci injekce 7
Často kladené otázky a odpovědi
1. Po smíchání přípravku, jak dlouho mohu čekat před aplikací injekce?
Přípravek Bydureon musíte aplikovat ihned po smíchání. Pokud si neaplikujete přípravek Bydureon okamžitě, začnou se ve stříkačce tvořit malé shluky. Tyto shluky mohou při aplikaci injekce ucpat jehlu (viz otázka 7).
2. Jak poznám, že je přípravek dobře promísen?
Pokud je přípravek dobře promíchán, měl by vypadat zakalený. Na stěnách nebo dně lahvičky nesmí být suchý prášek. Pokud uvidíte suchý prášek, důkladně třepejte a držte píst stále stisknutý palcem. (Tato otázka se vztahuje ke krokům popsaným v bodech 3a až 3d).
3. Jsem připraven(a) podat si injekci. Co mám udělat, pokud ve stříkačce uvidím vzduchové bubliny?
Vzduchové bubliny se ve stříkačce vyskytují normálně. Vzduchové bubliny Vám neublíží ani neovlivní dávku. Přípravek Bydureon je nyní aplikován do kůže (subkutánně). Při tomto typu aplikace nejsou vzduchové bubliny problém.
4. Co mám dělat, pokud mám potíže s nasazením jehly?
Nejdříve se ujistěte, že jste odtranili modrý kryt. Potom našroubujte jehlu na stříkačku do utažení. Abyste předešli ztrátám přípravku, vyvarujte se stisknutí pístu při nasazování jehly. Pro více informací týkajících se injekční techniky kontaktujte svého lékaře.
(Tato otázka se vztahuje ke kroku 4a)
5. Co mám dělat, pokud mám potíže se sundáním krytu jehly?
Jednou rukou držte stříkačku poblíž černě značené dávkovací rysky. Druhou rukou uchopte kryt jehly. Plynule stáhněte kryt jehly. Neotáčejte krytem. (Tato otázka se vztahuje ke kroku 4f)
6. Jsem u kroku 4c. Co mám dělat, když se vršek pístu dostal za černě označenou dávkovací rysku?
Černě označená dávkovací ryska označuje správnou dávku. Když se vršek pístu dostane za rysku, měli byste pokračovat krokem 4d a aplikovat si injekci. Před další injekcí v následujícím týdnu si pečlivě přečtěte pokyny v krocích 3 a až 4h.
7. Co mám dělat, pokud při aplikaci injekce nemohu píst stisknout úplně dolů?
Znamená to, že došlo k ucpání jehly. Odstraňte jehlu a vyměňte ji za náhradní jehlu ze soupravy. Potom vyberte odlišné místo pro aplikaci a dokončete injekci.
Pro kontrolu jak:
• Odstranit modrý kryt jehly, viz krok 1d
• Připojit jehlu, viz krok 4a
• Odstranit kryt jehly a podat si injekci, viz kroky 4e až 4g
Pokud stále nemůžete stisknout píst úplně dolů, vytáhněte jehlu. Přečtěte si v příbalové informaci (bod 3) co dělat, pokud si nejste jistá(ý), že jste si podal(a) celou dávku.
Abyste předešli ucpání jehly, vždy důkladně promíchejte přípravek a aplikujte jej ihned po promíchání.
Přípravek Bydureon je nutné podávat jednou týdně.
Udělejte si poznámku, že jste si dnes aplikoval(a) přípravek Bydureon, a poznamenejte si v kalendáři Vaši další injekci.
Kde se dozvědět více o přípravku Bydureon
• Promluvte si se svým lékařem, lékárníkem nebo diabetologickou sestrou Přečtěte si pečlivě příbalovou informaci
POKYNY PRO UŽIVATELE
Před použitím si přečtěte pečlivě tyto pokyny
Bydureon 2 mg prášek a rozpouštědlo pro injekční suspenzi s prodlouženým účinkem v předplněném peru
Jako používat Bydureon předplněné pero
Mísíd okénko Pruhovaný štítek Knoflík

Kryt Jehla pera Kontro|ní °kénk° Zelený štítek Oranžový štítek
Před použitím pera se doporučuje, abyste byl(a) proškolen(a) lékařem nebo diabetologickou sestrou o správném používání pera.
Nedoporučuje se, aby pero používali lidé, kteří nevidí či lidé slabozrací, pokud nemůže pomoci podat injekci proškolená osoba.
Krok 1: Příprava Vašeho pera
Nechte Vaše pero, aby se ohřálo. Vyndejte jedno pero z chladničky a ponechejte ho při pokojové teplotě po dobu alespoň 15 minut. NEPOUŽÍVEJTE pero po ukončení doby použitelnosti.
Umyjte si ruce, zatímco se pero ohřívá.
Otevřete plastovou vložku tahem za fólii od rohu. Vyjměte pero a jehlu. NEPOUŽÍVEJTE Vaše pero nebo jehlu, pokud je kterákoli část poškozená nebo chybí.

ZKONTROLUJTE kapalinu uvnitř kontrolního okénka. Musí být čirá a prostá pevných částic. Pokud v kapalině uvidíte vzduchové bubliny, je to normální.
Odstraňte papírový kryt z krytu jehly.
Upevněte jehlu na pero zatlačením a zašroubováním na horní část pera až na doraz. Zatím NESUNDÁVEJTE kryt jehly.
Krok 2: Smísení Vaší dávky Smísení léčivého přípravku. Držte pero ve svislé poloze s krytem jehly směrem nahoru a pomalu otáčejte knoflíkem proti směru hodinových ručiček. PŘESTAŇTE otáčet, jakmile uslyšíte kliknutí a zelený štítek zmizí.
Silně klepejte perem, aby došlo ke smísení.
Držte pero v místě oranžového štítku a klepejte jím silně do dlaně Vaší ruky. Po několika klepnutích vždy otočte poněkud pero bez toho, že byste otáčel(a) knoflíkem. Může být potřebné poklepat 80krát nebo i vícekrát.
Zkontrolujte suspenzi. Držte pero směrem vzhůru proti světlu a podívejte se z obou stran mísícího okénka. Tekutina nesmí obsahovat žádné SHLUKY a musí být stejnoměrně zakalená.
Léčivý přípravek musí být dobře smísený, abyste dostal(a) celou dávku. Pokud není dobře smísený, klepejte dále a silněji.
NEPOKRAČUJTE dále, dokud není léčivý přípravek dobře smísený.
Léčivý přípravek musí být dobře smísený, abyste dostal(a) celou dávku. Pokud není dobře smísený, klepejte dále a silněji. Pokud vidíte v tekutině vzduchové bubliny, je to normální a nepředstavují žádné
riziko.
Porovnejte obě strany mísícího okénka s fotografiemi níže tak, že podržíte pero proti této stránce. Věnujte pozornost povrchu dolní strany. Pokud nevidíte shluky, máte pero připraveno k podání injekce.
Vaše pero prohlížejte zde.
Krok 3: Podejte si Vaši dávku
DŮLEŽITÉ. Jakmile je léčivý přípravek dobře smísen, musíte ihned podat dávku injekcí.
Nemůžete přípravek schovat k pozdějšímu použití.
Vyberte si místo pro podání buďto na Vašem břiše, stehně, nebo zadní horní části paže. Každý týden můžete zvolit stejnou část Vašeho těla, ale použijte vždy jiné místo v této části. Lehce očistěte plochu mýdlem a vodou nebo vatou namočenou v lihu.
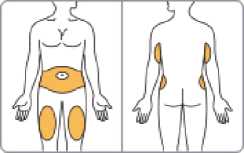
Otočte knoflíkem, abyste odjistil(a) injekční tlačítko. Držte pero ve svislé poloze s krytem jehly směrem nahoru a otáčejte knoflíkem proti směru hodinových ručiček až oranžový štítek zmizí a objeví se injekční tlačítko. Zatím injekční tlačítko NETISKNĚTE.
Odstraňte kryt jehly tahem směrem od pera. NEOTÁČEJTE. Můžete vidět několik kapek tekutiny na jehle nebo v krytu jehly.
Podejte si léčivý přípravek. Vpíchněte jehlu do Vaší kůže. Stlačte na injekční tlačítko Vaším palcem dokud neuslyšíte kliknutí. Držte takto po dobu 10 sekund, abyste podal(a) celou dávku.

sekund
Vaše pero správně zlikvidujte i s nasazenou jehlou v nádobě, kterou nelze propíchnout.
NESNAŽTE SE jehlu snímat nebo znovu používat.
Často kladené otázky a odpovědi
1. Jak zjistím, že je léčivý přípravek dobře smísen?
Léčivý přípravek je dobře smísen, když je tekutina zakalená z obou stran mísícího okénka. V tekutině nesmíte vidět žádné shluky. Může Vám pomoci, když se podíváte do okénka proti světlu. Pokud vidíte shluky jakékoliv velikosti, pokračujte v důkladném poklepávání perem proti dlani Vaší ruky až je dobře smísen.
2. Mám problém se smísením své dávky. Co bych měl(a) udělat?
Zapamatujte si: Před přípravou dávky vyndejte pero z chladničky na dobu alespoň 15 minut. Tím se pero ohřeje na teplotu místnosti. Když má pero teplotu místnosti, smísení léčivého přípravku bude snadnější.
Ověřte si, že pero držíte na konci za knoflík a oranžový štítek. Tak se Vám bude pero lépe držet a klepání proti dlani ruky bude silnější.
Může též pomoci, když budete klepat proti dlani ruky po obou stranách v místě mísícího okénka. Pokud vidíte shluky, pokračujte v klepání.
3. Jak dlouho mám čekat před podáním injekce, když mám léčivý přípravek smísený?
Injekci musíte podat hned po smísení přípravku. Pokud si nepodáte Vaši dávku hned, mohou se v peru vytvořit malé shluky a Vy si nepodáte celou dávku.
4. Jsem připraven si podat svou dávku. Co mám dělat, když v peru vidím vzduchové bubliny?
Vzduchové bubliny v peru jsou normální. Léčivý přípravek se podává pod kůži (subkutánně). Vzduchové bubliny Vám neublíží, ani nemají vliv na dávku u tohoto způsobu podání injekce.
5. Co bych měl(a) dělat v případě, že nemohu úplně stlačit injekční tlačítko při podávání své dávky?
Zkontrolujte, zda jste dobře dotáhl(a) injekční jehlu na pero. Ujistěte se také, že jste otočil(a) knoflíkem až do konce, oranžový štítek není vidět a injekční tlačítko je venku.
Pokud stále nemůžete stlačit injekční tlačítko, může být ucpaná injekční jehla. Vytáhněte jehlu z kůže a nahraďte ji náhradní jehlou z krabičky. Podívejte se znovu na způsob, jak nasazovat jehlu. Potom si vyberte jiné místo pro injekci a dokončete podání injekce.
Pokud stále nemůžete úplně stlačit injekční tlačítko, vytáhněte jehlu z Vaší kůže. Pero i s nasazenou jehlou vhoďte do nádoby odolné proti propíchnutí.
6. Jak se dozvím, že jsem si podal(a) injekcí celou dávku?
Abyste měl(a) jistotu, že si podáte celou dávku, stlačte injekční tlačítko Vaším palcem, až uslyšíte klapnutí. Po tomto klapnutí podržte jehlu ve Vaší kůži po dobu 10 sekund. To je dostatečná doba pro to, aby se veškerý léčivý přípravek dostal z pera pod Vaši kůži.
Jak mám zacházet s perem Bydureon?
7.
Budete potřebovat nádobu, která je odolná proti propíchnutí, která je dostatečně velká, aby se do ní vešlo pero i s nasazenou jehlou. Ujistěte se, že nádoba se dá uzavřít. Můžete použít nádobu na nebezpečný biologický odpad, jinou nádobu z tvrdého plastu nobo kovovou nádobu. Nádoba není součástí balení přípravku.
Zeptejte se svého lékárníka, jak bezpečně naložit s nádobou s použitými pery a jehlami.
Nevyhazujte nádobu do Vašeho domovního odpadu.
88