Briviact 75 Mg
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1.
NÁZEV PŘÍPRAVKU
Briviact 10 mg potahované tablety Briviact 25 mg potahované tablety Briviact 50 mg potahované tablety Briviact 75 mg potahované tablety Briviact 100 mg potahované tablety
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Briviact 10 mg potahované tablety
Jedna potahovaná tableta obsahuje brivaracetamum 10 mg. Briviact 25 mg potahované tablety
Jedna potahovaná tableta obsahuje brivaracetamum 25 mg. Briviact 50 mg potahované tablety
Jedna potahovaná tableta obsahuje brivaracetamum 50 mg. Briviact 75 mg potahované tablety
Jedna potahovaná tableta obsahuje brivaracetamum 75 mg. Briviact 100 mg potahované tablety
Jedna potahovaná tableta obsahuje brivaracetamum 100 mg. Pomocné látky se známým účinkem:
Briviact 10 mg potahované tablety
Jedna potahovaná tableta o síle 10 mg obsahuje 88 mg laktózy. Briviact 25 mg potahované tablety
Jedna potahovaná tableta o síle 25 mg obsahuje 94 mg laktózy. Briviact 50 mg potahované tablety
Jedna potahovaná tableta o síle 50 mg obsahuje 189 mg laktózy. Briviact 75 mg potahované tablety
Jedna potahovaná tableta o síle 75 mg obsahuje 283 mg laktózy. Briviact 100 mg potahované tablety
Jedna potahovaná tableta o síle 100 mg obsahuje 377 mg laktózy. Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Potahovaná tableta (tablety).
Briviact 10 mg potahované tablety
Bílé až téměř bílé, kulaté potahované tablety, průměr 6,5 mm, s vyraženým „u10“ na jedné straně.
Briviact 25 mg potahované tablety
Šedé, oválné potahované tablety o rozměrech 8,9 mm x 5,0 mm a s vyraženým „u25“ na jedné straně. Briviact 50 mg potahované tablety
Žluté, oválné potahované tablety o rozměrech 11,7 mm x 6,6 mm a s vyraženým „u50“ na jedné straně.
Briviact 75 mg potahované tablety
Nachové, oválné potahované tablety o rozměrech 13,0 mm x 7,3 mm a s vyraženým „u75“ na jedné straně.
Briviact 100 mg potahované tablety
Zelenošedé, oválné potahované tablety o rozměrech 14,5 mm x 8,1 mm a s vyraženým „u100“ na jedné straně.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Briviact je indikován jako přídatná terapie při léčbě parciálních záchvatů s nebo bez sekundární generalizace u dospělých a dospívajících pacientů s epilepsií ve věku od 16 let.
4.2 Dávkování a způsob podání
Dávkování
Doporučená počáteční dávka je buď 50 mg/den nebo 100 mg/den, potřebná ke snížení počtu záchvatů na základě posouzení lékaře oproti potenciálním nežádoucím účinkům. Dávka se podává ve dvou stejných rozdělených dávkách, jednou ráno a jednou večer. Na základě individuální odpovědi pacienta a snášenlivosti lze dávku upravit v dávkovém rozmezí 50 mg/den až 200 mg/den.
Opomenutá dávka
Jestliže pacienti opomenou užít jednu nebo více dávek, doporučuje se užít jednu dávku, jakmile si vzpomenou, a následující dávku vzít v obvyklé době ráno nebo večer. To může zabránit poklesu plazmatické koncentrace brivaracetamu pod účinnou hladinu, a tím znovupropuknutí záchvatů.
Ukončení léčby
Pokud je nutno podávání brivaracetamu ukončit, doporučuje se postupné snižování o 50 mg/den v týdenním intervalu. Po týdnu léčby dávkou 50 mg/den se doporučuje v posledním týdnu léčby dávka 20 mg/den.
Zvláštní populace
Starší pacienti (65 let a starší)
Není nutná žádná úprava dávky u starších pacientů (viz bod 5.2).
Klinická zkušenost u pacientů >65 let je omezená.
Porucha funkce ledvin
U pacientů s poruchou funkce ledvin není třeba dávku nijak upravovat (viz bod 5.2). Podávání brivaracetamu se nedoporučuje u pacientů v konečné fázi onemocnění ledvin, kteří jsou léčeni dialýzou, vzhledem k nedostatku údajů.
Porucha funkce jater
Expozice vůči brivaracetamu byla zvýšená u pacientů s chronickým onemocněním jater. Počáteční dávka 50 mg/den by měla být zvážena. Ve všech fázích poruchy funkce jater se doporučuje maximální
denní dávka 150 mg podávaná ve 2 rozdělených dávkách (viz body 4.4 a 5.2).
Pediatrická populace
Bezpečnost a účinnost brivaracetamu u dětí ve věku méně než 16 let nebyly dosud stanoveny.
V současné době dostupné údaje jsou uvedeny v bodech 4.8, 5.1, a 5.2, ale na jejich základě nelze učinit žádná doporučení ohledně dávkování.
Způsob podání
Potahované tablety brivaracetamu je nutno užívat perorálně, polykat celé a zapíjet tekutinou. Lze je užívat s jídlem i bez jídla (viz bod 5.2).
4.3 Kontraindikace
Hypersenzitivita na léčivou látku, jiné deriváty pyrrolidonu nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Sebevražedné myšlenky a chování
Sebevražedné myšlenky a chování byly hlášeny v několika indikacích u pacientů léčených antiepileptiky, včetně brivaracetamu. Metaanalýza randomizovaných placebem kontrolovaných studií antiepileptik prokázala mírně zvýšené riziko sebevražedných myšlenek a chování. Mechanismus vzniku není znám a dostupné údaje nevylučují možnost zvýšeného rizika u brivaracetamu.
Proto by u pacientů měly být sledovány příznaky sebevražedných představ a chování a zvážena vhodná léčba. Pacienti (a osoby poskytující pacientům péči) by měli být upozorněni na to, že v případě výskytu příznaků sebevražedného myšlení či chování, by měli vyhledat lékařskou pomoc.
Porucha funkce jater
Jsou k dispozici omezené klinické údaje o použití brivaracetamu u pacientů s již existující poruchou funkce jater. U pacientů s poruchou funkce jater se doporučuje úprava dávky (viz bod 4.2).
Intolerance laktózy
Potahované tablety brivaracetamu obsahují laktózu. Pacienti se vzácnými dědičnými problémy s intolerancí galaktózy, vrozeným deficitem laktázy nebo malabsorpcí glukózy a galaktózy by tento přípravek neměli užívat.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Formální studie interakce byly provedeny jen u dospělých pacientů.
Farmakodynamické interakce
Současná léčba s levetiracetamem
V klinických studiích (ačkoli počet je velmi omezený) nebyl pozorován žádný přínos brivaracetamu versus placebo u pacientů současně užívajících levetiracetam. Nebyly doloženy žádné další údaje ohledně bezpečnosti a tolerability (viz bod 5.1).
Interakce s alkoholem
Ve studii farmakokinetické a farmakodynamické interakce mezi brivaracetamem v jednorázové dávce 200 mg a ethanolem v kontinuální infuzi 0,6 g/l u zdravých subjektů nenastala žádná farmakokinetická interakce, ale brivaracetam přibližně zdvojnásobil účinky alkoholu na psychomotorické funkce, pozornost a paměť. Podávání brivaracetamu s alkoholem se nedoporučuje.
Farmakokinetické interakce
Účinky jiných látek na farmakokinatiku brivaracetamu
In vitro údaje naznačují, že brivaracetam má nízký interakční potenciál. Hlavní metabolickou cestou brivaracetamu je na CYP nezávislá hydrolýza. Druhá cesta zahrnuje hydroxylaci, která je zprostředkována CYP2C19 (viz bod 5.2).
Plazmatické koncentrace brivaracetamu se mohou zvýšit, je-li současně podáván se silnými inhibitory CYP2C19 (jako flukonazol, fluvoxamin), ale riziko klinicky významné interakce zprostředkované CYP2C19 je považováno za nízké.
Rifampicin
U zdravých dobrovolníků současné podávání silného induktoru enzymů rifampicinu (600 mg/den po dobu 5 dnů) snížilo plochu brivaracetamu pod křivkou (AUC) o 45 %. Předepisující lékaři musí zvážit úpravu dávky brivaracetamu u pacientů, u kterých se zahajuje nebo ukončuje léčba rifampicinem.
Antiepileptika se silnou indukcí enzymů
Plazmatické koncentrace brivaracetamu klesají při současném podávání s antiepileptiky silně indukující enzymy (karbamazepin, fenobarbital, fenytoin), ale není nutná žádná úprava dávky (viz tabulka 1).
Jiné induktory enzymů
Očekává se, že jiné silné induktory enzymů (jako třezalka tečkovaná (Hypericum perforatum)) mohou také snížit systémovou expozici brivaracetamu. Proto zahájení nebo ukončení léčby třezalkou tečkovanou má být provedeno s opatrností.
Účinek brivaracetamu na jiné léčivé přípravky
Brivaracetam podávaný v dávkách 50 mg nebo 150 mg/den neovlivňoval AUC midazolamu (metabolizován CYP3A4). Riziko klinicky relevantních CYP3A4 interakcí je považováno za nízké.
Studie in vitro prokázaly, že brivaracetam vykazuje malou nebo žádnou inhibici izoforem CYP450 s výjimkou CYP2C19. Brivaracetam může zvyšovat plazmatické koncentrace léčivých přípravků metabolizovaných CYP2C19 (např. lanzoprazol, omeprazol, diazepam). Při zkoušení in vitro brivaracetam neindukoval CYP1A1/2, ale indukoval CYP3A4 a CYP2B6. Žádná CYP3A4 indukce nebyla nalezena in vivo (viz midazolam výše). CYP2B6 indukce nebyla zkoumána in vivo a brivaracetam může snižovat plazmatické koncentrace léčivých přípravků metabolizovaných CYP2B6 (např. efavirenz). In vitro interakční studie k určení potenciálních inhibičních účinků na transportéry vedly k závěru, že nedochází k žádným klinicky významným účinkům s výjimkou pro OAT3. In vitro brivaracetam inhibuje OAT3 s poloviční maximální inhibiční koncentrací 42x vyšší, než je Cmax při nejvyšší klinické dávce. Brivaracetam v dávce 200 mg/den může zvýšit plazmatické koncentrace léčivých přípravků transportovaných OAT3.
Antiepileptika
Potenciální interakce mezi brivaracetamem (50 mg/den až 200 mg/den) a jinými antiepileptiky byly zkoumány v souhrnné analýze plazmatických lékových koncentrací ze všech studií fáze 2-3, v populační farmakokinetické analýze v placebem kontrolovaných studiích fáze 2-3 a ve vyhrazených studiích lékových interakcí (pro následující antiepileptika: karbamazepin, lamotrigin, fenytoin a topiramát). Účinek vzájemných interakcí na plazmatickou koncentraci je shrnut v tabulce 1 (zvýšení je označeno jako “t” a snížení jako “j” plocha pod křivkou plazmatické koncentrace v průběhu času jako „AUC“, maximální pozorovaná koncentrace jako „Cmax“).
|
Současně podávané antiepiletikum |
Vliv antiepiletika na koncentraci brivaracetamu v plazmě |
Vliv brivaracetamu na koncentraci antiepiletika v plazmě |
|
Karbamazepin |
AUC 29 % | Cmax 13 % 4 není nutná žádná úprava dávky |
karbamazepin - žádný epoxid karbamazepinu | (viz níže) není nutná žádná úprava dávky |
|
Klobazam |
žádné údaje |
žádný |
|
Klonazepam |
žádné údaje |
žádný |
|
Lacosamid |
žádné údaje |
žádný |
|
Lamotrigin |
žádný |
žádný |
|
Levetiracetam |
žádný |
žádný |
|
Oxkarbazepin |
žádný |
žádný (monohydroxyderivát, MHD) |
|
Fenobarbital |
AUC 19 % 4 není nutná žádná úprava dávky |
žádný |
|
Fenytoin |
AUC 21 % 4 není nutná žádná úprava dávky |
žádný aAUC 20 % t aCmax 20 % t |
|
Pregabalin |
žádné údaje |
žádný |
|
Topiramát |
žádný |
žádný |
|
Kyselina valproová |
žádný |
žádný |
|
Zonisamid |
žádné údaje |
žádný |
a na základě studie týkající se podávání supraterapeutické dávky brivaracetamu 400 mg/den
Karbamazepin
Brivaracetam je středně účinný reverzibilní inhibitor epoxidové hydrolázy vyvolávající zvýšenou koncentraci epoxidu karbamazepinu, aktivního metabolitu karbamazepinu. V kontrolovaných studiích vzrostla plazmatická koncentrace epoxidu karbamazepinu v průměru o 37 %, 62 % a 98 % s malou variabilitou při dávkách brivaracetamu odpovídajících 50 mg/den, 100 mg/den a 200 mg/den. Nebyla pozorována žádná bezpečnostní rizika. Nebyl žádný aditivní účinek brivaracetamu a valproátu na AUC u epoxidu karbamazepinu.
Perorální kontraceptiva
Současné podávání brivaracetamu (100 mg/den) spolu s perorálním kontraceptivem obsahujícím ethinylestradiol (0,03 mg) a levonorgestrel (0,15 mg) neovlivňovalo farmakokinetiku žádné látky. Když byl brivaracetam současně podáván v dávce 400 mg/den (dvojnásobek doporučené maximální denní dávky) současně s perorálním kontraceptivem obsahujícím ethinylestradiol (0,03 mg) a levonorgestrel (0,15 mg), bylo pozorováno snížení AUC estrogenu o 27 % a snížení AUC progestinu o 23 %, a to bez dopadu na potlačení ovulace. Obecně nenastala žádná změna v profilech koncentrací v čase u endogenních markerů estradiolu, progesteronu, luteinizačního hormonu (LH), folikuly stimulujícího hormonu a globulinu vázajícího pohlavní hormony (SHBG).
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku
Lékaři by měli prodiskutovat plánované rodičovství a antikoncepci se ženami ve fertilním věku, které užívají brivaracetam (viz Těhotenství).
Jestliže se žena rozhodne otěhotnět, užívání brivaracetamu by mělo být opět přehodnoceno. Těhotenství
Riziko spojené s epilepsií a s antiepileptiky obecně
U všech antiepileptik bylo prokázáno, že mezi potomky léčených žen s epilepsií je prevalence malformací dvakrát až třikrát vyšší, než je přibližně 3% výskyt malformací v běžné populaci. V léčené populaci byl pozorován nárůst malformací při polyterapii, ale rozsah, za který odpovídá léčba a/nebo základní onemocnění, nebyl objasněn. Přerušení antiepileptické léčby může vést k exacerbaci onemocnění, které může poškodit matku i plod.
Riziko spojené s brivaracetamem
K dispozici je omezené množství údajů o použití brivaracetamu u těhotných žen. Nejsou k dispozici žádné údaje o placentárním transferu u člověka, ale brivaracetam rychle prochází placentou u potkanů (viz bod 5.3). Potenciální riziko u člověka není známo. Studie na zvířatech neprokázaly žádný teratogenní potenciál brivaracetamu (viz bod 5.3).
Brivaracetam byl užíván v klinických studiích jako přídatná terapie a když byl užíván společně s karbamazepinem, vedl v závislosti na dávce k nárůstu koncentrace aktivního metabolitu, epoxidu karbamazepinu (viz bod 4.5). Nejsou k dispozici dostatečné údaje k určení klinického významu tohoto účinku v těhotenství.
Z preventivních důvodů by brivaracetam neměl být užíván během těhotenství, pokud to není klinicky nezbytně nutné (pokud přínos pro matku jasně převáží potenciální riziko pro plod).
Kojení
Není známo, zda se u člověka brivaracetam vylučuje do mateřského mléka. Studie u potkanů vylučování brivaracetamu do mateřského mléka prokázaly (viz bod 5.3). Je nutno učinit rozhodnutí, zda přerušit kojení nebo přerušit podávání brivaracetamu, přičemž je třeba zhodnotit přínos léku pro matku. V případě současného podávání brivaracetamu a epoxidu karbamazepinu by se mohlo vylučování množství epoxidu karbamazepinu do mateřského mléka zvýšit. Není k dispozici dostatek údajů k určení klinického významu.
Fertilita
Nejsou k dispozici žádné údaje o účinku brivaracetamu na fertilitu u člověka. U potkanů nebyl při léčbě brivaracetamem pozorován žádný účinek na fertilitu (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Brivaracetam má zanedbatelný nebo mírný vliv na schopnost řídit nebo obsluhovat stroje.
Vzhledem k možné rozdílné individuální citlivosti mohou někteří pacienti pociťovat somnolenci, závratě nebo jiné příznaky související s centrálním nervovým systémem (CNS). Pacientům je nutno doporučit neřídit vozidlo a neobsluhovat jiné potenciálně nebezpečné stroje, dokud se neseznámí s účinky brivaracetamu na vykonávání těchto aktivit.
4.8 Nežádoucí účinky
Souhrnný bezpečnostní profil
Ve všech kontrolovaných a nekontrolovaných studiích u pacientů s epilepsií dostávalo brivaracetam 2388 subjektů, z nichž 1740 bylo léčeno >6 měsíců, 1363 >12 měsíců, 923 ^24 měsíců a 569 >60 měsíců (5 let).
Nejčastěji hlášené nežádoucí účinky (>10 %) při léčbě brivaracetamem byly somnolence (14,3 %) a závrať (11,0 %). Byly obvykle mírné až střední intenzity. Somnolence a únava (8,2 %) byly hlášeny ve vyšší míře při zvyšující se dávce. Typy nežádoucích účinků hlášených během prvních 7 dnů léčby se podobaly těm, které byly hlášeny během celkové doby léčby.
Frekvence ukončení léčby z důvodu nežádoucích účinků byly 3,5 %, 3,4 % a 4,0 % u pacientů randomizovaných k užívání brivaracetamu v příslušné dávce 50 mg/den, 100 mg/den a 200 mg/den, a 1,7 % pro pacienty randomizované k užívání placeba. Nežádoucím účinkem, který vedl nejčastěji k ukončení léčby brivaracetamem, byly závrať (0,8 %) a konvulze (0,8 %).
Seznam nežádoucích účinků v tabulce
V níže uvedené tabulce jsou nežádoucí účinky, které byly identifikovány na základě přehledu úplné bezpečnostní databáze klinických studií brivaracetamu, uvedeny podle tříd orgánových systémů a
podle frekvence.
Četnosti jsou definovány následovně: velmi časté (>1/10), časté (>1/100 až <1/10), méně časté (>1/1 000 až <1/100). V každé skupině frekvence jsou nežádoucí účinky řazeny za sebou podle klesající závažnosti.
|
Třídy orgánových systémů |
Frekvence |
Nežádoucí účinky z klinických studií |
|
Infekce a infestace |
časté |
chřipka |
|
Poruchy krve a lymfatického systému |
méně časté |
neutropenie |
|
Poruchy metabolismu a výživy |
časté |
snížená chuť k jídlu |
|
Psychiatrické poruchy |
časté |
deprese, anxieta, insomnie, iritabilita |
|
méně časté |
sebevražedné představy, psychotické poruchy, agresivita, agitovanost | |
|
Poruchy nervového systému |
velmi časté |
závrať, somnolence |
|
časté |
konvulze, vertigo | |
|
Respirační, hrudní a mediastinální poruchy |
časté |
infekce horních cest dýchacích, kašel |
|
Gastrointestinální poruchy |
časté | |
|
Celkové poruchy a reakce v místě aplikace |
časté |
Popis vybraných nežádoucích účinků
Neutropenie byla hlášena u 0,5 % (6/1099) pacientů s brivaracetamem a u 0 % (0/459) pacientů s placebem. Čtyři z těchto subjektů měly snížený počet neutrofilů ve výchozím stavu a po zahájení léčby brivaracetamem došlo k dalšímu snížení počtu neutrofilů. Žádný z těchto 6 případů neutropenie nebyl závažný, nevyžadoval speciální léčbu ani nevedl k ukončení léčby brivaracetamem a žádný neměl přidružené infekce.
Sebevražedné představy byly hlášeny u 0,3 % (3/1099) pacientů s brivaracetamem a u 0,7 % (3/459) pacientů s placebem. V krátkodobých klinických studiích brivaracetamu u pacientů s epilepsií nedošlo k žádnému případu dokonané sebevraždy a sebevražedného pokusu, nicméně oboje bylo hlášeno v otevřených prodloužených studiích (viz bod 4.4).
Otevřené prodloužené studie
U pacientů, kteří byli sledováni v otevřených prodloužených studiích po dobu až 8 let, byl bezpečnostní profil podobný profilu pozorovanému v krátkodobých placebem kontrolovaných studiích.
Pediatrická populace
Jsou k dispozici omezené bezpečnostní údaje z otevřených studií u dětí ve věku od 1 měsíce do <16 let. Celkem 152 dětí (ve věku 1 měsíc až <16 let) bylo léčeno brivaracetamem ve farmakokinetické studii a v související navazující studii. Většina zkoušejícím často hlášených nežádoucích účinků spojených s léčbou (TEAE) považovaných za související s lékem byly somnolence (10 %), snížená chuť k jídlu (8 %), únava (5 %) a snížení tělesné hmotnosti (5 %). Bezpečnostní profil se zdá být v souladu s bezpečnostním profilem známým u dospělých pacientů. V současné době nejsou k dispozici žádné klinické údaje u novorozenců.
Starší pacienti:
Ze 130 starších subjektů zahrnutých do fáze 2/3 vývojového programu brivaracetamu (44 s epilepsií) bylo 100 ve věku 65-74 let a 30 ve věku 75-84 let. Bezpečnostní profil u starších pacientů se zdá být podobný bezpečnostnímu profilu pozorovanému u mladších dospělých pacientů.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Příznaky
Je k dispozici omezená klinická zkušenost s předávkováním brivaracetamu u člověka. U zdravých subjektů, které užily jednotlivou dávku 1400 mg brivaracetamu, byla hlášena somnolence a závrať.
Léčba předávkování
Není k dispozici žádné specifické antidotum pro předávkování brivaracetamem. Léčba předávkování zahrnuje obecná podpůrná opatření. Protože se močí vylučuje méně než 10 % brivaracetamu, neočekává se, že hemodialýza významně zvýší clearance brivaracetamu (viz bod 5.2).
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antiepileptika, jiná antiepileptika, ATC kód: N03AX23 Mechanismus účinku
Brivaracetam vykazuje vysokou a selektivní afinitu k 2A proteinu synaptických vezikul (SV2A), transmembránový glykoprotein byl nalezen na presynaptické úrovni v neuronech a v endokrinních buňkách. Ačkoli přesnou roli tohoto proteinu je ještě nutno objasnit, bylo prokázáno, že moduluje exocytózu neurotransmiterů. Má se za to, že vazba na SV2A představuje primární mechanismus protizáchvatové aktivity brivaracetamu.
Klinická účinnost a bezpečnost
Účinnost brivaracetamu při přídatné terapii parciálních záchvatů (partial onset seisures-POS) byla stanovena ve 3 randomizovaných dvojitě zaslepených placebem kontrolovaných multicentrických studiích s pevnou dávkou u subjektů ve věku 16 let a starších. Denní dávka brivaracetamu se v těchto studiích pohybovala v rozsahu 5 až 200 mg/den. Všechny studie začínaly základní periodou trvající 8 týdnů, následovanou 12 týdnů trvající léčebnou periodou bez titrace ve smyslu zvyšování dávky. 1558 pacientů dostávalo lék ze studie, z toho 1099 dostávalo brivaracetam. Kritéria pro zařazení do studie vyžadovala, aby měli pacienti nekontrolované parciální záchvaty navzdory léčbě buď 1, nebo 2 současně podávanými antiepileptiky. Bylo požadováno, aby pacienti prodělali přinejmenším 8 parciálních záchvatů během základní periody. Primárními cílovými parametry u studie fáze 3 bylo procento snížení frekvence POS oproti placebu a poměr respondérů s dosaženou 50% odpovědí založený na 50% snížení frekvence POS od výchozího stavu.
Nejčastěji užívanými antiepileptiky na začátku studie byly karbamazepin (40,6 %), lamotrigin (25,2 %), valproát (20,5 %), oxkarbazepin (16,0 %), topiramát (13,5 %), fenytoin (10,2 %) a levetiracetam (9,8 %). Medián výchozí frekvence záchvatů napříč 3 studiemi byl 9 záchvatů během 28 dní. Pacienti měli epilepsii v průměru přibližně 23 let.
Výsledky účinnosti jsou shrnuty v tabulce 2. Celkově byl brivaracetam účinný při přídatné terapii parciálních záchvatů u pacientů ve věku 16 let a starších v dávce mezi 50 mg/den a 200 mg/den.
|
Studie |
Placebo |
Brivaracetam * statisticky signifikantní (hodnota p) | ||
|
50 mg/den |
100 mg/den |
200 mg/den | ||
|
Studie N01253(1) | ||||
|
n = 96 |
n = 101 | |||
|
Dosažení 50% odpovědi respondéra |
16,73 |
32,7* (p=0,008) | ||
|
Procentuální snížení oproti placebu (%) |
NA |
22,0* (p=0,0040) | ||
|
Studie N01252(1) | ||||
|
n = 100 |
n = 99 |
n = 100 | ||
|
Dosažení 50% odpovědi respondéra |
20,0 |
27,3 (p=0,372) |
36,0(2) (p=0,023) | |
|
Procentuální snížení oproti placebu (%) |
NA |
9,2 (p=0,0274) |
20,5(2) (p=0,0097) | |
|
Studie N01358 | ||||
|
n = 259 |
n = 252 |
n = 249 | ||
|
Dosažení 50% odpovědi respondéra |
21,6 |
38,9 (p<0,001) |
37,8 (p<0,001) | |
|
Procentuální snížení oproti placebu (%) |
NA |
22,8* (p<0,001) |
23,2* (p<0,001) | |
n = randomizovaní pacienti, kteří dostali nejméně 1 dávku léčiva ve studii ~ dávka nebyla studována ‘statisticky významné
(1) Přibližně 20 % pacientů dostávalo současně levetiracetam
(2) Primární výsledek pro N01252 nedosáhl statistické významnosti na základě sekvenčního zkoušení. Dávka 100 mg/den byla nominálně významná.
V klinických studiích bylo vyšší snížení frekvence záchvatů oproti placebu při dávce 100 mg/den než při dávce 50 mg/den. Na rozdíl od na dávce závislého zvýšení výskytu somnolence a únavy měl brivaracetam v dávce 50 mg/den a 100 mg/den podobný bezpečnostní profil včetně nežádoucích účinků se vztahem k CNS a při dlouhodobém užívání.
Obrázek 1 ukazuje procento pacientů (s výjimkou pacientů současně užívajících levetiracetam) podle kategorie snížení frekvence POS během 28 dní od výchozího stavu ve všech 3 studiích. Pacienti s více než 25% zvýšením parciálních záchvatů jsou uvedeni zcela nalevo jako „horší“. Pacienti se zlepšením procentuálního snížení frekvence POS od výchozího stavu jsou uvedeni ve 4 kategoriích napravo. Procento pacientů s nejméně 50% snížením četnosti záchvatu bylo 20,3 %, 34,2 %, 39,5 %, a 37,8 % pro placebo, 50 mg/den, 100 mg/den a 200 mg/den (v tomto pořadí).
Obrázek 1: Podíl pacientů s brivaracetamem a placebem podle kategorie odpovědi záchvatů po dobu 12 týdnů napříč všemi třemi dvojitě zaslepenými pivotními studiemi
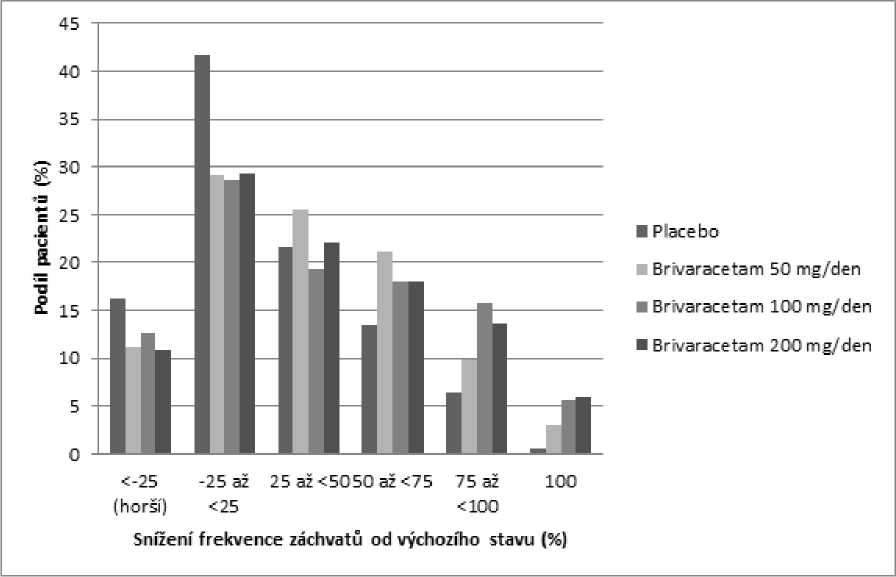
V souhrnné analýze tří pivotních studií nebyly pozorovány žádné rozdíly v účinnosti (měřené jako 50% odpověď respondérů) v rozmezí dávek 50 mg/den až 200 mg/den, když je brivaracetam zkombinován s antiepileptiky vyvolávajícími nebo nevyvolávajícími indukci enzymů.
V klinických studiích dosáhlo stavu bez záchvatů 2,5 % (4/161), 5,1 % (17/332) a 4,0 % (10/249) pacientů s brivaracetamem v dávce odpovídající 50 mg/den, 100 mg/den a 200 mg/den, a to během léčebné periody v trvání 12 týdnů ve srovnání s 0,5 % (2/418) pacientů s placebem.
Zlepšení v mediánu procentuálního snížení frekvence záchvatů od začátku léčby za 28 dní bylo pozorováno u pacientů s typem záchvatů IC (sekundární generalizované tonicko-klonické záchvaty) ve výchozím stavu léčených brivaracetamem, 66,6 % (n=62), 61,2 % (n=100) a 82,1 % (n=75) z pacientů s brivaracetamem v odpovídající dávce 50 mg/den, 100 mg/den a 200 mg/den ve srovnání s placebem 33,3 % (n=115).
Účinnost brivaracetamu v monoterapii nebyla ještě stanovena. Použití brivaracetamu v monoterapii se nedoporučuje.
Léčba levetiracetamem
Ve 2 randomizovaných placebem kontrolovaných studiích fáze 3 byl levetiracetam podáván jako současně podávané antiepileptikum asi u 20 % pacientů. Ačkoli je počet subjektů limitován, nebyl u pacientů, kteří současně užívali levetiracetam, pozorován žádný přínos brivaracetamu oproti placebu, který by reflektoval kompetici na vazebném místě SV2A. Nebyly zjištěny žádné další okolnosti týkající se bezpečnosti a snášenlivosti.
Ve třetí studii předem specifikovaná analýza prokázala účinnost vůči placebu pro dávky 100 mg/den a 200 mg/den u pacientů předtím užívajících levetiracetam. Nižší účinnost pozorovaná u těchto pacientů ve srovnání s pacienty neužívajícími levetiracetam byla pravděpodobně důsledkem vyššího počtu použitých předchozích antiepileptik a vyšší výchozí hodnoty frekvence záchvatů.
Starší pacienti (65 let a starší)
Tři pivotní dvojitě zaslepené placebem kontrolované studie zahrnovaly 38 starších pacientů ve věku mezi 65 a 80 roky. Třebaže jsou údaje omezené, účinnost byla srovnatelná s účinností u mladších subjektů.
Otevřené prodloužené studie
Napříč všemi studiemi bylo zahrnuto do dlouhodobých otevřených prodloužených studií 81,7 % pacientů, kteří dokončili randomizované studie. Od vstupu do randomizovaných studií bylo 5,3 % subjektů s brivaracetamem po dobu 6 měsíců (n=1500) bez záchvatů ve srovnání s 4,6 % a 3,7 % u subjektů exponovaných po dobu odpovídající 12 měsíců (n=1188) a 24 měsíců (n=847). Nicméně protože vysoký podíl subjektů (26 %) přerušil léčbu v otevřených studiích kvůli nedostatečné účinnosti, mohlo dojít ke zkreslení, subjekty, které zůstaly ve studii, reagovaly lépe než ty, které předčasně ukončily.
Pediatrická populace
Účinnost a snášenlivost brivaracetamu nebyla u pediatrických pacientů stanovena (viz bod 4.2). Brivaracetam byl hodnocen u těchto pacientů v krátkodobé otevřené farmakokinetické studii a probíhající otevřené prodloužené studii u 152 subjektů ve věku od 1 měsíce do 16 let (viz bod 5.2).
Evropská agentura pro léčivé přípravky odložila závazek předložit výsledky studií s brivaracetamem u jedné nebo více podskupin pediatrické populace s epilepsií s parciálními záchvaty.
5.2 Farmakokinetické vlastnosti
Brivaracetam potahované tablety, perorální roztok a roztok pro intravenózní injekci vykazují identickou AUC, zatímco maximální plazmatická koncentrace je mírně vyšší po intravenózním podání. Brivaracetam vykazuje lineární a na čase nezávislou farmakokinetiku s nízkou intra- a interindividuální variabilitou a dále úplnou absorpci, nízkou vazbu na proteiny, renální exkreci po rozsáhlé biotransformaci a farmakologicky inaktivní metabolity.
Absorpce
Brivaracetam je rychle a úplně absorbován po perorálním podání a absolutní biologická dostupnost je přibližně 100%. Medián tmax pro tablety užité bez jídla je 1 hodina (rozsah tmax je 0,25 až 3 h).
Současné podávání s jídlem s vysokým obsahem tuku zpomalilo rychlost absorpce (medián tnax 3 h) a snížilo maximální plazmatickou koncentraci (37% pokles) brivaracetamu, přičemž rozsah absorpce zůstal nezměněn.
Distribuce
Brivaracetam se slabě váže (<20 %) na plazmatické proteiny. Distribuční objem je 0,5 l/kg, což je hodnota blízká celkovému množství tělesné vody.
Buněčné membrány jsou pro brivaracetam vysoce permeabilní z důvodu jeho lipofilie (log P). Biotransformace
Brivaracetam je primárně metabolizován hydrolýzou své amidové části za vzniku odpovídající karboxylové kyseliny (přibližně 60% eliminace) a sekundárně hydroxylací propylového vedlejšího řetězce (přibližně 30% eliminace). Hydrolýza amidové části vedoucí ke vzniku metabolitu povahy karboxylové kyseliny (34 % dávky v moči) je podporována jaterní a mimojaterní amidázou. In vitro je hydroxylace brivaracetamu zprostředkována v první řadě CYP2C19. Oba metabolity jsou dále metabolizovány za vzniku běžné hydroxylované kyseliny. In vivo u člověka s neúčinnou mutací CYP2C19 se tvorba hydroxymetabolitu snižuje 10x, zatímco samotný brivaracetam se zvyšuje o 22 % nebo 42 % u jedinců s jednou nebo s oběma mutovanými alelami. Tři metabolity nejsou farmakologicky aktivní.
Eliminace
Brivaracetam je eliminován primárně metabolizací a vylučováním močí. Více než 95 % dávky včetně metabolitů se vylučuje do moči během 72 hodin po požití. Méně než 1 % dávky se vylučuje stolicí a méně než 10 % brivaracetamu se vylučuje beze změny močí. Terminální plazmatický poločas (ti/2) je přibližně 9 hodin. Celková plazmatická clearance byla u pacientů odhadnuta na 3,6 l/hod.
Linearita
Farmakokinetika je úměrná dávce od 10 do nejméně 600 mg.
Interakce s léčivými přípravky
Brivaracetam je eliminován více cestami, včetně vylučování ledvinami, na CYP nezávislou hydrolýzou a CYP zprostředkovanou oxidací. In vitro brivaracetam není u člověka substrátem lidského P-glykoproteinu (P-gp), vícečetná léková rezistence proteinů (MRP - multidrug rezistence proteins) 1 a 2 a pravděpodobně ne polypeptidového transportéru organických aniontů 1B1 (OATP1B1) a OATP1B3.
Testy in vitro ukázaly, že metabolismus brivaracetamu by neměl být významně ovlivněn žádným CYP inhibitorem (např. CYP1A, 2C8, 2C9, 2D6 a 3A4).
Brivaracetam in vitro nebyl inhibitorem CYP1A2, 2A6, 2B6, 2C8, 2C9, 2D6, 3A4 ani transportérů P-gp, BCRP, BSEP MRP2, MATE-K, MATE-1, OATP1B1, OATP1B3, OAT1 a OCT1 v klinicky relevantních koncentracích. In vitro brivaracetam neindukoval CYP1A2.
Farmakokinetika u zvláštních skupin pacientů
Starší pacienti (65 let a starší)
Ve studii u starších pacientů (ve věku 65 až 79 let; clearance kreatininu 53 až 98 ml/min/1,73 m2), kteří užívali brivaracetam v dávce 400 mg/den s podáváním 2x denně, byl plazmatický poločas brivaracetamu 7,9 hodiny u skupiny ve věku 65 až 75 let, a 9,3 hodiny u skupiny >75 let. Plazmatická clearance rovnovážného stavu brivaracetamu byla podobná (0,76 ml/min/kg) jako u mladých zdravých mužů (0,83 ml/min/kg) (viz bod 4.2).
Porucha funkce ledvin
Studie u subjektů s těžkou poruchou funkce ledvin (clearance kreatininu <30 ml/min/1,73 m2 bez nutnosti dialýzy) odhalila, že plazmatická AUC brivaracetamu byla středně zvýšená (+21 %) v poměru ke zdravým subjektům zatímco AUC kyseliny, hydroxymetabolitu a metabolitu hydroxykyseliny byly zvýšeny 3x, 4x, a 21x (v uvedeném pořadí). Renální clearance těchto neaktivních metabolitů byla snížena 10x. Metabolit hydroxykyseliny v neklinických studiích nevyvolal žádné obavy ze strany bezpečnosti. Brivaracetam nebyl studován u pacientů léčených hemodialýzou (viz bod 4.2).
Porucha funkce jater
Farmakokinetická studie u subjektů s cirhózou jater (Child-Pugh třídy A, B, a C) ukázala podobná zvýšení expozice vůči brivaracetamu bez ohledu na závažnost onemocnění (50 %, 57 % a 59 %) v poměru k odpovídajícím zdravým subjektům (viz bod 4.2).
Pediatrická populace
Ve farmakokinetické studii u 99 subjektů ve věku 1 měsíc až <16 let užívajících perorální roztok brivaracetamu bylo prokázáno, že plazmatické koncentrace jsou úměrné dávce ve všech věkových skupinách. Populační farmakokinetické modelování ukázalo, že dávka 2,0 mg/kg dvakrát denně vede ke stejné průměrné plazmatické koncentraci ustáleného stavu jako u dospělých užívajících 100 mg dvakrát denně.
Tělesná hmotnost
Byl odhadnut 40% pokles plazmatické koncentrace ustáleného stavu v rozsahu tělesné hmotnosti od 46 kg do 115 kg. To však není považováno za klinicky významný rozdíl ve farmakokinetice brivaracetamu.
Pohlaví
Nejsou žádné klinicky významné rozdíly ve farmakokinetice brivaracetamu mezi pohlavími.
Rasa
Farmakokinetika brivaracetamu nebyla významně ovlivněna rasou (kavkazská, asijská) při populačním farmakokinetickém modelování u pacientů s epilepsií. Počet pacientů s jiným etnickým pozadím byl omezený.
Farmakokinetické/ farmakodvnamické vztahy
EC50 (plazmatická koncentrace brivaracetamu odpovídající 50 % maximálního účinku) byla odhadnuta na 0,57 mg/l. Tato plazmatická koncentrace je mírně nad mediánem expozice po podávání brivaracetamu v dávkách 50 mg/den. Další snížení frekvence záchvatů se dostavuje při zvýšení dávky na 100 mg/den a dosahuje plató při dávce 200 mg/den.
5.3 Předklinické údaje vztahující se k bezpečnosti
Ve farmakologických studiích bezpečnosti měly převládající účinky souvislost s CNS (zejména přechodná deprese CNS a snížení spontánní lokomoční aktivity) a byly pozorovány při násobcích (vyšších než 50x) farmakologicky aktivní dávky brivaracetamu 2 mg/kg. Učení a funkce paměti nebyly neovlivněny.
Nálezy nepozorované v klinických studiích, ale pozorované v toxikologických studiích s opakovaným podáváním u psů v podobné expozici ke klinické plazmatické AUC, jednalo se o hepatotoxické účinky (zejména porfyrie). Toxikologické údaje shromážděné o brivaracetamu a o strukturálně příbuzných sloučeninách ale ukazují, že jaterní změny se u psů vyvinuly působením mechanismů, které nejsou pro člověka relevantní. Žádné nežádoucí změny jater nebyly pozorovány u potkanů a opic při dlouhodobém podávání brivaracetamu s expozicí jasně převyšující AUC expozici 5 až 42x. CNS příznaky u opic (pády, ztráta rovnováhy, těžkopádné pohyby) se vyskytly u 64násobků klinické Cmax. Tyto účinky byly méně patrné v průběhu času.
Studie genotoxicity neodhalily žádnou mutagenní nebo klastogenní aktivitu. Studie kancerogenity neprokázaly žádný onkogenní potenciál u potkanů, zatímco zvýšený výskyt hepatocelulárních nádorů u myších samců je považován za důsledek negenotoxického fenoménu, známého u hlodavců, jehož mechanismus účinku se vztahuje k indukci jaterních ezymů, podobnou jako po fenobarbitalu.
Brivaracetam neovlivnil fertilitu samic ani samců, neprokázal se žádný teratogenní potenciál u potkanů nebo králíků. Embryotoxicita byla pozorována u králíků při dávce brivaracetamu toxické pro matky s expozicí 8x vyšší než klinická AUC expozice při maximální doporučené dávce.
U potkanů bylo prokázáno, že brivaracetam snadno přestupuje transplacentárně a je vylučován do mateřského mléka u kojících samic potkanů v koncentracích podobným plazmatickým koncentracím u matek.
Brivaracetam u potkanů neprokázal žádný potenciál ke zneužívání.
Studie u nedospělých zvířat
U nedospělých potkanů expoziční hladiny 6-15násobné klinické AUC expozice brivaracetamu při maximální doporučené dávce vyvolávaly vývojové nežádoucí účinky (např. mortalitu, klinické projevy, snížení tělesné hmotnosti a pokles váhy mozku). Neobjevily se žádné nežádoucí účinky u funkcí CNS, žádné neuropatologické a histopatologické vyšetření mozku. U nedospělých psů byly brivaracetamem indukované změny spojené s 6násobným zvýšením hladin AUC, podobným změnám pozorovaným u dospělých zvířat. Nebyly pozorovány žádné nežádoucí účinky ve standardních ukazatelích vývoje nebo maturace.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Jádro tablety
sodná sůl kroskarmelózy
monohydrát laktózy
betadex
laktóza
magnesium-stearát
Potahová soustava
Briviact 10 mg potahované tablety
polyvinylalkohol
oxid titaničitý (E171)
makrogol 3350
mastek.
Briviact 25 mg potahované tablety
polyvinylalkohol
oxid titaničitý (E171)
makrogol 3350
mastek
žlutý oxid železitý (E172) černý oxid železitý (E172).
Briviact 50 mg potahované tablety
polyvinylalkohol
oxid titaničitý (E171)
makrogol 3350
mastek
žlutý oxid železitý (E172) červený oxid železitý (E172).
Briviact 75 mg potahované tablety
polyvinylalkohol
oxid titaničitý (E171)
makrogol 3350
mastek
žlutý oxid železitý (E172) červený oxid železitý (E172) černý oxid železitý (E172).
Briviact 100 mg potahované tablety
polyvinylalkohol
oxid titaničitý (E171)
makrogol 3350
mastek
žlutý oxid železitý (E172) černý oxid železitý (E172).
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
3 roky.
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
6.5 Druh obalu a obsah balení Briviact 10 mg potahované tablety
• Balení 14, 56 potahovaných tablet a multipack obsahující 168 (3 balení po 56) potahovaných tablet v PVC/PCTFE/Al blistrech
• Balení 14 x 1 a 100 x1 potahovaná tableta v PVC/PCTFE/Al blistrech Briviact 25 mg potahované tablety
• Balení 14, 56 potahovaných tablet a multipack obsahující 168 (3 balení po 56) potahovaných tablet v PVC/PCTFE/Al blistrech
• Balení 14 x 1 a 100 x1 potahovaná tableta v PVC/PCTFE/Al blistrech Briviact 50 mg potahované tablety
• Balení 14, 56 potahovaných tablet a multipack obsahující 168 (3 balení po 56) potahovaných tablet v PVC/PCTFE/Al blistrech
• Balení 14 x 1 a 100 x1 potahovaná tableta v PVC/PCTFE/Al blistrech Briviact 75 mg potahované tablety
• Balení 14, 56 potahovaných tablet a multipack obsahující 168 (3 balení po 56) potahovaných tablet v PVC/PCTFE/Al blistrech
• Balení 14 x 1 a 100 x1 potahovaná tableta v PVC/PCTFE/Al blistrech Briviact 100 mg potahované tablety
• Balení 14, 56 potahovaných tablet a multipack obsahující 168 (3 balení po 56) potahovaných tablet v PVC/PCTFE/Al blistrech
• Balení 14 x 1 a 100 x1 potahovaná tableta v PVC/PCTFE/Al blistrech Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku
Žádné zvláštní požadavky.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
UCB Pharma S.A.
Allée de la Recherche 60 B-1070 Brusel Belgie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/15/1073/001
EU/1/15/1073/002
EU/1/15/1073/003
EU/1/15/1073/004
EU/1/15/1073/005
EU/1/15/1073/006
EU/1/15/1073/007
EU/1/15/1073/008
EU/1/15/1073/009
EU/1/15/1073/010
EU/1/15/1073/011
EU/1/15/1073/012
EU/1/15/1073/013
EU/1/15/1073/014
EU/1/15/1073/015
EU/1/15/1073/016
EU/1/15/1073/017
EU/1/15/1073/018
EU/1/15/1073/019
EU/1/15/1073/020
EU/1/15/1073/023
EU/1/15/1073/024
EU/1/15/1073/025
EU/1/15/1073/026
EU/1/15/1073/027
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 14. ledna 2016
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu
▼ Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakékoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Briviact 10 mg/ml perorální roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden ml obsahuje brivaracetamum 10 mg.
Pomocné látky se známým účinkem:
Jeden ml perorálního roztoku obsahuje 239,8 mg sorbitolu (E420), 1 mg methylparabenu (E218) a 1,16 mg sodíku.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Perorální roztok
Mírně viskózní, čirá, bezbarvá až nažloutlá tekutina.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Briviact je indikován jako přídatná terapie při léčbě parciálních záchvatů s nebo bez sekundární generalizace u dospělých a dospívajících pacientů s epilepsií ve věku od 16 let.
4.2 Dávkování a způsob podání
Dávkování
Doporučená počáteční dávka je buď 50 mg nebo 100 mg/den, potřebná ke snížení počtu záchvatů na základě posouzení lékaře oproti potenciálním nežádoucím účinkům. Dávka se podává ve dvou stejných rozdělených dávkách, jednou ráno a jednou večer. Na základě individuální odpovědi pacienta a snášenlivosti lze dávku upravit v dávkovém rozmezí 50 mg/den až 200 mg/den.
Opomenutá dávka
Jestliže pacienti opomenou užít jednu nebo více dávek, doporučuje se užít jednu dávku, jakmile si vzpomenou, a následující dávku vzít v obvyklé době ráno nebo večer. To může zabránit poklesu plazmatické koncentrace brivaracetamu pod účinnou hladinu, a tím znovupropuknutí záchvatů.
Ukončení léčby
Pokud je nutno podávání brivaracetamu ukončit, doporučuje se postupné snižování o 50 mg/den v týdenním intervalu. Po týdnu léčby dávkou 50 mg/den se doporučuje v posledním týdnu léčby dávka 20 mg/den.
Zvláštní populace
Starší pacienti (65 let a starší)
Není nutná žádná úprava dávky u starších pacientů (viz bod 5.2).
Klinická zkušenost u pacientů >65 let je omezená.
Porucha funkce ledvin
U pacientů s poruchou funkce ledvin není třeba dávku nijak upravovat (viz bod 5.2). Podávání brivaracetamu se nedoporučuje u pacientů v konečné fázi onemocnění ledvin, kteří jsou léčeni dialýzou, vzhledem k nedostatku údajů.
Porucha funkce jater
Expozice vůči brivaracetamu byla zvýšená u pacientů s chronickým onemocněním jater. Počáteční dávka 50 mg/den by měla být zvážena. Ve všech fázích poruchy funkce jater se doporučuje maximální denní dávka 150 mg podávaná ve 2 rozdělených dávkách (viz body 4.4 a 5.2).
Pediatrická populace
Bezpečnost a účinnost brivaracetamu u dětí ve věku méně než 16 let nebyly dosud stanoveny.
V současné době dostupné údaje jsou uvedeny v bodech 4.8, 5.1, a 5.2, ale na jejich základě nelze učinit žádná doporučení ohledně dávkování.
Způsob podání
Perorální roztok brivaracetamu lze před spolyknutímím naředit vodou nebo džusem a může se užívat současně s jídlem nebo bez jídla (viz bod 5.2). Perorální roztok brivaracetamu lze podávat nazogastrickou nebo gastrostomickou sondou.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku, jiné deriváty pyrrolidonu nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Sebevražedné myšlenky a chování
Sebevražedné myšlenky a chování byly hlášeny v několika indikacích u pacientů léčených antiepileptiky, včetně brivaracetamu. Metaanalýza randomizovaných placebem kontrolovaných studií s antiepileptiky prokázala mírně zvýšené riziko sebevražedných myšlenek a chování. Mechanismus vzniku není znám a u brivaracetamu dostupné údaje nevylučují zvýšené riziko.
Proto by u pacientů měly být sledovány příznaky sebevražedných představ a chování a zvážena vhodná léčba. Pacienti (a osoby poskytující pacientům péči) by měli být upozorněni na to, že v případě výskytu příznaků sebevražedného myšlení či chování, by měli vyhledat lékařskou pomoc.
Porucha funkce jater
Jsou k dispozici omezené klinické údaje o použití brivaracetamu u pacientů s již existující poruchou funkce jater. U pacientů s poruchou funkce jater se doporučuje úprava dávky (viz bod 4.2).
Pomocné látky
Obsah sodíku
Perorální roztok brivaracetamu obsahuje sodík. Nutno vzít v úvahu u pacientů na dietě s nízkým obsahem sodíku.
Intolerance fruktózy
Perorální roztok obsahuje sorbitol (E420). Pacienti se vzácnými dědičnými problémy s intolerancí fruktózy by tento přípravek neměli užívat.
Pomocné látky, které mohou způsobit intoleranci
Perorální roztok obsahuje methylparaben (E218), který může způsobit alergické reakce (pravděpodobně opožděné).
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Formální studie interakce byly provedeny jen u dospělých pacientů.
Farmakodvnamické interakce
Současná léčba s levetiracetamem
V klinických studiích (ačkoli počet je velmi omezený) nebyl pozorován žádný přínos brivaracetamu versus placebo u pacientů současně užívajících levetiracetam. Nebyly doloženy žádné další údaje ohledně bezpečnosti a tolerability (viz bod 5.1).
Interakce s alkoholem
Ve studii farmakokinetické a farmakodynamické interakce mezi brivaracetamem v jednorázové dávce 200 mg a ethanolem v kontinuální infuzi 0,6 g/l u zdravých subjektů nenastala žádná farmakokinetická interakce, ale brivaracetam přibližně zdvojnásobil účinky alkoholu na psychomotorické funkce, pozornost a paměť. Podávání brivaracetamu s alkoholem se neporučuje.
Farmakokinetické interakce
Účinky jiných látek na farmakokinatiku brivaracetamu
In vitro údaje naznačují, že brivaracetam má nízký interakční potenciál. Hlavní metabolickou cestou brivaracetamu je na CYP nezávislá hydrolýza. Druhá cesta zahrnuje hydroxylaci, která je zprostředkována CYP2C19 (viz bod 5.2).
Plazmatické koncentrace brivaracetamu se mohou zvýšit, je-li současně podáván se silnými inhibitory CYP2C19 (jako flukonazol, fluvoxamin), ale riziko klinicky významné interakce zprostředkované CYP2C19 je považováno za nízké.
Rifampicin
U zdravých dobrovolníků současné podávání silného induktoru enzymů rifampicinu (600 mg/den po dobu 5 dnů) snížilo plochu brivaracetamu pod křivkou (AUC) o 45 %. Předepisující lékaři musí zvážit úpravu dávky brivaracetamu u pacientů, u kterých se zahajuje nebo ukončuje léčba rifampicinem.
Antiepileptika se silnou indukcí enzymů
Plazmatické koncentrace brivaracetamu klesají při současném podávání s antiepileptiky silně indukující enzymy (karbamazepin, fenobarbital, fenytoin), ale není nutná žádná úprava dávky (viz tabulka 1).
Jiné induktory enzymů
Očekává se, že jiné silné induktory enzymů (jako třezalka tečkovaná (Hypericum perforatum)) mohou také snížit systémovou expozici brivaracetamu. Proto zahájení nebo ukončení léčby třezalkou tečkovanou má být provedeno s opatrností.
Účinek brivaracetamu na jiné léčivé přípravky
Brivaracetam podávaný v dávkách 50 mg nebo 150 mg/den neovlivňoval AUC midazolamu (metabolizován CYP3A4). Riziko klinicky relevantních CYP3A4 interakcí je považováno za nízké.
Studie in vitro prokázaly, že brivaracetam vykazuje malou nebo žádnou inhibici izoforem CYP450 s výjimkou CYP2C19. Brivaracetam může zvyšovat plazmatické koncentrace léčivých přípravků metabolizovaných CYP2C19 (např. lanzoprazol, omeprazol, diazepam). Při zkoušení in vitro brivaracetam neindukoval CYP1A1/2, ale indukoval CYP3A4 a CYP2B6. Žádná CYP3A4 indukce nebyla nalezena in vivo (viz midazolam výše). CYP2B6 indukce nebyla zkoumána in vivo a brivaracetam může snižovat plazmatické koncentrace léčivých přípravků přípravků metabolizovaných CYP2B6 (např. efavirenz). In vitro interakční studie k určení potenciálních inhibičních účinků na transportéry vedly k závěru, že nedochází k žádným klinicky významným účinkům s výjimkou pro OAT3. In vitro brivaracetam inhibuje OAT3 s poloviční maximální inhibiční koncentrací 42x vyšší, než je Cmax při nejvyšší klinické dávce. Brivaracetam v dávce 200 mg/den může zvýšit plazmatické koncentrace léčivých přípravků transportovaných OAT3.
Antiepileptika
Potenciální interakce mezi brivaracetamem (50 mg/den až 200 mg/den) a jinými antiepileptiky byly zkoumány v souhrnné analýze plazmatických lékových koncentrací ze všech studií fáze 2-3 a v populační farmakokinetické analýze v placebem kontrolovaných studiích fáze 2-3 a ve vyhrazených studiích lékových interakcí (pro následující antiepileptika: karbamazepin, lamotrigin, fenytoin a topiramát). Účinek vzájemných interakcí na plazmatickou koncentraci je shrnut v tabulce 1 (zvýšení je označeno jako “t” a snížení jako “j” plocha pod křivkou plazmatické koncentrace v průběhu času jako „AUC“, maximální pozorovaná koncentrace jako „Cmax ).
Tabulka 1: Farmakokinetické interakce mezi brivaracetamem a^jinými antiepileptiky
|
Současně podávané antiepiletikum |
Vliv antiepiletika na koncentraci brivaracetamu v plazmě |
Vliv brivaracetamu na koncentraci antiepiletika v plazmě |
|
Karbamazepin |
AUC 29 % j Cmax 13 % j není nutná žádná úprava dávky |
karbamazepin - žádný epoxid karbamazepinu t (viz níže) není nutná žádná úprava dávky |
|
Klobazam |
žádné údaje |
žádný |
|
Klonazepam |
žádné údaje |
žádný |
|
Lacosamid |
žádné údaje |
žádný |
|
Lamotrigin |
žádný |
žádný |
|
Levetiracetam |
žádný |
žádný |
|
Oxkarbazepin |
žádný |
žádný (monohydroxyderivát, MHD) |
|
Fenobarbital |
AUC 19 % j není nutná žádná úprava dávky |
žádný |
|
Fenytoin |
AUC 21 % j není nutná žádná úprava dávky |
žádný aAUC 20 % t aCmax 20 % t |
|
Pregabalin |
žádné údaje |
žádný |
|
Topiramát |
žádný |
žádný |
|
Kyselina valproová |
žádný |
žádný |
|
Zonisamid |
žádné údaje |
žádný |
a na základě studie týkající se podávání supraterapeutické dávky brivaracetamu 400 mg/den
Karbamazepin
Brivaracetam je středně účinný reverzibilní inhibitor epoxidové hydrolázy vyvolávající zvýšenou koncentraci epoxidu karbamazepinu, aktivního metabolitu karbamazepinu. V kontrolovaných studiích vzrostla plazmatická koncentrace epoxidu karbamazepinu v průměru o 37 %, 62 % a 98 % s malou variabilitou při dávkách brivaracetamu odpovídajících 50 mg/den, 100 mg/den a 200 mg/den. Nebyla pozorována žádná bezpečnostní rizika. Nebyl žádný aditivní účinek brivaracetamu a valproátu na AUC u epoxidu karbamazepinu.
Perorální kontraceptiva
Současné podávání brivaracetamu (100 mg/den) spolu s perorálním kontraceptivem obsahujícím ethinylestradiol (0,03 mg) a levonorgestrel (0,15 mg) neovlivňovalo farmakokinetiku žádné látky. Když byl brivaracetam současně podáván v dávce 400 mg/den (dvojnásobek doporučené maximální denní dávky) současně s perorálním kontraceptivem obsahujícím ethinylestradiol (0,03 mg) a levonorgestrel (0,15 mg), bylo pozorováno snížení AUC estrogenu o 27 % a snížení AUC progestinu o 23 %, a to bez dopadu na potlačení ovulace. Obecně nenastala žádná změna v profilech koncentrací v čase u endogenních markerů estradiolu, progesteronu, luteinizačního hormonu (LH), folikuly stimulujícího hormonu a globulinu vázajícího pohlavní hormony (SHBG).
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku
Lékaři by měli prodiskutovat plánované rodičovství a antikoncepci se ženami ve fertilním věku, které užívají brivaracetam (viz Těhotenství).
Jestliže se žena rozhodne otěhotnět, užívání brivaracetamu by mělo být opět přehodnoceno. Těhotenství
Riziko spojené s epilepsií a s antiepileptiky obecně
U všech antiepileptik bylo prokázáno, že mezi potomky léčených žen s epilepsií je prevalence malformací dvakrát až třikrát vyšší, než je přibližně 3% výskyt malformací v běžné populaci. V léčené populaci byl pozorován nárůst malformací při polyterapii, ale rozsah, za který odpovídá léčba a/nebo základní onemocnění, nebyl objasněn. Přerušení antiepileptické léčby může vést k exacerbaci onemocnění, které může poškodit matku i plod.
Riziko spojené s brivaracetamem
K dispozici je omezené množství údajů o použití brivaracetamu u těhotných žen. Nejsou k dispozici žádné údaje o placentárním transferu u člověka, ale brivaracetam rychle prochází placentou u potkanů (viz bod 5.3). Potenciální riziko u člověka není známo. Studie na zvířatech neprokázaly žádný teratogenní potenciál brivaracetamu (viz bod 5.3).
Brivaracetam byl užíván v klinických studiích jako přídatná terapie a když byl užíván společně s karbamazepinem, vedl v závislosti na dávce k nárůstu koncentrace aktivního metabolitu, epoxidu karbamazepinu (viz bod 4.5). Nejsou k dispozici dostatečné údaje k určení klinického významu tohoto účinku v těhotenství.
Z preventivních důvodů by brivaracetam neměl být užíván během těhotenství, pokud to není klinicky nezbytně nutné (pokud přínos pro matku jasně převáží potenciální riziko pro plod).
Kojení
Není známo, zda se u člověka brivaracetam vylučuje do mateřského mléka. Studie u potkanů vylučování brivaracetamu do mateřského mléka prokázaly (viz bod 5.3). Je nutno učinit rozhodnutí, zda přerušit kojení nebo přerušit podávání brivaracetamu, přičemž je třeba zhodnotit přínos léku pro matku. V případě současného podávání brivaracetamu a epoxidu karbamazepinu by se mohlo vylučování množství epoxidu karbamazepinu do mateřského mléka zvýšit. Není k dispozici dostatek údajů k určení klinického významu.
Fertilita
Nejsou k dispozici žádné údaje o účinku brivaracetamu na fertilitu u člověka. U potkanů nebyl při léčbě brivaracetamem pozorován žádný účinek na fertilitu (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Brivaracetam má zanedbatelný nebo mírný vliv na schopnost řídit nebo obsluhovat stroje.
Vzhledem k možné rozdílné individuální citlivosti mohou někteří pacienti pociťovat somnolenci, závratě nebo jiné příznaky související s centrálním nervovým systémem (CNS). Pacientům je nutno doporučit neřídit vozidlo a neobsluhovat jiné potenciálně nebezpečné stroje, dokud se neseznámí s účinky brivaracetamu na vykonávání těchto aktivit.
4.8 Nežádoucí účinky
Souhrnný bezpečnostní profil
Ve všech kontrolovaných a nekontrolovaných studiích u pacientů s epilepsií dostávalo brivaracetam 2388 subjektů, z nichž 1740 bylo léčeno >6 měsíců, 1363 >12 měsíců, 923 ^24 měsíců a 569 >60 měsíců (5 let).
Nejčastěji hlášené nežádoucí účinky (>10 %) při léčbě brivaracetamem byly somnolence (14,3 %) a závrať (11,0 %). Byly obvykle mírné až střední intenzity. Somnolence a únava (8,2 %) byly hlášeny ve vyšší míře při zvyšující se dávce. Typy nežádoucích účinků hlášených během prvních 7 dnů léčby se podobaly těm, které byly hlášeny během celkové doby léčby.
Frekvence ukončení léčby z důvodu nežádoucích účinků byly 3,5 %, 3,4 % a 4,0 % u pacientů randomizovaných k užívání brivaracetamu v příslušné dávce 50 mg/den, 100 mg/den a 200 mg/den, a 1,7 % pro pacienty randomizované k užívání placeba. Nežádoucím účinkem, který vedl nejčastěji k ukončení léčby brivaracetamem, byly závrať (0,8 %) a konvulze (0,8 %).
Seznam nežádoucích účinků v tabulce
V níže uvedené tabulce jsou nežádoucí účinky, které byly identifikovány na základě přehledu úplné bezpečnostní databáze klinických studií brivaracetamu, uvedeny podle tříd orgánových systémů a podle frekvence.
Četnosti jsou definovány následovně: velmi časté (>1/10), časté (>1/100 až <1/10), méně časté (>1/1 000 až <1/100). V každé skupině frekvence jsou nežádoucí účinky řazeny za sebou podle klesající závažnosti.
|
Třídy orgánových systémů |
Frekvence |
Nežádoucí účinky z klinických studií |
|
Infekce a infestace |
časté |
chřipka |
|
Poruchy krve a lymfatického systému |
méně časté |
neutropenie |
|
Poruchy metabolismu a výživy |
časté |
snížená chuť k jídlu |
|
Psychiatrické poruchy |
časté |
deprese, anxieta, insomnie, iritabilita |
|
méně časté |
sebevražedné představy, psychotické poruchy, agresivita, agitovanost | |
|
Poruchy nervového systému |
velmi časté |
závrať, somnolence |
|
časté |
konvulze, vertigo | |
|
Respirační, hrudní a mediastinální poruchy |
časté |
infekce horních cest dýchacích, kašel |
|
Gastrointestinální poruchy |
časté | |
|
Celkové poruchy a reakce v místě aplikace |
časté |
Popis vybraných nežádoucích účinků
Neutropenie byla hlášena u 0,5 % (6/1099) pacientů s brivaracetamem a u 0 % (0/459) pacientů s placebem. Čtyři z těchto subjektů měly snížený počet neutrofilů ve výchozím stavu a po zahájení léčby brivaracetamem došlo k dalšímu snížení počtu neutrofilů. Žádný z těchto 6 případů neutropenie nebyl závažný, nevyžadoval speciální léčbu ani nevedl k ukončení léčby brivaracetamem a žádný neměl přidružené infekce.
Sebevražedné představy byly hlášeny u 0,3 % (3/1099) pacientů s brivaracetamem a u 0,7 % (3/459) pacientů s placebem. V krátkodobých klinických studiích brivaracetamu u pacientů s epilepsií nedošlo k žádnému případu dokonané sebevraždy a sebevražedného pokusu, nicméně oboje bylo hlášeno v otevřených prodloužených studiích (viz bod 4.4).
Otevřené prodloužené studie
U pacientů, kteří byli sledováni v otevřených prodloužených studiích po dobu až 8 let, byl bezpečnostní profil podobný profilu pozorovanému v krátkodobých placebem kontrolovaných studiích.
Pediatrická populace
Jsou k dispozici omezené bezpečnostní údaje z otevřených studií u dětí ve věku od 1 měsíce do <16 let. Celkem 152 dětí (ve věku 1 měsíc až <16 let) bylo léčeno brivaracetamem ve farmakokinetické studii a v související navazující studii. Většina zkoušejícím často hlášených nežádoucích účinků spojených s léčbou (TEAE) považovaných za související s lékem byly somnolence (10 %), snížená chuť k jídlu (8 %), únava (5 %) a snížení tělesné hmotnosti (5 %). Bezpečnostní profil se zdá být v souladu s bezpečnostním profilem známým u dospělých pacientů.
V současné době nejsou k dispozici žádné klinické údaje u novorozenců.
Starší pacienti:
Ze 130 starších subjektů zahrnutých do fáze 2/3 vývojového programu brivaracetamu (44 s epilepsií) bylo 100 ve věku 65-74 let a 30 ve věku 75-84 let. Bezpečnostní profil u starších pacientů se zdá být podobný bezpečnostnímu profilu pozorovanému u mladších dospělých pacientů.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Příznaky
Je k dispozici omezená klinická zkušenost s předávkováním brivaracetamu u člověka. U zdravých subjektů, které užily jednotlivou dávku 1400 mg brivaracetamu, byla hlášena somnolence a závrať.
Léčba předávkování
Není k dispozici žádné specifické antidotum pro předávkování brivaracetamem. Léčba předávkování zahrnuje obecná podpůrná opatření. Protože se močí vylučuje méně než 10 % brivaracetamu, neočekává se, že hemodialýza významně zvýší clearance brivaracetamu (viz bod 5.2).
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antiepileptika, jiná antiepileptika, ATC kód: N03AX23 Mechanismus účinku
Brivaracetam vykazuje vysokou a selektivní afinitu k 2A proteinu synaptických vezikul (SV2A), transmembránový glykoprotein byl nalezen na presynaptické úrovni v neuronech a v endokrinních buňkách. Ačkoli přesnou roli tohoto proteinu je ještě nutno objasnit, bylo prokázáno, že moduluje exocytózu neurotransmiterů. Má se za to, že vazba na SV2A představuje primární mechanismus protizáchvatové aktivity brivaracetamu.
Klinická účinnost a bezpečnost
Účinnost brivaracetamu při přídatné terapii parciálních záchvatů (partial onset seisures-POS) byla stanovena ve 3 randomizovaných dvojitě zaslepených placebem kontrolovaných multicentrických studiích s pevnou dávkou u subjektů ve věku 16 let a starších. Denní dávka brivaracetamu se v těchto studiích pohybovala v rozsahu 5 až 200 mg/den. Všechny studie začínaly základní periodou trvající 8 týdnů, následovanou 12 týdnů trvající léčebnou periodou bez titrace ve smyslu zvyšování dávky. 1558 pacientů dostávalo lék ze studie, z toho 1099 dostávalo brivaracetam. Kritéria pro zařazení do studie vyžadovala, aby měli pacienti nekontrolované parciální záchvaty navzdory léčbě buď 1, nebo 2 současně podávanými antiepileptiky. Bylo požadováno, aby pacienti prodělali přinejmenším 8 parciálních záchvatů během základní periody. Primárními cílovými parametry u studie fáze 3 bylo procento snížení frekvence POS oproti placebu a poměr respondérů s dosaženou 50% odpovědí založený na 50% snížení frekvence POS od výchozího stavu.
Nejčastěji užívanými antiepileptiky na začátku studie byly karbamazepin (40,6 %), lamotrigin (25,2 %), valproát (20,5 %), oxkarbazepin (16,0 %), topiramát (13,5 %), fenytoin (10,2 %) a levetiracetam (9,8 %). Medián výchozí frekvence záchvatů napříč 3 studiemi byl 9 záchvatů během
28 dní. Pacienti měli epilepsii v průměru přibližně 23 let.
Výsledky účinnosti jsou shrnuty v tabulce 2. Celkově byl brivaracetam účinný při přídatné terapii parciálních záchvatů u pacientů ve věku 16 let a starších v dávce mezi 50 mg/den a 200 mg/den.
Tabulka 2: Výsledky účinnosti pro ^ frekvenci parciálních záchvatů během 28 dní
|
Studie |
Placebo |
Brivaracetam * statisticky signifikantní (hodnota p) | ||
|
50 mg/den |
100 mg/den |
200 mg/den | ||
|
Studie N01253(1) | ||||
|
n = 96 |
n = 101 | |||
|
Dosažení 50% odpovědi respondéra |
16,7 |
32,7* (p=0,008) |
~ |
~ |
|
Procentuální snížení oproti placebu (%) |
NA |
22,0* (p=0,0040) |
~ |
~ |
|
Studie N01252(1) | ||||
|
n = 100 |
n = 99 |
n = 100 | ||
|
Dosažení 50% odpovědi respondéra |
20,0 |
27,3 (p=0,372) |
36,0(2) (p=0,023) |
~ |
|
Procentuální snížení oproti placebu (%) |
NA |
9,2 (p=0,0274) |
20,5(2) (p=0,0097) |
~ |
|
Studie N01358 | ||||
|
n = 259 |
n = 99 |
n = 252 |
n = 249 | |
|
Dosažení 50% odpovědi respondéra |
21,6 |
~ |
38,9 (p<0,001) |
37,8 (p<0,001) |
|
Procentuální snížení oproti placebu (%) |
NA |
~ |
22,8* (p<0,001) |
23,2* (p<0,001) |
|
n = randomizovaní pacienti, kteří dostali nejméně 1 dávku léčiva ve stuc |
ii | |||
~ dávka nebyla studována *statisticky významné
(1) Přibližně 20 % pacientů dostávalo současně levetiracetam
(2) Primární výsledek pro N01252 nedosáhl statistické významnosti na základě sekvenčního zkoušení. Dávka 100 mg/den byla nominálně významná.
V klinických studiích bylo vyšší snížení frekvence záchvatů oproti placebu při dávce 100 mg/den než při dávce 50 mg/den. Na rozdíl od na dávce závislého zvýšení výskytu somnolence a únavy měl brivaracetam v dávce 50 mg/den a 100 mg/den podobný bezpečnostní profil včetně nežádoucích účinků se vztahem k CNS a při dlouhodobém užívání.
Obrázek 1 ukazuje procento pacientů (s výjimkou pacientů současně užívajících levetiracetam) podle kategorie snížení frekvence POS během 28 dní od výchozího stavu ve všech 3 studiích. Pacienti s více než 25% zvýšením parciálních záchvatů jsou uvedeni zcela nalevo jako „horší“. Pacienti se zlepšením procentuálního snížení frekvence POS od výchozího stavu jsou uvedeni ve 4 kategoriích napravo. Procento pacientů s nejméně 50% snížením četnosti záchvatu bylo 20,3 %, 34,2 %, 39,5 %, a 37,8 % pro placebo, 50 mg/den, 100 mg/den a 200 mg/den (v tomto pořadí).
Obrázek 1: Podíl pacientů s brivaracetamem a placebem podle kategorie odpovědi záchvatů po dobu 12 týdnů napříč všemi třemi dvojitě zaslepenými pivotními studiemi
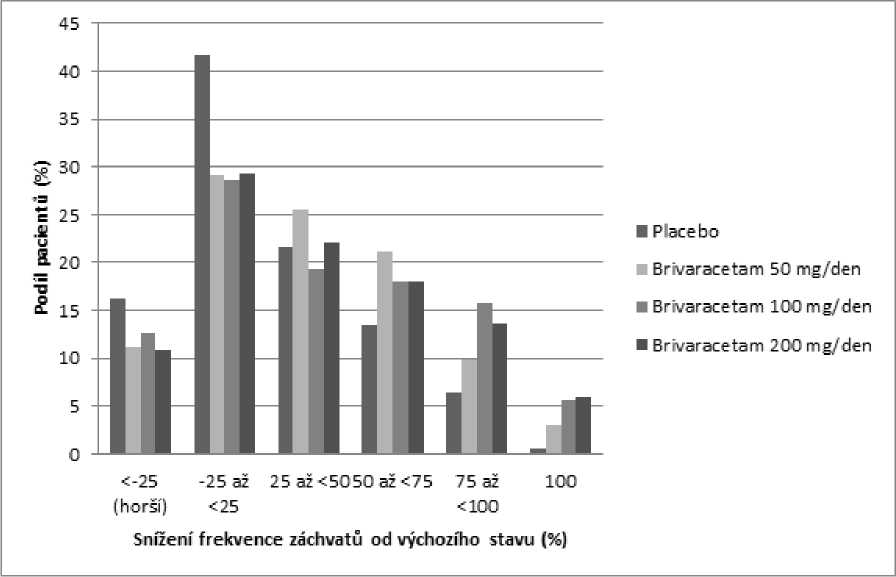
V souhrnné analýze tří pivotních studií nebyly pozorovány žádné rozdíly v účinnosti (měřené jako 50% odpověď respondérů) v rozmezí dávek 50 mg/den až 200 mg/den, když je brivaracetam zkombinován s antiepileptiky vyvolávajícími nebo nevyvolávajícími indukci enzymů.
V klinických studiích dosáhlo stavu bez záchvatů 2,5 % (4/161), 5,1 % (17/332) a 4,0 % (10/249) pacientů s brivaracetamem v dávce odpovídající 50 mg/den, 100 mg/den a 200 mg/den, a to během léčebné periody v trvání 12 týdnů ve srovnání s 0,5 % (2/418) pacientů s placebem.
Zlepšení v mediánu procentuálního snížení frekvence záchvatů od začátku léčby za 28 dní bylo pozorováno u pacientů s typem záchvatů IC (sekundární generalizované tonicko-klonické záchvaty) ve výchozím stavu léčených brivaracetamem, 66,6 % (n=62), 61,2 % (n=100) a 82,1 % (n=75) z pacientů s brivaracetamem v odpovídající dávce 50 mg/den, 100 mg/den a 200 mg/den ve srovnání s placebem 33,3 % (n=115).
Účinnost brivaracetamu v monoterapii nebyla ještě stanovena. Použití brivaracetamu v monoterapii se nedoporučuje.
Léčba levetiracetamem
Ve 2 randomizovaných placebem kontrolovaných studiích fáze 3 byl levetiracetam podáván jako současně podávané antiepileptikum asi u 20 % pacientů. Ačkoli je počet subjektů limitován, nebyl u pacientů, kteří současně užívali levetiracetam, pozorován žádný přínos brivaracetamu oproti placebu, který by reflektoval kompetici na vazebném místě SV2A. Nebyly zjištěny žádné další okolnosti týkající se bezpečnosti a snášenlivosti.
Ve třetí studii předem specifikovaná analýza prokázala účinnost vůči placebu pro dávky 100 mg/den a 200 mg/den u pacientů předtím užívajících levetiracetam. Nižší účinnost pozorovaná u těchto pacientů ve srovnání s pacienty neužívajícími levetiracetam byla pravděpodobně důsledkem vyššího počtu použitých předchozích antiepileptik a vyšší výchozí hodnoty frekvence záchvatů.
Starší pacienti (65 let a starší)
Tři pivotní dvojitě zaslepené placebem kontrolované studie zahrnovaly 38 starších pacientů ve věku mezi 65 a 80 roky. Třebaže jsou údaje omezené, účinnost byla srovnatelná s účinností u mladších subjektů.
Otevřené prodloužené studie
Napříč všemi studiemi bylo zahrnuto do dlouhodobých otevřených prodloužených studií 81,7 % pacientů, kteří dokončili randomizované studie. Od vstupu do randomizovaných studií bylo 5,3 % subjektů s brivaracetamem po dobu 6 měsíců (n=1500) bez záchvatů ve srovnání s 4,6 % a 3,7 % u subjektů exponovaných po dobu odpovídající 12 měsíců (n=1188) a 24 měsíců (n=847). Nicméně protože vysoký podíl subjektů (26 %) ukončil léčbu v otevřených studiích kvůli nedostatečné účinnosti, mohlo dojít ke zkreslení, subjekty, které zůstaly ve studii, reagovaly lépe než ty, které předčasně ukončily.
Pediatrická populace
Účinnost a snášenlivost brivaracetamu nebyla u pediatrických pacientů stanovena (viz bod 4.2). Brivaracetam byl hodnocen u těchto pacientů v krátkodobé otevřené farmakokinetické studii a probíhající otevřené prodloužené studii u 152 subjektů ve věku od 1 měsíce do 16 let (viz bod 5.2).
Evropská agentura pro léčivé přípravky odložila závazek předložit výsledky studií s brivaracetamem u jedné nebo více podskupin pediatrické populace s epilepsií s parciálními záchvaty.
5.2 Farmakokinetické vlastnosti
Brivaracetam potahované tablety, perorální roztok a roztok pro intravenózní injekci vykazují identickou AUC, zatímco maximální plazmatická koncentrace je mírně vyšší po intravenózním podání. Brivaracetam vykazuje lineární a na čase nezávislou farmakokinetiku s nízkou intra- a interindividuální variabilitou a dále úplnou absorpci, nízkou vazbu na proteiny, renální exkreci po rozsáhlé biotransformaci a farmakologicky inaktivní metabolity.
Absorpce
Brivaracetam je rychle a úplně absorbován po perorálním podání a absolutní biologická dostupnost je přibližně 100%. Medián tmax pro tablety užité bez jídla je 1 hodina (rozsah tmax je 0,25 až 3 h).
Současné podávání s jídlem s vysokým obsahem tuku zpomalilo rychlost absorpce (medián knax 3 h) a snížilo maximální plazmatickou koncentraci (37% pokles) brivaracetamu, přičemž rozsah absorpce zůstal nezměněn.
Distribuce
Brivaracetam se slabě váže (<20 %) na plazmatické proteiny. Distribuční objem je 0,5 l/kg, což je hodnota blízká celkovému množství tělesné vody.
Buněčné membrány jsou pro brivaracetam vysoce permeabilní z důvodu jeho lipofilie (log P). Biotransformace
Brivaracetam je primárně metabolizován hydrolýzou své amidové části za vzniku odpovídající karboxylové kyseliny (přibližně 60% eliminace) a sekundárně hydroxylací propylového vedlejšího řetězce (přibližně 30% eliminace). Hydrolýza amidové části vedoucí ke vzniku metabolitu povahy karboxylové kyseliny (34 % dávky v moči) je podporována jaterní a mimojaterní amidázou. In vitro je hydroxylace brivaracetamu zprostředkována v první řadě CYP2C19. Oba metabolity jsou dále metabolizovány za vzniku běžné hydroxylované kyseliny. In vivo u člověka s neúčinnou mutací CYP2C19 se tvorba hydroxymetabolitu snižuje 10x, zatímco samotný brivaracetam se zvyšuje o 22 % nebo 42 % u jedinců s jednou nebo s oběma mutovanými alelami. Tři metabolity nejsou farmakologicky aktivní.
Eliminace
Brivaracetam je eliminován primárně metabolizací a vylučováním močí. Více než 95 % dávky včetně metabolitů se vylučuje do moči během 72 hodin po požití. Méně než 1 % dávky se vylučuje stolicí a méně než 10 % brivaracetamu se vylučuje beze změny močí. Terminální plazmatický poločas (ti/2) je přibližně 9 hodin. Celková plazmatická clearance byla u pacientů odhadnuta na 3,6 l/hod.
Linearita
Farmakokinetika je úměrná dávce od 10 do nejméně 600 mg.
Interakce s léčivými přípravky
Brivaracetam je eliminován více cestami, včetně vylučování ledvinami, na CYP nezávislou hydrolýzou a CYP zprostředkovanou oxidací. In vitro brivaracetam není u člověka substrátem lidského P-glykoproteinu (P-gp), vícečetná léková rezistence proteinů (MRP - multidrug rezistence proteins) 1 a 2 a pravděpodobně ne polypeptidového transportéru organických aniontů 1B1 (OATP1B1) a OATP1B3.
Testy in vitro ukázaly, že metabolismus brivaracetamu by neměl být významně ovlivněn žádným CYP inhibitorem (např. CYP1A, 2C8, 2C9, 2D6 a 3A4).
Brivaracetam in vitro nebyl inhibitorem CYP1A2, 2A6, 2B6, 2C8, 2C9, 2D6, 3A4 ani transportérů P-gp, BCRP, BSEP MRP2, MATE-K, MATE-1, OATP1B1, OATP1B3, OAT1 a OCT1 v klinicky relevantních koncentracích. In vitro brivaracetam neindukoval CYP1A2.
Farmakokinetika u zvláštních skupin pacientů
Starší pacienti (65 let a starší)
Ve studii u starších pacientů (ve věku 65 až 79 let; clearance kreatininu 53 až 98 ml/min/1,73 m2), kteří užívali brivaracetam v dávce 400 mg/den s podáváním 2x denně, byl plazmatický poločas brivaracetamu 7,9 hodiny u skupiny ve věku 65 až 75 let, a 9,3 hodiny u skupiny >75 let. Plazmatická clearance rovnovážného stavu brivaracetamu byla podobná (0,76 ml/min/kg) jako u mladých zdravých mužů (0,83 ml/min/kg) (viz bod 4.2).
Porucha funkce ledvin
Studie u subjektů s těžkou poruchou funkce ledvin (clearance kreatininu <30 ml/min/1,73 m2 bez nutnosti dialýzy) odhalila, že plazmatická AUC brivaracetamu byla středně zvýšená (+21 %) v poměru ke zdravým subjektům zatímco AUC kyseliny, hydroxymetabolitu a metabolitu hydroxykyseliny byly zvýšeny 3x, 4x, a 21x (v uvedeném pořadí). Renální clearance těchto neaktivních metabolitů byla snížena 10x. Metabolit hydroxykyseliny v neklinických studiích nevyvolal žádné obavy ze strany bezpečnosti. Brivaracetam nebyl studován u pacientů léčených hemodialýzou (viz bod 4.2).
Porucha funkce jater
Farmakokinetická studie u subjektů s cirhózou jater (Child-Pugh třídy A, B, a C) ukázala podobná zvýšení expozice vůči brivaracetamu bez ohledu na závažnost onemocnění (50 %, 57 % a 59 %) v poměru k odpovídajícím zdravým subjektům (viz bod 4.2).
Pediatrická populace
Ve farmakokinetické studii u 99 subjektů ve věku 1 měsíc až <16 let užívajících perorální roztok brivaracetamu bylo prokázáno, že plazmatické koncentrace jsou úměrné dávce ve všech věkových skupinách. Populační farmakokinetické modelování ukázalo, že dávka 2,0 mg/kg dvakrát denně vede ke stejné průměrné plazmatické koncentraci ustáleného stavu jako u dospělých užívajících 100 mg dvakrát denně.
Tělesná hmotnost
Byl odhadnut 40% pokles plazmatické koncentrace ustáleného stavu v rozsahu tělesné hmotnosti od 46 kg do 115 kg. To však není považováno za klinicky významný rozdíl ve farmakokinetice brivaracetamu.
Pohlaví
Nejsou žádné klinicky významné rozdíly ve farmakokinetice brivaracetamu mezi pohlavími.
Rasa
Farmakokinetika brivaracetamu nebyla významně ovlivněna rasou (kavkazská, asijská) při populačním farmakokinetickém modelování u pacientů s epilepsií. Počet pacientů s jiným etnickým pozadím byl omezený.
Farmakokinetické/farmakodynamické vztahy
EC50 (plazmatická koncentrace brivaracetamu odpovídající 50 % maximálního účinku) byla odhadnuta na 0,57 mg/l. Tato plazmatická koncentrace je mírně nad mediánem expozice po podávání brivaracetamu v dávkách 50 mg/den. Další snížení frekvence záchvatů se dostavuje při zvýšení dávky na 100 mg/den a dosahuje plató při dávce 200 mg/den.
5.3 Předklinické údaje vztahující se k bezpečnosti
Ve farmakologických studiích bezpečnosti měly převládající účinky souvislost s CNS (zejména přechodná deprese CNS a snížení spontánní lokomoční aktivity) a byly pozorovány při násobcích (vyšších než 50x) farmakologicky aktivní dávky brivaracetamu 2 mg/kg. Učení a funkce paměti nebyly neovlivněny.
Nálezy nepozorované v klinických studiích, ale pozorované v toxikologických studiích s opakovaným podáváním u psů v podobné expozici ke klinické plazmatické AUC, jednalo se o hepatotoxické účinky (zejména porfyrie). Toxikologické údaje shromážděné o brivaracetamu a o strukturálně příbuzných sloučeninách ale ukazují, že jaterní změny se u psů vyvinuly působením mechanismů, které nejsou pro člověka relevantní. Žádné nežádoucí změny jater nebyly pozorovány u potkanů a opic při dlouhodobém podávání brivaracetamu s expozicí jasně převyšující AUC expozici 5 až 42x. CNS příznaky u opic (pády, ztráta rovnováhy, těžkopádné pohyby) se vyskytly u 64násobků klinické Cmax. Tyto účinky byly méně patrné v průběhu času.
Studie genotoxicity neodhalily žádnou mutagenní nebo klastogenní aktivitu. Studie kancerogenity neprokázaly žádný onkogenní potenciál u potkanů, zatímco zvýšený výskyt hepatocelulárních nádorů u myších samců je považován za důsledek negenotoxického fenoménu, známého u hlodavců, jehož mechanismus účinku se vztahuje k indukci jaterních ezymů, podobnou jako po fenobarbitalu.
Brivaracetam neovlivnil fertilitu samic ani samců, neprokázal se žádný teratogenní potenciál u potkanů nebo králíků. Embryotoxicita byla pozorována u králíků při dávce brivaracetamu toxické pro matky s expozicí 8x vyšší než klinická AUC expozice při maximální doporučené dávce.
U potkanů bylo prokázáno, že brivaracetam snadno přestupuje transplacentárně a je vylučován do mateřského mléka u kojících samic potkanů v koncentracích podobným plazmatickým koncentracím u matek.
Brivaracetam u potkanů neprokázal žádný potenciál ke zneužívání.
Studie u nedospělých zvířat
U nedospělých potkanů expoziční hladiny 6-15násobné klinické AUC expozice brivaracetamu při maximální doporučené dávce vyvolávaly vývojové nežádoucí účinky (např. mortalitu, klinické projevy, snížení tělesné hmotnosti a pokles váhy mozku). Neobjevily se žádné nežádoucí účinky u funkcí CNS, žádné neuropatologické a histopatologické vyšetření mozku. U nedospělých psů byly brivaracetamem indukované změny spojené s 6násobným zvýšením hladin AUC, podobným změnám pozorovaným u dospělých zvířat. Nebyly pozorovány žádné nežádoucí účinky ve standardních ukazatelích vývoje nebo maturace.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Dihydrát natrium-citrátu
bezvodá kyselina citronová (k úpravě pH)
methylparaben (E218)
sodná sůl karmelózy
sukralosa
krystalizující sorbitol 70% glycerol 85% (E422)
malinové aroma (propylenglykol 90% - 98%) čištěná voda.
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
3 roky.
Po prvním otevření: 5 měsíců
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
6.5 Druh obalu a obsah balení
300ml jantarově hnědá lahvička (třídy III) s bílým dětským bezpečnostním uzávěrem (z polypropylenu) v papírové krabičce s 10ml kalibrovanou perorální stříkačkou (polypropylen, polyethylen) a adaptérem na stříkačku (polyethylen).
6.6 Zvláštní opatření pro likvidaci přípravku
Žádné zvláštní požadavky.
Veškerý nepoužitý léčivý přípravek (neředěný i ředěný) nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
UCB Pharma S.A.
Allée de la Recherche 60 B-1070 Brusel Belgie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/15/1073/021
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 14. ledna 2016
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu
▼ Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakékoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Briviact 10 mg/ml injekční/infuzní roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden ml obsahuje brivaracetamum 10 mg.
Jedna 5ml lahvička obsahuje brivaracetamum 50 mg.
Pomocné látky se známým účinkem:
Jeden ml injekčního/infuzního roztoku obsahuje 3,8 mg sodíku. Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční/infuzní roztok (injekce/infuze) Čirý, bezbarvý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Briviact je indikován jako přídatná terapie při léčbě parciálních záchvatů s nebo bez sekundární generalizace u dospělých a dospívajících pacientů s epilepsií ve věku od 16 let.
4.2 Dávkování a způsob podání
Dávkování
Léčbu brivaracetamem lze zahájit buďintravenózní nebo perorální aplikací. Pokud se přechází z perorálního na intravenózní podávání nebo naopak, celková denní dávka a frekvence podávání zůstávají stejné. Injekční/infuzní roztok brivaracetamu je alternativou pro pacienty, u kterých není dočasně možné perorální podávání.
Doporučená počáteční dávka je buď 50 mg/den nebo 100 mg/den, potřebná ke snížení počtu záchvatů na základě posouzení lékaře oproti potenciálním nežádoucím účinkům. Dávka se podává ve dvou stejných rozdělených dávkách, jednou ráno a jednou večer. Na základě individuální odpovědi pacienta a snášenlivosti lze dávku upravit v dávkovém rozmezí 50 mg/den až 200 mg/den.
Nejsou žádné zkušenosti s intravenózním podáváním brivaracetamu trvajícím déle než 4 dny.
Opomenutá dávka
Jestliže pacienti opomenou jednu nebo více dávek, doporučuje se podat jednu dávku, jakmile si vzpomenou, a následující dávku podat v obvyklé době ráno nebo večer. To může zabránit poklesu plazmatické koncentrace brivaracetamu pod účinnou hladinu, a tím znovupropuknutí záchvatů.
Ukončení léčby
Pokud je nutno podávání brivaracetamu ukončit, doporučuje se postupné snižování o 50 mg/den
v týdenním intervalu. Po týdnu léčby dávkou 50 mg/den se doporučuje v posledním týdnu léčby dávka 20 mg/den.
Zvláštní populace
Starší pacienti (65 let a starší)
Není nutná žádná úprava dávky u starších pacientů (viz bod 5.2).
Klinická zkušenost u pacientů >65 let je omezená.
Porucha funkce ledvin
U pacientů s poruchou funkce ledvin není třeba dávku nijak upravovat (viz bod 5.2). Podávání brivaracetamu se nedoporučuje u pacientů v konečné fázi onemocnění ledvin, kteří jsou léčeni dialýzou, vzhledem k nedostatku údajů
Porucha funkce jater
Expozice vůči brivaracetamu byla zvýšená u pacientů s chronickým onemocněním jater. Počáteční dávka 50 mg/den by měla být zvážena. Ve všech fázích poruchy funkce jater se doporučuje maximální denní dávka 150 mg podávaná ve 2 rozdělených dávkách (viz body 4.4 a 5.2).
Pediatrická populace
Bezpečnost a účinnost brivaracetamu u dětí ve věku méně než 16 let nebyly dosud stanoveny.
V současné době dostupné údaje jsou uvedeny v bodech 4.8, 5.1, a 5.2, ale na jejich základě nelze učinit žádná doporučení ohledně dávkování.
Způsob podání
• Intravenózní bolus: brivaracetam nůže být podáván bez ředění jako intravenózní bolus.
• Intravenózní infuze: brivaracetam může být naředěn kompatibilním ředidlem a podáván 15minutovou intravenózní infuzí (viz bod 6.6). Tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky.
Podávání brivaracetamu bolusovou injekcí nebo intravenózní infuzí nebylo studováno při akutních stavech, např. status epilepticus, a proto se při takových stavech nedoporučuje.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku, jiné deriváty pyrrolidonu nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Sebevražedné myšlenky a chování
Sebevražedné myšlenky a chování byly hlášeny v několika indikacích u pacientů léčených antiepileptiky, včetně brivaracetamu. Metaanalýza randomizovaných placebem kontrolovaných studií s antiepileptiky prokázala mírně zvýšené riziko sebevražedných myšlenek a chování. Mechanismus vzniku není znám a dostupné údaje nevylučují možnost zvýšeného rizika u brivaracetamu.
Proto by u pacientů měly být sledovány příznaky sebevražedných představ a chování a zvážena vhodná léčba. Pacienti (a osoby poskytující pacientům péči) by měli být upozorněni na to, že v případě výskytu příznaků sebevražedného myšlení či chování, by měli vyhledat lékařskou pomoc.
Porucha funkce jater
Jsou k dispozici omezené klinické údaje o použití brivaracetamu u pacientů s již existující poruchou funkce jater. U pacientů s poruchou funkce jater se doporučuje úprava dávky (viz bod 4.2).
Obsah sodíku
Injekční/infuzní roztok obsahuje 0,83 mmol (odp. 19,14 mg) sodíku v injekční lahvičce. Nutno vzít v úvahu u pacientů na dietě s nízkým obsahem sodíku.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Formální studie interakce byly provedeny jen u dospělých pacientů. Farmakodvnamické interakce
Současná léčba s levetiracetamem
V klinických studiích (ačkoli počet je velmi omezený) nebyl pozorován žádný přínos brivaracetamu versus placebo u pacientů současně užívajících levetiracetam. Nebyly doloženy žádné další údaje ohledně bezpečnosti a tolerability (viz bod 5.1).
Interakce s alkoholem
Ve studii farmakokinetické a farmakodynamické interakce mezi brivaracetamem v jednorázové dávce 200 mg a ethanolem v kontinuální infuzi 0,6 g/l u zdravých subjektů nenastala žádná farmakokinetická interakce, ale brivaracetam přibližně zdvojnásobil účinky alkoholu na psychomotorické funkce, pozornost a paměť. Podávání brivaracetamu s alkoholem se nedoporučuje.
Farmakokinetické interakce
Účinky jiných látek na farmakokinatiku brivaracetamu
In vitro údaje naznačují, že brivaracetam má nízký interakční potenciál. Hlavní metabolickou cestou brivaracetamu je na CYP nezávislá hydrolýza. Druhá cesta zahrnuje hydroxylaci, která je zprostředkována CYP2C19 (viz bod 5.2).
Plazmatické koncentrace brivaracetamu se mohou zvýšit, je-li současně podáván se silnými inhibitory CYP2C19 (jako flukonazol, fluvoxamin), ale riziko klinicky významné interakce zprostředkované CYP2C19 je považováno za nízké.
Rifampicin
U zdravých dobrovolníků současné podávání silného induktoru enzymů rifampicinu (600 mg/den po dobu 5 dnů) snížilo plochu brivaracetamu pod křivkou (AUC) o 45 %. Předepisující lékaři musí zvážit úpravu dávky brivaracetamu u pacientů, u kterých se zahajuje nebo ukončuje léčba rifampicinem.
Antiepileptika se silnou indukcí enzymů
Plazmatické koncentrace brivaracetamu klesají při současném podávání s antiepileptiky silně indukující enzymy (karbamazepin, fenobarbital, fenytoin), ale není nutná žádná úprava dávky (viz tabulka 1).
Jiné induktory enzymů
Očekává se, že jiné silné induktory enzymů (jako třezalka tečkovaná (Hypericum perforatum)) mohou také snížit systémovou expozici brivaracetamu. Proto zahájení nebo ukončení léčby třezalkou tečkovanou má být provedeno s opatrností.
Účinek brivaracetamu na jiné léčivé přípravky
Brivaracetam podávaný v dávkách 50 mg nebo 150 mg/den neovlivňoval AUC midazolamu (metabolizován CYP3A4). Riziko klinicky relevantních CYP3A4 interakcí je považováno za nízké.
Studie in vitro prokázaly, že brivaracetam vykazuje malou nebo žádnou inhibici izoforem CYP450 s výjimkou CYP2C19. Brivaracetam může zvyšovat plazmatické koncentrace léčivých přípravků metabolizovaných CYP2C19 (např. lanzoprazol, omeprazol, diazepam). Při zkoušení in vitro brivaracetam neindukoval CYP1A1/2, ale indukoval CYP3A4 a CYP2B6. Žádná CYP3A4 indukce nebyla nalezena in vivo (viz midazolam výše). CYP2B6 indukce nebyla zkoumána in vivo a brivaracetam může snižovat plazmatické koncentrace léčivých přípravků metabolizovaných CYP2B6 (např. efavirenz). In vitro interakční studie k určení potenciálních inhibičních účinků na transportéry vedly k závěru, že nedochází k žádným klinicky významným účinkům s výjimkou pro OAT3. In vitro brivaracetam inhibuje OAT3 s poloviční maximální inhibiční koncentrací 42x vyšší, než je Cmax při nejvyšší klinické dávce. Brivaracetam v dávce 200 mg/den může zvýšit plazmatické koncentrace léčivých přípravků transportovaných OAT3.
Antiepileptika
Potenciální interakce mezi brivaracetamem (50 mg/den až 200 mg/den) a jinými antiepileptiky byly zkoumány v souhrnné analýze plazmatických lékových koncentrací ze všech studií fáze 2-3 a v populační farmakokinetické analýze v placebem kontrolovaných studiích fáze 2-3 a ve vyhrazených studiích lékových interakcí (pro následující antiepileptika: karbamazepin, lamotrigin, fenytoin a topiramát). Účinek vzájemných interakcí na plazmatickou koncentraci je shrnut v tabulce 1 (zvýšení je označeno jako “t” a snížení jako “j” plocha pod křivkou plazmatické koncentrace v průběhu času jako „AUC“, maximální pozorovaná koncentrace jako „Cmax ).
Tabulka 1: Farmakokinetické interakce mezi brivaracetamem a jinými antiepileptiky
|
Současně podávané antiepiletikum |
Vliv antiepiletika na koncentraci brivaracetamu v plazmě |
Vliv brivaracetamu na koncentraci antiepiletika v plazmě |
|
Karbamazepin |
AUC 29 % j Cmax 13 % j není nutná žádná úprava dávky |
karbamazepin - žádný epoxid karbamazepinu t (viz níže) není nutná žádná úprava dávky |
|
Klobazam |
žádné údaje |
žádný |
|
Klonazepam |
žádné údaje |
žádný |
|
Lacosamid |
žádné údaje |
žádný |
|
Lamotrigin |
žádný |
žádný |
|
Levetiracetam |
žádný |
žádný |
|
Oxkarbazepin |
žádný |
žádný (monohydroxyderivát, MHD) |
|
Fenobarbital |
AUC 19 % j není nutná žádná úprava dávky |
žádný |
|
Fenytoin |
AUC 21 % j není nutná žádná úprava dávky |
žádný aAUC 20 % t aCmax 20 % t |
|
Pregabalin |
žádné údaje |
žádný |
|
Topiramát |
žádný |
žádný |
|
Kyselina valproová |
žádný |
žádný |
|
Zonisamid |
žádné údaje |
žádný |
a na základě studie týkající se podávání supraterapeutické dávky brivaracetamu 400 mg/den
Karbamazepin
Brivaracetam je středně účinný reverzibilní inhibitor epoxidové hydrolázy vyvolávající zvýšenou koncentraci epoxidu karbamazepinu, aktivního metabolitu karbamazepinu. V kontrolovaných studiích vzrostla plazmatická koncentrace epoxidu karbamazepinu v průměru o 37 %, 62 % a 98 % s malou variabilitou při dávkách brivaracetamu odpovídajících 50 mg/den, 100 mg/den a 200 mg/den. Nebyla pozorována žádná bezpečnostní rizika. Nebyl žádný aditivní účinek brivaracetamu a valproátu na AUC u epoxidu karbamazepinu.
Perorální kontraceptiva
Současné podávání brivaracetamu (100 mg/den) spolu s perorálním kontraceptivem obsahujícím ethinylestradiol (0,03 mg) a levonorgestrel (0,15 mg) neovlivňovalo farmakokinetiku žádné látky. Když byl brivaracetam současně podáván v dávce 400 mg/den (dvojnásobek doporučené maximální denní dávky) současně s perorálním kontraceptivem obsahujícím ethinylestradiol (0,03 mg) a levonorgestrel (0,15 mg), bylo pozorováno snížení AUC estrogenu o 27 % a snížení AUC progestinu o 23 %, a to bez dopadu na potlačení ovulace. Obecně nenastala žádná změna v profilech koncentrací v čase u endogenních markerů estradiolu, progesteronu, luteinizačního hormonu (LH), folikuly stimulujícího hormonu a globulinu vázajícího pohlavní hormony (SHBG).
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku.
Lékaři by měli prodiskutovat plánované rodičovství a antikoncepci se ženami ve fertilním věku, které užívají brivaracetam (viz Těhotenství).
Jestliže se žena rozhodne otěhotnět, užívání brivaracetamu by mělo být opět přehodnoceno. Těhotenství
Riziko spojené s epilepsií a s antiepileptiky obecně
U všech antiepileptik bylo prokázáno, že mezi potomky léčených žen s epilepsií je prevalence malformací dvakrát až třikrát vyšší, než je přibližně 3% výskyt malformací v běžné populaci. V léčené populaci byl pozorován nárůst malformací při polyterapii, ale rozsah, za který odpovídá léčba a/nebo základní onemocnění, nebyl objasněn. Přerušení antiepileptické léčby může vést k exacerbaci onemocnění, které může poškodit matku i plod.
Riziko spojené s brivaracetamem
K dispozici je omezené množství údajů o použití brivaracetamu u těhotných žen. Nejsou k dispozici žádné údaje o placentárním transferu u člověka, ale brivaracetam rychle prochází placentou u potkanů (viz bod 5.3). Potenciální riziko u člověka není známo. Studie na zvířatech neprokázaly žádný teratogenní potenciál brivaracetamu (viz bod 5.3).
Brivaracetam byl užíván v klinických studiích jako přídatná terapie a když byl užíván společně s karbamazepinem, vedl v závislosti na dávce k nárůstu koncentrace aktivního metabolitu, epoxidu karbamazepinu (viz bod 4.5). Nejsou k dispozici dostatečné údaje k určení klinického významu tohoto účinku v těhotenství.
Z preventivních důvodů by brivaracetam neměl být užíván během těhotenství, pokud to není klinicky nezbytně nutné (pokud přínos pro matku jasně převáží potenciální riziko pro plod).
Kojení
Není známo, zda se u člověka brivaracetam vylučuje do mateřského mléka. Studie u potkanů vylučování brivaracetamu do mateřského mléka prokázaly (viz bod 5.3). Je nutno učinit rozhodnutí, zda přerušit kojení nebo přerušit podávání brivaracetamu, přičemž je třeba zhodnotit přínos léku pro matku. V případě současného podávání brivaracetamu a epoxidu karbamazepinu by se mohlo vylučování množství epoxidu karbamazepinu do mateřského mléka zvýšit. Není k dispozici dostatek údajů k určení klinického významu.
Fertilita
Nejsou k dispozici žádné údaje o účinku brivaracetamu na fertilitu u člověka. U potkanů nebyl při léčbě brivaracetamem pozorován žádný účinek na fertilitu (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Brivaracetam má zanedbatelný nebo mírný vliv na schopnost řídit nebo obsluhovat stroje.
Vzhledem k možné rozdílné individuální citlivosti mohou někteří pacienti pociťovat somnolenci, závratě nebo jiné příznaky související s centrálním nervovým systémem (CNS). Pacientům je nutno doporučit neřídit vozidlo a neobsluhovat jiné potenciálně nebezpečné stroje, dokud se neseznámí s účinky brivaracetamu na vykonávání těchto aktivit.
4.8 Nežádoucí účinky
Souhrnný bezpečnostní profil
Ve všech kontrolovaných a nekontrolovaných studiích u pacientů s epilepsií dostávalo brivaracetam 2388 subjektů, z nichž 1740 bylo léčeno >6 měsíců, 1363 >12 měsíců, 923 ^24 měsíců a 569 >60 měsíců (5 let).
Nejčastěji hlášené nežádoucí účinky (>10 %) při léčbě brivaracetamem byly somnolence (14,3 %) a závrať (11,0 %). Byly obvykle mírné až střední intenzity. Somnolence a únava (8,2 %) byly hlášeny ve vyšší míře při zvyšující se dávce. Typy nežádoucích účinků hlášených během prvních 7 dnů léčby se podobaly těm, které byly hlášeny během celkové doby léčby.
Frekvence ukončení léčby z důvodu nežádoucích účinků byly 3,5 %, 3,4 % a 4,0 % u pacientů randomizovaných k užívání brivaracetamu v příslušné dávce 50 mg/den, 100 mg/den a 200 mg/den, a
1,7 % pro pacienty randomizované k užívání placeba. Nežádoucím účinkem, který vedl nejčastěji k ukončení léčby brivaracetamem, byly závrať (0,8 %) a konvulze (0,8 %).
Seznam nežádoucích účinků v tabulce
V níže uvedené tabulce jsou nežádoucí účinky, které byly identifikovány na základě přehledu úplné bezpečnostní databáze klinických studií brivaracetamu, uvedeny podle tříd orgánových systémů a podle frekvence.
Četnosti jsou definovány následovně: velmi časté (>1/10), časté (>1/100 až <1/10), méně časté (>1/1 000 až <1/100). V každé skupině frekvence jsou nežádoucí účinky řazeny za sebou podle klesající závažnosti.
|
Třídy orgánových systémů |
Frekvence |
Nežádoucí účinky z klinických studií |
|
Infekce a infestace |
časté |
chřipka |
|
Poruchy krve a lymfatického systému |
méně časté |
neutropenie |
|
Poruchy metabolismu a výživy |
časté |
snížená chuť k jídlu |
|
Psychiatrické poruchy |
časté |
deprese, anxieta, insomnie, iritabilita |
|
méně časté |
sebevražedné představy, psychotické poruchy, agresivita, agitovanost | |
|
Poruchy nervového systému |
velmi časté |
závrať, somnolence |
|
časté |
konvulze, vertigo | |
|
Respirační, hrudní a mediastinální poruchy |
časté |
infekce horních cest dýchacích, kašel |
|
Gastrointestinální poruchy |
časté | |
|
Celkové poruchy a reakce v místě aplikace |
časté |
Popis vybraných nežádoucích účinků
Neutropenie byla hlášena u 0,5 % (6/1099) pacientů s brivaracetamem a u 0 % (0/459) pacientů s placebem. Čtyři z těchto subjektů měly snížený počet neutrofilů ve výchozím stavu a po zahájení léčby brivaracetamem došlo k dalšímu snížení počtu neutrofilů. Žádný z těchto 6 případů neutropenie nebyl závažný, nevyžadoval speciální léčbu ani nevedl k ukončení léčby brivaracetamem a žádný neměl přidružené infekce.
Sebevražedné představy byly hlášeny u 0,3 % (3/1099) pacientů s brivaracetamem a u 0,7 % (3/459) pacientů s placebem. V krátkodobých klinických studiích brivaracetamu u pacientů s epilepsií nedošlo k žádnému případu dokonané sebevraždy a sebevražedného pokusu, nicméně oboje bylo hlášeno v otevřených prodloužených studiích (viz bod 4.4).
Nežádoucí účinky spojené s intravenózním podáním se obecně jeví jako podobné těm, které jsou pozorovány po perorálním podání. Intravenózní podání bylo spojené s bolestí v místě aplikace u 2,8 % pacientů.
Otevřené prodloužené studie
U pacientů, kteří byli sledováni v otevřených prodloužených studiích po dobu až 8 let, byl bezpečnostní profil podobný profilu pozorovanému v krátkodobých placebem kontrolovaných studiích.
Pediatrická populace
Jsou k dispozici omezené bezpečnostní údaje z otevřených studií u dětí ve věku od 1 měsíce do <16 let. Celkem 152 dětí (ve věku 1 měsíc až <16 let) bylo léčeno brivaracetamem ve farmakokinetické studii a v související navazující studii. Většina zkoušejícím často hlášených nežádoucích účinků spojených s léčbou (TEAE) považovaných za související s lékem byly somnolence (10 %), snížená chuť k jídlu (8 %), únava (5%) a snížení tělesné hmotnosti (5 %). Bezpečnostní profil se zdá být v souladu s bezpečnostním profilem známým u dospělých pacientů.
V současné době nejsou k dispozici žádné klinické údaje u novorozenců.
Starší pacienti:
Ze 130 starších subjektů zahrnutých do fáze 2/3 vývojového programu brivaracetamu (44 s epilepsií) bylo 100 ve věku 65-74 let a 30 ve věku 75-84 let. Bezpečnostní profil u starších pacientů se zdá být podobný bezpečnostnímu profilu pozorovanému u mladších dospělých pacientů.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Příznaky
Je k dispozici omezená klinická zkušenost s předávkováním brivaracetamu u člověka. U zdravých subjektů, které užily jednotlivou dávku 1400 mg brivaracetamu, byla hlášena somnolence a závrať.
Léčba předávkování
Není k dispozici žádné specifické antidotum pro předávkování brivaracetamem. Léčba předávkování zahrnuje obecná podpůrná opatření. Protože se močí vylučuje méně než 10 % brivaracetamu, neočekává se, že hemodialýza významně zvýší clearance brivaracetamu (viz bod 5.2).
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antiepileptika, jiná antiepileptika, ATC kód: N03AX23 Mechanismus účinku
Brivaracetam vykazuje vysokou a selektivní afinitu k 2A proteinu synaptických vezikul (SV2A), transmembránový glykoprotein byl nalezen na presynaptické úrovni v neuronech a v endokrinních buňkách. Ačkoli přesnou roli tohoto proteinu je ještě nutno objasnit, bylo prokázáno, že moduluje exocytózu neurotransmiterů. Má se za to, že vazba na SV2A představuje primární mechanismus protizáchvatové aktivity brivaracetamu.
Klinická účinnost a bezpečnost
Účinnost brivaracetamu při přídatné terapii parciálních záchvatů (partial onset seisures-POS) byla stanovena ve 3 randomizovaných dvojitě zaslepených placebem kontrolovaných multicentrických studiích s pevnou dávkou u subjektů ve věku 16 let a starších. Denní dávka brivaracetamu se v těchto studiích pohybovala v rozsahu 5 až 200 mg/den. Všechny studie začínaly základní periodou trvající 8 týdnů, následovanou 12 týdnů trvající léčebnou periodou bez titrace ve smyslu zvyšování dávky. 1558 pacientů dostávalo lék ze studie, z toho 1099 dostávalo brivaracetam. Kritéria pro zařazení do studie vyžadovala, aby měli pacienti nekontrolované parciální záchvaty navzdory léčbě buď 1, nebo 2 současně podávanými antiepileptiky. Bylo požadováno, aby pacienti prodělali přinejmenším 8 parciálních záchvatů během základní periody. Primárními cílovými parametry u studie fáze 3 bylo procento snížení frekvence POS oproti placebu a poměr respondérů s dosaženou 50% odpovědí založený na 50% snížení frekvence POS od výchozího stavu.
Nejčastěji užívanými antiepileptiky na začátku studie byly karbamazepin (40,6 %), lamotrigin (25,2 %), valproát (20,5 %), oxkarbazepin (16,0 %), topiramát (13,5 %), fenytoin (10,2 %) a levetiracetam (9,8 %). Medián výchozí frekvence záchvatů napříč 3 studiemi byl 9 záchvatů během 28 dní. Pacienti měli epilepsii v průměru přibližně 23 let.
Výsledky účinnosti jsou shrnuty v tabulce 2. Celkově byl brivaracetam účinný při přídatné terapii parciálních záchvatů u pacientů ve věku 16 let a starších v dávce mezi 50 mg/den a 200 mg/den.
Tabulka 2: Výsledky účinnosti pro frekvenci parciálních záchvatů během 28 dní
|
Studie |
Placebo |
Brivaracetam * statisticky signifikantní (hodnota p) | ||
|
50 mg/den |
100 mg/den |
200 mg/den | ||
|
Studie N01253(1) | ||||
|
n = 96 |
n = 101 | |||
|
Dosažení 50% odpovědi respondéra |
16,7 |
32,7* (p=0,008) |
~ |
~ |
|
Procentuální snížení oproti placebu (%) |
NA |
22,0 (p=0,0040) |
~ |
~ |
|
Studie N01252(1) | ||||
|
n = 100 |
n = 99 |
n = 100 | ||
|
Dosažení 50% odpovědi respondéra |
20,0 |
27,3 (p=0,372) |
36,0(2) (p=0,023) |
~ |
|
Procentuální snížení oproti placebu (%) |
NA |
9,2 (p=0,0274) |
20,5(2) (p=0,0097) |
~ |
|
Studie N01358 | ||||
|
n = 259 |
n = 252 |
n=249 | ||
|
Dosažení 50% odpovědi respondéra |
21,6 |
~ |
38,9 (p<0,001) |
37,8 (p<0,001) |
|
Procentuální snížení oproti placebu (%) |
NA |
~ |
22,8* (p<0,001) |
23,2* (p<0,001) |
n = randomizovaní pacienti, kteří dostali nejméně 1 dávku léčiva ve studii ~ dávka nebyla studována *statisticky významné
(1) Přibližně 20 % pacientů dostávalo současně levetiracetam
(2) Primární výsledek pro N01252 nedosáhl statistické významnosti na základě sekvenčního zkoušení. Dávka 100 mg/den byla nominálně významná.
V klinických studiích bylo vyšší snížení frekvence záchvatů oproti placebu při dávce 100 mg/den než při dávce 50 mg/den. Na rozdíl od na dávce závislého zvýšení výskytu somnolence a únavy měl brivaracetam v dávce 50 mg/den a 100 mg/den podobný bezpečnostní profil včetně nežádoucích účinků se vztahem k CNS a při dlouhodobém užívání.
Obrázek 1 ukazuje procento pacientů (s výjimkou pacientů současně užívajících levetiracetam) podle kategorie snížení frekvence POS během 28 dní od výchozího stavu ve všech 3 studiích. Pacienti s více než 25% zvýšením parciálních záchvatů jsou uvedeni zcela nalevo jako „horší“. Pacienti se zlepšením procentuálního snížení frekvence POS od výchozího stavu jsou uvedeni ve 4 kategoriích napravo. Procento pacientů s nejméně 50% snížením četnosti záchvatu bylo 20,3 %, 34,2 %, 39,5 %, a 37,8 % pro placebo, 50 mg/den, 100 mg/den a 200 mg/den (v tomto pořadí).
Obrázek 1: Podíl pacientů s brivaracetamem a placebem podle kategorie odpovědi záchvatů po dobu 12 týdnů napříč všemi třemi dvojitě zaslepenými pivotními studiemi
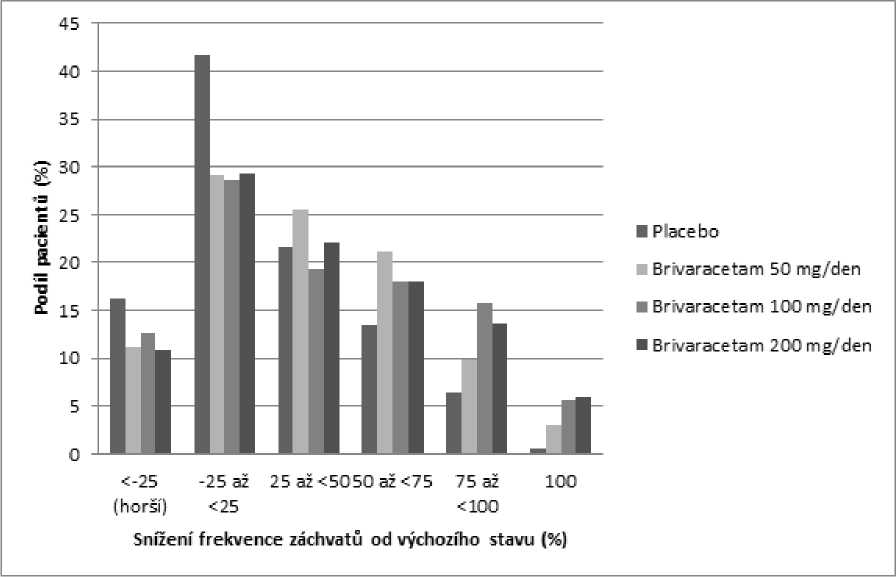
V souhrnné analýze tří pivotních studií nebyly pozorovány žádné rozdíly v účinnosti (měřené jako 50% odpověď respondérů) v rozmezí dávek 50 mg/den až 200 mg/den, když je brivaracetam zkombinován s antiepileptiky vyvolávajícími nebo nevyvolávajícími indukci enzymů.
V klinických studiích dosáhlo stavu bez záchvatů 2,5 % (4/161), 5,1 % (17/332) a 4,0 % (10/249) pacientů s brivaracetamem v dávce odpovídající 50 mg/den, 100 mg/den a 200 mg/den, a to během léčebné periody v trvání 12 týdnů ve srovnání s 0,5 % (2/418) pacientů s placebem.
Zlepšení v mediánu procentuálního snížení frekvence záchvatů od začátku léčby za 28 dní bylo pozorováno u pacientů s typem záchvatů IC (sekundární generalizované tonicko-klonické záchvaty) ve výchozím stavu léčených brivaracetamem, 66,6 % (n=62), 61,2 % (n=100) a 82,1 % (n=75) z pacientů s brivaracetamem v odpovídající dávce 50 mg/den, 100 mg/den a 200 mg/den ve srovnání s placebem
33,3 % (n=115).
Účinnost brivaracetamu v monoterapii nebyla ještě stanovena. V monoterapii se použití brivaracetamu nedoporučuje.
Léčba levetiracetamem
Ve 2 randomizovaných placebem kontrolovaných studiích fáze 3 byl levetiracetam podáván jako současně podávané antiepileptikum asi u 20 % pacientů. Ačkoli je počet subjektů limitován, nebyl u pacientů, kteří současně užívali levetiracetam, pozorován žádný přínos brivaracetamu oproti placebu, který by reflektoval kompetici na vazebném místě SV2A. Nebyly zjištěny žádné další okolnosti týkající se bezpečnosti a snášenlivosti.
Ve třetí studii předem specifikovaná analýza prokázala účinnost vůči placebu pro dávky 100 mg/den a 200 mg/den u pacientů předtím užívajících levetiracetam. Nižší účinnost pozorovaná u těchto pacientů ve srovnání s pacienty neužívajícími levetiracetam byla pravděpodobně důsledkem vyššího počtu použitých předchozích antiepileptik a vyšší výchozí hodnoty frekvence záchvatů.
Starší pacienti (65 let a starší)
Tři pivotní dvojitě zaslepené placebem kontrolované studie zahrnovaly 38 starších pacientů ve věku mezi 65 a 80 roky. Třebaže jsou údaje omezené, účinnost byla srovnatelná s účinností u mladších subjektů.
Otevřené prodloužené studie
Napříč všemi studiemi bylo zahrnuto do dlouhodobých otevřených prodloužených studií 81,7 % pacientů, kteří dokončili randomizované studie. Od vstupu do randomizovaných studií bylo 5,3 % subjektů s brivaracetamem po dobu 6 měsíců (n=1500) bez záchvatů ve srovnání s 4,6 % a 3,7 % u subjektů exponovaných po dobu odpovídající 12 měsíců (n=1188) a 24 měsíců (n=847). Nicméně protože vysoký podíl subjektů (26 %) přerušil léčbu v otevřených studiích kvůli nedostatečné účinnosti, mohlo dojít ke zkreslení, subjekty, které zůstaly ve studii, reagovaly lépe než ty, které předčasně ukončily.
Pediatrická populace
Účinnost a snášenlivost brivaracetamu nebyla u pediatrických pacientů stanovena (viz bod 4.2). Brivaracetam byl hodnocen u těchto pacientů v krátkodobé otevřené farmakokinetické studii a probíhající otevřené prodloužené studii u 152 subjektů ve věku od 1 měsíce do 16 let (viz bod 5.2).
Evropská agentura pro léčivé přípravky odložila závazek předložit výsledky studií s brivaracetamem u jedné nebo více podskupin pediatrické populace s epilepsií s parciálními záchvaty.
5.2 Farmakokinetické vlastnosti
Brivaracetam potahované tablety, perorální roztok a roztok pro intravenózní injekci vykazují identickou AUC, zatímco maximální plazmatická koncentrace je mírně vyšší po intravenózním podání. Brivaracetam vykazuje lineární a na čase nezávislou farmakokinetiku s nízkou intra- a interindividuální variabilitou a dále úplnou absorpci, nízkou vazbu na proteiny, renální exkreci po rozsáhlé biotransformaci a farmakologicky inaktivní metabolity.
Absorpce
Brivaracetam je rychle a úplně absorbován po perorálním podání a absolutní biologická dostupnost je přibližně 100%. Medián tmax pro tablety užité bez jídla je 1 hodina (rozsah tmax je 0,25 až 3 h).
Současné podávání s jídlem s vysokým obsahem tuku zpomalilo rychlost absorpce (medián tnax 3 h) a snížilo maximální plazmatickou koncentraci (37% pokles) brivaracetamu, přičemž rozsah absorpce zůstal nezměněn.
Distribuce
Brivaracetam se slabě váže (<20 %) na plazmatické proteiny. Distribuční objem je 0,5 l/kg, což je hodnota blízká celkovému množství tělesné vody.
Buněčné membrány jsou pro brivaracetam vysoce permeabilní z důvodu jeho lipofilie (log P). Biotransformace
Brivaracetam je primárně metabolizován hydrolýzou své amidové části za vzniku odpovídající karboxylové kyseliny (přibližně 60% eliminace) a sekundárně hydroxylací propylového vedlejšího řetězce (přibližně 30% eliminace). Hydrolýza amidové části vedoucí ke vzniku metabolitu povahy karboxylové kyseliny (34 % dávky v moči) je podporována jaterní a mimojaterní amidázou. In vitro je hydroxylace brivaracetamu zprostředkována v první řadě CYP2C19. Oba metabolity jsou dále metabolizovány za vzniku běžné hydroxylované kyseliny. In vivo u člověka s neúčinnou mutací CYP2C19 se tvorba hydroxymetabolitu snižuje 10x, zatímco samotný brivaracetam se zvyšuje o 22 % nebo 42 % u jedinců s jednou nebo s oběma mutovanými alelami. Tři metabolity nejsou farmakologicky aktivní.
Eliminace
Brivaracetam je eliminován primárně metabolizací a vylučováním močí. Více než 95 % dávky včetně metabolitů se vylučuje do moči během 72 hodin po požití. Méně než 1 % dávky se vylučuje stolicí a méně než 10 % brivaracetamu se vylučuje beze změny močí. Terminální plazmatický poločas (ti/2) je přibližně 9 hodin. Celková plazmatická clearance byla u pacientů odhadnuta na 3,6 l/hod.
Linearita
Farmakokinetika je úměrná dávce od 10 do nejméně 600 mg.
Interakce s léčivými přípravky
Brivaracetam je eliminován více cestami, včetně vylučování ledvinami, na CYP nezávislou hydrolýzou a CYP zprostředkovanou oxidací. In vitro brivaracetam není u člověka substrátem lidského P-glykoproteinu (P-gp), vícečetná léková rezistence proteinů (MRP - multidrug rezistence proteins) 1 a 2 a pravděpodobně ne polypeptidového transportéru organických aniontů 1B1 (OATP1B1) a OATP1B3.
Testy in vitro ukázaly, že metabolismus brivaracetamu by neměl být významně ovlivněn žádným CYP inhibitorem (např. CYP1A, 2C8, 2C9, 2D6 a 3A4).
Brivaracetam in vitro nebyl inhibitorem CYP1A2, 2A6, 2B6, 2C8, 2C9, 2D6, 3A4 ani transportérů P-gp, BCRP, BSEP MRP2, MATE-K, MATE-1, OATP1B1, OATP1B3, OAT1 a OCT1 v klinicky relevantních koncentracích. In vitro brivaracetam neindukoval CYP1A2.
Farmakokinetika u zvláštních skupin pacientů
Starší pacienti (65 let a starší)
Ve studii u starších pacientů (ve věku 65 až 79 let; clearance kreatininu 53 až 98 ml/min/1,73 m2), kteří užívali brivaracetam v dávce 400 mg/den s podáváním 2x denně, byl plazmatický poločas brivaracetamu 7,9 hodiny u skupiny ve věku 65 až 75 let, a 9,3 hodiny u skupiny >75 let. Plazmatická clearance rovnovážného stavu brivaracetamu byla podobná (0,76 ml/min/kg) jako u mladých zdravých mužů (0,83 ml/min/kg) (viz bod 4.2).
Porucha funkce ledvin
Studie u subjektů s těžkou poruchou funkce ledvin (clearance kreatininu <30 ml/min/1,73 m2 bez nutnosti dialýzy) odhalila, že plazmatická AUC brivaracetamu byla středně zvýšená (+21 %) v poměru ke zdravým subjektům zatímco AUC kyseliny, hydroxymetabolitu a metabolitu hydroxykyseliny byly zvýšeny 3x, 4x, a 21x (v uvedeném pořadí). Renální clearance těchto neaktivních metabolitů byla snížena 10x. Metabolit hydroxykyseliny v neklinických studiích nevyvolal žádné obavy ze strany bezpečnosti. Brivaracetam nebyl studován u pacientů léčených hemodialýzou (viz bod 4.2).
Porucha funkce jater
Farmakokinetická studie u subjektů s cirhózou jater (Child-Pugh třídy A, B, a C) ukázala podobná zvýšení expozice vůči brivaracetamu bez ohledu na závažnost onemocnění (50 %, 57 % a 59 %) v poměru k odpovídajícím zdravým subjektům (viz bod 4.2).
Pediatrická populace
Ve farmakokinetické studii u 99 subjektů ve věku 1 měsíc až <16 let užívajících perorální roztok brivaracetamu bylo prokázáno, že plazmatické koncentrace jsou úměrné dávce ve všech věkových skupinách. Populační farmakokinetické modelování ukázalo, že dávka 2,0 mg/kg dvakrát denně vede ke stejné průměrné plazmatické koncentraci ustáleného stavu jako u dospělých užívajících 100 mg dvakrát denně.
Tělesná hmotnost
Byl odhadnut 40% pokles plazmatické koncentrace ustáleného stavu v rozsahu tělesné hmotnosti od 46 kg do 115 kg. To však není považováno za klinicky významný rozdíl ve farmakokinetice brivaracetamu.
Pohlaví
Nejsou žádné klinicky významné rozdíly ve farmakokinetice brivaracetamu mezi pohlavími.
Rasa
Farmakokinetika brivaracetamu nebyla významně ovlivněna rasou (kavkazská, asijská) při populačním farmakokinetickém modelování u pacientů s epilepsií. Počet pacientů s jiným etnickým pozadím byl omezený.
Farmakokinetické/farmakodynamické vztahy
EC50 (plazmatická koncentrace brivaracetamu odpovídající 50 % maximálního účinku) byla odhadnuta na 0,57 mg/l. Tato plazmatická koncentrace je mírně nad mediánem expozice po podávání brivaracetamu v dávkách 50 mg/den. Další snížení frekvence záchvatů se dostavuje při zvýšení dávky na 100 mg/den a dosahuje plató při dávce 200 mg/den.
5.3 Předklinické údaje vztahující se k bezpečnosti
Ve farmakologických studiích bezpečnosti měly převládající účinky souvislost s CNS (zejména přechodná deprese CNS a snížení spontánní lokomoční aktivity) a byly pozorovány při násobcích (vyšších než 50x) farmakologicky aktivní dávky brivaracetamu 2 mg/kg. Učení a funkce paměti nebyly neovlivněny.
Nálezy nepozorované v klinických studiích, ale pozorované v toxikologických studiích s opakovaným podáváním u psů v podobné expozici ke klinické plazmatické AUC, jednalo se o hepatotoxické účinky (zejména porfyrie). Toxikologické údaje shromážděné o brivaracetamu a o strukturálně příbuzných sloučeninách ale ukazují, že se jaterní změny u psů vyvinuly působením mechanismů, které nejsou pro člověka relevantní. Žádné nežádoucí změny jater nebyly pozorovány u potkanů a opic při dlouhodobém podávání brivaracetamu s expozicí jasně převyšující AUC expozici 5 až 42x. CNS příznaky u opic (pády, ztráta rovnováhy, těžkopádné pohyby) se vyskytly u 64násobků klinické Cmax. Tyto účinky byly méně patrné v průběhu času.
Studie genotoxicity neodhalily žádnou mutagenní nebo klastogenní aktivitu. Studie kancerogenity neprokázaly žádný onkogenní potenciál u potkanů, zatímco zvýšený výskyt hepatocelulárních nádorů u myších samců je považován za důsledek negenotoxického fenoménu, známého u hlodavců, jehož mechanismus účinku se vztahuje k indukci jaterních ezymů, podobnou jako po fenobarbitalu.
Brivaracetam neovlivnil fertilitu samic ani samců, neprokázal se žádný teratogenní potenciál u potkanů nebo králíků. Embryotoxicita byla pozorována u králíků při dávce brivaracetamu toxické pro matky s expozicí 8x vyšší než klinická AUC expozice při maximální doporučené dávce.
U potkanů bylo prokázáno, že brivaracetam snadno přestupuje transplacentárně a je vylučován do mateřského mléka u kojících samic potkanů v koncentracích podobným plazmatickým koncentracím u matek.
Brivaracetam u potkanů neprokázal žádný potenciál ke zneužívání.
Studie u nedospělých zvířat
U nedospělých potkanů expoziční hladiny 6-15násobné klinické AUC expozice brivaracetamu při maximální doporučené dávce vyvolávaly vývojové nežádoucí účinky (např. mortalitu, klinické projevy, snížení tělesné hmotnosti a pokles váhy mozku). Neobjevily se žádné nežádoucí účinky u funkcí CNS, žádné neuropatologické a histopatologické vyšetření mozku. U nedospělých psů byly brivaracetamem indukované změny spojené s 6násobným zvýšením hladin AUC, podobným změnám pozorovaným u dospělých zvířat. Nebyly pozorovány žádné nežádoucí účinky ve standardních ukazatelích vývoje nebo maturace.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
trihydrát octanu sodného kyselina octová 99% (k úpravě pH) chlorid sodný voda na injekci
6.2 Inkompatibility
Tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti
3 roky.
Bylo zjištěno, že po naředění je injekční/infuzní roztok brivaracetamu fyzikálně kompatibilní a chemicky stabilní při mísení s ředidly uvedenými v bodě 6.6 po dobu 24 hodin a při uchovávání v PVC nebo polyolefinových vacích při teplotě až do 25°C. Z mikrobiologického hlediska má být přípravek použit okamžitě. Není-li použit okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou v odpovědnosti uživatele.
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
Podmínky uchovávání tohoto léčivého přípravku po jeho naředění jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
Injekční/infuzní roztok 10 mg/ml je balen skleněných injekčních lahvičkách (třídy I) o nominální kapacitě 6 ml se silikonizovanou bromobutylovou pryžovou zátkou a Al/polypropylenovým odtrhávacím víčkem. Jedna injekční lahvička na jednorázové podání obsahuje extrahovatelný objem minimálně 5,0 ml injekčního/infuzního roztoku.
Jedna krabička obsahuje 10 injekčních lahviček.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Tento přípravek je pouze pro jednorázové použití, nepoužitý roztok musí být zlikvidován.
Přípravek s výskytem částic nebo změnou barvy se nesmí použít.
Injekční/infuzní roztok briavaracetamu je fyzikálně kompatibilní a chemicky stabilní při mísení s následujícími ředidly:
Ředidla:
• injekční roztok chloridu sodného 9 mg/ml (0,9%)
• injekční roztok glukosy 50 mg/ml (5%)
• injekční Ringerův roztok s laktátem.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
UCB Pharma S.A.
Allée de la Recherche 60 B-1070 Brusel Belgie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/15/1073/022
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 14. ledna 2016
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu
PŘÍLOHA II
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného za propouštění šarží
UCB Pharma S.A.
Chemin du Foriest B-1420 Braine lAlleud Belgie
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis.
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl.
107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
Držitel rozhodnutí o registraci předloží první pravidelně aktualizovanou zprávu o bezpečnosti pro tento léčivý přípravek do 6 měsíců od jeho registrace.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
PŘÍLOHA III
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
Briviact 10 mg potahované tablety brivaracetamum
Jedna potahovaná tableta obsahuje brivaracetamum 10 mg.
Obsahuje sodnou sůl kroskarmelózy, monohydrát laktózy a laktózu.
Více údajů viz příbalová informace. (Neuvádí se na krabičkách pro 14potahovaných tablet)
14 potahovaných tablet 56 potahovaných tablet 100 x 1 potahovaná tableta 14 x 1 potahovaná tableta
Před použitím si přečtěte příbalovou informaci. Perorální podání
Uchovávejte mimo dohled a dosah dětí.
EXP
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
UCB Pharma S.A.
Allée de la Recherche 60 B-1070 Brusel Belgie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/15/1073/001 14 potahovaných tablet EU/1/15/1073/002 56 potahovaných tablet EU/1/15/1073/003 100 x 1 potahovaná tableta EU/1/15/1073/023 14 x 1 potahovaná tableta
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
Briviact 10 mg
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
<2D čárový kód s jedinečným identifikátorem>
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
PC:
SN:
NN:
Briviact 10 mg potahované tablety brivaracetamum
Jedna potahovaná tableta obsahuje brivaracetamum 10 mg.
Obsahuje sodnou sůl kroskarmelózy, monohydrát laktózy a laktózu.
Více údajů viz příbalová informace. (Neuvádí se na krabičkách pro 14 potahovaných tablet)
Multipack: 168 (3 balení po 56) potahovaných tablet
Před použitím si přečtěte příbalovou informaci. Perorální podání
Uchovávejte mimo dohled a dosah dětí.
EXP
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
UCB Pharma S.A.
Allée de la Recherche 60 B-1070 Brusel Belgie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/15/1073/004 168 (3 balení po 56) potahovaných tablet
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
Briviact 10 mg
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
<2D čárový kód s jedinečným identifikátorem>
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
PC:
SN:
NN:
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU
VNITŘNÍ KRABIČKA MULTIPACKU (3 BALENÍ PO 56 POTAHOVANÝCH TABLETÁCH) (BEZ BLUE BOXU)_
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Briviact 10 mg potahované tablety brivaracetamum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna potahovaná tableta obsahuje brivaracetamum 10 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje sodnou sůl kroskarmelózy, monohydrát laktózy a laktózu.
Více údajů viz příbalová informace. (Neuvádí se na krabičkách pro 14 potahovaných tablet)
4. LÉKOVÁ FORMA A OBSAH BALENÍ
56 potahovaných tablet. Součást balení multipack, není možné prodávat odděleně.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
UCB Pharma S.A.
Allée de la Recherche 60 B-1070 Brusel Belgie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Briviact 10 mg

Briviact 10 mg tablety brivaracetamum
UCB Pharma S.A. (logo)
EXP
Lot
Kalendářní dny: Po, Út, St, Čt, Pá, So, Ne
(neuvádí se u velikosti balení 14 x 1 a 100 x 1 potahovaná tableta)
Briviact 25 mg potahované tablety brivaracetamum
Jedna potahovaná tableta obsahuje brivaracetamum 25 mg.
Obsahuje sodnou sůl kroskarmelózy, monohydrát laktózy a laktózu..
Více údajů viz příbalová informace. (Neuvádí se na krabičkách pro 14 potahovaných tablet)
14 potahovaných tablet 56 potahovaných tablet 100 x 1 potahovaná tableta 14 x 1 potahovaná tableta
Před použitím si přečtěte příbalovou informaci. Perorální podání
Uchovávejte mimo dohled a dosah dětí.
EXP
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
UCB Pharma S.A.
Allée de la Recherche 60 B-1070 Brusel Belgie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/15/1073/005 14 potahovaných tablet EU/1/15/1073/006 56 potahovaných tablet EU/1/15/1073/007 100 x 1 potahovaná tableta EU/1/15/1073/024 14 x 1 potahovaná tableta
13. ČÍSLO ŠARŽE
Lot
|
14. |
KLASIFIKACE PRO VÝDEJ |
|
15. |
NÁVOD K POUŽITÍ |
|
16. |
INFORMACE V BRAILLOVĚ PÍSMU |
|
Briviact 25 mg | |
|
17. |
JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD |
|
<2D |
čárový kód s jedinečným identifikátorem> |
|
18. |
JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM |
PC:
SN:
NN:
Briviact 25 mg potahované tablety brivaracetamum
Jedna potahovaná tableta obsahuje brivaracetamum 25 mg.
Obsahuje sodnou sůl kroskarmelózy, monohydrát laktózy a laktózu.
Více údajů viz příbalová informace. (Neuvádí se na krabičkách pro 14potahovaných tablet)
Multipack: 168 (3 balení po 56) potahovaných tablet
Před použitím si přečtěte příbalovou informaci. Perorální podání.
Uchovávejte mimo dohled a dosah dětí.
EXP
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
UCB Pharma S.A.
Allée de la Recherche 60 B-1070 Brusel Belgie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/15/1073/008 168 (3 balení po 56) potahovaných tablet
13. ČÍSLO ŠARŽE
Lot
|
14. |
KLASIFIKACE PRO VÝDEJ |
|
15. |
NÁVOD K POUŽITÍ |
|
16. |
INFORMACE V BRAILLOVĚ PÍSMU |
|
Briviact 25 mg | |
|
17. |
JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD |
|
<2D |
čárový kód s jedinečným identifikátorem .> |
|
18. |
JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM |
PC:
SN:
NN:
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU
VNITŘNÍ KRABIČKA MULTIPACKU (3 BALENÍ PO 56 POTAHOVANÝCH TABLETÁCH) (BEZ BLUE BOXU)_
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Briviact 25 mg potahované tablety brivaracetamum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna potahovaná tableta obsahuje brivaracetamum 25 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje sodnou sůl kroskarmelózy, monohydrát laktózy a laktózu..
Více údajů viz příbalová informace. (Neuvádí se na krabičkách pro 14potahovaných tablet)
4. LÉKOVÁ FORMA A OBSAH BALENÍ
56 potahovaných tablet
Součást balení multipack, není možné prodávat odděleně.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
UCB Pharma S.A.
Allée de la Recherche 60 B-1070 Brusel Belgie
v , v ,
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
v v
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
Briviact 25 mg

Briviact 25 mg tablety brivaracetamum
UCB Pharma S.A. (logo)
EXP
Lot
Kalendářní dny: Po, Út, St, Čt, Pá, So, Ne
(neuvádí se u velikosti balení 14 x 1 a 100 x 1 potahovaná tableta)
Briviact 50 mg potahované tablety brivaracetamum
Jedna potahovaná tableta obsahuje brivaracetamum 50 mg.
Obsahuje sodnou sůl kroskarmelózy, monohydrát laktózy a laktózu..
Více údajů viz příbalová informace. (Neuvádí se na krabičkách pro 14potahovaných tablet)
14 potahovaných tablet 56 potahovaných tablet 100 x 1 potahovaná tableta 14 x 1 potahovaná tableta
Před použitím si přečtěte příbalovou informaci. Perorální podání
Uchovávejte mimo dohled a dosah dětí.
EXP
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
UCB Pharma S.A.
Allée de la Recherche 60 B-1070 Brusel Belgie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/15/1073/009 14 potahovaných tablet EU/1/15/1073/010 56 potahovaných tablet EU/1/15/1073/011 100 x 1 potahovaná tableta EU/1/15/1073/025 14 x 1 potahovaná tableta
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
Briviact 50 mg
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
<2D čárový kód s jedinečným identifikátorem>
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
PC:
SN:
NN:
Briviact 50 mg potahované tablety brivaracetamum
Jedna potahovaná tableta obsahuje brivaracetamum 50 mg.
Obsahuje sodnou sůl kroskarmelózy, monohydrát laktózy a laktózu.
Více údajů viz příbalová informace. (Neuvádí se na krabičkách pro 14 potahovaných tablet)
Multipack: 168 (3 balení po 56) potahovaných tablet
Před použitím si přečtěte příbalovou informaci. Perorální podání
Uchovávejte mimo dohled a dosah dětí.
EXP
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
UCB Pharma S.A.
Allée de la Recherche 60 B-1070 Brusel Belgie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/15/1073/012 168 (3 balení po 56) potahovaných tablet
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
Briviact 50 mg
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
<2D čárový kód s jedinečným identifikátorem>
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
PC:
SN:
NN:
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU
VNITŘNÍ KRABIČKA MULTIPACKU (3 BALENÍ PO 56 POTAHOVANÝCH TABLETÁCH) (BEZ BLUE BOXU)_
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Briviact 50 mg potahované tablety brivaracetamum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna potahovaná tableta obsahuje brivaracetamum 50 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje sodnou sůl kroskarmelózy, monohydrát laktózy a laktózu.
Více údajů viz příbalová informace. (Neuvádí se na krabičkách pro 14potahovaných tablet)
4. LÉKOVÁ FORMA A OBSAH BALENÍ
56 potahovaných tablet
Součást balení multipack, není možné prodávat odděleně.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
UCB Pharma S.A.
Allée de la Recherche 60 B-1070 Brusel Belgie
v , v ,
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
v v
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
Briviact 50 mg

Briviact 50 mg tablety brivaracetamum
UCB Pharma S.A. (logo)
EXP
Lot
Kalendářní dny: Po, Út, St, Čt, Pá, So, Ne
(neuvádí se u velikosti balení 14 x 1 a 100 x 1 potahovaná tableta)

Briviact 75 mg potahované tablety brivaracetamum
Jedna potahovaná tableta obsahuje brivaracetamum 75 mg.
Obsahuje sodnou sůl kroskarmelózy, monohydrát laktózy a laktózu.
Více údajů viz příbalová informace. (Neuvádí se na krabičkách pro 14 potahovaných tablet)
14 potahovaných tablet 56 potahovaných tablet 100 x 1 potahovaná tableta 14 x 1 potahovaná tableta
Před použitím si přečtěte příbalovou informaci. Perorální podání
Uchovávejte mimo dohled a dosah dětí.
EXP
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
UCB Pharma S.A.
Allée de la Recherche 60 B-1070 Brusel Belgie
12. REGISTRAČNÍ ČÍSLO/CÍSLA
EU/1/15/1073/013 14 potahovaných tablet EU/1/15/1073/014 56 potahovaných tablet EU/1/15/1073/015 100 x 1 potahovaná tableta EU/1/15/1073/026 14 x 1 potahovaná tableta
13. ČÍSLO ŠARŽE
Lot
|
14. |
KLASIFIKACE PRO VÝDEJ |
|
15. |
NÁVOD K POUŽITÍ |
|
16. |
INFORMACE V BRAILLOVĚ PÍSMU |
|
Briviact 75 mg | |
|
17. |
JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD |
|
<2D |
čárový kód s jedinečným identifikátorem> |
|
18. |
JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM |
PC:
SN:
NN:
Briviact 75 mg potahované tablety brivaracetamum
Jedna potahovaná tableta obsahuje brivaracetamum 75 mg.
Obsahuje sodnou sůl kroskarmelózy, monohydrát laktózy a laktózu.
Více údajů viz příbalová informace. (Neuvádí se na krabičkách pro 14 potahovaných tablet)
Multipack: 168 (3 balení po 56) potahovaných tablet.
Před použitím si přečtěte příbalovou informaci. Perorální podání
Uchovávejte mimo dohled a dosah dětí.
EXP
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
UCB Pharma S.A.
Allée de la Recherche 60 B-1070 Brusel Belgie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/15/1073/016 168 (3 balení po 56) potahovaných tablet
13. ČÍSLO ŠARŽE
Lot
|
14. |
KLASIFIKACE PRO VÝDEJ |
|
15. |
NÁVOD K POUŽITÍ |
|
16. |
INFORMACE V BRAILLOVĚ PÍSMU |
|
Briviact 75 mg | |
|
17. |
JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD |
|
<2D |
čárový kód s jedinečným identifikátorem> |
|
18. |
JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM |
PC:
SN:
NN:
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU
VNITŘNÍ KRABIČKA MULTIPACKU (3 BALENÍ PO 56 POTAHOVANÝCH TABLETÁCH) (BEZ BLUE BOXU)_
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Briviact 75 mg potahované tablety brivaracetamum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna potahovaná tableta obsahuje brivaracetamum 75 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje sodnou sůl kroskarmelózy, monohydrát laktózy a laktózu.
Více údajů viz příbalová informace. (Neuvádí se na krabičkách pro 14 potahovaných tablet)
4. LÉKOVÁ FORMA A OBSAH BALENÍ
56 potahovaných tablet
Součást balení multipack, není možné prodávat odděleně.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
UCB Pharma S.A.
Allée de la Recherche 60 B-1070 Brusel Belgie
v , v ,
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
v v
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
Briviact 75 mg

Briviact 75 mg tablety brivaracetamum
UCB Pharma S.A. (logo)
EXP
Lot
Kalendářní dny: Po, Út, St, Čt, Pá, So, Ne
(neuvádí se u velikosti balení 14 x 1 a 100 x 1 potahovaná tableta)

Briviact 100 mg potahované tablety brivaracetamum
Jedna potahovaná tableta obsahuje brivaracetamum 100 mg.
Obsahuje sodnou sůl kroskarmelózy, monohydrát laktózy a laktózu.
Více údajů viz příbalová informace. (Neuvádí se na krabičkách pro 14 potahovaných tablet)
14 potahovaných tablet 56 potahovaných tablet 100 x 1 potahovaná tableta 14 x 1 potahovaná tableta
Před použitím si přečtěte příbalovou informaci. Perorální podání
Uchovávejte mimo dohled a dosah dětí.
EXP
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
UCB Pharma S.A.
Allée de la Recherche 60 B-1070 Brusel Belgie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/15/1073/017 14 potahovaných tablet EU/1/15/1073/018 56 potahovaných tablet EU/1/15/1073/019 100 x 1 potahovaná tableta EU/1/15/1073/027 14 x 1 potahovaná tableta
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
Briviact 100 mg
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
<2D čárový kód s jedinečným identifikátorem>
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
PC:
SN:
NN:
Briviact 100 mg potahované tablety brivaracetamum
Jedna potahovaná tableta obsahuje brivaracetamum 100 mg.
Obsahuje sodnou sůl kroskarmelózy, monohydrát laktózy a laktózu.
Více údajů viz příbalová informace. (Neuvádí se na krabičkách pro 14 potahovaných tablet)
Multipack: 168 (3 balení po 56) potahovaných tablet
Před použitím si přečtěte příbalovou informaci. Perorální podání
Uchovávejte mimo dohled a dosah dětí.
EXP
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
UCB Pharma S.A.
Allée de la Recherche 60 B-1070 Brusel Belgie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/15/1073/020 168 (3 balení po 56) potahovaných tablet
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
Briviact 100 mg
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
<2D čárový kód s jedinečným identifikátorem>
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
PC:
SN:
NN:
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU
VNITŘNÍ KRABIČKA MULTIPACKU (3 BALENÍ PO 56 POTAHOVANÝCH TABLETÁCH) (BEZ BLUE BOXU)_
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Briviact 100 mg potahované tablety brivaracetamum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna potahovaná tableta obsahuje brivaracetamum 100 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje sodnou sůl kroskarmelózy, monohydrát laktózy a laktózu.
Více údajů viz příbalová informace. (Neuvádí se na krabičkách pro 14 potahovaných tablet)
4. LÉKOVÁ FORMA A OBSAH BALENÍ
56 potahovaných tablet
Součást balení multipack, není možné prodávat odděleně.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
UCB Pharma S.A.
Allée de la Recherche 60 B-1070 Brusel Belgie
v , v ,
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
v v
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
Briviact 100 mg

Briviact 100 mg tablety brivaracetamum
UCB Pharma S.A. (logo)
EXP
Lot
Kalendářní dny: Po, Út, St, Čt, Pá, So, Ne
(neuvádí se u velikosti balení 14 x 1 a 100 x 1 potahovaná tableta)
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Briviact 10 mg/ml perorální roztok brivaracetamum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jeden ml perorálního roztoku obsahuje brivaracetamum 10 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje dihydrát natrium-citrátu, sodnou sůl karmelosy, methylparaben (E218), krystalizující sorbitol 70% a glycerol 85% (E422).
Více údajů viz příbalová informace. (pouze na vnější krabičce)
4. LÉKOVÁ FORMA A OBSAH BALENÍ
300 ml perorálního roztoku
Použijte přiloženou 10ml perorální stříkačku.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
Po prvním otevření lahvičky spotřebujte do 5 měsíců. Datum otevření (pouze na vnější krabičce)
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
UCB Pharma S.A.
Allée de la Recherche 60 (adresa pouze na vnější krabičce)
B-1070 Brusel
Belgie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/15/1073/021 perorální roztok s 10ml perorální stříkačkou
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
Briviact 10 mg/ml (pouze na vnější krabičce)
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
<2D čárový kód s jedinečným identifikátorem>
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
PC:
SN:
NN:
Briviact 10 mg/ml injekční/infuzní roztok brivaracetamum
Jeden ml injekčního/mfuzního roztoku obsahuje brivaracetamum 10 mg. Jedna injekční lahvička o obsahu 5 ml obsahuje brivaracetamum 50 mg.
Dále obsahuje trihydrát octanu sodného, chlorid sodný. Více údajů viz příbalová informace.
50 mg/5 ml
10 injekčních lahviček s injekčmm/infuzním roztokem
Před použitím si přečtěte příbalovou informaci. Intravenózní podání.
Uchovávejte mimo dohled a dosah dětí.
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
UCB Pharma S.A.
Allée de la Recherche 60 B-1070 Brusel Belgie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/15/1073/022
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
Nevyžaduje se - odůvodnění přijato.
17. JEDINEČNÝ IDENTIFIKÁTOR - 2D ČÁROVÝ KÓD
<2D čárový kód s jedinečným identifikátorem>
18. JEDINEČNÝ IDENTIFIKÁTOR - DATA ČITELNÁ OKEM
PC:
SN:
NN:
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITRNÍM OBALU INJEKČNÍ LAHVIČKA
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Briviact 10 mg/ml injekce/infuze
brivaracetamum
i.v.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST OBJEM NEBO POČET
50 mg/5 ml
6. JINÉ
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro pacienta
Briviact 10 mg potahované tablety Briviact 25 mg potahované tablety Briviact 50 mg potahované tablety Briviact 75 mg potahované tablety Briviact 100 mg potahované tablety brivaracetamum
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí
ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je Briviact a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete Briviact užívat
3. Jak se Briviact užívá
4. Možné nežádoucí účinky
5. Jak Briviact uchovávat
6. Obsah balení a další informace
1. Co je Briviact a k čemu se používá
Co je Briviact
Briviact obsahuje léčivou látku brivaracetam. Ta patří do skupiny léků nazývaných „antiepileptika“. Tyto léky se používají k léčbě epilepsie.
K čemu se Briviact používá
Briviact se používá u dospělých a dospívajících ve věku od 16 let k léčbě určitých forem epilepsie, pro něž je charakteristický výskyt „parciálních záchvatů s nebo bez sekundární generalizace“ Parciální záchvaty jsou záchvaty, které začínají ovlivněním pouze jedné strany mozku. Tyto parciální záchvaty se mohou rozšířit a postihnout rozsáhlejší oblasti na obou stranách mozku - což se nazývá „sekundární generalizace“. Budete užívat tento přípravek ke snížení počtu Vašich záchvatů. Briviact se užívá současně s jinými léky na epilepsii.
2. Čemu musíte věnovat pozornost, než začnete Briviact užívat Neužívejte Briviact, jestliže:
- jste alergický(á) na brivaracetam, jiné deriváty pyrollidonu nebo na kteroukoli další složku
tohoto přípravku (uvedenou v bodě 6). Pokud se nejste jistý(á), poraďte se svým lékařem nebo lékárníkem před zahájením užívání přípravku Briviact.
Upozornění a opatření
Před užitím přípravku Briviact se poraďte se svým lékařem nebo lékárníkem, jestliže:
- máte myšlenky ublížit si nebo spáchat sebevraždu. U malého počtu osob léčených antiepileptiky, jako je Briviact, se vyskytly myšlenky na sebepoškození nebo sebevraždu. Pokud se kdykoli u Vás objeví podobné myšlenky, neprodleně kontaktujte svého lékaře.
- pokud máte problémy s játry, lékař může upravit Vaši dávku.
Děti a mladí lidé
Briviact se nedoporučuje k podávání dětem a mladým lidem ve věku pod 16 let.
Další léčivé přípravky a Briviact
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Váš lékař bude zejména potřebovat nastavit Vaši dávku přípravku Briviact, jestliže užíváte následující léčivé látky:
• rifampicin - léčivá látka používaná k léčbě bakteriálních infekcí
• třezalka tečkovaná (známá také jako Hypericum perforatum), rostlinný přípravek používaný k léčbě deprese, úzkosti a jiných stavů.
Briviact s alkoholem
• Kombinace tohoto přípravku s alkoholem se nedoporučuje.
• Jestliže při užívání přípravku Briviact pijete alkohol, negativní účinky alkoholu mohou být
zvýšeny.
Těhotenství a kojení
Nedoporučuje se užívat Briviact, jestliže jste těhotná nebo kojíte, protože účinek přípravku Briviact na těhotenství a nenarozené dítě nebo novorozeně není znám. Poraďte se neprodleně se svým lékařem, jestliže jste těhotná nebo plánujete otěhotnět.
Nepřerušujte léčbu bez porady se svým lékařem, protože by to mohlo vést ke zvýšení Vašich záchvatů a poškodit Vaše dítě.
Řízení dopravních prostředků a obsluha strojů
- Můžete se cítit ospale, pociťovat závratě nebo únavu při užívání přípravku Briviact.
- Tyto účinky jsou pravděpodobnější na začátku léčby nebo po zvýšení dávky.
- Neřiďte vozidla, nejezděte na kole a nepoužívejte žádné nástroje nebo stroje až do doby, kdy budete vědět, jakým způsobem na Vás lék působí.
Briviact obsahuje laktózu
Potahované tablety Briviact obsahují laktózu (druh cukru). Jestliže Vám lékař řekl, že nesnášíte některé cukry, poraďte se s ním, než začnete tento léčivý přípravek užívat.
3. Jak se Briviact užívá
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Budete užívat Briviact společně s jinými léky na epilepsii.
Jaké množství užívat
• Doporučená dávka je mezi 50 mg a 200 mg každý den. V tomto rozmezí dávek se může lékař rozhodnout a upravit pro Vás optimální dávku.
• Užívejte přípravek ve dvou stejných rozdělených dávkách - jednou ráno a jednou večer, každý den přibližně ve stejnou dobu.
Lidé s problémy s játry
Pokud máte problémy s játry, maximální dávka, kterou budete užívat, je 150 mg denně.
Jak se tablety Briviact užívají
• Tablety se polykají celé a zapíjejí se sklenicí tekutiny.
• Přípravek lze užívat současně s jídlem i bez jídla.
Jak dlouho se Briviact užívá
Briviact je určen k dlouhodobé léčbě - pokračujte v užívání přípravku Briviact, dokud Vám lékař nedoporučí jeho užívání ukončit.
Jestliže jste užil(a) více přípravku Briviact, než jste měl(a)
Pokud se domníváte, že jste užil(a) vyšší dávku přípravku Briviact, než jste měl(a), informujte prosím svého lékaře. Můžete pociťovat závrať a ospalost.
Jestliže jste zapomněl(a) užít Briviact
• Pokud zapomenete užít dávku, užijte ji, jakmile si vzpomenete.
• Potom si vezměte svou následující dávku v době, kdy byste ji normálně užil(a).
• Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku.
• Jestliže si nejste jistý(á), co dělat, kontaktujte svého lékaře nebo lékárníka.
Jestliže jste přestal(a) užívat Briviact
- Nepřestávejte užívat tento lék, pokud Vám to nedoporučil lékař. Ukončení léčby může zvýšit počet záchvatů, kterými trpíte.
- Pokud Vám lékař sdělí, že máte tento lék přestat užívat, bude Vám postupně snižovat užívanou dávku. To pomůže zabránit návratu záchvatů nebo jejich zhoršení.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Velmi časté: mohou postihnout více než 1 z 10 pacientů
- pocit ospalosti nebo závrati.
Časté: mohou postihnout až 1 z 10 pacientů
- chřipka
- pocit velké únavy
- křeče, pocit „točení hlavy” (vertigo)
- nevolnost a zvracení, zácpa
- deprese, úzkost, nespavost (insomnie), podrážděnost
- infekce nosu a hrdla (jako je „běžná rýma”), kašel
- snížená chuť k jídlu.
Méně časté: postihují 1 ze 100 pacientů
- psychotické poruchy, agresivita, myšlenky a pokusy o sebepoškození nebo na sebevraždu, nervózní vzrušení (agitovanost)
- pokles počtu bílých krvinek (nazývá se „neutropenie”) - zjistí se v testech krve.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, nebo lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak Briviact uchovávat
• Uchovávejte tento přípravek mimo dohled a dosah dětí.
• Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a blistru za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
• Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
• Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co Briviact obsahuje
Léčivou látkou je brivaracetamum.
Jedna potahovaná tableta obsahuje 10 mg, 25 mg, 50 mg, 75 mg nebo 100 mg brivaracetamu.
Dalšími složkami jsou:
Jádro tablety
sodná sůl kroskarmelózy, monohydrát laktózy, betadex, laktóza, magnesium-stearát.
Potahová soustava
• potahované tablety 10 mg: polyvinylalkohol, oxid titaničitý (E171), makrogol 3350, mastek.
• potahované tablety 25 mg: polyvinylalkohol, oxid titaničitý (E171), makrogol 3350, mastek, žlutý oxid železitý (E172), černý oxid železitý (E172).
• potahované tablety 50 mg: polyvinylalkohol, oxid titaničitý (E171), makrogol 3350, mastek, žlutý oxid železitý (E172), červený oxid železitý(E172).
• potahované tablety 75 mg: polyvinylalkohol, oxid titaničitý (E171), makrogol 3350, mastek, žlutý oxid železitý (E172), červený oxid železitý (E172), černý oxid železitý (E172).
• potahované tablety 100 mg: polyvinylalkohol, oxid titaničitý (E171), makrogol 3350, mastek, žlutý oxid železitý (E172), černý oxid železitý (E172).
Jak Briviact vypadá a co obsahuje toto balení
Briviact 10 mg jsou bílé až téměř bílé, kulaté potahované tablety, průměr 6,5 mm, s vyraženým „u10“ na jedné straně.
Briviact 25 mg jsou šedé, oválné potahované tablety o rozměrech 8,9 mm x 5,0 mm a s vyraženým „u25“ na jedné straně.
Briviact 50 mg jsou žluté, oválné potahované tablety o rozměrech 11,7 mm x 6,6 mm a s vyraženým „u50“ na jedné straně.
Briviact 75 mg jsou nachové, oválné potahované tablety o rozměrech 13,0 mm x 7,3 mm a s vyraženým „u75“ na jedné straně.
Briviact 100 mg jsou zelenošedé, oválné potahované tablety o rozměrech 14,5 mm x 8,1 mm a s vyraženým „u100“ na jedné straně.
Tablety Briviact jsou baleny v blistrech v krabičkách obsahujících 14, 56 potahovaných tablet nebo 14 x 1 nebo 100 x1 potahovanou tabletu nebo multipack (vícečetné balení) obsahující 168 (3 balení po 56) potahovaných tablet.
Všechny velikosti balení jsou dostupné v PVC/PCTFE/Al blistrech.
Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci
UCB Pharma S.A., Allée de la Recherche 60, B-1070 Brusel, Belgie.
Výrobce
UCB Pharma S.A., Chemin du Foriest, B-1420 Braine-l’Alleud, Belgie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci
|
Belgie/Belgique/Belgien UCB Pharma SA/NV Tél/Tel: + 32 / (0)2 559 92 00 |
Lietuva UCB Pharma Oy Finland Tel: + 358 10 234 6800 (Suomija) |
|
Btarapna ro CH EH Etarapua EOOfl Tea.: + 359 (0) 2 962 30 49 |
Luxembourg/Luxemburg UCB Pharma SA/NV Tél/Tel: + 32 / (0)2 559 92 00 |
|
Česká republika UCB s.r.o. Tel: + 420 221 773 411 |
Magyarország UCB Magyarország Kft. Tel.: + 36-(1) 391 0060 |
|
Danmark UCB Nordic A/S Tlf: + 45 / 32 46 24 00 |
Malta Pharmasud Ltd. Tel: + 356 / 21 37 64 36 |
|
Deutschland UCB Pharma GmbH Tel: + 49 /(0) 2173 48 4848 |
Nederland UCB Pharma B.V. Tel.: + 31 / (0)76-573 11 40 |
|
Eesti UCB Pharma Oy Finland Tel: + 358 10 234 6800 (Soome) |
Norge UCB Nordic A/S Tlf: + 45 / 32 46 24 00 |
|
EXXáda UCB A.E. Tqk + 30 /2109974000 |
Osterreich UCB Pharma GmbH Tel: + 43 (1) 291 80 00 |
|
Espaňa UCB Pharma, S.A. Tel: + 34 / 91 570 34 44 |
Polska UCB Pharma Sp. z o.o. Tel: + 48 22 696 99 20 |
|
France UCB Pharma S.A. Tél: + 33 / (0)1 47 29 44 35 |
Portugal UCB Pharma (Produtos Farmaceuticos), Lda Tel: + 351 / 21 302 5300 |
|
Hrvatska Medis Adria d.o.o. Tel: +385 (0) 1 230 34 46 |
Románia UCB Pharma Romania S.R.L. Tel: + 40 21 300 29 04 |
|
Ireland UCB (Pharma) Ireland Ltd. Tel: + 353 / (0)1-46 37 395 |
Slovenija Medis, d.o.o. Tel: + 386 1 589 69 00 |
|
Island Vistor hf. Simi: + 354 535 7000 |
Slovenská republika UCB s.r.o., organizačná zložka Tel: + 421 (0) 2 5920 2020 |
Italia
UCB Pharma S.p.A.
Tel: + 39 / 02 300 791
Kúrcpog
Lifepharma (Z.A.M.) Ltd Tqk + 357 22 34 74 40
Latvija
UCB Pharma Oy Finland Tel: + 358 10 234 6800 (Somija)
Suomi/Finland
UCB Pharma Oy Finland Puh/Tel: + 358 10 234 6800
Sverige
UCB Nordic A/S
Tel: + 46 / (0) 40 29 49 00
United Kingdom
UCB Pharma Ltd.
Tel : + 44 / (0)1753 534 655
Tato příbalová informace byla naposledy revidována {měsíc/RRRR}
Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu
Příbalová informace: informace pro pacienta
Briviact 10mg/ml perorální roztok
brivaracetamum
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí
ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je Briviact a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete Briviact užívat
3. Jak se Briviact užívá
4. Možné nežádoucí účinky
5. Jak Briviact uchovávat
6. Obsah balení a další informace
1. Co je Briviact a k čemu se používá
Co je Briviact
Briviact obsahuje léčivou látku brivaracetam. Ta patří do skupiny léků nazývaných „antiepileptika“. Tyto léky se užívají k léčbě epilepsie.
K čemu se Briviact používá
Briviact se používá u dospělých a dospívajících ve věku od 16 let k léčbě určitých forem epilepsie pro něž je charakteristický výskyt „parciálních záchvatů s nebo bez sekundární generalizace“. Parciální záchvaty jsou záchvaty, které začínají ovlivněním pouze jedné strany mozku. Tyto parciální záchvaty se mohou rozšířit a postihnout rozsáhlejší oblasti na obou stranách mozku - což se nazývá „sekundární generalizace“. Budete užívat tento přípravek ke snížení počtu záchvatů. Briviact se užívá současně s jinými léky na epilepsii.
2. Čemu musíte věnovat pozornost, než začnete Briviact užívat Neužívejte Briviact, jestliže:
- jste alergický(á) na brivaracetam, jiné deriváty pyrollidonu nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6). Pokud se nejste jistý(á), poraďte se svým lékařem nebo lékárníkem před zahájením užívání přípravku Briviact.
Upozornění a opatření
Před užitím přípravku Briviact se poraďte se svým lékařem nebo lékárníkem, jestliže:
- máte myšlenky ublížit si nebo spáchat sebevraždu. U malého počtu osob léčených antiepileptiky, jako je Briviact, se vyskytly myšlenky na sebepoškození nebo sebevraždu. Pokud se kdykoli u Vás objeví podobné myšlenky, neprodleně kontaktujte svého lékaře.
- pokud máte problémy s játry, lékař může upravit Vaši dávku.
Děti a mladí lidé
Briviact se nedoporučuje k podávání dětem a mladým lidem ve věku pod 16 let.
Další léčivé přípravky a Briviact
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Váš lékař bude zejména potřebovat nastavit Vaši dávku přípravku Briviact, jestliže užíváte následující léčivé látky:
• rifampicin - léčivá látka používaná k léčbě bakteriálních infekcí
• třezalka tečkovaná (známá také jako Hypericum perforatum), rostlinný přípravek používaný k léčbě deprese, úzkosti a jiných stavů.
Briviact s alkoholem
• Kombinace tohoto přípravku s alkoholem se nedoporučuje.
• Jestliže při užívání přípravku Briviact pijete alkohol, negativní účinky alkoholu mohou být
zvýšeny.
Těhotenství a kojení
Nedoporučuje se užívat Briviact, jestliže jste těhotná nebo kojíte, protože účinek přípravku Briviact na těhotenství a nenarozené dítě nebo novorozeně není znám. Poraďte se neprodleně se svým lékařem, jestliže jste těhotná nebo plánujete otěhotnět.
Nepřerušujte léčbu bez porady se svým lékařem, protože by to mohlo vést ke zvýšení Vašich záchvatů a poškodit Vaše dítě.
Řízení dopravních prostředků a obsluha strojů
- Můžete se cítit ospale, pociťovat závratě nebo únavu při užívání přípravku Briviact.
- Tyto účinky jsou pravděpodobnější na začátku léčby nebo po zvýšení dávky.
- Neřiďte vozidla, nejezděte na kole a nepoužívejte žádné nástroje nebo stroje až do doby, kdy budete vědět, jakým způsobem na Vás lék působí.
Perorální roztok Briviact obsahuje methylparaben, sodík a sorbitol
Perorální roztok Briviact obsahuje:
- methylparaben (E218) - může způsobit alergické reakce (pravděpodobně opožděné).
- 1,16 miligramu sodíku na mililitr. Nutno vzít v úvahu u pacientů na dietě s nízkým obsahem sodíku.
- sorbitol (druh cukru). Jestliže Vám lékař řekl, že nesnášíte některé cukry, poraďte se s ním, než začnete tento léčivý přípravek užívat.
3. Jak se Briviact užívá
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Budete užívat Briviact společně s jinými léky na epilepsii.
Jaké množství užívat
• Doporučená dávka je mezi 50 mg a 200 mg každý den. V tomto rozmezí dávek se může lékař rozhodnout upravit pro Vás optimální dávku.
• Užívejte přípravek ve dvou stejných rozdělených dávkách - jednou ráno a jednou večer, každý den přibližně ve stejnou dobu.
Lidé s problémys játry
Pokud máte problémy s játry, maximální dávka, kterou budete užívat, je 150 mg denně.
Jak se perorální roztok Briviact užívá
- Můžete užívat perorální roztok Briviact samotný, nebo jej těsně před spolknutím rozředit ve vodě nebo džusu
- Přípravek lze užívat současně s jídlem i bez jídla.
Návod na použití pro pacienty/ošetřující osoby:
- Otevřete lahvičku: stiskněte víčko a otočte jím proti směru hodinových ručiček (obr. 1)

Postupujte podle těchto kroků, když užíváte Briviact poprvé:
- Sejměte adaptér z perorální stříkačky (obr. 2).
- Nasaďte adaptér shora na lahvičku (obr. 3). Ujistěte se, že je na lahvičce dobře upevněn. Po použití není třeba adaptér z lahvičky sundávat.

Postupujte podle těchto kroků pokaždé, když užíváte Briviact:
- Vezměte perorální stříkačku a nasaďte ji na otvor v adaptéru (obr. 4).
- Otočte lahvičku dnem vzhůru (obr. 5).

Držte lahvičku dnem vzhůru v jedné ruce a druhou ruku použijte k plnění perorální stříkačky.
Naplňte perorální stříkačku malým množstvím roztoku tahem pístu směrem dolů (obr. 5A), Poté zatlačte píst směrem nahoru k odstranění možných bublin vzduchu (obr. 5B).
Táhněte pístem opět dolů až ke značce na perorální stříkačce, která odpovídá dávce předepsané Vaším lékařem - údaje jsou zde uvedeny v mililitrech (ml), (obr. 5C)

Odstraňte perorální stříkačku z adaptéru (obr. 6B).

Užijte lék - zatlačte píst do perorální stříkačky až na samotné dno. Můžete užít Briviact přímo z perorální stříkačky. Pokud užíváte lék naředěný, vyprázdněte obsah perorální stříkačky do sklenice s vodou nebo džusem a veškerou tekutinu vypijte (obr. 7).

Uzavřete lahvičku plastikovým šroubovacím uzávěrem.
Perorální stříkačku myjte jen čistou vodou (obr. 8).

Jak dlouho se Briviact užívá
Briviact je určen k dlouhodobé léčbě - pokračujte v užívání přípravku Briviact, dokud Vám lékař nedoporučí jeho užívání ukončit.
Jestliže jste užil(a) více přípravku Briviact, než jste měl(a)
Pokud se domníváte, že jste užil(a) vyšší dávku přípravku Briviact, než jste měl(a), informujte prosím svého lékaře. Můžete pociťovat závrať a ospalost.
Jestliže jste zapomněl(a) užít Briviact
- Pokud zapomenete užít dávku, užijte ji, jakmile si vzpomenete.
- Potom si vezměte svou následující dávku v době, kdy byste ji normálně užil(a).
- Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku.
- Jestliže si nejste jistý(á), co dělat, kontaktujte svého lékaře nebo lékárníka.
Jestliže jste přestal(a) užívat Briviact
- Nepřestávejte užívat tento lék, pokud Vám to nedoporučil lékař. Ukončení léčby může zvýšit počet záchvatů, kterými trpíte.
- Pokud Vám lékař sdělí, že máte tento lék přestat užívat, bude Vám postupně snižovat užívanou dávku. To pomůže zabránit návratu záchvatů nebo jejich zhoršení.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Velmi časté: mohou postihnout více než 1 z 10 pacientů
- pocit ospalosti nebo závrati.
Časté: mohou postihnout až 1 z 10 pacientů
- chřipka
- pocit velké únavy
- křeče, pocit „točení hlavy” (vertigo)
- nevolnost a zvracení, zácpa
- deprese, úzkost, nespavost (insomnie), podrážděnost
- infekce nosu a hrdla (jako je „běžná rýma”), kašel
- snížená chuť k jídlu.
Méně časté: mohou postihnout 1 ze 100 pacientů
- psychotické poruchy, agresivita, myšlenky a pokusy o sebepoškození nebo na sebevraždu, nervózní vzrušení (agitovanost)
- pokles počtu bílých krvinek (nazývá se „neutropenie”) - zjistí se v testech krve.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, nebo lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak Briviact uchovávat
- Uchovávejte tento přípravek mimo dohled a dosah dětí.
- Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a lahvičce za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
- Po prvním otevření lahvičky spotřebujte do 5 měsíců.
- Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co Briviact obsahuje
Léčivou látkou je brivaracetamum.
Jeden mililitr (ml) obsahuje 10 miligramů (mg) brivaracetamu.
Dalšími složkami jsou: dihydrát natrium-citrátu, bezvodá kyselina citronová, methylparaben (E218), sodná sůl kroskarmelózy, sukralóza, krystalizující sorbitol 70%, glycerol 85% (E422), malinové aroma, čištěná voda.
Jak Briviact vypadá a co obsahuje toto balení
Perorální roztok Briviact 10 mg/ml je lehce viskózní, čirá, bezbarvá až nažloutlá tekutina.
Skleněná lahvička Briviact o obsahu 300 ml je zabalena v papírové krabici, která obsahuje perorální stříkačku o obsahu 10 ml a adaptér na stříkačku.
Držitel rozhodnutí o registraci
UCB Pharma S.A., Allée de la Recherche 60, B-1070 Brusel, Belgie.
Výrobce
UCB Pharma S.A., Chemin du Foriest, B-1420 Braine-l’Alleud, Belgie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgie/Belgique/Belgien
UCB Pharma SA/NV Tél/Tel: + 32 / (0)2 559 92 00
Bt^rapnn
ro CH EH Etnrapna EOOfl Ten.: + 359 (0) 2 962 30 49
Lietuva
UCB Pharma Oy Finland
Tel: + 358 10 234 6800 (Suomija)
Luxembourg/Luxemburg
UCB Pharma SA/NV Tél/Tel: + 32 / (0)2 559 92 00
|
Česká republika UCB s.r.o. Tel: + 420 221 773 411 |
Magyarország UCB Magyarország Kft. Tel.: + 36-(1) 391 0060 |
|
Danmark UCB Nordic A/S Tlf: + 45 / 32 46 24 00 |
Malta Pharmasud Ltd. Tel: + 356 / 21 37 64 36 |
|
Deutschland UCB Pharma GmbH Tel: + 49 /(0) 2173 48 4848 |
Nederland UCB Pharma B.V. Tel.: + 31 / (0)76-573 11 40 |
|
Eesti UCB Pharma Oy Finland Tel: + 358 10 234 6800 (Soome) |
Norge UCB Nordic A/S Tlf: + 45 / 32 46 24 00 |
|
EXXáSa UCB A.E. Tr\l: + 30 / 2109974000 |
Osterreich UCB Pharma GmbH Tel: + 43 (1) 291 80 00 |
|
Espaňa UCB Pharma, S.A. Tel: + 34 / 91 570 34 44 |
Polska UCB Pharma Sp. z o.o. Tel: + 48 22 696 99 20 |
|
France UCB Pharma S.A. Tél: + 33 / (0)1 47 29 44 35 |
Portugal UCB Pharma (Produtos Farmaceuticos), Lda Tel: + 351 / 21 302 5300 |
|
Hrvatska Medis Adria d.o.o. Tel: +385 (0) 1 230 34 46 |
Románia UCB Pharma Romania S.R.L. Tel: + 40 21 300 29 04 |
|
Ireland UCB (Pharma) Ireland Ltd. Tel: + 353 / (0)1-46 37 395 |
Slovenija Medis, d.o.o. Tel: + 386 1 589 69 00 |
|
Island Vistor hf. Simi: + 354 535 7000 |
Slovenská republika UCB s.r.o., organizačná zložka Tel: + 421 (0) 2 5920 2020 |
|
Italia UCB Pharma S.p.A. Tel: + 39 / 02 300 791 |
Suomi/Finland UCB Pharma Oy Finland Puh/Tel: + 358 10 234 6800 |
|
Kórcpog Lifepharma (Z.A.M.) Ltd Tr\l: + 357 22 34 74 40 |
Sverige UCB Nordic A/S Tel: + 46 / (0) 40 29 49 00 |
|
Latvija UCB Pharma Oy Finland Tel: + 358 10 234 6800 (Somija) |
United Kingdom UCB Pharma Ltd. Tel : + 44 / (0)1753 534 655 |
Tato příbalová informace byla naposledy revidována {měsíc/RRRR} Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu
Příbalová informace: informace pro pacienta
Briviact 10mg/ml injekční/infuzní roztok
brivaracetamum
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí
ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je Briviact a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete Briviact používat
3. Jak se Briviact používá
4. Možné nežádoucí účinky
5. Jak Briviact uchovávat
6. Obsah balení a další informace
1. Co je Briviact a k čemu se používá
Co je Briviact
Briviact obsahuje léčivou látku brivaracetam. Ta patří do skupiny léků nazývaných „antiepileptika“. Tyto léky se užívají k léčbě epilepsie.
K čemu se Briviact používá
Briviact se používá u dospělých a dospívajících ve věku od 16 let k léčbě určitých forem epilepsie, pro něž je charakteristický výskyt „parciálních záchvatů s nebo bez sekundární generalizace“. Parciální záchvaty jsou záchvaty, které začínají ovlivněním pouze jedné strany mozku. Tyto parciální záchvaty se mohou rozšířit a postihnout rozsáhlejší oblasti na obou stranách mozku - což se nazývá „sekundární generalizace“. Budete užívat tento přípravek ke snížení počtu záchvatů. Briviact se používá současně s jinými léky na epilepsii.
2. Čemu musíte věnovat pozornost, než začnete Briviact používat
Nepoužívejte Briviact, jestliže:
- jste alergický(á) na brivaracetam, jiné deriváty pyrollidonu nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6). Pokud se nejste jistý(á), poraďte se svým lékařem nebo lékárníkem před zahájením používání přípravku Briviact.
Upozornění a opatření
Před použitím přípravku Briviact se poraďte se svým lékařem nebo lékárníkem, jestliže:
- máte myšlenky ublížit si nebo spáchat sebevraždu. U malého počtu osob léčených antiepileptiky, jako je Briviact, se vyskytly myšlenky na sebepoškození nebo sebevraždu. Pokud se kdykoli u Vás objeví podobné myšlenky, neprodleně kontaktujte svého lékaře.
- pokud máte problémy s játry, lékař může upravit Vaši dávku.
Děti a mladí lidé
Briviact se nedoporučuje k podávání dětem a mladým lidem ve věku pod 16 let.
Další léčivé přípravky a Briviact
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Váš lékař bude zejména potřebovat nastavit Vaši dávku přípravku Briviact, jestliže užíváte následující léčivé látky:
• rifampicin - léčivá látka používaná k léčbě bakteriálních infekcí
• třezalka tečkovaná (známá také jako Hypericum perforatum), rostlinný přípravek používaný k léčbě deprese, úzkosti a jiných stavů.
Briviact s alkoholem
• Kombinace tohoto přípravku s alkoholem se nedoporučuje.
• Jestliže při používání přípravku Briviact pijete alkohol, negativní účinky alkoholu mohou být
zvýšeny.
Těhotenství a kojení
Nedoporučuje se používat Briviact, jestliže jste těhotná nebo kojíte, protože účinek přípravku Briviact na těhotenství a nenarozené dítě nebo novorozeně není znám. Poraďte se neprodleně se svým lékařem, jestliže jste těhotná nebo plánujete otěhotnět.
Nepřerušujte léčbu bez porady se svým lékařem, protože by to mohlo vést ke zvýšení Vašich záchvatů a poškodit Vaše dítě.
Řízení dopravních prostředků a obsluha strojů
- Můžete se cítit ospale, pociťovat závratě nebo únavu při používání přípravku Briviact.
- Tyto účinky jsou pravděpodobnější na začátku léčby nebo po zvýšení dávky.
- Neřiďte vozidla, nejezděte na kole a nepoužívejte žádné nástroje nebo stroje až do doby, kdy budete vědět, jakým způsobem na Vás lék působí.
Briviact obsahuje sodík
Injekční/infuzní roztok Briviact obsahuje 0,83 mmol (odp.19,14 mg) sodíku v injekční lahvičce.
Nutno vzít v úvahu u pacientů na dietě s nízkým obsahem sodíku.
3. Jak se Briviact používá
Vždy používejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se
se svým lékařem nebo lékárníkem.
Budete používat Briviact společně s jinými léky na epilepsii.
• Když začnete užívat tento léčivý přípravek, budete užívat Briviact perorálně (ústy, jako tablety nebo perorální roztok) nebo Vám bude podán jako injekce nebo infuze.
• Injekční/infuzní roztok Briviact se podává po kratší dobu, když nemůžete užívat Briviact perorálně.
• Můžete přecházet z perorálního podávání přípravku Briviact na injekční/infuzní a opačně.
Jaké množství bude podáno
• Doporučená dávka je mezi 50 mg a 200 mg každý den. V tomto rozmezí dávek se může lékař rozhodnout upravit pro Vás optimální dávku.
• Přípravek se podává ve dvou stejných rozdělených dávkách - jednou ráno a jednou večer, každý den přibližně ve stejnou dobu.
Lidé s problémy s játry
Pokud máte problémy s játry, maximální dávka, která Vám bude podána, je 150 mg denně.
Jak se Briviact podává
Briviact je podáván lékařem nebo zdravotní sestrou jako injekce nebo infuze do žíly. Tento přípravek je podáván pomalou injekcí do Vaší žíly nebo infuzí (kapáním) po dobu 15 minut.
Jak dlouho se Briviact podává
• Váš lékař rozhodne, kolik dní Vám bude injekce nebo infuze podávána.
• Pro dlouhodobou léčbu přípravkem Briviact Vám lékař předepíše Briviact tablety nebo perorální roztok.
Jestliže Vám bylo podáno více přípravku Briviact, než mělo
Pokud se domníváte, že Vám bylo podáno více přípravku Briviact, než mělo, informujte prosím okamžitě svého lékaře.
Jestliže přerušíte podávání přípravku Briviact
- Nepřestávejte používat tento lék, pokud Vám to nedoporučil lékař. Ukončení léčby může zvýšit počet záchvatů, kterými trpíte.
- Pokud Vám lékař sdělí, že máte tento lék přestat užívat, bude Vám postupně snižovat používanou dávku. To pomůže zabránit návratu záchvatů nebo jejich zhoršení.
Máte-li jakékoli další otázky týkající se používání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Velmi časté: mohou postihnout více než 1 z 10 pacientů
- pocit ospalosti nebo závrati.
Časté: mohou postihnout až 1 z 10 pacientů
- chřipka
- pocit velké únavy
- křeče, pocit „točení hlavy” (vertigo)
- nevolnost a zvracení, zácpa
- bolest nebo nepříjemné pocity v místě vpichu
- deprese, úzkost, nespavost (insomnie), podrážděnost
- infekce nosu a hrdla (jako je „běžná rýma”), kašel
- snížená chuť k jídlu.
Méně časté: mohou postihnout 1 ze 100 pacientů
- psychotické poruchy, agresivita, myšlenky a pokusy o sebepoškození nebo na sebevraždu, nervózní vzrušení (agitovanost)
- pokles počtu bílých krvinek (nazývá se „neutropenie”) - zjistí se v testech krve.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, nebo lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
Jak Briviact uchovávat
5.
• Uchovávejte tento přípravek mimo dohled a dosah dětí.
• Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a lahvičce za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
• Briviact může být před podáním naředěn lékařem nebo sestrou. V tomto případě má být použit ihned po naředění.
• Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
• Lahvička s injekčním roztokem přípravku Briviact smí být použita jen jednou (jednorázové použití). Veškerý nepoužitý roztok musí být zlikvidován.
• Smí se použít pouze přípravek, který neobsahuje částice nebo nezměnil barvu.
• Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co Briviact obsahuje
Léčivou látkou je brivaracetamum.
• Jeden ml obsahuje brivaracetamum 10 mg.
• Jedna 5ml lahvička obsahuje brivaracetamum 50 mg.
Dalšími složkami jsou: trihydrát octanu sodného, kyselina octová 99%, chlorid sodný, voda na injekci.
Jak Briviact vypadá a co obsahuje toto balení
Briviact 10 mg/ml injekční/infuzní roztok je čirý, bezbarvý, sterilní roztok.
Briviact 10 mg/ml injekční/ infuzní roztok v 5ml lahvičkách je balen v krabičce po 10 injekčních lahvičkách.
Držitel rozhodnutí o registraci
UCB Pharma S.A., Allée de la Recherche 60, B-1070 Brusel, Belgie.
Výrobce
UCB Pharma S.A., Chemin du Foriest, B-1420 Braine-l’Alleud, Belgie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgie/Belgique/Belgien
UCB Pharma SA/NV Tél/Tel: + 32 / (0)2 559 92 00
Eunrapnu
ro CH EH Eunrapua EOOfl Ten.: + 359 (0) 2 962 30 49
Česká republika UCB s.r.o.
Tel: + 420 221 773 411
Danmark
UCB Nordic A/S Tlf: + 45 / 32 46 24 00
Deutschland
Lietuva
UCB Pharma Oy Finland
Tel: + 358 10 234 6800 (Suomija)
Luxembourg/Luxemburg
UCB Pharma SA/NV Tél/Tel: + 32 / (0)2 559 92 00
Magyarország
UCB Magyarország Kft.
Tel.: + 36-(1) 391 0060
Malta
Pharmasud Ltd.
Tel: + 356 / 21 37 64 36
Nederland
|
Eesti UCB Pharma Oy Finland Tel: + 358 10 234 6800 (Soome) |
Norge UCB Nordic A/S Tlf: + 45 / 32 46 24 00 |
|
EkXába UCB A.E. T^k: + 30 /2109974000 |
Osterreich UCB Pharma GmbH Tel: + 43 (1) 291 80 00 |
|
Espaňa UCB Pharma, S.A. Tel: + 34 / 91 570 34 44 |
Polska UCB Pharma Sp. z o.o. Tel: + 48 22 696 99 20 |
|
France UCB Pharma S.A. Tél: + 33 / (0)1 47 29 44 35 |
Portugal UCB Pharma (Produtos Farmaceuticos), Lda Tel: + 351 / 21 302 5300 |
|
Hrvatska Medis Adria d.o.o. Tel: +385 (0) 1 230 34 46 |
Románia UCB Pharma Romania S.R.L. Tel: + 40 21 300 29 04 |
|
Ireland UCB (Pharma) Ireland Ltd. Tel: + 353 / (0)1-46 37 395 |
Slovenija Medis, d.o.o. Tel: + 386 1 589 69 00 |
|
Island Vistor hf. Simi: + 354 535 7000 |
Slovenská republika UCB s.r.o., organizačná zložka Tel: + 421 (0) 2 5920 2020 |
|
Italia UCB Pharma S.p.A. Tel: + 39 / 02 300 791 |
Suomi/Finland UCB Pharma Oy Finland Puh/Tel: + 358 10 234 6800 |
|
Kónpoq Lifepharma (Z.A.M.) Ltd Tnk: + 357 22 34 74 40 |
Sverige UCB Nordic A/S Tel: + 46 / (0) 40 29 49 00 |
|
Latvija UCB Pharma Oy Finland Tel: + 358 10 234 6800 (Somija) |
United Kingdom UCB Pharma Ltd. Tel : + 44 / (0)1753 534 655 |
UCB Pharma GmbH Tel: + 49 /(0) 2173 48 4848
UCB Pharma B.V.
Tel.: + 31 / (0)76-573 11 40
Tato příbalová informace byla naposledy revidována {měsíc/RRRR}
Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu
Následující údaje jsou určeny pouze pro zdravotnické pracovníky:
Briviact mjekční/infuzm roztok může být podán jako injekční bolus nebo jako infuze:
• Intravenózní bolus: může být podán přímo bez ředění
Intravenózní infuze: může být podána po dobu 15 minut v kompatibilním ředidle.
Briviact může být ředěn následujícími roztoky: injekční roztok chloridu sodného 9 mg/ml (0,9%), injekční roztok glukózy 50 mg/ml (5%), injekční Ringerův roztok s laktátem.
Lahvička s injekčním/infuzním roztokem přípravku Briviact smí být použita jen jednou (jednorázové použití). Veškerý nepoužitý roztok musí být zlikvidován (viz bod 3).
109