Blincyto 38,5 Mikrogramu
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
BLINCYTO 38,5 mikrogramu prášek pro koncentrát a roztok pro infuzní roztok.
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička s práškem obsahuje blinatumomabum 38,5 mikrogramu.
Rekonstitucí s vodou na injekci se získá blinatumomabum o výsledné koncentraci 12,5 mikrogramu/ml.
Blinatumomab je vyráběn ovariálními buňkami čínských křečků pomocí rekombinantní DNA technologie.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek pro koncentrát a roztok pro infuzní roztok.
BLINCYTO prášek (prášek pro koncentrát): Bílý až téměř bílý prášek. Roztok (stabilizátor): Bezbarvý až světle žlutý, čirý roztok s pH 7,0.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
BLINCYTO je indikován k léčbě dospělých s Philadelphia chromozom negativní relabovanou nebo refrakterní B-prekurzorovou akutní lymfoblastickou leukemií (ALL).
4.2 Dávkování a způsob podání
Léčba se má zahájit pod vedením a dohledem lékaře se zkušenostmi v léčbě hematologických malignit.
Při zahájení a minimálně v prvních 9 dnech prvního cyklu a prvních 2 dnech druhého cyklu se doporučuje hospitalizace.
U pacientů s klinicky relevantní patologií centrálního nervového systému (CNS) v současné době nebo v anamnéze (viz bod 4.4) je doporučena hospitalizace minimálně prvních 14 dnů prvního cyklu. Ve druhém cyklu je doporučena hospitalizace minimálně 2 dny a klinické rozhodnutí má být založeno na toleranci přípravku BLINCYTO v prvním cyklu. Je třeba dbát opatrnosti, jelikož byly pozorovány případy pozdního výskytu prvních neurologických příhod ve druhém cyklu.
Zahájení všech dalších cyklů a opakované zahájení (např. jestliže je léčba přerušena na 4 a více hodin) se doporučuje provádět za dohledu zdravotnického pracovníka nebo během hospitalizace.
Pacienti mohou dostat 2 cykly léčby. Jeden cyklus léčby představuje 4 týdny kontinuální infuze. Cykly jsou odděleny dvoutýdenním intervalem bez léčby.
Pacienti, kteří dosáhli úplné remise (CR/CRh*) po dvou léčebných cyklech, mohou dostat až 3 další cykly konsolidační léčby přípravkem BLINCYTO, a to na základě individuálního vyhodnocení poměru přínosů a rizik.
Doporučená dávka (pro pacienty s tělesnou hmotností nejméně 45 kg):
|
1. cyklus |
Dvoutýdenní interval bez léčby (29. - 42. den) |
2. cyklus a následující cykly (1. - 28. den) | |
|
Zahajovací dávka 1. - 7. den |
Další dávka 8. - 28. den | ||
|
9 pg/den kontinuální infuzí |
28 pg/den kontinuální infuzí |
28 pg/den kontinuální infuzí | |
Doporučení pro premedikaci a další medikaci
Dexamethason 20 mg intravenózně se má podat 1 hodinu před zahájením každého cyklu léčby přípravkem BLINCYTO.
Doporučuje se použít antipyretikum (např. paracetamol) ke snížení pyrexie v prvních 48 hodinách každého léčebného cyklu.
Před léčbou a během léčby přípravkem BLINCYTO se doporučuje profylaxe intratekální chemoterapií k zabránění relapsu ALL v centrálním nervovém systému.
Předléčba u pacientů se silnou nádorovou zátěží
Pacienti s > 50 % leukemických blastů nebo > 15 000/mikrolitr leukemických blastů v periferní krvi se léčí dexamethasonem (maximálně 24 mg/den).
Úpravy dávky
Dočasné nebo trvalé vysazení přípravku BLINCYTO je třeba zvážit v případě těchto závažných (stupeň 3) nebo život ohrožujících (stupeň 4) toxicit (viz bod 4.4): syndrom z uvolnění cytokinů, syndrom nádorového rozpadu, neurologická toxicita, zvýšené hladiny jaterních enzymů a všechny další klinicky významné toxicity.
Jestliže přerušení léčby po nežádoucí příhodě netrvá déle než 7 dní, pokračujte stejným cyklem do celkové délky infuze 28 dní včetně dní před a po přerušení v daném cyklu. Jestliže přerušení léčby po nežádoucí příhodě trvá déle než 7 dní, zahajte nový cyklus. Jestliže úprava toxicity trvá déle než 14 dní, vysaďte BLINCYTO trvale, pokud to není uvedeno jinak v tabulce níže.
|
Toxicita |
Stupeň* |
Postup |
|
Syndrom |
Stupeň 3 |
Přerušte BLINCYTO až do úpravy, potom opět zahajte léčbu |
|
z uvolnění |
přípravkem BLINCYTO dávkou 9 pg/den. Pokud se toxicita | |
|
cytokinů, syndrom |
neopakuje, po 7 dnech zvyšujte dávku na 28 pg/den. | |
|
nádorového rozpadu |
Stupeň 4 |
Vysaďte BLINCYTO trvale. |
|
Neurologická |
Záchvat |
V případě, že se objeví více než 1 epileptický záchvat, vysaďte |
|
toxicita |
BLINCYTO trvale. | |
|
Stupeň 3 |
Přerušte BLINCYTO, dokud toxicita nedosáhne stupně 1 (mírná) a nejméně po dobu 3 dnů, potom opět zahajte léčbu přípravkem BLINCYTO dávkou 9 pg/den. Pokud se toxicita neopakuje, po 7 dnech zvyšujte dávku na 28 pg/den. Při opětovném zahájení podejte premedikaci dexamethasonem v dávce 24 mg. Potom dávku dexamethasonu postupně snižujte během 4 dnů. Jestliže se toxicita vyskytla při dávce 9 pg/den, nebo když k úpravě toxicity je třeba více než 7 dní, vysaďte BLINCYTO trvale. | |
|
Stupeň 4 |
Vysaďte BLINCYTO trvale. | |
|
Zvýšené hladiny |
Stupeň 3 |
Je-li zvýšení klinicky významné, přerušte BLINCYTO dokud |
|
jaterních enzymů |
toxicita nedosáhne stupně 1 (mírná), potom opět zahajte léčbu přípravkem BLINCYTO dávkou 9 pg/den. Pokud se toxicita neopakuje, po 7 dnech zvyšujte dávku na 28 pg/den. | |
|
Stupeň 4 |
Zvažte trvalé vysazení přípravku BLINCYTO. | |
|
Jiné klinicky |
Stupeň 3 |
Přerušte BLINCYTO dokud toxicita nedosáhne stupně 1 (mírná), |
|
významné (podle |
potom opět zahajte léčbu přípravkem BLINCYTO dávkou 9 | |
|
určení |
pg/den. Pokud se toxicita neopakuje, po 7 dnech zvyšujte dávku | |
|
ošetřujícího lékaře) nežádoucí |
na 28 pg/den. | |
|
účinky |
Stupeň 4 |
Zvažte trvalé vysazení přípravku BLINCYTO. |
*Na základě společných terminologických kritérií pro nežádoucí příhody (CTCAE) NCI, verze 4.0. Stupeň 3 je těžký a stupeň 4 je život ohrožující.
Zvláštní _ populace Starší pacienti
Úprava dávky u starších pacientů (> 65 let věku) není nutná, viz bod 5.1. Zkušenosti s přípravkem BLINCYTO u pacientů > 75 let věku jsou omezené.
Porucha funkce ledvin
Na základě farmakokinetických analýz úprava dávky není nutná u pacientů s lehkou až středně těžkou poruchou funkce ledvin (viz bod 5.2). Bezpečnost a účinnost přípravku BLINCYTO u pacientů s těžkou poruchou funkce ledvin nebyla hodnocena.
Porucha funkce jater
Na základě farmakokinetických analýz se nepředpokládá žádný vliv stavu jaterních funkcí při zahájení léčby na expozici blinatumomabu a úprava počáteční dávky není nutná (viz bod 5.2). Bezpečnost a účinnost přípravku BLINCYTO u pacientů s těžkou poruchou funkce jater nebyla hodnocena.
Pediatrická populace
Bezpečnost a účinnost přípravku BLINCYTO u pediatrických pacientů nebyla dosud stanovena.
V současnosti dostupné údaje jsou uvedeny v bodě 4.8, ale na jejich základě nelze učinit žádná doporučení ohledně dávkování.
Způsob podání
Důležitá poznámka: Neproplachujte infuzní linky vedoucí k pacientovi, protože to může způsobit neúmyslné podání bolusu přípravku BLINCYTO. BLINCYTO se má podávat infuzí přes zvolený lumen linky.
Návod na zacházení a přípravu léčivého přípravku před podáním je uveden v bodě 6.6.
BLINCYTO infuzní roztok se podává jako kontinuální intravenózní infuze konstantní rychlostí pomocí infuzní pumpy po dobu až 96 hodin.
BLINCYTO infuzní roztok se musí podat pomocí intravenózního setu, který obsahuje in-line, sterilní, nepyrogenní in-line filtr 0,2 mikrometrů s nízkou vazbou bílkovin.
Terapeutická dávka 9 pg/den nebo 28 pg/den se podá pacientovi v infuzi o celkovém objemu 240 ml přípravku BLINCYTO infuzní roztok jednou ze 4 konstantních rychlostí s příslušnou délkou trvání infuze:
• Rychlost infuze 10 ml/hod. při trvání infuze 24 hodin
• Rychlost infuze 5 ml/hod. při trvání infuze 48 hodin
• Rychlost infuze 3,3 ml/hod. při trvání infuze 72 hodin
• Rychlost infuze 2,5 ml/hod. při trvání infuze 96 hodin
Délku trvání infuze zvolí ošetřující lékař na základě zvážení frekvence výměn infuzního vaku. Cílová podaná terapeutická dávka přípravku BLINCYTO se nemění.
Výměna infuzního vaku
Infuzní vak musí zdravotnický pracovník vyměnit minimálně každých 96 hodin z důvodů zachování sterility.
4.3 Kontraindikace
- Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
- Kojení (viz bod 4.6).
4.4 Zvláštní upozornění a opatření pro použití
Neurologické příhody
Byly pozorovány neurologické příhody včetně příhod s fatálním zakončením. Neurologické příhody 3. stupně (CTCAE verze 4.0) nebo vyššího stupně (těžké nebo život ohrožující) po zahájení léčby blinatumomabem zahrnovaly encefalopatii, epileptické záchvaty, poruchy řeči, poruchy vědomí, zmatenost a dezorientaci a poruchy koordinace a rovnováhy.
Medián doby od zahájení léčby blinatumomabem do nástupu neurologické příhody byl 9 dní. Většina příhod odezněla po přerušení léčby.
U starších pacientů byl pozorován vyšší výskyt neurologických toxicit včetně kognitivní poruchy, encefalopatie a zmatenosti.
U pacientů s anamnézou neurologických známek a příznaků (jako jsou závratě, hypestézie, hyporeflexie, třes, dysestézie, parestézie a poruchy paměti) byl pozorován vyšší výskyt neurologických příhod (jako jsou třes, závratě, stavy zmatenosti, encefalopatie a ataxie). Medián doby do výskytu neurologické příhody u těchto pacientů byl 12 dní.
Zkušenosti u pacientů s klinicky významnou patologií centrálního nervového systému (CNS) přítomnou v současné době nebo v anamnéze (např. epilepsie, epileptické záchvaty, paréza, afázie, cévní mozková příhoda, těžké poranění mozku, demence, Parkinsonova choroba, onemocnění mozečku, organický mozkový syndrom, psychóza) jsou omezené, jelikož tito pacienti byli vyloučeni z klinických studií. U této populace existuje možnost vyššího rizika neurologických příhod. Je třeba pečlivě zvážit možné přínosy léčby oproti riziku neurologických příhod a při podávání přípravku BLINCYTO je třeba těmto pacientům věnovat zvýšenou pozornost.
Zkušenosti s blinatumomabem u pacientů s dokumentovanou aktivní ALL v CNS nebo mozkomíšním moku (CSF) jsou omezené. Avšak v klinických studiích byli pacienti léčeni blinatumomabem po odstranění blastů v CSF terapií cílenou na CNS (např. intratekální chemoterapie). Proto po vyčištění CSF může být zahájena léčba přípravkem BLINCYTO.
Doporučuje se, aby před zahájením léčby přípravkem BLINCYTO byli pacienti neurologicky vyšetřeni a aby byli pacienti klinicky sledováni z hlediska přítomnosti známek a příznaků neurologických příhod (např. test psaní). Léčba těchto známek a příznaků do jejich odeznění může vyžadovat buď dočasné přerušení nebo trvalé vysazení přípravku BLINCYTO (viz bod 4.2).
V případě epileptických záchvatů se doporučuje sekundární prevence vhodnými antikonvulzivy (např. levetiracetamem).
Infekce
U pacientů léčených blinatumomabem byly pozorovány těžké infekce včetně sepse, pneumonie, bakteriemie, oportunních infekcí a infekcí v místě katétru, z nichž některé byly život ohrožující nebo fatální. Pacienti s výkonnostním stavem 2 podle Eastern Cooperative Oncology Group (ECOG) před zahájením léčby měli vyšší výskyt těžkých infekcí v porovnání s pacienty s ECOG výkonnostním stavem < 2. Zkušenosti s léčbou přípravkem BLINCYTO u pacientů s aktivní nekontrolovanou infekcí jsou omezené.
Pacienti léčeni přípravkem BLINCYTO mají být klinicky sledováni z hlediska přítomnosti známek a příznaků infekce a příslušným způsobem léčeni. Léčba infekcí vyžaduje buď dočasné přerušení nebo trvalé vysazení přípravku BLINCYTO (viz bod 4.2).
Syndrom z uvolnění cytokinů a infuzní reakce
U pacientů léčených přípravkem BLINCYTO byl hlášen syndrom z uvolnění cytokinů (CRS), který může být život ohrožující nebo fatální (stupeň > 4) (viz bod 4.8).
Závažné nežádoucí příhody, které mohou být známkami a příznaky CRS, zahrnovaly pyrexii, astenii, bolest hlavy, hypotenzi, zvýšení hladiny celkového bilirubinu a nauzeu; méně často tyto příhody vedly k vysazení přípravku BLINCYTO. Medián doby do nástupu příhod CRS byl 2 dny. Pacienti mají být pečlivě sledováni z hlediska známek a příznaků těchto příhod.
S CRS se často vyskytovaly diseminovaná intravaskulární koagulace (DIC) a syndrom zvýšené permeability kapilár (CLS, tj. hypotenze, hypoalbuminemie, otoky a hemokoncentrace) (viz bod 4.8). Pacienty se syndromem zvýšené permeability kapilár je třeba okamžitě léčit.
Hemofagocytující lymfohistiocytóza/syndrom aktivovaných makrofágů (HLH/MAS) byly méně často zaznamenány jako součást CRS.
Infuzní reakce mohou být klinicky nerozlišitelné od manifestace CRS (viz bod 4.8). Infuzní reakce byly obecně rychlé, dostavovaly se do 48 hodin od zahájení infuze. Avšak někteří pacienti uváděli opožděný nástup infuzních reakcí nebo jejich výskyt v pozdějších cyklech. Pacienty je třeba pečlivě pozorovat z hlediska infuzních reakcí, a to zejména při zahájení prvního a druhého léčebného cyklu, a
řádně je léčit. Doporučuje se použití antipyretik (např. paracetamolu) v prvních 48 hodinách každého cyklu ke snížení pyrexie. Léčba těchto příhod může vyžadovat buď dočasné přerušení nebo vysazení léčby přípravkem BLINCYTO (viz bod 4.2).
Syndrom nádorového rozpadu
U pacientů léčených přípravkem BLINCYTO byl pozorován syndrom nádorového rozpadu (TLS), který může být život ohrožující nebo fatální (stupeň > 4).
V prevenci a léčbě TLS během léčby přípravkem BLINCYTO se mají použít vhodná preventivní opatření včetně razantní hydratace a antihyperurikemické léčby (jako alopurinol nebo rasburikáza), zejména u pacientů s vyšší leukocytózou nebo vysokou nádorovou zátěží. Pacienty je třeba v prvních 48 hodinách po první infuzi pečlivě sledovat z hlediska známek a příznaků TLS včetně funkce ledvin a rovnováhy tekutin. V klinických studiích byl pozorován zvýšený výskyt TLS u pacientů se středně těžkou poruchou funkce ledvin v porovnání s pacienty s lehkou poruchou funkce ledvin nebo normální funkcí ledvin. Léčba těchto příhod může vyžadovat buď dočasné přerušení nebo vysazení léčby přípravkem BLINCYTO (viz bod 4.2).
Neutropenie a febrilní neutropenie
U pacientů léčených přípravkem BLINCYTO byla pozorována neutropenie a febrilní neutropenie včetně život ohrožujících případů. Během infuze přípravku BLINCYTO je třeba rutinně monitorovat a řádně léčit laboratorní parametry (včetně, ale ne pouze počet leukocytů a absolutní počet neutrofilů), zejména v prvních 9 dnech prvního cyklu.
Zvýšené hladiny jaterních enzymů
Při léčbě přípravkem BLINCYTO bylo zaznamenáno přechodné zvýšení jaterních enzymů. Většina těchto příhod byla pozorována v prvním týdnu po zahájení léčby a nebylo nutné přerušení ani ukončení léčby přípravkem BLINCYTO (viz bod 4.8).
Před zahájením a během léčby přípravkem BLINCYTO se má provádět monitorování hladin alanin aminotransferázy (ALT), aspartát aminotransferázy (AST), gama-glutamyl transferázy (GGT) a celkového sérového bilirubinu, a to zejména v prvních 48 hodinách prvních 2 cyklů. Léčba těchto příhod může vyžadovat buď dočasné přerušení nebo vysazení léčby přípravkem BLINCYTO (viz bod 4.2).
Leukoencefalopatie včetně progresivní multifokální leukoencefalopatie
U pacientů léčených přípravkem BLINCYTO byly při vyšetření lebky magnetickou rezonancí (MRI) pozorovány změny svědčící pro leukoencefalopatii, a to zejména u pacientů s předchozím ozařováním lebky a antileukemickou chemoterapií (včetně vysokých systémových dávek methotrexátu nebo intratekálně podávaného cytarabinu). Klinický význam těchto změn při zobrazovacím vyšetření není znám.
Vzhledem k potenciálu léku pro progresivní multifokální leukoencefalopatii (PML) mají být pacienti sledováni z hlediska přítomnosti jejích známek a příznaků. V případě podezřelé příhody je třeba zvážit konziliární vyšetření neurologem, MRI mozku a vyšetření mozkomíšního moku (CSF), viz bod 4.8.
Imunizace
Bezpečnost imunizace vakcínami s živými viry během léčby nebo po léčbě přípravkem BLINCYTO nebyla studována. Očkování vakcínami s živými viry se nedoporučuje nejméně 2 týdny před zahájením léčby přípravkem BLINCYTO, během léčby a až do úpravy B-lymfocytů na normální hodnoty po posledním léčebném cyklu.
Vzhledem k možné depleci B buněk u novorozenců po expozici blinatumomabu během těhotenství mají být novorozenci sledováni pro depleci B buněk a očkování vakcínami s živými viry se má odložit až do obnovení počtu B buněk u novorozence (viz bod 4.6).
Antikoncepce
Ženy ve fertilním věku musí během léčby a nejméně 48 hodin po léčbě přípravkem BLINCYTO používat účinnou antikoncepci (viz bod 4.6).
Chyby medikace
Při léčbě přípravkem BLINCYTO byly pozorovány chyby medikace. Je velmi důležité přísně dodržovat návody k přípravě (včetně rekonstituce a ředění) a podání, aby chyby medikace byly minimalizovány (včetně poddávkování a předávkování) (viz bod 4.2).
Pomocná látka se známým účinkem
Tento léčivý přípravek poskytne méně než 1 mmol (23 mg) sodíku během 24 hodinové infuze, tj. je v podstatě „bez sodíku“.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné studie interakcí. Výsledky testu in vitro na lidských hepatocytech naznačují, že blinatumomab neovlivňuje aktivity enzymu CYP450.
Zahájení léčby přípravkem BLINCYTO způsobuje přechodné uvolnění cytokinů v prvních dnech léčby, které mohou způsobovat supresi enzymů CYP450. Pacienti, kteří dostávají léčivé přípravky s úzkým terapeutickým indexem, které jsou substráty CYP450 a transportérů, mají být během této doby sledováni z hlediska výskytu nežádoucích účinků (např. warfarin) nebo lékových koncentrací (např. cyklosporin). Dávka souběžně používaného léčivého přípravku se má podle potřeby upravit.
4.6 Fertilita, těhotenství a kojení
Studie reprodukční toxicity s blinatumomabem nebyly provedeny. Ve studii embryo-fetální vývojové toxicity provedené u myší procházely surogátní myší molekuly placentou a nevyvolávaly embryotoxicitu nebo teratogenitu (viz bod 5.3). U březích myší byla pozorována očekávaná deplece B a T buněk, ale hematologické účinky u plodů nebyly hodnoceny.
Nejsou k dispozici žádná data o použití blinatumomabu u těhotných žen.
Blinatumomab nemá být použit v těhotenství, pokud možný přínos léčby nepřevýší možné riziko pro plod.
Ženy ve fertilním věku musí během léčby a nejméně 48 hodin po léčbě blinatumomabem používat účinnou antikoncepci (viz bod 4.4).
V případě expozice během těhotenství se dá očekávat deplece B buněk u novorozenců vzhledem k farmakologickým vlastnostem přípravku. Proto novorozenci mají být sledováni pro depleci B buněk a očkování vakcínami s živými viry se má odložit až do obnovení počtu B buněk (viz bod 4.4).
Kojení
Není známo, zda se blinatumomab nebo jeho metabolity vylučují do lidského mateřského mléka. Na základě jeho farmakologických vlastností nemůže být vyloučeno riziko pro kojence. Proto je z preventivních důvodů kojení kontraindikováno během léčby a nejméně 48 hodin po léčbě blinatumomabem.
Fertilita
Nebyly provedeny žádné studie hodnotící účinky blinatumomabu na fertilitu. Ve 13týdenních studiích toxicity se surogátní myší molekulou nebyly zjištěny žádné nežádoucí účinky na samčí nebo samičí myší reprodukční orgány (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Blinatumomab má výrazný vliv na schopnost řídit nebo obsluhovat stroje. Může se vyskytnout zmatenost a dezorientace, poruchy koordinace a rovnováhy, riziko epileptických záchvatů a poruch vědomí (viz bod 4.4). Vzhledem k potenciálu neurologických příhod pacienti léčení blinatumomabem nesmějí řídit, pracovat v rizikovém povolání nebo provádět aktivity jako je řízení či obsluha těžkých nebo potenciálně nebezpečných strojů po dobu podávání blinatumomabu. Pacienti musí být poučeni, že se u nich mohou vyskytnout neurologické příhody.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Nežádoucí účinky popsané v tomto bodu byly identifikované v pivotní klinické studii (N = 189).
Nejzávažnější nežádoucí účinky, které se mohou vyskytnout během léčby blinatumomabem jsou: infekce (31,7 %), neurologické příhody (16,4 %), neutropenie/febrilní neutropenie (15,3 %), syndrom z uvolnění cytokinů (0,5 %) a syndrom nádorového rozpadu (0,5 %).
Nejčastější nežádoucí účinky byly: infuzní reakce (67,2 %), infekce (63,0 %), pyrexie (59,8 %), bolest hlavy (34,4 %), febrilní neutropenie (28 %), periferní otoky (25,9 %), nauzea (24,3 %), hypokalemie (23,8 %), zácpa (20,6 %), anemie (20,1 %), kašel (18,5 %), průjem (18,0 %), třes (17,5 %), neutropenie (17,5 %), bolest břicha (16,9 %), insomnie (15,3 %), únava (15,3 %) a zimnice (15,3 %).
Tabulkový seznam nežádoucích účinků
Nežádoucí účinky jsou uvedeny níže podle třídy orgánových systémů a kategorie frekvence. Kategorie frekvence byly stanoveny podle přibližné četnosti výskytu hlášené pro každý nežádoucí účinek v pivotní klinické studii (N = 189). U každé třídy orgánových systémů jsou nežádoucí účinky uvedené v sestupném pořadí závažnosti.
|
Třída orgánových systémů MedDRA |
Velmi časté (> 1/10) |
Časté (> 1/100 až < 1/10) |
Méně časté (> 1/1000 až < 1/100) |
|
Infekce a infestace |
Bakteriální infekcea, b Plísňové infekce3, b Virové infekce*1, b Infekce způsobené jinými patogenyb |
Sepse | |
|
Poruchy krve a lymfatického systému |
Febrilní neutropenie Anemie Neutropenie Trombocytopenie Leukopenie |
Leukocytóza Lymfopenie | |
|
Poruchy imunitního systému |
Syndrom z uvolnění cytokinů*1 |
Cytokinová bouře Hypersenzitivita |
|
Třída orgánových systémů MedDRA |
Velmi časté (> 1/10) |
Časté (> 1/100 až < 1/10) |
Méně časté (> 1/1000 až < 1/100) |
|
Poruchy metabolismu a výživy |
Hypokalemie Hypomagnesemie Hyperglykemie Snížená chuť k jídlu |
Hypofosfatemie Hypoalbuminemie Syndrom nádorového rozpadu | |
|
Psychiatrické poruchy |
Insomnie |
Stav zmatenostia Dezorientace | |
|
Poruchy nervového systému |
Třesa Závratě |
Encefalopatiea Afázie Parestézie Konvulze Kognitivní porucha Zhoršení paměti | |
|
Srdeční poruchy | |||
|
Cévní poruchy |
Syndrom zvýšené permeability kapilár | ||
|
Respirační, hrudní a mediastinální poruchy | |||
|
Gastrointestinální poruchy |
Nauzea Zácpa Průjem Bolest břicha Zvracení | ||
|
Poruchy kůže a podkožní tkáně | |||
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Bolest zad Bolest v končetině Artralgie Bolest kostí | ||
|
Celkové poruchy a reakce v místě aplikace |
Pyrexie Periferní otok Zimnice Únava |
Otok | |
|
Vyšetření |
Zvýšení alanin aminotransferázya Zvýšení aspartát aminotransferázya |
Snížení imunoglobulinů Zvýšení bilirubinu v krvi Zvýšení jaterních enzymů (gama-glutamyl transferázy) | |
|
Poranění, otravy a procedurální komplikace |
Infuzní reakce (a přidružené symptomy včetně sípotu, návalů, otoku obličeje, dušnosti, hypotenze a hypertenze) |
a Další informace jsou uvedeny v bodě Popis vybraných nežádoucích účinků b Termíny skupin s vysokou úrovní podle MedDRA (MedDRA verze 16.1)
Popis vybraných nežádoucích účinků
Neurologické příhody
V pivotní klinické studii (N=189) se u 51,9 % pacientů vyskytl jeden nebo více neurologických nežádoucích účinků (včetně psychiatrických poruch) postihujících hlavně centrální nervový systém.
Závažné neurologické účinky byly pozorovány u 16,4 % a nežádoucí neurologické účinky stupně > 3 byly pozorovány u 12,7 % pacientů, z nichž nejčastější byly encefalopatie, třes a stav zmatenosti. Byla hlášena fatální encefalopatie, avšak většina neurologických příhod (74,5 %) byla klinicky reverzibilní a odezněla po přerušení léčby přípravkem BLINCYTO. Medián doby do výskytu neurologické příhody byl 9 dní. Pro klinický postup u neurologických příhod odkazujeme na bod 4.4.
Infekce
U pacientů léčených přípravkem BLINCYTO byly hlášeny život ohrožující nebo fatální (stupeň > 4) virové, bakteriální a plísňové infekce. Kromě toho byly pozorovány případy reaktivace virové infekce (např. polyomavirová infekce). U pacientů s počátečním ECOG výkonnostním stavem 2 byl pozorován vyšší výskyt závažných infekcí v porovnání s pacienty s ECOG výkonnostním stavem < 2. Klinický postup u infekcí viz bod 4.4.
Syndrom z uvolnění cytokinů (CRS)
V pivotní klinické studii (N=189) byly hlášené závažné reakce CRS u 0,5 % pacientů s mediánem doby do výskytu 2 dny. Klinický postup u CRS viz bod 4.4.
Zvýšení jaterních enzymů
V pivotní klinické studii (N=189) bylo hlášeno zvýšení jaterních enzymů u 27,5 % pacientů. Závažné nežádoucí příhody a nežádoucí příhody stupně > 3 (jako zvýšení ALT, zvýšení AST a zvýšení bilirubinu v krvi) byly pozorovány u 2,1 %, resp. 15,3 % pacientů. Medián doby do výskytu první příhody byl 3 dny od zahájení léčby přípravkem BLINCYTO. Trvání hepatálních nežádoucích účinků bylo obecně krátké a rychle odeznělo, často při pokračování nepřerušené léčby přípravkem BLINCYTO. Klinický postup u zvýšených jaterních enzymů viz bod 4.4.
Leukoencefalopatie včetně progresivní multifokální encefalopatie
Byly hlášeny případy leukoencefalopatie. U pacientů s nálezem při MRI nebo CT vyšetření mozku odpovídajícím leukoencefalopatii se vyskytly souběžně závažné nežádoucí příhody včetně stavu zmatenosti, třesu, kognitivní poruchy, encefalopatie a konvulzí. I když existuje potenciál pro rozvoj progresivní multifokální leukoencefalopatie (PML), v pivotní studii nebyl hlášen žádný takový případ.
Pediatrická populace
U pediatrických pacientů jsou omezené zkušenosti. BLINCYTO byl hodnocen u pediatrických pacientů s relabovanou nebo refrakterní B-prekurzorovou ALL v dávkově eskalační studii fáze I/II..
Při vyšší dávce než je doporučená dávka pro dospělé pacienty byl hlášen případ fatálního srdečního selhání v souvislosti se syndromem z uvolnění cytokinů (CRS) a syndromem nádorového rozpadu, viz bod 4.4.
Jiné zvláštní populace
Zkušenosti s přípravkem BLINCYTO u pacientů ve věku > 75 let jsou omezené. Obecně byla bezpečnost obdobná u starších pacientů (věk > 65 let) a u pacientů mladších než 65 let léčených přípravkem BLINCYTO. Starší pacienti ale mohou být náchylnější k závažným neurologickým příhodám, jako je kognitivní porucha, encefalopatie a zmatenost.
Bezpečnost přípravku BLINCYTO nebyla studovaná u pacientů s těžkou poruchou funkce ledvin. Imunogenicita
V pivotní klinické studii (N=189) mělo méně než 1,4 % pacientů léčených blinatumomabem pozitivní testy na vazebné a neutralizační protilátky proti blinatumomabu. Všichni pacienti s pozitivitou na vazebné protilátky měli rovněž pozitivní testy na neutralizační protilátky proti blinatumomabu. Tvorba protilátek proti blinatumomabu může ovlivnit farmakokinetiku blinatumomabu.
Při podezření na tvorbu protilátek proti blinatumomabu s klinicky významným účinkem projednejte možnost testování na protilátky s držitelem rozhodnutí o registraci. Kontaktní údaje jsou uvedeny v bodě 6 příbalové informace.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Byly pozorovány případy předávkování včetně jednoho pacienta, který dostal 133x vyšší dávku, než je doporučená terapeutická dávka přípravku BLINCYTO podanou během krátké doby. Předávkování vedlo k nežádoucím účinkům, které byly v souladu s účinky pozorovanými při doporučené terapeutické dávce a zahrnovaly horečku, třes a bolest hlavy. V případě předávkování se má infuze dočasně přerušit a pacienti mají být sledováni. O opětovném zahájení podávání přípravku BLINCYTO ve správné terapeutické dávce lze uvažovat po odeznění všech toxicit a nejdříve za 12 hodin po přerušení infuze (viz bod 4.2).
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Cytostatika, jiná cytostatika, ATC kód: L01XC19. Mechanismus účinku
Blinatumomab je bispecifický konstrukt T-buněčného protilátkového fragmentu, který se specificky váže na CD19 exprimovaný na povrchu buněk B-linie a CD3 exprimovaný na povrchu T-buněk. Dochází k aktivaci endogenní T-buňky spojením CD3 v receptorovém komplexu T-buněk (TCR) s CD19 na benigních a maligních B-buňkách. Protinádorová aktivita imunoterapie blinatumomabem není závislá na T-buňkách nesoucích specifický TCR nebo na peptidových antigenech přítomných na nádorových buňkách, ale je svou povahou polyklonální a nezávislá na molekulách humánního leukocytárního antigenu (HLA) na cílových buňkách. Blinatumomab zprostředkovává tvorbu cytolytické synapse mezi T-buňkou a nádorovou buňkou, čímž se uvolňují proteolytické enzymy, které ničí proliferující i klidové cílové buňky. Blinatumomab se podílí na přechodném zvýšení buněčných adhezních molekul, tvorbě cytolytických proteinů, uvolňování zánětlivých cytokinů a proliferaci T-buněk, čímž dochází k eliminaci CD19+ buněk.
Farmakodynamické účinky
U studovaných pacientů byla pozorována konzistentní farmakodynamická odpověď. Farmakodynamická odpověď během kontinuální infuze trvající 4 týdny byla charakterizována aktivací a úvodní redistribucí T-buněk, rychlou deplecí periferních B-buněk a přechodným zvýšením cytokinů.
Redistribuce periferních T-buněk (tj. adheze T-buněk na endotel cév a/anebo jejich migrace do tkání) nastala po zahájení infuze nebo zvýšení dávky blinatumomabu. Počet T-buněk zpočátku během 1 - 2 dnů poklesl a poté se u většiny pacientů vrátil na výchozí hodnoty během 7 - 14 dní. U několika pacientů byl pozorován vzestup počtu T-buněk nad výchozí hodnoty (expanze T-buněk).
U většiny pacientů rychle poklesl počet periferních B-buněk na neměřitelné hodnoty při léčbě dávkami > 5 pg/m2/den nebo > 9 pg/den. Obnovení počtu periferních B-buněk nebylo pozorováno během dvoutýdenních období bez léčby mezi léčebnými cykly. Neúplná deplece B-buněk byla pozorovaná při dávkách 0,5 pg/m2/den a 1,5 pg/m2/den a u několika non-respondérů při vyšších dávkách.
Byly měřeny hladiny cytokinů včetně IL-2, IL-4, IL-6, IL-8, IL-10, IL-12, TNF-a a IFN-y a největší zvýšení bylo zjištěno u IL-6, IL-10 a IFN-y. Přechodné zvýšení hladin cytokinů bylo pozorováno v prvních dvou dnech po zahájení infuze blinatumomabu. Zvýšené hladiny cytokinů se vrátily na výchozí hodnoty během 24 až 48 hodin v průběhu infuze. V dalších léčebných cyklech se zvýšení hladin cytokinů objevilo u méně pacientů a s nižší intenzitou v porovnání s úvodními 48 hodinami v prvním léčebném cyklu.
Klinická účinnost a bezpečnost
V klinických studiích byl přípravek BLINCYTO podáván celkem 225 pacientům ve věku > 18 let s relabovanou nebo refrakterní B-prekurzorovou ALL.
BLINCYTO byl hodnocen v otevřené, multicentrické, jednoramenné studii fáze II, u 189 pacientů. Pro účast ve studii byli vhodní pacienti ve věku > 18 let s relabovanou nebo refrakterní B-prekurzorovou ALL s Philadelphia negativním chromozomem (k relapsu došlo při první remisi trvající < 12 měsíců v první záchranné léčbě, nebo šlo o relabovanou či refrakterní ALL po první záchranné léčbě, případně o relaps během 12 měsíců po alogenní HSCT a pacienti ve studii měli > 10 % blastů v kostní dřeni).
Pacienti byli premedikováni povinnou profylaxí mozkomíšního moku s použitím intratekálního režimu dle národních doporučení nebo doporučení daného pracoviště během 1 týdne před zahájením léčby blinatumomabem. BLINCYTO byl podáván v kontinuální intravenózní infuzi. V prvním cyklu byla úvodní dávka 9 pg/den v 1. týdnu a dále 28 pg/den ve zbylých 3 týdnech. Cílová dávka 28 pg/den byla podávána ve 2. cyklu a v dalších cyklech počínaje 1. dnem každého cyklu. V případě nežádoucích účinků byla možná úprava dávky. Léčená populace zahrnovala 189 pacientů, kteří dostali alespoň 1 infuzi přípravku BLINCYTO; průměrný počet cyklů u pacienta byl 1,6. Pacienti, kteří odpověděli na léčbu přípravkem BLINCYTO, ale později u nich došlo k relapsu, měli možnost opětovné léčby přípravkem BLINCYTO. Medián věku léčených pacientů byl 39 let (rozptyl 18 - 79 let včetně 25 pacientů ve věku > 65 let), 64 pacientů ze 189 (33,9 %) podstoupilo HSCT před léčbou přípravkem BLINCYTO a 32 pacientů ze 189 (16.9 %) dostalo předtím více než 2 záchranné léčby.
Primární cílový parametr byl výskyt úplné remise/úplné remise s částečnou hematologickou úpravou (CR/CRh*) během 2 léčebných cyklů s přípravkem BLINCYTO. Celkem 81 pacientů ze 189 (42,9 %) dosáhlo CR/CRh* během prvních 2 léčebných cyklů. Většina odpovědí (64 z 81) se vyskytla v 1. léčebném cyklu. U populace starších pacientů (> 65 let) dosáhlo 11 z 25 pacientů (44,0 %) CR/CRh* během prvních 2 léčebných cyklů (viz bod 4.8 pro bezpečnost u starších pacientů). Čtyři pacienti dosáhli CR během konsolidačních cyklů, takže kumulativní výskyt CR byl 35,4 % (67/189; 95% CI: 28,6 % - 42,7 %). 32 ze 189 (17 %) pacientů podstoupilo alogenní HSCT v CR/CRh* navozené léčbou přípravkem BLINCYTO (viz tabulka 1).
Tab. 1. Výsledky účinnosti u pacientů ve věku > 18 let s Philadelphia chromozom negativní relabovanou nebo refrakterní B-prekurzorovou akutní lymfoblastickou leukemií (ALL)
|
n (%) n = 189 |
95% CI | |
|
Úplná remise (CR)VÚplná remise s částečnou hematologickou úpravou (CRh*)2 |
81 (42,9 %) |
[35,7 % - 50,2 %] |
|
CR |
63 (33,3 %) |
[26,7 % - 40,5 %] |
|
CRh* |
18 (9,5 %) |
[5,7 % - 14,6 %] |
|
Hypoplastická nebo aplastická kostní dřeň bez blastů3 |
17 (9 %) |
[5,3 % - 14,0 %] |
|
Částečná remise4 |
5 (2,6 %) |
[0,9 % - 6,1 %] |
|
Doba přežití bez relapsů (RFS) pro CR/CRh* |
5,9 měsíců |
[4,8 až 8,3 měsíců] |
|
Celková doba přežití |
6,1 měsíců |
[4,2 až 7,5 měsíců] |
|
1 CR byla definována jako < 5 % blastů v kostní dřeni, bez průkazu nemoci a úplná úprava periferního krevního obrazu (trombocyty > 100 000/mikrolitr a absolutní počet neutrofilů [ANC] > 1000/mikrolitr). 2 CRh* byla definována jako < 5 % blastů v kostní dřeni, bez průkazu nemoci a částečná úprava periferního krevního obrazu (trombocyty > 50 000/mikrolitr a ANC > 500/mikrolitr). 3. Hypoplastická nebo aplastická kostní dřeň bez blastů byla definována jako blasty v kostní dřeni < 5 %, bez průkazu nemoci, nedostatečná úprava periferního krevního obrazu:(trombocyty < 50 000/mikrolitr a/nebo ANC < 500/mikrolitr 4. Částečná remise byla definována jako blasty v kostní dřeni 6 % až 25 % spolu s nejméně 50% snížením oproti výchozí hodnotě | ||
V předem specifikované explorativní analýze 60 ze 73 hodnotitelných pacientů s MRD CR/CRh*
(82,2 %) mělo rovněž odpověď MRD (definovanou jako MRD pomocí PCR < 1 x 10-4).
Pacienti s předchozí alogenní HSCT měli obdobný výskyt odpovědi jako pacienti bez předchozí HSCT, starší pacienti měli obdobný výskyt odpovědi jako mladší pacienti a nebyl pozorován podstatný rozdíl ve výskytu remisí na základě počtu linií předchozí záchranné léčby.
U pacientů s extramedulárním projevem nemoci bez postižení CNS/varlat (definovaným jako minimálně 1 léze > 1,5 cm) při screeningu (N = 8/189) byl výskyt klinických odpovědí (25 % [95% CI
3,2 - 65,1] nižší ve srovnání s pacienty bez průkazu extramedulárního projevu nemoci (N=181, 43,6 % [95% CI 36,3 - 51,2]) (viz obrázek 1).
Pacienti s nejvyšší nádorovou zátěží měřenou podle procenta blastů v kostní dřeni před zahájením léčby (> 90 %) měli stále ještě klinicky smysluplnou odpověď s výskytem CR/CRh* 21,6 % (CI
12,9 - 32,7; viz obrázek 1). Pacienti s nízkou nádorovou zátěží (< 50 %) odpovídali nejlépe na léčbu blinatumomabem s výskytem CR/CRh* 72,9 % (CI 59,7 - 83,6).
Obr. 1. Znázornění typu „Forest plot“ výskytu CR/CRh* v prvních dvou cyklech ve studii
MT103-211 (soubor primární analýzy)
Podskupina
Infiltrace kostní dřeně blasty (Centrální laboratoř)
<50%
s50 % až <75 %
275 % až <90 %
290 %
Extramedulární nemoc při vstupu do studie
Ano
Ne
Celkem
Výskyt a 95% interval spolehlivosti (IS)
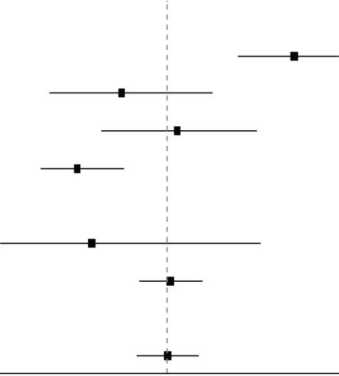
20 40 60 80
Procento dosahujících CR/CRh*
n/N Výskyt (95% IS)
43/59 72,9 % (59,7 %-83,6 %)
8/25 32,0 % (14,9 %-53,5 %)
14/31 45,2 % (27,3 %-64,0 %)
16/74 21,6% (12,9 %-32,7%)
2/8 25,0 % (3,2 %-65,1 %)
79/181 43,6 % (36,3 %-51,2 %)
81/189 42,9 % (35,7 %-50,2 %)
o
V otevřené multicentrické dávkově eskalační studii fáze II u 36 pacientů (> 18 let věku, s B-prekurzorovou ALL s relapsem nejméně po indukci a konsolidaci nebo s refraktemím onemocněním s > 5 % blastů v kostní dřeni, ECOG výkonnostním stavem < 2, očekávanou délkou života > 12 týdnů, kteří nepodstoupili autologní transplantaci krvetvorných kmenových buněk (HSCT) v období 6 týdnů před zahájením léčby blinatumomabem, alogenní HSCT v období 3 měsíců před zahájením léčby blinatumomabem nebo po předchozí léčbě blinatumomabem) byla hodnocena bezpečnost a účinnost blinatumomabu. Alogenní HSCT před léčbou přípravkem BLINCYTO podstoupilo 15 z 36 (41,7 %) pacientů. CR/CRh* poměr byl 69,4 % (25 z 36 pacientů: 15 [41,7 %; 95% CI: 25,5 % - 59,2 %] CR; 10 [27,8 %; 95% CI: 14,2 % - 45,2 %] CRh*). V populaci starších pacientů (> 65 let), 4 z 5 pacientů (80,0 %) dosáhlo CR/CRh* během 2 léčebných cyklů (viz bod 4.8 o bezpečnosti u starších pacientů). 22 z 25 (88 %) pacientů s úplnou hematologickou remisí mělo rovněž odpověď ve formě minimálního reziduálního onemocnění (MRD), která byla definována jako MRD pomocí PCR < 1 x 10-4. Medián trvání remise byl 8,9 měsíců a medián doby přežití bez relapsu (RFS) byl 7,6 měsíců. Medián celkového přežití (OS) byl 9,8 měsíců.
Existují pouze omezená data u pacientů s pozdním prvním relapsem B-prekurzorové ALL definovaným jako relaps po více než 12 měsíců trvající remisi nebo po více než 12 měsících od HSCT během první remise. V klinických studiích 88,9 % (8/9) pacientů s pozdním prvním relapsem, dle definice v jednotlivých studiích, dosáhlo CR/CRh* během prvních 2 cyklů léčby, 62,5 % (6/9) pacientů dosáhlo odpovědi MRD a 37,5 % (3/9) pacientů podstoupilo alogenní HSCT po léčbě blinatumomabem. Medián celkového přežití (OS) byl 17,7 měsíce (CI 3,1 - nedosažen).
Pediatrická populace
U pediatrických pacientů jsou pouze omezené zkušenosti, viz bod 4.8.
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s přípravkem BLINCYTO u dětí ve věku od 1 měsíce do méně než 18 let s akutní lymfoblastickou leukemií (informace o použití u dětí viz bod 4.2).
Tento léčivý přípravek byl registrován postupem tzv. podmíněného schválení. Znamená to, že jsou očekávány další důkazy o jeho přínosech. Evropská agentura pro léčivé přípravky nejméně jednou za rok vyhodnotí nové informace o tomto léčivém přípravku a tento souhrn údajů o přípravku bude podle potřeby aktualizován.
Farmakokinetika blinatumomabu u dospělých pacientů se v dávkovacím rozmezí od 5 do 90 ^g/m2/den (ekvivalent přibližně 9 - 162 ^g/den) jeví jako lineární. Po kontinuální infuzi byly sérové koncentrace v ustáleném stavu (Css) dosaženy v průběhu jednoho dne a zůstaly dále stabilní. Vzestup průměrných hodnot Css byl v testovaném dávkovacím rozmezí přibližně proporcionální. V klinických dávkách 9 ^g/den a 28 ^g/den použitých při léčbě relabované/refrakterní ALL byla průměrná Css (SD) 211 (258) pg/ml při podávání nižší dávky a 621 (502) pg/ml při podávání vyšší dávky.
Distribuce
Odhadovaný průměrný distribuční objem (SD) na základě terminální fáze (Vz) byl 4,52 (2,89) litrů při kontinuálním podávání intravenózních infuzí s blinatumomabem.
Biotransformace
Charakteristika metabolické dráhy blinatumomabu nebyla provedena. Stejně jako u ostatních proteinových látek se předpokládá, že blinatumomab je degradován katabolickými dráhami na malé peptidy a aminokyseliny.
Eliminace
Odhadovaná průměrná systémová clearance (SD) při kontinuální intravenózní infuzi u pacientů léčených blinatumomabem v klinických studiích byla 2,92 (2,83) litrů/hod. Průměrný poločas (SD) byl 2,11 (1,42) hod. Při testovaných klinických dávkách se do moče vylučovalo pouze zanedbatelné množství blinatumomabu.
Tělesná hmotnost, plocha tělesného povrchu, pohlaví a věk
Byla provedena populační farmakokinetická analýza s cílem vyhodnotit efekt charakteristických demografických znaků na farmakokinetiku blinatumomabu. Výsledky naznačují, že věk (18 - 80 let), pohlaví, tělesná hmotnost (44 - 134 kg) a plocha tělesného povrchu (1,39 - 2,57) neovlivňují farmakokinetiku blinatumomabu. Existují pouze velmi omezená data u dospělých pacientů s tělesnou hmotností nižší než 45 kg.
Porucha funkce ledvin
U pacientů s poruchou funkce ledvin nebyly provedené žádné formální farmakokinetické studie.
Farmakokinetické analýzy prokázaly přibližně dvojnásobný rozdíl průměrných hodnot clearance blinatumomabu mezi pacienty se středně těžkou poruchou funkce ledvin a pacienty s normální funkcí ledvin. Variabilita mezi pacienty byla nicméně stěží rozeznatelná (CV% až do 95,6 %) a hodnoty clearance u pacientů s poruchou funkce ledvin byly v podstatě ve stejném rozmezí jako u pacientů s normální funkcí ledvin. Proto se nepředpokládá klinicky významný dopad funkce ledvin na klinické výsledky.
Porucha funkce jater
U pacientů s poruchou funkce jater nebyly provedené žádné formální farmakokinetické studie.
K vyhodnocení účinku poruchy funkce jater na clearance blinatumomabu byly použity výchozí hodnoty ALT a AST. Populační farmakokinetická analýza naznačila, že neexistuje spojitost mezi hladinami ALT nebo AST a clearance blinatumomabu.
Pediatrická populace
U pediatrických pacientů jsou omezené zkušenosti.
Studie toxicity po opakovaném podávání provedené s blinatumomabem a u myšího surogátu odhalily předpokládané farmakologické účinky (včetně uvolnění cytokinů, poklesu počtu leukocytů, deplece B-buněk, poklesu T-buněk, poklesu celularity v lymfoidních tkáních). Po vysazení léčby se tyto změny vrátily zpět.
Studie reprodukční toxicity s blinatumomabem nebyly provedeny. Ve studii embryofetální vývojové toxicity provedené u myší, myší surogát procházel placentou v omezeném množství (poměr koncentrace v séru plodu a matky byl < 1 %) a nevyvolal embryofetální toxicitu nebo teratogenitu. U březích myší byla pozorována předpokládaná deplece B- a T-buněk, avšak u plodů nebyly hodnoceny hematologické účinky. Nebyly provedeny studie hodnotící účinky léčby na fertilitu. Ve studiích toxicity s myšími surogáty nebyly zjištěny žádné účinky na samčí nebo samičí reprodukční orgány.
6. FARMACEUTICKÉ ÚDAJE 6.1 Seznam pomocných látek
Prášek
Monohydrát kyseliny citronové (E330)
Dihydrát trehalosy Lysin-hydrochlorid Polysorbát 80
Hydroxid sodný (k úpravě pH)
Roztok (stabilizátor)
Monohydrát kyseliny citronové (E330)
Lysin-hydrochlorid Polysorbát 80
Hydroxid sodný (k úpravě pH)
Voda na injekci
6.2 Inkompatibility
Tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky s výjimkou těch, které jsou uvedeny v bodě 6.6.
6.3 Doba použitelnosti
Neotevřené injekční lahvičky 4 roky
Rekonstituovaný roztok
Chemická a fyzikální stabilita po otevření před použitím byla prokázána na dobu 24 hodin při teplotě 2 °C - 8 °C nebo 4 hodin při teplotě do 27 °C.
Z mikrobiologického hlediska, pokud způsob rekonstituce nevyloučí riziko mikrobiologické kontaminace, se má rekonstituovaný roztok okamžitě rozpustit. Jestliže se nerozpustí ihned, doba a podmínky uchovávání přípravku po otevření před použitím jsou v odpovědnosti uživatele.
Chemická a fyzikální stabilita po otevření před použitím byla prokázána na dobu 10 dní při teplotě 2 °C - 8 °C nebo na dobu 96 hodin při teplotě do 27 °C.
Z mikrobiologického hlediska se připravené infuzí vaky mají použít okamžitě.
Nejsou-li použity okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou v odpovědnosti uživatele a normálně by doba neměla být delší než 24 hodin při teplotě 2 °C - 8 °C, pokud ředění neproběhlo za kontrolovaných a validovaných aseptických podmínek.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte a převážejte chlazené (2 °C - 8 °C).
Chraňte před mrazem.
Uchovávejte injekční lahvičky v původním obalu, aby byl přípravek chráněn před světlem.
Podmínky uchovávání tohoto léčivého přípravku po rekonstituci a naředění jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
Jedno balení přípravku BLINCYTO obsahuje 1 injekční lahvičku prášku pro koncentrát pro infuzní roztok a 1 injekční lahvičku roztoku (stabilizátoru):
• 38,5 mikrogramu prášku blinatumomabu v injekční lahvičce (sklo třídy I) se zátkou (elastomerová pryž), závěrem (hliník) a odtrhovacím víčkem a
• 10 ml roztoku v injekční lahvičce (sklo třídy I) se zátkou (elastomerová pryž), závěrem (hliník) a odtrhovacím víčkem.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Aseptická příprava
Při přípravě infuze je třeba dodržovat aseptické podmínky. Příprava přípravku BLINCYTO se má:
- provádět za aseptických podmínek personálem zaškoleným v pravidlech správné praxe, zejména z hlediska aseptické přípravy parenterálních přípravků;
- připravovat v laminárním boxu nebo v boxu pro bezpečné zacházení s biologickým materiálem při použití standardních bezpečnostních opatření pro bezpečné zacházení s intravenózními látkami.
Je mimořádně důležité, aby se přísně dodržovaly pokyny pro přípravu a podání uvedené v tomto bodě, aby se tak minimalizovaly chyby medikace (včetně poddávkování a předávkování).
Zvláštní pokyny sloužící ke správné přípravě
• Roztok (stabilizátor) se dodává v balení přípravku BLINCYTO a používá se k potažení předplněného infuzního vaku před přidáním rekonstituovaného přípravku BLINCYTO. Nepoužívejte tento roztok (stabilizátor) k rekonstituci prášku pro koncentrát přípravku BLINCYTO.
• Celý objem rekonstituovaného a naředěného přípravku BLINCYTO bude větší než objem, který se má podat pacientovi (240 ml). Je třeba počítat se ztrátou použitím intravenózní infuzní linky a zajistit, že pacient dostane plnou dávku přípravku BLINCYTO.
• Při přípravě infuzního vaku z něho odstraňte všechen vzduch. Toto je mimořádně důležité při použití ambulantní infuzní pumpy.
• Použijte konkrétní objemy uvedené níže v návodu pro rekonstituci a ředění, aby se minimalizovaly chyby při výpočtu.
• BLINCYTO je kompatibilní s infuzními vaky/kazetami infuzních pump z polyolefinu, PVC non-diethylhexylftalátu (non-DEHP), nebo ethylvinylacetátu (EVA).
• Specifikace pump: Infuzní pumpa k podání přípravku BLINCYTO infuzní roztok musí být programovatelná, zamykatelná a mít alarm. Elastomerické infuzní pumpy se nesmějí používat.
• Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Příprava infíizního roztoku
Pro každou dávku a dobu infuze jsou uvedené přesné pokyny pro rekonstituci a naředění. Ověřte si předepsanou dávku a dobu infuze přípravku BLINCYTO a použijte příslušný bod níže pro přípravu správné dávky. Dodržujte postup pro rekonstituci přípravku BLINCYTO a pro přípravu infuzního vaku.
a) pro infuzi o koncentraci 9 ^g/den podanou během 24 hodin rychlostí 10 ml/hod.
b) pro infuzi o koncentraci 9 ^g/den podanou během 48 hodin rychlostí 5 ml/hod.
c) pro infuzi o koncentraci 9 ^g/den podanou během 72 hodin rychlostí 3,3 ml/hod.
d) pro infuzi o koncentraci 9 ^g/den podanou během 96 hodin rychlostí 2,5 ml/hod.
e) pro infuzi o koncentraci 28 ^g/den podanou během 24 hodin rychlostí 10 ml/hod.
f) pro infuzi o koncentraci 28 ^g/den podanou během 48 hodin rychlostí 5 ml/hod.
g) pro infuzi o koncentraci 28 ^g/den podanou během 72 hodin rychlostí 3,3 ml/hod.
h) pro infuzi o koncentraci 28 ^g/den podanou během 96 hodin rychlostí 2,5 ml/hod.
Před přípravou se ujistěte, že máte připraven následující materiál:
|
Dávka |
Trvání infuze (hod.) |
Rychlost infuze (ml/hod.) |
Počet balení přípravku BLINCYTO |
|
9 ^g/den |
24 |
10 |
1 |
|
48 |
5 |
1 | |
|
72 |
3,3 |
1 | |
|
96 |
2,5 |
2 | |
|
28 ^g/den |
24 |
10 |
1 |
|
48 |
5 |
2 | |
|
72 |
3,3 |
3 | |
|
96 |
2,5 |
4 |
Dále budete potřebovat tento materiál, který ale není součástí balení přípravku BLINCYTO
• Sterilní injekční stříkačky na jedno použití
• Jehly o rozměru 21 - 23 gauge (doporučeno)
• Voda na injekci
• Infuzní vak s 250 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9%);
o K minimalizaci počtu aseptických přenosů použijte 250 ml předplněný infuzní vak. Výpočty dávky přípravku BLINCYTO jsou založené na obvyklém přeplnění objemu 265 až 275 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9%).
o Použijte pouze infuzní vaky/kazety infuzních pump z polyolefinu, PVC non-diethylhexylftalátu (non-DEHP), nebo ethylvinylacetátu (EVA).
• Infuzní set z polyolefinu, PVC non-DEHP, nebo EVA se sterilním a nepyrogenním 0,2^m in-line filtrem s nízkou vazbou na bílkoviny
o Přesvědčte se, že infuzní set je kompatibilní s infuzní pumpou.
a) Příprava přípravku BLINCYTO 9 yg/den podaného infuzí během 24 hodin rychlostí 10 ml/hod.
1. Použijte infuzní vak předplněný 250 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9%), který obvykle obsahuje celkový objem 265 až 275 ml.
2. K potažení infuzního vaku asepticky přeneste pomocí stříkačky 5,5 ml roztoku (stabilizátoru) do infuzního vaku. Jemně zamíchejte obsahem vaku, aby se nevytvořila pěna. Injekční lahvičku se zbylým roztokem (stabilizátorem) vyhoďte.
3. Pomocí stříkačky rekonstituujte jednu injekční lahvičku přípravku BLINCYTO prášek pro koncentrát použitím 3 ml vody na injekci. Během rekonstituce nasměrujte vodu na injekci na boční stěnu injekční lahvičky. Injekční lahvičkou jemně otáčejte, aby se nevytvořilo nadměrné množství pěny. Injekční lahvičkou netřepejte.
• Nepoužívejte roztok (stabilizátor) k rekonstituci přípravku BLINCYTO prášek pro koncentrát.
• Přidáním vody na injekci k prášku pro koncentrát dosáhnete celkový objem 3,08 ml přípravku BLINCYTO o konečné koncentraci 12,5 ^g/ml.
4. Během rekonstituce a před podáním infuze prohlédněte rekonstituovaný roztok, zda se
v něm nevyskytují částice a nedošlo ke změně zbarvení. Výsledný roztok musí být čirý až lehce opalescentní a bezbarvý až nažloutlý. Roztok nepoužívejte, pokud je zkalený nebo obsahuje usazeniny.
5. Pomocí stříkačky asepticky přeneste 0,83 ml rekonstituovaného přípravku BLINCYTO do infuzního vaku. Jemně zamíchejte obsahem vaku, aby se nevytvořila pěna. Veškerý zbylý rekonstituovaný roztok přípravku BLINCYTO vyhoďte.
6. Za aseptických podmínek připevněte intravenózní set k infuznímu vaku se sterilním 0,2^ in-line filtrem.
7. Z infuzního vaku odstraňte vzduch a naplňte intravenózní linku pouze připraveným infuzním roztokem. Linku nenaplňujte injekčním roztokem chloridu sodného 9 mg/ml (0,9%).
8. Jestliže takto připravený roztok nepoužijete ihned, uchovávejte ho při teplotě 2 °C - 8 °C.
b) Příprava přípravku BLINCYTO 9 yg/den podaného infuzí během 48 hodin rychlostí 5 ml/hod.
1. Použijte infuzní vak předplněný 250 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9%), který obvykle obsahuje celkový objem 265 až 275 ml.
2. K potažení infuzního vaku asepticky přeneste pomocí stříkačky 5,5 ml roztoku (stabilizátor) do infuzního vaku. Jemně zamíchejte obsahem vaku, aby se nevytvořila pěna. Injekční lahvičku se zbylým roztokem (stabilizátor) vyhoďte.
3. Pomocí stříkačky rekonstituujte jednu injekční lahvičku přípravku BLINCYTO prášek pro koncentrát použitím 3 ml vody na injekci. Během rekonstituce nasměrujte vodu na injekci na boční stěnu injekční lahvičky. Injekční lahvičkou jemně otáčejte, aby se nevytvořilo nadměrné množství pěny. Injekční lahvičkou netřepejte.
• Nepoužívejte roztok (stabilizátor) k rekonstituci přípravku BLINCYTO prášek pro koncentrát.
• Přidáním vody na injekci k prášku pro koncentrát dosáhnete celkový objem 3,08 ml přípravku BLINCYTO o konečné koncentraci 12,5 ^g/ml.
4. Během rekonstituce a před podáním infuze prohlédněte rekonstituovaný roztok, zda se
v něm nevyskytují částice a nedošlo ke změně zbarvení. Výsledný roztok musí být čirý až lehce opalescentní a bezbarvý až nažloutlý. Roztok nepoužívejte, pokud je zkalený nebo obsahuje usazeniny.
5. Pomocí stříkačky asepticky přeneste 1,7 ml rekonstituovaného přípravku BLINCYTO do infuzního vaku. Jemně zamíchejte obsahem vaku, aby se nevytvořila pěna. Zbylý rekonstituovaný roztok přípravku BLINCYTO vyhoďte.
6. Za aseptických podmínek připevněte intravenózní set k infuznímu vaku se sterilním 0,2^ in-line filtrem.
7. Z infuzního vaku odstraňte vzduch a naplňte intravenózní linku pouze připraveným infuzním roztokem. Linku nenaplňujte injekčním roztokem chloridu sodného 9 mg/ml (0,9%).
8. Jestliže takto připravený roztok nepoužijete ihned, uchovávejte ho při teplotě 2 °C - 8 °C.
c) Příprava přípravku BLINCYTO 9 yg/den podaného infuzí během 72 hodin rychlostí 3,3 ml/hod.
1. Použijte infuzní vak předplněný 250 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9%), který obvykle obsahuje celkový objem 265 až 275 ml.
2. K potažení infuzního vaku asepticky přeneste pomocí stříkačky 5,5 ml roztoku (stabilizátor) do infuzního vaku. Jemně zamíchejte obsahem vaku, aby se nevytvořila pěna. Injekční lahvičku se zbylým roztokem (stabilizátor) vyhoďte.
3. Pomocí stříkačky rekonstituujte jednu injekční lahvičku přípravku BLINCYTO prášek pro koncentrát použitím 3 ml vody na injekci. Během rekonstituce nasměrujte vodu na injekci na boční stěnu injekční lahvičky. Injekční lahvičkou jemně otáčejte, aby se nevytvořilo nadměrné množství pěny. Injekční lahvičkou netřepejte.
• Nepoužívejte roztok (stabilizátor) k rekonstituci přípravku BLINCYTO prášek pro koncentrát.
• Přidáním vody na injekci k prášku pro koncentrát dosáhnete celkový objem 3,08 ml přípravku BLINCYTO o konečné koncentraci 12,5 ^g/ml.
4. Během rekonstituce a před podáním infuze prohlédněte rekonstituovaný roztok, zda se
v něm nevyskytují částice a nedošlo ke změně zbarvení. Výsledný roztok musí být čirý až lehce opalescentní a bezbarvý až nažloutlý. Roztok nepoužívejte, pokud je zkalený nebo obsahuje usazeniny.
5. Pomocí stříkačky asepticky přeneste 2,5 ml rekonstituovaného přípravku BLINCYTO do infuzního vaku. Jemně zamíchejte obsahem vaku, aby se nevytvořila pěna. Veškerý zbylý rekonstituovaný roztok přípravku BLINCYTO vyhoďte.
6. Za aseptických podmínek připevněte intravenózní set k infuznímu vaku se sterilním 0,2^ in-line filtrem.
7. Z infuzního vaku odstraňte vzduch a naplňte intravenózní linku pouze připraveným infuzním roztokem. Linku nenaplňujte injekčním roztokem chloridu sodného 9 mg/ml (0,9%).
8. Jestliže takto připravený roztok nepoužijete ihned, uchovávejte ho při teplotě 2 °C - 8 °C.
d) Příprava přípravku BLINCYTO 9 yg/den podaného infuzí během 96 hodin rychlostí 2,5 ml/hod.
1. Použijte infuzní vak předplněný 250 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9%), který obvykle obsahuje celkový objem 265 až 275 ml.
2. K potažení infuzního vaku asepticky přeneste pomocí stříkačky 5,5 ml roztoku (stabilizátoru) do infuzního vaku. Jemně zamíchejte obsahem vaku, aby se nevytvořila pěna. Injekční lahvičku se zbylým roztokem (stabilizátorem) vyhoďte.
3. Použijte dvě injekční lahvičky přípravku BLINCYTO prášek pro koncentrát. Pomocí stříkačky rekonstituujte každou lahvičku přípravku BLINCYTO prášek pro koncentrát použitím 3 ml vody na injekci. Během rekonstituce nasměrujte vodu na injekci na boční stěnu injekční lahvičky. Injekční lahvičkou jemně otáčejte, aby se nevytvořilo nadměrné množství pěny. Injekční lahvičkou netřepejte.
• Nepoužívejte roztok (stabilizátor) k rekonstituci přípravku BLINCYTO prášek pro koncentrát.
• Přidáním vody na injekci k prášku pro koncentrát dosáhnete celkový objem 3,08 ml přípravku BLINCYTO o konečné koncentraci 12,5 ^g/ml.
4. Během rekonstituce a před podáním infuze prohlédněte rekonstituovaný roztok, zda se
v něm nevyskytují částice a nedošlo ke změně zbarvení. Výsledný roztok musí být čirý až lehce opalescentní a bezbarvý až nažloutlý. Roztok nepoužívejte, pokud je zkalený nebo obsahuje usazeniny.
5. Pomocí stříkačky asepticky přeneste 3,3 ml rekonstituovaného přípravku BLINCYTO do infuzního vaku (2,0 ml z jedné injekční lahvičky a dalších 1,3 ml z druhé injekční lahvičky). Jemně zamíchejte obsahem vaku, aby se nevytvořila pěna. Veškerý zbylý rekonstituovaný roztok přípravku BLINCYTO vyhoďte.
6. Za aseptických podmínek připevněte intravenózní set k infuznímu vaku se sterilním 0,2^ in-line filtrem.
7. Z infuzního vaku odstraňte vzduch a naplňte intravenózní linku pouze připraveným infuzním roztokem. Linku nenaplňujte injekčním roztokem chloridu sodného 9 mg/ml (0,9%).
8. Jestliže takto připravený roztok nepoužijete ihned, uchovávejte ho při teplotě 2 °C - 8 °C.
e) Příprava přípravku BLINCYTO 28 pg/den podaného infuzí během 24 hodin rychlostí 10 ml/hod.
1. Použijte infuzní vak předplněný 250 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9%), který obvykle obsahuje celkový objem 265 až 275 ml.
2. K potažení infuzního vaku asepticky přeneste pomocí stříkačky 5,5 ml roztoku (stabilizátor) do infuzního vaku. Jemně zamíchejte obsahem vaku, aby se nevytvořila pěna. Injekční lahvičku se zbylým roztokem (stabilizátor) vyhoďte.
3. Pomocí stříkačky rekonstituujte jednu injekční lahvičku přípravku BLINCYTO prášek pro koncentrát použitím 3 ml vody na injekci. Během rekonstituce nasměrujte vodu na injekci na boční stěnu injekční lahvičky. Injekční lahvičkou jemně otáčejte, aby se nevytvořilo nadměrné množství pěny. Injekční lahvičkou netřepejte.
• Nepoužívejte roztok (stabilizátor) k rekonstituci přípravku BLINCYTO prášek pro koncentrát.
• Přidáním vody na injekci k prášku pro koncentrát dosáhnete celkový objem 3,08 ml přípravku BLINCYTO o konečné koncentraci 12,5 ^g/ml.
4. Během rekonstituce a před podáním infuze prohlédněte rekonstituovaný roztok, zda se
v něm nevyskytují částice a nedošlo ke změně zbarvení. Výsledný roztok musí být čirý až lehce opalescentní a bezbarvý až nažloutlý. Roztok nepoužívejte, pokud je zkalený nebo obsahuje usazeniny.
5. Pomocí stříkačky asepticky přeneste 2,6 ml rekonstituovaného přípravku BLINCYTO do infuzního vaku. Jemně zamíchejte obsahem vaku, aby se nevytvořila pěna. Zbylý rekonstituovaný roztok přípravku BLINCYTO vyhoďte.
6. Za aseptických podmínek připevněte intravenózní set k infuznímu vaku se sterilním 0,2^ in-line filtrem.
7. Z infuzního vaku odstraňte vzduch a naplňte intravenózní linku pouze připraveným infuzním roztokem. Linku nenaplňujte injekčním roztokem chloridu sodného 9 mg/ml (0,9%).
8. Jestliže takto připravený roztok nepoužijete ihned, uchovávejte ho při teplotě 2 °C - 8 °C.
f) Příprava přípravku BLINCYTO 28 pg/den podaného infuzí během 48 hodin rychlostí 5 ml/hod.
1. Použijte infuzní vak předplněný 250 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9%), který obvykle obsahuje celkový objem 265 až 275 ml.
2. K potažení infuzního vaku asepticky přeneste pomocí stříkačky 5,5 ml roztoku (stabilizátor) do infuzního vaku. Jemně zamíchejte obsahem vaku, aby se nevytvořila pěna. Injekční lahvičku se zbylým roztokem (stabilizátor) vyhoďte.
3. Použijte dvě injekční lahvičky přípravku BLINCYTO prášek pro koncentrát. Pomocí stříkačky rekonstituujte každou injekční lahvičku přípravku BLINCYTO prášek pro koncentrát použitím 3 ml vody na injekci. Během rekonstituce nasměřujte vodu na injekci na boční stěnu injekční lahvičky. Injekční lahvičkou jemně otáčejte, aby se nevytvořilo nadměrné množství pěny. Injekční lahvičkou netřepejte.
• Nepoužívejte roztok (stabilizátor) k rekonstituci přípravku BLINCYTO prášek pro koncentrát.
• Přidáním vody na injekci k prášku pro koncentrát dosáhnete celkový objem 3,08 ml přípravku BLINCYTO o konečné koncentraci 12,5 ^g/ml.
4. Během rekonstituce a před podáním infuze prohlédněte rekonstituovaný roztok, zda se
v něm nevyskytují částice a nedošlo ke změně zbarvení. Výsledný roztok musí být čirý až lehce opalescentní a bezbarvý až nažloutlý. Roztok nepoužívejte, pokud je zkalený nebo obsahuje usazeniny.
5. Pomocí stříkačky asepticky přeneste 5,2 ml rekonstituovaného přípravku BLINCYTO do infuzního vaku (2,7 ml z jedné injekční lahvičky a zbývajících 2,5 ml z druhé injekční lahvičky). Jemně zamíchejte obsahem vaku, aby se nevytvořila pěna. Zbylý rekonstituovaný roztok přípravku BLINCYTO vyhoďte.
6. Za aseptických podmínek připevněte intravenózní set k infuznímu vaku se sterilním 0,2^ in-line filtrem.
7. Z infuzního vaku odstraňte vzduch a naplňte intravenózní linku pouze připraveným infuzním roztokem. Linku nenaplňujte injekčním roztokem chloridu sodného 9 mg/ml (0,9%).
8. Jestliže takto připravený roztok nepoužijete ihned, uchovávejte ho při teplotě 2 °C - 8 °C.
g) Příprava přípravku BLINCYTO 28 pg/den podaného infuzí během 72 hodin rychlostí 3,3 ml/hod.
1. Použijte infuzní vak předplněný 250 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9%), který obvykle obsahuje celkový objem 265 až 275 ml.
2. K potažení infuzního vaku asepticky přeneste pomocí stříkačky 5,5 ml roztoku (stabilizátor) do infuzního vaku. Jemně zamíchejte obsahem vaku, aby se nevytvořila pěna. Injekční lahvičku se zbylým roztokem (stabilizátor) vyhoďte.
3. Použijte tři injekční lahvičky přípravku BLINCYTO prášek pro koncentrát. Pomocí stříkačky rekonstituujte každou injekční lahvičku přípravku BLINCYTO prášek pro koncentrát použitím 3 ml vody na injekci. Během rekonstituce nasměrujte vodu na injekci na boční stěnu injekční lahvičky. Injekční lahvičkou jemně otáčejte, aby se nevytvořilo nadměrné množství pěny. Injekční lahvičkou netřepejte.
• Nepoužívejte roztok (stabilizátor) k rekonstituci přípravku BLINCYTO prášek pro koncentrát.
• Přidáním vody na injekci k prášku pro koncentrát dosáhnete celkový objem 3,08 ml přípravku BLINCYTO o konečné koncentraci 12,5 ^g/ml.
4. Během rekonstituce a před podáním infuze prohlédněte rekonstituovaný roztok, zda se
v něm nevyskytují částice a nedošlo ke změně zbarvení. Výsledný roztok musí být čirý až lehce opalescentní a bezbarvý až nažloutlý. Roztok nepoužívejte, pokud je zkalený nebo obsahuje usazeniny.
5. Pomocí stříkačky asepticky přeneste 8 ml rekonstituovaného přípravku BLINCYTO do infuzního vaku (2,8 ml z každé z prvních dvou injekčních lahviček a zbývajících 2,4 ml z třetí injekční lahvičky). Jemně zamíchejte obsahem vaku, aby se nevytvořila pěna.
Zbylý rekonstituovaný roztok přípravku BLINCYTO vyhoďte.
6. Za aseptických podmínek připevněte intravenózní set k infuznímu vaku se sterilním 0,2^ in-line filtrem.
7. Z infuzního vaku odstraňte vzduch a naplňte intravenózní linku pouze připraveným infuzním roztokem. Linku nenaplňujte injekčním roztokem chloridu sodného 9 mg/ml (0,9%).
8. Jestliže takto připravený roztok nepoužijete ihned, uchovávejte ho při teplotě 2 °C - 8 °C.
h) Příprava přípravku BLINCYTO 28 pg/den podaného infuzí během 96 hodin rychlostí 2,5 ml/hod.
1. Použijte infuzní vak předplněný 250 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9%), který obvykle obsahuje celkový objem 265 až 275 ml.
2. K potažení infuzního vaku asepticky přeneste pomocí stříkačky 5,5 ml roztoku (stabilizátor) do infuzního vaku. Jemně zamíchejte obsahem vaku, aby se nevytvořila pěna. Injekční lahvičku se zbylým roztokem (stabilizátor) vyhoďte.
3. Použijte čtyři injekční lahvičky přípravku BLINCYTO prášek pro koncentrát. Pomocí stříkačky rekonstituujte každou injekční lahvičku přípravku BLINCYTO prášek pro koncentrát použitím 3 ml vody na injekci. Během rekonstituce nasměrujte vodu na injekci na boční stěnu injekční lahvičky. Injekční lahvičkou jemně otáčejte, aby se nevytvořilo nadměrné množství pěny. Injekční lahvičkou netřepejte.
• Nepoužívejte roztok (stabilizátor) k rekonstituci přípravku BLINCYTO prášek pro koncentrát.
• Přidáním vody na injekci k prášku pro koncentrát dosáhnete celkový objem 3,08 ml přípravku BLINCYTO o konečné koncentraci 12,5 ^g/ml.
4. Během rekonstituce a před podáním infuze prohlédněte rekonstituovaný roztok, zda se
v něm nevyskytují částice a nedošlo ke změně zbarvení. Výsledný roztok musí být čirý až lehce opalescentní a bezbarvý až nažloutlý. Roztok nepoužívejte, pokud je zkalený nebo obsahuje usazeniny.
5. Pomocí stříkačky asepticky přeneste 10,7 ml rekonstituovaného přípravku BLINCYTO do infuzního vaku (2,8 ml z každé z prvních tří injekčních lahviček a zbývajících 2,3 ml ze čtvrté injekční lahvičky). Jemně zamíchejte obsahem vaku, aby se nevytvořila pěna. Zbylý rekonstituovaný roztok přípravku BLINCYTO vyhoďte.
6. Za aseptických podmínek připevněte intravenózní set k infuznímu vaku se sterilním 0,2^ in-line filtrem.
7. Z infuzního vaku odstraňte vzduch a naplňte intravenózrn linku pouze připraveným infuzním roztokem. Linku nenaplňujte injekčním roztokem chloridu sodného 9 mg/ml (0,9%).
8. Jestliže takto připravený roztok nepoužijete ihned, uchovávejte ho při teplotě 2 °C - 8 °C. Pokyny pro podání léku jsou uvedeny v bodu 4.2.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Amgen Europe B.V.
Minervum 7061 4817 ZK Breda Nizozemsko
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/15/1047/001
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 23. listopad 2015
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
E. ZVLÁŠTNÍ POVINNOST USKUTEČNIT POREGISTRAČNÍ OPATŘENÍ PRO PODMÍNĚNOU REGISTRACI PŘÍPRAVKU
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce biologické léčivé látky
Lonza Biologics plc 228 Bath Road Slough
Berkshire, SL1 4DX Spojené království
Název a adresa výrobce odpovědného za propouštění šarží
Amgen Europe B.V.
Minervum 7061 4817ZK Breda Nizozemsko
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl.
107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
Držitel rozhodnutí o registraci předloží první pravidelně aktualizovanou zprávu o bezpečnosti pro tento léčivý přípravek do 6 měsíců od jeho registrace.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Další opatření k minimalizaci rizik
Před uvedením přípravku BLINCYTO na trh v každém členském státu se držitel rozhodnutí o registraci musí s příslušnými národními orgány dohodnout na obsahu a formě edukačního programu včetně komunikačních médií, způsobů distribuce a všech dalších aspektů programu.
Držitel rozhodnutí o registraci zabezpečí, aby v každém členském státu, kde je přípravek BLINCYTO obchodovaný, všichni zdravotničtí pracovníci a pacienti/pečovatelé, u nichž lze předpokládat, že budou přípravek BLINCYTO předepisovat, vydávat a používat, dostali následující edukační sady:
• Edukační materiál pro lékaře
• Edukační materiál pro lékárníky
• Edukační materiál pre zdravotní sestry
• Edukační materiál pro pacienty/pečovatele
• Karta pacienta
Edukační materiál pro lékaře musí obsahovat:
1. Souhrn údajů o přípravku (SPC)
2. Pokyny pro lékaře musí obsahovat tyto klíčové prvky:
• Upozornění na význam hlášení nežádoucích účinků léku
• Informace o léčbě přípravkem BLINCYTO, způsobu aplikace a dávkovaní, délce hospitalizace, přerušení a/nebo trvalém ukončení léčby
Chyby při medikaci (medication errors, ME)
• Údaje z klinických studií, příčiny ME, frekvence, závažnost a následky.
• Upozornění poradit pacientům, jak omezit riziko ME při používání infuzní pumpy. Neurologické příhody
• Údaje z klinických studií, frekvence a závažnost (byla pozorována neurologická toxicita stupně 3 a 4)
• Doporučení sledovat u pacientů projevy a příznaky neurotoxicity
• Postup při léčbě neurotoxicity (včetně úpravy dávky a přerušení léčby)
• Doporučení pacientům, aby během léčby přípravkem BLINCYTO neřídili motorová vozidla a aby se v případě výskytu neurologických příznaků okamžitě obrátili na ošetřujícícho lékaře
Edukační materiál pro lékárníky musí obsahovat:
1. Souhrn údajů o přípravku (SPC)
2. Pokyny pro lékárníky obsahující tyto klíčové prvky:
• Upozornění na význam hlášení nežádoucích účinků léku
• Podrobný popis postupů rekonstituce a přípravy infuzního roztoku BLINCYTO na intravenózní podání za aseptických podmínek a s použitím aseptických technik.
Edukační materiál pro zdravotní sestry musí obsahovat:
1. Souhrn údajů o přípravku (SPC)
2. Pokyny pro zdravotní sestry obsahující tyto klíčové prvky:
• Upozornění na význam hlášení nežádoucích účinků léku
• Popis postupů podávání přípravku BLINCYTO
• Popis postupů sledování pacienta a zvládání časných známek a příznaků neurologických příhod
• Doporučení pacientům, aby během léčby přípravkem BLINCYTO neřídili motorová vozidla a aby se v případě výskytu neurologických příznaků okamžitě obrátili na ošetřujícícho lékaře/zdravotní sestru
Edukační materiál pro pacienty (včetně pečovatelů) musí obsahovat:
1. Informační příručku pro pacienta včetně následujících klíčových prvků:
• Upozornění na význam hlášení nežádoucích účinků léku
• Popis postupů podávání přípravku BLINCYTO a pokyny jak snížit riziko ME při používání infuzní pumpy.
• Popis hlavních známek a/nebo příznaků neurologických příhod a význam okamžitého informování ošetřujícího lékaře nebo zdravotní sestry, pokud se příznaky vyskytnou
• Doporučení pacientům, aby během léčby přípravkem BLINCYTO neřídili motorová vozidla
2. Příbalovou informaci pro pacienta
Karta pacienta musí obsahovat:
• Upozornění pro zdravotnické pracovníky ošetřující pacienta, včetně naléhavých situací, že pacient užívá přípravek BLINCYTO
• Kontaktní údaje lékaře předepisujícího přípravek BLINCYTO
• Datum zahájení léčby přípravkem přípravkem BLINCYTO
• Upozornění na význam hlášení nežádoucích účinků léku
• Povinnost uskutečnit poregistrační opatření
Držitel rozhodnutí o registraci uskuteční v daném termínu níže uvedená opatření:
|
Popis |
Termín splnění |
|
Neintervenční poregistrační studie bezpečnosti (PASS): Studie 20150136: observační studie zaměřená na bezpečnost a účinnost blinatumomabu, jeho použití a léčebné postupy* |
Q42021 |
* Je třeba vypracovat protokol studie a předložit ho na posouzení výboru PRAC do 2 měsíců po rozhodnutí komise EU.
E. ZVLÁŠTNÍ POVINNOST USKUTEČNIT POREGISTRAČNÍ OPATŘENÍ PRO PODMÍNĚNOU REGISTRACI PŘÍPRAVKU
Tato registrace byla schválena postupem tzv. podmíněného schválení, a proto podle článku 14(7) nařízení (ES) č. 726/2004 držitel rozhodnutí o registraci uskuteční v daném termínu následující opatření:
|
Popis |
Termín splnění |
|
Poregistrační studie účinnosti (PAES):Studie 00103311 (TOWER): Studie hodnotící BITE protilátku blinatumomab ve srovnání se standardní chemoterapií u dospělých pacientů s relabovanou/refrakterní B-prekurzorovou akutní lymfoblastickou leukémií (ALL) |
Q12017 |
• Podmínky nebo omezení s ohledem na bezpečné a účinné používání léčivého přípravku, které mají členské státy zavést.
Neuplatňuje se.
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
BLINCYTO 38,5 mikrogramu prášek pro koncentrát a roztok pro infuzní roztok blinatumomabum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna injekční lahvička s práškem obsahuje blinatumomabum 38,5 mikrogramu.
Jedna injekční lahvička obsahuje po rozpuštění vodou na injekci blinatumomabum 12,5 mikrogramu/ml.
3. SEZNAM POMOCNÝCH LÁTEK
Prášek: monohydrát kyseliny citronové (E330), dihydrát trehalosy, lysin-hydrochlorid, polysorbát 80 a hydroxid sodný.
Roztok (stabilizátor): monohydrát kyseliny citronové (E330), lysin-hydrochlorid, polysorbát 80, hydroxid sodný a voda na injekci.
Další informace viz příbalová informace.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Prášek pro koncentrát a roztok pro infuzní roztok 1 injekční lahvička s práškem.
1 injekční lahvička s roztokem (stabilizátor). Přidat pouze do vaku s chloridem sodným.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Intravenózní podání po rekonstituci a naředění.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN
MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
Rozpuštěný roztok neprotřepávejte.
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte a převážejte chlazené.
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Amgen Europe B.V. Minervum 7061, 4817 ZK Breda, Nizozemsko
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/15/1047/001
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
Nevyžaduje se - odůvodnění přijato.
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
BLINCYTO 38,5 pg prášek pro koncentrát
blinatumomabum
i.v. po rekonstituci a naředění
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Roztok (stabilizátor)
BLINCYTO
|
2. |
ZPŮSOB PODÁNÍ |
|
3. |
POUŽITELNOST |
|
EXP | |
|
ČÍSLO ŠARŽE | |
|
4. | |
|
Lot | |
|
5. |
OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET |
|
10 ml | |
|
6. |
JINÉ |
Přidat pouze do vaku s chloridem sodným.
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro pacienta
BLINCYTO 38,5 mikrogramu prášek pro koncentrát a roztok pro infuzní roztok
blinatumomabum
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je BLINCYTO a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete BLINCYTO používat
3. Jak se BLINCYTO používá
4. Možné nežádoucí účinky
5 Jak BLINCYTO uchovávat
6. Obsah balení a další informace
1. Co je BLINCYTO a k čemu se používá
Léčivou látkou přípravku BLINCYTO je blinatumomab. Ten patří do skupiny léků nazývaných cytostatika, která se zaměřují na nádorové buňky.
BLINCYTO se používá k léčbě dospělých s akutní lymfoblastickou leukemií. Akutní lymfoblastická leukemie je nádorové onemocnění krve, při kterém nekontrolovaně roste určitý druh bílých krvinek zvaných B-lymfocyty. Tento lék účinkuje tak, že umožní imunitnímu systému útočit na tyto abnormální nádorové bílé krvinky a ničit je.
2. Čemu musíte věnovat pozornost, než začnete BLINCYTO používat Nepoužívejte BLINCYTO:
- jestliže jste alergický(á) na blinatumomab nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6);
- pokud kojíte.
Upozornění a opatření
Před použitím přípravku BLINCYTO se poraďte se svým lékařem, lékárníkem nebo zdravotní sestrou, pokud se Vás týká některá z níže uvedených situací. BLINCYTO pro vás nemusí být vhodný: • jestliže jste někdy měl(a) neurologické potíže, např. třes, abnormální pocity, epileptické
záchvaty, ztrátu paměti, zmatenost, dezorientaci, ztrátu rovnováhy nebo obtíže při mluvení. Jestli máte stále aktivní neurologické potíže nebo onemocnění, informujte o tom svého lékaře. Jestliže se u Vás leukemie rozšířila do mozku a/nebo míchy, lékař před zahájením léčby přípravkem BLINCYTO možná bude muset léčit nejdříve tento stav. Před rozhodnutím, zda byste měl(a) být léčen(a) přípravkem BLINCYTO, lékař vyšetří Váš nervový systém a provede určitá vyšetření. Může se stát, že Vám lékař během léčby přípravkem BLINCYTO bude muset věnovat zvláštní péči.
• jestliže máte aktivní infekci.
• jestliže jste v minulosti měl(a) infuzní reakci po používání přípravku BLINCYTO. Příznaky mohou zahrnovat sípot, návaly, otok obličeje, potíže s dýcháním, nízký nebo vysoký krevní tlak.
• jestliže se domníváte, že v blízké budoucnosti budete očkován(a) včetně očkování potřebných při cestě do zahraničí. Některé očkovací látky se nesmějí podat dva týdny před léčbou, během léčby nebo několik měsíců po léčbě přípravkem BLINCYTO. Váš lékař zkontroluje, zda máte být očkován(a).
Informujte ihned lékaře, lékárníka nebo zdravotní sestru, pokud se u Vás během léčby přípravkem BLINCYTO vyskytne některá z níže uvedených reakcí, protože tato reakce možná bude muset být léčena a dávka léku upravena:
• jestliže se u Vás vyskytnou epileptické záchvaty, obtíže při mluvení nebo špatné vyslovování, zmatenost a dezorientace, případně ztráta rovnováhy.
• jestliže máte zimnici nebo třesavku, případně pociťujete horkost. Změřte si teplotu, protože můžete mít horečku - toto mohou být projevy infekce.
• jestliže se u Vás kdykoliv během infuze vyskytne reakce, která se může projevovat jako závratě, mdloby, pocit na zvracení, otok obličeje, dechové obtíže, sípot nebo vyrážka.
Lékař nebo zdravotní sestra Vás budou sledovat z hlediska známek a příznaků těchto reakcí.
Informujte ihned svého lékaře, lékárníka nebo zdravotní sestru, pokud otěhotníte během léčby přípravkem BLINCYTO. Lékař s Vámi prodiskutuje opatření týkající se očkování Vašeho dítěte.
Před každým cyklem infuze s přípravkem BLINCYTO dostanete léky, které mohou pomoci snížit potenciálně život ohrožující komplikace zvané syndrom nádorového rozpadu, který je způsobený chemickými poruchami v krvi v důsledku rozpadu odumírajících nádorových buněk. Můžete rovněž dostat léky ke snížení horečky.
Během léčby, zejména v prvních několika dnech po zahájení léčby, u Vás může dojít k těžkému poklesu počtu bílých krvinek (neutropenie), těžkému poklesu počtu bílých krvinek spolu s horečkou (febrilní neutropenie), zvýšení jaterních enzymů nebo zvýšení hladiny kyseliny močové. Lékař Vám bude během léčby přípravkem BLINCYTO provádět pravidelná vyšetření krve s cílem sledovat Váš krevní obraz.
Děti a dospívající
BLINCYTO se nemá používat u dětí a dospívajících mladších 18 let.
Další léčivé přípravky a BLINCYTO
Informujte svého lékaře, lékárníka nebo zdravotní sestru o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Těhotenství a kojení
Pokud jste těhotná, kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo zdravotní sestrou dříve, než začnete tento přípravek používat.
Antikoncepce
Ženy, které mohou otěhotnět, musí během léčby a nejméně 48 hodin po léčbě přípravkem BLINCYTO používat účinnou antikoncepci. Poraďte se se svým lékařem nebo zdravotní sestrou o vhodné metodě antikoncepce.
Účinky přípravku BLINCYTO u těhotných žen nejsou známé, ale na základě jeho mechanismu účinku může přípravek BLINCYTO poškodit nenarozené dítě. Nepoužívejte přípravek BLINCYTO během těhotenství, ledaže by Váš lékař rozhodl, že to je pro Vás nejlepší léčba.
Jestliže během léčby přípravkem BLINCYTO otěhotníte, informujte o tom svého lékaře nebo zdravotní sestru. Lékař s Vámi prodiskutuje opatření týkající se očkování Vašeho dítěte.
Kojení
Nesmíte kojit během léčby a nejméně 48 hodin po poslední léčbě. Není známo, zda se BLINCYTO vylučuje do mateřského mléka, ale nemůže být vyloučeno riziko pro kojené dítě.
Řízení dopravních prostředků a obsluha strojů
Během léčby přípravkem BLINCYTO neřiďte dopravní prostředky, neobsluhujte těžké stroje ani se nezúčastňujte nebezpečných činností. BLINCYTO může způsobovat neurologické potíže, jako jsou závratě, epileptické záchvaty, zmatenost, poruchy koordinace a rovnováhy.
BLINCYTO obsahuje sodík
Tento přípravek obsahuje méně než 1 mmol (23 mg) sodíku v jedné dávce, tj., v podstatě je „bez sodíku“.
3. Jak se BLINCYTO používá
Vždy používejte tento přípravek přesně podle pokynů svého lékaře, lékárníka nebo zdravotní sestry. Pokud si nejste jistý(á), poraďte se se svým lékařem, lékárníkem nebo zdravotní sestrou.
Jak se BLINCYTO podává
BLINCYTO budete dostávat do žíly (intravenózně) nepřetržitě po dobu 4 týdnů pomocí infuzní pumpy (toto období představuje 1 léčebný cyklus). Poté budete mít dvoutýdenní přestávku, během které nebudete mít infuzi. Infuzní katetr (cévku) budete mít zavedený po celou dobu každého léčebného cyklu.
BLINCYTO se obvykle podává 2 léčebné cykly. Jestliže budete dobře reagovat na léčbu přípravkem BLINCYTO v prvních dvou cyklech, lékař se může rozhodnout pro podání až 3 dalších cyklů. Počet léčebných cyklů, které dostanete, bude záležet na tom, jak budete BLINCYTO snášet a na léčbu odpovídat. Lékař s Vámi probere, jak dlouho bude léčba trvat. Léčba může být rovněž přerušena v závislosti na tom, jak budete BLINCYTO snášet.
Doporučuje se, abyste v prvních 9 dnech léčby zůstal (a) v nemocnici pod dohledem lékaře nebo zdravotní sestry se zkušenostmi s používáním protinádorových léků. Pokud jste v minulosti měl(a) nebo máte neurologické obtíže, doporučuje se, abyste během prvních 14 dnů léčby zůstal(a) v nemocnici. Lékař s Vámi probere, zda po úvodním pobytu v nemocnici můžete pokračovat v léčbě doma. Léčba může zahrnovat výměnu vaku, kterou provede zdravotní sestra.
Lékař určí, kdy se má infuzní vak s přípravkem BLINCYTO vyměnit, což může být denně až každé 4 dny. Rychlost podávání infuze může být rychlejší nebo pomalejší v závislosti na tom, jak často se bude vak měnit.
První cyklus
Doporučená úvodní dávka v prvním cyklu je 9 mikrogramů denně po dobu 1 týdne. Poté lékař může rozhodnout o zvýšení dávky na 28 mikrogramů denně ve 2., 3. a 4. týdnu léčby.
Další cykly
Jestliže lékař určí, že máte dostat víc cyklů s přípravkem BLINCYTO, nastaví Vám infuzní pumpu na dávku 28 mikrogramů denně.
Léky, které dostanete před každým cyklem léčby přípravkem BLINCYTO
Před léčbou přípravkem BLINCYTO dostanete další léky (premedikace), které mají pomoci snížit infuzní reakce a další možné nežádoucí účinky. Mohou to být kortikosteroidy (např. dexamethason).
Infuzní katetr
Jestliže máte zavedený infuzní katetr, je velmi důležité, abyste udržoval(a) oblast kolem katetru čistou, jinak byste mohl(a) dostat infekci. Lékař nebo zdravotní sestra Vám ukáží, jak se máte starat o místo zavedení katetru.
Infuzní pumpa a nitrožilní sety
Neupravujte nastavení infuzní pumpy, i když se vyskytne problém s pumpou nebo alarm pumpy vydává zvuk. Změny nastavení pumpy mohou vést k tomu, že podávaná dávka bude příliš vysoká nebo příliš nízká.
Kontaktujte svého lékaře nebo zdravotní sestru ihned, když:
• se vyskytne problém s pumpou nebo alarm pumpy vydává zvuk
• se domníváte, že byste mohl(a) dostat více přípravku BLINCYTO než byste měl(a), např. když se infuzní vak vyprázdní dříve, než se má vyměnit
• se infuzní pumpa neočekávaně zastaví. Nepokoušejte se restartovat pumpu. Lékař rozhodne, kdy dostanete další dávku přípravku BLINCYTO.
Lékař nebo zdravotní sestra Vám poradí, jak provádět každodenní činnosti spolu s infuzní pumpou. Chcete-li se na něco zeptat, obraťte se na svého lékaře nebo zdravotní sestru.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého. Některé z těchto nežádoucích účinků mohou být závažné.
Informujte svého lékaře ihned, pokud se objeví některý z níže uvedených nežádoucích účinků nebo jejich kombinace:
• zimnice, třesavka, horečka, rychlá srdeční akce, pokles krevního tlaku, bolest svalů, pocit únavy, kašel, dechové obtíže, zmatenost, zarudnutí, otok nebo výtok z postižené oblasti nebo v místě infuzní linky - tyto potíže mohou být známky infekce
• neurologické příhody: třes, zmatenost, poruchy funkce mozku (encefalopatie), obtíže v komunikaci (afázie), epileptické záchvaty (křeče)
• horečka, otok, zimnice, pokles nebo vzestup krevního tlaku a voda na plicích. Tyto potíže mohou být závažné a může se se jednat o příznak tzv. syndromu z uvolnění cytokinů.
Léčba přípravkem BLINCYTO může způsobit snížení počtu některých bílých krvinek spolu s horečkou nebo i bez ní (febrilní neutropenie nebo neutropenie) nebo může vést ke zvýšení hladiny draslíku, kyseliny močové a fosfátu v krvi a snížení hladiny vápníku v krvi (syndrom nádorového rozpadu). Lékař Vám bude pravidelně provádět vyšetření krve během léčby přípravkem BLINCYTO.
Další nežádoucí účinky jsou:
Velmi časté nežádoucí účinky (mohou se vyskytnout u více než 1 z 10 léčených osob):
• infekce krve včetně bakteriálních, plísňových, virových anebo jiných typů infekce
• snížený počet určitých bílých krvinek s horečkou nebo bez ní ((febrilní) neutropenie, leukopenie), snížený počet červených krvinek, snížený počet krevních destiček
• horečka, otok, zimnice, pokles nebo vzestup krevního tlaku a voda na plicích, které mohou být závažné (syndrom z uvolnění cytokinů)
• nízká hladina draslíku v krvi, nízká hladina hořčíku v krvi, vysoká hladina cukru v krvi, snížená chuť k jídlu
• nespavost
• bolest hlavy, třes, závratě
• nízký krevní tlak
• kašel
• pocit na zvracení, zácpa, průjem, bolest břicha, zvracení, vyrážka
• bolest zad, bolest končetiny, bolestivé a oteklé klouby, bolest kostí
• horečka (pyrexie), otoky rukou, kotníků nebo nohou, zimnice, únava, bolest na hrudi
• zvýšené hladiny jaterních enzymů (ALT, AST)
• reakce v souvislosti s infuzí mohou být sípot, návaly, otok obličeje, dechové potíže, nízký krevní tlak, vysoký krevní tlak.
Časté nežádoucí účinky (mohou se vyskytnout až u 1 z 10 léčených osob):
• těžké infekce, které mohou vést k orgánovému selhání, šoku a mohou být i smrtelné (sepse)
• infekce plic (pneumonie)
• vyšší hodnoty počtu bílých krvinek, snížení počtu některých bílých krvinek (lymfopenie)
• horečka, otoky, zimnice, snížený nebo zvýšený krevní tlak a voda na plicích, které mohou být závažné a mohou být i smrtelné (cytokinová bouře), alergická reakce
• snížená hladina fosfátu v krvi
• snížená hladina bílkovin v krvi, která způsobuje zadržování vody
• komplikace vyskytující se po léčbě nádorových onemocnění vedoucí k vyšším hladinám draslíku, kyseliny močové a fosfátu v krvi a sníženým hladinám vápníku v krvi (syndrom nádorového rozpadu)
• zmatenost, dezorientace
• poruchy funkce mozku (encefalopatie), jako jsou potíže v komunikaci (afázie), mravenčení v kůži (parestezie), epileptické záchvaty (křeče), potíže s myšlením nebo zpracováním myšlenek, potíže se zapamatováním si
• rychlá srdeční akce (tachykardie)
• otok obličeje, rtů, úst, jazyka nebo hrdla, který může způsobit polykací nebo dechové obtíže
• snížené hladiny protilátek, zvaných imunoglobuliny, které pomáhají imunitnímu systému v boji s infekcí (pokles imunoglobulinů)
• změny v krvi včetně zvýšené hladiny bilirubinu
• zvýšené hladiny jaterních enzymů (GGT).
Méně časté nežádoucí účinky (mohou se se vyskytnout až u 1 ze 100 léčených osob):
• stav vedoucí k úniku tekutiny z drobných krevních cév do těla (syndrom zvýšené permeability kapilár).
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak BLINCYTO uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na štítku a na krabičce za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Neotevřené injekční lahvičky:
- Uchovávejte a převážejte chlazené (2 °C - 8 °C).
- Chraňte před mrazem.
- Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Rekonstituovaný roztok (BLINCYTO roztok):
- Při uchovávání v chladničce musí být rekonstituovaný roztok použit do 24 hodin. Jinak je možné uchovávat injekční lahvičky při pokojové teplotě (do 27 °C) po dobu maximálně
4 hodin.
Zředěný roztok (připravený infuzní vak):
Jestliže se infuzní vak mění doma:
- Infuzní vaky obsahující přípravek BLINCYTO infuzní roztok budou doručeny ve speciálním balení obsahujícím chladící sáčky.
• Neotvírejte balení.
• Uchovávejte balení při pokojové teplotě (do 27 °C).
• Neuchovávejte balení v chladničce nebo mrazničce.
- Balení otevře zdravotní sestra a infuzní vaky budou uchovávány v chladničce až do doby podání infuze.
- Při uchovávání v chladničce se musí infuzní vaky použít do 10 dnů od přípravy.
- Při pokojové teplotě (do 27 °C) se má roztok podat infuzí během 96 hodin.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co BLINCYTO obsahuje
- Léčivou látkou je blinatumomabum. Jedna injekční lahvička s práškem obsahuje blinatumomabum 38,5 mikrogramu. Po rekonstituci s vodou na injekci získáme blinatumomab o výsledné koncentraci 12,5 mikrogramu/ml.
- Dalšími složkami v prášku jsou: monohydrát kyseliny citronové (E330), dihydrát trehalosy, lysin-hydrochlorid, polysorbát 80 a hydroxid sodný.
- Roztok (stabilizátor) obsahuje monohydrát kyseliny citronové (E330), lysin-hydrochlorid, polysorbát 80, hydroxid sodný a vodu na injekci.
Jak BLINCYTO vypadá a co obsahuje toto balení
BLINCYTO je prášek pro koncentrát a roztok pro infuzní roztok.
Jedno balení přípravku BLINCYTO obsahuje:
• 1 skleněnou injekční lahvičku obsahující bílý až téměř bílý prášek.
• 1 skleněnou injekční lahvičku obsahující bezbarvý až nažloutlý čirý roztok.
Držitel rozhodnutí o registraci a výrobce
Amgen Europe B.V.
Minervum 7061 4817 ZK Breda Nizozemsko
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci
|
Belgie/Belgique/Belgien s.a. Amgen n.v. Tel/Tél: +32 (0)2 7752711 |
Lietuva Amgen Switzerland AG Vilniaus filialas Tel: +370 5 219 7474 |
|
BtnrapHH AMg^eH Etnrapua EOOfl Ten.: +359 (0)2 424 7440 |
Luxembourg/Luxemburg s.a. Amgen Belgique/Belgien Tel/Tél: +32 (0)2 7752711 |
|
Česká republika Amgen s.r.o. Tel: +420 221 773 500 |
Magyarország Amgen Kft. Tel.: +36 1 35 44 700 |
|
Danmark Amgen filial af Amgen AB, Sverige Tlf: +45 39617500 |
Malta Amgen B.V. The Netherlands Tel: +31 (0)76 5732500 |
|
Deutschland AMGEN GmbH Tel.: +49 89 1490960 |
Nederland Amgen B.V. Tel: +31 (0)76 5732500 |
|
Eesti Amgen Switzerland AG Vilniaus filialas Tel: +372 586 09553 |
Norge Amgen AB Tel: +47 23308000 |
|
EXláda Amgen ETAág OappaKaurixá E.n.E. TpL: +30 210 3447000 |
Osterreich Amgen GmbH Tel: +43 (0)1 50 217 |
|
Espaňa Amgen S.A. Tel: +34 93 600 18 60 |
Polska Amgen Biotechnologia Sp. z o.o. Tel.: +48 22 581 3000 |
|
France Amgen S.A.S. Tél: +33 (0)9 69 363 363 |
Portugal Amgen Biofarmaceutica, Lda. Tel: +351 21 4220550 |
|
Hrvatska Amgen d.o.o. Tel: +385 (1) 562 57 20 |
Románia Amgen Románia SRL Tel: +4021 527 3000 |
|
Ireland Amgen Limited United Kingdom Tel: +44 (0)1223 420305 |
Slovenija AMGEN zdravila d.o.o. Tel: +386 (0)1 585 1767 |
Suomi/Finland
Italia
Amgen S.r.l.
Tel: +39 02 6241121
Amgen AB, sivuliike Suomessa/Amgen AB, filial i Finland
Puh/Tel: +358 (0)9 54900500
Latvija United Kingdom
Amgen Switzerland AG Rlgas filiale Amgen Limited
Tel: +371 257 25888 Tel: +44 (0)1223 420305
Tato příbalová informace byla naposledy revidována v Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Následující informace jsou určeny pouze pro zdravotnické pracovníky:
BLINCYTO infuzní roztok se podává jako kontinuální intravenózní infuze konstantní rychlostí pomocí infuzní pumpy po dobu až 96 hodin.
Doporučená úvodní dávka přípravku BLINCYTO v prvním cyklu je 9 pg denně po dobu týdne 1 (prvních 7 dní) léčby.
Dávka se zvýší na 28 pg/den počínaje týdnem 2 do týdne 4 prvního cyklu. Ve všech dalších cyklech se podává dávka 28 pg/den po celé čtyřtýdenní léčebné období.
Terapeutická dávka 9 pg/den nebo 28 pg/den se podá pacientovi v infuzi o celkovém objemu 240 ml přípravku BLINCYTO infuzní roztok jednou ze 4 konstantních rychlostí a příslušnou délkou trvání infuze:
• Rychlost infuze 10 ml/hod. při trvání infuze 24 hodin
• Rychlost infuze 5 ml/hod. při trvání infuze 48 hodin
• Rychlost infuze 3,3 ml/hod. při trvání infuze 72 hodin
• Rychlost infuze 2,5 ml/hod. při trvání infuze 96 hodin
Délku trvání infuze zvolí ošetřující lékař na základě zvážení frekvence výměn infuzního vaku. Cílová podaná terapeutická dávka přípravku BLINCYTO se nemění.
Aseptická příprava
Při přípravě infuze je třeba dodržovat aseptické podmínky. Příprava přípravku BLINCYTO se má:
- provádět za aseptických podmínek personálem zaškoleným v pravidlech správné praxe, zejména z hlediska aseptické přípravy parenterálních přípravků;
- připravovat v laminárním boxu nebo v boxu pro bezpečné zacházení s biologickým materiálem při použití standardních bezpečnostních opatření na bezpečné zacházení s intravenózními látkami.
Je mimořádně důležité, aby se přísně dodržovaly pokyny pro přípravu a podání uvedené v tomto bodě, aby se tak minimalizovaly chyby medikace (včetně poddávkování a předávkování).
Speciální pokyny sloužící ke správné přípravě
• Roztok (stabilizátor) se dodává v balení přípravku BLINCYTO a používá se k potažení předplněného infuzního vaku před přidáním rekonstituovaného přípravku BLINCYTO. Nepoužívejte tento roztok (stabilizátor) k rekonstituci přípravku BLINCYTO prášku pro koncentrát.
• Celý objem rekonstituovaného a naředěného přípravku BLINCYTO bude větší než objem, který se má podat pacientovi (240 ml). Je třeba počítat se ztrátou použitím intravenózní infuzní linky a zajistit, že pacient dostane plnou dávku přípravku BLINCYTO.
• Při přípravě infuzního vaku z něho odstraňte všechen vzduch. Toto je mimořádně důležité při použití ambulantní infuzní pumpy.
• Použijte konkrétní objemy uvedené níže v návodu pro rekonstituci a ředění, aby se minimalizovaly chyby při výpočtu.
Další pokyny
• BLINCYTO je kompatibilní s infuzními vaky/kazetami infuzních pump z polyolefinu, PVC non-diethylhexylftalátu (non-DEHP), nebo ethylvinylacetátu (EVA).
• Specifikace pump: Infuzní pumpa k podání přípravku BLINCYTO infuzní roztok musí být programovatelná, zamykatelná a mít alarm. Elastomerické infuzní pumpy se nesmějí používat.
• Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Příprava infuzního roztoku
Pro každou dávku a dobu infuze jsou uvedené přesné pokyny pro rekonstituci a naředění. Ověřte si předepsanou dávku a dobu infuze přípravku BLINCYTO a použijte příslušný bod níže pro přípravu správné dávky. Dodržujte postup pro rekonstituci přípravku BLINCYTO a pro přípravu infuzního vaku.
a) pro infuzi o koncentraci 9 pg/den podanou během 24 hodin rychlostí 10 ml/hod.
b) pro infuzi o koncentraci 9 pg/den podanou během 48 hodin rychlostí 5 ml/hod.
c) pro infuzi o koncentraci 9 pg/den podanou během 72 hodin rychlostí 3,3 ml/hod.
d) pro infuzi o koncentraci 9 pg/den podanou během 96 hodin rychlostí 2,5 ml/hod.
e) pro infuzi o koncentraci 28 pg/den podanou během 24 hodin rychlostí 10 ml/hod.
f) pro infuzi o koncentraci 28 pg/den podanou během 48 hodin rychlostí 5 ml/hod.
g) pro infuzi o koncentraci 28 pg/den podanou během 72 hodin rychlostí 3,3 ml/hod.
h) pro infuzi o koncentraci 28 pg/den podanou během 96 hodin rychlostí 2,5 ml/hod.
Před přípravou se ujistěte, že máte připraven následující materiál:
|
Dávka |
Trvání infuze (hod.) |
Rychlost infuze (ml/hod.) |
Počet balení přípravku BLINCYTO |
|
9 pg/den |
24 |
10 |
1 |
|
48 |
5 |
1 | |
|
72 |
3,3 |
1 | |
|
96 |
2,5 |
2 | |
|
28 pg/den |
24 |
10 |
1 |
|
48 |
5 |
2 | |
|
72 |
3,3 |
3 | |
|
96 |
2,5 |
4 |
Dále budete potřebovat tento materiál, který ale není součástí balení přípravku BLINCYTO
• Sterilní injekční stříkačky na jedno použití
• Jehly o rozměru 21 - 23 gauge (doporučeno)
• Voda na injekci
• Infuzní vak s 250 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9%);
o K minimalizaci počtu aseptických přenosů použijte 250 ml předplněný infuzní vak. Výpočty dávky přípravku BLINCYTO jsou založené na obvyklém přeplněném objemu 265 až 275 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9%).
o Použijte pouze infuzní vaky/kazety infuzních pump z polyolefinu, PVC non-diethylhexylftalátu (non-DEHP), nebo ethylvinylacetátu (EVA).
• Infuzní set z polyolefinu, PVC non-DEHP, nebo EVA se sterilním a nepyrogenním 0,2^m in-line filtrem s nízkou vazbou na bílkoviny
o Přesvědčte se, že infuzní set je kompatibilní s infuzní pumpou.
a) Příprava přípravku BLINCYTO 9 yg/den podaného infuzí během 24 hodin rychlostí 10 ml/hod.
1. Použijte infuzní vak předplněný 250 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9%), který obvykle obsahuje celkový objem 265 až 275 ml.
2. K potažení infuzního vaku asepticky přeneste pomocí stříkačky 5,5 ml roztoku (stabilizátoru) do infuzního vaku. Jemně zamíchejte obsahem vaku, aby se nevytvořila pěna. Injekční lahvičku se zbylým roztokem (stabilizátorem) vyhoďte.
3. Pomocí stříkačky rekonstituujte jednu injekční lahvičku přípravku BLINCYTO prášek pro koncentrát použitím 3 ml vody na injekci. Během rekonstituce nasměrujte vodu na injekci na boční stěnu injekční lahvičky. Injekční lahvičkou jemně otáčejte, aby se nevytvořilo nadměrné množství pěny. Injekční lahvičkou netřepejte.
• Nepoužívejte roztok (stabilizátor) k rekonstituci přípravku BLINCYTO prášek pro koncentrát.
• Přidáním vody na injekci k prášku pro koncentrát dosáhnete celkový objem 3,08 ml přípravku BLINCYTO o konečné koncentraci 12,5 ^g/ml.
4. Během rekonstituce a před podáním infuze prohlédněte rekonstituovaný roztok, zda se
v něm nevyskytují částice a nedošlo ke změně zbarvení. Výsledný roztok musí být čirý až lehce opalescentní a bezbarvý až nažloutlý. Roztok nepoužívejte, pokud je zkalený nebo obsahuje usazeniny.
5. Pomocí stříkačky asepticky přeneste 0,83 ml rekonstituovaného přípravku BLINCYTO do infuzního vaku. Jemně zamíchejte obsahem vaku, aby se nevytvořila pěna. Zbylý rekonstituovaný roztok přípravku BLINCYTO vyhoďte.
6. Za aseptických podmínek připevněte intravenózní set k infuznímu vaku se sterilním 0,2^ in-line filtrem.
7. Z infuzního vaku odstraňte vzduch a naplňte intravenózní linku pouze připraveným infuzním roztokem. Linku nenaplňujte injekčním roztokem chloridu sodného 9 mg/ml (0,9%).
8. Jestliže takto připravený roztok nepoužijete ihned, uchovávejte ho při teplotě 2 °C - 8 °C.
b) Příprava přípravku BLINCYTO 9 yg/den podaného infuzí během 48 hodin rychlostí 5 ml/hod.
1. Použijte infuzní vak předplněný 250 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9%), který obvykle obsahuje celkový objem 265 až 275 ml.
2. K potažení infuzního vaku asepticky přeneste pomocí stříkačky 5,5 ml roztoku (stabilizátoru) do infuzního vaku. Jemně zamíchejte obsahem vaku, aby se nevytvořila pěna. Injekční lahvičku se zbylým roztokem (stabilizátorem) vyhoďte.
3. Pomocí stříkačky rekonstituujte jednu injekční lahvičku přípravku BLINCYTO prášek pro koncentrát použitím 3 ml vody na injekci. Během rekonstituce nasměrujte vodu na injekci na boční stěnu injekční lahvičky. Injekční lahvičkou jemně otáčejte, aby se nevytvořilo nadměrné množství pěny. Injekční lahvičkou netřepejte.
• Nepoužívejte roztok (stabilizátor) k rekonstituci přípravku BLINCYTO prášek pro koncentrát.
• Přidáním vody na injekci k prášku pro koncentrát dosáhnete celkový objem 3,08 ml přípravku BLINCYTO o konečné koncentraci 12,5 ^g/ml.
4. Během rekonstituce a před podáním infuze prohlédněte rekonstituovaný roztok, zda se
v něm nevyskytují částice a nedošlo ke změně zbarvení. Výsledný roztok musí být čirý až lehce opalescentní a bezbarvý až nažloutlý. Roztok nepoužívejte, pokud je zkalený nebo obsahuje usazeniny.
5. Pomocí stříkačky asepticky přeneste 1,7 ml rekonstituovaného přípravku BLINCYTO do infuzního vaku. Jemně zamíchejte obsahem vaku, aby se nevytvořila pěna. Zbylý rekonstituovaný roztok přípravku BLINCYTO vyhoďte.
6. Za aseptických podmínek připevněte intravenózní set k infuznímu vaku se sterilním 0,2p in-line filtrem.
7. Z infuzního vaku odstraňte vzduch a naplňte intravenózní linku pouze připraveným infuzním roztokem. Linku nenaplňujte injekčním roztokem chloridu sodného 9 mg/ml (0,9%).
8. Jestliže takto připravený roztok nepoužijete ihned, uchovávejte ho při teplotě 2 °C - 8 °C.
c) Příprava přípravku BLINCYTO 9 ug/den podaného infuzí během 72 hodin rychlostí 3,3 ml/hod.
1. Použijte infuzní vak předplněný 250 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9%), který obvykle obsahuje celkový objem 265 až 275 ml.
2. K potažení infuzního vaku asepticky přeneste pomocí stříkačky 5,5 ml roztoku (stabilizátoru) do infuzního vaku. Jemně zamíchejte obsahem vaku, aby se nevytvořila pěna. Injekční lahvičku se zbylým roztokem (stabilizátorem) vyhoďte.
3. Pomocí stříkačky rekonstituujte jednu injekční lahvičku přípravku BLINCYTO prášek pro koncentrát použitím 3 ml vody na injekci. Během rekonstituce nasměrujte vodu na injekci na boční stěnu injekční lahvičky. Injekční lahvičkou jemně otáčejte, aby se nevytvořilo nadměrné množství pěny. Injekční lahvičkou netřepejte.
• Nepoužívejte roztok (stabilizátor) k rekonstituci přípravku BLINCYTO prášek pro koncentrát.
• Přidáním vody na injekci k prášku pro koncentrát dosáhnete celkový objem 3,08 ml přípravku BLINCYTO o konečné koncentraci 12,5 pg/ml.
4. Během rekonstituce a před podáním infuze prohlédněte rekonstituovaný roztok, zda se
v něm nevyskytují částice a nedošlo ke změně zbarvení. Výsledný roztok musí být čirý až lehce opalescentní a bezbarvý až nažloutlý. Roztok nepoužívejte, pokud je zkalený nebo obsahuje usazeniny.
5. Pomocí stříkačky asepticky přeneste 2,5 ml rekonstituovaného přípravku BLINCYTO do infuzního vaku. Jemně zamíchejte obsahem vaku, aby se nevytvořila pěna. Zbylý rekonstituovaný roztok přípravku BLINCYTO vyhoďte.
6. Za aseptických podmínek připevněte intravenózní set k infuznímu vaku se sterilním 0,2p in-line filtrem.
7. Z infuzního vaku odstraňte vzduch a naplňte intravenózní linku pouze připraveným infuzním roztokem. Linku nenaplňujte injekčním roztokem chloridu sodného 9 mg/ml (0,9%).
8. Jestliže takto připravený roztok nepoužijete ihned, uchovávejte ho při teplotě 2 °C - 8 °C.
d) Příprava přípravku BLINCYTO 9 jug/den podaného infuzí během 96 hodin rychlostí 2,5 ml/hod.
1. Použijte infuzní vak předplněný 250 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9%), který obvykle obsahuje celkový objem 265 až 275 ml.
2. K potažení infuzního vaku asepticky přeneste pomocí stříkačky 5,5 ml roztoku (stabilizátoru) do infuzního vaku. Jemně zamíchejte obsahem vaku, aby se nevytvořila pěna. Injekční lahvičku se zbylým roztokem (stabilizátorem) vyhoďte.
3. Použijte dvě injekční lahvičky přípravku BLINCYTO prášek pro koncentrát. Pomocí stříkačky rekonstituujte každou injekční lahvičku přípravku BLINCYTO prášek pro koncentrát použitím 3 ml vody na injekci. Během rekonstituce nasměrujte vodu na injekci na boční stěnu injekční lahvičky. Injekční lahvičkou jemně otáčejte, aby se nevytvořilo nadměrné množství pěny. Injekční lahvičkou netřepejte.
• Nepoužívejte roztok (stabilizátor) k rekonstituci přípravku BLINCYTO prášek pro koncentrát.
• Přidáním vody na injekci k prášku pro koncentrát dosáhnete celkový objem 3,08 ml přípravku BLINCYTO o konečné koncentraci 12,5 pg/ml.
4. Během rekonstituce a před podáním infuze prohlédněte rekonstituovaný roztok, zda se
v něm nevyskytují částice a nedošlo ke změně zbarvení. Výsledný roztok musí být čirý až lehce opalescentní a bezbarvý až nažloutlý. Roztok nepoužívejte, pokud je zkalený nebo obsahuje usazeniny.
5. Pomocí stříkačky asepticky přeneste 3,3 ml rekonstituovaného přípravku BLINCYTO do infuzního vaku (2,0 ml z jedné injekční lahvičky a dalších 1,3 ml z druhé injekční lahvičky). Jemně zamíchejte obsahem vaku, aby se nevytvořila pěna. Zbylý rekonstituovaný roztok přípravku BLINCYTO vyhoďte.
6. Za aseptických podmínek připevněte intravenózní set k infuznímu vaku se sterilním 0,2p in-line filtrem.
7. Z infuzního vaku odstraňte vzduch a naplňte intravenózní linku pouze připraveným infuzním roztokem. Linku nenaplňujte injekčním roztokem chloridu sodného 9 mg/ml (0,9%).
8. Jestliže takto připravený roztok nepoužijete ihned, uchovávejte ho při teplotě 2 °C - 8 °C.
e) Příprava přípravku BLINCYTO 28 ug/den podaného infuzí během 24 hodin rychlostí 10 ml/hod.
1. Použijte infuzní vak předplněný 250 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9%), který obvykle obsahuje celkový objem 265 až 275 ml.
2. K potažení infuzního vaku asepticky přeneste pomocí stříkačky 5,5 ml roztoku (stabilizátoru) do infuzního vaku. Jemně zamíchejte obsahem vaku, aby se nevytvořila pěna. Injekční lahvičku se zbylým roztokem (stabilizátorem) vyhoďte.
3. Pomocí stříkačky rekonstituujte jednu injekční lahvičku přípravku BLINCYTO prášek pro koncentrát použitím 3 ml vody na injekci. Během rekonstituce nasměrujte vodu na injekci na boční stěnu injekční lahvičky. Injekční lahvičkou jemně otáčejte, aby se nevytvořilo nadměrné množství pěny. Injekční lahvičkou netřepejte.
• Nepoužívejte roztok (stabilizátor) k rekonstituci přípravku BLINCYTO prášek pro koncentrát.
• Přidáním vody na injekci k prášku pro koncentrát dosáhnete celkový objem 3,08 ml přípravku BLINCYTO o konečné koncentraci 12,5 pg/ml.
4. Během rekonstituce a před podáním infuze prohlédněte rekonstituovaný roztok, zda se
v něm nevyskytují částice a nedošlo ke změně zbarvení. Výsledný roztok musí být čirý až lehce opalescentní a bezbarvý až nažloutlý. Roztok nepoužívejte, pokud je zkalený nebo obsahuje usazeniny.
5. Pomocí stříkačky asepticky přeneste 2,6 ml rekonstituovaného přípravku BLINCYTO do infuzního vaku. Jemně zamíchejte obsahem vaku, aby se nevytvořila pěna. Zbylý rekonstituovaný roztok přípravku BLINCYTO vyhoďte.
6. Za aseptických podmínek připevněte intravenózní set k infuznímu vaku se sterilním 0,2p in-line filtrem.
7. Z infuzního vaku odstraňte vzduch a naplňte intravenózní linku pouze připraveným infuzním roztokem. Linku nenaplňujte injekčním roztokem chloridu sodného 9 mg/ml (0,9%).
8. Jestliže takto připravený roztok nepoužijete ihned, uchovávejte ho při teplotě 2 °C - 8 °C.
f) Příprava přípravku BLINCYTO 28 jug/den podaného infuzí během 48 hodin rychlostí 5 ml/hod.
1. Použijte infuzní vak předplněný 250 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9%), který obvykle obsahuje celkový objem 265 až 275 ml.
2. K potažení infuzního vaku asepticky přeneste pomocí stříkačky 5,5 ml roztoku (stabilizátoru) do infuzního vaku. Jemně zamíchejte obsahem vaku, aby se nevytvořila pěna. Injekční lahvičku se zbylým roztokem (stabilizátorem) vyhoďte.
3. Použijte dvě injekční lahvičky přípravku BLINCYTO prášek pro koncentrát. Pomocí stříkačky rekonstituujte každou injekční lahvičku přípravku BLINCYTO prášek pro koncentrát použitím 3 ml vody na injekci. Během rekonstituce nasměrujte vodu na injekci na boční stěnu injekční lahvičky. Injekční lahvičkou jemně otáčejte, aby se nevytvořilo nadměrné množství pěny. Injekční lahvičkou netřepejte.
• Nepoužívejte roztok (stabilizátor) k rekonstituci přípravku BLINCYTO prášek pro koncentrát.
• Přidáním vody na injekci k prášku pro koncentrát dosáhnete celkový objem 3,08 ml přípravku BLINCYTO o konečné koncentraci 12,5 pg/ml.
4. Během rekonstituce a před podáním infuze prohlédněte rekonstituovaný roztok, zda se
v něm nevyskytují částice a nedošlo ke změně zbarvení. Výsledný roztok musí být čirý až lehce opalescentní a bezbarvý až nažloutlý. Roztok nepoužívejte, pokud je zkalený nebo obsahuje usazeniny.
5. Pomocí stříkačky asepticky přeneste 5,2 ml rekonstituovaného přípravku BLINCYTO do infuzního vaku (2,7 ml z jedné injekční lahvičky a zbývajících 2,5 ml z druhé injekční lahvičky). Jemně zamíchejte obsahem vaku, aby se nevytvořila pěna. Zbylý rekonstituovaný roztok přípravku BLINCYTO vyhoďte.
6. Za aseptických podmínek připevněte intravenózní set k infuznímu vaku se sterilním 0,2p in-line filtrem.
7. Z infuzního vaku odstraňte vzduch a naplňte intravenózní linku pouze připraveným infuzním roztokem. Linku nenaplňujte injekčním roztokem chloridu sodného 9 mg/ml (0,9%).
8. Jestliže takto připravený roztok nepoužijete ihned, uchovávejte ho při teplotě 2 °C - 8 °C.
g) Příprava přípravku BLINCYTO 28 pg/den podaného infuzí během 72 hodin rychlostí 3,3 ml/hod.
1. Použijte infuzní vak předplněný 250 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9%), který obvykle obsahuje celkový objem 265 až 275 ml.
2. K potažení infuzního vaku asepticky přeneste pomocí stříkačky 5,5 ml roztoku (stabilizátoru) do infuzního vaku. Jemně zamíchejte obsahem vaku, aby se nevytvořila pěna. Injekční lahvičku se zbylým roztokem (stabilizátorem) vyhoďte.
3. Použijte tři injekční lahvičky přípravku BLINCYTO prášek pro koncentrát. Pomocí stříkačky rekonstituujte každou injekční lahvičku přípravku BLINCYTO prášek pro koncentrát použitím 3 ml vody na injekci. Během rekonstituce nasměrujte vodu na injekci na boční stěnu injekční lahvičky. Injekční lahvičkou jemně otáčejte, aby se nevytvořilo nadměrné množství pěny. Injekční lahvičkou netřepejte.
• Nepoužívejte roztok (stabilizátor) k rekonstituci přípravku BLINCYTO prášek pro koncentrát.
• Přidáním vody na injekci k prášku pro koncentrát dosáhnete celkový objem 3,08 ml přípravku BLINCYTO o konečné koncentraci 12,5 pg/ml.
4. Během rekonstituce a před podáním infuze prohlédněte rekonstituovaný roztok, zda se
v něm nevyskytují částice a nedošlo ke změně zbarvení. Výsledný roztok musí být čirý až lehce opalescentní a bezbarvý až nažloutlý. Roztok nepoužívejte, pokud je zkalený nebo obsahuje usazeniny.
5. Pomocí stříkačky asepticky přeneste 8 ml rekonstituovaného přípravku BLINCYTO do infuzního vaku (2,8 ml z každé z prvních dvou injekčních lahviček a zbývajících 2,4 ml z třetí injekční lahvičky). Jemně zamíchejte obsahem vaku, aby se nevytvořila pěna.
Zbylý rekonstituovaný roztok přípravku BLINCYTO vyhoďte.
6. Za aseptických podmínek připevněte intravenózní set k infuznímu vaku se sterilním 0,2p in-line filtrem.
7. Z infuzního vaku odstraňte vzduch a naplňte intravenózní linku pouze připraveným infuzním roztokem. Linku nenaplňujte injekčním roztokem chloridu sodného 9 mg/ml (0,9%).
8. Jestliže takto připravený roztok nepoužijete ihned, uchovávejte ho při teplotě 2 °C - 8 °C.
h) Příprava přípravku BLINCYTO 28 pg/den podaného infuzí během 96 hodin rychlostí 2,5 ml/hod.
1. Použijte infuzní vak předplněný 250 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9%), který obvykle obsahuje celkový objem 265 až 275 ml.
2. K potažení infuzního vaku asepticky přeneste pomocí stříkačky 5,5 ml roztoku (stabilizátoru) do infuzního vaku. Jemně zamíchejte obsahem vaku, aby se nevytvořila pěna. Injekční lahvičku se zbylým roztokem (stabilizátorem) vyhoďte.
3. Použijte čtyři injekční lahvičky přípravku BLINCYTO prášek pro koncentrát. Pomocí stříkačky rekonstituujte každou injekční lahvičku přípravku BLINCYTO prášek pro koncentrát použitím 3 ml vody na injekci. Během rekonstituce nasměrujte vodu na injekci na boční stěnu injekční lahvičky. Lahvičkou jemně otáčejte, aby se nevytvořilo nadměrné množství pěny. Injekční lahvičkou netřepejte.
• Nepoužívejte roztok (stabilizátor) k rekonstituci přípravku BLINCYTO prášek pro koncentrát.
• Přidáním vody na injekci k prášku pro koncentrát dosáhnete celkový objem 3,08 ml přípravku BLINCYTO o konečné koncentraci 12,5 pg/ml.
4. Během rekonstituce a před podáním infuze prohlédněte rekonstituovaný roztok, zda se
v něm nevyskytují částice a nedošlo ke změně zbarvení. Výsledný roztok musí být čirý až lehce opalescentní a bezbarvý až nažloutlý. Roztok nepoužívejte, pokud je zkalený nebo obsahuje usazeniny.
5. Pomocí stříkačky asepticky přeneste 10,7 ml rekonstituovaného přípravku BLINCYTO do infuzního vaku (2,8 ml z každé z prvních tří injekčních lahviček a zbývajících 2,3 ml ze čtvrté injekční lahvičky). Jemně zamíchejte obsahem vaku, aby se nevytvořila pěna. Zbylý rekonstituovaný roztok přípravku BLINCYTO vyhoďte.
6. Za aseptických podmínek připevněte intravenózní set k infuznímu vaku se sterilním 0,2p in-line filtrem.
7. Z infuzního vaku odstraňte vzduch a naplňte intravenózní linku pouze připraveným infuzním roztokem. Linku nenaplňujte injekčním roztokem chloridu sodného 9 mg/ml (0,9%).
8. Jestliže takto připravený roztok nepoužijete ihned, uchovávejte ho při teplotě 2 °C - 8 °C. Pokyny pro podání jsou uvedeny v Souhrnu údajů o přípravku v bodě 4.2.
Způsob podání
Důležitá poznámka: Neproplachujte infuzní linky vedoucí k pacientovi, protože to může způsobit neúmyslné podání bolusu přípravku BLINCYTO. BLINCYTO se má podávat infuzí přes zvolený lumen linky.
BLINCYTO infuzní roztok se podává jako kontinuální intravenózní infuze konstantní rychlostí pomocí infuzní pumpy po dobu až 96 hodin.
BLINCYTO infuzní roztok se musí podat pomocí intravenózního setu, který obsahuje in-line, sterilní, nepyrogenní 0,2 pm in-line filtr s nízkou vazbou bílkovin.
Infuzní vak musí zdravotnický pracovník vyměnit minimálně každých 96 hodin z důvodů zachování sterility.
Opatření pro uchovávání a doba použitelnosti
Neotevřené injekční lahvičky:
4 roky (2 °C - 8 °C)
Rekonstituovaný roztok
Chemická a fyzikální stabilita po otevření před použitím byla prokázána na dobu 24 hodin při teplotě 2 °C - 8 °C nebo 4 hodin při teplotě do 27 °C.
Z mikrobiologického hlediska, pokud způsob rekonstituce nevyloučí riziko mikrobiologické kontaminace, se má rekonstituovaný roztok okamžitě rozpustit. Jestliže se nerozpustí ihned, doba a podmínky uchovávání přípravku po otevření před použitím jsou v odpovědnosti uživatele
Rozpuštěný roztok (připravený infuzní vak)
Chemická a fyzikální stabilita po otevření před použitím byla prokázána na dobu 10 dní při teplotě 2 °C - 8 °C nebo na dobu 96 hodin při teplotě 27 °C a nižší.
Z mikrobiologického hlediska se připravené infuzní vaky mají použít okamžitě.
Nejsou-li použity okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou v odpovědnosti uživatele a normálně by doba neměla být delší než 24 hodin při teplotě 2 °C - 8 °C, pokud ředění neproběhlo za kontrolovaných a validovaných aseptických podmínek.
50