Binocrit 20000 Iu/0,5Ml
1. NÁZEV PŘÍPRAVKU
Binocrit 1 000 IU/0,5 ml injekční roztok v předplněné injekční stříkačce Binocrit 2 000 IU/1 ml injekční roztok v předplněné injekční stříkačce Binocrit 3 000 IU/0,3 ml injekční roztok v předplněné injekční stříkačce
Binocrit 4 000 IU/0,4 ml injekční roztok v předplněné injekční stříkačce
Binocrit 5 000 IU/0,5 ml injekční roztok v předplněné injekční stříkačce
Binocrit 6 000 IU/0,6 ml injekční roztok v předplněné injekční stříkačce
Binocrit 7 000 IU/0,7 ml injekční roztok v předplněné injekční stříkačce
Binocrit 8 000 IU/0,8 ml injekční roztok v předplněné injekční stříkačce
Binocrit 9 000 IU/0,9 ml injekční roztok v předplněné injekční stříkačce
Binocrit 10 000 IU/1 ml injekční roztok v předplněné injekční stříkačce Binocrit 20 000 IU/0,5 ml injekční roztok v předplněné injekční stříkačce Binocrit 30 000 IU/0,75 ml injekční roztok v předplněné injekční stříkačce Binocrit 40 000 IU/1 ml injekční roztok v předplněné injekční stříkačce
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Binocrit 1 000 IU/0,5 ml injekční roztok v předplněné injekční stříkačce Jeden ml roztoku obsahuje 2 000 IU epoetinum alfa*, což odpovídá 16,8 mikrogramům/ml Předplněná injekční stříkačka s obsahem 0,5 ml obsahuje 1 000 mezinárodních jednotek (IU), což odpovídá 8,4 mikrogramům epoetinum alfa. *
Binocrit 2 000 IU/1 ml injekční roztok v předplněné injekční stříkačce Jeden ml roztoku obsahuje 2 000 IU epoetinum alfa*, což odpovídá 16,8 mikrogramům/ml Předplněná injekční stříkačka s obsahem 1 ml obsahuje 2 000 mezinárodních jednotek (IU), což odpovídá 16,8 mikrogramům epoetinum alfa. *
Binocrit 3 000 IU/0,3 ml injekční roztok v předplněné injekční stříkačce Jeden ml roztoku obsahuje 10 000 IU epoetinum alfa*, což odpovídá 84,0 mikrogramům/ml Předplněná injekční stříkačka s obsahem 0,3 ml obsahuje 3 000 mezinárodních jednotek (IU), což odpovídá 25,2 mikrogramům epoetinum alfa. *
Binocrit 4 000 IU/0,4 ml injekční roztok v předplněné injekční stříkačce Jeden ml roztoku obsahuje 10 000 IU epoetinum alfa*, což odpovídá 84,0 mikrogramům/ml Předplněná injekční stříkačka s obsahem 0,4 ml obsahuje 4 000 mezinárodních jednotek (IU), což odpovídá 33,6 mikrogramům epoetinum alfa. *
Binocrit 5 000 IU/0,5 ml injekční roztok v předplněné injekční stříkačce Jeden ml roztoku obsahuje 10 000 IU epoetinum alfa*, což odpovídá 84,0 mikrogramům/ml Předplněná injekční stříkačka s obsahem 0,5 ml obsahuje 5 000 mezinárodních jednotek (IU), což odpovídá 42,0 mikrogramům epoetinum alfa. *
Binocrit 6 000 IU/0,6 ml injekční roztok v předplněné injekční stříkačce Jeden ml roztoku obsahuje 10 000 IU epoetinum alfa*, což odpovídá 84,0 mikrogramům/ml Předplněná injekční stříkačka s obsahem 0,6 ml obsahuje 6 000 mezinárodních jednotek (IU), což odpovídá 50,4 mikrogramům epoetinum alfa. *
Binocrit 7 000 IU/0,7 ml injekční roztok v předplněné injekční stříkačce Jeden ml roztoku obsahuje 10 000 IU epoetinum alfa*, což odpovídá 84,0 mikrogramům/ml Předplněná injekční stříkačka s obsahem 0,7 ml obsahuje 7 000 mezinárodních jednotek (IU), což odpovídá 58,8 mikrogramům epoetinum alfa. *
Binocrit 8 000 IU/0,8 ml injekční roztok v předplněné injekční stříkačce Jeden ml roztoku obsahuje 10 000 IU epoetinum alfa*, což odpovídá 84,0 mikrogramům/ml Předplněná injekční stříkačka s obsahem 0,8 ml obsahuje 8 000 mezinárodních jednotek (IU), což odpovídá 67,2 mikrogramům epoetinum alfa. *
Binocrit 9 000 IU/0,9 ml injekční roztok v předplněné injekční stříkačce Jeden ml roztoku obsahuje 10 000 IU epoetinum alfa*, což odpovídá 84,0 mikrogramům/ml Předplněná injekční stříkačka s obsahem 0,9 ml obsahuje 9 000 mezinárodních jednotek (IU), což odpovídá 75,6 mikrogramům epoetinum alfa. *
Binocrit 10 000 IU/1 ml injekční roztok v předplněné injekční stříkačce Jeden ml roztoku obsahuje 10 000 IU epoetinum alfa*, což odpovídá 84,0 mikrogramům/ml Předplněná injekční stříkačka s obsahem1 ml obsahuje 10 000 mezinárodních jednotek (IU), což odpovídá 84,0 mikrogramům epoetinum alfa. *
Binocrit 20 000 IU/0,5 ml injekční roztok v předplněné injekční stříkačce Jeden ml roztoku obsahuje 40 000 IU epoetinum alfa*, což odpovídá 336,0 mikrogramům/ml Předplněná injekční stříkačka s obsahem 0,5 ml obsahuje 20 000 mezinárodních jednotek (IU), což odpovídá 168,0 mikrogramům epoetinum alfa. *
Binocrit 30 000 IU/0,75 ml injekční roztok v předplněné injekční stříkačce Jeden ml roztoku obsahuje 40 000 IU epoetinum alfa*, což odpovídá 336,0 mikrogramům/ml Předplněná injekční stříkačka s obsahem 0,75 ml obsahuje 30 000 mezinárodních jednotek (IU), což odpovídá 252,0 mikrogramům epoetinum alfa. *
Binocrit 40 000 IU/1 ml injekční roztok v předplněné injekční stříkačce Jeden ml roztoku obsahuje 40 000 IU epoetinum alfa*, což odpovídá 336,0 mikrogramům/ml Předplněná injekční stříkačka s obsahem 1 ml obsahuje 40 000 mezinárodních jednotek (IU), což odpovídá 336,0 mikrogramům epoetinum alfa. *
* Připraveno v buněčné linii ovariálních buněk čínského krečka (CHO) technologií rekombinantní DNA
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok v předplněné injekční stříkačce (injekce) Čirý bezbarvý roztok
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Binocrit je indikován k léčbě symptomatické anémie spojené s chronickým renálním selháním (CRF):
- u dospělých a pediatrických hemodialyzovaných pacientů ve věku od 1 do 18 let a dospělých pacientů dialyzovaných peritoneálně (viz bod 4.4).
- u dospělých pacientů s renální insuficiencí, kteří dosud dialyzováni nejsou, k léčbě těžké anémie ledvinového původu doprovázené klinickými příznaky (viz bod 4.4).
Binocrit je indikován k léčbě anémie a snížení počtu transfuzí u dospělých absolvujících chemoterapii pro onemocnění solidními tumory, maligním lymfomem nebo mnohočetným myelomem, pokud je u nich transfuze vzhledem k celkovému zdravotnímu stavu riziková (například při onemocnění srdce nebo při anémii již před počátkem chemoterapie).
Binocrit je indikován ke zvýšení přínosu u dospělých pacientů, kteří si připravují vlastní (autologní) dávky krve v programu předoperačního autologního odběru. Léčbu je třeba omezit na pacienty se středně závažnou anémií (rozsah koncentrací hemoglobinu [Hb] 10 až 13 g/dl [6,2 až 8,1 mmol/l], bez deficitu železa), nejsou-li k dispozici procedury šetřící krev nebo jsou-li tyto procedury nedostatečné, pokud velká naplánovaná ortopedická operace vyžaduje velké množství krve (4 nebo více jednotek krve u žen nebo 5 nebo více jednotek krve u mužů).
Binocrit je indikován k omezení expozice alogenními krevními transfuzemi u dospělých, kteří netrpí deficitem železa a jsou již zařazeni do programu velké naplánované ortopedické operace, pokud při transfuzi hrozí vysoké riziko komplikací. Použití je třeba omezit na pacienty se středně závažnou anémií (např. rozsah koncentrací hemoglobinu Hb 10 až 13 g/dl nebo 6,2 až 8,1 mmol/l), kteří neměli možnost připravit si vlastní (autologní) krevní dávky předem a u nichž se očekává středně závažná ztráta krve (900 až 1800 ml).
4.2 Dávkování a způsob podání
Léčba Binocritem se musí zahájit pod dohledem lékařů, kteří mají zkušenosti s léčbou pacientů se shora uvedenými indikacemi.
Dávkování
Před nasazením epoetinu alfa a před rozhodnutím o zvýšení dávky je třeba zvážit a případně léčit všechny ostatní příčiny anémie (deficit železa, kyseliny listové nebo vitaminu B12; intoxikace hliníkem; infekce nebo záněty, ztráta krve; hemolýza, fibróza kostní dřeně různého původu).
K zajištění optimální odezvy na epoetin alfa by měly být zajištěny přiměřené zásoby železa a v případě potřeby prováděna suplementace železem (viz bod 4.4).
Léčba symptomatické anémie u dospělých _pacientů s chronickým renálním selháním
Symptomy a následky anémie se liší podle věku, pohlaví a současně probíhajících onemocnění; léčbu a stav jednotlivých pacientů musí vyhodnotit lékař.
Doporučený požadovaný rozsah koncentrací hemoglobinu je 10 g/dl až 12 g/dl (6,2 až 7,5 mmol/l). Binocrit má být podáván tak, aby se hemoglobin zvýšil nejvýše na hladinu 12 g/dl (7,5 mmol/l). Je třeba se vyhýbat tomu, aby došlo ke většímu zvýšení hemoglobinu než o 2 g/dl (1,25 mmol/l) za období čtyř týdnů. Jestliže k němu dojde, je třeba vhodným způsobem upravit dávku.
Vzhledem k intraindividuální variabilitě je možné pozorovat, že občas jsou individuální hodnoty hemoglobinu u pacienta vyšší nebo nižší než požadovaný rozsah koncentrací. Variabilitu hladiny hemoglobinu je vhodné řešit úpravami dávkování, s ohledem na cílový rozsah koncentrací hemoglobinu 10 g/dl (6,2 mmol/l) až 12 g/dl (7,5 mmol/l).
Je třeba se vyhýbat trvalému zvýšení hladiny hemoglobinu na hodnoty vyšší než 12 g/dl (7,5 mmol/l). Jestliže hemoglobin stoupá o více než 2 g/dl (1,25 mmol/l) za měsíc nebo jestliže trvalá hladina hemoglobinu přesáhne 12 g/dl (7,5 mmol/l), snižte dávku přípravku Binocrit o 25 %. Jestliže hladina hemoglobinu přesáhne 13 g/dl (8,1 mmol/l), přerušte terapii, dokud neklesne pod 12 g/dl (7,5 mmol/l) a poté terapii přípravkem Binocrit obnovte při dávce o 25 % nižší než předchozí dávka.
Pacienty je třeba pečlivě sledovat, aby byla zajištěna adekvátní kontrola symptomů anémie a současně udržení koncentrace hemoglobinu nižší nebo rovné 12 g/dl (7,45 mmol/l) při použití nejnižší vyzkoušené účinné dávky Binocrit.
Při zvyšování dávek Binocrit u pacientů s chronickým renálním selháním je nutná opatrnost.
U pacientů se slabou odpovědí hemoglobinu na Binocrit mají být zvážena alternativní vysvětlení pro tuto slabou odpověd (viz bod 4.4 a 5.1).
Léčba přípravkem Binocrit je rozdělena na dvě fáze - korekční a udržovací.
Dospělí hemodialyzovaní pacienti
U hemodialyzovaných pacientů, u nichž je k dispozici intravenózní přístup, se upřednostňuje intravenózní podání.
Korekční fáze
Počáteční dávka je 50 IU/kg 3x týdně.
Je-li třeba, zvyšujte nebo snižujte dávku o 25 IU/kg (3krát týdně), dokud nebude dosaženo požadovaného rozsahu koncentrací hemoglobinu 10 g/dl až 12 g/dl (6,2 až 7,5 mmol/l) (to je třeba provádět po stupních trvajících vždy alespoň čtyři týdny).
Udržovací fáze
Doporučená celková týdenní dávka je mezi 75 IU/kg a 300 IU/kg.
Je nutná vhodná úprava dávkování k udržení hodnot hemoglobinu v požadovaném rozsahu koncentrací mezi 10 g/dl až 12 g/dl (6,2 až 7,5 mmol/l).
Pacienti s velmi nízkými úvodními hodnotami hemoglobinu (< 6 g/dl čili < 3,75 mmol/l) mohou vyžadovat vyšší udržovací dávky než pacienti, jejichž počáteční anémie je méně těžká ( > 8 g/dl čili > 5 mmol/l).
Dospělí pacienti s renální insuficiencí, kteří dosud dialyzovaní nebyli
Pokud není intravenózní přístup k dispozici, lze přípravek Binocrit podávat subkutánně.
Korekční fáze
Počáteční dávka je 50 IU/kg 3x týdně. Poté je-li třeba, zvyšujte dávku v přírůstcích po 25 IU/kg (3krát týdně), dokud nebude dosaženo požadovaného cíle (to je třeba provádět po stupních trvajících vždy alespoň čtyři týdny).
Udržovací fáze
Během udržovací fáze lze přípravek Binocrit podávat buď 3krát týdně, a v případě subkutánního podání, jednou týdně, nebo jednou za 2 týdny.
Je nutná vhodná úprava dávky a intervalů dávkování k udržení hodnot hemoglobinu na požadované úrovni: Hb mezi 10 g/dl a 12 g/dl (6,2 až 7,5 mmol/l). Prodloužení intervalů mezi dávkami může vyžadovat zvýšení dávky.
Maximální dávka nemá přesáhnout 150 IU/kg 3x týdně, 240 IU/kg (do maxima 20 000 IU) jednou týdně nebo 480 IU/kg (do maxima 40 000 IU) jednou za 2 týdny.
Dospělí pacienti dialyzovaní peritoneálně
Pokud není intravenózní přístup k dispozici, lze přípravek Binocrit podávat subkutánně.
Korekční fáze:
Počáteční dávka je 50 IU/kg 2x týdně.
Udržovací fáze:
Doporučená udržovací dávka je mezi 25 IU/kg a 50 IU/kg 2krát týdně rozdělená do 2 stejně velkých injekčních dávek.
Je nutná vhodná úprava dávkování k udržení hodnot hemoglobinu na požadovaném rozsahu koncentrací mezi 10 g/dl a 12 g/dl (6,2 až 7,5 mmol/l).
Léčba dospělých _pacientů s anémií vyvolanou chemoterapií
Symptomy a následky anémie se liší podle věku, pohlaví a celkových obtíží způsobených onemocněním; léčbu a stav jednotlivých pacientů musí vyhodnotit lékař.
Binocrit se má podávat pacientům s anémií (např. koncentrace hemoglobinu < 10 g/dl (6,2 mmol/l)). Počáteční dávka je 150 IU/kg subkutánně 3krát týdně.
Alternativně lze Binocrit podávat v počáteční dávce 450 IU/kg subkutánně, a to jednou týdně.
Je nutná vhodná úprava dávky a intervalů dávkování k udržení hodnot hemoglobinu v požadovaném rozsahu koncentrací mezi 10 g/dl až 12 g/dl (6,2 až 7,5 mmol/l).
Vzhledem k intraindividuální variabilitě je možné pozorovat, že občas jsou individuální koncentrace hemoglobinu u pacienta vyšší nebo nižší než požadovaný rozsah koncentrací. Na variabilitu hladiny hemoglobinu je třeba reagovat úpravou dávek, přičemž požadovaný rozsah koncentrací hemoglobinu by měl být mezi 10 g/dl (6,2 mmol/l) a 12 g/dl (7,5 mmol/l). Je třeba se vyhnout tomu, aby koncentrace hemoglobinu byla trvale vyšší než 12 g/dl (7,5 mmol/l); pokyny k příslušné úpravě dávek pro případ, kdy koncentrace hemoglobinu převyšují 12 g/dl (7,5 mmol/l), jsou uvedeny níže.
- Pokud se po čtyřtýdenní léčbě koncentrace hemoglobinu zvýšila alespoň o 1 g/dl (0,62 mmol/l) anebo pokud počet retikulocytů stoupl o > 40 000 buněk/^l nad počáteční hodnoty, je třeba dávku udržovat na 150 IU/kg 3x týdně nebo 450 IU/kg jednou týdně.
- Pokud se koncentrace hemoglobinu zvýšila o < 1 g/dl (< 0,62 mmol/l) anebo počet retikulocytů stoupl o < 40 000 buněk/^l nad počáteční hodnoty, dávka se zvýší na 300 IU/kg 3x týdně. Pokud po dalších 4 týdnech léčby dávkami 300 IU/kg 3x týdně koncentrace hemoglobinu stoupne o > 1 g/dl (> 0,62 mmol/l) anebo počet retikulocytů stoupne o > 40 000 buněk/^l, dávkování se udržuje na 300 IU/kg 3x týdně.
- Pokud se koncentrace hemoglobinu zvýšila o <1 g/dl (< 0,62 mmol/l) anebo počet retikulocytů stoupl o < 40 000 buněk/^l nad počáteční hodnoty, odpověď na terapii je nepravděpodobná, a proto je třeba léčbu přerušit.
Úprava dávkování k udržení koncentrací hemoglobinu mezi 10 g/dl až 12 g/dl (6,2 až 7,5 mmol/l) Jestliže se koncentrace hemoglobinu zvýšila o více než 2 g/dl (1,25 mmol/l) za měsíc nebo jestliže úroveň koncentrace hemoglobinu přesáhne 12 g/dl (7,5 mmol/l), snižte dávku přípravku Binocrit přibližně o 25 až 50 %.
Jestliže úroveň koncentrace hemoglobinu přesáhne 13 g/dl (8,1 mmol/l), přerušte terapii, dokud koncentrace neklesne pod 12 g/dl (7,5 mmol/l) a poté terapii přípravkem Binocrit obnovte při dávce o 25 % nižší než byla předchozí dávka.
Doporučený dávkovací režim je popsán v následujícím diagramu:
150 IU/kg 3x/týden nebo 450 IU/kg jednou týdně
po 4 týdny
Zvýšení počtu retikulocytů o > 40 000/^l nebo zvýšení koncentrace Hb o > 1 g/dl
Zvýšení počtu retikulocytů o < 40 000/^1
a zvýšení koncentrace Hb o < 1 g/dl
1
300 IU/kg 3x/týden po 4 týdny
1
Cílová hladina Hb (<12 g/dl)
í
Zvýšení počtu retikulocytů o > 40 000/^l nebo zvýšení koncentrace Hb o > 1 g/dl
Zvýšení počtu retikulocytů o < 40 000/^l
a zvýšení koncentrace Hb o < 1 g/dl
1
Přerušte terapii
Pacienty je třeba pečlivě sledovat, aby se zajistilo, že je podávána nejnižší účinná dávka přípravku stimulujícího tvorbu erytrocytů (ESA) pro adekvátní kontrolu příznaků anémie.
Léčba epoetinem alfa musí pokračovat ještě měsíc po ukončení chemoterapie.
Léčba dospělých chirurgických pacientů v programu předoperačního autologního odběru Středně anemičtí pacienti (hematokrit 33 až 39 %) vyžadující předzásobení krví ve výši > 4 jednotky by měli být léčeni přípravkem Binocrit v dávce 600 IU/kg tělesné hmotnosti intravenózně 2x týdně po dobu 3 týdnů před operací. V případě dárcovství krve se přípravek Binocrit musí podat po ukončení procedury darování krve.
Léčba dospělých chirurgických _pacientů _před_plánovanou velkou ortopedickou operací
Doporučená dávka je 600 IU přípravku Binocrit na kg podávaných subkutánně týdně po tři týdny (21., 14. a 7. den) před výkonem, a v den výkonu (0. den).
V případech, kde je z lékařského hlediska nutno zkrátit čas do operace na méně než tři týdny, podává se 300 IU/kg přípravku Binocrit subkutánně denně po 10 po sobě jdoucích dní před výkonem, v den operace a po čtyři dny po ní.
Pokud při hematologickém hodnocení v předoperačním období hladina hemoglobinu dosáhne 15 g/dl (9,38 mmol/l) nebo více, podávání přípravku Binocrit je třeba zastavit a další dávku již nepodávat.
Pediatrická _ populace
Léčba symptomatické anémie u pacientů s chronickým renálním selháním na hemodialýze Symptomy a následky anémie se liší podle věku, pohlaví a současně probíhajících onemocnění; léčbu a stav jednotlivých pacientů musí vyhodnotit lékař.
U pediatrických pacientů je doporučený rozsah koncentrací hemoglobinu 9,5 g/dl až 11 g/dl (5,9 až 6,8 mmol/l). Binocrit má být podáván tak, aby se hemoglobin zvýšil nejvýše na hladinu 11 g/dl (6,8 mmol/l). Je třeba se vyhýbat tomu, aby došlo ke většímu zvýšení hemoglobinu než o 2 g/dl (1,25 mmol/l) za období čtyř týdnů. Jestliže k němu dojde, je třeba vhodným způsobem upravit dávku.
Pacienty je třeba pečlivě sledovat, aby byla zajištěna adekvátní kontrola symptomů anémie při použití nejnižší schválené dávky přípravku Binocrit.
Léčba přípravkem Binocrit je rozdělena na dvě fáze - korekční a udržovací.
U hemodialyzovaných pediatrických pacientů, u nichž je k dispozici intravenózní přístup, se upřednostňuje intravenózní podání.
Korekční fáze
Počáteční dávka je 50 IU/kg 3krát týdně intravenózně.
Je-li třeba, zvyšujte nebo snižujte dávku o 25 IU/kg (3krát týdně), dokud nebude dosaženo požadovaného rozsahu koncentrací hemoglobinu 9,5 g/dl až 11 g/dl (5,9 až 6,8 mmol/l) (to je třeba provádět po stupních trvajících vždy alespoň čtyři týdny).
Udržovací fáze
Je nutná vhodná úprava dávkování k udržení hladin hemoglobinu v požadovaném rozsahu koncentrací mezi 9,5 g/dl až 11 g/dl (5,9 až 6,8 mmol/l).
Děti s hmotností pod 30 kg většinou vyžadují vyšší udržovací dávky než děti s hmotností nad 30 kg a než dospělí.
Pediatričtí pacienti s velmi nízkými výchozími hodnotami hemoglobinu (< 6,8 g/dl nebo < 4,25 mmol/l) mohou vyžadovat vyšší udržovací dávky než pacienti, jejichž počáteční hodnota hemoglobinu je vyšší (> 6,8 g/dl nebo > 4,25 mmol/l).
Léčba pediatrických pacientů s anémií v důsledku chemoterapie
Bezpečnost a účinnost epoetinu alfa u pediatrických pacientů léčených chemoterapií nebyla stanovena.
Léčba pediatrických chirurgických pacientů v programu předoperačního autologního odběru Bezpečnost a účinnost epoetinu alfa u pediatrických pacientů nebyla stanovena. Nejsou dostupné žádné údaje.
Léčba pediatrických pacientů před plánovanou velkou ortopedickou operací
Bezpečnost a účinnost epoetinu alfa u pediatrických pacientů nebyla stanovena. Nejsou dostupné
žádné údaje.
Způsob podání
Opatření, která je nutno učinit před zacházením s léčivým přípravkem nebo před jeho podáním.
Před použitím nechte stříkačky přípravku Binocrit stát do dosažení pokojové teploty. To obvykle trvá 15 až 30 minut.
Jako u všech přípravků podávaných injekčně je třeba před aplikací vždy zkontrolovat kvalitu injekčního roztoku, zda neobsahuje částice nebo zda nedošlo ke změně barvy. Binocrit je sterilní výrobek neobsahující konzervační látky, určený pouze k jednorázovému použití. Podávejte požadované množství.
Léčba symptomatické anémie u dospělých pacientů s chronickým renálním selháním
U pacientů s chronickým renálním selháním, u nichž je k dispozici intravenózní přístup (hemodialyzovaní pacienti), se upřednostňuje intravenózní podání přípavku Binocrit.
Pokud není intravenózní přístup k dispozici (pacienti, kteří dosud nepodstoupili hemodialýzu, nebo pacienti dialyzovaní peritoneálně), lze přípravek Binocrit podávat subkutánní injekcí.
Léčba dospělých _pacientů s anémií vyvolanou chemoterapií Binocrit se musí podávat subkutánní injekcí.
Léčba dospělých chirurgických _pacientů v _ programu _předoperačního autologního odběru Binocrit se musí podávat intravenózně.
Léčba dospělých chirurgických pacientů před plánovanou velkou ortopedickou operací Binocrit se musí podávat subkutánní injekcí.
Léčba symptomatické anémie u pediatrických hemodialyzovaných pacientů s chronickým renálním selháním
Binocrit se musí podávat intravenózně.
Intravenózní podání
Aplikuje se po dobu nejméně jedné až pěti minut, v závislosti na celkové dávce. U hemodialyzovaných pacientů je možné podat během dialýzy bolus do vhodného žilního vstupu dialyzační linky. Alternativně lze injekci podat při ukončení dialýzy, hadičkou zavedené dialyzační jehly, a pak hadičku propláchnout 10 ml fyziologického roztoku pro zajištění uspokojivého vpravení přípravku do oběhu (viz Dávkování, „Dospělí hemodialyzovaní pacienti“).
Pomalejší podání je vhodnější u pacientů, kteří na léčbu reagují „chřipkovitými“ příznaky (viz bod 4.8).
Nepodávejte přípravek Binocrit intravenózní infuzí nebo spolu s roztoky jiných léčivých přípravků (další informace viz bod 6.6).
Subkutánní _ podání
Obecně se nemá překročit objem 1 ml na jedno místo vpichu. Větší objem je třeba rozdělit na aplikace do několika míst.
Injekce se mají podávat do končetin nebo do přední břišní stěny.
V případech, kdy lékař rozhodne, že je možné, aby pacient nebo jeho pečovatel bezpečně a účinně aplikoval subkutánní injekci přípravku Binocrit samostatně, je nutné pacientovi nebo pečovateli poskytnout přesné instrukce týkající se správné dávky a způsobu podání přípravku.
„Pokyny, jak samostatně podávat injekci přípravku“ jsou k dispozici na konci příbalové informace.
4.3 Kontraindikace
- Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
- Pacientům, u kterých se po léčbě jakýmkoli erytropoetinem vyvine čistá aplazie červené krevní řady (pure red cell aplasia, PRCA), se Binocrit ani jiný erytropoetin nesmí podávat (viz
bod 4.4).
- Nekontrolovaná hypertenze.
- U pacientů suplementovaných přípravkem Binocrit musí být respektovány všechny kontraindikace spojené s programem předoperačního autologního odběru.
U pacientů před velkou plánovanou ortopedickou operací, kteří neměli možnost připravit si vlastní (autologní) dávky krve, je použití přípravku Binocrit kontraindikováno, pokud pacient trpí závažným onemocněním věnčitých a periferních tepen, karotid či mozkových cév, včetně nedávno prodělaného infarktu myokardu anebo mozkové cévní příhody.
- Chirurgičtí pacienti, u kterých z jakéhokoli důvodu nelze zajistit přiměřenou antitrombotickou profylaxi.
4.4 Zvláštní upozornění a opatření pro použití
Celkové poruchy
U všech pacientů užívajících epoetin alfa je třeba pečlivě monitorovat krevní tlak, a podle potřeby jej regulovat. Epoetin alfa je nutno používat se zvýšenou opatrností u neléčené, nedostatečně léčené či špatně kontrolovatelné hypertenze. Může být nutné zavést či zintenzívnit léčbu antihypertenzivy. Pokud krevní tlak nelze udržet pod kontrolou, je třeba léčbu epoetinem alfa přerušit.
U pacientů s normálním nebo nízkým krevním tlakem léčených epoetinem alfa se objevily také tyto reakce: hypertenzní krize s encefalopatií a záchvaty, vyžadující okamžitý zásah lékaře a intenzivní lékařskou péči. Zvláštní pozornost je třeba věnovat náhlým bodavým migrenózním bolestem hlavy jako možnému varovnému signálu (viz bod 4.8).
Epoetin alfa má být podáván s opatrností pacientům s epilepsií, záchvaty v anamnéze nebo zdravotními stavy spojenými s predispozicí k záchvatové aktivitě, jako jsou infekce CNS a metastázy v mozku.
Epoetin alfa má být podáván s opatrností pacientům s chronickým selháním jater. Bezpečnost epoetinu alfa u pacientů s poruchou funkce jater nebyla stanovena.
U pacientů užívajících přípravky ESA byl hlášen zvýšený výskyt trombotických cévních příhod (TVE) (viz bod 4.8). Ty zahrnují žilní a arteriální trombózu a embolii (včetně několika fatálních případů), například hlubokou žilní trombózu, plicní embolii, retinální trombózu a infarkt myokardu. Dále byl hlášen výskyt cévních mozkových příhod (včetně mozkového infarktu, mozkového krvácení a tranzitorních ischemických atak).
Uvedené riziko těchto TVE je třeba pečlivě zvažovat oproti předpokládaným přínosům léčby epoetinem alfa, zejména u pacientů s existujícími rizikovými faktory TVE, včetně obezity a TVE v anamnéze (například s hlubokou žilní trombózou, plicní embolií a cévní mozkovou příhodou).
U všech pacientů je třeba pečlivě monitorovat hladiny hemoglobinu vzhledem k potencionálně zvýšenému riziku tromboembolických příhod a fatálních případů, když jsou pacienti léčeni s hladinou hemoglobinu nad rozsahem koncentrací stanoveným pro indikaci použití.
Během léčby epoetinem alfa se může vyvinout mírné a podané dávce úměrné zmnožení trombocytů, které však zůstává v rozsahu normálních hodnot. V dalším průběhu terapie toto zmnožení opět odezní. Kromě toho byly hlášeny i případy trombocytemie nad normálními hodnotami. Doporučuje se počty trombocytů pravidelně monitorovat během prvních 8 týdnů terapie.
Před nasazením epoetinu alfa a před rozhodnutím o zvýšení dávky je třeba uvážit a případně léčit všechny ostatní příčiny anémie (deficit železa, kyseliny listové nebo vitaminu B12; intoxikace hliníkem; infekce nebo záněty, ztráta krve; hemolýza, fibróza kostní dřeně různého původu). Ve většině případů hodnoty feritinu v séru klesají současně se zvýšením hodnot hematokritu. K zajištění optimální odezvy na epoetin alfa by měly být zajištěny přiměřené zásoby železa a v případě potřeby prováděna suplementace železem (viz bod 4.2):
- U pacientů s chronickým renálním selháním se suplementace železem (200 až 300 mg elementárního železa/den perorálně u dospělých a 100 až 200 mg/den perorálně u pediatrických pacientů) doporučuje při sérových hladinách feritinu pod 100 ng/ml.
- U pacientů s nádorovým onemocněním se suplementace železem (200 až 300 mg elementárního železa/den perorálně) doporučuje při saturací transferinem pod 20 %.
- U pacientů v programu předoperačního autologního odběru se provádí suplementace železem (200 mg elementárního železa/den perorálně) několik týdnů před zahájením programu předoperačního autologního odběru, aby se vytvořila dostatečně vysoká zásoba železa ještě před terapií epoetinem alfa, a po celou dobu léčby epoetinem alfa.
- U pacientů před plánovanou velkou ortopedickou operací se provádí suplementace železem (200 mg elementárního železa/den perorálně) po celou dobu léčby epoetinem alfa. Pokud je to možné, suplementace železem má být zahájena ještě před začátkem terapie epoetinem alfa, aby se vytvořila dostatečná zásoba železa.
U pacientů léčených epoetinem alfa byl velmi vzácně pozorován vývoj nebo exacerbace porfyrie. Epoetin alfa by měl být podáván s opatrností pacientům s porfyrií.
Za účelem zdokonalení sledovanosti přípravků stimulujících tvorbu erytrocytů (ESA), by se měly názvy podaných přípravků ESA zřetelně zaznamenávat (nebo uvádět) do dokumentace pacienta. Pacienti mají být převedeni z léčby jedním typem ESA na jiný pouze pod náležitým dohledem.
Čistá aplazie červené krevní řady (PRCA)
Protilátkami zprostředkovaná PRCA byla pozorována po několika měsících až letech subkutánního podávání epoetinu zejména u pacientů s chronickým selháním ledvin. Případy byly také hlášeny u pacientů s hepatitidou C léčených interferonem a ribavirinem, když byly současně užívány ESA. Epoetin alfa není schválen k léčbě anémie související s hepatitidou C.
U pacientů, u nichž se vyvine náhlá ztráta účinnosti charakterizovaná poklesem hemoglobinu (1 až 2 g/dl nebo 0,62 až 1,25 mmol/l na měsíc) a zvýšenou potřebou transfuzí, je třeba zjistit počet retikulocytů a přešetřit typické příčiny špatné odpovědi na léčbu (např. deficit železa, kyseliny listové či vitaminu B12, intoxikaci hliníkem, infekci či zánět, ztrátu krve, hemolýzu a fibrózu kostní dřeně různého původu).
Paradoxní pokles hemoglobinu a rozvoj závažné anémie související s nízkým počtem retikulocytů musí být podnětem k ukončení léčby epoetinem alfa a k provedení testu na anti-erytropoetinové protilátky. Při diagnóze PRCA by mělo být zváženo vyšetření kostní dřeně.
Pacienti by neměli být převáděni na jinou léčbu ESA vzhledem ke zkřížené reakci.
Léčba symptomatické anémie u dospělých a pediatrických pacientů s chronickým renálním selháním
U pacientů s chronickým renálním selháním léčených epoetinem alfa je třeba hladiny hemoglobinu pravidelně měřit, dokud není dosaženo stabilních hodnot, a v pravidelných intervalech pokračovat i potom.
U pacientů s chronickým renálním selháním se má hodnota hemoglobinu zvyšovat přibližně o 1 g/dl (0,62 mmol/l) měsíčně; přírůstek by však neměl přesáhnout 2 g/dl (1,25 mmol/l) měsíčně, aby se minimalizovalo riziko zvýšení hypertenze.
U pacientů s chronickým renálním selháním by neměla udržovací koncentrace hemoglobinu překročit horní mez rozsahu koncentrací hemoglobinu, která je doporučena v bodě 4.2. V klinických studiích bylo pozorováno zvýšení rizika úmrtí a závažných kardiovaskulárních příhod při podávání ESA za účelem dosažení úrovně koncentrací hemoglobinu vyšší než 12 g/dl (7,5 mmol/l).
Kontrolované klinické zkoušky neprokázaly výrazné přínosy odpovídající podávání epoetinů, pokud byla koncentrace hemoglobinu vyšší než hladina potřebná ke kontrole symptomů anémie a k tomu, aby nebylo potřeba transfuze krve.
Při zvyšování dávek Binocrit u pacientů s chronickým renálním selháním je nutná opatrnost, protože vysoké kumulující se dávky epoetinu mohou být souviset se zvýšeným rizikem mortality, závažnými kardiovaskulárními a cerebrovaskulárními příhodami. U pacientů se slabou odpovědí hemoglobinu na epoetiny mají být zvážena alternativní vysvětlení pro tuto slabou odpověď (viz bod 4.2 a 5.1).
U pacientů s chronickým renálním selháním léčených subkutánním podáváním epoetinu alfa je třeba pravidelně sledovat, zda nedochází ke ztrátě účinnosti, která je definována jako nepřítomná nebo snížená odezva na léčbu epoetinem alfa u pacientů, již na takovou léčbu dříve reagovali. Pro tuto situaci je typický setrvalý pokles hemoglobinu navzdory zvýšenému dávkování epoetinu alfa (viz bod 4.8).
Někteří pacienti s delšími dávkovacími intervaly podávání epoetinu alfa (delšími než jeden týden) si nemusí udržet adekvátní hladiny hemoglobinu (viz bod 5.1) a mohou potřebovat zvýšení dávky epoetinu alfa. Hladiny hemoglobinu musí být pravidelně sledovány.
U hemodialyzovaných pacientů se objevila trombóza arteriovenózní spojky, zejména u pacientů s tendencí k hypotenzi nebo s komplikacemi na arteriovenózních shuntech (například stenózy, výdutě atd.). U těchto pacientů je doporučena časná revize shuntu a prevence trombózy například podáváním kyseliny acetylsalicylové.
V ojedinělých případech byla zjištěna hyperkalémie, ačkoli příčinná souvislost nebyla zjištěna. U pacientů s chronickým renálním selháním je třeba monitorovat hladiny sérových elektrolytů. Pokud bude zjištěna zvýšená či stoupající sérová hladina draslíku, je třeba zvážit vysazení epoetinu alfa až do korekce sérových hodnot draslíku a kromě toho zvolit příslušnou léčbu hyperkalémie.
Zvýšené hodnoty hematokritu v průběhu terapie epoetinem alfa často přinutí zvýšit dávky heparinu během hemodialýzy. Jestliže heparinizace není optimální, dialyzační systém se může ucpat.
Dosud dostupné informace naznačují, že korekce anémie epoetinem alfa neurychluje progresi renální nedostatečnosti u dospělých pacientů s renální nedostatečností, kteří dosud dialyzováni nebyli.
Léčba pacientů s anémií v důsledku chemoterapie
U pacientů s rakovinou léčených epoetinem alfa je třeba hladiny hemoglobinu pravidelně měřit, dokud není dosaženo stabilních hodnot, a v pravidelných intervalech pokračovat i potom.
Epoetiny jsou růstové faktory, které primárně stimulují tvorbu erytrocytů. Erytropoetinové receptory mohou být exprimovány na povrchu různých nádorových buněk. Stejně jako u všech ostatních růstových faktorů i zde existují obavy, že by epoetiny mohly stimulovat růst některého typu tumorů. V několika kontrolovaných studiích nebylo prokázáno, že by epoetiny zlepšily celkovou dobu přežití, nebo že by snižovaly riziko nádorové progrese u pacientů s anémií související s rakovinou.
V kontrolovaných klinických studiích s užíváním epoetinu alfa a dalších přípravků ESA bylo prokázáno:
- že se u pacientů s rakovinou hlavy nebo krku v pokročilém stádiu léčených radiační terapií při podávání těchto přípravků za účelem dosažení úrovně koncentrace hemoglobinu vyšší než
14 g/dl (8,7 mmol/l) snížila lokoregionální kontrola,
- že se u pacientů s metastatickou rakovinou prsu léčených chemoterapií při podávání těchto přípravků za účelem dosažení rozsahu koncentrací hemoglobinu 12 až 14 g/dl (7,5 až
8,7 mmol/l) zkrátila celková doba přežití a zvýšil se počet úmrtí přičítaných progresi onemocnění během 4 měsíců,
- že se u pacientů s aktivním maligním onemocněním, kteří nebyli léčeni ani chemoterapií ani radiační terapií, při podávání těchto přípravků za účelem dosažení úrovně koncentrace hemoglobinu 12 g/dl (7,5 mmol/l) zvýšilo riziko úmrtí. ESA nejsou indikovány pro použití u této populace pacientů.
Vzhledem k výše uvedenému by v některých klinických situacích měla být upřednostňovanou léčbou při zvládání anémie u pacientů s rakovinou krevní transfuze. Rozhodnutí o léčbě rekombinantními erytropoetiny musí být provedeno po vyhodnocení přínosů a rizik se zapojením konkrétního pacienta, a při zvážení konkrétního klinického kontextu. Při takovémto vyhodnocení musí být zvažovány faktory, jako je typ nádoru a jeho stádium; stupeň anémie; pravděpodobnost přežití; prostředí, ve kterém je pacient léčen a pacientovy preference (viz bod 5.1).
U pacientů s nádorovým onemocněním léčených chemoterapií je třeba při posuzování vhodnosti terapie přípravky ESA (pacienti, u nichž transfuze představuje riziko) počítat se 2-3 týdenním zpožděním po podání léku, než se objeví erytropoetinem indukované erytrocyty.
Chirurgičtí pacienti v programech předoperačního autologního odběru
Musí se respektovat všechna zvláštní upozornění a zvláštní opatření spojená s programem autologní krve, zejména běžná náhrada objemu.
Pacienti před velkou naplánovanou ortopedickou operací
Při použití v perioperačním období je zapotřebí vždy dodržovat zásady správného postupu zacházení s krví a krevními deriváty.
Pacientům zařazeným do programu velkých plánovaných ortopedických operací je třeba zajistit přiměřenou antitrombotickou profylaxi - u operovaných pacientů se mohou vyvinout trombotické a cévní příhody, zejména pokud trpí základním kardiovaskulárním onemocněním. Mimoto je třeba zvláštní opatrnosti u pacientů s predispozicí k rozvoji hlubokých žilních trombóz (DVT, deep vein thrombosis). U pacientů s počáteční hodnotou hemoglobinu > 13 g/dl (> 8,1 mmol/l) také nelze vyloučit možnost, že léčba epoetinem alfa bude spojena se zvýšeným rizikem pooperačních trombotických a cévních příhod. Epoetin alfa se proto nepodává pacientům s počáteční koncentrací hemoglobinu > 13 g/dl (> 8,1 mmol/l).
Pomocné látky
Tento přípravek obsahuje méně než 1 mmol sodíku (23 mg) na 1 předplněnou injekční stříkačku, tj. v podstatě je „bez sodíku“.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Neexistují žádné důkazy, podle kterých by léčba epoetinem alfa ovlivňovala metabolismus jiných léčivých přípravků.
Léčivé přípravky, které snižují tvorbu červených krvinek, mohou snižovat odpověď na epoetin alfa.
Protože se však cyklosporin váže na červené krvinky (red blood cells, RBCs), existuje možnost interakce léčivého přípravku. Pokud se epoetin alfa podává současně s cyklosporinem, je třeba monitorovat hladinu cyklosporinu v krvi a upravit dávky tohoto cytostatika, jakmile se hodnoty hematokritu zvýší.
Neexistují žádné důkazy svědčící pro interakci mezi epoetinem alfa a granulocytární kolonie stimulujícím faktorem (G-CSF) či faktorem stimulujícím granulocytární a makrofágové kolonie (GM-CSF), která by měla vliv na hematologickou diferenciaci anebo proliferaci nádorových buněk bioptických vzorků tumorů in vitro.
U dospělých pacientek s metastatickým karcinomem prsu nemělo subkutánní podání 40 000 IU/ml epoetinu alfa současně s 6 mg/kg trastuzumabu žádný vliv na farmakokinetiku trastuzumabu.
4.6 Fertilita, těhotenství a kojení
Údaje o podávání epoetinu alfa těhotným ženám jsou omezené nebo nejsou k dispozici. Studie na zvířatech prokázaly reprodukční toxicitu (viz bod 5.3). Proto u pacientek se epoetin alfa v těhotenství má použít pouze tehdy, pokud potenciální přínos převáží potenciální rizika pro plod. Použití epoetinu alfa se nedoporučuje u těhotných chirurgických pacientek účastnících se programu přípravy autologní krve.
Kojení
Není známo, zda se exogenní epoetin alfa vylučuje do lidského mateřského mléka. Epoetin alfa by měl být užíván s opatrností u kojících žen. Na základě posouzení prospěšnosti kojení pro dítě a prospěčnosti léčby epoetinem alfa pro matku je nutno rozhodnout, zda přerušit kojení nebo ukončit/přerušit podávání epoetinu alfa.
Použití epoetinu alfa se nedoporučuje u kojících chirurgických pacientek účastnících se programu přípravy autologní krve.
Fertilita
Nejsou k dispozici žádné studie hodnotící možný účinek epoetinu alfa na mužskou či ženskou fertilitu.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Studie hodnotící účinky na schopnost řídit nebo obsluhovat stroje nebyly provedeny. Binocrit nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky
Souhrn bezpečnostního _profilu
Nejčastější nežádoucí účinek při léčbě epoetinem alfa je zvýšení krevního tlaku či zhoršení stávající hypertenze úměrné podané dávce. Musí se provádět monitorování krevního tlaku, především na počátku terapie (viz bod 4.4).
Nežádoucí účinky, které se nejčastěji objevily v klinických studiích s epoetinem alfa, jsou průjem, nauzea, zvracení, pyrexie,a bolest hlavy. Příznaky podobné chřipce se mohou vyskytnout zvláště na počátku léčby.
Ve studiích s delším intervalem dávkování u dospělých pacientů s renální insuficiencí, kteří dosud nepodstoupili dialýzu, byly hlášeny kogesce respiračního traktu, která zahrnuje příhody kongesce horních cest dýchacích, nosní kongesci a nasofaryngitidu.
U pacientů užívajících přípravky ESA byl hlášen zvýšený výskyt trombotických cévních příhod (TVE) (viz bod 4.4).
Tabulkově uspořádaný seznam nežádoucích účinků
Z celkem 3 262 pacientů ve 23 randomizovaných, dvojitě zaslepených studiích, kontrolovaných placebem nebo standardní léčbou, byl celkový bezpečnostní profil epoetinu alfa vyhodnocen u 1 992 anemických pacientů. Začleněno bylo 228 pacientů s chronickým renálním selháním (CRF) léčených epoetinem ze 4 studií chronického renálního selhání (2 studie pacientů před dialýzou [N = 131 pacientů s CRF, kteří přípravek užili] a 2 studie dialyzovaných pacientů [N = 97 pacientů s CRF, kteří přípravek užili]; 1 404 pacientů s rakovinou v 16 studiích pacientů s anémií vyvolanou chemoterapií; 147 pacientů, kteří přípravek užili, ve 2 studiích programu předoperačního autologního odběru; a 213 pacientů, kteří přípravek užili, v 1 studii perioperačního období. Nežádoucí účinky léků, které hlásilo > 1 % pacientů léčených epoetinem alfa v těchto klinických studiích, jsou uvedeny v tabulce níže.
Odhad frekvence: velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1 000 až <1 /100), vzácné (> 1/10 000 až < 1/1 000), velmi vzácné (< 1/10 000), není známo (z dostupných údajů nelze určit).
|
Třída orgánového systému |
Frekvence | |||||
|
Velmi časté |
Časté |
Méně časté |
Vzácné |
Velmi vzácné |
Není známo | |
|
Poruchy krve a |
Erytropoetinový | |||||
1 Identifikováno po uvedení na trh a kategorie frekvence odhadovaná ze spontánně hlášené míry výskytu
|
lymfatického systému |
mi protilátkami zprostředkovaná čistá aplazie červené krevní řady1,4, trombocytémie1 | |||||
|
Poruchy metabolismu a výživy |
Hyperkalémie2 | |||||
|
Poruchy imunitního systému |
Anafylaktická reakce4, Hypersenzitivita 4 | |||||
|
Poruchy nervového systému |
Křeče | |||||
|
Cévní poruchy |
Žilní a arteriální trombóza3, hypertenze |
Hypertenzní krize4 | ||||
|
Respirační, hrudní a mediastinální poruchy |
Kongesce dýchacích cest | |||||
|
Gastrointestinální poruchy | ||||||
|
Poruchy kůže a podkožní tkáně |
Angioneurotick ý edém4, kopřivka4 | |||||
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Artralgie, bolesti kostí, myalgie, bolesti v končetinách | |||||
|
Vrozené, familiální a genetické vady |
Porfyrie4 | |||||
|
Celkové poruchy a reakce v místě aplikace |
Pyrexie |
příznaky podobné chřipce, reakce v místě vpichu injekce, periferní edém |
Neúčinnost léčivých přípravků4 |
2 Časté při dialýze
3 Zahrnuje arteriální a žilní, fatální a nefatální příhody, například hlubokou žilní trombózu, plicní embolii, retinální trombózu, arteriální trombózu (včetně infarktu myokardu), cévní mozkové příhody (včetně mozkového infarktu a mozkového krvácení), tranzitorní ischemické ataky a trombózu v shuntu (včetně dialyzačního systému) a trombózu ve výduti arteriovenózního shuntu
4 Popsáno v podbodu níže a/nebo v bodu 4.4.
Popis vybraných nežádoucích účinků
Byly hlášeny hypersenzitivní reakce, včetně případů vyrážky (včetně kopřivky), anafylaktické reakce a angioneurotického edému (viz bod 4.4).
U pacientů s normálním nebo nízkým krevním tlakem léčených epoetinem alfa se objevily také tyto reakce: hypertenzní krize s encefalopatií a záchvaty, vyžadující okamžitý zásah lékaře a intenzivní lékařskou péči. Zvláštní pozornost je třeba věnovat náhlým bodavým migrenózním bolestem hlavy jako možnému varovnému signálu (viz bod 4.4).
Protilátkami zprostředkovaná čistá aplazie červené krevní řady byla hlášena velmi vzácně (u < 1/10 000 případů na pacienta za rok) po několika měsících až letech léčby epoetinem alfa (viz bod 4.4).
Pediatrická _populace s chronickým renálním selháním na hemodialýze
Expozice pediatrických pacientů s chronickým renálním selháním na hemodialýze v klinických studiích a zkušenost po uvedení na trh je omezená. V této populaci nebyly hlášeny žádné nežádoucí účinky specifické pro pediatrické pacienty, které již nebyly uvedeny v tabulce výše, nebo které neodpovídaly základnímu onemocnění.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Terapeutické rozmezí epoetinu alfa je široké. Předávkování epoetinem alfa může vyvolat zesílené farmakologické účinky tohoto hormonu. Dosáhne-li hemoglobin příliš vysokých hodnot, je možné provést flebotomii. Další podpůrnou péči je třeba zajistit podle potřeby.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Jiná antianemika, erytropoetin, ATC kód: B03XA01
Binocrit je tzv. podobným biologickým léčivým přípravkem („biosimilar“). Podrobné informace jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Mechanismus účinku
Erytropoetin (EPO) je glykoproteinový hormon, který je vytvářen zejména ledvinami jako odpověď na hypoxii a který je klíčovým regulátorem produkce červených krvinek (RBC). EPO se účastní všech fází vývoje červené krvinky a hlavním místem jeho účinku je úroveň prekurzorů červené krevní řady. Když se EPO naváže na receptory na buněčném povrchu, aktivuje dráhy pro přenos signálu, které interferují s apoptózou, a stimuluje proliferaci buněk červené krevní řady.
Rekombinantní lidský EPO (epoetin alfa), exprimovaný ovariálními buňkami čínského křečka, má 165 aminokyselinových sekvencí identických s EPO v lidské moči; tyto 2 látky jsou podle funkčních testů nerozlišitelné. Zdánlivá molekulární hmotnost erytropoetinu je 32 000-40 000 daltonů.
Erytropoetin je růstový faktor, který primárně stimuluje tvorbu erytrocytů. Erytropoetinové receptory mohou být exprimovány na povrchu různých nádorových buněk.
Farmakodynamické účinky
Zdraví dobrovolníci
Po podání jedné dávky (20 000 až 160 000 IU subkutánně) epoetinu alfa byla pozorována na dávce závislá odpověď vyšetřovaných farmakodynamických markerů zahrnujících retikulocyty, červené krvinky a hemoglobin. Změny v procentech retikulocytů měly zřetelný profil závislosti koncentrace na čase, vyznačující se vrcholem a návratem k výchozí hodnotě. Méně zřetelný profil byl zjištěn pro červené krvinky a hemoglobin. Obecně se hodnoty všech farmakodynamických markerů lineárně zvýšily, přičemž po dávce na nejvyšší dávkovací úrovni byla pozorována maximální odpověď.
Další farmakodynamické studie zkoumaly dávku 40 000 IU jednou týdně a porovnávaly ji s dávkou 150 IU/kg 3krát týdně. I přes rozdílné profily závislosti koncentrace na čase byla farmakodynamická odpověď (jejímž měřítkem byly změny v procentech retikulocytů, hemoglobinu a celkovém počtu červených krvinek) při těchto režimech podobná. Další studie porovnávaly režim se subkutánním podáváním 40 000 IU epoetinu alfa jednou týdně s dávkami v rozsahu od 80 000 do 120 000 IU dvakrát týdně. Celkově se na základě výsledků těchto farmakodynamických studií na zdravých dobrovolnících jeví, že dávkovací režim s podáváním 40 000 IU jednou týdně má na vytváření červených krvinek větší účinek, než to režimy s podáváním dvakrát týdně, a to i přes pozorovanou podobnost produkce retikulocytů v režimu s podáváním jednou týdně a režimu s podáváním dvakrát týdně.
Chronické renální selhání
Bylo prokázáno, že epoetin alfa stimuluje erytropoezu u anemických pacientů s chronickým renálním selháním, včetně dialyzovaných pacientů a pacientů ve fázi před dialýzou. První známkou odpovědi na epoetin alfa je zvýšení počtu retikulocytů do 10 dní, po kterém následuje zvýšení počtu červených krvinek, hemoglobinu a hematokritu, zpravidla do 2 až 6 týdnů. Odpověď hemoglobinu se u různých pacientů liší a mohou na ni mít vliv zásoby železa a přítomnost dalších zdravotních problémů.
Anémie vyvolaná chemoterapií
Bylo prokázáno, že epoetin alfa podávaný 3krát týdně nebo jednou týdně zvyšoval hodnotu hemoglobinu a snižoval požadavky na transfuze po prvním měsíci léčby u anemických pacientů s rakovinou užívajících chemoterapii.
Ve studii srovnávající dávkovací režim 150 IU/kg 3krát týdně a dávkovací režim 40 000 IU jednou týdně u zdravých osob a u anemických pacientů s rakovinou byly časové profily změn v procentech retikulocytů, hemoglobinu a celkovém počtu červených krvinek u zdravých osob a u anemických pacientů s rakovinou při těchto dvou dávkovacích režimech podobné. Hodnoty AUC jednotlivých farmakodynamických parametrů při dávkovacích režimech 150 IU/kg 3krát týdně a 40 000 IU jednou týdně u zdravých osob a u anemických pacientů s rakovinou byly podobné.
Dospělí chirurgičtí_pacienti v _ programu _předoperačního autologního odběru Bylo prokázáno, že epoetin alfa stimuluje produkci červených krvinek, což umožňuje použít jej k podpoře programu předoperačního autologního odběru krve a k omezení poklesu hemoglobinu u dospělých pacientů, kteří mají naplánovanou velkou operaci a nepředpokládá se, že by si udělali zásobu vlastní krve pokrývající celou perioperační potřebu. Největší efekt bývá pozorován u pacientů s nízkým hemoglobinem (< 13 g/dl).
Léčba dospělých _pacientů _před_plánovanou velkou ortopedickou operací
Bylo prokázáno, že u pacientů, kteří mají naplánovanou velkou ortopedickou operaci a před léčbou mají hodnotu hemoglobinu > 10 až < 13 g/dl, epoetin alfa snižuje riziko podání alogenních transfuzí a urychluje obnovu červeného krevního obrazu (zvyšuje hladiny hemoglobinu, hematokritu a počty retikulocytů).
Klinická účinnost a bezpečnost
Chronické renální selhání
Epoetin alfa byl hodnocen v klinických studiích u dospělých anemických pacientů s chronickým renálním selháním, včetně dialyzovaných pacientů a pacientů ve fázi před dialýzou, jako přípravek používaný k léčbě anémie a udržení hematokritu v cílovém rozsahu koncentrací od 30 do 36 %.
V klinických studiích s počátečními dávkami 50 až 150 IU/kg třikrát týdně přibližně 95 % všech pacientů odpovědělo klinicky významným zvýšením hematokritu. Přibližně po dvou měsících léčby byli prakticky všichni pacienti nezávislí na transfuzích. Jakmile bylo dosaženo cílového hematokritu, byla udržovací dávka pro každého pacienta individualizována.
Ve třech největších klinických studiích prováděných u dospělých dialyzovaných pacientů, byl medián udržovací dávky nezbytné pro zachování hematokritu mezi 30 a 36 % přibližně 75 IU/kg podávaných 3krát týdně.
V dvojitě zaslepené placebem kontrolované multicentrické studii kvality života u hemodialyzovaných pacientů s chronickým renálním selháním bylo prokázáno klinicky a statisticky významné zlepšení
u pacientů léčených epoetinem alfa ve srovnání se skupinou užívající placebo při měření únavy, tělesných příznaků, vztahů a deprese (dotazník pro onemocnění ledvin) po šesti měsících léčby. Pacienti ze skupiny léčené epoetinem alfa byli také zařazeni do otevřené prodloužené studie, která prokázala zlepšení kvality jejich života, jež se udrželo po dobu dalších 12 měsíců.
Dospělí _pacienti s renální insuficiencí, kteří dosud dialyzovaní nebyli
V klinických studiích prováděných u nedialyzovaných pacientů s chronickým renálním selháním léčených epoetinem alfa byla průměrná délka trvání léčby téměř pět měsíců. Tito pacienti odpověděli na léčbu epoetinem alfa podobným způsobem, jaký byl pozorován u dialyzovaných pacientů. Nedialyzovaní pacienti s chronickým renálním selháním vykazovali po intravenózním nebo subkutánním podání epoetinu alfa setrvalý a na dávce závisející nárůst hematokritu. Ať již byl epoetin alfa podán kterýmkoli z těchto způsobů, byly zaznamenány podobné míry zvýšení hematokritu. Navíc bylo prokázáno, že dávky epoetinu alfa od 75 do 150 IU/kg týdně udrží hematokrit v rozmezí 36 až 38 % po dobu až šesti měsíců.
Ve 2 studiích s prodlouženým dávkovacím intervalem epoetinu alfa (3krát týdně, jednou týdně, jednou za každé 2 týdny a jednou za každé 4 týdny) si někteří pacienti s delšími dávkovacími intervaly neudrželi dostatečné hladiny hemoglobinu a dosáhli protokolem definovaných kritérií vysazení z důvodu hladiny hemoglobinu (0 % ve skupině s podáváním jednou týdně, 3,7 % ve skupině s podáváním jednou za 2 týdny a 3,3 % ve skupině s podáváním jednou za 4 týdny).
Randomizovaná prospektivní klinická studie posuzovala 1 432 nedialyzovaných anemických pacientů s chronickým renálním selháním. Pacientům byla přidělena léčba epoetinem alfa, jejímž cílem bylo udržovat hladinu hemoglobinu 13,5 g/dl (která je vyšší než doporučená úroveň koncentrace hemoglobinu) nebo 11,3 g/dl. K závažným kardiovaskulárním příhodám (smrt, infarkt myokardu, cévní mozková příhoda nebo hospitalizace z důvodu městnavého srdečního selhání) došlo u 125 (18 %) ze 715 pacientů ve skupině s vyšší hodnotou hemoglobinu oproti 97 (14 %) pacientům ze 717 ve skupině s nižší hodnotou hemoglobinu (poměr rizik [HR] 1,3, 95 % CI: 1,0, 1,7, p = 0,03).
Byly provedeny sdružené dodatečné analýzy klinických studií ESA u pacientů s chronickým renálním selháním (na dialýze, nedialyzovaných, diabetiků a nediabetiků). Byla pozorována tendence ke zvýšeným hodnotám odhadů rizik mortality ze všech příčin a kardiovaskulárních a cévních mozkových příhod spojených s vyššími kumulativními dávkami ESA, nezávisle na zjištěném stavu diabetu nebo dialýzy (viz bod 4.2 a bod 4.4).
Léčba pacientů s anémií vyvolanou chemoterapií
Epoetin alfa byl hodnocen v klinických studiích u dospělých anemických pacientů s rakovinou, kteří měli lymfoidní a solidní tumory, a pacientů na různých režimech chemoterapie, zahrnujících režimy obsahující platinu i režimy bez platiny. V těchto studiích bylo prokázáno, že epoetin alfa podávaný 3krát týdně a jednou týdně po prvním měsíci léčby zvyšuje hemoglobin a snižuje požadavky na transfuze u anemických pacientů s rakovinou. V některých studiích po dvojitě zaslepené fázi následovala otevřená fáze, během které všichni pacienti užívali epoetin alfa a sledovalo se, zda bude zachován účinek.
Dostupné důkazy nasvědčují, že pacienti s hematologickými malignitami a solidními tumory odpovídají na léčbu epoetinem alfa stejně a že pacienti s nádorovou infiltrací kostní dřeně nebo bez ní odpovídají na léčbu epoetinem alfa stejně. V klinických studiích s chemoterapií byla prokázána srovnatelná intenzita chemoterapie ve skupině s epoetinem alfa a s placebem podobností plochy pod křivkou závislosti počtu neutrofilů na čase u pacientů léčených epoetinem alfa a placebem a také podobným podílem pacientů ve skupině léčené epoetinem alfa a skupině léčené placebem, u nichž absolutní počty neutrofilů byly nižší než 1 000 a 500 buněk/^l.
V prospektivní, randomizované, dvojitě zaslepené a placebem kontrolované klinické studii, které se účastnilo 375 anemických pacientů s různými nemyeloidními malignitami, absolvujícími chemoterapii neplatinovými přípravky, se významně snížily následky anémie (např. únava, úbytek energie a omezení aktivity), a to podle měření za použití těchto dotazníků a hodnotících škál: funkční hodnocení protinádorové terapie-anémie (FACT-An) (a to jak na všeobecné hodnotící škále, tak i škále únavy) a nádorová lineární analogová škála (CLAS). Dvě jiné menší, randomizované a placebem kontrolované studie nevykázaly významné zlepšení u parametrů kvality života na škále EORTC-QLQ-C30 resp. CLAS.
Přežití a doba progrese byla zkoumána v pěti velkých kontrolovaných studiích zahrnujících celkem 2 833 pacientů, čtyři studie byly dvojitě zaslepené, kontrolované placebem a jedna studie byla otevřená. Studie zahrnovaly buď pacienty, kteří byli léčeni chemoterapií (dvě studie) nebo populaci pacientů, pro kterou nejsou indikovány přípravky ESA: anémie u pacientů s rakovinou, kteří nejsou léčeni chemoterapií, a pacienti s rakovinou hlavy a krku léčených radioterapií. Požadovaná úroveň koncentrací hemoglobinu byla ve dvou studiích > 13 g/dl (8,1 mmol/l); ve zbývajících studiích 12 až 14 g/dl (7,5 až 8,7 mmol/l). V otevřené studii nebyl prokázán žádný rozdíl v celkové době přežití mezi pacienty léčenými rekombinantním lidským erytropoetinem a kontrolami. Ve čtyřech studiích kontrolovaných placebem se pohybovala míra rizika celkového přežití mezi 1,25 a 2,47 ve prospěch kontrol. Tyto studie ukázaly konzistentní nevysvětlitelné, statisticky významné zvýšení mortality u pacientů s anémií spojenou s různými typy obvyklých nádorových onemocnění, kterým se podával rekombinantní lidský erytropoetin v porovnání s kontrolními skupinami. Výsledné celkové přežití v těchto zkouškách není možné dostatečně vysvětlit rozdíly ve výskytu trombózy a příbuzných komplikací mezi pacienty, kteří dostávali rekombinantní lidský erytropoetin a těmi z kontrolní skupiny.
Byla rovněž provedena analýza údajů z 53 kontrolovaných klinických studií s několika epoetiny u více než 13 900 pacientů s rakovinou (chemoterapie, radioterapie, chemoradioterapie nebo neléčení). Meta-analýza údajů celkového přežití vygenerovala odhad míry rizika přežití na 1,06 ve prospěch kontrolních skupin (95 % CI: 1,00, 1,12; 53 zkoušek a 13 933 pacientů) a pro pacienty s rakovinou léčené chemoterapií byla míra celkové doby přežití 1,04 (95 % CI: 0,97, 1,11; 38 zkoušek a 10 441 pacientů). Meta-analýzy rovněž ukázaly konzistentní a významné zvýšení relativního rizika tromboembolických příhod u pacientů s rakovinou léčených rekombinantním lidským erytropoetinem (viz bod 4.4).
Program vředoveračního autologního odběru
Podpůrný vliv epoetinu alfa na program předoperačního autologního odběru u pacientů s nízkým hematokritem (< 39 % při nepřítomnosti anémie v důsledku deficitu železa), u nichž byla naplánována velká ortopedická operace, byl hodnocen v dvojitě zaslepené placebem kontrolované studii, prováděné u 204 pacientů, a v jednoduše zaslepené placebem kontrolované studii u 55 pacientů.
V dvojitě zaslepené placebem kontrolované studii byli pacienti léčeni dávkou 600 IU/kg epoetinu alfa nebo placebem, podávanými intravenózně jednou denně každé 3 až 4 dny po dobu 3 týdnů (celkem
6 dávek). V průměru bylo možné odebrat a uskladnit pacientům léčených epoetinem alfa signifikantně více dárcovských jednotek krve (4,5 jednotky) než pacientům léčených placebem (3,0 jednotky).
V dvojitě zaslepené placebem kontrolované studii byli pacienti léčeni dávkou 300 IU/kg epoetinu alfa nebo 600 IU/kg epoetinu alfa nebo placebem, podávanými intravenózně jednou denně každé 3 až
4 dny po dobu 3 týdnů (celkem 6 dávek). Pacientům léčeným epoetinem alfa bylo také možné odebrat a uskladnit signifikantně více dárcovských jednotek krve (epoetin alfa 300 IU/kg = 4,4 jednotky; epoetin alfa 600 IU/kg = 4,7 jednotky) než pacientům léčeným placebem (2,9 jednotky).
Léčba epoetinem alfa snížila riziko expozice alogenní krvi o 50 % ve srovnání s pacienty, kteří epoetin alfa nedostávali.
Plánovaná velká ortopedická operace
Vliv epoetinu alfa (300 IU/kg nebo 100 IU/kg) na transfuzi alogenní krve byl hodnocen v placebem kontrolované dvojitě zaslepené klinické studii u dospělých pacientů bez deficitu železa, u nichž byla naplánována velká elektivní ortopedická operace kyčle nebo kolena. Epoetin alfa byl podáván subkutánně po dobu 10 dní před operací, v den operace a po dobu čtyř dní po operaci. Pacienti byli zařazeni do skupin podle výchozí hodnoty hemoglobinu (< 10 g/dl, > 10 až < 13 g/dl a > 13 g/dl).
Epoetin alfa 300 IU/kg signifikantně snížil riziko alogenní transfuze u pacientů s hodnotou hemoglobinu před léčbou > 10 až < 13 g/dl. 16 % pacientů léčených dávkou 300 IU/kg epoetinu alfa, 23 % pacientů léčených dávkou 100 IU/kg epoetinu alfa a 45 % pacientů léčených placebem vyžadovalo transfuzi.
V otevřené klinické studii s paralelními skupinami u dospělých pacientů bez deficitu železa s hodnotou hemoglobinu před léčbou > 10 až < 13 g/dl, u nichž byla naplánována velká ortopedická operace kyčle nebo kolena, se porovnávala dávka 300 IU/kg epoetinu alfa subkutánně denně po dobu 10 dní před operací, v den operace a čtyř dní po operaci s dávkou 600 IU/kg epoetinu alfa subkutánně jednou týdně po dobu 3 týdnů před operací a v den operace.
Mezi okamžikem před léčbou a okamžikem před operací bylo střední zvýšení hemoglobinu ve skupině, která dostávala dávku 600 IU/kg jednou týdně (1,44 g/dl), dvojnásobné oproti skupině, která dostávala dávku 300 IU/kg denně (0,73 g/dl). Po celé pooperační období byly střední hladiny hemoglobinu v obou léčených skupinách podobné.
Odpověď erytropoezy pozorovaná v obou léčených skupinách vedla k podobným mírám podání transfuzí (16 % ve skupině, která dostávala dávku 600 IU/kg týdně, a 20 % ve skupině, která dostávala dávku 300 IU/kg denně).
Pediatrická populace
Chronické renální selhání
Epoetin alfa byl hodnocen v otevřené, nerandomizované, 52týdenní klinické studii s otevřeným dávkovacím rozmezím u hemodialyzovaných pediatrických pacientů s chronickým renálním selháním. Průměrná hodnota věku pacientů zařazených do studie byl 11,6 roku (při rozsahu 0,5 až 20,1 roku).
Epoetin alfa byl podáván intravenózně po dialýze v dávce 75 IU/kg/týden rozděleně do 2 nebo 3 dávek a titrován na dávku75 IU/kg/týden v intervalech 4 týdnů (do maxima300 IU/kg/týden), aby bylo dosaženo zvýšení hladiny hemoglobinu o 1 g/dl/měsíc. Žádoucí rozsah koncentrací hemoglobinu byl 9,6 až 11,2 g/dl. 81 % pacientů dosáhlo této úrovně koncentrací hemoglobinu. Medián doby do dosažení cíle byl 11 týdnů, medián dávky při dosažení cíle byl 150 IU/kg/týden. Z pacientů, kteří dosáhli cíle, jich ho 90 % dosáhlo při režimu s podáním dávky 3krát týdně.
Po 52 týdnech 57 % pacientů zůstávalo ve studii a průměrná hodnota podávané dávky byl 200 IU/kg/týden.
5.2 Farmakokinetické vlastnosti
Absorpce
Po subkutánní injekci dosáhnou sérové hladiny epoetinu alfa vrcholu mezi 12 a 18 hodinami po podání dávky. Po opakované subkutánně podané dávce 600 IU/kg jednou týdně nedocházelo k akumulaci.
Absolutní biologická dostupnost subkutánně podaného epoetinu alfa je u zdravých jedinců přibližně 20 %.
Distribuce
Střední distribuční objem po intravenózních dávkách 50 a 100 IU/kg u zdravých osob byl 49,3 ml/kg. Po intravenózním podání epoetinu alfa osobám s chronickým renálním selháním byl rozsah distribučního objemu 57-107 ml/kg po podání jedné dávky (12 IU/kg) až 42-64 ml/kg po podání více dávek (48 až 192 IU/kg). Proto je distribuční objem o něco větší než plazmatický prostor.
Eliminace
Poločas eliminace epoetinu alfa u zdravých osob po opakované intravenózně podané dávce je přibližně 4 hodiny.
Poločas eliminace u subkutánního podání zdravým osobám se odhaduje přibližně na 24 hodiny.
Střední hodnota CL/F pro režimy podávání 150 IU/kg 3krát týdně a 40 000 IU jednou týdně u zdravých osob byla 31,2, resp. 12,6 ml/h/kg. Střední hodnota CL/F pro režimy podávání 150 IU/kg, 3krát týdně a 40 000 IU jednou týdně u anemických pacientů s rakovinou byla 45,8, resp.
11.3 ml/h/kg. U většiny anemických pacientů s rakovinou, kteří dostávali cyklickou chemoterapii, byl poměr CL/F nižší po subkutánních dávkách 40 000 IU jednou týdně a 150 IU/kg 3krát týdně oproti hodnotám u zdravých osob.
Linearita/nelinearita
U zdravých osob bylo po intravenózním podávání dávky 150 a 300 IU/kg 3krát týdně pozorováno zvýšení sérových koncentrací epoetinu alfa přímo úměrné dávce. Subkutánní podání jednotlivé dávky epoetinu alfa 300 až 2 400 IU/kg vedlo k lineárnímu vztahu mezi střední hodnotou Cmax a dávkou a mezi střední hodnotou AUC a dávkou. U zdravých osob byl pozorován vztah nepřímé úměry mezi zdánlivou clearancí a dávkou.
Ve studiích, které zkoumaly prodloužený dávkovací interval (40 000 IU jednou týdně a 80 000,
100 000 a 120 000 IU dvakrát týdně), byl v ustáleném stavu pozorován lineární vztah mezi střední hodnotou Cmax a dávkou a mezi střední hodnotou AUC a dávkou, nikoli však přímo úměrný dávce.
Farmakokinetické/farmakodynamické vztahy
Epoetin alfa má na hematologické parametry účinek závislý na dávce, který nezávisí na způsobu podání.
Pediatrická _ populace
U pediatrických pacientů s chronickým renálním selháním byl po opakované intravenózně podané dávce epoetinu alfa hlášen poločas přibližně 6,2 až 8,7 hodiny. Farmakokinetický profil epoetinu alfa u dětí a dospívajících je podobný profilu u dospělých.
Porucha funkce ledvin
U pacientů s chronickým renálním selháním je poločas intravenózně podávaného epoetinu alfa mírně prodloužen oproti zdravým osobám, přibližně na 5 hodin.
5.3 Předklinické údaje vztahující se k bezpečnosti
V toxikologických studiích po opakovaném podávání u psů a potkanů, ne však u opic, byla terapie epoetinem alfa spojena se subklinickou fibrózou kostní dřeně. Fibróza kostní dřeně je známou komplikací chronického renálního selhání u lidí a může souviset se sekundárním hyperparatyroidismem anebo s neznámými faktory. Výskyt této fibrózy se v jiné studii nezvýšil u dialyzovaných pacientů, kteří byli léčeni epoetinem alfa po 3 roky - v porovnání s kontrolní skupinou dialyzovaných pacientů, kteří epoetinem alfa léčeni nebyli.
Epoetin alfa nevyvolává mutace bakteriálních genů (Amesův test), chromozomové aberace u savčích buněk, vznik mikrojader u myší, ani mutace genů v lokusu HGPRT.
Dlouhodobé studie karcinogenity provedeny nebyly. V literatuře existují na základě nálezů získaných in vitro na vzorcích lidské tumorózní tkáně protichůdné názory na to, zda erytropoetin může hrát nějakou roli při proliferaci tumoru. Význam pro klinickou situaci je však nejasný.
V kulturách lidských buněk kostní dřeně se podařilo dokázat, že epoetin alfa stimuluje specificky tvorbu erytrocytů a neovlivní leukopoezu. Cytotoxické účinky epoetinu alfa na kostní dřeň nebylo možné zjistit.
Ve studiích na zvířatech epoetin alfa při podávání v týdenních dávkách přibližně 20x vyšších než odpovídá doporučeným týdenním dávkám u člověka, průkazně snižoval tělesnou hmotnost plodů, opožďoval osifikaci a zvyšoval mortalitu plodu. Tyto změny byly vyhodnoceny jako sekundární, závislé na zpomalení přírůstku hmotnosti matky, a jejich význam pro člověka při daných úrovních terapeutických dávek je nejasný.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Dihydrát dihydrogenfosforečnanu sodného Dihydrát hydrogenfosforečnanu sodného Chlorid sodný Glycin
Polysorbát 80 Voda na injekci
Kyselina chlorovodíková (k úpravě pH)
Hydroxid sodný (k úpravě pH)
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti
2 roky
6.4 Zvláštní opatření pro uchovávání
Uchovávejte a převážejte chlazené (2 °C až 8 °C). Tento rozsah teplot musí být pečlivě dodržován až do podání pacientovi.
Pro účely ambulantního použití lze léčivý přípravek vyjmout z chladničky a uchovávat jej mimo chladničku maximálně po dobu 3 dní při teplotě do 25 °C. Pokud se léčivý přípravek nespotřebuje do konce tohoto období, musí se zlikvidovat.
Chraňte před mrazem. Netřepejte.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
6.5 Druh obalu a obsah balení
Předplněné injekční stříkačky (sklo I. typu), s bezpečnostním krytem jehly anebo bez něho, se zátkou s pístem (pryž potažená teflonem), uzavřené v blistru.
Binocrit 1 000 IU/0,5 ml injekční roztok v předplněné injekční stříkačce Jedna předplněná injekční stříkačka obsahuje 0,5 ml injekčního roztoku.
Balení po 1 nebo 6 kusech.
Binocrit 2 000 IU/1 ml injekční roztok v předplněné injekční stříkačce Jedna předplněná injekční stříkačka obsahuje 1 ml injekčního roztoku.
Balení po 1 nebo 6 kusech.
Binocrit 3 000 IU/0,3 ml injekční roztok v předplněné injekční stříkačce Jedna předplněná injekční stříkačka obsahuje 0,3 ml injekčního roztoku.
Balení po 1 nebo 6 kusech.
Binocrit 4 000 IU/0,4 ml injekční roztok v předplněné injekční stříkačce Jedna předplněná injekční stříkačka obsahuje 0,4 ml injekčního roztoku.
Balení po 1 nebo 6 kusech.
Binocrit 5 000 IU/0,5 ml injekční roztok v předplněné injekční stříkačce Jedna předplněná injekční stříkačka obsahuje 0,5 ml injekčního roztoku.
Balení po 1 nebo 6 kusech.
Binocrit 6 000 IU/0,6 ml injekční roztok v předplněné injekční stříkačce Jedna předplněná injekční stříkačka obsahuje 0,6 ml injekčního roztoku.
Balení po 1 nebo 6 kusech.
Binocrit 7 000 IU/0,7 ml injekční roztok v předplněné injekční stříkačce Jedna předplněná injekční stříkačka obsahuje 0,7 ml injekčního roztoku.
Balení po 1 nebo 6 kusech.
Binocrit 8 000 IU/0,8 ml injekční roztok v předplněné injekční stříkačce Jedna předplněná injekční stříkačka obsahuje 0,8 ml injekčního roztoku.
Balení po 1 nebo 6 kusech.
Binocrit 9 000 IU/0,9 ml injekční roztok v předplněné injekční stříkačce Jedna předplněná injekční stříkačka obsahuje 0,9 ml injekčního roztoku.
Balení po 1 nebo 6 kusech.
Binocrit 10 000 IU/1 ml injekční roztok v předplněné injekční stříkačce Jedna předplněná injekční stříkačka obsahuje 1 ml injekčního roztoku.
Balení po 1 nebo 6 kusech.
Binocrit 20 000 IU/0,5 ml injekční roztok v předplněné injekční stříkačce Jedna předplněná injekční stříkačka obsahuje 0,5 ml injekčního roztoku.
Balení po 1, 4 nebo 6 kusech.
Binocrit 30 000 IU/0,75 ml injekční roztok v předplněné injekční stříkačce Jedna předplněná injekční stříkačka obsahuje 0,75 ml injekčního roztoku.
Balení po 1, 4 nebo 6 kusech.
Binocrit 40 000 IU/1 ml injekční roztok v předplněné injekční stříkačce Jedna předplněná injekční stříkačka obsahuje 1 ml injekčního roztoku.
Balení po 1, 4 nebo 6 kusech.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Binocrit by neměl být použit a měl by být zlikvidován:
- pokud je tekutina zabarvena či v ní jsou plovoucí viditelné částice,
- je-li poškozená pečeť,
- pokud víte nebo se domníváte, že roztok byl nedopatřením zmražen nebo
- při poruše chladničky.
Předplněné injekční stříkačky jsou připraveny k použití (viz bod 4.2). Předplněnou injekční stříkačku neprotřepávejte. Injekční stříkačky mají výstupky s prstenci stupnice, aby bylo možné částečné použití v případě potřeby. Jeden prstenec stupnice odpovídá objemu 0,1 ml. Tento přípravek je určen pouze k jednorázovému použití. Aplikujte pouze jednu dávku přípravku Binocrit z jedné injekční stříkačky, z které byl před aplikací injekce odstraněn nepotřebný roztok.
Použití předplněné injekční stříkačky s bezpečnostním krytem jehly
Bezpečnostní kryt jehly překrývá jehlu po injekci, aby zabránil zranění píchnutím jehlou. Tím nijak neruší normální zacházení se stříkačkou.Tlačte na píst pomalu až do té doby, kdy je podána celá dávka a kdy se píst již nedá dále stlačit. Zatímco udržujete tlak na píst, vytáhněte stříkačku s jehlou z pacientova těla. Bezpečnostní kryt zakryje jehlu, jakmile povolíte tlak na píst.
Použití předplněné injekční stříkačky bez ochranného krytu jehly Podejte dávku standardním způsobem.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Sandoz GmbH Biochemiestr. 10 A-6250 Kundl Rakousko
8. REGISTRAČNÍ ČÍSLO(A)
Binocrit 1 000 IU/0,5 ml injekční roztok v předplněné injekční stříkačce
EU/1/07/410/001
EU/1/07/410/002
EU/1/07/410/027
EU/1/07/410/028
Binocrit 2 000 IU/1 ml injekční roztok v předplněné injekční stříkačce
EU/1/07/410/003
EU/1/07/410/004
EU/1/07/410/029
EU/1/07/410/030
Binocrit 3 000 IU/0,3 ml injekční roztok v předplněné injekční stříkačce
EU/1/07/410/005
EU/1/07/410/006
EU/1/07/410/031
EU/1/07/410/032
Binocrit 4 000 IU/0,4 ml injekční roztok v předplněné injekční stříkačce
EU/1/07/410/007
EU/1/07/410/008
EU/1/07/410/033
EU/1/07/410/034
Binocrit 5 000 IU/0,5 ml injekční roztok v předplněné injekční stříkačce
EU/1/07/410/009
EU/1/07/410/010
EU/1/07/410/035
EU/1/07/410/036
Binocrit 6 000 IU/0,6 ml injekční roztok v předplněné injekční stříkačce
EU/1/07/410/011
EU/1/07/410/012
EU/1/07/410/037
EU/1/07/410/038
Binocrit 7 000 IU/0,7 ml injekční roztok v předplněné injekční stříkačce
EU/1/07/410/017
EU/1/07/410/018
EU/1/07/410/039
EU/1/07/410/040
Binocrit 8 000 IU/0,8 ml injekční roztok v předplněné injekční stříkačce
EU/1/07/410/013
EU/1/07/410/014
EU/1/07/410/041
EU/1/07/410/042
Binocrit 9 000 IU/0,9 ml injekční roztok v předplněné injekční stříkačce
EU/1/07/410/019
EU/1/07/410/020
EU/1/07/410/043
EU/1/07/410/044
Binocrit 10 000 IU/1 ml injekční roztok v předplněné injekční stříkačce
EU/1/07/410/015
EU/1/07/410/016
EU/1/07/410/045
EU/1/07/410/046
Binocrit 20 000 IU/0,5 ml injekční roztok v předplněné injekční stříkačce
EU/1/07/410/021
EU/1/07/410/022
EU/1/07/410/047
EU/1/07/410/053
EU/1/07/410/048
Binocrit 30 000 IU/0,75 ml injekční roztok v předplněné injekční stříkačce
EU/1/07/410/023
EU/1/07/410/024
EU/1/07/410/049
EU/1/07/410/054
EU/1/07/410/050
Binocrit 40 000 IU/1 ml injekční roztok v předplněné injekční stříkačce
EU/1/07/410/025
EU/1/07/410/026
EU/1/07/410/051
EU/1/07/410/055
EU/1/07/410/052
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 28. srpna 2007
Datum posledního prodloužení registrace: 18. června 2012
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/.
PŘÍLOHA II
A. VÝROBCI BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCI BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobců biologické léčivé látky
Rentschler Biotechnologie GmbH Erwin-Rentschler-Strasse 21 D-88471 Laupheim Německo
Lek Pharmaceuticals d.d.
Kolodvorska 27 SI-1234 Menges Slovinsko
Název a adresa výrobce odpovědného za propouštění šarží
Sandoz GmbH Biochemiestr. 10 A-6250 Kundl Rakousko
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
TTV 1 r V ; V f V V r
• Úřední propouštění sarzí
Podle článku 114 směrnice 2001/83/ES bude úřední propouštění šarží provádět některá státní laboratoř nebo laboratoř k tomuto účelu určená.
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
PŘÍLOHA III
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
Binocrit 1 000 IU/0,5 ml injekční roztok v předplněné injekční stříkačce Epoetinum alfa
1 předplněná injekční stříkačka s obsahem 0,5 ml obsahuje 1 000 mezinárodních jednotek (IU), což odpovídá 8,4 mikrogramům epoetinum alfa.
Pomocné látky: dihydrát dihydrogenfosforečnanu sodného, dihydrát hydrogenfosforečnanu sodného, chlorid sodný, glycin, polysorbát 80, kyselina chlorovodíková (pro úpravu pH), hydroxid sodný (pro úpravu pH) a voda na injekci.
Další informace naleznete v příbalové informaci.
Injekční roztok v předplněné injekční stříkačce.
1 předplněná injekční stříkačka s 0,5 ml 6 předplněných injekčních stříkaček s 0,5 ml
1 předplněná injekční stříkačka s 0,5 ml s bezpečnostním krytem jehly 6 předplněných injekčních stříkaček s 0,5 ml s bezpečnostním krytem jehly
Pro subkutánní a intravenózní podání.
Před použitím si přečtěte příbalovou informaci. Netřepejte.
Uchovávejte mimo dohled a dosah dětí.
Uchovávejte a převážejte chlazené (2°C-8°C).
Chraňte před mrazem.
Předplněné stříkačky uchovávejte ve vnějším obalu (v krabičce), aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Sandoz GmbH, Biochemiestr. 10, A-6250 Kundl, Rakousko
EU/1/07/410/001
EU/1/07/410/002
EU/1/07/410/027
EU/1/07/410/028
č.š.:
Binocrit 1 000 IU/0,5 ml
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Binocrit 1 000 IU/0,5 ml injekce
Epoetinum alfa i.v./s.c.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
Binocrit 2 000 IU/1 ml injekční roztok v předplněné injekční stříkačce Epoetinum alfa
1 předplněná injekční stříkačka s obsahem 1 ml obsahuje 2 000 mezinárodních jednotek (IU), což odpovídá 16,8 mikrogramům epoetinum alfa.
Pomocné látky: dihydrát dihydrogenfosforečnanu sodného, dihydrát hydrogenfosforečnanu sodného, chlorid sodný, glycin, polysorbát 80, kyselina chlorovodíková (pro úpravu pH), hydroxid sodný (pro úpravu pH) a voda na injekci.
Další informace naleznete v příbalové informaci.
Injekční roztok v předplněné injekční stříkačce.
1 předplněná injekční stříkačka s 1 ml 6 předplněných injekčních stříkaček s 1 ml
1 předplněná injekční stříkačka s 1 ml s bezpečnostním krytem jehly 6 předplněných injekčních stříkaček s 1 ml s bezpečnostním krytem jehly
Pro subkutánní a intravenózní podání.
Před použitím si přečtěte příbalovou informaci. Netřepejte.
Uchovávejte mimo dohled a dosah dětí.
Uchovávejte a převážejte chlazené (2°C-8°C).
Chraňte před mrazem.
Předplněné stříkačky uchovávejte ve vnějším obalu (v krabičce), aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Sandoz GmbH, Biochemiestr. 10, A-6250 Kundl, Rakousko
EU/1/07/410/003
EU/1/07/410/004
EU/1/07/410/029
EU/1/07/410/030
č.š.:
Binocrit 2 000 IU/1 ml
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Binocrit 2 000 IU/1 ml injekce
Epoetinum alfa i.v./s.c.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
Binocrit 3 000 IU/0,3 ml injekční roztok v předplněné injekční stříkačce Epoetinum alfa
1 předplněná injekční stříkačka s obsahem 0,3 ml obsahuje 3 000 mezinárodních jednotek (IU), což odpovídá 25,2 mikrogramům epoetinum alfa.
Pomocné látky: dihydrát dihydrogenfosforečnanu sodného, dihydrát hydrogenfosforečnanu sodného, chlorid sodný, glycin, polysorbát 80, kyselina chlorovodíková (pro úpravu pH), hydroxid sodný (pro úpravu pH) a voda na injekci.
Další informace naleznete v příbalové informaci.
Injekční roztok v předplněné injekční stříkačce.
1 předplněná injekční stříkačka s 0,3 ml 6 předplněných injekčních stříkaček s 0,3 ml
1 předplněná injekční stříkačka s 0,3 ml s bezpečnostním krytem jehly 6 předplněných injekčních stříkaček s 0,3 ml s bezpečnostním krytem jehly
Pro subkutánní a intravenózní podání.
Před použitím si přečtěte příbalovou informaci. Netřepejte.
Uchovávejte mimo dohled a dosah dětí.
Uchovávejte a převážejte chlazené (2°C-8°C).
Chraňte před mrazem.
Předplněné stříkačky uchovávejte ve vnějším obalu (v krabičce), aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Sandoz GmbH, Biochemiestr. 10, A-6250 Kundl, Rakousko
EU/1/07/410/005
EU/1/07/410/006
EU/1/07/410/031
EU/1/07/410/032
č.š.:
Binocrit 3 000 IU/0,3 ml
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Binocrit 3 000 IU/0,3 ml injekce
Epoetinum alfa i.v./s.c.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
Binocrit 4 000 IU/0,4 ml injekční roztok v předplněné injekční stříkačce Epoetinum alfa
1 předplněná injekční stříkačka s obsahem 0,4 ml obsahuje 4 000 mezinárodních jednotek (IU), což odpovídá 33,6 mikrogramům epoetinum alfa.
Pomocné látky: dihydrát dihydrogenfosforečnanu sodného, dihydrát hydrogenfosforečnanu sodného, chlorid sodný, glycin, polysorbát 80, kyselina chlorovodíková (pro úpravu pH), hydroxid sodný (pro úpravu pH) a voda na injekci.
Další informace naleznete v příbalové informaci.
Injekční roztok v předplněné injekční stříkačce.
1 předplněná injekční stříkačka s 0,4 ml 6 předplněných injekčních stříkaček s 0,4 ml
1 předplněná injekční stříkačka s 0,4 ml s bezpečnostním krytem jehly 6 předplněných injekčních stříkaček s 0,4 ml s bezpečnostním krytem jehly
Pro subkutánní a intravenózní podání.
Před použitím si přečtěte příbalovou informaci. Netřepejte.
Uchovávejte mimo dohled a dosah dětí.
Uchovávejte a převážejte chlazené (2°C-8°C).
Chraňte před mrazem.
Předplněné stříkačky uchovávejte ve vnějším obalu (v krabičce), aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Sandoz GmbH, Biochemiestr. 10, A-6250 Kundl, Rakousko
EU/1/07/410/007
EU/1/07/410/008
EU/1/07/410/033
EU/1/07/410/034
č.š.:
Binocrit 4 000 IU/0,4 ml
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Binocrit 4 000 IU/0,4 ml injekce
Epoetinum alfa i.v./s.c.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
Binocrit 5 000 IU/0,5 ml injekční roztok v předplněné injekční stříkačce Epoetinum alfa
1 předplněná injekční stříkačka s obsahem 0,5 ml obsahuje 5 000 mezinárodních jednotek (IU), což odpovídá 42,0 mikrogramům epoetinum alfa.
Pomocné látky: dihydrát dihydrogenfosforečnanu sodného, dihydrát hydrogenfosforečnanu sodného, chlorid sodný, glycin, polysorbát 80, kyselina chlorovodíková (pro úpravu pH), hydroxid sodný (pro úpravu pH) a voda na injekci.
Další informace naleznete v příbalové informaci.
Injekční roztok v předplněné injekční stříkačce.
1 předplněná injekční stříkačka s 0,5 ml 6 předplněných injekčních stříkaček s 0,5 ml
1 předplněná injekční stříkačka s 0,5 ml s bezpečnostním krytem jehly 6 předplněných injekčních stříkaček s 0,5 ml s bezpečnostním krytem jehly
Pro subkutánní a intravenózní podání.
Před použitím si přečtěte příbalovou informaci. Netřepejte.
Uchovávejte mimo dohled a dosah dětí.
Uchovávejte a převážejte chlazené (2°C-8°C).
Chraňte před mrazem.
Předplněné stříkačky uchovávejte ve vnějším obalu (v krabičce), aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Sandoz GmbH, Biochemiestr. 10, A-6250 Kundl, Rakousko
EU/1/07/410/009
EU/1/07/410/010
EU/1/07/410/035
EU/1/07/410/036
č.š.:
Binocrit 5 000 IU/0,5 ml
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Binocrit 5 000 IU/0,5 ml injekce
Epoetinum alfa i.v./s.c.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
Binocrit 6 000 IU/0,6 ml injekční roztok v předplněné injekční stříkačce Epoetinum alfa
1 předplněná injekční stříkačka s obsahem 0,6 ml obsahuje 6 000 mezinárodních jednotek (IU), což odpovídá 50,4 mikrogramům epoetinum alfa.
Pomocné látky: dihydrát dihydrogenfosforečnanu sodného, dihydrát hydrogenfosforečnanu sodného, chlorid sodný, glycin, polysorbát 80, kyselina chlorovodíková (pro úpravu pH), hydroxid sodný (pro úpravu pH) a voda na injekci.
Další informace naleznete v příbalové informaci.
Injekční roztok v předplněné injekční stříkačce.
1 předplněná injekční stříkačka s 0,6 ml 6 předplněných injekčních stříkaček s 0,6 ml
1 předplněná injekční stříkačka s 0,6 ml s bezpečnostním krytem jehly 6 předplněných injekčních stříkaček s 0,6 ml s bezpečnostním krytem jehly
Pro subkutánní a intravenózní podání.
Před použitím si přečtěte příbalovou informaci. Netřepejte.
Uchovávejte mimo dohled a dosah dětí.
Uchovávejte a převážejte chlazené (2°C-8°C).
Chraňte před mrazem.
Předplněné stříkačky uchovávejte ve vnějším obalu (v krabičce), aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Sandoz GmbH, Biochemiestr. 10, A-6250 Kundl, Rakousko
EU/1/07/410/011
EU/1/07/410/012
EU/1/07/410/037
EU/1/07/410/038
č.š.:
Binocrit 6 000 IU/0,6 ml
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Binocrit 6 000 IU/0,6 ml injekce
Epoetinum alfa i.v./s.c.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
Binocrit 7 000 IU/0,7 ml injekční roztok v předplněné injekční stříkačce Epoetinum alfa
1 předplněná injekční stříkačka s obsahem 0,7 ml obsahuje 7 000 mezinárodních jednotek (IU), což odpovídá 58,8 mikrogramům epoetinum alfa.
Pomocné látky: dihydrát dihydrogenfosforečnanu sodného, dihydrát hydrogenfosforečnanu sodného, chlorid sodný, glycin, polysorbát 80, kyselina chlorovodíková (pro úpravu pH), hydroxid sodný (pro úpravu pH) a voda na injekci.
Další informace naleznete v příbalové informaci.
Injekční roztok v předplněné injekční stříkačce.
1 předplněná injekční stříkačka s 0,7 ml 6 předplněných injekčních stříkaček s 0,7 ml
1 předplněná injekční stříkačka s 0,7 ml s bezpečnostním krytem jehly 6 předplněných injekčních stříkaček s 0,7 ml s bezpečnostním krytem jehly
Pro subkutánní a intravenózní podání.
Před použitím si přečtěte příbalovou informaci. Netřepejte.
Uchovávejte mimo dohled a dosah dětí.
Uchovávejte a převážejte chlazené (2°C-8°C).
Chraňte před mrazem.
Předplněné stříkačky uchovávejte ve vnějším obalu (v krabičce), aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Sandoz GmbH, Biochemiestr. 10, A-6250 Kundl, Rakousko
EU/1/07/410/017
EU/1/07/410/018
EU/1/07/410/039
EU/1/07/410/040
č.š.:
Binocrit 7 000 IU/0,7 ml
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Binocrit 7 000 IU/0,7 ml injekce
Epoetinum alfa i.v./s.c.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
Binocrit 8 000 IU/0,8 ml injekční roztok v předplněné injekční stříkačce Epoetinum alfa
1 předplněná injekční stříkačka s obsahem 0,8 ml obsahuje 8 000 mezinárodních jednotek (IU), což odpovídá 67,2 mikrogramům epoetinum alfa.
Pomocné látky: dihydrát dihydrogenfosforečnanu sodného, dihydrát hydrogenfosforečnanu sodného, chlorid sodný, glycin, polysorbát 80, kyselina chlorovodíková (pro úpravu pH), hydroxid sodný (pro úpravu pH) a voda na injekci.
Další informace naleznete v příbalové informaci.
Injekční roztok v předplněné injekční stříkačce.
1 předplněná injekční stříkačka s 0,8 ml 6 předplněných injekčních stříkaček s 0,8 ml
1 předplněná injekční stříkačka s 0,8 ml s bezpečnostním krytem jehly 6 předplněných injekčních stříkaček s 0,8 ml s bezpečnostním krytem jehly
Pro subkutánní a intravenózní podání.
Před použitím si přečtěte příbalovou informaci. Netřepejte.
Uchovávejte mimo dohled a dosah dětí.
Uchovávejte a převážejte chlazené (2°C-8°C).
Chraňte před mrazem.
Předplněné stříkačky uchovávejte ve vnějším obalu (v krabičce), aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Sandoz GmbH, Biochemiestr. 10, A-6250 Kundl, Rakousko
EU/1/07/410/013
EU/1/07/410/014
EU/1/07/410/041
EU/1/07/410/042
č.š.:
Binocrit 8 000 IU/0,8 ml
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Binocrit 8 000 IU/0,8 ml injekce
Epoetinum alfa i.v./s.c.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
Binocrit 9 000 IU/0,9 ml injekční roztok v předplněné injekční stříkačce Epoetinum alfa
1 předplněná injekční stříkačka s obsahem 0,9 ml obsahuje 9 000 mezinárodních jednotek (IU), což odpovídá 75,6 mikrogramům epoetinum alfa.
Pomocné látky: dihydrát dihydrogenfosforečnanu sodného, dihydrát hydrogenfosforečnanu sodného, chlorid sodný, glycin, polysorbát 80, kyselina chlorovodíková (pro úpravu pH), hydroxid sodný (pro úpravu pH) a voda na injekci.
Další informace naleznete v příbalové informaci.
Injekční roztok v předplněné injekční stříkačce.
1 předplněná injekční stříkačka s 0,9 ml 6 předplněných injekčních stříkaček s 0,9 ml
1 předplněná injekční stříkačka s 0,9 ml s bezpečnostním krytem jehly 6 předplněných injekčních stříkaček s 0,9 ml s bezpečnostním krytem jehly
Pro subkutánní a intravenózní podání.
Před použitím si přečtěte příbalovou informaci. Netřepejte.
Uchovávejte mimo dohled a dosah dětí.
Uchovávejte a převážejte chlazené (2°C-8°C).
Chraňte před mrazem.
Předplněné stříkačky uchovávejte ve vnějším obalu (v krabičce), aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Sandoz GmbH, Biochemiestr. 10, A-6250 Kundl, Rakousko
EU/1/07/410/019
EU/1/07/410/020
EU/1/07/410/043
EU/1/07/410/044
č.š.:
Binocrit 9 000 IU/0,9 ml
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Binocrit 9 000 IU/0,9 ml injekce
Epoetinum alfa i.v./s.c.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
Binocrit 10 000 IU/1 ml injekční roztok v předplněné injekční stříkačce Epoetinum alfa
1 předplněná injekční stříkačka s obsahem 1 ml obsahuje 10 000 mezinárodních jednotek (IU), což odpovídá 84,0 mikrogramům epoetinum alfa.
Pomocné látky: dihydrát dihydrogenfosforečnanu sodného, dihydrát hydrogenfosforečnanu sodného, chlorid sodný, glycin, polysorbát 80, kyselina chlorovodíková (pro úpravu pH), hydroxid sodný (pro úpravu pH) a voda na injekci.
Další informace naleznete v příbalové informaci.
Injekční roztok v předplněné injekční stříkačce.
1 předplněná injekční stříkačka s 1 ml 6 předplněných injekčních stříkaček s 1 ml
1 předplněná injekční stříkačka s 1 ml s bezpečnostním krytem jehly 6 předplněných injekčních stříkaček s 1 ml s bezpečnostním krytem jehly
Pro subkutánní a intravenózní podání.
Před použitím si přečtěte příbalovou informaci. Netřepejte.
Uchovávejte mimo dohled a dosah dětí.
Uchovávejte a převážejte chlazené (2°C-8°C).
Chraňte před mrazem.
Předplněné stříkačky uchovávejte ve vnějším obalu (v krabičce), aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Sandoz GmbH, Biochemiestr. 10, A-6250 Kundl, Rakousko
EU/1/07/410/015
EU/1/07/410/016
EU/1/07/410/045
EU/1/07/410/046
č.š.:
Binocrit 10 000 IU/1 ml
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Binocrit 10 000 IU/1 ml injekce
Epoetinum alfa i.v./s.c.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
Binocrit 20 000 IU/0,5 ml injekční roztok v předplněné injekční stříkačce Epoetinum alfa
1 předplněná injekční stříkačka s obsahem 0,5 ml obsahuje 20 000 mezinárodních jednotek (IU), což odpovídá 168,0 mikrogramům epoetinum alfa.
Pomocné látky: dihydrát dihydrogenfosforečnanu sodného, dihydrát hydrogenfosforečnanu sodného, chlorid sodný, glycin, polysorbát 80, kyselina chlorovodíková (pro úpravu pH), hydroxid sodný (pro úpravu pH) a voda na injekci.
Další informace naleznete v příbalové informaci.
Injekční roztok v předplněné injekční stříkačce.
1 předplněná injekční stříkačka s 0,5 ml 6 předplněných injekčních stříkaček s 0,5 ml
1 předplněná injekční stříkačka s 0,5 ml s bezpečnostním krytem jehly 4 předplněných injekčních stříkaček s 0,5 ml s bezpečnostním krytem jehly 6 předplněných injekčních stříkaček s 0,5 ml s bezpečnostním krytem jehly
Pro subkutánní a intravenózní podání.
Před použitím si přečtěte příbalovou informaci. Netřepejte.
Uchovávejte mimo dohled a dosah dětí.
Použitelné do:
Uchovávejte a převážejte chlazené (2°C-8°C).
Chraňte před mrazem.
Předplněné stříkačky uchovávejte ve vnějším obalu (v krabičce), aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
Sandoz GmbH, Biochemiestr. 10, A-6250 Kundl, Rakousko
EU/1/07/410/021
EU/1/07/410/022
EU/1/07/410/047
EU/1/07/410/053
EU/1/07/410/048
č.š.:
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Binocrit 20 000 IU/0,5 ml injekce
Epoetinum alfa i.v./s.c.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
Binocrit 30 000 IU/0,75 ml injekční roztok v předplněné injekční stříkačce Epoetinum alfa
1 předplněná injekční stříkačka s obsahem 0,75 ml obsahuje 30 000 mezinárodních jednotek (IU), což odpovídá 252,0 mikrogramům epoetinum alfa.
Pomocné látky: dihydrát dihydrogenfosforečnanu sodného, dihydrát hydrogenfosforečnanu sodného, chlorid sodný, glycin, polysorbát 80, kyselina chlorovodíková (pro úpravu pH), hydroxid sodný (pro úpravu pH) a voda na injekci.
Další informace naleznete v příbalové informaci.
Injekční roztok v předplněné injekční stříkačce.
1 předplněná injekční stříkačka s 0,75 ml 6 předplněných injekčních stříkaček s 0,75 ml
1 předplněná injekční stříkačka s 0,75 ml s bezpečnostním krytem jehly 4 předplněných injekčních stříkaček s 0,75 ml s bezpečnostním krytem jehly 6 předplněných injekčních stříkaček s 0,75 ml s bezpečnostním krytem jehly
Pro subkutánní a intravenózní podání.
Před použitím si přečtěte příbalovou informaci. Netřepejte.
Uchovávejte mimo dohled a dosah dětí.
Použitelné do:
Uchovávejte a převážejte chlazené (2°C-8°C).
Chraňte před mrazem.
Předplněné stříkačky uchovávejte ve vnějším obalu (v krabičce), aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
Sandoz GmbH, Biochemiestr. 10, A-6250 Kundl, Rakousko
EU/1/07/410/023
EU/1/07/410/024
EU/1/07/410/049
EU/1/07/410/054
EU/1/07/410/050
č.š.:
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Binocrit 30 000 IU/0,75 ml injekce
Epoetinum alfa i.v./s.c.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
Binocrit 40 000 IU/1 ml injekční roztok v předplněné injekční stříkačce Epoetinum alfa
1 předplněná injekční stříkačka s obsahem 1 ml obsahuje 40 000 mezinárodních jednotek (IU), což odpovídá 336,0 mikrogramům epoetinum alfa.
Pomocné látky: dihydrát dihydrogenfosforečnanu sodného, dihydrát hydrogenfosforečnanu sodného, chlorid sodný, glycin, polysorbát 80, kyselina chlorovodíková (pro úpravu pH), hydroxid sodný (pro úpravu pH) a voda na injekci.
Další informace naleznete v příbalové informaci.
Injekční roztok v předplněné injekční stříkačce.
1 předplněná injekční stříkačka s 1 ml 6 předplněných injekčních stříkaček s 1 ml
1 předplněná injekční stříkačka s 1 ml s bezpečnostním krytem jehly 4 předplněných injekčních stříkaček s 1 ml s bezpečnostním krytem jehly 6 předplněných injekčních stříkaček s 1 ml s bezpečnostním krytem jehly
Pro subkutánní a intravenózní podání.
Před použitím si přečtěte příbalovou informaci. Netřepejte.
Uchovávejte mimo dohled a dosah dětí.
Použitelné do:
Uchovávejte a převážejte chlazené (2°C-8°C).
Chraňte před mrazem.
Předplněné stříkačky uchovávejte ve vnějším obalu (v krabičce), aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
Sandoz GmbH, Biochemiestr. 10, A-6250 Kundl, Rakousko
EU/1/07/410/025
EU/1/07/410/026
EU/1/07/410/051
EU/1/07/410/055
EU/1/07/410/052
č.š.:
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Binocrit 40 000 IU/1 ml injekce
Epoetinum alfa i.v./s.c.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro pacienta Binocrit 1 000 IU/0,5 ml injekční roztok v předplněné injekční stříkačce Binocrit 2 000 IU/1 ml injekční roztok v předplněné injekční stříkačce Binocrit 3 000 IU/0,3 ml injekční roztok v předplněné injekční stříkačce Binocrit 4 000 IU/0,4 ml injekční roztok v předplněné injekční stříkačce Binocrit 5 000 IU/0,5 ml injekční roztok v předplněné injekční stříkačce Binocrit 6 000 IU/0,6 ml injekční roztok v předplněné injekční stříkačce Binocrit 7 000 IU/0,7 ml injekční roztok v předplněné injekční stříkačce Binocrit 8 000 IU/0,8 ml injekční roztok v předplněné injekční stříkačce Binocrit 9 000 IU/0,9 ml injekční roztok v předplněné injekční stříkačce Binocrit 10 000 IU/1 ml injekční roztok v předplněné injekční stříkačce Binocrit 20 000 IU/0,5 ml injekční roztok v předplněné injekční stříkačce Binocrit 30 000 IU/0,75 ml injekční roztok v předplněné injekční stříkačce
Binocrit 40 000 IU/1 ml injekční roztok v předplněné injekční stříkačce
Epoetinum alfa
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Binocrit a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Binocrit používat
3. Jak se přípravek Binocrit používá
4. Možné nežádoucí účinky
5. Jak přípravek Binocrit uchovávat
6. Obsah balení a další informace
1. Co je přípravek Binocrit a k čemu se používá
Binocrit obsahuje léčivou látku epoetin alfa, bílkovinu, která povzbuzuje kostní dřeň ke zvýšené tvorbě červených krvinek, které obsahují hemoglobin (látku, která zajišťuje přenos kyslíku). Epoetin alfa je kopií lidské bílkoviny erytropoetinu a účinkuje stejným způsobem.
Binocrit je určen k léčbě příznaků anémie způsobené ledvinovým onemocněním:
• u dětí podstupujících hemodialýzu,
• u dospělých podstupujících hemodialýzu nebo peritoneální dialýzu,
• u dospělých trpících závažnou anémií, kteří dosud nepodstupují dialýzu.
Jestliže trpíte ledvinovým onemocněním, je možné, že máte nedostatek červených krvinek, protože Vaše ledviny netvoří dostatek erytropoetinu (nutného k tvorbě červených krvinek). Přípravek Binocrit je předepisován k povzbuzení kostní dřeně ke zvýšené tvorbě červených krvinek.
Binocrit je určen k léčbě anémie, pokud podstupujete chemoterapii k léčbě solidních nádorů, maligního lymfomu nebo mnohočetného lymfomu (nádor kostní dřeně) a Váš lékař se rozhodne, že je pro Vás nutná krevní transfuze. Binocrit může snížit potřebu krevní transfuze.
Binocrit se používá u osob se středně závažnou anémií, kteří darují krev před chirurgickým výkonem, aby jim mohla být vrácena během nebo po operaci. Protože Binocrit stimuluje tvorbu červených krvinek, mohou lékaři odebrat od těchto lidí více krve.
Binocrit se používá u dospělých se středně závažnou formou anémie, kteří mají podstoupit velkou ortopedickou operaci (např. náhradu kyčelního nebo kolenního kloubu) ke snížení případné potřeby krevních transfuzí.
2. Čemu musíte věnovat pozornost, než začnete přípravek Binocrit používat Nepoužívejte přípravek Binocrit:
• jestliže jste alergický(á) na epoetin alfa nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
• pokud u Vás byla zjištěna čistá aplazie červené krevní řady (kostní dřeň nevytváří dostatek červených krvinek) po předchozí léčbě jakýmkoli přípravkem, který povzbuzuje tvorbu červených krvinek (včetně přípravku Binocrit). Viz bod 4.
• jestliže trpíte vysokým krevním tlakem, který není náležitě léčen léčivými přípravky.
• ke zvýšení tvorby červených krvinek (aby Vám lékař mohl odebrat větší množství krve), pokud nemůžete podstoupit transfuzi vlastní krve během operace nebo po operaci.
• jestliže máte podstoupit plánovanou velkou elektivní ortopedickou operaci (např. operaci kyčlí nebo kolen) a jestliže:
• trpíte závažným srdečním onemocněním,
• trpíte závažnými žilními a tepennými poruchami,
• jste nedávno prodělal(a) srdeční infarkt nebo cévní mozkovou příhodu,
• nemůžete užívat léky na ředění krve.
Binocrit nemusí být pro Vás vhodný. Poraďte se se svým lékařem. Při léčbě přípravkem Binocrit potřebují někteří lidé užívat léky snižující riziko tvorby krevních sraženin. Pokud nemůžete užívat léky zabraňující tvorbě krevních sraženin, nesmíte používat Binocrit.
Upozornění a opatření
Před použitím přípravku Binocrit se poraďte se svým lékařem, lékarníkem nebo zdravotní sestrou.
Binocrit a další přípravky, které stimulují produkci červených krvinek, mohou zvyšovat riziko vzniku krevních sraženin u všech pacientů. Toto riziko může být vyšší, pokud jsou u Vás přítomny jiné rizikové faktory tvorby krevních sraženin(například pokud jste měl(a) krevní sraženinu v minulosti, při nadměrné tělesné hmotnosti, cukrovce, srdečním onemocnění nebo jste-li dlouho upoután(a) na lůžko z důvodu operace nebo nemoci). Prosím, informujte svého lékaře o kterémkoli z těchto stavů. Váš lékař Vám pomůže s rozhodnutím, zda je pro Vás přípravek Binocrit vhodný.
Pokud se Vás týkají některé z následujících skutečností, je důležité o nich informovat lékaře. Je možné, že Vaše léčba přípravkem Binocrit může nadále pokračovat, ale poraďte se nejdříve se svým lékařem.
Pokud trpíte nebo jste trpěl(a):
• vysokým krevním tlakem;
• epileptickými záchvaty nebo křečemi;
onemocnění jater; anémií z jiných příčin; porfyrií (vzácná porucha krve).
V případě, že trpíte nádorovým onemocněním, je zapotřebí si být vědom(a), že přípravky, které povzbuzují tvorbu červených krvinek, jako přípravek Binocrit, mohou působit jako růstový faktor, a proto teoreticky mohou ovlivnit růst nádoru.
V závislosti na Vašem osobním stavu může být vhodnější krevní transfuze. Poraďte se o tom, prosím, se svým lékařem.
Jestliže jste pacient trpící hepatitidou C a dostáváte interferon a ribavirin, měl(a) byste se poradit se svým lékařem, protože kombinace epoetinu alfa s interferonem a ribavirinem vedla ve vzácných případech ke ztrátě účinku a vzniku stavu nazývaného jako čistá aplazie červené krevní řady (PRCA), což je závažná forma anémie. Přípravek Binocrit není schválen k léčbě anémie související s hepatitidou C.
Jestliže jste pacient/ka s chronickým selháním ledvin a zejména pokud neodpovídáte dostatečně na podávání přípravku Binocrit, Váš lékař Vaši dávku přípravku Binocrit zkontroluje, protože opakované zvyšování dávky přípravku Binocrit při nepřítomnosti odpovědi na léčbu by mohlo zvýšit riziko problémů se srdcem nebo krevními cévami a mohlo by vést ke zvýšení rizika infarktu myokardu, cévní mozkové příhody a úmrtí.
Je zapotřebí zvláštní opatrnosti při použití jiných přípravků, které stimulují tvorbu červených krvinek:
Binocrit patří do skupiny přípravků, které stimulují tvorbu červených krvinek, stejně jako je tomu u lidského proteinu erytropoetinu. Váš lékař si vždy poznamená, který přípravek přesně užíváte.
Jestliže je Vám během léčby podáván jiný přípravek z této skupiny než Binocrit, než jej užijete, sdělte to svému lékaři nebo lékárníkovi.
Další léčivé přípravky a přípravek Binocrit
Přípravek Binocrit obvykle neovlivňuje účinek jiných léků, informujte však svého lékaře o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat, a to i o lécích, které jsou dostupné bez lékařského předpisu.
Pokud užíváte lék obsahující léčivou látku cyklosporin (užívaný např. po transplantaci ledvin), lékař může v průběhu léčby přípravkem Binocrit požadovat vyšetření krve ke zjištění hladiny cyklosporinu v krvi.
Přípravky doplňující železo nebo podporující krvetvorbu mohou zvýšit účinek přípravku Binocrit. O jejich užívání rozhodne lékař.
Při návštěvě nemocnice, polikliniky nebo praktického lékaře sdělte, že jste léčen(a) přípravkem Binocrit. Může to mít vliv na jinou léčbu nebo výsledky testů.
Těhotenství a kojení
Pokud se Vás cokoli z níže uvedeného týká, je důležité o tom informovat Vašeho lékaře. Možná budete moci používat Binocrit, ale poraďte se o tom předem se svým lékařem:
• jestliže jste těhotná nebo se domníváte, že můžete být těhotná.
• jestliže kojíte.
Přípravek Binocrit obsahuje sodík
Binocrit obsahuje méně než 1 mmol sodíku (23 mg) v jedné dávce, tj. v podstatě je „bez sodíku“.
Vždy používejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem.
Váš lékař provedl vyšetření krve a rozhodl, že potřebujete přípravek Binocrit.
Binocrit může být podán injekčně:
• buď do žíly, nebo hadičky vedoucí do žíly (intravenózně)
• nebo pod kůži (subkutánně).
Váš lékař rozhodne, jak Vám bude Binocrit podáván. Obvykle Vám budou injekce podávány lékařem, zdravotní sestrou nebo jiným zdravotníkem. Někteří lidé, v závislosti na důvodu, proč Binocrit potřebují, se mohou později naučit podávat si podkožní injekci sami: viz Pokyny, jak si podávat Binocrit samostatně.
Binocrit nesmí být používán:
• po datu použitelnosti uvedeném na štítku nebo vnější krabičce
• jestliže víte nebo se domníváte, že mohl být omylem vystaven působení mrazu, nebo
• jestliže došlo k selhání chladničky.
Dávka přípravku Binocrit je založena na Vaší tělesné hmotnosti v kilogramech. Příčina anémie je rovněž faktorem, podle kterého lékař stanoví správnou dávku.
V průběhu léčby přípravkem Binocrit Vám bude pravidelně kontrolován krevní tlak.
Osoby s ledvinovým onemocněním
• Váš lékař bude udržovat koncentraci hemoglobinu ve Vaší krvi mezi 10 a 12 g/dl, protože vysoká koncentrace hemoglobinu může zvýšit riziko tvorby krevních sraženin a úmrtí.
• Obvyklá úvodní dávka přípravku Binocrit činí u dospělých i dětí 50 mezinárodních jednotek (IU) na kilogram (/kg) tělesné hmotnosti podávaných třikrát týdně. U nemocných podstupujících peritoneální dialýzu se Binocrit může podávat dvakrát týdně.
• Dospělým i dětem se Binocrit podává injekcí buď do žíly (intravenózně), nebo hadičkou vedoucí do žíly. Pokud tento přístup (žilou nebo hadičkou) není k dispozici, lékař může rozhodnout, že se přípravek Binocrit má podávat pod kůži (subkutánně). To platí pro dialyzované pacienty a pro pacienty, kteří dialýzu dosud nepodstoupili.
• Váš lékař zajistí pravidelné vyšetření krve, aby bylo jisté, že lék stále správně působí, a může upravovat dávku, obvykle ne častěji než každý čtvrtý týden.
• Jakmile se anémie zlepší, lékař u Vás bude pokračovat v pravidelných kontrolách krve. Může Vám dále upravit Vaši dávku přípravku Binocrit a četnost podávání léku tak, aby zůstala zachována odpověď na léčbu. Váš lékař použije nejnižší účinnou dávku umožňující kontrolu příznaků anémie.
• Pokud neodpovídáte dostatečně na podávání přípravku Binocrit, Váš lékař Vaši dávku zkontroluje a informuje Vás, jestliže bude potřeba dávkování přípravku Binocrit změnit.
• Jestliže máte delší dávkovací interval podávání přípravku Binocrit (delší než jeden týden), nemusíte si udržet adekvátní hladiny hemoglobinu a můžete potřebovat zvýšení dávky přípravku Binocrit nebo jeho častější podávání.
• K zajištění vyšší účinnosti můžete před léčbou a v průběhu léčby přípravkem Binocrit dostávat doplňky železa.
• Pokud podstupujete dialýzu v době zahájení léčby přípravkem Binocrit, může být zapotřebí upravit dialyzační režim. O tom rozhodne lékař.
Dospělí podstupující chemoterapii
• Váš lékař může u Vás zahájit léčbu přípravkem Binocrit, jestliže máte koncentraci hemoglobinu v krvi 10 g/dl nebo nižší.
• Váš lékař bude udržovat koncentraci hemoglobinu ve Vaší krvi mezi 10 a 12 g/dl, protože vysoká koncentrace hemoglobinu může zvýšit riziko tvorby krevních sraženin a úmrtí.
• Obvyklá úvodní dávka činí buď 150 IU/kg tělesné hmotnosti třikrát týdně, nebo 450 IU/kg tělesné hmotnosti jednou týdně.
• Binocrit se podává injekcí pod kůži.
• Váš lékař zajistí vyšetření krve a může upravovat dávku podle odpovědi anémie na léčbu přípravkem Binocrit.
• K zajištění vyšší účinnosti léčby můžete před léčbou a v průběhu léčby přípravkem Binocrit dostávat doplňky železa.
• Léčba přípravkem Binocrit obvykle pokračuje ještě jeden měsíc po ukončení chemoterapie. Dospělí v programu odběrů a shromažďování vlastní krve
• Obvyklá dávka činí 600 IU/kg tělesné hmotnosti podávaných dvakrát týdně.
• Binocrit se podává injekcí do žíly ihned po odběru krve po dobu 3 týdnů před operací.
• K zajištění vyšší účinnosti léčby můžete před léčbou a v průběhu léčby přípravkem Binocrit dostávat doplňky železa.
Dospělí před plánovanou velkou ortopedickou operací
• Doporučená dávka přípravku je 600 IU/kg podkožně jednou týdně.
• Binocrit se podává injekcí pod kůži každý týden po dobu tří týdnů před operací a v den operace.
• Při potřebě zkrátit z léčebných důvodů předoperační období činí obvyklá denní dávka 300 IU/kg deset dnů před operací, v den operace a po čtyři dny bezprostředně po operaci.
• Pokud vyšetření krve před operací prokáží příliš vysokou hodnotu hemoglobinu (krevního barviva), bude léčba ukončena.
• K zajištění vyšší účinnosti léčby můžete před léčbou a v průběhu léčby přípravkem Binocrit dostávat doplňky železa.
Pokyny k podání přípravku Binocrit pod kůži
Na začátku léčby obvykle podává injekce přípravku Binocrit zdravotnický pracovník nebo sestra. Později může lékař navrhnout, že buď Vy sám (sama), nebo osoba, která o Vás pečuje, se můžete naučit podávat injekce přípravku Binocrit pod kůži (subkutánně).
• Bez vyškolení lékařem nebo zdravotní sestrou si nezkoušejte podávat injekci samostatně.
• Binocrit podávejte vždy přesně dle pokynů lékaře nebo zdravotní sestry.
• Ujistěte se, že vždy podáváte pouze množství roztoku odpovídající pokynům lékaře nebo zdravotní sestry.
• Binocrit používejte, pouze pokud byl správně uchováván - viz bod 5, Jak přípravek Binocrit uchovávat.
• Před použitím nechte stříkačky přípravku Binocrit stát do dosažení pokojové teploty. To obvykle trvá 15 až 30 minut. Přípravek použijte do 3 dnů od vyjmutí z chladničky.
Z každé předplněné injekční stříkačky přípravku Binocrit použijte vždy pouze jednu dávku.
Jestliže je Binocrit podáván injekčně pod kůži (subkutánně), objem podaný do jednoho místa by neměl běžně překročit 1 ml pro jednorázovou injekci.
Binocrit se podává samostatně, nikoli spolu s dalšími injekčními roztoky.
Injekční stříkačky s přípravkem Binocrit neprotřepávejte. Dlouhé silné třepání může přípravek poškodit. Pokud byl přípravek silně protřepáván, nepoužívejte jej.
Instrukce, jak si sám (sama) podáte injekci přípravku Binocrit, naleznete na konci této příbalové informace.
Pokud se domníváte, že dávka přípravku Binocrit byla příliš vysoká, informujte neprodleně lékaře nebo zdravotní sestru. Nežádoucí účinky při předávkování jsou nepravděpodobné.
Jestliže jste zapomněl(a) použít přípravek Binocrit
Podejte příští injekci, co nejdříve si vzpomenete. Pokud zbývá do Vaší příští injekce jeden den, zapomenutou injekci vynechejte a pokračujte v původním plánu pro dávkování. Dávku nezdvojnásobujte.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře, zdravotní sestry nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí
vyskytnout u každého.
Jestliže zaznamenáte kterýkoli účinek v seznamu níže, okamžitě informujte svého lékaře nebo
zdravotní sestru.
Velmi časté nežádoucí účinky
Tyto nežádoucí účinky se mohou vyskytnout u více než 1 z 10 osob užívajících přípravek Binocrit.
• Průjem
• Žaludeční nevolnost
• Zvracení
• Horečka
• Překrvení sliznice dýchacích cest, například ucpaný nos nebo bolesti v krku, byla hlášena u dosud nedialyzovaných pacientů s onemocněním ledvin.
Časté nežádoucí účinky
Tyto nežádoucí účinky se mohou vyskytnout až u 1 z 10 osob užívajících přípravek Binocrit.
• Zvýšený krevní tlak. Bolesti hlavy, zvláště náhlé a bodavé migrenózní bolesti hlavy, zmatenost nebo záchvaty křečí, mohou to být varovné známky náhlého vzestupu krevního tlaku, který vyžaduje neodkladnou léčbu. Zvýšený krevní tlak si může vyžádat nasazení dalších léčivých přípravků (anebo úpravu dávky přípravků, které už k léčbě vysokého krevního tlaku užíváte).
• Krevní sraženiny (včetně hluboké žilní trombózy a embolie), které mohou vyžadovat neodkladnou léčbu. Jako příznaky můžete mít bolest na hrudi, dýchavičnost a bolestivé otoky a zarudnutí, obvykle na nohou.
• Kašel.
• Kožní vyrážky, které mohou být důsledkem alergické reakce.
• Bolest kostí nebo svalů.
• Chřipce podobné příznaky, například bolesti hlavy a kloubů, slabost, zimnice, únava, závratě. Tyto příznaky se mohou často objevit na počátku léčby. Jestliže máte tyto příznaky během intravenózní injekce, je možné, že pomalé podání injekce do žíly pomůže odstranit tyto příznaky v budoucnu.
• Zarudnutí, pálení a bolest v místě vpichu injekce.
• Otoky kotníků, chodidel nebo prstů.
Méně časté nežádoucí účinky
Tyto nežádoucí účinky se mohou vyskytnout až u 1 z 100 osob užívajících přípravek Binocrit.
• Vysoké hladiny draslíku v krvi, které mohou způsobovat abnormální srdeční rytmus (velmi častý nežádoucí účinek u dialyzovaných pacientů).
• Záchvaty.
• Zduření sliznice nosu nebo dýchacích cest.
Velmi vzácné nežádoucí účinky
Tyto nežádoucí účinky se mohou vyskytnout až u 1 z 10 000 osob užívajících přípravek Binocrit.
• Příznaky čisté aplazie červené krevní řady (PRCA)
PRCA představuje neschopnost kostní dřeně vytvářet dostatek červených krvinek. PRCA způsobuje náhlou a závažnou anémii. Příznaky jsou:
• neobvyklá únava,
• pocity zmatení,
• dušnost.
PRCA byla velmi vzácně pozorována většinou u pacientů s onemocněním ledvin po několika měsících až letech léčby epoetinem alfa a dalších přípravků stimulujících tvorbu červených krvinek.
• Může se u Vás objevit zvýšený počet malých krevních buněk (nazývaných krevní destičky), které se za normálních okolností podílejí na tvorbě krevní sraženiny. Lékař tuto situaci zkontroluje.
Jestliže jste hemodialyzováni:
• Mohou se tvořit krevní sraženiny (trombóza) v arteriovenózní píštěli pro dialýzu. To je pravděpodobnější, pokud máte nízký krevní tlak, nebo když jsou v píštěli komplikace.
• Krevní sraženiny se mohou tvořit rovněž v hemodialyzačním systému. Lékař může rozhodnout, aby se během dialýzy zvýšily dávky heparinu.
Ihned informujte svého lékaře nebo zdravotní sestru, jestliže si všimnete kteréhokoli z těchto účinků nebo jestliže si během léčby přípravkem Binocrit všimnete jakýchkoli jiných účinků.
Pokud se kterýkoli z nežádoucích účinků vyskytne v závažné míře, nebo pokud si všimnete jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci, prosím, sdělte to svému lékaři, zdravotní sestře nebo lékárníkovi.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Binocrit uchovávat Uchovávejte tento přípravek mimo dohled a dosah dětí.
• Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce za slovy „Použitelné do“.
• Uchovávejte a převážejte chlazené (2°C-8°C).
• Binocrit můžete vyjmout z chladničky a uchovávat při pokojové teplotě (do 25°C) až 3 dny. Jakmile je stříkačka vyjmuta z chladničky a ohřála se na pokojovou teplotu (max. 25°C), musíte ji do 3 dní buď použít, nebo zlikvidovat.
• Chraňte před mrazem. Netřepejte.
• Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Nepoužívejte tento přípravek, pokud si všimnete
• že roztok byl nedopatřením zmražen nebo
• při poruše chladničky,
• pokud je tekutina zabarvena nebo obsahuje viditelné částice,
• je-li poškozena pečeť.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co přípravek Binocrit obsahuje
- Léčivou látkou je: epoetinum alfa (množství je uvedeno v tabulce níže).
- Dalšími složkami jsou: dihydrát dihydrogenfosforečnanu sodného, dihydrát hydrogenfosforečnanu sodného, chlorid sodný, glycin, polysorbát 80, kyselina chlorovodíková (k úpravě pH), hydroxid sodný (k úpravě pH) a voda na injekci.
Jak přípravek Binocrit vypadá a co obsahuje toto balení
Binocrit je dodáván jako čirý, bezbarvý injekční roztok v předplněné injekční stříkačce. Stříkačky jsou uzavřeny v blistrech.
|
Balení přípravku |
Odpovídající balení přípravku pro každou sílu - množství/objem |
Množství epoetinu alfa |
|
Předplněné injekční |
2 000 IU/ml: | |
|
stříkačky |
1 000 IU/0,5 ml |
8,4 mikrogramů |
|
2 000 IU/1 ml |
16,8 mikrogramů | |
|
10 000 IU/ml: | ||
|
3 000 IU/0,3 ml |
25,2 mikrogramů | |
|
4 000 IU/0,4 ml |
33,6 mikrogramů | |
|
5 000 IU/0,5 ml |
42,0 mikrogramů | |
|
6 000 IU/0,6 ml |
50,4 mikrogramů | |
|
7 000 IU/0,7 ml |
58,8 mikrogramů | |
|
8 000 IU/0,8 ml |
67,2 mikrogramů | |
|
9 000 IU/0,9 ml |
75,6 mikrogramů | |
|
10 000 IU/1 ml |
84,0 mikrogramů | |
|
40 000 IU/ml: | ||
|
20 000 IU/0,5 ml |
168,0 mikrogramů | |
|
30 000 IU/0,75 ml |
252,0 mikrogramů | |
|
40 000 IU/1 ml |
336,0 mikrogramů |
*Balení obsahuje 1, 4 nebo 6 předplněných injekčních stříkaček s bezpečnostním krytem jehly anebo bez něho.
Na trhu nemusí být k dispozici všechny velikosti balení.
Držitel rozhodnutí o registraci a výrobce
Sandoz GmbH Biochemiestr. 10 A-6250 Kundl Rakousko
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Lietuva
Belgie/Belgique/Belgien
Sandoz nv-sa Tél/Tel: +32 2 722 97 97
Sandoz Pharmaceuticals d.d filialas Tel: +370 5 2636 037
|
BtnrapHH TtproBCKO npegcTaBHTeacTBO CaHgo3 g.g. Ten.: +359 2 970 47 47 |
Luxembourg/Luxemburg Sandoz GmbH Tél/Tel: +43 5338 2000 |
|
Česká republika Sandoz s.r.o. Tel: +420 221 421 611 |
Magyarország Sandoz Hungária Kft. Tel.: +36 1 430 2890 |
|
Danmark/Ísland/Norge/Sverige Sandoz A/S Tlf/Sími/Tel: +45 63 95 10 00 |
Malta Sandoz GmbH Tel: +43 5338 2000 |
|
Deutschland Hexal AG Tel: +49 8024 908 0 |
Nederland Sandoz B.V. Tel: +31 36 52 41 600 |
|
Eesti Sandoz d.d. Eesti filiaal Tel: +372 6652 400 |
Osterreich Sandoz GmbH Tel: +43 5338 2000 |
|
EXXáSa Sandoz Pharmaceuticals d.d, Tn^: +30 216 600 5000 n DEMO Avróvu^og Bio^nxaviKq Kai E^nopiKq Exaipsía TnX: +30 210 816 18 02 |
Polska Sandoz Polska Sp. z o.o. Tel.: +48 22 209 70 00 |
|
Espaňa Sandoz Farmacéutica, S.A. Tel: +34 900 456 856 |
Portugal Sandoz Farmacéutica Lda. Tel: +351 21 924 19 11 |
|
France Sandoz SAS Tél: + 33 1 49 64 48 00 |
Románia S.C. Sandoz S.R.L. Tel: +40 265 208 120 |
|
Hrvatska Sandoz d.o.o. Tel: +385 1 23 53 111 |
Slovenija Lek farmacevtska družba d.d. Tel: +386 1 580 21 11 |
|
Ireland Novartis Ireland Limited Tel: + 353 1 260 12 55 |
Slovenská republika Sandoz d.d. - organizačná zložka Tel: +421 2 48 200 600 |
|
Italia Sandoz S.p.A. Tel: +39 02 96541 |
Suomi/Finland Sandoz A/S Puh/Tel: +358 10 6133 415 |
|
Kúrcpog P.T.Hadjigeorgiou co ltd TnX: +357 25372425 |
United Kingdom Sandoz Ltd Tel: + 44 1276 69 8020 |
Latvija
Sandoz d.d. Párstávnieciba Latvija Tel: +371 67 892 006
Tato příbalová informace byla naposledy revidována {MM/RRRR}.
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/.
Instrukce, jak si sám (sama) podáte injekci (pouze pro pacienty podstupující chemoterapii nebo dospělé pacienty před plánovanou ortopedickou operací)
Tento oddíl obsahuje informace, jak si sám (sama) podáte injekci přípravku Binocrit. Je důležité, abyste se nesnažil(a) si injekci sám (sama) podat, pokud to s Vámi Váš lékař nebo zdravotní sestra zvlášť nenatrénovali. Přípravek Binocrit je anebo není vybaven ochranným krytem jehly a Váš lékař nebo ošetřovatel/ka Vám ukáží, jak jej používat. Jestliže si nejste jistý(á), jak si injekci podávat, anebo máte-li jakékoli další otázky, prosím, požádejte svého lékaře anebo zdravotní sestru o pomoc.
1. Umyjte si ruce.
2. Vyjměte jednu stříkačku z balení a sejměte ochranný kryt z jehly. Injekční stříkačky mají výstupky s prstenci stupnice, aby bylo možné částečné použití v případě potřeby. Jeden prstenec stupnice odpovídá objemu 0,1 ml. Jestliže je vyžadováno pouze částečné použití injekce, odstraňte před aplikací nepotřebný roztok.
3. Očistěte kůži na místě vpichu alkoholovým tampónem.
4. Vytvořte kožní záhyb stisknutím kůže mezi palec a ukazováček.
5. Rychlým a rozhodným vbodnutím vpíchněte jehlu do záhybu. Vstříkněte roztok přípravku Binocrit podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
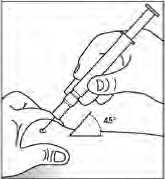
Předplněná stříkačka bez ochranného krytu injekční jehly
6. Stále držte kůži zřasenou mezi palcem a ukazováčkem a tlačte na píst pomalu a stejnoměrně.
7. Po injekci tekutiny vytáhněte injekční jehlu a uvolněte kůži. Na místo vpichu zatlačte suchým a sterilním tampónem.
8. Všechen nepoužitý přípravek nebo odpad zlikvidujte. Každou stříkačku použijte pouze pro jednu injekci.
Předplněná stříkačka s ochranným krytem injekční jehly
6.
7.
8. 9.
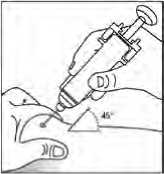
Stále držte kůži zřasenou mezi palcem a ukazováčkem a tlačte na píst pomalu a stejnoměrně, dokud jste nepodal(a) celou dávku a dokud se píst již nedá dále zatlačit. Neuvolňujte tlak na píst!
Po ukončené injekci tekutiny vytáhněte injekční jehlu za stálého tlaku na píst, a potom uvolněte kůži. Na místo vpichu zatlačte suchým a sterilním tampónem.
Uvolněte píst. Chránič injekční jehly se rychle vysune a přikryje injekční jehlu.
Všechen nepoužitý přípravek nebo odpad zlikvidujte. Každou stříkačku použijte pouze pro jednu injekci.
79