Benepali 50 Mg
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Benepali 50 mg, injekční roztok v předplněné injekční stříkačce. Benepali 50 mg, injekční roztok v předplněném peru.
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
50 mg, injekční roztok v předplněné injekční stříkačce
Jedna předplněná injekční stříkačka obsahuje etanerceptum 50 mg v celkovém objemu 1 ml.
50 mg, injekční roztok v předplněném peru
Jedno předplněné pero obsahuje etanerceptum 50 mg v celkovém objemu 1 ml.
Etanercept je fúzní protein složený z receptoru p 75 tumor nekrotizujícího faktoru a Fc proteinu, vyráběný technologií rekombinace DNA v expresním systému ovárií čínského křečka (CHO). Etanercept je dimer chimerického proteinu, vzniklého genetickým inženýrstvím, který vzniká spojením extracelulární vazebné domény lidského receptoru 2 tumor nekrotizujícího faktoru (TNFR2/p75) s Fc oblastí lidského IgG1. Tato Fc složka obsahuje pantovou oblast, CH2 a CH3 regiony ale nemá CH1 region IgG1. Etanercept obsahuje 934 aminokyselin a má molekulovou hmotnost přibližně 150 kilodaltonů. Specifická aktivita etanerceptu je 1,7 * 106 jednotek/mg.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok (injekce).
Roztok je čirý, bezbarvý nebo světle žlutý a jeho pH je nastaveno na 6,2 ± 0,3. Osmolalita roztoku je 325 ± 35 mOsm/kg.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Revmatoidní artritida
Benepali v kombinaci s methotrexátem je indikován k léčbě středně těžké až těžké aktivní revmatoidní artritidy u dospělých v případech, kdy po podání jiných chorobu modifikujících léků, včetně methotrexátu (pokud není kontraindikován), nebylo dosaženo adekvátní terapeutické odpovědi.
Benepali může být podáván jako monoterapie v případě intolerance methotrexátu nebo pokud je pokračující léčba methotrexátem nevhodná.
Benepali je také indikován k léčbě závažné aktivní a progresivní revmatoidní artritidy u dospělých, kteří nebyli předtím léčeni methotrexátem.
Benepali použitý samostatně nebo v kombinaci s methotrexátem prokázal rentgenologicky měřitelné snížení míry progrese poškození kloubů a fyzické funkční zlepšení.
Psoriatická artritida
Léčení aktivní a progresivní psoriatické artritidy u dospělých v případech, kdy po předchozím podání jiných chorobu modifikujících léků nebylo dosaženo adekvátní terapeutické odpovědi. Etanercept prokázal fyzické funkční zlepšení u pacientů s psoriatickou artritidou a rentgenologicky měřitelné snížení míry progrese poškození periferních kloubů u pacientů se symetrickými polyartikulárními podtypy choroby.
Axiální spondylartritidy
Ankylozující spondylitida
Léčení dospělých se závažnou aktivní ankylozující spondylitidou, jestliže nebylo dosaženo adekvátní odpovědi konvenční léčbou.
Radiograficky neprokazatelná axiální spondylartritida
Léčba dospělých se závažnou radiograficky neprokazatelnou axiální spondylartritidou s objektivními známkami zánětu indikovanými zvýšenou hladinou C-reaktivního proteinu (CRP) a/nebo prokázanými při zobrazení magnetickou rezonancí (MR), kteří měli nedostatečnou odpověď na nesteroidní antirevmatika (NSAID-nonsteroidal anti-inflammatory drugs).
Ložisková psoriáza
Léčení dospělých se středně těžkou až těžkou ložiskovou psoriázou, kteří neodpovídají na jinou celkovou terapii zahrnující cyklosporin, methotrexát nebo psoralen a ultrafialové světlo A (PUVA)
(viz bod 5.1), nebo tato terapie je pro ně kontraindikována, nebo ji netolerují.
4.2 Dávkování a způsob podání
Léčení přípravkem Benepali má být zahájeno a prováděno pod dohledem lékaře specializovaného na diagnostikování a léčbu revmatoidní artritidy, psoriatické artritidy, ankylozující spondylitidy, radiograficky neprokazatelné axiální spondylartritidy nebo ložiskové psoriázy. Pacienti léčení přípravkem Benepali mají dostat pohotovostní kartu pacienta.
Dávkování
Revmatoidní artritida
Doporučená dávka je 50 mg etanerceptu podaná jednou týdně (viz bod 5.1).
Psoriatická artritida, ankylozující spondylitida a radiograficky neprokazatelná axiální spondylartri tida
Doporučená dávka je 50 mg etanerceptu podaná jednou týdně.
Dostupné údaje naznačují, že u všech výše uvedených indikací je klinické odpovědi obvykle dosaženo v průběhu 12 týdnů léčby. Je třeba pečlivě zvážit pokračování léčby u pacienta, který na ni během této doby nevykazuje odpověď.
Ložisková psoriáza
Doporučená dávka je 50 mg etanerceptu podaná jednou týdně. Alternativně může být podáváno 50 mg 2x týdně až 12 týdnů a dále, je-li to nezbytné, dávka 50 mg jednou týdně. Léčení přípravkem Benepali má pokračovat až do dosažení remise, až po dobu 24 týdnů. U některých dospělých pacientů může být vhodné pokračování v terapii po 24 týdnech (viz bod 5.1). U pacientů, kteří nevykazují odpověď po 12 týdnech, má být léčení ukončeno. Je-li indikována opakovaná léčba přípravkem Benepali, má se dodržet výše uvedená délka léčení. Dávka má být 50 mg jednou týdně.
Zvláštní populace
Porucha funkce ledvin a jater Nevyžaduje se úprava dávek.
Starší osoby
Nevyžaduje se žádná úprava dávek. Dávkování a způsob podání jsou stejné jako u dospělých ve věku od 18 do 64 let.
Pediatrická populace
Přípravek Benepali není indikován k použití u dětí mladších 18 let. Přípravek Benepali je k dispozici pouze jako předplněná injekční stříkačka 50 mg a předplněné pero 50 mg, zatímco doporučená dávka etanerceptu pro pediatrické pacienty je 0,8 mg/kg podávaných jednou týdně v lyofilizovaném preparátu.
Pro děti jsou k dispozici jiné přípravky s obsahem etanerceptu s vhodnými lékovými formami pro děti. Způsob podání
Přípravek Benepali je podáván subkutánní injekcí (viz bod 6.6).
Úplný návod na přípravu a podání rozpuštěného přípravku Benepali je k dispozici v příbalové informaci, bod 7 „Návod k přípravě a podání“.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Sepse nebo riziko sepse.
Léčba přípravkem Benepali nesmí být zahájena u pacientů s aktivní infekcí včetně chronických a lokálních infekcí.
4.4 Zvláštní upozornění a opatření pro použití
V zájmu zlepšení vysledovatelnosti biologických léčivých přípravků je nutné, aby ochranná známka a číslo šarže podávaného přípravku byly jasně zaznamenány (nebo uvedeny) v dokumentaci pacienta.
Infekce
Pacienti mají být vyšetřeni na infekce před zahájením, v průběhu a po ukončení léčení přípravkem Benepali, přičemž je třeba brát v úvahu, že průměrný poločas vylučování etanerceptu je přibližně 70 hodin (rozmezí 7 -300 hodin).
Při používání etanerceptu byly pozorovány závažné infekce, sepse, tuberkulóza a oportunní infekce, včetně invazivních plísňových infekcí, listerióza a legionelóza (viz bod 4.8). Příčinou těchto infekcí byly bakterie, mykobakterie, plísně, viry a parazité (včetně protozoí). V některých případech, obzvláště u plísňových a dalších oportunních infekcí, nebyly tyto infekce rozpoznány, což vedlo k opožděnému nasazení příslušné léčby a někdy k úmrtí. Při vyšetření pacientů na přítomnost infekce je třeba brát v úvahu riziko pacienta vzhledem k významným oportunním infekcím (např. expozice endemickým mykózám).
Pacienty, u kterých se v průběhu podávání přípravku Benepali rozvine infekce, je třeba přísně monitorovat. Jestliže se u pacienta rozvíjí závažná infekce, musí být přípravek Benepali vysazen. Bezpečnost a účinnost etanerceptu nebyla u pacientů s chronickými infekcemi hodnocena. Lékaři mají pečlivě zvažovat podání přípravku Benepali pacientům, kteří mají v anamnéze opakované nebo chronické infekce nebo trpí stavy, které mohou vytvářet dispozici k infekcím, např. pokročilý nebo špatně kompenzovaný diabetes.
Tuberkulóza
U pacientů léčených etanerceptem byly hlášeny případy aktivní tuberkulózy včetně miliární a extrapulmonální tuberkulózy.
Před zahájením léčení přípravkem Benepali musí být všichni pacienti vyšetřeni na aktivní a inaktivní (latentní) tuberkulózu. Toto vyšetření by mělo obsahovat podrobnou lékařskou anamnézu a osobní anamnézu tuberkulózy, možný předchozí kontakt s tuberkulózou a předchozí a/nebo současnou imunosupresivní terapii. U všech pacientů musí být (v souladu s lokálními doporučeními) provedeny vhodné screeningové testy - tj. tuberkulinový kožní test a rentgen hrudníku. Doporučuje se zaznamenat výsledky těchto vyšetření do pohotovostní karty pacienta. Předepisující lékařům se připomíná riziko falešně negativních výsledků tuberkulinových testů, zvláště u pacientů těžce nemocných nebo se sníženou imunitou.
Při pozitivní diagnóze aktivní tuberkulózy se nesmí terapie přípravkem Benepali zahájit. Pokud je diagnostikována inaktivní (latentní) tuberkulóza, antituberkulózní léčba latentní tuberkulózy musí být zahájena před začátkem terapie přípravkem Benepali a v souladu s místními doporučeními. V takovém případě je třeba velmi pečlivě zhodnotit poměr přínosu/rizika terapie přípravkem Benepali.
Všechny pacienty je třeba informovat, že pokud se během nebo po ukončení léčby přípravkem Benepali objeví známky/příznaky naznačující tuberkulózu (např. přetrvávající kašel, chřadnutí/ztráta hmotnosti, zvýšená teplota), mají vyhledat lékařskou pomoc.
Reaktivace hepatitidy B
U pacientů, kteří byli v minulosti infikováni virem hepatitidy B (HBV) a léčeni souběžně antagonisty TNF včetně etanerceptu, byla hlášena reaktivace hepatitidy B. Patří sem i zprávy o reaktivaci hepatitidy B u pacientů, kteří byli anti-HBc pozitivní, avšak HBsAg negativní. Před zahájením léčby přípravkem Benepali se musí u pacientů provést test na infekci HBV. U pacientů s pozitivním výsledkem testu na infekci HBV se doporučuje konzultace s lékařem, který je odborníkem v léčbě hepatitidy B. Při podávání přípravku Benepali pacientům, kteří byli v minulosti infikováni HBV, je třeba opatrnosti. Tito pacienti musí být po celou dobu léčby a několik týdnů po jejím ukončení sledováni na známky a příznaky aktivní infekce HBV. Nejsou k dispozici dostatečné údaje o léčbě HBV infikovaných pacientů antivirovou terapií společně s terapií antagonisty TNF. U pacientů, u nichž dojde k rozvoji infekce HBV, by se měla léčba přípravkem Benepali ukončit a zahájit léčba účinnou antivirovou terapií spolu s vhodnou podpůrnou léčbou.
Zhoršení hepatitidy C
U pacientů léčených etanerceptem bylo hlášeno zhoršení hepatitidy C. Přípravek Benepali má být podáván s opatrností u pacientů s hepatitidou C v anamnéze.
Současná léčba s anakinrou
Současné podávání etanerceptu s anakinrou bylo spojeno se zvýšeným rizikem závažných infekcí a s neutropenií ve srovnání s podáváním samotného etanerceptu. Tato kombinace neprokázala zvýšení klinického prospěchu. Proto se současné podávání přípravku Benepali s anakinrou nedoporučuje (viz bod 4.5 a 4.8).
Současná léčba s abataceptem
V klinických studiích vedlo současné podávání abataceptu a etanerceptu ke zvýšené incidenci závažných nežádoucích příhod. Tato kombinace neprokázala lepší klinický benefit; takové použití se nedoporučuje (viz bod 4.5).
Alergické reakce
Často byly hlášeny alergické reakce spojené s podáváním etanerceptu. Alergické reakce zahrnovaly angioedém a kopřivku jakožto závažné reakce. Vyskytne-li se závažná alergická nebo anafylaktická reakce, léčení přípravkem Benepali musí být okamžitě přerušeno a má být zahájena adekvátní terapie.
Imunosuprese
Protože TNF ovlivňuje protizánětlivý proces a moduluje buněčnou imunitní odpověď, je možné, že antagonisté TNF včetně etanerceptu, ovlivní přirozenou ochranu proti infekcím a malignitám. Ve studii se 49 dospělými pacienty s revmatoidní artritidou léčenou etanerceptem nebylo pozorováno žádné potlačení opožděné přecitlivělosti nebo hladiny imunoglobulinů nebo změny počtu efektorových buněčných populací.
U dvou pacientů s juvenilní idiopatickou artritidou se rozvinula infekce varicely se známkami a příznaky aseptické meningitidy, která byla vyléčena bez následků. Pacienti s výraznou expozicí viru varicely musí dočasně přerušit léčení přípravkem Benepali a je třeba u nich zvážit profylaktickou léčbu imunoglobulinem proti varicella zoster.
Bezpečnost a účinnost etanerceptu u pacientů se sníženou imunitou nebyla hodnocena.
Maligní nádory a lymfoproliferativní onemocnění
Solidní a hematopoetické maligní nádory (s vyloučením karcinomů kůže)
V postmarketingovém období se vyskytla hlášení o různých nádorových onemocněních (včetně karcinomu prsu, plic a lymfomu) (viz bod 4.8).
V kontrolovaných částech klinických studií s antagonisty TNF bylo u pacientů léčených antagonisty TNF pozorováno více případů lymfomů než u kontrolních pacientů. Jejich výskyt však byl vzácný a období sledování pacientů léčených placebem bylo kratší než u pacientů, léčených antagonisty TNF. Po uvedení přípravku na trh byly u pacientů léčených antagonisty TNF hlášeny případy leukémie. Zvýšené riziko vzniku lymfomu a leukémie je u pacientů s revmatoidní artritidou při dlouhotrvajícím průběhu a vysoké aktivitě zánětlivého onemocnění předpokládané, což komplikuje odhad rizika.
Na základě současného stavu znalostí nelze u pacientů léčených antagonisty TNF vyloučit možné riziko rozvoje lymfomů, leukémie či jiných hematopoetických nebo solidních malignit. Při zvažování terapie antagonisty TNF u pacientů s maligním nádorovým onemocněním v anamnéze nebo při zvažování pokračování léčby u pacientů, u kterých se rozvine maligní nádor, je třeba zvýšené opatrnosti.
Zhoubná nádorová onemocnění, některá končící úmrtím, byla po uvedení přípravku hlášena u dětí, dospívajících a mladých dospělých (do 22 let) léčených antagonisty TNF (zahájení terapie < 18 let), včetně etanerceptu. Lymfomy tvořily přibližně polovinu případů. Ostatní případy představovaly různé typy malignit a zahrnovaly vzácné, s imunosupresí typicky spojené malignity. Riziko rozvoje malignit u dětí a dospívajících léčených antagonisty TNF nelze vyloučit.
Karcinomy kůže
U pacientů léčených antagonisty TNF, včetně etanerceptu, byly hlášeny případy melanomu a nemelanomového karcinomu kůže (NMSC). U pacientů léčených etanerceptem byly v postmarketingovém období velmi zřídka hlášeny případy Merkelova buněčného karcinomu. U všech pacientů, zejména u těch s rizikovými faktory pro vznik karcinomu kůže, se doporučuje pravidelné vyšetření kůže.
Ze společných výsledků kontrolovaných klinických studií bylo pozorováno více případů NMSC u pacientů léčených etanerceptem v porovnání s kontrolními pacienty, obzvláště u pacientů s psoriázou.
Vakcinace
Současně s přípravkem Benepali nesmí být podávány živé vakcíny. Údaje o sekundárním přenosu infekce živými vakcínami u pacientů léčených etanerceptem nejsou k dispozici. Ve dvojitě zaslepené placebem kontrolované randomizované klinické studii na dospělých pacientech s psoriatickou artritidou dostalo 184 pacientů ve 4. týdnu také multivalentní pneumokokovou polysacharidovou vakcínu. V této studii většina pacientů s psoriatickou artritidou, kteří dostávali etanercept, byla schopna vykázat účinnou imunitní odpověď B-lymfocytů na pneumokokovou polysacharidovou vakcínu, ale titry byly v úhrnu mírně nižší, přičemž několik pacientů mělo dvojnásobně vyšší titry v porovnání s pacienty, kteří nedostávali etanercept. Klinický význam tohoto jevu není známý.
Tvorba autoprotilátek
Léčení přípravkem Benepali může vést k tvorbě autoprotilátek (viz bod 4.8).
Hematologické reakce
U pacientů léčených etanerceptem byly vzácně zjištěny případy pancytopenie a velmi vzácně případy aplastické anémie, některé s fatálním koncem. Pozornost musí být věnována pacientům léčeným přípravkem Benepali, kteří mají v anamnéze krevní dyskrazii. Všichni pacienti léčení přípravkem Benepali a rodiče/pečovatelé musí být upozorněni, aby neprodleně vyhledali lékařskou pomoc v případě rozvoje známek a příznaků připomínajících krevní dyskrazii nebo infekci u pacienta (např. přetrvávání horečky, bolest v krku, podlitiny, krvácení a bledost). Tito pacienti musí být bezodkladně vyšetřeni, včetně vyšetření úplného krevního obrazu; při potvrzení krevní dyskrazie musí být přípravek Benepali vysazen.
Neurologické poruchy
Vzácně byly u pacientů léčených etanerceptem zaznamenány demyelinizační poruchy CNS (viz bod 4.8). Dodatečně byly vzácně hlášeny případy periferní demyelinizační polyneuropatie (včetně syndromu Guillain-Barré, chronické zánětlivé demyelinizační polyneuropatie, demyelinizační polyneuropatie a multifokální neuropatie motoneuronů). Přestože nebyly provedeny klinické studie podávání etanerceptu pacientům s roztroušenou sklerózou, klinické studie jiných antagonistů TNF u pacientů s roztroušenou sklerózou prokázaly zvýšení aktivity onemocnění. Při zvažování podávání přípravku Benepali pacientům se stávající nebo nově objevenou demyelinizační poruchou nebo pacientům, u nichž je možno uvažovat o zvýšeném riziku rozvoje demyelinizační poruchy, je třeba pečlivě vyhodnotit možná rizika a výhody terapie, včetně neurologického posouzení.
Kombinovaná terapie
Ve dvouleté kontrolované klinické studii u pacientů s revmatoidní artritidou nevedla kombinace etanerceptu a methotrexátu k neočekávaným bezpečnostním nálezům, a bezpečnostní profil etanerceptu byl při podávání v kombinaci s methotrexátem podobný profilům zaznamenaným ve studiích, kde byl podáván etanercept nebo samotný methotrexát. Dlouhodobé klinické studie na posouzení bezpečnosti podání této kombinace stále pokračují. Dlouhodobá bezpečnost podávání etanerceptu v kombinaci s jinými chorobu modifikujícími léky (DMARD) nebyla sledována.
Použití etanerceptu při terapii psoriázy v kombinaci s jinými způsoby celkové léčby nebo s fototerapií nebylo studováno.
Porucha funkce ledvin a jater
Na základě farmakokinetických údajů (viz bod 5.2) není potřeba upravovat dávku u pacientů s poruchou funkce ledvin nebo jater. Klinické zkušenosti s těmito pacienty jsou omezené.
Městnavé srdeční selhání
Lékaři musí být opatrní při používání přípravku Benepali u pacientů s městnavým srdečním selháním. Existují postmarketingové zprávy o zhoršení městnavého srdečního selhání s identifikovatelnými vyvolávajícími faktory i bez nich u pacientů užívajících etanercept. Vzácně (< 0,1 %) byly také hlášeny případy nového nástupu městnavého srdečního selhání, a to včetně městnavého srdečního selhání u pacientů bez známého preexistujícího kardiovaskulárního onemocnění. Někteří z těchto pacientů byli mladší 50 let. Dvě rozsáhlé klinické studie hodnotící podávání etanerceptu při léčení městnavého srdečního selhání byly předčasně ukončeny z důvodu nedostatečné účinnosti. Údaje z jedné z těchto studií, i když nepřesvědčivé, naznačují možnou tendenci ke zhoršení městnavého srdečního selhání u pacientů léčených etanerceptem.
Alkoholická hepatitida
V randomizované placebem kontrolované studii fáze II bylo 48 hospitalizovaných pacientů léčeno etanerceptem nebo placebem pro středně těžkou až těžkou alkoholickou hepatitidu. Etanercept nebyl účinný a míra mortality po 6 měsících byla u pacientů léčených etanerceptem signifikantně vyšší. Proto se přípravek Benepali nesmí podávat pacientům k léčbě alkoholické hepatitidy. Lékaři musí být opatrní při podávání přípravku Benepali pacientům, kteří mají také středně těžkou až těžkou alkoholickou hepatitidu.
Wegenerova granulomatóza
Placebem kontrolovaná studie, v níž bylo 89 dospělých pacientů léčeno etanerceptem a standardní terapií (zahrnující cyklofosfamid nebo methotrexát a glukokortikoidy) v průměru po dobu 25 měsíců, neprokázala účinnost etanerceptu při léčení Wegenerovy granulomatózy. U pacientů léčených etanerceptem byl signifikantně vyšší výskyt různých typů malignit (jiných než kožních) než v kontrolní skupině. Přípravek Benepali se nedoporučuje k léčení Wegenerovy granulomatózy.
Hypoglykémie u pacientů s léčbou diabetu
U pacientů léčených antidiabetiky byly po zahájení léčby etanerceptem hlášeny případy hypoglykémie. U některých z těchto pacientů bylo nutné snížit dávky antidiabetik.
Zvláštní populace
Starší osoby
U pacientů ve věku 65 let a starších, kterým byl podáván etanercept, ve srovnání s mladšími pacienty, ve studiích fáze 3 u revmatoidní artritidy, psoriatické artritidy a ankylozující spondylitidy, nebyly pozorovány žádné celkové rozdíly v nežádoucích účincích, závažných nežádoucích účincích a závažných infekcích. Léčbě starších pacientů musí být však věnována zvýšená pozornost, zvláště pak s důrazem na výskyt infekcí.
Pediatrická populace
Přípravek Benepali není u dětí indikován. Dostupné údaje o léčbě etanerceptem u pediatrické populace jsou shrnuty níže:
Vakcinace
Doporučuje se, aby pediatričtí pacienti absolvovali všechna očkování podle platných očkovacích schémat pokud možno před zahájením terapie etanerceptem (viz Očkování uvedené výše).
Zánětlivé onemocnění střev (IBD) a uveitida u pacientů s juvenilní idiopatickou artritidou (JIA)
U pacientů s JIA a s uveitidou léčených etanerceptem byly hlášeny případy IBD (viz bod 4.8).
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Současná léčba s anakinrou
U dospělých pacientů léčených současně etanerceptem a anakinrou, byla pozorována vyšší četnost závažných infekcí ve srovnání s pacienty léčenými samotným etanerceptem nebo samotnou anakinrou (starší data).
Navíc ve dvojitě zaslepené placebem kontrolované studii dospělých pacientů na základní léčbě methotrexátem, byla u pacientů léčených současně etanerceptem a anakinrou pozorována vyšší četnost závažných infekcí (7 %) a neutropenie než u pacientů, léčených samotným etanerceptem (viz bod 4.4 a 4.8). Etanercept v kombinaci s anakinrou neprokázal zvýšení klinického prospěchu, a proto se nedoporučuje.
Současná léčba s abataceptem
V klinických studiích vedlo současné podávání abataceptu a etanerceptu ke zvýšené incidenci závažných nežádoucích příhod. Tato kombinace neprokázala lepší klinický benefit; takové použití se nedoporučuje (viz bod 4.4).
Současná léčba s sulfasalazinem
V klinické studii na dospělých pacientech, kteří dostávali pevně stanovené dávky sulfasalazinu, ke kterým byl přidán etanercept, měli pacienti statisticky významný pokles průměrného počtu bílých krvinek ve srovnání se skupinou, léčenou buď samotným etanerceptem, nebo samotným sulfasalazinem. Klinický význam této interakce není známý. Při zvažování kombinované terapie se sulfasalazinem musí být lékaři opatrní.
Žádné interakce
V klinických studiích při podávání etanerceptu s glukokortikoidy, salicyláty (kromě sulfasalazinu), nesteroidními antirevmatiky (NSAID), analgetiky nebo methotrexátem nebyly pozorovány žádné interakce. Pokyny ohledně vakcinace viz bod 4.4 Očkování.
Žádné klinicky významné farmakokinetické interakce nebyly pozorovány ve studiích s methotrexátem, digoxinem ani s warfarinem.
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku
Ženy ve fertilním věku musí být upozorněny, aby v průběhu léčby a tři týdny po ukončení léčby přípravkem Benepali používaly vhodnou antikoncepci k zabránění otěhotnění.
Toxikologické studie na potkanech a králících neodhalily žádné poškození plodu nebo narozených mláďat etanerceptem. Vyšší frekvence závažných vrozených vad byla pozorována v observační studii porovnávající těhotenství vystavená etanerceptu během prvního trimestru s těhotenstvími nevystavenými etanerceptu či jiným antagonistům TNF (adjustovaný poměr šancí [odds ratio] 2,4, 95% CI: 1,0—5,5). Nejčastěji hlášené typy závažných vrozených vad se shodovaly s těmi nejčastěji hlášenými u běžné populace a nebyl identifikován žádný zvláštní obraz abnormalit. Studie nezjistila vyšší míru spontánního potratu, porodu mrtvého plodu nebo menších malformací. Podávání přípravku Benepali během těhotenství se nedoporučuje.
Etanercept prostupuje placentou a byl zjištěn v séru novorozenců narozených pacientkám léčeným během těhotenství etanerceptem. Klinický dopad této skutečnosti není znám, u novorozenců však může existovat zvýšené riziko infekce. Podávání živé vakcíny novorozencům po dobu 16 týdnů od poslední dávky přípravku Benepali matce se obecně nedoporučuje.
Kojení
Bylo hlášeno, že se etanercept po subkutánním podání vylučuje do mateřského mléka. U kojících potkanů se etanercept po subkutánním podání vylučoval do mléka a objevil se v séru mláďat. Protože imunoglobuliny mohou být společně s mnoha dalšími léčivy vylučovány do mateřského mléka, je na základě posouzení prospěšnosti kojení pro dítě a prospěšnosti léčby pro matku nutno rozhodnout, zda přerušit kojení nebo přerušit podávání přípravku Benepali.
Fertilita
Preklinické údaje o perinatální a postnatální toxicitě etanerceptu a o jeho účincích na plodnost a celkovou reprodukční schopnost nejsou k dispozici.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Studie hodnotící účinky na schopnost řídit a obsluhovat stroje nebyly provedeny.
4.8 Nežádoucí účinky Shrnutí bezpečnostního profilu
Nejčastěji hlášenými nežádoucími účinky jsou reakce v místě aplikace (jako jsou bolest, otoky, svědění, zarudnutí a krvácení v místě vpichu), infekce (např. infekce horních cest dýchacích, zánět průdušek, infekce močového měchýře a kožní infekce), alergické reakce, tvorba autoprotilátek, svědění a horečka.
U pacientů léčených etanerceptem byly také hlášeny závažné nežádoucí účinky. Antagonisté TNF, jako je etanercept, působí na imunitní systém a jejich podávání může ovlivnit přirozenou ochranu organismu proti infekcím a vzniku malignit. Závažné infekce postihují méně než 1 ze 100 pacientů léčených etanerceptem. Hlášení zahrnovala fatální a život ohrožující infekce a sepsi. U pacientů léčených etanerceptem se také vyskytla hlášení o různých typech malignit včetně karcinomu prsu, plic a lymfatických uzlin (lymfom).
Byly také hlášeny případy závažných hematologických, neurologických a autoimunitních reakcí. Mezi ně také patří vzácná hlášení pancytopenie a velmi vzácného výskytu aplastické anémie. Při podávání etanerceptu se objevily vzácně, respektive velmi vzácně, případy centrální a periferní demyelinizace. Vzácně byly hlášeny případy lupusu, s lupusem souvisejících onemocnění a vaskulitidy.
Tabulkový seznam nežádoucích účinků
Následující seznam nežádoucích účinků je založen na zkušenostech z klinických studií u dospělých a postmarketingových zkušenostech u dospělých.
Ve skupinách tříděných podle orgánových systémů jsou nežádoucí účinky uvedeny vedle označení jejich frekvence (počtu pacientů, u nichž se tato reakce očekává) na základě těchto kategorií: Velmi časté (> 1/10); časté (> 1/100 až < 1/10); méně časté (> 1/1 000 až < 1/100); vzácné (> 1/10 000 až < 1/1 000); velmi vzácné (< 1/10 000); není známo (z dostupných údajů nelze určit).
|
Infekce a infestace: Velmi časté: Méně časté: Vzácné: Není známo: |
Infekce (včetně infekcí horních cest dýchacích, bronchitidy, cystitidy, infekce kůže)* Závažné infekce (včetně pneumonie, celulitidy, septické artritidy, sepse a parazitárních infekcí)* Tuberkulóza, oportunní infekce (včetně invazivních plísňových, protozoálních, bakteriálních, atypických mykobakteriálních, virových infekcí a legionelózy)* Listerióza, reaktivace hepatitidy B |
|
Novotvary benigní, maligní a blíže neurčené (zahrnující cysty apolypy): | |
|
Méně časté: |
Nemelanomový karcinom kůže* (viz bod 4.4) |
|
Vzácné: |
Lymfom, melanom (viz bod 4.4) |
|
Není známo: |
Leukémie, Merkelův buněčný karcinom (viz bod 4.4) |
|
Poruchy krve a lymfatického systému: | |
|
Méně časté: |
Trombocytopenie |
|
Vzácné: |
Anémie, leukopenie, neutropenie, pancytopenie* |
|
Velmi vzácné: |
Aplastická anémie* |
|
Poruchy imunitního systému: | |
|
Časté: |
Alergické reakce (viz poruchy kůže a podkožní tkáně), tvorba autoprotilátek* |
|
Méně časté: |
Systémová vaskulitida (včetně vaskulitidy s pozitivními anti-neutrofilními cytoplazmatickými protilátkami) |
|
Vzácné: |
Závažné alergické/anafylaktické reakce (včetně angioedému, bronchospasmu), sarkoidóza |
|
Není známo: |
Syndrom aktivace makrofágů*, zhoršení příznaků dermatomyositidy |
|
Poruchy nervového systému: | |
|
Vzácné: |
Demyelinizace CNS připomínající roztroušenou sklerózu nebo lokalizované demyelinizační stavy, jako např. zánět n. opticus nebo transverzální myelitida (viz bod 4.4) |
|
Velmi vzácné: |
Periferní demyelinizační stavy včetně syndromu Guillain-Barré, chronická zánětlivá demyelinizační polyneuropatie, demyelinizační polyneuropatie a multifokální neuropatie motoneuronů (viz bod 4.4) |
|
Poruchy oka: | |
|
Méně časté: |
Uveitida, skleritida |
|
Srdeční poruchy: | |
|
Vzácné: |
Městnavé srdeční selhání (viz bod 4.4) |
|
Respirační, hrudní a mediastinální poruchy: | |
|
Méně časté: |
Intersticiální onemocnění plic (včetně pneumonitidy a plicní |
|
fibrózy)* | |
|
Poruchy jater a žlučových cest: Vzácné: |
Zvýšené hladiny jaterních enzymů, autoimunitní hepatitida |
Poruchy kůže a podkožní tkáně: Časté:
Méně časté:
* Viz Popis vybraných nežádoucích účinků níže. Popis vybraných nežádoucích účinků
Maligní nádory a lymfoproliferativní onemocnění
V klinických studiích zahrnujících 4 114 pacientů s revmatoidní artritidou léčených etanerceptem přibližně až 6 let, včetně 231 pacientů léčených etanerceptem v kombinaci s methotrexátem ve 2roční aktivním komparátorem kontrolované studii, bylo zjištěno sto dvacet devět (129) nových maligních nádorů různých typů. Zjištěná míra incidence v těchto studiích byla podobná očekávané incidenci v obdobném vzorku běžné populace. V klinických studiích zahrnujících 240 pacientů s psoriatickou artritidou léčených etanerceptem přibližně 2 roky byly hlášeny celkem 2 malignity. V klinických studiích zahrnujících 351 pacientů s ankylozující spondylitidou trvající déle než 2 roky bylo u pacientů léčených etanerceptem hlášeno 6 malignit. Ve skupině 2 711 pacientů s ložiskovou psoriázou léčených etanerceptem bylo ve dvojitě zaslepených a otevřených klinických studiích v trvání až 2,5 roku hlášeno 30 maligních nádorů a 43 nemelanomových karcinomů kůže.
V klinických studiích zahrnujících 7 416 pacientů s revmatoidní artritidou, psoriatickou artritidou, ankylozující spondylitidou a psoriázou léčených etanerceptem bylo celkem hlášeno 18 lymfomů.
Zprávy o různých maligních nádorech (včetně karcinomu prsu, plic a lymfomu) byly rovněž získány v postmarketingovém období (viz bod 4.4).
Reakce v místě aplikace injekce
Ve srovnání s placebem měli pacienti s revmatickými chorobami léčení etanerceptem signifikantně vyšší incidenci reakcí v místě aplikace injekce (36 % versus 9 %). Lokální reakce byly pozorovány obvykle během prvního měsíce podávání. Přetrvávaly v průměru přibližně 3-5 dnů. Většina lokálních reakcí po injekci etanerceptu nevyžadovala léčbu. Pacienti, kteří byli léčeni, dostali převážně lokální léčbu, například kortikosteroidy, nebo perorálně antihistaminika. Dodatečně se u některých pacientů rozvinula opožděná reakce v posledním místě aplikace se současným projevem reakce v předchozím místě aplikace. Tyto reakce byly všeobecně přechodného rázu a po odléčení se neopakovaly.
V kontrolovaných studiích u pacientů s ložiskovou psoriázou se rozvinula v průběhu prvních 12 týdnů lokální reakce v místě injekce u 13,6 % pacientů léčených etanerceptem v porovnání s 3,4 % pacientů léčených placebem.
Závažné infekce
V placebem kontrolovaných studiích nebyla pozorována zvýšená incidence vážných infekcí (fatálních, život ohrožujících nebo vyžadujících hospitalizaci nebo podání intravenózních antibiotik). Závažné infekce se vyskytly u 6,3 % pacientů s revmatoidní artritidou, léčených etanerceptem až po dobu
48 měsíců. Tyto zahrnovaly absces (různě lokalizovaný), bakteriémii, bronchitidu, bursitidu, celulitidu, cholecystitidu, průjem, divertikulitidu, endokarditidu (suspektní), gastroenteritidu, hepatitidu B, herpes zoster, bércové vředy, infekce dutiny ústní, osteomyelitidu, otitidu, peritonitidu, pneumonii, pyelonefritidu, sepsi, septickou artritidu, sinusitidu, infekce kůže, vředy na kůži, infekce močových cest, vaskulitidu a infekce ran. Ve dvouleté aktivním komparátorem kontrolované studii, kde byli pacienti léčeni buď jenom etanerceptem, nebo jenom methotrexátem, nebo etanerceptem v kombinaci s methotrexátem, byl podíl vážných infekcí v léčených skupinách podobný. Nelze však vyloučit, že by kombinace etanerceptu s methotrexátem mohla být spojena se zvýšením podílu infekcí.
V placebem kontrolovaných studiích trvajících až 24 týdnů nebyl u pacientů s ložiskovou psoriázou léčených etanerceptem a pacientů léčených placebem pozorován rozdíl v četnosti infekcí. Závažné infekce u pacientů léčených etanerceptem zahrnovaly celulitidu, gastroenteritidu, pneumonii, cholecystitidu, osteomyelitidu, gastritidu, apendicitidu, streptokokovou fasciitidu, myositidu, septický šok, divertikulitidu a absces. Ve dvojitě zaslepených i otevřených studiích u pacientů s psoriatickou artritidou byla u jednoho pacienta hlášena závažná infekce (pneumonie).
Během používání etanerceptu byly hlášeny závažné a fatální infekce; zjištěné patogeny zahrnovaly baktérie, mykobaktérie (včetně tuberkulózy), viry a plísně. Některé z nich se objevily v průběhu několika týdnů po zahájení terapie etanerceptem u pacientů s revmatoidní artritidou, kteří současně měli takové choroby jako např. diabetes, městnavé srdeční selhání, aktivní nebo chronickou infekci v anamnéze (viz bod 4.4). Terapie přípravkem Benepali může zvýšit mortalitu u pacientů se sepsí.
V souvislosti s podáváním etanerceptu byly hlášeny oportunní infekce, včetně invazivních plísňových, parazitárních (zahrnující protozoální), virových (zahrnující herpes zoster), bakteriálních (zahrnující Listerie a Legionelly) a atypických mykobakteriálních infekcí. V souhrnných údajích získaných z klinických studií byla celková incidence oportunních infekcí u 15 402 subjektů léčených etanerceptem 0,09 %. Míra četnosti vzhledem k expozici byla 0,06 příhod na 100 paciento-roků. V postmarketingovém používání tvořily invazivní plísňové infekce přibližně polovinu všech hlášených případů oportunních infekcí v celosvětovém měřítku. Nejčastěji hlášené invazivní plísňové infekce zahrnovaly Candidu, Pneumocystis, Aspergillus a Histoplasmu. U pacientů, kteří prodělali oportunní infekce, činily invazivní plísňové infekce více než polovinu fatálních případů. Většinu případů s fatálním zakončením tvořili pacienti s pneumonií vyvolanou Pneumocystis, nespecifickými systémovými plísňovými infekcemi a aspergilózou (viz bod 4.4).
Autoprotilátky
Vzorky séra dospělých léčených pacientů byly v několika časových obdobích testovány na autoprotilátky. U pacientů s revmatoidní artritidou bylo z hlediska hodnocení antinukleárních protilátek (ANA) zjištěno, že ve skupině pacientů léčených etanerceptem bylo vyšší procento pacientů (11 %) s nově vyvinutými pozitivními protilátkami (> 1:40) než ve skupině léčené placebem (5 %). Vyšší procento pacientů s nově vyvinutými protilátkami proti anti-dvouvláknové DNA bylo také stanoveno radioimunoanalýzou (15 % pacientů léčených etanerceptem versus 4 % v placebové skupině) a stanovením Crithidia luciliae esejí (3 % pacientů léčených etanerceptem ve srovnání s 0 % ve skupině placeba). Podobně se v porovnání s pacienty léčenými placebem zvýšil ve skupině pacientů léčených etanerceptem počet nemocných s nově vyvinutými antikardiolipinovými protilátkami. Vliv dlouhodobé terapie etanerceptem na rozvoj autoimunních onemocnění není znám.
Vzácně se u některých pacientů, včetně pacientů s pozitivním revmatoidním faktorem, vyskytly případy vzniku autoprotilátek ve spojení s lupus-like syndromem nebo exantémem, které klinicky a biopticky odpovídají subakutnímu kožnímu lupusu nebo diskoidnímu lupusu.
Pancytopenie a aplastická anémie
V postmarketingových hlášeních existují případy pancytopenie a aplastické anémie, některé z nich s fatálním koncem (viz bod 4.4).
Intersticiální plicní onemocnění
V postmarketingových hlášeních bylo zaznamenáno intersticiální onemocnění plic (včetně pneumonitidy a plicní fibrózy), některá z nich byla fatální.
Současná léčba s anakinrou
Ve studiích byla u dospělých pacientů léčených současně etanerceptem a anakinrou pozorována vyšší četnost závažných infekcí než u pacientů léčených samotným etanerceptem, a u 2 % pacientů (3 ze 139) došlo k neutropenii (absolutní počet neutrofilů pod < 1000/mm3). U jednoho pacienta s neutropenií se rozvinula celulitida, která byla zvládnuta hospitalizací (viz bod 4.4 a 4.5).
Pediatrická populace
Přípravek Benepali není u dětí indikován. Dostupné údaje o léčbě etanerceptem u pediatrické populace jsou shrnuty níže:
Nežádoucí účinky u dětských pacientů s juvenilní idiopatickou artritidou
Nežádoucí účinky u dětských pacientů s juvenilní idiopatickou artritidou byly obdobné, co do typu a frekvence, jako u dospělých pacientů. Rozdíly a další speciální úvahy jsou diskutovány v následujících odstavcích.
Infekce pozorované v klinických studiích u pacientů ve věku 2-18 let s juvenilní idiopatickou artritidou byly obecně lehkého až středně těžkého typu a odpovídaly infekcím běžně pozorovaným u ambulantních pacientů dětské populace. Hlášené závažné nežádoucí účinky zahrnovaly plané neštovice se známkami a příznaky aseptické meningitidy, vyléčené bez následků (viz také bod 4.4), apendicitidu, gastroenteritidu, deprese/poruchy osobnosti, kožní vředy, ezofagitidu/gastritidu, septický šok vyvolaný streptokokem skupiny A, diabetes mellitus 1. typu, infekce měkkých tkání a pooperační infekce ran.
V jedné studii 43 z 69 dětí (tj. 62 %) s juvenilní idiopatickou artritidou ve věku 4-17 let prodělalo infekci v průběhu tříměsíčního léčení etanerceptem (část I, otevřená fáze), frekvence a závažnost infekcí byla podobná jako u 58 pacientů, kteří dokončili 12měsíční otevřené pokračování terapie. Druhy a poměr nežádoucích účinků (NÚ) u pacientů s idiopatickou juvenilní artritidou byly podobné těm, které byly pozorovány ve studiích podávání etanerceptu u dospělých pacientů s revmatoidní artritidou, a většinou byly mírné. Závažné nežádoucí účinky byly hlášeny častěji u 69 pacientů s juvenilní idiopatickou artritidou léčenou po 3 měsíce etanerceptem než u 349 dospělých pacientů s revmatoidní artritidou. Zahrnovaly bolesti hlavy (19 % pacientů, tj. 1,7 NÚ/pac./rok), nevolnost (9 %, tj. 1,0 NÚ/pac./rok), bolest břicha (19 %, 0,74 NÚ/pac./rok) a zvracení (13 %, 0,74 NÚ/pac./rok).
V klinických studiích s juvenilní idiopatickou artritidou byla 4 hlášení syndromu aktivace makrofágů.
Z postmarketingových zdrojů byly hlášeny případy zánětlivého onemocnění střev a uveitidy u pacientů s juvenilní idiopatickou artritidou léčených etanerceptem včetně velmi malého počtu případů indikujících vyprovokování nemoci positivní stimulací (viz bod 4.4).
Nežádoucí účinky u dětských pacientů s ložiskovou psoriázou
Ve studii trvající 48 týdnů s 211 dětmi ve věku od 4 do 17 let s ložiskovou psoriázou byly hlášeny podobné nežádoucí účinky jako v předchozích studiích u dospělých s ložiskovou psoriázou.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
V průběhu klinického hodnocení léčení revmatoidní artritidy nebyla pozorována toxicita limitující dávku. Nejvyšší hodnocená dávka byla zátěžová intravenózní dávka 32 mg/m2, po níž následovala subkutánní dávka 16 mg/ m2 podávaná 2x týdně. Jeden pacient s revmatoidní artritidou si omylem aplikoval subkutánně 62 mg etanerceptu 2x týdně po dobu 3 týdnů bez nežádoucích účinků. Není známo žádné antidotum etanerceptu.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Imunosupresiva, inhibitory tumor nekrotizujícího faktoru alfa (TNF-a) ATC kód: L04AB01
Benepali je tzv. podobným biologickým léčivým přípravkem („biosimilar“). Podrobné informace jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Tumor nekrotizující faktor (TNF) je dominantním cytokinem v zánětlivém procesu revmatoidní artritidy. Zvýšené hladiny TNF byly také nalezeny v synovii a psoriatických placích u pacientů s psoriatickou artritidou a v séru i synoviální tkáni u pacientů s ankylozující spondylitidou. U ložiskové psoriázy vede infiltrace zánětlivými buňkami, včetně T-lymfocytů, ke zvýšení hladin TNF v psoriatických lézích v porovnání s jeho hladinami v nezasažené kůži. Etanercept je kompetitivním inhibitorem vazby TNF na jeho buněčné povrchové receptory, čímž inhibuje biologickou aktivitu TNF. TNF a lymfotoxin jsou pro-zánětlivé cytokiny, které se vážou ke dvěma odlišným povrchovým buněčným receptorům: TNF receptor p55 (55 kilodaltonů) a p75 (75 kilodaltonů). Oba TNF receptory se přirozeně vyskytují buď jako membránově vázané, nebo ve volné formě. Rozpustné TNF receptory jsou považovány za regulátory biologické aktivity TNF.
TNF a lymfotoxin existují převážně jako homotrimery, jejichž biologická aktivita je závislá na zkříženém navázání (cross-linking) na TNF receptory vázané na povrchu buněk. Dimery volných receptorů jako je etanercept mají vyšší afinitu k TNF než monomerní receptory a jsou zjevně silnějšími kompetitivními inhibitory vazby TNF na jeho buněčné receptory. Navíc použití Fc regionu imunoglobulinu jako spojujícího elementu v konstrukci dimerického receptoru mu dodává delší poločas v séru.
Mechanismus účinku
Většina patologických procesů v kloubu s revmatoidní artritidou a ankylozující spondylitidou a v kůži u ložiskové psoriázy je ovlivňována prozánětlivými molekulami, které jsou součástí procesů řízených TNF. Má se za to, že mechanismus účinku etanerceptu je kompetitivní inhibice vazby TNF na jeho povrchové buněčné receptory, vytvoření biologicky neaktivního TNF a tím zabránění buněčné odpovědi. Etanercept může také ovlivňovat buněčné odpovědi řízené dalšími molekulami, které jsou indukovány prostřednictvím TNF (např. cytokiny, adhezivní molekuly nebo proteinázy).
Klinická účinnost a bezpečnost
Tato část popisuje údaje ze čtyř randomizovaných kontrolovaných studií u dospělých s revmatoidní artritidou, z jedné studie u dospělých s psoriatickou artritidou, z jedné studie u dospělých s ankylozující spondylitidou, z jedné studie u dospělých s radiograficky neprokazatelnou axiální spondylartritidou, ze čtyř studií u dospělých s ložiskovou psoriázou.
Dospělí pacienti s revmatoidní artritidou
Účinnost etanerceptu byla hodnocena v randomizované dvojitě zaslepené placebem kontrolované klinické studii. Studie hodnotila 234 dospělých pacientů s aktivní revmatoidní artritidou, u nichž selhala předchozí terapie minimálně jedním, ale ne více než čtyřmi chorobu modifikujícími léky (DMARD). Etanercept v dávkách 10 mg nebo 25 mg nebo placebo byly 2x týdně subkutánně podávány po dobu 6 po sobě jdoucích měsíců. Výsledky této kontrolované studie byly vyjádřeny v procentech zlepšení revmatoidní artritidy s použitím kritérií odpovědi podle ACR (American College of Rheumatology).
Odpověď ACR 20 a 50 byly vyšší u pacientů léčených etanerceptem po dobu 3 a 6 měsíců než u pacientů léčených placebem (ACR 20: etanercept 62 % a 59 %, placebo 23 % a 11 % po 3 a 6 měsících: ACR 50: etanercept 41 % a 40 %, placebo 8 % a 5 % po 3 a 6 měsících; p<0,01 etanercept vs. placebo ve všech časových úsecích pro obě odpovědi ACR 20 a ACR 50).
Přibližně u 15 % pacientů léčených etanerceptem bylo dosaženo odpovědi ACR 70 ve 3. a 6. měsíci ve srovnání s méně než 5 % pacientů ve skupině léčené placebem. Ve skupině léčené etanerceptem se klinická odpověď obyčejně dostavila během 1-2 týdnů od zahájení a téměř vždy k ní došlo do 3 měsíců. Byla pozorována závislost na dávce: výsledky léčení 10 mg etanerceptu byly uprostřed mezi placebem a dávkou 25 mg. Výsledky léčby etanerceptem byly signifikantně lepší než podávání placeba ve všech ukazatelích kritérií ACR, jakož i v dalších ukazatelích aktivity revmatoidní artritidy, které nebyly součástí hodnocení odpovědi na léčbu dle ACR, jako např. ranní ztuhlost. V průběhu klinického hodnocení pacienti každé 3 měsíce vyplňovali dotazník zdravotního hodnocení (HAQ), který zahrnoval otázky na invaliditu, vitalitu, duševní zdraví, celkový zdravotní stav a oblasti zdravotního stavu, které mají spojitost s artritidou. U pacientů léčených etanerceptem došlo po 3 a 6 měsících ke zlepšení ve všech oblastech hodnocených v dotazníku ve srovnání s kontrolní skupinou.
Po ukončení podávání etanerceptu se příznaky artritidy objevily znovu všeobecně do jednoho měsíce. Výsledky otevřené studie prokázaly, že opětovné zahájení léčby po přerušení, trvajícím až 24 měsíců, vedlo ke stejné výsledné odpovědi, jaké bylo dosaženo u pacientů, kterým byl etanercept podáván bez přerušení. V otevřené studii dlouhodobého podávání etanerceptu bez přerušení byla pozorována přetrvávající odpověď po dobu až 10 let.
Účinnost etanerceptu byla porovnávána s methotrexátem v randomizované, aktivním komparátorem kontrolované studii, mající jako primární kritérium hodnocení zaslepené radiografické hodnocení u 632 dospělých pacientů s aktivní revmatoidní artritidou (trvání kratší než 3 roky), kteří předtím nebyli nikdy léčeni methotrexátem. Etanercept byl podáván subkutánně (s.c.) v dávkách 10 nebo 25 mg 2krát týdně po dobu až 24 měsíců. Dávky methotrexátu byly zvyšovány od 7,5 mg/týden až maximálně na 20 mg/týden po dobu prvních 8 týdnů studie a pokračovaly až 24 měsíců. Klinické zlepšení, včetně nástupu účinku do 2 týdnů při dávkách etanerceptu 25 mg, bylo podobné tomu, jež bylo pozorováno u předchozích studií, a přetrvávalo až 24 měsíců. Při zahájení měli pacienti střední stupeň neschopnosti s průměrným skóre HAQ 1,4-1,5. Léčba etanerceptem 25 mg vedla po 12 měsících k podstatnému zlepšení a k dosažení normálního skóre HAQ (méně než 0,5) u 44 % pacientů. Tento přínos přetrvával ve 2. roce studie.
V této studii byly radiograficky vyhodnoceny strukturální změny kloubů a byly vyjádřeny jako změna TSS (Total Sharp Score) a jeho komponent, tj. skóre erozí a Joint Space Narrowing (JSN) Score. Radiogramy rukou/zápěstí a nohou byly hodnoceny při zahájení, po 6, 12 a 24 měsících. Etanercept v dávce 10 mg měl konzistentně nižší účinek na strukturální poškození než dávka 25 mg. Ve skóre erozí byl po 12 a 24 měsících etanercept v dávce 25 mg signifikantně lepší než methotrexát. Rozdíly mezi etanerceptem v dávce 25 mg a methotrexátem v TSS a JSN nebyly statisticky významné. Výsledky jsou znázorněny na následujícím grafu.
Radiografická progrese: porovnání etanercept vs. metotrexát u pacientů s revmatoidní artritidou v trvání do 3 let
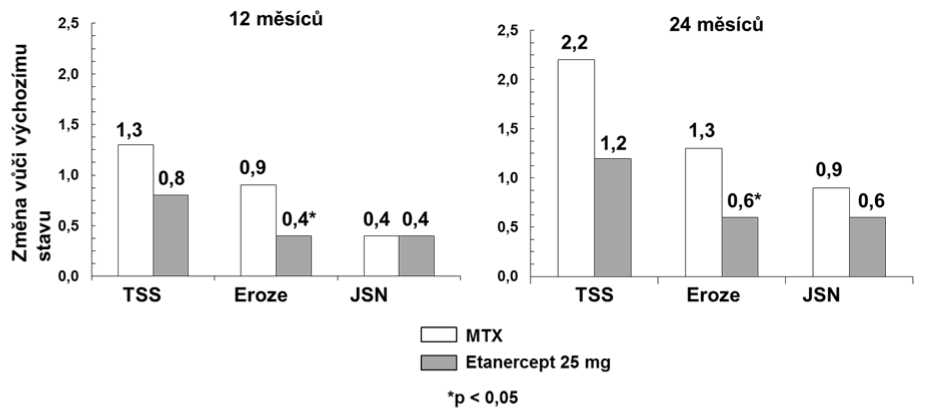
V jiné dvojitě zaslepené randomizované studii kontrolované aktivním komparátorem na 682 dospělých pacientech s aktivní revmatoidní artritidou, v trvání od 6 měsíců do 20 let (průměrně 5 let), kteří měli méně než uspokojivou odpověď nejméně na jedno chorobu modifikující antirevmatikum (DMARD), jiné než methotrexát, byla porovnávána klinická účinnost, bezpečnost a radiografický vývoj u pacientů léčených buď samotným etanerceptem (2x týdně 25 mg), nebo samotným methotrexátem (7,5-20 mg za týden, střední dávka 20 mg) nebo kombinací etanercept a methotrexát.
Pacienti léčení kombinací etanercept a methotrexát měli signifikantně vyšší odpovědi ACR 20, ACR 50 a ACR 70 a zlepšení ve skóre DAS a HAQ po 24 i po 52 týdnech než pacienti ve skupinách léčených monoterapií jedné z látek (výsledky jsou uvedeny v tabulce níže). Signifikantní výhody pro etanercept v kombinaci s methotrexátem v porovnání s monoterapií etanerceptem a monoterapií methotrexátem byly pozorovány také po 24 měsících.
Výsledky klinické účinnosti po 12 měsících: porovnání etanercept vs. methotrexát vs. etanercept v kombinaci s methotrexátem u pacientů s revmatoidní artritidou v trvání od 6 měsíců do 20 let
|
Cílový ukazatel |
Methotrexát (n = 228) |
Etanercept (n = 223) |
Etanercept + Methotrexát (n = 231) | |
|
Odpovědi ACRa |
ACR 20 |
58,8 % |
65,5 % |
74,5 %f, ® |
|
ACR 50 |
36,4 % |
43,0 % |
63,2 %f, ® | |
|
ACR 70 |
16,7 % |
22,0 % |
39,8 %f, ® | |
|
DAS |
Výchozí |
5,5 |
5,7 |
5,5 |
|
(Skóreb) v 52. týdnu |
3,0 |
3,0 |
2,3 Ť’ ° | |
|
(Skóreb) Remisec |
14 % |
18 % |
37 %f, ® | |
|
HAQ |
Výchozí |
1,7 |
1,7 |
1,8 |
|
52. týden |
1,1 |
1,0 |
0,8 Ť’ ° | |
a Pacienti, kteří nedokončili 12 měsíců ve studii, byli považováni za na léčbu neodpovídající. b Hodnoty DAS představují střední hodnoty. c Remise je definována jako DAS < 1,6
Párové porovnání p-hodnot: f = p < 0,05 k porovnání etanercept + methotrexát vs. methotrexát a ® = p < 0,05 k porovnání etanercept + methotrexát vs. etanercept.
Radiografický vývoj po 12 měsících byl ve skupině léčené etanerceptem signifikantně nižší než ve skupině léčené methotrexátem, zatímco kombinovaná terapie byla ve zpomalení radiografického vývoje signifikantně lepší než obě monoterapie (viz obrázek níže).
Radiografická progrese: porovnání etanercept vs. methotrexát vs. etanercept v kombinaci s methotrexátem u pacientů s revmatoidní artritidou v trvání od 6 měsíců do 20 let (12měsíční výsledky)
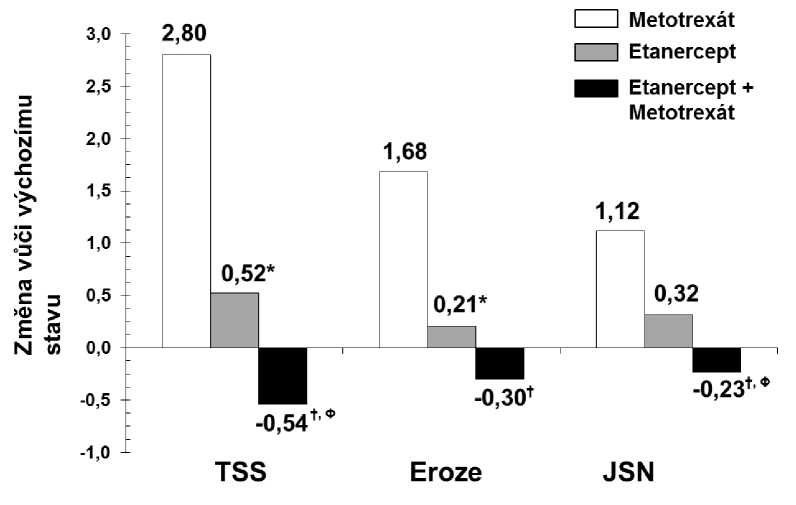
Párové porovnání p-hodnot: * = p < 0,05 k porovnání etanercept vs. methotrexát, f = p < 0,05 k porovnání etanercept + methotrexát vs. methotrexát, ® = p < 0,05 k porovnání etanercept + methotrexát vs. etanercept.
Signifikantní výhody pro etanercept v kombinaci s methotrexátem v porovnání s monoterapií etanerceptem a monoterapií methotrexátem byly pozorovány také po 24 měsících. Obdobně signifikantní výhody pro monoterapii etanerceptem v porovnání s monoterapií methotrexátem byly pozorovány také po 24 měsících.
V analýze, v níž všichni pacienti, kteří z jakéhokoli důvodu ze studie odešli, byli považováni za pacienty s progresí, bylo ve skupině pacientů léčených etanerceptem v kombinaci s methotrexátem po dobu 24 měsíců vyšší procento pacientů bez progrese (změna TSS < 0,5) v porovnání se skupinou léčenou samotným etanerceptem a samotným methotrexátem (62 %, 50 % a 36 %, p < 0,05). Rozdíl mezi monoterapií etanerceptem a monoterapií methotrexátem byl také signifikantní (p < 0,05). Mezi pacienty, kteří dokončili celých 24 měsíců léčení ve studii, byly podíly pacientů bez progrese v jednotlivých skupinách 78 %, 70 % a 61 %.
Bezpečnost a účinnost 50 mg etanerceptu (2 subkutánní injekce po 25 mg) podaných jednou týdně byly vyhodnoceny ve dvojitě zaslepené placebem kontrolované studii na 420 pacientech s aktivní revmatoidní artritidou. V této studii 53 pacientů dostávalo placebo, 214 pacientů dostávalo 50 mg etanerceptu jednou týdně a 153 pacientů dostávalo 25 mg etanerceptu 2x týdně. Profil bezpečnosti a účinnosti těchto dvou léčebných režimů etanerceptu byly porovnatelné v 8. týdnu v ovlivnění známek a příznaků revmatoidní artritidy; údaje z 16. týdne ukázaly komparabilitu (non-inferiorita) těchto dvou režimů. Jednotlivá injekce etanerceptu 50 mg/ml byla shledána bioekvivalentní dvěma simultánně podaným injekcím po 25 mg/ml.
Dospělí pacienti s psoriatickou artritidou
Účinnost etanerceptu byla hodnocena v randomizované, dvojitě zaslepené, placebem kontrolované studii na 205 pacientech s psoriatickou artritidou. Pacienti byli ve věku mezi 18 a 70 lety a měli aktivní psoriatickou artritidu (minimálně 3 oteklé klouby a minimálně 3 bolestivé klouby) nejméně v některé z následujících forem: (1) Distální interfalangeální postižení (DIP); (2) Polyartikulární artritida (absence revmatoidních uzlíků a přítomnost psoriázy); (3) Arthritis mutilans; (4) Asymetrická psoriatická artritida; (5) Ankylóza podobná spondylitidě. Pacienti také měli psoriatická ložiska o minimálním průměru 2 cm a větším.
Pacienti byli předtím léčeni nesteroidními antirevmatiky (86 %), DMARD (80 %) a kortikosteroidy (24 %). Pacienti, kteří byli současně léčeni methotrexátem (alespoň 2 měsíce), mohli pokračovat při stabilním dávkování methotrexátu < 25 mg/týden. Dávky 25 mg etanerceptu (podloženo studiemi na stanovení dávek na pacientech s revmatoidní artritidou) nebo placebo byly podávány subkutánně 2x týdně po dobu 6 měsíců. Na konci dvojitě zaslepené studie mohli pacienti vstoupit do rozšířené dlouhodobé otevřené studie v celkové délce až 2 roky.
Klinické odpovědi byly vyjádřeny jako % pacientů, u nichž bylo dosaženo odpovědi ACR 20, 50 a 70 a % pacientů se zlepšením podle kritérií odpovědi u psoriatické artritidy (PsARC). Souhrn výsledků ukazuje následující tabulka:
Odpovědi pacientů s psoriatickou artritidou v placebem kontrolované studii
|
Odpověď psoriatické artritidy |
Procento pacientů | ||
|
Placebo n = 104 |
Etanercepta n = 101 | ||
|
ACR 20 |
3. měsíc |
15 |
79 |
|
6. měsíc |
13 |
“50 | |
|
ACR 50 |
3. měsíc |
4 |
"38 |
|
6. měsíc |
4 |
77 | |
|
ACR 70 |
3. měsíc |
0 |
11b |
|
6. měsíc |
1 |
9c | |
|
PsARC |
3. měsíc |
31 |
77 |
|
6. měsíc |
23 |
^1 O cr | |
a 25 mg etanerceptu 2x týdně s.c. b p < 0,001, etanercept versus placebo c p < 0,01, etanercept versus placebo
U pacientů s psoriatickou artritidou léčených etanerceptem, byla klinická odpověď zřejmá od první návštěvy (po 4 týdnech) a přetrvávala po 6 měsíců léčení. Etanercept byl signifikantně lepší než placebo ve všech charakteristikách aktivity onemocnění (p < 0,001) a odpovědi na samotnou léčbu etanerceptem a na společné podávání etanerceptu s methotrexátem byly podobné. Kvalita života pacientů s psoriatickou artritidou byla vždy hodnocena za použití HAQ indexu invalidity. Skóre indexu invalidity se signifikantně zlepšilo ve všech hodnocených obdobích u pacientů s psoriatickou artritidou léčených etanerceptem ve srovnání s pacienty léčenými placebem (p < 0,001).
Ve studii u pacientů s psoriatickou artritidou byly hodnoceny radiografické změny. Radiogramy rukou a zápěstí byly pořízeny při zahájení a po 6, 12 a 24 měsících. Hodnoty upraveného TSS po 12 měsících jsou uvedeny v následující tabulce. V analýze, ve které všichni pacienti, z jakéhokoli důvodu vyřazení ze studie, byli považováni za pacienty s progresí, bylo po 12 měsících ve skupině léčené etanerceptem vyšší procento pacientů bez progrese (změna TSS < 0,5), než ve srovnávací skupině léčené placebem (73 % vs. 47 %, p < 0,001). Účinek etanerceptu na radiografickou progresi přetrvával u pacientů, kteří pokračovali v léčení během druhého roku. Zpomalení poškození periferních kloubů bylo pozorováno u pacientů se symetrickým polyartikulárním kloubním postižením.
Střední hodnota (SE) roční změny výchozích hodnot v celkovém Sharpově skóre
|
Čas |
Placebo (n = 104) |
Etanercept (n = 101) |
|
12. měsíc |
1,00 (0,29) |
-0,03 (0,09)a |
SE = standardní chyba
a p = 0,0001.
Léčení etanerceptem vedlo k fyzickému funkčnímu zlepšení ve dvojitě zaslepené etapě studie a tento přínos přetrvával po dobu dlouhodobější expozice až po 2 roky.
Pro nízký počet pacientů ve studii není dostatečný průkaz účinnosti etanerceptu u pacientů s chorobou podobnou ankylozující spondylitidě a s mutilující artritidou doprovázející psoriatické artropatie.
U pacientů s psoriatickou artritidou nebyla provedena žádná studie pro použití dávkovacího režimu 50 mg jednou týdně. Průkaz účinnosti pro dávkovací režim jedenkrát týdně byl u této populace pacientů založen na údajích ze studie u pacientů s ankylozující spondylitidou.
Dospělí pacienti s ankylozující spondylitidou
Účinnost etanerceptu u ankylozující spondylitidy byla hodnocena ve 3 randomizovaných, dvojitě zaslepených studiích, porovnávajících podávání etanerceptu 25 mg dvakrát týdně s placebem. Celkem bylo zařazeno 401 pacientů, z nichž 203 bylo léčeno etanerceptem. Nejrozsáhlejší z těchto studií (n = 277) zahrnovala pacienty ve věku od 18 do 70 let, kteří měli aktivní ankylozující spondylitidu definovanou na základě stupnice „visual analog scale“ (VAS) se skóre > 30 u průměrné délky trvání a intenzity ranní ztuhlosti a VAS skóre > 30 u nejméně dvou ze tří následujících parametrů: pacientovo celkové hodnocení; průměr VAS hodnot pro noční bolest zad a celkovou bolest zad; průměr 10 u otázek ze stupnice „Bath Ankylosing Spondylitis Functional Index“ (BASFI). Pacienti léčení DMARD, NSAID nebo kortikosteroidy mohli pokračovat v jejich podávání ve stabilních dávkách. Do studie nebyli zařazeni pacienti s úplnou ankylózou páteře. 138 pacientům byla podávána dávka 25 mg etanerceptu (na základě studií pro stanovení dávky u pacientů s revmatoidní artritidou) nebo placeba subkutánně 2x týdně po dobu 6 měsíců.
Primárním měřítkem účinnosti (ASAS 20) bylo zlepšení o 20 a více % v alespoň 3 ze 4 domén (pacientovo celkové hodnocení, bolest zad, BASFI a zánět) „Assessment in Ankylosing Spondylitis (ASAS) a žádné zhoršení ve zbývající doméně. U ASAS 50 a 70 odpovědí byla pro 50 procentní, resp. 70 procentní zlepšení použita stejná kritéria.
Léčba etanerceptem ve srovnání s placebem vedla k signifikantnímu zlepšení v ASAS 20, ASAS 50 a ASAS 70 již 2 týdny po zahájení terapie.
Odpovědi pacientů s ankylozující spondylitidou v placebem kontrolované studii
|
Procento pacientů | ||
|
Odpověď ankylozující spondylitidy |
Placebo n = 139 |
Etanercept n = 138 |
|
ASAS 20 | ||
|
2 týdny |
22 |
46a |
|
3 měsíce |
27 |
60a |
|
6 měsíců |
23 |
58a |
|
ASAS 50 | ||
|
2 týdny |
7 |
24a |
|
3 měsíce |
13 |
45a |
|
6 měsíců |
10 |
42a |
|
ASAS 70 | ||
|
2 týdny |
2 |
12 |
|
3 měsíce |
7 |
19 |
|
6 měsíců |
5 |
18 |
p < 0,001, etanercept versus placebo b p = 0,002, etanercept versus placebo
U pacientů s ankylozující spondylitidou léčených etanerceptem byla klinická odpověď zřejmá od první návštěvy (po 2 týdnech) a přetrvávala po 6 měsíců léčení. Klinická odpověď byla podobná jak u pacientů léčených při zahájení dalšími léčivy, tak u těch, kteří žádnou další terapii nedostávali.
Obdobné výsledky byly zjištěny i ve dvou menších studiích u pacientů s ankylozující spondylitidou.
Ve čtvrté studii, která byla dvojitě zaslepená a placebem kontrolovaná, byly hodnoceny bezpečnost a účinnost 50 mg etanerceptu (dvě s.c. injekce po 25 mg) podaných jednou týdně a 25 mg etanerceptu podaných dvakrát týdně 356 pacientům s aktivní ankylozující spondylitidou. Profily bezpečnosti a účinnosti dávkovacích režimů 50 mg jednou týdně a 25 mg dvakrát týdně byly podobné.
Dospělí pacienti s radiograficky neprokazatelnou axiální spondylartritidou
Účinnost etanerceptu u pacientů s radiograficky neprokazatelnou axiální spondylartritidou (nr-AxSpa) byla hodnocena v randomizované, 12 týdenní, dvojitě zaslepené, placebem kontrolované studii. Studie hodnotila 215 dospělých pacientů (modifikovaná populace určená k léčbě „intent-to-treat“)s aktivním onemocněním nr-AxSpa (ve věku 18 až 49 let), definovaných jako pacienti, kteří splňovali kritéria klasifikace ASAS pro axiální spondylartritidu, avšak nesplňovali modifikovaná New York kritéria pro AS. Dále se vyžadovalo, aby měli pacienti nedostatečnou odpověď nebo intoleranci na dvě či více NSAID. V dvojitě zaslepeném období dostávali pacienti etanercept v dávce 50 mg týdně nebo placebo po dobu 12 týdnů. Primárním měřítkem účinnosti (ASAS 40) bylo 40% zlepšení v alespoň třech ze čtyř domén ASAS a žádné zhoršení ve zbývající doméně. Po dvojitě zaslepeném období následovalo nezaslepené období, během něhož všichni pacienti dostávali etanercept v dávce 50 mg týdně až po dalších 92 týdnů. Při zahájení a ve 12. a 104. týdnu se provedlo zobrazení sakroiliakálního kloubu a páteře pomocí MR ke zhodnocení zánětu.
V porovnání s placebem vedla léčba etanerceptem k statisticky významnému zlepšení v ASAS 40, ASAS 20 a ASAS 5/6. Významné zlepšení bylo pozorováno také u ASAS částečné remise a BASDAI 50. Výsledky z 12. týdne jsou uvedeny v tabulce níže.
Odpověď z hlediska účinnosti v placebem kontrolované studii nr-AxSpa: procento pacientů, kteří dosáhli cílových parametrů
|
Dvojitě zaslepené klinické odpovědi v 12. týdnu |
Placebo n = 106 až 109* |
Etanercept n = 103 až 105* |
|
ASAS** 40 |
15,7 |
32,4b |
|
ASAS 20 |
36,1 |
52,4c |
|
ASAS 5/6 |
10,4 |
33,0a |
|
ASAS částečná remise |
11,9 |
24,8c |
|
BASDAI***50 |
23,9 |
43,8b |
*Někteří pacienti neposkytli úplné údaje pro každý cílový parametr **ASAS=Assessments in Spondyloarthritis International Society ***Bath Ankylosing Spondylitis Disease Activity Index a: p < 0,001, b: < 0,01 a c: < 0,05, v daném pořadí, mezi etanerceptem a placebem
V 12. týdnu došlo u pacientů užívajících etanercept k statisticky významnému zlepšení skóre SPARCC (Spondyloarthritis Research Consortium of Canada) u sakroiliakálního kloubu (SIJ), na základě měření pomocí zobrazení MR. Upravená průměrná změna od výchozí hodnoty činila 3,8 u pacientů léčených etanerceptem (n = 95) oproti 0,8 u pacientů léčených placebem (n = 105)
(p < 0,001). V týdnu 104 byla průměrná změna od výchozí hodnoty ve skóre SPARCC na základě měření pomocí zobrazení MR u všech pacientů užívajících přípravek etanercept 4,64 u SIJ (n=153) a 1,40 u páteře (n= 154).
Etanercept vykazoval statisticky významně větší zlepšení oproti placebu za období od zahájení studie do 12. týdne u většiny hodnocení kvality života souvisejících se zdravím a tělesnými funkcemi, včetně BASFI (Bath Ankylosing Spondylitis Functional Index), EuroQol 5D Overall Health State Score a SF-36 Physical Component Score.
Klinická odpověď u pacientů s onemocněním nr-AxSpa, kterým byl podáván etanercept, byla zjevná při první návštěvě (2 týdny) a přetrvala po celou dobu 2leté léčby. Zlepšení kvality života související se zdravím a tělesnými funkcemi rovněž přetrvala po celou dobu 2leté léčby. Údaje z těchto 2 let neukázaly žádná nová bezpečnostní zjištění. V týdnu 104 progredovalo 8 pacientů do stupně 2 bilaterálního skóre RTG páteře podle modifikovaných Newyorských kritérií, svědčící pro axiální formu spondyloartropatie.
Dospělí pacienti s ložiskovou psoriázou
Etanercept se doporučuje podávat pacientům definovaným v bodu 4.1. Pacienti „bez léčebné odpovědi“ jsou v cílové populaci definováni nedostatečnou odpovědí (PASI < 50 nebo PGA méně než dobrý), nebo zhoršením nemoci v průběhu léčení, pokud byli léčení minimálně třemi nejvýznamnějšími dostupnými způsoby celkové terapie, a dostávali přiměřené dávky po dostatečnou dobu k vyhodnocení odpovědi.
Účinnost etanerceptu v porovnání s jinými způsoby celkové terapie u pacientů se středně těžkou až těžkou psoriázou (reagující na jiné způsoby celkové terapie) nebyla ve studiích vyhodnocována přímo porovnáním etanerceptu s jinými způsoby celkovými terapiemi. Místo toho byly vyhodnoceny bezpečnost a účinnost ve čtyřech randomizovaných dvojitě zaslepených, placebem kontrolovaných studiích. Primárním kritériem účinnosti ve všech čtyřech studiích byl podíl pacientů v každé léčebné skupině, kteří dosáhli po 12 týdnech PASI 75 (tj. minimálně 75% zlepšení ve skóre Psoriasis Area and Severity Index oproti výchozí hodnotě).
Studie 1 byla studií 2. fáze klinického hodnocení u pacientů ve věku > 18 let s aktivní, ale klinicky stabilní, ložiskovou psoriázou na ploše > 10% povrchu těla (BSA). 112 pacientů bylo randomizováno do skupiny, která dostávala dávku 25 mg etanerceptu (n = 57) nebo placebo (n = 55) 2x týdně po dobu
24 týdnů.
Studie 2 hodnotila 652 pacientů s chronickou ložiskovou psoriázou při použití stejných zařazovacích kritérií jako studie 1 spolu s minimální psoriatickou plochou a indexem závažnosti (PASI) 10 při screeningu. Etanercept byl podáván po dobu 6 po sobě jdoucích měsíců v dávkách 25 mg 1 x týdně,
25 mg 2x týdně nebo 50 mg 2x týdně. V období prvních 12 týdnů dvojitě zaslepené terapie dostávali pacienti placebo nebo jednu z výše uvedených dávek etanerceptu. Po 12 týdnech léčení začali dostávat pacienti v placebové skupině zaslepený etanercept (25 mg 2x týdně); pacienti v aktivně léčených skupinách pokračovali až do 24. týdne v dávkách podle skupin, do nichž byli původně randomizováni.
Studie 3 hodnotila 583 pacientů a měla stejná zařazovací kritéria jako studie 2. Pacienti v této studii dostávali po dobu 12 týdnů 2x týdně etanercept v dávkách 25 mg, nebo 50 mg, nebo placebo. Potom po dobu následujících 24 týdnů dostávali všichni pacienti nezaslepený etanercept v dávkách 25 mg 2x týdně.
Studie 4 hodnotila 142 pacientů a měla podobná zařazovací kritéria jako studie 2 a 3. Pacienti v této studii dostávali po dobu 12 týdnů jednou týdně dávku 50 mg etanerceptu nebo placebo, a potom po dobu následujících 12 týdnů dostávali všichni pacienti nezaslepený etanercept v dávce 50 mg jednou týdně.
Ve studii 1 měla skupina léčená etanerceptem signifikantně vyšší podíl pacientů s klinickou odpovědí PASI 75 ve 12. týdnu (30 %) v porovnání se skupinou léčenou placebem (2 %) (p < 0,0001). Po 24 týdnech dosáhlo PASI 75 56 % pacientů ve skupině léčené etanerceptem v porovnání s 5 % pacientů léčených placebem. Klíčové výsledky studií 2, 3 a 4 jsou uvedeny níže.
Vzácné:
Velmi vzácné:
Pruritus
Angioedém, kopřivka, vyrážka, psoriatiformní vyrážka, psoriáza (včetně nově začínající nebo zhoršující se a pustulózní, zejména dlaní a chodidel)
Kožní vaskulitida (včetně leukocystoklastické vaskulitidy), Stevensonův-Johnsonův syndrom, erythema multiforme Toxická epidermální nekrolýza_
Poruchy svalové a kosterní soustavy a pojivové tkáně:
Vzácné: Subakutní kožní lupus erythematodes, diskoidní lupus
erythematodes, lupus-like syndrom
Celkové poruchy a reakce v místě aplikace:_
Velmi časté: Reakce v místě injekce (včetně krvácení, podlitin, erytému,
svědění, bolesti, otoku)*
Časté: Horečka
Odpovědi pacientů s psoriázou ve studiích 2, 3 a 4
|
Odpově ď (%) |
Studie 2 |
Studie 3 |
Studie 4 | ||||||||
|
Placeb o |
Etanercept |
Placeb o |
Etanercept |
Placeb o |
Etanercept | ||||||
|
25 mg dvakrát týdně |
50 mg dvakrát týdně |
25 mg dvakrá t týdně |
50 mg dvakrá t týdně |
50 mg jedno u týdně |
50 mg jedno u týdně | ||||||
|
n = 166 |
n = 162 |
n = 162 |
n = 164 |
n = 164 |
n = 193 |
n = 196 |
n = 196 |
n = 46 |
n = 96 |
n = 90 | |
|
12. týden |
12. týde n |
24. týden a |
12. týde n |
24. týden a |
12. týden |
12. týden |
12. týden |
12. týden |
12. týden |
24. týdena | |
|
PASI 50 |
14 |
58* |
70 |
74* |
77 |
9 |
64* |
77* |
9 |
69* |
83 |
|
PASI 75 |
4 |
34* |
44 |
49* |
59 |
3 |
34* |
49* |
2 |
38* |
71 |
|
DSGAb, čistá nebo téměř čistá |
5 |
34* |
39 |
49* |
55 |
4 |
39* |
57* |
4 |
39* |
64 |
p < 0,0001 porovnáváno s placebem
a Ve studii 2 a 4 nebylo ve 24. týdnu provedeno statistické porovnání s placebem, protože skupina původně léčená placebem začala dostávat od 13. do 24. týdne etanercept v dávce 25 mg 2x týdně nebo 50 mg jednou týdně. b Dermatologist Static Global Assessment. Čistá nebo téměř čistá je definována jako 0 nebo 1 na stupnici 0 až 5.
U pacientů s ložiskovou psoriázou, kteří dostávali etanercept, byly zjevné signifikantní odpovědi v porovnání s placebem v době první kontroly (2 týdny) a přetrvávaly po 24 týdnů léčení.
Studie 2 měla také období po vysazení terapie, kdy bylo léčení ukončeno ve 24. týdnu u pacientů, kteří dosáhli zlepšení PASI minimálně o 50 %. U pacientů byl sledován výskyt rebound fenoménu (PASI > 150 % od úvodní hodnoty) a doba do relapsu (definován jako ztráta nejméně poloviny zlepšení dosaženého ve 24. týdnu v porovnání s úvodní hodnotou). V období po ukončení léčení se symptomy psoriázy postupně vrátily s průměrnou dobou 3 měsíce do relapsu onemocnění. Nebyl pozorován rebound fenomén ani závažné nežádoucí účinky spojené s psoriázou. Existují některé důkazy podporující prospěšnost opakované terapie etanerceptem u pacientů, kteří od počátku odpovídají na léčení.
Ve studii 3 si většina pacientů (77 %), kteří byli původně randomizováni do skupiny léčené 50 mg etanerceptu 2x týdně a měli od 12. týdne snížené dávky na 25 mg 2x týdně, udržela odpověď PASI 75 do 36. týdne. U pacientů léčených po celou dobu studie dávkami 25 mg 2x týdně, se mezi 12. a 36. týdnem odpověď PASI 75 postupně zlepšovala.
Ve studii 4 měla etanerceptem léčená skupina pacientů vyšší podíl pacientů s klinickou odpovědí PASI 75 ve 12. týdnu (38 %) v porovnání se skupinou léčenou placebem (2 %) (p < 0,0001). U pacientů léčených po celou dobu studie dávkami 50 mg jednou týdně, se odpověď i nadále zlepšovala a ve 24. týdnu dosáhla u 71 % PASI 75.
V dlouhodobých otevřených studiích (až 34 měsíců), v nichž byl etanercept podáván nepřetržitě, byly udrženy klinické odpovědi a bezpečnost byla srovnatelná s krátkodobými studiemi.
Analýza údajů z klinických studií neprokázala žádné charakteristiky základního onemocnění, které by napomohly klinikům ve výběru nejvhodnější možnosti dávkování (přerušované nebo kontinuální). Proto musí být výběr přerušované nebo kontinuální terapie založen na lékařském posouzení a na individuálních potřebách pacienta.
Protilátky proti etanerceptu
V séru některých osob léčených etanerceptem byly zjištěny protilátky proti etanerceptu. Všechny tyto protilátky byly non-neutralizační a jejich výskyt byl zpravidla dočasný. Zdá se, že mezi vznikem protilátek, klinickou odpovědí ani nežádoucími účinky není žádná souvislost.
5.2 Farmakokinetické vlastnosti
Hodnoty etanerceptu v séru byly určovány metodou enzymatické imunoanalýzy (ELISA), která může detekovat ELISA-reaktivní degradační produkty, jakož i původní látku.
Absorpce
Etanercept je pomalu absorbován z místa subkutánní injekce a po jednotlivé dávce dosahuje maximální koncentraci přibližně po 48 hodinách. Absolutní biologická dostupnost je 76 %. Při dávkování 2x týdně se očekávají ustálené koncentrace, které jsou přibližně dvakrát tak vysoké než koncentrace zjištěné po jednotlivé dávce. Průměrná maximální sérová koncentrace po podání jednotlivé subkutánní dávky 25 mg etanerceptu zdravým dobrovolníkům dosahovala 1,65 ± 0,66 pg/ml a plocha pod křivkou byla 235 ± 96,6 pg*h/ml.
Profily středních hodnot koncentrací v ustáleném stavu u pacientů s revmatoidní artritidou, léčených 50 mg etanerceptu jednou týdně (n = 21) vs. 25 mg etanercept 2x týdně (n = 16) byly Cmax 2,4 mg/l vs. 2,6 mg/l, Cmin byly 1,2 mg/l vs. 1,4 mg/l a parciální AUC byla 297 mg*h/l vs. 316 mg*h/l. V otevřené jednodávkové dvourežimové zkřížené studii na zdravých dobrovolnících byl shledán etanercept podávaný v jednotlivé injekci 50 mg/ml bioekvivalentním se dvěma jednotlivými injekcemi po 25 mg/ml podanými současně.
V populační farmakokinetické analýze pacientů s ankylozující spondylitidou byly ustálené stavy AUCs etanerceptu 466 pg*h/ml pro 50 mg etanerceptu jednou týdně (n = 154) a 474 pg*h/ml pro 25 mg etanerceptu dvakrát týdně (n = 148).
Distribuce
Závislost koncentrace etanerceptu na čase zobrazuje biexponenciální křivka. Celkový distribuční objem etanerceptu je 7,6 l, zatímco distribuční objem v ustáleném stavu je 10,4 l.
Eliminace
Etanercept je pomalu vylučován z organismu. Poločas eliminace je dlouhý, přibližně 70 hodin. Clearance je u pacientů s revmatoidní artritidou přibližně 0,066 l/h, tj. o něco nižší než hodnota 0,11 l/h pozorovaná u zdravých dobrovolníků. Navíc farmakokinetika etanerceptu u pacientů s revmatoidní artritidou, u pacientů s ankylozující spondylitidou a u pacientů s ložiskovou psoriázou je podobná.
Není zjevný rozdíl ve farmakokinetice u žen a mužů.
Linearita
Závislost na dávce nebyla formálně hodnocena, ale v dávkovém rozmezí není zřejmé nasycení clearance.
Zvláštní populace
Porucha funkce ledvin
I když po podání radioaktivně značeného etanerceptu zdravým dobrovolníkům i pacientům byla radioaktivita vylučována močí, u pacientů s akutním renálním selháním nebyla pozorována zvýšená koncentrace etanerceptu. Renální porucha by tedy neměla vyžadovat změnu dávkování.
Porucha funkce jater
U pacientů s akutním jaterním selháním nebyla pozorována zvýšená koncentrace etanerceptu. Jaterní porucha by tedy neměla vyžadovat změnu dávkování.
Starší pacienti
Vliv pokročilého věku na koncentrace etanerceptu v séru byl zkoumán populační farmakokinetickou analýzou. Clearance a objem zjištěné u pacientů ve věku od 65 do 87 let byly podobné hodnotám stanoveným u pacientů, kteří byli mladší než 65 let.
5.3 Předklinické údaje vztahující se k bezpečnosti
V toxikologických studiích s etanerceptem nebyla zjevná žádná dávku limitující toxicita ani toxicita v cílových orgánech. Podle výsledků celé řady studií in vitro a in vivo je etanercept považován za negenotoxický. Studie karcinogenity a standardní hodnocení fertility a postnatální toxicity nebyly s etanerceptem provedeny v důsledku zjištění vzniku neutralizujících protilátek u hlodavců.
Etanercept nevyvolal úmrtí ani zjevné příznaky toxicity u myší nebo potkanů po podání jednotlivé dávky 2 000 mg/kg subkutánně nebo jednotlivé dávky 1 000 mg/kg intravenózně. Etanercept nevykazoval toxicitu limitující velikost dávky nebo orgánovou toxicitu u opic druhu cynomolgus po podávání 2x týdně subkutánně po 4 nebo 26 po sobě jdoucích týdnů v dávce 15 mg/kg, což vedlo k sérovým koncentracím podle velikosti plochy pod křivkou více než 27x vyšším než zjištěným u lidí po doporučené dávce 25 mg.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Sacharóza Chlorid sodný
Monohydrát dihydrogenfosforečnanu sodného Heptahydrát hydrogenfosforečnanu sodného Voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti
30 měsíců.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Uchovávejte předplněné injekční stříkačky nebo pera v krabičce, aby byl přípravek chráněn před světlem.
Přípravek Benepali lze jednorázově po dobu až čtyř týdnů uchovávat při teplotě maximálně do 25 °C; po této době nesmí být znovu uchováván v chladu (v chladničce). Přípravek Benepali musí být zlikvidován, pokud není použit do čtyř týdnů po vyjmutí z chladničky.
6.5 Druh obalu a velikost balení
50 mg, injekční roztok v předplněné injekční stříkačce
Injekční stříkačka z čirého skla (typ I) s jehlou z nerezové oceli, krytkou jehly z pryže a táhla z plastické hmoty obsahující 0,98 ml roztoku.
Přípravek Benepali je dostupný v baleních obsahujících 4 předplněné injekční stříkačky a baleních s více kusy obsahujících 12 (3 balení po 1) předplněných injekčních stříkaček. Na trhu nemusí být všechny velikosti balení.
50 mg, injekční roztok v předplněném peru
Předplněné pero obsahuje předplněnou stříkačku přípravku Benepali. Injekční stříkačka uvnitř pera je vyrobena z čirého skla typu 1 s jehlou z nerezové oceli o velikosti 27, krytkou jehly z pryže a táhla z plastické hmoty.
Přípravek Benepali je dostupný v baleních obsahujících 4 předplněná pera a baleních s více kusy obsahujících 12 (3 balení po 4) předplněných per. Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
50 mg, injekční roztok v předplněné injekční stříkačce
Před použitím počkejte, než přípravek Benepali v předplněné injekční stříkačce pro jedno použití dosáhne pokojové teploty (přibližně 30 minut). Po dobu než předplněná injekční stříkačka dosáhne pokojové teploty, se nemá snímat kryt jehly. Roztok má být čirý až mírně opalescentní, bezbarvý nebo světle žlutý a může obsahovat malé průsvitné nebo bílé částice proteinu.
Úplný návod na podání je k dispozici v příbalové informaci, bod 7 „Návod k použití“.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
50 mg, injekční roztok v předplněném peru
Před použitím počkejte, než přípravek Benepali v předplněných perech pro jedno použití dosáhne pokojové teploty (přibližně 30 minut). Po dobu než předplněné pero dosáhne pokojové teploty, se nemá snímat kryt jehly. Při pohledu skrz kontrolní okénko má být roztok čirý až mírně opalescentní, bezbarvý nebo světle žlutý a může obsahovat malé průsvitné nebo bílé částice proteinu.
Úplný návod na podání je k dispozici v příbalové informaci, bod 7 „Návod k použití“.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Samsung Bioepis UK Limited 3000 Hillswood Drive Chertsey Surrey KT16 0RS Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/15/1074/001
EU/1/15/1074/002
EU/1/15/1074/003
EU/1/15/1074/004
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 14 leden 2016
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce biologické léčivé látky Biogen (Denmark) Manufacturing ApS Biogen Allé 1 3400 Hillered Dánsko
Název a adresa výrobce odpovědného za propouštění šarží
Biogen (Denmark) Manufacturing ApS
Biogen Allé 1
3400 Hillered
Dánsko
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl.
107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
Držitel rozhodnutí o registraci předloží první pravidelně aktualizovanou zprávu o bezpečnosti pro tento léčivý přípravek do 6 měsíců od jeho registrace.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
• Další opatření k minimalizaci rizik
1. Před uvedením na trh v každém členském státě odsouhlasí držitel rozhodnutí o registraci konečný vzdělávací materiál s kompententním orgánem v členském státě, kdy tento bude obsahovat informace poskytované všem zdravotníkům, u nichž se očekává, že budou předepisovat přípravek pro správné a
bezpečné používání předplněného pera/předplněných injekčních stříkaček, aby je informoval o tom, že přípravek není určen k použití u dětí, a dále pohotovostní kartu pacienta, která bude vydána pacientům užívajícím přípravek Benepali.
2. Vzdělávací materiál pro zdravotníky by měl obsahovat následující klíčové prvky:
• Průvodce výukou, který umožní školení pacientů v bezpečném používání předplněného pera/předplněných injekčních stříkaček
• Ukázkové zařízení bez jehly
• Materiál, který bude připomínat zdravotníkům, že přípravek Benepali není určen pro použití u dětí
• Materiály s pokyny určenými pro pacienty.
3. Pohotovostní karty pacienta bude obsahovat následující klíčové prvky určené pacientům léčeným přípravkem Benepali:
• Riziko oportunních infekcí a tuberkulózy (TB)
• Riziko městnavého srdečního selhání (MSS)
• Přípravek Benepali není určen k použití u dětí.
Benepali 50 mg, injekční roztok v předplněné injekční stříkačce etanerceptum
Jedna předplněná injekční stříkačka obsahuje etanerceptum 50 mg v 1 ml.
Pomocné látky:
Sacharóza, chlorid sodný, monohydrát dihydrogenfosforečnanu sodného, heptahydrát hydrogenfosforečnanu sodného, voda na injekci.
Injekční roztok v předplněné injekční stříkačce 4 předplněné injekční stříkačky
Před použitím si přečtěte příbalovou informaci.
Subkutánní podání
Před použitím vyjměte injekční stříkačku z chladničky a nechte při pokojové teplotě 30 minut. Otevřete vytažením
Uchovávejte mimo dohled a dosah dětí.
Uchovávejte v chladničce.
Chraňte před mrazem.
Uchovávejte předplněné injekční stříkačky v krabičce, aby byl přípravek chráněn před světlem. Dodatečné podmínky uchovávání viz příbalová informace.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Samsung Bioepis UK Limited 3000 Hillswood Drive Chertsey
Surrey KT16 0RS Velká Británie
EU/1/15/1074/001
č.s.:
Benepali 50 mg
Benepali 50 mg, injekční roztok v předplněné injekční stříkačce etanerceptum
Jedna předplněná injekční stříkačka obsahuje etanerceptum 50 mg v 1 ml.
Pomocné látky:
Sacharóza, chlorid sodný, monohydrát dihydrogenfosforečnanu sodného, heptahydrát hydrogenfosforečnanu sodného, voda na injekci.
Injekční roztok v předplněné injekční stříkačce
Balení s více kusy: 12 (3 balení po 4) předplněných injekčních stříkaček
Před použitím si přečtěte příbalovou informaci.
Subkutánní podání
Před použitím vyjměte injekční stříkačku z chladničky a nechte při pokojové teplotě 30 minut.
Uchovávejte mimo dohled a dosah dětí.
Uchovávejte v chladničce.
Chraňte před mrazem.
Uchovávejte předplněné injekční stříkačky v krabičce, aby byl přípravek chráněn před světlem. Dodatečné podmínky uchovávání viz příbalová informace.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
Samsung Bioepis UK Limited 3000 Hillswood Drive Chertsey
Surrey KT16 0RS Velká Británie
EU/1/15/1074/003
č.s.:
Benepali 50 mg
Benepali 50 mg, injekční roztok v předplněné injekční stříkačce etanerceptum
Jedna předplněná injekční stříkačka obsahuje etanerceptum 50 mg v 1 ml.
Pomocné látky:
Sacharóza, chlorid sodný, monohydrát dihydrogenfosforečnanu sodného, heptahydrát hydrogenfosforečnanu sodného, voda na injekci.
Injekční roztok v předplněné injekční stříkačce 4 předplněné injekční stříkačky
Součást balení s více kusy, nesmí být prodáváno samostatně
Před použitím si přečtěte příbalovou informaci.
Subkutánní podání
Před použitím vyjměte injekční stříkačku z chladničky a nechte při pokojové teplotě 30 minut. Otevřete vytažením
Uchovávejte mimo dohled a dosah dětí.
EXP:
Uchovávejte v chladničce.
Chraňte před mrazem.
Uchovávejte předplněné injekční stříkačky v krabičce, aby byl přípravek chráněn před světlem. Dodatečné podmínky uchovávání viz příbalová informace.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Samsung Bioepis UK Limited 3000 Hillswood Drive Chertsey
Surrey KT16 0RS Velká Británie
EU/1/15/1074/003
č.š.:
Benepali 50 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Benepali 50 mg injekce etanerceptum Subkutánní podání <s.c.>
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP:
4. ČÍSLO ŠARŽE
c.s.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
1 ml
6. JINÉ
Benepali 50 mg, injekční roztok v předplněném peru etanerceptum
Jedno předplněné pero obsahuje etanerceptum 50 mg v 1 ml.
Pomocné látky:
Sacharóza, chlorid sodný, monohydrát dihydrogenfosforečnanu sodného, heptahydrát hydrogenfosforečnanu sodného, voda na injekci.
Injekční roztok v předplněném peru 4 předplněná pera
Před použitím si přečtěte příbalovou informaci.
Subkutánní podání
Před použitím vyjměte předplněné pero z chladničky a nechte při pokojové teplotě 30 minut. Otevřete vytažením
Uchovávejte mimo dohled a dosah dětí.
Uchovávejte v chladničce.
Chraňte před mrazem.
Uchovávejte předplněná pera v krabičce, aby byl přípravek chráněn před světlem. Dodatečné podmínky uchovávání viz příbalová informace.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Samsung Bioepis UK Limited 3000 Hillswood Drive Chertsey
Surrey KT16 0RS Velká Británie
EU/1/15/1074/002
č.s.:
Benepali 50 mg
Benepali 50 mg, injekční roztok v předplněném peru etanerceptum
Jedno předplněné pero obsahuje etanerceptum 50 mg v 1 ml.
Pomocné látky:
Sacharóza, chlorid sodný, monohydrát dihydrogenfosforečnanu sodného, heptahydrát hydrogenfosforečnanu sodného, voda na injekci.
Injekční roztok v předplněném peru
Balení s více kusy: 12 (3 balení po 4) předplněná pera
Před použitím si přečtěte příbalovou informaci.
Subkutánní podání
Před použitím vyjměte předplněné pero z chladničky a nechte při pokojové teplotě 30 minut.
Uchovávejte mimo dohled a dosah dětí.
Uchovávejte v chladničce.
Chraňte před mrazem.
Uchovávejte předplněná pera v krabičce, aby byl přípravek chráněn před světlem. Dodatečné podmínky uchovávání viz příbalová informace.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
Samsung Bioepis UK Limited 3000 Hillswood Drive Chertsey
Surrey KT16 0RS Velká Británie
EU/1/15/1074/004
č.s.:
Benepali 50 mg
Benepali 50 mg, injekční roztok v předplněném peru etanerceptum
Jedno předplněné pero obsahuje etanerceptum 50 mg v 1 ml.
Pomocné látky:
Sacharóza, chlorid sodný, monohydrát dihydrogenfosforečnanu sodného, heptahydrát hydrogenfosforečnanu sodného, voda na injekci.
Injekční roztok v předplněném peru 4 předplněná pera
Součást balení s více kusy, nesmí být prodáváno samostatně
Před použitím si přečtěte příbalovou informaci.
Subkutánní podání
Před použitím vyjměte předplněné pero z chladničky a nechte při pokojové teplotě 30 minut. Otevřete vytažením
Uchovávejte mimo dohled a dosah dětí.
EXP:
Uchovávejte v chladničce.
Chraňte před mrazem.
Uchovávejte předplněná pera v krabičce, aby byl přípravek chráněn před světlem. Dodatečné podmínky uchovávání viz příbalová informace.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Samsung Bioepis UK Limited 3000 Hillswood Drive Chertsey
Surrey KT16 0RS Velká Británie
EU/1/15/1074/004
č.š.:
Benepali 50 mg
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITŘNÍM OBALU ŠTÍTEK (PŘEDPLNĚNÉ PERO)_
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Benepali 50 mg, injekční roztok v předplněném peru
etanerceptum
Subkutánní podání <s.c.>
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP:
4. ČÍSLO ŠARŽE
č.s.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
1 ml
6. JINÉ
Pohotovostní karta pacienta Přípravku Benepali etanerceptum
Tato karta obsahuje důležité bezpečnostní informace, které musíte vzít na vědomí předtím, než budete přípravek Benepali užívat, a během léčby přípravkem Benepali. Pokud těmto informacím nebudete rozumět, požádejte svého lékaře, aby Vám je vysvětlil. Tento přípravek je určen pro dospělé ve věku 18 let a starší.
• Ukažte tuto kartu každému lékaři, který se podílí na Vaší léčbě.
Použití u dětí a dospívajících (ve věku nižším než 18 let)
Přípravek Benepali není indikován k použití u dětí a dospívajících. Pokud máte jakékoliv dotazy ohledně tohoto, pohovořte si o tom se svým lékařem.
Infekce
Přípravek Benepali může u Vás zvýšit riziko vzniku infekcí, které by mohly být závažné.
• Pokud máte infekci, nepoužívejte přípravek Benepali. Pokud si nejste jisti, zeptejte se svého lékaře.
• Pokud se u Vás rozvinou příznaky naznačující přítomnost infekce, jako je horečka, přetrvávající kašel, úbytek tělesné hmotnosti nebo malátnost, ihned vyhledejte lékařskou pomoc.
• Měli byste být vyšetřeni na tuberkulózu (TBC). Požádejte svého lékaře, aby zaznamenal data a výsledky Vašeho posledního screeningového vyšetření na TBC níže:
Test:_Test:_
Datum:_Datum:_
Výsledky:_Výsledky:_
• Požádejte svého lékaře, aby Vám vypsal seznam dalších léků, které u Vás mohou zvyšovat riziko infekce. 1 2
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro uživatele
Benepali 50 mg, injekční roztok v předplněné injekční stříkačce
etanerceptum
'VTento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Lékař Vám rovněž vydá informační kartu pacienta, která obsahuje důležité bezpečnostní informace, které musíte vzít před léčbou přípravkem Benepali a během ní na vědomí.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Benepali a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Benepali používat
3. Jak se přípravek Benepali používá
4. Možné nežádoucí účinky
5. Jak přípravek Benepali uchovávat
6. Obsah balení a další informace
7. Návod k použití přípravku Benepali (viz druhá strana)
1. Co je přípravek Benepali a k čemu se používá
Přípravek Benepali je biologické léčivo, které se vyrábí ze dvou lidských bílkovin. Blokuje aktivitu jiné bílkoviny v těle, která způsobuje zánět. Přípravek Benepali snižuje zánět spojený s určitými onemocněními. Přípravek Benepali obsahuje léčivou látku etanercept.
U dospělých (věk 18 let a více) se může přípravek Benepali použít k léčení:
• středně těžkého až těžkého revmatoidního zánětu kloubů (revmatoidní artritida),
• revmatoidního zánětu kloubů s lupénkou (psoriatická artritida),
• závažných axiálních spondylartritid (záněty kloubů v oblasti páteře) včetně ankylozující spondylitidy (Bechtěrevova nemoc),
• středně těžké až těžké lupénky (psoriáza).
Ve všech případech se přípravek Benepali obvykle používá, pokud běžně používané léčebné postupy nebyly dostatečně účinné nebo nejsou pro Vás vhodné.
U revmatoidní artritidy se přípravek Benepali obvykle používá v kombinaci s methotrexátem, i když se může použít také samotný, jestliže léčení methotrexátem pro Vás není vhodné. Přípravek Benepali použitý samostatně nebo v kombinaci s methotrexátem může zpomalit poškození kloubů, způsobené revmatoidní artritidou, a zlepšit Vaši schopnost provádět normální denní činnosti.
U pacientů s psoriatickou artritidou postihující více kloubů může přípravek Benepali zlepšit schopnost provádět normální denní činnosti.
U pacientů se symetrickým postižením kloubů bolestí nebo otokem (např. ruce, zápěstí a nohy) může přípravek Benepali zpomalit strukturální poškození těchto kloubů způsobená chorobou.
2. Čemu musíte věnovat pozornost, než začnete přípravek Benepali používat
Nepoužívejte přípravek Benepali
• jestliže jste alergický(á) na etanercept nebo kteroukoli další složku přípravku Benepali
(uvedenou v bodě 6);
• jestliže Vám hrozí rozvoj závažné krevní infekce zvané sepse (otrava krve). Jestliže si nejste jist(a), poraďte se s lékařem;
• jestliže máte jakýkoli druh infekce. Jestliže si nejste jist(a), poraďte se s lékařem.
Upozornění a opatření
Před použitím přípravku Benepali se poraďte se svým lékařem.
• Alergické reakce: Jestliže pocítíte příznaky alergické reakce, jako svírání na prsou, sípání, závratě nebo vyrážku, nepokračujte v podávání přípravku Benepali a ihned vyhledejte lékaře.
• Infekce/operace: Jestliže se u Vás rozvíjí nová infekce nebo jste před velkou chirurgickou operací, lékař může chtít sledovat léčbu přípravkem Benepali.
• Infekce/cukrovka: Informujte lékaře, jestliže jste měl(a) opakující se infekce nebo trpíte cukrovkou nebo jinými stavy, při nichž se zvyšuje riziko infekce.
• Infekce/sledování: Informujte svého lékaře o každé cestě mimo Evropu, kterou jste absolvoval(a) v poslední době. Pokud se u Vás rozvinou příznaky infekce, jako je horečka, zimnice nebo kašel, neprodleně informujte svého lékaře. Lékař může rozhodnout, že Vás bude nadále sledovat na přítomnost infekcí i poté, co přestanete používat přípravek Benepali.
• Tuberkulóza: U pacientů léčených přípravkem Benepali byly hlášeny případy tuberkulózy. Lékař vyšetří známky a příznaky tuberkulózy předtím, než zahájí léčení přípravkem Benepali. To může zahrnovat podrobnou lékařskou anamnézu, rentgen hrudníku a tuberkulinový test. Výsledky těchto vyšetření musí být zaznamenány do pohotovostní karty pacienta. Je velmi důležité, abyste informoval(a) lékaře, pokud jste měl(a), nebo pokud jste byl(a) v kontaktu s někým, kdo ji měl. Pokud by se vyskytly příznaky tuberkulózy (jako přetrvávající kašel, ztráta hmotnosti, netečnost, mírná horečka), nebo jiná infekce v průběhu léčení nebo po jeho ukončení, informujte neprodleně lékaře.
• Hepatitida B: Informujte lékaře, pokud máte nebo jste měl/a v minulosti hepatitidu B. Lékař by měl provést vyšetření na přítomnost infekce hepatitidy B předtím, než se začnete léčit přípravkem Benepali. Léčba přípravkem Benepali může vést k reaktivaci hepatitidy B u pacientů, kteří byli v minulosti nakaženi virem hepatitidy B. Pokud k tomu dojde, musíte přestat přípravek Benepali používat.
• Hepatitida C: Informujte lékaře, pokud máte hepatitidu C. Lékař bude sledovat léčení přípravkem Benepali, pokud se infekce zhorší.
• Krevní poruchy: Jestliže se u Vás objeví známky nebo příznaky jako přetrvávající horečka, bolest v krku, podlitiny, krvácení nebo bledost, vyhledejte neodkladně lékařskou pomoc.
Takové příznaky mohou svědčit o možném život ohrožujícím onemocnění krve, které si může vyžádat přerušení podávání přípravku Benepali.
• Poruchy nervového systému a oční poruchy: Informujte lékaře, jestliže máte roztroušenou sklerózu, optickou neuritidu (zánět zrakových nervů) nebo transverzální myelitidu (zánět míchy). Lékař určí, zda je přípravek Benepali vhodnou léčbou.
• Městnavé srdeční selhání: Informujte lékaře, jestliže jste měl(a) v minulosti městnavé srdeční selhání, neboť za těchto okolností je třeba opatrnosti při podávání přípravku Benepali.
• Zhoubná nádorová onemocnění: Než Vám bude předepsán přípravek Benepali, informujte lékaře, pokud máte nebo Vám byl někdy diagnostikován lymfom (typ zhoubného onemocnění krve) či jiný typ zhoubného nádorového onemocnění/rakoviny. U pacientů s těžkou revmatoidní artritidou, kteří trpí tímto onemocněním delší dobu, je riziko vzniku lymfomu vyšší než průměrné. U některých dětí a dospívajících pacientů, kterým byl podáván etanercept nebo jiné léčivé přípravky působící stejně jako etanercept, se rozvinula zhoubná nádorová onemocnění/rakovina včetně neobvyklých typů, což někdy vedlo k úmrtí. Někteří pacienti užívající přípravek Benepali onemocněli rakovinou kůže. Informujte lékaře, pokud u Vás dojde k rozvoji jakékoli změny vzhledu kůže nebo výrůstků na kůži.
• Plané neštovice: Upozorněte lékaře, jestliže jste byli v kontaktu s planými neštovicemi v průběhu podávání přípravku Benepali. Lékař stanoví, zda je vhodné preventivní opatření proti planým neštovicím.
• Zneužívání alkoholu: Přípravek Benepali by se neměl použít u pacientů, kteří se léčí s hepatitidou spojenou se zneužíváním alkoholu. Informujte, prosím, lékaře, pokud máte zneužívání alkoholu v anamnéze.
• Wegenerova granulomatóza: Přípravek Benepali se nedoporučuje k léčení Wegenerovy granulomatózy, vzácného zánětlivého onemocnění. Informujte lékaře, máte-li Wegenerovu granulomatózu.
• Antidiabetické přípravky: Informujte svého lékaře, trpíte-li cukrovkou či užíváte-li léky k léčbě cukrovky. Váš lékař stanoví, zda je u Vás nutné během léčby přípravkem Benepali snížit dávku léků k léčbě cukrovky.
• Očkování: Některá očkování by neměla proběhnout v době podávání přípravku Benepali, např. vakcína proti poliomyelitidě, tj. proti dětské obrně. Prosím, poraďte se s lékařem před případným očkováním.
Děti a dospívající
Přípravek Benepali není indikován k použití u dětí a dospívajících mladších 18 let.
Přípravek Benepali je k dispozici jako 50 mg předplněná injekční stříkačka nebo 50 mg předplněné pero, které jsou nevhodné pro použití u dětí nebo dospívajících.
Další léčivé přípravky a přípravek Benepali
Neměl/a byste používat přípravek Benepali s léky, které obsahují léčivou látku anakinru nebo abatacept.
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat (včetně sulfasalazinu), a to i o lécích, které jsou dostupné bez lékařského předpisu.
Těhotenství a kojení
Podávání přípravku Benepali v průběhu těhotenství se nedoporučuje. Pokud jste těhotná, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem.
Pokud Vám byl podáván během těhotenství přípravek Benepali, může u Vašeho novorozence existovat vyšší riziko infekce. Jedna studie ukázala, že když byl matkám v těhotenství podáván etanercept, vyskytlo se více vrozených vad v porovnání s těhotenstvími matek, kterým nebyl podáván etanercept ani jiné podobné léky (antagonisté TNF), avšak nebyl hlášen žádný zvláštní druh vrozených vad. Je důležité, abyste dětského lékaře a další zdravotnické pracovníky informovala o užívání přípravku Benepali v těhotenství ještě před tím, než novorozenec dostane jakoukoli vakcínu (další informace viz bod 2, „Očkování “).
Ženy užívající přípravek Benepali nesmějí kojit, protože přípravek Benepali přechází do mateřského mléka.
Řízení dopravních prostředků a obsluha strojů
Nejsou k dispozici žádné informace o tom, že přípravek Benepali ovlivňuje schopnost řídit nebo obsluhovat stroje.
Jak se přípravek Benepali používá
3.
Vždy používejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Jestliže máte pocit, že účinek přípravku Benepali je příliš silný nebo naopak příliš slabý, informujte o tom svého lékaře nebo lékárníka.
Dávkování u dospělých pacientů (věk 18 let a více)
Revmatoidní artritida, psoriatická artritida a axiální spondvlartritidv včetně ankvlozující spondvlitidv Obvyklá dávka je 50 mg jednou týdně ve formě injekce pod kůži.
Lékař však může stanovit odlišnou frekvenci podávání přípravku Benepali.
Ložisková lupénka
Obvyklá dávka je 50 mg jednou týdně.
Alternativně se může podávat 50 mg dvakrát týdně až po dobu 12 týdnů a potom pokračovat 50 mg jednou týdně.
Podle Vaší odpovědi na léčbu lékař rozhodne, jak dlouho máte přípravek Benepali používat a zda je třeba léčení opakovat. Pokud nevvkazuje přípravek Benepali účinek na Váš stav po 12 týdnech, může lékař zastavit podávání tohoto léku.
Způsob a cesta podání
Přípravek Benepali se podává injekcí pod kůži (subkutánní injekce).
Přípravek Benepali se může užívat s jídlem nebo pitím nebo i bez nich.
Podrobný návod k injekci přípravku Benepali je k dispozici v bodu 7 „Návod k přípravě a podání injekce přípravku Benepali“. Roztok přípravku Benepali se nesmí mísit s žádným jiným lékem.
Pro zapamatování je dobré si zapsat do kalendáře dnv v týdnu, kdv má být přípravek Benepali podán. Jestliže jste použil(a) více přípravku Benepali, než jste měl(a)
Jestliže jste použil(a) větší dávku přípravku Benepali, než bvste měl(a) (buď injekcí příliš velké dávkv, nebo příliš častým podáním), informujte neprodleně lékaře nebo lékárníka. Vždv si vezměte vnější obal od léku s sebou, a to i tehdy, je-li prázdný.
Jestliže jste zapomněl(a) podat přípravek Benepali
Jestliže zapomenete podat injekci, měl(a) byste ji aplikovat ihned, jakmile si vzpomenete, ledaže by následující pravidelná dávka měla být podána následující den. V tom případě zapomenutou dávku vvnechejte. Potom pokračujte v injekcích v obvvklé dnv. Pokud si nevzpomenete až do dne, kdy má být podána následující injekce, nezdvojnásobujte dávku (dvě dávkv ve stejný den), abvste nahradil(a) vynechanou dávku.
Jestliže jste přestal(a) používat přípravek Benepali
Po přerušení se mohou vrátit příznakv onemocnění.
Máte-li jakékoli další otázky týkající se používání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechnv lékv, může mít i tento přípravek nežádoucí účinkv, které se ale nemusí vyskytnout u každého.
Alergické reakce
Jestliže se vyskytne některý z následujících stavů, nepoužívejte dále přípravek Benepali. Informujte neprodleně lékaře, nebo navštivte oddělení pohotovosti nejbližšího zdravotnického zařízení.
• Potíže s polykáním nebo dýcháním
• Otok obličeje, krku, rukou nebo nohou.
• Pocit nervozity nebo úzkosti, pocit bušení srdce, nebo náhlé zrudnutí kůže a/nebo pocit horka.
• Těžká vyrážka, svědění, nebo kopřivka (vyvýšené červené nebo bledé skvrny na kůži, které často svědí).
Závažné alergické reakce jsou vzácné, avšak kterýkoliv z příznaků uvedených výše může znamenat alergickou reakci na přípravek Benepali, proto byste měl(a) neprodleně vyhledat lékařskou pomoc.
T-W r V r Vil i i V3 1
Závažné nežádoucí ucmky
Pokud si všimnete jakéhokoliv z následujících stavů, může být zapotřebí urgentní lékařský zásah.
• Známky závažné infekce, jako vysoká horečka, která může být spojená s kašlem, dušností, zimnicí, slabostí, nebo vznikem horkých červených citlivých bolestivých oblastí na kůži nebo v kloubech.
• Známky krevních chorob, jako krvácení, podlitiny nebo bledost.
• Známky nervových poruch, jako necitlivost nebo mravenčení, změny vidění, bolest očí, nebo nástup slabosti v horní nebo dolní končetině.
• Známky srdečního selhání nebo zhoršení srdečního selhání, jako únava nebo dýchavičnost při aktivitě, otok kotníků, pocit plnosti v krku nebo v břiše, noční dýchavičnost nebo kašel, zmodrání nehtových lůžek nebo okolí rtů.
• Známky zhoubného nádorového onemocnění: zhoubné nádorové onemocnění může postihnout všechny části těla včetně kůže a krve a onemocnění se může projevovat v závislosti na typu a místě zhoubného nádorového onemocnění. Tyto známky onemocnění mohou zahrnovat úbytek hmotnosti, horečku, otoky (bolestivé či nebolestivé), přetrvávající kašel, přítomnost boule na těle nebo výrůstky na kůži.
• Známky autoimunitní reakce (nově vytvořené protilátky mohou poškodit normální tkáně v těle) jako jsou bolest, svědění, slabost a poruchy dýchání, myšlení, vnímání nebo zraku.
• Známky lupusu nebo syndromu podobnému lupusu, jako jsou změny hmotnosti, přetrvávající vyrážka, horečka, bolest kloubů nebo svalů, únava.
• Známky zánětu cévní stěny, jako j sou bolest, horečka, zarudnutí nebo horkost kůže, svědění.
Tyto nežádoucí účinky jsou vzácné nebo méně časté, ale představují závažné stavy (některé z nich mohou vzácně skončit i smrtí). Pokud se u Vás vyskytne kterýkoli z výše uvedených nežádoucích účinků, neprodleně informujte lékaře nebo navštivte nejbližší zdravotnické zařízení.
Známé nežádoucí účinky přípravku Benepali zahrnují níže uvedené ve skupinách s klesající četností:
• Velmi časté (mohou postihnout více než 1 z 10 pacientů):
Infekce (včetně nachlazení, zánětu vedlejších nosních dutin, zánětu průdušek, infekce močových cest, zánětu kůže); reakce v místě injekce (včetně krvácení, podlitin, zčervenání, svědění, bolesti a otoku).
Reakce v místě injekce (nedostavují se tak často po prvním měsíci léčení).
U některých pacientů se rozvinula reakce v předchozím místě injekce.
• Časté (mohou postihnout až 1 z 10 pacientů):
Alergické reakce; horečka; svědění; protilátky proti normální tkáni (tvorba autoprotilátek).
infekce v různých místech); nízký počet krevních destiček; rakovina kůže (s výjimkou melanomu); lokalizované otoky kůže (angioedém); kopřivka (vyvýšené červené nebo bledé skvrny na kůži, které často svědí); oční zánět; lupénka (nová nebo zhoršující se); vyrážka; zánět nebo zjizvení plic; zánět cévní stěny postihující více orgánů.
• Vzácné (mohou postihnout až 1 z 1 000 pacientů):
Závažné alergické reakce (včetně závažných lokalizovaných otoků kůže a sípání); lymfom (typ nádorového onemocnění krve); melanom (typ rakoviny kůže); kombinace nízkého počtu krevních destiček, červených a bílých krvinek; poruchy nervového systému (se závažnou svalovou slabostí a známkami a příznaky podobnými roztroušené skleróze nebo zánětu očních nervů nebo míchy); tuberkulóza; městnavé srdeční selhání; křeče; lupus erythematodes, nebo onemocnění, které jej klinicky připomíná (příznaky mohou zahrnovat přetrvávající vyrážku, horečku, bolest kloubů a únavu); nízký počet červených krvinek, nízký počet bílých krvinek, nízký počet neutrofilů (typ bílých krvinek); zvýšené hodnoty jaterních testů v krvi; kožní vyrážka, která může vést k závažným puchýřům a k olupování kůže; zánětlivé onemocnění jater vyvolané poruchou imunitního systému (autoimunitní hepatitida); porucha imunity, která může postihnout plíce, kůži a lymfatické uzliny (sarkoidóza).
• Velmi vzácné (mohou postihnout až 1 z 10 000 pacientů):
Porucha tvorby krevních buněk v kostní dřeni.
• Není známo (z dostupných údajů nelze určit):
Leukémie (nádorové onemocnění postihující krev a kostní dřeň); Merkelův buněčný karcinom (typ rakoviny kůže); nadměrná aktivace bílých krvinek spojená se zánětem (syndrom aktivace makrofágů); recidiva (opakování) hepatitidy B (jaterní infekce); zhoršení stavu dermatomyositidy (zánět svalů a svalová slabost spojená s kožní vyrážkou).
Nežádoucí účinky u dětí a dospívajících
Přípravek Benepali není indikován k použití u dětí a dospívajících mladších 18 let. Nežádoucí účinky a jejich četnost pozorované u dětí a dospívajících jsou podobné těm, které jsou uvedeny výše.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Benepali uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na obalu a předplněné injekční stříkačce za „EXP“. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v chladničce (2 °C - 8 °C). Chraňte před mrazem.
Uchovávejte předplněné injekční stříkačky v krabičce, aby byl přípravek chráněn před světlem.
Po vyjmutí injekční stříkačky z chladničky počkejte přibližně 30 minut, až dosáhne roztok přípravku Benepali v injekční stříkačce pokojové teploty. Neohřívejte žádným jiným způsobem. Pak se doporučuje přípravek Benepali ihned použít.
Přípravek Benepali lze jednorázově mimo chladničku po dobu až čtyř týdnů uchovávat při teplotě maximálně do 25 °C; po této době nesmí být znovu uchováván v chladu. Přípravek Benepali musí být zlikvidován, pokud není použit do čtyř týdnů po vyjmutí z chladničky. Doporučuje se zaznamenat si datum, kdy byl přípravek Benepali vyjmut z chladničky, a datum, po kterém musí být přípravek Benepali zlikvidován (ne déle než 4 týdny po vyjmutí z chladničky).
Zkontrolujte roztok v injekční stříkačce. Roztok by měl být čirý až mírně opalescentní, bezbarvý nebo světle žlutý a může obsahovat malé bílé nebo téměř průhledné částice proteinu. Toto je normální vzhled přípravku Benepali. Nepoužívejte roztok, jestliže změnil barvu, je zakalený, nebo jestliže jsou v něm přítomny jiné než výše popsané částice. Jestliže si nejste jist(a) vzhledem roztoku, požádejte lékárníka o spolupráci.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co přípravek Benepali obsahuje
- Léčivou látkou v přípravku Benepali je etanerceptum. Jedna předplněná injekční stříkačka obsahuje 1,0 ml roztoku obsahujícího etanerceptum 50 mg.
- Pomocnými látkami j sou: sacharóza, chlorid sodný, monohydrát dihydrogenfosforečnanu sodného, heptahydrát hydrogenfosforečnanu sodného, voda na injekci.
Jak přípravek Benepali vypadá a co obsahuje toto balení
Přípravek Benepali se dodává jako předplněné injekční stříkačky obsahující čirý, bezbarvý nebo světle žlutý injekční roztok.
Přípravek Benepali je dostupný v baleních obsahujících 4 předplněné injekční stříkačky a baleních s více kusy obsahujících 3 papírové krabičky po 4 předplněných injekčních stříkačkách. Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci
Samsung Bioepis UK Limited 3000 Hillswood Drive Chertsey
Surrey KT16 0RS Velká Británie
Výrobce
Biogen (Denmark) Manufacturing ApS Biogen Allé 1 3400 Hillerod Dánsko
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci.
Belgie/Belgique/Belgien
Biogen Belgium NV/S.A Tél/Tel: + 32 (0)2 808 5947
Bt^rapnn
Biogen Idec Limited (UK) Ten.: + 359 249 176 81
Lietuva
Biogen Idec Limited (UK) Tel: +370 52 14 02 60
Luxembourg/Luxemburg
Biogen Belgium NV/SA Tél/Tel: +35 227 772 038
|
Česká republika Biogen (Czech Republic) s.r.o. Tel: + 420 228 884 152 |
Magyarország Biogen Hungary Kft. Tel.: + 36 (0)6 1 848 04 64 |
|
Danmark Biogen (Denmark) A/S Tlf: + 45 78 79 37 53 |
Malta Biogen Idec Limited (UK) Tel: + 356 27 78 15 79 |
|
Deutschland Biogen GmbH Tel: + 49 (0)30 223 864 72 |
Nederland Biogen Netherlands B.V. Tel: + 31 (0)20 808 02 70 |
|
Eesti Biogen Idec Limited (UK) Tel: + 372 6 68 30 56 |
Norge Biogen Norway AS Tlf: + 47 21 93 95 87 |
|
Eklába Biogen Idec Limited (UK) Tr(k: + 30 211 176 8555 |
Osterreich Biogen Austria GmbH Tel: + 43 (0)1 267 51 42 |
|
Espaňa Biogen Spain, S.L. Tel: + 34 931 790 519 |
Polska Biogen Poland Sp. z o.o. Tel.: + 48 22 116 86 94 |
|
France Biogen France SAS Tél: + 33 (0)1 776 968 14 |
Portugal Biogen Portugal Sociedade Farmaceutica, Unipessoal, Lda Tel: + 351 308 800 792 |
|
Hrvatska Biogen Idec Limited (UK) Tel: + 385 (0)1 777 64 37 |
Románia Biogen Idec Limited (UK) Tel: + 40 377 881 045 |
|
Ireland Biogen Idec (Ireland) Ltd. Tel: +353 (0)1 513 33 33 |
Slovenija Biogen Pharma d.o.o. Tel: + 386 (0)1 888 81 07 |
|
Ísland Biogen Idec Limited (UK) Sími: + 354 800 9836 |
Slovenská republika Biogen Slovakia s.r.o. Tel: + 421 (0)2 333 257 10 |
|
Italia Biogen Italia s.r.l. Tel: + 39 (0)6 899 701 50 |
Suomi/Finland Biogen Finland Oy Puh/Tel: + 358 (0)9 427 041 08 |
|
Kúnpoq Biogen Idec Limited (UK) Tr(k: + 357 22 00 04 93 |
Sverige Biogen Sweden AB Tel: +46 (0)8 525 038 36 |
|
Latvija Biogen Idec Limited (UK) Tel: + 371 66 16 40 32 |
United Kingdom Biogen Idec Limited Tel: +44 (0)20 360 886 22 |
Tato příbalová informace byla naposledy revidována
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Návod k použití
7.
Předtím, než začnete přípravek Benepali používat a pokaždé, když obdržíte nové balení na základě lékařského předpisu, si přečtěte návod k použití. Mohou v něm být nové informace.
• Nepokoušejte si podat injekci sám/a, pokud Vám lékař nebo zdravotní sestra neukázali, jak se injekce podává.
Jednorázová předplněná injekční stříkačka obsahuje jednu dávku 50 mg přípravku Benepali.
Zvolte si dobře osvětlenou, čistou pracovní plochu a shromážděte na ní veškeré vybavení, které budete potřebovat.
• Nová předplněná injekční stříkačka přípravku Benepali

Benepali
injectíon

o Předplněnou injekční stříkačku neprotřepávejte. Balení neobsahuje:
• 1 tampón namočený v alkoholu

• Nádobu na likvidaci ostrých předmětů

A. Než začnete s podáním injekce
1. Zkontrolujte předplněnou injekční stříkačku:
zkontrolujte dobu použitelnosti na obale předplněné injekční stříkačky.
• Nepoužívejte předplněnou injekční stříkačku poté, co vypršela doba použitelnosti.
• Nepoužívejte předplněnou injekční stříkačku, pokud spadla na tvrdý povrch. Může dojít k rozbití součástí uvnitř předplněné injekční stříkačky.
• Nepoužívejte předplněnou injekční stříkačku, pokud chybí krytka jehly, nebo když není bezpečně připevněna.
2. Zkontrolujte roztok:
Prohlédněte si léčivý přípravek v předplněné injekční stříkačce.
Léčivý přípravek má být čirý až mírně opalescentní, bezbarvý nebo světle žlutý a může obsahovat
malé bílé nebo téměř průhledné částice bílkoviny.
• Nepoužívejte roztok, jestliže změnil barvu, je zakalený, nebo jestliže jsou v něm přítomny jiné než výše popsané částice.
3. Nechte léčivý přípravek dosáhnout pokojové teploty:
Z chladničky vyjměte jednu předplněnou injekční stříkačku a nechte ji před injekčním podáním při pokojové teplotě nejméně 30 minut.
Je to důležité, protože pak bude injekční podání léčivého přípravku snadnější a pohodlnější.
• Nesnímejte kryt jehly, dokud nebudete připraven/a injekci podat.
• K ohřátí přípravku Benepali nepoužívejte tepelné zdroje, jako je například mikrovlnná trouba nebo horká voda.
4. Vyberte místo vpichu injekce:
Předplněná injekční stříkačka s přípravkem Benepali je určena pro podkožní injekční podání. Podává se do stehna, krajiny břišní nebo zadní strany horní části paže (viz obrázek vlevo).
Pro každou injekci si vyberte vždy jiné místo.
Pokud injekci podáváte do krajiny břišní, vyberte si místo, které je nejméně 5 cm od pupku.
• Nepodávejte injekci do místa, které je zarudlé, tvrdé, pohmožděné nebo bolestivé.
• Nepodávejte injekci do jizev nebo strií.
• Jestliže máte lupénku, nesmíte podat injekci do jakkoli vyvýšených, tlustých, červených nebo šupinatých kožních polí ani do lézí.
B. Kroky při podání injekce
Krok 1:
Krok 2:
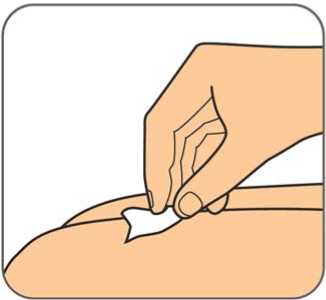
Krok 3:
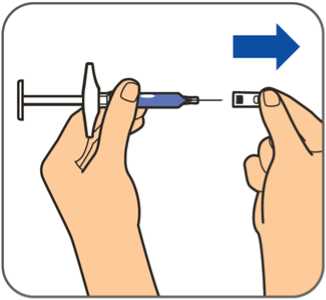
Krok 4:
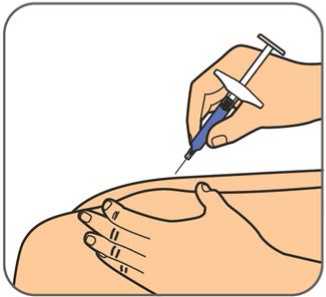
Omyjte si ruce mýdlem a vodou.
Otřete kůži v místě vpichu tampónem namočeným v alkoholu.
Viz „Vyberte místo vpichu injekce“, kde jsou pokyny pro výběr místa vpichu.
• Před podáním injekce se tohoto místa již nedotýkejte.
Stáhněte kryt jehly přímým tahem a zlikvidujte jej odhodzením do koše nebo nádoby na ostré předměty.
Viz „Vyberte místo vpichu injekce“, kde jsou pokyny pro výběr místa vpichu.
• Neotáčejte ani neohýbejte kryt j ehly, když j ej odstraňujete, protože to může jehlu poškodit.
• Nedotýkejte se táhla pístu při odstraňování krytu jehly.
• Nikdy kryt na jehlu znovu nenasazujte.
Jemným stiskem sevřete kožní řasu v očištěném místě pro vpich injekce. Předplněnou injekční stříkačku držte v úhlu 45 stupňů vůči kůži. Rychlým pohybem, jako když házíte šipkou, zasuňte jehlu celou do kůže.
Jakmile bude celá jehla uvnitř, pusťte kůži, kterou svíráte rukou.
Pak pomalu stlačujte píst a ustálenou rychlostí injikujte veškerý roztok Benepali.
Krok 5:
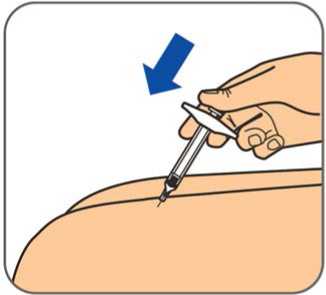
Krok 6:
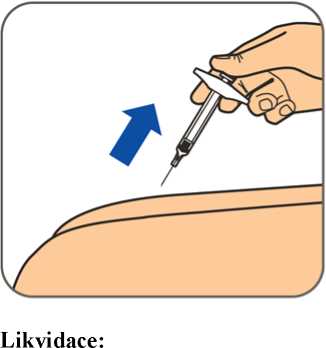
Když je injekční stříkačka prázdná, vytáhněte jehlu z kůže a držte jehlu stále pod stejným úhlem jako při vpichu.
• Nikdy znovu nezakrývejte jehlu krytem. Opakované zakrytí by mohlo způsobit poranění jehlou.
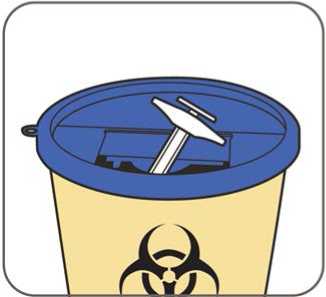
Celou injekční stříkačku zlikvidujte ve schválené nádobě na ostré předměty.
Informujte se u svého poskytovatele zdravotní péče na pokyny, jak správně likvidovat plné odpadní nádoby na ostré předměty. Nádoby na likvidaci ostrých předmětů můžete zakoupit v místní lékárně.
• Neodhazujte odpadní nádobu na ostré předměty do domácího odpadu.
• Nerecyklujte.
• Předplněnou injekční stříkačku přípravku Benepali nepoužívejte opakovaně.
• Uchovávejte tuto nádobu mimo dohled a dosah dětí.
C. Péče o místo vpichu injekce
Pokud dojde ke krvácení v místě vpichu, přitlačte na místo vpichu gázový tampón.
• Místo vpichu netřete.
Podle potřeby místo vpichu přelepte náplastí.
Příbalová informace: informace pro uživatele
Benepali 50 mg, injekční roztok v předplněném peru
etanerceptum
'VTento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Lékař Vám rovněž vydá informační kartu pacienta, která obsahuje důležité bezpečnostní informace, které musíte vzít před léčbou přípravkem Benepali a během ní na vědomí.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Benepali a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Benepali používat
3. Jak se přípravek Benepali používá
4. Možné nežádoucí účinky
5. Jak přípravek Benepali uchovávat
6. Obsah balení a další informace
7. Návod k použití přípravku Benepali (viz druhá strana)
1. Co je přípravek Benepali a k čemu se používá
Přípravek Benepali je biologické léčivo, které se vyrábí ze dvou lidských bílkovin. Blokuje aktivitu jiné bílkoviny v těle, která způsobuje zánět. Přípravek Benepali snižuje zánět spojený s určitými onemocněními. Přípravek Benepali obsahuje léčivou látku, etanercept.
U dospělých (věk 18 let a více) se může přípravek Benepali použít k léčení:
• středně těžkého až těžkého revmatoidního zánětu kloubů (revmatoidní artritida),
• revmatoidního zánětu kloubů s lupénkou (psoriatická artritida),
• závažných axiálních spondylartritid (záněty kloubů v oblasti páteře) včetně ankylozující spondylitidy (Bechtěrevova nemoc),
• středně těžké až těžké lupénky (psoriáza).
Ve všech případech se přípravek Benepali obvykle používá, pokud běžně používané léčebné postupy nebyly dostatečně účinné nebo nejsou pro Vás vhodné.
U revmatoidní artritidy se přípravek Benepali obvykle používá v kombinaci s methotrexátem, i když se může použít také samotný, jestliže léčení methotrexátem pro Vás není vhodné. Přípravek Benepali použitý samostatně nebo v kombinaci s methotrexátem může zpomalit poškození kloubů, způsobené revmatoidní artritidou, a zlepšit Vaši schopnost provádět normální denní činnosti.
U pacientů s psoriatickou artritidou postihující více kloubů může přípravek Benepali zlepšit schopnost provádět normální denní činnosti.
U pacientů se symetrickým bolestivým nebo otokovým postižením více kloubů (např. ruce, zápěstí a nohy) může přípravek Benepali zpomalit strukturální poškození těchto kloubů způsobená chorobou.
2. Čemu musíte věnovat pozornost, než začnete přípravek Benepali používat
Nepoužívejte přípravek Benepali
• jestliže jste alergický(á) na etanercept nebo kteroukoli další složku přípravku Benepali
(uvedenou v bodě 6);
• jestliže Vám hrozí rozvoj závažné krevní infekce zvané sepse (otrava krve). Jestliže si nejste jist(a), poraďte se s lékařem;
• jestliže máte jakýkoli druh infekce. Jestliže si nejste jist(a), poraďte se s lékařem.
Upozornění a opatření
Před použitím přípravku Enbrel se poraďte se svým lékařem.
• Alergické reakce: Jestliže se u Vás objeví alergické reakce, jako je tíha na hrudi, sípání, závratě nebo vyrážka, nepodávejte více přípravku Benepali a kontaktujte ihned lékaře.
• Infekce/operace: Jestliže se u Vás rozvíjí nová infekce nebo jste před velkou chirurgickou operací, lékař může chtít sledovat léčbu přípravkem Benepali.
• Infekce/cukrovka: Informujte lékaře, jestliže jste měl(a) opakující se infekce nebo trpíte cukrovkou nebo jinými stavy, při nichž se zvyšuje riziko infekce.
• Infekce/sledování: Informujte svého lékaře o každé cestě mimo Evropu, kterou jste absolvoval(a) v poslední době. Pokud se u Vás rozvinou příznaky infekce, jako je horečka, zimnice nebo kašel, neprodleně informujte svého lékaře. Lékař může rozhodnout, že Vás bude nadále sledovat na přítomnost infekcí i poté, co přestanete používat přípravek Benepali.
• Tuberkulóza: U pacientů léčených přípravkem Benepali byly hlášeny případy tuberkulózy. Lékař vyšetří známky a příznaky tuberkulózy předtím, než zahájí léčení přípravkem Benepali. To může zahrnovat podrobnou lékařskou anamnézu, rentgen hrudníku a tuberkulinový test. Výsledky těchto vyšetření musí být zaznamenány do pohotovostní karty pacienta. Je velmi důležité, abyste informoval(a) lékaře, pokud jste měl(a), nebo pokud jste byl(a) v kontaktu s někým, kdo ji měl. Pokud by se vyskytly příznaky tuberkulózy (jako přetrvávající kašel, ztráta hmotnosti, netečnost, mírná horečka), nebo jiná infekce v průběhu léčení nebo po jeho ukončení, informujte neprodleně lékaře.
• Hepatitida B: Informujte lékaře, pokud máte nebo jste měl(a) v minulosti hepatitidu B. Lékař by měl provést vyšetření na přítomnost infekce hepatitidy B předtím, než se začnete léčit přípravkem Benepali. Léčba přípravkem Benepali může vést k reaktivaci hepatitidy B u pacientů, kteří byli v minulosti nakaženi virem hepatitidy B. Pokud k tomu dojde, musíte přestat přípravek Benepali používat.
• Hepatitida C: Informujte lékaře, pokud máte hepatitidu C. Lékař bude sledovat léčení přípravkem Benepali, pokud se infekce zhorší.
• Krevní poruchy: Jestliže se u Vás objeví známky nebo příznaky jako přetrvávající horečka, bolest v krku, podlitiny, krvácení nebo bledost, vyhledejte neodkladně lékařskou pomoc.
Takové příznaky mohou svědčit o možném život ohrožujícím onemocnění krve, které si může vyžádat přerušení podávání přípravku Benepali.
• Poruchy nervového systému a oční poruchy: Informujte lékaře, jestliže máte roztroušenou sklerózu, optickou neuritidu (zánět zrakových nervů) nebo transverzální myelitidu (zánět míchy). Lékař určí, zda je přípravek Benepali vhodnou léčbou.
• Městnavé srdeční selhání: Informujte lékaře, jestliže jste měl(a) v minulosti městnavé srdeční selhání, neboť za těchto okolností je třeba opatrnosti při podávání přípravku Benepali.
• Zhoubná nádorová onemocnění: Než Vám bude předepsán přípravek Benepali, informujte lékaře, pokud máte nebo Vám byl někdy diagnostikován lymfom (typ zhoubného onemocnění krve) či jiný typ zhoubného nádorového onemocnění/rakoviny. U pacientů s těžkou revmatoidní artritidou, kteří trpí tímto onemocněním delší dobu, je riziko vzniku lymfomu vyšší než průměrné. U některých dětí a dospívajících pacientů, kterým byl podáván etanercept nebo jiné léčivé přípravky působící stejně jako etanercept, se rozvinula zhoubná nádorová onemocnění/rakovina včetně neobvyklých typů, což někdy vedlo k úmrtí. Někteří pacienti užívající přípravek Benepali onemocněli rakovinou kůže. Informujte lékaře, pokud u Vás dojde k rozvoji jakékoli změny vzhledu kůže nebo výrůstků na kůži.
• Plané neštovice: Upozorněte lékaře, jestliže jste byli v kontaktu s planými neštovicemi v průběhu podávání přípravku Benepali. Lékař stanoví, zda je vhodné preventivní opatření proti planým neštovicím.
• Zneužívání alkoholu: Přípravek Benepali by se neměl použít u pacientů, kteří se léčí s hepatitidou spojenou se zneužíváním alkoholu. Informujte, prosím, lékaře, pokud máte zneužívání alkoholu v anamnéze.
• Wegenerova granulomatóza: Přípravek Benepali se nedoporučuje k léčení Wegenerovy granulomatózy, vzácného zánětlivého onemocnění. Informujte lékaře, máte-li Wegenerovu granulomatózu.
• Antidiabetické přípravky: Informujte svého lékaře, trpíte-li cukrovkou či užíváte-li léky k léčbě cukrovky. Váš lékař stanoví, zda je u Vás nutné během léčby přípravkem Benepali snížit dávku léků k léčbě cukrovky.
• Očkování: Některá očkování by neměla proběhnout v době podávání přípravku Benepali, např. vakcína proti poliomyelitidě, tj. proti dětské obrně. Prosím, poraďte se s lékařem před případným očkováním.
Děti a dospívající
Přípravek Benepali není indikován k použití u dětí a dospívajících mladších 18 let.
Přípravek Benepali je k dispozici jako 50 mg předplněná injekční stříkačka nebo 50 mg předplněné pero, které jsou nevhodné pro použití u dětí nebo dospívajících.
Další léčivé přípravky a přípravek Benepali
Neměl/a byste používat přípravek Benepali s léky, které obsahují léčivou látku anakinru nebo abatacept.
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat (včetně sulfasalazinu), a to i o lécích, které jsou dostupné bez lékařského předpisu.
Těhotenství a kojení
Podávání přípravku Benepali v průběhu těhotenství se nedoporučuje. Pokud jste těhotná, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem.
Pokud Vám byl podáván během těhotenství přípravek Benepali, může u Vašeho novorozence existovat vyšší riziko infekce. Jedna studie ukázala, že když byl matkám v těhotenství podáván etanercept, vyskytlo se více vrozených vad v porovnání s těhotenstvími matek, kterým nebyl podáván etanercept ani jiné podobné léky (antagonisté TNF), avšak nebyl hlášen žádný zvláštní druh vrozených vad. Je důležité, abyste dětského lékaře a další zdravotnické pracovníky informovala o užívání přípravku Benepali v těhotenství ještě před tím, než novorozenec dostane jakoukoli vakcínu (další informace viz bod 2, „Očkování “).
Ženy užívající přípravek Benepali nesmějí kojit, protože přípravek Benepali přechází do mateřského mléka.
Řízení dopravních prostředků a obsluha strojů
Nejsou k dispozici žádné informace o tom, že přípravek Benepali ovlivňuje schopnost řídit nebo obsluhovat stroje.
Jak se přípravek Benepali používá
3.
Vždy používejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Jestliže máte pocit, že účinek přípravku Benepali je příliš silný nebo naopak příliš slabý, informujte o tom svého lékaře nebo lékárníka.
Dávkování u dospělých pacientů (věk 18 let a více)
Revmatoidní artritida, psoriatická artritida a axiální spondvlartritidv včetně ankylozující spondvlitidv Obvyklá dávka je 50 mg jednou týdně ve formě injekce pod kůži.
Lékař však může stanovit odlišnou frekvenci podávání přípravku Benepali.
Ložisková lupénka
Obvyklá dávka je 50 mg jednou týdně.
Alternativně se může podávat 50 mg dvakrát týdně až po dobu 12 týdnů a potom pokračovat 50 mg jednou týdně.
Podle Vaší odpovědi na léčbu lékař rozhodne, jak dlouho máte přípravek Benepali používat a zda je třeba léčení opakovat. Pokud nevykazuje přípravek Benepali účinek na Váš stav po 12 týdnech, může lékař zastavit podávání tohoto léku.
Způsob a cesta podání
Přípravek Benepali se podává injekcí pod kůži (subkutánní injekce).
Přípravek Benepali se může používat s tekutinou nebo s jídlem nebo bez nich.
Podrobný návod k injekci přípravku Benepali je k dispozici v bodu 7 „Návod k přípravě a podání injekce přípravku Benepali“. Roztok přípravku Benepali se nesmí mísit s žádným jiným lékem.
Pro zapamatování je dobré si zapsat do kalendáře dny v týdnu, kdy má být přípravek Benepali podán. Jestliže jste použil(a) více přípravku Benepali, než jste měl(a)
Jestliže jste použil(a) větší dávku přípravku Benepali, než byste měl(a) (buď injekcí příliš velké dávky, nebo příliš častým podáním), informujte neprodleně lékaře nebo lékárníka. Vždy si vezměte vnější obal od léku s sebou, a to i tehdy, je-li prázdný.
Jestliže jste zapomněl(a) podat přípravek Benepali
Jestliže zapomenete podat injekci, měl(a) byste ji aplikovat ihned, jakmile si vzpomenete, ledaže by následující pravidelná dávka měla být podána následující den. V tom případě zapomenutou dávku vynechejte. Potom pokračujte v injekcích v obvyklé dny. Pokud si nevzpomenete až do dne, kdy má být podána následující injekce, nezdvojnásobujte dávku (dvě dávky ve stejný den), abyste nahradil(a) vynechanou dávku.
Jestliže jste přestal(a) používat přípravek Benepali
Po přerušení se mohou vrátit příznaky onemocnění.
Máte-li jakékoli další otázky týkající se používání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Alergické reakce
Jestliže se vyskytne některý z následujících stavů, nepoužívejte dále přípravek Benepali. Informujte neprodleně lékaře, nebo navštivte oddělení pohotovosti nejbližšího zdravotnického zařízení.
• Potíže s polykáním nebo dýcháním
• Otok obličeje, krku, rukou nebo nohou.
• Pocit nervozity nebo úzkosti, pocit bušení srdce, nebo náhlé zrudnutí kůže a/nebo pocit horka.
• Těžká vyrážka, svědění, nebo kopřivka (vyvýšené červené nebo bledé skvrny na kůži, které často svědí).
Závažné alergické reakce jsou vzácné, avšak kterýkoliv z příznaků uvedených výše může znamenat alergickou reakci na přípravek Benepali, proto byste měl(a) neprodleně vyhledat lékařskou pomoc.
T-W r V r Vil i i V4 1
Závažné nežádoucí ucmky
Pokud si všimnete jakéhokoliv z následujících stavů, může být zapotřebí urgentní lékařský zásah.
• Známky závažné infekce, jako vysoká horečka, která může být spojená s kašlem, dušností, zimnicí, slabostí, nebo vznikem horkých červených citlivých bolestivých oblastí na kůži nebo v kloubech.
• Známky krevních chorob, jako krvácení, podlitiny nebo bledost.
• Známky nervových poruch, jako necitlivost nebo mravenčení, změny vidění, bolest očí, nebo nástup slabosti v horní nebo dolní končetině.
• Známky srdečního selhání nebo zhoršení srdečního selhání, jako únava nebo dýchavičnost při aktivitě, otok kotníků, pocit plnosti v krku nebo v břiše, noční dýchavičnost nebo kašel, zmodrání nehtových lůžek nebo okolí rtů.
• Známky zhoubného nádorového onemocnění: zhoubné nádorové onemocnění může postihnout všechny části těla včetně kůže a krve a onemocnění se může projevovat v závislosti na typu a místě zhoubného nádorového onemocnění. Tyto známky onemocnění mohou zahrnovat úbytek hmotnosti, horečku, otoky (bolestivé či nebolestivé), přetrvávající kašel, přítomnost boule na těle nebo výrůstky na kůži.
• Známky autoimunitní reakce (nově vytvořené protilátky mohou poškodit normální tkáně v těle) jako jsou bolest, svědění, slabost a poruchy dýchání, myšlení, vnímání nebo zraku.
• Známky lupusu nebo syndromu podobnému lupusu, jako jsou změny hmotnosti, přetrvávající vyrážka, horečka, bolest kloubů nebo svalů, únava.
• Známky zánětu cévní stěny, jako j sou bolest, horečka, zarudnutí nebo horkost kůže, svědění.
Tyto nežádoucí účinky jsou vzácné nebo méně časté, ale představují závažné stavy (některé z nich mohou vzácně skončit i smrtí). Pokud se u Vás vyskytne kterýkoli z výše uvedených nežádoucích účinků, neprodleně informujte lékaře nebo navštivte nejbližší zdravotnické zařízení.
Známé nežádoucí účinky přípravku Benepali zahrnují níže uvedené ve skupinách s klesající četností:
• Velmi časté (mohou postihnout více než 1 z 10 pacientů):
Infekce (včetně nachlazení, zánětu vedlejších nosních dutin, zánětu průdušek, infekce močových cest, zánětu kůže); reakce v místě injekce (včetně krvácení, podlitin, zčervenání, svědění, bolesti a otoku).
Reakce v místě injekce (nedostavují se tak často po prvním měsíci léčení).
U některých pacientů se rozvinula reakce v předchozím místě injekce.
• Časté (mohou postihnout až 1 z 10 pacientů):
Alergické reakce; horečka; svědění; protilátky proti normální tkáni (tvorba autoprotilátek).
melanomu); lokalizované otoky kůže (angioedém); kopřivka (vyvýšené červené nebo bledé skvrny na kůži, které často svědí); oční zánět; lupénka (nová nebo zhoršující se); vyrážka; zánět nebo zjizvení plic; zánět cévní stěny postihující více orgánů.
• Vzácné (mohou postihnout až 1 z 1000 pacientů):
Závažné alergické reakce (včetně závažných lokalizovaných otoků kůže a sípání); lymfom (typ nádorového onemocnění krve); melanom (typ rakoviny kůže); kombinace nízkého počtu krevních destiček, červených a bílých krvinek; poruchy nervového systému (se závažnou svalovou slabostí a známkami a příznaky podobnými roztroušené skleróze nebo zánětu očních nervů nebo míchy); tuberkulóza; městnavé srdeční selhání; křeče; lupus erythematodes, nebo onemocnění, které jej klinicky připomíná (příznaky mohou zahrnovat přetrvávající vyrážku, horečku, bolest kloubů a únavu); nízký počet červených krvinek, nízký počet bílých krvinek, nízký počet neutrofilů (typ bílých krvinek); zvýšené hodnoty jaterních testů v krvi; kožní vyrážka, která může vést k závažným puchýřům a k olupování kůže; zánětlivé onemocnění jater vyvolané poruchou imunitního systému (autoimunitní hepatitida); porucha imunity, která může postihnout plíce, kůži a lymfatické uzliny (sarkoidóza).
• Velmi vzácné (mohou postihnout až 1 z 10000 pacientů):
Porucha tvorby krevních buněk v kostní dřeni.
• Není známo (z dostupných údajů nelze určit):
Leukémie (nádorové onemocnění postihující krev a kostní dřeň); Merkelův buněčný karcinom (typ rakoviny kůže); nadměrná aktivace bílých krvinek spojená se zánětem (syndrom aktivace makrofágů); recidiva (opakování) hepatitidy B (jatemí infekce); zhoršení stavu dermatomyositidy (zánět svalů a svalová slabost spojená s kožní vyrážkou).
Nežádoucí účinky u dětí a dospívajících
Přípravek Benepali není indikován k použití u dětí a dospívajících mladších 18 let. Nežádoucí účinky a jejich četnost pozorované u dětí a dospívajících jsou podobné těm, které jsou uvedeny výše.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Benepali uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené obalu a předplněném peru za „EXP“. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v chladničce (2 °C - 8 °C). Chraňte před mrazem.
Uchovávejte předplněná pera v krabičce, aby byl přípravek chráněn před světlem.
Po vyjmutí předplněného pera z chladničky počkejte přibližně 30 minut, až dosáhne roztok přípravku Benepali v peru pokojové teploty. Neohřívejte žádným jiným způsobem. Pak se doporučuje přípravek Benepali ihned použít.
Přípravek Benepali lze jednorázově mimo chladničku po dobu až čtyř týdnů uchovávat při teplotě maximálně do 25 °C; po této době nesmí být znovu uchováván v chladu. Přípravek Benepali musí být zlikvidován, pokud není použit do čtyř týdnů po vyjmutí z chladničky. Doporučuje se zaznamenat si datum, kdy byl přípravek Benepali vyjmut z chladničky, a datum, po kterém musí být přípravek Benepali zlikvidován (ne déle než 4 týdny po vyjmutí z chladničky).
Zkontrolujte roztok v peru pohledem skrz kontrolní okénko. Roztok by měl být čirý až mírně opalescentní, bezbarvý nebo světle žlutý a může obsahovat malé bílé nebo téměř průhledné částice proteinu. Toto je normální vzhled přípravku Benepali. Nepoužívejte roztok, jestliže změnil barvu, je zakalený, nebo jestliže jsou v něm přítomny jiné než výše popsané částice. Jestliže si nejste jist(a) vzhledem roztoku, požádejte lékárníka o spolupráci.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co přípravek Benepali obsahuje
- Léčivou látkou v přípravku Benepali je etanerceptum. Jedno předplněné pero obsahuje 50 mg etanerceptu.
- Pomocnými látkami j sou: sacharóza, chlorid sodný, monohydrát dihydrogenfosforečnanu sodného, heptahydrát hydrogenfosforečnanu sodného, voda na injekci.
Jak přípravek Benepali vypadá a co obsahuje toto balení
Přípravek Benepali se dodává jako injekční roztok v předplněném peru (injekční roztok). Pero obsahuje čirý, bezbarvý nebo slabě žlutý injekční roztok.
Přípravek Benepali je dostupný v baleních obsahujících 4 předplněná pera a baleních s více kusy obsahujících 3 papírové krabičky po 4 předplněných perech. Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci
Samsung Bioepis UK Limited
3000 Hillswood Drive
Chertsey
Surrey KT16 0RS
Velká Británie
Výrobce
Biogen (Denmark) Manufacturing ApS Biogen Allé 1 3400 Hillerod Dánsko
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci.
Belgie/Belgique/Belgien
Biogen Belgium NV/S.A Tél/Tel: + 32 (0)2 808 5947
Btnrapnn
Biogen Idec Limited (UK) Ten.: + 359 249 176 81
Česká republika
Biogen (Czech Republic) s.r.o. Tel: + 420 228 884 152
Lietuva
Biogen Idec Limited (UK) Tel: +370 52 14 02 60
Luxembourg/Luxemburg
Biogen Belgium NV/SA Tél/Tel: +35 227 772 038
Magyarország
Biogen Hungary Kft.
Tel.: + 36 (0)6 1 848 04 64
|
Danmark Biogen (Denmark) A/S Tlf: + 45 78 79 37 53 |
Malta Biogen Idec Limited (UK) Tel: + 356 27 78 15 79 |
|
Deutschland Biogen GmbH Tel: + 49 (0)30 223 864 72 |
Nederland Biogen Netherlands B.V. Tel: + 31 (0)20 808 02 70 |
|
Eesti Biogen Idec Limited (UK) Tel: + 372 6 68 30 56 |
Norge Biogen Norway AS Tlf: + 47 21 93 95 87 |
|
EXlába Biogen Idec Limited (UK) TpA,: + 30 211 176 8555 |
Osterreich Biogen Austria GmbH Tel: + 43 (0)1 267 51 42 |
|
Espaňa Biogen Spain, S.L. Tel: + 34 931 790 519 |
Polska Biogen Poland Sp. z o.o. Tel.: + 48 22 116 86 94 |
|
France Biogen France SAS Tél: + 33 (0)1 776 968 14 |
Portugal Biogen Portugal Sociedade Farmaceutica, Unipessoal, Lda Tel: + 351 308 800 792 |
|
Hrvatska Biogen Idec Limited (UK) Tel: + 385 (0)1 777 64 37 |
Románia Biogen Idec Limited (UK) Tel: + 40 377 881 045 |
|
Ireland Biogen Idec (Ireland) Ltd. Tel: +353 (0)1 513 33 33 |
Slovenija Biogen Pharma d.o.o. Tel: + 386 (0)1 888 81 07 |
|
Ísland Biogen Idec Limited (UK) Sími: + 354 800 9836 |
Slovenská republika Biogen Slovakia s.r.o. Tel: + 421 (0)2 333 257 10 |
|
Italia Biogen Italia s.r.l. Tel: + 39 (0)6 899 701 50 |
Suomi/Finland Biogen Finland Oy Puh/Tel: + 358 (0)9 427 041 08 |
|
Kúrcpog Biogen Idec Limited (UK) TpA,: + 357 22 00 04 93 |
Sverige Biogen Sweden AB Tel: +46 (0)8 525 038 36 |
|
Latvija Biogen Idec Limited (UK) Tel: + 371 66 16 40 32 |
United Kingdom Biogen Idec Limited Tel: +44 (0)20 360 886 22 |
Tato příbalová informace byla naposledy revidována
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Návod k použití
7.
Předtím, než začnete přípravek Benepali používat a pokaždé, když obdržíte nové balení na základě lékařského předpisu, si přečtěte návod k použití. Mohou v něm být nové informace.
• Nepokoušejte si podat injekci sám/a, pokud Vám lékař nebo zdravotní sestra neukázali, jak se injekce podává.
Jednorázové předplněné pero obsahuje jednu dávku 50 mg přípravku Benepali.
Zvolte si dobře osvětlenou, čistou pracovní plochu a shromážděte na ní veškeré vybavení, které budete potřebovat.
Nová předplněné pero přípravku Benepali

o Předplněné pero neprotřepávejte.
Balení neobsahuje:
• 1 tampón namočený v alkoholu
tampon namočený v alkoholu
• Nádobu na likvidaci ostrých předmětů

A. Než začnete s podáním injekce
1. Zkontrolujte předplněné pero:
Zkontrolujte dobu použitelnosti na obale předplněného pera.
• Nepoužívejte předplněné pero poté, co vypršela doba použitelnosti.
• Nepoužívejte předplněné pero, pokud spadlo na tvrdý povrch. Může dojít k rozbití součástí uvnitř předplněného pera.
• Nepoužívejte předplněné pero, pokud chybí krytka jehly, nebo když není bezpečně připevněna.
2. Zkontrolujte roztok:
Prohlédněte si léčivý přípravek kontrolním okénkem.
Léčivý přípravek má být čirý až mírně opalescentní, bezbarvý nebo světle žlutý a může obsahovat malé bílé nebo téměř průhledné částice bílkoviny.
• Nepoužívejte roztok, jestliže změnil barvu, je zakalený, nebo jestliže jsou v něm přítomny jiné než výše popsané částice.
3. Nechte léčivý přípravek dosáhnout pokojové teploty:
Z krabičky uložené v chladničce vyjměte jedno předplněné pero a nechte jej před injekčním podáním při pokojové teplotě nejméně 30 minut.
Je to důležité, protože pak bude injekční podání léčivého přípravku snadnější a pohodlnější.
• Nesnímejte krytku jehly, dokud nebudete připraven/a injekci podat.
• K ohřátí přípravku Benepali nepoužívejte tepelné zdroje, jako je například mikrovlnná trouba nebo horká voda.
4. Vyberte místo vpichu injekce:

Předplněné pero s přípravkem Benepali je určeno pro podkožní injekční podání. Podává se do stehna, krajiny břišní nebo zadní strany horní části paže (viz obrázek vlevo).
Pro každou injekci si vyberte vždy jiné místo.
Pokud injekci podáváte do krajiny břišní, vyberte si místo, které je nejméně 5 cm od pupku.
• Nepodávejte injekci do místa, které je zarudlé, tvrdé, pohmožděné nebo bolestivé.
• Nepodávejte injekci do jizev nebo strií.
• Jestliže máte lupénku, nesmíte podat injekci do jakkoli vyvýšených, tlustých, červených nebo šupinatých kožních polí ani do lézí.
B. Kroky při podání injekce
Krok 1:

Omyjte si ruce mýdlem a vodou.
Otřete kůži v místě vpichu tampónem namočeným v alkoholu.

Krok 3:

Krok 4:


Viz „Vyberte místo vpichu injekce“, kde jsou pokyny pro výběr místa vpichu.
• Před podáním injekce se tohoto místa již nedotýkejte.
Stáhněte krytku jehly přímým tahem a zlikvidujte jej odhozením do koše nebo nádoby na ostré předměty.
• Neotáčejte ani neohýbejte kryt j ehly, když j ej odstraňujete, protože to může jehlu poškodit.
• Nikdy kryt na jehlu znovu nenasazujte.
Jemně roztáhněte kůži v očištěném místě pro vpich injekce. Předplněné pero držte v úhlu 90 stupňů vůči kůži.
• Kůži nesvírejte.
• Roztažením kůže vytvoříte pevný povrch.
Pevně přitlačte předplněné pero dolů do místa určeného k podání injekce.
Zařízení klikne, když začne injekční podání.
Držte i nadále předplněné pero pevně přitlačené k místu vpichu.
Zařízení klikne podruhé.
Po druhém kliknutí pomalu počítejte do 15, aby bylo zajištěno úplné injekční podání.



• Neuvolňujte tlak proti místu vpichu před dokončením injekce.
• Nepohybujte předplněným perem v průběhu injekce.
Odstraňte prázdné pero z kůže.
Kryt jehly zcela jehlu zakryje.
Zkontrolujte žlutý píst pera v okénku, abyste si ověřili, že byla podána plná dávka.
Prázdné pero zlikvidujte ve schválené nádobě na ostré předměty.
Informujte se u svého poskytovatele zdravotní péče na pokyny, jak správně likvidaci plné odpadní nádoby na ostré předměty.
Nádoby na likvidaci ostrých předmětů můžete zakoupit v místní lékárně.
• Neodhazujte odpadní nádobu na ostré předměty do domácího odpadu.
• Nerecyklujte.
• Uchovávejte tuto nádobu mimo dohled a dosah dětí.
C. Péče o místo vpichu injekce
Pokud dojde ke krvácení v místě vpichu, přitlačte na místo vpichu gázový tampón. • Místo vpichu netřete.
Podle potřeby místo vpichu přelepte náplastí.
72
Městnavé srdeční selhání
Pokud máte městnavé srdeční selhání a myslíte si, že se Vaše příznaky (např. dušnost nebo otok dolních končetin) zhoršují, vyhledejte neprodleně lékařskou pomoc.
Další informace (vyplňte prosím)
Jméno pacienta:
Jméno lékaře:
Telefon lékaře: _
• Noste tuto kartu u sebe 2 měsíce po poslední dávce přípravku Benepali, protože po poslední dávce přípravku Benepali se mohou objevit nežádoucí účinky.
Méně časté (mohou postihnout až 1 ze 100 pacientů):
Závažné infekce (včetně zápalu plic, hlubokých infekcí kůže, infekcí kloubů, infekcí krve a
Méně časté (mohou postihnout až 1 z 100 pacientů):
Závažné infekce (včetně zápalu plic, hlubokých infekcí kůže, infekcí kloubů, infekcí krve a infekce v různých místech); nízký počet krevních destiček; rakovina kůže (s výjimkou