Benefix 1000 Iu
SOUHRN ÚDAJŮ O PŘÍPRAVKU
PŘÍLOHA I
BeneFIX 250 IU prášek a rozpouštědlo pro injekční roztok BeneFIX 500 IU prášek a rozpouštědlo pro injekční roztok BeneFIX 1000 IU prášek a rozpouštědlo pro injekční roztok BeneFIX 2000 IU prášek a rozpouštědlo pro injekční roztok BeneFIX 3000 IU prášek a rozpouštědlo pro injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
BeneFIX 250 IU prášek a rozpouštědlo pro injekční roztok
Jedna injekční lahvička obsahuje nominálně 250 IU látky nonacogum alfa (rekombinantní koagulační faktor IX). Po rekonstituci v 5 ml přibaleného (0,234%) injekčního roztoku chloridu sodného obsahuje jeden ml roztoku přibližně 50 IU nonacogum alfa.
BeneFIX 500 IU prášek a rozpouštědlo pro injekční roztok
Jedna injekční lahvička obsahuje nominálně 500 IU látky nonacogum alfa (rekombinantní koagulační faktor IX). Po rekonstituci v 5 ml přibaleného (0,234%) injekčního roztoku chloridu sodného obsahuje jeden ml roztoku přibližně 100 IU nonacogum alfa.
BeneFIX 1000 IU prášek a rozpouštědlo pro injekční roztok
Jedna injekční lahvička obsahuje nominálně 1000 IU látky nonacogum alfa (rekombinantní koagulační faktor IX). Po rekonstituci v 5 ml přibaleného (0,234%) injekčního roztoku chloridu sodného obsahuje jeden ml roztoku přibližně 200 IU nonacogum alfa.
BeneFIX 2000 IU prášek a rozpouštědlo pro injekční roztok
Jedna injekční lahvička obsahuje nominálně 2000 IU látky nonacogum alfa (rekombinantní koagulační faktor IX). Po rekonstituci v 5 ml přibaleného (0,234%) injekčního roztoku chloridu sodného obsahuje jeden ml roztoku přibližně 400 IU nonacogum alfa.
BeneFIX 3000 IU prášek a rozpouštědlo pro injekční roztok
Jedna injekční lahvička obsahuje nominálně 3000 IU látky nonacogum alfa (rekombinantní koagulační faktor IX). Po rekonstituci v 5 ml přibaleného (0,234%) injekčního roztoku chloridu sodného obsahuje jeden ml roztoku přibližně 600 IU nonacogum alfa.
Účinnost (IU) je určena pomocí jednostupňového testu srážení krve podle Evropského lékopisu. Specifická aktivita přípravku BeneFIX je minimálně 200 IU/mg proteinu.
Přípravek BeneFIX obsahuje rekombinantní koagulační faktor IX (INN název nonacogum alfa). Nonakog alfa je přečištěný protein, který se skládá ze 415 aminokyselin v jednom řetězci. Má primární aminokyselinovou sekvenci srovnatelnou s Ala148 alelickou formou faktoru IX získaného z plazmy, zatímco některé posttranslační modifikace rekombinantní molekuly se od přírodního faktoru liší. Rekombinantní koagulační faktor IX je glykoprotein vylučovaný geneticky upravenými savčími buňkami odvozenými z linie ovariálních buněk křečíka čínského (CHO).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
BeneFIX 250 IU, 500 IU, 1000 IU, 2000 IU, 3000 IU prášek a rozpouštědlo pro injekční roztok
Prášek a rozpouštědlo pro injekční roztok
Bílý / téměř bílý prášek a čiré, bezbarvé rozpouštědlo.
KLINICKÉ ÚDAJE
4.
4.1 Terapeutické indikace
Léčba a profylaxe krvácení u pacientů s hemofilií typu B (vrozená deficience faktoru IX).
Přípravek BeneFIX lze podávat všem věkovým skupinám.
4.2 Dávkování a způsob podání
Léčba musí probíhat pouze pod dohledem lékaře se zkušenostmi s léčbou hemofilie.
Monitorování léčby
Během léčby se doporučuje provádět vhodné stanovení hladin faktoru IX a podle výsledků upravovat podávané dávky a četnost opakovaných infuzí. Odpovědi jednotlivých pacientů na faktor IX se mohou lišit, a to jak různými poločasy, tak hodnotami recovery. U dávky vycházející z tělesné hmotnosti může být nutná úprava v případě pacientů s nízkou váhou nebo nadváhou. Zejména v případě velkých chirurgických zákroků je nezbytné přesné monitorování průběhu substituční terapie pomocí koagulační analýzy (test aktivity faktoru IX v plazmě).
Pokud se ke stanovení aktivity faktoru IX ve vzorcích krve pacientů používá in vitro jednostupňový test srážení krve založený na tromboplastinovém čase (aPTT), mohou být výsledky aktivity faktoru IX v plazmě významně ovlivněny jak typem reakčního činidla aPTT, tak referenčním standardem použitým při analýze. Tato skutečnost je důležitá zejména v případě, kdy se mění laboratoř a/nebo reakční činidla používaná při analýze.
Dávkování
Dávka a délka substituční terapie závisí na závažnosti deficience faktoru IX, na lokalizaci a rozsahu krvácení, a také na klinickém stavu pacienta.
Počet podávaných jednotek faktoru IX je vyjadřován v mezinárodních jednotkách (IU), odvozených ze současného standardu WHO pro přípravky s faktorem IX. Aktivita faktoru IX v plazmě je vyjadřována jako procento (vzhledem k normální lidské plazmě) nebo v mezinárodních jednotkách (vzhledem k mezinárodnímu standardu pro faktor IX v plazmě).
Jedna mezinárodní jednotka (IU) aktivity faktoru IX je ekvivalentní množství faktoru IX v jednom ml normální lidské plazmy.
Léčba v případě potřeby (on demand)
Výpočet potřebné dávky přípravku BeneFIX lze stanovit na základě předpokladu, že jedna jednotka aktivity faktoru IX na kg tělesné hmotnosti zvýší hladinu faktoru IX v krevním oběhu v průměru o 0,8 IU/dl (v rozmezí 0,4 až 1,4 IU/dl) u pacientů > 12 let (další informace naleznete v bodě 5.2).
Potřebná dávka se určuje pomocí následujícího vzorce:
|
Potřebné |
= tělesná hmotnost (v |
X požadovaný nárůst |
X převrácená hodnota |
|
množství IU |
kg) |
hladiny faktoru IX (%) |
zjištěné recovery |
|
faktoru IX |
nebo (IU/dl) |
Příklad: Pro zotavení (recovery) 0,8 IU/dl platí vzorec:
|
Potřebné |
= tělesná hmotnost (v |
X požadovaný nárůst X 1,3 IU/kg |
|
množství IU |
kg) |
hladiny faktoru IX (%) |
|
faktoru IX |
nebo (IU/dl) |
Množství a četnost podávání se stanovuje s ohledem na klinickou účinnost v konkrétním případě.
V případě následujících typů krvácení by aktivita faktoru IX neměla v příslušném období poklesnout pod udané hodnoty aktivity v plazmě (v % normální aktivity nebo v IU/dl). Následující tabulku lze použit jako vodítko ke stanovení dávek při krvácení a operacích:
|
Stupeň krvácení/typ |
Potřebná hladina |
Četnost dávek (hod.)/délka terapie |
|
chirurgického zákroku |
faktoru IX (%) nebo (IU/dl) |
(dní) |
|
Krvácení | ||
|
Časný hemartros, svalové |
20-40 |
Opakujte každých 24 hodin. Alespoň |
|
krvácení nebo krvácení z úst |
1 den, dokud se krvácení, které se vyznačuje bolestí, nezastaví nebo nedojde ke zhojení. | |
|
Intenzivnější hemartros, |
30-60 |
Opakujte infuzi každých 24 hodin po |
|
svalové krvácení nebo |
3-4 dny nebo déle, dokud bolest a | |
|
hematom |
akutní potíže neustoupí. | |
|
Život ohrožující krvácení |
60-100 |
Opakujte infuzi každých 8 až 24 hodin, dokud ohrožení nepomine. |
|
Operace | ||
|
Menší |
30-60 |
Každých 24 hodin, alespoň |
|
Včetně extrakce zubu |
1 den, dokud nedojde ke zhojení. | |
|
Větší |
80-100 |
Opakujte infuzi každých 8-24 hodin, |
|
(před a po operaci) |
dokud nedojde k přiměřenému zhojení rány, poté pokračujte v terapii po alespoň dalších 7 dní a udržujte aktivitu faktoru IX mezi 30% až 60% (IU/dl) |
Profylaxe
Přípravek BeneFIX lze podávat při dlouhodobé profylaxi krvácení u pacientů se závažnou hemofilií B. V klinické studii zkoumající rutinní sekundární profylaxi činila průměrná dávka u dříve léčených pacientů (DLP) 40 IU/kg (v rozmezí 13 až 78 IU/kg) při intervalech dávek 3 až 4 dny.
V některých případech, zejména u mladších pacientů, může být nutné použít kratší intervaly dávek nebo vyšší dávky.
Pediatrická populace
K dispozici je omezená dokumentace léčby v případě podle potřeby (on-demand) a operací u dětí mladších 6 let věku léčených přípravkem BeneFIX.
Střední dávka (± standardní odchylka) pro profylaxi byla 63,7 (± 19,1) IU/kg v intervalech 3 až 7 dní. U mladších pacientů může být nutné použít kratší intervaly dávek nebo vyšší dávky. Spotřeba FIX pro rutinní profylaxi u 22 vyhodnocovaných pacientů byla 4607 (± 1849) IU/kg za rok a 378 (± 152) IU/kg za měsíc.
V souladu s klinickou indikací je třeba zajistit pečlivé sledování aktivity faktoru IX v plazmě a výpočet farmakokinetických parametrů jako recovery a poločas, aby bylo možné přiměřeně dávky upravovat.
Starší osoby
Do klinických studií přípravku BeneFIX nebyl zařazen dostatečný počet pacientů ve věku 65 a více let, aby bylo možno stanovit, zda reagují odlišně než mladší subjekty. Stejně jako u všech pacientů, jimž je podáván přípravek BeneFIX, je třeba stanovit dávku pro staršího pacienta individuálně.
Způsob podání
Přípravek BeneFIX je podáván intravenózní infuzí po rekonstituci lyofilizovaného prášku pro injekční roztok ve sterilním 0,234% roztoku chloridu sodného (viz bod 6.6).
Přípravek BeneFIX je třeba podávat pomalou infuzí. Ve většině případů byla používána rychlost podání infuze až 4 ml za minutu. Rychlost podání by měla být stanovena tak, aby se pacient cítil pohodlně.
V případě podezření na hypersenzitivní reakci, u níž se má za to, že souvisí s podáváním přípravku BeneFIX, se má snížit počet infuzí nebo se má infuze zastavit (viz body 4.4 a 4.8).
Aglutinace červených krvinek v kanyle/stříkačce
Některé zdroje uvádějí, že při podávání přípravku BeneFIX docházelo v kanyle/stříkačce k aglutinaci červených krvinek. Ve spojitosti s tímto jevem nebyly hlášeny žádné nežádoucí příhody.
K minimalizaci možnosti vzniku aglutinace je důležité omezit množství krve, které se vrací do kanyly. Krev se nemá dostat do injekční stříkačky. Pokud dojde k aglutinaci červených krvinek v infuzním setu / stříkačce, je třeba zlikvidovat veškerý materiál (infuzní set, stříkačku a naředěný přípravek BeneFIX) a pokračovat v podávání s novým balením.
Kontinuální infuze
Podávání přípravku BeneFIX kontinuální infuzí nebylo schváleno a nedoporučuje se (viz také body 4.4 a 6.6).
Návod na rekonstituci léčivého přípravku před jeho podáním je uveden v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoliv pomocnou látku uvedenou v bodě 6.1.
Známá alergická reakce na křeččí proteiny.
4.4 Zvláštní upozornění a opatření pro použití Hypersenzitivita
Jsou možné hypersenzitivní reakce alergického typu ve spojení s přípravkem BeneFIX. Přípravek obsahuje stopy křeččích proteinů. Při používání přípravků s faktorem IX včetně přípravku BeneFIX došlo v minulosti k anafylaktickým/anafylaktoidním reakcím potenciálně ohrožujícím život pacienta. Pokud se objeví příznaky hypersenzitivity, je třeba pacientům doporučit, aby okamžitě ukončili používání léčivého přípravku a vyhledali svého lékaře. Pacienta je nutné informovat, jak vypadají časné známky hypersenzitivní reakce včetně obtíží s dechem, dušnosti, otoku, kopřivky, generalizované kopřivky, svědění, tísnivého pocitu na prsou, bronchospasmu, laryngospasmu, sípotu, hypotenze, rozmazaného vidění a anafylaxe.
V některých případech tyto reakce vyústily až v závažnou anafylaxi. V případě šoku je třeba postupovat podle současných lékařských zásad terapie šoku. V případě závažných alergických reakcí je třeba zvážit alternativní opatření k podpoře hemostázy.
Inhibitory
Inhibitory jsou méně častou příhodou u dříve léčených pacientů (DLP) dostávajících přípravky s faktorem IX. Vzhledem k tomu, že se u jednoho DLP léčeného přípravkem BeneFIX během klinických studií vyvinul klinicky relevantní slabě reagující inhibitor, a že zkušenosti s antigenicitou rekombinantního faktoru IX jsou dosud omezené, je třeba u pacientů léčených přípravkem BeneFIX
5
pečlivě sledovat rozvoj inhibitorů faktoru IX, které by měly být titrovány v Bethesda jednotkách pomocí vhodného biologického testu.
Některé odkazy v literatuře prokazují korelaci mezi výskytem inhibitoru faktoru IX a alergickou reakcí. Proto by pacienti trpící alergickými reakcemi měli být vyšetřeni na přítomnost inhibitoru. Je třeba upozornit, že pacientům s inhibitory faktoru IX může hrozit zvýšené riziko vzniku anafylaxe při následnému vystavení faktoru IX. Předběžné informace nasvědčují tomu, že může existovat vztah mezi přítomností významných delečních mutací v genu pro faktor IX a zvýšeným rizikem tvorby inhibitoru a akutních reakcí přecitlivělosti. Pacienty, o kterých je známo, že mají významné deleční mutace v genu pro faktor IX, je nutné pozorně sledovat na výskyt příznaků a symptomů akutních reakcí přecitlivělosti, obzvláště během časných fází iniciální expozice přípravku.
Vzhledem k riziku alergických reakcí u koncentrátů faktoru IX je nutné úvodní podávání faktoru IX, dle uvážení ošetřujícího lékaře, provádět pod lékařským dohledem tam, kde je možné poskytnout adekvátní léčbu alergických reakcí.
Trombóza
Přestože přípravek BeneFIX obsahuje pouze faktor IX, je třeba pamatovat na možné riziko trombózy a diseminované intravaskulární koagulace (DIC). Vzhledem k tomu, že bylo použití koncentrátů komplexu faktoru IX v minulosti spojováno s rozvojem tromboembolických komplikací, použití přípravků s faktorem IX může být potenciálně nebezpečné u pacientů se známkami fibrinolýzy a u pacientů s diseminovanou intravaskulární koagulací (DIC). Vzhledem k potenciálnímu riziku trombotických komplikací je třeba při podávání tohoto přípravku pacientům s onemocněním jater, po operaci, novorozeným dětem, pacientům s rizikem trombózních jevů nebo DIC zahájit klinické sledování časných známek trombotické a konzumpční koagulopatie provedením příslušných biologických testů. V každé z těchto situací je třeba zvážit výhody léčby přípravkem BeneFIX proti riziku uvedených komplikací.
Bezpečnost a účinnost podávání přípravku BeneFIX kontinuální infuzí nebyly hodnoceny (viz také body 4.2 a 4.8). Po podávání přípravku BeneFIX kontinuální infuzí centrálním venózním katetrem (viz také bod 4.8) byly v post-marketingovém období hlášeny trombotické příhody, včetně život ohrožujícího syndromu vena cava superior (SVC) u kriticky nemocných novorozenců.
Kardiovaskulární příhody
U pacientů s existujícími kardiovaskulárními rizikovými faktory může substituční terapie pomocí FIX zvýšit kardiovaskulární riziko.
Nefrotický syndrom
Nefrotický syndrom byl hlášen při pokusech o navození imunitní tolerance u pacientů s hemofilií B s inhibitory faktoru IX a anamnézou alergické reakce. Bezpečnost a účinnost přípravku BeneFIX při indukci imunitní tolerance nebyla dosud stanovena.
Zvláštní populace
Klinické studie zatím neposkytly dostatečné údaje o léčbě přípravkem BeneFIX u dosud neléčených pacientů (DNP).
Záznam o podání
Důrazně se doporučuje zaznamenat název a číslo šarže při každém podání přípravku BeneFIX pacientovi, aby se zachovala informace propojující pacienta a šarži léčivého přípravku. Pacienti mohou nalepit jeden z odlepovacích štítků na injekční lahvičce do svého deníku ke zdokumentování čísla šarže nebo ke hlášení jakýchkoli nežádoucích účinků.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly hlášeny žádné interakce přípravků obsahujících lidský koagulační faktor IX (rDNA) s jinými léčivými přípravky.
4.6 Fertilita, těhotenství a kojení
Studie reprodukce na zvířatech s faktorem IX nebyly realizovány. Vzhledem ke vzácnosti výskytu hemofilie B u žen nejsou zkušenosti s použitím faktoru IX během těhotenství a kojení. Faktor IX se má proto během těhotenství a kojení použít pouze v případě, je-li jasně indikován.
Účinek přípravku BeneFIX na fertilitu nebyl stanoven.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek BeneFIX nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky Souhrn bezpečnostního profilu
Byly pozorovány hypersenzitivní nebo alergické reakce (jež mohou zahrnovat angioedém, pálení a bodavý pocit v místě infuze, zimnici, prchavé zrudnutí, generalizovanou kopřivku, bolest hlavy, kopřivku, hypotenzi, letargii, nauzeu, neklid, tachykardii, tísnivý pocit na prsou, mravenčení, zvracení, sípot), které se mohou v některých případech rozvinout až do závažné anafylaxe (včetně šoku). V některých případech se tyto reakce rozvinuly až do závažné anafylaxe a vyskytly se v těsné časové souvislosti se vznikem inhibitorů faktoru IX (viz také bod 4.4). Nefrotický syndrom byl hlášen při pokusech o navození imunitní tolerance u pacientů s hemofilií B s inhibitory faktoru IX a anamnézou alergické reakce.
Velmi vzácně byl pozorován vznik protilátek proti křeččímu proteinu se souvisejícími hypersenzitivními reakcemi.
U pacientů s hemofilií B může dojít k tvorbě neutralizujících protilátek (inhibitorů) proti faktoru IX. Pokud se takové inhibitory vyskytnou, projeví se tento stav nedostatečnou klinickou odpovědí na léčbu. V takových případech se doporučuje kontaktovat specializované centrum pro hemofiliky.
Existuje potenciální riziko tromboembolických příhod po podání přípravků obsahujících faktor IX, viz bod 4.4.
Tabulkový seznam nežádoucích účinků
Níže uvedená tabulka byla sestavena podle klasifikace orgánových systémů MedDRA (třída orgánového systému a preferovaný termín). Četnosti byly vyhodnoceny podle následujícího pravidla: velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1000 až < 1/100), není známo (z dostupných údajů nelze určit). V tabulce jsou uvedeny nežádoucí účinky hlášené během klinických studií dříve léčených pacientů a zjištěné během používání po uvedení přípravku na trh. Četnosti vycházejí ze všech nežádoucích příhod vzniklých během léčby s prokázanou kauzalitou ve sloučených klinických studiích s 224 subjekty.
V jednotlivých skupinách četnosti výskytu jsou nežádoucí účinky seřazeny v pořadí podle klesající závažnosti.
|
Třída orgánového systému |
Velmi časté >1/10 |
Časté > 1/100 až < 1/10 |
Méně časté > 1/1000 až < 1/100 |
Četnost není známa (z dostupných údajů nelze určit) |
|
Infekce a infestace |
Celulitida v místě infuzea | |||
|
Poruchy krve a lymfatického systému |
Inhibice faktoru IXb | |||
|
Poruchy imunitního |
Hypersenzitivitac |
Anafylaktická reakce* |
|
Třída orgánového systému |
Velmi časté >1/10 |
Časté > 1/100 až < 1/10 |
Méně časté > 1/1000 až < 1/100 |
Četnost není známa (z dostupných údajů nelze určit) |
|
systému | ||||
|
Poruchy nervového systému |
Bolest hlavyd |
Závratě, dysgeuzie |
Somnolence, tremor | |
|
Poruchy oka |
Postižení zrakue | |||
|
Srdeční poruchy | ||||
|
Cévní poruchy |
Flebitida, prchavé zrudnutí8 |
Syndrom horní duté žílyi*, hluboká žilní trombóza*, trombóza*, tromboflebitida* | ||
|
Respirační, hrudní a mediastinální poruchy | ||||
|
Gastrointestinální poruchy | ||||
|
Poruchy kůže a podkožní tkáně |
Vyrážkak, kopřivka | |||
|
Poruchy ledvin a močových cest |
Renální infarkt' | |||
|
Celkové poruchy a reakce v místě aplikace |
Pyrexie |
Hrudní dyskomforť, reakce v místě infuzen, bolest v místě infuzem |
Nedostatečná odpověď na léčbu* | |
|
Vyšetření |
Nedostatečná hodnota recovery faktoru IX p * | |||
|
* Nežádoucí účinek léčivého přípravku zjištěný po jeho uvedení na trh a včetně celulitidy b tvorba nízkého titru přechodného inhibitoru c včetně hypersenzitivity na lék, angioedému, bronchospasmu, sípotu, dyspnoe a laryngospasmu d včetně migrény, sinusální bolesti hlavy e včetně scintilačního skotomu a rozmazaného vidění f včetně zvýšené srdeční frekvence a sinusové tachykardie 8 včetně návalu horka, pocitu horka a teplé kůže h včetně sníženého krevního tlaku 1 syndrom horní duté žíly (SHDŽ) u kriticky nemocných novorozenců, kteří dostávali kontinuální infuzi přípravku BeneFIX centrálním žilním katetrem J včetně produktivního kašle k včetně makulózní vyrážky, papulózní vyrážky a makulopapulózní vyrážky l rozvinul se u pacienta pozitivního na přítomnost protilátek proti hepatitidě C 12 dnů po podání dávky přípravku BeneFIX na ošetření krvácivé příhody m včetně bolesti v místě injekce, nepříjemných pocitů v místě infuze n včetně pruritu v místě infuze, erytému v místě infuze o včetně bolesti na hrudi a tísnivého pocitu na prsou p Toto je doslovný výraz. Není k dispozici preferovaný termín dle MedDRA 17.1. | ||||
Popis vybraných nežádoucích účinků
Přecitlivělost/alergické reakce
V případě podezření na hypersenzitivní reakci, u níž se má za to, že souvisí s podáváním přípravku BeneFIX viz body 4.2 a 4.4.
Tvorba inhibitoru
Klinicky relevantní slabě reagující inhibitor byl zjištěn u jednoho z 65 pacientů (včetně 9 pacientů účastnících se pouze chirurgické studie) dostávajících přípravek BeneFIX, kteří dříve dostávali přípravky odvozené z plazmy. Tento pacient byl schopen pokračovat v léčbě přípravkem BeneFIX, přičemž se v anamnéze neobjevil nárůst hladin inhibitoru nebo anafylaxe (viz bod 4.4).
Pediatrická populace
U dětí mohou být alergické reakce častější než u dospělých.
K dispozici nejsou dostatečné údaje k poskytnutí informací o výskytu inhibitorů u DNP (viz též bod 5.1).
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Nebyly hlášeny žádné příznaky předávkování při podávání přípravků obsahujících rekombinantní koagulační faktor IX.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antikoagulancia, koagulační faktor IX; ATC kód: B02BD09 Mechanismus účinku
Přípravek BeneFIX obsahuje rekombinantní koagulační faktor IX (nonacog alfa). Rekombinantní koagulační faktor IX je glykoprotein s jedním řetězcem o přibližné molekulové hmotnosti 55 kDa, který je členem rodiny vitamin K-dependentních koagulačních faktorů typu serinových proteáz. Rekombinantní koagulační faktor IX je léčivý bílkovinný přípravek připravený pomocí rekombinantní DNA, který má strukturální a funkční charakteristiky srovnatelné s endogenním faktorem IX. Faktor IX je aktivován faktorem VII a komplexem tkáňového faktoru vnější cestou, a také faktorem XIa vnitřní cestou koagulace. Aktivovaný faktor IX v kombinaci s aktivovaným faktorem VIII aktivují faktor X. Konečným výsledkem je konverze protrombinu na trombin. Trombin poté konvertuje fibrinogen na fibrin a vzniká krevní sraženina. Aktivita faktoru IX u pacientů trpících hemofilií B chybí nebo je značně snížená a může být nutná substituční terapie.
Farmakodynamické účinky
Hemofilie B je dědičná, na pohlaví vázaná porucha krevní srážlivosti způsobená sníženou hladinou faktoru IX, jejímž výsledkem je profuzní krvácení do kloubů, svalů nebo vnitřních orgánů, a to buď spontánně nebo v důsledku náhodného nebo chirurgického poranění. Substituční léčba zvyšuje hladinu faktoru IX v plazmě, a dočasně tak napravuje nedostatek faktoru a sklon ke krvácení.
Pediatrická populace
Analýza účinnosti ve studii 3090A1-301-WW byla založena na 22 vyhodnocovaných pediatrických pacientech v profylaktickém režimu včetně 4 pacientů s léčbou podle potřeby (on-demand), kteří zakrátko přešli na profylaxi. Dva pacienti podstoupili chirurgické procedury (cirkumcize a zavedení port-a-cathu). Analýza bezpečnosti 25 vyhodnocovaných pacientů prokázala očekávaný bezpečnostní profil. Jediný zdokumentovaný závažný nežádoucí účinek související s přípravkem BeneFIX byl hlášen u jediného DNP, u něhož došlo k přecitlivělosti a rozvoji inhibitorů.
Ve dvou otevřených studiích bylo zjištěno, že přípravek BeneFIX lze bezpečně podávat v dávce 100 IU/kg jednou týdně. Avšak poločas přípravku (viz bod 5.2) a omezené farmakokinetické údaje pro režim s podáváním jednou týdně neumožňují doporučit tento režim všeobecně k dlouhodobé profylaxi u pacientů se závažnou hemofilií B.
5.2 Farmakokinetické vlastnosti
V randomizované zkřížené farmakokinetické studii bylo prokázáno, že přípravek BeneFIX rekonstituovaný v 0,234% roztoku chloridu sodného je farmakokineticky ekvivalentní dříve prodávanému přípravku BeneFIX (rekonstituovanému sterilní vodou) u 24 dříve léčených pacientů (> 12 let) při dávce 75 IU/kg. Dále byly farmakokinetické parametry sledovány u 23 z těchto pacientů po opakovaném podávání přípravku BeneFIX po dobu 6 měsíců a v porovnání se stejnými parametry získanými při počátečním vyhodnocení bylo zjištěno, že zůstávají nezměněné. V tabulce 1 je uvedeno shrnutí farmakokinetických údajů.
|
Tabulka 1. Odhad farmakokinetických parametrů u přípravku BeneFIX (75 IU/kg) při zahájení studie a v 6. měsíci u dříve léčených pacientů s hemofilií B | ||
|
Parametr |
Při zahájení studie n = 24 Průměr ± SD |
6. měsíc n = 23 Průměr ± SD |
|
Cmax (IU/dl) |
54,5 ± 15,0 |
57,3 ± 13,2 |
|
AUC*. (IU-h/dl) |
940 ± 237 |
923 ± 205 |
|
ti/2 (h) |
22,4 ± 5,3 |
23,8 ± 6,5 |
|
CL (ml/h/kg) |
8,47 ± 2,12 |
8,54 ± 2,04 |
|
Biologická dostupnost (IU/dl na IU/kg) |
0,73 ± 0,20 |
0,76 ± 0,18 |
|
Zkratky: AUCm = plocha pod křivkou plazmatické koncentrace v čase od času nula do nekonečna; Cmax = maximální koncentrace; t1/2 = poločas eliminace z plazmy; CL = clearance; SD = směrodatná odchylka. | ||
Farmakokinetický model populace byl vytvořen pomocí dat shromážděných od 73 pacientů ve věku 7 měsíců až 60 let. Tabulka 2 ukazuje parametry odhadnuté pomocí finálního dvoukompartmentového modelu. Batolata a děti měly vyšší clearance, větší distribuční objem, kratší poločas eliminace a nižší zotavení (recovery) než dospívající a dospělí. Konečná fáze nebyla jednoznačně pokryta v důsledku nedostatku dat od vyhodnocovaných dětských pacientů mladších 6 let pro období po 24 hodinách.
|
Tabulka 2. Střední farmakokinetické parametry ± směrodatná odchylka založené na jednotlivých Bayesových odhadech z farmakokinetické analýzy populace | |||||
|
Věková skupina (roky) |
Batolata < 2 |
Děti 2 až < 6 |
Děti 6 až < 12 |
Dospívající 12 až < 18 |
Dospělí 18 až 60 |
|
Počet subjektů |
7 |
16 |
1 |
19 |
30 |
|
Clearance (ml/h/kg) |
13,1 ± 2,1 |
13,1 ± 2,9 |
15,5 |
9,2 ± 2,3 |
8,0 ± 0,6 |
|
Vss (ustálený objem, ml/kg) |
252 ± 35 |
257 ± 25 |
303 |
234 ± 49 |
225 ± 59 |
|
Poločas eliminace (h) |
15,6 ± 1,2 |
16,7 ± 1,9 |
16,3 |
21,5 ± 5,0 |
23,9 ± 4,5 |
|
Biologická dostupnost (IU/dl na |
0,61 ± 0,10 |
0,60 ± 0,08 |
0,47 |
0,69 ± 0,16 |
0,74 ± 0,20 |
|
IU/kg) |
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje, získané na základě konvenčních studií genotoxicity, neodhalily žádné zvláštní riziko pro člověka.
Studie kancerogenity a účinků na plodnost a narušení vývoje plodu nebyly realizovány.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek Sacharosa Glycin Histidin Polysorbát 80
Rozpouštědlo Roztok chloridu sodného
6.2 Inkompatibility
Protože chybí studie kompatibility, nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky. Používejte pouze dodanou infuzní soupravu. Léčba může selhat v důsledku adsorpce faktoru IX na vnitřní povrch některé části infuzního zařízení.
6.3 Doba použitelnosti
2 roky
Rekonstituovaný přípravek neobsahuje protimikrobní přísadu a má být použit okamžitě, avšak ne později než do 3 hodin po rekonstituci. Chemická a fyzikální stabilita po otevření před použitím byla prokázána na dobu 3 hodin při teplotě do 25 °C.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte při teplotě do 30°C. Chraňte před mrazem.
6.5 Druh obalu a obsah balení
BeneFIX 250 IU, 500 IU, 1000 IU, 2000 IU, 3000 IU prášek a rozpouštědlo pro injekční roztok BeneFIX 250 IU, 500 IU, 1000 IU, 2000 IU, 3000 IU prášek v 10 ml injekční lahvičce (sklo typ 1) se zátkou (chlorobutyl) a odtrhávacím uzávěrem (hliník) a 5 ml čirého, bezbarvého rozpouštědla v předplněné injekční stříkačce (sklo typ 1) s pístovou zátkou (bromobutyl), kryt hrotu (bromobutyl) a sterilní adaptér na injekční lahvičku k rozpouštění, sterilní infuzní souprava, dva alkoholem napuštěné tampony, náplast a gázový polštářek.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Přípravek BeneFIX se podává intravenózní infuzí po rekonstituci lyofilizovaného prášku pro injekční roztok v dodaném rozpouštědle (0,234% vodný roztok chloridu sodného) v předplněné injekční stříkačce (viz bod 3 pokyny k rekonstituci léčivého přípravku uvedené v Příbalové informaci).
Přípravek BeneFIX po rekonstituci obsahuje polysorbát-80, o němž je známo, že zvyšuje rychlost extrakce di-(2-ethyl hexyl)ftalátu (DEHP) z polyvinylchloridu (PVC). Toto je třeba vzít v úvahu během přípravy a podávání přípravku BeneFIX. Je důležité, aby byla bedlivě dodržována doporučení uvedená v bodu 4.2.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Protože použití přípravku BeneFIX kontinuální infuzí nebylo hodnoceno, přípravek BeneFIX nesmí být mísen s infuzními roztoky ani nesmí být podán v infuzi.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/97/047/004
EU/1/97/047/005
EU/1/97/047/006
EU/1/97/047/007
EU/1/97/047/008
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 27. srpna 1997
Datum posledního prodloužení registrace: 20. července 2012
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu.
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce biologické léčivé látky
Wyeth BioPharma
Division of Wyeth Pharmaceuticals Inc., a subsidiary of Pfizer Inc.
One Burtt Road Andover MA 01810 USA
Název a adresa výrobce odpovědného za propouštění šarží Wyeth Farma S.A.
Autovia del Norte.A-1, Km. 23. Desvio Algete, Km. 1, 28700 San Sebastian de los Reyes, Madrid Španělsko
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz Příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU KRABIČKA
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU_
BeneFIX 250 IU prášek a rozpouštědlo pro injekční roztok BeneFIX 500 IU prášek a rozpouštědlo pro injekční roztok BeneFIX 1000 IU prášek a rozpouštědlo pro injekční roztok BeneFIX 2000 IU prášek a rozpouštědlo pro injekční roztok BeneFIX 3000 IU prášek a rozpouštědlo pro injekční roztok Nonacogum alfa (rekombinantní koagulační faktor IX)
2. OBSAH LÉČIVÉ LÁTKY/LÁTEK)
1 injekční lahvička: 250 IU nonacogum alfa (cca 50 IU/ml po rekonstituci).
1 injekční lahvička: 500 IU nonacogum alfa (cca 100 IU/ml po rekonstituci)
1 injekční lahvička: 1000 IU nonacogum alfa (cca 200 IU/ml po rekonstituci)
1 injekční lahvička: 2000 IU nonacogum alfa (cca 400 IU/mlpo rekonstituci)
1 injekční lahvička: 3000 IU nonacogum alfa (cca 600 IU/ml po rekonstituci)
3. SEZNAM POMOCNÝCH LÁTEK
Sacharóza, glycin, histidin, chlorid sodný, polysorbát 80.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Prášek a rozpouštědlo pro injekční roztok
1 injekční lahvička s 250 IU látky nonacogum alfa 1 injekční lahvička s 500 IU látky nonacogum alfa 1 injekční lahvička s 1000 IU látky nonacogum alfa 1 injekční lahvička s 2000 IU látky nonacogum alfa 1 injekční lahvička s 3000 IU látky nonacogum alfa
1 předplněná stříkačka s 5 ml rozpouštědla 1 sterilní adaptér na injekční lahvičku k rozpouštění
1 sterilní infuzní souprava
2 alkoholem napuštěné tampony
1 náplast
1 gázový polštářek
5. ZPŮSOB A CESTA PODÁNÍ
Intravenózní podání, jen pro jednorázové použití. Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
K rozpuštění použijte pouze rozpouštědlo v předplněné injekční stříkačce dodávané v krabičce.
8. POUŽITELNOST
EXP
Použijte ihned nebo nejpozději do 3 hodin po rozpuštění.
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte při teplotě do 30 °C. Chraňte před mrazem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Velká Británie
12. REGISTRAČNÍ ČÍSLO(A)
EU/1/97/047/004
EU/1/97/047/005
EU/1/97/047/006
EU/1/97/047/007
EU/1/97/047/008
13. ČÍSLO ŠARŽE
c.s.:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVÉ PÍSMU
BeneFIX 250 BeneFIX 500 BeneFIX 1000 BeneFIX 2000 BeneFIX 3000
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA(Y) PODÁNÍ
BeneFIX 250 IU prášek pro injekční roztok
BeneFIX 500 IU prášek pro injekční roztok
BeneFIX 1000 IU prášek pro injekční roztok
BeneFIX 2000 IU prášek pro injekční roztok
BeneFIX 3000 IU prášek pro injekční roztok
Nonacogum alfa (rekombinantní koagulační faktor IX)
Intravenózní podání
2. ZPŮSOB PODÁNÍ
Jednorázová injekce
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE č.š.:
Viz štítek na přední straně (č.š., EXP)
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET DÁVEK
250 IU 500 IU 1000IU 2000 IU 3000IU
6. JINÉ
Uchovávejte při teplotě do 30 °C. Chraňte před mrazem.
K rozpuštění použijte pouze rozpouštědlo v předplněné injekční stříkačce dodávané v krabičce.
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA(Y) PODÁNÍ
Rozpouštědlo pro BeneFIX Intravenózní podání
2. ZPŮSOB PODÁNÍ
Použijte celý obsah.
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE c.š.:
Pfizer Limited
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET DÁVEK
Obsahuje 5 ml sterilního 0,234% roztoku chloridu sodného na injekci.
6. JINÉ
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: Informace pro uživatele
BeneFIX 250 IU prášek a rozpouštědlo pro injekční roztok BeneFIX 500 IU prášek a rozpouštědlo pro injekční roztok BeneFIX 1000 IU prášek a rozpouštědlo pro injekční roztok BeneFIX 2000 IU prášek a rozpouštědlo pro injekční roztok BeneFIX 3000 IU prášek a rozpouštědlo pro injekční roztok Nonacogum alfa (rekombinantní koagulační faktor IX)
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek BeneFIX a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek BeneFIX používat
3. Jak se přípravek BeneFIX používá
4. Možné nežádoucí účinky
5. Jak přípravek BeneFIX uchovávat
6. Obsah balení a další informace
1. Co je přípravek BeneFIX a k čemu se používá
Přípravek BeneFIX je injekční koagulační faktor IX získávaný rekombinantní DNA technologií. Léčivou látkou přípravku BeneFIX je nonakog alfa. Lidé, kteří se narodili s hemofilií B (Christmasova nemoc), nemají dostatek koagulačního faktoru IX ke kontrole krvácení. Přípravek BeneFIX účinkuje tak, že u pacientů s hemofilií B doplňuje hladinu faktoru IX a umožňuje tak srážení krve.
Přípravek BeneFIX se používá k léčbě a prevenci krvácení u pacientů s hemofilií B (vrozený nedostatek faktoru IX) ve všech věkových skupinách.
2. Čemu musíte věnovat pozornost, než začnete přípravek BeneFIX užívat Přípravek BeneFIX nepoužívejte
- Pokud jste alergický(á) na nonakog nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
- Pokud jste alergický(á) na křeččí proteiny.
Upozornění a opatření
- Pokud krvácení neustane podle očekávání, ihned vyhledejte lékaře.
- Alergické reakce jsou možné. Přípravek může obsahovat stopy křečcích proteinů (viz Přípravek BeneFIX nepoužívejte). Při užívání přípravků s faktorem IX, včetně přípravku BeneFIX, došlo v minulosti k anafylaktickým reakcím (závažné alergické reakce) potenciálně ohrožujícím život pacienta. Časné známky alergické reakce zahrnují dechové potíže, dušnost, otok, kopřivku, generalizovanou kopřivku, svědění, tísnivý pocit na prsou, sípot, nízký krevní tlak, rozmazané vidění a anafylaxi (těžká alergická reakce, která může způsobit obtíže při polykání a/nebo dýchání, zrudnutí či otok tváře a/nebo rukou).
- Pokud k alergické či anafylaktické reakci dojde, ihned zastavte infuzi a spojte se s lékařem či vyhledejte neodkladnou lékařskou pomoc. V případě závažných alergických reakcí je třeba zvážit alternativní léčbu.
- Protilátky neutralizující aktivitu přípravku (inhibitory) jsou méně častým jevem u pacientů, kteří dříve podstoupili léčbu přípravky obsahující faktor IX. Stejně jako u všech přípravků
s faktorem IX byste ale měl(a) být pečlivě sledován(a) na rozvoj inhibitorů faktoru IX po dobu léčení přípravkem BeneFIX.
- Výzkum prokázal souvislost mezi výskytem inhibitoru faktoru IX a alergickými reakcemi. Pokud tedy zaznamenáte alergické reakce, které jsou uvedené výše, měl(a) byste být vyšetřen(a) na přítomnost inhibitoru. Je nutno uvést, že pacientům s inhibitory faktoru IX může hrozit zvýšené riziko vzniku anafylaxe během následující léčby přípravkem BeneFIX.
- Produkce faktoru IX v těle je řízena genem pro faktor IX. U pacientů se specifickou mutací genu faktoru IX, jako je např. významná deleční mutace, je vyšší pravděpodobnost vzniku inhibitoru faktoru IX a/nebo výskytu alergických reakcí. Proto Vás, pokud víte, že máte takovou mutaci, může lékař pozorněji sledovat na výskyt příznaků alergické reakce, zejména když začínáte užívat přípravek BeneFIX poprvé.
- Vzhledem k riziku alergických reakcí u faktoru IX by měl být přípravek BeneFIX podáván v počáteční fázi pod lékařským dozorem, aby byla poskytnuta vhodná léčba v případě alergických reakcí.
- I v nepřítomnosti inhibitoru faktoru IX však mohou být zapotřebí vyšší dávky přípravku BeneFIX, než bylo zapotřebí pro jiné plazmatické deriváty faktoru IX dříve podávané. Je tedy nutné pečlivé sledovat plazmatickou aktivitu faktoru IX (která měří schopnost Vaší krve vytvářet krevní sraženiny) a provést přiměřenou úpravu dávky. Pokud se krvácení s podáním doporučené dávky neupraví, kontaktujte lékaře.
- Pokud trpíte onemocněním jater nebo onemocněním srdce nebo jste v nedávné době podstoupil(a) operaci, hrozí Vám zvýšené riziko tvorby krevních sraženin.
- Porucha funkce ledvin (nefrotický syndrom) byla hlášena po podání vysokých dávek faktoru IX u pacientů s hemofilií B, inhibitorem faktoru IX a anamnézou alergických reakcí.
- Klinické studie zatím neposkytly dostatečné údaje o léčbě přípravkem BeneFIX u dosud neléčených pacientů (pacienti, kteří nebyli nikdy dříve léčeni infuzí faktoru IX).
- Při každém použití přípravku BeneFIX se doporučuje zaznamenat název a číslo šarže přípravku. Můžete použít odlepovací štítky na injekční lahvičce ke zdokumentování čísla šarže ve svém deníku nebo ke hlášení jakýchkoli nežádoucích účinků.
Další léčivé přípravky a přípravek BeneFIX
Prosím, informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné
době užíval(a) nebo které možná budete užívat.
Těhotenství, kojení a fertilita
Pokud jste těhotná či kojíte, domníváte se, že můžete být těhotná nebo plánujete otěhotnět, můžete používat přípravek BeneFIX pouze podle konkrétních pokynů lékaře. Není známo, zda může podávání přípravku BeneFIX těhotným ženám způsobit poškození nenarozeného dítěte. Lékař Vám může poradit, abyste léčbu přípravkem BeneFIX přerušila, pokud kojíte či otěhotníte.
Poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek používat.
Řízení dopravních prostředků a obsluha strojů
Přípravek BeneFIX nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
3. Jak se přípravek BeneFIX používá
Vždy používejte tento přípravek přesně podle pokynů svého lékaře nebo lékárníka. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
O dávce přípravku BeneFIX, kterou dostanete, rozhodne lékař. Dávka a délka léčby závisí na Vaší specifické potřebě náhradní léčby faktorem IX, a jak rychle Vaše tělo spotřebovává faktor IX, tyto údaje musí být pravidelně kontrolovány. Pokud jste vyměnil(a) přípravek obsahující faktor IX derivovaný z plazmy za přípravek BeneFIX, můžete pozorovat rozdíl v dávce, kterou dostáváte.
Lékař může během léčby rozhodnout o změně dávky přípravku BeneFIX.
Rekonstituce a podávání léku
Níže uvedené postupy mají sloužit jako návod při rekonstituci a podávání přípravku BeneFIX. Pacienti musí dodržovat konkrétní postup určený jejich lékařem.
Přípravek BeneFIX se podává nitrožilní (i.v.) infuzí po rekonstituci prášku pro injekční roztok přiloženým rozpouštědlem (roztoku chloridu sodného) v předplněné injekční stříkačce.
Před následujícími kroky si vždy umyjte ruce. Při rozpouštění přípravku pracujte asepticky (tj. dbejte na čistotu a zamezení choroboplodných zárodků).
Rekonstituce léku:
Přípravek BeneFIX se po rekonstituci ve sterilním rozpouštědle na injekci podává nitrožilní (i.v.) infuzí.
1. Lyofilizovaný (vysušený mrazením) přípravek BeneFIX v předplněné injekční stříkačce nechte zahřát na pokojovou teplotu.
2. Sejměte pojistný plastový kryt z injekční lahvičky přípravku BeneFIX a odkryjte tak centrální část gumové zátky.
3. Otřete horní část lahvičky dodaným tamponem namočeným v alkoholu, či použijte jiný antiseptický roztok a nechte zaschnout. Po očištění se nedotýkejte rukou pryžové zátky a dbejte, aby se zátka nedotkla žádného povrchu.
4. Sejměte kryt z plastické hmoty z průhledného adaptéru injekční lahvičky. Nevytahujte adaptér z obalu.
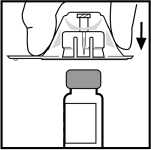
5. Postavte injekční lahvičku na plochý rovný povrch. Držte adaptér v obalu a současně jej umístěte na lahvičku. Stlačte pevně dolů, až adaptér zapadne do místa na vrchu lahvičky s hrotem adaptéru, pronikajícím zátkou lahvičky.
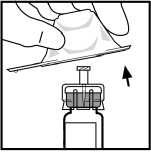
6. Odstraňte obal z adaptéru a zlikvidujte obal.
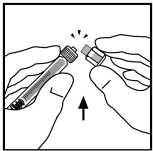
7. Pevným tlačením a otáčením připojte píst s táhlem k injekční stříkačce s rozpouštědlem.
8. Odlomte plastový konec krytu ze stříkačky s ředidlem tak, že zlomíte perforaci krytu. Toho dosáhnete kýváním krytu nahoru a dolů až do zlomení perforace. Nedotýkejte se vnitřku krytu nebo konce stříkačky. Možná budete muset kryt znovu nasadit na původní místo (pokud rozpuštěný roztok nebude okamžitě podán), proto si jej položte vedle otočený odlomenou částí nahoru.
9. Postavte injekční lahvičku na plochý rovný povrch. Spojte injekční stříkačku s ředidlem
s adaptérem vložením konce injekční stříkačky do otvoru adaptéru. Za stálého tlaku a otáčení stříkačky ve směru hodinových ručiček, až je spoj pevný.
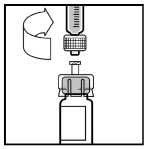
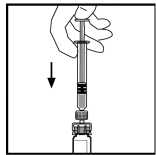
10. Pomalu tlačte na táhlo pístu, až injikujete všechno rozpouštědlo do injekční lahvičky přípravku BeneFIX.
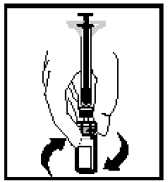
11. S injekční stříkačkou stále spojenou s adaptérem jemně otáčejte až do úplného rozpuštění prášku.
12. Finální roztok má být před podáním vizuálně zkontrolován na přítomnost jemných částic. Roztok by měl být čirý a bezbarvý.
Poznámka: Pokud na infuzi použijete více než jednu injekční lahvičku přípravku BeneFIX, každá injekční lahvička by měla být rekonstituována podle předcházejících pokynů. Stříkačka s ředidlem se má odstranit, zatímco adaptér injekční lahvičky má zůstat na svém místě a ke stažení rekonstituovaného obsahu každé jednotlivé injekční lahvičky může být použita samostatná velká stříkačka Luer (zařízení, které spojuje injekční stříkačku s injekční lahvičkou).
13. Přesvědčte se, že táhlo pístu stříkačky je úplně stlačeno a obraťte lékovku dnem vzhůru. Pomalu natáhněte roztok do stříkačky.
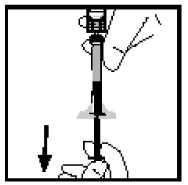
14. Odpojte injekční stříkačku od adaptéru injekční lahvičky jemným tahem a otáčením stříkačky ve směru hodinových ručiček. Zlikvidujte injekční lahvičku s připojeným adaptérem.
Poznámka: Když nepoužijete roztok ihned, nasaďte opatrně na stříkačku kryt. Nedotýkejte se konce injekční stříkačky ani vnitřku krytu.
Přípravek BeneFIX je třeba podat okamžitě nebo do 3 hodin po rekonstituci. Rekonstituovaný roztok lze před podáním uchovávat při pokojové teplotě.
Podávání (intravenózní injekce)
Přípravek BeneFIX se podává přiloženou předplněnou injekční stříkačkou s rozpouštědlem nebo jedinou jednorázovou sterilní plastovou stříkačkou s kónusem luer. Rozpuštěný přípravek z injekční lahvičky se nasaje pomocí adaptéru na injekční lahvičku.
Přípravek BeneFIX musí být podán intravenózně v průběhu několika minut. Váš lékař může změnit rychlost infuze tak, aby byla infuze pohodlnější.
Použití přípravku BeneFIX v kontinuální infuzi nebylo hodnoceno, proto se přípravek BeneFIX nesmí mísit s infuzními roztoky a ani podávat v infuzi.
Veškeré nespotřebované roztoky, prázdné injekční lahvičky a použité jehly a stříkačky před vhozením do odpadu zabalte, aby nedošlo k poranění jiných osob.
Některé zdroje uvádějí, že při podávání přípravku BeneFIX docházelo v hadičce/stříkačce ke shlukování (aglutinaci) červených krvinek. Ve spojitosti s tímto jevem nebyly hlášeny žádné nežádoucí účinky. K minimalizaci možnosti shlukování červených krvinek je důležité omezit množství krve, které se vrací do hadičky. Krev se nemá dostat do stříkačky. Pokud dojde ke shlukování červených krvinek v hadičce/stříkačce, zlikvidujte veškerý materiál (hadičku, stříkačku a roztok přípravku BeneFIX) a pokračujte v podávání s novým balením.
Jestliže jste použil(a) více přípravku BeneFIX, než jste měl(a)
Pokud jste podal(a) více přípravku BeneFIX, než Vám lékař doporučil, kontaktujte ihned svého lékaře.
Jestliže jste přestal(a) používat přípravek BeneFIX
Nepřestávejte používat přípravek BeneFIX bez konzultace s lékařem.
Máte-li jakékoli další otázky týkající se používání tohoto přípravku, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Přecitlivělost/alergické reakce
Při používání přípravku BeneFIX se mohou objevit alergické reakce přecitlivělosti. Tyto reakce mohou zahrnovat otok obličeje nebo hrdla, pálení a bodavý pocit v místě infuze, zimnici, prchavé zrudnutí, svědění, bolest hlavy, kopřivku, nízký krevní tlak, letargii, pocit na zvracení, neklid, zrychlený tep, tísnivý pocit na prsou, mravenčení, zvracení a sípání. V některých případech vyústily tyto reakce až v závažnou anafylaxi. K alergickým reakcím došlo v těsné spojitosti s rozvojem inhibitoru faktoru IX (viz také „Upozornění a opatření“).
Tyto reakce mohou potenciálně ohrozit život pacienta. Pokud k alergické / anafylaktické reakci dojde, ihned zastavte infuzi a spojte se ihned se svým lékařem či vyhledejte neodkladnou lékařskou pomoc. Potřebná léčba závisí na povaze a závažnosti nežádoucích účinků (viz také „ Upozornění a opatření“).
Tvorba inhibitoru
U pacientů s hemofilií B se mohou vytvořit neutralizující protilátky (inhibitory) proti faktoru IX. Pokud se takové inhibitory vytvoří, příznakem může být nárůst množství přípravku BeneFIX obvykle nutného k léčbě krvácení a/nebo pokračující krvácení po léčbě. V takových případech se doporučuje kontaktovat specializované centrum pro hemofiliky. Váš lékař Vás může chtít monitorovat s ohledem na tvorbu inhibitoru (viz „Upozornění a opatření“).
Porucha funkce ledvin byla hlášena po podání vysokých dávek faktoru IX odvozeného z plazmy, který měl navodit imunitní toleranci u pacientů s hemofilií B, inhibitorem faktoru IX a anamnézou alergických reakcí (viz také „ Upozornění a opatření“).
Trombotické příhody
Přípravek BeneFIX může zvýšit riziko trombózy (abnormální krevní sraženina) ve Vašem těle, pokud máte rizikové faktory pro rozvoj krevních sraženin, včetně dlouhodobě zavedeného žilního katétru.
Při podávání přípravku BeneFIX kontinuální infuzí centrálním žilním katetrem byly hlášeny závažné příhody krevních sraženin, včetně život ohrožujících krevních sraženin u kriticky nemocných novorozenců. Byly hlášeny případy periferní tromboflebitidy (zánět žil) (bolest a zčervenání žil) a hluboké žilní trombózy (krevní sraženiny v končetinách); ve většině těchto případů byl přípravek BeneFIX podáván kontinuální infuzí, což je nepovolený způsob podání.
Velmi časté nežádoucí účinky (mohou se vyskytnut u více než 1 osoby z 10)
• bolest hlavy
• kašel
• horečka
Časté nežádoucí účinky (mohou se vyskytnut až u 1 osoby z 10)
• přecitlivělost / alergické reakce
• zimnice, změny chuti
• zánět žil (bolest a zčervenání žil), prchavé zrudnutí
• zvracení, pocit na zvracení
• vyrážka, kopřivka
• nepříjemné pocity na hrudi (včetně bolesti na hrudi)
• reakce v místě infuze (včetně svědění a zarudnutí v místě infuze), bolest a nepříjemné pocity v místě infuze
Méně časté nežádoucí účinky (mohou se vyskytnut až u 1 osoby ze 100)
• vytvoření neutralizujících protilátek (inhibitory)
• celulitida v místě infuze (bolest a zčervenání kůže)
• ospalost, třes
• postižení zraku (včetně rozmazaného vidění, vidění teček/světel)
• zrychlený tep, nízký krevní tlak
• infarkt ledvin (přerušení krevního zásobení ledvin)
Nežádoucí účinky s neznámou četností (četnost nelze z dostupných údajů určit)
• anafylaktická reakce
• trombotické příhody (abnormální krevní sraženiny)
• nedostatečná odpověď na léčbu (neschopnost zastavit nebo zabránit krvácení)
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek BeneFIX uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a etiketě injekční lahvičky. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Přípravek BeneFIX musí být uchováván při teplotě do 30 °C a musí být použit do uplynutí doby použitelnosti vyznačené na obalu.
Chraňte před mrazem, aby se předešlo poškození předplněné injekční stříkačky.
Rekonstituovaný roztok použijte ihned nebo do 3 hodin.
Nepoužívejte přípravek BeneFIX, pokud není roztok čirý nebo bezbarvý.
K rekonstituci a podávání léku použijte pouze předplněnou injekční stříkačku přibalenou v krabici. K podání je možno použít jiné jednorázové injekční stříkačky.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co přípravek BeneFIX obsahuje
- Léčivou látkou je nonakogum alfa (rekombinantní koagulační faktor IX). Jedna injekční lahvička přípravku BeneFIX obsahuje nominálně 250, 500, 1000, 2000 nebo 3000 IU látky nonakog alfa.
- Pomocnými látkami jsou sacharosa, glycin, histidin a polysorbát 80. K rozpouštění je též přiloženo rozpouštědlo (0,234 % roztok chloridu sodného).
- Po rozpuštění v dodaném rozpouštědle (0,234 % roztok chloridu sodného) jedna injekční lahvička obsahuje 50, 100, 200, 400 nebo 600 IU/ ml (viz. Tab. 1).
Tabulka 1. Síla přípravku BeneFIX na ml připraveného roztoku
|
Množství BeneFIXu v injekční lahvičce |
Množství BeneFIXu na 1 ml připraveného injekčního roztoku |
|
250 IU |
50 IU |
|
500 IU |
100 IU |
|
1000IU |
200 IU |
|
2000 IU |
400 IU |
|
3000 IU |
600 IU |
Jak přípravek BeneFIX vypadá a co obsahuje toto balení
Přípravek BeneFIX se dodává jako prášek pro injekční roztok ve skleněné lahvičce a rozpouštědlo v předplněné injekční stříkačce.
Balení obsahuje:
• Jednu injekční lahvičku s práškem BeneFIX 250, 500, 1000, 2000 nebo 3000 IU
• Jednu předplněnou injekční stříkačku s rozpouštědlem, 5 ml 0,234% roztoku chloridu sodného na injekci k rozpouštění s jedním pístovým táhlem
• Jeden sterilní adaptér na injekční lahvičku k rozpouštění
• Jednu sterilní infuzní soupravu
• Dva alkoholem napuštěné tampony
• Jednu náplast
• Jeden gázový polštářek
Držitel rozhodnutí o registraci
Pfizer Limited Ramsgate Road Sandwich Kent CT13 9NJ Velká Británie
Výrobce
Wyeth Farma S.A.
Autovia del Norte. A-1, Km. 23. Desvio Algete, Km. 1, 28700 San Sebastian de los Reyes, Madrid, Španělsko
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci.
Belgie /Belgique / Belgien
Pfizer S.A./N.V.
Tél/Tel: +32 (0)2 554 62 11
EtnrapHH
n$aň3ep HroKceMÓypr CAPH, Kuoh Etnrapna
Ten.: +359 2 970 4333
Česká republika
Pfizer PFE, spol. s .r.o.
Tel: +420 283 004 111
Danmark
Pfizer ApS
Tlf: +45 44 20 11 00
Deutschland
Pfizer Pharma GmbH Tel: +49 (0)30 550055 51000
Eesti
Pfizer Luxembourg SARL Eesti filiaal
Tel: +372 666 7500
Lietuva
Pfizer Luxembourg SARL filialas Lietuvoje
Tel. +3705 2514000
Luxembourg/Luxemburg
Pfizer S.A.
Tél/Tel: +32 (0)2 554 62 11
Magyarország
Pfizer Kft..
Tel.: + 36 1 488 37 00 Malta
Vivian Corporation Ltd. Tel: +35621 344610
Nederland
Pfizer bv
Tel: +31 (0)10 406 43 01 Norge
Pfizer Norge AS.
Tlf: +47 67 52 61 00
|
EIIá8a PFIZER EAAAI A.E. Tni: +30 210 67 85 800 |
Osterreich Pfizer Corporation Austria Ges.m.b.H.o., Tel: +43 (0)1 521 15-0 |
|
Espaňa Pfizer S.L. Tel: +34 91 490 99 00 |
Polska Pfizer Polska Sp. z o.o.,. Tel.: +48 22 335 61 00 |
|
France Pfizer |
Portugal Pfizer Biofarmaceutica, Sociedade Unipessoal Lda |
|
Tél: +33 (0)1 58 07 34 40 |
Tel: +351 21 423 5500 |
|
Hrvatska Pfizer Croatia d.o.o. Tel: + 385 1 3908 777 |
Románia Pfizer Románia S.R.L Tel: +40 21 207 28 00 |
|
Ireland Pfizer Healthcare Ireland Tel: 1800 633 363 (toll free) +44 (0)1304 616161 |
Slovenija Pfizer Luxembourg SARL Pfizer, podružnica za svetovanje s področja farmacevtske dejavnosti, Ljubljana Tel: + 386 (0) 1 52 11 400 |
|
Ísland Icepharma hf. Sími: + 354 540 8000 |
Slovenská republika Pfizer Luxembourg SARL, organizačná zložka Tel: +421-2-3355 5500 |
|
Italia Pfizer S.r.l. Tel: +39 06 33 18 21 |
Suomi/Finland Pfizer Oy Puh/Tel: +358 (0)9 43 00 40 |
|
Kúnpoq PFIZER EAAAI A.E. (CYPRUS BRANCH), Tni: +357 22 817690 |
Sverige Pfizer Innovations AB Tel: + 46 (0)8 550 520 00 |
|
Latvija Pfizer Luxembourg SARL filiale Latvija Tel: +371 670 35 775 |
United Kingdom Pfizer Limited Tel: +44 (0)1304 616161 |
Tato příbalová informace byla naposledy revidována
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europea.eu. Na těchto stránkách naleznete též odkazy na další webové stránky týkající se vzácných onemocnění a jejich léčby.
33