Avamys 27,5 Mikrogramů
SOUHRN ÚDAJŮ O PŘÍPRAVKU
PŘÍLOHA I
AVAMYS 27,5 mikrogramů/vstřik, nosní sprej, suspenze
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden vstřik obsahuje fluticasoni furoas 27,5 mikrogramů. Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Nosní sprej, suspenze.
Bílá suspenze.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Avamys je indikovaný dospělým, dospívajícím a dětem (6 let a starším).
Avamys je indikovaný k léčbě příznaků alergické rýmy.
4.2 Dávkování a způsob podání
Dávkování
Dospělí a dospívající (starší než 12 let)
Doporučenou úvodní dávkou jsou dva vstřiky (27,5 mikrogramů flutikason-furoátu v jednom vstřiku) podané do každé nosní dírky jednou denně (celková denní dávka je 110 mikrogramů).
Jakmile příznaky jsou pod kontrolou, je možné dávku snížit a podat jeden vstřik do každé nosní dírky (celková denní dávka je 55 mikrogramů), ve které je možné dále pokračovat. Dávka má být titrována na nejnižší účinnou dávku, která udržuje příznaky pod kontrolou.
Děti (od 6 do 11 let)
Doporučenou úvodní dávkou je 1 vstřik (27,5 mikrogramů flutikason-furoátu v jednom vstřiku) podaný do každé nosní dírky jednou denně (celková denní dávka je 55 mikrogramů).
Pacientům, kteří nedostatečně reagují na jeden vstřik podaný do každé nosní dírky jednou denně (celková denní dávka je 55 mikrogramů), se mohou podat dva vstřiky do každé nosní dírky jednou denně (celková denní dávka je 110 mikrogramů). Jakmile se dosáhne odpovídající kontroly příznaků, doporučuje se snížit dávku na jeden vstřik podaný do každé nosní dírky jednou denně (celková denní dávka je 55 mikrogramů).
Pro dosažení plného terapeutického přínosu je nezbytné, aby byl přípravek podáván pravidelně podle předpisu. Nástup účinku byl pozorován již po 8 hodinách po úvodní dávce. Pacientovi se musí vysvětlit, že účinek se nedostaví ihned a maximální úlevy může být dosaženo až po několika dnech pravidelné léčby (viz bod 5.1). Délka léčby by měla být omezena na dobu výskytu alergenů.
Děti do 6 let
Bezpečnost a účinnost přípravku Avamys u dětí mladších 6 let nebyla stanovena. Běžně dostupná data jsou popsána v bodech 5.1 a 5.2, ale bez doporučení dávkování.
Starší pacienti
U této skupiny není potřeba dávku upravovat (viz bod 5.2).
Porucha renálních funkcí
U této skupiny není potřeba dávku upravovat (viz bod 5.2).
Porucha jaterních funkcí
U pacientů s poruchou jaterních funkcí není potřeba dávku upravovat (viz bod 5.2).
Způsob podání
Avamys nosní sprej je určen výhradně pro intranazální podání.
Před použitím je třeba intranazální zdravotnický prostředek dobře protřepat. Pro přípravu zdravotnického prostředku k použití jej podržte ve svislé poloze a stiskněte nejméně šestkrát tlačítko uvolňující aerosol (dokud se neobjeví jemný aerosol). Nové připravení (přibližně 6 vstřiků, pokud se neobjeví jemná mlha) je zapotřebí pouze tehdy, jestliže je kryt odstraněn po dobu 5 dnů, nebo nebyl-li nosní sprej používán po dobu 30 dnů, nebo déle.
Po každém použití je třeba zdravotnický prostředek utřít a kryt dát zpět.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku tohoto přípravku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Účinky systémových kortikosteroidů
Může se vyskytnout systémový účinek nosních kortikosteroidů, zvláště při dlouhodobém podávání vysokých dávek. Tyto nežádoucí účinky se vyskytují méně často než u perorálních kortikosteroidů a mohou být různé pro jednotlivé pacienty a různé kortikosteroidové přípravky. Mezi možné systémové účinky patří: Cushingův syndrom, cushingoidní rysy, adrenální suprese, růstová retardace u dětí a dospívajících, katarakta, glaukom a velmi vzácně řada psychologických nežádoucích účinků nebo poruchy chování, např.: psychomotorická hyperaktivita, poruchy spánku, úzkost, deprese a agresivita (především u dětí).
Léčba vyššími než doporučenými dávkami intranazálních kortikosteroidů může vést ke klinicky signifikantní adrenální supresi. Je-li zřejmé, že jsou podávány vyšší dávky, než je doporučeno, pak by mělo být zváženo podání dodatečných systémových kortikosteroidů během stresových situací nebo při chirurgických výkonech. Dávka 110 mikrogramů flutikason-furoátu jednou denně nebyla spojena se supresí osy hypotalamo-hypofýzo-adrenální (HPA) u dospělých, dospívajících nebo dětských pacientů. Avšak dávka intranazálního flutikason-furoátu by se měla snížit na nejnižší dávku, která účinně kontroluje příznaky rinitidy. Jako u ostatních intranazálních kortikosteroidů by se měla zvážit celková systémová zátěž kortikosteroidy, kdykoliv jsou souběžně předepisovány jiné lékové formy kortikosteroidů.
Zvláštní péči je nutné věnovat pacientům, u nichž se přechází ze systémových kortikosteroidů na flutikason-furoát, zvláště tehdy, je-li podezření, že pacient má narušenou adrenální funkci.
Poruchy oka
Intranazální a inhalační kortikosteroidy mohou vést k vývoji glaukomu a/nebo katarakty.
Z tohoto důvodu vyžadují pečlivé sledování pacienti, u kterých došlo ke změně vizu, nebo mají v anamnéze výskyt zvýšeného nitroočního tlaku, glaukomu a/nebo katarakty.
Poruchy růstu
U dětí, kterým byly podávány povolené dávky intranazálních kortikosteroidů, byla pozorována růstová retardace. U dětí léčených flutikason-furoátem v denní dávce 110 mikrogramů po dobu jednoho roku, bylo pozorováno snížení rychlosti růstu (viz body 4.8 a 5.1). Dětem by proto měla být podávána nejnižší možná udržovací účinná dávka, která zajistí adekvátní kontrolu symptomů (viz bod 4.2).
Doporučuje se, aby u dětí dlouhodobě léčených intranazálními kortikosteroidy byl pravidelně sledován růst. Pokud je růst zpomalený, léčba by měla být přehodnocena s cílem snížení dávky intranazálního kortikosteroidu, je-li to možné, a to na nejnižší dávku, která účinně kontroluje příznaky. Navíc by se mělo zvážit odeslání pacienta dětskému odborníkovi (viz bod 5.1).
Pacienti užívající ritonavir
Současné podávání s ritonavirem se nedoporučuje vzhledem k riziku zvýšené systémové expozice flutikason-furoátu (viz bod 4.5).
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Interakce s inhibitory CYP3A4
Flutikason-furoát je rychle odstraňován z organismu během prvního přechodu játry (first pass metabolism) cytochromem P450 3A4.
Na základě údajů s jinými glukokortikoidy (flutikason-propionát) metabolizovanými cestou CYP3A4, současné podávání s ritonavirem se nedoporučuje vzhledem k riziku zvýšené systémové expozice flutikason-furoátem.
Při souběžném užívání flutikason-furoátu se silnými inhibitory CYP3A4 se doporučuje opatrnost, jelikož není možné vyloučit zvýšenou systémovou expozici. Ve studii lékové interakce intranazálního flutikason-furoátu se silným inhibitorem CYP3A4 ketokonazolem byly zjištěni jedinci s měřitelnou koncentrací flutikason-furoátu v ketokonazolové skupině (6 z 20 jedinců) ve srovnání s placebovou skupinou (1 z 20 jedinců). Toto malé zvýšení expozice nebylo statisticky významně rozdílné u hladiny kortizolu během 24 hodin v obou skupinách (viz bod 4.4).
Údaje o enzymové indukci a inhibici naznačují, že není teoretický základ pro očekávané metabolické interakce mezi flutikason-furoátem a cytochromem P450 zprostředkovaným metabolismem jiných složek v klinicky odpovídajících intranazálních dávkách. Proto nebyly provedeny žádné klinické studie zkoumající interakce flutikason-furoátu s jinými léčivy.
4.6 Fertilita, těhotenství a kojení
Nejsou dostupné dostatečné údaje o užití flutikason-furoátu u těhotných žen. Ve studiích glukokortikoidů u zvířat bylo pozorováno, že dochází ke vzniku malformací, včetně rozštěpů patra a retardace nitroděložního růstu. Není pravděpodobné, že by tyto údaje byly relevantní pro lidí užívající doporučené intranazální dávky, které vedou k minimální systémové expozici (viz bod 5.2). Flutikason-furoát může být podán těhotným ženám pouze tehdy, jestliže přínos pro matku převáží možné riziko pro plod nebo dítě.
Kojení
Není známo, zda intranazálně podávaný flutikason-furoát se vylučuje do mateřského mléka. Podávání flutikason-furoátu ženám v období kojení může být zvažováno pouze tehdy, jestliže přínos pro matku je vyšší než možné riziko pro dítě.
Fertilita
Nejsou dostupná data týkající se fertility u lidí.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Avamys nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky Shrnutí bezpečnostního profilu
Obvyle nejčastěji hlášenými nežádoucími reakcemi vyskytujícími se v průběhu léčby flutikason-furoátem jsou epistaxe, nosní ulcerace a bolest hlavy. Nejzávažnější nežádoucí účinky jsou vzácně hlášené reakce přecitlivělosti, včetně anafylaxe (méně než 1 případ na 1 000 pacientů)
Souhrn nežádoucích účinků v tabulce
Více než 2 700 pacientů bylo léčeno flutikason-furoátem ve studiích bezpečnosti a účinnosti sezónní a celoroční alergické rinitidy. Pediatrická expozice flutikason-furoátem ve studiích bezpečnosti a účinnosti sezónní a celoroční alergické rinitidy zahrnovala 243 pacientů ve věku 12 až < 18 let,
790 pacientů ve věku 6 až < 12 let a 241 pacientů ve věku 2 až < 6 let.
Údaje z velkých klinických studií byly použity k určení četnosti výskytu nežádoucích reakcí. Následující hodnoty byly použity v klasifikaci frekvence výskytu: velmi časté > 1/10; časté > 1/100 až < 1/10; méně časté > 1/1 000 až < 1/100; vzácné > 1/10 000 až < 1/1 000; velmi vzácné < 1/10 000.
|
Poruchy imunitního systému | |
|
vzácné |
hypersenzitivní reakce, včetně anafylaxe, angioedému, kožní vyrážky (rash) a kopřivky |
|
Poruchy nervového systému | |
|
časté | |
|
Poruchy oka | |
|
není známo |
přechodné oční změny (viz klinické zkušenosti) |
|
Respirační, hrudní a mediastinálníporuchy | |
|
velmi časté |
*epistaxe |
|
časté |
nosní ulcerace |
|
neznámé |
bolest nosu, nosní dyskomfort (včetně pocitu pálení v nose, podráždění nosu a bolest nosu), pocit sucha v nose. |
|
Velmi vzácné |
perforace nosní přepážky |
|
Poruchy svalové a kosterní soustavy a po jivové tkáně | |
|
není známo |
**retardace růstu (viz klinické zkušenosti) |
Popis jednotlivých nežádoucích reakcí Epistaxe
*Epistaxe byly obecně mírné nebo střední intenzity. U dospělých a dospívajících byl výskyt epistaxe vyšší při delším užívání (více než 6 týdnů), než při krátkodobém užívání (až do 6 týdnů).
Systémové účinky
Systémové účinky intranasálních kortikosteroidů se mohou vyskytnout zvláště tehdy, jestliže byly podávány dlouhodobě vysoké dávky (viz bod 4.4). U dětí léčených nosními kortikosteroidy byla hlášena retardace růstu.
Pediatrická populace
U dětí mladších 6 let nebyla bezpečnost stanovena. Četnost, typ a závažnost nežádoucích reakcí pozorovaných u pediatrické populace je stejná s těmi u dospělé populace.
Epistaxe
*V pediatrických klinických studiích trvajících až 12 týdnů byl výskyt epistaxí stejný mezi pacienty užívajícími flutikason-furoát a pacienty užívajícími placebo.
Porucha růstu
**V klinické studii trvající jeden rok a posuzující růst dětí v prepubertálním období užívajících 110 mikrogramů flutikason-furoátu jednou denně byl pozorován průměrný rozdíl při léčbě rovnající se -0,27 cm rychlosti růstu za rok ve srovnání se skupinou užívající placebo (viz Klinická účinnost a bezpečnost).
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Ve studiích biologické dostupnosti byly intranazální dávky až 2 640 mikrogramů/den podány více než tři dny bez známek systémových nežádoucích reakcí (viz bod 5.2).
Není pravděpodobné, že by akutní předávkování vyžadovalo jinou léčbu než sledování.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: nosní přípravek, kortikosteroid, ATC kód: R01AD12 Mechanismus účinku
Flutikason-furoát je syntetický trifluoridovaný kortikosteroid, který má velmi vysokou afinitu ke glukokortikoidnímu receptoru a má silný protizánětlivý účinek.
Klinická účinnost a bezpečnost
Sezónní alergická rýma u dospělých a dospívajících
Ve srovnání s placebem zlepšil intranazálně podaný flutikason-furoát v dávce 110 mikrogramů jednou denně významně nosní příznaky (zahrnující vodnatý výtok z nosu, nosní kongesci, kýchání a svědění nosu) a oční příznaky (zahrnující svědění/pálení, slzení/zvlhnutí a zarudnutí očí) ve všech 4 studiích. Účinnost trvala více než celých 24 hodin dávkovacího období při podávání jednou denně.
Začátek léčebného účinku byl pozorován již po 8 hodinách po úvodním podání s dalším zlepšením pozorovaným později po několika dnech.
Flutikason-furoát nosní sprej významně zlepšuje pacientovu vnímavost na celkovou odpověď na léčbu a pacientovu, s chorobou související, kvalitu života (Rhinoconjunctivitis Quality of Life Questionnaire - RQLQ) ve všech 4 studiích.
Celoroční alergická rýma u dospělých a dospívajících
Flutikason-furoát nosní sprej v dávce 110 mikrogramů jednou denně významně zlepšuje nosní příznaky právě tak, jako pacientovu vnímavost na celkovou odpověď na léčbu ve srovnání s placebem ve třech studiích.
Flutikason-furoát nosní sprej v dávce 110 mikrogramů jednou denně významně zlepšuje oční příznaky právě tak, jako zlepšující se pacientovu s chorobou související kvalitu života (RQLQ) ve srovnání s placebem v jedné studii.
Účinnost trvala více než celých 24 hodin dávkovacího období při podávání jednou denně.
Ve dvouleté klinické studii hodnotící roční bezpečnost při užívání flutikason-furoátu (intranazální sprej v dávce 110 mikrogramů jednou denně) u dospělých a dospívajících s celoroční alergickou rýmou užívajících buď flutikason-furoát (n = 367) nebo placebo (n = 181). Primární výsledky [doba do zhoršení opacity zadní subkapsulární vrstvy čočky (> 0,3 od výchozí hodnoty podle LOCS III (Lens Opacities Classification Systém, version III)) a čas do zvýšení nitroočního tlaku (IOP; > 7 mmHg od výchozí hodnoty)] nebyly statisticky významně rozdílné mezi oběma skupinami. Zhoršení opacity zadní subkapsulární vrstvy čočky (> 0,3 od výchozí hodnoty) bylo častější u jedinců léčených 110 mikrogramy flutikason-furoátem [14 (4 %)] ve srovnání s placebem [4 (2 %)] a byly přechodné u deseti jedinců ve skupině užívající flutikason-furoát a u dvou jedinců ve skupině s placebem. Zvýšení u IOP (> 7 mmHg od výchozí hodnoty) bylo častější u jedinců léčených 110 mikrogramy flutikason-furoátem: 7 (2 %) 110 mikrogramy flutikason-furoát jednou denně a 1 (< 1 %) u placeba. Tyto jevy byly přechodné u šesti jedinců ve skupině užívající flutikason-furoát a u jednoho jedince ve skupině s placebem. Po 52 a 104 týdnech mělo 95 % jedinců v obou léčebných skupinách zhoršení opacity zadní subkapsulární vrstvy čočky ± 0,1 výchozí hodnoty na každém oku a ve 104. týdnu mělo < 1 % jedinců v obou léčených skupinách zhoršení opacity zadní subkapsulámí vrstvy čočky > 0,3 od výchozí hodnoty. Po 52 a 104 týdnech měla většina jedinců (> 95 %) hodnoty IOP v rozmezí ± 5 mmHg od základní hodnoty. Zhoršení opacity zadní subkapsulární vrstvy čočky nebo IOP nebylo doprovázeno žádnými nežádoucími účinky jako je katarakta nebo glaukom.
Pediatrická populace
Sezónní a celoroční alergická rýma u dětí
Studium dávkování léků v pediatrii je založeno na hodnocení dat účinnosti napříč populací dětí s alergickou rýmou.
U sezónní alergické rýmy byl flutikason-furoát nosní sprej v dávce 110 mikrogramů jednou denně účinný, ale nebyly pozorovány významné rozdíly mezi flutikason-furoátem nosním sprejem v dávce 55 mikrogramů jednou denně a placebem v žádném cílovém parametru.
U celoroční alergické rýmy ukázal flutikason-furoát nosní sprej v dávce 55 mikrogramů jednou denně vyváženější profil účinnosti než flutikason-furoát nosní sprej v dávce 110 mikrogramů jednou denně po 4 týdny léčby. Post-hoc analýza po 6 a 12 týdnech ve stejné studii právě tak jako 6týdenní bezpečnostní studie zkoumající osu HPA podpořily účinnost flutikason-furoátu nosního spreje v dávce 110 mikrogramů jednou denně.
6týdenní studie, která hodnotila účinnost flutikason-furoátu nosního spreje v dávce 110 mikrogramů jednou denně na adrenální funkci u dětí ve věku od 2 do 11 let prokázala, že nedošlo k významnému vlivu na 24 hodinové profily sérového kortizolu ve srovnání s placebem.
Randomizovaná, dvojitě zaslepená, paralelní skupiny obsahující, multicentrická jeden rok placebem kontrolovaná růstová klinická studie hodnotila účinek denně podávaného flutikason-furoátu nosního spreje v dávce 110 mikrogramů na rychlost růstu 474 prepubescentních dětí (dívky ve věku 5 až 7,5 let a chlapci ve věku 5 až 8,5 let) měřením jejich výšky. Průměrná rychlost růstu byla u léčby po dobu 52 týdnů nižší u pacientů užívajících flutikason-furoát (5,19 cm/rok) ve srovnání s placebem (5,46 cm/rok). Průměrný rozdíl byl při léčbě -0,27 cm za rok [95% CI -0,48 až -0,06].
Sezónní a celoroční alergická rýma u dětí (mladších než 6 let)
Studie bezpečnosti a účinnosti byly provedeny u 271 pacienta ve věku od 2 do 5 let se sezonní a celoroční alergickou rýmou, z nichž bylo 176 vystaveno flutikason-furoátu.
Bezpečnost a účinnost v této skupině nebyly dobře stanoveny.
5.2 Farmakokinetické vlastnosti
Absorpce
Flutikason-furoát podléhá neúplnému vstřebávání a významné metabolizaci při prvním průchodu játry a střevem vedoucí k zanedbatelné systémové expozici. Intranazální dávkování 110 mikrogramů jednou denně nevede typicky k měřitelné plazmatické koncentraci (< 10 pg/ml). Absolutní biologická dostupnost intranazálního flutikason-furoátu je 0,50 %, takže méně než 1 mikrogram flutikason-furoátu by mohl být systémově dostupný po podání 110 mikrogramů (viz bod 4.9).
Distribuce v organismu
Vazba flutikason-furoátu na plazmatické proteiny je vyšší než 99 %. Flutikason-furoát má velký distribuční objem v ustáleném stavu, v průměru 608 l.
Biotransformace
Flutikason-furoát je ze systémové cirkulace vyloučen velmi rychle (celková plazmatická clearance je 58,7 l/h), hlavně játry biotransformací na neúčinný metabolit 17p-karboxylový metabolit (GW694301X) prostřednictvím izoenzymu cytochromu P450 CYP3A4. Základní cestou metabolické přeměny byla hydrolýza S-fluoromethyl karbothioatu s tvorbou metabolitu kyseliny 17p-karboxylové. Studie in vitro neodhalily výskyt štěpení furoátové skupiny s tvorbou flutikasonu.
Eliminace z organismu
Po perorálním a nitrožilním podání probíhá vylučování flutikason-furoátu primárně stolicí a jeho metabolitů žlučí. Po nitrožilním podání je průměrný poločas fáze eliminace 15,1 hodin. Vylučování ledvinami je přibližně 1 % po perorálním podání a 2 % po nitrožilním podání.
Pediatrická populace
Po intranazálním podání dávky 110 mikrogramů jednou denně není u většiny pacientů flutikason-furoát v plazmě detekovatelný (< 10 pg/ml). Detekovatelné množství bylo zjištěno u 15,1 % pediatrických pacientů užívajících intranazální dávku 110 mikrogramů jednou denně a pouze 6,8 % pediatrických pacientů užívajících 55 mikrogramů jednou denně. Nebyly prokázány vyšší detekovatelné hladiny flutikason-furoátu u mladších dětí (mladších než 6 let). Střední koncentrace flutikason-furoátu u těchto jedinců s detekovatelnými hladinami u 55 mikrogramů byly 18,4 pg/ml pro 2 - 5 let a 18,9 pg/ml pro 6 - 11 let. U 110 mikrogramů byly střední koncentrace u těchto jedinců s detekovatelnými hladinami 14,3 pg/ml pro 2 - 5 let a 14,4 pg/ml pro 6 - 11 let. Tyto hodnoty jsou podobné hodnotám pozorovaným u dospělých (12+), kde byly střední koncentrace u těchto jedinců s detekovatelnými hladinami 15,4 pg/ml u 55 mikrogramů a 21,8 pg/ml u 110 mikrogramů.
Starší pacienti
Pouze malý počet starších pacientů (> 65 let, n = 23/872; 2,6 %) poskytl farmakokinetické údaje.
U starších pacientů ve srovnání s mladšími pacienty se neprokázal vyšší výskyt pacientů s detekovatelnou koncentrací flutikason-furoátu.
Renální selhání
Flutikason-furoát není po intranazálním podání detekovatelný v moči u zdravých dobrovolníků. Méně než 1 % s dávkou souvisejícího množství látky je vylučováno močí, a proto by nebyla farmakokinetika flutikason-furoátu ovlivněna při renálním selhání.
Jaterní selhání
U pacientů s jaterním selháním nejsou dostupná data související s podáním intranazálního flutikason-furoátu. Jsou k dispozici data po inhalačním podání flutikason-furoátu (jako flutikason-furoát nebo flutikason-furoátu/vilanterolu) jedincům s jaterním selháním, která také platí pro intranasální podání. Studie s jednou dávkou 400 mikrogramů perorálně inhalovaného flutikason-furoátu u pacientů se středně těžkou formou jaterního onemocnění (Child-Pugh B) vedlo ke zvýšení Cmax (42 %) a AUC (0-w) (172 %) a mírným (v průměru 23 %) snížením hladin kortizolu u pacientů srovnávaných se zdravými jedinci. Po opakovaném perorálním podávání inhalačního flutikason-furoátu/vilanterolu po dobu 7 dní, došlo k nárůstu systémové expozice fluticason-fúroátu [v průměru dvojnásobně, měřeno pomocí AUC(0_24)] u jedinců se středním nebo těžkým poškozením jater (Child Pugh B nebo C), ve srovnání se zdravými jedinci. Zvýšení systémové expozice fluticason-íuroátu u jedinců se středně těžkou poruchou funkce jater (flutikason-furoátu/vilanterol 200/25 mikrogramů) bylo spojeno s průměrným snížením kortizolu o 34 % v séru ve srovnání se zdravými jedinci. Nebyl pozorován žádný vliv na sérovou hladinu kortizolu u jedinců s těžkým poškozením jater (flutikason-luroát/vilanterol 100/12,5 mikrogramů). Na základě těchto nálezů by předpokládaná průměrná expozice 110 mikrogramy intranazálního flutikason-furoátu u této populace pacientů nevedla k supresi kortizolu.
5.3 Předklinické údaje vztahující se k bezpečnosti
Nálezy ze všeobecných toxikologických studií byly podobné jako po podání jiných kortikosteroidů a jsou spojovány s přehnanou farmakologickou aktivitou. Tyto nálezy nejsou pravděpodobně relevantní u lidí užívajících doporučené intranazální dávky, které vedou k minimální systémové expozici. V běžných testech genotoxicity nebyly pozorovány žádné genotoxické účinky flutikason-furoátu. Dále ve dvouleté inhalační studii u potkanů a myší nebyl pozorován zvýšený výskyt nádorů souvisejících s léčbou.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
glukosa
disperzní celulosa
polysorbát 80 benzalkonium-chlorid dihydrát dinatrium-edetátu čištěná voda
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
3 roky
Doba použitelnosti po otevření: 2 měsíce
6.4 Zvláštní opatření pro uchovávání
Chraňte před chladem a mrazem.
Uchovávejte ve svislé poloze.
Kryt vždy ponechte uzavřený.
6.5 Druh obalu a obsah balení
14,2 ml v lahvičce z jantarově zbarveného skla typu I nebo typu III, vybavené dávkovací rozprašovací pumpou.
Přípravek je dostupný na trhu ve třech velikostech balení: 1 lahvička s 30, 60 nebo 120 vstřiky.
Na trhu nemusí být k dispozici všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Žádné zvláštní požadavky pro likvidaci.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Glaxo Group Ltd
980 Great West Road, Brentford, Middlesex, TW8 9GS Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/07/434/001
EU/1/07/434/002
EU/1/07/434/003
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 11. ledna 2008
Datum posledního prodloužení registrace: 17. prosince 2012
10. DATUM REVIZE TEXTU 17.12.2015
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/
PŘÍLOHA II
A. VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného/výrobců odpovědných za propouštění šarží
Glaxo Operations UK, Ltd (trading as Glaxo Wellcome Operations)
Harmire Road Barnard Castle Country Durham DL 12 8DT
Glaxo Wellcome S.A.
Avenida de Extremadura 3 09400 Aranda de Duero Burgos Španělsko
V příbalové informaci k léčivému přípravku musí být uveden název a adresa výrobce odpovědného za propouštění dané šarže.
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis.
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
Systém farmakovigilance
Držitel rozhodnutí o registraci musí zajistit, aby byl zaveden funkční systém farmakovigilance uvedený v modulu 1.8.1 schválené registrace předtím, než bude léčivý přípravek uveden na trh, a dále po celou dobu, kdy bude léčivý přípravek na trhu.
Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční činnosti v oblasti farmakovigilance podrobně uvedené v plánu farmakovigilance tak, jak byly schváleny v RMP uvedeném v modulu 1.8.2 schválené registrace, a dle případných aktualizací RMP schválených Výborem pro humánní léčivé přípravky (CHMP).
V souladu s pokynem Výboru pro humánní léčivé přípravky (CHMP) k systémům řízení rizik pro humánní přípravky má být aktualizovaný RMP předložen současně s příští periodicky aktualizovanou zprávou o bezpečnosti (PSUR).
Dále má být aktualizovány RMP předložen:
• jestliže byly obdrženy nové informace, které mohou mít dopad na současné specifikace bezpečnosti, farmakovigilanční plán nebo na činnosti k minimalizaci rizik,
• do 60 dní po dosažení důležitého milníku (týkajícího se farmakovigilance nebo minimalizace rizik),
• na žádost Evropské agentury pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ TOHOTO LÉČIVÉHO PŘÍPRAVKU
Neuplatňuje se.
PŘÍLOHA III
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU KRABIČKA
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Avamys 27,5 mikrogramů/vstřik, nosní sprej, suspenze Fluticasoni furoas
2 OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jeden vstřik obsahuje fluticasoni furoas 27,5 mikrogramů.
3. SEZNAM POMOCNÝCH LÁTEK
Rovněž obsahuje: glukosu, disperzní celulosu, polysorbát 80, benzalkonium-chlorid, dihydrát dinatrium-edetátu, čištěnou vodu.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Nosní sprej, suspenze 1 lahvička - 30 vstřiků 1 lahvička - 60 vstřiků 1 lahvička - 120 vstřiků
5 ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím dobře protřepejte.
Před použitím si přečtěte příbalovou informaci. Nosní podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
Doba použitelnosti po otevření: 2 měsíce
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Chraňte před chladem a mrazem.
Uchovávejte ve svislé poloze. Kryt vždy ponechte nasazený.
10 ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Glaxo Group Ltd.
980 Great West Road, Brentford, Middlesex, TW8 9GS Velká Británie
12 REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/07/434/001
EU/1/07/434/002
EU/1/07/434/003
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16 INFORMACE V BRAILLOVĚ PÍSMU
avamys
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITŘNÍM OBALU INTRANAZÁLNÍ SPREJ/ŠTÍTEK NA ZDRAVOTNICKÝ PROSTŘEDEK
1 NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Avamys 27,5 mikrogramů/vstřik, nosní sprej, suspenze Fluticasoni furoas
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci.
3 POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
30 vstřiků 60 vstřiků 120 vstřiků
6. JINÉ
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro uživatele
Avamys 27,5 mikrogramů/vstřik, nosní sprej, suspenze
Fluticasoni furoas
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka nebo zdravotní sestry.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí
ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je Avamys a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete Avamys užívat
3. Jak se Avamys užívá
4. Možné nežádoucí účinky
5. Jak Avamys uchovávat
6. Obsah balení a další informace
Pokyny k používání nosního spreje krok za krokem.
1. Co je Avamys a k čemu se používá
Avamys (flutikason-furoát) patří do skupiny léčiv, která se nazývají glukokortikoidy. Avamys snižuje zánět způsobený alergií (rýmou) a proto snižuje příznaky alergie.
Avamys nosní sprej je užíván k léčbě příznaků alergické rýmy včetně neprůchodnosti nebo sekrece z nosu, kýchání, slzení a svědění očí u dospělých a dětí ve věku 6 let a starších.
Příznaky mohou být očekávány v určitých obdobích roku a jsou způsobeny alergií na pyl z trávy nebo stromů (senná rýma), nebo je můžete očekávat během celého roku a jsou způsobeny alergií na zvířata, domácí prach nebo plísně, ty jsou nejčastější.
2. Čemu musíte věnovat pozornost, než začnete Avamys užívat
Neužívejte Avamys:
• Jestliže jste alergický(á) na flutikason-furoát nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Upozornění a opatření
Děti a dospívající
Nepodávejte přípravek dětem mladším než 6 let.
Užívání přípravku Avamys:
• může při dlouhodobém užívání u dětí způsobit zpomalení růstu. Lékař bude pravidelně sledovat růst Vašeho dítěte, a ujistí se, zda Vaše dítě užívá nejnižší možnou účinnou dávku.
• může způsobit poruchu zraku, jako glaukom (zvýšení nitroočního tlaku), nebo kataraktu (zákal oční čočky). Informujte svého lékaře, pokud jste již tato onemocnění v minulosti měl(a), nebo pozorujete nějaké změny vizu při užívání přípravku Avamys.
Další léčivé přípravky a Avamys
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a).
Je zvláště důležité, abyste informoval(a) svého lékaře užíváte-li, nebo v nedávné době jste užíval(a) některé z následujících přípravků:
• steroidy v tabletách nebo v injekcích;
• krémy obsahující steroidy;
• přípravky k léčbě astmatu;
• ritonavir, který se užívá k léčbě HIV;
• ketokonazol, který se užívá k léčbě plísňových onemocnění.
Váš lékař zhodnotí, zda můžete užívat Avamys s těmito přípravky.
Avamys nesmíte používat současně s jinými nosními spreji obsahujícími kortikosteroidy. Těhotenství a kojení
Pokud jste těhotná, nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem dříve, než začnete tento přípravek užívat.
Jste-li těhotná, nebo plánujete těhotenství, neužívejte přípravek Avamys dříve, než se poradíte se svým lékařem nebo lékárníkem.
Neužívejte přípravek Avamys během kojení, dokud se neporadíte se svým lékařem nebo lékárníkem.
Řízení dopravních prostředků a obsluha strojů
Není pravděpodobné, že by přípravek Avamys ovlivňoval schopnost řídit a obsluhovat stroje. Avamys obsahuje benzalkonium-chlorid
U některých pacientů může benzalkonium-chlorid způsobit podráždění uvnitř v nose. V případě, že během užívání spreje cítíte nepříjemný pocit, informujte o tom svého lékaře nebo lékárníka.
3. Jak se Avamys užívá
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře nebo lékárníka. Doporučenou dávku nepřekračujte. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Kdy použít Avamys
• používá se jednou denně;
• používá se ve stejnou dobu každý den.
Tímto způsobem tak předejdete příznakům, které se mohou objevit během celého dne.
Jak dlouho trvá, než Avamys začne působit
Někteří lidé budou cítit plný účinek až po několika dnech od začátku užívání přípravku Avamys. Je však obvykle účinný během 8 až 24 hodin užívání.
Kolik přípravku se užívá
Dospělí a dospívající starší než 12 let
• Obvyklou úvodní dávkou jsou 2 vstřiky do každé nosní dírky jednou denně.
• Jakmile jsou Vaše příznaky pod kontrolou, můžete snížit dávkování na 1 vstřik do každé nosní dírky.
Děti ve věku 6 až 11 let
• Obvyklou úvodní dávkou je 1 vstřik do každé nosní dírky jednou denně.
• Pokud jsou příznaky velmi těžké, lékař může zvýšit dávkování na 2 vstřiky do každé nosní dírky, dokud se příznaky nezlepší. Pak se může dávkování snížit na 1 vstřik do každé nosní dírky j ednou denně.
Jak se nosní sprej používá
Avamys je prakticky bez chutě nebo zápachu. Podává se ve spreji do nosní dírky jako jemná mlha. Buďte opatrný(á), abyste si nenastříkal(a) sprej do očí. Pokud se tak stane, vypláchněte si oči vodou.
Pokyny k používání nosního spreje krok za krokem najdete za bodem 6 této příbalové informace. Pečlivě postupujte podle pokynů, aby byl přípravek Avamys pro Vás přínosem.
^Pokyny k používání nosního spreje krok za krokem, za bodem 6.
Jestliže jste užil(a) více přípravku Avamys, než jste měl(a)
Užijete-li větší množství přípravku, informujte o tom svého lékaře nebo lékárníka.
Jestliže jste zapomněl(a) užít přípravek Avamys
Zapomenete-li si vzít dávku přípravku, užijte ji ihned, jakmile si vzpomenete.
Blíží-li se však doba pro Vaši další pravidelnou dávku, vyčkejte na ni. Nezdvojujte následující dávku, abyste nahradil(a) vynechanou dávku.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, nebo pozorujete-li nějaké potíže při užívání nosního spreje, zeptejte se svého lékaře nebo lékárníka nebo zdravotní sestry.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Alergické reakce: ihned vyhledejte lékařskou pomoc
Alergické reakce na Avamys jsou vzácné a vznikají u méně než 1 pacienta z 1 000. Pokud tento stav není léčen, mohou mít alergické reakce u malého počtu pacientů závažný, až život ohrožující průběh. Příznaky zahrnují:
- nástup závažného sípání, kašle nebo potíží s dýcháním;
- náhlý pocit slabosti nebo závratí (točení hlavy) (které mohou vést ke kolapsu nebo ztrátě vědomí);
- otok tváře;
- kožní vyrážky nebo zarudnutí.
V mnoha případech se tyto příznaky projeví jako méně závažné nežádoucí účinky. Musíte si však být vědom(a) toho, že mohou být potenciálně závažné - proto pokud jste si vědom(a), že máte některý z těchto příznaků: ihned kontaktujte lékaře.
Velmi časté nežádoucí účinky (mohou se vyskytnout u více než 1 z 10 pacientů)
• Krvácení z nosu (většinou mírné) zvláště užíváte-li Avamys déle než 6 týdnů nepřerušeně.
Časté nežádoucí účinky (mohou se vyskytnout až u 1 z 10 pacientů)
• Nosní vředy - které mohou způsobit podráždění nebo nepříjemný pocit v nose. Při smrkání můžete rovněž pozorovat stopu krve.
• Bolest hlavy.
Méně časté nežádoucí účinky (mohou se vyskytnout až u 1 ze 100 pacientů)
• Bolest, pálení, podráždění, bolest nebo suchost v nose.
Velmi vzácné nežádoucí účinky (mohou se vyskytnout až u 1 z 10 000 pacientů)
• Malé otvory (perforace) v nosní přepážce oddělující obě nosní dírky od sebe.
Není známo (četnost nelze z dostupných údajů určit)
• Zpomalení růstu u dětí;
• Přechodná změna vízu při dlouhodobém používání.
Nosní kortikosteroidy mohou ve Vašem těle ovlivnit normální produkci steroidních hormonů, zvláště pokud užíváte dlouhodobě vysoké dávky. U dětí tento nežádoucí účinek může vést ke zpomalení jejich růstu ve srovnání s ostatními dětmi.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak Avamys uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Uchovávejte Avamys nejlépe ve svislé poloze. Vždy ponechte kryt na přípravku.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na štítku a krabičce. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce. Po prvním otevření se přípravek Avamys nosní sprej má používat 2 měsíce.
Chraňte před chladem a mrazem.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co Avamys obsahuje
Léčivou látkou je fluticasoni furoas. Jeden vstřik obsahuje 27,5 mikrogramů fluticasoni furoas. Pomocnými látkami jsou glukosa, disperzní celulosa, polysorbát 80, benzalkonium-chlorid, dihydrát dinatrium-edetátu, čištěná voda (viz bod 2).
Jak Avamys vypadá a co obsahuje toto balení
Přípravek nosní sprej je bílou suspenzí obsaženou v lahvičce z jantarově zbarveného skla, vybavené dávkovací rozprašovací pumpou. Téměř bílý plastikový obal se světle modrým krytem a postranní ovládací pákou. Obal má okénko pro sledování obsahu lahvičky. Avamys je dostupný v baleních po 30, 60 a 120 vstřicích. Na trhu nemusí být k dispozici všechny velikosti balení.
Držitel rozhodnutí o registraci a výrobci
Držitel rozhodnutí o registraci:
Glaxo Group Ltd
980 Great West Road, Brentford, Middlesex, TW8 9GS Velká Británie
Výrobci:
Glaxo Operations UK, Ltd (trading as Glaxo Wellcome Operations)
Harmire Road
Barnard Castle
Country Durham
DL 12 8DT
Velká Británie
Glaxo Wellcome S.A.
Avenida de Extremadura 3 09400 Aranda de Duero Burgos Španělsko
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Btarapna
TaaKCoCMHTKaaHH EOOfl Tea.: + 359 2 953 10 34
Česká republika
GlaxoSmithKline s.r.o.
Tel: + 420 222 001 111 cz.info@gsk.com
Danmark
GlaxoSmithKline Pharma A/S Tlf: + 45 36 35 91 00 dk-info@gsk.com
Deutschland
GlaxoSmithKline GmbH & Co. KG Tel.: + 49 (0)89 36044 8701 produkt.info@gsk.com
Eesti
GlaxoSmithKline Eesti OU Tel: + 372 6676 900 estonia@gsk.com
EkXáda
GlaxoSmithKline A.E.B.E.
Tr|U + 30 210 68 82 100
Espaňa
GlaxoSmithKline, S.A.
Tel: + 34 902 202 700 es-ci@gsk.com
Luxembourg/Luxemburg
GlaxoSmithKline Pharmaceuticals s.a./n.v.
Belgique/Belgien
Tél/Tel: + 32 (0) 10 85 52 0
Magyarország
GlaxoSmithKline Kft.
Tel.: + 36 1 225 5300
Malta
GlaxoSmithKline (Malta) Limited Tel: + 356 21 238131
Nederland
GlaxoSmithKline BV Tel: + 31 (0)30 6938100 nlinfo@gsk.com
Norge
GlaxoSmithKline AS Tlf: + 47 22 70 20 00 firmapost@gsk.no
Osterreich
GlaxoSmithKline Pharma GmbH Tel: + 43 (0)1 97075 0 at.info@gsk.com
Polska
GSK Services Sp. z o.o.
Tel.: + 48 (0)22 576 9000
Portugal
GlaxoSmithKline - Produtos Farmaceuticos, Lda.
Tel: + 351 21 412 95 00
Tél.: + 33 (0)1 39 17 84 44 diam@gsk.com
Tel: + 4021 3028 208
Slovenija
GlaxoSmithKline d.o.o. Tel: + 386 (0)1 280 25 00 medical.x.si@gsk.com
Slovenská republika
GlaxoSmithKline Slovakia s. r. o. Tel: + 421 (0)2 48 26 11 11 recepcia.sk@gsk.com
Suomi/Finland
GlaxoSmithKline Oy Puh/Tel: + 358 (0)10 30 30 30 Finland.tuoteinfo@gsk.com
Kúnpoq
GlaxoSmithKline (Cyprus) Ltd T@: + 357 22 39 70 00 gskcyprus@gsk.com
Sverige
GlaxoSmithKline AB Tel: + 46 (0)8 638 93 00 info.produkt@gsk.com
United Kingdom
GlaxoSmithKline UK Ltd Tel: + 44 (0)800 221441 customercontactuk@gsk.com
Latvija
GlaxoSmithKline Latvia SIA Tel: + 371 67312687 lv-epasts@gsk.com
Lietuva
GlaxoSmithKline Lietuva UAB Tel: + 370 5 264 90 00 info.lt@gsk.com
Tato příbalová informace byla naposledy revidována 12/2015
Podrobné informace o tomto léčivém přípravku j sou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky: http://www.ema.europa.eu
POKYNY K POUŽÍVÁNÍ NOSNÍHO SPREJE KROK ZA KROKEM
Jak nosní sprej vypadá
Nosní sprej se dodává v jantarově zbarvené skleněné lahvičce uzavřené v plastovém obalu - viz obrázek a. Lahvička obsahuje buď 30, 60 nebo 120 vstřiků v závislosti na velikosti balení, které Vám bylo předepsáno.
přední část
zadní část
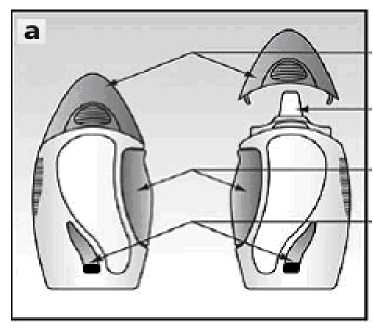
kryt
vystřikovací tryska
postranní tlačítko okénko
Okénko na obalu Vám poskytuje informaci, kolik přípravku Avamys zbývá v lahvičce. V okénku uvidíte hladinu roztoku obsaženého v nové 30 nebo 60 dávkové lahvičce spreje, ale ne v nové 120 dávkové lahvičce spreje, protože hladina roztoku je nad okénkem.
Šest důležitých věcí, které musíte vědět při používání nosního spreje
• Avamys se dodává v jantarově zbarvené skleněné lahvičce. Pokud potřebujete zkontrolovat, kolik jste již přípravku vypotřeboval(a), držte nosní sprej kolmo proti jasnému světlu. Uvidíte v okénku hladinu přípravku.
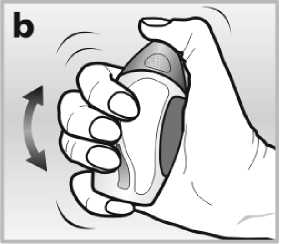
• Při prvním použití nosního spreje jím budete muset 10 vteřin důkladně třepat
s ponechaným krytem. Toto je důležité, jelikož Avamys je hustá suspenze, která se při dobře provedeném protřepání změní v tekutinu - viz obrázek b. Bude ho možné použít jako sprej jen tehdy, stane-li se tekutým.
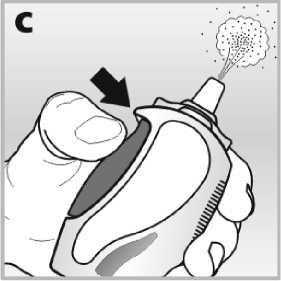
• Zmáčkněte pevně postranní tlačítko v celém rozsahu tak, aby došlo k uvolnění jemné mlhy vystřikovací tryskou - viz obrázek c.
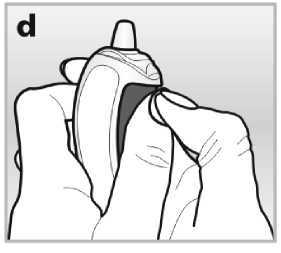
Budete-li mít potíže se stlačením postranního tlačítka palcem, můžete použít obě ruce - viz obrázek d.
• V případě, že sprej nepoužíváte, ponechte na něm kryt. Kryt chrání před zaprášením, udržuje vnitřní tlak a brání zanesení trysky. Pokud je kryt na svém místě, nelze náhodně vystříknout žádnou dávku.
• K čištění vystřikovací trysky nesmíte použít špendlík, nebo jakýkoliv jiný ostrý předmět. Došlo by k poškození nosního spreje.
Příprava nosního spreje k použití
Nosní sprej musíte připravit následovně:
• před prvním použitím;
• jestliže jste ho ponechal(a) bez krytu 5 dní nebo jste nosní sprej nepoužíval(a) 30 dní nebo déle.
Příprava nosního spreje Vám zajistí, že obdržíte celou dávku přípravku.
Sledujte tyto kroky:
1. S nasazeným krytem důkladně protřepejte nosní sprej přibližně po dobu 10 vteřin.
2. Jemným stlačením odstraňte kryt tak, že uchopíte boční části krytu mezi palec a ukazováček -viz obrázek e.
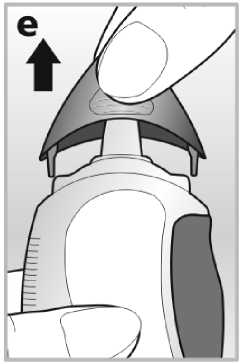
3. Držte nosní sprej svisle, pak jej nakloňte a natočte vystřikovací trysku od sebe.
4. Pevně stlačte postranní tlačítko v celém rozsahu. Zmáčkněte ho alespoň šestkrát, dokud nedojde k uvolnění jemné mlhy do ovzduší - viz obrázek f.
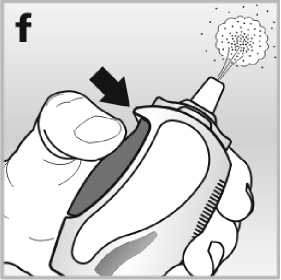
Nosní sprej je nyní připraven k použití.
Užití nosního spreje
1. Nosní sprej důkladně protřepejte.
2. Odstraňte kryt.
3. Vyčistěte si nos a mírně nakloňte hlavu vpřed.
4. Vsuňte vystřikovací trysku do jedné z nosních dírek - viz obrázek g. Otočte konec vystřikovací trysky mírně stranou od nosní přepážky. Toto Vám pomůže podat přípravek do správného místa v nose.
5. Během vdechu stlačte pevně postranní tlačítko tak, aby se do nosu uvolnila jedna dávka přípravku - viz obrázek h.
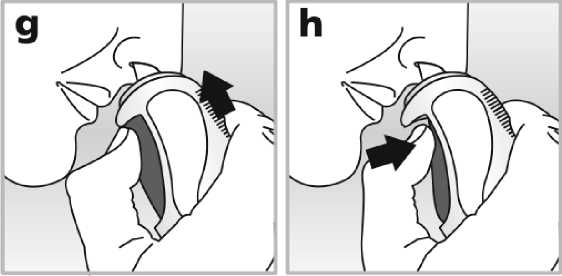
6. Vyjměte vystřikovací trysku z nosu a vydechněte ústy.
7. Pokud máte podat 2 vstřiky do každé nosní dírky, opakujte kroky 4 až 6.
8. Kroky 4 až 7 opakujte v druhé nosní dírce.
9. Nasaďte kryt na nosní sprej.
Čištění nosního spreje Po každém použití:
1. Po každém použití otřete vystřikovací trysku čistou suchou tkaninou - viz obrázek i a j.

2. Při čištění nepoužívejte vodu.
3. K čištění vystřikovací trysky neužívejte špendlík, nebo jiné ostré předměty.
4. Po ukončení, vraťte kryt na původní místo.
Pokud se Vám zdá, že nosní sprej nepracuje:
• Zkontrolujte, zda ještě máte v nádobce přípravek. Podívejte se na hladinu v okénku. Je-li hladina velmi nízko, nemuselo se uvolnit dost nosního spreje.
• Zkontrolujte, zda nedošlo k poškození nosního spreje.
• Domníváte-li se, že došlo k ucpání vystřikovací trysky, k vyčištění nepoužijte špendlík, nebo jiný ostrý předmět.
• Zkuste opravit přístroj opakováním návodu od začátku „Příprava nosního spreje k použití“.
Pokud nadále nosní sprej nepracuje, nebo z něho tryská tekutina, vraťte nosní sprej zpět do lékárny, kde Vám poradí, jak dál.
29