Arzerra 100 Mg
SOUHRN ÚDAJŮ O PŘÍPRAVKU
PŘÍLOHA I
Arzerra 100 mg koncentrát pro infuzní roztok.
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden ml koncentrátu obsahuje ofatumumabum 20 mg.
Jedna lahvička obsahuje ofatumumabum 100 mg v 5 ml.
Ofatumumab je lidská monoklonální protilátka tvořená rekombinantními myšími buněčnými liniemi (NS0).
Pomocné látky se známým účinkem:
Tento léčivý přípravek obsahuje 34,8 mg sodíku v 300 mg dávce, 116 mg sodíku v 1000 mg dávce a 232 mg sodíku v 2000 mg dávce.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Koncentrát pro infuzní roztok (sterilní koncentrát).
Čirý až opalizující, bezbarvý až světle žlutý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Dříve neléčená chronická lymfocytární leukémie (CLL):
Přípravek Arzerra je v kombinaci s chlorambucilem nebo bendamustinem indikován k léčbě pacientů s CLL bez předchozí léčby, kteří nemohou být léčeni terapií založenou na fludarabinu.
Další informace viz bod 5.1.
Refrakterní CLL:
Přípravek Arzerra je indikován k léčbě CLL u pacientů, u kterých selhává léčba fludarabinem a alemtuzumabem.
Další informace viz bod 5.1.
4.2 Dávkování a způsob podání
Přípravek Arzerra má být podáván pod dohledem lékaře, který má zkušenosti s onkologickou léčbou, a v prostředí, které je plně vybavené pro okamžitou resuscitaci.
Monitorování
Pacienti mají být v průběhu podávání ofatumumabu, zejména v průběhu první infuze, pečlivě monitorováni z hlediska výskytu reakcí souvisejících s infuzí, včetně syndromu z uvolnění cytokinů.
Premedikace
Pacient má být vždy premedikován 30 minut až 2 hodiny před aplikací infuze přípravku Arzerra podle následujících dávkovacích schémat:
Dříve neléčená CLL:
• perorální paracetamol 1000 mg (nebo ekvivalent), plus
• perorální nebo intravenózní antihistaminikum (difenhydramin 50 mg nebo cetirizin 10 mg nebo ekvivalent), plus
• intravenózní kortikoid (prednisolon 50 mg nebo ekvivalent).
Pokud se u pacienta po první a druhé infuzi neobjeví závažné nežádoucí účinky, může být před dalšími infuzemi premedikace kortikoidy snížena nebo vynechána podle uvážení lékaře.
Refrakterní CLL:
• perorální paracetamol 1000 mg (nebo ekvivalent), plus
• perorální nebo intravenózní antihistaminikum (difenhydramin 50 mg nebo cetirizin 10 mg nebo ekvivalent), plus
• intravenózní kortikoid (prednisolon 100 mg nebo ekvivalent).
Pokud je druhá týdenní infuze ukončena bez závažných nežádoucích účinků, může být pro infuze 3 až 8 dávka kortikoidů snížena podle uvážení lékaře.
Před devátou infuzí (první infuze v měsíčním intervalu) musí být pacientům podána kompletní premedikace popsaná výše. Pokud je devátá infuze ukončena bez závažných nežádoucích účinků, může být před dalšími infuzemi dávka snížena na ekvivalent 50 mg prednisolonu podle uvážení lékaře.
Dávkování
Dříve neléčená CLL:
Doporučená dávka a schéma podávání je 300 mg v den 1, následuje 1000 mg za 1 týden, tj. v den 8 (cyklus 1), poté následuje 1000 mg v den 1 následujících cyklů. Tyto cykly mají být nejméně 3 a mají se opakovat do dosažení nejlepší odpovědi na léčbu nebo do celkového maximálního počtu 12 cyklů (každých 28 dní).
Nejlepší odpověď na léčbu je klinická odpověď, která se nezlepší během dalších 3 cyklů léčby.
První infuze
Úvodní rychlost první infuze přípravku Arzerra má být 12 ml/h. V průběhu infuze má být rychlost zvyšována každých 30 minut až do maximální rychlosti 400 ml/h (viz bod 6.6).
Následující infuze
Pokud byla první infuze dokončena bez závažných nežádoucích účinků souvisejících s infuzí, je možné následující infuze zahájit rychlostí 25 ml/h a jejich rychlost má být zvyšována každých 30 minut až do maximální rychlosti 400 ml/h (viz bod 6.6).
Refrakterní CLL:
Doporučená dávka je 300 mg pro první infuzi a 2000 mg pro všechny následující infuze. Léčba sestává z 8 infuzí podaných v týdenních intervalech, po kterých následuje 4 - 5 týdenní pauza. Léčba dále pokračuje 4 infuzemi podanými v měsíčních intervalech (tj. každé 4 týdny).
První a druhá infuze
Úvodní rychlost první a druhé infuze přípravku Arzerra je 12 ml/hodinu. V průběhu infuze má být její rychlost zvyšována každých 30 minut až do maximální rychlosti 200 ml/hodinu (viz bod 6.6).
Následující infuze
Pokud byla druhá infuze dokončena bez závažných nežádoucích účinků souvisejících s infuzí, je možné zbývající infuze zahájit rychlostí 25 ml/hodinu a jejich rychlost má být zvyšována každých 30 minut až do maximální rychlosti 400 ml/hodinu (viz bod 6.6).
Úprava dávkování a opakované zahájení léčby při závažných nežádoucích účincích souvisejících s infuzí - u pacientů s dříve neléčenou CLL a refrakterní CLL
Infuzi je třeba přerušit při nežádoucích účincích souvisejících s infuzí jakékoli intenzity. Léčba může být znovu zahájena podle uvážení ošetřujícího lékaře. Následující úpravy rychlosti infuze mohou být použity jako návod:
• V případě mírných nebo středně závažných nežádoucích účinků by měla být infuze přerušena a znovu zahájena poloviční rychlostí, než jaká byla v čase přerušení, až když je stav pacienta stabilizovaný. Pokud rychlost infuze nebyla od úvodní rychlosti 12 ml/hodinu před přerušením z důvodu nežádoucích účinků souvisejících s infuzí zvýšena, infuze by měla být znovu zahájena standardní rychlostí 12 ml/hodinu. Rychlost infuze může být zvyšována podle standardního postupu, uvážení lékaře a tolerance pacienta (nemá být překročeno zvyšování rychlosti infuze každých 30 minut).
• V případě závažných nežádoucích účinků má být infuze přerušena a znovu zahájena rychlostí 12 ml/hodinu, až pokud je stav pacienta již stabilizovaný. Rychlost infuze může být zvyšována podle standardního postupu, uvážení lékaře a tolerance pacienta (nemá být překročeno zvyšování rychlosti infuze každých 30 minut).
Pediatrická populace
Vzhledem k nedostatečným údajům o bezpečnosti a/nebo účinnosti není přípravek Arzerra doporučen pro užívání u dětí mladších 18 let.
Starší pacienti
V souvislosti s věkem nebyly pozorovány žádné rozdíly v bezpečnosti a účinnosti (viz bod 5.1). Na základě dostupných údajů o bezpečnosti a účinnosti u starších pacientů není nutná úprava dávky (viz bod 5.2).
Porucha funkce ledvin
U pacientů s poruchou funkce ledvin nebyly provedeny žádné formální studie s přípravkem Arzerra.
U mírné až středně závažné poruchy funkce ledvin (clearance kreatininu > 30 ml/min) není nutná žádná úprava dávky (viz bod 5.2).
Porucha funkce jater
U pacientů s poruchou funkce jater nebyly provedeny žádné formální studie s přípravkem Arzerra. Není ale pravděpodobné, že by u pacientů s poruchou funkce jater byla nutná úprava dávky (viz bod 5.2).
Způsob podání
Přípravek Arzerra je k intravenózní infuzi a musí být před podáním naředěn. Návod k naředění léčivého přípravku před podáním je uveden v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na ofatumumab nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Reakce související s infuzí
Intravenózně podávaný ofatumumab je spojován s reakcemi na infuzi. Tyto reakce mohou vést k dočasnému přerušení nebo ukončení léčby. Premedikace zmírňuje reakce související s infuzí, ale ty se mohou přesto objevit, zvláště v průběhu první infuze. Reakce související s infuzí mohou zahrnovat, ale nejsou omezeny pouze na, anafylaktoidní reakce, bronchospasmus, srdeční příhody (např. ischemii/infarkt myokardu, bradykardii), zimnici/ztuhlost, kašel, syndrom z uvolnění cytokinů (cytokine release syndrom), průjem, dušnost, únavu, návaly, hypertenzi, hypotenzi, nauzeu, bolest, edém plic, pruritus, pyrexii, vyrážku a kopřivku. Ve vzácných případech mohou tyto reakce vést k úmrtí. I s premedikací mohou být po podání ofatumumabu zaznamenány závažné reakce, včetně syndromu z uvolnění cytokinů. V případě závažné reakce související s infuzí musí být infuze přípravku Arzerra okamžitě přerušena a zahájena symptomatická léčba (viz bod 4.2).
Reakce související s infuzí se mohou objevit častěji první den infuze a mají tendenci se s každou následující infuzí zmírňovat. Pacienti s anamnézou snížené plicní funkce mohou mít vyšší riziko plicních komplikací od závažných reakcí a měli by být v průběhu infuze ofatumumabu pečlivě monitorováni.
Syndrom lýzy tumoru
U pacientů s CLL se může při podávání ofatumumabu objevit syndrom lýzy tumoru (TLS, tumour lysis syndrome). Rizikovými faktory pro TLS jsou velký objem nádoru, vysoká koncentrace cirkulujících buněk (> 25000/mm3), hypovolémie, renální insuficience, zvýšená hladina kyseliny močové před léčbou a zvýšené hladiny laktát-dehydrogenázy. Léčba TLS zahrnuje korekci elektrolytových abnormalit, monitorování renálních funkcí, udržování bilance tekutin a podpůrnou léčbu.
Progresivní multifokální leukoencefalopatie
U pacientů s CLL, kteří podstupují cytotoxickou farmakoterapii (včetně ofatumumabu), byla zaznamenána progresivní multifokální leukoencefalopatie (PML) i úmrtí. Diagnózu PML je třeba zvážit u každého pacienta užívajícího přípravek Arzerra, u kterého se objeví nové neurologické příznaky a symptomy nebo dojde ke změně již existujících neurologických příznaků a symptomů. Pokud je podezření na diagnózu PML, je třeba léčbu přípravkem Arzerra přerušit a předat pacienta k neurologickému vyšetření.
Imunizace
Bezpečnost imunizace a schopnost vytvářet primární nebo anamnestickou odpověď na imunizaci živou atenuovanou nebo inaktivovanou vakcínou v průběhu léčby ofatumumabem nebyla studována. Odpověď na vakcinaci může být při depleci B buněk narušena. Vzhledem k riziku infekce je třeba se vyvarovat podávání živých atenuovaných vakcín v průběhu a po ukončení léčby ofatumumabem, dokud nedojde k normalizaci B buněk. V průběhu léčby ofatumumabem je třeba rizika a přínosy očkování pečlivě zvážit.
Hepatitida B
U pacientů léčených léčivy klasifikovanými jako cytolytické protilátky proti CD20, včetně přípravku Arzerra, se vyskytly případy infekce virem hepatitidy B (HBV) a reaktivace HBV, které v některých případech vyústily ve fulminantní hepatitidu, selhání jater a úmrtí. Tyto případy byly hlášeny u pacientů s pozitivním povrchovým antigenem viru hepatitidy B (HBsAg) a také u pacientů s pozitivními protilátkami proti core antigenu hepatitidy B (anti-HBc), ale negativním HBsAg. Reaktivace HBV se rovněž vyskytla u pacientů, u kterých byla infekce HBV považována za zvládnutou (tj. u pacientů HBsAg negativních, anti-HBc pozitivních a s pozitivními protilátkami proti povrchovému antigenu viru hepatitidy B [anti-HBs]).
Reaktivace viru hepatitidy B je definována jako náhlé zvýšení replikace viru hepatitidy B, projevující se jako rychlý vzestup hladiny HBV DNA, nebo detekce HBsAg u osob, které byly dříve HBsAg negativní a anti-HBc pozitivní. Po reaktivaci HBV často následuje hepatitida, tj. zvýšení hladin transamináz a, v závažných případech, zvýšení hladiny bilirubinu, selhání jater a úmrtí.
U všech pacientů má být před zahájením léčby přípravkem Arzerra proveden screening HBV infekce stanovením HBsAg a anti-HBc. U pacientů s prokázanou předchozí infekcí hepatitidou B (HBsAg negativní, anti-HBc pozitivní) má být monitorování a zahájení antivirové HBV terapie konzultováno s lékařem kvalifikovaným k léčbě hepatitidy B. Léčba přípravkem Arzerra nemá být zahajována u pacientů s prokázanou současnou infekcí virem hepatitidy B (HBsAg pozitivních), dokud není tato infekce odpovídajícím způsobem zvládnuta.
Pacienti s prokázanou předchozí infekcí HBV mají být během léčby a 6 - 12 měsíců po poslední infuzi přípravku Arzerra monitorováni z hlediska klinických a laboratorních příznaků hepatitidy nebo reaktivace HBV. Reaktivace HBV byla hlášena až 12 měsíců po ukončení léčby. Ukončení antivirové terapie HBV má být konzultováno s lékařem kvalifikovaným k léčbě hepatitidy B.
Pokud u pacientů dojde během léčby přípravkem Arzerra k reaktivaci HBV, má být Arzerra a veškerá současná chemoterapie okamžitě přerušena a zahájena odpovídající léčba. K dispozici jsou pouze nedostatečné údaje týkající se bezpečnosti opětovného zahájení podávání přípravku Arzerra u pacientů, u kterých došlo k reaktivaci HBV. Opětovné zahájení podávání přípravku Arzerra pacientům, u kterých byla reaktivace HBV vyřešena, má být konzultováno s lékařem kvalifikovaným k léčbě hepatitidy B.
Kardiovaskulární účinky
Pacienti s anamnézou srdečního onemocnění by měli být pečlivě sledováni. Léčba přípravkem Arzerra má být ukončena u pacientů se závažnou nebo život ohrožující srdeční arytmií.
Vliv opakovaných dávek přípravku Arzerra na interval QTc byl hodnocen v souhrnné analýze tří otevřených studií u pacientů s CLL (N = 85). V této souhrnné analýze byla pozorována prodloužení mediánu/průměru intervalů QT/QTc nad 5 ms. Nebyly zjištěny žádné velké změny průměrného intervalu QTc (tj. > 20 milisekund). U žádného pacienta nedošlo k prodloužení QTc > 500 ms. Prodloužení intervalu QTc závislé na koncentraci nebylo zjištěno. Doporučuje se, aby pacientům byly před zahájením podávání a v průběhu podávání ofatumumabu měřeny hladiny iontů, jako jsou draslík a hořčík. Abnormality v hladinách iontů mají být upraveny. Vliv ofatumumabu na pacienty s prodloužením intervalu QT (získaným nebo vrozeným) není znám.
Střevní obstrukce
U pacientů, kteří podstupují léčbu monoklonálními protilátkami proti CD20, včetně ofatumumabu, byla zaznamenána střevní obstrukce. Pacienti, u kterých se objeví bolesti břicha, zvláště časně v průběhu léčby ofatumumabem, by měli být vyšetřeni a měla by být zahájena vhodná léčba.
Laboratorní vyšetření
Během léčby ofatumumabem byly hlášeny cytopenie, včetně dlouhodobé neutropenie a neutropenie s opožděným nástupem. V průběhu léčby ofatumumabem je třeba v pravidelných intervalech vyšetřit kompletní krevní obraz, včetně počtu neutrofilů a krevních destiček, častěji pak u pacientů, u kterých se rozvíjí cytopenie.
Obsah sodíku
Tento léčivý přípravek obsahuje 34,8 mg sodíku v dávce 300 mg, 116 mg sodíku v dávce 1000 mg a 232 mg sodíku v dávce 2000 mg. To by mělo být vzato v úvahu u pacientů na řízené sodíkové dietě.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Ačkoli u ofatumumabu je k dispozici pouze omezené množství dat týkajících se lékových interakcí, nejsou známé žádné klinicky významné interakce s dalšími léčivými přípravky. Ofatumumab nemá klinicky významný vliv na farmakokinetiku chlorambucilu nebo jeho aktivního metabolitu, kyseliny 4-[bis(2-chlorethyl)amino]fenyloctové.
Účinnost živých atenuovaných nebo inaktivovaných vakcín může být ofatumumabem narušena, proto je vhodné se vyvarovat užití takových látek spolu s ofatumumabem. Pokud je společné podání považováno za nezbytné, mělo by být zváženo riziko a prospěch vakcinace v průběhu léčby ofatumumabem (viz bod 4.4).
4.6 Fertilita, těhotenství a kojení
Údaje o podávání ofatumumabu těhotným ženám nejsou k dispozici. Studie na zvířatech nenaznačují žádné přímé nebo nepřímé škodlivé účinky s ohledem na reprodukční toxicitu (viz bod 5.3). Ofatumumab nemá být podáván těhotným ženám, pokud jeho možný přínos pro matku nepřevýší možné riziko pro plod.
Ženy ve fertilním věku musí v průběhu léčby ofatumumabem a 12 měsíců po jejím ukončení používat účinnou antikoncepci.
Kojení
Není známo, zda je ofatumumab vylučován do mateřského mléka, nicméně lidské IgG do mateřského mléka vylučovány jsou. Bezpečnost užití ofatumumabu u lidí v průběhu kojení nebyla stanovena. Vylučování ofatumumabu do mateřského mléka nebylo u zvířat hodnoceno. Publikované údaje naznačují, že spotřeba mateřského mléka u novorozence ani kojence nevede k významné absorpci těchto mateřských protilátek do krevního oběhu. Riziko pro novorozence/kojence nelze vyloučit. Během léčby ofatumumabem a 12 měsíců po jejím ukončení má být kojení přerušeno.
Fertilita
O účincích na fertilitu u člověka nejsou k dispozici žádné údaje. Ve studiích na zvířatech nebyly účinky na fertilitu samců ani samic hodnoceny.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Žádné studie účinku přípravku Arzerra na schopnost řídit a obsluhovat stroje nebyly provedeny.
Žádné zásadní účinky na takové aktivity nejsou z farmakologie ofatumumabu očekávány. Pokud je zvažována pacientova schopnost provádět úkony, které vyžadují rozhodování, motorické nebo kognitivní schopnosti, je třeba vzít v úvahu jeho klinický stav a profil nežádoucích účinků (viz bod 4.8).
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Celkový bezpečnostní profil ofatumumabu u CLL (dříve neléčené a relabující nebo refrakterní) je založen na údajích od 511 pacientů v klinických studiích (viz bod 5.1). Ty zahrnují 250 pacientů léčených samotným ofatumumabem (u pacientů s relabující nebo refrakterní CLL) a 261 pacienta léčeného ofatumumabem v kombinaci s alkylačním činidlem (u pacientů s dříve neléčenou CLL, kteří nemohou být léčeni terapií založenou na fludarabinu).
Profil nežádoucích účinků ofatumumabu u pacientů s CLL s lymfadenopatií, kteří jsou refrakterní k fludarabinu a u kterých došlo k selhání nejméně 2 předchozích terapií, byl konzistentní s celkovým bezpečnostním profilem stanoveným z dalších CLL studií, jak je popsáno v níže uvedeném tabulkovém seznamu.
Tabulkový seznam nežádoucích účinků
Nežádoucí účinky hlášené při léčbě dříve neléčených pacientů, pacientů s relapsem onemocnění nebo pacientů s refraktemí CLL buď samotným ofatumumabem, nebo ofatumumabem v kombinaci s alkylační látkou jsou shrnuty níže podle MedDRA tříd orgánových systémů a podle frekvence. Velmi časté (> 1/10); časté (> 1/100 až < 1/10); méně časté (> 1/1000 až < 1/100); vzácné (> 1/10000 až < 1/1000); velmi vzácné (< 1/10000), není známo (z dostupných údajů nelze určit). V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
|
MedDRA třídy orgánových systémů |
Velmi časté |
Časté |
Méně časté |
Vzácné |
|
Infekce a infestace |
Infekce dolních cest dýchacích, včetně pneumonie, infekce horních cest dýchacích |
Sepse, včetně neutropenické sepse a septického šoku, infekce herpetickými viry, infekce močových cest |
Infekce virem hepatitidy B a reaktivace viru hepatitidy B | |
|
Poruchy krve a lymfatického systému |
Neutropenie, |
Febrilní neutropenie, trombocytopenie, leukopenie |
Agranulocytóza, koagulopatie, aplazie červených krvinek, lymfopenie | |
|
Poruchy imunitního systému |
Anafylaktoidní reakce*, hypersenzitivita* |
Anafylaktický šok* | ||
|
Poruchy metabolismu a výživy |
Syndrom lýzy tumoru | |||
|
Srdeční poruchy |
Tachykardie* |
Bradykardie* | ||
|
Cévní poruchy |
Hypotenze*, hypertenze* | |||
|
Respirační, hrudní a mediastinální poruchy |
Bronchospasmus*, hypoxie*, dyspnoe*, hrudní diskomfort*, faryngolaryngeální bolest*, kašel*, nazální kongesce* |
Edém plic* | ||
|
Gastrointestinální poruchy |
Nauzea* |
Průjem* |
Obstrukce tenkého střeva | |
|
Poruchy kůže a podkožní tkáně |
Vyrážka* |
Kopřivka*, pruritus*, zčervenání (flushing)* |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Bolest zad* | |||
|
Celkové poruchy a reakce v místě aplikace |
Pyrexie* |
Syndrom z uvolnění cytokinů*, rigor*, zimnice*, hyperhidróza*, únava* |
* Tyto nežádoucí účinky pravděpodobně souvisejí s podáním ofatumumabu a jsou reakcí na infuzi. Typicky se objevují po zahájení infuze a během 24 hodin po ukončení infuze (viz bod 4.4).
Popis vybraných nežádoucích účinků
Reakce na infuzi
Nejčastěji pozorovanými nežádoucími účinky u pacientů v klinických studiích, kterým byl podáván ofatumumab, byly reakce související s infuzí, které se vyskytly u 68 % (348/511) pacientů kdykoli v průběhu léčby. Většina reakcí na infuzi odpovídala závažností stupni 1 nebo stupni 2. U 8 % pacientů se kdykoli během léčby vyskytly reakce související s infuzí > 3. stupně. Ve 2 % vedly reakce související s infuzí k ukončení léčby. Žádná z reakcí na infuzi nevedla k úmrtí (viz bod 4.4).
Infekce
Z celkového počtu 511 pacientů, kterým byl podáván ofatumumab v klinických studiích, se u 300 pacientů (59 %) vyskytla infekce. Tyto infekce byly bakteriální, virové nebo mykotické. U 104 (20 %) z 511 pacientů se vyskytly infekce > 3. stupně. U 28 (5 %) z 511 pacientů byla infekce smrtelná.
Neutropenie
Z celkového počtu 511 pacientů, kterým byl podáván ofatumumab v klinických studiích, se u 139 pacientů (27 %) vyskytl nežádoucí účinek související se sníženým počtem neutrofilů; u 188 (23 %) z 511 pacientů se vyskytly nežádoucí účinky související se sníženým počtem neutrofilů > 3. stupně. U 42 pacientů (8 %) se vyskytl závažný nežádoucí účinek související se sníženým počtem neutrofilů.
V pivotní studii s dříve neléčenou CLL (OMB110911) byla u 41 pacienta (u 23 pacientů léčených ofatumumabem a chlorambucilem a u 18 pacientů léčených samotným chlorambucilem) hlášena dlouhodobá neutropenie (definovaná jako neutropenie stupně 3 nebo 4, u které mezi 24. až 42. dnem od poslední léčby nedošlo ke zlepšení). U 9 pacientů léčených ofatumumabem a chlorambucilem a u 3 pacientů léčených samotným chlorambucilem došlo k opožděnému výskytu neutropenie definované jako neutropenie stupně 3 nebo 4, která se projevila nejméně 42 dní po poslední léčbě.
Kardiovaskulární účinky
Vliv opakovaných dávek přípravku Arzerra na interval QTc byl hodnocen v souhrnné analýze tří otevřených studií u pacientů s CLL (N = 85). V této souhrnné analýze byla pozorována prodloužení mediánu/průměru intervalů QT/QTc nad 5 ms. Nebyly zjištěny žádné velké změny průměrného intervalu QTc (tj. > 20 milisekund). U žádného pacienta nedošlo k prodloužení QTc > 500 ms. Prodloužení intervalu QTc závislé na koncentraci nebylo zjištěno.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Nebyl hlášen žádný případ předávkování.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: monoklonální protilátky, ATC kód: L01XC10 Mechanismus účinku
Ofatumumab je lidská monoklonální protilátka (IgG1), která se specificky váže na určité epitopy zahrnující jak malé, tak velké extracelulární kličky molekuly CD20. Molekula CD20 je transmembranózní fosfoprotein exprimovaný na B lymfocytech od stadia pre-B až po zralé B lymfocyty, a na nádorových B buňkách. Nádorové B buňky zahrnují CLL (obecně spojovanou s nižší mírou genové exprese povrchové molekuly CD20) a non-Hodgkinské lymfomy, což j sou tumory, u kterých je ve více než v 90 % případů genová exprese povrchové molekuly CD20 vysoká. Molekuly CD20 se neodlučují od buněčného povrchu ani nejsou po navázání protilátky internalizovány.
Vazba ofatumumabu na proximální membránový epitop molekuly CD20 indukuje nábor a aktivaci metabolické cesty komplementu na povrchu buňky, která vede k cytotoxicitě závislé na komplementu a následné lýze nádorových buněk. Je prokázáno, že ofatumumab způsobuje významnou lýzu buněk s vysokou hladinou exprese ochranných molekul komplementu. Ofatumumab rovněž vykazoval indukci lýzy buněk s vysokou i nízkou expresí CD20, a buněk rezistentních na rituximab. Navíc vazba ofatumumabu umožnila nábor NK buněk (natural killer), které umožňují navození buněčné smrti prostřednictvím buňkami mediované na protilátkách závislé cytotoxicity.
Farmakodynamické účinky
Počet periferních B buněk po první infuzi ofatumumabu u pacientů s hematologickými malignitami klesal. U pacientů s refrakterní CLL byl medián poklesu počtu B buněk 22 % po podání první infuze a 92 % při osmé týdenní infuzi. Počet periferních B buněk zůstával v průběhu zbývající léčby u většiny pacientů nízký a u pacientů s odpovědí na léčbu zůstával nižší než výchozí hodnota až 15 měsíců po podání poslední dávky.
U pacientů s dříve neléčenou CLL byl medián poklesu počtu B buněk po prvním cyklu 94 % při léčbě ofatumumabem v kombinaci s chlorambucilem a 73 % při léčbě samotným chlorambucilem, před šestým měsíčním cyklem byl > 99 % při léčbě ofatumumabem v kombinaci s chlorambucilem a 97 % při léčbě samotným chlorambucilem. Šest měsíců po poslední dávce byl medián poklesu počtu B buněk > 99 % při léčbě ofatumumabem v kombinaci s chlorambucilem a 94 % při léčbě samotným chlorambucilem.
Imunogenita
U terapeutických proteinů, jako je ofatumumab, je potenciální riziko imunogenity. Vzorky séra od více než 440 pacientů napříč celým klinickým programem pro CLL byly testovány na přítomnost protilátek proti ofatumumabu (buď s použitím metody ELISA, nebo elektrochemiluminiscence) v průběhu léčby a po ukončení léčby v rozmezí od 4 do 45 týdnů. U pacientů s CLL nedocházelo po léčbě ofatumumabem k tvorbě protilátek proti ofatumumabu.
Klinická účinnost a bezpečnost
Účinnost přípravku Arzerra byla hodnocena ve dvou klinických studiích (OMB110911 a OMB115991) u pacientů s dříve neléčenou CLL, kteří nemohli být léčeni terapií založenou na fludarabinu, a ve dvou klinických studiích (Hx-CD20-406 a Hx-CD20-402) u pacientů s relabující nebo refrakterní CLL.
Dříve neléčená CLL:
Studie OMB110911 (randomizovaná, otevřená, s paralelními rameny, multicentrická) hodnotila účinnost přípravku Arzerra v kombinaci s chlorambucilem v porovnání se samotným chlorambucilem u 447 pacientů s dříve neléčenou CLL, kteří nemohli být léčeni terapií založenou na fludarabinu (např. kvůli pokročilému věku nebo přítomnosti dalších onemocnění), s aktivním onemocněním a indikovaných k léčbě. Pacienti dostávali buď přípravek Arzerra v měsíčních intravenózních infuzích (1. cyklus: 300 mg v den 1 a 1000 mg v den 8. Následující cykly: 1000 mg v den 1 každých 28 dní) v kombinaci s chlorambucilem (10 mg/m2 perorálně ve dnech 1 - 7 každých 28 dní), nebo samotný chlorambucil (10 mg/m2 perorálně ve dnech 1 - 7 každých 28 dní). Pacienti dostávali léčbu minimálně 3 měsíce do dosažení nejlepší odpovědi nebo do maximálně 12 cyklů. Medián věku činil 69 let (rozmezí 35 až 92 let), 27 % pacientů bylo ve věku > 75 let, 63 % byli muži a 89 % byli běloši.
Medián hodnoty na škále souhrnného hodnocení multimorbidity pro geriatrii (CIRS-G, cumulative illness rating score for geriatrics) byl 9 a 31 % pacientů mělo CIRS-G > 10. Medián hodnoty clearance kreatininu (CrCl) vypočtené pomocí Cockroft-Gaultova vzorce byl 70 ml/min a 48 % pacientů mělo CrCl < 70 ml/min. Do studie byli zařazeni pacienti s ECOG výkonnostním stavem 0 až 2, přičemž 91 % pacientů mělo ECOG výkonnostní stav 0 nebo 1. Přibližně 60 % pacientů podstoupilo 3 -6 cyklů léčby přípravkem Arzerra a 32 % podstoupilo 7 - 12 cyklů. Medián počtu dokončených cyklů byl 6 (celková dávka přípravku Arzerra 6300 mg).
Primárním cílovým parametrem byl medián přežití bez progrese (PFS) hodnocený zaslepenou nezávislou komisí (IRC, Independent Review Committee) podle pokynů International Workshop for Chronic Lymphocytic Leukaemia (IWCLL) updated National Cancer Institute-sponsored Working Group (NCI-WG) z roku 2008. Celkový výskyt odpovědí na léčbu (ORR), včetně kompletní odpovědi (CR), byl rovněž hodnocen IRC podle pokynů IWCLL.
Přípravek Arzerra v kombinaci s chlorambucilem prokázal statisticky významné, 71%, zlepšení mediánu PFS v porovnání se samotným chlorambucilem (HR: 0,57, 95% IS: 0,45, 0,72), (viz tabulka 1, obrázek 1). Zlepšení PFS přidáním přípravku Arzerra bylo pozorováno u všech pacientů, včetně těch s rizikovými biologickými faktory (jako jsou delece 17p nebo 11q, nemutovaný IGHV, P2M > 3500 pg/l a exprese ZAP-70).
Tabulka 1 Souhrn porovnání PFS u přípravku Arzerra v kombinaci s chlorambucilem se samotným chlorambucilem u dříve neléčené CLL
|
Primární analýza PFS a analýzy podskupin hodnocené IRC, měsíce |
Chlorambucil (N = 226) |
Arzerra a chlorambucil (N = 221) |
|
Medián, všichni pacienti 95% IS Poměr rizik P hodnota |
13,1 22,4 (10,6, 13,8) (19,0, 25,2) 0,57 (0,45, 0,72) p < 0,001 | |
|
Věk > let (n = 119) |
12,2 |
23,8 |
|
Další onemocnění 0 nebo 1 (n = 126) |
10,9 |
23,0 |
|
Další onemocnění 2 nebo více (n = 321) |
13,3 |
21,9 |
|
ECOG 0, 1 (n = 411) |
13,3 |
23,0 |
|
ECOG 2 (n = 35) |
7,9 |
20,9 |
|
CIRS-G < 10 (n = 310) |
13,1 |
21,7 |
|
CIRS-G > 10 (n = 137) |
12,2 |
23,2 |
|
CrCl < 70 ml/min (n = 214) |
10,9 |
23,1 |
|
CrCl > 70 ml/min (n = 227) |
14,5 |
22,1 |
|
Delece 17p nebo 11q (n = 90) |
7,9 |
13,6 |
|
Mutovaný IGHV (< 98 %) (n = 177) |
12,2 |
30,5 |
|
Nemutovaný IGHV (> 98 %) (n = 227) |
11,7 |
17,3 |
|
p2M < 3500 pg/l (n = 109) |
13,8 |
25,5 |
|
P2M > 3500 pg/l (n = 322) |
11,6 |
19,6 |
|
Pozitivní ZAP-70 (n = 161) |
9,7 |
17,7 |
|
Přechodný ZAP-70 (n = 160) |
13,6 |
25,3 |
|
Negativní ZAP-70 (n = 100) |
13,8 |
25,6 |
|
Mutovaný IGHV & negativní ZAP-70 (n = 60) |
10,5 |
NR |
|
Mutovaný IGHV & pozitivní ZAP-70 (n = 35) |
7,9 |
27,2 |
|
Nemutovaný IGHV & negativní ZAP-70 (n = 27) |
16,7 |
16,2 |
|
Nemutovaný IGHV & pozitivní ZAP-70 (n =122) |
11,2 |
16,2 |
|
Zkratky: P2M = Beta-2-mikroglobulin, I |
S = interval spolehlivosti (confidence interval), CIRS-G = | |
škála souhrnného hodnocení multimorbidity pro geriatrii, CLL = chronická lymfocytární leukémie, CrCl = clearance kreatininu, ECOG = Eastern Cooperative Oncology Group, IGHV = variabilní doména těžkého řetězce imunoglobulinu, IRC = nezávislá komise, N = počet, NR = nebylo dosaženo, PFS = přežití bez progrese, ZAP-70 = zeta-asociovaný protein 70
U heterogenní nebělošské populace a u pacientů s ECOG výkonnostním stavem = 2 jsou k dispozici pouze omezené údaje.
Obrázek 1 Kaplan-Meierův odhad PFS hodnoceného IRC
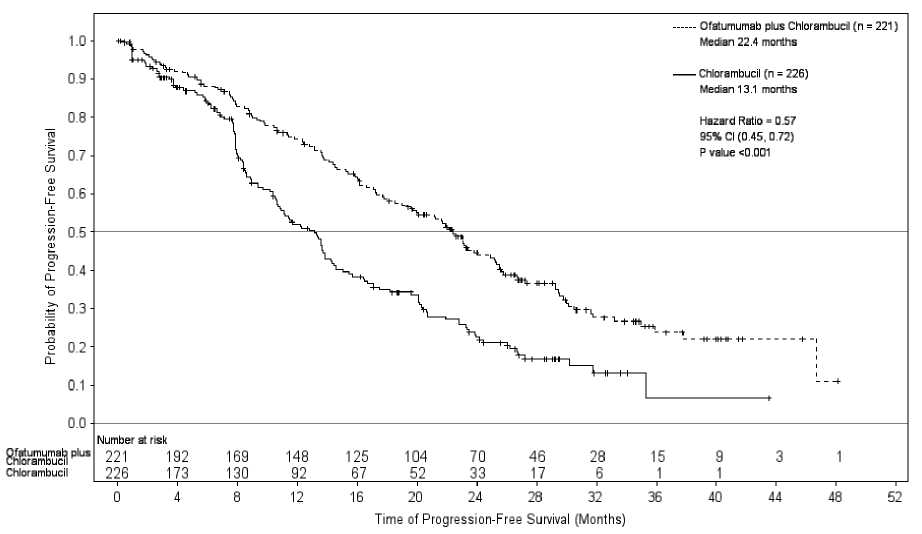
Překlad popisu obrázku: Probability of Progression-Free Survival = Pravděpodobnost přežití bez progrese; Time of Progression-Free Survival = Doba přežití bez progrese; Ofatumumab plus chlorambucil = ofatumumab s chlorambucilem; Chlorambucil = chlorambucil, Median = medián; months = měsíce; Hazard Ratio = poměr rizik; CI = IS (interval spolehlivosti), P value = P hodnota
Tabulka 2 Souhrn porovnání sekundárních výstupů u přípravku Arzerra v kombinaci s chlorambucilem se samotným chlorambucilem u dříve neléčené CLL
|
Sekundární výstupy hodnocené IRC |
Chlorambucil (N = 226) |
Arzerra a Chlorambucil (N = 221) |
|
ORR (%) |
69 |
82 |
|
95% IS |
(62,1, 74,6) |
(76,7, 87,1) |
|
P hodnota |
p < 0,001 | |
|
CR (%) |
1 |
12 |
|
CR s negativní MRD (% of CR) |
0 |
37 |
|
Medián trvání odpovědi, všichni pacienti, |
13 2 |
22 1 |
|
měsíce | ||
|
95% IS |
(10,8, 16,4) |
(19,1, 24,6) |
|
P hodnota |
p < 0,001 | |
Zkratky: IS = interval spolehlivosti (confidence interval), CLL = chronická lymfocytární leukémie, CR = kompletní odpověď, ICR = nezávislá komise, MRD = minimální reziduální nemoc, N = počet, ORR = celkový výskyt odpovědí na léčbu
Ve studii OMB115991 byla hodnocena účinnost přípravku Arzerra v kombinaci s bendamustinem u 44 pacientů s dříve neléčenou CLL, kteří nemohli být léčeni terapií založenou na fludarabinu. Pacienti dostávali přípravek Arzerra ve formě měsíčně podávaných intravenózních infuzí (1. cyklus: 300 mg v den 1 a 1000 mg v den 8, následující cykly: 1000 mg v den 1 každých 28 dní) v kombinaci s intravenózně podávaným bendamustinem v dávce 90 mg/m2 ve dnech 1 a 2 každých 28 dní. Pacienti dostali minimálně 3 cykly léčby a pacienti se stabilizovaným onemocněním nebo s odpovědí na léčbu po 3 cyklech pokračovali v léčbě dalšími 3 cykly do maximálně 6 cyklů. Medián počtu dokončených cyklů byl 6 (celková dávka přípravku Arzerra 6300 mg).
Primárním cílovým parametrem byl ORR hodnocený investigátorem podle pokynů IWCLL z roku 2008.
Výsledky této studie prokázaly, že přípravek Arzerra v kombinaci s bendamustinem je efektivní léčbou, ORR byl 95 % (95% IS: 85, 99) a CR byla 43 %. Více než polovina (56 %) pacientů s kompletní odpovědí byla po dokončení léčby v rámci studie MRD negativní.
K dispozici nejsou žádné údaje týkající se porovnání léčby přípravkem Arzerra v kombinaci s bendamustinem nebo chlorambucilem a léčebného režimu, jehož základem je rituximab, např. léčba rituximabem v kombinaci s chlorambucilem. Přínos této nové kombinace proti léčebnému režimu založenému na rituximabu proto není znám.
Refrakterní CLL:
Přípravek Arzerra byl podáván v monoterapii 223 pacientům s refrakterní CLL (studie Hx-CD20-406). Medián věku pacientů byl 64 let (rozmezí 41 až 87 let), většina byli muži (73 %) bílé rasy (96 %). Medián počtu podstoupených předchozích terapií byl u těchto pacientů 5, včetně rituximabu (57 %).
Z těchto 223 pacientů bylo 95 pacientů rezistentních na fludarabin a alemtuzumab (selhání léčby bylo definováno jako nedosažení alespoň částečné odpovědi na léčbu fludarabinem nebo alemtuzumabem či jako progrese onemocnění v průběhu 6 měsíců od poslední dávky fludarabinu nebo alemtuzumabu) Výchozí cytogenetické údaje (FISH) byly dostupné u 209 pacientů. 36 pacientů mělo normální karyotyp a chromozomální aberace byly detekovány u 174 pacientů, 47 pacientů mělo deleci 17p,
73 pacientů mělo deleci 11q, 23 pacientů trisomii 12q a 31 pacientů deleci13q jako jedinou aberaci.
ORR byl 49 % ve skupině rezistentní na fludarabin a alemtuzumab (viz tabulka 3, shrnutí údajů účinnosti ze studie). Pacienti, kteří již podstoupili terapii rituximabem, ať už v monoterapii nebo v kombinaci s dalšími léčivými přípravky, reagovali na léčbu ofatumumabem podobně jako ti, kteří léčbu rituximabem nepodstoupili.
Tabulka 3: Souhrn odpovědí na přípravek Arzerra u pacientů s refrakterní CLL
|
(Primární) cílový parametr 1 |
Pacienti rezistentní na fludarabin a alemtuzumab n = 95 |
|
Celkový výskyt odpovědi na léčbu Reagující na léčbu, n (%) |
47 (49) |
|
95,3% IS (%) |
39; 60 |
|
Výskyt odpovědi u pacientů s předchozí léčbou rituximabem Reagující na léčbu, n (%) |
25/56 (45) |
|
95% IS (%) |
31; 59 |
|
Výskyt odpovědi u pacientů s chromozomálními abnormalitami 17p delece Reagující na léčbu, n (%) |
10/27 (37) |
|
95% IS (%) |
19; 58 |
|
11q delece Reagující na léčbu, n (%) |
15/32 (47) |
|
95% IS (%) |
29; 65 |
|
Medián celkového přežití Měsíce |
13,9 |
|
95% IS |
9,9; 18,6 |
|
Přežití bez progrese Měsíce |
4,6 |
|
95% IS |
3,9; 6,3 |
|
Medián přetrvávání odpovědi Měsíce |
5,5 |
|
95% IS |
3,7; 7,2 |
|
Medián doby k další léčbě CLL Měsíce |
8,5 |
|
95% IS |
7,2; 9,9 |
|
1 Celková odpověď byla stanovena nezávislou komisí (Independent Response Committee) s použitím | |
|
pokynů NCI-WG pro CLL z roku 1996. IS - interval spolehlivosti (confidence interval) | |
Zlepšení bylo rovněž demonstrováno v jednotlivých kritériích podle NCI-WG. Ta zahrnovala zlepšení týkající se celkových příznaků, lymfadenopatie, organomegalie nebo cytopenie (viz tabulka 4).
Tabulka 4: Shrnutí klinického zlepšení s trváním alespoň 2 měsíce u pacientů s refrakterní
CLL s abnormalitami na počátku léčby
|
Cílový parametr účinnosti nebo hematologický parametr3 |
Pacienti benefitující z léčby/pacienti s iniciálními abnormalitami (%) |
|
Pacienti rezistentní na fludarabin a alemtuzumab | |
|
Počet lymfocytů > 50% pokles Normalizace (< 4x109/l) |
49/71(69) 36/71 (51) |
|
Kompletní vymizení celkových příznakůb |
21/47 (45) |
|
Lymfadenopatie3 > 50% zlepšení Kompletní úprava |
51/88 (58) 17/88 (19) |
|
Splenomegalie > 50% zlepšení Kompletní úprava |
27/47 (57) 23/47 (49) |
|
Hepatomegalie | |
|
> 50% zlepšení |
14/24 (58) |
|
Kompletní úprava |
11/24 (46) |
|
Hemoglobin < 11 g/dl na počátku léčby proti > 11 g/dl po začátku |
12/49 (24) |
|
Počet krevních destiček < 100x109/l na počátku léčby proti > 50% zvýšení nebo > 100x109/l po začátku |
19/50 (38) |
|
Neutrofily < 1x109/l na počátku léčby proti > 1,5x109/l |
1/17 (6) |
|
a Vyřazení údajů pacientů od doby první transfuze, léčby erytropoetinem nebo léčby růstovými faktory. U pacientů, u kterých chybí výchozí údaje, byly za výchozí považovány poslední údaje o krevním obrazu u pacienta. b Kompletní vymizení celkových příznaků (horečka, noční pocení, únava, pokles hmotnosti) definované jako přítomnost kteréhokoli příznaku na začátku léčby, ale následně bez příznaku. c Lymfadenopatie hodnocená jako výsledek součinu maximálních průměrů (SPD) podle fyzikálního vyšetření. | |
Přípravek Arzerra byl rovněž podáván skupině pacientů (n = 112) s lymfadenopatií (definovanou jako lymfadenopatie v případě, že alespoň 1 lymfatická uzlina byla větší než 5 cm), kteří byli rezistentní na léčbu fludarabinem. ORR přípravku Arzerra v této skupině pacientů byl 43 % (95,3% IS: 33; 53). Medián přežití bez progrese onemocnění byl 5,5 měsíců (95% IS: 4,6; 6,4) a medián celkové doby přežití byl 17,4 měsíců (95% IS: 15,0; 24,0). Výskyt odpovědi na léčbu byl u pacientů, kteří byli předtím léčeni rituximabem, 38 % (95%IS: 23; 61). U těchto pacientů bylo pozorováno rovněž srovnatelné zlepšení klinického stavu podle cílových parametrů účinnosti léčby a hematologických parametrů upřesněných výše, a to u pacientů rezistentních jak na léčbu fludarabinem, tak na léčbu alemtuzumabem.
Navíc byla provedena studie s podáváním přípravku Arzerra skupině pacientů (n = 16), kteří netolerovali léčbu fludarabinem nebo alemtuzumabem. Celkový výskyt léčebné odpovědi na farmakoterapii přípravkem Arzerra byl 63 % (95,3% IS: 35; 85).
V otevřené, randomizované, dvouramenné studii (OMB114242) provedené u pacientů s CLL s lymfadenopatií refrakterní k fludarabinu, u kterých došlo k selhání nejméně 2 předchozích terapií (n = 122) byla porovnávána monoterapie přípravkem Arzerra (n = 79) s terapií podle volby lékaře (n = 43). V primárním cílovém parametru PFS hodnoceném nezávislou komisí (IRC) nebyl statisticky významný rozdíl (5,4 vs. 3,6 měsíců, HR = 0,79, p = 0,27). PFS v rameni s přípravkem Arzerra v monoterapii byl srovnatelný s výsledky pozorovanými při monoterapii přípravkem Arzerra ve studii Hx-CD20-406.
Studie zaměřená na hledání dávky (Hx-CD20-402) byla provedena u 33 pacientů s relabující nebo refrakterní CLL. Medián věku pacientů byl 61 let (rozmezí: 27 až 82 let), většina byli muži (58 %) a všichni byli běloši. Léčba ofatumumabem (podávaným v infuzi 1x týdně 4 po sobě následující týdny) vedla k 50% výskytu objektivní odpovědi ve skupině s nejvyšší dávkou (1. dávka: 500 mg, 2., 3., a 4. dávka 2000 mg) a zahrnovala 12 pacientů s parciální remisí a jednoho s nodulární parciální remisí. Ve skupině s nejvyšší dávkou byl medián doby do progrese 15,6 týdnů (95% IS: 15; 22,6) v analýze všech pacientů a 23 týdnů (IS: 20; 31) u pacientů reagujících na léčbu. Trvání odpovědi bylo 16 týdnů (IS: 13; 19) a čas do další léčby CLL byl 52,4 týdnů (IS: 36,9; nelze stanovit).
Pediatrická populace
Evropská léková agentura rozhodla o zproštění povinnosti předložit výsledky klinických studií s přípravkem Arzerra u všech podskupin pediatrické populace s chronickou lymfatickou leukémií (viz bod 4.2 pro bližší informace týkající se použití přípravku Arzerra v pediatrii).
5.2 Farmakokinetické vlastnosti
Absorpce
Ofatumumab je podáván prostřednictvím intravenózní infuze, proto se absorpce neuplatňuje. Maximální sérové koncentrace ofatumumabu byly obvykle pozorovány krátce po ukončení infuze. Farmakokinetické údaje byly dostupné od 215 pacientů s refrakterní CLL. Geometrický průměr hodnoty Cmax byl 61 pg/ml po první infuzi (300 mg), po osmé infuzi 1x v týdnu (sedmé infuzi s dávkou 2000 mg) byl geometrický průměr hodnoty Cmax 1391 pg/ml a geometrický průměr hodnoty AUC(0_<x,) 463418 pg.h/ml. Po 12. infuzi (čtvrté infuzi 1x za měsíc s dávkou 2000 mg) byl geometrický průměr hodnoty Cmax 827 pg/ml a geometrický průměr hodnoty AUC(0_<») 203536 pg.h/ml. U pacientů s dříve neléčenou CLL, kteří dostávali ofatumumab a chlorambucil, byl geometrický průměr hodnoty Cmax po první infuzi (s dávkou 300 mg) 52 pg/ml, po infuzi v den 8 s dávkou 1000 mg 241 pg/ml a po infuzi ve čtvrtém měsíčním cyklu s dávkou 1000 mg 285 pg/ml; geometrický průměr hodnoty AUC(0-X) ve čtvrtém cyklu byl 65100 pg.h/ml.
Distribuce
Ofatumumab má malý distribuční objem, s průměrnou hodnotou Vss v rozmezí od 1,7 do 8,1 litru ve všech studiích, bez ohledu na dávku nebo počet infuzí.
Biotransformace
Ofatumumab je protein, jehož očekávaná cesta metabolismu je degradace na malé peptidy a jednotlivé aminokyseliny všudypřítomnými proteolytickými enzymy. Klasické studie biotransformace nebyly provedeny.
Eliminace
Ofatumumab je eliminován dvěma způsoby: na cíli nezávislá cesta jako u dalších IgG molekul a cílem-mediovaná cesta, která souvisí s vazbou na B buňky. Po první infuzi ofatumumabu následuje rychlá a setrvalá deplece CD20+ B buněk, která vede ke snížení počtu CD20+ buněk dostupných pro protilátky z následující infuze. Výsledkem je velmi nízká hodnota clearance ofatumumabu a významně vyšší hodnoty t12 po následných infuzích v porovnání s úvodní infuzí. Po opakovaných týdenních infuzích stoupaly hodnoty AUC a Cmax více, než by se dalo očekávat podle kumulace odvozené na základě údajů získaných z první infuze.
Ve studiích u pacientů s relabující nebo refraktemí CLL byly geometrické průměry hodnot pro CL a ti/2 64 ml/h (rozmezí 4,3 - 1122 ml/h) a 1,3 dne (rozmezí 0,2 - 6,0 dnů) po první infuzi; 8,5 ml/h (rozmezí 1,3 - 41,5 ml/h) a 11,5 dnů (rozmezí 2,3 - 30,6 dne) po čtvrté infuzi; 11,7 ml/h (rozmezí 3,9 -
54,2 ml/h) a 13,6 dne (rozmezí 2,4 - 36,0 dnů) po osmé infuzi a 12,1 ml/h (rozmezí 3,0 - 233 ml/h) a
11.5 dne (rozmezí 1,8 - 36,4 dne) po dvanácté infuzi.
U pacientů s dříve neléčenou CLL, kteří dostávali ofatumumab a chlorambucil, byly geometrické průměry hodnot CL a t1/2 po čtvrté infuzi 15,4 ml/h (rozmezí 4,1 - 146 ml/h) a 18,5 dne (rozmezí 2,7 -
82.6 dne).
Starší pacienti (ve věku 65 let nebo starší)
Neprokázalo se, že by věk byl významným faktorem, který by ovlivňoval farmakokinetiku ofatumumabu ve zkřížené analýze farmakokinetiky populací pacientů ve věku od 21 do 87 let.
Děti a dospívající
U pediatrických pacientů nejsou k dispozici žádné údaje o farmakokinetice.
Pohlaví
Pohlaví mělo ve zkřížených populačních analýzách jen mírný vliv (12 %) na centrální distribuční objem ofatumumabu, u pacientek (48 % pacientů byli muži, 52 % byly ženy) byly pozorovány vyšší hodnoty Cmax a AUC; tyto účinky se nepovažují za klinicky významné a nevycházejí z nich žádná doporučení pro dávkování.
Porucha funkce ledvin
Bylo zjištěno, že počáteční vypočtená clearance kreatininu nebyla významným faktorem, který by měl vliv na farmakokinetiku ofatumumabu ve zkřížených analýzách populací u pacientů s vypočítanými hodnotami clearance kreatininu v rozmezí od 26 do 287 ml/min. U pacientů s mírnou až středně závažnou poruchou funkce ledvin (clearance kreatininu > 30 ml/min) není nutná žádná úprava dávky. U pacientů se závažnou poruchou funkce ledvin (clearance kreatininu < 30 ml/min) jsou k dispozici pouze omezené farmakokinetické údaje.
Porucha funkce jater
Nebyly provedeny žádné formální studie hodnotící vliv poruchy funkce jater. Molekuly IgG1, jako je ofatumumab, jsou odbourávány všudypřítomnými proteolytickými enzymy, které nejsou vázány na jaterní tkáň, proto není pravděpodobné, že by změny jaterních funkcí měly účinek na eliminaci ofatumumabu.
5.3 Předklinické údaje vztahující se k bezpečnosti
Preklinické údaje neukazují na žádné zvláštní riziko pro člověka.
Intravenózní a subkutánní podání opicím vedlo k očekávané depleci počtu periferních B buněk i B buněk lymfoidních tkání bez dalších toxikologických nálezů. Podle předpokladu byl zaznamenán pokles humorální imunitní odpovědi IgG na klíčovou část hemocyaninu, ale nebyly prokázány žádné účinky na reakci hypersenzitivity opožděného typu. U několika zvířat došlo ke zvýšené destrukci červených krvinek, která se pravděpodobně objevila jako následek obalení červených krvinek opic protilátkami proti léku. U těchto opic bylo pozorováno odpovídající zvýšení počtu retikulocytů, což ukazovalo na regenerační odpověď kostní dřeně.
Intravenózní podání ofatumumabu březím opicím druhu cynomolgus (makak) v dávce 100 mg/kg jednou týdně od 20. dne do 50. dne gestace nezpůsobilo mateřskou ani fetální toxicitu ani teratogenitu. Stý den gestace byla pozorována deplece B buněk odpovídající farmakologické aktivitě ofatumumabu v pupečníkové krvi plodu a fetální tkáni sleziny. Studie pre- a postnatálního vývoje nebyly provedeny. Postnatální úprava proto nebyla demonstrována.
Protože ofatumumab je monoklonální protilátka, studie genotoxicity a kancerogenity nebyly s ofatumumabem provedeny.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Arginin
Natrium-acetát (E262)
Chlorid sodný Polysorbát 80 (E433)
Dihydrát dinatrium-edetátu (E386)
Kyselina chlorovodíková (E507) (k úpravě pH)
Voda na injekci
6.2 Inkompatibility
Tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky s výjimkou těch, které jsou uvedeny v bodě 6.6.
6.3 Doba použitelnosti
Injekční lahvička 3 roky.
Naředěná infuze
Chemická a fyzikální stabilita po otevření byla prokázána po dobu 48 hodin při teplotě do 25 °C.
Z mikrobiologického hlediska má být léčivý přípravek použit okamžitě. Není-li použit okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou v odpovědnosti uživatele a obvykle by doba neměla být delší než 24 hodin při 2 až 8 °C, pokud rekonstituce/ředění neproběhlo za kontrolovaných a validovaných aseptických podmínek.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte a převážejte chlazené (2 °C - 8 °C).
Chraňte před mrazem.
Injekční lahvičku uchovávejte v krabičce, aby byl přípravek chráněn před světlem.
Podmínky uchovávání léčivého přípravku po naředění viz bod 6.3.
6.5 Druh obalu a obsah balení
Injekční lahvička z čirého skla typu I s bromobutylovou zátkou neobsahující latex a hliníkovou pertlí obsahující 5 ml koncentrátu pro infuzní roztok.
Přípravek Arzerra je dostupný v balení po 3 injekčních lahvičkách.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Přípravek Arzerra koncentrát pro infuzní roztok neobsahuje konzervační látky, proto by ředění mělo být prováděno za aseptických podmínek. Naředěný roztok pro infuzi musí být použit do 24 hodin od přípravy. Jakýkoli zbývající naředěný roztok musí být po tomto čase zlikvidován.
• Před naředěním přípravku Arzerra
Zkontrolujte koncentrát přípravku Arzerra na přítomnost jakýchkoli částic a změny barvy před jeho naředěním. Ofatumumab má být bezbarvý až světle žlutý roztok. Neužívejte koncentrát přípravku Arzerra, pokud je přítomná jakákoli změna barvy.
Netřeste lahvičkou s ofatumumabem při této kontrole.
• Jak roztok pro infuzi naředit
Koncentrát přípravku Arzerra se musí před podáním naředit injekčním roztokem chloridu sodného 9 mg/ml (0,9%), a to za použití aseptické techniky.
300 mg dávka - Použijte 3 lahvičky (15 ml celkově, 5 ml v jedné lahvičce)
- odeberte a znehodnoťte 15 ml z vaku s 1000 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9%),
- odeberte 5 ml ofatumumabu z každé ze 3 lahviček a aplikujte je do 1000 ml vaku,
- neprotřepávejte, zamíchejte roztok opatrným převrácením vaku.
• Jak podávat naředěný roztok
Přípravek Arzerra nesmí být podáván jako intravenózní bolus. Podává se prostřednictvím intravenózní infuzní pumpy.
Infuze musí být podána do 24 hodin od přípravy roztoku. Jakýkoli nepoužitý roztok musí být po uplynutí tohoto času znehodnocen.
Přípravek Arzerra nesmí být smíchán, nebo podáván infuzí s dalšími léčivými přípravky nebo intravenózními roztoky. Propláchněte linku před a po podání ofatumumabu injekčním roztokem chloridu sodného 9 mg/ml (0,9%), abyste se tohoto vyvarovali.
Dříve neléčená CLL:
První infuzi podávejte po dobu minimálně 4,5 hodiny (viz bod 4.2) prostřednictvím periferního žilního přístupu nebo dlouhodobým žilním katetrem podle schématu níže:
Infuze 1: schéma
|
Čas (minuty) |
ml/hodina |
|
0 - 30 |
12 |
|
31 - 60 |
25 |
|
61 - 90 |
50 |
|
91 - 120 |
100 |
|
121 - 150 |
200 |
|
151 - 180 |
300 |
|
180 + |
400 |
Pokud byla první infuze dokončena bez závažných nežádoucích účinků, zbývající infuze (2 - 13) s dávkou 1000 mg mají být podávány po dobu minimálně 4 hodin (viz bod 4.2) prostřednictvím periferního žilního přístupu nebo dlouhodobým žilním katetrem podle schématu níže:
Tnfuze 2 až 13: schéma
|
Cas (minuty) |
ml/hodina |
|
0 - 30 |
25 |
|
31 - 60 |
50 |
|
61 - 90 |
100 |
|
91 - 120 |
200 |
|
121 + |
400 |
Refrakterní CLL:
První a druhou infuzi podávejte po dobu minimálně 6,5 hodiny (viz bod 4.2) prostřednictvím periferního žilního přístupu nebo dlouhodobým žilním katetrem podle schématu níže:
Tnfuze 1 a 2: schéma
|
Cas (minuty) |
ml/hodina |
|
0 - 30 |
12 |
|
31 - 60 |
25 |
|
61 - 90 |
50 |
|
91 - 120 |
100 |
|
121 + |
200 |
Pokud byla druhá infuze dokončena bez závažných nežádoucích účinků, zbývající infuze (3 - 12) mají být podávány po dobu minimálně 4 hodin (viz bod 4.2) prostřednictvím periferního žilního přístupu nebo dlouhodobým žilním katetrem podle schématu níže:
Tnfuze 3 až 12: schéma
|
Cas (minuty) |
ml/hodina |
|
0 - 30 |
25 |
|
31 - 60 |
50 |
|
61 - 90 |
100 |
|
91 - 120 |
200 |
|
121 + |
400 |
Pokud se objeví jakékoli nežádoucí účinky, rychlost infuze je třeba zpomalit (viz bod 4.2).
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/10/625/001
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 19/04/2010
Datum posledního prodloužení registrace: 16/01/2014
10. DATUM REVIZE TEXTU
Podrobné informace o tomto přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/.
NÁZEV PŘÍPRAVKU
1.
Arzerra 1000 mg koncentrát pro infuzní roztok.
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden ml koncentrátu obsahuje ofatumumabum 20 mg.
Jedna lahvička obsahuje ofatumumabum 1000 mg v 50 ml.
Ofatumumab je lidská monoklonální protilátka tvořená rekombinantními myšími buněčnými liniemi (NS0).
Pomocné látky se známým účinkem:
Tento léčivý přípravek obsahuje 34,8 mg sodíku v 300 mg dávce, 116 mg sodíku v 1000 mg dávce a 232 mg sodíku v 2000 mg dávce.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Koncentrát pro infuzní roztok (sterilní koncentrát).
Čirý až opalizující, bezbarvý až světle žlutý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Dříve neléčená chronická lymfocytární leukémie (CLL):
Přípravek Arzerra je v kombinaci s chlorambucilem nebo bendamustinem indikován k léčbě pacientů s CLL bez předchozí léčby, kteří nemohou být léčeni terapií založenou na fludarabinu.
Další informace viz bod 5.1.
Refrakterní CLL:
Přípravek Arzerra je indikován k léčbě CLL u pacientů, u kterých selhává léčba fludarabinem a alemtuzumabem.
Další informace viz bod 5.1.
4.2 Dávkování a způsob podání
Přípravek Arzerra má být podáván pod dohledem lékaře, který má zkušenosti s onkologickou léčbou, a v prostředí, které je plně vybavené pro okamžitou resuscitaci.
Monitorování
Pacienti mají být v průběhu podávání ofatumumabu, zejména v průběhu první infuze, pečlivě monitorováni z hlediska výskytu reakcí souvisejících s infuzí, včetně syndromu z uvolnění cytokinů.
Premedikace
Pacient má být vždy premedikován 30 minut až 2 hodiny před aplikací infuze přípravku Arzerra podle následujících dávkovacích schémat:
Dříve neléčená CLL:
• perorální paracetamol 1000 mg (nebo ekvivalent), plus
• perorální nebo intravenózní antihistaminikum (difenhydramin 50 mg nebo cetirizin 10 mg nebo ekvivalent), plus
• intravenózní kortikoid (prednisolon 50 mg nebo ekvivalent).
Pokud se u pacienta po první a druhé infuzi neobjeví závažné nežádoucí účinky, může být před dalšími infuzemi premedikace kortikoidy snížena nebo vynechána podle uvážení lékaře.
Refrakterní CLL:
• perorální paracetamol 1000 mg (nebo ekvivalent), plus
• perorální nebo intravenózní antihistaminikum (difenhydramin 50 mg nebo cetirizin 10 mg nebo ekvivalent), plus
• intravenózní kortikoid (prednisolon 100 mg nebo ekvivalent).
Pokud je druhá týdenní infuze ukončena bez závažných nežádoucích účinků, může být pro infuze 3 až 8 dávka kortikoidů snížena podle uvážení lékaře.
Před devátou infuzí (první infuze v měsíčním intervalu) musí být pacientům podána kompletní premedikace popsaná výše. Pokud je devátá infuze ukončena bez závažných nežádoucích účinků, může být před dalšími infuzemi dávka snížena na ekvivalent 50 mg prednisolonu podle uvážení lékaře.
Dávkování
Dříve neléčená CLL:
Doporučená dávka a schéma podávání je 300 mg v den 1, následuje 1000 mg za 1 týden, tj. v den 8 (cyklus 1), poté následuje 1000 mg v den 1 následujících cyklů. Tyto cykly mají být nejméně 3 a mají se opakovat do dosažení nejlepší odpovědi na léčbu nebo do celkového maximálního počtu 12 cyklů (každých 28 dní).
Nejlepší odpověď na léčbu je klinická odpověď, která se nezlepší během dalších 3 cyklů léčby.
První infuze
Úvodní rychlost první infuze přípravku Arzerra má být 12 ml/h. V průběhu infuze má být rychlost zvyšována každých 30 minut až do maximální rychlosti 400 ml/h (viz bod 6.6).
Následující infuze
Pokud byla první infuze dokončena bez závažných nežádoucích účinků souvisejících s infuzí, je možné následující infuze zahájit rychlostí 25 ml/h a jejich rychlost má být zvyšována každých 30 minut až do maximální rychlosti 400 ml/h (viz bod 6.6).
Refrakterní CLL:
Doporučená dávka je 300 mg pro první infuzi a 2000 mg pro všechny následující infuze. Léčba sestává z 8 infuzí podaných v týdenních intervalech, po kterých následuje 4 - 5 týdenní pauza. Léčba dále pokračuje 4 infuzemi podanými v měsíčních intervalech (tj. každé 4 týdny).
První a druhá infuze
Úvodní rychlost první a druhé infuze přípravku Arzerra je 12 ml/hodinu. V průběhu infuze má být její rychlost zvyšována každých 30 minut až do maximální rychlosti 200 ml/hodinu (viz bod 6.6).
Následující infuze
Pokud byla druhá infuze dokončena bez závažných nežádoucích účinků souvisejících s infuzí, je možné zbývající infuze zahájit rychlostí 25 ml/hodinu a jejich rychlost má být zvyšována každých 30 minut až do maximální rychlosti 400 ml/hodinu (viz bod 6.6).
Úprava dávkování a opakované zahájení léčby při závažných nežádoucích účincích souvisejících s infuzí - u pacientů s dříve neléčenou CLL a refrakterní CLL
Infuzi je třeba přerušit při nežádoucích účincích souvisejících s infuzí jakékoli intenzity. Léčba může být znovu zahájena podle uvážení ošetřujícího lékaře. Následující úpravy rychlosti infuze mohou být použity jako návod:
• V případě mírných nebo středně závažných nežádoucích účinků by měla být infuze přerušena a znovu zahájena poloviční rychlostí, než jaká byla v čase přerušení, až když je stav pacienta stabilizovaný. Pokud rychlost infuze nebyla od úvodní rychlosti 12 ml/hodinu před přerušením
z důvodu nežádoucích účinků souvisejících s infuzí zvýšena, infuze by měla být znovu zahájena standardní rychlostí 12 ml/hodinu. Rychlost infuze může být zvyšována podle standardního postupu, uvážení lékaře a tolerance pacienta (nemá být překročeno zvyšování rychlosti infuze každých 30 minut).
• V případě závažných nežádoucích účinků má být infuze přerušena a znovu zahájena rychlostí 12 ml/hodinu, až pokud je stav pacienta již stabilizovaný. Rychlost infuze může být zvyšována podle standardního postupu, uvážení lékaře a tolerance pacienta (nemá být překročeno zvyšování rychlosti infuze každých 30 minut).
Pediatrická populace
Vzhledem k nedostatečným údajům o bezpečnosti a/nebo účinnosti není přípravek Arzerra doporučen pro užívání u dětí mladších 18 let.
Starší pacienti
V souvislosti s věkem nebyly pozorovány žádné rozdíly v bezpečnosti a účinnosti (viz bod 5.1). Na základě dostupných údajů o bezpečnosti a účinnosti u starších pacientů není nutná úprava dávky (viz bod 5.2).
Porucha funkce ledvin
U pacientů s poruchou funkce ledvin nebyly provedeny žádné formální studie s přípravkem Arzerra.
U mírné až středně závažné poruchy funkce ledvin (clearance kreatininu > 30 ml/min) není nutná žádná úprava dávky (viz bod 5.2).
Porucha funkce jater
U pacientů s poruchou funkce jater nebyly provedeny žádné formální studie s přípravkem Arzerra. Není ale pravděpodobné, že by u pacientů s poruchou funkce jater byla nutná úprava dávky (viz bod 5.2).
Způsob podání
Přípravek Arzerra je k intravenózní infuzi a musí být před podáním naředěn. Návod k naředění léčivého přípravku před podáním je uveden v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na ofatumumab nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Reakce související s infuzí
Intravenózně podávaný ofatumumab je spojován s reakcemi na infuzi. Tyto reakce mohou vést k dočasnému přerušení nebo ukončení léčby. Premedikace zmírňuje reakce související s infuzí, ale ty se mohou přesto objevit, zvláště v průběhu první infuze. Reakce související s infuzí mohou zahrnovat, ale nejsou omezeny pouze na, anafylaktoidní reakce, bronchospasmus, srdeční příhody (např. ischemii/infarkt myokardu, bradykardii), zimnici/ztuhlost, kašel, syndrom z uvolnění cytokinů (cytokine release syndrom), průjem, dušnost, únavu, návaly, hypertenzi, hypotenzi, nauzeu, bolest, edém plic, pruritus, pyrexii, vyrážku a kopřivku. Ve vzácných případech mohou tyto reakce vést k úmrtí. I s premedikací mohou být po podání ofatumumabu zaznamenány závažné reakce, včetně syndromu z uvolnění cytokinů. V případě závažné reakce související s infuzí musí být infuze přípravku Arzerra okamžitě přerušena a zahájena symptomatická léčba (viz bod 4.2).
Reakce související s infuzí se mohou objevit častěji první den infuze a mají tendenci se s každou následující infuzí zmírňovat. Pacienti s anamnézou snížené plicní funkce mohou mít vyšší riziko plicních komplikací od závažných reakcí a měli by být v průběhu infuze ofatumumabu pečlivě monitorováni.
Syndrom lýzy tumoru
U pacientů s CLL se může při podávání ofatumumabu objevit syndrom lýzy tumoru (TLS, tumour lysis syndrome). Rizikovými faktory pro TLS jsou velký objem nádoru, vysoká koncentrace cirkulujících buněk (> 25000/mm3), hypovolémie, renální insuficience, zvýšená hladina kyseliny močové před léčbou a zvýšené hladiny laktát-dehydrogenázy. Léčba TLS zahrnuje korekci elektrolytových abnormalit, monitorování renálních funkcí, udržování bilance tekutin a podpůrnou léčbu.
Progresivní multifokální leukoencefalopatie
U pacientů s CLL, kteří podstupují cytotoxickou farmakoterapii (včetně ofatumumabu), byla zaznamenána progresivní multifokální leukoencefalopatie (PML) i úmrtí. Diagnózu PML je třeba zvážit u každého pacienta užívajícího přípravek Arzerra, u kterého se objeví nové neurologické příznaky a symptomy nebo dojde ke změně již existujících neurologických příznaků a symptomů. Pokud je podezření na diagnózu PML, je třeba léčbu přípravkem Arzerra přerušit a předat pacienta k neurologickému vyšetření.
Imunizace
Bezpečnost imunizace a schopnost vytvářet primární nebo anamnestickou odpověď na imunizaci živou atenuovanou nebo inaktivovanou vakcínou v průběhu léčby ofatumumabem nebyla studována. Odpověď na vakcinaci může být při depleci B buněk narušena. Vzhledem k riziku infekce je třeba se vyvarovat podávání živých atenuovaných vakcín v průběhu a po ukončení léčby ofatumumabem, dokud nedojde k normalizaci B buněk. V průběhu léčby ofatumumabem je třeba rizika a přínosy očkování pečlivě zvážit.
Hepatitida B
U pacientů léčených léčivy klasifikovanými jako cytolytické protilátky proti CD20, včetně přípravku Arzerra, se vyskytly případy infekce virem hepatitidy B (HBV) a reaktivace HBV, které v některých případech vyústily ve fulminantní hepatitidu, selhání jater a úmrtí. Tyto případy byly hlášeny u pacientů s pozitivním povrchovým antigenem viru hepatitidy B (HBsAg) a také u pacientů s pozitivními protilátkami proti core antigenu hepatitidy B (anti-HBc), ale negativním HBsAg. Reaktivace HBV se rovněž vyskytla u pacientů, u kterých byla infekce HBV považována za zvládnutou (tj. u pacientů HBsAg negativních, anti-HBc pozitivních a s pozitivními protilátkami proti povrchovému antigenu viru hepatitidy B [anti-HBs]).
Reaktivace viru hepatitidy B je definována jako náhlé zvýšení replikace viru hepatitidy B, projevující se jako rychlý vzestup hladiny HBV DNA, nebo detekce HBsAg u osob, které byly dříve HBsAg negativní a anti-HBc pozitivní. Po reaktivaci HBV často následuje hepatitida, tj. zvýšení hladin transamináz a, v závažných případech, zvýšení hladiny bilirubinu, selhání jater a úmrtí.
U všech pacientů má být před zahájením léčby přípravkem Arzerra proveden screening HBV infekce stanovením HBsAg a anti-HBc. U pacientů s prokázanou předchozí infekcí hepatitidou B (HBsAg negativní, anti-HBc pozitivní), má být monitorování a zahájení antivirové HBV terapie konzultováno s lékařem kvalifikovaným k léčbě hepatitidy B. Léčba přípravkem Arzerra nemá být zahajována u pacientů s prokázanou současnou infekcí virem hepatitidy B (HBsAg pozitivních), dokud není tato infekce odpovídajícím způsobem zvládnuta.
Pacienti s prokázanou předchozí infekcí HBV mají být během léčby a 6 - 12 měsíců po poslední infuzi přípravku Arzerra monitorováni z hlediska klinických a laboratorních příznaků hepatitidy nebo reaktivace HBV. Reaktivace HBV byla hlášena až 12 měsíců po ukončení léčby. Ukončení antivirové terapie HBV má být konzultováno s lékařem kvalifikovaným k léčbě hepatitidy B.
Pokud u pacientů dojde během léčby přípravkem Arzerra k reaktivaci HBV, má být Arzerra a veškerá současná chemoterapie okamžitě přerušena a zahájena odpovídající léčba. K dispozici jsou pouze nedostatečné údaje týkající se bezpečnosti opětovného zahájení podávání přípravku Arzerra u pacientů, u kterých došlo k reaktivaci HBV. Opětovné zahájení podávání přípravku Arzerra pacientům, u kterých byla reaktivace HBV vyřešena, má být konzultováno s lékařem kvalifikovaným k léčbě hepatitidy B.
Kardiovaskulární účinky
Pacienti s anamnézou srdečního onemocnění by měli být pečlivě sledováni. Léčba přípravkem Arzerra má být ukončena u pacientů se závažnou nebo život ohrožující srdeční arytmií.
Vliv opakovaných dávek přípravku Arzerra na interval QTc byl hodnocen v souhrnné analýze tří otevřených studií u pacientů s CLL (N = 85). V této souhrnné analýze byla pozorována prodloužení mediánu/průměru intervalů QT/QTc nad 5 ms. Nebyly zjištěny žádné velké změny průměrného intervalu QTc (tj. > 20 milisekund). U žádného pacienta nedošlo k prodloužení QTc > 500 ms. Prodloužení intervalu QTc závislé na koncentraci nebylo zjištěno. Doporučuje se, aby pacientům byly před zahájením podávání a v průběhu podávání ofatumumabu měřeny hladiny iontů, jako jsou draslík a hořčík. Abnormality v hladinách iontů mají být upraveny. Vliv ofatumumabu na pacienty s prodloužením intervalu QT (získaným nebo vrozeným) není znám.
Střevní obstrukce
U pacientů, kteří podstupují léčbu monoklonálními protilátkami proti CD20, včetně ofatumumabu, byla zaznamenána střevní obstrukce. Pacienti, u kterých se objeví bolesti břicha, zvláště časně v průběhu léčby ofatumumabem, by měli být vyšetřeni a měla by být zahájena vhodná léčba.
Laboratorní vyšetření
Během léčby ofatumumabem byly hlášeny cytopenie, včetně dlouhodobé neutropenie a neutropenie s opožděným nástupem. V průběhu léčby ofatumumabem je třeba v pravidelných intervalech vyšetřit kompletní krevní obraz, včetně počtu neutrofilů a krevních destiček, častěji pak u pacientů, u kterých se rozvíjí cytopenie.
Obsah sodíku
Tento léčivý přípravek obsahuje 34,8 mg sodíku v dávce 300 mg, 116 mg sodíku v dávce 1000 mg a 232 mg sodíku v dávce 2000 mg. To by mělo být vzato v úvahu u pacientů na řízené sodíkové dietě.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Ačkoli u ofatumumabu je k dispozici pouze omezené množství dat týkajících se lékových interakcí, nejsou známé žádné klinicky významné interakce s dalšími léčivými přípravky. Ofatumumab nemá klinicky významný vliv na farmakokinetiku chlorambucilu nebo jeho aktivního metabolitu, kyseliny 4-[bis(2-chlorethyl)amino]fenyloctové.
Účinnost živých atenuovaných nebo inaktivovaných vakcín může být ofatumumabem narušena, proto je vhodné se vyvarovat užití takových látek spolu s ofatumumabem. Pokud je společné podání považováno za nezbytné, mělo by být zváženo riziko a prospěch vakcinace v průběhu léčby ofatumumabem (viz bod 4.4).
4.6 Fertilita, těhotenství a kojení
Údaje o podávání ofatumumabu těhotným ženám nejsou k dispozici. Studie na zvířatech nenaznačují žádné přímé nebo nepřímé škodlivé účinky s ohledem na reprodukční toxicitu (viz bod 5.3). Ofatumumab nemá být podáván těhotným ženám, pokud jeho možný přínos pro matku nepřevýší možné riziko pro plod.
Ženy ve fertilním věku musí v průběhu léčby ofatumumabem a 12 měsíců po jejím ukončení používat účinnou antikoncepci.
Kojení
Není známo, zda je ofatumumab vylučován do mateřského mléka, nicméně lidské IgG do mateřského mléka vylučovány jsou. Bezpečnost užití ofatumumabu u lidí v průběhu kojení nebyla stanovena. Vylučování ofatumumabu do mateřského mléka nebylo u zvířat hodnoceno. Publikované údaje naznačují, že spotřeba mateřského mléka u novorozence ani kojence nevede k významné absorpci těchto mateřských protilátek do krevního oběhu. Riziko pro novorozence/kojence nelze vyloučit. Během léčby ofatumumabem a 12 měsíců po jejím ukončení má být kojení přerušeno.
Fertilita
O účincích na fertilitu u člověka nejsou k dispozici žádné údaje. Ve studiích na zvířatech nebyly účinky na fertilitu samců ani samic hodnoceny.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Žádné studie účinku přípravku Arzerra na schopnost řídit a obsluhovat stroje nebyly provedeny.
Žádné zásadní účinky na takové aktivity nejsou z farmakologie ofatumumabu očekávány. Pokud je zvažována pacientova schopnost provádět úkony, které vyžadují rozhodování, motorické nebo kognitivní schopnosti, je třeba vzít v úvahu jeho klinický stav a profil nežádoucích účinků (viz bod 4.8).
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Celkový bezpečnostní profil ofatumumabu u CLL (dříve neléčené a relabující nebo refrakterní) je založen na údajích od 511 pacientů v klinických studiích (viz bod 5.1). Ty zahrnují 250 pacientů léčených samotným ofatumumabem (u pacientů s relabující nebo refrakterní CLL) a 261 pacienta léčeného ofatumumabem v kombinaci s alkylačním činidlem (u pacientů s dříve neléčenou CLL, kteří nemohou být léčeni terapií založenou na fludarabinu).
Profil nežádoucích účinků ofatumumabu u pacientů s CLL s lymfadenopatií, kteří jsou refrakterní k fludarabinu a u kterých došlo k selhání nejméně 2 předchozích terapií, byl konzistentní s celkovým bezpečnostním profilem stanoveným z dalších CLL studií, jak je popsáno v níže uvedeném tabulkovém seznamu.
Tabulkový seznam nežádoucích účinků
Nežádoucí účinky hlášené při léčbě dříve neléčených pacientů, pacientů s relapsem onemocnění nebo pacientů s refrakterní CLL buď samotným ofatumumabem, nebo ofatumumabem v kombinaci s alkylační látkou, jsou shrnuty níže podle MedDRA tříd orgánových systémů a podle frekvence. Velmi časté (> 1/10); časté (> 1/100 až < 1/10); méně časté (> 1/1000 až < 1/100); vzácné (> 1/10000 až < 1/1000); velmi vzácné (< 1/10000), není známo (z dostupných údajů nelze určit). V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
|
MedDRA třídy orgánových systémů |
Velmi časté |
Časté |
Méně časté |
Vzácné |
|
Infekce a infestace |
Infekce dolních cest dýchacích, včetně pneumonie, infekce horních cest dýchacích |
Sepse, včetně neutropenické sepse a septického šoku, infekce herpetickými viry, infekce močových cest |
Infekce virem hepatitidy B a reaktivace viru hepatitidy B | |
|
Poruchy krve a lymfatického systému |
Neutropenie, anémie |
Febrilní neutropenie, trombocytopenie, leukopenie |
Agranulocytóza, koagulopatie, aplazie červených krvinek, lymfopenie | |
|
Poruchy imunitního systému |
Anafylaktoidní reakce*, hypersenzitivita* |
Anafylaktický šok* | ||
|
Poruchy metabolismu a výživy |
Syndrom lýzy tumoru | |||
|
Srdeční poruchy |
Tachykardie* |
Bradykardie* | ||
|
Cévní poruchy |
Hypotenze*, hypertenze* | |||
|
Respirační, hrudní a mediastinální poruchy |
Bronchospasmus*, hypoxie*, dyspnoe*, hrudní diskomfort*, faryngolaryngeální bolest*, kašel*, nazální kongesce* |
Edém plic* | ||
|
Gastrointestinální poruchy |
Nauzea* |
Průjem* |
Obstrukce tenkého střeva | |
|
Poruchy kůže a podkožní tkáně |
Vyrážka* |
Kopřivka*, pruritus*, zčervenání (flushing)* | ||
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Bolest zad* | |||
|
Celkové poruchy a reakce v místě aplikace |
Pyrexie* |
Syndrom z uvolnění cytokinů*, rigor*, zimnice*, hyperhidróza*, únava* |
* Tyto nežádoucí účinky pravděpodobně souvisejí s podáním ofatumumabu a jsou reakcí na infuzi. Typicky se objevují po zahájení infuze a během 24 hodin po ukončení infuze (viz bod 4.4).
Popis vybraných nežádoucích účinků
Reakce na infuzi
Nejčastěji pozorovanými nežádoucími účinky u pacientů v klinických studiích, kterým byl podáván ofatumumab, byly reakce související s infuzí, které se vyskytly u 68 % (348/511) pacientů kdykoli v průběhu léčby. Většina reakcí na infuzi odpovídala závažností stupni 1 nebo stupni 2. U 8 % pacientů se kdykoli během léčby vyskytly reakce související s infuzí > 3. stupně. Ve 2 % vedly reakce související s infuzí k ukončení léčby. Žádná z reakcí na infuzi nevedla k úmrtí (viz bod 4.4).
Infekce
Z celkového počtu 511 pacientů, kterým byl podáván ofatumumab v klinických studiích, se u 300 pacientů (59 %) vyskytla infekce. Tyto infekce byly bakteriální, virové nebo mykotické. U 104 (20 %) z 511 pacientů se vyskytly infekce > 3. stupně. U 28 (5 %) z 511 pacientů byla infekce smrtelná.
Neutropenie
Z celkového počtu 511 pacientů, kterým byl podáván ofatumumab v klinických studiích, se u 139 pacientů (27 %) vyskytl nežádoucí účinek související se sníženým počtem neutrofilů; u 188 (23 %) z 511 pacientů se vyskytly nežádoucí účinky související se sníženým počtem neutrofilů > 3. stupně. U 42 pacientů (8 %) se vyskytl závažný nežádoucí účinek související se sníženým počtem neutrofilů.
V pivotní studii s dříve neléčenou CLL (OMB110911) byla u 41 pacienta (u 23 pacientů léčených ofatumumabem a chlorambucilem a u 18 pacientů léčených samotným chlorambucilem) hlášena dlouhodobá neutropenie (definovaná jako neutropenie stupně 3 nebo 4, u které mezi 24. až 42. dnem od poslední léčby nedošlo ke zlepšení). U 9 pacientů léčených ofatumumabem a chlorambucilem a u 3 pacientů léčených samotným chlorambucilem došlo k opožděnému výskytu neutropenie definované jako neutropenie stupně 3 nebo 4, která se projevila nejméně 42 dní po poslední léčbě.
Kardiovaskulární účinky
Vliv opakovaných dávek přípravku Arzerra na interval QTc byl hodnocen v souhrnné analýze tří otevřených studií u pacientů s CLL (N = 85). V této souhrnné analýze byla pozorována prodloužení mediánu/průměru intervalů QT/QTc nad 5 ms. Nebyly zjištěny žádné velké změny průměrného intervalu QTc (tj. > 20 milisekund). U žádného pacienta nedošlo k prodloužení QTc > 500 ms. Prodloužení intervalu QTc závislé na koncentraci nebylo zjištěno.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Nebyl hlášen žádný případ předávkování.
FARMAKOLOGICKÉ VLASTNOSTI
5.
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: monoklonální protilátky, ATC kód: L01XC10 Mechanismus účinku
Ofatumumab je lidská monoklonální protilátka (IgG1), která se specificky váže na určité epitopy zahrnující jak malé, tak velké extracelulární kličky molekuly CD20. Molekula CD20 je transmembranózní fosfoprotein exprimovaný na B lymfocytech od stadia pre-B až po zralé B lymfocyty, a na nádorových B buňkách. Nádorové B buňky zahrnují CLL (obecně spojovanou s nižší mírou genové exprese povrchové molekuly CD20) a non-Hodgkinské lymfomy, což jsou tumory, u kterých je ve více než v 90 % případů genová exprese povrchové molekuly CD20 vysoká. Molekuly CD20 se neodlučují od buněčného povrchu ani nejsou po navázání protilátky internalizovány.
Vazba ofatumumabu na proximální membránový epitop molekuly CD20 indukuje nábor a aktivaci metabolické cesty komplementu na povrchu buňky, která vede k cytotoxicitě závislé na komplementu a následné lýze nádorových buněk. Je prokázáno, že ofatumumab způsobuje významnou lýzu buněk s vysokou hladinou exprese ochranných molekul komplementu. Ofatumumab rovněž vykazoval indukci lýzy buněk s vysokou i nízkou expresí CD20, a buněk rezistentních na rituximab. Navíc vazba ofatumumabu umožnila nábor NK buněk (natural killer), které umožňují navození buněčné smrti prostřednictvím buňkami mediované na protilátkách závislé cytotoxicity.
Farmakodynamické účinky
Počet periferních B buněk po první infuzi ofatumumabu u pacientů s hematologickými malignitami klesal. U pacientů s refrakterní CLL byl medián poklesu počtu B buněk 22 % po podání první infuze a 92 % při osmé týdenní infuzi. Počet periferních B buněk zůstával v průběhu zbývající léčby u většiny pacientů nízký a u pacientů s odpovědí na léčbu zůstával nižší než výchozí hodnota až 15 měsíců po podání poslední dávky.
U pacientů s dříve neléčenou CLL byl medián poklesu počtu B buněk po prvním cyklu 94 % při léčbě ofatumumabem v kombinaci s chlorambucilem a 73 % při léčbě samotným chlorambucilem, před šestým měsíčním cyklem byl > 99 % při léčbě ofatumumabem v kombinaci s chlorambucilem a 97 % při léčbě samotným chlorambucilem. Šest měsíců po poslední dávce byl medián poklesu počtu B buněk > 99 % při léčbě ofatumumabem v kombinaci s chlorambucilem a 94 % při léčbě samotným chlorambucilem.
Imunogenita
U terapeutických proteinů, jako je ofatumumab, je potenciální riziko imunogenity. Vzorky séra od více než 440 pacientů napříč celým klinickým programem pro CLL byly testovány na přítomnost protilátek proti ofatumumabu (buď s použitím metody ELISA, nebo elektrochemiluminiscence) v průběhu léčby a po ukončení léčby v rozmezí od 4 do 45 týdnů. U pacientů s CLL nedocházelo po léčbě ofatumumabem k tvorbě protilátek proti ofatumumabu.
Klinická účinnost a bezpečnost
Účinnost přípravku Arzerra byla hodnocena ve dvou klinických studiích (OMB110911 a OMB115991) u pacientů s dříve neléčenou CLL, kteří nemohli být léčeni terapií založenou na fludarabinu, a ve dvou klinických studiích (Hx-CD20-406 a Hx-CD20-402) u pacientů s relabující nebo refrakterní CLL.
Studie OMB110911 (randomizovaná, otevřená, s paralelními rameny, multicentrická) hodnotila účinnost přípravku Arzerra v kombinaci s chlorambucilem v porovnání se samotným chlorambucilem u 447 pacientů s dříve neléčenou CLL, kteří nemohli být léčeni terapií založenou na fludarabinu (např. kvůli pokročilému věku nebo přítomnosti dalších onemocnění), s aktivním onemocněním a indikovaných k léčbě. Pacienti dostávali buď přípravek Arzerra v měsíčních intravenózních infuzích (1. cyklus: 300 mg v den 1 a 1000 mg v den 8. Následující cykly: 1000 mg v den 1 každých 28 dní) v kombinaci s chlorambucilem (10 mg/m2 perorálně ve dnech 1 - 7 každých 28 dní), nebo samotný chlorambucil (10 mg/m2 perorálně ve dnech 1 - 7 každých 28 dní). Pacienti dostávali léčbu minimálně 3 měsíce do dosažení nejlepší odpovědi nebo do maximálně 12 cyklů. Medián věku činil 69 let (rozmezí 35 až 92 let), 27 % pacientů bylo ve věku > 75 let, 63 % byli muži a 89 % byli běloši.
Medián hodnoty na škále souhrnného hodnocení multimorbidity pro geriatrii (CIRS-G, cumulative illness rating score for geriatrics) byl 9 a 31 % pacientů mělo CIRS-G > 10. Medián hodnoty clearance kreatininu (CrCl) vypočtené pomocí Cockroft-Gaultova vzorce byl 70 ml/min a 48 % pacientů mělo CrCl < 70 ml/min. Do studie byli zařazeni pacienti s ECOG výkonnostním stavem 0 až 2, přičemž 91 % pacientů mělo ECOG výkonnostní stav 0 nebo 1. Přibližně 60 % pacientů podstoupilo 3 -6 cyklů léčby přípravkem Arzerra a 32 % podstoupilo 7 - 12 cyklů. Medián počtu dokončených cyklů byl 6 (celková dávka přípravku Arzerra 6300 mg).
Primárním cílovým parametrem byl medián přežití bez progrese (PFS) hodnocený zaslepenou nezávislou komisí (IRC, Independent Review Committee) podle pokynů International Workshop for Chronic Lymphocytic Leukaemia (IWCLL) updated National Cancer Institute-sponsored Working Group (NCI-WG) z roku 2008. Celkový výskyt odpovědí na léčbu (ORR), včetně kompletní odpovědi (CR), byl rovněž hodnocen IRC podle pokynů IWCLL.
Přípravek Arzerra v kombinaci s chlorambucilem prokázal statisticky významné, 71%, zlepšení mediánu PFS v porovnání se samotným chlorambucilem (HR: 0,57, 95% IS: 0,45, 0,72), (viz tabulka 1, obrázek 1). Zlepšení PFS přidáním přípravku Arzerra bylo pozorováno u všech pacientů, včetně těch s rizikovými biologickými faktory (jako jsou delece 17p nebo 11q, nemutovaný IGHV, P2M > 3500 pg/l a exprese ZAP-70).
Tabulka 1 Souhrn porovnání PFS u přípravku Arzerra v kombinaci s chlorambucilem se samotným chlorambucilem u dříve neléčené CLL
|
Primární analýza PFS a analýzy podskupin hodnocené IRC, měsíce |
Chlorambucil (N = 226) |
Arzerra a chlorambucil (N = 221) |
|
Medián, všichni pacienti 95% IS Poměr rizik P hodnota |
13,1 22,4 (10,6, 13,8) (19,0, 25,2) 0,57 (0,45, 0,72) p < 0,001 | |
|
Věk > let (n = 119) |
12,2 |
23,8 |
|
Další onemocnění 0 nebo 1 (n = 126) |
10,9 |
23,0 |
|
Další onemocnění 2 nebo více (n = 321) |
13,3 |
21,9 |
|
ECOG 0, 1 (n = 411) |
13,3 |
23,0 |
|
ECOG 2 (n = 35) |
7,9 |
20,9 |
|
CIRS-G < 10 (n = 310) |
13,1 |
21,7 |
|
CIRS-G > 10 (n = 137) |
12,2 |
23,2 |
|
CrCl < 70 ml/min (n = 214) |
10,9 |
23,1 |
|
CrCl > 70 ml/min (n = 227) |
14,5 |
22,1 |
|
Delece 17p nebo 11q (n = 90) |
7,9 |
13,6 |
|
Mutovaný IGHV (< 98 %) (n = 177) |
12,2 |
30,5 |
|
Nemutovaný IGHV (> 98 %) (n = 227) |
11,7 |
17,3 |
|
p2M < 3500 pg/l (n = 109) |
13,8 |
25,5 |
|
P2M > 3500 pg/l (n = 322) |
11,6 |
19,6 |
|
Pozitivní ZAP-70 (n = 161) |
9,7 |
17,7 |
|
Přechodný ZAP-70 (n = 160) |
13,6 |
25,3 |
|
Negativní ZAP-70 (n = 100) |
13,8 |
25,6 |
|
Mutovaný IGHV & negativní ZAP-70 (n = 60) |
10,5 |
NR |
|
Mutovaný IGHV & pozitivní ZAP-70 (n = 35) |
7,9 |
27,2 |
|
Nemutovaný IGHV & negativní ZAP-70 (n = 27) |
16,7 |
16,2 |
|
Nemutovaný IGHV & pozitivní ZAP-70 (n =122) |
11,2 |
16,2 |
|
Zkratky: P2M = Beta-2-mikroglobulin, I |
S = interval spolehlivosti (confidence interval), CIRS-G = | |
škála souhrnného hodnocení multimorbidity pro geriatrii, CLL = chronická lymfocytární leukémie, CrCl = clearance kreatininu, ECOG = Eastern Cooperative Oncology Group, IGHV = variabilní doména těžkého řetězce imunoglobulinu, IRC = nezávislá komise, N = počet, NR = nebylo dosaženo, PFS = přežití bez progrese, ZAP-70 = zeta-asociovaný protein 70
U heterogenní nebělošské populace a u pacientů s ECOG výkonnostním stavem = 2 jsou k dispozici pouze omezené údaje.
Obrázek 1 Kaplan-Meierův odhad PFS hodnoceného IRC
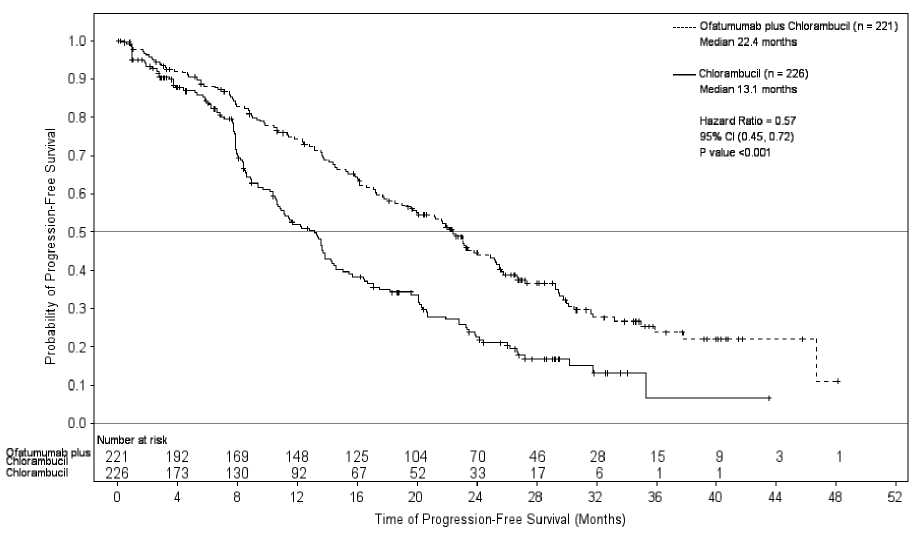
Překlad popisu obrázku: Probability of Progression-Free Survival = Pravděpodobnost přežití bez progrese; Time of Progression-Free Survival = Doba přežití bez progrese; Ofatumumab plus chlorambucil = ofatumumab s chlorambucilem; Chlorambucil = chlorambucil, Median = medián; months = měsíce; Hazard Ratio = poměr rizik; CI = IS (interval spolehlivosti), P value = P hodnota
Tabulka 2 Souhrn porovnání sekundárních výstupů u přípravku Arzerra v kombinaci s chlorambucilem se samotným chlorambucilem u dříve neléčené CLL
|
Sekundární výstupy hodnocené IRC |
Chlorambucil (N = 226) |
Arzerra a Chlorambucil (N = 221) |
|
ORR (%) |
69 |
82 |
|
95% IS |
(62,1, 74,6) |
(76,7, 87,1) |
|
P hodnota |
p < 0,001 | |
|
CR (%) |
1 |
12 |
|
CR s negativní MRD (% of CR) |
0 |
37 |
|
Medián trvání odpovědi, všichni pacienti, |
13 2 |
22 1 |
|
měsíce | ||
|
95% IS |
(10,8, 16,4) |
(19,1, 24,6) |
|
P hodnota |
p < 0,001 | |
Zkratky: IS = interval spolehlivosti (confidence interval), CLL = chronická lymfocytární leukémie, CR = kompletní odpověď, ICR = nezávislý hodnotící výbor, MRD = minimální reziduální nemoc, N = počet, ORR = celkový výskyt odpovědí na léčbu
Ve studii OMB115991 byla hodnocena účinnost přípravku Arzerra v kombinaci s bendamustinem u 44 pacientů s dříve neléčenou CLL, kteří nemohli být léčeni terapií založenou na fludarabinu. Pacienti dostávali přípravek Arzerra ve formě měsíčně podávaných intravenózních infuzí (1. cyklus: 300 mg v den 1 a 1000 mg v den 8, následující cykly: 1000 mg v den 1 každých 28 dní) v kombinaci s intravenózně podávaným bendamustinem v dávce 90 mg/m2 ve dnech 1 a 2 každých 28 dní. Pacienti dostali minimálně 3 cykly léčby a pacienti se stabilizovaným onemocněním nebo s odpovědí na léčbu po 3 cyklech pokračovali v léčbě dalšími 3 cykly do maximálně 6 cyklů. Medián počtu dokončených cyklů byl 6 (celková dávka přípravku Arzerra 6300 mg).
Primárním cílovým parametrem byl ORR hodnocený investigátorem podle pokynů IWCLL z roku 2008.
Výsledky této studie prokázaly, že přípravek Arzerra v kombinaci s bendamustinem je efektivní léčbou, ORR byl 95 % (95% IS: 85, 99) a CR byla 43 %. Více než polovina (56 %) pacientů s kompletní odpovědí byla po dokončení léčby v rámci studie MRD negativní.
K dispozici nejsou žádné údaje týkající se porovnání léčby přípravkem Arzerra v kombinaci s bendamustinem nebo chlorambucilem a léčebného režimu, jehož základem je rituximab, např. léčba rituximabem v kombinaci s chlorambucilem. Přínos této nové kombinace proti léčebnému režimu založenému na rituximabu proto není znám.
Přípravek Arzerra byl podáván v monoterapii 223 pacientům s refraktemí CLL (studie Hx-CD20-406). Medián věku pacientů byl 64 let (rozmezí 41 až 87 let), většina byli muži (73 %) bílé rasy (96 %). Medián počtu podstoupených předchozích terapií byl u těchto pacientů 5, včetně rituximabu (57 %).
Z těchto 223 pacientů bylo 95 pacientů rezistentních na fludarabin a alemtuzumab (selhání léčby bylo definováno jako nedosažení alespoň částečné odpovědi na léčbu fludarabinem nebo alemtuzumabem či jako progrese onemocnění v průběhu 6 měsíců od poslední dávky fludarabinu nebo alemtuzumabu) Výchozí cytogenetické údaje (FISH) byly dostupné u 209 pacientů. 36 pacientů mělo normální karyotyp a chromozomální aberace byly detekovány u 174 pacientů, 47 pacientů mělo deleci 17p,
73 pacientů mělo deleci 11q, 23 pacientů trisomii 12q a 31 pacientů deleci13q jako jedinou aberaci.
ORR byl 49 % ve skupině rezistentní na fludarabin a alemtuzumab (viz tabulka 3, shrnutí údajů účinnosti ze studie). Pacienti, kteří již podstoupili terapii rituximabem, ať už v monoterapii nebo v kombinaci s dalšími léčivými přípravky, reagovali na léčbu ofatumumabem podobně jako ti, kteří léčbu rituximabem nepodstoupili.
Tabulka 3 Souhrn odpovědí na přípravek Arzerra u pacientů s refrakterní CLL
|
(Primární) cílový parametr 1 |
Pacienti rezistentní na fludarabin a alemtuzumab n = 95 |
|
Celkový výskyt odpovědi na léčbu Reagující na léčbu, n (%) |
47 (49) |
|
95,3% IS (%) |
39; 60 |
|
Výskyt odpovědi u pacientů s předchozí léčbou rituximabem Reagující na léčbu, n (%) |
25/56 (45) |
|
95% IS (%) |
31; 59 |
|
Výskyt odpovědi u pacientů s chromozomálními abnormalitami 17p delece Reagující na léčbu, n (%) |
10/27 (37) |
|
95% IS (%) |
19; 58 |
|
11q delece Reagující na léčbu, n (%) |
15/32 (47) |
|
95% IS (%) |
29; 65 |
|
Medián celkového přežití Měsíce |
13,9 |
|
95% IS |
9,9; 18,6 |
|
Přežití bez progrese Měsíce |
4,6 |
|
95% IS |
3,9; 6,3 |
|
Medián přetrvávání odpovědi Měsíce |
5,5 |
|
95% IS |
3,7; 7,2 |
|
Medián doby k další léčbě CLL Měsíce |
8,5 |
|
95% IS |
7,2; 9,9 |
|
1 Celková odpověď byla stanovena nezávislou komisí (Independent Response Committee) s použitím | |
|
pokynů NCI-WG pro CLL z roku 1996. IS - interval spolehlivosti (confidence interval) | |
Zlepšení bylo rovněž demonstrováno v jednotlivých kritériích podle NCI-WG. Ta zahrnovala zlepšení týkající se celkových příznaků, lymfadenopatie, organomegalie nebo cytopenie (viz tabulka 4).
Tabulka 4: Shrnutí klinického zlepšení s trváním alespoň 2 měsíce u pacientů s refrakterní CLL s abnormalitami na počátku léčby
|
Cílový parametr účinnosti nebo hematologický parametr3 |
Pacienti benefitující z léčby/pacienti s iniciálními abnormalitami (%) |
|
Pacienti rezistentní na fludarabin a alemtuzumab | |
|
Počet lymfocytů > 50% pokles Normalizace (< 4x109/l) |
49/71(69) 36/71 (51) |
|
Kompletní vymizení celkových příznakůb |
21/47 (45) |
|
Lymfadenopatie3 > 50% zlepšení Kompletní úprava |
51/88 (58) 17/88 (19) |
|
Splenomegalie > 50% zlepšení Kompletní úprava |
27/47 (57) 23/47 (49) |
|
Hepatomegalie | |
|
> 50% zlepšení |
14/24 (58) |
|
Kompletní úprava |
11/24 (46) |
|
Hemoglobin < 11 g/dl na počátku léčby proti > 11 g/dl po začátku |
12/49 (24) |
|
Počet krevních destiček < 100x109/l na počátku léčby proti > 50% zvýšení nebo > 100x109/l po začátku |
19/50 (38) |
|
Neutrofily < 1x109/l na počátku léčby proti > 1,5x109/l |
1/17 (6) |
|
a Vyřazení údajů pacientů od doby první transfuze, léčby erytropoetinem nebo léčby růstovými faktory. U pacientů, u kterých chybí výchozí údaje, byly za výchozí považovány poslední údaje o krevním obrazu u pacienta. b Kompletní vymizení celkových příznaků (horečka, noční pocení, únava, pokles hmotnosti) definované jako přítomnost kteréhokoli příznaku na začátku léčby, ale následně bez příznaku. c Lymfadenopatie hodnocená jako výsledek součinu maximálních průměrů (SPD) podle fýzikálního vyšetření. | |
Přípravek Arzerra byl rovněž podáván skupině pacientů (n = 112) s lymfadenopatií (definovanou jako lymfadenopatie v případě, že alespoň 1 lymfatická uzlina byla větší než 5 cm), kteří byli rezistentní na léčbu fludarabinem. ORR přípravku Arzerra v této skupině pacientů byl 43 % (95,3% IS: 33; 53). Medián přežití bez progrese onemocnění byl 5,5 měsíců (95% IS:4,6; 6,4) a medián celkové doby přežití byl 17,4 měsíců (95% IS: 15,0; 24,0). Výskyt odpovědi na léčbu byl u pacientů, kteří byli předtím léčeni rituximabem, 38 % (95% IS: 23; 61). U těchto pacientů bylo pozorováno rovněž srovnatelné zlepšení klinického stavu podle cílových parametrů účinnosti léčby a hematologických parametrů upřesněných výše, a to u pacientů rezistentních jak na léčbu fludarabinem, tak na léčbu alemtuzumabem.
Navíc byla provedena studie s podáváním přípravku Arzerra skupině pacientů (n = 16), kteří netolerovali léčbu fludarabinem nebo alemtuzumabem. Celkový výskyt léčebné odpovědi na farmakoterapii přípravkem Arzerra byl 63 % (95,3% IS: 35; 85).
V otevřené, randomizované studii se dvěma rameny (OMB114242) provedené u pacientů s refraktemí CLL rezistentní na fludarabin, u kterých došlo k selhání nejméně 2 předchozích terapií (n = 122) byla porovnávána monoterapie přípravkem Arzerra (n = 79) s terapií podle volby lékaře (n = 43).
V primárním cílovém parametru PFS hodnoceném nezávislým hodnotícím výborem (IRC) nebyl statisticky významný rozdíl (5,4 vs. 3,6 měsíců, HR = 0,79, p = 0,27). PFS v rameni s přípravkem Arzerra v monoterapii byl srovnatelný s výsledky pozorovanými při monoterapii přípravkem Arzerra ve studii Hx-CD20-406.
Studie zaměřená na hledání dávky (Hx-CD20-402) byla provedena u 33 pacientů s relabující nebo refraktemí CLL. Medián věku pacientů byl 61 let (rozmezí: 27 až 82 let), většina byli muži (58 %) a všichni byli běloši. Léčba ofatumumabem (podávaným v infuzi 1x týdně 4 po sobě následující týdny) vedla k 50% výskytu objektivní odpovědi ve skupině s nejvyšší dávkou (1. dávka: 500 mg, 2., 3., a 4. dávka 2000 mg) a zahrnovala 12 pacientů s parciální remisí a jednoho s nodulární parciální remisí. Ve skupině s nejvyšší dávkou byl medián doby do progrese 15,6 týdnů (95% IS: 15; 22,6) v analýze všech pacientů a 23 týdnů (IS: 20; 31) u pacientů reagujících na léčbu. Trvání odpovědi bylo 16 týdnů (IS: 13; 19) a čas do další léčby CLL byl 52,4 týdnů (IS: 36,9; nelze stanovit).
Pediatrická populace
Evropská léková agentura rozhodla o zproštění povinnosti předložit výsledky klinických studií s přípravkem Arzerra u všech podskupin pediatrické populace s chronickou lymfatickou leukémií (viz bod 4.2 pro bližší informace týkající se použití přípravku Arzerra v pediatrii).
5.2 Farmakokinetické vlastnosti
Absorpce
Ofatumumab je podáván prostřednictvím intravenózní infuze, proto se absorpce neuplatňuje. Maximální sérové koncentrace ofatumumabu byly obvykle pozorovány krátce po ukončení infuze. Farmakokinetické údaje byly dostupné od 215 pacientů s refraktemí CLL. Geometrický průměr hodnoty Cmax byl 61 pg/ml po první infuzi (300 mg), po osmé infuzi 1x v týdnu (sedmé infuzi s dávkou 2000 mg) byl geometrický průměr hodnoty Cmax 1391 pg/ml a geometrický průměr hodnoty AUC(0_<x,) 463418 pg.h/ml. Po 12. infuzi (čtvrté infuzi 1x za měsíc s dávkou 2000 mg) byl geometrický průměr hodnoty Cmax 827 pg/ml a geometrický průměr hodnoty AUC(0_<») 203536 pg.h/ml. U pacientů s dříve neléčenou CLL, kteří dostávali ofatumumab a chlorambucil, byl geometrický průměr hodnoty Cmax po první infuzi (s dávkou 300 mg) 52 pg/ml, po infuzi v den 8 s dávkou 1000 mg 241 pg/ml a po infuzi ve čtvrtém měsíčním cyklu s dávkou 1000 mg 285 pg/ml; geometrický průměr hodnoty AUC(0-X) ve čtvrtém cyklu byl 65100 pg.h/ml.
Distribuce
Ofatumumab má malý distribuční objem, s průměrnou hodnotou Vss v rozmezí od 1,7 do 8,1 litru ve všech studiích, bez ohledu na dávku nebo počet infuzí.
Biotransformace
Ofatumumab je protein, jehož očekávaná cesta metabolismu je degradace na malé peptidy a jednotlivé aminokyseliny všudypřítomnými proteolytickými enzymy. Klasické studie biotransformace nebyly provedeny.
Eliminace
Ofatumumab je eliminován dvěma způsoby: na cíli nezávislá cesta jako u dalších IgG molekul a cílem-mediovaná cesta, která souvisí s vazbou na B buňky. Po první infuzi ofatumumabu následuje rychlá a setrvalá deplece CD20+ B buněk, která vede ke snížení počtu CD20+ buněk dostupných pro protilátky z následující infuze. Výsledkem je velmi nízká hodnota clearance ofatumumabu a významně vyšší hodnoty t12 po následných infuzích v porovnání s úvodní infuzí. Po opakovaných týdenních infuzích stoupaly hodnoty AUC a Cmax více, než by se dalo očekávat podle kumulace odvozené na základě údajů získaných z první infuze.
Ve studiích u pacientů s relabující nebo refraktemí CLL byly geometrické průměry hodnot pro CL a ti/2 64 ml/h (rozmezí 4,3 - 1122 ml/h) a 1,3 dne (rozmezí 0,2 - 6,0 dnů) po první infuzi; 8,5 ml/h (rozmezí 1,3 - 41,5 ml/h) a 11,5 dnů (rozmezí 2,3 - 30,6 dne) po čtvrté infuzi; 11,7 ml/h (rozmezí 3,9 -
54,2 ml/h) a 13,6 dne (rozmezí 2,4 - 36,0 dnů) po osmé infuzi a 12,1 ml/h (rozmezí 3,0 - 233 ml/h) a
11.5 dne (rozmezí 1,8 - 36,4 dne) po dvanácté infuzi.
U pacientů s dříve neléčenou CLL, kteří dostávali ofatumumab a chlorambucil, byly geometrické průměry hodnot CL a t1/2 po čtvrté infuzi 15,4 ml/h (rozmezí 4,1 - 146 ml/h) a 18,5 dne (rozmezí 2,7 -
82.6 dne).
Starší pacienti (ve věku 65 let nebo starší)
Neprokázalo se, že by věk byl významným faktorem, který by ovlivňoval farmakokinetiku ofatumumabu ve zkřížené analýze farmakokinetiky populací pacientů ve věku od 21 do 87 let.
Děti a dospívající
U pediatrických pacientů nejsou k dispozici žádné údaje o farmakokinetice.
Pohlaví
Pohlaví mělo ve zkřížených populačních analýzách jen mírný vliv (12 %) na centrální distribuční objem ofatumumabu, u pacientek (48 % pacientů byli muži, 52 % byly ženy) byly pozorovány vyšší hodnoty Cmax a AUC; tyto účinky se nepovažují za klinicky významné a nevycházejí z nich žádná doporučení pro dávkování.
Porucha funkce ledvin
Bylo zjištěno, že počáteční vypočtená clearance kreatininu nebyla významným faktorem, který by měl vliv na farmakokinetiku ofatumumabu ve zkřížených analýzách populací u pacientů s vypočítanými hodnotami clearance kreatininu v rozmezí od 26 do 287 ml/min. U pacientů s mírnou až středně závažnou poruchou funkce ledvin (clearance kreatininu > 30 ml/min) není nutná žádná úprava dávky. U pacientů se závažnou poruchou funkce ledvin (clearance kreatininu < 30 ml/min) jsou k dispozici pouze omezené farmakokinetické údaje.
Porucha funkce jater
Nebyly provedeny žádné formální studie hodnotící vliv poruchy funkce jater. Molekuly IgG1, jako je ofatumumab, jsou odbourávány všudypřítomnými proteolytickými enzymy, které nejsou vázány na jaterní tkáň, proto není pravděpodobné, že by změny jaterních funkcí měly účinek na eliminaci ofatumumabu.
5.3 Předklinické údaje vztahující se k bezpečnosti
Preklinické údaje neukazují na žádné zvláštní riziko pro člověka.
Intravenózní a subkutánní podání opicím vedlo k očekávané depleci počtu periferních B buněk i B buněk lymfoidních tkání bez dalších toxikologických nálezů. Podle předpokladu byl zaznamenán pokles humorální imunitní odpovědi IgG na klíčovou část hemocyaninu, ale nebyly prokázány žádné účinky na reakci hypersenzitivity opožděného typu. U několika zvířat došlo ke zvýšené destrukci červených krvinek, která se pravděpodobně objevila jako následek obalení červených krvinek opic protilátkami proti léku. U těchto opic bylo pozorováno odpovídající zvýšení počtu retikulocytů, což ukazovalo na regenerační odpověď kostní dřeně.
Intravenózní podání ofatumumabu březím opicím druhu cynomolgus (makak) v dávce 100 mg/kg jednou týdně od 20. dne do 50. dne gestace nezpůsobilo mateřskou ani fetální toxicitu ani teratogenitu. Stý den gestace byla pozorována deplece B buněk odpovídající farmakologické aktivitě ofatumumabu v pupečníkové krvi plodu a fetální tkáni sleziny. Studie pre- a postnatálního vývoje nebyly provedeny. Postnatální úprava proto nebyla demonstrována.
Protože ofatumumab je monoklonální protilátka, studie genotoxicity a kancerogenity nebyly s ofatumumabem provedeny.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Arginin
Natrium-acetát (E262)
Chlorid sodný Polysorbát 80 (E433)
Dihydrát dinatrium-edetátu (E386)
Kyselina chlorovodíková (E507) (k úpravě pH)
Voda na injekci
6.2 Inkompatibility
Tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky s výjimkou těch, které jsou uvedeny v bodě 6.6.
6.3 Doba použitelnosti
Injekční lahvička 3 roky.
Naředěná infuze
Chemická a fyzikální stabilita po otevření byla prokázána po dobu 48 hodin při teplotě do 25 °C.
Z mikrobiologického hlediska má být léčivý přípravek použit okamžitě. Není-li použit okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou v odpovědnosti uživatele a obvykle by doba neměla být delší než 24 hodin při 2 až 8 °C, pokud rekonstituce/ředění neproběhlo za kontrolovaných a validovaných aseptických podmínek.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte a převážejte chlazené (2 °C - 8 °C).
Chraňte před mrazem.
Injekční lahvičku uchovávejte v krabičce, aby byl přípravek chráněn před světlem.
Podmínky uchovávání léčivého přípravku po naředění viz bod 6.3.
6.5 Druh obalu a obsah balení
Injekční lahvička z čirého skla typu I s bromobutylovou zátkou neobsahující latex a hliníkovou pertlí obsahující 50 ml koncentrátu pro infuzní roztok.
Přípravek Arzerra je dostupný v balení po 1 injekční lahvičce.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Přípravek Arzerra koncentrát pro infuzní roztok neobsahuje konzervační látky, proto by ředění mělo být prováděno za aseptických podmínek. Naředěný roztok pro infuzi musí být použit do 24 hodin od přípravy. Jakýkoli zbývající naředěný roztok musí být po tomto čase zlikvidován.
• Před naředěním přípravku Arzerra
Zkontrolujte koncentrát přípravku Arzerra na přítomnost jakýchkoli částic a změny barvy před jeho naředěním. Ofatumumab má být bezbarvý až světle žlutý roztok. Neužívejte koncentrát přípravku Arzerra, pokud je přítomná jakákoli změna barvy.
Netřeste lahvičkou s ofatumumabem při této kontrole.
• Jak roztok pro infuzi naředit
Koncentrát přípravku Arzerra se musí před podáním naředit injekčním roztokem chloridu sodného 9 mg/ml (0,9%), a to za použití aseptické techniky.
1000 mg dávka - Použijte 1 lahvičku 1000 mg/50 ml (50 ml celkem, 50 ml v jedné lahvičce)
- odeberte a znehodnoťte 50 ml z vaku s 1000 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9%),
- odeberte 50 ml ofatumumabu z 1000 mg lahvičky a aplikujte je do 1000 ml vaku,
- neprotřepávejte, zamíchejte roztok opatrným převrácením vaku.
2000 mg dávka - Použijte 2 lahvičky (100 ml celkem, 50 ml v jedné lahvičce)
- odeberte a znehodnoťte 100 ml z vaku s 1000 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9%)
- odeberte 50 ml ofatumumabu z každé ze 2 lahviček a aplikujte je do 1000 ml vaku
- neprotřepávejte, zamíchejte roztok opatrným převrácením vaku.
• Jak podávat naředěný roztok
Přípravek Arzerra nesmí být podáván jako intravenózní bolus. Podává se prostřednictvím intravenózní infuzní pumpy.
Infuze musí být podána do 24 hodin od přípravy roztoku. Jakýkoli nepoužitý roztok musí být po uplynutí tohoto času znehodnocen.
Přípravek Arzerra nesmí být smíchán, nebo podáván infuzí s dalšími léčivými přípravky nebo intravenózními roztoky. Propláchněte linku před a po podání ofatumumabu injekčním roztokem chloridu sodného 9 mg/ml (0,9%), abyste se tohoto vyvarovali.
Dříve neléčená CLL:
První infuzi podávejte po dobu minimálně 4,5 hodiny (viz bod 4.2) prostřednictvím periferního žilního přístupu nebo dlouhodobým žilním katetrem podle schématu níže:
Infuze 1: schéma
|
Cas (minuty) |
ml/hodina |
|
0 - 30 |
12 |
|
31 - 60 |
25 |
|
61 - 90 |
50 |
|
91 - 120 |
100 |
|
121 - 150 |
200 |
|
151 - 180 |
300 |
|
180 + |
400 |
Pokud byla první infuze dokončena bez závažných nežádoucích účinků, zbývající infuze (2 - 13) s dávkou 1000 mg mají být podávány po dobu minimálně 4 hodin (viz bod 4.2) prostřednictvím periferního žilního přístupu nebo dlouhodobým žilním katetrem podle schématu níže:
Infuze 2 až 13: schéma
|
Cas (minuty) |
ml/hodina |
|
0 - 30 |
25 |
|
31 - 60 |
50 |
|
61 - 90 |
100 |
|
91 - 120 |
200 |
|
121 + |
400 |
Refrakterní CLL:
První a druhou infuzi podávejte po dobu minimálně 6,5 hodiny (viz bod 4.2) prostřednictvím periferního žilního přístupu nebo dlouhodobým žilním katetrem podle schématu níže:
Infuze 1 a 2: schéma
|
Cas (minuty) |
ml/hodina |
|
0 - 30 |
12 |
|
31 - 60 |
25 |
|
61 - 90 |
50 |
|
91 - 120 |
100 |
|
121 + |
200 |
Pokud byla druhá infuze dokončena bez závažných nežádoucích účinků, zbývající infuze (3 - 12) mají být podávány po dobu minimálně 4 hodin (viz bod 4.2) prostřednictvím periferního žilního přístupu nebo dlouhodobým žilním katetrem podle schématu níže:
Tnfuze 3 až 12: schéma
|
Cas (minuty) |
ml/hodina |
|
0 - 30 |
25 |
|
31 - 60 |
50 |
|
61 - 90 |
100 |
|
91 - 120 |
200 |
|
121 + |
400 |
Pokud se objeví jakékoli nežádoucí účinky, rychlost infuze je třeba zpomalit (viz bod 4.2).
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/10/625/003
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 19/04/2010
Datum posledního prodloužení registrace: 16/01/2014
10. DATUM REVIZE TEXTU
Podrobné informace o tomto přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/.
A. VÝROBCI BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCI BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobců biologické léčivé látky
Lonza Biologics plc.
228 Bath Road Slough Berks SL1 4DX Velká Británie
Lonza Biologics Inc.
101 International Drive Portsmouth, NH 03801-2815 Spojené státy americké
Název a adresa výrobců odpovědných za propouštění šarží
Glaxo Operations UK Ltd.
Harmire Road Barnard Castle Durham, DL 12 8DT Velká Británie
Novartis Pharmaceuticals UK Limited
Frimley Business Park
Frimley
Camberley, Surrey GU16 7SR Velká Británie
Novartis Pharma GmbH Roonstrasse 25 D-90429 Norimberk Německo
V příbalové informaci k léčivému přípravku musí být uveden název a adresa výrobce odpovědného za propouštění dané šarže.
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz Příloha I: Souhrn údajů o přípravku, bod 4.2)
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace, a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Arzerra 100 mg koncentrát pro infuzní roztok Ofatumumabum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jeden ml obsahuje 20 mg ofatumumabum.
Jedna injekční lahvička obsahuje 100 mg ofatumumabu v 5 ml.
3. SEZNAM POMOCNÝCH LÁTEK
Arginin, natrium-acetát (E262), chlorid sodný, polysorbát 80 (E433), dihydrát dinatrium-edetátu (E386), kyselina chlorovodíková (E507), voda na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Koncentrát pro infuzní roztok 100 mg/5 ml 3 injekční lahvičky
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Intravenózní podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
Uchovávejte a převážejte chlazené.
Chraňte před mrazem.
Injekční lahvičku uchovávejte v krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/10/625/001 13 ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
Nevyžaduje se - odůvodnění přijato
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Arzerra 100 mg sterilní koncentrát
Ofatumumabum
i.v.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
100 mg/5 ml
6. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Arzerra 1000 mg koncentrát pro infuzní roztok Ofatumumabum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jeden ml obsahuje 20 mg ofatumumabum.
Jedna injekční lahvička obsahuje 1000 mg ofatumumabu v 50 ml.
3. SEZNAM POMOCNÝCH LÁTEK
Arginin, natrium-acetát (E262), chlorid sodný, polysorbát 80 (E433), dihydrát dinatrium-edetátu (E386), kyselina chlorovodíková (E507), voda na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Koncentrát pro infuzní roztok 1000 mg/50 ml 1 injekční lahvička
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Intravenózní podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
Uchovávejte a převážejte chlazené.
Chraňte před mrazem.
Injekční lahvičku uchovávejte v krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKU NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/10/625/003 13 ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVE PÍSMU
Nevyžaduje se - odůvodnění přijato
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Arzerra 1000 mg sterilní koncentrát
Ofatumumabum
i.v.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
1000 mg/50 ml
6. JINÉ
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro uživatele
Arzerra 100 mg koncentrát pro infuzní roztok Arzerra 1000 mg koncentrát pro infuzní roztok
Ofatumumabum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo zdravotní sestry.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Arzerra a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Arzerra používat
3. Jak se přípravek Arzerra používá
4. Možné nežádoucí účinky
5. Jak přípravek Arzerra uchovávat
6. Obsah balení a další informace
1. Co je přípravek Arzerra a k čemu se používá
Přípravek Arzerra obsahuje ofatumumab, který patří do skupiny léků nazývaných monoklonální protilátky.
Přípravek Arzerra je určen k léčbě chronické lymfocytární leukémie (CLL). CLL je zhoubné nádorové onemocnění krve, které postihuje určitý typ bílých krvinek nazývaných lymfocyty. Lymfocyty se množí velmi rychle a žijí velmi dlouho, proto je jejich počet v krvi vysoký. Toto onemocnění může rovněž ovlivnit další orgány v těle. Protilátky v přípravku Arzerra rozpoznají látky na povrchu lymfocytů a způsobí jejich smrt.
2. Čemu musíte věnovat pozornost, než začnete přípravek Arzerra používat Nepoužívejte přípravek Arzerra:
• jestliže jste alergický(á) (přecitlivělý(á)) na ofatumumab nebo na kteroukoli další složku přípravku Arzerra (uvedenou v bodě 6. Obsah balení a další informace).
Pokud si myslíte, že se Vás to týká, poraďte se se svým lékařem.
Upozornění a opatření
Před použitím přípravku Arzerra se poraďte se svým lékařem nebo zdravotní sestrou:
• pokud máte onemocnění srdce,
• pokud máte onemocnění plic.
Pokud si myslíte, že se Vás to týká, poraďte se se svým lékařem. Při léčbě přípravkem Arzerra může být nutné provést více lékařských prohlídek.
Váš lékař může před zahájením léčby přípravkem Arzerra provést vyšetření ke stanovení množství solí (iontů), např. hořčíku a draslíku, ve Vaší krvi. Váš lékař může léčit nerovnováhu solí v těle.
Očkování a přípravek Arzerra
Pokud máte podstoupit jakékoli očkování, řekněte svému lékaři nebo osobě, která Vám bude vakcínu podávat, že se léčíte přípravkem Arzerra. Odpověď na očkování u Vás může být oslabená a nemusí Vás plně ochránit.
Hepatitida B
Před zahájením léčby přípravkem Arzerra by Vám měly být provedeny testy na hepatitidu B (žloutenku typu B, onemocnění jater). Pokud jste v minulosti měl(a) hepatitidu B, může přípravek Arzerra způsobit opětovné propuknutí hepatitidy B. Váš lékař tomu může předcházet tím, že Vám podá odpovídající antivirové léky.
Pokud máte nebo jste někdy měl(a) hepatitidu B, sdělte to svému lékaři předtím, než Vám bude přípravek Arzerra podán.
Reakce související s infuzí
Léky tohoto typu (monoklonálníprotilátky), pokud jsou aplikovány (podány) do těla, mohou způsobovat reakce související s infuzí. Dostanete léky, jako jsou antihistaminika (léky proti alergii), steroidy (hormony) nebo analgetika (léky proti bolesti), které pomohou tuto reakci zmírnit. Viz bod 4.
Možné nežádoucí účinky.
Pokud si myslíte, že jste podobnou reakci měl(a) již v minulosti, řekněte to svému lékaři dříve, než Vám bude přípravek Arzerra podán.
Progresivní multifokální leukoencefalopatie (PML)
Progresivní multifokální leukoencefalopatie (PML) je závažný a život ohrožující stav postihující mozek, který byl hlášen v souvislosti s podáváním léčivých přípravků podobných přípravku Arzerra. Řekněte okamžitě svému lékaři, pokud pociťujete ztrátu paměti, máte obtíže s myšlením, potíže s chůzí nebo pozorujete ztrátu zraku. Pokud jste tyto příznaky měl (a) v době před započetím léčby přípravkem Arzerra, informujte svého lékaře ihned o změnách v těchto příznacích.
Děti a dospívající
Není známo, jak přípravek Arzerra působí u dětí a dospívajících. Proto není použití přípravku Arzerra u dětí a dospívajících doporučeno.
Další léčivé přípravky a přípravek Arzerra
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat. Toto se týká i rostlinných léčivých přípravků a přípravků dostupných bez lékařského předpisu.
Těhotenství, kojení a fertilita
Přípravek Arzerra se obvykle nedoporučuje v průběhu těhotenství. O bezpečnosti přípravku Arzerra u těhotných žen nejsou k dispozici žádné údaje.
• Pokud jste těhotná, nebo těhotenství plánujete, řekněte to svému lékaři. Lékař zváží prospěch užívání přípravku Arzerra v těhotenství pro Vás oproti riziku pro Vaše dítě.
• Používejte vhodnou metodu antikoncepce, abyste se těhotenství vyvarovala v době, kdy jste léčena přípravkem Arzerra a 12 měsíců po ukončení léčby.
• Pokud v průběhu léčby přípravkem Arzerra otěhotníte, řekněte to svému lékaři.
Není známo, zda složky přípravku Arzerra procházejí do mateřského mléka. V průběhu léčby přípravkem Arzerra a 12 měsíců po jeho ukončení se kojení nedoporučuje.
Řízení dopravních prostředků a obsluha strojů
Není pravděpodobné, že by přípravek Arzerra ovlivňoval schopnost řídit a obsluhovat stroje.
Přípravek Arzerra obsahuje sodík
Přípravek Arzerra obsahuje 34,8 mg sodíku v jedné dávce 300 mg, 116 mg sodíku v jedné dávce 1000 mg a 232 mg sodíku v jedné dávce 2000 mg. Toto je potřeba vzít v úvahu u pacientů s řízenou sodíkovou dietou.
3. Jak se přípravek Arzerra podává
Jestliže máte jakékoli otázky týkající se podávání přípravku Arzerra, zeptejte se lékaře, který Vám bude infuzi podávat.
Obvyklá dávka
Obvyklá dávka přípravku Arzerra při první infuzi je 300 mg. Tato dávka bude zvýšena obvykle na 1000 mg nebo na 2000 mg u dalších infuzí.
Jak se podává
Přípravek Arzerra se podává do žíly (intravenózně) pomocí infuze v průběhu několika hodin.
Jestliže jste se dříve neléčil(a) s CLL, podstoupíte léčbu maximálně 13 infuzemi. Bude Vám podána první infuze, po které bude o 7 dní později následovat druhá infuze. Zbývající infuze Vám budou podávány jednou měsíčně, po dobu nejdéle 11 měsíců.
Jestliže jste se již dříve léčil(a) s CLL, obvykle podstoupíte léčbu 12 infuzemi. Infuze Vám budou podávány jednou týdně po dobu osmi týdnů. Poté bude následovat 4 - 5týdenní pauza. Zbývající infuze se podávají jednou měsíčně po dobu následujících 4 měsíců.
Léky podávané před každou infuzí
Před každou infuzí přípravku Arzerra Vám bude podána premedikace - léky, které pomáhají snížit jakoukoli reakci na infuzi. To mohou být antihistaminika, steroidy a analgetika. Budete pečlivě sledován(a) a pokud se u Vás objeví jakákoli reakce, bude se léčit.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Reakce související s infuzí
Léky tohoto typu (monoklonálníprotilátky) mohou způsobovat reakce související s infuzí, které jsou občas závažné a mohou být příčinou úmrtí. Nejpravděpodobněji se objeví při první léčbě.
Velmi časté nežádoucí účinky související s infuzí (mohou se objevit u více než 1 osoby z 10):
• pocit nevolnosti (nauzea),
• vysoká teplota,
• kožní vyrážka.
Časté nežádoucí účinky související s infuzí (mohou se objevit až u 1 osoby z 10):
• alergické reakce, občas závažné s otokem obličeje nebo úst, které způsobují potíže s dechem
(anafylaktoidní reakce),
• dušnost, zkrácený dech, tíseň (pocit tlaku) na hrudi, kašel,
• nízký krevní tlak (může způsobit závratě při vstávání),
• návaly,
• výrazné pocení,
• třes a zimnice,
• zrychlená srdeční frekvence,
• průjem,
• bolest zad,
• vysoký krevní tlak,
• svědivá vystouplá vyrážka (kopřivka),
• bolest nebo podráždění v krku,
• nedostatek energie,
• ucpaný nos.
Méně časté příznaky reakce související s infuzí (mohou se objevit až u 1 osoby ze 100):
• tekutina v plicích (edémplic) způsobující ztížené dýchání,
• pomalý srdeční rytmus.
Pokud se u Vás objeví kterýkoli z těchto příznaků, řekněte to okamžitě lékaři nebo zdravotní sestře.
Velmi časté nežádoucí účinky
Mohou objevit u více než 1 osoby z 10:
• infekce plic nebo dýchacích cest, jako např. pneumonie (zápal plic),
• infekce ucha, nosu nebo krku.
Velmi časté nežádoucí účinky, které se mohou objevit v krevních testech:
• nízký počet bílých krvinek (neutropenie),
• nízký počet červených krvinek (anémie).
Časté nežádoucí účinky
Mohou objevit až u 1 osoby z 10:
• horečka způsobená infekcí a nízkým počtem bílých krvinek,
• infekce v krvi,
• infekce močových cest,
• pásový opar,
• opar.
Časté nežádoucí účinky, které se mohou objevit v krevních testech:
• nízký počet krevních destiček v krvi (buňky, které pomáhají srážení krve).
Méně časté nežádoucí účinky
Mohou se objevit až u 1 osoby ze 100:
• zablokování střeva, které můžete vnímat jako bolest břicha.
Pokud u Vás přetrvává bolest břicha, vyhledejte co nejdříve svého lékaře.
• zvýšení hladin draslíku, fosfátu a kyseliny močové v krvi, což může způsobit potíže s ledvinami (syndrom lýzy tumoru).
Příznaky tohoto onemocnění zahrnují:
• sníženou tvorbu moči,
• svalové křeče.
Pokud zaznamenáte jakýkoli z těchto příznaků, vyhledejte co nejdříve svého lékaře.
Méně časté nežádoucí účinky, které se mohou projevit v krevních testech:
• poruchy srážlivosti krve,
• selhání tvorby červených a bílých krvinek v kostní dřeni.
Vzácné nežádoucí účinky
Mohou se objevit až u 1 osoby z 1000:
• infekce virem hepatitidy B nebo reaktivace viru hepatitidy B.
Pokud se u Vás objeví nežádoucí účinky
Pokud se kterýkoli z nežádoucích účinků vyskytne v závažné míře, nebo pokud si všimnete jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci, prosím, sdělte to svému lékaři nebo zdravotní sestře.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Arzerra uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a štítku injekční lahvičky. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte a převážejte chlazené (2 °C - 8 °C).
Chraňte před mrazem.
Injekční lahvičku uchovávejte v krabičce, aby byl přípravek chráněn před světlem.
Naředěný infuzní roztok uchovávejte mezi 2 °C až 8 °C a použijte do 24 hodin. Jakýkoli nespotřebovaný infuzní roztok musí být za 24 hodin od přípravy vyřazen.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak máte likvidovat přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co přípravek Arzerra obsahuje
- Léčivou látkou je ofatumumabum. Jeden ml koncentrátu obsahuje 20 mg ofatumumabu.
- Pomocnými látkami jsou arginin, natrium-acetát (E262), chlorid sodný, polysorbát 80 (E433), dihydrát dinatrium-edetátu (E386), kyselina chlorovodíková (E507) (k úpravě pH), voda na injekci.
Jak přípravek Arzerra vypadá a co obsahuje toto balení
Arzerra je bezbarvý až světle žlutý koncentrát pro infuzní roztok.
Přípravek Arzerra 100 mg je dostupný v balení obsahujícím 3 injekční lahvičky. Každá skleněná injekční lahvička je uzavřena gumovou zátkou neobsahující latex a hliníkovou pertlí, a obsahuje 5 ml koncentrátu (100 mg ofatumumabu).
Přípravek Arzerra 1000 mg je dostupný v balení obsahujícím 1 injekční lahvičku. Každá skleněná injekční lahvička je uzavřena gumovou zátkou neobsahující latex a hliníkovou pertlí a obsahuje 50 ml koncentrátu (1000 mg ofatumumabu).
Držitel rozhodnutí o registraci
Novartis Europharm Limited Frimley Business Park Camberley GU16 7SR Velká Británie
Výrobce
Glaxo Operations UK Limited (Trading as Glaxo Wellcome Operations), Harmire Road, Barnard Castle, County Durham, DL12 8DT, Velká Británie.
Novartis Pharmaceuticals UK Limited, Frimley Business Park, Frimley, Camberley, Surrey GU16 7SR, Velká Británie
Novartis Pharma GmbH, Roonstrasse 25, D-90429 Norimberk, Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgie/Belgique/Belgien
Novartis Pharma N.V.
Tél/Tel: +32 2 246 16 11
EtnrapHH
Novartis Pharma Services Inc. Ten: +359 2 489 98 28
Česká republika
Novartis s.r.o.
Tel: +420 225 775 111
Danmark
Novartis Healthcare A/S Tlf: +45 39 16 84 00
Deutschland
Novartis Pharma GmbH Tel: +49 911 273 0
Lietuva
Novartis Pharma Services Inc. Tel: +370 5 269 16 50
Luxembourg/Luxemburg
Novartis Pharma N.V.
Tél/Tel: +32 2 246 16 11
Magyarország
Novartis Hungária Kft. Pharma Tel.: +36 1 457 65 00
Malta
Novartis Pharma Services Inc. Tel: +356 2122 2872
Nederland
Novartis Pharma B.V.
Tel: +31 26 37 82 555
Eesti
Norge
Novartis Norge AS Tlf: +47 23 05 20 00
Novartis Pharma Services Inc. Tel: +372 66 30 810
|
EXXáSa Novartis (Hellas) A.E.B.E. Tn^: +30 210 281 17 12 |
Osterreich Novartis Pharma GmbH Tel: +43 1 86 6570 |
|
Espaňa Novartis Farmacéutica, S.A. Tel: +34 93 306 42 00 |
Polska Novartis Poland Sp. z o.o. Tel.: +48 22 375 4888 |
|
France Novartis Pharma S.A.S. Tél: +33 1 55 47 66 00 |
Portugal Novartis Farma - Produtos Farmaceuticos, S.A Tel: +351 21 000 8600 |
|
Hrvatska Novartis Hrvatska d.o.o. Tel. +385 1 6274 220 |
Románia Novartis Pharma Services Romania SRL Tel: +40 21 31299 01 |
|
Ireland Novartis Ireland Limited Tel: +353 1 260 12 55 |
Slovenija Novartis Pharma Services Inc. Tel: +386 1 300 75 50 |
|
Ísland Vistor hf. Sími: +354 535 7000 |
Slovenská republika Novartis Slovakia s.r.o. Tel: +421 2 5542 5439 |
|
Italia Novartis Farma S.p.A. Tel: +39 02 96 54 1 |
Suomi/Finland Novartis Finland Oy Puh/Tel: +358 (0)10 6133 200 |
|
Kúnpoq Novartis Pharma Services Inc. Tn^: +357 22 690 690 |
Sverige Novartis Sverige AB Tel: +46 8 732 32 00 |
|
Latvija Novartis Pharma Services Inc. Tel: +371 67 887 070 |
United Kingdom Novartis Pharmaceuticals UK Ltd. Tel: +44 1276 698370 |
Tato příbalová informace byla naposledy revidována
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/.
Následující informace jsou určeny pouze pro zdravotnické pracovníky:
1) Před naředěním přípravku Arzerra
Zkontrolujte přítomnost jakýchkoli částic a změnu zbarvení koncentrátu přípravku Arzerra před jeho naředěním. Ofatumumab má být bezbarvý až světle žlutý roztok. Neužívejte koncentrát přípravku Arzerra, pokud je přítomná jakákoli změna barvy.
Netřeste lahvičkou s ofatumumabem při této kontrole.
2) Jak roztok pro infuzi naředit
Koncentrát přípravku Arzerra se musí před podáním naředit injekčním roztokem chloridu sodného 9 mg/ml (0,9%), a to za použití aseptické techniky.
300 mg dávka - Použijte 3 lahvičky 100 mg/5 ml (15 ml celkově, 5 ml v jedné lahvičce)
• odeberte a znehodnoťte 15 ml z vaku s 1000 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9%),
• odeberte 5 ml ofatumumabu z každé ze 3 100 mg lahviček a aplikujte je do 1000 ml vaku,
• neprotřepávejte, zamíchejte roztok opatrným převrácením vaku.
1000 mg dávka - Použijte 1 lahvičku 1000 mg/50 ml (50 ml celkem, 50 ml v jedné lahvičce)
• odeberte a znehodnoťte 50 ml z vaku s 1000 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9%),
• odeberte 50 ml ofatumumabu z 1000 mg lahvičky a aplikujte je do 1000 ml vaku,
• neprotřepávejte, zamíchejte roztok opatrným převrácením vaku.
2000 mg dávka - Použijte 2 lahvičky 1000 mg/50 ml (100 ml celkem, 50 ml v jedné lahvičce)
• odeberte a znehodnoťte 100 ml z vaku s 1000 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9%),
• odeberte 50 ml ofatumumabu z každé ze 2 1000 mg lahviček a aplikujte je do 1000 ml vaku,
• neprotřepávejte, zamíchejte roztok opatrným převrácením vaku.
3) Jak podávat naředěný roztok
Přípravek Arzerra nesmí být podáván jako intravenózní bolus. Podává se prostřednictvím intravenózní infuzní pumpy.
Infuze musí být podána do 24 hodin od přípravy roztoku. Jakýkoli nepoužitý roztok musí být po uplynutí tohoto času znehodnocen.
Přípravek Arzerra nesmí být smíchán ani podáván infuzí s dalšími léčivými přípravky nebo
intravenózními roztoky. Před a po podání ofatumumabu propláchněte linku infuzním roztokem chloridu sodného 9 mg/ml (0,9%), abyste se tohoto vyvarovali.
Dříve neléčená CLL:
První infuzi podávejte po dobu minimálně 4,5 hodiny (viz bod 4.2 SPC) prostřednictvím periferního žilního přístupu nebo dlouhodobým žilním katetrem podle schématu níže:
Infuze 1: schéma
|
Cas (minuty) |
ml/hodina |
|
0 - 30 |
12 |
|
31 - 60 |
25 |
|
61 - 90 |
50 |
|
91 - 120 |
100 |
|
121 - 150 |
200 |
|
151 - 180 |
300 |
|
180 + |
400 |
Pokud byla první infuze dokončena bez závažných nežádoucích účinků, zbývající infuze (2 - 13) s dávkou 1000 mg mají být podávány po dobu minimálně 4 hodin (viz bod 4.2 SPC) prostřednictvím periferního žilního přístupu nebo dlouhodobým žilním katetrem podle schématu níže:
Infuze 2 až 13: schéma
|
Cas (minuty) |
ml/hodina |
|
0 - 30 |
25 |
|
31 - 60 |
50 |
|
61 - 90 |
100 |
|
91 - 120 |
200 |
|
121 + |
400 |
Refrakterní CLL:
První a druhou infuzi podávejte po dobu minimálně 6,5 hodiny (viz bod 4.2 SPC) prostřednictvím periferního žilního přístupu nebo dlouhodobým žilním katetrem podle schématu níže:
Infuze 1 a 2: schéma
|
Cas (minuty) |
ml/hodina |
|
0 - 30 |
12 |
|
31 - 60 |
25 |
|
61 - 90 |
50 |
|
91 - 120 |
100 |
|
121 + |
200 |
Pokud byla druhá infuze dokončena bez závažných nežádoucích účinků, zbývající infuze (3 - 12) mohou být podávány po dobu minimálně 4 hodin (viz bod 4.2 SPC) prostřednictvím periferního žilního přístupu nebo dlouhodobým žilním katetrem podle schématu níže:
Infuze 3 až 12: schéma
|
Cas (minuty) |
ml/hodina |
|
0 - 30 |
25 |
|
31 - 60 |
50 |
|
61 - 90 |
100 |
|
91 - 120 |
200 |
|
121 + |
400 |
Pokud se objeví jakékoli nežádoucí účinky, rychlost infuze má být zpomalena (viz bod 4.2 SPC).
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
65