Apidra Solostar 100 Jednotek/Ml
SOUHRN ÚDAJŮ O PŘÍPRAVKU
PŘÍLOHA I
Apidra 100 jednotek/ml injekční roztok v injekční lahvičce
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden ml obsahuje 100 jednotek insulinum glulisinum (odpovídá 3,49 mg).
Jedna injekční lahvička obsahuje 10 ml injekčního roztoku, což odpovídá 1000 jednotek.
Inzulín glulisin se vyrábí technologií rekombinace DNA využitím kmenů bakterie Escherichia coli. Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok v injekční lahvičce.
Čirý bezbarvý vodný roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba dospělých, mladistvých a dětí starších 6 let s diabetes mellitus, kde je nutná léčba inzulínem.
4.2 Dávkování a způsob podání
Dávkování
Síla tohoto přípravku se uvádí v jednotkách. Tyto jednotky se vztahují výhradně k přípravku Apidra a liší se od m.j. (IU) nebo jednotek používaných k vyjádření síly jiných inzulínových analogů (viz bod 5.1).
Apidra by se měla podávat v režimech, které zahrnují středně nebo dlouhodobě účinkující inzulín nebo analog bazálního inzulínu a může být podávána s perorálními antidiabetiky.
Dávka přípravku Apidra by měla být upravena individuálně.
Zvláštní populace
Porucha funkce ledvin
Farmakokinetické vlastnosti inzulínu glulisin jsou obecně u pacientů s renálním poškozením zachovány. Potřeba inzulínu však může být při poškození renálních funkcí snížena (viz bod 5.2).
Porucha funkce jater
Farmakokinetické vlastnosti inzulínu glulisin nebyly zkoumány u pacientů se sníženou funkcí jater.
U pacientů se zhoršením funkce jater může být potřeba inzulínu snížena z důvodu snížené kapacity glukoneogeneze a poklesu inzulínového metabolizmu.
Starší pacienti
U starších pacientů s diabetes mellitus jsou dostupné omezené farmakokinetické údaje. Zhoršení renálních funkcí může vést k poklesu potřeby inzulínu.
Pediatrická populace
Nej sou dostatečné klinické informace o použití přípravku Apidra u dětí mladších 6 let.
Způsob podání
Intravenózní podání
Apidra může být podávána intravenózně. Toto podání by měl provádět zdravotník.
Apidra se nesmí míchat s glukózou, Ringerovým roztokem ani s žádným jiným inzulínem.
Subkutánní podání
Apidra by se měla podávat subkutánní injekcí krátce (0-15 min) před jídlem nebo brzy po jídle, nebo kontinuální subkutánní infuzní pumpou.
Apidra by se měla podávat subkutánně do břišní stěny, stehna, deltového svalu nebo kontinuální infuzí do břišní stěny. Místa vpichu injekce a infuze v rámci jedné injekční oblasti (břicho, stehno nebo deltový sval) je nutno s každou injekcí střídat. Míra absorpce a následně nástup a trvání účinku mohou být ovlivněny místem vpichu, cvičením a dalšími faktory. Subkutánní injekce do břišní stěny zajišťuje o něco rychlejší absorpci než ostatní injekční místa (viz bod 5.2).
Musí se dávat pozor, aby bylo zajištěno, že nebyla propíchnuta krevní céva. Místo vpichu se po injekci nesmí masírovat. Pacienti musí být poučeni, aby používali správné injekční techniky.
Pokud je použita do inzulínové infuzní pumpy, nesmí se Apidra mísit s rozpouštědlem nebo s jakýmkoliv jiným inzulínem.
Míchání s inzulíny
Při podávání v subkutánní injekci se Apidra nesmí mísit s žádnými jinými léčivými přípravky s výjimkou NPH inzulínu.
Další podrobnosti o zacházení viz bod 6.6.
Kontinuální subkutánní infuze inzulínu
Přípravek Apidra je možné podávat pomocí systému pumpy (CSII) (Continuous Subcutaneous Insulin Infusion, kontinuální subkutánní infuze inzulínu), která je s příslušnými katetry a zásobníky vhodná pro infuzi inzulínu. Pacienti používající CSII mají být důkladně poučeni o používání systému pumpy.
Infuzní set a zásobník používaný pro přípravek Apidra musí být aseptickou technikou vyměňován nejméně každých 48 hodin. Tyto instrukce se mohou lišit od obecných pokynů uvedených v návodu k pumpě. Je důležité, aby se pacienti užívající přípravek Apidra řídili pouze instrukcemi specifickými pro přípravek Apidra. Nedodržení specifických pokynů pro podávání přípravku Apidra může vést k závažným nežádoucím účinkům.
Při používání v subkutánní infuzní inzulinové pumpě se Apidra nesmí mísit s ředicími roztoky ani s žádným jiným inzulínem.
Pacienti, jimž je přípravek Apidra podáván pomocí CSII, musí mít k dispozici alternativní systém pro aplikaci inzulínu pro případ selhání pumpy (viz bod 4.4 a 4.8).
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1. Hypoglykémie.
4.4 Zvláštní upozornění a opatření pro použití
Převedení pacienta na jiný typ nebo značku inzulínu musí být prováděno za přísného lékařského dohledu. Změny síly, značky (výrobce), typu (rozpustný rychle účinkující, neutral protamin Hagedorn [NPH], lente, dlouhodobě působící, atd.), původu (zvířecí, lidský, analog lidského inzulinu) a/nebo způsobu výroby mohou vést k nutnosti změnit dávku. Současná léčba perorálními antidiabetiky může vyžadovat úpravu dávkování.
Hyperglykémie
Podávání neadekvátních dávek nebo přerušení léčby zejména u inzulíndependentních diabetiků může vést k hyperglykémii a diabetické ketoacidóze; tedy k okolnostem, které jsou potenciálně letální.
Hypoglykémie
Doba výskytu hypoglykémie závisí na profilu účinku podaného inzulínu, a proto se může měnit, pokud se mění režim léčby.
Okolnosti, za kterých mohou být včasné varovné symptomy pozměněny nebo být méně výrazné, zahrnují dlouhou anamnézu diabetu, zintenzivnění léčby inzulínem, diabetickou neuropatii, léčivé přípravky jako betablokátory nebo převedení ze zvířecího inzulínu na lidský inzulín.
Úprava dávky může být nezbytná u pacientů, kteří zvýší svou fyzickou aktivitu nebo změní svůj obvyklý stravovací řád. Cvičení prováděné okamžitě po jídle může zvýšit riziko hypoglykémie.
V porovnání s rozpustným lidským inzulínem, pokud se po injekci analogu rychlého inzulínu objeví hypoglykémie, tato se může objevit dříve.
Nekontrolovaná hypoglykemická nebo hyperglykemická reakce může způsobit bezvědomí, kóma nebo smrt.
Potřeba inzulínu může být pozměněna během nemoci nebo emočního rozrušení.
Chyby v medikaci
Byly hlášeny chyby v léčbě, kdy byly omylem podány jiné, zejména dlouhodobě působící, inzulíny namísto inzulínu glulisin. Před každým podáním injekce musí být vždy zkontrolován štítek inzulínu, aby nedošlo k záměně inzulínu glulisin a jiných inzulínů.
Kontinuální subkutánní infuze inzulínu
Závada inzulinové pumpy nebo infuzního setu nebo chyby při manipulaci mohou rychle přivodit hyperglykémii, ketózu a diabetickou ketoacidózu. Je nutné včas zjistit a odstranit příčinu hyperglykémie nebo ketózy nebo diabetické ketoacidózy.
Při podávání přípravku Apidra systémem skubkutánní inzulinové infuzní pumpy byly hlášeny případy diabetické ketoacidózy. Většina z těchto případů souvisela s nesprávnou manipulací nebo se selháním systému pumpy.
V mezidobí může být nutné podávat přípravek Apidra ve formě subkutánní injekce. Pacienti, kteří používají k léčbě kontinuální subkutánní infuzní pumpu, musí být zaškoleni v injekčním podávání inzulínu a musí mít k dispozici alternativní systém pro aplikaci inzulínu pro případ selhání pumpy (viz body 4.2 a 4.8).
Pomocné látky
Tento léčivý přípravek obsahuje méně než 1 mmol (23 mg) sodíku v jedné dávce, tj. v podstatě sodík neobsahuje.
Apidra obsahuje metakresol, který může způsobovat alergické reakce.
Kombinace přípravku Apidra s pioglitazonem
Zejména u pacientů s rizikovými faktory pro vznik srdečního selhání byly při podávání pioglitazonu v kombinaci s inzulínem hlášeny případy srdečního selhání. To je třeba mít na paměti, pokud je zvažována léčba přípravkem Apidra v kombinaci s pioglitazonem. Jestliže je tato kombinace použita, je třeba pacienty sledovat, zda se u nich neobjevují známky a příznaky srdečního selhání, zvýšení hmotnosti a edém. Dojde-li k jakémukoli zhoršení srdečních příznaků, je zapotřebí léčbu pioglitazonem ukončit.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Studie sledující farmakokinetické interakce nebyly prováděny. Na základě empirických znalostí u podobných léčivých přípravků je pravděpodobné, že se neobjeví klinicky relevantní farmakokinetické interakce.
Rada látek ovlivňuje metabolizmus glukózy a může vyžadovat úpravu dávky inzulínu glulisin a zvláště pečlivé sledování.
Mezi látky, které mohou zvýšit hypoglykemizující účinek a zvýšit náchylnost k hypoglykémii, patří perorální antidiabetika, ACE inhibitory, disopyramid, fibráty, fluoxetin, inhibitory monoaminooxidázy (MAOI), pentoxifylin, propoxyfen, salicyláty a sulfonamidová antibiotika.
Mezi látky, které mohou snižovat hypoglykemizující účinek, patří kortikosteroidy, danazol, diazoxid, diuretika, glukagon, isoniazid, deriváty fenothiazinu, somatotropin, sympatomimetika (např. epinefrin [adrenalin], salbutamol, terbutalin), tyreoidální hormony, estrogeny a progestogeny (např. perorální kontraceptiva), inhibitory proteáz a atypické antipsychotické léky (např. olanzapin a klozapin).
Betablokátory, klonidin, soli lithia nebo alkohol mohou zesílit nebo zeslabit hypoglykemizující účinek inzulínu. Pentamidin může vyvolat hypoglykémii, po které může někdy následovat hyperglykémie.
Navíc vlivem sympatolytických léčivých přípravků jako jsou betablokátory, klonidin, guanethidin a reserpin, mohou být známky adrenergní kontraregulace sníženy nebo mohou zcela chybět.
4.6 Těhotenství a kojení
Nejsou k dispozici žádné nebo jen omezené údaje (výsledky u méně než 300 těhotenství) o použití inzulínu glulisin u těhotných žen.
Reprodukční studie na zvířatech neodhalily žádné rozdíly mezi inzulínem glulisin a lidským inzulínem co se týče těhotenství, embryofetálního vývoje, porodu nebo postnatálního vývoje (viz bod 5.3).
Při předepisování těhotným ženám je nutno postupovat opatrně. Pečlivé monitorování kompenzace glykémie je nutné.
Je nutné, aby u pacientek s preexistujícím diabetem nebo s těhotenským diabetem byla udržována v průběhu těhotenství dobrá metabolická kontrola. Požadavky na inzulín mohou klesat během prvního trimestru a obecně se zvyšují během druhého a třetího trimestru. Okamžitě po porodu potřeba inzulínu rapidně poklesne.
Kojení
Není známo, zda je inzulín glulisin vylučován do mateřského mléka, ale obecně inzulín nepřestupuje do mateřského mléka a není absorbován po perorálním podání.
Kojící matky mohou vyžadovat úpravu dávky inzulínu a diety.
Fertilita
Reprodukční studie u zvířat neodhalily žádné nežádoucí účinky inzulínu glulisinu na fertilitu.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Pacientova schopnost koncentrace a reakce může být zhoršena následkem hypoglykémie, hyperglykémie nebo například následkem poruchy zraku. To může představovat riziko v situacích, kde jsou tyto schopnosti zvláště důležité (např. při řízení vozidla nebo obsluze strojů).
Pacient by měl být obeznámen s opatřeními, zabraňujícími vzniku hypoglykémie během řízení. To je důležité zejména u těch pacientů, kteří mají sníženou nebo chybějící vnímavost k varovným příznakům hypoglykémie nebo mají časté epizody hypoglykémie. Je třeba zvážit, zda řízení nebo obsluha strojů jsou za těchto okolností vhodné.
4.8 Nežádoucí účinky
Souhrnný bezpečnostní profil
Hypoglykémie, nejčastější nežádoucí účinek inzulínové terapie, se může objevit, jestliže dávka inzulínu je vzhledem k jeho potřebě příliš vysoká.
Tabulkový přehled nežádoucích účinků
Následující nežádoucí účinky, které se vyskytly během klinických hodnocení, byly seřazeny níže systematicky podle skupiny orgánů a podle snižujícího se výskytu (velmi časté: >1/10; časté: >1/100 až <1/10; méně časté: >1/1 000 až <1/100; vzácné >1/10 000 až <1/1 000; velmi vzácné: <1/10 000), není známo (z dostupných údajů nelze určit).
V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
|
MedDRA Třídy orgánových systémů |
Velmi časté |
Časté |
Méně časté |
Vzácné |
Není známo |
|
Poruchy metabolismu a výživy |
Hypoglykémie |
Hyperglykémie (potenciálně vedoucí k diabetické ketoacidóze (1)) | |||
|
Poruchy kůže a podkožní tkáně |
Reakce v místě vpichu Lokální hypersenzitivní reakce |
Lipodystrofie | |||
|
Celkové poruchy a reakce v místě aplikace |
Systémová hypersenzitivní reakce |
(1) Většina případů souvisela s chybnou manipulací nebo se selháním systému pumpy, když byl přípravek Apidra podáván pomocí CSII.
Popis vybraných nežádoucích účinků
Poruchy metabolismu a výživy
Symptomy hypoglykémie se obvykle objeví náhle. Mohou zahrnovat studený pot, chladnou bledou kůži, únavu, nervozitu nebo třes, úzkost, neobvyklou únavu nebo slabost, zmatenost, poruchy koncentrace, ospalost, vlčí hlad, poruchy vidění, bolest hlavy, nauseu a palpitace.
Hypoglykémie se může stát závažnou a může vést k bezvědomí a/nebo křečím a k přechodnému nebo trvalému poškození mozkových funkcí nebo dokonce ke smrti.
U přípravku Apidra podávaného pomocí CSII byly hlášeny případy výskytu hyperglykémie (viz bod 4.4), která vedla k diabetické ketoacidóze (DKA); většina případů souvisela s nesprávnou manipulací nebo se selháním systému pumpy. Pacient se má vždy řídit specifickými pokyny pro používání přípravku Apidra a vždy má být vybaven alternativním systémem pro aplikaci inzulínu pro případ selhání pumpy.
Poruchy kůže a podkožní tkáně
Lokální reakce z přecitlivělosti (zčervenání, otok a svědění v místě vpichu) se mohou objevit v průběhu léčby inzulínem. Tyto reakce jsou obvykle přechodné a v průběhu pokračující léčby vymizí.
Lipodystrofie se může objevit v místě vpichu následkem nedodržení plynulého střídání místa vpichu v rámci oblasti aplikace.
Celkové poruchy a reakce v místě aplikace
Systémové reakce z přecitlivělosti mohou zahrnovat kopřivku, pocit tíhy na hrudi, dušnost, alergickou dermatitidu a svědění. Těžké případy generalizované alergické reakce včetně anafylaktické reakce mohou být život ohrožující.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Příznaky
Hypoglykémie se může objevit jako výsledek nadměrné inzulínové aktivity vzhledem k příjmu potravy a výdeji energie.
Nejsou k dispozici specifické údaje týkající se předávkování inzulínem glulisin. Hypoglykémie se však může rozvinout přes následující stádia:
Léčba
Mírné hypoglykemické epizody mohou být zvládnuty perorálním podáním glukózy nebo produktů obsahujících cukr. Doporučuje se proto, aby diabetici u sebe neustále nosili kostky cukru, sladkosti, sušenky nebo sladkou ovocnou šťávu.
Těžké hypoglykemické epizody, kdy pacient upadne do bezvědomí, mohou být léčeny glukagonem (0,5 mg až 1 mg) podaným intramuskulárně nebo subkutánně někým, kdo dostal patřičné instrukce, nebo glukózou podanou intravenózně zdravotnickým pracovníkem. Glukóza musí být podána intravenózně také tehdy, pokud u pacienta nenastane odpověď na glukagon během 10 až 15 minut.
Jakmile pacient znovu nabude vědomí, doporučuje se z důvodu prevence relapsu podání sacharidů perorálně.
Po injekci glukagonu by měl být pacient sledován v nemocnici z důvodu zjištění příčiny tak těžké hypoglykémie a z důvodu prevence dalších podobných epizod.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Léčiva k terapii diabetu, inzulíny a analogy rychle působící, k injekční aplikaci, ATC kód: A10AB06.
Mechanismus účinku
Inzulín glulisin je rekombinantní analog lidského inzulínu, který je ekvipotentní k normálnímu lidskému inzulínu. Inzulín glulisin má rychlejší nástup účinku a kratší trvání účinku než běžný lidský inzulín.
Primárním účinkem inzulínu a inzulínových analog včetně inzulínu glulisin je regulace metabolismu glukózy. Inzulíny snižují hladinu glukózy v krvi stimulací periferního vychytávání glukózy zvláště kosterními svaly a tukem a inhibicí glukoneogeneze v játrech. Inzulín inhibuje lipolýzu v tukových buňkách, inhibuje proteolýzu a podporuje syntézu proteinů.
Studie u zdravých dobrovolníků a pacientů s diabetem ukázaly, že inzulín glulisin, je-li podán subkutánně, je rychlejší v nástupu a kratší v trvání účinku než běžný lidský inzulín. Pokud je inzulín glulisin podán subkutánní injekcí, hypoglykemizující účinek nastupuje během 10 - 20 minut. Po intravenózním podání byl při srovnání se subkutánním podáním pozorován rychlejší nástup a kratší trvání účinku, stejně jako větší maximální účinek. Účinky na snížení glykémie jsou u inzulínu glulisin a běžného lidského inzulínu při podání intravenózní cestou ekvipotentní. Jedna jednotka inzulínu glulisin má stejný hypoglykemizující účinek jako jedna dávka běžného lidského inzulínu.
Proporcionalita dávky
Ve studii s 18 muži s diabetes mellitus 1. typu ve věku od 21 do 50 let snižoval inzulín glulisin hladinu glukózy úměrně podané dávce v terapeuticky relevantním dávkovém rozmezí 0,075 až 0,15 jednotky/kg. Při dávkách 0,3 jednotky/kg nebo vyšších byl hypoglykemický účinek inzulínu glulisinu nižší, než by úměrně odpovídalo dávce, stejně jako u lidského inzulínu.
Nástup účinku inzulínu glulisin je dvakrát rychlejší než u běžného lidského inzulínu a jeho hypoglykemický efekt končí asi o 2 hodiny dříve než u běžného lidského inzulínu.
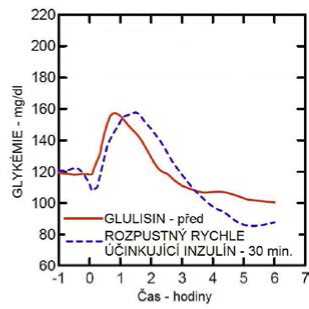
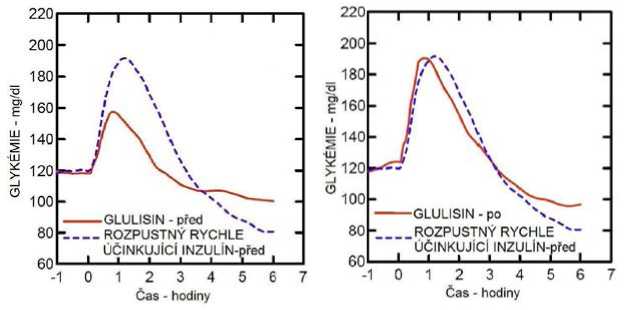
Studie I. fáze u pacientů s diabetes mellitus 1. typu hodnotily hypoglykemizující profily inzulínu glulisin a rozpustného rychle účinkujícího inzulínu podaného subkutánně v dávce 0,15 jednotky/kg v různé době ve vztahu k 15minutovému standardnímu jídlu. Data ukázala, že inzulín glulisin podaný 2 minuty před jídlem, poskytuje v porovnání s rozpustným rychle účinkujícím lidským inzulínem podaným 30 minut před jídlem podobnou postprandiální kompenzaci. Je-li podán 2 minuty před jídlem, poskytuje inzulín glulisin lepší postprandiální kompenzaci než rozpustný rychle účinkující lidský inzulín podaný 2 minuty před jídlem. Inzulín glulisin podaný 15 minut po začátku jídla poskytuje podobnou glykemickou kompenzaci jako rozpustný rychle účinkující lidský inzulín podaný 2 minuty před jídlem (viz obrázek 1).
í í í
Obrázek 1 A Obrázek 1B Obrázek 1C
Obrázek 1: Průměrný hypoglykemizující účinek po dobu 6 hodin u 20 pacientů s diabetes mellitus 1. typu. Inzulín glulisin podaný 2 minuty (Glulisin před) před začátkem jídla v porovnání s běžným lidským inzulínem podaným 30 minut (Rozpustný rychle účinkující inzulín-30 min) před začátkem jídla (obrázek 1A) a v porovnání s běžným lidským inzulínem podaným 2 minuty (Rozpustný rychle účinkující inzulín před) před jídlem (obrázek 1B). Inzulín glulisin podaný 15 minut (Glulisin po) po začátku jídla v porovnání s běžným inzulínem podaným 2 minuty (Rozpustný rychle účinkující inzulín před) před začátkem jídla (obrázek 1C). Na ose x je nula (šipka) začátek jídla trvajícího 15 minut.
Obezita
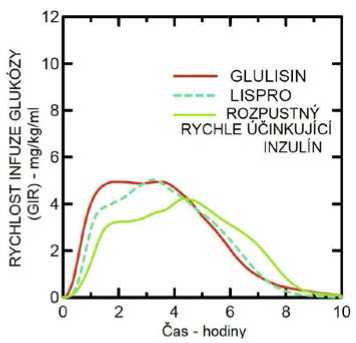
Studie I. fáze prováděná s inzulínem glulisin, inzulínem lispro a běžným lidským inzulínem u obézní populace ukázala, že inzulín glulisin si zachovává své rychle účinkující vlastnosti. V této studii byl čas představující časnou hypoglykemizující aktivitu až 20 % celkové AUC a AUC (0 -2 h) a sice 114 minut a 427 mg/kg" u inzulínu glulisin, 121 minut a 354 mg/kg" pro lispro a 150 minut a 197 mg/kg pro běžný lidský inzulín (viz obrázek 2).
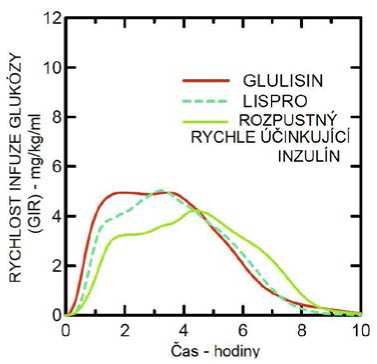
Obrázek 2: Rychlost infuze glukózy (GIR) po subkutánní injekci 0,3 jednotky/kg inzulínu glulisin (GLULISIN) nebo inzulínu lispro (LISPRO) nebo normálního lidského inzulínu (Rozpustný rychle účinkující inzulín) u obézní populace.
Další studie I. fáze s inzulínem glulisin a inzulínem lispro u nediabetické populace 80 pacientů s širokým rozmezím indexů tělesné hmotnosti (18-46 kg/m2) ukázala, že k rychlému nástupu účinku docházelo napříč širokým rozmezím indexů tělesné hmotnosti (BMI), zatímco celkový hypoglykemický efekt klesá se zvyšující se obezitou.
Průměrná plocha pod křivkou (AUC) pro celkovou GIR (glucose infusion rate- rychlost infuze glukózy) mezi 0-1. hodinou byla 102±75 mg/kg s 0,2 jednotky/kg inzulínu glulisin a 158±100 mg/kg s 0,4 jednotky/kg inzulínu glulisin a 83,1±72,8 mg/kg s 0,2 jednotky/kg inzulínu lispro a 112,3±70,8 mg/kg s 0,4 jednotky/kg inzulínu lispro.
Studie I. fáze u 18 obézních pacientů s diabetem mellitus 2. typu (BMI mezi 35 a 40 kg/m2) s inzulínem glulisin a inzulínem lispro [90% CI: 0,81, 0,95 (p=<0,01)] ukázala, že inzulín glulisin efektivně kontroluje diurnální postprandiální výkyvy hladiny glukózy v krvi.
Klinická účinnost a bezpečnost
Diabetes mellitus 1. typu - dospělí
Ve 26týdenní klinické studii fáze III srovnávající inzulín glulisin s inzulínem lispro podávanými subkutánně krátce (0-15 minut) před jídlem u pacientů s diabetes mellitus 1. typu užívajících inzulín glargin jako bazální inzulín, byl inzulín glulisin, co do kompenzace glykémie, srovnatelný s inzulínem lispro, jak ukazují změny glykovaného hemoglobinu od začátku do konce studie (vyjádřené jako HbA1c ekvivalent). Při selfmonitoringu byly pozorovány srovnatelné hodnoty glykémie. Na rozdíl od inzulínu lispro nebylo potřeba při terapii inzulínem glulisin zvyšovat dávku bazálního inzulínu.
12týdenní klinická studie fáze III prováděná u pacientů s diabetes mellitus 1. typu, kteří dostávali jako bazální terapii inzulín glargin, ukazuje, že podání inzulínu glulisin bezprostředně po jídle poskytuje srovnatelnou účinnost jako inzulín glulisin bezprostředně (0-15 minut) před jídlem nebo jako rozpustný rychle účinkující inzulín (30-45 minut).
V „per protokol“ populaci bylo ve skupině s glulisinem před jídlem pozorováno signifikantně větší snížení hodnot glykovaného hemoglobinu než ve skupině s rozpustným rychle účinkujícím inzulínem.
Diabetes mellitus 1. typu - děti
26týdenní klinická studie fáze III srovnávala subkutánní aplikaci inzulínu glulisin s inzulínem lispro podanými krátce (0 - 15 minut) před jídlem u dětí (4 - 5 let: n = 9; 6 - 7 let: n = 32 a 8 - 11 let: n = 149) a mladistvých (12-17 let: n = 382) s diabetes mellitus 1. typu, kterým byl aplikován jako bazální inzulín, inzulín glargin nebo NPH inzulín. S inzulínem glulisin bylo dosaženo srovnatelné glykemické kontroly hodnocené změnou glykovaného hemoglobinu (GHb vyjádřený jako ekvivalent HbA1c) od zahájení studie do jejího ukončení a hodnotami glukózy v krvi zjištěnými při selfmonitoringu jako s inzulínem lispro.
Nejsou dostatečné klinické informace o použití přípravku Apidra u dětí mladších 6 let.
Diabetes mellitus 2. typu - dospělí
26týdenní klinická studie fáze III následovaná rozšířením o 26týdenní studii bezpečnosti byla prováděna za účelem srovnání inzulínu glulisin (0-15 minut před jídlem) s rozpustným rychle účinkujícím lidským inzulínem (30-45 minut před jídlem) při subkutánním podání u pacientů s diabetes mellitus 2. typu užívajících NPH inzulín jako bazální inzulín. Průměrný body mass index (BMI) u pacientů byl 34,55 kg/m2. Inzulín glulisin ukázal, že je srovnatelný s rozpustným rychle účinkujícím lidským inzulínem, pokud se týče změn glykovaného hemoglobinu (vyjádřené jako HbA1c ekvivalent) od začátku do konce 6-měsíční studie (- 0,46 % u inzulínu glulisin a - 0,30 % u rozpustného rychle účinkujícího lidského inzulínu) a od začátku do konce 12-měsíční studie (- 0,23 % u inzulínu glulisin a - 0,13 % u rozpustného rychle účinkujícího lidského inzulínu, rozdíl není významný). V této studii většina pacientů (79 %) mísila jejich rychle účinkující inzulín s NPH inzulínem bezprostředně před injekcí a 58 % pacientů užívalo v době randomizace perorální antidiabetika. Tito pacienti byli instruováni, aby v jejich užívání pokračovali ve stejných dávkách.
Rasa a pohlaví
V kontrolovaných klinických studiích u dospělých neprokázal inzulín glulisin rozdíly v bezpečnosti a účinnosti v podskupinové analýze založené na rase a pohlaví.
5.2 Farmakokinetické vlastnosti
Náhrada aminokyseliny asparagin na pozici B3 lidského inzulínu lysinem a lysinu na pozici B29 kyselinou glutamovou zajišťuje u inzulínu glulisin rychlejší absorpci.
Ve studii s 18 muži s diabetes mellitus 1. typu ve věku od 21 do 50 let vykazuje inzulín glulisin pro časnou, maximální a celkovou expozici v dávkovém rozmezí 0,075 až 0,4 jednotky/kg dávkovou proporcionalitu.
Absorpce a biologická dostupnost
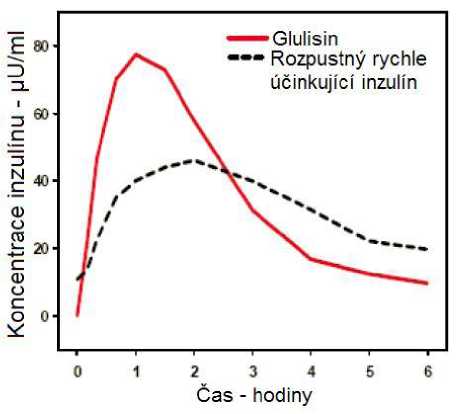
Farmakokinetické profily u zdravých dobrovolníků a diabetiků 1. a 2. typu ukázaly, že absorpce inzulínu glulisin byla asi dvakrát rychlejší s přibližně dvakrát tak vysokou vrcholovou koncentrací ve srovnání s rozpustným rychle účinkujícím inzulínem.
Ve studii u pacientů s diabetes mellitus 1. typu po subkutánním podání 0,15 jednotky/kg byl pro inzulín glulisin Tmax 55 minut a C max 82 ± 1,3^ jednotky/ml ve srovnání s Tmax 82 minut a C max 46 ±
1.3 ^jednotky/ml pro rozpustný rychle účinkující inzulín. Průměrný čas výskytu v organismu u inzulínu glulisin byl kratší (98 minut) než pro rozpustný rychle účinkující inzulín (161 minut) (viz obrázek 3).
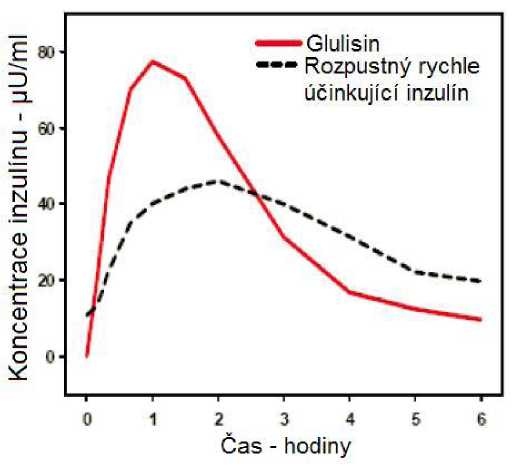
Obrázek 3: Farmakokinetický profil inzulínu glulisin a rozpustného rychle účinkujícího inzulínu u pacientů s diabetes mellitus 1. typu po dávce 0,15 jednotky/kg.
Ve studii u pacientů s diabetes mellitus 2. typu po subkutánním podání 0,2 jednotky/kg inzulínu glulisin byla Cmax 91 pjednotky/ml s rozmezím mezi kvartily od 78 do 104 pjednotky/ml.
Pokud byl inzulín glulisin aplikován subkutánně do břišní stěny, deltového svalu a stehna, profily koncentrace-čas byly podobné s mírně rychlejší absorpcí při podání do břišní stěny ve srovnání s podáním do stehna. Absorpce z deltového svalu byla střední (viz bod 4.2). Absolutní biologická dostupnost (70%) inzulínu glulisin byla v rámci míst aplikace podobná a s nízkou variabilitou mezi jednotlivými jedinci (11% CV). V porovnání se subkutánní injekcí vedlo podání intravenózního bolusu inzulinu glulisinu k vyšší systémové expozici, hodnota Cmax byla přibližně 40krát vyšší.
Obezita
Další studie I. fáze s inzulínem glulisin a inzulínem lispro u nediabetické populace 80 pacientů s širokým rozmezím indexů tělesné hmotnosti (18-46 kg/m2) ukázala, že k rychlému nástupu účinku dochází napříč širokým rozmezím indexů tělesné hmotnosti a že celková expozice je napříč širokým rozmezím indexů tělesné hmotnosti zachována.
Doba do 10 % celkové INS expozice byla s inzulínem glulisin dosažena o asi 5-6 minut dříve. Distribuce a eliminace
Distribuce a eliminace inzulínu glulisin a rozpustného rychle účinkujícího inzulínu po intravenózním podání je podobná s distribučními objemy 13 l a 22 l a poločasem eliminace 13 a 18 minut.
Po subkutánním podání je inzulín glulisin eliminován rychleji než rozpustný rychle účinkující inzulín s poločasem eliminace 42 minut ve srovnání s 86 minutami. Ve zkřížené analýze u zdravých jedinců nebo u diabetiků 1. nebo 2. typu byl poločas eliminace v rozmezí od 37 do 75 minut (rozmezí mezi kvartily).
Inzulín glulisin vykazuje nízkou vazbu na plazmatické proteiny stejně jako lidský inzulín.
Zvláštní populace
Porucha funkce ledvin
V klinické studii prováděné u nediabetiků, pokrývající široké rozmezí renálních funkcí
(CrCl > 80 ml/min, 30-50 ml/min, < 30 ml/min), byly obecně zachovány rychle účinkující vlastnosti
inzulínu glulisin. U pacientů s poškozením ledvin však mohou být nároky na inzulín sníženy.
Porucha funkce jater
Farmakokinetické vlastnosti nebyly zkoumány u pacientů se sníženou funkcí jater.
Starší pacienti
U starších pacientů s diabetes mellitus jsou dostupné pouze velmi omezené farmakokinetické údaje.
Děti a mladiství
Farmakokinetické a farmakodynamické vlastnosti inzulínu glulisin byly zkoumány u dětí (7-11 let) a mladistvých (12-16 let) s diabetes mellitus 1. typu. Inzulín glulisin byl rychle absorbován v obou věkových skupinách, s podobným Tmax a Cmax jako u dospělých (viz bod 4.2). Při podání těsně před testovaným jídlem poskytuje inzulín glulisin stejně jako u dospělých lepší postprandiální kompenzaci než rozpustný rychle účinkující lidský inzulín (viz bod 5.1). Výchylky glukózy (AUC 0-6h) byly 641 mg.h/dl pro insulin glulisin a 801 mg.h/dl pro rozpustný rychle účinkující lidský inzulín.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje neodhalily jiné nálezy toxicity než ty, které jsou spojené s hypoglykemizující farmakodynamickou aktivitou (hypoglykémie), odlišné od rozpustného rychle účinkujícího inzulínu nebo s klinickým významem pro člověka.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Metakresol Chlorid sodný Trometamol Polysorbát 20
Kyselina chlorovodíková 35%
Hydroxid sodný Voda na injekci
6.2 Inkompatibility
Subkutánní podání
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky s výjimkou NPH lidských inzulínů.
Pokud je užívána v inzulínové pumpě, nesmí být Apidra mísena s žádnými dalšími léčivými přípravky.
Intravenózní podání
Bylo zjištěno, že Apidra není kompatibilní s 5% roztokem glukózy a s Ringerovým roztokem, tudíž se s těmito roztoky nesmí používat. Použití jiných roztoků dosud nebylo studováno.
6.3 Doba použitelnosti
2 roky
Doba použitelnosti po prvním použití injekční lahvičky
Přípravek může být uchováván až čtyři týdny při teplotě do 25°C mimo přímé teplo nebo světlo. Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
Je doporučeno, aby datum prvního použití z injekční lahvičky bylo zaznamenáno na štítek.
Doba použitelnosti pro intravenózní podání
Inzulin glulisin k intravenóznímu podání v koncentraci 1 jednotka/ml je stabilní při teplotě 15 °C až 25 °C po dobu 48 hodin (viz bod 6.6).
6.4 Zvláštní opatření pro uchovávání
Neotevřené injekční lahvičky
Uchovávejte v chladničce (2°C - 8°C).
Chraňte před mrazem.
Neukládejte přípravek Apidra do blízkosti mrazicího boxu nebo mrazicí vložky.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
Otevřené injekční lahvičky
Podmínky uchovávání tohoto léčivého přípravku po jeho prvním otevření jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
10 ml roztoku v injekční lahvičce (bezbarvé sklo typu I) se zátkou (hliníkový pertl, pružná chlorobutylová guma) a polypropylenovým odtrhávacím víčkem. Dostupná jsou balení s 1, 2, 4 a 5 injekčními lahvičkami.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Subkutánní podání
Apidra v injekčních lahvičkách je pro použití do inzulínových stříkaček s odpovídající stupnicí jednotek a pro použití v inzulínové pumpě (viz bod 4.2).
Injekční lahvičku před použitím prohlédněte. Smí být použita pouze tehdy, je-li roztok čirý, bezbarvý a bez viditelných pevných částic. Vzhledem k tomu, že Apidra je roztok, není potřeba ho před použitím znovu protřepávat.
Před každým podáním injekce musí být vždy zkontrolován štítek inzulínu, aby nedošlo k záměně inzulínu glulisin a jiných inzulínů (viz bod 4.4).
Míchání s inzulíny
Pokud se Apidra mísí s NPH lidským inzulínem, měla by být natažena do stříkačky jako první. Injekce by se měla podat okamžitě po smíchání, protože údaje týkající se směsí připravených značnou dobu před injekcí nejsou dostupné.
Kontinuální subkutánní infuzní pumpa Informace viz body 4.2 a 4.8.
Intravenózní podání
Apidra by měla být používána v koncentraci 1 jednotka inzulínu glulisinu /ml v infuzních systémech s infuzním roztokem chloridu sodného 9 mg/ml (0,9%) s chloridem draselným o koncentraci 40 mmol/l nebo bez něj, v koextrudovaných polyolefinových/polyamidových umělohmotných infuzních vacích s jednorázovými infuzními sety. Inzulin glulisin k intravenóznímu podání v koncentraci 1 jednotka/ml je stabilní při pokojové teplotě po dobu 48 hodin.
Po naředění k intravenóznímu podání musí být roztok před podáním vizuálně zkontrolován, zda neobsahuje částice. Smí být použit pouze čirý bezbarvý roztok, který není zkalený a neobsahuje žádné viditelné částice.
Bylo zjištěno, že Apidra není kompatibilní s 5% roztokem glukózy a s Ringerovým roztokem, tudíž se s těmito roztoky nesmí používat. Použití jiných roztoků dosud nebylo studováno.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Sanofi-Aventis Deutschland GmbH D-65926 Frankfurt am Main Německo.
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/04/285/001-004
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 27. září 2004
Datum posledního prodloužení registrace: 20. srpna 2009
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku j sou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/
NÁZEV PŘÍPRAVKU
1.
Apidra 100 jednotek/ml injekční roztok v zásobní vložce
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden ml obsahuje 100 jednotek insulinum glulisinum (odpovídá 3,49 mg).
Jedna zásobní vložka obsahuje 3 ml injekčního roztoku, což odpovídá 300 jednotek.
Inzulín glulisin se vyrábí technologií rekombinace DNA využitím kmenů bakterie Escherichia coli. Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok v zásobní vložce.
Čirý bezbarvý vodný roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba dospělých, mladistvých a dětí starších 6 let s diabetes mellitus, kde je nutná léčba inzulínem.
4.2 Dávkování a způsob podání
Dávkování
Síla tohoto přípravku se uvádí v jednotkách. Tyto jednotky se vztahují výhradně k přípravku Apidra a liší se od m.j. (IU) nebo jednotek používaných k vyjádření síly jiných inzulínových analogů (viz bod 5.1).
Apidra by se měla podávat v režimech, které zahrnují středně nebo dlouhodobě účinkující inzulín nebo analog bazálního inzulínu a může být podávána s perorálními antidiabetiky.
Dávka přípravku Apidra by měl být upravena individuálně.
Zvláštní populace
Porucha funkce ledvin
Farmakokinetické vlastnosti inzulínu glulisin jsou obecně u pacientů s renálním poškozením zachovány. Potřeba inzulínu však může být při poškození renálních funkcí snížena (viz bod 5.2).
Porucha funkce jater
Farmakokinetické vlastnosti inzulínu glulisin nebyly zkoumány u pacientů se sníženou funkcí jater.
U pacientů se zhoršením funkce jater může být potřeba inzulínu snížena z důvodu snížené kapacity glukoneogeneze a poklesu inzulínového metabolizmu.
Starší pacienti
U starších pacientů s diabetes mellitus jsou dostupné omezené farmakokinetické údaje. Zhoršení renálních funkcí může vést k poklesu potřeby inzulínu.
Pediatrická populace
Nej sou dostatečné klinické informace o použití přípravku Apidra u dětí mladších 6 let.
Způsob podání
Subkutánní podání
Apidra by se měla podávat subkutánní injekcí krátce (0-15 min) před jídlem nebo brzy po jídle, nebo kontinuální subkutánní infuzní pumpou.
Apidra by se měla podávat subkutánně do břišní stěny, stehna, deltového svalu nebo kontinuální infuzí do břišní stěny. Místa vpichu injekce a infuze v rámci jedné injekční oblasti (břicho, stehno nebo deltový sval) je nutno s každou injekcí střídat. Míra absorpce a následně nástup a trvání účinku mohou být ovlivněny místem vpichu, cvičením a dalšími faktory. Subkutánní injekce do břišní stěny zajišťuje o něco rychlejší absorpci než ostatní injekční místa (viz bod 5.2).
Musí se dávat pozor, aby bylo zajištěno, že nebyla propíchnuta krevní céva. Místo vpichu se po injekci nesmí masírovat. Pacienti musí být poučeni, aby používali správné injekční techniky.
Míchání s inzulíny
Při podávání v subkutánní injekci se Apidra nesmí mísit s žádnými jinými léčivými přípravky s výjimkou NPH inzulínu.
Další podrobnosti o zacházení viz bod 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1. Hypoglykémie.
4.4 Zvláštní upozornění a opatření pro použití
Převedení pacienta na jiný typ nebo značku inzulínu musí být prováděno za přísného lékařského dohledu. Změny síly, značky (výrobce), typu (rozpustný rychle účinkující, neutral protamin Hagedorn [NPH], lente, dlouhodobě působící, atd.), původu (zvířecí, lidský, analog lidského inzulinu) a/nebo způsobu výroby mohou vést k nutnosti změnit dávku. Současná léčba perorálními antidiabetiky může vyžadovat úpravu dávkování.
Hyperglykémie
Podávání neadekvátních dávek nebo přerušení léčby zejména u inzulíndependentních diabetiků může vést k hyperglykémii a diabetické ketoacidóze; tedy k okolnostem, které jsou potenciálně letální.
Hypoglykémie
Doba výskytu hypoglykémie závisí na profilu účinku podaného inzulínu, a proto se může měnit, pokud se mění režim léčby.
Okolnosti, za kterých mohou být včasné varovné symptomy pozměněny nebo být méně výrazné, zahrnují dlouhou anamnézu diabetu, zintenzivnění léčby inzulínem, diabetickou neuropatii, léčivé přípravky jako betablokátory nebo převedení ze zvířecího inzulínu na lidský inzulín.
Úprava dávky může být nezbytná u pacientů, kteří zvýší svou fyzickou aktivitu nebo změní svůj obvyklý stravovací řád. Cvičení prováděné okamžitě po jídle může zvýšit riziko hypoglykémie.
V porovnání s rozpustným lidským inzulínem, pokud se po injekci analogu rychlého inzulínu objeví hypoglykémie, tato se může objevit dříve.
Nekontrolovaná hypoglykemická nebo hyperglykemická reakce může způsobit bezvědomí, kóma nebo smrt.
Potřeba inzulínu může být pozměněna během nemoci nebo emočního rozrušení.
Pera, která se mají používat se zásobními vložkami Apidra
Zásobní vložky Apidra je možné použít pouze s následujícími pery:
- JuniorSTAR, které dávkuje přípravek Apidra po 0,5 jednotky
- OptiPen, ClikSTAR, Tactipen, Autopen 24 a AllStar, která dávkují přípravek Apidra po 1 jednotce. Tyto zásobní vložky se nemají používat s žádnými jinými pery pro opakované použití, protože přesnost dávky byla ověřena jen pro výše uvedená pera.
Všechna uvedená pera nemusí být ve Vaší zemi na trhu.
Chyby v medikaci
Byly hlášeny chyby v léčbě, kdy byly omylem podány jiné, zejména dlouhodobě působící, inzulíny namísto inzulínu glulisin. Před každým podáním injekce musí být vždy zkontrolován štítek inzulínu, aby nedošlo k záměně inzulínu glulisin a jiných inzulínů.
Pomocné látky
Tento léčivý přípravek obsahuje méně než 1 mmol (23 mg) sodíku v jedné dávce, tj. v podstatě sodík neobsahuje.
Apidra obsahuje metakresol, který může způsobovat alergické reakce.
Kombinace přípravku Apidra s pioglitazonem
Zejména u pacientů s rizikovými faktory pro vznik srdečního selhání byly při podávání pioglitazonu v kombinaci s inzulínem hlášeny případy srdečního selhání. To je třeba mít na paměti, pokud je zvažována léčba přípravkem Apidra v kombinaci s pioglitazonem. Jestliže je tato kombinace použita, je třeba pacienty sledovat, zda se u nich neobjevují známky a příznaky srdečního selhání, zvýšení hmotnosti a edém. Dojde-li k jakémukoli zhoršení srdečních příznaků, je zapotřebí léčbu pioglitazonem ukončit.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Studie sledující farmakokinetické interakce nebyly prováděny. Na základě empirických znalostí u podobných léčivých přípravků je pravděpodobné, že se neobjeví klinicky relevantní farmakokinetické interakce.
Rada látek ovlivňuje metabolizmus glukózy a může vyžadovat úpravu dávky inzulínu glulisin a zvláště pečlivé sledování.
Mezi látky, které mohou zvýšit hypoglykemizující účinek a zvýšit náchylnost k hypoglykémii, patří perorální antidiabetika, ACE inhibitory, disopyramid, fibráty, fluoxetin, inhibitory monoaminooxidázy (MAOI), pentoxifylin, propoxyfen, salicyláty a sulfonamidová antibiotika.
Mezi látky, které mohou snižovat hypoglykemizující účinek patří kortikosteroidy, danazol, diazoxid, diuretika, glukagon, isoniazid, deriváty fenothiazinu, somatotropin, sympatomimetika (např. epinefrin [adrenalin], salbutamol, terbutalin), tyreoidální hormony, estrogeny a progestogeny (např. perorální kontraceptiva), inhibitory proteáz a atypické antipsychotické léky (např. olanzapin a klozapin).
Betablokátory, klonidin, soli lithia nebo alkohol mohou zesílit nebo zeslabit hypoglykemizující účinek inzulínu. Pentamidin může vyvolat hypoglykémii, po které může někdy následovat hyperglykémie.
Navíc vlivem sympatolytických léčivých přípravků jako jsou betablokátory, klonidin, guanethidin a reserpin, mohou být známky adrenergní kontraregulace sníženy nebo mohou zcela chybět.
4.6 Těhotenství a kojení
Nejsou k dispozici žádné nebo jen omezené údaje (výsledky u méně než 300 těhotenství) o použití inzulínu glulisin u těhotných žen.
Reprodukční studie na zvířatech neodhalily žádné rozdíly mezi inzulínem glulisin a lidským inzulínem co se týče těhotenství, embryofetálního vývoje, porodu nebo postnatálního vývoje (viz bod 5.3).
Při předepisování těhotným ženám je nutno postupovat opatrně. Pečlivé monitorování kompenzace glykémie je nutné.
Je nutné, aby u pacientek s preexistujícím diabetem nebo s těhotenským diabetem byla udržována v průběhu těhotenství dobrá metabolická kontrola. Požadavky na inzulín mohou klesat během prvního trimestru a obecně se zvyšují během druhého a třetího trimestru. Okamžitě po porodu potřeba inzulínu rapidně poklesne.
Kojení
Není známo, zda je inzulín glulisin vylučován do mateřského mléka, ale obecně inzulín nepřestupuje do mateřského mléka a není absorbován po perorálním podání.
Kojící matky mohou vyžadovat úpravu dávky inzulínu a diety.
Fertilita
Reprodukční studie u zvířat neodhalily žádné nežádoucí účinky inzulínu glulisinu na fertilitu.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Pacientova schopnost koncentrace a reakce může být zhoršena následkem hypoglykémie nebo hyperglykémie nebo například následkem poruchy zraku. To může představovat riziko v situacích, kde jsou tyto schopnosti zvláště důležité (např. při řízení vozidla nebo při obsluze strojů).
Pacient by měl být obeznámen s opatřeními, zabraňujícími vzniku hypoglykémie během řízení. To je důležité zejména u těch pacientů, kteří mají sníženou nebo chybějící vnímavost k varovným příznakům hypoglykémie nebo mají časté epizody hypoglykémie. Je třeba zvážit, zda řízení nebo obsluha strojů jsou za těchto okolností vhodné.
4.8 Nežádoucí účinky
Souhrnný bezpečnostní profil
Hypoglykémie, nejčastější nežádoucí účinek inzulínové terapie, se může objevit, jestliže dávka inzulínu je vzhledem k jeho potřebě příliš vysoká.
Tabulkový přehled nežádoucích účinků
Následující nežádoucí účinky, které se vyskytly během klinických hodnocení, byly seřazeny níže systematicky podle skupiny orgánů a podle snižujícího se výskytu (velmi časté: >1/10; časté: >1/100 až <1/10; méně časté: >1/1 000 až <1/100; vzácné >1/10 000 až <1/1 000; velmi vzácné: <1/10 000), není známo (z dostupných údajů nelze určit).
V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
|
MedDRA Třídy orgánových systémů |
Velmi časté |
Časté |
Méně časté |
Vzácné |
|
Poruchy metabolismu a výživy |
Hypoglykémie | |||
|
Poruchy kůže a podkožní tkáně |
Reakce v místě vpichu Lokální hypersenzitivní reakce |
Lipodystrofie | ||
|
Celkové poruchy a reakce v místě aplikace |
Systémová hypersenzitivní reakce |
Popis vybraných nežádoucích účinků
Poruchy metabolismu a výživy
Symptomy hypoglykémie se obvykle objeví náhle. Mohou zahrnovat studený pot, chladnou bledou kůži, únavu, nervozitu nebo třes, úzkost, neobvyklou únavu nebo slabost, zmatenost, poruchy koncentrace, ospalost, vlčí hlad, poruchy vidění, bolest hlavy, nauseu a palpitace.
Hypoglykémie se může stát závažnou a může vést k bezvědomí a/nebo křečím a k přechodnému nebo trvalému poškození mozkových funkcí nebo dokonce ke smrti.
Poruchy kůže a podkožní tkáně
Lokální reakce z přecitlivělosti (zčervenání, otok a svědění v místě vpichu) se mohou objevit v průběhu léčby inzulínem. Tyto reakce jsou obvykle přechodné a v průběhu pokračující léčby vymizí.
Lipodystrofie se může objevit v místě vpichu následkem nedodržení plynulého střídání místa vpichu v rámci oblasti aplikace.
Celkové poruchy a reakce v místě aplikace
Systémové reakce z přecitlivělosti mohou zahrnovat kopřivku, pocit tíhy na hrudi, dušnost, alergickou dermatitidu a svědění. Těžké případy generalizované alergické reakce včetně anafylaktické reakce mohou být život ohrožující.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Příznaky
Hypoglykémie se může objevit jako výsledek nadměrné inzulínové aktivity vzhledem k příjmu potravy a výdeji energie.
Nejsou k dispozici specifické údaje týkající se předávkování inzulínem glulisin. Hypoglykémie se však může rozvinout přes následující stádia:
Léčba
Mírné hypoglykemické epizody mohou být zvládnuty perorálním podáním glukózy nebo produktů obsahujících cukr. Doporučuje se proto, aby diabetici u sebe neustále nosili kostky cukru, sladkosti, sušenky nebo sladkou ovocnou šťávu.
Těžké hypoglykemické epizody, kdy pacient upadne do bezvědomí, mohou být léčeny glukagonem (0,5 mg až 1 mg) podaným intramuskulárně nebo subkutánně někým, kdo dostal patřičné instrukce, nebo glukózou podanou intravenózně zdravotnickým pracovníkem. Glukóza musí být podána intravenózně také tehdy, pokud u pacienta nenastane odpověď na glukagon během 10 až 15 minut.
Jakmile pacient znovu nabude vědomí, doporučuje se z důvodu prevence relapsu podání sacharidů perorálně.
Po injekci glukagonu by měl být pacient sledován v nemocnici z důvodu zjištění příčiny tak těžké hypoglykémie a z důvodu prevence dalších podobných epizod.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Léčiva k terapii diabetu, inzulíny a analogy rychle působící, k injekční aplikaci ATC kód: A10AB06.
Mechanismus účinku
Inzulín glulisin je rekombinantní analog lidského inzulínu, který je ekvipotentní k normálnímu lidskému inzulínu. Inzulín glulisin má rychlejší nástup účinku a kratší trvání účinku než běžný lidský inzulín.
Primárním účinkem inzulínu a inzulínových analog včetně inzulínu glulisin je regulace metabolismu glukózy. Inzulíny snižují hladinu glukózy v krvi stimulací periferního vychytávání glukózy zvláště kosterními svaly a tukem a inhibicí glukoneogeneze v játrech. Inzulín inhibuje lipolýzu v tukových buňkách, inhibuje proteolýzu a podporuje syntézu proteinů.
Studie u zdravých dobrovolníků a pacientů s diabetem ukázaly, že inzulín glulisin, je-li podán subkutánně, je rychlejší v nástupu a kratší v trvání účinku než běžný lidský inzulín. Pokud je inzulín glulisin podán subkutánní injekcí, hypoglykemizující účinek nastupuje během 10 - 20 minut. Účinky na snížení glykémie j sou u inzulínu glulisin a běžného lidského inzulínu při podání intravenózní cestou ekvipotentní. Jedna jednotka inzulínu glulisin má stejný hypoglykemizující účinek jako jedna dávka běžného lidského inzulínu.
Proporcionalita dávky
Ve studii s 18 muži s diabetes mellitus 1. typu ve věku od 21 do 50 let snižoval inzulín glulisin hladinu glukózy úměrně podané dávce v terapeuticky relevantním dávkovém rozmezí 0,075 až 0,15 jednotky/kg. Při dávkách 0,3 jednotky/kg nebo vyšších byl hypoglykemický účinek inzulínu glulisinu nižší, než by úměrně odpovídalo dávce, stejně jako u lidského inzulínu.
Nástup účinku inzulínu glulisin je dvakrát rychlejší než u běžného lidského inzulínu a jeho hypoglykemický efekt končí asi o 2 hodiny dříve než u běžného lidského inzulínu.
Studie I. fáze u pacientů s diabetes mellitus 1. typu hodnotily hypoglykemizující profily inzulínu glulisin a rozpustného rychle účinkujícího inzulínu podaného subkutánně v dávce 0,15 jednotky/kg v různé době ve vztahu k 15minutovému standardnímu jídlu. Data ukázala, že inzulín glulisin podaný 2 minuty před jídlem, poskytuje v porovnání s rozpustným rychle účinkujícím lidským inzulínem podaným 30 minut před jídlem podobnou postprandiální kompenzaci. Je-li podán 2 minuty před jídlem, poskytuje inzulín glulisin lepší postprandiální kompenzaci než rozpustný rychle účinkující lidský inzulín podaný 2 minuty před jídlem. Inzulín glulisin podaný 15 minut po začátku jídla poskytuje podobnou glykemickou kompenzaci jako rozpustný rychle účinkující lidský inzulín podaný 2 minuty před jídlem (viz obrázek 1).
220
220
180
60
140
£
tu
ž 120
100
GLULISIN - před N___.
ROZPUSTNÝ RYCHLE ’
účinkující inzulín - 30 min
GLULISIN - před
ROZPUSTNÝ RYCHLE
Účinkující iNZULiN-eřed
|
220 200 |
—r |
—i—i—i—i—i—r | |
|
180 |
ú\\ | ||
|
£160 |
L \ ' Jf \ \ P \ \ | ||
|
^140 |
I \' F \V |
- | |
|
UJ £ 120 |
_ | ||
|
—I O 100 |
V _ |
_ | |
|
80 |
-GLULISIN - po ROZPUSTNÝ RYCHLE |
_ | |
|
60 |
_L |
ŮČINKUJiCMNZUUN-pfed f |
-1 01234567
Čas - hodiny
Čas - hodiny
Obrázek 1 A
2 3 4
Čas - hodiny
Obrázek 1B
Obrázek 1C
Obrázek 1: Průměrný hypoglykemizující účinek po dobu 6 hodin u 20 pacientů s diabetes mellitus 1. typu. Inzulín glulisin podaný 2 minuty (Glulisin před) před začátkem jídla v porovnání s běžným lidským inzulínem podaným 30 minut (Rozpustný rychle účinkující inzulín-30 min) před začátkem jídla (obrázek 1A) a v porovnání s běžným lidským inzulínem podaným 2 minuty (Rozpustný rychle účinkující inzulín před) před jídlem (obrázek 1B). Inzulín glulisin podaný 15 minut (Glulisin po) po začátku jídla v porovnání s běžným inzulínem podaným 2 minuty (Rozpustný rychle účinkující inzulín před) před začátkem jídla (obrázek 1C). Na ose x je nula (šipka) začátek jídla trvajícího 15 minut.
Obezita
Studie I. fáze prováděná s inzulínem glulisin, inzulínem lispro a běžným lidským inzulínem u obézní populace ukázala, že inzulín glulisin si zachovává své rychle účinkující vlastnosti. V této studii byl čas představující časnou hypoglykemizující aktivitu až 20 % celkové AUC a AUC (0 -2 h) a sice 114 minut a 427 mg/kg" u inzulínu glulisin, 121 minut a 354 mg/kg" pro lispro a 150 minut a 197 mg/kg pro běžný lidský inzulín (viz obrázek 2).
Obrázek 2: Rychlost infuze glukózy (GIR) po subkutánní injekci 0,3 jednotky/kg inzulínu glulisin (GLULISIN) nebo inzulínu lispro (LISPRO) nebo normálního lidského inzulínu (Rozpustný rychle účinkující inzulín) u obézní populace.
Další studie I. fáze s inzulínem glulisin a inzulínem lispro u nediabetické populace 80 pacientů s širokým rozmezím indexů tělesné hmotnosti (18-46 kg/m2) ukázala, že k rychlému nástupu účinku docházelo napříč širokým rozmezím indexů tělesné hmotnosti (BMI), zatímco celkový hypoglykemický efekt klesá se zvyšující se obezitou.
Průměrná plocha pod křivkou (AUC) pro celkovou GIR (glucose infusion rate- rychlost infuze glukózy) mezi 0-1. hodinou byla 102±75 mg/kg s 0,2 jednotky/kg inzulínu glulisin a 158±100 mg/kg s 0,4 jednotky/kg inzulínu glulisin a 83,1±72,8 mg/kg s 0,2 jednotky/kg inzulínu lispro a 112,3±70,8 mg/kg s 0,4 jednotky/kg inzulínu lispro.
Studie I. fáze u 18 obézních pacientů s diabetem mellitem 2. typu (BMI mezi 35 a 40 kg/m2) s inzulínem glulisin a inzulínem lispro [90% CI:0,81, 0,95 (p=<0,01)] ukázala, že inzulín glulisin efektivně kontroluje diurnální postprandiální výkyvy hladiny glukózy v krvi.
Klinická účinnost a bezpečnost
Diabetes mellitus 1. typu - dospělí
Ve 26týdenní klinické studii fáze III srovnávající inzulín glulisin s inzulínem lispro podávanými subkutánně krátce (0-15 minut) před jídlem u pacientů s diabetes mellitus 1. typu užívajících inzulín glargin jako bazální inzulín, byl inzulín glulisin, co do kompenzace glykémie, srovnatelný s inzulínem lispro, jak ukazují změny glykovaného hemoglobinu od začátku do konce studie (vyjádřené jako HbAJc ekvivalent). Při selfmonitoringu byly pozorovány srovnatelné hodnoty glykémie. Na rozdíl od inzulínu lispro nebylo potřeba při terapii inzulínem glulisin zvyšovat dávku bazálního inzulínu.
12týdenní klinická studie fáze III prováděná u pacientů s diabetes mellitus 1. typu, kteří dostávali jako bazální terapii inzulín glargin, ukazuje, že podání inzulínu glulisin bezprostředně po jídle poskytuje srovnatelnou účinnost jako inzulín glulisin bezprostředně (0-15 minut) před jídlem nebo jako rozpustný rychle účinkující inzulín (30-45 minut).
V „per protokol“ populaci bylo ve skupině s glulisinem před jídlem pozorováno signifikantně větší snížení hodnot glykovaného hemoglobinu než ve skupině s rozpustným rychle účinkujícím inzulínem.
Diabetes mellitus 1. typu - děti
26týdenní klinická studie fáze III srovnávala subkutánní aplikaci inzulínu glulisin s inzulínem lispro podanými krátce (0 - 15minut) před jídlem u dětí (4 - 5 let: n = 9; 6 - 7 let: n = 32 a 8 - 11 let: n = 149) a mladistvých (12-17 let: n = 382) s diabetem mellitem 1.typu, kterým byl aplikován jako bazální inzulín glargin nebo NPH inzulín. S inzulínem glulisin bylo dosaženo srovnatelné glykemické kontroly hodnocené změnou glykovaného hemoglobinu (GHb vyjádřený jako ekvivalent HbAJc) od zahájení studie do jejího ukončení a hodnotami glukózy v krvi zjištěnými při selfmonitoringu jako s inzulínem lispro.
Nej sou dostatečné klinické informace o použití přípravku Apidra u dětí mladších 6 let.
Diabetes mellitus 2. typu - dospělí
26týdenní klinická studie fáze III následovaná rozšířením o 26týdenní studii bezpečnosti byla prováděna za účelem srovnání inzulínu glulisin (0-15 minut před jídlem) s rozpustným rychle účinkujícím lidským inzulínem (30-45 minut před jídlem) při subkutánním podání u pacientů s diabetem mellitem 2. typu užívajících NPH inzulín jako bazální inzulín. Průměrný body mass index (BMI) u pacientů byl 34,55 kg/m2. Inzulín glulisin ukázal, že je srovnatelný s rozpustným rychle účinkujícím lidském inzulínem, pokud se týče změn glykovaného hemoglobinu (vyjádřené jako HbAJc ekvivalent) od začátku do konce 6-měsíční studie (- 0,46 % u inzulínu glulisin a - 0,30 % u rozpustného rychle účinkujícího lidského inzulínu) a od začátku do konce 12-měsíční studie (- 0,23 % u inzulínu glulisin a - 0,13 % u rozpustného rychle účinkujícího lidského inzulínu, rozdíl není významný). V této studii většina pacientů (79 %) mísila jejich rychle účinkující inzulín s NPH inzulínem bezprostředně před injekcí a 58 % pacientů užívalo v době randomizace perorální antidiabetika. Tito pacienti byli instruováni, aby v jejich užívání pokračovali ve stejných dávkách.
Rasa a pohlaví
V kontrolovaných klinických studiích u dospělých neprokázal inzulín glulisin rozdíly v bezpečnosti a účinnosti v podskupinové analýze založené na rase a pohlaví.
5.2 Farmakokinetické vlastnosti
Náhrada aminokyseliny asparagin na pozici B3 lidského inzulínu lysinem a lysinu na pozici B29 kyselinou glutamovou zajišťuje u inzulínu glulisin rychlejší absorpci.
Ve studii s 18 muži s diabetes mellitus 1. typu ve věku od 21 do 50 let vykazuje inzulín glulisin pro časnou, maximální a celkovou expozici v dávkovém rozmezí 0,075 až 0,4 jednotky/kg dávkovou proporcionalitu.
Absorpce a biologická dostupnost
Farmakokinetické profily u zdravých dobrovolníků a diabetiků 1. a 2. typu ukázaly, že absorpce inzulínu glulisin byla asi dvakrát rychlejší s přibližně dvakrát tak vysokou vrcholovou koncentrací ve srovnání s rozpustným rychle účinkujícím inzulínem.
Ve studii u pacientů s diabetes mellitus 1. typu po subkutánním podání 0,15 jednotky/kg byl pro inzulín glulisin Tmax 55 minut a Cmax 82 ± 1,3 pjednotky/ml ve srovnání s Tmax 82 minut a Cmax 46 ±
1.3 pjednotky/ml pro rozpustný rychle účinkující inzulín. Průměrný čas výskytu v organismu u inzulínu glulisin byl kratší (98 minut) než pro rozpustný rychle účinkující inzulín (161 minut) (viz obrázek 3).
Obrázek 3: Farmakokinetický profil inzulínu glulisin a rozpustného rychle účinkujícího inzulínu u pacientů s diabetes mellitus 1. typu po dávce 0,15 jednotky/kg.
Ve studii u pacientů s diabetes mellitus 2. typu po subkutánním podání 0,2 jednotky/kg inzulínu glulisin byla Cmax 91 pjednotky/ml s rozmezím mezi kvartily od 78 do 104 pjednotky/ml.
Pokud byl inzulín glulisin aplikován subkutánně do břišní stěny, deltového svalu a stehna, profily koncentrace-čas byly podobné s mírně rychlejší absorpcí při podání do břišní stěny ve srovnání s podáním do stehna. Absorpce z deltového svalu byla střední (viz bod 4.2). Absolutní biologická dostupnost (70%) inzulínu glulisin byla v rámci míst aplikace podobná a s nízkou variabilitou mezi jednotlivými jedinci (11% CV). V porovnání se subkutánní injekcí vedlo podání intravenózního bolusu inzulínu glulisinu k vyšší systémové expozici, hodnota Cmax byla přibližně 40krát vyšší.
Obezita
Další studie I. fáze s inzulínem glulisin a inzulínem lispro u nediabetické populace 80 pacientů s širokým rozmezím indexů tělesné hmotnosti (18-46 kg/m2) ukázala, že k rychlému nástupu účinku dochází napříč širokým rozmezím indexů tělesné hmotnosti a že celková expozice je napříč širokým rozmezím indexů tělesné hmotnosti zachována.
Doba do 10% celkové INS expozice byla s inzulínem glulisin dosažena o asi 5-6 minut dříve. Distribuce a eliminace
Distribuce a eliminace inzulínu glulisin a rozpustného rychle účinkujícího inzulínu po intravenózním podání je podobná s distribučními objemy 13 l a 22 l a poločasem eliminace 13 a 18 minut.
Po subkutánním podání je inzulín glulisin eliminován rychleji než rozpustný rychle účinkující inzulín s poločasem eliminace 42 minut ve srovnání s 86 minutami. Ve zkřížené analýze u zdravých jedinců nebo u diabetiků 1. nebo 2. typu byl poločas eliminace v rozmezí od 37 do 75 minut (rozmezí mezi kvartily).
Inzulín glulisin vykazuje nízkou vazbu na plazmatické proteiny stejně jako lidský inzulín.
Porucha funkce ledvin
V klinické studii prováděné u nediabetiků, pokrývající široké rozmezí renálních funkcí
(CrCl > 80 ml/min, 30-50 ml/min, < 30 ml/min), byly obecně zachovány rychle účinkující vlastnosti
inzulínu glulisin. U pacientů s poškozením ledvin však mohou být nároky na inzulín sníženy.
Porucha funkce jater
Farmakokinetické vlastnosti nebyly zkoumány u pacientů se sníženou funkcí jater.
Starší pacienti
U starších pacientů s diabetes mellitus jsou dostupné pouze velmi omezené farmakokinetické údaje.
Děti a mladiství
Farmakokinetické a farmakodynamické vlastnosti inzulínu glulisin byly zkoumány u dětí (7-11 let) a mladistvých (12-16 let) s diabetes mellitus 1. typu. Inzulín glulisin byl rychle absorbován v obou věkových skupinách, s podobným Tmax a Cmax jako u dospělých (viz bod 4.2).
Při podání těsně před testovaným jídlem poskytuje inzulín glulisin stejně jako u dospělých lepší postprandiální kompenzaci než rozpustný rychle účinkující lidský inzulín (viz bod 5.1). Výchylky glukózy (AUC 0-6h) byly 641 mg.h/dl pro insulin glulisin a 801 mg.h/dl pro rozpustný rychle účinkující lidský inzulín.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje neodhalily jiné nálezy toxicity než ty, které jsou spojené s hypoglykemizující farmakodynamickou aktivitou (hypoglykémie), odlišné od rozpustného rychle účinkujícího inzulínu nebo s klinickým významem pro člověka.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Metakresol Chlorid sodný Trometamol Polysorbát 20
Kyselina chlorovodíková 35%
Hydroxid sodný Voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky s výjimkou NPH lidských inzulínů.
6.3 Doba použitelnosti
2 roky
Doba použitelnosti po prvním použití zásobní vložky
Přípravek může být uchováván až 4 týdny při teplotě do 25°C mimo přímé teplo nebo světlo. Pero obsahující zásobní vložku nesmí být skladováno v chladničce.
Víčko pera musí být vráceno na pero po každé injekci, aby byl přípravek chráněn před světlem.
Neotevřené zásobní vložky
Uchovávejte v chladničce (2°C - 8°C).
Chraňte před mrazem.
Neukládejte přípravek Apidra do blízkosti mrazicího boxu nebo mrazicí vložky. Uchovávejte zásobní vložku v krabičce, aby byl přípravek chráněn před světlem.
Používané zásobní vložky
Podmínky uchovávání tohoto léčivého přípravku po jeho prvním otevření jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
3 ml roztoku v zásobní vložce (bezbarvé sklo typu I) se zátkou (pružnou bromobutylovou gumovou) a pertlem (hliníkovým) a těsněním (pružným bromobutylovým gumovým). Dostupná jsou balení s 1, 3, 4, 5, 6, 8, 9 a 10 zásobními vložkami.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Zásobní vložky Apidra se mají používat pouze s inzulínovými pery: OptiPen, ClikSTAR, Autopen 24 Tactipen, AllStar nebo JuniorSTAR (viz bod 4.4). Všechna uvedená pera nemusí být ve Vaší zemi na trhu.
Pero je třeba používat podle doporučení v informaci, kterou poskytuje výrobce.
Pokyny výrobce na použití pera musí být pečlivě dodržovány při vložení zásobní vložky, upevnění jehly a podání injekce inzulínu. Zásobní vložku před použitím prohlédněte. Smí být použita pouze tehdy, je-li roztok čirý, bezbarvý a bez viditelných pevných částic. Před vložením zásobní vložky do pera k opakovanému použití ponechejte zásobní vložku 1 až 2 hodiny při pokojové teplotě. Před injekcí musí být ze zásobní vložky odstraněny vzduchové bubliny (viz návod na použití pera). Prázdné zásobní vložky nesmí být znovu plněny.
Pokud pero selže (viz návod pro použití inzulínového pera), může být roztok natažen ze zásobní vložky do stříkačky (vhodné pro inzulín 100 jednotek/ml) a injikován. Pokud je inzulinové pero poškozeno nebo nefunguje správně (z důvodu mechanické závady), musí být zlikvidováno a použito nové pero.
Pero pro opakované použití smí být používáno výhradně jedním pacientem, aby se zabránilo jakémukoliv druhu kontaminace.
Před každým podáním injekce musí být vždy zkontrolován štítek inzulínu, aby nedošlo k záměně inzulínu glulisin a jiných inzulínů (viz bod 4.4).
Míchání s inzulíny
Pokud se Apidra mísí s NPH lidským inzulínem, měla by být natažena do stříkačky jako první. Injekce by se měla podat okamžitě po smíchání, protože údaje týkající se směsí připravených značnou dobu před injekcí nejsou dostupné.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Sanofi-Aventis Deutschland GmbH D-65926 Frankfurt am Main Německo.
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/04/285/005-012
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 27. září 2004
Datum posledního prodloužení registrace: 20. srpna 2009
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku j sou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/
NÁZEV PŘÍPRAVKU
1.
Apidra 100 jednotek/ml injekční roztok v zásobní vložce pro OptiClik
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden ml obsahuje 100 jednotek insulinum glulisinum (odpovídá 3,49 mg).
Jedna zásobní vložka obsahuje 3 ml injekčního roztoku, což odpovídá 300 jednotek.
Inzulín glulisin se vyrábí technologií rekombinace DNA využitím kmenů bakterie Escherichia coli. Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok v zásobní vložce pro OptiClik.
Čirý, bezbarvý, vodný roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba dospělých, mladistvých a dětí starších 6 let s diabetes mellitus, kde je nutná léčba inzulínem.
4.2 Dávkování a způsob podání
Dávkování
Síla tohoto přípravku se uvádí v jednotkách. Tyto jednotky se vztahují výhradně k přípravku Apidra a liší se od m.j. (IU) nebo jednotek používaných k vyjádření síly jiných inzulínových analogů (viz bod 5.1).
Apidra by se měla podávat v režimech, které zahrnují středně nebo dlouhodobě účinkující inzulín nebo analog bazálního inzulínu a může být podávána s perorálními antidiabetiky.
Dávka přípravku Apidra by měla být upravena individuálně.
Zvláštní populace
Porucha funkce ledvin
Farmakokinetické vlastnosti inzulínu glulisin jsou obecně u pacientů s renálním poškozením zachovány. Potřeba inzulínu však může být při poškození renálních funkcí snížena (viz bod 5.2).
Porucha funkce jater
Farmakokinetické vlastnosti inzulínu glulisin nebyly zkoumány u pacientů se sníženou funkcí jater.
U pacientů se zhoršením funkce jater může být potřeba inzulínu snížena z důvodu snížené kapacity glukoneogeneze a poklesu inzulínového metabolizmu.
Starší pacienti
U starších pacientů s diabetes mellitus jsou dostupné omezené farmakokinetické údaje. Zhoršení renálních funkcí může vést k poklesu potřeby inzulínu.
Pediatrická populace
Nej sou dostatečné klinické informace o použití přípravku Apidra u dětí mladších 6 let.
Způsob podání
Subkutánní podání
Apidra by se měla podávat subkutánní injekcí krátce (0-15 min) před jídlem nebo brzy po jídle, nebo kontinuální subkutánní infuzní pumpou.
Apidra by se měla podávat subkutánně do břišní stěny, stehna, deltového svalu nebo kontinuální infuzí do břišní stěny. Místa vpichu injekce a infuze v rámci jedné injekční oblasti (břicho, stehno nebo deltový sval) je nutno s každou injekcí střídat. Míra absorpce a následně nástup a trvání účinku mohou být ovlivněny místem vpichu, cvičením a dalšími faktory. Subkutánní injekce do břišní stěny zajišťuje o něco rychlejší absorpci než ostatní injekční místa (viz bod 5.2).
Musí se dávat pozor, aby bylo zajištěno, že nebyla propíchnuta krevní céva. Místo vpichu se po injekci nesmí masírovat. Pacienti musí být poučeni, aby používali správné injekční techniky.
Míchání s inzulíny
Při podávání v subkutánní injekci se Apidra nesmí mísit s žádnými jinými léčivými přípravky s výjimkou NPH inzulínu.
Další podrobnosti o zacházení viz bod 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1. Hypoglykémie.
4.4 Zvláštní upozornění a opatření pro použití
Převedení pacienta na jiný typ nebo značku inzulínu musí být prováděno za přísného lékařského dohledu. Změny síly, značky (výrobce), typu (rozpustný rychle účinkující, neutral protamin Hagedorn [NPH], lente, dlouhodobě působící, atd.), původu (zvířecí, lidský, analog lidského inzulinu) a/nebo způsobu výroby mohou vést k nutnosti změnit dávku. Současná léčba perorálními antidiabetiky může vyžadovat úpravu dávkování.
Hyperglykémie
Podávání neadekvátních dávek nebo přerušení léčby zejména u inzulíndependentních diabetiků může vést k hyperglykémii a diabetické ketoacidóze; tedy k okolnostem, které jsou potenciálně letální.
Hypoglykémie
Doba výskytu hypoglykémie závisí na profilu účinku podaného inzulínu, a proto se může měnit, pokud se mění režim léčby.
Okolnosti, za kterých mohou být včasné varovné symptomy pozměněny nebo být méně výrazné, zahrnují dlouhou anamnézu diabetu, zintenzivnění léčby inzulínem, diabetickou neuropatii, léčivé přípravky jako betablokátory nebo převedení ze zvířecího inzulínu na lidský inzulín.
Úprava dávky může být nezbytná u pacientů, kteří zvýší svou fyzickou aktivitu nebo změní svůj obvyklý stravovací řád. Cvičení prováděné okamžitě po jídle může zvýšit riziko hypoglykémie.
V porovnání s rozpustným lidským inzulínem, pokud se po injekci analogu rychlého inzulínu objeví hypoglykémie, tato se může objevit dříve.
Nekontrolovaná hypoglykemická nebo hyperglykemická reakce může způsobit bezvědomí, kóma nebo smrt.
Potřeba inzulínu může být pozměněna během nemoci nebo emočního rozrušení.
Chyby v medikaci
Byly hlášeny chyby v léčbě, kdy byly omylem podány jiné, zejména dlouhodobě působící, inzulíny namísto inzulínu glulisin. Před každým podáním injekce musí být vždy zkontrolován štítek inzulínu, aby nedošlo k záměně inzulínu glulisin a jiných inzulínů.
Pomocné látky
Tento léčivý přípravek obsahuje méně než 1 mmol (23 mg) sodíku v jedné dávce, tj. v podstatě sodík neobsahuje.
Apidra obsahuje metakresol, který může způsobovat alergické reakce.
Kombinace přípravku Apidra s pioglitazonem
Zejména u pacientů s rizikovými faktory pro vznik srdečního selhání byly při podávání pioglitazonu v kombinaci s inzulínem hlášeny případy srdečního selhání. To je třeba mít na paměti, pokud je zvažována léčba přípravkem Apidra v kombinaci s pioglitazonem. Jestliže je tato kombinace použita, je třeba pacienty sledovat, zda se u nich neobjevují známky a příznaky srdečního selhání, zvýšení hmotnosti a edém. Dojde-li k jakémukoli zhoršení srdečních příznaků, je zapotřebí léčbu pioglitazonem ukončit.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Studie sledující farmakokinetické interakce nebyly prováděny. Na základě empirických znalostí u podobných léčivých přípravků je pravděpodobné, že se neobjeví klinicky relevantní farmakokinetické interakce.
Rada látek ovlivňuje metabolizmus glukózy a může vyžadovat úpravu dávky inzulínu glulisin a zvláště pečlivé sledování.
Mezi látky, které mohou zvýšit hypoglykemizující účinek a zvýšit náchylnost k hypoglykémii, patří perorální antidiabetika, ACE inhibitory, disopyramid, fibráty, fluoxetin, inhibitory monoaminooxidázy (MAOI), pentoxifylin, propoxyfen, salicyláty a sulfonamidová antibiotika.
Mezi látky, které mohou snižovat hypoglykemizující účinek, patří kortikosteroidy, danazol, diazoxid, diuretika, glukagon, isoniazid, deriváty fenothiazinu, somatotropin, sympatomimetika (např. epinefrin [adrenalin], salbutamol, terbutalin), tyreoidální hormony, estrogeny a progestogeny (např. perorální kontraceptiva), inhibitory proteáz a atypické antipsychotické léky (např. olanzapin a klozapin).
Betablokátory, klonidin, soli lithia nebo alkohol mohou zesílit nebo zeslabit hypoglykemizující účinek inzulínu. Pentamidin může vyvolat hypoglykémii, po které může někdy následovat hyperglykémie.
Navíc vlivem sympatolytických léčivých přípravků jako jsou betablokátory, klonidin, guanethidin a reserpin, mohou být známky adrenergní kontraregulace sníženy nebo mohou zcela chybět.
4.6 Těhotenství a kojení
Nejsou k dispozici žádné nebo jen omezené údaje (výsledky u méně než 300 těhotenství) o použití inzulínu glulisin u těhotných žen.
Reprodukční studie na zvířatech neodhalily žádné rozdíly mezi inzulínem glulisin a lidským inzulínem co se týče těhotenství, embryofetálního vývoje, porodu nebo postnatálního vývoje (viz bod 5.3).
Při předepisování těhotným ženám je nutno postupovat opatrně. Pečlivé monitorování kompenzace glykémie je nutné.
Je nutné, aby u pacientek s preexistujícím diabetem nebo s těhotenským diabetem byla udržována v průběhu těhotenství dobrá metabolická kontrola. Požadavky na inzulín mohou klesat během prvního trimestru a obecně se zvyšují během druhého a třetího trimestru. Okamžitě po porodu potřeba inzulínu rapidně poklesne.
Kojení
Není známo, zda je inzulín glulisin vylučován do mateřského mléka, ale obecně inzulín nepřestupuje do mateřského mléka a není absorbován po perorálním podání.
Kojící matky mohou vyžadovat úpravu dávky inzulínu a diety.
Fertilita
Reprodukční studie u zvířat neodhalily žádné nežádoucí účinky inzulínu glulisinu na fertilitu.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Pacientova schopnost koncentrace a reakce může být zhoršena následkem hypoglykémie nebo hyperglykémie nebo například následkem poruchy zraku. To může představovat riziko v situacích, kde jsou tyto schopnosti zvláště důležité (např. při řízení vozidla nebo při obsluze strojů).
Pacient by měl být obeznámen s opatřeními, zabraňujícími vzniku hypoglykémie během řízení. To je důležité zejména u těch pacientů, kteří mají sníženou nebo chybějící vnímavost k varovným příznakům hypoglykémie nebo mají časté epizody hypoglykémie. Je třeba zvážit, zda řízení nebo obsluha strojů jsou za těchto okolností vhodné.
4.8 Nežádoucí účinky
Souhrnný bezpečnostní profil
Hypoglykémie, nejčastější nežádoucí účinek inzulínové terapie, se může objevit, jestliže dávka inzulínu je vzhledem k jeho potřebě příliš vysoká.
Tabulkový přehled nežádoucích účinků
Následující nežádoucí účinky, které se vyskytly během klinických hodnocení, byly seřazeny níže systematicky podle skupiny orgánů a podle snižujícího se výskytu (velmi časté: >1/10; časté: >1/100 až <1/10; méně časté: >1/1 000 až <1/100; vzácné >1/10 000 až <1/1 000; velmi vzácné: <1/10 000), není známo (z dostupných údajů nelze určit).
V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
|
MedDRA Třídy orgánových systémů |
Velmi časté |
Časté |
Méně časté |
Vzácné |
|
Poruchy metabolismu a výživy |
Hypoglykémie | |||
|
Poruchy kůže a podkožní tkáně |
Reakce v místě vpichu Lokální hypersenzitivní reakce |
Lipodystrofie | ||
|
Celkové poruchy a reakce v místě aplikace |
Systémová hypersenzitivní reakce |
Popis vybraných nežádoucích účinků
Poruchy metabolismu a výživy
Symptomy hypoglykémie se obvykle objeví náhle. Mohou zahrnovat studený pot, chladnou bledou kůži, únavu, nervozitu nebo třes, úzkost, neobvyklou únavu nebo slabost, zmatenost, poruchy koncentrace, ospalost, vlčí hlad, poruchy vidění, bolest hlavy, nauseu a palpitace.
Hypoglykémie se může stát závažnou a může vést k bezvědomí a/nebo křečím a k přechodnému nebo trvalému poškození mozkových funkcí nebo dokonce ke smrti.
Poruchy kůže a podkožní tkáně
Lokální reakce z přecitlivělosti (zčervenání, otok a svědění v místě vpichu) se mohou objevit v průběhu léčby inzulínem. Tyto reakce jsou obvykle přechodné a v průběhu pokračující léčby vymizí.
Lipodystrofie se může objevit v místě vpichu následkem nedodržení plynulého střídání místa vpichu v rámci oblasti aplikace.
Celkové poruchy a reakce v místě aplikace
Systémové reakce z přecitlivělosti mohou zahrnovat kopřivku, pocit tíhy na hrudi, dušnost, alergickou dermatitidu a svědění. Těžké případy generalizované alergické reakce včetně anafylaktické reakce mohou být život ohrožující.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Příznaky
Hypoglykémie se může objevit jako výsledek nadměrné inzulínové aktivity vzhledem k příjmu potravy a výdeji energie.
Nejsou k dispozici specifické údaje týkající se předávkování inzulínem glulisin. Hypoglykémie se však může rozvinout přes následující stádia:
Léčba
Mírné hypoglykemické epizody mohou být zvládnuty perorálním podáním glukózy nebo produktů obsahujících cukr. Doporučuje se proto, aby diabetici u sebe neustále nosili kostky cukru, sladkosti, sušenky nebo sladkou ovocnou šťávu.
Těžké hypoglykemické epizody, kdy pacient upadne do bezvědomí, mohou být léčeny glukagonem (0,5 mg až 1 mg) podaným intramuskulárně nebo subkutánně někým, kdo dostal patřičné instrukce, nebo glukózou podanou intravenózně zdravotnickým pracovníkem. Glukóza musí být podána intravenózně také tehdy, pokud u pacienta nenastane odpověď na glukagon během 10 až 15 minut.
Jakmile pacient znovu nabude vědomí, doporučuje se z důvodu prevence relapsu podání sacharidů perorálně.
Po injekci glukagonu by měl být pacient sledován v nemocnici z důvodu zjištění příčiny tak těžké hypoglykémie a z důvodu prevence dalších podobných epizod.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Léčiva k terapii diabetu, inzulíny a analogy rychle působící, k injekční aplikaci, ATC kód: A10AB06.
Mechanismus účinku
Inzulín glulisin je rekombinantní analog lidského inzulínu, který je ekvipotentní k normálnímu lidskému inzulínu. Inzulín glulisin má rychlejší nástup účinku a kratší trvání účinku než běžný lidský inzulín.
Primárním účinkem inzulínu a inzulínových analog včetně inzulínu glulisin je regulace metabolismu glukózy. Inzulíny snižují hladinu glukózy v krvi stimulací periferního vychytávání glukózy zvláště kosterními svaly a tukem a inhibicí glukoneogeneze v játrech. Inzulín inhibuje lipolýzu v tukových buňkách, inhibuje proteolýzu a podporuje syntézu proteinů.
Studie u zdravých dobrovolníků a pacientů s diabetem ukázaly, že inzulín glulisin, je-li podán subkutánně, je rychlejší v nástupu a kratší v trvání účinku než běžný lidský inzulín. Pokud je inzulín glulisin podán subkutánní injekcí, hypoglykemizující účinek nastupuje během 10 - 20 minut. Účinky na snížení glykémie jsou u inzulínu glulisin a běžného lidského inzulínu při podání intravenózní cestou ekvipotentní. Jedna jednotka inzulínu glulisin má stejný hypoglykemizující účinek jako jedna dávka běžného lidského inzulínu.
Proporcionalita dávky
Ve studii s 18 muži s diabetes mellitus 1. typu ve věku od 21 do 50 let snižoval inzulín glulisin hladinu glukózy úměrně podané dávce v terapeuticky relevantním dávkovém rozmezí 0,075 až 0,15 jednotky/kg. Při dávkách 0,3 jednotky/kg nebo vyšších byl hypoglykemický účinek inzulínu glulisinu nižší, než by úměrně odpovídalo dávce, stejně jako u lidského inzulínu.
Nástup účinku inzulínu glulisin je dvakrát rychlejší než u běžného lidského inzulínu a jeho hypoglykemický efekt končí asi o 2 hodiny dříve než u běžného lidského inzulínu.
Studie I. fáze u pacientů s diabetes mellitus 1. typu hodnotily hypoglykemizující profily inzulínu glulisin a rozpustného rychle účinkujícího inzulínu podaného subkutánně v dávce 0,15 jednotky/kg
v různé době ve vztahu k 15minutovému standardnímu jídlu. Data ukázala, že inzulín glulisin podaný 2 minuty před jídlem, poskytuje v porovnání s rozpustným rychle účinkujícím lidským inzulínem podaným 30 minut před jídlem podobnou postprandiální kompenzaci. Je-li podán 2 minuty před jídlem, poskytuje inzulín glulisin lepší postprandiální kompenzaci než rozpustný rychle účinkující lidský inzulín podaný 2 minuty před jídlem. Inzulín glulisin podaný 15 minut po začátku jídla poskytuje podobnou glykemickou kompenzaci jako rozpustný rychle účinkující lidský inzulín podaný 2 minuty před jídlem (viz obrázek 1).
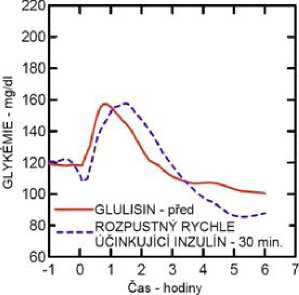
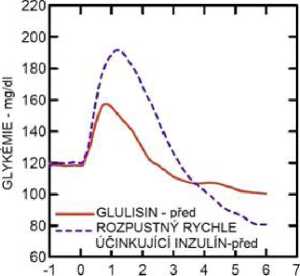
Čas - hodiny
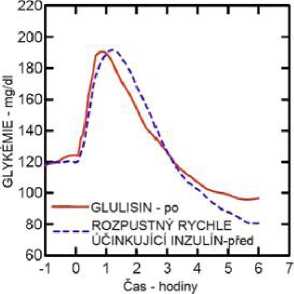
Obrázek 1 A
t
Obrázek 1B
Obrázek 1C
Obrázek 1: Průměrný hypoglykemizující účinek po dobu 6 hodin u 20 pacientů s diabetes mellitus 1. typu. Inzulín glulisin podaný 2 minuty (Glulisin před) před začátkem jídla v porovnání s běžným lidským inzulínem podaným 30 minut (Rozpustný rychle účinkující inzulín-30 min) před začátkem jídla (obrázek 1A) a v porovnání s běžném lidským inzulínem podaným 2 minuty (Rozpustný rychle účinkující inzulín před) před jídlem (obrázek 1B). Inzulín glulisin podaný 15 minut (Glulisin po) po začátku jídla v porovnání s běžným inzulínem podaným 2 minuty (Rozpustný rychle účinkující inzulín před) před začátkem jídla (obrázek 1C). Na ose x je nula (šipka) začátek jídla trvajícího 15 minut.
Obezita
Studie I. fáze prováděná s inzulínem glulisin, inzulínem lispro a běžným lidským inzulínem u obézní populace ukázala, že inzulín glulisin si zachovává své rychle účinkující vlastnosti. V této studii byl čas představující časnou hypoglykemizující aktivitu až 20 % celkové AUC a AUC (0 -2 h) a sice 114 minut a 427 mg/kg- u inzulínu glulisin, 121 minut a 354 mg/kg- pro lispro a 150 minut a 197 mg/kg pro běžný lidský inzulín (viz obrázek 2).
- GLULISIN
LISPRO ROZPUSTNÝ RYCHLE ÚČINKUJÍCÍ INZULÍN
Cas - hodiny
Obrázek 2: Rychlost infuze glukózy (GIR) po subkutánní injekci 0,3 jednotky/kg inzulínu glulisin (GLULISIN) nebo inzulínu lispro (LISPRO) nebo normálního lidského inzulínu (Rozpustný rychle účinkující inzulín) u obézní populace.
Další studie I. fáze s inzulínem glulisin a inzulínem lispro u nediabetické populace 80 pacientů s širokým rozmezím indexů tělesné hmotnosti (18-46 kg/m2) ukázala, že k rychlému nástupu účinku docházelo napříč širokým rozmezím indexů tělesné hmotnosti (BMI), zatímco celkový hypoglykemický efekt klesá se zvyšující se obezitou.
Průměrná plocha pod křivkou (AUC) pro celkovou GIR (glucose infusion rate- rychlost infuze glukózy) mezi 0-1. hodinou byla 102±75 mg/kg s 0,2 jednotky/kg inzulínu glulisin a 158±100 mg/kg s0,4 jednotky/kg inzulínu glulisin a 83,1±72,8 mg/kg s 0,2 jednotky/kg inzulínu lispro a 112,3±70,8 mg/kg s 0,4 jednotky/kg inzulínu lispro.
Studie I. fáze u 18 obézních pacientů s diabetem mellitus 2. typu (BMI mezi 35 a 40 kg/m2) s inzulínem glulisin a inzulínem lispro [90% CI: 0,81, 0,95 (p=<0,01)] ukázala, že inzulín glulisin efektivně kontroluje diurnální postprandiální výkyvy hladiny glukózy v krvi.
Klinická účinnost a bezpečnost
Diabetes mellitus 1. typu - dospělí
Ve 26týdenní klinické studii fáze III srovnávající inzulín glulisin s inzulínem lispro podávanými subkutánně krátce (0-15 minut) před jídlem u pacientů s diabetes mellitus 1. typu užívajících inzulín glargin jako bazální inzulín, byl inzulín glulisin, co do kompenzace glykémie, srovnatelný s inzulínem lispro, jak ukazují změny glykovaného hemoglobinu od začátku do konce studie (vyjádřené jako HbA1c ekvivalent). Při selfmonitoringu byly pozorovány srovnatelné hodnoty glykémie. Na rozdíl od inzulínu lispro nebylo potřeba při terapii inzulínem glulisin zvyšovat dávku bazálního inzulínu.
12týdenní klinická studie fáze III prováděná u pacientů s diabetes mellitus 1. typu, kteří dostávali jako bazální terapii inzulín glargin, ukazuje, že podání inzulínu glulisin bezprostředně po jídle poskytuje srovnatelnou účinnost jako inzulín glulisin bezprostředně (0-15 minut) před jídlem nebo jako rozpustný rychle účinkující inzulín (30-45minut).
V „per protokol“ populaci bylo ve skupině s glulisinem před jídlem pozorováno signifikantně větší snížení hodnot glykovaného hemoglobinu než ve skupině s rozpustným rychle účinkujícím inzulínem.
Diabetes mellitus 1. typu - děti
26týdenní klinická studie fáze III srovnávala subkutánní aplikaci inzulínu glulisin s inzulínem lispro podanými krátce (0 - 15 minut) před jídlem u dětí (4 - 5 let: n = 9; 6 - 7 let: n = 32 a 8 - 11 let: n = 149) a mladistvých (12-17 let: n = 382) s diabetes mellitus l.typu, kterým byl aplikován jako bazální inzulín glargin nebo NPH inzulín. S inzulínem glulisin bylo dosaženo srovnatelné glykemické kontroly hodnocené změnou glykovaného hemoglobinu (GHb vyjádřený jako ekvivalent HbAJc) od zahájení studie do jejího ukončení a hodnotami glukózy v krvi zjištěnými při selfmonitoringu jako s inzulínem lispro.
Nej sou dostatečné klinické informace o použití přípravku Apidra u dětí mladších 6 let.
Diabetes mellitus 2. typu - dospělí
26týdenní klinická studie fáze III následovaná rozšířením o 26týdenní studii bezpečnosti byla prováděna za účelem srovnání inzulínu glulisin (0-15 minut před jídlem) s rozpustným rychle účinkujícím lidským inzulínem (30-45 minut před jídlem) při subkutánním podání u pacientů s diabetes mellitus 2. typu užívajících NPH inzulín jako bazální inzulín. Průměrný body mass index (BMI) u pacientů byl 34,55 kg/m2. Inzulín glulisin ukázal, že je srovnatelný s rozpustným rychle účinkujícím lidském inzulínem, pokud se týče změn glykovaného hemoglobinu (vyjádřené jako HbAJc ekvivalent) od začátku do konce 6-měsíčná studie (- 0,46 % u inzulínu glulisin a - 0,30 % u rozpustného rychle účinkujícího lidského inzulínu) a od začátku do konce 12-měsíční studie (- 0,23 % u inzulínu glulisin a - 0,13 % u rozpustného rychle účinkujícího lidského inzulínu, rozdíl není významný). V této studii většina pacientů (79 %) mísila jejich rychle účinkující inzulín s NPH inzulínem bezprostředně před injekcí a 58 % pacientů užívalo v době randomizace perorální antidiabetika. Tito pacienti byli instruováni, aby v jejich užívání pokračovali ve stejných dávkách.
Rasa a pohlaví
V kontrolovaných klinických studiích u dospělých neprokázal inzulín glulisin rozdíly v bezpečnosti a účinnosti v podskupinové analýze založené na rase a pohlaví.
5.2 Farmakokinetické vlastnosti
Náhrada aminokyseliny asparagin na pozici B3 lidského inzulínu lysinem a lysinu na pozici B29 kyselinou glutamovou zajišťuje u inzulínu glulisin rychlejší absorpci.
Ve studii s 18 muži s diabetes mellitus 1. typu ve věku od 21 do 50 let inzulín glulisin vykazuje pro časnou, maximální a celkovou expozici v dávkovém rozmezí 0,075 až 0,4 jednotky/kg dávkovou proporcionalitu.
Absorpce a biologická dostupnost
Farmakokinetické profily u zdravých dobrovolníků a diabetiků 1. a 2. typu ukázaly, že absorpce inzulínu glulisin byla asi dvakrát rychlejší s přibližně dvakrát tak vysokou vrcholovou koncentrací ve srovnání s rozpustným rychle účinkujícím inzulínem.
Ve studii u pacientů s diabetes mellitus 1. typu po subkutánním podání 0,15 jednotky/kg byl pro inzulín glulisin Tmax 55 minut a C max 82 ± 1,3 pjednotky/ml ve srovnání s Tmax 82 minut a C max 46 ±
1.3 pjednotky/ml pro rozpustný rychle účinkující inzulín. Průměrný čas výskytu v organismu
u inzulínu glulisin byl kratší (98 minut) než pro rozpustný rychle účinkující inzulín (161 minut) (viz obrázek 3).
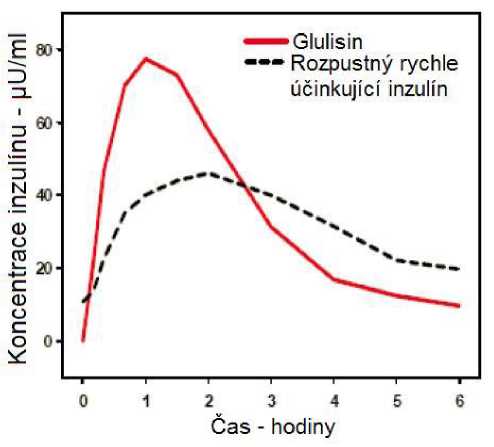
Obrázek 3: Farmakokinetický profil inzulínu glulisin a rozpustného rychle účinkujícího inzulínu u pacientů s diabetes mellitus 1. typu po dávce 0,15 jednotky/kg.
Ve studii u pacientů s diabetes mellitus 2. typu po subkutánním podání 0,2 jednotky/kg inzulínu glulisin byla Cmax 91 pjednotky/ml s rozmezím mezi kvartily od 78 do 104 pjednotky/ml.
Pokud byl inzulín glulisin aplikován subkutánně do břišní stěny, deltového svalu a stehna, profily koncentrace-čas byly podobné s mírně rychlejší absorpcí při podání do břišní stěny ve srovnání s podáním do stehna. Absorpce z deltového svalu byla střední (viz bod 4.2). Absolutní biologická dostupnost (70%) inzulínu glulisin byla v rámci míst aplikace podobná a s nízkou variabilitou mezi jednotlivými jedinci (11% CV). V porovnání se subkutánní injekcí vedlo podání intravenózního bolusu inzulínu glulisinu k vyšší systémové expozici, hodnota Cmax byla přibližně 40krát vyšší.
Obezita
Další studie I. fáze s inzulínem glulisin a inzulínem lispro u nediabetické populace 80 pacientů s širokým rozmezím indexů tělesné hmotnosti (18-46 kg/m2) ukázala, že k rychlému nástupu účinku dochází napříč širokým rozmezím indexů tělesné hmotnosti a že celková expozice je napříč širokým rozmezím indexů tělesné hmotnosti zachována.
Doba do 10% celkové INS expozice byla s inzulínem glulisin dosažena o asi 5-6 minut dříve. Distribuce a eliminace
Distribuce a eliminace inzulínu glulisin a rozpustného rychle účinkujícího inzulínu po intravenózním podání je podobná s distribučními objemy 13 l a 22 l a poločasem eliminace 13 a 18 minut.
Po subkutánním podání je inzulín glulisin eliminován rychleji než rozpustný rychle účinkující inzulín s poločasem eliminace 42 minut ve srovnání s 86 minutami. Ve zkřížené analýze u zdravých jedinců nebo u diabetiků 1. nebo 2. typu byl poločas eliminace v rozmezí od 37 do 75 minut (rozmezí mezi kvartily).
Inzulín glulisin vykazuje nízkou vazbu na plazmatické proteiny stejně jako lidský inzulín.
Porucha funkce ledvin
V klinické studii prováděné u nediabetiků, pokrývající široké rozmezí renálních funkcí
(CrCl > 80 ml/min, 30-50 ml/min, < 30 ml/min), byly obecně zachovány rychle účinkující vlastnosti
inzulínu glulisin. U pacientů s poškozením ledvin však mohou být nároky na inzulín sníženy.
Porucha funkce jater
Farmakokinetické vlastnosti nebyly zkoumány u pacientů se sníženou funkcí jater.
Starší pacienti
U starších pacientů s diabetes mellitus jsou dostupné pouze velmi omezené farmakokinetické údaje.
Děti a mladiství
Farmakokinetické a farmakodynamické vlastnosti inzulínu glulisin byly zkoumány u dětí (7-11 let) a mladistvých (12-16 let) s diabetes mellitus 1. typu. Inzulín glulisin byl rychle absorbován v obou věkových skupinách, s podobným Tmax a Cmax jako u dospělých (viz bod 4.2).
Při podání těsně před testovaným jídlem poskytuje inzulín glulisin stejně jako u dospělých lepší postprandiální kompenzaci než rozpustný rychle účinkující lidský inzulín (viz bod 5.1). Výchylky glukózy (AUC 0-6h) byly 641 mg.h/dl pro insulin glulisin a 801 mg.h/dl pro rozpustný rychle účinkující lidský inzulín.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje neodhalily jiné nálezy toxicity než ty, které jsou spojené s hypoglykemizující farmakodynamickou aktivitou (hypoglykémie), odlišné od rozpustného rychle účinkujícího inzulínu nebo s klinickým významem pro člověka.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Metakresol Chlorid sodný Trometamol Polysorbát 20
Kyselina chlorovodíková 35%
Hydroxid sodný Voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky s výjimkou NPH lidských inzulínů.
6.3 Doba použitelnosti
2 roky
Doba použitelnosti po prvním použití zásobní vložky
Přípravek může být uchováván až 4 týdny při teplotě do 25°C mimo přímé teplo nebo světlo. Pero obsahující zásobní vložku nesmí být skladováno v chladničce. Víčko pera musí být vráceno na pero po každé injekci, aby byl přípravek chráněn před světlem.
Neotevřené zásobní vložky
Uchovávejte v chladničce (2°C - 8°C).
Chraňte před mrazem.
Neukládejte přípravek Apidra do blízkosti mrazicího boxu nebo mrazicí vložky. Uchovávejte v krabičce, aby byl přípravek chráněn před světlem.
Používané zásobní vložky
Podmínky uchovávání tohoto léčivého přípravku po jeho prvním otevření jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
3 ml roztoku v zásobní vložce (bezbarvé sklo typu I) se zátkou (pružnou bromobutylovou gumovou) a pertlem (hliníkovým) a těsněním (pružným bromobutylovým gumovým).
Skleněná zásobní vložka je nevratně včleněna do průhledného pouzdra a spojena na jedné straně s plastovým mechanizmem šroubovým pístem.
Dostupná jsou balení s 1, 3, 4, 5, 6, 8, 9 a 10 zásobními vložkami pro OptiClik. Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Zásobní vložky pro OptiClik jsou určeny pouze k užití ve spojení s OptiClik, jak je doporučeno v informaci od výrobce.
Instrukce od výrobce pro použití pera musí být pečlivě dodržovány pro správné vsunutí zásobní vložky, připojení jehly a aplikaci inzulínové injekce.
Pokud je OptiClik poškozen nebo správně nefunguje (kvůli mechanické závadě), je třeba jej vyřadit a použít nový OptiClik.
Před vložením do pera pro více použití musí být zásobní vložka skladována 1-2 hodiny při pokojové teplotě. Zásobní vložku před použitím prohlédněte. Smí být použita pouze tehdy, je-li neporušená a roztok je čirý, bezbarvý, bez viditelných pevných částic.
Vzduchové bubliny musí být před použitím odstraněny (viz návod na použití pera). Prázdné zásobní vložky nesmí být znovu plněny.
Při špatné funkčnosti pera pro opakované použití (viz návod na použití pera) může být roztok ze zásobní vložky natažen do injekční stříkačky (vhodné pouze pro inzulín 100 jednotek /ml) a aplikován.
Jedno pero smí být používáno výhradně jedním pacientem, aby se zabránilo jakémukoliv druhu kontaminace.
Před každým podáním injekce musí být vždy zkontrolován štítek inzulínu, aby nedošlo k záměně inzulínu glulisin a jiných inzulínů (viz bod 4.4).
Míchání s inzulíny
Pokud se Apidra mísí s NPH lidským inzulínem, měla by být natažena do stříkačky jako první. Injekce by se měla podat okamžitě po smíchání, protože údaje týkající se směsí připravených značnou dobu před injekcí nejsou dostupné.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Sanofi-Aventis Deutschland GmbH D-65926 Frankfurt am Main Německo.
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/04/285/021-028
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 27. září 2004
Datum posledního prodloužení registrace: 20. srpna 2009
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku j sou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/
NÁZEV PŘÍPRAVKU
1.
Apidra 100 jednotek/ml injekční roztok v předplněném peru
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden ml obsahuje 100 jednotek insulinum glulisinum (odpovídá 3,49 mg).
Jedno pero obsahuje 3 ml injekčního roztoku, což odpovídá 300 jednotek.
Inzulín glulisin se vyrábí technologií rekombinace DNA využitím kmenů bakterie Escherichia coli. Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok v předplněném peru. OptiSet.
Čirý bezbarvý vodný roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba dospělých, mladistvých a dětí starších 6 let s diabetes mellitus, kde je nutná léčba inzulínem.
4.2 Dávkování a způsob podání
Dávkování
Síla tohoto přípravku se uvádí v jednotkách. Tyto jednotky se vztahují výhradně k přípravku Apidra a liší se od m.j. (IU) nebo jednotek používaných k vyjádření síly jiných inzulínových analogů (viz bod 5.1).
Apidra by se měla podávat v režimech, které zahrnují středně nebo dlouhodobě účinkující inzulín nebo analog bazálního inzulínu a může být podávána s perorálními antidiabetiky.
Dávka přípravku Apidra by měla být upravena individuálně.
Zvláštní populace
Porucha funkce ledvin
Farmakokinetické vlastnosti inzulínu glulisin jsou obecně u pacientů s renálním poškozením zachovány. Potřeba inzulínu však může být při poškození renálních funkcí snížena (viz bod 5.2).
Porucha funkce jater
Farmakokinetické vlastnosti inzulínu glulisin nebyly zkoumány u pacientů se sníženou funkcí jater.
U pacientů se zhoršením funkce jater může být potřeba inzulínu snížena z důvodu snížené kapacity glukoneogeneze a poklesu inzulínového metabolizmu.
Starší pacienti
U starších pacientů s diabetes mellitus jsou dostupné omezené farmakokinetické údaje. Zhoršení renálních funkcí může vést k poklesu potřeby inzulínu.
Pediatrická populace
Nej sou dostatečné klinické informace o použití přípravku Apidra u dětí mladších 6 let.
Způsob podání
Subkutánní podání
Apidra by se měla podávat subkutánní injekcí krátce (0-15 min) před jídlem nebo brzy po jídle, nebo kontinuální subkutánní infuzní pumpou.
Apidra by se měla podávat subkutánně do břišní stěny, stehna, deltového svalu nebo kontinuální infuzí do břišní stěny. Místa vpichu injekce a infuze v rámci jedné injekční oblasti (břicho, stehno nebo deltový sval) je nutno s každou injekcí střídat. Míra absorpce a následně nástup a trvání účinku mohou být ovlivněny místem vpichu, cvičením a dalšími faktory. Subkutánní injekce do břišní stěny zajišťuje o něco rychlejší absorpci než ostatní injekční místa (viz bod 5.2).
Musí se dávat pozor, aby bylo zajištěno, že nebyla propíchnuta krevní céva. Místo vpichu se po injekci nesmí masírovat. Pacienti musí být poučeni, aby používali správné injekční techniky.
Míchání s inzulíny
Při podávání v subkutánní injekci se Apidra nesmí mísit s žádnými jinými léčivými přípravky s výjimkou NPH inzulínu.
Předtím, než se OptiSet začne používat, je nutné pozorně přečíst návod na použití, který je součástí příbalové informace (viz bod 6.6).
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1. Hypoglykémie.
4.4 Zvláštní upozornění a opatření pro použití
Převedení pacienta na jiný typ nebo značku inzulínu musí být prováděno za přísného lékařského dohledu. Změny síly, značky (výrobce), typu (rozpustný rychle účinkující, neutral protamin Hagedorn [NPH], lente, dlouhodobě působící, atd.), původu (zvířecí, lidský, analog lidského inzulínu) a/nebo způsobu výroby mohou vést k nutnosti změnit dávku. Současná léčba perorálními antidiabetiky může vyžadovat úpravu dávkování.
Hyperglykémie
Podávání neadekvátních dávek nebo přerušení léčby zejména u inzulíndependentních diabetiků může vést k hyperglykémii a diabetické ketoacidóze; tedy k okolnostem, které jsou potenciálně letální.
Hypoglykémie
Doba výskytu hypoglykémie závisí na profilu účinku podaného inzulínu, a proto se může měnit, pokud se mění režim léčby.
Okolnosti, za kterých mohou být včasné varovné symptomy pozměněny nebo být méně výrazné, zahrnují dlouhou anamnézu diabetu, zintenzivnění léčby inzulínem, diabetickou neuropatii, léčivé přípravky jako betablokátory nebo převedení ze zvířecího inzulínu na lidský inzulín.
Úprava dávky může být nezbytná u pacientů, kteří zvýší svou fyzickou aktivitu nebo změní svůj obvyklý stravovací řád. Cvičení prováděné okamžitě po jídle může zvýšit riziko hypoglykémie.
V porovnání s rozpustným lidským inzulínem, pokud se po injekci analogu rychlého inzulínu objeví hypoglykémie, tato se může objevit dříve.
Nekontrolovaná hypoglykemická nebo hyperglykemická reakce může způsobit bezvědomí, kóma nebo smrt.
Potřeba inzulínu může být pozměněna během nemoci nebo emočního rozrušení.
Chyby v medikaci
Byly hlášeny chyby v léčbě, kdy byly omylem podány jiné, zejména dlouhodobě působící, inzulíny namísto inzulínu glulisin. Před každým podáním injekce musí být vždy zkontrolován štítek inzulínu, aby nedošlo k záměně inzulínu glulisin a jiných inzulínů.
Pomocné látky
Tento léčivý přípravek obsahuje méně než 1 mmol (23 mg) sodíku v jedné dávce, tj. v podstatě sodík neobsahuje.
Apidra obsahuje metakresol, který může způsobovat alergické reakce.
Kombinace přípravku Apidra s pioglitazonem
Zejména u pacientů s rizikovými faktory pro vznik srdečního selhání byly při podávání pioglitazonu v kombinaci s inzulínem hlášeny případy srdečního selhání. To je třeba mít na paměti, pokud je zvažována léčba přípravkem Apidra v kombinaci s pioglitazonem. Jestliže je tato kombinace použita, je třeba pacienty sledovat, zda se u nich neobjevují známky a příznaky srdečního selhání, zvýšení hmotnosti a edém. Dojde-li k jakémukoli zhoršení srdečních příznaků, je zapotřebí léčbu pioglitazonem ukončit.
Zacházení s perem
Předtím, než se OptiSet začne používat, je nutné pozorně přečíst návod na použití, který je součástí příbalové informace. OptiSet se musí používat tak, jak je doporučeno v tomto návodu na použití (viz bod 6.6).
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Studie sledující farmakokinetické interakce nebyly prováděny. Na základě empirických znalostí u podobných léčivých přípravků je pravděpodobné, že se neobjeví klinicky relevantní farmakokinetické interakce.
Rada látek ovlivňuje metabolizmus glukózy a může vyžadovat úpravu dávky inzulínu glulisin a zvláště pečlivé sledování.
Mezi látky, které mohou zvýšit hypoglykemizující účinek a zvýšit náchylnost k hypoglykémii, patří perorální antidiabetika, ACE inhibitory, disopyramid, fibráty, fluoxetin, inhibitory monoaminooxidázy (MAOI), pentoxifylin, propoxyfen, salicyláty a sulfonamidová antibiotika.
Mezi látky, které mohou snižovat hypoglykemizující účinek patří kortikosteroidy, danazol, diazoxid, diuretika, glukagon, isoniazid, deriváty fenothiazinu, somatotropin, sympatomimetika (např. epinefrin [adrenalin], salbutamol, terbutalin), tyreoidální hormony, estrogeny a progestogeny (např. perorální kontraceptiva), inhibitory proteáz a atypické antipsychotické léky (např. olanzapin a klozapin).
Betablokátory, klonidin, soli lithia nebo alkohol mohou zesílit nebo zeslabit hypoglykemizující účinek inzulínu. Pentamidin může vyvolat hypoglykémii, po které může někdy následovat hyperglykémie.
Navíc vlivem sympatolytických léčivých přípravků jako jsou betablokátory, klonidin, guanethidin a reserpin, mohou být známky adrenergní kontraregulace sníženy nebo mohou zcela chybět.
4.6 Těhotenství a kojení
Nejsou k dispozici žádné nebo jen omezené údaje (výsledky u méně než 300 těhotenství) o použití inzulínu glulisin u těhotných žen.
Reprodukční studie na zvířatech neodhalily žádné rozdíly mezi inzulínem glulisin a lidským inzulínem co se týče těhotenství, embryofetálního vývoje, porodu nebo postnatálního vývoje (viz bod 5.3).
Při předepisování těhotným ženám je nutno postupovat opatrně. Pečlivé monitorování kompenzace glykémie je nutné.
Je nutné, aby u pacientek s preexistujícím diabetem nebo s těhotenským diabetem byla udržována v průběhu těhotenství dobrá metabolická kontrola. Požadavky na inzulín mohou klesat během prvního trimestru a obecně se zvyšují během druhého a třetího trimestru. Okamžitě po porodu potřeba inzulínu rapidně poklesne.
Kojení
Není známo, zda je inzulín glulisin vylučován do mateřského mléka, ale obecně inzulín nepřestupuje do mateřského mléka a není absorbován po perorálním podání.
Kojící matky mohou vyžadovat úpravu dávky inzulínu a diety.
Fertilita
Reprodukční studie u zvířat neodhalily žádné nežádoucí účinky inzulínu glulisinu na fertilitu.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Pacientova schopnost koncentrace a reakce může být zhoršena následkem hypoglykémie nebo hyperglykémie nebo například následkem poruchy zraku. To může představovat riziko v situacích, kde jsou tyto schopnosti zvláště důležité (např. při řízení vozidla nebo při obsluze strojů).
Pacient by měl být obeznámen s opatřeními, zabraňujícími vzniku hypoglykémie během řízení. To je důležité zejména u těch pacientů, kteří mají sníženou nebo chybějící vnímavost k varovným příznakům hypoglykémie nebo mají časté epizody hypoglykémie. Je třeba zvážit, zda řízení nebo obsluha strojů jsou za těchto okolností vhodné.
4.8 Nežádoucí účinky
Souhrnný bezpečnostní profil
Hypoglykémie, nejčastější nežádoucí účinek inzulínové terapie, se může objevit, jestliže dávka inzulínu je vzhledem k jeho potřebě příliš vysoká.
Tabulkový přehled nežádoucích účinků
Následující nežádoucí účinky, které se vyskytly během klinických hodnocení, byly seřazeny níže systematicky podle skupiny orgánů a podle snižujícího se výskytu (velmi časté: >1/10; časté: >1/100 až <1/10; méně časté: >1/1 000 až <1/100; vzácné >1/10 000 až <1/1 000; velmi vzácné: <1/10 000), není známo (z dostupných údajů nelze určit).
V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
|
MedDRA Třídy orgánových systémů |
Velmi časté |
Časté |
Méně časté |
Vzácné |
|
Poruchy metabolismu a výživy |
Hypoglykémie | |||
|
Poruchy kůže a podkožní tkáně |
Reakce v místě vpichu Lokální hypersenzitivní reakce |
Lipodystrofie | ||
|
Celkové poruchy a reakce v místě aplikace |
Systémová hypersenzitivní reakce |
Popis vybraných nežádoucích účinků
Poruchy metabolismu a výživy
Symptomy hypoglykémie se obvykle objeví náhle. Mohou zahrnovat studený pot, chladnou bledou kůži, únavu, nervozitu nebo třes, úzkost, neobvyklou únavu nebo slabost, zmatenost, poruchy koncentrace, ospalost, vlčí hlad, poruchy vidění, bolest hlavy, nauseu a palpitace.
Hypoglykémie se může stát závažnou a může vést k bezvědomí a/nebo křečím a k přechodnému nebo trvalému poškození mozkových funkcí nebo dokonce ke smrti.
Poruchy kůže a podkožní tkáně
Lokální reakce z přecitlivělosti (zčervenání, otok a svědění v místě vpichu) se mohou objevit v průběhu léčby inzulínem. Tyto reakce jsou obvykle přechodné a v průběhu pokračující léčby vymizí.
Lipodystrofie se může objevit v místě vpichu následkem nedodržení plynulého střídání místa vpichu v rámci oblasti aplikace.
Celkové poruchy a reakce v místě aplikace
Systémové reakce z přecitlivělosti mohou zahrnovat kopřivku, pocit tíhy na hrudi, dušnost, alergickou dermatitidu a svědění. Těžké případy generalizované alergické reakce včetně anafylaktické reakce mohou být život ohrožující.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Příznaky
Hypoglykémie se může objevit jako výsledek nadměrné inzulínové aktivity vzhledem k příjmu potravy a výdeji energie.
Nejsou k dispozici specifické údaje týkající se předávkování inzulínem glulisin. Hypoglykémie se však může rozvinout přes následující stádia:
Léčba
Mírné hypoglykemické epizody mohou být zvládnuty perorálním podáním glukózy nebo produktů obsahujících cukr. Doporučuje se proto, aby diabetici u sebe neustále nosili kostky cukru, sladkosti, sušenky nebo sladkou ovocnou šťávu.
Těžké hypoglykemické epizody, kdy pacient upadne do bezvědomí, mohou být léčeny glukagonem (0,5 mg až 1 mg) podaným intramuskulárně nebo subkutánně někým, kdo dostal patřičné instrukce, nebo glukózou podanou intravenózně zdravotnickým pracovníkem. Glukóza musí být podána intravenózně také tehdy, pokud u pacienta nenastane odpověď na glukagon během 10 až 15 minut.
Jakmile pacient znovu nabude vědomí, doporučuje se z důvodu prevence relapsu podání sacharidů perorálně.
Po injekci glukagonu by měl být pacient sledován v nemocnici z důvodu zjištění příčiny tak těžké hypoglykémie a z důvodu prevence dalších podobných epizod.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Léčiva k terapii diabetu, inzulíny a analogy rychle působící, k injekční aplikaci, ATC kód: A10AB06.
Mechanismus účinku
Inzulín glulisin je rekombinantní analog lidského inzulínu, který je ekvipotentní k normálnímu lidskému inzulínu. Inzulín glulisin má rychlejší nástup účinku a kratší trvání účinku než běžný lidský inzulín.
Primárním účinkem inzulínu a inzulínových analog včetně inzulínu glulisin je regulace metabolismu glukózy. Inzulíny snižují hladinu glukózy v krvi stimulací periferního vychytávání glukózy zvláště kosterními svaly a tukem a inhibicí glukoneogeneze v játrech. Inzulín inhibuje lipolýzu v tukových buňkách, inhibuje proteolýzu a podporuje syntézu proteinů.
Studie u zdravých dobrovolníků a pacientů s diabetem ukázaly, že inzulín glulisin, je-li podán subkutánně, je rychlejší v nástupu a kratší v trvání účinku než běžný lidský inzulín. Pokud je inzulín glulisin podán subkutánní injekcí, hypoglykemizující účinek nastupuje během 10 - 20 minut. Účinky na snížení glykémie jsou u inzulínu glulisin a běžného lidského inzulínu při podání intravenózní cestou ekvipotentní. Jedna jednotka inzulínu glulisin má stejný hypoglykemizující účinek jako jedna dávka běžného lidského inzulínu.
Proporcionalita dávky
Ve studii s 18 muži s diabetes mellitus 1. typu ve věku od 21 do 50 let snižoval inzulín glulisin hladinu glukózy úměrně podané dávce v terapeuticky relevantním dávkovém rozmezí 0,075 až 0,15 jednotky/kg. Při dávkách 0,3 jednotky/kg nebo vyšších byl hypoglykemický účinek inzulínu glulisinu nižší, než by úměrně odpovídalo dávce, stejně jako u lidského inzulínu.
Nástup účinku inzulínu glulisin je dvakrát rychlejší než u běžného lidského inzulínu a jeho hypoglykemický efekt končí asi o 2 hodiny dříve než u běžného lidského inzulínu.
Studie I. fáze u pacientů s diabetes mellitus 1. typu hodnotily hypoglykemizující profily inzulínu glulisin a rozpustného rychle účinkujícího inzulínu podaného subkutánně v dávce 0,15 jednotky/kg v různé době ve vztahu k 15minutovému standardnímu jídlu. Data ukázala, že inzulín glulisin podaný 2 minuty před jídlem, poskytuje v porovnání s rozpustným rychle účinkujícím lidským inzulínem podaným 30 minut před jídlem podobnou postprandiální kompenzaci. Je-li podán 2 minuty před jídlem, poskytuje inzulín glulisin lepší postprandiální kompenzaci než rozpustný rychle účinkující lidský inzulín podaný 2 minuty před jídlem. Inzulín glulisin podaný 15 minut po začátku jídla poskytuje podobnou glykemickou kompenzaci jako rozpustný rychle účinkující lidský inzulín podaný 2 minuty před jídlem (viz obrázek 1).
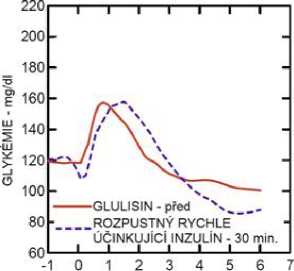
Čas - hodiny
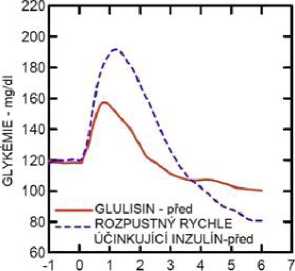
Čas - hodiny
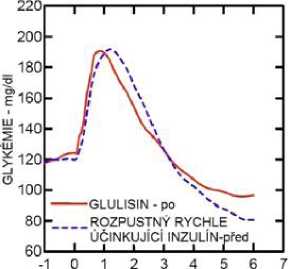
Čas - hodiny
Obrázek 1: Průměrný hypoglykemizující účinek po dobu 6 hodin u 20 pacientů s diabetes mellitus 1. typu. Inzulín glulisin podaný 2 minuty (Glulisin před) před začátkem jídla v porovnání s běžným lidským inzulínem podaným 30 minut (Rozpustný rychle účinkující inzulín-30 min) před začátkem jídla (obrázek 1A) a v porovnání s běžným lidským inzulínem podaným 2 minuty (Rozpustný rychle účinkující inzulín před) před jídlem (obrázek 1B). Inzulín glulisin podaný 15 minut (Glulisin po) po začátku jídla v porovnání s běžným inzulínem podaným 2 minuty (Rozpustný rychle účinkující inzulín před) před začátkem jídla (obrázek 1C). Na ose x je nula (šipka) začátek jídla trvajícího 15 minut.
Obezita
Studie I. fáze prováděná s inzulínem glulisin, inzulínem lispro a běžným lidským inzulínem u obézní populace ukázala, že inzulín glulisin si zachovává své rychle účinkující vlastnosti. V této studii byl čas představující časnou hypoglykemizující aktivitu až 20 % celkové AUC a AUC (0 -2 h) a sice 114 minut a 427 mg/kg" u inzulínu glulisin, 121 minut a 354 mg/kg" pro lispro a 150 minut a 197 mg/kg pro běžný lidský inzulín (viz obrázek 2).
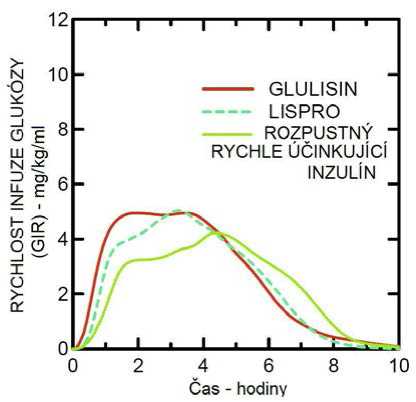
Obrázek 2: Rychlost infuze glukózy (GIR) po subkutánní injekci 0,3 jednotky/kg inzulínu glulisin (GLULISIN) nebo inzulínu lispro (LISPRO) nebo normálního lidského inzulínu (Rozpustný rychle účinkující inzulín) u obézní populace.
Další studie I. fáze s inzulínem glulisin a inzulínem lispro u nediabetické populace 80 pacientů s širokým rozmezím indexů tělesné hmotnosti (18-46 kg/m2) ukázala, že k rychlému nástupu účinku docházelo napříč širokým rozmezím indexů tělesné hmotnosti (BMI), zatímco celkový hypoglykemický efekt klesá se zvyšující se obezitou.
Průměrná plocha pod křivkou (AUC) pro celkovou GIR (glucose infusion rate- rychlost infuze glukózy) mezi 0-1. hodinou byla 102±75 mg/kg s 0,2 jednotky/kg inzulínu glulisin a 158±100 mg/kg s 0,4 jednotky/kg inzulínu glulisin a 83,1±72,8 mg/kg s 0,2 jednotky/kg inzulínu lispro a 112,3±70,8 mg/kg s 0,4 jednotky/kg inzulínu lispro.
Studie I. fáze u 18 obézních pacientů s diabetem mellitus 2. typu (BMI mezi 35 a 40 kg/m2) s inzulínem glulisin a inzulínem lispro [90% CI: 0,81, 0,95 (p=<0,01)] ukázala, že inzulín glulisin efektivně kontroluje diurnální postprandiální výkyvy hladiny glukózy v krvi.
Klinická účinnost a bezpečnost
Diabetes mellitus 1. typu - dospělí
Ve 26týdenní klinické studii fáze III srovnávající inzulín glulisin s inzulínem lispro podávanými subkutánně krátce (0-15 minut) před jídlem u pacientů s diabetes mellitus 1. typu užívajících inzulín glargin jako bazální inzulín, byl inzulín glulisin, co do kompenzace glykémie, srovnatelný s inzulínem lispro, jak ukazují změny glykovaného hemoglobinu od začátku do konce studie (vyjádřené jako HbA1c ekvivalent). Při selfmonitoringu byly pozorovány srovnatelné hodnoty glykémie. Na rozdíl od inzulínu lispro nebylo potřeba při terapii inzulínem glulisin zvyšovat dávku bazálního inzulínu.
12týdenní klinická studie fáze III prováděná u pacientů s diabetes mellitus 1. typu, kteří dostávali jako bazální terapii inzulín glargin, ukazuje, že podání inzulínu glulisin bezprostředně po jídle poskytuje srovnatelnou účinnost jako inzulín glulisin bezprostředně (0-15 minut) před jídlem nebo jako rozpustný rychle účinkující inzulín (30-45 minut).
V „per protokol“ populaci bylo ve skupině s glulisinem před jídlem pozorováno signifikantně větší snížení hodnot glykovaného hemoglobinu než ve skupině s rozpustným rychle účinkujícím inzulínem.
Diabetes mellitus 1. typu - děti
26týdenní klinická studie fáze III srovnávala subkutánní aplikaci inzulínu glulisin s inzulínem lispro podanými krátce (0-15 minut) před jídlem u dětí (4 - 5 let: n = 9; 6 - 7 let: n = 32 a 8 - 11 let: n = 149) a mladistvých (12-17 let: n = 382) s diabetes mellitus 1. typu, kterým byl aplikován jako bazální inzulín glargin nebo NPH inzulín. S inzulínem glulisin bylo dosaženo srovnatelné glykemické kontroly hodnocené změnou glykovaného hemoglobinu (GHb vyjádřený jako ekvivalent HbA1c) od zahájení studie do jejího ukončení a hodnotami glukózy v krvi zjištěnými při selfmonitoringu jako s inzulínem lispro.
Nejsou dostatečné klinické informace o použití přípravku Apidra u dětí mladších 6 let.
Diabetes mellitus 2. typu - dospělí
26týdenní klinická studie fáze III následovaná rozšířením o 26týdenní studii bezpečnosti byla prováděna za účelem srovnání inzulínu glulisin (0-15 minut před jídlem) s rozpustným rychle účinkujícím lidským inzulínem (30-45 minut před jídlem) při subkutánním podání u pacientů s diabetes mellitus 2. typu užívajících NPH inzulín jako bazální inzulín. Průměrný body mass index (BMI) u pacientů byl 34,55 kg/m2. Inzulín glulisin ukázal, že je srovnatelný s rozpustným rychle účinkujícím lidským inzulínem, pokud se týče změn glykovaného hemoglobinu (vyjádřené jako HbA1c ekvivalent) od začátku do konce 6-měsíční studie (- 0,46 % u inzulínu glulisin a - 0,30 % u rozpustného rychle účinkujícího lidského inzulínu) a od začátku do konce 12-měsíční studie (- 0,23 % u inzulínu glulisin a - 0,13 % u rozpustného rychle účinkujícího lidského inzulínu, rozdíl není významný). V této studii většina pacientů (79 %) mísila jejich rychle účinkující inzulín s NPH inzulínem bezprostředně před injekcí a 58 % pacientů užívalo v době randomizace perorální antidiabetika. Tito pacienti byli instruováni, aby v jejich užívání pokračovali ve stejných dávkách.
Rasa a pohlaví
V kontrolovaných klinických studiích u dospělých neprokázal inzulín glulisin rozdíly v bezpečnosti a účinnosti v podskupinové analýze založené na rase a pohlaví.
5.2 Farmakokinetické vlastnosti
Náhrada aminokyseliny asparagin na pozici B3 lidského inzulínu lysinem a lysinu na pozici B29 kyselinou glutamovou zajišťuje u inzulínu glulisin rychlejší absorpci.
Ve studii s 18 muži s diabetes mellitus 1. typu ve věku od 21 do 50 let inzulín glulisin vykazuje pro časnou, maximální a celkovou expozici v dávkovém rozmezí 0,075 až 0,4 jednotky/kg dávkovou proporcionalitu.
Absorpce a biologická dostupnost
Farmakokinetické profily u zdravých dobrovolníků a diabetiků (1. a 2. typu) ukázaly, že absorpce inzulínu glulisin byla asi dvakrát rychlej ší s přibližně dvakrát tak vysokou vrcholovou koncentrací ve srovnání s rozpustným rychle účinkujícím inzulínem.
Ve studii u pacientů s diabetes mellitus 1. typu po subkutánním podání 0,15 U/kg byl pro inzulín glulisin Tmax 55 minut a Cmax 82 ± 1,3 gU/ml ve srovnání s Tmax 82 minut a Cmax 46 ± 1,3 gU/ml pro rozpustný rychle účinkující inzulín. Průměrný čas výskytu v organismu u inzulínu glulisin byl kratší (98 minut) než pro rozpustný rychle účinkující inzulín (161 minut) (viz obrázek 3).
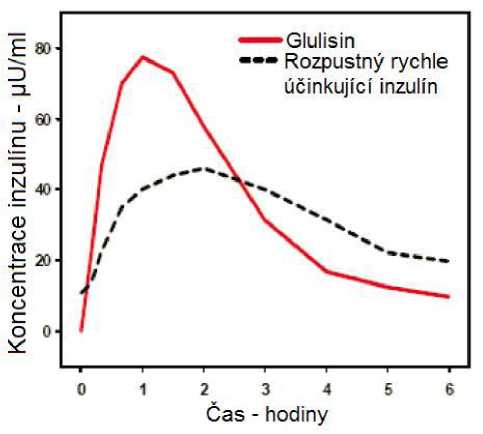
Obrázek 3: Farmakokinetický profil inzulínu glulisin a rozpustného rychle účinkujícího inzulínu u pacientů s diabetes mellitus 1. typu po dávce 0,15 jednotky/kg.
Ve studii u pacientů s diabetes mellitus 2. typu po subkutánním podání 0,2 jednotky/kg inzulínu glulisin byla Cmax 91 gjednotky/ml s rozmezím mezi kvartily od 78 do 104 gjednotky/ml.
Pokud byl inzulín glulisin aplikován subkutánně do břišní stěny, deltového svalu a stehna, profily koncentrace-čas byly podobné s mírně rychlejší absorpcí při podání do břišní stěny ve srovnání s podáním do stehna. Absorpce z deltového svalu byla střední (viz bod 4.2). Absolutní biologická dostupnost (70%) inzulínu glulisin byla v rámci míst aplikace podobná a s nízkou variabilitou mezi jednotlivými jedinci (11% CV). V porovnání se subkutánní injekcí vedlo podání intravenózního bolusu inzulínu glulisinu k vyšší systémové expozici, hodnota Cmax byla přibližně 40krát vyšší.
Obezita
Další studie I. fáze s inzulínem glulisin a inzulínem lispro u nediabetické populace 80 pacientů s širokým rozmezím indexů tělesné hmotnosti (18-46 kg/m2) ukázala, že k rychlému nástupu účinku dochází napříč širokým rozmezím indexů tělesné hmotnosti a že celková expozice je napříč širokým rozmezím indexů tělesné hmotnosti zachována.
Doba do 10% celkové INS expozice byla s inzulínem glulisin dosažena o asi 5-6 minut dříve. Distribuce a eliminace
Distribuce a eliminace inzulínu glulisin a rozpustného rychle účinkujícího inzulínu po intravenózním podání je podobná s distribučními objemy 13 l a 22 l a poločasem eliminace 13 a 18 minut.
Po subkutánním podání je inzulín glulisin eliminován rychleji než rozpustný rychle účinkující inzulín s poločasem eliminace 42 minut ve srovnání s 86 minutami. Ve zkřížené analýze u zdravých jedinců nebo u diabetiků 1. nebo 2. typu byl poločas eliminace v rozmezí od 37 do 75 minut (rozmezí mezi kvartily).
Inzulín glulisin vykazuje nízkou vazbu na plazmatické proteiny stejně jako lidský inzulín.
Zvláštní populace
Porucha funkce ledvin
V klinické studii prováděné u nediabetiků, pokrývající široké rozmezí renálních funkcí
(CrCl > 80 ml/min, 30-50 ml/min, < 30 ml/min), byly obecně zachovány rychle účinkující vlastnosti
inzulínu glulisin. U pacientů s poškozením ledvin však mohou být nároky na inzulín sníženy.
Porucha funkce jater
Farmakokinetické vlastnosti nebyly zkoumány u pacientů se sníženou funkcí jater.
Starší pacienti
U starších pacientů s diabetes mellitus jsou dostupné pouze velmi omezené farmakokinetické údaje.
Děti a mladiství
Farmakokinetické a farmakodynamické vlastnosti inzulínu glulisin byly zkoumány u dětí (7-11 let) a mladistvých (12-16 let) s diabetes mellitus 1. typu. Inzulín glulisin byl rychle absorbován v obou věkových skupinách, s podobným Tmax a Cmax jako u dospělých (viz bod 4.2).
Při podání těsně před testovaným jídlem poskytuje inzulín glulisin stejně jako u dospělých lepší postprandiální kompenzaci než rozpustný rychle účinkující lidský inzulín (viz bod 5.1). Výchylky glukózy (AUC 0-6h) byly 641 mg.h/dl pro insulin glulisin a 801 mg.h/dl pro rozpustný rychle účinkující lidský inzulín.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje neodhalily jiné nálezy toxicity než ty, které jsou spojené s hypoglykemizující farmakodynamickou aktivitou (hypoglykémie), odlišné od rozpustného rychle účinkujícího inzulínu nebo s klinickým významem pro člověka.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Metakresol Chlorid sodný Trometamol Polysorbát 20
Kyselina chlorovodíková 35%
Hydroxid sodný Voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být léčivý přípravek mísen s jinými léčivými přípravky s výjimkou NPH lidských inzulínů.
6.3 Doba použitelnosti
2 roky
Doba použitelnosti po prvním použití pera
Přípravek může být uchováván až 4 týdny při teplotě do 25°C mimo přímé teplo nebo světlo. Používaná pera nesmí být skladována v chladničce. Víčko pera musí být vráceno na pero po každé injekci, aby byl přípravek chráněn před světlem.
6.4 Zvláštní opatření pro uchovávání
Nepoužívaná pera
Uchovávejte v chladničce (2°C - 8°C).
Chraňte před mrazem.
Neukládejte přípravek Apidra do blízkosti mrazicího boxu nebo mrazicí vložky.
Uchovávejte v krabičce, aby byl přípravek chráněn před světlem.
Používaná pera
Podmínky uchovávání tohoto léčivého přípravku po jeho prvním otevření jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
3 ml roztoku v zásobní vložce (bezbarvé sklo) se zátkou (pružná bromobutylová guma) a pertlem (hliník) s těsněním (pružná bromobutylová guma). Zásobní vložka je uzavřena v předplněném peru k jednorázovému použití. Dostupná jsou balení s 1, 3, 4, 5, 6, 8, 9 a 10 pery.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Před prvním použitím musí být pero skladováno 1-2 hodiny při pokojové teplotě.
Zásobní vložku před použitím prohlédněte. Smí být použita pouze tehdy, je-li roztok čirý, bezbarvý, bez viditelných pevných částic a má-li konzistenci vody. Vzhledem k tomu, že Apidra je roztok, není potřeba ho před použitím znovu protřepávat.
Prázdná pera nesmí být znovu použita a musí být zlikvidována odpovídajícím způsobem.
Jedno pero smí být používáno výhradně jedním pacientem, aby se zabránilo jakémukoliv druhu kontaminace.
Před každým podáním injekce musí být vždy zkontrolován štítek inzulínu, aby nedošlo k záměně inzulínu glulisin a jiných inzulínů (viz bod 4.4).
Míchání s inzulíny
Pokud se Apidra mísí s NPH lidským inzulínem, měla by být natažena do stříkačky jako první. Injekce by se měla podat okamžitě po smíchání, protože údaje týkající se směsí připravených značnou dobu před injekcí nejsou dostupné.
Zacházení s perem
Pacientovi je třeba doporučit, aby si před použitím OptiSet pozorně přečetl návod, který je součástí příbalové informace.
Schematický nákres pera
Důležité informace pro použití OptiSet:
• Před každým použitím musí být vždy nasazena nová jehla. Smí být použity pouze jehly určené pro OptiSet.
• Před každou injekcí musí být vždy provedena kontrola bezpečnosti.
• Pokud je používán nový OptiSet, musí být provedena počáteční kontrola bezpečnosti s 8 jednotkami přednastavenými výrobcem.
• Voličem dávky j e možné otáčet pouze j edním směrem.
• Nikdy se nesmí otáčet voličem dávky (měnit dávku) poté, co bylo vysunuto injekční tlačítko.
• Toto pero je určeno pouze pro použití určitým pacientem. Nesmí být sdíleno s nikým dalším.
• Pokud je injekce dávána jinou osobou, musí si tato osoba počínat zvlášť opatrně, aby se
náhodně jehlou neporanila a nedošlo k přenosu infekce.
• OptiSet se nesmí nikdy použít, pokud je poškozený, nebo není jisté, že funguje správně.
• Pacient musí mít vždy k dispozici náhradní OptiSet pro případy, že se OptiSet ztratí nebo bude poškozen.
Pokyny pro uchovávání
Prosím, pro informace o uchovávání OptiSet si přečtěte bod 6.4 tohoto SPC.
Pokud je OptiSet skladován v chladu, je třeba ho vyjmout 1 až 2 hodiny před použitím, aby se mohl ohřát na pokojovou teplotu. Injikování studeného inzulínu je více bolestivé.
Použitý OptiSet musí být zlikvidován podle pokynů místních úřadů.
Údržba
OptiSet musí být chráněn před prachem a špínou.
OptiSet je možné čistit zvenčí otíráním vlhkým hadříkem.
Pero se nesmí máčet, mýt ani promazávat, mohlo by tím být poškozeno.
OptiSet je navržen tak, aby pracoval přesně a bezpečně. Musí s ním být zacházeno s opatrností. Pacienti by se měli vyhnout situacím, kdy může být OptiSet poškozen. Pokud se pacient domnívá, že je jeho OptiSet poškozen, musí použít nový.
Krok 1 Kontrola inzulínu
Po odstranění krytu pera je nutné zkontrolovat označení na peru a na zásobníku inzulínu, aby bylo jisté, že obsahuje správný inzulín.
Vzhled inzulínu musí být také zkontrolován: roztok inzulínu musí být čirý, bezbarvý, bez viditelných pevných částic a musí mít konzistenci vody.
Nepoužívejte tento OptiSet, pokud je inzulín zakalený, zabarvený, nebo obsahuje viditelné částice.
Krok 2 Upevnění jehly
Jehla se opatrně upevní přímo na pero.
Krok 3 Provedení kontroly bezpečnosti
Před každou injekcí je třeba provést kontrolu bezpečnosti.
U nového a nepoužitého OptiSet je dávka 8 jednotek již předvolena výrobcem pro provedení první kontroly bezpečnosti.
U používaného OptiSet musí být navolena dávka 2 jednotky otočením voliče dávky dopředu, dokud ukazatel dávky neukazuje na 2. Volič dávky se bude otáčet pouze v jednom směru.
Pro natažení dávky je zapotřebí úplně vytáhnout injekční tlačítko. Nikdy se nesmí otáčet voličem dávky poté, co bylo injekční tlačítko vytaženo.
Vnější a vnitřní kryt jehly se musí odstranit. Vnější kryt uchovejte pro odstranění použité jehly.
Zatímco se pero drží tak, aby jehla směřovala nahoru, poklepe se prsty na zásobník inzulínu, tak, že všechny vzduchové bubliny vystoupají nahoru k jehle.
Potom se stiskne injekční tlačítko nadoraz.
Jestliže byl inzulín protlačen hrotem jehly, pak pero a jehla fungují správně.
Pokud se žádný inzulín neobjeví na hrotu jehly, je třeba opakovat ještě dvakrát krok 3, dokud se inzulín na hrotu jehly neobjeví. Pokud se ani poté inzulín neobjeví, vyměňte jehlu, protože může být ucpaná, a postup opakujte. Pokud se inzulín neobjeví ani po výměně jehly, OptiSet může být poškozen. Tento OptiSet se nesmí použít.
Krok 4 Nastavení dávky
Dávka se může nastavit v krocích po 2 jednotkách, od minimální dávky 2 jednotky až po maximální dávku 40 jednotek. Pokud je požadována dávka větší než 40 jednotek, musí se podat jako dvě a více injekcí.
Pacient musí vždy zkontrolovat, zda má dostatek inzulínu pro dávku.
Stupnice zbývajícího inzulínu viditelná na průhledném inzulínovém zásobníku přibližně ukazuje, kolik inzulínu zbývá v OptiSet. Tato stupnice nesmí být používána pro nastavení dávky inzulínu.
Jestliže je černý píst na začátku barevného proužku, zbývá ještě přibližně 40 jednotek inzulínu.
Jestliže je černý píst na konci barevného proužku, zbývá ještě přibližně 20 jednotek inzulínu.
Voličem dávky se otáčí dopředu, dokud ukazatel dávky nesměřuje na požadovanou dávku.
Krok 5 Natažení dávky
Aby se pero naplnilo, musí být injekční tlačítko co nejvíce vytaženo.
Pacient musí zkontrolovat, zda se natáhla celá zvolená dávka. Injekční tlačítko jde vytáhnout jen tak daleko, kolik inzulínu zbývá v zásobníku.
Injekční tlačítko dovoluje kontrolu aktuálně natažené dávky. Injekční tlačítko musí být během kontroly pod tlakem vytaženo. Poslední tlustá viditelná čárka na injekčním tlačítku ukazuje množství nataženého inzulínu. Když je injekční tlačítko vytaženo, je vidět pouze horní část této tlusté čáry.
Krok 6 Injekce dávky
Pacient musí být svým lékařem informován o injekčních technikách.
Jehla se vpíchne do kůže.
Injekční tlačítko se úplně stiskne. Bude slyšet klapavý zvuk, který ustane, jakmile je injekční tlačítko úplně stisknuté. Potom se drží tlačítko stisknuté 10 vteřin, než se jehla vytáhne z kůže. Tak se zajistí, že byla podána celá dávka inzulínu.
Krok 7 Odstranění a likvidace jehly
Jehla se musí odstranit po každé injekci a vyhodit. To pomáhá zabránit kontaminaci a/nebo infekci, stejně jako proniknutí vzduchu do zásobníku inzulínu a vytékání inzulínu, což by mohlo způsobit nepřesné dávkování. Jehly nesmí být znovu použity.
Na pero se znovu nasadí kryt pera.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Sanofi-Aventis Deutschland GmbH D-65926 Frankfurt am Main Německo.
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/04/285/013-020
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 27. září 2004
Datum posledního prodloužení registrace: 20. srpna 2009
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/
NÁZEV PŘÍPRAVKU
1.
Apidra SoloStar 100 jednotek/ml injekční roztok v předplněném peru
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden ml obsahuje 100 jednotek insulinum glulisinum (odpovídá 3,49 mg).
Jedno pero obsahuje 3 ml injekčního roztoku, což odpovídá 300 jednotek.
Inzulín glulisin se vyrábí technologií rekombinace DNA využitím kmenů bakterie Escherichia coli. Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok v předplněném peru.
Čirý bezbarvý vodný roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba dospělých, mladistvých a dětí starších 6 let s diabetes mellitus, kde je nutná léčba inzulínem.
4.2 Dávkování a způsob podání
Dávkování
Síla tohoto přípravku se uvádí v jednotkách. Tyto jednotky se vztahují výhradně k přípravku Apidra a liší se od m.j. (IU) nebo jednotek používaných k vyjádření síly jiných inzulínových analogů (viz bod 5.1).
Apidra by se měla podávat v režimech, které zahrnují středně nebo dlouhodobě účinkující inzulín nebo analog bazálního inzulínu a může být podávána s perorálními antidiabetiky.
Dávka přípravku Apidra by měla být upravena individuálně.
Zvláštní populace
Porucha funkce ledvin
Farmakokinetické vlastnosti inzulínu glulisin jsou obecně u pacientů s renálním poškozením zachovány. Potřeba inzulínu však může být při poškození renálních funkcí snížena (viz bod 5.2).
Porucha funkce jater
Farmakokinetické vlastnosti inzulínu glulisin nebyly zkoumány u pacientů se sníženou funkcí jater. U pacientů se zhoršením funkce jater může být potřeba inzulínu snížena z důvodu snížené kapacity glukoneogeneze a poklesu inzulínového metabolizmu.
Starší pacienti
U starších pacientů s diabetes mellitus jsou dostupné omezené farmakokinetické údaje. Zhoršení renálních funkcí může vést k poklesu potřeby inzulínu.
Pediatrická populace
Nej sou dostatečné klinické informace o použití přípravku Apidra u dětí mladších 6 let.
Způsob podání
Subkutánní podání
Apidra by se měla podávat subkutánní injekcí krátce (0-15 min) před jídlem nebo brzy po jídle, nebo kontinuální subkutánní infuzní pumpou.
Apidra by se měla podávat subkutánně do břišní stěny, stehna, deltového svalu nebo kontinuální infuzí do břišní stěny. Místa vpichu injekce a infuze v rámci jedné injekční oblasti (břicho, stehno nebo deltový sval) je nutno s každou injekcí střídat. Míra absorpce a následně nástup a trvání účinku mohou být ovlivněny místem vpichu, cvičením a dalšími faktory. Subkutánní injekce do břišní stěny zajišťuje o něco rychlejší absorpci než ostatní injekční místa (viz bod 5.2).
Musí se dávat pozor, aby bylo zajištěno, že nebyla propíchnuta krevní céva. Místo vpichu se po injekci nesmí masírovat. Pacienti musí být poučeni, aby používali správné injekční techniky.
Míchání s inzulíny
Při podávání v subkutánní injekci se Apidra nesmí mísit s žádnými jinými léčivými přípravky s výjimkou NPH inzulínu.
Předtím, než se SoloStar začne používat, je nutné pozorně přečíst návod na použití, který je součástí příbalové informace (viz bod 6.6).
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1. Hypoglykémie.
4.4 Zvláštní upozornění a opatření pro použití
Převedení pacienta na jiný typ nebo značku inzulínu musí být prováděno za přísného lékařského dohledu. Změny síly, značky (výrobce), typu (rozpustný rychle účinkující, neutral protamin Hagedorn [NPH], lente, dlouhodobě působící, atd.), původu (zvířecí, lidský, analog lidského inzulínu) a/nebo způsobu výroby mohou vést k nutnosti změnit dávku. Současná léčba perorálními antidiabetiky může vyžadovat úpravu dávkování.
Hyperglykémie
Podávání neadekvátních dávek nebo přerušení léčby zejména u inzulíndependentních diabetiků může vést k hyperglykémii a diabetické ketoacidóze; tedy k okolnostem, které jsou potenciálně letální.
Hypoglykémie
Doba výskytu hypoglykémie závisí na profilu účinku podaného inzulínu, a proto se může měnit, pokud se mění režim léčby.
Okolnosti, za kterých mohou být včasné varovné symptomy pozměněny nebo být méně výrazné, zahrnují dlouhou anamnézu diabetu, zintenzivnění léčby inzulínem, diabetickou neuropatii, léčivé přípravky jako betablokátory nebo převedení ze zvířecího inzulínu na lidský inzulín.
Úprava dávky může být nezbytná u pacientů, kteří zvýší svou fyzickou aktivitu nebo změní svůj obvyklý stravovací řád. Cvičení prováděné okamžitě po jídle může zvýšit riziko hypoglykémie.
V porovnání s rozpustným lidským inzulínem, pokud se po injekci analogu rychlého inzulínu objeví hypoglykémie, tato se může objevit dříve.
Nekontrolovaná hypoglykemická nebo hyperglykemická reakce může způsobit bezvědomí, kóma nebo smrt.
Potřeba inzulínu může být pozměněna během nemoci nebo emočního rozrušení.
Chyby v medikaci
Byly hlášeny chyby v léčbě, kdy byly omylem podány jiné, zejména dlouhodobě působící, inzulíny namísto inzulínu glulisin. Před každým podáním injekce musí být vždy zkontrolován štítek inzulínu, aby nedošlo k záměně inzulínu glulisin a jiných inzulínů.
Pomocné látky
Tento léčivý přípravek obsahuje méně než 1 mmol (23 mg) sodíku v jedné dávce, tj. v podstatě sodík neobsahuje.
Apidra obsahuje metakresol, který může způsobovat alergické reakce.
Kombinace přípravku Apidra s pioglitazonem
Zejména u pacientů s rizikovými faktory pro vznik srdečního selhání byly při podávání pioglitazonu v kombinaci s inzulínem hlášeny případy srdečního selhání. To je třeba mít na paměti, pokud je zvažována léčba přípravkem Apidra v kombinaci s pioglitazonem. Jestliže je tato kombinace použita, je třeba pacienty sledovat, zda se u nich neobjevují známky a příznaky srdečního selhání, zvýšení hmotnosti a edém. Dojde-li k jakémukoli zhoršení srdečních příznaků, je zapotřebí léčbu pioglitazonem ukončit.
Zacházení s perem
Předtím, než se SoloStar začne používat, je nutné pozorně přečíst návod na použití, který je součástí příbalové informace. SoloStar se musí používat tak, jak je doporučeno v tomto návodu na použití (viz bod 6.6).
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Studie sledující farmakokinetické interakce nebyly prováděny. Na základě empirických znalostí u podobných léčivých přípravků je pravděpodobné, že se neobjeví klinicky relevantní farmakokinetické interakce.
Rada látek ovlivňuje metabolizmus glukózy a může vyžadovat úpravu dávky inzulínu glulisin a zvláště pečlivé sledování.
Mezi látky, které mohou zvýšit hypoglykemizující účinek a zvýšit náchylnost k hypoglykémii, patří perorální antidiabetika, ACE inhibitory, disopyramid, fibráty, fluoxetin, inhibitory monoaminooxidázy (MAOI), pentoxifylin, propoxyfen, salicyláty a sulfonamidová antibiotika.
Mezi látky, které mohou snižovat hypoglykemizující účinek patří kortikosteroidy, danazol, diazoxid, diuretika, glukagon, isoniazid, deriváty fenothiazinu, somatotropin, sympatomimetika (např. epinefrin [adrenalin], salbutamol, terbutalin), tyreoidální hormony, estrogeny a progestogeny (např. perorální kontraceptiva), inhibitory proteáz a atypické antipsychotické léky (např. olanzapin a klozapin).
Betablokátory, klonidin, soli lithia nebo alkohol mohou zesílit nebo zeslabit hypoglykemizující účinek inzulínu. Pentamidin může vyvolat hypoglykémii, po které může někdy následovat hyperglykémie.
Navíc vlivem sympatolytických léčivých přípravků jako jsou betablokátory, klonidin, guanethidin a reserpin, mohou být známky adrenergní kontraregulace sníženy nebo mohou zcela chybět.
4.6 Těhotenství a kojení
Nejsou k dispozici žádné nebo jen omezené údaje (výsledky u méně než 300 těhotenství) o použití inzulínu glulisin u těhotných žen.
Reprodukční studie na zvířatech neodhalily žádné rozdíly mezi inzulínem glulisin a lidským inzulínem co se týče těhotenství, embryofetálního vývoje, porodu nebo postnatálního vývoje (viz bod 5.3).
Při předepisování těhotným ženám je nutno postupovat opatrně. Pečlivé monitorování kompenzace glykémie je nutné.
Je nutné, aby u pacientek s preexistujícím diabetem nebo s těhotenským diabetem byla udržována v průběhu těhotenství dobrá metabolická kontrola. Požadavky na inzulín mohou klesat během prvního trimestru a obecně se zvyšují během druhého a třetího trimestru. Okamžitě po porodu potřeba inzulínu rapidně poklesne.
Kojení
Není známo, zda je inzulín glulisin vylučován do mateřského mléka, ale obecně inzulín nepřestupuje do mateřského mléka a není absorbován po perorálním podání.
Kojící matky mohou vyžadovat úpravu dávky inzulínu a diety.
Fertilita
Reprodukční studie u zvířat neodhalily žádné nežádoucí účinky inzulínu glulisinu na fertilitu.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Pacientova schopnost koncentrace a reakce může být zhoršena následkem hypoglykémie nebo hyperglykémie nebo například následkem poruchy zraku. To může představovat riziko v situacích, kde jsou tyto schopnosti zvláště důležité (např. při řízení vozidla nebo při obsluze strojů).
Pacient by měl být obeznámen s opatřeními, zabraňujícími vzniku hypoglykémie během řízení. To je důležité zejména u těch pacientů, kteří mají sníženou nebo chybějící vnímavost k varovným příznakům hypoglykémie nebo mají časté epizody hypoglykémie. Je třeba zvážit, zda řízení nebo obsluha strojů jsou za těchto okolností vhodné.
4.8 Nežádoucí účinky
Souhrnný bezpečnostní profil
Hypoglykémie, nejčastější nežádoucí účinek inzulínové terapie, se může objevit, jestliže dávka inzulínu je vzhledem k jeho potřebě příliš vysoká.
Tabulkový přehled nežádoucích účinků
Následující nežádoucí účinky, které se vyskytly během klinických hodnocení, byly seřazeny níže systematicky podle skupiny orgánů a podle snižujícího se výskytu (velmi časté: >1/10; časté: >1/100 až <1/10; méně časté: >1/1 000 až <1/100; vzácné >1/10 000 až <1/1 000; velmi vzácné: <1/10 000), není známo (z dostupných údajů nelze určit).
V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
|
MedDRA Třídy orgánových systémů |
Velmi časté |
Časté |
Méně časté |
Vzácné |
|
Poruchy metabolismu a výživy |
Hypoglykémie | |||
|
Poruchy kůže a podkožní tkáně |
Reakce v místě vpichu Lokální hypersenzitivní reakce |
Lipodystrofie | ||
|
Celkové poruchy a reakce v místě aplikace |
Systémová hypersenzitivní reakce |
Popis vybraných nežádoucích účinků
Poruchy metabolismu a výživy
Symptomy hypoglykémie se obvykle objeví náhle. Mohou zahrnovat studený pot, chladnou bledou kůži, únavu, nervozitu nebo třes, úzkost, neobvyklou únavu nebo slabost, zmatenost, poruchy koncentrace, ospalost, vlčí hlad, poruchy vidění, bolest hlavy, nauseu a palpitace.
Hypoglykémie se může stát závažnou a může vést k bezvědomí a/nebo křečím a k přechodnému nebo trvalému poškození mozkových funkcí nebo dokonce ke smrti.
Poruchy kůže a podkožní tkáně
Lokální reakce z přecitlivělosti (zčervenání, otok a svědění v místě vpichu) se mohou objevit v průběhu léčby inzulínem. Tyto reakce jsou obvykle přechodné a v průběhu pokračující léčby vymizí.
Lipodystrofie se může objevit v místě vpichu následkem nedodržení plynulého střídání místa vpichu v rámci oblasti aplikace.
Celkové poruchy a reakce v místě aplikace
Systémové reakce z přecitlivělosti mohou zahrnovat kopřivku, pocit tíhy na hrudi, dušnost, alergickou dermatitidu a svědění. Těžké případy generalizované alergické reakce včetně anafylaktické reakce mohou být život ohrožující.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Příznaky
Hypoglykémie se může objevit jako výsledek nadměrné inzulínové aktivity vzhledem k příjmu potravy a výdeji energie.
Nejsou k dispozici specifické údaje týkající se předávkování inzulínem glulisin. Hypoglykémie se však může rozvinout přes následující stádia:
Léčba
Mírné hypoglykemické epizody mohou být zvládnuty perorálním podáním glukózy nebo produktů obsahujících cukr. Doporučuje se proto, aby diabetici u sebe neustále nosili kostky cukru, sladkosti, sušenky nebo sladkou ovocnou šťávu.
Těžké hypoglykemické epizody, kdy pacient upadne do bezvědomí, mohou být léčeny glukagonem (0,5 mg až 1 mg) podaným intramuskulárně nebo subkutánně někým, kdo dostal patřičné instrukce, nebo glukózou podanou intravenózně zdravotnickým pracovníkem. Glukóza musí být podána intravenózně také tehdy, pokud u pacienta nenastane odpověď na glukagon během 10 až 15 minut.
Jakmile pacient znovu nabude vědomí, doporučuje se z důvodu prevence relapsu podání sacharidů perorálně.
Po injekci glukagonu by měl být pacient sledován v nemocnici z důvodu zjištění příčiny tak těžké hypoglykémie a z důvodu prevence dalších podobných epizod.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Léčiva k terapii diabetu, inzulíny a analogy rychle působící, k injekční aplikaci, ATC kód: A10AB06.
Mechanismus účinku
Inzulín glulisin je rekombinantní analog lidského inzulínu, který je ekvipotentní k normálnímu lidskému inzulínu. Inzulín glulisin má rychlejší nástup účinku a kratší trvání účinku než běžný lidský inzulín.
Primárním účinkem inzulínu a inzulínových analog včetně inzulínu glulisin je regulace metabolismu glukózy. Inzulíny snižují hladinu glukózy v krvi stimulací periferního vychytávání glukózy zvláště kosterními svaly a tukem a inhibicí glukoneogeneze v játrech. Inzulín inhibuje lipolýzu v tukových buňkách, inhibuje proteolýzu a podporuje syntézu proteinů.
Studie u zdravých dobrovolníků a pacientů s diabetem ukázaly, že inzulín glulisin, je-li podán subkutánně, je rychlejší v nástupu a kratší v trvání účinku než běžný lidský inzulín. Pokud je inzulín glulisin podán subkutánní injekcí, hypoglykemizující účinek nastupuje během 10 - 20 minut. Účinky na snížení glykémie jsou u inzulínu glulisin a běžného lidského inzulínu při podání intravenózní cestou ekvipotentní. Jedna jednotka inzulínu glulisin má stejný hypoglykemizující účinek jako jedna dávka běžného lidského inzulínu.
Proporcionalita dávky
Ve studii s 18 muži s diabetes mellitus 1. typu ve věku od 21 do 50 let snižoval inzulín glulisin hladinu glukózy úměrně podané dávce v terapeuticky relevantním dávkovém rozmezí 0,075 až 0,15 jednotky/kg. Při dávkách 0,3 jednotky/kg nebo vyšších byl hypoglykemický účinek inzulínu glulisinu nižší, než by úměrně odpovídalo dávce, stejně jako u lidského inzulínu.
Nástup účinku inzulínu glulisin je dvakrát rychlejší než u běžného lidského inzulínu a jeho hypoglykemický efekt končí asi o 2 hodiny dříve než u běžného lidského inzulínu.
Studie I. fáze u pacientů s diabetes mellitus 1. typu hodnotily hypoglykemizující profily inzulínu glulisin a rozpustného rychle účinkujícího inzulínu podaného subkutánně v dávce 0,15 jednotky/kg v různé době ve vztahu k 15minutovému standardnímu jídlu. Data ukázala, že inzulín glulisin podaný 2 minuty před jídlem, poskytuje v porovnání s rozpustným rychle účinkujícím lidským inzulínem podaným 30 minut před jídlem podobnou postprandiální kompenzaci. Je-li podán 2 minuty před jídlem, poskytuje inzulín glulisin lepší postprandiální kompenzaci než rozpustný rychle účinkující lidský inzulín podaný 2 minuty před jídlem. Inzulín glulisin podaný 15 minut po začátku jídla poskytuje podobnou glykemickou kompenzaci jako rozpustný rychle účinkující lidský inzulín podaný 2 minuty před jídlem (viz obrázek 1).

Čas - hodiny

Čas - hodiny

Čas - hodiny
Obrázek 1: Průměrný hypoglykemizující účinek po dobu 6 hodin u 20 pacientů s diabetes mellitus 1. typu. Inzulín glulisin podaný 2 minuty (Glulisin před) před začátkem jídla v porovnání s běžným lidským inzulínem podaným 30 minut (Rozpustný rychle účinkující inzulín-30 min) před začátkem jídla (obrázek 1A) a v porovnání s běžným lidským inzulínem podaným 2 minuty (Rozpustný rychle účinkující inzulín před) před jídlem (obrázek 1B). Inzulín glulisin podaný 15 minut (Glulisin po) po začátku jídla v porovnání s běžným inzulínem podaným 2 minuty (Rozpustný rychle účinkující inzulín před) před začátkem jídla (obrázek 1C). Na ose x je nula (šipka) začátek jídla trvajícího 15 minut.
Obezita
Studie I. fáze prováděná s inzulínem glulisin, inzulínem lispro a běžným lidským inzulínem u obézní populace ukázala, že inzulín glulisin si zachovává své rychle účinkující vlastnosti. V této studii byl čas představující časnou hypoglykemizující aktivitu až 20 % celkové AUC a AUC (0 -2 h) a sice 114 minut a 427 mg/kg- u inzulínu glulisin, 121 minut a 354 mg/kg- pro lispro a 150 minut a 197 mg/kg pro běžný lidský inzulín (viz obrázek 2).
- GLULISIN
LISPRO ROZPUSTNÝ ' RYCHLE ÚČINKUJÍCÍ INZULÍN
Čas - hodiny
Obrázek 2: Rychlost infuze glukózy (GIR) po subkutánní injekci 0,3 jednotky/kg inzulínu glulisin (GLULISIN) nebo inzulínu lispro (LISPRO) nebo normálního lidského inzulínu (Rozpustný rychle účinkující inzulín) u obézní populace.
Další studie I. fáze s inzulínem glulisin a inzulínem lispro u nediabetické populace 80 pacientů s širokým rozmezím indexů tělesné hmotnosti (18-46 kg/m2) ukázala, že k rychlému nástupu účinku docházelo napříč širokým rozmezím indexů tělesné hmotnosti (BMI), zatímco celkový hypoglykemický efekt klesá se zvyšující se obezitou.
Průměrná plocha pod křivkou (AUC) pro celkovou GIR (glucose infusion rate- rychlost infuze glukózy) mezi 0-1. hodinou byla 102±75 mg/kg s 0,2 jednotky/kg inzulínu glulisin a 158±100 mg/kg s 0,4 jednotky/kg inzulínu glulisin a 83,1±72,8 mg/kg s 0,2 jednotky/kg inzulínu lispro a 112,3±70,8 mg/kg s 0,4 jednotky/kg inzulínu lispro.
Studie I. fáze u 18 obézních pacientů s diabetem mellitus 2. typu (BMI mezi 35 a 40 kg/m2) s inzulínem glulisin a inzulínem lispro [90% CI: 0,81, 0,95 (p=<0,01)] ukázala, že inzulín glulisin efektivně kontroluje diurnální postprandiální výkyvy hladiny glukózy v krvi.
Klinická účinnost a bezpečnost
Diabetes mellitus 1. typu - dospělí
Ve 26týdenní klinické studii fáze III srovnávající inzulín glulisin s inzulínem lispro podávanými subkutánně krátce (0-15 minut) před jídlem u pacientů s diabetes mellitus 1. typu užívajících inzulín glargin jako bazální inzulín, byl inzulín glulisin, co do kompenzace glykémie, srovnatelný s inzulínem lispro, jak ukazují změny glykovaného hemoglobinu od začátku do konce studie (vyjádřené jako HbA1c ekvivalent). Při selfmonitoringu byly pozorovány srovnatelné hodnoty glykémie. Na rozdíl od inzulínu lispro nebylo potřeba při terapii inzulínem glulisin zvyšovat dávku bazálního inzulínu.
12týdenní klinická studie fáze III prováděná u pacientů s diabetes mellitus 1. typu, kteří dostávali jako bazální terapii inzulín glargin, ukazuje, že podání inzulínu glulisin bezprostředně po jídle poskytuje
srovnatelnou účinnost jako inzulín glulisin bezprostředně (0-15 minut) před jídlem nebo jako rozpustný rychle účinkující inzulín (30-45 minut).
V „per protokol“ populaci bylo ve skupině s glulisinem před jídlem pozorováno signifikantně větší snížení hodnot glykovaného hemoglobinu než ve skupině s rozpustným rychle účinkujícím inzulínem.
Diabetes mellitus 1. typu - děti
26týdenní klinická studie fáze III srovnávala subkutánní aplikaci inzulínu glulisin s inzulínem lispro podanými krátce (0 - 15 minut) před jídlem u dětí (4 - 5 let: n = 9; 6 - 7 let: n = 32 a 8 - 11 let: n =
149) a mladistvých (12-17 let: n = 382) s diabetes mellitus 1.typu, kterým byl aplikován jako bazální inzulín glargin nebo NPH inzulín. S inzulínem glulisin bylo dosaženo srovnatelné glykemické kontroly hodnocené změnou glykovaného hemoglobinu (GHb vyjádřený jako ekvivalent HbAJc) od zahájení studie do jejího ukončení a hodnotami glukózy v krvi zjištěnými při selfmonitoringu jako s inzulínem lispro.
Nejsou dostatečné klinické informace o použití přípravku Apidra u dětí mladších 6 let.
Diabetes mellitus 2. typu - dospělí
26týdenní klinická studie fáze III následovaná rozšířením o 26týdenní studii bezpečnosti byla prováděna za účelem srovnání inzulínu glulisin (0-15 minut před jídlem) s rozpustným rychle účinkujícím lidským inzulínem (30-45 minut před jídlem) při subkutánním podání u pacientů s diabetes mellitus 2. typu užívajících NPH inzulín jako bazální inzulín. Průměrný body mass index (BMI) u pacientů byl 34,55 kg/m2. Inzulín glulisin ukázal, že je srovnatelný s rozpustným rychle účinkujícím lidským inzulínem, pokud se týče změn glykovaného hemoglobinu (vyjádřené jako HbAJc ekvivalent) od začátku do konce 6-měsíční studie (- 0,46 % u inzulínu glulisin a - 0,30 % u rozpustného rychle účinkujícího lidského inzulínu) a od začátku do konce 12-měsíční studie (- 0,23 % u inzulínu glulisin a - 0,13 % u rozpustného rychle účinkujícího lidského inzulínu, rozdíl není významný). V této studii většina pacientů (79 %) mísila jejich rychle účinkující inzulín s NPH inzulínem bezprostředně před injekcí a 58 % pacientů užívalo v době randomizace perorální antidiabetika. Tito pacienti byli instruováni, aby v jejich užívání pokračovali ve stejných dávkách.
Rasa a pohlaví
V kontrolovaných klinických studiích u dospělých neprokázal inzulín glulisin rozdíly v bezpečnosti a účinnosti v podskupinové analýze založené na rase a pohlaví.
5.2 Farmakokinetické vlastnosti
Náhrada aminokyseliny asparagin na pozici B3 lidského inzulínu lysinem a lysinu na pozici B29 kyselinou glutamovou zajišťuje u inzulínu glulisin rychlejší absorpci.
Ve studii s 18 muži s diabetes mellitus 1. typu ve věku od 21 do 50 let inzulín glulisin vykazuje pro časnou, maximální a celkovou expozici v dávkovém rozmezí 0,075 až 0,4 jednotky/kg dávkovou proporcionalitu.
Absorpce a biologická dostupnost
Farmakokinetické profily u zdravých dobrovolníků a diabetiků 1. a 2. typu ukázaly, že absorpce inzulínu glulisin byla asi dvakrát rychlejší s přibližně dvakrát tak vysokou vrcholovou koncentrací ve srovnání s rozpustným rychle účinkujícím inzulínem.
Ve studii u pacientů s diabetes mellitus 1. typu po subkutánním podání 0,15 jednotky/kg byl pro inzulín glulisin Tmax 55 minut a Cmax 82 ± 1,3 pjednotky/ml ve srovnání s Tmax 82 minut a Cmax 46 ±
1,3 pjednotky/ml pro rozpustný rychle účinkující inzulín. Průměrný čas výskytu v organismu u inzulínu glulisin byl kratší (98 minut) než pro rozpustný rychle účinkující inzulín (161 minut) (viz obrázek 3).

Obrázek 3: Farmakokinetický profil inzulínu glulisin a rozpustného rychle účinkujícího inzulínu u pacientů s diabetes mellitus 1. typu po dávce 0,15 jednotky/kg.
Ve studii u pacientů s diabetes mellitus 2. typu po subkutánním podání 0,2 jednotky/kg inzulínu glulisin byla Cmax 91 pjednotky/ml s rozmezím mezi kvartily od 78 do 104 pjednotky/ml.
Pokud byl inzulín glulisin aplikován subkutánně do břišní stěny, deltového svalu a stehna, profily koncentrace-čas byly podobné s mírně rychlejší absorpcí při podání do břišní stěny ve srovnání s podáním do stehna. Absorpce z deltového svalu byla střední (viz bod 4.2). Absolutní biologická dostupnost (70%) inzulínu glulisin byla v rámci míst aplikace podobná a s nízkou variabilitou mezi jednotlivými jedinci (11% CV). V porovnání se subkutánní injekcí vedlo podání intravenózního bolusu inzulínu glulisinu k vyšší systémové expozici, hodnota Cmax byla přibližně 40krát vyšší.
Obezita
Další studie I. fáze s inzulínem glulisin a inzulínem lispro u nediabetické populace 80ti pacientů s širokým rozmezím indexů tělesné hmotnosti (18-46 kg/m2) ukázala, že k rychlému nástupu účinku dochází napříč širokým rozmezím indexů tělesné hmotnosti a že celková expozice je napříč širokým rozmezím indexů tělesné hmotnosti zachována.
Doba do 10% celkové INS expozice byla s inzulínem glulisin dosažena o asi 5-6 minut dříve. Distribuce a eliminace
Distribuce a eliminace inzulínu glulisin a rozpustného rychle účinkujícího inzulínu po intravenózním podání je podobná s distribučními objemy 13 l a 22 l a poločasem eliminace 13 a 18 minut.
Po subkutánním podání je inzulín glulisin eliminován rychleji než rozpustný rychle účinkující inzulín s poločasem eliminace 42 minut ve srovnání s 86 minutami. Ve zkřížené analýze u zdravých jedinců nebo u diabetiků 1. nebo 2. typu byl poločas eliminace v rozmezí od 37 do 75 minut (rozmezí mezi kvartily).
Inzulín glulisin vykazuje nízkou vazbu na plazmatické proteiny stejně jako lidský inzulín.
Zvláštní populace
Porucha funkce ledvin
V klinické studii prováděné u nediabetiků, pokrývající široké rozmezí renálních funkcí
(CrCl > 80 ml/min, 30-50 ml/min, < 30 ml/min), byly obecně zachovány rychle účinkující vlastnosti
inzulínu glulisin. U pacientů s poškozením ledvin však mohou být nároky na inzulín sníženy.
Porucha funkce jater
Farmakokinetické vlastnosti nebyly zkoumány u pacientů se sníženou funkcí jater.
Starší pacienti
U starších pacientů s diabetes mellitus jsou dostupné pouze velmi omezené farmakokinetické údaje.
Děti a mladiství
Farmakokinetické a farmakodynamické vlastnosti inzulínu glulisin byly zkoumány u dětí (7-11 let) a mladistvých (12-16 let) s diabetes mellitus 1. typu. Inzulín glulisin byl rychle absorbován v obou věkových skupinách, s podobným Tmax a Cmax jako u dospělých (viz bod 4.2).
Při podání těsně před testovaným jídlem poskytuje inzulín glulisin stejně jako u dospělých lepší postprandiální kompenzaci než rozpustný rychle účinkující lidský inzulín (viz bod 5.1). Výchylky glukózy (AUC 0-6h) byly 641 mg.h/dl pro insulin glulisin a 801 mg.h/dl pro rozpustný rychle účinkující lidský inzulín.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje neodhalily jiné nálezy toxicity než ty, které jsou spojené s hypoglykemizující farmakodynamickou aktivitou (hypoglykémie), odlišné od rozpustného rychle účinkujícího inzulínu nebo s klinickým významem pro člověka.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Metakresol Chlorid sodný Trometamol Polysorbát 20
Kyselina chlorovodíková 35%
Hydroxid sodný Voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky s výjimkou NPH lidských inzulínů.
6.3 Doba použitelnosti
2 roky
Doba použitelnosti po prvním použití pera
Přípravek může být uchováván až 4 týdny při teplotě do 25°C mimo přímé teplo nebo světlo. Používaná pera nesmí být skladována v chladničce. Víčko pera musí být vráceno na pero po každé injekci, aby byl přípravek chráněn před světlem.
6.4 Zvláštní opatření pro uchovávání
Nepoužívaná pera
Uchovávejte v chladničce (2°C - 8°C).
Chraňte před mrazem.
Neukládejte přípravek Apidra do blízkosti mrazicího boxu nebo mrazicí vložky.
Uchovávejte předplněné pero v krabičce, aby byl přípravek chráněn před světlem.
Používaná pera
Podmínky uchovávání tohoto léčivého přípravku po jeho prvním otevření jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
3 ml roztoku v zásobní vložce (bezbarvé sklo) se zátkou (pružná brombutylová guma) a pertlem (hliník) s těsněním (pružná brombutylová guma). Zásobní vložka je uzavřena v předplněném peru k jednorázovému použití. Dostupná jsou balení s 1, 3, 4, 5, 6, 8, 9 a 10 pery.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Před prvním použitím musí být pero skladováno 1-2 hodiny při pokojové teplotě.
Zásobní vložku před použitím prohlédněte. Smí být použita pouze tehdy, je-li roztok čirý, bezbarvý, bez viditelných pevných částic a má-li konzistenci vody. Vzhledem k tomu, že Apidra je roztok, není potřeba ho před použitím znovu protřepávat.
Prázdná pera nesmí být znovu použita a musí být zlikvidována odpovídajícím způsobem.
Jedno pero smí být používáno výhradně jedním pacientem, aby se zabránilo jakémukoliv druhu kontaminace.
Před každým podáním injekce musí být vždy zkontrolován štítek inzulínu, aby nedošlo k záměně inzulínu glulisin a jiných inzulínů (viz bod 4.4).
Míchání s inzulíny
Pokud se Apidra mísí s NPH lidským inzulínem, měla by být natažena do stříkačky jako první. Injekce by se měla podat okamžitě po smíchání, protože údaje týkající se směsí připravených značnou dobu před injekcí nejsou dostupné.
Pacientovi je třeba doporučit, aby si před použitím SoloStar pozorně přečetl návod, který je součástí příbalové informace.
Pero
Kryt pera
Jehla (není přiložena)
|
c—crTlf |
Ochranný uzávěr li D—d 1 |
|
Vnější kryt jehly |
'-' \J Vnitřní kryt ehly Jehla |
Okénko ukazující dávku
Zásobník inzulínu I

Injekční
tlačítko
Gumový uzávěr
Schematický nákres pera Důležité informace pro použití SoloStar:
• Před použitím se musí vždy nejprve opatrně upevnit jehla a musí se provést kontrola bezpečnosti. Bez nasazené jehly neotáčejte voličem dávky ani nestlačujte injekční tlačítko.
• Používejte pouze jehly, které jsou určeny pro použití se SoloStar.
• Je třeba zvláštní opatrnosti, aby se předešlo náhodnému zranění jehlou a přenosu infekce.
• SoloStar se nesmí nikdy použít, pokud je poškozen, nebo pokud není jisté, že funguje správně.
• Pacient musí mít vždy k dispozici náhradní SoloStar pro případ, že se SoloStar ztratí nebo poškodí.
Pokyny pro uchovávání
Prosím, přečtěte si pokyny pro uchovávání SoloStar, které jsou uvedeny v bodě 6.4 tohoto SPC.
Pokud je SoloStar skladován v chladu, je třeba ho vyjmout 1 až 2 hodiny před použitím, aby se mohl ohřát. Injikování studeného inzulínu je bolestivější.
Použitý SoloStar musí být zlikvidován podle pokynů místních úřadů.
Údržba
SoloStar musí být chráněn před prachem a špínou.
SoloStar je možné čistit zvenčí otíráním vlhkým textilem.
Pero se nesmí máčet, mýt ani promazávat, mohlo by tím být poškozeno.
SoloStar je navržen tak, aby pracoval přesně a bezpečně. Musí s ním být zacházeno s opatrností. Pacienti by se měli vyhnout situacím, kdy může být SoloStar poškozen. Pokud se pacient domnívá, že je jeho SoloStar poškozen, musí použít nový.
Krok 1. Kontrola inzulínu
Je nutné zkontrolovat označení na štítku pera, aby bylo jisté, že obsahuje správný inzulín. Apidra SoloStar je modrý. Má tmavě modré injekční tlačítko s vyvýšeným kroužkem na konci. Po odstranění krytu pera musí být také zkontrolován vzhled inzulínu: roztok inzulínu musí být čirý, bezbarvý, bez viditelných pevných částic a musí mít konzistenci vody.
Krok 2. Upevnění jehly
Použít by se měly pouze jehly, které byly schváleny pro použití se SoloStar.
Pro každou injekci musí být použita nová sterilní jehla. Po odstranění krytu by se jehla měla opatrně upevnit přímo na pero.
Krok 3. Kontrola bezpečnosti
Před každou injekcí je třeba provést kontrolu bezpečnosti pro ujištění, že pero a jehla řádně fungují, a odstranit vzduchové bubliny.
Musí být zvolena dávka 2 jednotky.
Vnější a vnitřní kryt jehly se musí odstranit.
Zatímco se pero drží tak, aby jehla směřovala nahoru, poklepe se jemně prsty na zásobník inzulínu, aby všechny vzduchové bubliny vystoupaly nahoru k jehle.
Potom by se mělo injekční tlačítko stlačit, co nejvíc to půjde.
Jestliže byl inzulín protlačen hrotem jehly, pak pero a jehla fungují správně. Pokud se žádný inzulín na hrotu j ehly neobj eví, je třeba opakovat krok 3, dokud se inzulín na hrotu j ehly neobj eví.
Krok 4. Volba dávky
Dávka se může nastavit v krocích po 1 jednotce, od minimální dávky 1 jednotky až po maximální dávku 80 jednotek. Pokud je požadována dávka větší než 80 jednotek, musí se podat jako dvě a více injekcí.
Okénko ukazující dávku musí po kontrole bezpečnosti ukazovat „0“. Poté může být zvolena dávka. Krok 5. Injekce dávky
Pacient musí být svým lékařem informován o injekčních technikách.
Jehla se vpíchne do kůže.
Injekční tlačítko se úplně stiskne. Potom se drží tlačítko stisknuté 10 vteřin, než se jehla vytáhne. Tak se zajistí, že bude podána celá dávka inzulínu.
Krok 6. Odstranění a zlikvidování jehly
Jehla se musí odstranit po každé injekci a vyhodit. To zabrání kontaminaci a/nebo infekci, opětovnému vniknutí vzduchu do zásobníku inzulínu a vytékání inzulínu. Jehly nesmí být znovu použity.
Při odstraňování a likvidaci jehly je třeba zvláštní opatrnosti. Aby se předešlo riziku náhodného zranění jehlou a přenosu infekčních chorob, řiďte se doporučenými bezpečnostními opatřeními pro odstranění a likvidaci jehel (například nasazení krytu jednou rukou “one handed capping technique”).
Na pero se znovu nasadí kryt pera.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Sanofi-Aventis Deutschland GmbH D-65926 Frankfurt am Main Německo.
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/04/285/ 029-036
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 27. září 2004
Datum posledního prodloužení registrace: 20. srpna 2009
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku j sou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/
A. VÝROBCE/VÝROBCI BIOLOGICKÉ LÉČIVÉ LÁTKY/BIOLOGICKÝCH LÉČIVÝCH LÁTEK
A VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE/VÝROBCI BIOLOGICKÉ LÉČIVÉ LÁTKY/LÁTEK A VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce/výrobců biologické léčivé látky/biologických léčivých látek
Sanofi-Aventis Deutschland GmbH Industriepark Hochst, D-65926 Frankfurt Německo
Název a adresa výrobce odpovědného/vvrobců odpovědných za propouštění šarží
Sanofi-Aventis Deutschland GmbH Industriepark Hochst, D-65926 Frankfurt Německo
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis.
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
• Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ TOHOTO LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Apidra 100 jednotek/ml, injekční roztok v injekční lahvičce Insulinům glulisinum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jeden ml obsahuje 100 jednotek inzulínu glulisin (odpovídá 3,49 mg).
Jedna injekční lahvička obsahuje 10 ml injekčního roztoku, což odpovídá 1000 jednotkám.
3. SEZNAM POMOCNÝCH LÁTEK
Dále obsahuje: metakresol, chlorid sodný (další údaje viz příbalová informace), trometamol, polysorbát 20, kyselina chlorovodíková 35%, hydroxid sodný (další údaje viz příbalová informace), voda na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Injekční roztok v injekční lahvičce.
1 injekční lahvička s 10 ml.
2 injekční lahvičky s 10 ml.
4 injekční lahvičky s 10 ml.
5 injekčních lahviček s 10 ml.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Subkutánní nebo intravenózní podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
Používejte pouze čiré a bezbarvé roztoky.
8. POUŽITELNOST
EXP
Neotevřené injekční lahvičky:
Uchovávejte v chladničce.
Chraňte před mrazem.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
Po prvním použití: Přípravek může být uchováván až 4 týdny při teplotě do 25°C. Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Sanofi-Aventis Deutschland GmbH D-65926 Frankfurt am Main, Německo
12. REGISTRAČNÍ ČÍSLO(A)
EU/1/04/285/001 1 injekční lahvička s 10 ml
EU/1/04/285/002 2 injekční lahvičky s 10 ml
EU/1/04/285/003 4 injekční lahvičky s 10 ml
EU/1/04/285/004 5 injekčních lahviček s 10 ml
13. ČÍSLO ŠARŽE
č.S.
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Apidra
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA(Y) PODÁNÍ
Apidra 100 jednotek/ml, injekční roztok Insulinům glulisinum.
Subkutánní nebo intravenózní podání.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
10 ml
6. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Apidra 100 jednotek/ml, injekční roztok v zásobní vložce. Insulinům glulisinum.
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jeden ml obsahuje 100 jednotek inzulínu glulisin (odpovídá 3,49 mg).
3. SEZNAM POMOCNÝCH LÁTEK
Dále obsahuje: metakresol, chlorid sodný, trometamol, polysorbát 20, kyselina chlorovodíková 35%, hydroxid sodný, voda na injekci (další údaje viz příbalová informace).
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Injekční roztok v zásobní vložce. 1 zásobní vložka se 3 ml.
3 zásobní vložky se 3 ml.
4 zásobní vložky se 3 ml.
5 zásobních vložek se 3 ml.
6 zásobních vložek se 3 ml.
8 zásobních vložek se 3 ml.
9 zásobních vložek se 3 ml.
10 zásobních vložek se 3 ml.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Zásobní vložky Apidra se mají používat pouze s inzulínovými pery: OptiPen, ClikSTAR, Tactipen Autopen 24, AllStar, JuniorSTAR.
Všechna uvedená pera nemusí být ve Vaší zemi na trhu.
Před použitím si přečtěte příbalovou informaci.
Subkutánní podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ZE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
Používejte pouze čiré a bezbarvé roztoky.
Pokud je inzulínové pero poškozeno nebo nefunguje správně (kvůli mechanické závadě), je třeba je vyřadit a použít nové inzulínové pero.
8. POUŽITELNOST
EXP 9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Neotevřené zásobní vložky:
Uchovávejte v chladničce.
Chraňte před mrazem.
Uchovávejte zásobní vložku v krabičce, aby byl přípravek chráněn před světlem.
Po prvním použití: Přípravek může být uchováván až 4 týdny při teplotě do 25°C. Neuchovávejte v chladničce. Uchovávejte zásobní vložku vloženou v peru, aby byla chráněna před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Sanofi-Aventis Deutschland GmbH D-65926 Frankfurt am Main, Německo
12. REGISTRAČNÍ ČÍSLO(A)
EU/1/04/285/005
EU/1/04/285/006
EU/1/04/285/007
EU/1/04/285/008
EU/1/04/285/009
EU/1/04/285/010
EU/1/04/285/011
EU/1/04/285/012
1 zásobní vložka se 3 ml
3 zásobní vložky se 3 ml
4 zásobní vložky se 3 ml
5 zásobních vložek se 3 ml
6 zásobních vložek se 3 ml
8 zásobních vložek se 3 ml
9 zásobních vložek se 3 ml
10 zásobních vložek se 3 ml
13. ČÍSLO ŠARŽE
č.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Apidra
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA(Y) PODÁNÍ
Apidra 100 jednotek /ml, injekční roztok Insulinům glulisinum.
Subkutánní podání Zkratka schválená pro vícejazyčné balení.
2. ZPŮSOB PODÁNÍ
Použití s konkrétními pery: viz příbalová informace.
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
3 ml
6. JINÉ
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA BLISTRECH NEBO STRIPECH
TEXT NA HLINÍKOVÉ FÓLII, KTERÁ JE POUŽITA K UZAVŘENÍ PRŮHLEDNÉHO PLASTOVÉHO ZÁSOBNÍKU OBSAHUJÍCÍHO ZÁSOBNÍ VLOŽKY
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Apidra 100 jednotek /ml, injekční roztok v zásobní vložce. Insulinům glulisinum.
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Sanofi-Aventis Deutschland GmbH
3. POUŽITELNOST
4. ČÍSLO ŠARŽE
5. JINÉ
Po vložení nové zásobní vložky:
Před podáním první dávky musíte zkontrolovat, zda Vaše pero funguje správně. Další podrobnosti, jak s tímto perem zacházet, se dozvíte v návodu pro použití, který je přiložen u pera.
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Apidra 100 jednotek /ml, injekční roztok v zásobní vložce pro OptiClik. Insulinům glulisinum.
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jeden ml obsahuje 100 jednotek inzulínu glulisin (odpovídá 3,49 mg).
3. SEZNAM POMOCNÝCH LÁTEK
Dále obsahuje: metakresol, chlorid sodný (další údaje viz příbalová informace), trometamol, polysorbát 20, kyselina chlorovodíková 35%, hydroxid sodný (další údaje viz příbalová informace), voda na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Injekční roztok v zásobní vložce pro OptiClik. 1 zásobní vložka o obsahu 3 ml.
3 zásobní vložky o obsahu 3 ml
4 zásobní vložky o obsahu 3 ml
5 zásobních vložek o obsahu 3 ml
6 zásobních vložek o obsahu 3 ml
8 zásobních vložek o obsahu 3 ml
9 zásobních vložek o obsahu 3 ml
10 zásobních vložek o obsahu 3 m .
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Subkutánní podání.
Pouze k použití s OptiClik.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
Používejte pouze čiré a bezbarvé roztoky.
Pokud je OptiClik poškozen nebo nefunguje správně (kvůli mechanické závadě), je třeba jej vyřadit a použít nový OptiClik.
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Neotevřené zásobní vložky:
Uchovávejte v chladničce.
Chraňte před mrazem.
Uchovávejte zásobní vložku v krabičce, aby byl přípravek chráněn před světlem.
Po prvním použití: Přípravek může být uchováván až 4 týdny při teplotě do 25°C. Neuchovávejte v chladničce. Uchovávejte zásobní vložku vloženou v peru, aby byla chráněna před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Sanofi-Aventis Deutschland GmbH D-65926 Frankfurt am Main, Německo
12. REGISTRAČNÍ ČÍSLO(A)
EU/1/04/285/021
EU/1/04/285/022
EU/1/04/285/023
EU/1/04/285/024
EU/1/04/285/025
EU/1/04/285/026
EU/1/04/285/027
EU/1/04/285/028
1 zásobní vložka o obsahu 3 ml
3 zásobní vložky o obsahu 3 ml
4 zásobní vložky o obsahu 3 ml
5 zásobních vložek o obsahu 3 ml
6 zásobních vložek o obsahu 3 ml
8 zásobních vložek o obsahu 3 ml
9 zásobních vložek o obsahu 3 ml
10 zásobních vložek o obsahu 3 ml.
13. ČÍSLO ŠARŽE
č.s.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Apidra
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA(Y) PODÁNÍ
Apidra 100 jednotek /ml, injekční roztok Insulinům glulisinum Subkutánní podání.
2. ZPŮSOB PODÁNÍ
Pouze k použití s OptiClik.
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
3 ml
6. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Apidra, 100 jednotek /ml injekční roztok v předplněném peru. Insulinům glulisinum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jeden ml obsahuje 100 jednotek inzulínu glulisin (odpovídá 3,49 mg).
3. SEZNAM POMOCNÝCH LÁTEK
Dále obsahuje: metakresol, chlorid sodný (další údaje viz příbalová informace), trometamol, polysorbát 20, kyselina chlorovodíková 35%, hydroxid sodný (další údaje viz příbalová informace), voda na injekci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Injekční roztok v předplněném peru. OptiSet. 1 pero se 3 ml.
3 pera se 3 ml.
4 pera se 3 ml.
5 per se 3 ml.
6 per se 3 ml.
8 per se 3 ml.
9 per se 3 ml.
10 per se 3 ml.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Subkutánní podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
Používejte pouze čiré a bezbarvé roztoky.
Používejte pouze jehly schválené pro použití s OptiSet.
DŮLEŽITÁ INFORMACE
Před použitím OptiSet vždy nejprve nasaďte novou jehlu.
Před použitím OptiSet vždy proveďte kontrolu bezpečnosti.
Před prvním použitím OptiSet si přečtěte celou příbalovou informaci.
Nové informace pro použití:
• Název inzulínu j e vytištěn na peru
• Voličem dávky lze otáčet pouze v jednom směru
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Nepoužívaná pera:
Uchovávejte v chladničce.
Chraňte před mrazem.
Uchovávejte předplněné pero v krabičce, aby byl přípravek chráněn před světlem.
Po prvním použití: Přípravek může být uchováván až 4 týdny při teplotě do 25°C. Neuchovávejte v chladničce.
Uchovávejte předplněné pero ve vnějším obalu, aby bylo chráněno před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Sanofi-Aventis Deutschland GmbH D-65926 Frankfurt am Main, Německo
12. REGISTRAČNÍ ČÍSLO(A)
1 pero se 3 ml
3 pera se 3 ml
4 pera se 3 ml
5 per se 3 ml
6 per se 3 ml
8 per se 3 ml
9 per se 3 ml
10 per se 3 ml
EU/1/04/285/013
EU/1/04/285/014
EU/1/04/285/015
EU/1/04/285/016
EU/1/04/285/017
EU/1/04/285/018
EU/1/04/285/019
EU/1/04/285/020
13. ČÍSLO ŠARŽE
č.š.:
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Apidra OptiSet
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA(Y) PODÁNÍ
Apidra, 100 jednotek /ml injekční roztok Insulinům glulisinum Subkutánní podání.

4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
3 ml
6. JINÉ
OptiSet
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Apidra SoloStar 100 jednotek /ml, injekční roztok v předplněném peru. Insulinům glulisinum.
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jeden ml obsahuje 100 jednotek inzulínu glulisin (odpovídá 3,49 mg).
3. SEZNAM POMOCNÝCH LÁTEK
Dále obsahuje: metakresol, chlorid sodný, trometamol, polysorbát 20, kyselina chlorovodíková 35%, hydroxid sodný, voda na injekci (další údaje viz příbalová informace).
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Injekční roztok v předplněném peru. 1 pero se 3 ml.
3 pera se 3 ml.
4 pera se 3 ml.
5 per se 3 ml.
6 per se 3 ml.
8 per se 3 ml.
9 per se 3 ml.
10 per se 3 ml.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Subkutánní podání.
Před použitím si přečtěte příbalovou informaci.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
Používejte pouze čiré a bezbarvé roztoky.
Používejte pouze jehly schválené pro použití se SoloStar.
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Nepoužívaná pera:
Uchovávejte v chladničce.
Chraňte před mrazem.
Uchovávejte předplněné pero v krabičce, aby byl přípravek chráněn před světlem.
Po prvním použití: Přípravek může být uchováván až 4 týdny při teplotě do 25°C. Neuchovávejte v chladničce.
Uchovávejte pero ve vnějším obalu, aby bylo chráněno před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ_
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Sanofi-Aventis Deutschland GmbH D-65926 Frankfurt am Main, Německo 12. REGISTRAČNÍ ČÍSLO(A)
1 pero se 3 ml
3 pera se 3 ml
4 pera se 3 ml
5 per se 3 ml
6 per se 3 ml
8 per se 3 ml
9 per se 3 ml
10 per se 3 ml
EU/1/04/285/029
EU/1/04/285/030
EU/1/04/285/031
EU/1/04/285/032
EU/1/04/285/033
EU/1/04/285/034
EU/1/04/285/035
EU/1/04/285/036
13. ČÍSLO ŠARŽE
č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
Zde otevřít
Apidra SoloStar
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA(Y) PODÁNÍ
Apidra SoloStar 100 jednotek/ml injekční roztok.
Insulinům glulisinum Subkutánní podání.

4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
3 ml
6. JINÉ
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro uživatele
Apidra 100 jednotek/ml injekční roztok v injekční lahvičce
Insulinům glulisinum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je Apidra a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Apidra používat
3. Jak se přípravek Apidra používá
4. Možné nežádoucí účinky
5. Jak přípravek Apidra uchovávat
6. Obsah balení a další informace
1. Co je Apidra a k čemu se používá
Apidra je antidiabetikum používané ke snížení hladiny cukru v krvi u pacientů s diabetes mellitus; může být podána dospělým, mladistvým a dětem ve věku 6 a více let. Diabetes mellitus je onemocnění, při kterém Váš organismus neprodukuje dostatek inzulínu pro kontrolu hladiny cukru v krvi.
Přípravek je vyroben biotechnologicky. Inzulín glulisin má rychlý nástup účinku během 10-20 minut a krátké trvání účinku přibližně 4 hodiny.
2. Čemu musíte věnovat pozornost, než začnete přípravek Apidra používat Nepoužívejte přípravek Apidra
- Jestliže jste alergický(á) na inzulín glulisin nebo na kteroukoliv další složku tohoto přípravku (uvedenou v bodě 6).
- Jestliže máte hladinu cukru v krvi příliš nízkou (hypoglykémie). Postupujte podle pokynů pro hypoglykémii (viz rámeček na konci této příbalové informace).
Upozornění a opatření
Před užitím přípravku Apidra se poraďte se svým lékařem, lékarníkem nebo zdravotní sestrou. Dodržujte pečlivě pokyny pro dávkování, sledování (testy krve), dietu a fyzickou aktivitu (fyzickou práci a cvičení), které jste projednali se svým lékařem.
Zvláštní skupiny pacientů
Pokud máte problémy s játry nebo s ledvinami, upozorněte svého lékaře, protože můžete potřebovat nižší dávku.
Nejsou dostatečné klinické informace o použití přípravku Apidra u dětí mladších 6 let.
Cestování
Před cestováním se poraďte se svým lékařem. Možná bude třeba se poradit o
- dostupnosti Vašeho inzulínu v zemi, kterou navštívíte,
- dodávkách inzulínu, injekčních stříkaček, atd.,
- správném uchovávání Vašeho inzulínu během cestování,
- časové dostupnosti jídla a podávání inzulínu během cestování,
- možných účincích přesunu v různých časových pásmech,
- možných zdravotních rizicích v zemích, které navštívíte,
- tom, jak se zachovat v mimořádných situacích, tj. pokud se Vám udělá špatně nebo onemocníte. Onemocnění a úrazy
V následujících situacích může léčba Vašeho diabetu vyžadovat zvláštní péči:
- pokud jste nemocný/á nebo máte větší úraz, může dojít ke zvýšení hladiny cukru v krvi (hyperglykémie),
- pokud dostatečně nejíte, může se Vám hladina cukru v krvi velmi snížit (hypoglykémie).
Ve většině případů budete potřebovat lékařskou péči. Zajistěte možnost rychlého kontaktu lékaře.
Pokud máte diabetes typu 1 (na inzulínu závislý diabetes), nepřestávejte užívat svůj inzulín a pokračujte v příjmu dostatečného množství cukrů. Vždy řekněte lidem, kteří Vás ošetřují nebo léčí, že potřebujete inzulín.
U některých pacientů, kteří dlouhodobě trpěli cukrovkou typu 2 a onemocněním srdce, nebo prodělali mozkovou příhodu, a kteří byli léčeni pioglitazonem a inzulínem, byl zaznamenán výskyt srdečního selhání. Jestliže zjistíte známky srdečního selhání jako je neobvyklá dušnost nebo náhlé zvýšení tělesné hmotnosti nebo lokalizovaný otok (edém), informujte co nejrychleji svého lékaře.
Další léčivé přípravky a přípravek Apidra
Některé léky způsobují změnu hladiny cukru v krvi (snížení, zvýšení nebo obojí v závislosti na situaci). V každém případě může být nezbytné upravit Vaši dávku inzulínu, aby se zabránilo hladinám cukru v krvi, které jsou buď příliš nízké, nebo příliš vysoké. Buďte opatrní, když začínáte nebo přestáváte užívat nějaký další lék.
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat. Zeptejte se svého lékaře předtím, než si lék vezmete, zda a jak jím může být ovlivněna Vaše hladina cukru v krvi, popř. co je třeba dělat.
Mezi léky, které mohou vyvolat pokles hladiny cukru v krvi (hypoglykémii), patří:
- všechny další léky pro léčbu diabetu,
- inhibitory angiotenzin konvertujícího enzymu (ACE inhibitory) (používané k léčbě některých srdečních onemocnění, vysokého krevního tlaku),
- disopyramid (používaný k léčbě některých srdečních onemocnění),
- fluoxetin (používaný k léčbě depresí),
- fibráty (používané ke snížení vysokých hladin krevních lipidů),
- inhibitory monoaminooxidázy (MAO inhibitory) (používané k léčbě depresí),
- pentoxifylin, propoxyfen, salicyláty (např. aspirin, používaný ke zmírnění bolesti a snížení horečky),
- sulfonamidová antibiotika.
Mezi léky, které mohou vyvolat zvýšení hladiny cukru v krvi (hyperglykémii), patří:
- kortikosteroidy (např. „kortizon“ používaný pro léčbu zánětu),
- danazol (lék pro navození ovulace),
- diazoxid (používaný pro léčbu vysokého krevního tlaku),
- diuretika (používaná pro léčbu vysokého krevního tlaku nebo nadměrného zadržování tekutin),
- glukagon (hormon slinivky břišní používaný pro léčbu závažné hypoglykémie),
- isoniazid (používaný pro léčbu tuberkulózy),
- estrogeny a progestogeny (např. v antikoncepčních pilulkách užívaných k zamezení početí),
- deriváty fenothiazinu (používané pro léčbu duševních poruch),
- somatropin (růstový hormon),
- sympatomimetika (např. epinefrin [adrenalin], salbutamol, terbutalin užívané k léčbě astmatu),
- hormony štítné žlázy (užívané k léčbě onemocnění štítné žlázy),
- inhibitory proteáz (užívané k léčbě HIV),
- atypická antipsychotika (např. olanzapin a klozapin).
Vaše hladina cukru v krvi může buď poklesnout, nebo vzrůst, pokud užíváte:
- betablokátory (používané pro léčbu vysokého krevního tlaku),
- klonidin (používaný pro léčbu vysokého krevního tlaku),
- soli lithia (používané pro léčbu duševních poruch).
Pentamidin (používaný pro léčbu některých infekcí způsobených parazity) může vyvolat hypoglykémii, po které může někdy následovat hyperglykémie.
Betablokátory stejně jako ostatní sympatolytika (např. klonidin, guanetidin a reserpin) mohou oslabit nebo úplně potlačit první varovné příznaky, které Vám mohou pomoci rozpoznat hypoglykémii.
Pokud si nejste jisti, zda neužíváte některý z takových léků, zeptejte se svého lékaře nebo lékárníka.
Užívání přípravku Apidra s alkoholem
Hladiny Vašeho cukru v krvi mohou buď vzrůst, nebo poklesnout, pokud konzumujete alkohol. Těhotenství a kojení
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek užívat.
Informujte svého lékaře, pokud plánujete těhotenství nebo pokud již jste těhotná. Je možné, že bude třeba měnit dávkování inzulínu během těhotenství a po porodu. Zvláště pečlivá kontrola diabetu a prevence hypoglykémie jsou důležité pro zdraví Vašeho dítěte.
Nejsou k dispozici žádné nebo jen omezené údaje o použití přípravku Apidra u těhotných žen.
Pokud kojíte, poraďte se se svým lékařem, neboť může být nezbytné upravit Vám dávku inzulínu a Vaši dietu.
Řízení dopravních prostředků a obsluha strojů
Vaše schopnost soustředit se nebo reagovat může být snížena, jestliže:
- máte hypoglykémii (nízkou hladinu cukru v krvi)
- máte hyperglykémii (vysokou hladinu cukru v krvi).
Myslete na tento možný problém ve všech situacích, kde byste mohli dostat sebe nebo ostatní do nebezpečí (např. při řízení vozidla nebo při obsluze strojů).
Měl(a) byste se poradit se svým lékařem o vhodnosti řízení vozidel, pokud:
- máte časté epizody hypoglykémie,
- jsou první varovné příznaky, které Vám pomohou rozpoznat hypoglykémii, omezené nebo chybějí.
Důležité informace o některých složkách přípravku Apidra
Tento léčivý přípravek obsahuje méně než 1 mmol (23 mg) sodíku v jedné dávce, tj. v podstatě sodík neobsahuje.
Apidra obsahuje metakresol
Apidra obsahuje metakresol, který může způsobovat alergické reakce.
3. Jak se přípravek Apidra používá Dávkování
Na základě Vašeho životního stylu a Vašich výsledků testů cukru (glukózy) v krvi a Vašeho předchozího užívání inzulínu Váš lékař určí, jakou dávku přípravku Apidra budete potřebovat.
Apidra je rychle účinkující inzulín. Je možné, že Váš lékař Vám řekne, abyste ji užívali v kombinaci se střednědobě nebo dlouhodobě působícím inzulínem nebo bazálním inzulínem nebo s tabletami na snížení vysoké hladiny cukru v krvi.
Jestliže přecházíte z jiného inzulínu na inzulín glulisin, Vaše dávkování může vyžadovat úpravu lékařem.
Hladinu cukru v krvi může ovlivnit mnoho faktorů. Měli byste znát tyto faktory, abyste byli schopni reagovat správně na změny hladiny cukru v krvi a předejít jejich přílišnému zvýšení nebo přílišnému snížení. Další informace si přečtěte na konci této příbalové informace.
Způsob podání
Apidra se podává injekcí pod kůži (subkutánně). Zdravotník Vám ji může podat rovněž nitrožilně, za přísného dozoru lékaře.
Lékař Vám ukáže, do které oblasti kůže máte přípravek Apidra podávat. Injekci přípravku Apidra lze podávat do břišní stěny, do stehna nebo horní části paže nebo kontinuální infuzí do břišní stěny. Účinek bude o něco rychlejší, jestliže si injekci inzulínu aplikujete do břišní stěny. Tak jako u všech ostatních inzulínů musíte měnit s každou injekcí místo vpichu injekce a místo infuze v rámci vybrané oblasti (břišní stěna, stehno, horní část paže), kam injekci podáváte.
Frekvence podávání
Apidra by se měla užívat krátce (0 - 15 minut) před jídlem nebo brzy po jídle.
Pokyny pro správné použití
Jak se zachází s injekčními lahvičkami
Lahvičky s přípravkem Apidra jsou určeny pro použití s inzulínovými stříkačkami s odpovídající stupnicí jednotek a pro použití v inzulínové pumpě.
Injekční lahvičku si prohlédněte ještě předtím, než ji použijete. Použijte pouze ty injekční lahvičky, ve kterých je roztok čirý, bezbarvý, bez viditelných částic. Před použitím přípravek nemíchejte, ani s ním netřepejte.
Vždy použijte novou injekční lahvičku, pokud si všimnete, že se neočekávaně zhoršuje kontrola hladiny Vašeho cukru v krvi. To je proto, že účinnost inzulínu se může do určité míry snižovat. Pokud si budete myslet, že máte problémy s přípravkem Apidra, nechte jej zkontrolovat svým lékařem nebo lékárníkem.
Jestliže musíte míchat dva typy inzulínu_
Apidra se nesmí míchat s žádnými jinými přípravky kromě NPH lidského inzulínu.
Jestliže je Apidra míchána s NPH lidským inzulínem, měla by být natažena do stříkačky jako první. Injekce by se měla podat okamžitě po smíchání.
Jak zachází s inzulínovou pumpou_
Před použitím přípravku Apidra v inzulínové pumpě byste měl(a) dostat kompletní pokyny pro používání systému pumpy. Navíc byste měl(a) dostat informace o tom, co podniknout v případě nemoci, v případě příliš vysoké nebo příliš nízké hladiny cukru v krvi nebo při selhání pumpy. Používejte ten typ pumpy, který Vám doporučil lékař. Přečtěte si a dodržujte pokyny, které jsou dodány s Vaší inzulínovou infuzní pumpou. Dodržujte pokyny Vašeho lékaře o základní infuzní rychlosti a inzulínovém bolusu, který se užívá v době jídla.Pravidelně si měřte hladinu cukru v krvi, abyste měl(a) z inzulínové pumpy prospěch a pro ověření, že pumpa pracuje správně.
Vyměňte infuzní set a rezervoár alespoň každých 48 hodin za použití aseptických technik. Tyto instrukce se mohou lišit od pokynů uvedených v návodu k inzulinové pumpě. Používáte-li přípravek Apidra v pumpě, je důležité, abyste se vždy řídil(a) těmito specifickými instrukcemi. Nedodržení specifických pokynů může vést k závažným nežádoucím účinkům.
Při používání v pumpě se Apidra nesmí mísit s ředicími roztoky ani s žádným jiným inzulínem.
Co dělat, pokud pumpa selže nebo při nesprávném použití inzulínové pumpy
Problémy s pumpou nebo infuzním setem nebo nesprávné používání pumpy mohou vést k situaci, kdy dostanete málo inzulínu. To může rychle způsobit vysokou hladinu cukru v krvi a diabetickou ketoacidózu (nahromadění kyseliny v krvi, protože organismus štěpí místo cukru tuk).
Pokud Vám začne hladina cukru v krvi stoupat, kontaktujte co možná nejdříve svého lékaře, lékárníka nebo zdravotní sestru. Poradí Vám, co je třeba dělat.
Může být zapotřebí aplikovat přípravek Apidra injekčními stříkačkami nebo pery. Měl(a) byste mít vždy k dispozici náhradní systém pro podání inzulínu pod kůži pro případ selhání inzulinové pumpy.
Jestliže jste užil(a) více přípravku Apidra, než jste měl(a)
- Pokud jste podali příliš mnoho přípravku Apidra, může se Vám příliš snížit hladina cukru
v krvi (hypoglykémie). Často si kontrolujte hladinu cukru v krvi. Obecně platí, že pro prevenci hypoglykémie musíte jíst více jídla a sledovat hladinu cukru v krvi. Pro informace o léčbě hypoglykémie viz rámeček s odkazem na konci této příbalové informace.
Jestliže jste zapomněl(a) užít přípravek Apidra
- Pokud jste vynechali dávku přípravku Apidra nebo jste podali nedostatečné množství inzulínu, může se Vám příliš zvýšit hladina cukru v krvi (hyperglykémie). Často si kontrolujte hladinu cukru v krvi. Pro informace o léčbě hyperglykémie viz rámeček na konci této příbalové informace.
- Nezdvojujte následující dávku, abyste nahradil(a) vynechanou dávku.
Jestliže jste přestal(a) užívat přípravek Apidra
Ukončení užívání přípravku Apidra by mohlo vést k závažné hyperglykémii (velmi vysoká hladina cukru v krvi) a ketoacidóze (hromadění kyseliny v krvi, protože tělo rozkládá tuk místo cukru).
Neukončujte léčbu přípravkem Apidra bez konzultace s lékařem, který Vám řekne, co byste měl(a) udělat.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
Záměna inzulínů
Před každou injekcí musíte vždy zkontrolovat štítek na inzulínu, aby nedošlo k záměně léčivého přípravku Apidra za jiné inzulíny.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
T-W r V r Vil i i V* 1
Závažné nežádoucí účinky
Hypoglykémie (nízká hladina cukru v krvi) může být velmi závažná. Hypoglykémie je velmi často hlášeným nežádoucím účinkem (může postihovat více než 1 z 10 osob). Hypoglykémie (nízká hladina cukru v krvi) znamená, že v krvi není dost cukru. Jestliže Vám příliš klesne hladina cukru v krvi, mohli byste ztratit vědomí. Těžká hypoglykémie může způsobit poškození mozku a může být život ohrožující. Pokud máte příznaky nízké hladiny cukru v krvi, okamžitě se snažte hladinu cukru v krvi zvýšit. Přečtěte si další důležité informace o hypoglykémii a její léčbě na konci této příbalové informace.
Pokud se u Vás vyskytnou následující příznaky, ihned kontaktujte svého lékaře:
Systémové alergické reakce jsou méně častými nežádoucími účinky (mohou postihovat až 1 ze 100 osob).
Generalizovaná alergie na inzulín: Přidružené symptomy mohou zahrnovat rozsáhlé kožní reakce (vyrážka a svědění po celém těle), závažný otok kůže nebo sliznic (angioedém), zkrácený dech, pokles krevního tlaku se zrychleným tepem a pocení. Může se jednat o příznaky závažného případu generalizované alergie na inzulín, včetně anafylaktické reakce, která může být život ohrožující.
Hyperglykémie (vysoká hladina cukru v krvi) znamená, že máte v krvi příliš mnoho cukru.
Četnost výskytu hyperglykémie nelze určit. Pokud máte příliš vysokou hladinu cukru v krvi, vypovídá to o tom, že možná budete potřebovat více inzulínu, než jste si aplikoval(a) v injekci.
Hyperglykémie může zapříčinit diabetickou ketoacidózu (nahromadění kyseliny v krvi v důsledku toho, že organismus odbourává tuk místo cukru).
Jde o závažné nežádoucí účinky.
K těmto situacím může dojít při potížích s infuzní pumpou, nebo pokud není pumpa používána správně.
Znamená to, že jste možná dostal(a) málo inzulínu k léčbě cukrovky.
Pokud k tomu dojde, musíte neodkladně vyhledat lékařskou pomoc.
Vždy mějte k dispozici alternativní možnost aplikace inzulínu pod kůži (viz bod 3, odstavec „Jak se zachází s inzulinovou pumpou“ a „Co dělat, pokud pumpa selže nebo při nesprávném použití inzulínové pumpy“.
Další nežádoucí účinky
Časté nežádoucí účinky (mohou postihovat až 1 z 10 osob)
• Kožní a alergické reakce v místě vpichu injekce
Může se objevit reakce v místě vpichu (např. zarudnutí, neobvykle intenzívní bolestivost injekce, svědění, vyrážka, otok nebo zánět). Tyto reakce se mohou také rozšířit kolem místa vpichu. Většina mírných reakcí na inzulín obvykle odezní během několika dnů až několika týdnů.
Vzácné nežádoucí účinky (mohou postihovat až 1 z 1000 osob)
• Změny kůže v místě aplikace injekce (lipodystrofie)
Pokud podáváte inzulín příliš často do stejného místa na kůži, může se tuková tkáň pod kůží v tomto místě buď ztenčit, nebo zesílit (lipodystrofie). Obojí snižuje účinnost inzulínu podávaného do takto změněného místa. Takovým změnám na kůži lze zabránit střídáním místa vpichu při každé injekci.
Nežádoucí účinky, jejichž frekvenci nelze z dostupných údajů určit
• Oční reakce
Výrazné změny (zlepšení nebo zhoršení) kompenzace Vašeho krevního cukru mohou přechodně narušit Váš zrak. Pokud máte proliferativní retinopatii (oční onemocnění vztahující se k diabetu), mohou závažné hypoglykemické ataky způsobit přechodnou ztrátu vidění.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Apidra uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti, uvedené na krabičce a na štítku injekční lahvičky za „Použitelné do:“ nebo „EXP“. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Neotevřené injekční lahvičky Uchovávejte v chladničce (2°C - 8°C).
Chraňte před mrazem.
Neukládejte přípravek Apidra do blízkosti mrazicího boxu nebo mrazicí vložky.
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
Otevřené injekční lahvičky
Po prvním použití může být injekční lahvička uchovávána až 4 týdny při teplotě do 25°C v krabičce mimo přímé teplo nebo světlo. Nepoužívejte injekční lahvičky po uplynutí této doby použitelnosti. Doporučuje se poznamenat na obal datum prvního použití z lahvičky.
Nepoužívejte tento přípravek, pokud není roztok čirý a bezbarvý.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co Apidra obsahuje
- Léčivou látkou je insulinum glulisinum. Jeden mililitr roztoku obsahuje 100 jednotek léčivé látky insulinum glulisinum (odpovídá množství 3,49 mg). Jedna injekční lahvička obsahuje 10 ml injekčního roztoku, což odpovídá 1000 jednotkám.
- Pomocné látky jsou: metakresol (viz bod 2 „Apidra obsahuje metakresol“), chlorid sodný (viz bod 2 „Důležité informace o některých složkách přípravku Apidra“), trometamol, polysorbát 20, kyselina chlorovodíková 35%, hydroxid sodný, voda pro injekci.
Jak Apidra vypadá a co obsahuje toto balení
Apidra 100 jednotek/ml injekční roztok v injekční lahvičce je čirý, bezbarvý, vodný roztok bez viditelných částic.
Jedna injekční lahvička obsahuje 10 ml roztoku (1000 jednotek). Dostupná jsou balení s 1, 2, 4 a 5 injekčními lahvičkami. Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci a výrobce
Držitel rozhodnutí o registraci:
Sanofi-Aventis Deutschland GmbH D-65926 Frankfurt am Main Německo
Výrobce:
Sanofi-Aventis Deutschland GmbH Industriepark Hochst, D-65926 Frankfurt Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci.
Belgie/Belgique/Belgien
Sanofi Belgium
Tél/Tel : +32 (0)2 710 54 00
Etarapnu
sanofi-aventis Bulgaria EOOD Tea.: +359 (0)2 970 53 00
Lietuva
UAB “SANOFI-AVENTIS LIETUVA” Tel: +370 5 2755224
Luxembourg/Luxemburg
Sanofi Belgium
Tél/Tel : +32 (0)2 710 54 00 (Belgique/Belgien)
Magyarország SANOFI-AVENTIS Zrt. Tel.: +36 1 505 0050
Malta
Sanofi Malta Ltd. Tel: +356 21493022
Danmark
sanofi-aventis Denmark A/S Tlf: +45 45 16 70 00
Deutschland
Sanofi-Aventis Deutschland GmbH Tel: +49 (0)180 2 222010
Eesti
sanofi-aventis Estonia OU Tel: +372 627 34 88
EkXúSa
sanofi-aventis AEBE Tr|k: +30 210 900 16 00
Nederland
sanofi-aventis Netherlands B.V. Tel: +31 (0)182 557 755
Norge
sanofi-aventis Norge AS Tlf: +47 67 10 71 00
Osterreich
sanofi-aventis GmbH Tel: +43 1 80 185 - 0
|
Espaňa sanofi-aventis, S.A. Tel: +34 93 485 94 00 |
Polska sanofi-aventis Sp. z o.o. Tel. : +48 22 280 00 00 |
|
France sanofi-aventis France Tél: 0 800 222 555 Appel depuis l’étranger : +33 1 57 63 23 23 |
Portugal Sanofi - Produtos Farmaceuticos, Lda. Tel : +351 21 35 89 400 |
|
Hrvatska sanofi-aventis Croatia d.o.o. Tel: +385 1 600 34 00 |
Románia Sanofi Romania SRL Tel: +40 (0) 21 317 31 36 |
|
Ireland sanofi-aventis Ireland Ltd. T/A SANOFI Tel : +353 (0) 1 403 56 00 |
Slovenija sanofi-aventis d.o.o. Tel: +386 1 560 48 00 |
|
Ísland Vistor hf. Sími: +354 535 7000 |
Slovenská republika sanofi-aventis Pharma Slovakia s.r.o. Tel: +421 2 33 100 100 |
|
Italia Sanofi S.p.A. Tel: 800 13 12 12 (domande di tipo tecnico) 800 536389 (altre domande) |
Suomi/Finland Sanofi Oy Puh/Tel: +358 (0) 201 200 300 |
|
Kúnpoq sanofi-aventis Cyprus Ltd. Tr^: +357 22 871600 |
Sverige Sanofi AB Tel: +46 (0)8 634 50 00 |
|
Latvija sanofi-aventis Latvia SIA Tel: +371 67 33 24 51 |
United Kingdom Sanofi Tel: +44 (0) 845 372 7101 |
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku j sou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky: http://www.ema.europa.eu/
HYPERGLYKÉMIE A HYPOGLYKÉMIE
Vždy u sebe mějte nějaký cukr (minimálně 20 gramů).
Noste u sebe nějakou informaci, která ukazuje, že máte cukrovku.
HYPERGLYKÉMIE (vysoká hladina cukru v krvi)
Pokud je Vaše hladina cukru v krvi příliš vysoká (hyperglykémie), možná jste si aplikoval(a) málo inzulínu.
Proč vzniká hyperglykémie?
Příklady:
- nepodali jste si injekci inzulínu nebo jste ho nepodali dostatečné množství nebo se inzulín stal méně účinným, např. z důvodu nesprávného uchovávání,
- máte méně pohybu než obvykle, jste ve stresu (emocionální úzkost, rozrušení) nebo máte úraz, j ste po operaci, máte infekci nebo horečku,
- užíváte nebo jste užíval(a) některé jiné léky (viz bod 2 „Další léčivé přípravky a Apidra“). Varovné příznaky hyperglykémie
Žízeň, zvýšené močení, únava, suchá pokožka, zčervenání obličeje, nechutenství, nízký krevní tlak, zrychlený tep a přítomnost glukózy a ketolátek v moči. Bolest žaludku, rychlé a hluboké dýchání, ospalost nebo dokonce ztráta vědomí mohou být známkami vážného stavu (ketoacidózy) vyplývajícího z nedostatku inzulínu.
Co dělat v případě hyperglykémie?
Změřte si hladinu cukru v krvi a ketolátek v moči co nejdříve, jakmile se výše uvedené příznaky objeví. Těžká hyperglykémie nebo ketoacidóza musí být vždy léčena lékařem, obvykle v nemocnici.
HYPOGLYKÉMIE (nízká hladina cukru v krvi)
Pokud je hladina Vašeho cukru v krvi příliš nízká, můžete ztratit vědomí. Závažná hypoglykémie může vyvolat srdeční záchvat nebo poškození mozku a může být život ohrožující. Za normálních okolností byste měl(a) být schopen/schopna rozpoznat přílišné snižování hladiny cukru v krvi, a proto můžete správně reagovat.
Proč vzniká hypoglykémie?
Příklady:
- aplikujete si příliš mnoho inzulínu,
- vynecháte nebo odložíte jídlo,
- nejíte dostatečně, nebo snězené jídlo obsahuje méně sacharidů než normálně (cukr a látky podobné cukru se nazývají sacharidy; umělá sladidla však NEJSOU sacharidy),
- dochází u Vás ke ztrátě sacharidů zvracením nebo průjmem,
- pijete alkohol, zvláště pokud nejíte dostatečně,
- cvičíte více než obvykle nebo vykonáváte jiný typ fyzické činnosti,
- zotavujete se ze zranění, operace, nebo z jiných forem stresu,
- zotavujete se po nemoci nebo z horečky,
- užíváte nebo jste přestali užívat některé jiné léky (viz bod 2 „Další léčivé přípravky a Apidra“). Výskyt hypoglykémie je pravděpodobnější, pokud:
- právě zahajujete léčbu inzulínem nebo přecházíte na jiný inzulínový přípravek,
- Vaše hladiny cukru v krvi j sou téměř normální nebo j sou nestálé,
- změníte oblast kůže, kam podáváte injekci inzulínu (např. ze stehna na horní část paže),
- trpíte závažným onemocněním ledvin nebo j ater nebo některou j inou chorobou, j ako j e hypotyreóza (snížená funkce štítné žlázy).
Varovné příznaky hypoglykémie
- Ve Vašem těle:
Příklady příznaků, které naznačují, že hladina cukru v krvi klesá příliš nízko nebo příliš rychle: pocení, vlhká pokožka, úzkost, zrychlený tep, vysoký krevní tlak, bušení srdce a nepravidelný srdeční rytmus. Často se tyto příznaky vyvinou dříve, než příznaky spojené s nízkou hladinou cukru v mozku.
- Ve Vašem mozku:
Příklady příznaků naznačujících nízkou hladinu cukru v mozku: bolest hlavy, intenzivní hlad, nevolnost, zvracení, únava, ospalost, poruchy spánku, neklid, agresivní chování, pokles koncentrace, zhoršené reakce, deprese, zmatenost, poruchy řeči (někdy úplná ztráta řeči), poruchy vidění, třes, paralýza, pocity mravenčení (parestézie), brnění a pocity necitlivosti v oblasti úst, závrať, ztráta sebekontroly, neschopnost postarat se o sebe, křeče a ztráta vědomí.
První příznaky, které Vás varují před hypoglykémií („varovné příznaky“), mohou být pozměněny, mohou být slabší nebo mohou zcela chybět, pokud:
- jste starší;
- máte diabetes již dlouho,
- trpíte určitým typem nervového onemocnění (diabetická autonomní neuropatie),
- Vás hypoglykémie trápila nedávno (např. předchozí den) nebo se vyvíjí pomalu,
- máte téměř normální nebo alespoň hodně zlepšenou hladinu cukru v krvi,
- užíváte nebo jste užíval(a) některé jiné léky (viz bod 2 „Další léčivé přípravky a Apidra“).
V takových případech se u Vás může rozvinout těžká hypoglykémie (a dokonce mdloba), aniž byste si toho všimli včas. Seznamte se co nejlépe se svými varovnými příznaky hypoglykémie. Je-li to nutné, častější měření cukru v krvi může pomoci odhalit mírné hypoglykemické epizody, které by jinak mohly být přehlédnuty. Pokud se nemůžete spolehnout na rozpoznání svých varovných příznaků, vyhněte se takovým situacím (např. řízení vozidla), při kterých by hypoglykémie pro Vás nebo pro ostatní představovala riziko.
Co dělat v případě hypoglykémie?
1. Nepodávejte inzulín. Okamžitě si vezměte asi 10 až 20 g cukru, např. glukózu, kostky cukru nebo cukrem oslazený nápoj. Upozornění: umělá sladidla a potraviny obsahující umělá sladidla (např. dietní nápoje) při hypoglykémii nepomáhají.
2. Poté snězte něco, co má dlouhotrvající účinek na nárůst cukru v krvi (např. chléb nebo těstoviny). Dbejte na to, abyste tyto záležitosti předem prohovořili s lékařem nebo sestrou.
3. Pokud se hypoglykémie znovu vrací, vezměte si dalších 10 až 20 g cukru.
4. Informujte okamžitě lékaře, jestliže hypoglykémii nebo její opakování nejste schopni zvládnout.
Oznamte svým příbuzným, přátelům a blízkým kolegům následující:
Pokud nemůžete polykat nebo upadnete do bezvědomí, budete potřebovat injekci glukózy nebo glukagonu (lék, který zvyšuje hladinu cukru v krvi). Tyto injekce jsou opodstatněné dokonce i tehdy, není-li jisté, že máte hypoglykémii.
Je vhodné změřit si hladinu cukru v krvi okamžitě po požití glukózy, abyste zkontrolovali, že máte skutečně hypoglykémii.
NÁSLEDUJÍCÍ INFORMACE JE URČENA POUZE PRO ZDRAVOTNICKÉ PRACOVNÍKY:
Přípravek Apidra smí být podáván intravenózně, podání musí provádět zdravotník.
Pokyny pro intravenózní podání
Přípravek Apidra by měl být používán v koncentraci inzulínu glulisinu 1 jednotka/ml v infuzních systémech s infuzním roztokem chloridu sodného 9 mg/ml (0,9%) s chloridem draselným o koncentraci 40 mmol/l nebo bez něj, v koextrudovaných polyolefinových/polyamidových umělohmotných infuzních vacích s jednorázovými infuzními sety. Inzulin glulisin k intravenóznímu podání v koncentraci 1 jednotka/ml je stabilní při pokojové teplotě po dobu 48 hodin.
Po naředění k intravenóznímu podání musí být roztok před podáním vizuálně zkontrolován, zda neobsahuje částice. Nikdy nepoužívejte roztok, který je zkalený nebo obsahuje částice; používejte pouze čirý bezbarvý roztok.
Bylo zjištěno, že Apidra není kompatibilní s 5% roztokem glukózy a s Ringerovým roztokem, tudíž se s těmito roztoky nesmí používat. Použití jiných roztoků dosud nebylo studováno.
Příbalová informace: informace pro uživatele
Apidra 100 jednotek/ml injekční roztok v zásobní vložce
Insulinům glulisinum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje. Návod na použití inzulínového pera je přiložen k Vašemu inzulínovému peru. Před použitím léčivého přípravku si oba návody přečtěte.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je Apidra a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Apidra používat
3. Jak se přípravek Apidra používá
4. Možné nežádoucí účinky
5. Jak přípravek Apidra uchovávat
6. Obsah balení a další informace
1. Co je Apidra a k čemu se používá
Apidra je antidiabetikum používané ke snížení hladiny cukru v krvi u pacientů s diabetes mellitus; může být podána dospělým, mladistvým a dětem ve věku 6 a více let. Diabetes mellitus je onemocnění, při kterém Váš organismus neprodukuje dostatek inzulínu pro kontrolu hladiny cukru v krvi.
Přípravek je vyroben biotechnologicky. Inzulín glulisin má rychlý nástup účinku během 10-20 minut a krátké trvání účinku přibližně 4 hodiny.
2. Čemu musíte věnovat pozornost, než začnete přípravek Apidra používat Nepoužívejte přípravek Apidra
- Jestliže jste alergický(á) na inzulín glulisin nebo na kteroukoliv další složku tohoto přípravku (uvedenou v bodě 6).
- Jestliže máte hladinu cukru v krvi příliš nízkou (hypoglykémie). Postupujte podle pokynů pro hypoglykémii (viz rámeček na konci této příbalové informace).
Upozornění a opatření
Před užitím přípravku Apidra se poraďte se svým lékařem, lékarníkem nebo zdravotní sestrou. Dodržujte pečlivě pokyny pro dávkování, sledování (testy krve), dietu a fyzickou aktivitu (fyzickou práci a cvičení), které jste projednali se svým lékařem.
Zvláštní skupiny pacientů
Pokud máte problémy s játry nebo s ledvinami, upozorněte svého lékaře, protože můžete potřebovat nižší dávku.
Nejsou dostatečné klinické informace o použití přípravku Apidra u dětí mladších 6 let.
Cestování
Před cestováním se poraďte se svým lékařem. Možná bude třeba se poradit o
- dostupnosti Vašeho inzulínu v zemi, kterou navštívíte,
- dodávkách inzulínu, injekčních stříkaček, atd.,
- správném uchovávání Vašeho inzulínu během cestování,
- časové dostupnosti jídla a podávání inzulínu během cestování,
- možných účincích přesunu v různých časových pásmech,
- možných zdravotních rizicích v zemích, které navštívíte,
- tom, jak se zachovat v mimořádných situacích, tj. pokud se Vám udělá špatně nebo onemocníte. Onemocnění a úrazy
V následujících situacích může léčba Vašeho diabetu vyžadovat zvláštní péči:
- pokud jste nemocný/á nebo máte větší úraz, může dojít ke zvýšení hladiny Vašeho cukru v krvi (hyperglykémie),
- pokud dostatečně nejíte, může se Vám hladina cukru v krvi velmi snížit (hypoglykémie).
Ve většině případů budete potřebovat lékařskou péči. Zajistěte možnost rychlého kontaktu lékaře.
Pokud máte diabetes typu 1 (na inzulínu závislý diabetes), nepřestávejte užívat svůj inzulín a pokračujte v příjmu dostatečného množství cukrů. Vždy řekněte lidem, kteří Vás ošetřují nebo léčí, že potřebujete inzulín.
U některých pacientů, kteří dlouhodobě trpěli cukrovkou typu 2 a onemocněním srdce, nebo prodělali mozkovou příhodu, a kteří byli léčeni pioglitazonem a inzulínem, byl zaznamenán výskyt srdečního selhání. Jestliže zjistíte známky srdečního selhání jako je neobvyklá dušnost nebo náhlé zvýšení tělesné hmotnosti nebo lokalizovaný otok (edém), informujte co nejrychleji svého lékaře.
Další léčivé přípravky a přípravek Apidra
Některé léky způsobují změnu hladiny cukru v krvi (snížení, zvýšení nebo obojí v závislosti na situaci). V každém případě může být nezbytné upravit Vaši dávku inzulínu, aby se zabránilo hladinám cukru v krvi, které jsou buď příliš nízké, nebo příliš vysoké. Buďte opatrní, když začínáte nebo přestáváte užívat nějaký další lék.
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat. Zeptejte se svého lékaře předtím, než si lék vezmete, zda a jak jím může být ovlivněna Vaše hladina cukru v krvi, popř. co je třeba dělat.
Mezi léky, které mohou vyvolat pokles hladiny cukru v krvi (hypoglykémii), patří:
- všechny další léky pro léčbu diabetu,
- inhibitory angiotenzin konvertujícího enzymu (ACE inhibitory) (používané k léčbě některých srdečních onemocnění, vysokého krevního tlaku),
- disopyramid (používaný k léčbě některých srdečních onemocnění),
- fluoxetin (používaný k léčbě depresí),
- fibráty (používané ke snížení vysokých hladin krevních lipidů),
- inhibitory monoaminooxidázy (MAO inhibitory) (používané k léčbě depresí),
- pentoxifylin, propoxyfen, salicyláty (např. aspirin, používaný ke zmírnění bolesti a snížení horečky),
- sulfonamidová antibiotika.
Mezi léky, které mohou vyvolat zvýšení hladiny cukru v krvi (hyperglykémii), patří:
- kortikosteroidy (např. „kortizon“ používaný pro léčbu zánětu),
- danazol (lék pro navození ovulace),
- diazoxid (používaný pro léčbu vysokého krevního tlaku),
- diuretika (používaná pro léčbu vysokého krevního tlaku nebo nadměrného zadržování tekutin),
- glukagon (hormon slinivky břišní používaný pro léčbu závažné hypoglykémie),
- isoniazid (používaný pro léčbu tuberkulózy),
- estrogeny a progestogeny (např. v antikoncepčních pilulkách užívaných k zamezení početí),
- deriváty fenothiazinu (používané pro léčbu duševních poruch),
- somatropin (růstový hormon),
- sympatomimetika (např. epinefrin [adrenalin], salbutamol, terbutalin užívané k léčbě astmatu),
- hormony štítné žlázy (užívané k léčbě onemocnění štítné žlázy),
- inhibitory proteáz (užívané k léčbě HIV),
- atypická antipsychotika (např. olanzapin a klozapin).
Vaše hladina cukru v krvi může buď poklesnout, nebo vzrůst, pokud užíváte:
- betablokátory (používané pro léčbu vysokého krevního tlaku),
- klonidin (používaný pro léčbu vysokého krevního tlaku),
- soli lithia (používané pro léčbu duševních poruch).
Pentamidin (používaný pro léčbu některých infekcí způsobených parazity) může vyvolat hypoglykémii, po které může někdy následovat hyperglykémie.
Betablokátory stejně jako ostatní sympatolytika (např. klonidin, guanetidin a reserpin) mohou oslabit nebo úplně potlačit první varovné příznaky, které Vám mohou pomoci rozpoznat hypoglykémii.
Pokud si nejste jisti, zda neužíváte některý z takových léků, zeptejte se svého lékaře nebo lékárníka.
Užívání přípravku Apidra s alkoholem
Hladiny Vašeho cukru v krvi mohou buď vzrůst, nebo poklesnout, pokud konzumujete alkohol. Těhotenství a kojení
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek užívat.
Informujte svého lékaře, pokud plánujete těhotenství nebo pokud již jste těhotná. Je možné, že bude třeba měnit dávku inzulínu během těhotenství a po porodu. Zvláště pečlivá kontrola diabetu a prevence hypoglykémie jsou důležité pro zdraví Vašeho dítěte.
Nejsou k dispozici žádné nebo jen omezené údaje o použití přípravku Apidra u těhotných žen.
Pokud kojíte, poraďte se se svým lékařem, neboť může být nezbytné upravit Vám dávku inzulínu a Vaši dietu.
Řízení dopravních prostředků a obsluha strojů
Vaše schopnost soustředit se nebo reagovat může být snížena, jestliže:
- máte hypoglykémii (nízkou hladinu cukru v krvi)
- máte hyperglykémii (vysokou hladinu cukru v krvi).
Myslete na tento možný problém ve všech situacích, kde byste mohli dostat sebe nebo ostatní do nebezpečí (např. při řízení vozidla nebo při obsluze strojů).
Měl(a) byste se poradit se svým lékařem o vhodnosti řízení vozidel, pokud:
- máte časté epizody hypoglykémie,
- jsou první varovné příznaky, které Vám pomohou rozpoznat hypoglykémii, omezené nebo chybějí.
Důležité informace o některých složkách přípravku Apidra
Tento léčivý přípravek obsahuje méně než 1 mmol (23 mg) sodíku v jedné dávce, tj. v podstatě sodík neobsahuje.
Apidra obsahuje metakresol
Apidra obsahuje metakresol, který může způsobovat alergické reakce.
3. Jak se přípravek Apidra používá Dávkování
Vždy užívejte přípravek Apidra přesně tak, jak Vám doporučil lékař. Pokud si nejste jist(a), poraďte se s lékařem nebo s lékárníkem.
Na základě Vašeho životního stylu a Vašich výsledků testů cukru (glukózy) v krvi a Vašeho předchozího užívání inzulínu lékař určí, jakou dávku přípravku Apidra budete potřebovat.
Apidra je rychle účinkující inzulín. Je možné, že Váš lékař Vám řekne, abyste ji užívali v kombinaci se střednědobě nebo dlouhodobě působícím inzulínem nebo bazálním inzulínem nebo s tabletami na snížení vysoké hladiny cukru v krvi.
Jestliže přecházíte z jiného inzulínu na inzulín glulisin, Vaše dávkování může vyžadovat úpravu lékařem.
Hladinu cukru v krvi může ovlivnit mnoho faktorů. Měli byste znát tyto faktory, abyste byli schopni reagovat správně na změny hladiny cukru v krvi a předejít jejich přílišnému zvýšení nebo přílišnému snížení. Další informace si přečtěte na konci této příbalové informace.
Způsob podání
Apidra se podává injekcí pod kůži (subkutánně).
Lékař Vám ukáže, do které oblasti kůže máte přípravek Apidra podávat. Injekci přípravku Apidra lze podávat do břišní stěny, do stehna nebo horní části paže nebo kontinuální infuzí do břišní stěny. Účinek bude o něco rychlejší, jestliže si injekci inzulínu aplikujete do břišní stěny. Tak jako u všech ostatních inzulínů musíte měnit s každou injekcí místo vpichu injekce a místo infuze v rámci vybrané oblasti (břišní stěna, stehno, horní část paže), kam injekci podáváte.
Frekvence podávání
Přípravek Apidra by se měl užívat krátce (0 - 15 minut) před jídlem nebo brzy po jídle.
Pokyny pro správné použití
Jak se zachází se zásobními vložkami
Pro zajištění přesné dávky přípravku Apidra je možné používat zásobní vložky pouze s následujícími pery:
- JunioSTAR, které odměřuje dávku v krocích po 0,5 jednotky
- OptiPen, ClikSTAR, Tactipen Autopen 24 nebo AllStar, která odměřují dávku v krocích po 1 jednotce. Všechna uvedená pera nemusí být ve Vaší zemi na trhu.
Pera je třeba používat podle doporučení v informaci, kterou poskytuje výrobce.
Pokyny výrobce na použití pera musí být pečlivě dodržovány při vložení zásobní vložky, upevnění jehly a podání injekce inzulínu.
Před vložením zásobní vložky do pera k opakovanému použití ponechejte zásobní vložku 1 až 2 hodiny při pokojové teplotě.
Zásobní vložku si prohlédněte ještě předtím, než ji použijete. Použijte pouze ty zásobní vložky, ve kterých je roztok čirý, bezbarvý, bez viditelných částic.
Před použitím přípravek nemíchejte ani s ním netřepejte.
Zvláštní upozornění před aplikací injekce
Před injekcí musí být ze zásobní vložky odstraněny vzduchové bubliny (viz návod na použití pera). Prázdné zásobní vložky nesmí být znovu plněny.
Pero pro opakované použití smíte používat pouze Vy, aby se tak zabránilo jakémukoliv druhu kontaminace.
Problémy s inzulínovým perem?
Při použití inzulínového pera se řiďte pokyny výrobce.
Pokud je inzulínové pero poškozeno nebo nefunguje správně (v důsledku mechanické závady), musí být zlikvidováno a musí být použito nové inzulínové pero.
Pokud pero selže, může být roztok natažen ze zásobní vložky do stříkačky. Proto si ponechte injekční stříkačky a rovněž jehly v zásobě. Používejte však pouze injekční stříkačky, které jsou určeny pro koncentrace inzulínu 100 jednotek/ml.
Jestliže jste užil(a) více přípravku Apidra, než jste měl(a)
- Pokud jste podali příliš mnoho přípravku Apidra, může se Vám příliš snížit hladiny cukru v krvi (hypoglykémie). Často si kontrolujte hladinu cukru v krvi. Obecně platí, že pro prevenci hypoglykémie musíte jíst více jídla a sledovat hladinu cukru v krvi. Informace o léčbě hypoglykémie si přečtěte v rámečku s odkazem na konci této příbalové informace.
Jestliže jste zapomněl(a) užít přípravek Apidra
- Pokud jste vynechali dávku přípravku Apidra nebo jste podali nedostatečné množství
inzulínu, může se Vám příliš zvýšit hladina cukru v krvi (hyperglykémie). Často si kontrolujte hladinu cukru v krvi. Pro informace o léčbě hyperglykémie viz rámeček na konci této příbalové informace.
- Nezdvojujte následující dávku, abyste nahradil(a) vynechanou dávku.
Jestliže jste přestal(a) užívat přípravek Apidra
Ukončení užívání přípravku Apidra by mohlo vést k závažné hyperglykémii (velmi vysoká hladina cukru v krvi) a ketoacidóze (hromadění kyseliny v krvi, protože tělo rozkládá tuk místo cukru). Neukončujte léčbu přípravkem Apidra bez konzultace s lékařem, který Vám řekne, co byste měl(a) udělat.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
Záměna inzulínů
Před každou injekcí musíte vždy zkontrolovat štítek na inzulínu, aby nedošlo k záměně léčivého přípravku Apidra za jiné inzulíny.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
T-W r V r V ř I r r v» 1
Závažné nežádoucí účinky
Hypoglykémie (nízká hladina cukru v krvi) může být velmi závažná. Hypoglykémie je velmi často hlášeným nežádoucím účinkem (může postihovat více než 1 z 10 osob). Hypoglykémie (nízká hladina cukru v krvi) znamená, že v krvi není dost cukru. Jestliže Vám příliš klesne hladina cukru v krvi, mohli byste ztratit vědomí. Těžká hypoglykémie může způsobit poškození mozku a může být život ohrožující. Pokud máte příznaky nízké hladiny cukru v krvi, okamžitě se snažte hladinu cukru v krvi zvýšit. Přečtěte si další důležité informace o hypoglykémii a její léčbě na konci této příbalové informace.
Pokud se u Vás vyskytnou následující příznaky, ihned kontaktujte svého lékaře:
Systémové alergické reakce jsou méně častými nežádoucími účinky (mohou postihovat až 1 ze 100 osob).
Generalizovaná alergie na inzulín: Přidružené symptomy mohou zahrnovat rozsáhlé kožní reakce (vyrážka a svědění po celém těle), závažný otok kůže nebo sliznic (angioedém), zkrácený dech, pokles krevního tlaku se zrychleným tepem a pocení. Může se jednat o příznaky závažného případu generalizované alergie na inzulín, včetně anafylaktické reakce, která může být život ohrožující.
Hyperglykémie (vysoká hladina cukru v krvi) znamená, že máte v krvi příliš mnoho cukru.
Četnost výskytu hyperglykémie nelze určit. Pokud máte příliš vysokou hladinu cukru v krvi, vypovídá to o tom, že možná budete potřebovat více inzulínu, než jste si aplikoval(a) v injekci. Může jít o závažný stav, pokud se hladina glukózy velmi zvýší.
Přečtěte si další informace o známkách a příznacích hyperglykémie v rámečku na konci této příbalové informace.
Jiné nežádoucí účinky
Časté nežádoucí účinky (mohou postihovat až 1 z 10 osob)
• Kožní a alergické reakce v místě vpichu injekce
Může se objevit reakce v místě vpichu (např. zarudnutí, neobvykle intenzívní bolestivost injekce, svědění, vyrážka, otok nebo zánět). Tyto reakce se mohou také rozšířit kolem místa vpichu. Většina mírných reakcí na inzulín obvykle odezní během několika dnů až několika týdnů.
Vzácné nežádoucí účinky (mohou postihovat až 1 z 1000 osob)
• Změny kůže v místě aplikace injekce (lipodystrofie)
Pokud podáváte inzulín příliš často do stejného místa na kůži, může se tuková tkáň pod kůží v tomto místě buď ztenčit, nebo zesílit (lipodystrofie). Obojí snižuje účinnost inzulínu podávaného do takto změněného místa. Takovým změnám na kůži lze zabránit střídáním místa vpichu při každé injekci.
Nežádoucí účinky, jejichž frekvenci nelze z dostupných údajů určit
• Oční reakce
Výrazné změny (zlepšení nebo zhoršení) kompenzace Vašeho krevního cukru mohou přechodně narušit Váš zrak. Pokud máte proliferativní retinopatii (oční onemocnění vztahující se k diabetu), mohou závažné hypoglykemické ataky způsobit přechodnou ztrátu vidění.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Apidra uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti, uvedené na krabičce a na štítku zásobní vložky za „Použitelné do:“ nebo „EXP“. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Neotevřené zásobní vložky Uchovávejte v chladničce (2°C - 8°C).
Chraňte před mrazem.
Neukládejte přípravek Apidra do blízkosti mrazicího boxu nebo mrazicí vložky.
Uchovávejte zásobní vložku v krabičce, aby byl přípravek chráněn před světlem.
Používané zásobní vložky
Používané zásobní vložky (v insulinovém peru) mohou být uchovávány až 4 týdny při teplotě do 25°C mimo přímé teplo nebo světlo a nesmí být uchovávány v chladničce. Nepoužívejte je po uplynutí této doby použitelnosti.
Nepoužívejte tento přípravek, pokud není roztok čirý a bezbarvý.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co Apidra obsahuje
- Léčivou látkou je insulinum glulisinum. Jeden mililitr roztoku obsahuje 100 jednotek léčivé látky insulinum glulisinum (odpovídá množství 3,49 mg). Jedna zásobní vložka obsahuje 3 ml injekčního roztoku, což odpovídá 300 jednotkám.
- Pomocné látky jsou: metakresol (viz bod 2 „Apidra obsahuje metakresol“), chlorid sodný (viz bod 2 „Důležité informace o některých složkách přípravku Apidra“), trometamol, polysorbát 20, kyselina chlorovodíková 35%, hydroxid sodný, voda pro injekci.
Jak Apidra vypadá a co obsahuje toto balení
Apidra 100 jednotek/ml injekční roztok v zásobní vložce je čirý, bezbarvý, vodný injekční roztok bez
viditelných částic.
Jedna zásobní vložka obsahuje 3 ml roztoku (300 jednotek). Dostupná jsou balení s 1, 3, 4, 5, 6, 8, 9 a
10 zásobními vložkami. Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci a výrobce
Držitel rozhodnutí o registraci: Sanofi-Aventis Deutschland GmbH
D-65926 Frankfurt am Main Německo
Výrobce:
Sanofi-Aventis Deutschland GmbH Industriepark Hochst, D-65926 Frankfurt Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci
|
Belgie/Belgique/Belgien Sanofi Belgium Tél/Tel : +32 (0)2 710 54 00 |
Lietuva UAB “SANOFI-AVENTIS LIETUVA” Tel: +370 5 2755224 |
|
BtnrapHH sanofi-aventis Bulgaria EOOD Ten.: +359 (0)2 970 53 00 |
Luxembourg/Luxemburg Sanofi Belgium Tél/Tel : +32 (0)2 710 54 00 (Belgique/Belgien) |
|
Česká republika sanofi-aventis, s.r.o. Tel: +420 233 086 111 |
Magyarország SANOFI-AVENTIS Zrt. Tel.: +36 1 505 0050 |
|
Danmark sanofi-aventis Denmark A/S Tlf: +45 45 16 70 00 |
Malta Sanofi Malta Ltd. Tel: +356 21493022 |
|
Deutschland Sanofi-Aventis Deutschland GmbH Tel: +49 (0)180 2 222010 |
Nederland sanofi-aventis Netherlands B.V. Tel: +31 (0)182 557 755 |
|
Eesti sanofi-aventis Estonia OU Tel: +372 627 34 88 |
Norge sanofi-aventis Norge AS Tlf: +47 67 10 71 00 |
|
EXXáSa sanofi-aventis AEBE Tr^: +30 210 900 16 00 |
Osterreich sanofi-aventis GmbH Tel: +43 1 80 185 - 0 |
|
Espaňa sanofi-aventis, S.A. Tel: +34 93 485 94 00 |
Polska sanofi-aventis Sp. z o.o. Tel. : +48 22 280 00 00 |
|
France sanofi-aventis France Tél: 0 800 222 555 Appel depuis l’étranger : +33 1 57 63 23 23 |
Portugal Sanofi - Produtos Farmaceuticos, Lda. Tel : +351 21 35 89 400 |
|
Hrvatska sanofi-aventis Croatia d.o.o. Tel: +385 1 600 34 00 |
Románia Sanofi Romania SRL Tel: +40 (0) 21 317 31 36 |
|
Ireland sanofi-aventis Ireland Ltd. T/A SANOFI Tel : +353 (0) 1 403 56 00 |
Slovenija sanofi-aventis d.o.o. Tel: +386 1 560 48 00 |
Ísland
Slovenská republika
sanofi-aventis Pharma Slovakia s.r.o. Tel: +421 2 33 100 100
Suomi/Finland
Sanofi Oy
Puh/Tel: +358 (0) 201 200 300
Sverige
Sanofi AB
Tel: +46 (0)8 634 50 00
United Kingdom
Sanofi
Tel: +44 (0) 845 372 7101
Vistor hf.
Sími: +354 535 7000 Italia
Sanofi S.p.A.
Tel: 800 13 12 12 (domande di tipo tecnico) 800 536389 (altre domande)
Kúrcpog
sanofi-aventis Cyprus Ltd.
Tr^: +357 22 871600
Latvija
sanofi-aventis Latvia SIA Tel: +371 67 33 24 51
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku j sou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/
HYPERGLYKÉMIE A HYPOGLYKÉMIE
Vždy u sebe mějte nějaký cukr (minimálně 20 gramů).
Noste u sebe nějakou informaci, která ukazuje, že máte cukrovku.
HYPERGLYKÉMIE (vysoká hladina cukru v krvi)
Pokud je Vaše hladina cukru v krvi příliš vysoká (hyperglykémie), možná jste si aplikoval(a) málo inzulínu.
Proč vzniká hyperglykémie?
Příklady:
- nepodali jste si injekci inzulínu nebo jste ho nepodali dostatečné množství nebo se inzulín stal méně účinným, např. z důvodu nesprávného uchovávání,
- máte méně pohybu než obvykle, jste ve stresu (emocionální úzkost, rozrušení) nebo máte úraz, j ste po operaci, máte infekci nebo horečku,
- užíváte nebo jste užíval(a) některé jiné léky (viz bod 2 „Další léčivé přípravky a Apidra“). Varovné příznaky hyperglykémie
Žízeň, zvýšené močení, únava, suchá pokožka, zčervenání obličeje, nechutenství, nízký krevní tlak, zrychlený tep a přítomnost glukózy a ketolátek v moči. Bolest žaludku, rychlé a hluboké dýchání, ospalost nebo dokonce ztráta vědomí mohou být známkami vážného stavu (ketoacidózy) vyplývajícího z nedostatku inzulínu.
Co dělat v případě hyperglykémie?
Změřte si hladinu cukru v krvi a ketolátek v moči co nejdříve, jakmile se výše uvedené příznaky objeví. Těžká hyperglykémie nebo ketoacidóza musí být vždy léčena lékařem, obvykle v nemocnici.
HYPOGLYKÉMIE (nízká hladina cukru v krvi)
Pokud je hladina Vašeho cukru v krvi příliš nízká, můžete ztratit vědomí. Závažná hypoglykémie může vyvolat srdeční záchvat nebo poškození mozku a může být život ohrožující. Za normálních okolností byste měl(a) být schopen/schopna rozpoznat přílišné snižování hladiny cukru v krvi, a proto můžete správně reagovat.
Proč vzniká hypoglykémie?
Příklady:
- aplikujete si příliš mnoho inzulínu,
- vynecháte nebo odložíte jídlo,
- nejíte dostatečně, nebo snězené jídlo obsahuje méně sacharidů než normálně (cukr a látky podobné cukru se nazývají sacharidy; umělá sladidla však NEJSOU sacharidy),
- dochází u Vás ke ztrátě sacharidů zvracením nebo průjmem,
- pijete alkohol, zvláště pokud nejíte dostatečně,
- cvičíte více než obvykle nebo vykonáváte jiný typ fyzické činnosti,
- zotavujete se ze zranění, operace, nebo z jiných forem stresu,
- zotavujete se po nemoci nebo z horečky,
- užíváte nebo jste přestali užívat některé jiné léky (viz bod 2 „Další léčivé přípravky a Apidra“). Výskyt hypoglykémie je pravděpodobnější, pokud:
- právě zahajujete léčbu inzulínem nebo přecházíte na jiný inzulínový přípravek,
- Vaše hladiny cukru v krvi j sou téměř normální nebo j sou nestálé,
- změníte oblast kůže, kam podáváte injekci inzulínu (např. ze stehna na horní část paže),
- trpíte závažným onemocněním ledvin nebo j ater nebo některou j inou chorobou, j ako j e hypotyreóza (snížená funkce štítné žlázy).
Varovné příznaky hypoglykémie
- Ve Vašem těle:
Příklady příznaků, které naznačují, že hladina cukru v krvi klesá příliš nízko nebo příliš rychle: pocení, vlhká pokožka, úzkost, zrychlený tep, vysoký krevní tlak, bušení srdce a nepravidelný srdeční rytmus. Často se tyto příznaky vyvinou dříve než příznaky spojené s nízkou hladinou cukru v mozku.
- Ve Vašem mozku:
Příklady příznaků naznačujících nízkou hladinu cukru v mozku: bolest hlavy, intenzivní hlad, nevolnost, zvracení, únava, ospalost, poruchy spánku, neklid, agresivní chování, pokles koncentrace, zhoršené reakce, deprese, zmatenost, poruchy řeči (někdy úplná ztráta řeči), poruchy vidění, třes, paralýza, pocity mravenčení (parestézie), brnění a pocity necitlivosti v oblasti úst, závrať, ztráta sebekontroly, neschopnost postarat se o sebe, křeče a ztráta vědomí.
První příznaky, které Vás varují před hypoglykémií („varovné příznaky“), mohou být pozměněny, mohou být slabší nebo mohou zcela chybět, pokud:
- jste starší;
- máte diabetes již dlouho,
- trpíte určitým typem nervového onemocnění (diabetická autonomní neuropatie),
- Vás hypoglykémie trápila nedávno (např. předchozí den) nebo se vyvíjí pomalu,
- máte téměř normální nebo alespoň hodně zlepšenou hladinu cukru v krvi,
- užíváte nebo jste užíval(a) některé jiné léky (viz bod 2 „Další léčivé přípravky a Apidra“).
V takových případech se u Vás může rozvinout těžká hypoglykémie (a dokonce mdloba), aniž byste si toho všimli včas. Seznamte se co nejlépe se svými varovnými příznaky hypoglykémie. Je-li to nutné, častější měření cukru v krvi může pomoci odhalit mírné hypoglykemické epizody, které by jinak mohly být přehlédnuty. Pokud se nemůžete spolehnout na rozpoznání svých varovných příznaků, vyhněte se takovým situacím (např. řízení vozidla), při kterých by hypoglykémie pro Vás nebo pro ostatní představovala riziko.
Co dělat v případě hypoglykémie?
1. Nepodávejte inzulín. Okamžitě si vezměte asi 10 až 20 g cukru, např. glukózu, kostky cukru nebo cukrem oslazený nápoj. Upozornění: umělá sladidla a potraviny obsahující umělá sladidla (např. dietní nápoje) při hypoglykémii nepomáhají.
2. Poté snězte něco, co má dlouhotrvající účinek na nárůst cukru v krvi (např. chléb nebo těstoviny). Dbejte na to, abyste tyto záležitosti předem prohovořili s lékařem nebo sestrou.
3. Pokud se hypoglykémie znovu vrací, vezměte si dalších 10 až 20 g cukru.
4. Informujte okamžitě lékaře, jestliže hypoglykémii nebo její opakování nejste schopni zvládnout.
Oznamte svým příbuzným, přátelům a blízkým kolegům následující:
Pokud nemůžete polykat nebo upadnete do bezvědomí, budete potřebovat injekci glukózy nebo glukagonu (lék, který zvyšuje hladinu cukru v krvi). Tyto injekce jsou opodstatněné dokonce i tehdy, není-li jisté, že máte hypoglykémii.
Je vhodné změřit si hladinu cukru v krvi okamžitě po požití glukózy, abyste zkontrolovali, že máte skutečně hypoglykémii.
Příbalová informace: informace pro uživatele
Apidra 100 jednotek/ml injekční roztok v zásobní vložce pro OptiClik
Insulinům glulisinum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje. Návod na použití OptiClik je přiložen k Vašemu inzulínovému peru. Před použitím léčivého přípravku si oba návody přečtěte.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je Apidra a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Apidra používat
3. Jak se přípravek Apidra používá
4. Možné nežádoucí účinky
5. Jak přípravek Apidra uchovávat
6. Obsah balení a další informace
1. Co je Apidra a k čemu se používá
Apidra je antidiabetikum používané ke snížení hladiny cukru v krvi u pacientů s diabetes mellitus; může být podána dospělým, mladistvým a dětem ve věku 6 a více let. Diabetes mellitus je onemocnění, při kterém Váš organismus neprodukuje dostatek inzulínu pro kontrolu hladiny cukru v krvi.
Přípravek je vyroben biotechnologicky. Inzulín glulisin má rychlý nástup účinku během 10-20 minut a krátké trvání účinku přibližně 4 hodiny.
2. Čemu musíte věnovat pozornost, než začnete přípravek Apidra používat Nepoužívejte přípravek Apidra
- Jestliže jste alergický(á) na inzulín glulisin nebo na kteroukoliv další složku tohoto přípravku (uvedenou v bodě 6).
- Jestliže máte hladinu cukru v krvi příliš nízkou (hypoglykémie). Postupujte podle pokynů pro hypoglykémii (viz rámeček na konci této příbalové informace).
Upozornění a opatření
Před užitím přípravku Apidra se poraďte se svým lékařem, lékarníkem nebo zdravotní sestrou. Dodržujte pečlivě pokyny pro dávkování, sledování (testy krve), dietu a fyzickou aktivitu (fyzickou práci a cvičení), které jste projednali se svým lékařem.
Zvláštní skupiny pacientů
Pokud máte problémy s játry nebo s ledvinami, upozorněte svého lékaře, protože můžete potřebovat nižší dávku.
Nejsou dostatečné klinické informace o použití přípravku Apidra u dětí mladších 6 let.
Cestování
Před cestováním se poraďte se svým lékařem. Možná bude třeba se poradit o
- dostupnosti Vašeho inzulínu v zemi, kterou navštívíte,
- dodávkách inzulínu, injekčních stříkaček, atd.,
- správném uchovávání Vašeho inzulínu během cestování,
- časové dostupnosti jídla a podávání inzulínu během cestování,
- možných účincích přesunu v různých časových pásmech,
- možných zdravotních rizicích v zemích, které navštívíte,
- tom, jak se zachovat v mimořádných situacích, tj. pokud se Vám udělá špatně nebo onemocníte. Onemocnění a úrazy
V následujících situacích může léčba Vašeho diabetu vyžadovat zvláštní péči:
- pokud jste nemocný/á nebo máte větší úraz, může dojít ke zvýšení hladiny cukru v krvi (hyperglykémie),
- pokud dostatečně nejíte, může se Vám hladina cukru v krvi velmi snížit (hypoglykémie).
Ve většině případů budete potřebovat lékařskou péči. Zajistěte možnost rychlého kontaktu lékaře.
Pokud máte diabetes typu 1 (na inzulínu závislý diabetes), nepřestávejte užívat svůj inzulín a pokračujte v příjmu dostatečného množství cukrů. Vždy řekněte lidem, kteří Vás ošetřují nebo léčí, že potřebujete inzulín.
U některých pacientů, kteří dlouhodobě trpěli cukrovkou typu 2 a onemocněním srdce, nebo prodělali mozkovou příhodu, a kteří byli léčeni pioglitazonem a inzulínem, byl zaznamenán výskyt srdečního selhání. Jestliže zjistíte známky srdečního selhání jako je neobvyklá dušnost nebo náhlé zvýšení tělesné hmotnosti nebo lokalizovaný otok (edém), informujte co nejrychleji svého lékaře.
Další léčivé přípravky a přípravek Apidra
Některé léky způsobují změnu hladiny cukru v krvi (snížení, zvýšení nebo obojí v závislosti na situaci). V každém případě může být nezbytné upravit Vaši dávku inzulínu, aby se zabránilo hladinám cukru v krvi, které jsou buď příliš nízké, nebo příliš vysoké. Buďte opatrní, když začínáte nebo přestáváte užívat nějaký další lék.
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat. Zeptejte se svého lékaře předtím, než si lék vezmete, zda a jak jím může být ovlivněna Vaše hladina cukru v krvi, popř. co je třeba dělat.
Mezi léky, které mohou vyvolat pokles hladiny cukru v krvi (hypoglykémii), patří:
- všechny další léky pro léčbu diabetu,
- inhibitory angiotenzin konvertujícího enzymu (ACE inhibitory) (používané k léčbě některých srdečních onemocnění, vysokého krevního tlaku),
- disopyramid (používaný k léčbě některých srdečních onemocnění),
- fluoxetin (používaný k léčbě depresí),
- fibráty (používané ke snížení vysokých hladin krevních lipidů),
- inhibitory monoaminooxidázy (MAO inhibitory) (používané k léčbě depresí),
- pentoxifylin, propoxyfen, salicyláty (např. aspirin, používaný ke zmírnění bolesti a snížení horečky),
- sulfonamidová antibiotika.
Mezi léky, které mohou vyvolat zvýšení hladiny cukru v krvi (hyperglykémii), patří:
- kortikosteroidy (např. „kortizon“ používaný pro léčbu zánětu),
- danazol (lék pro navození ovulace),
- diazoxid (používaný pro léčbu vysokého krevního tlaku),
- diuretika (používaná pro léčbu vysokého krevního tlaku nebo nadměrného zadržování tekutin),
- glukagon (hormon slinivky břišní používaný pro léčbu závažné hypoglykémie),
- isoniazid (používaný pro léčbu tuberkulózy),
- estrogeny a progestogeny (např. v antikoncepčních pilulkách užívaných k zamezení početí),
- deriváty fenothiazinu (používané pro léčbu duševních poruch),
- somatropin (růstový hormon),
- sympatomimetika (např. epinefrin [adrenalin], salbutamol, terbutalin užívané k léčbě astmatu),
- hormony štítné žlázy (užívané k léčbě onemocnění štítné žlázy),
- inhibitory proteáz (užívané k léčbě HIV),
- atypická antipsychotika léky (např. olanzapin a klozapin).
Vaše hladina cukru v krvi může buď poklesnout, nebo vzrůst, pokud užíváte:
- betablokátory (používané pro léčbu vysokého krevního tlaku),
- klonidin (používaný pro léčbu vysokého krevního tlaku),
- soli lithia (používané pro léčbu duševních poruch).
Pentamidin (používaný pro léčbu některých infekcí způsobených parazity) může vyvolat hypoglykémii, po které může někdy následovat hyperglykémie.
Betablokátory stejně jako ostatní sympatolytika (např. klonidin, guanetidin a reserpin) mohou oslabit nebo úplně potlačit první varovné příznaky, které Vám pomohou rozpoznat hypoglykémii
Pokud si nejste jisti, zda neužíváte některý z takových léků, zeptejte se svého lékaře nebo lékárníka.
Užívání přípravku Apidra s alkoholem
Hladiny Vašeho cukru v krvi mohou buď vzrůst, nebo poklesnout, pokud konzumujete alkohol. Těhotenství a kojení
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek užívat.
Informujte svého lékaře, pokud plánujete těhotenství nebo pokud již jste těhotná. Je možné, že bude třeba měnit dávkování inzulínu během těhotenství a po porodu. Zvláště pečlivá kontrola diabetu a prevence hypoglykémie jsou důležité pro zdraví Vašeho dítěte.
Nejsou k dispozici žádné nebo jen omezené údaje o použití přípravku Apidra u těhotných žen.
Pokud kojíte, poraďte se se svým lékařem, neboť může být nezbytné upravit Vám dávku inzulínu a Vaši dietu.
Řízení dopravních prostředků a obsluha strojů
Vaše schopnost soustředit se nebo reagovat může být snížena, jestliže:
- máte hypoglykémii (nízkou hladinu cukru v krvi)
- máte hyperglykémii (vysokou hladinu cukru v krvi).
Myslete na tento možný problém ve všech situacích, kde byste mohli dostat sebe nebo ostatní do nebezpečí (např. při řízení vozidla nebo při obsluze strojů).
Měl(a) byste se poradit se svým lékařem o vhodnosti řízení vozidel, pokud:
- máte časté epizody hypoglykémie,
- jsou první varovné příznaky, které Vám pomohou rozpoznat hypoglykémii, omezené nebo chybějí.
Tento léčivý přípravek obsahuje méně než 1 mmol (23 mg) sodíku v jedné dávce, tj. v podstatě sodík neobsahuje.
Apidra obsahuje metakresol
Apidra obsahuje metakresol, který může způsobovat alergické reakce.
3. JAK SE PŘÍPRAVEK APIDRA POUŽÍVÁ Dávkování
Vždy užívejte přípravek Apidra přesně tak, jak Vám doporučil lékař. Pokud si nejste jist(a), poraďte se s lékařem.
Na základě Vašeho životního stylu a Vašich výsledků testů cukru (glukózy) v krvi a Vašeho předchozího užívání inzulínu lékař určí, jakou dávku přípravku Apidra budete potřebovat.
Apidra je rychle účinkující inzulín. Je možné, že Váš lékař Vám řekne, abyste ji užívali v kombinaci se střednědobě nebo dlouhodobě působícím inzulínem nebo bazálním inzulínem nebo s tabletami na snížení vysoké hladiny cukru v krvi.
Jestliže přecházíte z jiného inzulínu na inzulín glulisin, Vaše dávkování může vyžadovat úpravu lékařem.
Hladinu cukru v krvi může ovlivnit mnoho faktorů. Měli byste znát tyto faktory, abyste byli schopni reagovat správně na změny hladiny cukru v krvi a předejít jejich přílišnému zvýšení nebo přílišnému snížení. Další informace si přečtěte na konci této příbalové informace.
Způsob podání
Přípravek Apidra se podává podkožně (subkutánně).
Váš lékař Vám ukáže, do které oblasti kůže máte přípravek Apidra podávat. Injekci přípravku Apidra lze podávat do břišní stěny, do stehna nebo horní části paže nebo kontinuální infuzí do břišní stěny. Účinek bude o něco rychlejší, jestliže si injekci inzulínu aplikujete do břišní stěny. Tak jako u všech ostatních inzulínů musíte měnit s každou injekcí místo vpichu injekce a místo infuze v rámci vybrané oblasti (břišní stěna, stehno, horní část paže), kam injekci podáváte.
Frekvence podávání
Apidra by se měla užívat krátce (0 - 15 minut) před jídlem nebo brzy po jídle.
Pokyny pro správné použití
Jak se zachází se zásobními vložkami pro OptiClik
Zásobní vložky s přípravkem Apidra pro OptiClik jsou určeny pouze k užití ve spojení s OptiClik. Instrukce od výrobce pro použití pera musí být pečlivě dodržovány pro správné vsunutí zásobní vložky, připojení jehly a aplikaci inzulínové injekce.
Před vložením zásobní vložky do pera k opakovanému použití ponechejte zásobní vložku 1 až 2 hodiny při pokojové teplotě. Před injekcí musí být ze zásobní vložky odstraněny vzduchové bubliny (viz návod na použití pera). Prázdné zásobní vložky nesmí být znovu plněny.
Zásobní vložku si prohlédněte ještě předtím, než ji použijete. Použijte pouze ty zásobní vložky, ve kterých je roztok čirý, bezbarvý, bez viditelných částic.
Před použitím přípravek nemíchejte ani s ním netřepejte.
Máte problémy s použitím OptiClik?
Při použití inzulínového pera se řiďte pokyny výrobce.
Pokud je OptiClik poškozen nebo nefunguje správně (kvůli mechanické závadě), je třeba jej vyřadit a použít nový OptiClik.
Pero pro opakované použití smíte používat pouze Vy, aby se tak zabránilo jakémukoliv druhu kontaminace.
Pokud OptiClik selže, může být roztok natažen ze zásobní vložky do injekční stříkačky a injikován. Proto vždy mějte při sobě i injekční stříkačky a jehly. Používejte pouze injekční stříkačky vyrobené pro inzulín o koncentraci 100 jednotek/ml.
Jestliže jste užil(a) více přípravku Apidra, než jste měl(a)
- Pokud jste podali příliš mnoho přípravku Apidra, může se Vám příliš snížit hladina cukru
v krvi (hypoglykémie). Často si kontrolujte hladinu cukru v krvi. Obecně platí, že pro prevenci hypoglykémie musíte jíst více jídla a sledovat hladinu cukru v krvi. Pro informace o léčbě hypoglykémie viz rámeček s odkazem na konci této příbalové informace.
Jestliže jste zapomněl(a) užít přípravek Apidra
- Pokud jste vynechali dávku přípravku Apidra nebo jste podali nedostatečné množství
inzulínu, může se Vám příliš zvýšit hladina cukru v krvi (hyperglykémie). Často si kontrolujte hladinu cukru v krvi. Pro další informace o léčbě hyperglykémie viz rámeček na konci této příbalové informace.
- Nezdvojujte následující dávku, abyste nahradil(a) vynechanou dávku.
Jestliže jste přestal(a) užívat přípravek Apidra
Ukončení užívání přípravku Apidra by mohlo vést k závažné hyperglykémii (velmi vysoká hladina cukru v krvi) a ketoacidóze (hromadění kyseliny v krvi, protože tělo rozkládá tuk místo cukru). Neukončujte léčbu přípravkem Apidra bez konzultace s lékařem, který Vám řekne, co byste měl(a) udělat.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
Záměna inzulínů
Před každou injekcí musíte vždy zkontrolovat štítek na inzulínu, aby nedošlo k záměně léčivého přípravku Apidra za jiné inzulíny.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
r-g r V r V a I r r V* *
Závažné nežádoucí účinky
Hypoglykémie (nízka hladina cukru v krvi) může být velmi zavažna. Hypoglykémie je velmi často hlášeným nežádoucím účinkem (může postihovat více než 1 z 10 osob). Hypoglykémie (nízká hladina cukru v krvi) znamená, že v krvi není dost cukru. Jestliže Vám příliš klesne hladina cukru v krvi, mohli byste ztratit vědomí. Těžká hypoglykémie může způsobit poškození mozku a může být život ohrožující. Pokud máte příznaky nízké hladiny cukru v krvi, okamžitě se snažte hladinu cukru v krvi zvýšit. Přečtěte si další důležité informace o hypoglykémii a její léčbě na konci této příbalové informace.
Pokud se u Vás vyskytnou následující příznaky, ihned kontaktujte svého lékaře:
Systémové alergické reakce jsou méně častými nežádoucími účinky (mohou postihovat až 1 ze 100 osob).
Generalizovaná alergie na inzulín: Přidružené symptomy mohou zahrnovat rozsáhlé kožní reakce (vyrážka a svědění po celém těle), závažný otok kůže nebo sliznic (angioedém), zkrácený dech, pokles krevního tlaku se zrychleným tepem a pocení. Může se jednat o příznaky závažného případu generalizované alergie na inzulín, včetně anafylaktické reakce, která může být život ohrožující.
Hyperglykémie (vysoká hladina cukru v krvi) znamená, že máte v krvi příliš mnoho cukru.
Četnost výskytu hyperglykémie nelze určit. Pokud máte příliš vysokou hladinu cukru v krvi, vypovídá to o tom, že možná budete potřebovat více inzulínu, než jste si aplikoval(a) v injekci. Může jít o závažný stav, pokud se hladina glukózy velmi zvýší.
Přečtěte si další informace o známkách a příznacích hyperglykémie v rámečku na konci této příbalové informace.
Jiné nežádoucí účinky
Časté nežádoucí účinky (mohou postihovat až 1 z 10 osob)
• Kožní a alergické reakce v místě vpichu injekce
Může se objevit reakce v místě vpichu (např. zarudnutí, neobvykle intenzívní bolestivost injekce, svědění, vyrážka, otok nebo zánět). Tyto reakce se mohou také rozšířit kolem místa vpichu. Většina mírných reakcí na inzulín obvykle odezní během několika dnů až několika týdnů.
Vzácné nežádoucí účinky (mohou postihovat až 1 z 1000 osob)
• Změny kůže v místě aplikace injekce (lipodystrofie)
Pokud podáváte inzulín příliš často do stejného místa na kůži, může se tuková tkáň pod kůží v tomto místě buď ztenčit, nebo zesílit (lipodystrofie). Obojí snižuje účinnost inzulínu podávaného do takto změněného místa. Takovým změnám na kůži lze zabránit střídáním místa vpichu při každé injekci.
Nežádoucí účinky, jejichž frekvenci nelze z dostupných údajů určit
• Oční reakce
Výrazné změny (zlepšení nebo zhoršení) kompenzace Vašeho krevního cukru mohou přechodně narušit Váš zrak. Pokud máte proliferativní retinopatii (oční onemocnění vztahující se k diabetu), mohou závažné hypoglykemické ataky způsobit přechodnou ztrátu vidění.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
Uchovávejte mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti, uvedené na krabičce a štítku zásobní vložky za „Použitelné do:“ nebo „EXP“. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Neotevřené zásobní vložky Uchovávejte v chladničce (2°C - 8°C).
Chraňte před mrazem.
Neukládejte přípravek Apidra do blízkosti mrazicího boxu nebo mrazicí vložky.
Uchovávejte zásobní vložku v krabičce, aby byla chráněna před světlem.
Používané zásobní vložky
Používané zásobní vložky (v inzulínovém peru) mohou být uchovávány až 4 týdny při teplotě do 25°C mimo přímé teplo nebo světlo a nesmí být uchovávány v chladničce. Nepoužívejte po uplynutí této doby použitelnosti.
Nepoužívejte tento přípravek, pokud není roztok čirý a bezbarvý.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co Apidra obsahuje
- Léčivou látkou je insulinum glulisinum. Jeden mililitr roztoku obsahuje 100 jednotek léčivé látky insulinum glulisinum (odpovídá množství 3,49 mg). Jedna zásobní vložka obsahuje 3 ml injekčního roztoku, což odpovídá 300 jednotkám.
- Pomocné látky jsou: metakresol (viz bod 2 „Apidra obsahuje metakresol“), chlorid sodný (viz bod 2 „Důležité informace o některých složkách přípravku Apidra“), trometamol, polysorbát 20, kyselina chlorovodíková 35%, hydroxid sodný, voda pro injekci.
Jak Apidra vypadá a co obsahuje toto balení
Apidra 100 jednotek/ml injekční roztok v zásobní vložce pro OptiClik je čirý, bezbarvý, vodný roztok bez viditelných částic. Tato zásobní vložka je pouze pro použití s OptiClik.
Jedna zásobní vložka obsahuje 3 ml roztoku (300 U). Dostupná jsou balení s 1, 3, 4, 5, 6, 8, 9 a 10 zásobními vložkami. Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci a výrobce
Držitel rozhodnutí o registraci:
Sanofi-Aventis Deutschland GmbH D-65926 Frankfurt am Main Německo
Výrobce:
Sanofi-Aventis Deutschland GmbH Industriepark Hochst, D-65926 Frankfurt Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci.
|
Belgie/Belgique/Belgien Sanofi Belgium Tél/Tel: +32 (0)2 710 54 00 |
Lietuva UAB “SANOFI-AVENTIS LIETUVA” Tel: +370 5 2755224 |
|
Btarapna sanofi-aventis Bulgaria EOOD Ten.: +359 (0)2 970 53 00 |
Luxembourg/Luxemburg Sanofi Belgium Tél/Tel: +32 (0)2 710 54 00 (Belgique/Belgien) |
|
Česká republika sanofi-aventis, s.r.o. Tel: +420 233 086 111 |
Magyarország SANOFI-AVENTIS Zrt. Tel.: +36 1 505 0050 |
|
Danmark sanofi-aventis Denmark A/S Tlf: +45 45 16 70 00 |
Malta Sanofi Malta Ltd. Tel: +356 21493022 |
|
Deutschland Sanofi-Aventis Deutschland GmbH Tel: +49 (0)180 2 222010 |
Nederland sanofi-aventis Netherlands B.V. Tel: +31 (0)182 557 755 |
|
Eesti sanofi-aventis Estonia OU Tel: +372 627 34 88 |
Norge sanofi-aventis Norge AS Tlf: +47 67 10 71 00 |
|
EXXáSa sanofi-aventis AEBE Tr^: +30 210 900 16 00 |
Osterreich sanofi-aventis GmbH Tel: +43 1 80 185 - 0 |
|
Espaňa sanofi-aventis, S.A. Tel: +34 93 485 94 00 |
Polska sanofi-aventis Sp. z o.o. Tel.: +48 22 280 00 00 |
|
France sanofi-aventis France Tél: 0 800 222 555 Appel depuis l’étranger : +33 1 57 63 23 23 |
Portugal Sanofi - Produtos Farmaceuticos, Lda. Tel: +351 21 35 89 400 |
|
Hrvatska sanofi-aventis Croatia d.o.o. Tel: +385 1 600 34 00 |
Románia Sanofi Romania SRL Tel: +40 (0) 21 317 31 36 |
|
Ireland sanofi-aventis Ireland Ltd. T/A SANOFI Tel: +353 (0) 1 403 56 00 |
Slovenija sanofi-aventis d.o.o. Tel: +386 1 560 48 00 |
|
Ísland Vistor hf. Sími: +354 535 7000 |
Slovenská republika sanofi-aventis Pharma Slovakia s.r.o. Tel: +421 2 33 100 100 |
Italia Suomi/Finland
Sanofi S.p.A. Sanofi Oy
Tel: 800 13 12 12 (domande di tipo tecnico) Puh/Tel: +358 (0) 201 200 300
800 536389 (altre domande)
Kúrcpog
sanofi-aventis Cyprus Ltd. Tr^: +357 22 871600
Sverige
Sanofi AB
Tel: +46 (0)8 634 50 00
Latvija
sanofi-aventis Latvia SIA Tel: +371 67 33 24 51
United Kingdom
Sanofi
Tel: +44 (0) 845 372 7101
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku j sou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/
HYPERGLYKÉMIE A HYPOGLYKÉMIE
Vždy u sebe mějte nějaký cukr (minimálně 20 gramů).
Noste u sebe nějakou informaci, která ukazuje, máte cukrovku.
HYPERGLYKÉMIE (vysoká hladina cukru v krvi)
Pokud je Vaše hladina cukru v krvi příliš vysoká (hyperglykémie), možná jste si aplikoval(a) málo inzulínu.
Proč vzniká hyperglykémie?
Příklady:
- nepodali jste si injekci inzulínu nebo jste ho nepodali dostatečné množství nebo se inzulín stal méně účinným, např. z důvodu nesprávného uchovávání,
- máte méně pohybu než obvykle, jste ve stresu (emocionální úzkost, rozrušení) nebo máte úraz, j ste po operaci, máte infekci nebo horečku,
- užíváte nebo jste užíval(a) některé jiné léky (viz bod 2 „Další léčivé přípravky a Apidra“). Varovné příznaky hyperglykémie
Žízeň, zvýšené močení, únava, suchá pokožka, zčervenání obličeje, nechutenství, nízký krevní tlak, zrychlený tep a přítomnost glukózy a ketolátek v moči. Bolest žaludku, rychlé a hluboké dýchání, ospalost nebo dokonce ztráta vědomí mohou být známkami vážného stavu (ketoacidózy) vyplývajícího z nedostatku inzulínu.
Co dělat v případě hyperglykémie?
Změřte si hladinu cukru v krvi a ketolátek v moči co nejdříve, jakmile se výše uvedené příznaky objeví. Těžká hyperglykémie nebo ketoacidóza musí být vždy léčena lékařem, obvykle v nemocnici.
HYPOGLYKÉMIE (nízká hladina cukru v krvi)
Pokud je hladina Vašeho cukru v krvi příliš nízká, můžete ztratit vědomí. Závažná hypoglykémie může vyvolat srdeční záchvat nebo poškození mozku a může být život ohrožující. Za normálních okolností byste měl(a) být schopen/schopna rozpoznat přílišné snižování hladiny cukru v krvi, a proto můžete správně reagovat.
Proč vzniká hypoglykémie?
Příklady:
- aplikujete si příliš mnoho inzulínu,
- vynecháte nebo odložíte jídlo,
- nejíte dostatečně, nebo snězené jídlo obsahuje méně sacharidů než normálně (cukr a látky podobné cukru se nazývají sacharidy; umělá sladidla však NEJSOU sacharidy),
- dochází u Vás ke ztrátě sacharidů zvracením nebo průjmem,
- pijete alkohol, zvláště pokud nejíte dostatečně,
- cvičíte více než obvykle nebo vykonáváte jiný typ fyzické činnosti,
- zotavujete se ze zranění, operace, nebo z jiných forem stresu,
- zotavujete se po nemoci nebo z horečky,
- užíváte nebo jste přestali užívat některé jiné léky (viz bod 2 „Další léčivé přípravky a Apidra“). Výskyt hypoglykémie je pravděpodobnější, pokud:
- právě zahajujete léčbu inzulínem nebo přecházíte na jiný inzulínový přípravek,
- Vaše hladiny cukru v krvi j sou téměř normální nebo j sou nestálé,
- změníte oblast kůže, kam podáváte injekci inzulínu (např. ze stehna na horní část paže),
- trpíte závažným onemocněním ledvin nebo j ater nebo některou j inou chorobou, j ako j e hypotyreóza (snížená funkce štítné žlázy).
Varovné příznaky hypoglykémie
- Ve Vašem těle:
Příklady příznaků, které naznačují, že hladina cukru v krvi klesá příliš nízko nebo příliš rychle: pocení, vlhká pokožka, úzkost, zrychlený tep, vysoký krevní tlak, bušení srdce a nepravidelný srdeční rytmus. Často se tyto příznaky vyvinou dříve než příznaky spojené s nízkou hladinou cukru v mozku.
- Ve Vašem mozku:
Příklady příznaků naznačujících nízkou hladinu cukru v mozku: bolest hlavy, intenzivní hlad, nevolnost, zvracení, únava, ospalost, poruchy spánku, neklid, agresivní chování, pokles koncentrace, zhoršené reakce, deprese, zmatenost, poruchy řeči (někdy úplná ztráta řeči), poruchy vidění, třes, paralýza, pocity mravenčení (parestézie), brnění a pocity necitlivosti v oblasti úst, závrať, ztráta sebekontroly, neschopnost postarat se o sebe, křeče a ztráta vědomí.
První příznaky, které Vás varují před hypoglykémií („varovné příznaky“), mohou být pozměněny, mohou být slabší nebo mohou zcela chybět, pokud:
- jste starší;
- máte diabetes již dlouho,
- trpíte určitým typem nervového onemocnění (diabetická autonomní neuropatie),
- Vás hypoglykémie trápila nedávno (např. předchozí den) nebo se vyvíjí pomalu,
- máte téměř normální nebo alespoň hodně zlepšenou hladinu cukru v krvi,
- užíváte nebo jste užíval(a) některé jiné léky (viz bod 2 „Další léčivé přípravky a Apidra“).
V takových případech se u Vás může rozvinout těžká hypoglykémie (a dokonce mdloba), aniž byste si toho všimli včas. Seznamte se co nejlépe se svými varovnými příznaky hypoglykémie. Je-li to nutné, častější měření cukru v krvi může pomoci odhalit mírné hypoglykemické epizody, které by jinak mohly být přehlédnuty. Pokud se nemůžete spolehnout na rozpoznání svých varovných příznaků, vyhněte se takovým situacím (např. řízení vozidla), při kterých by hypoglykémie pro Vás nebo pro ostatní představovala riziko.
Co dělat v případě hypoglykémie?
1. Nepodávejte inzulín. Okamžitě si vezměte asi 10 až 20 g cukru, např. glukózu, kostky cukru nebo cukrem oslazený nápoj. Upozornění: umělá sladidla a potraviny obsahující umělá sladidla (např. dietní nápoje) při hypoglykémii nepomáhají.
2. Poté snězte něco, co má dlouhotrvající účinek na nárůst cukru v krvi (např. chléb nebo těstoviny). Dbejte na to, abyste tyto záležitosti předem prohovořili s lékařem nebo sestrou.
3. Pokud se hypoglykémie znovu vrací, vezměte si dalších 10 až 20 g cukru.
4. Informujte okamžitě lékaře, jestliže hypoglykémii nebo její opakování nejste schopni zvládnout.
Oznamte svým příbuzným, přátelům a blízkým kolegům následující:
Pokud nemůžete polykat nebo upadnete do bezvědomí, budete potřebovat injekci glukózy nebo glukagonu (lék, který zvyšuje hladinu cukru v krvi). Tyto injekce jsou opodstatněné dokonce i tehdy, není-li jisté, že máte hypoglykémii.
Je vhodné změřit si hladinu cukru v krvi okamžitě po požití glukózy, abyste zkontrolovali, že máte skutečně hypoglykémii.
Příbalová informace: informace pro uživatele
Apidra 100 jednotek/ml injekční roztok v předplněném peru
Insulinům glulisinum
Přečtěte si pozorně celou příbalovou informaci včetně Návodu na použití přípravku Apidra, předplněné pero OptiSet dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité informace.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci.Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je Apidra a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Apidra používat
3. Jak se přípravek Apidra používá
4. Možné nežádoucí účinky
5. Jak přípravek Apidra uchovávat
6. Obsah balení a další informace
1. Co je Apidra a k čemu se používá
Apidra je antidiabetikum používané ke snížení hladiny cukru v krvi u pacientů s diabetes mellitus; může být podána dospělým, mladistvým a dětem ve věku 6 a více let. Diabetes mellitus je onemocnění, při kterém Váš organismus neprodukuje dostatek inzulínu pro kontrolu hladiny cukru v krvi.
Přípravek je vyroben biotechnologicky. Inzulín glulisin má rychlý nástup účinku během 10-20 minut a krátké trvání účinku přibližně 4 hodiny.
2. Čemu musíte věnovat pozornost, než začnete přípravek Apidra používat Nepoužívejte přípravek Apidra
- Jestliže jste alergický(á) (přecitlivělý/á) na inzulín glulisin nebo na kteroukoliv další složku přípravku Apidra.
- Jestliže máte hladinu cukru v krvi příliš nízkou (hypoglykémie). Postupujte podle pokynů pro hypoglykémii (viz rámeček na konci této příbalové informace).
Upozornění a opatření
Před užitím přípravku Apidra se poraďte se svým lékařem, lékarníkem nebo zdravotní sestrou. Dodržujte pečlivě pokyny pro dávkování, sledování (testy krve), dietu a fyzickou aktivitu (fyzickou práci a cvičení), které jste projednali se svým lékařem.
Zvláštní skupiny pacientů
Pokud máte problémy s játry nebo s ledvinami, upozorněte svého lékaře, protože můžete potřebovat nižší dávku.
Nejsou dostatečné klinické informace o použití přípravku Apidra u dětí mladších 6 let.
Cestování
Před cestováním se poraďte se svým lékařem. Možná bude třeba se poradit o
- dostupnosti Vašeho inzulínu v zemi, kterou navštívíte,
- dodávkách inzulínu, injekčních stříkaček, atd.,
- správném uchovávání Vašeho inzulínu během cestování,
- časové dostupnosti jídla a podávání inzulínu během cestování,
- možných účincích přesunu v různých časových pásmech,
- možných zdravotních rizicích v zemích, které navštívíte,
- tom, jak se zachovat v mimořádných situacích, tj. pokud se Vám udělá špatně nebo onemocníte. Onemocnění a úrazy
V následujících situacích může léčba Vašeho diabetu vyžadovat zvláštní péči:
- pokud jste nemocný/á nebo máte větší úraz, může dojít ke zvýšení hladiny Vašeho cukru v krvi (hyperglykémie),
- pokud dostatečně nejíte, může se Vám hladina cukru v krvi velmi snížit (hypoglykémie).
Ve většině případů budete potřebovat lékařskou péči. Zajistěte možnost rychlého kontaktu lékaře.
Pokud máte diabetes typu 1 (na inzulínu závislý diabetes), nepřestávejte užívat svůj inzulín a pokračujte v příjmu dostatečného množství cukrů. Vždy řekněte lidem, kteří Vás ošetřují nebo léčí, že potřebujete inzulín
U některých pacientů, kteří dlouhodobě trpěli cukrovkou typu 2 a onemocněním srdce, nebo prodělali mozkovou příhodu, a kteří byli léčeni pioglitazonem a inzulínem, byl zaznamenán výskyt srdečního selhání. Jestliže zjistíte známky srdečního selhání jako je neobvyklá dušnost nebo náhlé zvýšení tělesné hmotnosti nebo lokalizovaný otok (edém), informujte co nejrychleji svého lékaře.
Další léčivé přípravky a přípravek Apidra
Některé léky způsobují změnu hladiny cukru v krvi (snížení, zvýšení nebo obojí v závislosti na situaci). V každém případě může být nezbytné upravit Vaši dávku inzulínu, aby se zabránilo hladinám cukru v krvi, které jsou buď příliš nízké, nebo příliš vysoké. Buďte opatrní, když začínáte nebo přestáváte užívat nějaký další lék.
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat. Zeptejte se svého lékaře předtím, než si lék vezmete, zda a jak jím může být ovlivněna Vaše hladina cukru v krvi, popř. co je třeba dělat.
Mezi léky, které mohou vyvolat pokles hladiny cukru v krvi (hypoglykémii), patří:
- všechny další léky pro léčbu diabetu,
- inhibitory angiotenzin konvertujícího enzymu (ACE inhibitory) (používané k léčbě některých srdečních onemocnění, vysokého krevního tlaku),
- disopyramid (používaný k léčbě některých srdečních onemocnění),
- fluoxetin (používaný k léčbě depresí),
- fibráty (používané ke snížení vysokých hladin krevních lipidů),
- inhibitory monoaminooxidázy (MAO inhibitory) (používané k léčbě depresí),
- pentoxifylin, propoxyfen, salicyláty (např. aspirin, používaný ke zmírnění bolesti a snížení horečky),
- sulfonamidová antibiotika.
Mezi léky, které mohou vyvolat zvýšení hladiny cukru v krvi (hyperglykémii), patří:
- kortikosteroidy (např. „kortizon“ používaný pro léčbu zánětu),
- danazol (lék pro navození ovulace),
- diazoxid (používaný pro léčbu vysokého krevního tlaku),
- diuretika (používaná pro léčbu vysokého krevního tlaku nebo nadměrného zadržování tekutin),
- glukagon (hormon slinivky břišní používaný pro léčbu závažné hypoglykémie),
- isoniazid (používaný pro léčbu tuberkulózy),
- estrogeny a progestogeny (např. v antikoncepčních pilulkách užívaných k zamezení početí),
- deriváty fenothiazinu (používané pro léčbu duševních poruch),
- somatropin (růstový hormon),
- sympatomimetika (např. epinefrin [adrenalin], salbutamol, terbutalin užívané k léčbě astmatu),
- hormony štítné žlázy (užívané k léčbě onemocnění štítné žlázy),
- inhibitory proteáz (užívané k léčbě HIV),
- atypická antipsychotika (např. olanzapin a klozapin).
Vaše hladina cukru v krvi může buď poklesnout, nebo vzrůst, pokud užíváte:
- betablokátory (používané pro léčbu vysokého krevního tlaku),
- klonidin (používaný pro léčbu vysokého krevního tlaku),
- soli lithia (používané pro léčbu duševních poruch).
Pentamidin (používaný pro léčbu některých infekcí způsobených parazity) může vyvolat hypoglykémii, po které může někdy následovat hyperglykémie.
Betablokátory stejně jako ostatní sympatolytika (např. klonidin, guanetidin a reserpin) mohou oslabit nebo úplně potlačit první varovné příznaky, které Vám mohou pomoci rozpoznat hypoglykémii.
Pokud si nejste jisti, zda neužíváte některý z takových léků, zeptejte se svého lékaře nebo lékárníka.
Užívání přípravku Apidra s alkoholem
Hladiny Vašeho cukru v krvi mohou buď vzrůst, nebo poklesnout, pokud konzumujete alkohol. Těhotenství a kojení
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek užívat.
Informujte svého lékaře. pokud plánujete těhotenství nebo pokud již jste těhotná. Je možné, že bude třeba měnit dávku inzulínu během těhotenství a po porodu. Zvláště pečlivá kontrola diabetu a prevence hypoglykémie jsou důležité pro zdraví Vašeho dítěte.
Nejsou k dispozici žádné nebo jen omezené údaje o použití přípravku Apidra u těhotných žen.
Pokud kojíte, poraďte se se svým lékařem, neboť může být nezbytné upravit Vám dávku inzulínu a Vaši dietu.
Řízení dopravních prostředků a obsluha strojů
Vaše schopnost soustředit se nebo reagovat může být snížena, jestliže:
- máte hypoglykémii (nízkou hladinu cukru v krvi)
- máte hyperglykémii (vysokou hladinu cukru v krvi).
Myslete na tento možný problém ve všech situacích, kde byste mohli dostat sebe nebo ostatní do nebezpečí (např. při řízení vozidla nebo při obsluze strojů).
Měl(a) byste se poradit se svým lékařem o vhodnosti řízení vozidel, pokud:
- máte časté epizody hypoglykémie,
- jsou první varovné příznaky, které Vám pomohou rozpoznat hypoglykémii, omezené nebo chybějí.
Důležité informace o některých složkách přípravku Apidra
Tento léčivý přípravek obsahuje méně než 1 mmol (23 mg) sodíku v jedné dávce, tj. v podstatě sodík neobsahuje.
Apidra obsahuje metakresol
Apidra obsahuje metakresol, který může způsobovat alergické reakce.
3. Jak se přípravek Apidra používá Dávkování
Na základě Vašeho životního stylu a Vašich výsledků testů cukru (glukózy) v krvi a Vašeho předchozího užívání inzulínu lékař určí, jakou dávku přípravku Apidra budete potřebovat.
Vždy užívejte přípravek Apidra tak, jak Vám doporučil lékař. Pokud si nejste jist(a), poraďte se s lékařem.
Apidra je rychle účinkující inzulín. Je možné, že Váš lékař Vám řekne, abyste ji užívali v kombinaci se střednědobě nebo dlouhodobě působícím inzulínem nebo bazálním inzulínem nebo s tabletami na snížení vysoké hladiny cukru v krvi.
Jestliže přecházíte z jiného inzulínu na inzulín glulisin, Vaše dávkování může vyžadovat úpravu lékařem.
Hladinu cukru v krvi může ovlivnit mnoho faktorů. Měli byste znát tyto faktory, abyste byli schopni reagovat správně na změny hladiny cukru v krvi a předejít jejich přílišnému zvýšení nebo přílišnému snížení. Další informace si přečtěte na konci této příbalové informace.
Způsob podání
Injekce přípravku Apidra se podává pod kůži (subkutánně).
Váš lékař Vám ukáže, do které oblasti kůže máte přípravek Apidra podávat. Injekci přípravku Apidra lze podávat do břišní stěny, do stehna nebo horní části paže nebo kontinuální infuzí do břišní stěny. Účinek bude o něco rychlejší, jestliže si injekci inzulínu aplikujete do břišní stěny. Tak jako u všech ostatních inzulínů musíte měnit s každou injekcí místo vpichu injekce a místo infuze v rámci vybrané oblasti (břišní stěna, stehno, horní část paže), kam injekci podáváte.
Frekvence podávání
Apidra by se měla užívat krátce (0 - 15 minut) před jídlem nebo brzy po jídle.
Pokyny pro správné použití
Jak se zachází s perem OptiSet
OptiSet je předplněné pero pro jednorázové použití, které obsahuje inzulín glulisin.
Pečlivě si přečtěte „Návod na použití OptiSet“ uvedený na konci této příbalové informace. Pero musíte používat tak, jak je popsáno v tomto návodu na použití.
Z důvodu prevence přenosu nemoci musí být každé pero používáno pouze jedním pacientem.
Před použitím vždy nasaďte novou jehlu a proveďte zkoušku bezpečnosti. Používejte pouze jehly, které byly schváleny pro použití s OptiSet.
Zásobní vložku si prohlédněte ještě předtím, než ji použijete. Použijte pouze ty zásobní vložky, ve kterých je roztok čirý, bezbarvý, bez viditelných částic.
Před použitím přípravek nemíchejte ani s ním netřepejte.
Použijte nové pero pokaždé, když si všimnete, že kontrola hladiny cukru v krvi se nečekaně zhoršila. Domníváte-li se, že problém může být s OptiSet, podívejte se, prosím do kapitoly Otázky a odpovědi v přiloženém návodu pro použití OptiSet, nebo požádejte o kontrolu Vašeho lékaře nebo lékárníka.
Jestliže jste užil(a) více přípravku Apidra, než jste měl(a)
- Pokud jste podali příliš mnoho přípravku Apidra, může se Vám příliš snížit hladina cukru
v krvi (hypoglykémie). Často si kontrolujte hladinu cukru v krvi. Obecně platí, že pro prevenci hypoglykémie musíte jíst více jídla a sledovat hladinu cukru v krvi. Pro informace o léčbě hypoglykémie viz rámeček na konci této příbalové informace
Jestliže jste zapomněl(a) užít přípravek Apidra
- Pokud jste vynechali dávku přípravku Apidra nebo jste podali nedostatečné množství inzulínu, může se Vám příliš zvýšit hladina cukru v krvi (hyperglykémie). Často si kontrolujte hladinu cukru v krvi. Pro další informace o léčbě hyperglykémie viz rámeček na konci této příbalové informace.
- Nezdvojujte následující dávku, abyste nahradil(a) vynechanou dávku.
Jestliže jste přestal(a) užívat přípravek Apidra
Ukončení užívání přípravku Apidra by mohlo vést k závažné hyperglykémii (velmi vysoká hladina cukru v krvi) a ketoacidóze (hromadění kyseliny v krvi, protože tělo rozkládá tuk místo cukru). Neukončujte léčbu přípravkem Apidra bez konzultace s lékařem, který Vám řekne, co byste měl(a) udělat.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
Záměna inzulínů
Před každou injekcí musíte vždy zkontrolovat štítek na inzulínu, aby nedošlo k záměně léčivého přípravku Apidra za jiné inzulíny.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Závažné nežádoucí účinky
Hypoglykémie (nízká hladina cukru v krvi) může být velmi závažná. Hypoglykémie je velmi často hlášeným nežádoucím účinkem (může postihovat více než 1 z 10 osob). Hypoglykémie (nízká hladina cukru v krvi) znamená, že v krvi není dost cukru. Jestliže Vám příliš klesne hladina cukru v krvi, mohli byste ztratit vědomí. Těžká hypoglykémie může způsobit poškození mozku a může být život ohrožující. Pokud máte příznaky nízké hladiny cukru v krvi, okamžitě se snažte hladinu cukru v krvi zvýšit. Přečtěte si další důležité informace o hypoglykémii a její léčbě na konci této příbalové informace.
Pokud se u Vás vyskytnou následující příznaky, ihned kontaktujte svého lékaře:
Systémové alergické reakce jsou méně častými nežádoucími účinky (mohou postihovat až 1 ze 100 osob).
Generalizovaná alergie na inzulín: Přidružené symptomy mohou zahrnovat rozsáhlé kožní reakce (vyrážka a svědění po celém těle), závažný otok kůže nebo sliznic (angioedém), zkrácený dech, pokles krevního tlaku se zrychleným tepem a pocení. Může se jednat o příznaky závažného případu generalizované alergie na inzulín, včetně anafylaktické reakce, která může být život ohrožující.
Hyperglykémie (vysoká hladina cukru v krvi) znamená, že máte v krvi příliš mnoho cukru.
Četnost výskytu hyperglykémie nelze určit. Pokud máte příliš vysokou hladinu cukru v krvi, vypovídá to o tom, že možná budete potřebovat více inzulínu, než jste si aplikoval(a) v injekci. Může jít o závažný stav, pokud se hladina glukózy velmi zvýší.
Přečtěte si další informace o známkách a příznacích hyperglykémie v rámečku na konci této příbalové informace.
Jiné nežádoucí účinky
Časté nežádoucí účinky (mohou postihovat až 1 z 10 osob)
• Kožní a alergické reakce v místě vpichu injekce
Může se objevit reakce v místě vpichu (např. zarudnutí, neobvykle intenzívní bolestivost injekce, svědění, vyrážka, otok nebo zánět). Tyto reakce se mohou také rozšířit kolem místa vpichu. Většina mírných reakcí na inzulín obvykle odezní během několika dnů až několika týdnů.
Vzácné nežádoucí účinky (mohou postihovat až 1 z 1000 osob)
• Změny kůže v místě aplikace injekce (lipodystrofie)
Pokud podáváte inzulín příliš často do stejného místa na kůži, může se tuková tkáň pod kůží v tomto místě buď ztenčit, nebo zesílit (lipodystrofie). Obojí snižuje účinnost inzulínu podávaného do takto změněného místa. Takovým změnám na kůži lze zabránit střídáním místa vpichu při každé injekci.
Nežádoucí účinky, jejichž frekvenci nelze z dostupných údajů určit
• Oční reakce
Výrazné změny (zlepšení nebo zhoršení) kompenzace Vašeho krevního cukru mohou přechodně narušit Váš zrak. Pokud máte proliferativní retinopatii (oční onemocnění vztahující se k diabetu), mohou závažné hypoglykemické ataky způsobit přechodnou ztrátu vidění.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Apidra uchovávat
Uchovávejte mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti, uvedené na krabičce a štítku zásobní vložky za „Použitelné do:“ nebo „EXP“. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Nepoužívaná pera
Uchovávejte v chladničce (2°C - 8°C). Chraňte před mrazem. Neukládejte předplněné pero do blízkosti mrazicího boxu nebo mrazicí vložky.
Uchovávejte předplněné pero v krabičce, aby byl přípravek chráněn před světlem.
Používaná pera
Používaná předplněná pera mohou být uchovávána až 4 týdny při teplotě do 25°C mimo přímé teplo a světlo a nesmí být uchovávána v chladničce. Nepoužívejte je po uplynutí této doby použitelnosti.
Nepoužívejte tento přípravek, pokud není roztok čirý a bezbarvý.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co Apidra obsahuje
- Léčivou látkou je insulinum glulisinum. Jeden mililitr roztoku obsahuje 100 jednotek léčivé látky insulinum glulisinum (odpovídá množství 3,49 mg).
- Pomocné látky jsou: metakresol (viz bod 2 „Apidra obsahuje metakresol“), chlorid sodný (viz bod 2 „Důležité informace o některých složkách přípravku Apidra“), trometamol, polysorbát 20, kyselina chlorovodíková 35%, hydroxid sodný, voda pro injekci.
Jak Apidra vypadá a co obsahuje toto balení
Apidra 100 jednotek/ml injekční roztok v předplněném peru OptiSet. Čirý, bezbarvý, vodný roztok bez viditelných částic.
Jedno pero obsahuje 3 ml roztoku, což odpovídá 300 jednotkám. Dostupná jsou balení s 1, 3, 4, 5, 6, 8, 9 a 10 předplněnými pery. Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci a výrobce
Držitel rozhodnutí o registraci:
Sanofi-Aventis Deutschland GmbH D-65926 Frankfurt am Main Německo
Výrobce:
Sanofi-Aventis Deutschland GmbH Industriepark Hochst, D-65926 Frankfurt Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci.
Belgie/Belgique/Belgien
Sanofi Belgium Tél/Tel: +32 (0)2 710 54 00
Bturapun
sanofi-aventis Bulgaria EOOD Ten.: +359 (0)2 970 53 00
Lietuva
UAB “SANOFI-AVENTIS LIETUVA” Tel: +370 5 2755224
Luxembourg/Luxemburg
Sanofi Belgium
Tél/Tel: +32 (0)2 710 54 00 (Belgique/Belgien)
Magyarország SANOFI-AVENTIS Zrt. Tel.: +36 1 505 0050
Malta
Sanofi Malta Ltd. Tel: +356 21493022
Danmark
sanofi-aventis Denmark A/S Tlf: +45 45 16 70 00
|
Deutschland Sanofi-Aventis Deutschland GmbH Tel: +49 (0)180 2 222010 |
Nederland sanofi-aventis Netherlands B.V. Tel: +31 (0)182 557 755 |
|
Eesti sanofi-aventis Estonia OU Tel: +372 627 34 88 |
Norge sanofi-aventis Norge AS Tlf: +47 67 10 71 00 |
|
EXXáSa sanofi-aventis AEBE Tn^: +30 210 900 16 00 |
Osterreich sanofi-aventis GmbH Tel: +43 1 80 185 - 0 |
|
Espaňa sanofi-aventis, S.A. Tel: +34 93 485 94 00 |
Polska sanofi-aventis Sp. z o.o. Tel.: +48 22 280 00 00 |
|
France sanofi-aventis France Tél: 0 800 222 555 Appel depuis l’étranger : +33 1 57 63 23 23 |
Portugal Sanofi-aventis - Produtos Farmaceuticos, Lda. Tel: +351 21 35 89 400 |
|
Hrvatska sanofi-aventis Croatia d.o.o. Tel: +385 1 600 34 00 |
Románia Sanofi Romania SRL Tel: +40 (0) 21 317 31 36 |
|
Ireland sanofi-aventis Ireland Ltd. T/A SANOFI Tel: +353 (0) 1 403 56 00 |
Slovenija sanofi-aventis d.o.o. Tel: +386 1 560 48 00 |
|
Ísland Vistor hf. Sími: +354 535 7000 |
Slovenská republika sanofi-aventis Pharma Slovakia s.r.o. Tel: +421 2 33 100 100 |
|
Italia Sanofi S.p.A. Tel: 800 13 12 12 (domande di tipo tecnico) 800 536389 (altre domande) |
Suomi/Finland Sanofi Oy Puh/Tel: +358 (0) 201 200 300 |
|
Kúnpoq sanofi-aventis Cyprus Ltd. Tn^: +357 22 871600 |
Sverige Sanofi AB Tel: +46 (0)8 634 50 00 |
|
Latvija sanofi-aventis Latvia SIA Tel: +371 67 33 24 51 |
United Kingdom Sanofi Tel: +44 (0) 845 372 7101 |
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku j sou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/
HYPERGLYKÉMIE A HYPOGLYKÉMIE
Vždy u sebe mějte nějaký cukr (minimálně 20 gramů).
Noste u sebe nějakou informaci, která ukazuje, že máte cukrovku.
HYPERGLYKÉMIE (vysoká hladina cukru v krvi)
Pokud je Vaše hladina cukru v krvi příliš vysoká (hyperglykémie), možná jste si aplikoval(a) málo inzulínu.
Proč vzniká hyperglykémie?
Příklady:
- nepodali jste si injekci inzulínu nebo jste ho nepodali dostatečné množství nebo se inzulín stal méně účinným, např. z důvodu nesprávného uchovávání,
- máte méně pohybu než obvykle, jste ve stresu (emocionální úzkost, rozrušení) nebo máte úraz, j ste po operaci, máte infekci nebo horečku,
- užíváte nebo jste užíval(a) některé jiné léky (viz bod 2 „Další léčivé přípravky a Apidra“). Varovné příznaky hyperglykémie
Žízeň, zvýšené močení, únava, suchá pokožka, zčervenání obličeje, nechutenství, nízký krevní tlak, zrychlený tep a přítomnost glukózy a ketolátek v moči. Bolest žaludku, rychlé a hluboké dýchání, ospalost nebo dokonce ztráta vědomí mohou být známkami vážného stavu (ketoacidózy) vyplývajícího z nedostatku inzulínu.
Co dělat v případě hyperglykémie?
Změřte si hladinu cukru v krvi a ketolátek v moči co nejdříve, jakmile se výše uvedené příznaky objeví. Těžká hyperglykémie nebo ketoacidóza musí být vždy léčena lékařem, obvykle v nemocnici.
HYPOGLYKÉMIE (nízká hladina cukru v krvi)
Pokud je hladina Vašeho cukru v krvi příliš nízká, můžete ztratit vědomí. Závažná hypoglykémie může vyvolat srdeční záchvat nebo poškození mozku a může být život ohrožující. Za normálních okolností byste měl(a) být schopen/schopna rozpoznat přílišné snižování hladiny cukru v krvi, a proto můžete správně reagovat.
Proč vzniká hypoglykémie?
Příklady:
- aplikujete si příliš mnoho inzulínu,
- vynecháte nebo odložíte jídlo,
- nejíte dostatečně, nebo snězené jídlo obsahuje méně sacharidů než normálně (cukr a látky podobné cukru se nazývají sacharidy; umělá sladidla však NEJSOU sacharidy),
- dochází u Vás ke ztrátě sacharidů zvracením nebo průjmem,
- pijete alkohol, zvláště pokud nejíte dostatečně,
- cvičíte více než obvykle nebo vykonáváte jiný typ fyzické činnosti,
- zotavujete se ze zranění, operace, nebo z jiných forem stresu,
- zotavujete se po nemoci nebo z horečky,
- užíváte nebo jste přestali užívat některé jiné léky (viz bod 2 „Další léčivé přípravky a Apidra“). Výskyt hypoglykémie je pravděpodobnější, pokud:
- právě zahajujete léčbu inzulínem nebo přecházíte na jiný inzulínový přípravek,
- Vaše hladiny cukru v krvi j sou téměř normální nebo j sou nestálé,
- změníte oblast kůže, kam podáváte injekci inzulínu (např. ze stehna na horní část paže),
- trpíte závažným onemocněním ledvin nebo j ater nebo některou j inou chorobou, j ako j e hypotyreóza (snížená funkce štítné žlázy).
Varovné příznaky hypoglykémie
- Ve Vašem těle:
Příklady příznaků, které naznačují, že hladina cukru v krvi klesá příliš nízko nebo příliš rychle: pocení, vlhká pokožka, úzkost, zrychlený tep, vysoký krevní tlak, bušení srdce a nepravidelný srdeční rytmus. Často se tyto příznaky vyvinou dříve než příznaky spojené s nízkou hladinou cukru v mozku.
- Ve Vašem mozku:
Příklady příznaků naznačujících nízkou hladinu cukru v mozku: bolest hlavy, intenzivní hlad, nevolnost, zvracení, únava, ospalost, poruchy spánku, neklid, agresivní chování, pokles koncentrace, zhoršené reakce, deprese, zmatenost, poruchy řeči (někdy úplná ztráta řeči), poruchy vidění, třes, paralýza, pocity mravenčení (parestézie), brnění a pocity necitlivosti v oblasti úst, závrať, ztráta sebekontroly, neschopnost postarat se o sebe, křeče a ztráta vědomí.
První příznaky, které Vás varují před hypoglykémií („varovné příznaky“), mohou být pozměněny, mohou být slabší nebo mohou zcela chybět, pokud:
- jste starší;
- máte diabetes již dlouho,
- trpíte určitým typem nervového onemocnění (diabetická autonomní neuropatie),
- Vás hypoglykémie trápila nedávno (např. předchozí den) nebo se vyvíjí pomalu,
- máte téměř normální nebo alespoň hodně zlepšenou hladinu cukru v krvi,
- užíváte nebo jste užíval(a) některé jiné léky (viz bod 2 „Další léčivé přípravky a Apidra“).
V takových případech se u Vás může rozvinout těžká hypoglykémie (a dokonce mdloba), aniž byste si toho všimli včas. Seznamte se co nejlépe se svými varovnými příznaky hypoglykémie. Je-li to nutné, častější měření cukru v krvi může pomoci odhalit mírné hypoglykemické epizody, které by jinak mohly být přehlédnuty. Pokud se nemůžete spolehnout na rozpoznání svých varovných příznaků, vyhněte se takovým situacím (např. řízení vozidla), při kterých by hypoglykémie pro Vás nebo pro ostatní představovala riziko.
Co dělat v případě hypoglykémie?
1. Nepodávejte inzulín. Okamžitě si vezměte asi 10 až 20 g cukru, např. glukózu, kostky cukru nebo cukrem oslazený nápoj. Upozornění: umělá sladidla a potraviny obsahující umělá sladidla (např. dietní nápoje) při hypoglykémii nepomáhají.
2. Poté snězte něco, co má dlouhotrvající účinek na nárůst cukru v krvi (např. chléb nebo těstoviny). Dbejte na to, abyste tyto záležitosti předem prohovořili s lékařem nebo sestrou.
3. Pokud se hypoglykémie znovu vrací, vezměte si dalších 10 až 20 g cukru.
4. Informujte okamžitě lékaře, jestliže hypoglykémii nebo její opakování nejste schopni zvládnout.
Oznamte svým příbuzným, přátelům a blízkým kolegům následující:
Pokud nemůžete polykat nebo upadnete do bezvědomí, budete potřebovat injekci glukózy nebo glukagonu (lék, který zvyšuje hladinu cukru v krvi). Tyto injekce jsou opodstatněné dokonce i tehdy, není-li jisté, že máte hypoglykémii.
Je vhodné změřit si hladinu cukru v krvi okamžitě po požití glukózy, abyste zkontrolovali, že máte skutečně hypoglykémii.
OptiSet je předplněné pero určené k injekci inzulínu. Můžete nastavit dávku od 2 do 40 jednotek po 2 jednotkách. Jedno pero obsahuje více dávek.
Před použitím OptiSet se poraďte se svým lékařem, lékárníkem nebo zdravotní sestrou o správných injekčních technikách.
Před prvním použitím Vašeho OptiSet si pečlivě přečtěte tento návod na použití. V případě, že nejste schopen/schopna sám/sama dodržet instrukce, použijte OptiSet pouze v případě, že Vám pomáhá osoba, která je plně schopna postupovat podle instrukcí. Uchopte pero tak, jak je ukázáno v tomto návodu. Pro ujištění, že správně čtete velikost dávky, držte pero vodorovně, s jehlou nalevo a voličem dávky napravo, tak, jak ukazují níže uvedené obrázky.
Dodržujte všechny tyto pokyny pokaždé, když používáte OptiSet, aby bylo zajištěno, že dostanete přesnou dávku. Nebudete-li dodržovat všechny pokyny, můžete dostat příliš mnoho nebo příliš málo inzulínu, což může mít vliv na hladinu glukózy v krvi.
Pokud máte nějaké otázky týkající se OptiSet nebo cukrovky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry nebo zavolejte na číslo místní pobočky sanofi-aventis, které je uvedeno na přední straně této příbalové informace.
Uschovejte si tento návod pro budoucí použití pokaždé, když užíváte OptiSet.
Kryt pera
Jehla (není přiložena)
Pero

Schématický nákres pera
Nové informace _pro _použití:
• Název inzulínu je vytištěn na peru.
• Voličem dávky j e možné otáčet pouze j edním směrem.
Důležité informace _pro _použití OptiSet:
• Před každým použitím vždy nasaďte novou jehlu. Používejte pouze jehly, které byly schváleny k použití s OptiSet.
• Před každou injekcí proveďte kontrolu bezpečnosti (viz Krok 3).
• Pokud používáte nový OptiSet, musí být provedena počáteční kontrola bezpečnosti s 8 jednotkami přednastavenými výrobcem.
• Voličem dávky je možné otáčet pouze jedním směrem.
• Nikdy se nesmí otáčet voličem dávky (měnit dávku) poté, co bylo vysunuto injekční tlačítko.
• Toto pero je pouze pro Vaše použití. Nesdílejte jej s nikým dalším.
• Pokud Vám bude přípravek injikovat jiná osoba, musí si počínat zvlášť opatrně, aby se náhodně jehlou neporanila a nedošlo k přenosu infekce.
• Nikdy nepoužívejte OptiSet, pokud je poškozený nebo si nejste jisti, že funguje správně.
• Vždy s sebou noste náhradní OptiSet pro případy, že se Váš OptiSet ztratí nebo bude poškozen.
A. Odstraňte kryt pera.
B. Zkontrolujte štítek na OptiSet a inzulínovém zásobníku, abyste se ujistili, že máte správný inzulín.
C. Zkontrolujte vzhled inzulínu. Apidra je čirý inzulín. Neužívejte tento OptiSet, jestliže je inzulín zakalený, zbarvený nebo obsahuje částice.
Krok 2. Upevněte jehlu
Pro každou injekci musí být použita nová sterilní jehla. Toto opatření pomáhá předejít kontaminaci a
případnému ucpání jehly.
Před použitím jehly si pečlivě přečtěte „Návod k použití“, přiložený k jehlám.
Poznámka: Zde vyobrazené jehly mají pouze ilustrativní účel.
A. Z nové jehly odstraňte ochranný obal.
B. Přiložte jehlu k peru a při nasazování ji držte přímo (podle typu jehly ji zašroubujte nebo zatlačte).

• Pokud není jehla držena při nasazování přímo, může se poškodit gumový uzávěr a dojít k úniku inzulínu, nebo se může poškodit jehla.

Krok 3. Proveďte kontrolu bezpečnosti
Před každou injekcí vždy proveďte kontrolu bezpečnosti. Tím se zajistí, že dostanete přesnou dávku:
• Ujistěte se, že pero i jehla pracují správně
• Odstraňte vzduchové bubliny
Pokud používáte nový OptiSet, musí být provedena počáteční kontrola bezpečnosti s 8 jednotkami přednastavenými výrobcem, jinak nebude pero správně fungovat.
A. Ujistěte se, že injekční tlačítko je stlačeno.
B. Zvolte dávku pro kontrolu bezpečnosti.
• Nový a nepoužitý OptiSet: dávka 8 jednotek je již přednastavena výrobcem pro provedení první kontroly bezpečnosti.
Používaný OptiSet: zvolte dávku 2 jednotky otočením voliče dávky dopředu, dokud ukazatel dávky neukazuje na 2. Volič dávky se bude otáčet pouze v jednom směru.

C.
Úplně vytáhněte injekční tlačítko, abyste natáhli dávku. Nikdy neotáčejte voličem dávky poté, co bylo vytaženo injekční tlačítko.

D. Odstraňte vnější kryt jehly a ponechte si ho pro odstranění použité jehly po injekci. Odstraňte
vnitřní kryt jehly a vyhoďte ho.

E. Držte pero tak, aby j ehla směřovala nahoru.
F. Poklepejte na zásobník inzulínu, aby všechny vzduchové bubliny vystoupaly nahoru k jehle.
G. Úplně stlačte injekční tlačítko. Zkontrolujte, zda inzulín vytéká z hrotu jehly.

Možná budete muset opakovat kontrolu bezpečnosti několikrát, dokud se neobjeví inzulín.
• Pokud žádný insulin j ehlou nevytéká, zkontrolujte, zda nej sou uvnitř vzduchové bubliny a zopakujte kontrolu bezpečnosti ještě dvakrát, abyste je odstranili.
• Pokud stále žádný inzulín nelze jehlou vytlačit, jehla může být ucpaná. Vyměňte jehlu a zkuste to znovu.
• Pokud nevytéká inzulín ani po výměně jehly, Váš OptiSet může být poškozen. Tento OptiSet nepoužívejte.
Dávku můžete nastavit po 2 jednotkách, od minimální dávky 2 jednotky až po maximální dávku 40 jednotek. Jestliže potřebujete dávku větší než 40 jednotek, musíte ji podat jako dvě a více injekcí.
A. Ověřte, zda máte pro svou dávku dostatek inzulínu.
• Stupnice zbývajícího inzulínu viditelná na průhledném inzulínovém zásobníku přibližně ukazuje, kolik inzulínu zbývá v OptiSet. Tato stupnice nesmí být používána pro nastavení dávky.
• Jestliže je černý píst na začátku barevného proužku, zbývá ještě přibližně 40 jednotek inzulínu.
• Jestliže je černý píst na konci barevného proužku, zbývá ještě přibližně 20 jednotek inzulínu.

B. Zvolte Vámi požadovanou dávku otáčením voliče dávky dopředu.
Pokud jste navolil/a vyšší dávku než požadovanou,
• a ještě jste nevytáhli injekční tlačítko, můžete otáčet voličem dávky dopředu, dokud znovu nenastavíte Vaši dávku.
• a už jste vytáhli injekční tlačítko, musíte dávku, která byla natažena, odstranit předtím, než začnete znovu otáčet voličem dávky.

Krok 5. Natáhnete dávku
A. Úplně vytáhněte injekční tlačítko, abyste natáhli dávku.
B. Zkontrolujte, zda je zvolená dávka úplně natažena. Všimněte si, že injekční tlačítko jde vytáhnout jen tak daleko, kolik inzulínu zbývá v zásobníku.
• Injekční tlačítko musí být během této kontroly pod tlakem vytaženo.
• Poslední tlustá viditelná čárka na injekčním tlačítku ukazuje množství nataženého inzulínu. Když je injekční tlačítko vytaženo, je vidět pouze horní část této tlusté čáry.
• V tomto případě j e nataženo 12 j ednotek.
o Pokud jste si zvolili 12 jednotek, můžete si svou dávku injikovat.
o Pokud jste si zvolili více než 12 jednotek, můžete si tímto perem injikovat pouze 12
jednotek z Vaší celkové dávky inzulínu.

Co máte v tomto případě dělat:
• buď si můžete injikovat zbývající inzulín v peru a doplnit Vaši dávku z nového OptiSet
• nebo pro Vaši plnou dávku použijte nový OptiSet.
Krok 6. Injikujte dávku
A. Používejte injekční techniku, kterou Vám doporučil Váš lékař, lékárník nebo zdravotní sestra.
B. Vpíchněte jehlu do kůže.

C.
Úplným stisknutím injekčního tlačítka zajistíte vstříknutí dávky. Uslyšíte klapavý zvuk, který přestane, když dojde k úplnému stisknutí injekčního tlačítka.

D. Držte injekční tlačítko úplně stisknuté a pomalu počítejte do 10, než vytáhnete jehlu z kůže. Tím se zajistí vstříknutí celé dávky.
Píst se pohybuje s každou dávkou. Po spotřebování 300 jednotek inzulínu dosáhne píst konce zásobní vložky.
Krok 7. Sejměte jehlu a vyhoďte ji
Po každé injekci sundejte jehlu a skladujte OptiSet bez nasazené jehly. To pomáhá zabránit:
• Kontaminaci a/nebo infekci
• Proniknutí vzduchu do zásobníku inzulínu a vytékání inzulínu, což by mohlo způsobit nepřesné dávkování.
A. Nasaďte znovu na jehlu vnější kryt jehly a použijte ho k jejímu odšroubování z pera., Aby nedošlo k náhodnému poranění jehlou, nikdy nenasazujte zpět vnitřní kryt jehly.
• Jestliže Vám dává injekci jiná osoba, nebo pokud dáváte injekci někomu jinému Vy sám/sama, musíte při sejmutí a odstraňování jehly dbát zvláštní opatrnosti. Aby se předešlo náhodnému zranění jehlou a přenosu infekce, řiďte se doporučenými bezpečnostními opatřeními (kontaktujte svého lékaře, lékárníka nebo zdravotní sestru).
B. Vyhoďte jehlu podle návodu Vašeho lékaře, lékárníka nebo zdravotní sestry tak, aby to bylo bezpečné.
C. Vždy na pero nasaďte zpět jeho kryt a skladujte tak pero do další injekce.
Pokyny pro uchovávání
Pro informace o uchovávání OptiSet si, prosím, prohlédněte bod 5 - Uchovávání přípravku Apidra -na zadní (inzulínové) straně této příbalové informace.
Pokud je Váš OptiSet skladován v chladu, je třeba ho vyjmout 1 až 2 hodiny před použitím, aby se mohl ohřát na pokojovou teplotu. Injikování studeného inzulínu je bolestivější.
Použitý OptiSet musí být zlikvidován podle pokynů místních úřadů.
Údržba
Chraňte OptiSet před prachem a špínou.
OptiSet můžete čistit zvenčí otíráním vlhkým hadříkem.
Pero nemáčejte, nemyjte nebo nepromazávejte, může tím být poškozeno.
Váš OptiSet je navržen tak, aby pracoval přesně a bezpečně. Musí s ním být zacházeno s opatrností. Vyhněte se situacím, kdy může být OptiSet poškozen. Pokud se domníváte, že je Váš OptiSet poškozen, použijte nový.
Otázky a odpovědi
|
Byla zvolena špatná dávka. |
• Pro zvolení správné dávky následujte instrukce v Kroku 4. |
|
Dávka byla zvolena a injekční tlačítko bylo vytaženo a znovu stisknuto bez upevněné jehly. |
1. Připevněte novou j ehlu. 2. Stiskněte injekční tlačítko nadoraz a odstraňte inzulín. 3. Proveďte kontrolu bezpečnosti. Pokud je kontrola bezpečnosti úspěšná, OptiSet je připraven k použití. Pokud kontrola bezpečnosti není úspěšná, pero může být poškozeno. Použijte nový OptiSet. Pokud jsou jakékoliv pochybnosti, že pero pracuje přesně, použijte nový OptiSet. |
|
Volič dávky se neotáčí. |
• Otáčíte špatným směrem. Tímto voličem dávky lze otáčet pouze dopředu. • Otáčíte dopředu, zatímco je injekční tlačítko vytaženo. Stiskněte úplně injekční tlačítko k odstranění navolené dávky a navolte ji znovu. |
|
Injekční tlačítko ukazuje vyšší množství, než je zvolená dávka. |
• Rozdíl j sou 2 j ednotky. Vypusťte inzulín, potom nastavte svou dávku a znovu zkontrolujte. Vyskytne-li se znovu stejná chyba, OptiSet může být poškozen, použijte nový OptiSet. • Rozdíl je více než 2 jednotky. OptiSet je poškozen, použijte nový OptiSet. |
|
Injekční tlačítko ukazuje nižší množství, než je požadovaná dávka. |
V zásobníku není dostatek inzulínu. Můžete si vybrat jednu z následujících možností: • můžete si injikovat zbývající inzulín v peru a doplnit Vaši dávku z nového OptiSet • nebo můžete pro Vaši plnou dávku použít nový OptiSet. |
|
Injekční tlačítko nelze stisknout. |
1. Ujistěte se, že jste injekční tlačítko vytáhli úplně. 2. Upevněte novou jehlu. 3. Stiskněte injekční tlačítko nadoraz, aby se odstranil inzulín. 4. Proveďte kontrolu bezpečnosti. |
|
Během injekce neslyšíte klapání. |
OptiSet je poškozen, použijte nový OptiSet. |
|
Inzulín vytéká z pera. |
Jehla byla upevněna nepřesně (např. šikmo). Odstraňte jehlu a nahraďte ji novou jehlou, kterou připevníte rovně (viz Krok 2). Proveďte kontrolu bezpečnosti (viz Krok 3). |
|
V zásobníku jsou vzduchové bubliny. |
Při běžném používání může být v jehle a v zásobníku inzulínu přítomno malé množství vzduchových bublin. Vzduchové bubliny musíte odstranit provedením kontroly bezpečnosti (viz Krok 3). Malé vzduchové bubliny v zásobníku, které nelze odstranit jemným poklepáváním, nevadí injekci a dávkování. |
|
OptiSet je poškozen nebo nepracuje správně. |
Nepoužívejte sílu. Nepokoušejte se OptiSet opravit ani nepoužívejte nářadí. Použijte nový OptiSet. |
|
OptiSet upadl nebo byl vystaven nárazu. |
Pokud jsou jakékoliv pochybnosti, že pero pracuje přesně, použijte nový OptiSet. |
Příbalová informace: informace pro uživatele
Apidra SoloStar 100 jednotek/ml injekční roztok v předplněném peru
Insulinům glulisinum
Přečtěte si pozorně celou příbalovou informaci včetně Návodu na použití přípravku Apidra SoloStar, předplněné pero dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je Apidra a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Apidra používat
3. Jak se přípravek Apidra používá
4. Možné nežádoucí účinky
5. Jak přípravek Apidra uchovávat
6. Obsah balení a další informace
1. Co je Apidra a k čemu se používá
Apidra je antidiabetikum používané ke snížení hladiny cukru v krvi u pacientů s diabetes mellitus; může být podána dospělým, mladistvým a dětem ve věku 6 a více let. Diabetes mellitus je onemocnění, při kterém Váš organismus neprodukuje dostatek inzulínu pro kontrolu hladiny cukru v krvi.
Přípravek je vyroben biotechnologicky. Inzulín glulisin má rychlý nástup účinku během 10-20 minut a krátké trvání účinku přibližně 4 hodiny.
2. Čemu musíte věnovat pozornost, než začnete přípravek Apidra používat Nepoužívejte přípravek Apidra
- Jestliže jste alergický(á) na inzulín glulisin nebo na kteroukoliv další složku tohoto přípravku (uvedenou v bodě 6).
- Jestliže máte hladinu cukru v krvi příliš nízkou (hypoglykémie). Postupujte podle pokynů pro hypoglykémii (viz rámeček na konci této příbalové informace).
Upozornění a opatření
Před užitím přípravku Apidra se poraďte se svým lékařem, lékarníkem nebo zdravotní sestrou. Dodržujte pečlivě pokyny pro dávkování, sledování (testy krve), dietu a fyzickou aktivitu (fyzickou práci a cvičení), které jste projednali se svým lékařem.
Zvláštní skupiny pacientů
Pokud máte problémy s játry nebo s ledvinami, upozorněte svého lékaře, protože můžete potřebovat nižší dávku.
Nejsou dostatečné klinické informace o použití přípravku Apidra u dětí mladších 6 let.
Cestování
Před cestováním se poraďte se svým lékařem. Možná bude třeba se poradit o
- dostupnosti Vašeho inzulínu v zemi, kterou navštívíte,
- dodávkách inzulínu, injekčních stříkaček, atd.,
- správném uchovávání Vašeho inzulínu během cestování,
- časové dostupnosti jídla a podávání inzulínu během cestování,
- možných účincích přesunu v různých časových pásmech,
- možných zdravotních rizicích v zemích, které navštívíte,
- tom, jak se zachovat v mimořádných situacích, tj. pokud se Vám udělá špatně nebo onemocníte. Onemocnění a úrazy
V následujících situacích může léčba Vašeho diabetu vyžadovat zvláštní péči:
- pokud jste nemocný/á nebo máte větší úraz, může dojít ke zvýšení hladiny Vašeho cukru v krvi (hyperglykémie),
- pokud dostatečně nejíte, může se Vám hladina cukru v krvi velmi snížit (hypoglykémie).
Ve většině případů budete potřebovat lékařskou péči. Zajistěte možnost rychlého kontaktu lékaře.
Pokud máte diabetes typu 1 (na inzulínu závislý diabetes), nepřestávejte užívat svůj inzulín a pokračujte v příjmu dostatečného množství cukrů. Vždy řekněte lidem, kteří Vás ošetřují nebo léčí, že potřebujete inzulín.
U některých pacientů, kteří dlouhodobě trpěli cukrovkou typu 2 a onemocněním srdce, nebo prodělali mozkovou příhodu, a kteří byli léčeni pioglitazonem a inzulínem, byl zaznamenán výskyt srdečního selhání. Jestliže zjistíte známky srdečního selhání jako je neobvyklá dušnost nebo náhlé zvýšení tělesné hmotnosti nebo lokalizovaný otok (edém), informujte co nejrychleji svého lékaře.
Další léčivé přípravky a přípravek Apidra
Některé léky způsobují změnu hladiny cukru v krvi (snížení, zvýšení nebo obojí v závislosti na situaci). V každém případě může být nezbytné upravit Vaši dávku inzulínu, aby se zabránilo hladinám cukru v krvi, které jsou buď příliš nízké, nebo příliš vysoké. Buďte opatrní, když začínáte nebo přestáváte užívat nějaký další lék.
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat. Zeptejte se svého lékaře předtím, než si lék vezmete, zda a jak jím může být ovlivněna Vaše hladina cukru v krvi, popř. co je třeba dělat.
Mezi léky, které mohou vyvolat pokles hladiny cukru v krvi (hypoglykémii), patří:
- všechny další léky pro léčbu diabetu,
- inhibitory angiotenzin konvertujícího enzymu (ACE inhibitory) (používané k léčbě některých srdečních onemocnění, vysokého krevního tlaku),
- disopyramid (používaný k léčbě některých srdečních onemocnění),
- fluoxetin (používaný k léčbě depresí),
- fibráty (používané ke snížení vysokých hladin krevních lipidů),
- inhibitory monoaminooxidázy (MAO inhibitory) (používané k léčbě depresí),
- pentoxifylin, propoxyfen, salicyláty (např. aspirin, používaný ke zmírnění bolesti a snížení horečky),
- sulfonamidová antibiotika.
Mezi léky, které mohou vyvolat zvýšení hladiny cukru v krvi (hyperglykémii), patří:
- kortikosteroidy (např. „kortizon“ používaný pro léčbu zánětu),
- danazol (lék pro navození ovulace),
- diazoxid (používaný pro léčbu vysokého krevního tlaku),
- diuretika (používaná pro léčbu vysokého krevního tlaku nebo nadměrného zadržování tekutin),
- glukagon (hormon slinivky břišní používaný pro léčbu závažné hypoglykémie),
- isoniazid (používaný pro léčbu tuberkulózy),
- estrogeny a progestogeny (např. v antikoncepčních pilulkách užívaných k zamezení početí),
- deriváty fenothiazinu (používané pro léčbu duševních poruch),
- somatropin (růstový hormon),
- sympatomimetika (např. epinefrin [adrenalin], salbutamol, terbutalin užívané k léčbě astmatu),
- hormony štítné žlázy (užívané k léčbě onemocnění štítné žlázy),
- inhibitory proteáz (užívané k léčbě HIV),
- atypická antipsychotika (např. olanzapin a klozapin).
Vaše hladina cukru v krvi může buď poklesnout, nebo vzrůst, pokud užíváte:
- betablokátory (používané pro léčbu vysokého krevního tlaku),
- klonidin (používaný pro léčbu vysokého krevního tlaku),
- soli lithia (používané pro léčbu duševních poruch).
Pentamidin (používaný pro léčbu některých infekcí způsobených parazity) může vyvolat hypoglykémii, po které může někdy následovat hyperglykémie.
Betablokátory stejně jako ostatní sympatolytika (např. klonidin, guanetidin a reserpin) mohou oslabit nebo úplně potlačit první varovné příznaky, které Vám mohou pomoci rozpoznat hypoglykémii.
Pokud si nejste jisti, zda neužíváte některý z takových léků, zeptejte se svého lékaře nebo lékárníka.
Užívání přípravku Apidra s alkoholem
Hladiny Vašeho cukru v krvi mohou buď vzrůst, nebo poklesnout, pokud konzumujete alkohol. Těhotenství a kojení
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek užívat.
Informujte svého lékaře, pokud plánujete těhotenství nebo pokud již jste těhotná. Je možné, že bude třeba měnit dávku inzulínu během těhotenství a po porodu. Zvláště pečlivá kontrola diabetu a prevence hypoglykémie jsou důležité pro zdraví Vašeho dítěte.
Nejsou k dispozici žádné nebo jen omezené údaje o použití přípravku Apidra u těhotných žen.
Pokud kojíte, poraďte se se svým lékařem, neboť může být nezbytné upravit Vám dávku inzulínu a Vaši dietu.
Řízení dopravních prostředků a obsluha strojů
Vaše schopnost soustředit se nebo reagovat může být snížena, jestliže:
- máte hypoglykémii (nízkou hladinu cukru v krvi)
- máte hyperglykémii (vysokou hladinu cukru v krvi).
Myslete na tento možný problém ve všech situacích, kde byste mohli dostat sebe nebo ostatní do nebezpečí (např. při řízení vozidla nebo při obsluze strojů). Měl(a) byste se poradit se svým lékařem o vhodnosti řízení vozidel, pokud:
- máte časté epizody hypoglykémie,
- jsou první varovné příznaky, které Vám pomohou rozpoznat hypoglykémii, omezené nebo chybějí.
Důležité informace o některých složkách přípravku Apidra
Tento léčivý přípravek obsahuje méně než 1 mmol (23 mg) sodíku v jedné dávce, tj. v podstatě sodík neobsahuje.
Apidra obsahuje metakresol
Apidra obsahuje metakresol, který může způsobovat alergické reakce.
3. Jak se přípravek Apidra používá Dávkování
Vždy užívejte přípravek Apidra přesně tak, jak Vám doporučil lékař. Pokud si nejste jist(a), poraďte se s lékařem nebo lékárníkem.
Na základě Vašeho životního stylu a Vašich výsledků testů cukru (glukózy) v krvi a Vašeho předchozího užívání inzulínu lékař určí, jakou dávku přípravku Apidra budete potřebovat.
Apidra je rychle účinkující inzulín. Je možné, že Váš lékař Vám řekne, abyste ji užívali v kombinaci se střednědobě nebo dlouhodobě působícím inzulínem nebo bazálním inzulínem nebo s tabletami na snížení vysoké hladiny cukru v krvi.
Jestliže přecházíte z jiného inzulínu na inzulín glulisin, Vaše dávkování může vyžadovat úpravu lékařem.
Hladinu cukru v krvi může ovlivnit mnoho faktorů. Měli byste znát tyto faktory, abyste byli schopni reagovat správně na změny hladiny cukru v krvi a předejít jejich přílišnému zvýšení nebo přílišnému snížení. Další informace si přečtěte na konci této příbalové informace.
Způsob podání
Injekce přípravku Apidra se podává pod kůži (subkutánně).
Váš lékař Vám ukáže, do které oblasti kůže máte přípravek Apidra podávat. Injekci přípravku Apidra lze podávat do břišní stěny, do stehna nebo horní části paže nebo kontinuální infuzí do břišní stěny. Účinek bude o něco rychlejší, jestliže si injekci inzulínu aplikujete do břišní stěny. Tak jako u všech ostatních inzulínů musíte měnit s každou injekcí místo vpichu injekce a místo infuze v rámci vybrané oblasti (břišní stěna, stehno, horní část paže), kam injekci podáváte.
Frekvence podávání
Apidra by se měla užívat krátce (0 - 15 minut) před jídlem nebo brzy po jídle.
Pokyny ke správnému použití
Jak se zachází s perem SoloStar
SoloStar je předplněné pero pro jednorázové použití, které obsahuje inzulín glulisin.
Pečlivě si přečtěte „Návod na použití SoloStar“ zahrnutý v této příbalové informaci. Pero musíte používat tak, jak je popsáno v tomto návodu na použití.
Z důvodu prevence přenosu nemoci musí být každé pero používáno pouze jedním pacientem.
Před použitím vždy nasaďte novou jehlu a proveďte zkoušku bezpečnosti. Používejte pouze jehly, které byly schváleny pro použití se SoloStar.
Zásobní vložku si prohlédněte ještě předtím, než ji použijete. Použijte pouze ty zásobní vložky, ve kterých je roztok čirý, bezbarvý, bez viditelných částic. Před použitím přípravek nemíchejte ani s ním netřepejte.
Použijte nové pero pokaždé, když si všimnete, že kontrola hladiny cukru v krvi se nečekaně zhoršila. Domníváte-li se, že problém může být se SoloStar, požádejte o kontrolu Vašeho lékaře nebo lékárníka.
Jestliže jste užil(a) více přípravku Apidra, než jste měl(a)
- Pokud jste podali příliš mnoho přípravku Apidra, může se Vám příliš snížit hladina cukru
v krvi (hypoglykémie). Často si kontrolujte hladinu cukru v krvi. Obecně platí, že pro prevenci hypoglykémie musíte jíst více jídla a sledovat hladinu cukru v krvi. Pro informace o léčbě hypoglykémie viz rámeček na konci této příbalové informace.
Jestliže jste zapomněl(a) užít přípravek Apidra
- Pokud jste vynechali dávku přípravku Apidra nebo jste podali nedostatečné množství inzulínu, může se Vám příliš zvýšit hladina cukru v krvi (hyperglykémie). Často si kontrolujte hladinu cukru v krvi. Pro další informace o léčbě hyperglykémie viz rámeček na konci této příbalové informace.
- Nezdvojujte následující dávku, abyste nahradil(a) vynechanou dávku.
Jestliže jste přestal(a) užívat přípravek Apidra
Ukončení užívání přípravku Apidra by mohlo vést k závažné hyperglykémii (velmi vysoká hladina cukru v krvi) a ketoacidóze (hromadění kyseliny v krvi, protože tělo rozkládá tuk místo cukru). Neukončujte léčbu přípravkem Apidra bez konzultace s lékařem, který Vám řekne, co byste měl(a) udělat.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
Záměna inzulínů
Před každou injekcí musíte vždy zkontrolovat štítek na inzulínu, aby nedošlo k záměně léčivého přípravku Apidra za jiné inzulíny.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Závažné nežádoucí účinky
Hypoglykémie (nízká hladina cukru v krvi) může být velmi závažná. Hypoglykémie je velmi často hlášeným nežádoucím účinkem (může postihovat více než 1 z 10 osob). Hypoglykémie (nízká hladina cukru v krvi) znamená, že v krvi není dost cukru. Jestliže Vám příliš klesne hladina cukru v krvi, mohli byste ztratit vědomí. Těžká hypoglykémie může způsobit poškození mozku a může být život ohrožující. Pokud máte příznaky nízké hladiny cukru v krvi, okamžitě se snažte hladinu cukru v krvi zvýšit. Přečtěte si další důležité informace o hypoglykémii a její léčbě na konci této příbalové informace.
Pokud se u Vás vyskytnou následující příznaky, ihned kontaktujte svého lékaře:
Systémové alergické reakce jsou méně častými nežádoucími účinky (mohou postihovat až 1 ze 100 osob).
Generalizovaná alergie na inzulín: Přidružené symptomy mohou zahrnovat rozsáhlé kožní reakce (vyrážka a svědění po celém těle), závažný otok kůže nebo sliznic (angioedém), zkrácený dech, pokles krevního tlaku se zrychleným tepem a pocení. Může se jednat o příznaky závažného případu generalizované alergie na inzulín, včetně anafylaktické reakce, která může být život ohrožující.
Hyperglykémie (vysoká hladina cukru v krvi) znamená, že máte v krvi příliš mnoho cukru.
Četnost výskytu hyperglykémie nelze určit. Pokud máte příliš vysokou hladinu cukru v krvi, vypovídá to o tom, že možná budete potřebovat více inzulínu, než jste si aplikoval(a) v injekci. Může jít o závažný stav, pokud se hladina glukózy velmi zvýší.
Přečtěte si další informace o známkách a příznacích hyperglykémie v rámečku na konci této příbalové informace.
Jiné nežádoucí účinky
Časté nežádoucí účinky (mohou postihovat až 1 z 10 osob)
• Kožní a alergické reakce v místě vpichu injekce
Může se objevit reakce v místě vpichu (např. zarudnutí, neobvykle intenzívní bolestivost injekce, svědění, vyrážka, otok nebo zánět). Tyto reakce se mohou také rozšířit kolem místa vpichu. Většina mírných reakcí na inzulín obvykle odezní během několika dnů až několika týdnů.
Vzácné nežádoucí účinky (mohou postihovat až 1 z 1000 osob)
• Změny kůže v místě aplikace injekce (lipodystrofie)
Pokud podáváte inzulín příliš často do stejného místa na kůži, může se tuková tkáň pod kůží v tomto místě buď ztenčit, nebo zesílit (lipodystrofie). Obojí snižuje účinnost inzulínu podávaného do takto změněného místa. Takovým změnám na kůži lze zabránit střídáním místa vpichu při každé injekci.
Nežádoucí účinky, jejichž frekvenci nelze z dostupných údajů určit
• Oční reakce
Výrazné změny (zlepšení nebo zhoršení) kompenzace Vašeho krevního cukru mohou přechodně narušit Váš zrak. Pokud máte proliferativní retinopatii (oční onemocnění vztahující se k diabetu), mohou závažné hypoglykemické ataky způsobit přechodnou ztrátu vidění.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Apidra uchovávat
Uchovávejte mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti, uvedené na krabičce a na štítku pera za „Použitelné do:“ nebo „EXP“. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Nepoužívaná pera
Uchovávejte v chladničce (2°C - 8°C).
Chraňte před mrazem.
Neukládejte předplněné pero SoloStar do blízkosti mrazicího boxu nebo mrazicí vložky.
Uchovávejte předplněné pero v krabičce, aby byl přípravek chráněn před světlem.
Používaná pera
Používaná předplněná pera (nebo nošená jako náhradní) mohou být uchovávána až 4 týdny při teplotě do 25°C mimo přímé teplo nebo světlo. Používané pero nesmí být skladováno v chladničce. Nepoužívejte pero po uplynutí této doby použitelnosti.
Nepoužívejte tento přípravek, pokud není roztok čirý a bezbarvý.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co Apidra obsahuje
- Léčivou látkou je insulinum glulisinum. Jeden mililitr roztoku obsahuje 100 jednotek léčivé látky insulinum glulisinum (odpovídá množství 3,49 mg).
- Pomocné látky jsou: metakresol (viz bod 2 „Apidra obsahuje metakresol“), chlorid sodný (viz bod 2 „Důležité informace o některých složkách přípravku Apidra“), trometamol, polysorbát 20, kyselina chlorovodíková 35%, hydroxid sodný, voda pro injekci.
Jak Apidra vypadá a co obsahuje toto balení
Apidra SoloStar 100 jednotek/ml injekční roztok v předplněném peru. Čirý, bezbarvý, vodný roztok bez viditelných částic.
Jedno pero obsahuje 3 ml roztoku, což odpovídá 300 jednotkám. Dostupná jsou balení s 1, 3, 4, 5, 6, 8, 9 a 10 předplněnými pery. Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci a výrobce
Držitel rozhodnutí o registraci:
Sanofi-Aventis Deutschland GmbH D-65926 Frankfurt am Main Německo
Výrobce:
Sanofi-Aventis Deutschland GmbH Industriepark Hochst, D-65926 Frankfurt Německo
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci. Belgie/Belgique/Belgien Lietuva
Sanofi Belgium UAB “SANOFI-AVENTIS LIETUVA”
Tél/Tel: +32 (0)2 710 54 00 Tel: +370 5 2755224
|
Btarapna sanofi-aventis Bulgaria EOOD Ten.: +359 (0)2 970 53 00 |
Luxembourg/Luxemburg Sanofi Belgium Tél/Tel: +32 (0)2 710 54 00 (Belgique/Belgien) |
|
Česká republika sanofi-aventis, s.r.o. Tel: +420 233 086 111 |
Magyarország SANOFI-AVENTIS Zrt. Tel.: +36 1 505 0050 |
|
Danmark sanofi-aventis Denmark A/S Tlf: +45 45 16 70 00 |
Malta Sanofi Malta Ltd. Tel: +356 21493022 |
|
Deutschland Sanofi-Aventis Deutschland GmbH Tel: +49 (0)180 2 222010 |
Nederland sanofi-aventis Netherlands B.V. Tel: +31 (0)182 557 755 |
|
Eesti sanofi-aventis Estonia OU Tel: +372 627 34 88 |
Norge sanofi-aventis Norge AS Tlf: +47 67 10 71 00 |
|
EXXáSa sanofi-aventis AEBE Tn^: +30 210 900 16 00 |
Osterreich sanofi-aventis GmbH Tel: +43 1 80 185 - 0 |
|
Espaňa sanofi-aventis, S.A. Tel: +34 93 485 94 00 |
Polska sanofi-aventis Sp. z o.o. Tel.: +48 22 280 00 00 |
|
France sanofi-aventis France Tél: 0 800 222 555 Appel depuis l’étranger : +33 1 57 63 23 23 |
Portugal Sanofi - Produtos Farmaceuticos, Lda. Tel: +351 21 35 89 400 |
|
Hrvatska sanofi-aventis Croatia d.o.o. Tel: +385 1 600 34 00 |
Románia Sanofi Romania SRL Tel: +40 (0) 21 317 31 36 |
|
Ireland sanofi-aventis Ireland Ltd. T/A SANOFI Tel: +353 (0) 1 403 56 00 |
Slovenija sanofi-aventis d.o.o. Tel: +386 1 560 48 00 |
|
Ísland Vistor hf. Sími: +354 535 7000 |
Slovenská republika sanofi-aventis Pharma Slovakia s.r.o. Tel: +421 2 33 100 100 |
|
Italia Sanofi S.p.A. Tel: 800 13 12 12 (domande di tipo tecnico) 800 536389 (altre domande) |
Suomi/Finland Sanofi Oy Puh/Tel: +358 (0) 201 200 300 |
|
Kúnpoq sanofi-aventis Cyprus Ltd. Tn^: +357 22 871600 |
Sverige Sanofi AB Tel: +46 (0)8 634 50 00 |
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku j sou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/
HYPERGLYKÉMIE A HYPOGLYKÉMIE
Vždy u sebe mějte nějaký cukr (minimálně 20 gramů).
Noste u sebe nějakou informaci, která ukazuje, že máte cukrovku.
HYPERGLYKÉMIE (vysoká hladina cukru v krvi)
Pokud je Vaše hladina cukru v krvi příliš vysoká (hyperglykémie), možná jste si aplikoval(a) málo inzulínu.
Proč vzniká hyperglykémie?
Příklady:
- nepodali jste si injekci inzulínu nebo jste ho nepodali dostatečné množství nebo se inzulín stal méně účinným, např. z důvodu nesprávného uchovávání,
- máte méně pohybu než obvykle, jste ve stresu (emocionální úzkost, rozrušení) nebo máte úraz, j ste po operaci, máte infekci nebo horečku,
- užíváte nebo jste užíval(a) některé jiné léky (viz bod 2 „Další léčivé přípravky a Apidra“). Varovné příznaky hyperglykémie
Žízeň, zvýšené močení, únava, suchá pokožka, zčervenání obličeje, nechutenství, nízký krevní tlak, zrychlený tep a přítomnost glukózy a ketolátek v moči. Bolest žaludku, rychlé a hluboké dýchání, ospalost nebo dokonce ztráta vědomí mohou být známkami vážného stavu (ketoacidózy) vyplývajícího z nedostatku inzulínu.
Co dělat v případě hyperglykémie?
Změřte si hladinu cukru v krvi a ketolátek v moči co nejdříve, jakmile se výše uvedené příznaky objeví. Těžká hyperglykémie nebo ketoacidóza musí být vždy léčena lékařem, obvykle v nemocnici.
HYPOGLYKÉMIE (nízká hladina cukru v krvi)
Pokud je hladina Vašeho cukru v krvi příliš nízká, můžete ztratit vědomí. Závažná hypoglykémie může vyvolat srdeční záchvat nebo poškození mozku a může být život ohrožující. Za normálních okolností byste měl(a) být schopen/schopna rozpoznat přílišné snižování hladiny cukru v krvi, a proto můžete správně reagovat.
Proč vzniká hypoglykémie?
Příklady:
- aplikujete si příliš mnoho inzulínu,
- vynecháte nebo odložíte jídlo,
- nejíte dostatečně, nebo snězené jídlo obsahuje méně sacharidů než normálně (cukr a látky podobné cukru se nazývají sacharidy; umělá sladidla však NEJSOU sacharidy),
- dochází u Vás ke ztrátě sacharidů zvracením nebo průjmem,
- pijete alkohol, zvláště pokud nejíte dostatečně,
- cvičíte více než obvykle nebo vykonáváte jiný typ fyzické činnosti,
- zotavujete se ze zranění, operace, nebo z jiných forem stresu,
- zotavujete se po nemoci nebo z horečky,
- užíváte nebo jste přestali užívat některé jiné léky (viz bod 2 „Další léčivé přípravky a Apidra“). Výskyt hypoglykémie je pravděpodobnější, pokud:
- právě zahajujete léčbu inzulínem nebo přecházíte na jiný inzulínový přípravek,
- Vaše hladiny cukru v krvi j sou téměř normální nebo j sou nestálé,
- změníte oblast kůže, kam podáváte injekci inzulínu (např. ze stehna na horní část paže),
- trpíte závažným onemocněním ledvin nebo j ater nebo některou j inou chorobou, j ako j e hypotyreóza (snížená funkce štítné žlázy).
Varovné příznaky hypoglykémie
- Ve Vašem těle:
Příklady příznaků, které naznačují, že hladina cukru v krvi klesá příliš nízko nebo příliš rychle: pocení, vlhká pokožka, úzkost, zrychlený tep, vysoký krevní tlak, bušení srdce a nepravidelný srdeční rytmus. Často se tyto příznaky vyvinou dříve než příznaky spojené s nízkou hladinou cukru v mozku.
- Ve Vašem mozku:
Příklady příznaků naznačujících nízkou hladinu cukru v mozku: bolest hlavy, intenzivní hlad, nevolnost, zvracení, únava, ospalost, poruchy spánku, neklid, agresivní chování, pokles koncentrace, zhoršené reakce, deprese, zmatenost, poruchy řeči (někdy úplná ztráta řeči), poruchy vidění, třes, paralýza, pocity mravenčení (parestézie), brnění a pocity necitlivosti v oblasti úst, závrať, ztráta sebekontroly, neschopnost postarat se o sebe, křeče a ztráta vědomí.
První příznaky, které Vás varují před hypoglykémií („varovné příznaky“), mohou být pozměněny, mohou být slabší nebo mohou zcela chybět, pokud:
- jste starší;
- máte diabetes již dlouho,
- trpíte určitým typem nervového onemocnění (diabetická autonomní neuropatie),
- Vás hypoglykémie trápila nedávno (např. předchozí den) nebo se vyvíjí pomalu,
- máte téměř normální nebo alespoň hodně zlepšenou hladinu cukru v krvi,
- užíváte nebo jste užíval(a) některé jiné léky (viz bod 2 „Další léčivé přípravky a Apidra“).
V takových případech se u Vás může rozvinout těžká hypoglykémie (a dokonce mdloba), aniž byste si toho všimli včas. Seznamte se co nejlépe se svými varovnými příznaky hypoglykémie. Je-li to nutné, častější měření cukru v krvi může pomoci odhalit mírné hypoglykemické epizody, které by jinak mohly být přehlédnuty. Pokud se nemůžete spolehnout na rozpoznání svých varovných příznaků, vyhněte se takovým situacím (např. řízení vozidla), při kterých by hypoglykémie pro Vás nebo pro ostatní představovala riziko.
Co dělat v případě hypoglykémie?
1. Nepodávejte inzulín. Okamžitě si vezměte asi 10 až 20 g cukru, např. glukózu, kostky cukru nebo cukrem oslazený nápoj. Upozornění: umělá sladidla a potraviny obsahující umělá sladidla (např. dietní nápoje) při hypoglykémii nepomáhají.
2. Poté snězte něco, co má dlouhotrvající účinek na nárůst cukru v krvi (např. chléb nebo těstoviny). Dbejte na to, abyste tyto záležitosti předem prohovořili s lékařem nebo sestrou.
3. Pokud se hypoglykémie znovu vrací, vezměte si dalších 10 až 20 g cukru.
4. Informujte okamžitě lékaře, jestliže hypoglykémii nebo její opakování nejste schopni zvládnout.
Oznamte svým příbuzným, přátelům a blízkým kolegům následující:
Pokud nemůžete polykat nebo upadnete do bezvědomí, budete potřebovat injekci glukózy nebo glukagonu (lék, který zvyšuje hladinu cukru v krvi). Tyto injekce jsou opodstatněné dokonce i tehdy, není-li jisté, že máte hypoglykémii.
Je vhodné změřit si hladinu cukru v krvi okamžitě po požití glukózy, abyste zkontrolovali, že máte skutečně hypoglykémii.
Apidra SoloStar injekční roztok v předplněném peru. NÁVOD NA POUŽITÍ
SoloStar je předplněné pero pro injekce inzulínu. Váš lékař Vám doporučil používání SoloStar, protože jste podle něj schopen/schopna toto pero používat. Před použitím SoloStar se poraďte se svým lékařem, lékárníkem nebo zdravotní sestrou o správných injekčních technikách.
Před prvním použitím Vašeho SoloStar si pečlivě přečtěte tento návod na použití. V případě, že nejste schopen/schopna sám/sama dodržet instrukce, smíte použít SoloStar pouze v případě, že Vám pomáhá osoba, která je plně schopna postupovat podle instrukcí. Držte pero tak, jak je uvedeno v tomto návodu na použití. Abyste mohli správně odečíst dávku, držte pero vodorovně tak, aby jehla byla vlevo a volič dávky na pravé straně, jak ukazují obrázky níže.
Můžete nastavit dávku od 1 do 80 jednotek po jedné jednotce. Jedno pero obsahuje více dávek. Uschovejte si tento návod pro budoucí použití.
Pokud budete mít jakékoli otázky týkající se SoloStar nebo cukrovky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry nebo zavolejte na telefonní číslo společnosti sanofi-aventis uvedené na přední straně této příbalové informace.

Jehla
|
t—crTlf |
Ochranný uzávěr li D—d 1 |
|
Vnější kryt jehly |
'-' \J Vnitřní kryt ehly Jehla |
Pero
Okénko ukazující dávku

Volič
Gumový uzávěr dávky
Injekční
tlačítko
Schématický nákres pera
Důležité informace pro použití SoloStar:
• Před každým použitím se musí vždy nasadit nová jehla. Používejte pouze jehly určené k použití se SoloStar.
• Bez nasazené jehly neotáčejte voličem dávky ani nestlačujte injekční tlačítko.
• Před každou injekcí musí být proveden test bezpečnosti (viz Krok 3).
• Toto pero je pouze pro Vaše použití. Nesdílejte jej s nikým dalším.
• Pokud Vám bude přípravek injikovat jiná osoba, musí si počínat zvlášť opatrně, aby se náhodně jehlou neporanila a nedošlo k přenosu infekce.
• Nikdy nepoužívejte SoloStar, pokud je poškozený nebo si nejste jisti, že funguje správně.
• Vždy s sebou noste náhradní SoloStar pro případy, že se Váš SoloStar ztratí nebo bude poškozen.
Krok 1. Zkontrolujte inzulín
A. Zkontrolujte označení na peru a na zásobníku inzulínu, abyste se ujistili, že máte správný insulin. Apidra SoloStar je modrý. Má tmavě modré injekční tlačítko s vyvýšeným kroužkem na konci.
B. Odstraňte kryt pera.
C. Zkontrolujte vzhled Vašeho inzulínu. Apidra je čirý inzulín. Nepoužívejte tento SoloStar, jestliže je inzulín zakalený, zbarvený nebo obsahuje částice.
Krok 2. Upevněte jehlu
Pro každou injekci musí být použita nová sterilní jehla. Toto opatření pomáhá předejít kontaminaci a případnému ucpání jehly.
A. Odstraňte ochranné pouzdro z jehly.
B. Přiložte jehlu k peru a při nasazování ji držte přímo proti peru (podle typu jehly ji zašroubujte nebo zatlačte).

• Pokud není jehla držena při nasazování přímo proti peru, může se poškodit gumový uzávěr a dojít k úniku inzulínu, nebo se může poškodit jehla.

Krok 3. Proveďte kontrolu bezpečnosti
Před každou injekcí proveďte kontrolu bezpečnosti. Tím se zajistí, že dostanete přesnou dávku:
• Ujistěte se, že pero i jehla pracují správně
• Odstraňte vzduchové bubliny
A. Zvolte dávku 2 jednotky otáčením voliče dávky.

B. Odstraňte vnější kryt jehly a ponechte si ho pro odstranění použité jehly po injekci. Odstraňte vnitřní kryt jehly a vyhoďte ho.
Ponechat Vyhodit
C. Držte pero tak, aby j ehla směřovala nahoru.
D. Poklepte na zásobník inzulínu, aby všechny vzduchové bubliny vystoupaly nahoru k jehle.
E. Úplně stlačte injekční tlačítko. Zkontrolujte, zda inzulín vytéká z hrotu jehly.

Možná budete muset opakovat kontrolu bezpečnosti několikrát, dokud se neobjeví inzulín.
• Pokud žádný insulin j ehlou nevytéká, zkontrolujte, zda nej sou uvnitř vzduchové bubliny a zopakujte kontrolu bezpečnosti ještě dvakrát, abyste je odstranil/a.
• Pokud stále žádný inzulín nelze jehlou vytlačit, jehla může být ucpaná. Vyměňte jehlu a zkuste to znovu.
• Pokud nevytéká inzulín ani po výměně jehly, Váš SoloStar může být poškozen. Tento SoloStar nepoužívejte.
Krok 4. Nastavte dávku
Dávku můžete nastavit po 1 jednotce, od minimální dávky 1 jednotky až po maximální dávku 80 jednotek. Jestliže potřebujete dávku větší než 80 jednotek, musíte ji podat jako dvě a více injekcí.
A. Ověřte, že okénko ukazující dávku po kontrole bezpečnosti ukazuje „0“.
B. Zvolte Vámi požadovanou dávku (v příkladu níže je zvolená dávka 30 jednotek). Pokud jste navolil/a vyšší dávku než požadovanou, můžete otáčet voličem dávky zpět.

• Nestlačujte injekční tlačítko, zatímco otáčíte voličem dávky, jinak inzulín vyteče.
• Neotáčejte voličem dávky za číslo jednotek zbývajících v peru. K otáčení voliče dávky nepoužívejte sílu. Buď injikujte zbývající obsah v peru a doplňte Vaši dávku z nového SoloStar, nebo použijte pro injikování celé dávky nový SoloStar.
Krok 5. Injikujte dávku
A. Používejte injekční techniku, kterou Vám doporučil Váš lékař, lékárník nebo zdravotní sestra
B. Vpíchněte jehlu do kůže.

C.
Úplným stisknutím injekčního tlačítka zajistíte vstříknutí dávky. Číslo v okénku ukazujícím dávku se během injekce vrátí na „0“

D. Držte injekční tlačítko úplně stisknuté. Než vytáhnete jehlu z kůže, pomalu počítejte do 10. Tím se zajistí vstříknutí celé dávky.
Píst se pohybuje s každou dávkou. Po spotřebování 300 jednotek inzulínu dosáhne píst konce zásobní vložky.
Krok 6. Sejměte jehlu a vyhoďte ji
Po každé injekci sundejte jehlu a skladujte SoloStar bez nasazené jehly.
To pomáhá zabránit:
• Kontaminaci a/nebo infekci
• Proniknutí vzduchu do zásobníku inzulínu a vytékání inzulínu, což by mohlo způsobit nepřesné dávkování.
A. Nasaďte znovu na jehlu vnější kryt jehly a použijte ho k jejímu odšroubování z pera. Nikdy nenasazujte zpět vnitřní kryt jehly, abyste se vyhnuli náhodnému poranění jehlou.
B. Jestliže Vám dává injekci jiná osoba, nebo pokud dáváte injekci někomu jinému Vy sám/sama, musíte při sejmutí a odstraňování jehly dbát zvláštní opatrnosti. Aby se předešlo náhodnému zranění jehlou a přenosu infekce, řiďte se doporučenými bezpečnostními opatřeními (kontaktujte svého lékaře, lékárníka nebo zdravotní sestru).
C. Vyhoďte jehlu podle návodu Vašeho lékaře, lékárníka nebo zdravotní sestry tak, aby to bylo bezpečné.
D. Vždy na pero nasaďte zpět jeho kryt a skladujte tak pero do další injekce.
Pokyny pro uchovávání
Prohlédněte si, prosím, zadní (inzulínovou) stranu této příbalové informace, kde jsou uvedeny pokyny
pro uchovávání pera SoloStar.
Pokud je SoloStar skladován v chladu, je třeba ho vyjmout 1 až 2 hodiny před použitím, aby se mohl
ohřát na pokojovou teplotu. Injikování studeného inzulínu je bolestivější.
Použitý SoloStar musí být zlikvidován podle pokynů Vašich místních úřadů.
Údržba
Chraňte SoloStar před prachem a špínou.
SoloStar můžete čistit zvenčí otíráním vlhkým hadříkem.
Pero nemáčejte, nemyjte nebo nepromazávejte, může tím být poškozeno.
Váš SoloStar je navržen tak, aby pracoval přesně a bezpečně. Musí s ním být zacházeno s opatrností. Vyhněte se situacím, kdy může být SoloStar poškozen. Pokud se domníváte, že je Váš SoloStar poškozen, použijte nový.
164